sexdif
DESCRIPTION
diferentiereTRANSCRIPT

DETERMINAREA SEXULUI
Gene implicate in creerea gonadei nediferentiate
WT1 – gena : anomaliile sale se asociaza cu imposibilitatea diferentierii gonadei
Sindromul Deny-Drash:
- lipsa diferentierii gonadei
- nefropatie
- tumora Wilms
Sindromul Frasier:
- lipsa diferentierii gonadei
- gonadoblastom
Sindromul WAGR:
- tumora Wilms
- aniridie
- anomalii genitale, mental retardare mentalaHiort O., Holterus P-M: The mollecular bases of male sexual diffrentiation. Eur.J.Endocrinol. 2000, v. 142, 101-110

DETERMINAREA SEXUALA
Gene implicate in the crearea gonadei nediferentiate
gena LIM-1: deletia homozigota: lipsa de dezvoltare a gonadelor si rinichiului
La oameni s-au raportat anomalii renale, gonadale asociate cu anomalii ale creierului
FTZ-1-F1 – gena pt. SF-1 – steroidogenic factor 1 codifica un receptor orfan situat in nucleu . mARN pentru SF-1 a fost detectat in creasta genitala, suprarenale si hipotalamus
Deletia genei FTZ-1 –F1 la soarece determina lipsa de dezvoltare a gonadelor, suprarenalelor si a hipotalamusului
Roluri ale SF-1:
• differentierea gonadei, suparenalelor si a hipotalamului
• reglarea enzimelor implicate in steroidogeneza
• reglarea transcriptiei factorului antimullerian (AMH)
Hiort O., Holterus P-M: The mollecular bases of male sexual diffrentiation. Eur.J.Endocrinol. 2000, v. 142, 101-110

SEXUAL DETERMINATION
Testis determining factor (TDF) = SRY – sex-determining region de pe cromozomul Y
Gena cu un singur exon:
- se leaga de promoterul genei pentru factorul antimullerian si induce expresia factorului antimullerian inhibind dezvoltarea canalelor Muller
- manipulare genetica: femelele xx transfectate cu sry dezvolta fenotip masculin normal
- mutatiile genei SRY sunt asociate cu enversarea sexual completa a subiecii cu cariotip 46 ,xy
- mutatii ale genei SRY au fost descrise in hermafroditismul adevarat la care exista dezvoltarea concomitenta a tesutului ovarian si testicular
Hiort O., Holterus P-M: The mollecular bases of male sexual diffrentiation. Eur.J.Endocrinol. 2000, v. 142, 101-110

DETERMINAREA SEXUALA
Gene autosomale implicate in diferentierea sexului masculin
SOX-9 (SRY-box related) este transcrisa dupa SRY in tesuturile genitale masculine. Aceasta gena este si activator al genei care codifica colagenul de tip 2. Defectele genei SOX-9 determina inversarea sexului la subiectii 46,XY si o serie de malformatii scheletice cunoscute sub denumirea de campomelic dysplasia
Chromosome 10q: deletia terminala a acestuia determinamalformatii genitale si retardare mentala
Hiort O., Holterus P-M: The molecular bases of male sexual differentiation. Eur.J.Endocrinol. 2000, v.
142, 101-110

SEXUAL DETERMINATION
Cromosomul 9p are gene determinate ale sexului
Deletia acestei gene determina inversarea sexului la subiectii 46,xy asociata cu anomalii faciale: inchiderea prematura a suturii frontale, hidronefroza si dezvoltarea sexuala intirziata la subiectii 46, xy
• Locusul 9p24.3: genele DMRT1, DMRT2 si DMRT3 au deletie in inversiunea de sex asocata cu anomalii 9p sugerind faptul ca disgeneziile gonadale 46, xy sunt determinate de hemizigotismul combinat a DMTR1 si DMRT2
• Monosomia 9p se asociaza cu diverse anomalii ale dezvoltarii sexuale la subiectii 46,xy de la o dezvoltarea aproape normala pina la inversarea competa a sexului cu lipsa de dezvoltare a gonadei
• Anomaliile de dezvoltare nu sunt legate de dimensiunile regiunii afectate sau de originea parentala a deletiei cromozomului 9p
Hiort O., Holterus P-M: The mollecular bases of male sexual diffrentiation. Eur.J.Endocrinol. 2000, v. 142, 101-110
Ogata T., Muroya K., Ohashi H., Mochizuchi H., Hasegawa T., Kaji M.: Female gonadal development in XX patients with distal 9p monosomy, Eur.J.Endocrinol. 2001, 145, 4, 613-616.

SEXUAL DETERMINATION
DAX-1 : dosage sensitive sex reversal locus-adrenal hypoplasia congenita- critical region on the X chromosome, gene-1. (Xp21)
DAX-1: expresia acestei gene apare in tesutul ovarian in timpul dezvoltarii ovarului dar activitatea sa este suspendata in timpul formarii testiculului. Gena DAX-1 este gena critica pentru formarea ovarului
DAX-1 este reprezata de catre sry in timpul dezvoltarii testiculului. DAX-1 cind este in duplicat interfereaza cu dezvoltarea testiculului si determina inversarea sexului la subiectii 46. Xy
Mutatiile genei DAX-1 diminua activitatea genei si determina afectiunea cunoscuta drept: hipoplasia drenalis congenita si hipogonadism hipogonadotrop
Hiort O., Holterus P-M: The mollecular bases of male sexual diffrentiation. Eur.J.Endocrinol. 2000, v. 142, 101-110

SEXUAL DIFFERENTIATION
MEZODERM Bipotential gonad
WT-1
LIM-1
SF-1
TESTIS
Sertoli cells
AMH
Leydig cells
Testosterone
OVARY
Estrogens
Progesterone
DAX-1
DMRT1,2
SOX9
SRY

DIFERENTIEREA ORGANELOR
GENITALE INTERNE

TESTICULUL- ORGANELE GENITALE INTERNE MASCULINE
Testosteronul si dihidrotestosteronul stimuleaza dezvoltarea ductelor lui Wolff din care rezulta epididimul, deferentul si canalul ejaculator prin actiune directa de partea in care exista testiculul
La subiectii cu un singur testicul nu au ducte decit de partea in care exista gonada
Prostata se dezvolta sub actiunea DHT

DEZVOLTAREA ORGANELOR GENITALE INTERNE

OVARUL - ORGANELE GENITALE INTERNE FEMININE
Organele genitale interne feminine se dezvolta pornind de la primordiul comun in baza unui program genetic independent de hormonii gonadali
• in absenta AMH ductele lui Muller fuzioneaza pe linia mediana formind trompele, uterul, colul uterin si 2/3 superioare ale vaginului
• in absenta testosteronului ductele lui Wolff nu se pot dezvolta

DEZVOLTAREA ORGANELOR GENITALE EXTERNE
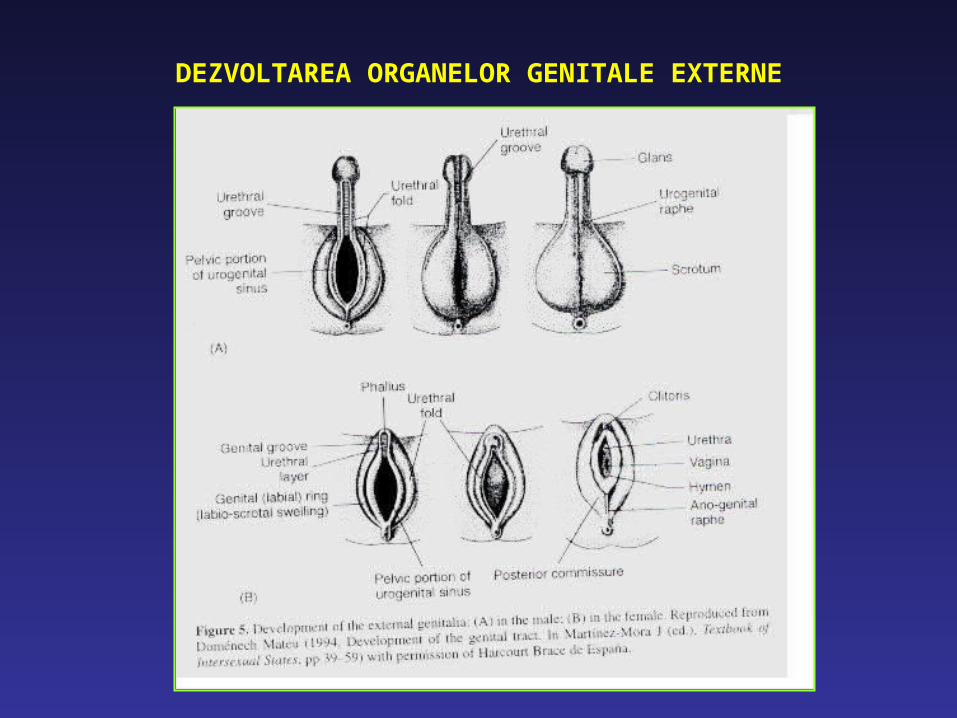
DEZVOLTAREA ORGANELOR GENITALE EXTERNE


DEZVOLTAREA ORGANELOR GENITALE EXTERNE MASCULINE

DEZVOLTAREA ORGANELOR GENITALE EXTERNE MASCULINE

DEZVOLTAREA ORGANELOR GENITALE EXTERNE MASCULINE
Presupune:
existenta gonadei masculine XY-sry
integritatea secretiei prenatale de testosteron la nivelul testiculului, a steroidogenezei
stimularea corecta a testiculului prin LH -hCG
existenta unor receptori normali pentru LH-RH/hCG (LHR) la nivelul celulelor Leydig
conversia normala a testosteronului in dihidrotestosteron (DHT) prin 5 reductaza
receptori functionali pentru T si DHT

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA- Conte, Melvin,
Grumbach
I. Tulburari de diferentiere ale gonadei
A. Disgenezia tubilor seminiferi (sindromul Klinefelter si variante)
B. Sindromul de disgenezie gonadala si variantele sale (sd.Turner)
C. Forme complete si incomplete ale disgeneziei gonadice XX sau XY
D. Hermafroditismul adevarat
II. Pseudohermafrotisme feminine
A. Hiperplazia congenitala virilizanta a suprarenalei
B. Deficitul p450 aromatazei placentare
C. Androgeni sau progestageni sintetici transferati de la mama
D. Asociat cu malformaii intestinale si ale tractusului urinar (pseudohermafrotismul feminin non-androgen dependent)
E. Alti factori teratogeni

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA
III. Pseudohermafroditisme masculine
A. Lipsa de raspuns testicular la LH/hCG- agenezia sau hipoplazia celulelor Leydig determinata de mutatii inactivamte ale receptorului de LH/nCG
B. Erori inascute ale biosintezei de testosteron:
1. Enzime care afecteaza sinteza costicosteroiszilor si testosteronului
a. Defectul genei StAR – hiperplazia lipoidica suparrenalei
b. defectul de 3-hidroxisteroid dehidrogenaza
c. Defectul de P450 c17: 17- hidroxilaza
2. Defecte acre afecteaza biosinteza de steroizi la nivelul testiculului
a. Defectul P450 c17: 17,20 liaza
b. Defectul de 17 hidroxisteroid oxido reductaza

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA
C. Defecte ale structurilor tinta androgen-dependente
1. Defetce ale receptorului de androgeni si defcte postreceptor
a. Sindromul de insensibilitate (rezistanta) completa la androgeni si variantele sale(feminizare testiculara si variante)
b. sindromul de insensibilitate (rezistenta) incompelta la androgeni si variantele sale (sd.Reifenstein si variantele sale)
c. insensibilitatea la androgeni la barbatul infertil
d. insensibilitatea la androgeni la barbatul fertil
2. Defecte ale metabolismului testosteronului la nivelul structurilor tinta
a. Defectul de 5 reductaza: hipospadiasul pseudovaginal perineo-scrotal

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA
D. Psudohermafroditismul masculin disgenetic
1. Disgenezia gonadica XY incompleta
2. mazaicism XO/XY, cromozom Y structural anormal, Xp+. 9p-, 10q-
3. Sindromul Denys Drash (mutatie WT-1)
4. WAGR – deletie WT-1
5. displazia campomelica (mutatie SOX 9)
6. Mutatie SF-1?
7. Sindromul de regresie testiculara
E. Defecte ale sintezei, secretiei su raspunsului la AMH
1. Sindromul de persistenta a ductelor mulleriene –” hernia uteri inguinale”
F. Disruptori chimici din mediu

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA
IV. Forme neclasificabile de dezvoltare sexuala anormal
A. La barbati
1. Hipospadias
2. Organe genitale ambigui la subiecti XY, asociate cu multiple anomalii congenitale
B. La femei
1. Absenta sau dezvoltarea anormal a vaginului, uterului sau trompelor uterine – sindromul Rokitansky - Kuster

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA
1. ERORI DE DETERMINARE PRIMARA A SEXULUI
A. Anomalii ale cromozomilor sexuali
1. Cu testiculi si fenotip masculin
a. Sindromul Klinefelter 47,XXY si variante
b. sindromul XYY
c. barbatii xx (interschimbari X-Y)
2. Cu ovare si fenotip feminin
a. trisomia XXX si variante
3. Cu gonade bisexuale
a. Hermafroditism adevarat (interschimbare X-Y)
b. Hermafroditismul adevarat cu himerism XX/XY
c. Hermafroditismul adevarat cu mozaic XX/XXY
4. Cu gonade disgenetice
a. anomalii ale cromozomului Y (Xyp-)
b. disgenezia gonadica mixta: 45X/46 XY
c. Sindromul Turner 45,X si variantele sale

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA
B. Anomalii ale gonadogenezei
1. Defecte ale dezvoltarii ovarului
a. disgenezia gonadica(pura) cu cariotip 46, XX
2. Defecte ale dezvoltarii testiculului
a. disgenezia gonadica (pura) 46, XY
b. pseudohrmafroditismul masculin disgenetic
c. sindromul de regresie testiculara ( agonadism, testiculi rudimentari, anorhia

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA
II. ERORI ALE DIFERENTERII SEXUALE
A. Masculinizarea inadecvata a fatului de sex masculin
1. Defecte de regresia a ductelor paramezonefrotic:
a. persistenta ductelor paramezonefrotice
2. Defecte de virilizare genitala
a. Hipoplazia celulelor Leydig- defetc de receptor pentru LH/hCG
b. Defecte inascute de sinteza a steroizilor suprarenali si gonadali
1. Defectul de 20-22 desmolaza
2. Defectul de 3 hidroxisteroid dehidrogenaza
3. defectul de 17 hidroxilaza (cyp 17)
c. Defecte inascute ale steroidogenezei testiculare
1. Defectul de 17,20 desmolaza
2. Defectul de 17 hidroxisteoir dehidrogenaza
d. Defecte ale metabolimsului testosteronului
1. Defectul de 5 reductaza
e. Defectul raspunsului celuleor tinta:
1. insensibilitatea completa si incompleta la androgeni si variante
f. Feminizarea datorata factorilor de mediu (disruptori)
1. Ingestia de estrogeni si progestageni sintetici in timpul sarcinii
g. Multigenic/multifactorial: hipospadiasul izolat si criptohidia. Sindromul de anomalii congenitale multiple

CLASIFICAREA ANOMALIILOR DE DIFERENTIERE SEXUALA
B. VIRILIZAREA FATULUI CU SEX GENETIC FEMININ
1. Hiperplazia congenitala a suprarenalei
a. Defectul de 21 hidroxilaza
b. Defectul de 11 hidroxilaza
c. Defectul de 3 hidroxisteroid dehidrogenaza
2. Hormoni din mediu
a. Tulburari cu virilizare materna –defectul de aromataza placentara
b. Virilizare iatrogena
3. Malformatii teratologice
a. Anomalii de dezvoltare ale uterului si vaginului
b. Sindromul de anomalii congenitale multiple

TULBURARI DE DIFERENTIERE ALE GONADEI
A. Disgenezia tubilor seminiferi (sindromul Klinefelter si variante) Este una dintre principalele forme de disgenezie ganadala si cauza de
infertilitate masculina
Incidenta: 1/1000 de nou nascuti, mult mai mare printre infertili si detinuti
Cariotip: 47,XXY, 48,XXXY, 48,XXYY, 49,XXXYY, 49, XXYYY
Nondisjunctia este in 60 % din cazuri de origine materna
Caracteristici testiculare si hormonale:
• sclerohialinizarea tubilor seminiferi cu testiculi mici 3.5 ± 1,5 mL
• hiperplazia celulelor Leydig si cresterea secretiei de estrogeni la pubertate
• azoospermie si infertilitate constanta cu exceptia mozaicismelor cu linii 46XY
• testosteron la limita inferioara a normalului
• estradiol la limita suiperioara a normalulul
• FSH si LH crescute mult in primul an de viata si apoi net crescute de la pubertate

ERORI ALE DETERMINARII
SEXULUI

Sindromul Klinefelter si variantele sale• in copilarie dificultati scolare si tulburari comportamentale, talia este mai inalta decit normal, iar dimensiunilr organelor genitale la nivelul inferior normal, coeficient intelectula verbal la limita inferioara a normalului, iar pubertatea este tardiva si incompleta
• Morfotipul adult:
• talie inalta si fenotip “ginoid”
• ginecomastie cvasiconstanta
• disociatie peno-orhitica: penis de dimenisuni normale, testiculi mici
• infertilitate
• tulburari de dinamica sexuala care debuteaza de la aprox. 30 ani si se agraveaza repede
• tendinta la dificultati de adaptare sociala si delincventa
• cu cit exista mia multi cromozomi X cu atit se noteaza retardare mentala si malformatii;sinostoza radio-cubitala, displazie epifizara, canal arterial patent
Explorare:
Spermograma: azoospermie
Cromatina sexuala –pozitiva/eventual multipla
Testosteron normal scazut, estradiol uosr crescut
FGS/HL crescuti
Tratament: testosteron enantat, undecanoat200 mg/luna, corectia ginecomastiei, suport psihologic



VARIANTE ALE SINDROMULUI KLINEFELTER
BARBATII XX
Incidenta: 1/20.000 de nou nascuti
Sunt de fapt subiecti cu XXY cu translocatii ale ADN-ului cromozomului Y care contine TDZ (SRI) de pe bratul scurt al comozomului Y pe bratul scurt al cromozomului X cuplata eventual cu pierderea materialului genetic de pe bratul scurt al cromozomului X
Fenotip:
• asemanator cu sd. Klinefelter dar cu talie mai mica – medie 168 cm.
• pot apare ambiguitati sexuale in 10 % din cazuri ca si hipospadias si criptorhidie
• sclerohialinizarea tubiloe seminiferi si azoospermia sunt prezente
Diagnostic citogenetic:
46 XX + trasnlocatie Y-X
Tratament :
Similar cu cel al sindromului Klinefelter

Barbat 46 ,XX

47, XYY
Incidenta: 1/1000 de barbati
Nu este recunoscut la nastere
Elemente comune: hipotonia, vorbirea tardiva si tulburari de coordonare neuro-musculara
10 % pot avea volum testicular redus
Biopsiile pot arata modificari importante ale spermatogenezei
Sindromul a devenit notoriu atunci cind a fost descris la subiectii cu comportament agresiv, dar se recunoaste in prezent ca multiple anomalii cromozomiale se pot asocia cu un comportament agresiv si nu numai 47,XYY

SINDROMUL DE DISGENEZIE GONADALA TURNER SI VARIANTELE SALE
INCIDENTA: 1/5000 nou nascuti de sex feminin au 45,X
99 % dintre fetusii 45,X nu supravietuiesc peste 28 saptamini de gestatie
15 % dintre avorturile spontane din primul trimestru au 45,X
In 60-70 % originea cromozomului X normal este materna
LA NASTERE: limfedemul generalizat sau al extremitatilor este patognomonic
DATELE CLINICE:
HIPOSTATURA <145 cm in forma 45,X, mai mare in mozaicuri cu 46 ,XX sau mai mica de –2,5 SD fata de normalul pentru virsta
Sindromul plurimalformativ: 200 malformatii somatice si viscerale
Amenoreea primara si lipsa de dezvoltare a caraterelor sexuale secundare
Sindromul plurimalformativ: epicantus, urechi jos implantate, micrognatie, ptosis, bolta ogivala, aspectde “gura de peste”, pterigium coli ( git palmat), gir scurt, torace larg, bazin ingust, nevi pigmentari, “cubitus valgus” scurtarea metacarpienelor IV,V si a metatarsienelor IV.V, “rinichi in potcoava”, coarctatie de aorta++++

SINDROMUL DE DISGENEZIE GONADALA TURNER SI VARIANTELE SALE
Genetica: 45,X, 45, X/46,XX, 46, X Xqi, 46, X X “in inel”
Absenta alelelor de pe bratul scurt al cromozomului X este determinanta a disgeneziei gonadice si a deficitului stattural
In prezenta 45,X ovarul se dezvolta in viata intrauterina si involueaza pina na nastere devenind “streak gonada”
Diagnostic:• cromatina sexuala negativa ( sau slab pozitiva in mozaicisme)
• cromatina sexuala poate fi pozitiva in izocromozom pentru brat lung sau cromozom X “ in inel”
• estradiol scazut
• FSH/LH mult crescuti ( in domeniul valorilor de menopauza la adulte)
• inventarul malformatiilor viscerale echografic , echicardiografic






Tipul anomaliei cromozomilor sexuali
Cariotip Fenotip Infantilism sexual
Hipostatura
Anomalii somatice de tip Turner
Pierderea cromozomului X sau YDeletia bratului scurt al cr.X(xp-)
Deletia bratului lung al unui XDeletia portiunii terminale a ambelor brate ale unui cr.X
Deletia bratului scurt al cr.Y
45,X46,Xxqi46,XXp-
46, XXq-
46,XXr
46,Xyp-
Feminin Feminin Feminin
Feminin
Feminin
Feminin
++(±)
+, ±, -
+
- sau +
+
++
+ (-)
- (+)
++
++
+ (-)
- sau -/+
+ sau ±+
Relatia dintre anomaliile cromozomilor X sau Y si manifestarile clinice ale sindromului de disgenezie
gonadala

SINDROMUL DE DISGENEZIE GONADALA TURNER SI VARIANTELE SALE
TRATAMENT obiective:
Cresterea
• hrGH: 0,1-0,14 UI /kcorp/zi sau 0,375 mg/kg.copr/saptamina cu o crestere de 8-10 cm fata de talia prezumata dupa 5-7 ani de terapie
• ± oxandrolon: 0,0625 mg/kg/zi
• doze mici de estrogeni
• asocierea estrogeni – hrGH nu aduce avantaje
Dezvoltarea caracterelor sexuale secundare + menstre: de la 12 –13 ani
• estrogeni conjugati: 0,3 mg /zi 21 zile sau EE2 =5 microg /zi 21 zile
• doze se creste progresiv pina la 0,625-1,250 mg estrogeni conjugati sau 10 microg. EE2
• asocierea MPA sau alt progestativ pentru ultimele 5 zile din cele 21
• se administreaza doza minima de E2 care produce menstre si evita osteoporoza
Sarcini : mame surogat pentru embrioni donati

VARIANTE X-CROMATIN NEGATIVE ALE SINDROMULUI DE DISGENEZIE GONADICA
Mozaicisme care contin linii 45,X in diferite combinatii:
45X/46 XY, 45 X/47 XXY, 45 X/46XY/47 XYY si/sau anomalii structurale ale cromozomului Y
Gonada:
• streak gonada bilateral
• streak gonada de o parte si testicul disgenetic de cealalta parte
• rar testicul aparent normal bilateral - disgenezie gonadica mixta 45x/46xy
Fenotip
• Feminin cu stigmate de Turner
• Intersexualitate
• Masculin cu putine stigmate de Turner, o raportare cu fenotip masculin si stigamte de Turner si fertilitate pastrata

VARIANTE X-CROMATIN NEGATIVE ALE SINDROMULUI DE DISGENEZIE GONADICA
Diagnostic citogenetic:
• demonstrarea mozaicismului in gonada, piele, singe
• determinarea fragmentului Y analiza FISH sau moleculara
• identificarea unei zone de Y se impune pentru ca tumorile se raporteaza si la cei cu SRY negativ
Management
• gen feminin: rezectia gonadei masculine si coretarea OGE + estrogeni la pubertate
• gen masculin: corectarea OGE si testosteron, testiculul se conserva numai daca este normal si situat in scrot
• integritatea testiculului se certifica prin nivelul de gonadotrofine si RMN

DISGENEZIA GONADICA 46 ,XY
DISGENEZIA GONADICA COMPLETA 46,XY (Sd.Swyer)Gonada: streak gonada bilaterala
Uneori gonada poate contine foliculi si s-a raporta un caz cu fucntie gonadica normala cu multi ani inainte de diagnostic
Fenotip feminin cu infantilism genital, talie inalta, eunucoida, fara stigmate de Turner, amenorree primara
OGE: feminine, cu frecventa hipertrofie clitoridiana
OGI: derivate mullerine normale
Citogenetica:
• mutatii ale HMG box pentru SRY care impiedica legarea proteinei codificate de SRY si afecteaza nivelul sau sincronizarea expresiei SRY cu formarea testiculului
• deletii ale bratului scrt Y se asociaza cu stigmate de Turner
• posibile mutatii ale unor gene de pe X sau de pe autosomi
• 70 % dintre cazuri sunt recesive legate de X
• 20 subiecti prezentau mutatii ale locusului Xp21.2-22.11 care contine 2 locusuri care se suprapun partial DAX-1 (hipoplazia adrenalis congenita) si DSS (dossage - sensitive sex reversal) a carei duplicare determina lipsa de dezvoltare a testiculului si fenotip feminin

DISGENEZIA GONADICA COMPLETA 46,XY (Sd.Swyer)
Alte anomalii cromozomiale:• displazia camptomelica cu determinata de mutatii ale genei SOX9, cromozom 17 cu nanism
• deletii: 9p-, 10q-
• nefrita interstitiala sau insuficienta renala
• Anomalii ectodermale sau cardiace
Diagnostic:• citogenetic
Management:• Rezectia gonadei independent de gradul de diferentiere
• riscul de neoplasm: gonadoblastom, disgerminon sau seminon este de pina la 30 %
• administrare de estrogeni si progesteron pentru inducerea si mentinerea caracterelor sexuale secundare
• exceptional la subiectii cu intersexualitate cu identitate de gen masculina: testosteron

Disgenezie gonadica completa 46,XY

DISGENEZIA GONADICA PARTIALA 46,XY
Determinarea incompleta a testiculului:
• streak gonada de o parte si testicul disgenetic cu histologie variabila de alta
• tubii seminiferi sunt rau formati si uneori exista stroma adiacenta de tip ovarian
• frecventa tumorilor pe gonada disgenetica este de 16 %
• au fost descrise tumori de la virsta de 15 luni
• unii subiecti au mutatii WT-1
Atentie la gonadele disgenetice XY:
• apar la subiectii cu cromozomi “marker” de origine Y fara SRY
• gonadectomia profilactica este indicata mai ales in cazurile cu virilizare
• palparea unei mase testiculare este un semn tardiv pentru ca seminoamele dau metastaze mult inainte de aparitia tumorilor intrascrotale
• in cazul in care se decide pastrarea gonadei se impun controale clinice si RMN periodice pentru surprinderea aparitiei tumorilor

PSEUDOHERMAFRODITISMUL MASCULIN DISGENETIC
Este o entitate polimorfa in care se include subiecti cu genotip/ fenotip variabil
• testicul disgenetic bilateral
• ambiguitate genitala variabila
• persistenta variabila a ductelor mulleriene
• tulburarea nu are caracter familial
Citogenetica:
• anomalii subtile ale cromozomului Y
• mozaicuri 46XYp-, sau
• forme nerecunoscute de 45,X/46 XY pentru ca uneori liniile 45.x nu supravietuiesc in singele periferic
• rare familii in care unii aveau disgnezie gonadica completa 46,XY si altii aveau caracterisiticile descrise pentru PHM disgenetic
• riscul de gonadoblastom este important, gonadectomia profilactica este necesara
Alte situatii:
• unii pot avea: hipertensiune, proteinurie, sindrom nefrotic, tumora Wilms
• aniridie (cataracte sau glaucom)PHM cu hipospadias, criptorhidie, ambiguitate genitala si gonadoblastom

PSEUDOHERMAFRODITISMUL MASCULIN DISGENETIC
Florea Otilia, 14 ani
Fenotip Turner-like
Amenoree primara
Absenta caracterelor sexuale secundare
Interventie in mica copilarie pentru hipertrofie clitoridiana si laparotomie cu “scoaterea unui ovar”
Cariotip 46,XY
Tumora pelvina operata ulterior(echo)
AP: gonadoblastom cu zone de seminom
Revazuta permanent postoprator si dupa chimioterapie fara recidiva sayu diseminari
Menstre sub EP

PSEUDOHERMAFRODITISMUL MASCULIN DISGENETIC

HERMFRODITISMUL ADEVARAT
DEFINITIE: o persoana este definita drept hermafrodit numai daca se poate pune in evidenta in aceeasi gonada sau in gonade diferite tesut testicular cu tubuli si tesut ovarian cu foliculi si copri albicans
In cazul in care se pun in evidenta in cadrul unei gonade disgenetice elemente testiculare+ stroma fibroasa fara foliculi, aceasta situatie nu poate fi classificataa drept hermfroditism adevarat
Forme: • altern sau lateral: testicul de o parte si ovar de partea opusa: 30 %
• ovotestis de o parte: 50 – 60 %
• ovotestis bilateral cu elemente testiculare si ovariene distincte
• ovotestis-ul este plasat: in labie de o parte: 30 %, in canalul inghinal 30 % sau in abdomen
• uterul este prezent, frecvent hipoplasitc si unicorn
• testiculul este rareori functional, cel mai adesea disgenetic

HERMFRODITISMUL ADEVARAT
Clinica:• La pubertate se dezvolta sinii si in 50 % dintre cazuri apar menstrele
• functia testiculului este frecvent insuficienta
• in cazul in care tesutul testicular este complet rezecat poate apare secretie ciclica de gonadotropi si fertilitate in sens feminin
• spermatogeneza este deseori insuficienta si fertilitatea in sens masculin nu a fost documentata
• la copii care au fost crescuti in sex masculin, numai daca testiculul se alfa in scrot acesta se retine
• 2 % dintre hermafroditii adevarati pot dezvolta tumori: gonadoblastom, seminom sau disgerminom

HERMFRODITISMUL ADEVARAT
Genetica este heterogena:
• 46, XX in 60 % 46, XY in 20 % chimerism: 46,XX/46XY: 20 %
Patogenia:
• translocatie a unei portiuni din Y pe X sau pe un autosom
• o parte sunt SRY pozitivi, dar majoritatea sunt SRY negativi
• translocatii Y-X pot avea diferite expresii fenotipice:
• barbatii 46,XX
• hermafroditism adevarat 46, XX
• disgenezie gonadica XX , in functie de cantitatea de material determinant de testicul care este translocata
• au fost descrise familii in care coexistau diferite forme ale translocatiei
• in cazurile SRY-negative se pot incrimina:
• mutatii ale genelor autosomale implicate in determinarea testiculara,
• mutatii, deleti, anomalii ale unui locus de pe X implicat in determinarea testiculara
• himerism circumscris la nivelul gonadei
• himerism rezultat din fertilizarea unui ou cu dublu nucleu de catre un Y si un X cu fuziunea precoce a zigotilor

HERMFRODITISMUL ADEVARAT
Diagnostic:
• poate fi luat in consideratie in toate cazurile deambiguitate sexuala
• gasirea cariotipului 46 XX/46 XY
• gasirea unei gonade bilobate care este ovotestis
• testosteronul este crescut la subiectii peste 6 luni si este stimulabil prin hCG
• laparotomia si biopsia gonadei, dupa excluderea diferitelor forme de psudoH
Management:
Depinde de:
• virsta la diagnostic
• capacitatea functionala apreciata prospectiv la gonadei
• ductelor genitale si OGE
• in general sexul masculin se da la subeictii care au virilizare buna si nu au uter







SINDROMUL DE REGRESIUNE A TESTICULELOR“VANISHING TESTES”, ANORHIA CONGENITALA
Termenul desemneazaun grup heterogen de sindroame caractrizate prin regresia testiculelor in cursul vietii fetale dar are probabil relatii cu anomalii ale cromozomului Ysi PHM disgenetic
In unele familii au fost raportate cazuri de PHM disgenetic si anorhie congenitala
Caritipul 46, XY
Gonada a fost probabil testicul partial functional pentru ca se produce virilizare si regresia canalelor Muller
Fenotipul variabil:
• ambuiguitate genitala cu falus de marimea unui clitoris mai mare
• fuziune partiala a labiilor
• sinus urogenital prezent
• absenta canalelor Muller care semnifica functie testiculara pina la cel putin 14 saptamini
• testul la hCG poate produce o usoara crestere a testosteronului chiar daca nu se identifica testicule
• mecanimsul regresiei testiculare: torsiune prenatala sau ocluzia vaselor testiculare
Diagnostic: FSH foarte crescut permite diferentierea de criptorhidia bilaterala
Tratament: androgeni de la pubertate



ERORI ALE DIFERENTIERII SEXUALE

TESTICULUL- ORGANELE GENITALE INTERNE
Anti-mullerian hormone (AMH- Mullerian inhibiting hormone) inhiba dezvoltarea ductelor Muller prin legarea de un receptor de pe suprafata celulelor (este un factor de crestere) si pe membrana celulelor din vecinatatea acestor canale. AMH este produs de celulele Sertoli
Gena AMH este controlata de alte gene de tipul : SRYsi SF-1
Persistent Mullerian ducts syndrome (PMDS)- sindromul de persistenta a ductelor Muller cu prezenta trompelor si a uterului poate fi produsa de urmatoarele situatii:
- mutatii ale genelor care codifica AMH – cu nivele reduse ale AMH
- mutatii ale genelor care codifica pentru receptorulu de tip II pentru AMH in care receptorul este mutant iar nivelul de AMH este noramal sau crescut AMH-receptor gene is localizata pe cromosomul 12
Ambele forme sunt mostenite in maniera autosomal recesiva
Hiort O., Holterus P-M: The mollecular bases of male sexual diffrentiation. Eur.J.Endocrinol. 2000, v. 142, 101-110

PSEUDOHERMAFROTISMUL MASCULINDEFECTE ALE VIRILIZARII SUBIECTILOR GENOTIPIC 46, XY
Defecte de virilizare genitala
a. Hipoplazia celulelor Leydig- defect de receptor pentru LH/hCG
b. Defecte innascute de sinteza a steroizilor suprarenali si gonadali
1. Defectul de 20-22 desmolaza
2. Defectul de 3 hidroxisteroid dehidrogenaza
3. defectul de 17 hidroxilaza (cyp 17)
c. Defecte inascute ale steroidogenezei testiculare
1. Defectul de 17,20 desmolaza
2. Defectul de 17 hidroxisteroid dehidrogenaza
d. Defecte ale metabolimsului testosteronului
1. Defectul de 5 reductaza
e. Defectul raspunsului celuleor tinta:
1. insensibilitatea completa si incompleta la androgeni si variante
f. Feminizarea datorata factorilor de mediu (disruptori)
1. Ingestia de estrogeni si progestageni sintetici in timpul sarcinii
g. Multigenic/multifactorial: hipospadiasul izolat si criptohidia. Sindromul de anomalii congenitale multiple


RECEPTORUL PENTRU LH/hCG

Hipoplazia celulelor Leydig- defect de receptor pentru LH/hCG
LH-receptor (LHR)- is stimulated by both LH and hCG. The Leydig cell development and function: the testosterone synthesis is controlled during fetal life by LH and hCG
LHR belongs to the family of G-protein- coupled receptors with six transmembrane helices
LHR is encoded by a gene localised on chromosome 2p21.
Loss of function mutations are located in the transmembrane domains but also on the extra cellular domain
LH-R Germline
chromosome 2p21
Inactivating mutation determine Leydig cell agenesis
XY male psudohermaphroditism with Leydig cell hypoplasia
Complete female phenotype
Male undervirilisation, micropenis
Kremer H et all 1995
LH-R Germline
or somatic
chromosome 2p21
Gain in function mutation
Gonadotropin-independent male limited precocious puberty
Familial or sporadic male precocious puberty
(testotoxicosis)
Shenher et all.
1993

Hipoplazia celulelor Leydig- defect de receptor pentru LH/hCG

Hipoplazia celulelor Leydig- defect de receptor pentru LH/hCG
Mollecular genetics – mutations that inactivates LH-R leading to Leydig cell hypoplasia were found in seven unrelated kindreds. The majority were found in affected homozygotes.
• single base substitution
• exon deletions
• in-frame insertions
In some compound heterozygotes was found:partial gene deletion at exon 8
Mutations impaires ligand binding and no response is elicit by both LH and hCGLeydig cell hypoplasia – autosomal recessive disorder
Severe form: XY undervirilised male with phenotypically female genitalia
male pseudohermaphroditism
Mild form: male hypogonadism ( the severity of the disease is correlated with residual activity of the mutant receptor)
Laboratory findings:
High LH, normal FSH, low testosterone which does not respond to hCG stimulation

Hipoplazia celulelor Leydig- defect de receptor pentru LH/hCG

Defecte innascute de sinteza a steroizilor suprarenali si gonadali
1. Defectul de 20-22 desmolaza
2. Defectul de 3 hidroxisteroid dehidrogenaza
3. defectul de 17 hidroxilaza (cyp 17)

STEROIDOGENEZA – zone afectate in PHM masculine
+ StAR

HIPERPLAZIA LIPOIDICA A SUPRARENALEIDefectul de 20-22 desmolaza, P450 scc
P450 scc produce hidroxilarea la C20 apoi la C22 si clivarea catenei laterale
Cod genetic: 15q23-q24
StAR gena care codifica o proteina care determina transportul rapid la colestrolului in mitocondrii unde incepe steroidogeneza ( numai o mica parte din steroidogeenza este independenta de StAR – 14 %)
HIPERPLAZIA LIPOIDICA A SUPRARENALEI
• este un defect de steroidogeneza autosomal recesiv care este dat de afectarea genei StAR
• defectul nu este la nivelul 20-22 desmolazei, desi numele initial al bolii s-a conservat, deoarece un atare defect ar afecta si steroidogeenza placentara de progesteron si ar determina avortul in primul trimestru al sarcinii
• denumirea de hiperplazie lipoidica este legata de aspectul intens hiperplazia al suparenalelor care sunt “sufocate” de colesterolul care nu mai urmeaza calei steroidogenezei
• steroidogenza poate continua in mica masura dupa nastere ceea ce explica supravietuirea celor netratati, dupa care depozitele de colesterol perturba complet fiziologia si distrug efectiv suparenalele aparind insuficienta suprarenala severa si decesul in cazurile netratate
• este ipoteza “celor 2 lovituri-two hit”: initial perturbarea tseroidogeenzei, apoi distrugerea celulelor

HIPERPLAZIA LIPOIDICA A SUPRARENALEI
Evolutie Sexul masculin Sexul feminin
Prenatal Lipsa sintezei de cortizol, mineralocorticoizi si androgeni cu defect complet de virilizare prenatala
Lipsa sintezei de cortizol, mineralocorticoiziLipsa de androgeni nu influenteaza sexualizarea prenatala feminina
La nastere
Pseudohermafroditism masculinPierdere de sare uneori dupa prima luna, hiperpigmentatie ACTH,FSH,LH, renina crescuteTratament : cortizol, 9 fluorocortizol
OGE normalePierdere de sare uneori dupa prima luna,hiperpigmentatie ACTH,FSH,LH, renina crescuteTratament : cortizol, 9 fluorocortizol
Pubertate
Fenotip feminin, lipsa uteruluiAmenoree primaraLipsa de dezvoltare a caracterelor sexualeSe induc c.sex. sec cu EP
Fenotip femininAmenoree primaraLipsa de dezvoltare a caracterelor sexualeSe induc c.sex. sec cu EP
Adult Fenotip femininCresterea sinilor este posibilaAmenoree primara
Fenotip femininDezvoltarea sinilorCicluri ca raspuns la EP

DEFECTUL DE 3 HIDROXISTEROID DEHIDROGENAZA, 5
IZOMERAZA
Codul enzomei pe bratul scurt al cromozomului 1
Codifica 2 enzime 3 hidroxisteroid dehidrogenaza I din placenta si
tesuturile periferice si 3 hidroxisteroid dehidrogenaza II din suprarenala si gonade
Fiziopatologie: incapacitatea de conversie a steroizilor 3 beta OH 5
in 4 steroizi
Defectul determina incapacitate de sinteza a cortizolului, aldosteronului, testisteronului si estradiolului si afecteaza suprarenala si gonadele
Exista:
forme complete cu “pieredere de sare”
forme usoare fara pierdere de sare
forme cu debut tardiv care se manifesta numai prin pubarha prematura

DEFECTUL DE 3 HIDROXISTEROID DEHIDROGENAZA, 5 IZOMERAZA
Date clinice si de laborator, tratament:
Sex genetic masculin 46,XY Sex genetic feminin 46, XX
Defect de productie a cortizolului, aldosteronului , androgenilor suprarenali si testiculari si estrogenilorDefect de productie a androgenilor la puberateLa nastere: pseudohermafroditism masculin cu OGE feminine sau ambuguiLa pubertate: infantilism sexual si puberha
Laborator: pregnenolon crescut,17 –OH pregnenolon crescut, DHEA crescut si raport crescut DHEA/ 4 crescut dupa ACTH
Tratament: cortizolul administrat de la nastere inhiba ACTH, productia de DHEA/DHEA-S ideala este corectia OGE in sens feminin +EPIn formele usoare: tratament cu androgeni la pubertate
Defect de productie a cortizolului, aldosteronului , androgenilor suprarenali si testiculari si estrogeniloDefect de productie a androgenilor la puberateLa nastere:hipertrofie clitoriadiana in formele severe, normale in formele usoare sau cu debit tardivLa pubertate: infantilism sexual sau pubarha precoceLaborator:pregnenolon crescut,17 –OH pregnenolon crescut, DHEA crescut si raport crescut DHEA/ 4 crescut dupa ACTHTratament: cortizolul administrat de la nastere inhiba ACTH, si productia de DHEAEP se adauga de la pubertate

Defectul de 17 hidroxilaza
17 hidroxilaza este enzima cheie care orienteaza soarta 17 OH pregnenolon:
• cind este prezenta – in zona fasciculata se produc C21 hdroxisteroizi de tipul cortizolului si C19 steroizi – androgenii
• cind este absenta – in zona glomerulata se produc mineralocorticoizii
17 hidroxilaza are doua actiuni principale:
• in calitate de 17 hidroxilaza
• in calitate de 17-20 liaza
Efectul de 17 hidroxilaza se manifesta in tot timpul vietii
Efectul de 17-20 liaza incepe in momentul adrenarhai, odata cu dezvoltarea zonei glomeruloza si continua dupa pubertate pina la 30 ani cind nivelul de DHEA-S este crescut
Nu se poate explica motivul pentru care efectul de 17-20 liaza se manifesta intr-un anumit moment al vietii, dar nu este exclus ca cele doua activitati ale enzimei sa fie controlate separat
Este posibil ca efectul hidroxilaza sau liaza sa fie modulat de o proteina din grupa flavoproteinelor care denumita p450 oxidoreductaza
La om 17 –20 liaza catalizeaza de 30 de ori mai bine conversia substratelor 5 (pregnenolon-DHEA) decit a substratelor 4 (Progesteron - 4 androestendion si majoritatea 4 androstendion la om provine din DHEA sub actiunea 3OH dehidrogenazei

Defectul de 17 hidroxilaza
Genetica : enzima cu dubla actiune este codificata pentru enzima CSR si gonadala pe cromozomul 10
Clinica si laborator in forma clasica a deficitului:
Sex genetic masculin 46,XY Sex genetic feminin 46, XX
Defect de productie a androgenilor suprarenali si testiculariDefect de productie a androgenilor la puberateLa nastere: pseudohermafroditism masculin cu OGE feminine sau ambuguiLa pubertate: infantilism sexual
Laborator: pregnenolon crescut, progesteron crescut, DOC, corticosteron so 18-OH cortcostron crescut, activitatea renine plasmatice si aldosteronul sunt scazute
Tratament: cortizolul adminsitrat de la nastere inhiba ACTH, productia de mineralocroticoizi si evita instalarea HTAideala este corectia OGE in sens feminin +EP
Defect de productie a androgenilor suprarenali si testiculariDefect de productie a androgenilor la puberateLa nastere: OGE feminine normale
La pubertate: infantilism sexual
Laborator: pregnenolon crescut, progesteron crescut, DOC, corticosteron so 18-OH cortcostron crescut, activitatea renine plasmatice si aldosteronul sunt scazute
Tratament: cortizolul administrat de la nastere inhiba ACTH, productia de mineralocroticoizi si evita instalarea HTAEP se adauga de la pubertate

Defectul de 17 hidroxilaza ca 17-20 liaza
S-a descris un dublu heterozigot pentru defectul de 17 hidroxilaza predominent pe forma liaza care reduce formarea de 19 C steroizi: testosteron din c21 steroizi cu nivele reduse de DHEA, androstendion, testosteron si estrogeni
Fenotipul organelor genitale externe este feminin
Nu se poate explica prezenta ductelor wollfiene
Ductele mulleriene sunt absente prin AHM
Defectul poate fi pus in evidenta la nivelul gonadelor prin lipsa productiei de androgeni testiculari la admistrarea de hGC
Tratament: in formele cu deficit sever si pseudohermafroditism masculin se indica atribuirea sexului feminin si rezectia gonadelor nefunctionale , urmata de administrarea de estrogeni la pubertate

DEFECTUL DE 17 HIDROXISTEROID DEHIDROGENAZA
Exista 5 gene care codifica enzime, 2 situate la nivelul bratului luing la cromozomului 17 (pseudogene) si una situata la nivelul cromozomului 9q22 (pt.izoenzima din testicul)
Enzima este responsabila de ultimul episod al conversiei androstendion-testosteron si a estronei in estradiol prin oxido-reducere
Rezultatul este PHM (46,XY) (prima descriere 1985):
• organe genitale externe ambigui cu deficit sever de virilizare si hipospadias perineal
• in forme mai putin severe apare micropenis
• OGI sunt masculine cu prezenta derivatelor wollfiene si ansenta celor mulleriene
• sexul civil este frecvent cel feminin cu identitate de gen initial feminina
• la pubertate survine virilizarea organelor genitale externe si aparitia pilozitatii sexuale
• la pubertate identitatea de gen si rolul sexual se schimba spontan car=tre cel masculin ( asemanator cu situatia din deficitul de 5 alpha reductaza)

DEFECTUL DE 17 HIDROXISTEROID DEHIDROGENAZA
TIP 1 TIP2 TIP3 TIP4 TIP5
Clonare Anticorpi expresie expresie cADN cADN
Dimensiuni AA
327 387 310 736 323
Gena -exoni 6 5 11 - 9
Cromozom 17q21 16q24 9q22 - 10p14,15
Distributie tisulara
Ovar, palcenta, prostata, sin
Ficat, placenta, endometru
Testicul generala Ficat, muschi
Localizare celulara
Citozol microsomi microsomi peroxisomi Citosol
Co-factor NADH, NADPH
NAD+ NADPH NAD+ NADPH
Preferinte catalitice
Reducere Oxidare reducere Oxidare reducere
Defect de 17 HSD
Normal Normal Mutanta - -

DEFECTUL DE 17 HIDROXISTEROID DEHIDROGENAZA


DEFECTUL DE 5 REDUCTAZA
Tipul I cr.5 p
Tipul II cr. 2 p23

DEFECTUL DE 5 REDUCTAZA
TIPUL I TIPUL 2
Structura genei 5 exoni, 4 introni 5 exoni, 4 introni
Localizarea genei pe cromozomi
SRD5A 1, 5p15 SRD5A 2, 2p23
Dimensiuni 259 AA, Mr = 29462 254 AA, Mr = 28 398
Distributie tisulara Ficat, pielea non-genitala, prostata
Prostata, epididim, vezicule seminale pielea genitala, uter, ficat, piele, sin, folicul pilos, placenta, cortex cerebral, ovar?, testicul?, rinichi?, suprarenale?
pH optim Neutru sau bazic Acid sau neutru
Nivelul enzimei in prostata Scazut crescut
Activitatea in deficitul de 5 reductaza
Normal Mutanta
Inhibitia prin finasterid K1>/+ 300nM K1 >/+ 3-5 nM







SINDROAME DE INSENSIBILITATE LA ANDROGENI
CODUL GENETIC penru repcetorul de androgeni: cromozomul Xq11-12
TIPURI DE SINDROAME DE INSENSIBILITATE LA ANDROGENI:
tip 1: fenotip masculin fara ambiguitati dar cu anomalii de virilizare pubertara sau ale spermatogenezei
tip 2 a: fenotip masculin cu hipospadias izolat
tip 2 b: fenotip masculin cu hipospadias = scrot bifid si micropenis
tip 3 a: intersexualitate evidenta cu micropenis (asemanaror cu o hipertrofie clitoridiana si orificiu uretral perineal
tip 3 b: intersexualitate evidenta cu micropenis (asemanaror cu o hipertrofie clitoridiana si orificiu uretral care face parte din sinus uro-genital asociat cu un mic vagin inchis “ in fund de sac”
tip 4 a: fenotip predominant feminin cu discreta virilizare: hipertrofie clitoridiana, fuziune labiala si sinus uro-genital
tip 4 b: fenotip predominant feminin cu discreta virilizare: hipertrofie clitoridiana, fuziune labiala partiala si deschidere separata a uretrei si vaginului
5 fenotip feminin fara ambiguitati

SINDROAME DE INSENSIBILITATE LA ANDROGENI
Diagnosticul clinic se stabileste in diferite etape ale existentei
• tipul 1: la pubertate prin virilizare insuficienta si ca adult prin infertiltate
• tipul 1 poate dezvolta ca adult de virsta medie sau mai tirziu atrofia musculaturii spinale – sindromul Kennedy ( au o anomalie particulara a AR)
• tipul 5: la pubertate prin amenoree primara sau la nastere prin discrepanta dintre fenotip si genotipul stabilit prin amniocenteza, sau prepubertar prin identificarea testiculului in labii
• tipurile 2b-4a sunt urgente pediatrice deoarece impun stabilirea relatiei genotip fenotip ca in orice intersexualiatte si stabilirea sexului civil in raport cu sansele de dezvoltare ulterioara a gonadei

SINDROAME DE INSENSIBILITATE LA ANDROGENI
DIAGNOSTICUL BIOLOGIC
Inainte de pubertate: administrarea de hCG determina cresterea exploziva a testosteronului si DHT si permite diagnosticul diferential cu deficite ale steroidogenezei
dupa pubertate:
• LH, FSH net crescuti (lipsa feed-back negativ)
• testosteronul este crescut, iar produsul T x LH crescut pledeza pentru AIS
Teste in vivo specifice pentru AIS:
• Stimularea productiei de testosteron cu hCG si determinarea gradului de reducere a nivelului de SHBG
• administrarea uni steroid anabolic: stanozol 0,2 mg/zi oral seara 3 zile si determinarea SHBG in zilele: 5,6,7, sau 8. Raportul dintre cel mai redus nivel al SHBG si nivelul bazal dinainte de test da informatii asupra existentei insensibilitatii la androgeni dar si asupra severitatii acestui deficit. Testul nu are sensibilitate suficienta inainte de 6 luni

SINDROAME DE INSENSIBILITATE LA ANDROGENI
DEFECTE GENETICE ALE RECEPTORULUI DE ANDROGENI:
• au fost descrise peste 300 de mutatii ale AR si baza de date asupra acestora este in permanenta “updatare”
• Nu exista o relatie stricta intre defectul genetic si fenotip
• anomalii: deletii complete ale unuia sau mai multi exoni
• deletia competa a exonului 4 in AIS tip 1
• subtitutia unui singur nucleotid
• mutatii “missense” care a;tereaza secventa normala de amino acizi
• substitutii ale unor amino acizi
• muatii afecteaza domeniul de legare la AND si mia rar domeniul de legare a hormonului
• mutatii ale domeniului N-terminal codat de exonul 1au fost decrise in atrofia musculara spinala sau bulbara si in alte boli degenerative
• s-a descris o mutatie a domeniului de lagare la AND care se asociaza cu aparitia cancerului de sin pe ginecomastia din AIS
• exista muatii ale receptorului de androgeni care determina progresiunea cancerului de prostata

SINDROAME DE INSENSIBILITATE LA ANDROGENI
CERCETAREA “ IN VITRO” A ACTIVITATII REEPTORULUI DE ANDROGENI
• Legarea androgenilor marcati de fibroblastii din pielea genitala
• absenta legarii este observata in insensibilitatea completa la androgeni
• expresia mARN pentru receptorul androgeni prin Northern blot analiza
• determinarea activitatii de transactivare a receptorului androgenic in celule co-transfectate cu receptorul mutant

SINDROMUL DE INSENSIBILITATE COMPLETA LA ANDROGENI







PSEUDOHERMAFRODTISME FEMININE

PSEUDOHERMAFRODTISME FEMININE 46 xx
• Hiperplazii congenitale ale suprarenalei
• Defectul de aromataza placentara• Androgeni administrati la mama

DEFECTUL DE 21 OH FORMA CLASICA ASPECT CLINIC
Sex genetic
Aspectul OGE la nastere
Copilarie si adolescenta
Adult
Feminin Pseudohermafroditism femininIntersexualitate tipuriPrader I – IVTipul II – pierdere de sare
Pseudo-pubertate precoce heterosexuala:AdrenarhaAbsenta dezvoltarii mamareDezvoltarea musculaturii dupa fenotip masculin
Virilizare somaticaPCOSTalie finala redusa
MasculinMacrogenitosomie
Tipul II – pierdere de sare
Pseudo-pubertate precoce izosexualaAdrenarhaDezvoltarea musculaturii dupa fenotip masculinAbsenta dezvoltarii testiculare
Virilizare somaticaInfertilitate Talie finala redusa










DEFECTUL DE 11 OH ASPECT CLINIC
Sex genetic
Aspectul OGE la nastere
Copilarie si adolescenta
Adult
Feminin Formal clasicaPseudohermafroditism femininIntersexualitate tipuriPrader I – IV
Forma non clasica
Pseudo-pubertate precoce heterosexuala: adrenarha precoceAbsenta dezvoltarii mamareDezvoltarea musculaturii dupa fenotip masculin, talie redusaHTA cu hipokaliemie
Adrenarha precoce, amenoree primara, hirsutism, PCOS
Virilizare somaticaPCOSTalie finala redusaHTA cu hipokaliemie
PCOSHTA
MasculinMacrogenitosomie
Forma non-clasica
Pseudo-pubertate precoce izosexuala: adrenarha precoceDezvoltarea musculaturii dupa fenotip masculinAbsenta dezvoltarii testiculareHTA cu hipokaliemie
Adrenarha precoce, talie redusa
Virilizare somaticaInfertilitate Talie finala redusaHTA cu hipokaliemie
HTA



HIPERPLAZIA CONGENITALA A CSR DIAGNOSTIC PARACLINIC: 21 OH
DEFECT INCIDENTA PRODUSI
PRE-BLOC
DOZABILI IN SINGE
PRODUSI
PRE-BLOC
DOZABILI
IN URINA
TESTE DINAMICE
ALTE DETERMI
NARITERAPIE
TIP I
21OH
forma clasica
Forma non-clasica
Forma criptica
1/ 5000
1/ 20.000
1/ 1000
17-OH
Progesteron
DHEA
Androstendion
17 OH – P
17 OH-P
17 KS
Complexul
CPG
17 OH-P/ACTH
17 OH-P / DXM
17 OH-P / ACTH
17 OH-P / ACTH
ACTH crescut
ARP crescuta
Cortizon
12-20 mg/m2/zi
In stress: HSHC
40 mg sub 4 ani
100mg dupa 4 ani
Corectie OGE dupa 3 ani
TIP II
Forma cu pierdere de sare
1 / 10.000 17-OH
Progesteron
DHEA
17 KS
Complexul
CPG
17 OH-P/ACTH
17 OH-P / DXM
ACTH crescut
ARP crescuta
Cortizon
12-20 mg/m2/zi
9 FHC
0.1-0.2 mg
Corectie OGE dupa 3 ani

EFECTE ALE DISRUPTORILOR ENDOCRINI
Efectele”disruptorilr” endocrini se manifesta la mai multe nivele:
• sinteza hormonilor
• metabolism
• excretie
• pentru diferentierea sexuala sunt cruciale efectele androgenice sau estrogenice ale compusilor
Hipospadiasul, criptorhidismul, cancerul tiroidian si calitatea slaba a spermei sunt corelate. Cancerul testicular este corelat cu testiculul necoborit si acesta la rindul sau se coreleaza cu hipospadiasul. Aceste situatii au cauze comune printre acre: greutatea mica la nastere si nivelul crescut al estrogenilor materni. Cresterea tendintei de aparitie si faptul ca acestea manifesta anumite variatii regionale ( mai mare in Danemarca si reduse in Finlanda) in aparitia acestor anomalii ale tractului reproductiv masculin pune problema interventiei unor factori de mediu

CRIPTORHIDISMUL INCIDENTA:
• 0.03 – 13,4 % pina la 1 an incluzind copiii prematuri (Anglia)
• 0,16 – 13,3 % in scoli sau la incorporare (Anglia)
• 2 – 4,7 % bazat pe studii asupra diagnosticului de externare a copiilor (Anglia)
• orhidopexia inainte de 15 ani: 1,4 % pe cohorta 1951 si 2,9 % pe cohorta 1977 (Anglia)
• exista o neta tendinta la crestere a incidentei criptorhidismului (Anglia)
• la copiii < 2500 g: 1,74 %, iar la cei peste 2500 g: 0,91 % (New York)
• incidenta este mai mica la afroamericani decit la caucazieni

HIPOSPADIAS
INCIDENTA:
Anglia: dublarea incidente de la 7,3 /10.000 ( 1960) la 16/10.000 (1980)
Ungaria: crestere de la 5,5 la 23,9 /10.000 intre 1960 si 1970
Danemarca: crestere de la 7,5 la 12/10.000 intre 1970-1980
USA:
• Birth Defects Monitoring Program: 20,2/10.000 (1970) – 39,6 /10.000 (1993) cu cresterea raportului dintre formele severe si cele usoase
• Metropoitan Atlanta Congenital Defects Program: dublarea incidentei si cresterea raportului intre formele severe si cele usoare de 3-5 ori (1968-1993)
• prevalenta este mai mare la caucaziei si la nativii americani
• Finlanda are o net mai mica incidenta decit Danemarca, Norvegia si Suedia, tari in care si rata de cancer testicular este de 4 ori mai mare decit in Finlanda

EFECTELE EXPUNERII “IN UTERO” LA DIETILSTILBESTROL SI ALTI ESTROGENI
Copiii de sex masculin nascuti de aceste mame aveau multiple anomalii ale tractusului genital
• chisti epididimari : 20,8 % fata de 4,9 % neexpusi
• stenoza de meat: 12,9 % fata de 1,8 %
• hipospadias: 4,4 % fata de 0 %
• cei expusi inainte de 11 saptaini aveau semnificativ mai multe anomalii
• calitatea spermei a fost semnificativ mai modesta la cei expusi
• riscul de cancer testicular 2,6
Copii de sex feminin (DES daughters):
• adenoza vaginala, creste transversale, deformari ale labiilor, ectropin extensiv, hipoplazie vaginala (90%)
• cancerul cu celule clare: 1,4/1000-1,4/10.000
• deformarea cavitatii uterine: “in T” (69%) si incompetenta cervicala
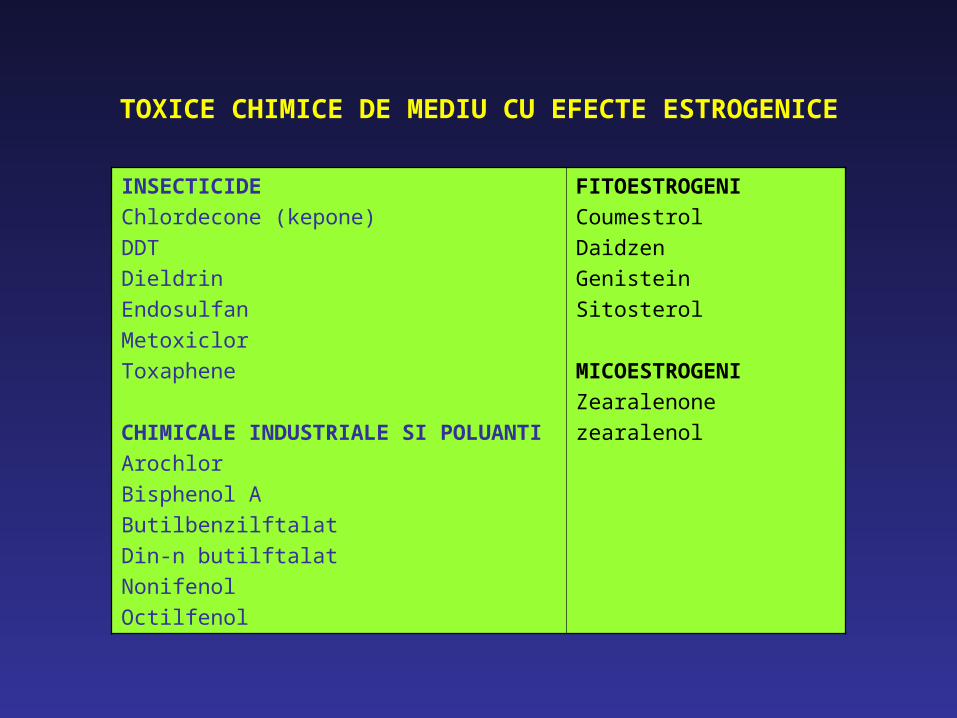
TOXICE CHIMICE DE MEDIU CU EFECTE ESTROGENICE
INSECTICIDEChlordecone (kepone)DDTDieldrinEndosulfanMetoxiclorToxaphene
CHIMICALE INDUSTRIALE SI POLUANTIArochlorBisphenol AButilbenzilftalatDin-n butilftalatNonifenolOctilfenol
FITOESTROGENICoumestrolDaidzenGenisteinSitosterol
MICOESTROGENIZearalenonezearalenol

EFECTELE EXPUNERII “IN UTERO” LA PROGESTAGENI
Unele studii au demosntrat incidentaai mare a hipospadiasului dupa expunerea la medroxiprogestron la icneputul sarcinii
Anumite progestine au efecte antiandrogenice la sexul masculin si androgenice la sexul feminin determinid masculinzare

EFECTELE EXPUNERII “IN UTERO” LA ANTIANDROGENI
CHIMICALE CU EFECTE ANTIANDROGENICE
p.p’-DDE9,10,dihidrofenantrenLinuronMetabolitii M1 si M2 de vinclozolinAcetatul de cyproteron ( farmaceutic)Flutamida ( farmaceutic)Hidroxiflutamida ( farmaceutic)

EFECTELE EXPUNERII “IN UTERO” LA ALTI “DISRUPTORI” ENDOCRINI
Disruptorii activitatii tiroidiene determina anomalii de dezvoltare a testiculului
• bifenili policlorinati disrup activitatea tiroidiana si au efecte adverse asupra dezvoltarii testiculare
• doxinele , cea mai toxica este 2,3,7,8 tetracloro-benzo p-dioxina rezulta ca produsi toxici din fabricile de hidrocarburi policlorinate, din arderi, din produsii de albire pentru hirtie sau emisia de gaze de automobil
• expunerea la dioxine determia anomalii ale regiunii ano-genitale , testicul necoborit si greutate mai mica a tuturor organelor din sfera genitala la animalele de experienta
• efectele par determinate de supresia productiei de androgeni perinatali

B. VIRILIZAREA FATULUI CU SEX GENETIC FEMININ
1. Hiperplazia congenitala a suprarenalei
a. Defectul de 21 hidroxilaza
b. Defectul de 11 hidroxilaza
c. Defectul de 3 hidroxisteroid dehidrogenaza
2. Hormoni din mediu
a. Tulburari cu virilizare materna – defectul de aromataza placentara
b. Virilizare iatrogena
3. Malformatii teratologice
a. Anomalii de dezvoltare ale uterului si vaginului
b. Sindromul de anomalii congenitale multiple

EXPLORAREA STARILOR DE INTERSEXUALITATE I
INVESTIGATII DE SCREENING PENTRU INTERSEXUALITATI
CariotipElectroliti si uree sanguinaGlucoza17-OH progesteron plasmaticTestosteron plasmaticSteroizi urinari ( 12, 24 ore0Ultrasonografie pelvina

EXPLORAREA STARILOR DE INTERSEXUALITATE II
INVESTIGATII DE ORDINUL II IN EXPLORAREA INTERSEXUALITATILOR
BAZALE• LH si FSH• Testosteron• Androestendion• dihidrotestosteron Testul de stimulare la LH-RH• virful de FSH si LH Test de stimulare la hCG•Testosteron• Androestendion• dihidrotestosteron
• stroizi urinari inainte si dupa hCG• imagerie pentru localizarea gonadelor

EXPLORAREA STARILOR DE INTERSEXUALITATE III
HORMONALE• AMH seric• inhibina serica• SHBG serica GENETICE• genele de determinare testiculara: SRY, SOX9, WT-1, SF-1, DAX-1• genele steroidogenetice: StAR, CYP17, 3HSD, 17 HSD, 5R• genele receptorilor: receptor de androgeni, receptor de LH
CHIRURGICALE• histologia gonadei• Histochimia gonadei• biopsia de piele pentru stabilirea liniei de fibroblasti
Nu se efectueaza Clinica Endo

EXPLORAREA STARILOR DE INTERSEXUALITATE
Ambiguitate genitala
Gonade palpabile Gonade nepalpabile
Structuri mulleriene
RMN Echo, CT
17-OH-P
Cariotip
Absente Prezente
Normal Normal
XYPoli X
+Y
XY
XO/XY
PHMDisg.tubi semniferi
PHMdisgenetic
Prezente absente
Normal Crescut Normal Normal
XX XX XO/XY XX,XY XY
XY XX/XYP450 ARO
CAHPHMdisgenetic
HER
PHM

MANAGEMENTUL STARILOR DE INTERSEXUALITATE
Stabilirea sexului civil se efecteaza functie de diagnostic:
• in cazul pseudohermafroditismelor feminine se poate stabili sexul civil feminin, la 6-12 luni se intervine pentru corectia OGE in sens feminin cu prezervarea clitorisului prin operatie de reductie
• in cazul pseudohermafroditismelor masculine sexul civil se stabileste in functie de dimensiunile falusului, sansele de dezvoltare prin tratament a acestuia si sansele asumare a identitatii de gen si rol masculin
• pentru cazurile in care se apreciaza ca terapia nu va permite o dezvoltare sexuala compatibila cu o existenta cvasinormala sub dentitate de gen masculina ested e preferat stabilirea , cu concursul informat al familiei a sexului civil feminin si corectia in consecinta a organelor genitale si educatie in sens feminin

THE TESTISHypogonadotropic hypogonadism
DAX-1 mutation causes hypogonadotropic hypogonadism and hypoplasia adrenalis congenita linked to the X chromosome
Frequency 1/12500
At birth: acute adrenal insufficiency with hypoglicaemia and salt –loosing syndrome
- low cortizol and mineralocorticoid levels are not stimulated by ACTH
- the adrenals have fetal histological appearance - the first sigh of hypogonadotropic hypogonadism could be
the undescended testis- the level of gonadotropins are low or normal during childhood
but definitely low at puberty suggesting a progressive hypothalamic-pituitary disease
- there is a combined hypothalamic and pituitary deficiency- most of the patients were diagnosed with 21 hydroxilase
deficiency with salt- loosing syndrome, were well treated and reached their pubertal age and than their gonadotropin deficiency became obvious

Hypogonadotropic hypogonadismKallmann’s syndrome
Hypogonadotropic hypogonadism with anosmia or hypoosmia
Frequency:
1/10,000 males and 5-7 times less frequent in females
- at birth an undescended testis or micropenis evokes the syndrome
Familial forms:
- X-linked (KAL-1)
- autosomal dominant (KAL-2)
- autosomal recessif (KAL-3)
Other associated abnormalities:
- neurological: controlateral imitation syncinesis ( mirror movements, visual attention abnormality, abnormalities of ocular movements, ptosis, deafness,
- non-neurological: aplastic kidney, labial or palatinal dehiscence, dental agenesis

Kallmann’s syndrome
During fetal development Gn-RH secreting neurons have a migration from the medial part of the olfactive epithelium to the hypothalamus in close relationship with olfactory accessory nerves: vomero- nasal and terminal. Than these neurons enter the brain just behind the olfactory bulbs and finally reach the hypothalamus.
In a 19 week fetus with x-linked Kallmann’s syndrome the Gn-RH secreting neurons do not reach the hypothalamus and accumulate outside the brain in the nasal region Thus the final way of Gn-RH secreting neurons is dependent of KAL-1 gene.
KAL-1 gene encodes a protein named ANOSMINE -1 a nerve-adhesion protein that is not present in nasal epithelium and hypothalamus in Kallmann’s syndrome