role of caspases in apoptosis and disease
TRANSCRIPT

Indian J Physiol Pharmacol 1999; 43 (2) : 151-159
REVIEW ARTICLE
ROLE OF CASPASES IN APOPTOSIS AND DISEASE
NEERU KHANNA AND NEETA SINGH*
Department of Biochemistry,All India Institute of Medical Sciences,Ansari Nagar, New Delhi - 110 029
(Received on July 15, 1998)
Abstract: Apoptosis, a genetically governed process of eliminating cellsin response to avariety of stimuli provides protection against cancer andviral infections as well as maintains homeostasis. Recent studies usingboth molecular and cloning approaches, and in vitro systems have identifieda class of highly specific proteases, termed caspases, that appear to havean important role in apoptotic execution. Caspases are synthesized asprecursor molecules that require processing at specific aspartate residuesto produce the active enzyme which in turn leads to the cleavage of variousdeath substrates that lead to morphological changes typical of apoptosis.This review discusses caspases, their inhibitors and regulators. Sincecytotoxic drugs used in chemotherapy of leukemia's and solid tumorscause apoptosis in target cells, elucidating the consequences of proteolyticactivity occupies a central role for understanding of the molecularmechanism of apoptosis which can help us to use the caspase inhibitors astargets of therapy.
Key words: caspases apoptosis Bcl-2
INTRODUCTION
Caspase mediated apoptotic cell deathplays an important role in both physiologicaland pathological processes. During embryodevelopment, surplus cells die as part of theprocess that balances growth anddifferentiation with death. In case of centraland peripheral nervous systems, apoptosisoccurs <3xtensively during development.Inappropriate apoptosis however, underliesthe etiology of many of the most intractableof human diseases, for e.g. suppression ofapoptosis is involved in the proliferation ofpreneoplastic and/or neoplastic cells and in
*Corresponding Author
the development of autoimmune diseases.Increased apoptosis may be involved inneurodegenerative diseases, duringretroviral infection (HIV-1), and perhapsin the development of diabetes mellitus(1-6) .
Apoptosis, which is conservedthroughout evolution comprises of fourstages. Priming is triggered either byremoval of survival factors or by a cellularinsult such as genotoxic damage caused byirradiation, mitotic defect or by reactiveoxygen species. A commitment. step follows,when cells activate the apoptotic machinery

152 Khanna and Singh
e.g. cell death induced by Fas/TNF fJ..
Commitment renders the processirreversible. The cell then enters anexecution phase where the morphologicalcharacteristics of apoptosis becomeapparent: i.e, membrane blebbing,cytoplasmic and nuclear shrinkage,internucleosomal DNA fragmentation andchromatin condensation. Finally, theremoval of apoptotic cells occurs by eithermacrophages or neighboring cells withoutinitiating an inflammatory response (7-8).The biochemical machinery involved in thekilling and degradation of the cells isexpressed constitutively and is accessible foractivation by various signals. It appears thatalthough distinct pathways leading toapoptosis are induced by different signalsthey finally converge to a common effectorpathway. At the heart of this pathwayare a family of cysteine proteases, the"caspases" that are related to mammalianICE CInterleukin 1-~ converting enzyme)and Ced-3 (cell death abnormal), the productof a gene that is absolutely necessary forapoptotic suicide in the nematode C. elegans(9-12).
As ICE - like cysteine proteases do playa fundamental role in apoptosis, it may bepossible to regula~e cell death by usingspecific protease inhibitors. This may haveimportant therapeutic implications in thetreatment of the diseases that arise fromexcess or premature cell death.
CASPASES
The cysteine proteases, caspases whichcleave after aspartic acid appear to playacritical role in initiating and sustaining thebiochemical events that result in apoptotic
Indian J Physiol Pharmacol 1999; 43(2)
cell death. Much of the knowledge on thegenetic regulation of cell death has emergedfrom C. elegans as a model organism.Threegenes Ced-3, Ced-4 and Ced-9 play crucialroles in executing and regulating cell deathwhile others are needed for engulfment anddisposal of dead cells (13-15). Ced-3 wasfound to exhibit significant homology to ICE(16), a cysteine protease which converts the33 kD protease form of IL-1~ to active 17.5kDa form, once again by cleaving afteraspartate residues. These observations havebeen closely followed by the discovery ofseveral more ICE /Ced-3 homologs whoseover expression in various cell typesunder the influence of diverse stimuliresults in apoptosis, thereby suggesting thatprogrammed cell death is conserved widelyin phylogeny from nematode C. elegans tohumans.
In humans, the caspase family ofproteases consists of 10 members that canbe subdivided into 3 subfamilies: ICEsubfamily (caspase 1, 4, 5), CPP32 subfamily(caspase 3, 6, 7, 8, 10) and Ich-1 subfamily(caspase 2 and caspase 9) (17). All thesecaspases are normally present in cells ascatalytically inactive forms (zymogens) andcontain cysteine residues and the conservedpentapeptide QACRG at the active site.They are proteolytic ally processed andactivated through cleavage at aspartic acidby other caspases or possibly throughautoprocessing in a proteolytic cascadesimilar to complement activation or bloodclotting (18-22).
The currently known family members inhuman, participate in one of the twodistinct signalling pathways.

Indian J Physiol Pharmacol 1999; 43(2)
1) activation of proinflammatory cytokinesand
2) promotion of apoptotic cell death.
Caspase 1 ( and probably 4 and 5 ) which1S required for activation of IL-1~ andIFN-y IF belongs to procytokine activationfamily and other caspases notably caspase2, 3, 6, 7, 8, and 10 are considered topromote pathways to apoptosis. Thisconclusion is based on the fact that thesecaspases endoproteolytically cleave a selectgroup of cellular proteins involved in DNArepair, replication, RNA splicing (PARP,lamin, a fodrin, 70 kDa U1 snRNP, gelsolin)etc. thereby causing nuclear andcytoplasmic alterations that typify apoptosis(22-25).
CASPASE INHIBITORS
Based on their substrate specificity,synthetic substrates and inhibitors (withYVAD specific for the ICE subfamily andDEVD specific for CPP32 subfamily) havebeen designed. Aldehyde derivatives ofthese peptides are reversible inhibitorswhereas chloromethyl, flouromethyl andacyloxy methyl ketones are irreversibleinhibitors (26).
In addition, several proteins encoded byviral genes are known to inhibit membersof the ICE family. These include crmA, acytokine response modifier gene encoded bycowpox virus and p35 encoded bybaculovirus (27-30). These viral proteinsseem to inhibit protease activity by forminga stable complex. p35 has a broaderspecificity for ICE family members thancrmA, i.e. crmA preferentially inhibits
Caspases in Apoptosis and Disease 153
caspase lover caspase 3 while p35 inhibitsboth caspase 1 and caspase 3 equally well.lAP (inhibitor of apoptosis) proteins wereidentified by their ability to compensate forloss of function of caspase inhibitor proteinp35 in Autographa Californica nuclearpolyhedrosis virus (AcNPV) mutants. Lastyear, several cellular homologues of thevirus iap were reported namely XIAP,clAP1, clAP2 etc. whose overexpression cansuppress cell death induced by a variety ofstimuli. The first mammalian homologueXIAP found was identified as the productof a gene mutated in several forms of spinalmuscular atrophy, a disease in which motorneurons die prematurely, whereas it hasbeen reported that clAP1, clAP2 (murinehomologues) are rapidly induced in cellswhich are resistant to TNF a. mediateddeath but not in cells sensitive to TNFa..
Although new proteases and theirmechanisms in bringing about cell deathhave emerged, basic issues relating to theconnection between ICE family of proteasesand the Bcl-2 family of proteins still remainunresolved. Overexpression of Bcl-2 andBclxL in Jurkat cells inhibited staurosporineinduced apoptosis and abrogated activationof CPP32 and ICE-LAP3 but had no effecton Fas mediated apoptosis therebysuggesting that two apoptosis pathways arepresent in Jurkat cells with Bcl-2 and BclXL functioning upstream of ICE likeproteases in one of the pathways. On theother hand E1B 19k and Bcl-2 preventprocessing of CPP32 and cleavage ofdownstream substrates during E1A inducedapoptosis. Thus cellular context and celltype specificity may be factors in theregulation of apoptosis. Recently a modelhas been proposed to explain the

154 Khanna and Singh
relationship between Bcl-2 and ICE likeproteases. According to this model Bcl-2molecules transduce constitutive survivalsignals by forming ion channels or by someother mechanism to suppress ICE likeprotease activation (31-34). This activity isantagonized by Bax like proteins whichactivate ICE like proteases by formingdimers and inducing death promotingsignal. Both pathways could be workingsimultaneously in a cell to activate orinhibit apoptosis as may be required.Although Bcl-2 is presumed to inhibitcaspase activation by acting upstream ofcaspases, a report by Cheng et al (35)suggested that Bcl-2 can also be adownstream death substrate of caspases,suggesting the existence of a feedback loopbetween caspases and Bcl-2. Theobservation that Bcl-2 cannot inhibitapoptosis in some situations implies thatspecific caspases may by pass the pathwayinhibited by Bcl-2. In this scenario,activation of a subset of caspases that areinsensitive to Bcl-2 may also promotecleavage of Bcl-2, not only inactivating itsantiapoptotic function but also enhancingcell death.
Apoptosis is of critical importance bothto pathogenesis of cancers and to theirlikelihood of resistance to conventionalantineoplastic treatments. Mutations in p53gene and its regulators (mdm2) areextremely common, occuring in perhaps 5575% of human cancers. In response to DNAdamage, the p53 protein induces apoptosisby acting as a transcription factor,activating expression of numerous apoptosismediating genes. A current model proposesthat DNA damage causes the p53 proteinto turn on genes whose products generate
Indian J Physiol Pharmacol 1999; 43(2)
free radicals that, in turn damage the cellsmitochondria, whose contents (such ascytochrome c) leak out into the cytoplasmand activate apoptotic caspases. Apartfrom this, the p53 protein can also induceapoptosis by upregulating expression of Bax,a proapoptotic Bcl-2 family member. Tumorsexhibit varying numbers of apoptotic cells;a high proportion of apoptotic cellscorrelates with slower tumor growth.Conversely, mice that lack the genes for Baxdevelop fast growing tumors than similartumors from mice with normal Bax genes.In summary, mutations in genes that leaddirectly or indirectly to reduced apoptosisare generally associated with poor prognosisin a variety of tumor types (36), sinceconventional chemotherapy and radiationtherapy rely primarily on induction ofapoptosis in cancer cells for therapeuticeffect. New cancer therapies that aim toinduce apoptosis specifically in cancer cellsare the source of much excitement andrenewed hopes for cures.
CASPASES AND APOPTOSIS
The caspases implicated III apoptosisare currently divided into initiators andexecutioners. The exact order of theexecutioners and the place of other caspasesin the pathway are still controversial butat least in Fas pathway, signalling of deathis transmitted in part by sequential caspaseactivators.
Receptor aggregation either by Fasligand or by antibody crosslinking inducesthe formation of a death inducing signallingcomplex (DISC) of proteins comprising Fasitself, an adaptor protein FADD (Fasassociated death domain protein) and the
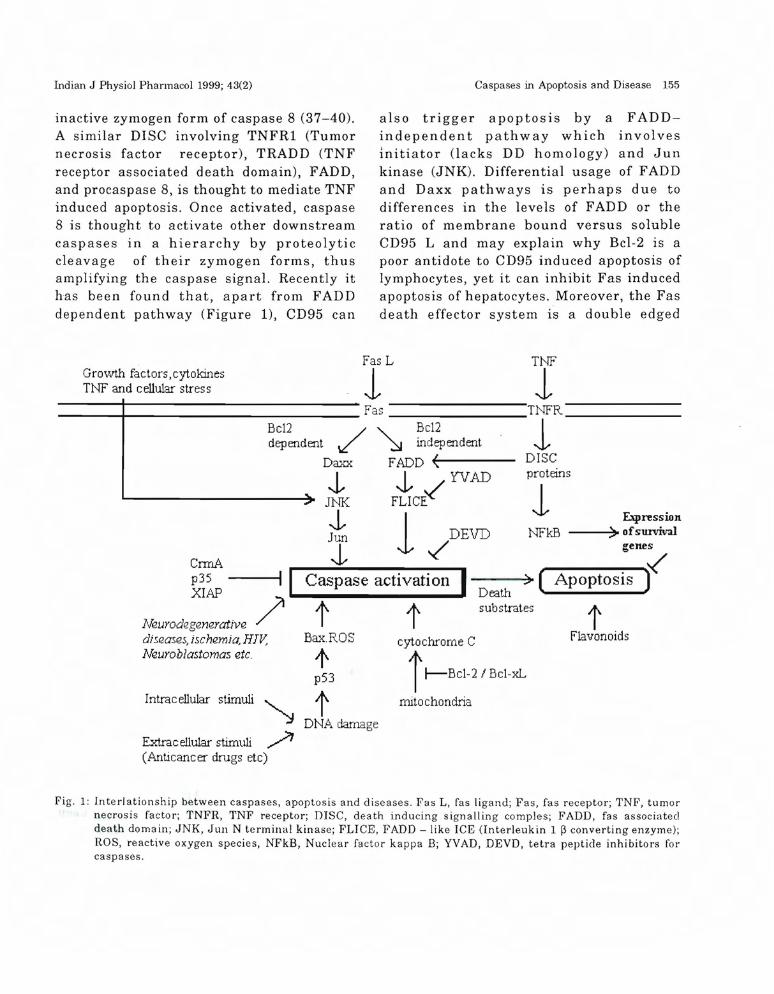
Indian J Physiol Pharmacol 1999; 43(2) Caspases in Apoptosis and Disease 155
inactive zymogen form of caspase 8 (37-40).A similar DISC involving TNFRl (Tumornecrosis factor receptor), TRADD (TNFreceptor associated death domain), FADD,and procaspase 8, is thought to mediate TNFinduced apoptosis. Once activated, caspase8 is thought to activate other downstreamcaspases in a hierarchy by proteolyticcleavage of their zymogen forms, thusamplifying the caspase signal. Recently ithas been found that, apart from FADDdependent pathway (Figure 1), CD95 can
also trigger apoptosis by a FADDindependent pathway which involvesinitiator (lacks DD homology) and Junkinase (JNK). Differential usage of FADDand Daxx pathways is perhaps due todifferences in the levels of FADD or theratio of membrane bound versus solubleCD95 L and may explain why Bcl-2 is apoor antidote to CD95 induced apoptosis oflymphocytes, yet it can inhibit Fas inducedapoptosis of hepatocytes. Moreover, the Fasdeath effector system is a double edged
tFlavonoids
Expression--->~ ofsurvivalNFkB
1
Death > (substratest
cytochrome C
i f--Bd-2/ Bd-xL
mitochondria
/ "'" Bc12o,!' ~ independent
fADD ~(----
J. ~ YVADfLICE
1-t'DEVD
I
Bc12dep endent
Daxx
J.JNK.j,Jun
J.CmJAp35XIAP
I I Caspase activation
/ tBax.ROS
tp53
Intrac el1ular stimuli t~ DNA damage
Extrac el1u1ar stimuli ?(Anticancer drugs etc)
l'!eurodegenerativediseases, ischemia, HIV,1'leuroblastomas etc.
Growth factors,cytokinesTNF and cellular stress
TNF
1---.......jl-------------- fas TNFR _
1DISCproteins
fas L
1----+-------------
Fig. 1: Interlationship between caspases, apoptosis and diseases. Fas L, fas ligand; Fas, fas receptor; TNF, tumornecrosis factor; TNFR, TNF receptor; DISC, death inducing signalling comples; FADD, fas associateddeath domain; JNK, Jun N terminal kinase; FLICE, FADD - like ICE (Interleukin 1 ~ converting enzyme);ROS, reactive oxygen species, NFkB, Nuclear factor kappa B; YVAD, DEVD, tetra peptide inhibitors forcaspases.

156 Kharma and Singh
sword. If this system is properly regulated,it is useful for downregulating the immunereaction and for removing virally infectedas well as cancerous cells; but, if this systemis exaggerated, it can cause tissuedestruction. It means this system can bemodulated to be applied to human diseases.It can be applied in the killing of tumorcells, since some cancer cells, particularlysome lymphoid tumors, express functionalFas. However, since the systemic treatmentof patients with Fas L will cause deleteriousside affects, methods of local administrationand/or proper targeting of Fas L to thetumor should be devised. Fas L can also beused as immunosuppressive agent since therejection of grafts is mediated by activatedT cells. If a transplanted tissue isengineered to express Fas L or iscotransplanted with Fas L- expressing cells,the transplant may be tolerated. The otherapplication of this system is to block Fas Linduced tissue destruction. As Fas is shownto playa role in human diseases, thereforeneutralizing antibodies against Fas or FasL, or other inhibitors of Fas mediatedapoptosis, would have potential astherapeutic agents.
Literature available suggests that TNFreceptor shares a signal cascade with Fasin one apoptotic pathway, but it alsoactivates additional signalling pathwaysincluding one that activates a survivalsignal. It will be interesting to examinewhat kinds of survival genes are activatedby NF-kB and how these molecules inhibitapoptosis. Identification of these survivalgenes may provide clues as to why sometumor cells are resistant to variousapoptosis inducing agents including Fas L,TNF, and other anticancer drugs. Report
lndian J Physiol Pharmacol 1999; 43(2)
by Wissing et al (41) has proved that TNFinduced activation of caspases resultsin the cleavage and activation of cytosolicphospholipase Az (cPLAz) and the activatedcPLA z contributes to apoptosis (amechanism similar to the one used by nonsteroidal anti inflammatory drugs,(NSAIDj, which are widely being used inmedications).
DYSREGULATED CELL DEATH AND APOPTOSIS
BASED THERAPIES.
There are several diseases/disordersIn which apoptosis is dysfunctional.Huntington's disease (HD) was the firstdisorder in which caspase cleavage of aspecific substrate has been linked to adisease pathology. In HD polyglutaminetract of Huntington (ubiquitously expressedprotein of unknown function) is cleaved incells or apoptotic extracts by caspase 3 andthe rate of cleavage increases with thelength of its polyglutamine tract.
Apoptotic cell death and its attendantmolecular mediators also appear to playarole in many neurodegenerative disorders,including Alzheimer's disease. Mutationsin the presenillin 2 gene have recently beenassociated with familial Alzheimer diseaseand is hypothesized to function in a pathwaydownstream of Fas. In primary cultures ofhuman neurons, peptide fragments ofamyloid ~ can down regulate antiapoptoticBcl-2 and up regulate proapoptotic Baxexpression, thus making the neurons moreprone to die especially in response tooxidative stress. Apart from this,involvement of ICE like proteases has beenproved in the bone marrow of patients withMyelodysplastic syndrome, neuroblastomas,

Indian J Physiol Pharmacol 1999; 43(2)
myelomas, heart failure, autoimmune /lymphoproliferative syndromes (ALPS) etc.(42-45).
Thus the ability to block apoptosisrepresents an exciting possibility forextending the productive lifetimes of cells.Experiments have already been performedin some cases for e.g. : it has been reportedthat the use of ICE like and CPP32 likeprotease inhibitors reduces ischemic andexcitoxic neuronal damage which isimplicated in stroke and neurodegenerativedisorders (46-47). T Leukemia cells andperipheral blood mononuclear cells exposedto HIV-l undergo enhanced viral replicationin the presence of cell death inhibitor(z VAD-fmk), thereby sugges bng thatprogrammed cell death may serve as abeneficial host mechanism to limit HIVspread (48).
Apoptosis also plays an important rolein cancer therapy. Most leukemic cells arewell known to undergo apoptosis by severalevents, and the basic strategy of leukemiatherapy is the induction of apoptosis. Forexample, many chemotherapeutic agentssuch as cytosine arabinoside and VP16induce apoptosis and kill leukemic cells.On the other hand, Fas ligand and TNF (1.,
which physiologically exist in the body, arealso inducers of apoptosis in leukemia cellsand other cells. Although previous reportshave shown that Bcr-Abl expression confersresistance against the antileukemicdrug induced DNA fragmentation andmorphological features of apoptosis, arecent report by Amarante et al (49) usingAML (acute myelogenous leukemia) HL-60cells has proved that treatment withantileukemic drugs causes the release of
Caspases in Apoptosis and Disease 157
cytochrome C, induces the activity of DFF(DNA fragmentation factor), while Bcr-Ablexpression results in the inhibition of thepreapoptotic mitochondrial perturbationthereby blocking the generation of caspaseactivity and apoptosis.
Many of the events in Fas signalling areregulated by protein - protein interactions.In a recent report by MacCorkle et al (50),they have used caspases (caspase 1 andcaspase 3) as ideal targets for designingconditional alleles based on chemicallyinduced dimerization. In both casesaggregation of the target protein is achievedby a nontoxic lipid permeable dimeric FK506analog that binds to the attached FK506binding proteins, FKBPs. It has also beenfound that crosslinking of caspase-land caspase-3 is sufficient to triggerrapid apoptosis in a Bcl -XL independentmanner, suggesting that these conditionalproapoptotic molecules can bypass intracellular checkpoint genes, such as Bel-xLthat limit apoptosis. Because these chimericmolecu les are derived from autologousproteins, they are nonimmunogenic andthus ideal for long-lived gene therapyvectors, which will be useful fordevelopmental studies, for treatinghyperproliferative disorders, and fordeveloping animal models to a wide varietyof diseases.
With new information on programmedcell death constantly being elucidated, celldeath promises to be a lively area of futureinnovation. The long term challenge in basicresearch is to understand the course ofcaspase activity during animal development,homeostasis and pathology, in therapy to

158 Khanna and Singh
develop drugs targeted to specific cells andtissues, and to block acute and chronic
Indian J Physiol Pharmacol 1999; 43(2)
diseases through intervention in caspasenetwork.
REFERENCES
1. Kerr JFR, Wyllie AH, Currie AR. Apoptosis: Abasic biological phenomenon with wide-rangingimplications in tissue kinetics. Br J Cancer 1972;26: 239-257.
2. Jacobson MD, Weil M, Raff MC. Programmed celldeath in animal development. Cell 1997; 88: 347354.
3. Benoist C, Mathis D. Cell death mediators inautoimmune diabetes-no shortage of suspects. Cell1997; 89: 1-3.
4. Stellar, H. Mechanisms and genes of cellularsuicide. Science 1995; 267: 1445-1449.
5. Azmi S, Bhatia L, Khanna N, Dhawan D, Singh N.Adriamycin induces Apoptosis in rat thymocytes.Cancer Letters 1997; Ill: 225-231.
6. Jacobson MD, Evan GI. Apoptosis and EIV disease.Nat Med 1995; 1: 386.
7. Petisch MC, Polzar B, Stellar H, Crompton T,MacDonald HR, Mannherz HG, Tschopp J.Characterization of the deoxyribonuclease involvedin nuclear DNA degradation during apoptosis(Programmed cel! death). Embo J 1993; 12: 371377.
8. Barry M. Eastman A. Identification ofdeoxyribonuclease II as an endonuclease involvedin apoptosis. Arch Biochem Biophys 1993; 300:440-450.
9. Chinnaiyan AM, Dixit VM. The cell death machine.CUI"!" Bioi 1996 ; 6 : 555-562.
10. Vaux DL, Strasser A. The molecular biology ofapoptosis. Proc. Natl. Acad Sci 1996; 83: 2239-2294.
11. White E. Life, death and pursuit of apoptosis.Genes Dev 1996; 10: 1-15.
12. Nicholson DW, Thornberry NA, Caspases-KillerProteases TIBS 1997; 22: 299-306.
13. Ellis RF, Yuan Y, Horvitz R, Mechanisms andfunctions of cell death. Annu Rev Cell Bioi 1991;7: 663-698.
14. Horvitz HR, Shaham S, Hengartner MO. Thegenetics of programmed cell death in the nematodeCaenorhabditis elegans. Cold Spring Harbor SympQuant Biol 1994; 56: 377-385.
15. Hengartner MO, Ellis RE, Horvitz RH.Caenorhabditis elegans gene Ced-9 protects cells
from programmed cell death. Nature 1992; 35G:484-499.
16. Yuan J, Shaham S, S. Ledoux S, Ellis lIM, HorvitzHR. The C. elegans cell death gene Ced-3 encodesa protein similar to mammalian interleukin-1-pconverting enzyme. Cell 1993; 75: 641-652.
17. Alnemri ES, Livington DJ, Nicholson DW, G.Thornbery NA, Wong WW, J. Human ICE/CED-3protease nomenclature. Cell 1996; 87: 171.
18. Martin SJ, Green DE. Protease activation duringapoptosis : Death by a thousand cuts? Cell 1995;82: 349-352.
19. Chinnaiyan AM, Ortho K, O'Rourke K. Duan H,Poirer GG, Dixit VM. Molecular ordering of thecell death pathway:' BcI-2 and Bcl-xL functionupstream of the Ced-3 like apoptotic proteases JBiol Chern 1996; 271: 4573-4576.
20. Fraser A, Evan G. A license to kill. Cell 1996; 85:781-784.
21. Henkart PA. ICE family proteases: mediators ofall apoptotic cell death? Immunity 1996; 4: 195201.
22. Patel T, Gores GJ, Kaufmann SH. The role ofproteases during apoptosis. FASEB J 1996; 10:587-597.
23. Salvesan GS, Dixit VM. Caspases: Intracellularsignalling by proteolysis. Cell 1997; 91: 443-446.
24. POl'ter AG, Patrick NG, Janicke RU. Deathsubstrate comes alive. Bioassays 1997; 19: 501-507.
25. Cohen GM. Caspases : the executioners ofapoptosis. Biochem J 1997; 326: 1-16.
26. Vilta P, Kaufmann SH, Earnshaw WC. Caspasesand caspase inhibitors. TIBS 1997; 22: 388-392.
27. Robertson NM, Zangrilli J, Alnemri TF, FriesenPD, Litwack G, Alnemri ES BaculovirusP35 inhibits the glucocorticoid-mediated pathwayofcel! death. Cancer Res 1997; 57: 43-47.
28. Ray CA, Black R, Kronheim SR, Greenstreet TA,Sleath PR, Salves an GS, Pickup DJ. Viralinhibition of inl1ammation : cowpox virus encodesan inhibitor of the interlukin 1-P convertingenzyme. Cell 1992; 69: 597-604.

Indian J Physiol Pharmacol 1999; 43(2)
29. Bump NJ, Hackett M, Hugunin M, Seshagiri S,Broady K, Chen 1', Frerenz C, Franklin S, GhayurT, Li 1', Lican 1', Mankovich NSL, Greenberg AB,Miller LK, Wong WW. Inhibition of ICE familyproteases by baculovirus antiapoptotic protein 1'35.Science 1995; 269: 1885-1888.
30. Beidler DR, Tewari M, Friesen I'D, Poirier G, DixitVM. The baculovirus 1'35 protein inhibits fas-andtumor necrosis factor induced apoptosis. J BiolChern 1995; 270: 16526-16528.
31. Deveraux QL, Takahashi R, Salvesen GS, ReedJC. X-linked lAP is a direct inhibitor of cell deathproteases. Nature 1997; 388: 300-304.
32. Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL,Korsmeyer SJ. Bcl-2 functions in an antioxidantpathway to prevent apoptosis. Cell 1993; 75: 241251.
33. Kane DJ, Sarafian TA, Anton R, Hahn H, GrallaEB, Valentine JS, Ord T, Bredesen DE. BcI-2inhibition of neural death: decreased generationof reactive oxygen species. Science 1993; 262: 12741277.
34. Kroemer G. The protooncogone Bcl-2 and its rolein regulating apoptosis. Nature Med 1997; 3: 614620.
35. Cheng EHY, Kirsch DH, Clem RJ, Ravi R, KastanMB, Bedi A, Uneo K, Hardwick JM. Conversion ofBcl-2 to a Bax-like death effector by caspases.Science 1997; 278: 1966-1968.
36. Thompson CB. Apoptosis in the pathogenesis andtreatment of disease. Science 1995; 267: 1456-1462.
37. l30Jdin MP, Goncharov TM, Goltsev YV, Walach D.Involvement of MACH, a novel MORT l/FADDinteracting protease in Fas/APO-1 and TNFreceptor induced cell death. Cell 1996; 85: 803815.
38. Muzio M, Chinnaiyan AM, Kishkel FC, 0' RourkeK, Shevchenko A, Scaffidi C, Zhang M, Ni J, GentzR, Mann M, Krammer I'll, Peter l'vIE, Dixit VM,FLICE : A novel FADD-homologous ICE/CED-3like protease is recruited to the CD95 (Fas/APO-1)death inducing signalling complex. Cell 1996; 85:817-827.
39. Nagata S. Apoptosis by Death Factor. Cell 1997;88: 355-365.
40. Krajewski M, Wang HG, Krajewski S. Zapata JM,Shabaik A, Gascoyne R, Reed ,rc. Immunohistochemical analysis of invivo patterns of expressionof CPP32 (caspase 3), a cell death protease. CancerResearch 1997; 57: 1605-1613.
Caspases in Apoptosis and Disease 159
41. Wissing D, Mouritzen H, Egeblad M, Poirier GG,Jaatela M. Involvement of caspase-dependentactivation of cytosolic phospholipase A2 in tumornecrosis factor-induced apoptosis. Proc Nail AcadSci USA 1997; 94: 5073-5077.'"
42. Mundie S, Venugopal 1', Cartlidge JD, Pandav DV,Robinson L, Gezer S, Robin EL, Rifkin SR, KleinM, Alston DE, Hernandez BM, Rosi D, Alvi S,Shetty VT, Gregory SA, Raza A. Indication of aninvolvement of Interlukin 1-~ converting enzymelinked protease in intramedullary apoptotic celldeath III the bone marrow of patientswith Myelodysplastic syndromes. Blood 1996; 88:2640-2647.
43. Nakagawara A, Nakamura Y, Ikeda T, Kuida K,Michael S-S. Su, Zhao H, Cnaan A, and SakiyamaS. High Levels of Expression and NuclearLocalization oflnterleukin-I ~. Converting Enzyme(lCE) and CPP32 in Favorable HumanNeuroblastomas. Cancer Res 1997; 57: 4578-4584.
44. Marlangue CC, Zouaoui A, Represa A, Ari BY.Apoptotic features of selective neuronal death inischemia, epilepsy and gp 120 toxicity. Trends inNeuro Sci 1996; 19: 109-114.
45. Nicholson DW. ICE/CED-3 like proteases as targetsfor the control of inappropriate apoptosis. NatBiotech 1996; 14: 297-301.
46. Hara H, Friedlander RM, Gagliardini V, Ayata C,Fink K, Huang W, Sasamate MS, Yuan J.Mosk owi tz MA. Inh i bi tion 0 f in te rlellk i n 1 ~
converting enzyme family proteases reducesischemic and excitotoxic neuronal damage. ProcNail Acad Sci USA 197; 94: 2007-2012.
47. Holtzman DM, Deshmllkh M. Caspases : Atreatment target for nellrodegenerative diseases.Nat Med 1997; 3: 954-955.
48. Chinnaiyan AM, Dixit VM, Nabel GJ. The inhibitionof pro-apoptotic ICE-like proteases enhances HIVreplication. Nat Med 1997; 3: 333-337.
49. Mendes GPA, Kim CN, Huang Y, Perkins CL,Green DR, Bhalla K. Bcr-Abl exerts itsantiapoptotic effect against diverse apoptoticstimuli through blockage of mitochondrial releaseof cytochrome c and activation of caspse3. Blood1998; 91: 1700-1705.
50. MacCorkle RA, Freeman KW, Spencer DM,Synthetic activation of caspases: Artificial deathswitches. Proc Nail Acad Sci USA 1998; 95: 3655
3660.