síndrome de rett
TRANSCRIPT

Síndrome de Rett
Centro de Investigaciones CerebralesDoctorado en Investigaciones Cerebrales
M.C. Eloy Gasca Pérez

• Trastorno generalizado del desarrollo.
– Características principales • Retraso o perdida de habilidades sociales apropiadas, lenguaje y
conducta.• Afecta el desarrollo de los individuos, de inicio temprano y
persistencia a través de la vida.
– Ejemplos• Síndrome de Rett, Autismo, Desorden desintegrativo infantil,
Síndrome de Asperger, Trastorno generalizado del desarrollo por otras causas no especificadas.
Antecedentes

Historia• 1966-Dr. Andreas Rett en Austria observó 2 pacientes del sexo femenino
con movimientos de apretón de manos inusuales.• 1983- Dr. Bengt Hagberg de Suecia publica la primera revisión sobre el
Síndrome de Rett en una revista de neurología inglesa.• 1984-Primera Conferencia Internacional sobre Síndrome de Rett en Viena.• 1985-Dr. Hugo Moser organizó la primera Conferencia Norteamericana
sobre Síndrome de Rett en Baltimore y se establece la Asociación Internacional del Síndrome de Rett.

DSM-IV: Diagnóstico
• Todos los siguientes:– Desarrollo prenatal y perinatal aparentemente normal.– Desarrollo psicomotor aparentemente normal a lo largo de los
primero 5 meses de vida.– Circunferencia del cráneo normal al nacer.

DSM-IV: Diagnóstico
• Aparición de todas estas características después del periodo de crecimiento normal:
– Retraso en el crecimiento de la cabeza entre los 5 y 48 meses de edad.
– Pérdida de las habilidades manuales intencionadas adquiridas previamente entre los 5 y 30 meses de edad con el subsecuente desarrollo de movimientos de las manos estereotipados. (hand-wringing o hand washing).
– Pérdida de la participación social en las primeras etapas (aunque a menudo la interacción social se desarrolla más tarde)
– Aparición de mala coordinación al caminar o movimientos del tronco.
– Severa afectación en el desarrollo del lenguaje expresivo y receptivo con retraso psicomotor severo.

Diagnóstico diferencial
• Síndrome de Rett
– Sobre todo las mujeres– El deterioro de los indicadores del
desarrollo, la circunferencia de la cabeza, el crecimiento en general
– Pérdida de los movimientos manuales intencionales
– Movimientos estereotipados de las manos (apretones de manos, lavado de manos, la mano a la boca)
– Falta de coordinación, ataxia, apraxia– Pérdida de verbalización– Irregularidad respiratoria– convulsiones tempranas– Factor de crecimiento nervioso bajo en
LCR
• Autismo
– Sobre todo los hombres– Anomalías presentes desde el
nacimiento– Movimientos estereotipados de las
manos no se presentan siempre– Poca o ninguna pérdida de la función
motora gruesa– Idioma aberrante, pero no existe una
pérdida completa– No hay irregularidad respiratoria– Las convulsiones son raras, si se
producen, se desarrollan en la adolescencia
– Factor de crecimiento nervioso normal en LCR
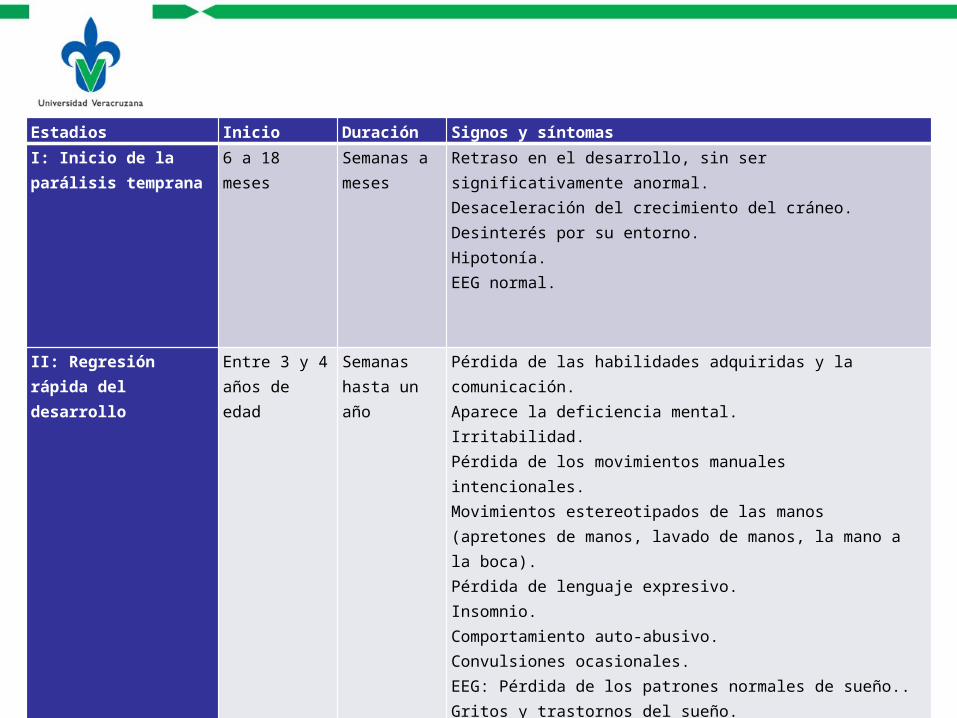
Estadios Inicio Duración Signos y síntomas
I: Inicio de la parálisis temprana
6 a 18 meses Semanas a meses
Retraso en el desarrollo, sin ser significativamente anormal.Desaceleración del crecimiento del cráneo.Desinterés por su entorno.Hipotonía.EEG normal.
II: Regresión rápida del desarrollo
Entre 3 y 4 años de edad
Semanas hasta un año
Pérdida de las habilidades adquiridas y la comunicación.Aparece la deficiencia mental.Irritabilidad.Pérdida de los movimientos manuales intencionales.Movimientos estereotipados de las manos (apretones de manos, lavado de manos, la mano a la boca).Pérdida de lenguaje expresivo.Insomnio.Comportamiento auto-abusivo.Convulsiones ocasionales.EEG: Pérdida de los patrones normales de sueño..Gritos y trastornos del sueño.

Estadios Inicio Duración Signos y síntomas
III: Periodo pseudoestacionario
Después del estadio III
Años a décadas
Restitución de la comunicaciónDeambulación Aumento de la ataxia, hiperreflexia, y la rigidezHiperventilación cuando se despiertaApnea del sueñoBruxismoPérdida de pesoEscoliosisEEG: alguna actividad epileptiformeDisposición feliz, disfrutan el contacto físicoAtaxia troncal
IV: Deterioro motor tardío
Cese de la deambulación
Décadas Dependencia completa silla de ruedasDiscapacidad grave y distorsionadoAtrofia progresiva muscular, espasticidad y EscoliosisRetraso en el crecimientoEnfriamiento en las extremidades debido a la Estasis venosaEstreñimientoMenos Convulsiones

Variantes del Síndrome de Rett
• Atípica o “Forme fruste”– Las primeras caracteristicas aparecen en la infancia tardía.
• Regresión de la infancia tardía– Retraso psicomotor temprano, regresión en la niñez
• Congénito– Carece período inicial de desarrollo normal
• Familiar• Trastorno de Rett en los hombres

Genética del Síndrome de Rett
• Trastorno dominante ligado al cromosoma X, letal en el 46
• Prueba de la base genética del Síndrome de Rett
• Confirmado sólo en mujeres y hombres con un cromosoma X adicional
• Completa concordancia en gemelos monocigóticos
• Identificado por primera vez la transmisión vertical: 1989

• 1990: los Dres. Zoghbi, Percy y Schultz descubrieron inactivación aleatoria del cromosoma X en la madre de dos medias hermanas con el Síndrome de Rett
• 1998: los Dres. Sirianni, Naidu y Pereira confirmmaron herencia dominante ligada a X, la localización de genes a Xq28
• 1999: los Dres. Amir, Van den Veyver y Wan vincularon al Síndrome de Rett a mutaciones en el gen MECP2 ligado al gen X, que codifica la proteína 2 ligada a metil-CpG que por lo general inactiva a otros genes.

Implicaciones neurológicas
• Crisis convulsivas en el 75 % de los casos, más severas en la edad temprana.
• EEG anormal en el 100 % de los casos.
• Ataxia troncal.
• Tratamiento a base de carbamazepina para las crisis y dieta cetogénica para aminorar las crisis y controlar la función motora.

Implicaciones sistémicas
• Pérdida de peso• Constipación• Bruxismo• Reflujo• Dificultad para masticar• Deficiencia de calcio• Hiperventilación• Periodos cianóticos• Apnea• Incremento del sueño en el día• Incapacidad de conciliar el
sueño por las noches
• Ataxia troncal temprana• Agitación• Piernas abducidas• Hipotonía temprana• Hiperreflexia• Rigidez tardía• Escoliosis (64 % de
prevalencia)• Usualmente inicio normal de la
pubertad• Perdida de la menarca• Infecciones por Candida sp.
Finalmente falla cardio-respiratoria o la aparición de status epilepticus conducen a la muerte


Introducción
• El Síndrome de Rett es un desorden del neurodesarrollo
– Incidencia de 1:10000 nacidas vivas y es una de las principales causas de retardo mental y conducta tipo autista en mujeres.
– Mutaciones que generan perdida de la función en la codificación del gen de la proteína 2 enlazada a metil-CpG (MeCP2) causa la mayoría de los casos del RTT.
– MeCP2 es un proteína de regulación transcripcional, y en su ausencia, un gran número de genes se expresan de forma anormal con implicaciones en el balance entre excitación e inhibición sináptica.

• La proteína MeCP2 constituye una cadena sencilla de 486 aminoácidos. Forma parte de una familia de proteínas (MBD1, MBD2, MBD3, MBD4 y MeCP1)
• Poseen un dominio funcional característico formado por 80 aminoácidos, conocido como methyl CpG binding domain o dominio para la unión con CpG metilados.
• Muchos tipos de genes dentro de diferentes cromosomas tienen regiones particulares denominadas Islotes CpG, que son secuencias con un alto porcentaje de peldaños o pares de base C-G (la “p” indica la unión fosfodiester entre C y G).
Toledo , 2007
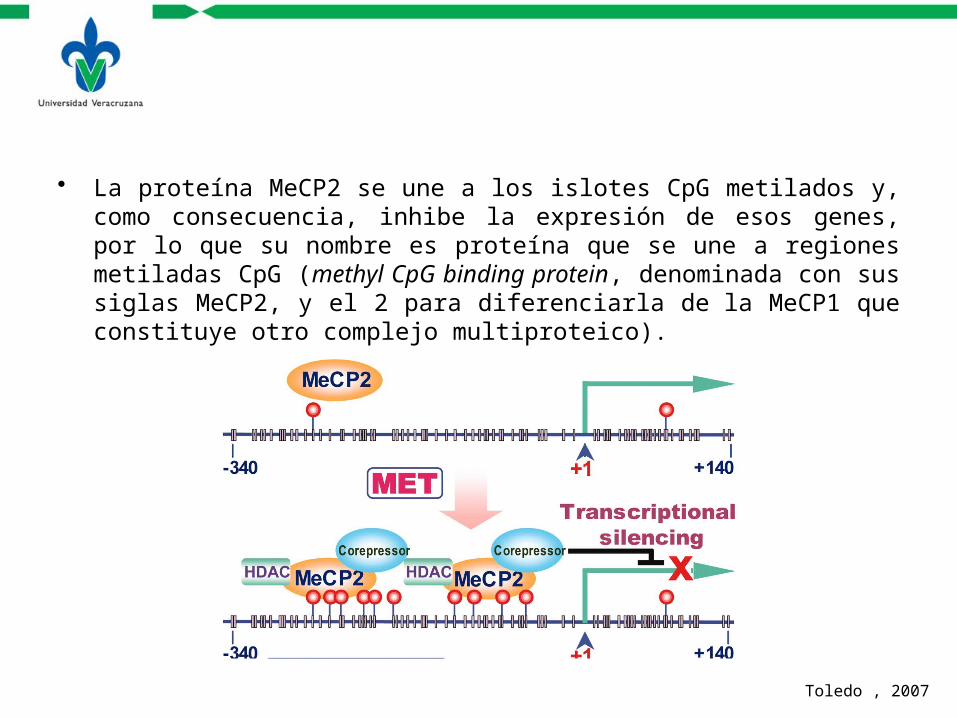
• La proteína MeCP2 se une a los islotes CpG metilados y, como consecuencia, inhibe la expresión de esos genes, por lo que su nombre es proteína que se une a regiones metiladas CpG (methyl CpG binding protein, denominada con sus siglas MeCP2, y el 2 para diferenciarla de la MeCP1 que constituye otro complejo multiproteico).
Toledo , 2007

• MeCP2 es particularmente importante en la función GABAérgica.– 50 % más expresión que en neuronas no GABAérgicas
• Ratones con deleción de MeCP2 en neuronas GABAérgicas– Inicialmente muestran conducta normal– Posteriormente aparecen movimientos estereotipados en las patas,
acicalamiento compulsivo, incapacidad de coordinación motora, deficit de memoria/aprendizaje, EEG anormales, hiperexcitabilidad, disritmias respiratorias y letalidad prematura.

Cootransportadores catión-cloruro
• NKCC1- Cotransportador Na+, K+, 2Cl- (Acumula Cl- en la célula)
• KCC2- Cotransportador K+, Cl- (Extrae Cl- de la célula)
• Receptores GABAA postsinápticos

• BNDF implicado en la maduración del sistema GABAérgico al inducir la expresión de KCC2.
• Se hipotetiza que cambios en la expresión de BNDF interfiere en la expresión normal de NKCC1 y KCC2, desencadenando una reducción en la proporción KCC2/NKCC1, característico de un sistema GABAérgico inmaduro.
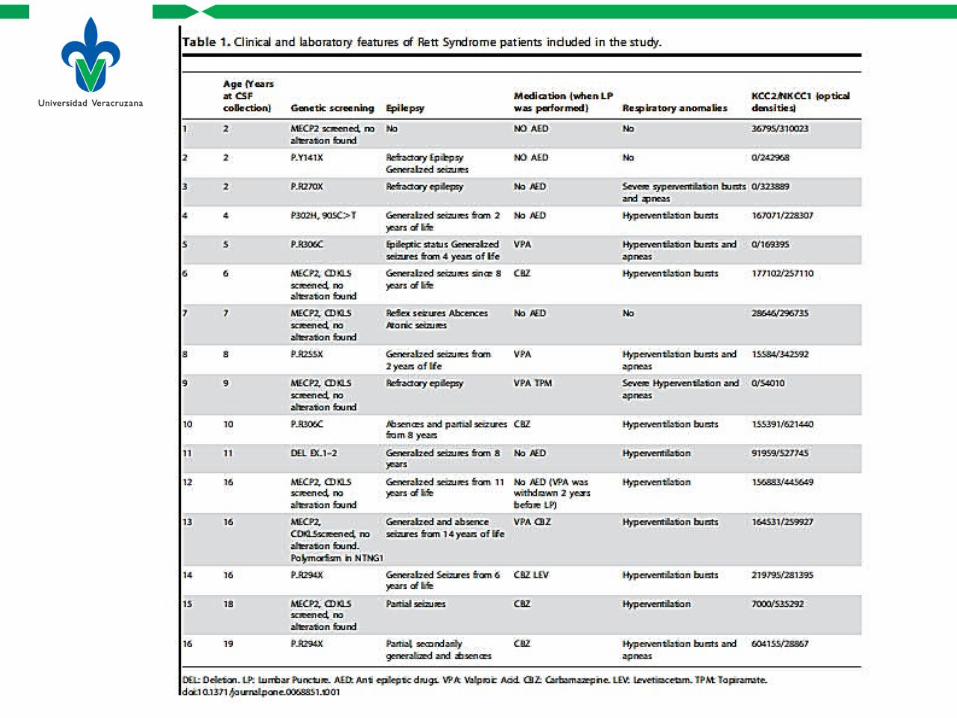
• Una comparación de los niveles de las proteínas NKCC1 y KCC2 en líquido cerebroespinal de pacientes con RTT y una población control fue realizada.

Pacientes y controles
• 16 pacientes con RTT– 2-19 años de edad– Pacientes sin una mutación documentada cumplen los criterios clínicos
para el RTT de acuerdo con la última revisión actualizada.
• 67 Controles– 1 día a 14 años de edad (promedio de 740 días, 27 mujeres, 40
hombres)
• Criterios de exclusión– Diagnostico de meningitis viral o bacteriana, enfermedad neurológica, y
CFS hemático o xantocrómico.

Muestras de CSF
• Colectadas por punción lumbar– Primeras 10 gotas
• Estudios de citoquímica y microbiológicos• 4 alícuotas almacenadas a -80 °C
– Siguientes 20 gotas• Análisis de metabolitos de aminas biogenas• Análisis de proteínas sinápticas

Análisis de CFS
• Western Blot
– PAGE-SDS– Transferencia a membrana de polivinilideno difloruro– Anti-NKCC1 y Anti-KCC2– Ac IgG (anti-rabbit)– Cuantificación por medición de densidades ópticas (OD)

Resultados



Discusión
• Se demuestra una reducción en los niveles de KCC2 y de la proporción KCC2/NKCC1 en CSF de pacientes con RTT.– Esto puede estar ligado a la aparición de epileptogénesis por una
alteración de la función de GABA.
• Primer reporte de detección de cotransportadores catión-cloruro por inmunoblot.
• El índice de flujo de CSF y la extensa muerte y poda celular pudiera afectar los niveles de cotransportadores con respecto a la edad.– Tasa alta de sinaptogénesis en el primer año de vida.

• El desbalance entre las funciones excitatorias e inhibitorias en pacientes con RTT pueden estar asociadas a:
– Reducción de los niveles de BDNF– Reducción de los niveles de GABA– Decremento en la expresión de subunidades del receptor a GABA– Reducción de las enzimas glutamato descarboxilasa 67 y glutamato
descarboxilasa 65– Reducción del número de sinapsis glutamatérgicas– Reducción de los ritmos inhibitorios basales
• Los resultados sugieren un patrón inmaduro de la neurotransmisión GABAérgica en pacientes con RTT.

Conclusiones
• Los hallazgos de esta investigación impactan en el entendimiento de la patofisiología del RTT.
• La mutación de neuronas en MeCP2 altera la proteína KCC2, que juega un rol en la función y estructura eléctrica neuronal.
• Los resultados pudieran llevar a nuevas alternativas terapeuticas, enfocadas a la manipulación farmacológica de estos cotransportadores.

GRACIAS