resonances in molecular photoionization. iv. theory of one-color and two-color near-threshold...
TRANSCRIPT

Resonances in molecular photoionization. IV. Theory of onecolor and twocolor nearthreshold photoionization of moleculesW. Domcke, A. L. Sobolewski, and S. H. Lin Citation: The Journal of Chemical Physics 89, 6209 (1988); doi: 10.1063/1.455438 View online: http://dx.doi.org/10.1063/1.455438 View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/89/10?ver=pdfcov Published by the AIP Publishing Articles you may be interested in Near-threshold shape resonance in the photoionization of 2-butyne J. Chem. Phys. 136, 154303 (2012); 10.1063/1.3701762 Near-threshold photoionization of hot isopropyl radicals J. Chem. Phys. 124, 114312 (2006); 10.1063/1.2172611 Model study of near-threshold photoionization of large molecules: The effect of vibrational relaxation J. Chem. Phys. 106, 3174 (1997); 10.1063/1.473059 Twocolor resonance photoionization of aromatic molecules in solid argon J. Chem. Phys. 76, 5005 (1982); 10.1063/1.442847 Twocolor photoionization of naphthalene and benzene at threshold J. Chem. Phys. 75, 2118 (1981); 10.1063/1.442315
This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

Resonances in molecular photoionization. IV. Theory of one-color and twocolor near-threshold photoionization of molecules
W.Domcke Institute 0/ Physical and Theoretical Chemistry. Technical University 0/ Munich. D-8046 Garching. West Germany
A. l. Sobolewski Institute 0/ Physics. Polish Academy o/Sciences. 02-668 Warsaw. Poland
S. H. Lin Department o/Chemistry. Arizona State University. Tempe. Arizona 85287-1604
(Received 14 June 1988; accepted 5 August 1988)
A relatively comprehensive theoretical description of one-color and two-color photoionization of molecules by strong laser fields is developed. The molecular system is modeled by a number of discrete electronic configurations and a number of electronic ionization continua in a diabatic representation, allowing for intramolecular coupling of the discrete states and the continua. The vibrational degrees of freedom are included in compact operator notation without invoking the Bom-Oppenheimer approximation. The relevant radiative dipole couplings are treated nonperturbatively on an equal footing with the intramolecular couplings. The important Coulomb threshold effects (accumulating Rydberg series at the electronicvibrational ionization thresholds) are included via a threshold expansion of the complex levelshift operators representing intramolecular and radiative couplings. The weak-field, long-time ionization rate (golden-rule formula) is rederived from the general theory both in the onecolor and the two-color case. In the two-color case, strong-field effects caused by either one of the two lasers are briefly discussed. We derive a simplified multichannel-quantum-defect model from the general two-color formalism which nicely reproduces a number of characteristic features of vibronic autoionization recently observed in several polyatomic molecules.
I. INTRODUCTION
Recent experimental work has demonstrated the versatility of two-color resonance-enhanced photoionization spectroscopy for probing highly excited neutral states as well as the lowest ionic states of polyatomic molecules. I- 7 In these experiments, a laser with frequency WI excites the molecular system to a specific vibrational level of a low-lying excited electronic state (usually the S I state). A second laser with tunable frequency W 2 ionizes the excited species. In the near-threshold region, one often observes pronounced autoionization structure caused by high members of Rydberg series converging to higher-lying vibrational ionization thresholds.3
•s-7 Recent work has revealed a surprising mode
specificity of vibronic autoionization in some polyatomic molecules, that is, only very few vibrational modes appear to be active in promoting vibrational autoionization.s.7
Clearly, any theoretical model devised to describe these spectroscopic results has to include the infinite Rydberg series converging to the various ionization thresholds. It is well known, moreover, that in molecules the Rydberg series and the adjoining ionization continua are often strongly perturbed by doubly excited valence configurations or shape resonances, see, e.g., Refs. 8 and 9. The standard theoretical scheme to describe the perturbation and autoionization of Rydberg series is the well-known multichannel-quantumdefect theory (MQDT), see Refs. 10-13 for reviews. While the MQDT has been extensively and very successfully applied to describe vibrational and rotational autoionization as
well as predissociation of Rydberg series in diatomic molecules, 14-16 it has so far found only limited application for the Rydberg spectra of poly atomic molecules. 17.18
Recently, two of us have developed a somewhat different theoretical formulation of near-threshold photoionization based on the projection-operator formalism of scattering theory.19.20 In its most general form,20 the theory includes an arbitrary number of discrete (valence) electronic configurations and an arbitrary number of electronic ionization continua with associated infinite Rydberg series, as well as the vibrational degrees offreedom. While the radiation-matter interaction is treated in lowest order (goldenrule formula), the discrete-continuum interaction and the resulting indirect Rydberg-Rydberg, Rydberg-continuum, and continuum-continuum couplings are included to infinite order without invoking the Bom-Oppenheimer (BO) approximation. The equivalence of the projection-operator formulation with the MQDT has been explicitly established in this rather general context. 20
In the present work, we wish to extend the multistatemultichannel formalism of Ref. 20 towards a nonperturbative treatment of the radiative couplings. Since the existing weak-field formulation20 is based on an extensive use of projection operators and resolvent operators, it can be straightforwardly merged with the well-established nonperturbative resolvent-operator formalism of matter-field interactions.21•22 The radiative couplings are thus treated on an equal footing with the strong intramolecular couplings.
The present formulation extends the basic work of Lam-
J. Chern. Phys. 89 (10). 15 November 1988 0021-9606/88/226209-11 $02.1 0 © 1988 American Institute of Physics 6209 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

6210 Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV
bropoulos and Zoller3 for strong-field ionization of atoms by the inclusion of the vibrational degrees of freedom of molecules as well as by a proper treatment of the important Coulomb threshold effects. The present theory can also be viewed as an extension of recent work on the strong-field ionization of the hydrogen atom where threshold effects have been properly included. 24
-29 The extension in this case
consists in the inclusion of many electronic ionization channels, their coupling with many discrete configurations, as well as the inclusion of the vibrational motion beyond the BO approximation. Finally, the present theory extends recent theoretical work on the multiphoton ionization of molecules based on the density-matrix method3
O-32 by the proper
treatment of Coulomb threshold effects. It is convenient for the presentation of the theory to
consider first the (hypothetical) case of one-color strongfield photoionization. It will be shown how the long-time weak-field ionization rate can be derived from the general strong-field ionization amplitude, thus recovering previous results. 19
,20 In a second step, we shall slightly extend the model to derive a fairly comprehensive description of twocolor resonance-enhanced near-threshold photoionization. Compact formal expressions for the ionization rate will be derived in the various weak-field limits, that is, low intensity of either of the two lasers or of both lasers.
The practical applicability of the theoretical scheme will be demonstrated by modeling the recently discovered phenomenon of strong vibrational autoionization via specific nontotally symmetric modes in polyatomic molecules.5
•7
For this purpose, the molecular multistate multichannel model is reformulated in the language of the MQDT, introducing a phenomenological reactance matrix which couples electronic ionization channels. It will be shown that an electronic two-channel model with vibronic coupling can satisfactorily account for the vibrational autoionization phenomena recently observed in aniline and pyrazine. 5,7
II. ONE-COLOR PHOTOIONIZATION
A. The Hamiltonian
The molecular Hamiltonian and the interaction of the molecule with the radiation field are represented in a diabatic electronic basis. Here the term "diabatic" means that the action of the nuclear kinetic-energy operator on the electronic wave functions can be neglected for all practical purposes.33
-35 The electronic basis states required for a general
model of one-color photoionization are the electronic ground state Ig) of the molecule, a manifold of N discrete (valence) excited states 1 d ), and a manifold of M ionization continua (plus associated Rydberg series) Vk). Depending on the specific situation, the discrete states Id) may represent doubly excited valence configurations, low members of Rydberg series converging to higher ionic thresholds, or shape resonances. The molecular Hamiltonian is assumed to be prediagonalized within the discrete manifold and within the ionization continua. There remains, however, an electronic coupling between the discrete excited states Id) and the ionization continua (and Rydberg series) Vk), represented by the coupling matrix element Vjk,d' We assume, for
simplicity, that the ground state Ig) is not coupled to other states by intramolecular interactions.
The molecular Hamiltonian thus reads
(2.la) N
Ho = Ig)Hg (gl + L Id )iId (d 1 d=1
M
+ L L Vk )(ilj + Ek)Vk I, (2.lb) j= 1 k
v = L Vk) Vjk,d(d 1 + h.c. jkd
(2.lc)
The basis states Vk) representing the ionization continua and the Rydberg series are defined as products of the ion-core states V) and the single-particle Coulombic states Ik ), i.e.,
Hion V) = EjV),
Helk) =Eklk).
(2.2a)
(2.2b)
Hion and He are the electronic Hamiltonian of the ion and the Coulomb Hamiltonian, respectively. The total electronic energy of the ionization continuum Vk ) is
Ejk = Ej + Ek, (2.2c)
where Ej is the electronic energy of the ion core (still a function of the nuclear coordinates) and E k represents the energy of a continuum electron for Ek > 0, or stands for the energy of a Rydberg state relative to the ionization threshold for E k < O. The phase of the Coulombic wave functions and the energies of the Rydberg states may include a constant (that is, independent of energy and internuclear distances) quantum defect as specified below.
The Hamiltonians
iIi = TN + Ei> i = g, d,j (2.3)
in Eq. (2.lb) are the Hamiltonians for nuclear motion in the corresponding electronic states. TN is the nuclear kineticenergy operator, and Ei is the electronic potential energy of state I i) which is a function of the nuclear coordinates.
We describe the radiation field quantum mechanically and take as the total (time-independent) Hamiltonian
H=HM + H rad +D, (2.4)
where Hrad is the Hamiltonian of the radiation field and D represents the molecule-field interaction. The eigenstates of H rad are the usual photon-number states In). Only one mode of the field with frequency OJ is considered.
To describe one-photon ionization of molecules initially in the electronic ground state Ig), it is sufficient to consider the dipole coupling of Ig) with the excited electronic configurations Id) and Vk ). We therefore neglect radiative transitions within the discrete manifold {Id ) }, within the Rydberg series, as well as within the electronic continua. The molecule-field coupling D is thus given by
D= L Vk)Djk,g(gl + L Id)Ddg(gl + h.c., (2.5) jk d
where Djk,g and D dg are the dipole transition moments for transitions from the electronic ground state Ig) to the ionization continua (plus Rydberg series) and to the discrete states, respectively.
J. Chem. Phys., Vol. 89, No. 10, 15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV 6211
Ij k> Ij'k>
FIG. 1. Schematic electronic energy-level scheme of the model for one-color photoionization. The level designated by Ig) represents the electronic ground state, {I d ) } represents the highly excited discrete electronic manifold, and {lik ) } are electronic ionization continua with associated Rydberg series. Dipole couplings are indicated by arrows, intramolecular electronic couplings by wavy lines.
Figure 1 indicates schematically the electronic level scheme (for a fixed nuclear configuration) and the intramolecular and radiative couplings considered in the present model.
B. Calculation of the Ionization amplitude
The initial state of the system is Ig)ln)IO), where (the superscript enumerates the individual vibrational modes)
10) = 10(1),0(2), ... )
denotes the vibrational ground state of the molecule in the electronic ground state Ig) (the generalization of the formalism to consider initially vibrationally excited molecules is trivial and not further elaborated here). In the well-known rotating-wave approximation36 (R W A), the states connected to the initial state via a single-photon process are Id) In - 1) 10) and lik) In - 1) 10) (for a discussion of nonRWA effects, see for example, Ref. 37). Assuming that the interaction with the field is turned on instantaneously at time t = 0, the full time-dependent wave function can thus be expanded as21,23
1'I1(t» = Ugg(t)lg)ln)IO) + L Udg(t)ld)ln -1)10) d
+ L Ujk,g (t) lik) In - 1) 10), jk
where (Ii = 1)
Uab(t) = (ale-iHtlb), a,b=g,d,jk.
Introducing, as usual,21 the full resolvent
G(z) = (z - H)-I, the matrix elements Uab (t) may be expressed as
Uab(t) =~Joo dEe-iE'Gab(E+i'TJ) 2'm - 00
(2.6a)
(2.6b)
(2.7)
(2.8a)
with
Gab (Z) = (aIG(z) Ib ) (2.8b)
and 'TJ a positive infinitesimal. Note that the matrix elements Uab (t) and Gab (z) are still operators acting in the Hilbert space of the vibrational degrees of freedom.
The amplitude of finding the system at the time t > 0 in the asymptotic continuum state lik ) with associated vibrational state Ivj ) = Ivj'),v]2), ... ) of the ion core is given by the projection
u;f.g(t) = (vjIUjk,g(t)IO) = (vjl(n - II(jk 1'I1(t»· (2.9)
The probability for ionization into continuum state lik ) and vibrational state IVj) by a rectangular pulse of duration Tis
Pjk,Vj (n = JU;f.g (n 12. (2.10)
The problem is thus reduced to the calculation of the (electronic and vibrational) matrix elements of the resolvent G(z) and the evaluation of the Laplace inversion integral (2.8a).
The electronic matrix elements of G(z) can immediatelybe calculated22,23 from the identify (z - H)G(z) = 1. For the present model, one obtains the system of operator equations
-Djk,gGgg - L ~k,d,Gd'g + (Z-Ek -Hj)Gjk,g =0. d'
As usual, the energy of the field containing n - 1 photons has been put to zero, since only energy differences matter.
Equations (2.11) can be solved for the matrix elements Ggg , Gdg , and Gjk,g which determine the wave function 1'I1(t» according to Eqs. (2.6) and (2.8). To express the solutions in compact form, we define the complex level-shift operators
- -I Fdd , (z) = L VdJk (z - Ek - Hj ) ~k,d"
jk
- -I Fgg(z) = LDgJk(z-Ek -Hj ) Djk,g'
jk
(2.12a)
(2.12b)
(2.12c)
(2.12d)
The summation over the continuum index k in Eq. (2.12) implies integration over the continuous spectrum as well as summation over the infinite Rydberg series. It is convenient, moreover, to introduce matrix-vector notation for the Ndimensional discrete electronic manifold {Id)}, which we shall call "Q space" in what follows (the complementary "P space" is spanned by the continuum states [jk ) ). Denoting the N XN matrix with elements Fdd , (z) by FQ(z), we define20
(2.13 )
J. Chem. Phys., Vol. 89, No. 10,15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

6212 Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV
where 1 is the N-dimensional unit matrix and HQ is a diagonal matrix with elements Hd • Introducing, furthermore,
A A
theN-dimensio~al vectors V~jk,DQ,g(z), and Dg,Q(z) with elements Vdjk , D dg (z), and Dgd (z), respectively, where
A
Ddg(z) =Ddg + Fdg(z), (2.14a) A
Dgd(z) =Dgd + Fgd(z), (2.14b)
we have the results
Ggg(z) = [z-m-Hg -Fgg(z)
-T G A ] 1 - Dg,Q(Z) Q(z)DQ,g(z) -, (2.15 ) - -I
Gjk,g (z) = (z - Ek - Hj ) ~k,g (z)Ggg (z), (2.16)
where
~k,g(Z) = Djk,g + VrukGQ(z)DQ,g(z). (2.17)
The ionization amplitude is finally given by
U vp (n =-I-f"" dEe-jET 1 ,k,g 2 . E IE'
7I'l - 00 - j - k - EYj + 1'TJ
X (vj 1 ~k,g (E + i'TJ)Ggg (E + i'TJ) 10), (2.18)
where we have defined
HJlvj) = (~ + Ev) Ivj ). (2.19)
~ is the (adiabatic) ionization potential of channelj and EYj
is the energy of vibrational state 1 Vj ). Equation (2.19) implies that we take the minimum of the potential energy surface of Ig) as the zero of the energy scale. It should be kept in mind that all energy-dependent quantities in Eqs. (2.12)(2.18) are still operators in vibrational Hilbert space.
While Eq. (2.18) is an essentially exact expression for the ionization amplitUde within the present model, its evaluation is far from trivial. It requires a careful examination of the energy dependence of the various resolvent matrix elements, in particular the complex level shifts of Eq. (2.12). They have the general structure
Fab (E + i'TJ) = fr (alX lik) (E - Ek -Hj + i'TJ)-1
X (jk 1 Ylb), (2.20)
where la), Ib) stand for either Id) or Ig) and X, Yrepresent either Vor D. The sum over the continuum can be evaluated in closed form by making use of the fact that Eq. (2.20) contains essentially the spectral resolution of the Coulomb Green's function in each channel. From a threshold expansion of the latter, taking account of the singularities introduced by the long-range Coulomb potential, one obtains the simple expression I9,20,38-40 (in atomic units)
M
Fab (E + i'TJ) = L Xaj{cj + 11" cot(i1T'iij + 11"ILjD»} ljb' j= 1
(2.21 )
where cj is a constant, ILJO) is a zero-order quantum defect for channel j, and
(2.22)
is still an operator in vibrational Hilbert space. The matrix elements X aj and ljb in Eq. (2.21) are defined as angular
averages, i.e.,
(2.23 )
and are essentially independent of energy near threshold (that is, within approximately one Rydberg of the ionization threshold). All essential energy dependences of the matrix element (2.21 ) are thus contained in the cotangent function. The constants cj in Eq. (2.21) represent energy-independent shifts and couplings of the discrete electronic states via their coupling (either radiative or intramolecular) to the electronic continua. Since these constant shifts and couplings do not alter the structure of the theory, they can be neglected for the present purposes. As discussed in detail in Ref. 20, the approximation (2.21) for the level-shift operators is equivalent to the well-known MQDT, which has been shown to be a very efficient scheme to parametrize Rydberg series and ionization continua in atoms and diatomic molecules. 10-16
With the approximation (2.21) all energy dependences in Eqs. (2.13 )-(2.18) are explicitly defined and the strong-
field ionization amplitUde U;t.g (n is calculable in principle. However, GQ(E + i'TJ) in Eqs. (2.15) and (2.17) contains multiply infinite series of poles representing autoionizing Rydberg series converging to the electronicvibrational ionization thresholds I j + EyJ' which contribute to the Laplace inversion integral in Eq. (2.18). Moreover, the long-time (T - (0) limit of the ionization probability does not exist and Pjk'
Yj (n has to be considered as a time
dependent quantity. The strong-field near-threshold ionization signal has been calculated for various models of the hydrogen atom by adopting a smoothing procedure for the Rydberg contribution. 2
4-29 Such calculations are probably not of primary interest for molecules with their much more complex level structures. Therefore, we confine ourselves in the following to the derivation of the weak-field long-time ionization rate from the general expression (2.18).
c. Weak-field limit
In the weak-field case, we may approximate Ggg (E + i'TJ) in Eq. (2.18) in zeroth order in the moleculefield interaction D, noting that ~k.g (E + i'TJ) defined in Eq. (2.17) is already of first order in D,
Ggg(z)::::;G~)(z) = (z-m-Hg)-I. (2.24)
We thus obtain
U~p (n =_I_foo dEe-jET 1 ,k.g 2' EI E . 7I'l - 00 - j - k - EYj + 1'TJ
where we have defined
HgIO) = EoIO),
Eo being the zero-point vibrational energy in the electronic ground state Ig).
The integral in Eq. (2.25) is now evaluated by standard residue integration.21 The path of integration may be closed in the lower half of the energy plane, and we obtain contribu-
J. Chern. Phys., Vol. 89, No. 10, 15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

Domcke, Sobolewski, and lin: Resonance in molecular photoionization.IV 6213
tions from the poles at I j + Ek + EYj
- hI, Ci.I + Eo - i1], as well as from the poles of ~k,g (z). The latter are given by the polXS of the resolvent GQ (z) [it can be shown that the poles ofDQ,g(z) are canceled by zeros ofGQ(z) in Eq. (2.17) J. GQ (z) possesses a very complicated pole structure, given explicitly by Eqs. (2.13) and (2.2 I ), with infinite series by Rydberg poles converging to the electronic-vibrational ionization thresholds as well as complex resonance poles associated with vibrational levels of the Id ) manifold. In the limit T --+ 00, however, we may neglect the contribution of all complex poles of ~k,g (z), since they are exponentially vanishing
contributions to U;t,g (n. Real poles of 1}k,8 (z) can only appear below the lowest electronic-vibrational ionization threshold. Since, in the case of photoionization, the total energy E exceeds the lowest ionization threshold, the real poles of ~k'8 (z) cannot lead to vanishing denominators in the limit 1]--+0 and their contribution can be neglected (see, e.g., Refs. 27 and 28 for detailed numerical studies of this effect for hydrogen-atom models). Neglecting, furthermore, as usual,21 the contribution of the branch cuts of ~k.g (z), we have
- 6.-1e-i(Ek-~+E",j)T{(vjl~k,g(Ek + ~ + Ev)IO)
- eit.T (vj l1}k,g (lU + Eo) 10)}, (2.26a)
where
6. = Ek + I j + EYj - Ci.I - Eo. (2.26b)
Applying the usual limiting procedure21
lim [1 - cos(6.T) ]/6.2 = 11"To(6.) , (2.27) T- oo
we obtain for the weak-field long-time ionization rate, defined as
_ r d IU VP 12 Yjk,vj - /.~ dT jk,8 ( n , the final result
Yjk'Vj = 211"1 (Vj I ~k,g (Ek + I j + Ev) lOW
Xo(Ek + I j + EVj - Ci.I - Eo).
(2.28)
(2.29)
The photoionization cross section is obtained by dividing through the photon flux. The transition matrix element in Eq. (2.29) is identical with Eq. (2.26) of Ref. 20.
The rate (2.29) is valid through lowest order in the molecule-field interaction, but is exact with respect to the intramolecular interactions included in the Hamiltonian (2.1). It includes the infinite Rydberg series, their interaction with the discrete states, as well as the resulting indirect coupling of Rydberg series and ionization continua to all orders. If the vibrational matrix elements of the operator ~k,g are exactly evaluated (see below), the theory includes the vibrational motion beyond the BO approximation and describes, nonperturbatively, the effect of vibrational autoionization of Rydberg series.
III. TWO-COLOR PHOTOIONIZATION
A. The Hamiltonian
We now extend the model developed in Sec. II such that we can describe the common experimental procedure of exciting a bound molecular energy level (typically a low vibrationallevel of SI) with a first laser of frequency Ci.l1, from which ionization is achieved with a second laser with tunable frequency Ci.l2•
For this purpose, we extend the electronic manifold introduced in Sec. II by an additional state la) which is radiativelycoupled to Ig) by Dga , as well as to Id) and lik) by Dad and D aJk' respectively. The total Hamiltonian is thus written as
H=Ho+ V+D+Hrad , ( 3.1a) N
Ho= Ig)Hg(gl + la)Ha(al + L Id)Hd(dl d=1
M
+ L L lik )(H) + Ek)(jk I, (3.1b) j= I k
v = L lik ) Jjk,d (d I + h.c., (3.1c) jkd
D = la)Dag (gl + Lid )Dda (al + L lik )Djk,a (al + h.c. d jk
( 3.1d)
The electronic level scheme is schematically indicated in Fig. 2. Brad now represents two field modes with frequencies Ci.I I and Ci.l2, respectively . We assume that Ci.I I is near resonance with the la)-Ig) electronic transition such that the ionization rate is dominated by this resonance. This allows us to neglect the radiative couplings of Ig) with the high-lying
Vjk/d (\ d» Vj'k,d "VVVV'o ------""""""
Ij k> Ij'k> ------ """"
D"k J ,a __ _ Djk,a ---
la>
__ --L-__ Ig>
FIG. 2. Schematic electronic energy-level scheme of the model for two-color photoionization. In addition to the levels and continua introduced in Fig. I, the level designated by 10> represents a low-lying excited electronic state which is resonantly excited by laser I with frequency llJ ,. The excited species are then photoionized by laser 2 with frequency llJ2' Dipole couplings are indicated by arrows, intramolecular electronic couplings by wavy lines.
J. Chem. Phys., Vol. 89, No. 10,15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

6214 Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV
excited electronic states and the ionization continua. As before, we neglect radiative transitions within the excited discrete manifold {Id)}, the Rydberg series, and the ionization continua. These approximations are appropriate for the laser intensities usually employed in multiphoton spectroscopy of molecules.
B. Calculation of the ionization amplitude
The initial state of the system is Ig) In\> n2 ) 10), where n I' n2 are the photon occupation numbers ofthe modes with frequency WI> W2' respectively. Within our model and the RWA, this state interacts with la) Inl - 1, n2 ) 10), Id) InJ -1, n2 -1) 10), and lik) In l -l, n2 -1) 10). The state la) Inl' n2 -0 10) in principle also interacts with the initial state in the RWA; however, the corresponding transition is assumed to be strongly off resonance and thus negligible compared to ionization via la) Inl - 1, n2 ) 10).
The operator equations for the electronic matrix elements of the full resolvent G(z) read
(z - Jig - WI - (2)Ggg - DgaGag = 1,
- DagGgg + (z - Jia - (2)Gag
- L DadGdg - '5' DaJkGjk,g = 0, d tt
- DdaGag + (z - Jid )Gdg - L VdJkGjk,g = 0, jk
(3.2)
In addition to the level-shift operators F dd' (z) defined in Eq. (2.12a), we introduce
(3.3a)
(3.3b)
(3.3c)
To write down the formal solutions to Eqs. (3.2) in compact form, it is useful to define the auxiliary Green's function [the bar indicates that this Green's function is not identical with the matrix element Gaa (z) = (aIG(z) la) J
A A
where Da,Q (z) is defined analogous to Dg.Q(z) in Eq. (2.14). We then have
where, in analogy to Eq. (2.17),
and
A
1jk,a(Z) = Djk.a + VQJkGQ(z)DQ,a(z) (3.6)
Ggg(z) = [z - Jig - WI - W2 - DgaGaa (z)Dag ] -I.
(3.7)
An alternative exact expression for Gjk,g (z) which is useful for the ensuing analysis, is obtained by application of the operator identity
- - -I Gaa (z)Ggg (z) = Gaa (z) (z - WI - W2 - Hg) ,(3.8)
where
Gaa(z) = [Gaa(z)-I- Taa(z)]-I (3.9)
and - -I Taa(z) =Dag (Z-W I -W2 -Hg) Dga' (3.10)
Note that Gaa (z) = (aIG(z) la) differs from the previously introduced Gaa (z) by the additional level-shift operator Taa (z). From Eqs. (3.5) and (3.8), we obtain
Gjk,g (z) = (z - Ek - ~) l1jk,a (z) Gaa (z)Dag - I X (z - WI - W2 - Hg)- • (3.11)
From either Eq. (3.5) or (3.11), we obtain the two-color photoionization amplitude (2) u;f.g (n by inverse Laplace transformation, i.e.,
(2)U~f' (n Jk,g
1 f"" =_ dEe iET ________ _
21Ti - '" E - ~ - Ek - EVj + i'YJ
X (vj 11jk,a (E + i'YJ)Gaa (E + i'YJ)DagGgg (E + i'YJ) 10)
(3.12)
and, equivalently,
(2)u~f'(n=-I-f"" dEe-iET 1 Jk,g 2' EI E .
'TTl - "" - j - k - EVj + I'YJ
X (Vj !1jk,a (E + i'YJ)Gaa (E + i'YJ)DagIO)
1 X----------------E - WI - W2 - Eo + i'YJ
(3.13 )
The strong-field two-color ionization probability for pulses of duration Tis
(3.14 )
With the threshold expansion (2.21) for the various levelshift operators, all energy dependencies are explicitly defined and Eqs. (3.12) and (3.13) represent calculable expressions.
C. Weak-field limits
Ifboth laser fields are strong such that their interaction with the molecule has to be treated nonperturbatively, it is not possible to define a time-independent long-time ionization rate. In this case, either Eq. (3.12) or (3.13) has to be evaluated directly as a function of time T. Such an analysis has been performed recently for a model of the hydrogen atom.27 A corresponding evaluation ofEqs. (3.12) or (3.13) for molecules appears to be too complicated to be of particular interest at present. We prefer to consider the various weak-field limits contained in the general expressions, that is, low intensity of either laser 1 (with frequency WI) or laser 2 (with frequency (2), or low intensity of both lasers.
J. Chern. Phys., Vol. 89, No. 10, 15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV 6215
1. Low intensity of laser 1
In this case it is convenient to start from Eq. (3.12) and approximate Ggg (z) in zeroth order in D,
Ggg(z) = Gi~l(Z) = (Z-W I -W2 -Hg)-I (3.15)
yielding
(2)U vJl (T) = _1_ J dE e - iET 1 Jk,g 2 . E I E
TTl - j - k - EVj + i7]
1 X __ --------------E - WI - w2 - Eo + i7]
X (vj I Tj~i (E + i7]) 10), (3.16)
where
(3.17)
The integration over energy now proceeds exactly as discussed in Sec. II. In the long-time limit, we obtain the twocolor ionization rate
rJf.!j = 217'1 (vj I TJf.i (Ek + Ij + Ev) lOW (3.18 )
The existence of a (time-independent) ionization rate is guaranteed by the fact that the population of the intermediate state 1 a) acts as a bottleneck for two-photon ionization.23
Strong-field effects caused by laser 2 are still included in the expression (3.18). These are represented by the radiative level-shift operator
_ A A
Faa (z) = Faa (z) + D~Q(z)GQ(z)DQ,a (z) (3.19)
contained in the resolvent Gao (z), reflecting the direct and indirect (via the discrete electronic manifold) radiative coupling of la) to the ionization continua.
Near ionization thresholds the amplitude (vj 1 T Jf.i (E + i7])IO) is a rapidly energy dependent quantity. It is explicitly calculable, however, with the same techniques which have been used to calculate (Vj l1)k,g (E + hJ) 10) in the one-color weak-field case. 19.20 Therefore, Eqs. (3.17) and (3.18) provide a useful extension of the weak-field theory to near-threshold photoionization by a strong laser field. It is obvious that the theoretical analysis is greatly simplified by the fact that all complex level-shift functions Fab (E + i7]) exhibit identical analytic structures near threshold, as given by Eq. (2.21).
2. Low intensity of laser 2
In this case, it is convenient to start from Eq. (3.13) and introduce the approximation
- -I Gaa (z)::::: [z - Ha - W2 - Taa (z)]. (3.20)
This implies neglect of the radiative level shift (3.19) caused by laser 2. Within the present model, where spontaneous emission and other (intramolecular) decay processes of la) are neglected, Gaa (z) ofEq. (3.20) possesses real poles. The level-shift operator Taa (z) describes the mixing of the vibrationallevels of Ig) and la) by the strong laser 1. The ionization amplitude (3.13) can be evaluated by diagonalizing the operator Haa + Taa (z) in the complete vibrational basis {Ivg )} ofHg [note that Tao (z) is diagonal in this basis in the Condon approximation]. In analogy to the atomic case,21.27
it can be shown that a long-time ionization rate exists and that the photoelectron spectrum reflects the splitting of the vibrational levels of la) owing to their strong radiative coupling with the Ig) levels by laser 1. At present, we shall not explore this type of strong-field mixing of discrete electronic-vibrationallevels any further.
3. Low intensity of both lasers
In this case, we immediately obtain from either Eq. ( 3 .12) or (3.13) the two-colorionization rate
rJf.!j = 217'1 (vj ITJf.i (Ek + ~ + Ev) lOW XD(~ + Ek + Ev. - WI - W 2 - Eo), (3.21)
J
where the bar on the transition amplitude indicates that it is taken in lowest order in both fields
TJf.i(z) = 1)k.a(z)(z-Ha -(2 )-IDag (3.22)
with 1)k.a(Z) defined in Eq. (3.6). Introducing the vibrational eigenstates IVa) of Ha,
HalvQ) = (Eo + Ev)lva ), (3.23)
we have the final result for ionization via the intermediate vibrational level Va of electronic state la) (assuming the Condon approximation for the vibrational matrix element of Dag ),
rJ:~al = 217'1 (vj l1)k.a (Ek + ~ + EVj) IVa >J2
X IDag 12 2 1 (Va 10) 12 (WI + EO - Ea - EVa)
XD(lj + Ek + EVj - WI - W2 - EO)' (3.24)
Equation (3.24) exhibits explicitly the resonance condition for laser 1 (for vanishing detuning, the usual phenomenologicallinewidth parameter ra of the vibrational levels of la) has to be introduced). It gives, moreover, the ionization rate as a function of the frequency W 2 of the second laser, as it is tuned through the ionization thresholds. It is seen that for fixed frequency WI' the dependence of t 2l on the frequency W2 is given by the one-color rate expression ofEq. (2.29).
Equation (3.24) provides the basis for our analysis of near-threshold laser photoionization spectra of polyatomic molecules presented in the next section.
IV. MQDT MODEL FOR VIBRONIC AUTOIONIZATION IN POL YATOMIC MOLECULES
A. Definition of the model
As mentioned in the Introduction, rather regular and mode-specific vibrational autoionization phenomena have recently been observed at the lowest electronic ionization threshold of several polyatomic molecules with two-color photoionization.5-
7 Aniline and pyrazine, in particular, show a strong propensity for vibrational autoionization via a specific non-totally symmetric mode.s.7
Let us try to specify the simplest molecular model which can account for the experimental observations. This model has to comprise at least one electronic ionization continuum and some particular non-totally symmetric vibrational mode. Let us assume that the ionization continuum (de-
J. Chem. Phys., Vol. 89, No. 10. 15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

6216 Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV
noted by I/,k ) ) transforms according to the irreducible representation r I in the molecular point group and that the vibrational mode is of symmetry r 0' The overall symmetry of the ionization continuum with excitation of one vibrational quantum is thus r I ® r ° = r 2' Therefore, the v = I ionization continuum will generally interact with discrete (valence) molecular states {Id)} of symmetry species r 2 via vibronically induced coupling, which in the lowest order expansion in vibrational normal coordinate q is given by V1k,d
= V7k,dq. On the other hand, there is also at least one electronic ionization continuum based on the same state of the ion, but transforming according to r 2 (we denote this continuum by 12,k». This continuum will interact with the abovementioned set of discrete states of r 2 symmetry due to electronic coupling V2k,d = V~k,d' but in general also withasetofdiscretestates {Id ')} of symmetry r 2 ® r o = r l
via vibronic coupling V2k.d. = V~k.d' q. The discrete states of symmetry r I are in turn electronically coupled to the ionization continuum of this same symmetry with the coupling element V1k,d' = V7k,d" The interaction cycle is thus closed and we see that a minimal model of polyatomic vibronic autoionization should contain at least two ionization continua of different orbital symmetry, two sets of discrete molecular states of the appropriate symmetries, and a non-totally symmetric vibrational mode which induces the coupling between the two symmetry spaces. To complete the model, we have to include the electronic ground state Ig) and the intermediate electronic state la). To simplify the analysis, we assume that radiative transitions from la) are only allowed to electronic states of symmetry r I in the present model, i.e., the set {Id)} and the continuum 12,k) are assumed to be dark in absorption from the intermediate state la).
Since there are no indications of resonant discrete interlopers in the experimental spectra,5-7 a simplified MQDTtype description of the problem is appropriate, where the impact of the discrete manifold on the Rydberg series and ionization continua is condensed into an approximately energy-independent reactance matrix.
The reformulation of the projection-operator theory in the language of MQDT has been discussed in detail in Ref. 20. It is convenient for this purpose to introduce matrixvector notation also for the ionic channel manifold enumerated by the index j. We recall, first of all, that the matrix elements D jk.a, fjk.d' etc. are essentially independent of the electron wave number k near threshold. By confining ourselves, furthermore, to the calculation of angle-integrated photoionization cross sections, it is sufficient to consider angle-averaged matrix elements as defined in Eq. (2.23).20 We thus introduce the M-dimensional vector D P.a with (angleaveraged) elements Djk,a,j = 1, ... M, the N XM matrix V with (angle-averaged) elements fjk.d' j = 1, ... M, d = 1, ... N, as well as the M-dimensional vector T(z) with (angle-averaged) elements 1}k.a (z), j = 1, ... M. We then have the compact expression20
T(z) = C-I(z)Gp(z)Dp,a(z) (4.1)
with
(4.2)
A - -I DP,a(z)=Dp,a+V(zl-HQ) D Q.a, (4.3)
where HQ and DQ,a have been introduced in Sec. II B. C(z) is the M X M diagonal matrix with elements [cf. Eqs. (2.21) and (2.22)]
Cii(z) = 17' cot (hrvj + 1T,uJO» (4.4)
and R(z) is the electronic reactance matrix given by
R(z) = V(zl- HQ)-IV+. (4.5)
All rapid energy dependences owing to accumulating Rydberg series are now contained in the resolvent matrices C(z) and Gp (z) which are still operators in vibrational Hilbert space. The vectors D p,a,D Q,a and the matrix V are possibly dependent on the vibrational coordinates, but are energy independent to a good approximation.
For the minimal two channel model introduced above, Eq. (4.1) reduces to
[T1(Z)] = [17'-1 tan(i1Tii) 0 ] T2 (z) 0 17'-1 tan(i1Tii)
X [Gl1 (Z) GI2 (Z)][D I (Z)] (4.6) G21 (Z} G22 (Z) 0 '
where the Gij (z) are the elements of the matrix Gp (z) defined in Eq. (4.2), and v = [2(z - il)] -1/2. il is the vibrational Hamiltonian of the ion (note that VI = V2 = v since the two electronic continua considered here have a common threshold). The zero-order quantum defects ,uJO) have been put to zero for simplicity.
In the electronic two-channel case the elements Gij (z) and thus TI (z) and T2 (z) can be given in closed form
TI(z) = 17'-1 tan(i1Tii) Gll (z)D1(z), (4.7a)
T2(z) = 17'-1 tan(i1Tii)[ 17'-1 tan(i1Tv) - R 22 (Z)]-1
A
xR2! (z)Gl1 (z)D1 (z), (4.7b)
where G IJ (z) = {1T- I tan(i1Tii) - R lI (z) - R 12(z)
X [17'-1 tan(i1Tv) - R 22 (Z)] -I R 21 (z)}-I.
(4.7c) A
In principle, the Rij (z) and D! (z) are energy-dependent operators. It follows from the definitions (4.3) and (4.5), however, that they reduce to energy-independent c numbers if all discrete electronic states are well separated from the spectral region under consideration. According to the standard description ofvibronic coupling in a diabatic electronic basis, we retain the dependence of the R ij on the normal coordinate q of the vibronically active vibrational mode only up to the first non vanishing term in the expansion about q = O. For the model described above, we have
R lI =R?I'
R22 = R~2' RI2 = R21 = R ?2q,
(4.8)
where the Rj are now parameters of the theory. By the same procedure, DI in Eq. (4.6) is replaced by a (q-independent) parameter D? After this simplification, the model is thus specified by only few phenomenological parameters.
J. Chem. Phys., Vol. 89, NO.10,15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV 6217
B. Calculation of photolonlzation cross sections
In the common situation that laser I is tuned to a fixed vibrational level v Q of the intermediate state I a) and laser 2 tuned through the ionization thresholds, the partial photoionization cross sections as a function of W2 are proportional to the absolute square of the amplitudes [cf. Eq. (3.24)]
TIV,ava = (vITIk•a(E+l7])lvo)'
T2v.oVa = (vIT2k.o(E+i17)lvo)'
(4.9a)
(4.9b)
where Iva} denotes the vibrational level in the intermediate electronic state and Iv) is a vibrational state of the ion. Introducing the complete set of vibrational states {I v'} } of the ion, the operator expressions (4.7) are more explicitly written as
Tlv.ova = tv ~ <vIGl1lv')<v'lvo)D~, v
T2v.ova = tvR ~2 (tv - R ~2 )-1 L v'v"
x (vlqlv') (v'IGlllv") (v" Iva }D~,
tv = 11"-1 tan{i11"[2(E + i17 - E"v)] -1/2}
(4.9c)
(4.9d)
(4.ge)
and E" v is the energy of the vth vibrational level of the ion. The calculation of the matrix element (vIG ll lv') be
comes particularly simple if we introduce the harmonic approximation for the vibrational Hamiltonian of the ion. Then the representation of q in the vibrational basis is tridiagonal and it follows from the definition (4.7c) that the nonvanishing matrix elements of G it l are
(VIG1,llv) =tv -R~2 -~(R~2)2[V(tv_I -R~2)-1
+ (v+ 1)(1v+1 -R~2)-I],
(viG 1,llv + 2) = - ~(R ~2 )2~(V + l)(v + 2)
X(tv+1 -R~2)-I,
(4.10a)
(4. lOb)
(VIGi,llv-2) = -!(R~2)2~V(V_1)(tv_1 -R~2)-I. (4.lOc)
It is seen that the matrix representation ofG it l is of banded form and decouples into two tridiagonal matrices for odd and even v, respectively. The individual tridiagonal matrices can be very efficiently inverted with the help of three-term recursion relations and continued fractions, as discussed, for example, in Ref. 19. This computational procedure amounts to an exact non-Born-Oppenheimer treatment of the vibronic dynamics including infinite Rydberg series and the ionization continua.
In the present model, there is no shift of the equilibrium geometry of the molecule upon ionization, since the vibrational mode considered is non-totally symmetric. There will, however, in general be a difference between the vibrational frequency w[,iOn) ofthe ion core and the frequency w[,O) of the intermediate state la). This effect enters via the Condon overlaps (v'lvo ) in Eq. (4.9).
A final remark concerns the handling of the tangent functions of Eq. (4. ge) which carry the rapid energy dependencies characteristic for Coulomb thresholds. When evaluating these functions we assign the parameter 17 a small, but finite, positive value, typically a few cm- I
. The finite
value of 17 introduces a zero-order energetical broadening of the Rydberg levels, thus accounting for effects which are not explicitly included within the model, e.g., the finite lifetime due to fluorescence, predissociation, or generally radiationless decay in polyatomic molecules. Putting 17 finite also allows us to simulate qualitatively the effects of inhomogeneous rotational broadening and the finite resolution of experimental spectra.
C. Discussion of the results
We choose the model parameters such as to qualitatively reproduce recently observed autoionization features in two-color photoionization of aniline and pyrazine.5
•7 The in
termediate state in both cases is SI' The vibronically active mode is the inversion mode v I (B I symmetry) in aniline5 and the out-of-plane mode VlOa (Big symmetry) in pyrazine,1 respectively. In both cases, the vibrational frequency in the SI state is notably lower than the frequency ofthe ion.5
•7 As
an initial guess, we choose w[,ion) = 500cm- 1 andw[,O) = 400 cm- I
, which is close to the observed values in pyrazine.7
The diagonal reactance matrix elements R ~l ,R ~2 are put to zero to reduce the number of parameters to a minimum. We assume a strong effective Rydberg-valence vibronic coupling represented by the parameter R ~2 = 0.05 a. u. The linewidth parameter 17 is taken as 2 cm -I.
Figure 3 shows a representative set of calculated photoionization cross sections as a function of energy above the lowest threshold for various intermediate levels va. Despite the 20% reduction of Wo in the intermediate state, the direct ionization shows a strong propensity for conservation of the vibrational state, i.e., the direct ionization signal exhibit a vertical onset of the threshold 1+ E"v with v = vo' For all va )0, we observe a strong and regular series of autoionizing Rydberg levels which converge to the direct-ionization threshold. It is seen that this autoionization process follows a
n :;0 o Ul Ul
Ul m
8 o z
o ~ C ::J ,
o 500 1000 1500
ENERGY [em-1 J
FIG. 3. Integral two-color photoionization cross section as a function of energy above the lowest ionization threshold for various choices of the vibrationallevel Va in the intermediate state 10), calculated with the simplified two-channel MQDT model (see the text). The vibrational ionization thresholds are situated at multiples of wgon
) = 500 cm - I.
J. Chem. Phys .• Vol. 89. No. 10. 15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46
6218 Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV
n :;0 0 tn tn
tn ", n -t ...... 0 z
C "1 tT
C ::J ..... ~ c.o
a
o 500 ENERGY [em-'1
1000
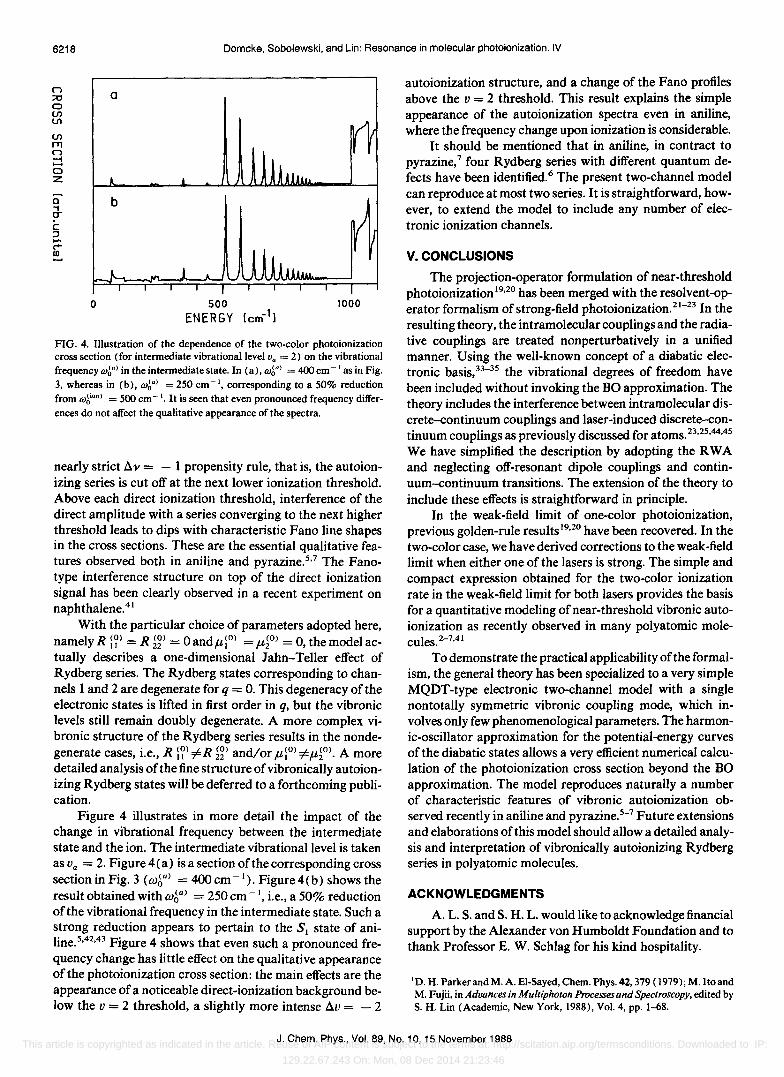
FIG. 4. Illustration of the dependence of the two-color photoionization cross section (for intermediate vibrational level Va = 2) on the vibrational frequency (d~a) in the intermediate state. In (a), (d~a) = 400 cm -I as in Fig. 3, whereas in (b), (d~a) = 250 cm- I
, corresponding to a 50% reduction from (d~ion) = 500 cm - I. It is seen that even pronounced frequency differences do not affect the qualitative appearance of the spectra.
nearly strict Av = - 1 propensity rule, that is, the autoionizing series is cut off at the next lower ionization threshold. Above each direct ionization threshold, interference of the direct amplitude with a series converging to the next higher threshold leads to dips with characteristic Fano line shapes in the cross sections. These are the essential qualitative features observed both in aniline and pyrazine.5.7 The Fanotype interference structure on top of the direct ionization signal has been clearly observed in a recent experiment on naphthalene.41
With the particular choice of parameters adopted here, namely R ~?) = R i~) = 0 and IL ~o) = lLiO) = 0, the model actually describes a one-dimensional Jahn-Teller effect of Rydberg series. The Rydberg states corresponding to channels 1 and 2 are degenerate for q = O. This degeneracy of the electronic states is lifted in first order in q, but the vibronic levels still remain doubly degenerate. A more complex vibronic structure of the Rydberg series results in the nondegenerate cases, i.e., R ~?) #R i~) and/or IL~O) #lLiO). A more detailed analysis of the fine structure ofvibronically autoionizing Rydberg states will be deferred to a forthcoming publication.
Figure 4 illustrates in more detail the impact of the change in vibrational frequency between the intermediate state and the ion. The intermediate vibrational level is taken as Va = 2. Figure 4 (a) is a section of the corresponding cross section in Fig. 3 (W6a
) = 400 cm- I). Figure 4(b) shows the
result obtained with W6Q) = 250 cm -I, i.e., a 50% reduction
ofthe vibrational frequency in the intermediate state. Such a strong reduction appears to pertain to the SI state of aniline.5,42,43 Figure 4 shows that even such a pronounced frequency change has little effect on the qualitative appearance of the photoionization cross section: the main effects are the appearance of a noticeable direct-ionization background below the V = 2 threshold, a slightly more intense Av = - 2
autoionization structure, and a change of the Fano profiles above the v = 2 threshold. This result explains the simple appearance of the autoionization spectra even in aniline, where the frequency change upon ionization is considerable.
It should be mentioned that in aniline, in contract to pyrazine,7 four Rydberg series with different quantum defects have been identified.6 The present two-channel model can reproduce at most two series. It is straightforward, however, to extend the model to include any number of electronic ionization channels.
v. CONCLUSIONS
The projection-operator formulation of near-threshold photoionizationl9,20 has been merged with the resolvent-operator formalism of strong-field photoionization. 21-23 In the resulting theory, the intramolecular couplings and the radiative couplings are treated nonperturbatively in a unified manner. Using the well-known concept of a diabatic electronic basis,33-35 the vibrational degrees of freedom have been included without invoking the BO approximation. The theory includes the interference between intramolecular discrete-continuum couplings and laser-induced discrete-continuum couplings as previously discussed for atoms. 23.25.44,45 We have simplified the description by adopting the RWA and neglecting off-resonant dipole couplings and continuum-continuum transitions. The extension of the theory to include these effects is straightforward in principle.
In the weak-field limit of one-color photoionization, previous golden-rule results 19,20 have been recovered. In the two-color case, we have derived corrections to the weak-field limit when either one of the lasers is strong. The simple and compact expression obtained for the two-color ionization rate in the weak-field limit for both lasers provides the basis for a quantitative modeling of near-threshold vibronic autoionization as recently observed in many polyatomic molecules. 2-7.4 I
To demonstrate the practical applicability of the formalism, the general theory has been specialized to a very simple MQDT-type electronic two-channel model with a single non totally symmetric vibronic coupling mode, which involves only few phenomenological parameters. The harmonic-oscillator approximation for the potential-energy curves of the diabatic states allows a very efficient numerical calculation of the photoionization cross section beyond the BO approximation. The model reproduces naturally a number of characteristic features of vibronic autoionization observed recently in aniline and pyrazine.5
-7 Future extensions
and elaborations of this model should allow a detailed analysis and interpretation of vibronically autoionizing Rydberg series in polyatomic molecules.
ACKNOWLEDGMENTS
A. L. S. and S. H. L. would like to acknowledge financial support by the Alexander von Humboldt Foundation and to thank Professor E. W. Schlag for his kind hospitality.
I D. H. Parker and M. A. El-Sayed, Chern. Phys. 42, 379 ( 1979); M. Ito and M. Fujii, in Advances in Multiphoton Processes and Spectroscopy, edited by S. H. Lin (Academic, New York, 1988), Vol. 4, pp. 1-68.
J. Chem. Phys., Vol. 89, No.1 0, 15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46

Domcke, Sobolewski, and Lin: Resonance in molecular photoionization. IV 6219
2M. A. Duncan, T. G. Dietz, and R. E. Smalley, J. Chem. Phys. 75, 2118 (1981); K. H. Fung, W. E. Henke, T. R. Hays, H. L. Selzle, and E. W. Schlag, J. Phys. Chem. 60,161 (1981).
3G. J. Fisanick, T. S. Eichelberger, M. B. Robin, and N. A. Kuebler, J. Phys. Chem. 87, 2240 (1983).
4M. A. Smith, J. W. Hager, and S. C. Wallace, J. Phys. Chem. 88, 2250 (1984).
5J. Hager, M. A. Smith,andS. C. Wallace,J. Chem. Phys. 84, 6771 (1986). 6M. Fujii, T. Ebata, N. Mikami, and M. Ito, J. Phys. Chem. 88, 4265 (1984); M. Fujii, N. Mikami, andM. Ito, Chem. Phys. 99,193 (1985); M. Fujii, T. Kakinuma, N. Mikami, and M. Ito, Chem. Phys. Lett. 127, 297 (1986).
7A. Goto, M. Fujii, and M. Ito, J. Phys. Chem. 91, 2268 (1987). 8J. Berkowitz, Photoabsorption, Photoionization and Photoelectron Spectroscopy (Academic, New York, 1979).
9J. L. Dehmer, D. Dill, and A. C. Parr, in Photo physics and Photochemistry in the Vacuum Ultraviolet, edited by S. McGlynn, G. Findley, and R. Huebner (Reidel, Dordrecht, 1983).
10M. J. Seaton, Rep. Progr. Phys. 46,167 (1983). "C. H. Greene, U. Fano, and G. Strinati, Phys. Rev. A 19, 1485 (1979). 12c. H. Greene and C. Jungen, Adv. At. Mol. Phys. 21, 51 (1985). I3G. V. Golubkov and G. K. Ivanov, Zh. Eksp. Teor. Fiz. 80,1321 (1980)
[SOy. Phys. JETP 53,674 (1981)]. 14C. Jungen and O. Atabek, J. Chem. Phys. 66, 5584 (1977). 15C. Jungen and M. Raoult, Faraday Discuss. Chem. Soc. 71, 253 (1981). lOA. Giusti-Suzor and C. Jungen, J. Chem. Phys. SO, 986 (1984). I7J. A. Dagata, G. L. Findley, S. P. McGlynn, J. P. Connerade, and M. A.
Baig, Phys. Rev. A 24, 2485 (1981); J. A. Dagata, M. A. Scott, and S. P. McGlynn, J. Chem. Phys. 88, 9 (1988).
IRM. A. Baig, J. P. Connerade, and J. Hormes, J. Phys. B 19, L343 (1986). 19A. L. Sobolewski and W. Domcke, J. Chem. Phys. 86,176 (1987). 2(
lA. L. Sobolewski and W. Domcke, J. Chem. Phys. 88, 5571 (1988). 21M. L. Goldberger and K. M. Watson, Collision Theory (Wiley, New
York, 1964), Sect. 8.
22L. Mower, Phys. Rev. 142, 799 (1966); 165,145 (1988); Phys. Rev. A 22, 882 (1980).
23p. Lambropoulos and P. Zoller, Phys. Rev. A 24,379 (1981). 24A. I. Andryushin, A. E. Kazakov, and M. V. Fedorov, Zh. Eksp. Teor.
Fiz.76, 1907 (1979) [SOy. Phys. JETP 49, 966 (1979)]. 25p. T. Greenland, J. Phys. B 15, 3191 (1982). 26J. Javanainen, Opt. Commun. 46, 175 (1983). 27A. Raczynski and J. Zaremba, J. Phys. B 19, 3895 (1986); 20, 1919
(1987); Phys. Rev. A 36,5079 (1987). 28J. GrochmaJicki, J. Mostowski, and K. Rzazewski, Phys. Rev. A 36, 2718
(1987). 29G. Alber and P. Zoller, Phys. Rev. A 37,377 (1988). 3OY. Fujimura and S. H. Lin, J. Chem. Phys. 75, 5110 (1981). 31A. Boeglin, B. Fain, and S. H. Lin, J. Chem. Phys. 84, 4838 (1986). 32S. H. Lin, A. BoegJin, and S. M. Lin, J. Photochem. 39, 173 (1987). 33H. C. Longuet-Higgins, in Advances in Spectroscopy, edited by H. W.
Thompson (Interscience, New York, 1961), Vol. II, p. 429. 34F. T. Smith, Phys. Rev. 179, III (1969). 35T. F. O'Malley, Adv. At. Mol. Phys. 7, 223 (1971). 36F. Bloch and A. Siegert, Phys. Rev. 57, 522 (1940); M. P. Silverman and
F. M. Pipkin, J. Phys. B 5, 1844 (1972). 37J. Javanainen, J. Phys. B 16,1343 (1983). 38y' N. Demkov and I. V. Komarov, Zh. Eksp. Theor. Fiz. SO, 286 (1966)
[SOy. Phys. JETP 23, 189 (1966)]. 39W. Domcke, J. Phys. B 16, 359 (1983). 40G. V. Golubkov and G. K. Ivanov, J. Phys. B 17, 747 (1984). 41J. A. Syage and J. E. Wessel, J. Chem. Phys. 87, 6207 (1987). 42N. Mikami, A. Hiraya, I. Fujiwara, and M. Ito, Chem. Phys. Lett. 74,531
(1980). 43J. T. Meek, E. Sekreta, W. Wilson, K. S. Viswanathan, and J. P. Reilly, J.
Chem. Phys. 82,1741 (1985). 44p. E. Coleman and P. L. Knight, J. Phys. B 15, L235 (1982). 45J. Zakrzewski, J. Phys. B 17, 719 (1984).
J. Chern. Phys., Vol. 89, No. 10, 15 November 1988 This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
129.22.67.243 On: Mon, 08 Dec 2014 21:23:46