proteinas: en la interaccion entre las matematicas, la fisica, la quimica y la biologia
DESCRIPTION
La comprensión de la forma en que las proteínas se pliegan—estructura tridimensional— y la forma en que interactúanentre ellas de manera secuencial en una red de elementos—cuyo número actualmente se desconoce—, son problemasde gran interés en el área de la biología, la química, la físicay las matemáticas, y tienen importantes implicaciones paralas ciencias médicas y la ingeniería.TRANSCRIPT




PROTEÍNASEN LA INTERSECCIÓN ENTRE LAS MATEMÁTICAS,
LA FÍSICA, LA QUÍMICA Y LA BIOLOGÍA


PROTEÍNASEN LA INTERSECCIÓN
ENTRE LAS MATEMÁTICAS,LA FÍSICA, LA QUÍMICA
Y LA BIOLOGÍA
Samuel GitlerEusebio Juaristi
Luis Fernando LaraAlicia Ortega(Coordinadores)
EL COLEGIO NACIONALMéxico, 2015

QD431P573 2015
Proteínas : en la intersección entre las matemáticas, la física, la química y labiología. -- Primera edición. -- México: El Colegio Nacional, 2015viii, 292 páginas; 22 centímetrosISBN 978-607-724-103-4
1. Lara, Luis Fernando. 2. Juaristi, Eusebio. 3. Gitler, Samuel.I. Título. II. El Colegio Nacional.
D. R. © 2015. EL COLEGIO NACIONALLuis González Obregón núm. 23, Centro HistóricoC. P. 06020, México, D. F.Teléfonos 57 02 17 79 y 57 89 43 30
ISBN: 978-607-724-103-4
Impreso y hecho en MéxicoPrinted and made in Mexico
Correo electrónico: [email protected]@mx.inter.net
Sitio web: http://www.colegionacional.org.mx

CONTENIDO
Prólogo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xiii
LUIS FERNANDO LARAProteínas, códigos y semiótica . . . . . . . . . . . . . . . . . . . . . . . . . 1
EUSEBIO JUARISTI, CLAUDIA GABRIELA ÁVILA ORTIZ
Síntesis asimétrica de a- y b-aminoácidos: acercando a la química con la biología y la medicina . . . . 13
GABRIEL DEL RÍOEstudio de la relación estructura-función de las proteínas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
ABEL MORENO, EUGENIO DE LA MORA, NURIA SÁNCHEZ-PUIGLa influencia de los campos magnéticos en la cristalización y estructura tridimensional de las proteínas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
XIM BOKHIMIFuentes de rayos X compactas y brillantes para estudiar la distribución atómica en las proteínas . . . . 67
JOSÉ ANTONIO DE LA PEÑARedes de residuos proteínicos. Estudio de un caso de gráficas grandes y su entropía . . . . . 85
ALICIA ORTEGA, JOEL MEDINA
Energética de la transición plegamiento/desplegamiento de las proteínas. Una herramienta para aprender sobre la conformación de proteínas en un sistema biológico . . 121
vii

JAIME MAS OLIVA
Transiciones estructurales del tipo desorden-al-orden en proteínas transferidoras de lípidos y su asociación a la función . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 143
PÁVEL VÁZQUEZProteómica de la Tiolación. De la modificación de tioles en la proteína a la disfunción celular . . . . . . . . . 167
SAMUEL GITLER
El cambio topológico de la membrana causado por las proteínas transmembranales . . . . . . . . . . . . . . . . . . 185
CHRISTIAN SOHLENKAMP, ROSA LIDIA SOLÍS OVIEDO, OTTO GEIGER
Estudiando la síntesis de fosfatidilcolina en bacterias: de lípidos de membrana a proteínas de membrana . . . . . 193
AURA MATILDE JIMÉNEZ-GARDUÑO
Agrupaciones proteicas. Cuando las proteínas forman un equipo para trabajar . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
DAVID E. GARCÍA, KARINA BERMEO, HÉCTOR CASTROBiofísica de canales iónicos sensibles al voltaje: modulación de canales de calcio por proteínas G . . . . . . 213
PABLO PADILLA LONGORIA, YURIRIA CORTÉS POZAMarcadores epigenéticos . . . . . . . . . . . . . . . . . . . . . . . . . . . 243
SUSANA CASTRO¿Cómo es una proteína capaz de promover la proliferación, la diferenciación y la muerte celular? . . . 261
HÉCTOR RASGADO-FLORESLa simplicidad del cerebro y la percepción musical.¿Tienen algo que ver con las proteínas? . . . . . . . . . . . . . . . 277
viii

Luis Fernando Lara, Jaime Mas, M
arcelo Galván, Xim Bokhimi, Pável Vázquez, H
éctor Rasgado,
Christian Sohlenkamp, Abel M
oreno, Gabriel del Río y Hubertus von Rom
er.
Aura Jim
énez, Eusebio Juaristi, Alicia Ortega, Robert H
uber, Susana Castro, Sam
uel G
itler y José Antonio de la Peña.




PRÓLOGO
La comprensión de la forma en que las proteínas se pliegan—estructura tridimensional— y la forma en que interactúanentre ellas de manera secuencial en una red de elementos—cuyo número actualmente se desconoce—, son problemasde gran interés en el área de la biología, la química, la físicay las matemáticas, y tienen importantes implicaciones paralas ciencias médicas y la ingeniería.Las proteínas son secuencias irrepetibles con significado
específico en la función celular; construidas con 20 elemen -tos que conforman el alfabeto químico (los aminoácidos).Éstos se combinan en secuencias de longitudes variablesy tienen representaciones espaciales y energéticas con coli -nas y valles que determinan su estructura funcional. El cómoesta estructura peculiar encuentra su forma, cómo reaccionaa su entorno y cómo sus elementos interactúan entre sí, hansido las interrogantes objeto de la investigación científicamás intensa que se ha hecho en lo que va del siglo. El estudiode la forma y la función de las proteínas está en la encruci-jada de la atención multidisciplinaria.El descubrimiento de la estructura de muchas proteínas ha
proporcionado información a la industria farmacéutica paradiseñar moléculas que interfieran de manera específica conla función de una determinada proteína, modificando suestructura. Hasta hoy, cada una de las proteínas que actual-mente se investigan, utilizando las herramientas teóricas decada una de las áreas del conocimiento, está implicada en
xiii

planteamientos de la investigación biomédica, ya sea paraentender el desarrollo de enfermedades y su tratamientoo para mejorar la calidad de vida del ser humano. La com-prensión de la forma en que se pliegan las proteínas y la for-ma en que interactúan en una compleja red de proteínas setraduciría en una nueva comprensión del estado de enfer-medad y nuevas medicinas con menores efectos colaterales.Pero sobre todo, nos dirige hacia un entendimiento de losmecanismos básicos de la vida. El objetivo de este simposio (Humboldt Kolleg) en el
marco de la colaboración científica multinstitucional entreMéxico y Alemania, a través de la vinculación con la Funda-ción Alexander von Humboldt, es establecer una comuni-cación entre especialistas en diferentes áreas interesados enel tema de las proteínas. Este simposio representa un desa-fío para la colaboración interdisciplinaria. El uso de un len-guaje accesible en cada una de las exposiciones de las áreasde conocimiento aquí representadas, dará la posibilidad a unacercamiento al tema de las proteínas al más alto nivel decomprensión. La conferencia introductoria, una contribución de las
ciencias sociales a cargo del lingüista Luis Fernando Lara, esuna reflexión sobre las similitudes de la construcción de laslenguas y la síntesis de proteínas (proteómica) —ésta últimacomo el lenguaje de la vida.La quiralidad es una característica muy importante de los
aminoácidos y por lo tanto de las proteínas presentes enlos seres vivos, ya que de ella depende su actividad biológica.En el capítulo 2, Eusebio Juaristi y Claudia Gabriela Ávila Ortizdiscuten algunas estrategias para la síntesis en el laboratoriode aminoácidos de una sola configuración. Las proteínas participan en muy diversas funciones fun-
damentales para los seres vivos, desde el movimiento muscu-lar, la comunicación de las células, el crecimiento y la dife-
xiv

xv
renciación celular, entre muchas otras. Gabriel del Río explicaen el capítulo 3 cómo logran las proteínas generar tan diver-sos fenómenos. Por su parte, en el capítulo 4, Abel Moreno, Eugenio de la
Mora yNuria Sánchez-Puig describen el uso de un campo mag-nético intenso sobre el crecimiento de cristales de proteínasmodelo y proteínas nuevas. Esto se llevó a cabo a través dedifracción de rayos X. En el capítulo 5, Xim Bokhimi describe nuevas fuentes de
rayos X compactas y brillantes que son actualmente accesiblespara el estudio de la distribución atómica de las proteínas.Su potencia es mayor que la de los tubos sellados de rayos Xy similar a la de los sincrotrones tradicionales; por supuestosu costo y mantenimiento es mucho más económico.Luego de una introducción corta sobre terminología
y algu nos resultados esenciales de la teoría espectral de grá-ficas, José Antonio de la Peña presenta en el capítulo 6 el con-cepto de “entropía de caminos”, que permite entender algu-nos hechos de la bioquímica de las proteínas mediante laestructura de las gráficas asociadas a ellas. En el capítulo 7, Alicia Ortega y Joel Medina muestran cómo
esfuerzos multidisciplinarios ayudan a resolver uno de losretos actuales más importantes en la biología: entender las re -glas que determinan cómo un polipéptido naciente adquiereuna estructura tridimensional específica y con ello una fun-ción fisiológica definida.Recientemente, se ha establecido que el plegamiento de
proteínas está directamente relacionado con los mecanismosde las transiciones reversibles “desorden-al-orden”, por elcual una cadena polipeptídica desplegada se pliega en unaestructura nativa funcional específica. Estos procesos sontratados por Jaime Mas Oliva en el capítulo 8.En este contexto, Pável Vázquez discute en el capítulo 9 la
forma como algunas cadenas laterales de los residuos de

aminoácidos, que constituyen la proteína, pueden sufrirmodificaciones químicas. En particular, la incorporacióndel grupo tiol -SH, (tiolación) desempeña un papel muyimportante en gran parte de las funciones celulares induci-das por proteínas.La relevancia de los estudios matemáticos para explicar el
comportamiento conformacional de las proteínas se apreciatambién en la contribución de Samuel Gitler, quien presentaen el capítulo 10 la clasificación de las superficies orientablesbidimensionales, lo que tiene relación con la forma en quemuchas proteínas cruzan más de una vez la membrana,haciendo “asas” en el interior y exterior de la célula.Christian Sohlenkamp, Rosa Lidia Solís Oviedo y Otto Geiger
abordan en el capítulo 11 diversos avances recientes relacio-nados con la síntesis de fosfatidilcolina en bacterias. Asimis-mo, se presenta aquí información acerca de la identifica-ción de los aminoácidos esenciales para la actividad en vivode la fosfatidilcolina sintasa.En el capítulo 12, Aura Matilde Jiménez-Garduño discute la
evidencia que muestra cómo la preservación de las agrupa-ciones en los organismos evolutivamente más antiguos, comolas bacterias, hasta los más complejos juega un papel muyimportante en la preservación de la vida. David E. García, Karina Bermeo y Héctor Castro, en el capí-
tulo 13, discuten el origen de las corrientes iónicas mediantela teoría del movimiento de carga y describen la aplicacióndel método en la comprensión de los procesos subyacentesa la regulación de los canales iónicos. Posteriormente, pre-sentan un ejemplo notable de un modelo de regulación delos canales de calcio mediado por proteínas G.En el capítulo 14 “marcadores epigenéticos”, Pablo Padilla
Longoria y Yuriria Cortés Poza comentan que el concepto depaisaje epigenético fue propuesto como una metáforade los procesos evolutivos y del desarrollo en biología. Esta
xvi

metodología se ilustra con el destino celular durante losprimeros estadios del desarrollo de un organismo o de laformación de órganos.En el capítulo 15, Susana Castro aborda el tema ¿cómo es
una proteína capaz de promover la proliferación, la diferen-ciación y la muerte celular? De nuestra capacidad para resol-ver esta pregunta puede depender el evitar el surgimientode padecimientos como el cáncer, o modular su efecto en elmetabolismo, importante para controlar la obesidad y resis-tencia a la insulina.Finalmente, Héctor Rasgado-Flores en el capítulo 16 se hace
la pregunta: “¿Tiene algo que ver la percepción musicalcon las proteínas?” En esta presentación se trata de descubrircómo es que oír música afecta las proteínas pertinentes ennuestro cerebro.
SAMUEL GITLEREUSEBIO JUARISTI
LUIS FERNANDO LARAALICIA ORTEGA
xvii


PROTEÍNAS, CÓDIGOS Y SEMIÓTICA
LUIS FERNANDO LARAEl Colegio de México
Miembro de El Colegio Nacional
En diversas ocasiones, frente a temas que desconocía, el his-toriador Edmundo O’Gorman aclaraba que su ignoranciaera enciclopédica. En relación con los temas que se discuti-rán durante los tres días de este simposio, afirmaré lo mis-mo: mi ignorancia de la genética y de todos los temas queaquí entroncan, es enciclopédica. Alicia Ortega, presidentadel Club Humboldt, me insistió en que participara yo enesta reunión, y acepté debido a su poder de convencimien-to pero ¿qué puede aportar un lingüista a las discusiones entorno a ese fenómeno complejo y maravilloso que son el ADNy las proteínas? Quizá —y eso espero que me justifique—la visión de alguien que no deja de asombrarse a su vez de lamaravilla de los lenguajes, particularmente los que creamoslos seres humanos y, entre éstos, las lenguas como el español,el inglés, el tzeltal o el náhuatl.Gastón Bachelard, en su conocido libro La formación del
espíritu científico examina con su conocida perspicacia lo quellama “el obstáculo verbal” que se presenta al conocimientocientífico. Tal obstáculo consiste en el hecho de que la únicamanera que tenemos los humanos de conocer el mundoque nos rodea y del que somos parte es la formación deconceptos acerca de los fenómenos en estudio, que necesa-
1

riamente formulamos con nuestra lengua materna. En efecto:aun cuando pensemos que hemos entendido perfectamenteun fenómeno, en tanto no seamos capaces de elaborar eseentendimiento con una lengua y comunicarlo a los demás,ese concepto que creemos tener todavía no se materializay no se puede someter a verificación. No quiero decir que lainteligencia sea verbal, pero sí que la inteligencia sólo puedeexpresarse mediante la lengua y mediante otros lenguajescomo la matemática, la música, la pintura, etcétera. De todoslos lenguajes de que disponemos los seres humanos, el másamplio, dúctil e ilimitado es la lengua. Pero precisamente poreso, puesto que la lengua nos ofrece medios de expresión quevuelven inteligibles a los demás nuestros pensamientos, esosmismos medios pueden llegar a obstaculizar la compren-sión de un fenómeno, atribuyéndole características que noson suyas, sino del significado de las palabras y la significa-ción con que nos referimos a él. El instrumento primordial de nuestra comprensión de
los fenómenos y su comunicación a los demás es la metáfora.Podemos definir la metáfora como aquel dispositivo de cono-cimiento que establece relaciones de significado entre lo yaconocido y lo que pretendemos conocer, dándole una sig-nificación que lo vuelva inteligible; la metáfora nos ofrece unesquema de conocimiento ya establecido en la lengua común,con el que podemos asimilar una experiencia absolutamentenueva. Pero, señala Bachelard, cuando se crean metáforascuya plasticidad es sorprendente y parecen develar la natu-raleza de los fenómenos estudiados, hay una tendenciaa atribuir al fenómeno novedoso las propiedades de aque-llo ya nombrado con una metáfora, con lo que se terminaatribuyendo al fenómeno características o propiedades queno son suyas, sino de lo previamente significado. Cuandouno considera por primera vez, como lingüista, el discursode la genética, tiende a pensar que aquí también hay un
2

conjunto de metáforas acerca de un fenómeno cuya natura-leza es muy diferente de los fenómenos lingüísticos. Si lasletras que representan los nucleótidos, por ser letras, parecenun alfabeto, y si los codones formados por tres letras pare-cen palabras, parece que estamos frente a un fenómeno lin-güístico. Tanto más cuando nos enteramos de que los codo-nes del ADN se transcriben en el ARN mensajero y que éste setraduce en una proteína. Esos términos: alfabeto, palabra,transcripción y traducción parecen metáforas. Uno tenderíaa pensar que la metáfora lingüística en genética solamentesirve para facilitar la comunicación acerca de un conjunto defenómenos que son exclusivamente químicos y no de unlenguaje (en español, aunque, a diferencia de otras lenguas,distinguimos entre lengua y lenguaje, sólo disponemos de unadjetivo para ambos sustantivos: lingüístico; en francés, el adje-tivo para langue o lengua es langagière y el adjetivo para lan-gage o lenguaje es linguistique. Al hablar de los fenómenos atri-buidos a un lenguaje genético, convendría decir lingüísticos,con el significado del francés).La semiótica es una disciplina que no ha terminado de
construirse a causa de todo lo que pretende abarcar: las len-guas, la literatura, los códigos, las señales, la música, las mate-máticas, la pintura, la danza y todas aquellas expresionesmateriales con las cuales un individuo transmite a otros desu especie o de otras, señales o signos que amplían su hori-zonte de percepción, una ampliación que lleva a sus recep-tores a modificar su conducta y a actuar en consecuencia.Forman parte de los intereses de la semiótica no solamentelos lenguajes humanos, como las lenguas o la mú sica, sinotambién los animales, como la llamada “danza de las abe-jas”, los trinos de los pájaros o las ondas sonoras que trans-miten ballenas y delfines por las profundidades marinas.La semiótica ha dependido en sus conceptos y en sus méto-dos de la lingüística, pues es esta ciencia la que ha logrado
3

avanzar en una comprensión adecuada y verificable de losfenómenos de la significación.Para entrar en materia, comenzaré por establecer una
diferencia central entre un código y lenguajes más comple-jos, como el de la música y la lengua. Un código es un con-junto de señales organizadas con cierto sistema, que sirvepara transmitir una información específica entre dos o másindividuos. Un código muy sencillo es el de los semáforosen el tráfico urbano: hay tres señales de colores y una deposición en un orden vertical, generalmente, o de izquierdaa derecha. Mediante esos tres colores y su posición sabemoscuándo se puede cruzar una avenida, cuándo hay quecomenzar a detenerse y cuándo hay que hacer un alto (laposición de las lámparas es redundante y contribuye a lograrla informatividad del código). Igualmente, la “danza de lasabejas” es un código sencillo: el mensaje consiste en indicaral panal en qué dirección y a qué distancia de él se encuentranlas flores. Sus elementos son la manera en que vuela la abeja,formando círculos u ochos, que indica la distancia; el ánguloformado por el plano de su danza con los rayos del sol y laposición de su abdomen indican la dirección. Los códigos sir-ven para transmitir ciertos mensajes, pero el tipo de mensajeque pueden transmitir es limitado. En el código de banderasde la navegación marina, a cada bandera y el orden en queaparecen colgadas entre los mástiles, corresponde un solosignificado y es imposible utilizarlo para transmitir otros sig-nificados que no estén previamente estipulados en un librode códigos fijado de antemano. Quizá valga la pena aclararque los mensajes cifrados, por complejos que sean, son códi-gos sólo en el sentido de que siguen ciertas reglas, pero enrealidad son instrumentos de transliteración de mensajes emi-tidos en una lengua particular o en un pequeño conjuntode elementos de ésta y, en consecuencia, las reglas profun-das de su sintaxis son las de la lengua en cuestión.
4

El lenguaje de la música es diferente de los códigos: nose estipula previamente el valor de cada uno de sus elemen-tos en relación con cierta clase de mensajes predeterminadosque uno quiera transmitir y el número de expresiones musi-cales que se pueden hacer con él es ilimitado. Hay que seña-lar que lo que transmite la música no son mensajes (a menosque se quiera entender por mensaje el paso de una sensa-ción auditiva a la corteza cerebral); la música no dice nada,no transmite información en el sentido en que lo hace un có -digo o una lengua, sino que da origen a una serie complejade sensaciones a las que, si se quiere, cada sociedad o cadapersona puede atribuir cierto sentido ; por ejemplo, el bluespuede despertar melancolía; los modos griegos, según Pitá-goras, se podían dividir entre los apolíneos que tendían a lacalma y la elevación, y los dionisíacos, dedicados a la excita-ción y el entusiasmo, pero ese valor moral de la música en -tre los griegos no constituía mensajes. Sin duda, la músicacomunica, pero no informa. El lenguaje musical consta deun conjunto limitado de elementos —las notas— que se orga-nizan de acuerdo con ciertos principios, como el ritmo, lamelodía, la armonía, etcétera, en una sintaxis compleja. Esasintaxis se produce a partir de las diferentes estructuras musi-cales que el músico aprende a hacer y desarrollar durantesu formación. El número de expresiones que se pueden crearmediante esos principios, como digo, es ilimitado, incluso siuno se atiene a la tradición de Bach a Wagner, sin aventu-rarse en las concepciones dodecafónicas o seriales.Una lengua cuenta con un número relativamente reducido
de elementos materiales: los fonemas, que se manifiestancon sonidos o, en las escrituras alfabéticas, con las letras, queson visuales, cuyas combinaciones no son exhaustivas, sinoque tienen limitaciones definidas por las relaciones distin-tivas que se establecen entre ellos, de acuerdo con su estruc-tura fonológica, así como por las características de la sílaba
5

en cada lengua. A estas unidades, el lingüista francés AndréMartinet las llamó “unidades de segunda articulación”, pueshay una “primera articulación”, que es la formada por laspalabras y su organización sintáctica, cuyas actualizacionesdependen del mensaje con sentido que uno quiera transmitir.Todas las lenguas tienen estas características, pero los modosen que se estructuran son particulares a cada una de ellas,aunque hay características compartidas, que dan lugar a fami-lias de lenguas, como las romance descendientes del latín,y a troncos como el indoeuropeo, del que forman parte ellatín y sus descendientes. Lo único y extraordinario de laslenguas es que sirven para transmitir cualquier mensaje, sinestipulación previa, de manera ilimitada. Por eso afirmamosque las lenguas no tienen parangón con el resto de los len-guajes que existen en el planeta. Se puede afirmar que lo quedistingue a las lenguas de los demás lenguajes y los códigos esesa doble articulación, que les da una capacidad ilimitadapara formar mensajes nuevos.Cuando un lingüista se acerca como verdadero y sincero
lego a los textos que hablan de genética y de proteínas,observa que las letras A, C, G, T y U representan —al igualque lo hacen las letras en las escrituras alfabéticas— otracosa, en este caso, nucleótidos; son tan arbitrarias como elalfabeto de las lenguas en relación con sus fonemas, aunqueen este caso haya una tradición histórica que diluye su con-vencionalismo; esos nucleótidos se combinan en codones enel ADN y en el ARN. Si en cuanto a los fonemas las combina-ciones se seleccionan a partir de posibilidades e imposibili-dades del aparato fonador de los seres humanos, lo que nose puede soslayar es que esos elementos de “segunda arti-culación” sólo tienen valor gracias a la “primera articulación”,en la cual el sentido se organiza a partir de dos núcleos: unodenominativo de objetos y relaciones y otro de acciones:nominales y verbos. Denominar y predicar, la utilidad que
6

define la existencia de las lenguas, no forman parte del sistemalingüístico, sino de la relación de la lengua con la experien-cia humana de la vida. Dicho de otra manera, las cadenasde letras o de fonemas pronunciados en una lengua, que enla realidad aparecen como un continuo sonoro o gráfico, sesegmentan, ante todo, gracias a la primera articulación y, ensegundo lugar, a las reglas de combinación determinadaspor la estructura fonológica. En cambio, si una cadena decodones del ADN como GGGAAACCC puede segmentarse apartir del primero en tripletes: GGG, AAA, CCC, pero tam-bién en GGA, AAC, etcétera, surge la pregunta: ¿qué deter-mina la segmentación? Leo que una cadena de ADN puedesegmentarse de tres maneras diferentes, de acuerdo con lainformación que ofrecen ciertos codones que suelen indicarel principio de la segmentación, aunque la informaciónque proveen no sea suficiente para comenzarla. En cambio,hay otros codones que marcan el fin de la segmentación:los llamados, convencionalmente, ámbar UAG, ópalo UGAy ocre UAA. Si nos asomamos a las lenguas particulares,cada una de ellas puede tener ciertas combinaciones defonemas que sólo se dan al comienzo de la palabra y otrosal final. Por ejemplo, en español, el fonema “erre” sólo seda al comienzo de la palabra o después de “ene”; igualmente,ninguna palabra del español puede terminar, por ejemplo,con “eñe” o con “gt”. Estas reglas, como decía antes, estándeterminadas por las capacidades del aparato fonador hu -mano, limitadas por la evolución de cada lengua particular. Salta a la atención el hecho de que los codones de inicio
y de fin de una segmentación informan cuándo segmentar;es decir, hay ciertas reglas que determinan las segmentacio-nes y se comportan como un lenguaje; esas reglas ¿son deorden exclusivamente químico y de la “misma articulación”—para usar los conceptos de la lingüística— que los codo-nes?, si fuera así, el lenguaje genético en este punto, es decir,
7

exclusivamente en las relaciones entre ADN y ARN, sería deuna sola articulación, como muchos, entre ellos el de lamúsica, si no ¿en qué consiste su “primera articulación”, aque-lla que da sentido a la segmentación? Lo que me sorprendees el complejo juego de interacciones que se produce entre elADN y el ARN con el concurso del ribosoma; entre el ARNmensajero y las enzimas del ARN de transferencia o de trans-cripción, etcétera. Tengo la impresión de que estos comple-jos procesos no se parecen a ninguno de los lenguajes conque tenemos familiaridad los lingüistas y mucho menos alos códigos. Concluyo entonces, provisionalmente, que el lla-mado código genético es mucho más que eso y parece unverdadero lenguaje. Es verdad que la lingüística todavía no acaba de entender
el funcionamiento de las lenguas cuando se produce unaoración y un conjunto encadenado de oraciones; el sistemaes extremadamente complejo y debe serlo aún más si pode-mos pasar de lo que observamos en el uso de una lenguaa lo que ocurre en la corteza cerebral cuando hablamos,fenómeno cuyas características todavía está lejos la neurofi-siología de llegar a comprender. Sólo ha habido un esfuerzopor modelar lo que sucede cuando se construye una oración:la gramática generativo-transformacional de Noam Chomsky.A partir de una analogía entre la lengua y los lenguajes arti-ficiales derivados de los lenguajes de Turing, Chomsky intentódar cuenta de la dinámica de construcción de las oracionessobre la base exclusiva de las relaciones sintácticas entre suspartes, mediante un conjunto de reglas de transformación,primero, y después generativas. Las reglas chomskyanas sonrealmente reglas de transcripción: a partir de un nodo lla-mado oración, se desarrolla un árbol jerárquico que va, diga-mos, desenvolviendo la oración y transcribiendo sus com-ponentes en diferentes niveles de la jerarquía, con la inter-vención de ciertas reglas recursivas que operan en diferentes
8

momentos. En principio, el origen de ese nodo inicial de laoración debería proceder de una significación elaborada enla mente; más tarde, Chomsky buscó afianzar esa idea atri-buyéndole características innatas, a la manera de Descartes,que en última instancia llevarían a la impronta genética delos seres humanos. Por eso pretendía que la gramática, cons-tituida por sustantivos y verbos, por reglas de predicación,etcétera, es universal. Lamentablemente, el esfuerzo de Chomsky no fructificó:
los sistemas de las lenguas son más complicados de lo quecreía. Me parece una ilusión lo que algunos genetistas hanestado proponiendo, haciendo otras metáforas, esta vez a par-tir de las gramáticas de Chomsky. Quizá la ilusión provengano de la naturaleza de las lenguas y el supuesto lenguajegenético, sino del modo en que analizamos las lenguas; esdecir, del modo en que la lingüística trata de descubrir elsistema dinámico y complejo de las lenguas. Para Chomsky,la gramática generativo-transformacional era, al principio, unahipótesis de análisis inspirada en los programas de compu-tación descendientes de Turing, por eso había un conceptoabstracto de oración al comienzo del procesamiento, comoinsumo, que se desarrollaba mediante reglas hasta llegara una oración como estas que estoy pronunciando. En tornoal programa sintáctico, Chomsky estableció dos módulossecundarios: uno fonológico, que atribuía fonemas a las ca -denas sintácticas terminales y otro semántico, que atribuíasignificado a los vocablos de las cadenas. Cuando optó poratribuir al insumo inicial un carácter innato el programa deanálisis se convirtió en la hipótesis de descubrimiento de unprograma de funcionamiento de las lenguas.Creo que, por lo pronto, puedo afirmar que los procesos
que se producen en el paso del ADN a la proteína es muchomás que un código, tal como lo definí antes, pues están cons-tituidos por una serie de pasos que ponen en juego amino-
9

ácidos, ribosomas, enzimas, tanto en la transcripción del ADNal ARN como en el funcionamiento de los dos ARN—el men-sajero y el de transferencia—, lo que sí parece dar lugara un programa ¿dirigido finalistamente o simplemente abier-to a la intervención del medio en que viven las células?Lo sorprendente para mí es que la hipótesis de un pro-
grama en el paso del ADN a la proteína no parece descabe-llada: el biólogo francés Henri Atlan, en su libro Entre lecristal et la fumée. Essai sur l’organisation du vivant (1979) pro-pone que la vida obedece a programas de autoorganización.El complicado concurso de las enzimas en los ribosomas, laselección que operan sobre los nucleótidos del ADN paratranscribirlos en ARN, y la traducción que lleva de éstemediante la intervención del ARN de transferencia hacia lasíntesis de las proteínas parece corresponder a un progra-ma de autoorganización. Las lenguas, hasta donde llega micomprensión de ellas, no obedecen a programas de autoor-ganización, al menos en el nivel en que las investigamos loslingüistas. La relación entre fonemas y palabras, entre laspalabras en la oración y el modo en que llegan a transmitirun mensaje no parece ser de autoorganización, aunque esténdeterminadas por el sentido que deseamos comunicar; esdecir, que se guían por una finalidad del orden semántico. Encambio, si los procesos genéticos son un lenguaje y su natu-raleza es la de un programa de autoorganización, estamosfrente a un tipo muy diferente de lenguaje. Si se trata de unaestructura con determinadas reglas de transcripción en elARN y de traducción a las proteínas, ¿en qué consisten esasreglas?, si se trata de un programa de autoorganización, ¿enqué consisten y qué determina los pasos y las reglas quehacen funcionar el programa?Por lo que me he enterado, en los diversos momentos de
funcionamiento del hipotético programa genético hayintervenciones determinantes del ambiente, lo cual indica
10

que el programa no está cerrado en sí mismo, sino que tienemúltiples momentos de apertura a la vida en el planeta, locual determina su capacidad de adaptación, el elementocentral de la evolución. Las lenguas, a su vez, están perma-nentemente abiertas a la experiencia humana y también seadaptan a la vida de las sociedades que las hablan y a lasnuevas experiencias de la inteligencia, pero el funciona-miento de su estructura se abre, ante todo, por el léxico, quees el que provee y crea las palabras necesarias para nombrar,formar conceptos; la evolución de su fonología y de su sinta-xis, en cambio, depende de las posibilidades, a veces debili-dades de sus estructuras, y de las tradiciones discursivas quese han ido elaborando en la necesidad de discurrir acerca delas experiencias de la vida.No puedo concluir de alguna manera que pueda aportar
algo interesante a las discusiones que se llevarán a cabo desdeahora y durante los próximos dos días. Quizá pueda aspirara convencerlos de que los procesos genéticos no son un merocódigo y que, si constituyen un lenguaje, es muy diferente detodos los demás, entre ellos las lenguas; también que no haymetáforas que se hayan convertido en un obstáculo verbal:la suposición de que los procesos genéticos obedecen a uncódigo se enriquece si se les considera un lenguaje particu-lar, constituido por un programa de autoorganización. Perono quiero dejar pasar la oportunidad de voltear la cuestióny ahora plantear qué será lo que da lugar a la aparición delos sistemas de las lenguas en los seres humanos. Una dispo-sición genética, sin duda, pero ¿de qué clase? El lingüistaespañol Ángel López García, en un libro titulado Fundamentosgenéticos del lenguaje (2002) propone la atractiva hipótesis deque las lenguas aparecieron en la historia de la humanidadcomo una especie de emergencia precisamente de las carac-terísticas formales del programa genético. La hipótesis es pordemás sugerente aunque quizá, por ahora, no pueda sustan-
11

ciarse; sin embargo, en el ámbito de las discusiones de la lin-güística contemporánea, a propósito de si existe una gramá-tica universal, como decía Chomsky, o una disposición ge -nética a construir gramáticas de las lenguas particulares, enque la vida social, como la epigenética, tiene un papel deter-minante, podría uno inclinarse hacia la segunda posibilidadcomo más prometedora que la primera; Chomsky no ten-dría razón; en cambio, Jean Piaget estaría de acuerdo.
12

SÍNTESIS ASIMÉTRICA DE �a- Y b-AMINOÁCIDOS: ACERCANDO A LA QUÍMICA CON LA BIOLOGÍA
Y LA MEDICINA
EUSEBIO JUARISTI*CLAUDIA GABRIELA ÁVILA ORTIZ
Departamento de Química Centro de Investigación y de Estudios
Avanzados del Instituto Politécnico Nacional* Miembro de El Colegio Nacional
La síntesis asimétrica tiene su fundamento en la quiralidad,que es una propiedad presente en un número importantede moléculas con actividad biológica. Existen cuatro aspec-tos que definen la estructura molecular [1]:1) Constitución, que se refiere a los átomos presentes en la
molécula.2) Conectividad, que nos indica cómo están unidos dichos
átomos.3) Configuración, que describe la disposición que guardan
en el espacio los átomos sustituyentes alrededor de un centrode quiralidad, es decir, en un átomo de carbono con cuatrosustituyentes diferentes se encontrarán dos formas posiblesde acomodo. Estas dos configuraciones son de imágenes enel espejo no superponibles una respecto de la otra. A lasmoléculas que guardan esta relación de imágenes especu-lares se les llama enantiómeros. En la figura 1 se puede observar
13

el aminoácido alanina en sus dos formas (S) y (R), enantió-mero que al igual que las manos izquierda y derecha resul-tan ser imágenes en el espejo. Con base en esta analogía, sedice que estas moléculas son quirales (de quir = mano, engriego) y que los átomos de carbono responsables de estaasimetría son centros de quiralidad.
4) Conformación, que se refiere a la orientación de los áto-mos de una molécula en el espacio, debida a giros alrededorde enlaces sencillos.La quiralidad, sin embargo, es un aspecto muy impor-
tante, presente en proteínas, aminoácidos, péptidos, etcé-tera, ya que de ella depende su actividad biológica [2]. Estose debe a que los receptores biológicos son quirales tam-bién, es decir, presentan una orientación determinada en elespacio, que se acopla únicamente con el sustrato (enantió-mero) correcto.La obtención de compuestos enantiopuros es vital en el
desarrollo de nuevos fármacos; es por ello que la síntesis asi-métrica constituye una herramienta importantísima en estecampo. La definición de síntesis asimétrica fue propuesta porMorrison y Mosher [3] quienes indican: La síntesis asimétrica
14
FIGURA 1. Existen moléculas asimétricas, como los aminoácidos, que se presentanen dos formas isoméricas (enantiómeros) que poseen diferente configuración.
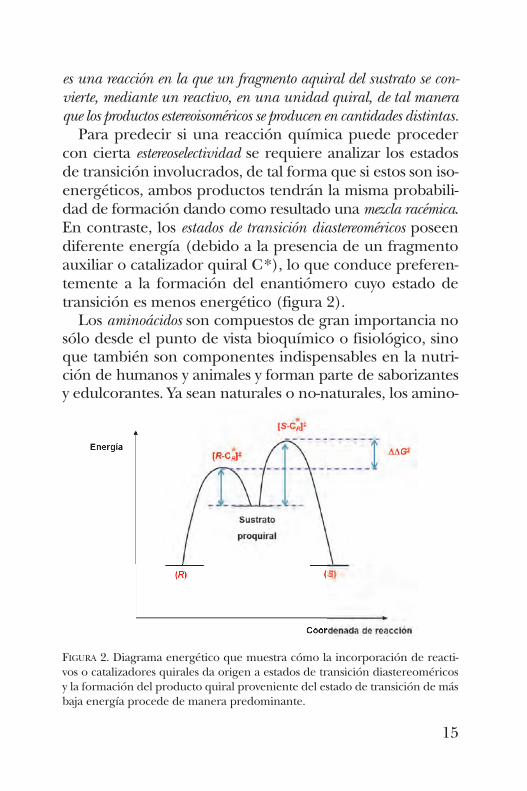
es una reacción en la que un fragmento aquiral del sustrato se con-vierte, mediante un reactivo, en una unidad quiral, de tal maneraque los productos estereoisoméricos se producen en cantidades distintas.Para predecir si una reacción química puede proceder
con cierta estereoselectividad se requiere analizar los estadosde transición involucrados, de tal forma que si estos son iso-energéticos, ambos productos tendrán la misma probabili-dad de formación dando como resultado una mezcla racémica.En contraste, los estados de transición diastereoméricos poseendiferente energía (debido a la presencia de un fragmentoauxiliar o catalizador quiral C*), lo que conduce preferen-temente a la formación del enantiómero cuyo estado detransición es menos energético (figura 2).Los aminoácidos son compuestos de gran importancia no
sólo desde el punto de vista bioquímico o fisiológico, sinoque también son componentes indispensables en la nutri-ción de humanos y animales y forman parte de saborizantesy edulcorantes. Ya sean naturales o no-naturales, los amino-
15
##
r
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
FIGURA 2. Diagrama energético que muestra cómo la incorporación de reacti-vos o catalizadores quirales da origen a estados de transición diastereoméricosy la formación del producto quiral proveniente del estado de transición de másbaja energía procede de manera predominante.

ácidos son componentes también de agentes terapéuticos,cosméticos, agroquímicos, etcétera. Como resultado de sugran variedad de aplicaciones, estas moléculas poseen unimpacto económico significativo y es así que se han desa-rrollado diferentes metodologías, ya sea para su extracciónde fuentes naturales o bien, para su síntesis.Debido a la naturaleza quiral de los aminoácidos, es impor-
tante recordar que las mejores rutas sintéticas serán aquellasque conduzcan al producto deseado de manera enantiopura. En 1987, Seebach y colaboradores [4] reportaron una sín-
tesis asimétrica de a-aminoácidos a través de la alquilacióndiastereoselectiva del enolato de una imidazolidinona impe-dida estéricamente, como se muestra en el esquema 1. Laimidazolidinona enantioméricamente pura fue obtenidaa partir del aminoácido glicina (que es un compuesto aquiraly muy económico) y mediante una resolución con ácido man-délico (quiral). Este compuesto forma un enolato que sealquila de manera altamente diastereoselectiva, por la caraopuesta al fragmento de t -butilo.Uno de los alcances de esta metodología lo constituye la
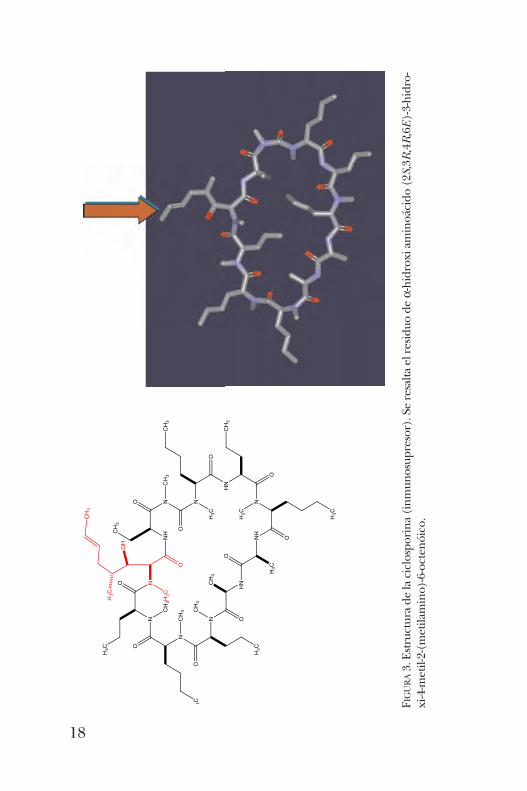
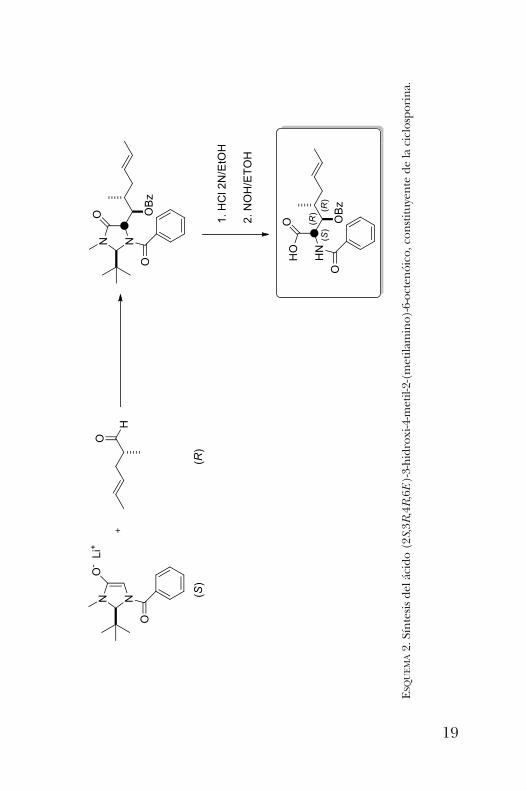
síntesis del ácido (2S,3R,4R,6E)-3-hidroxi-4-metil-2-(metila-mino)-6-octenóico, un a-hidroxi aminoácido constituyentede la ciclosporina (figura 3). La ciclosporina es un undeca-péptido cíclico con actividad inmunosupresora. La metodología seguida para la obtención de este impor-
tante aminoácido consistió, primeramente, en la formacióndel enolato de la imidazolidinona y su reacción con un alde-hído quiral. Este paso procede de manera altamente dias-tereoselectiva como consecuencia del impedimento estéricodel grupo t -butilo, que bloquea una de las caras del enolatodirigiendo el ataque del enolato por la cara opuesta. Las pos-teriores reacciones de hidrólisis permiten la obtención dela-hidroxi ácido deseado (esquema 2) [4].Unos análogos de estas imidazolidinonas son los derivados
que incorporan en lugar del N-metilo en la posición 3, un
16

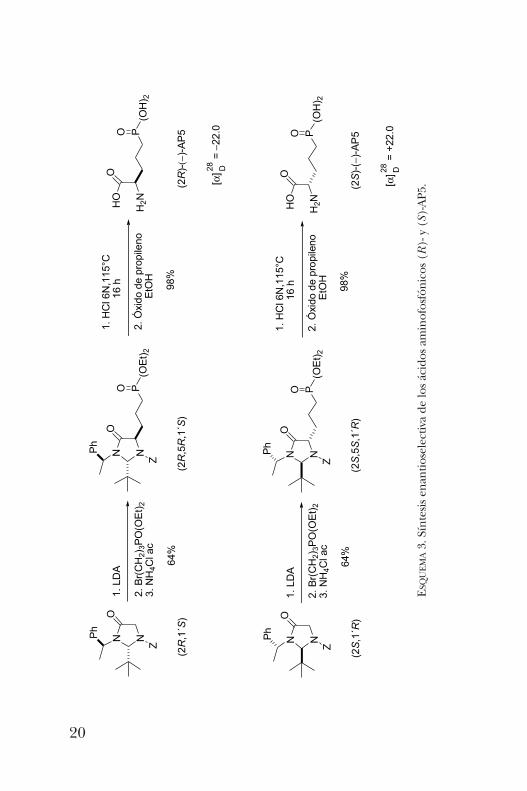
fragmento de feniletilo quiral. Este centro quiral adicionalen el sustrato permitió la obtención del ácido aminofosfó-nico AP5 (de importancia en el tratamiento de la epilepsia)en sus dos formas enantioméricas (esquema 3) [5].Las imidazolidinonas (2R,1’S) y (2S,1’R) se hicieron reac-
cionar con LDA y los enolatos correspondientes fueron adicio-nados al 3-bromopropilfosfonato de dietilo. Los análisis deRMN de 1H y 13C dieron evidencia de la formación del pro-ducto de alquilación con diastereoselectividad mayor al 98%.Los productos de alquilación se obtuvieron en ambos casosen un rendimiento del 64% y su hidrólisis en medio ácidoy posterior tratamiento con óxido de propileno permitióobtener los dos enantiómeros del AP5 con rendimientosexcelentes y sin racemización.Con base en los resultados anteriores, nos hicimos la pre-
gunta: ¿se pueden preparar b -aminoácidos de manera efi-ciente a partir de la b-alanina? Los b-aminoácidos no son tancomunes como sus análogos a-; sin embargo, pueden encon-
17 ##
#
ESQUEMA 1. Síntesis enantioselectiva de a-aminoácidos quirales a partir de laglicina [4].
18
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
FIGURA3. Estructura de la ciclosporina (inm
unosupresor). Se resalta el residuo de a-hidroxi aminoácido (2S,3R,4R,6E)-3-hidro-
xi-4-metil-2-(m
etilamino)-6-octenóico.
19
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
""
"
"
"
ESQ
UEMA2. Síntesis del ácido (2S,3R,4R,6E)-3-hidroxi-4-metil-2-(m
etilamino)-6-octenóico, constituyente de la ciclosporina.
20
##
N N
O
(2R
,1´S
)
Ph
Z
1. L
DA
2. B
r(C
H2)
3PO
(OE
t) 23.
NH
4Cl a
c
6
4%
N N
O
(2R
,5R
,1´S
)
Ph
Z
PO
(OEt
) 2
1. H
Cl 6
N,1
15°C
1
6 h
2. Ó
xido
de
prop
ileno
E
tOH
98%
HO
H2N
O
(2R
)-(!)
-AP
5PO
(OH
) 2
["]28 D
= !2
2.0
N N
O
(2S,
1´R
)
Ph
Z
1. L
DA
2. B
r(C
H2)
3PO
(OEt
) 23.
NH
4Cl a
c
6
4%
N N
O
(2S,
5S,1
´R)
Ph
Z
PO
(OE
t) 2
1. H
Cl 6
N,1
15°C
1
6 h
2. Ó
xido
de
prop
ileno
E
tOH
98%
HO
H2N
O
(2S)
-(!)
-AP
5PO
(OH
) 2
["]28 D
= +2
2.0
ESQ
UEMA3. Síntesis enantioselectiva de los ácidos aminofosfónicos (R)- y (S)-AP5.

trarse en la naturaleza, ya sea de manera libre o como parteconstituyente de otras moléculas como los péptidos, porejemplo. Estas moléculas tienen importancia no sólo bioló-gica, sino también en la industria alimentaria, agropecuariay farmacéutica, entre otras; uno de sus principales usos escomo precursores de b-lactamas, de las cuales es conocidasu actividad como antibióticos.Con esto en mente, se planteó la síntesis del análogo de
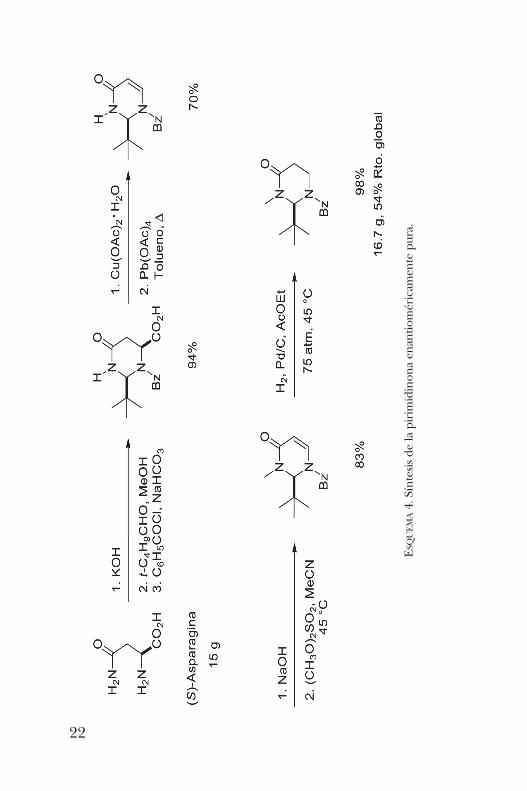
6 miembros de la imidazolidinona previamente estudiada, la1-benzoíl-2-t -butil-3-metil-pirimidin-4-ona [6]. Este hetero-ciclo fue preparado a partir de la (S)-asparagina, un ami-noácido comercialmente disponible en sus dos formas enan-tioméricas, y relativamente barato. Siguiendo la metodologíapresentada en el esquema 4, se logra llegar a la pirimidinonaesperada en cuatro pasos de síntesis con un rendimientoglobal del 54%.Esta pirimidinona es un sólido cristalino del cual se pudo
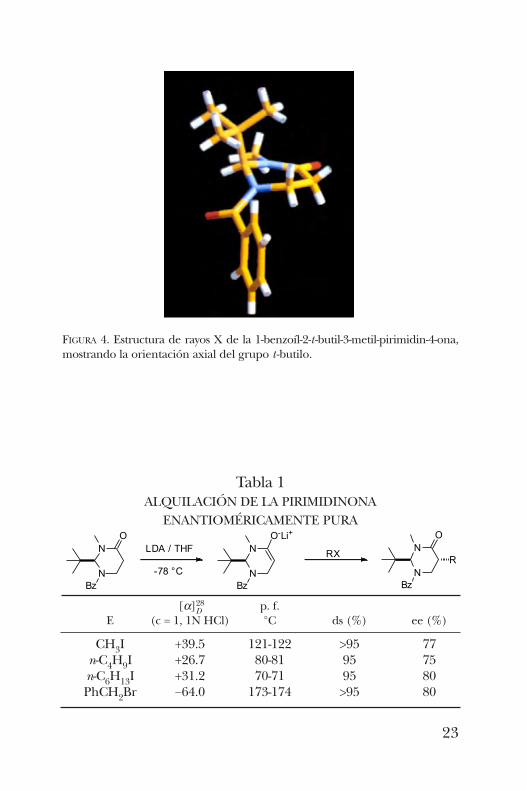
obtener una muestra adecuada para la difracción de rayos X,misma que presenta al grupo t -butilo en posición axial comoresultado de un efecto de tensión alílica (figura 4). Esto per-mitió predecir que la alquilación de este sustrato procederíapor la cara opuesta con alta selectividad.La alquilación de la pirimidinona procedió de manera
altamente diastereoselectiva con diferentes electrófilos y enbuenos rendimientos. Los diastereómeros obtenidos fueronaquellos de configuración relativa trans entre el grupo t-butiloy la cadena alquilada. La tabla 1 resume los resultados de lasreacciones de alquilación que se llevaron a cabo con cuatrodiferentes electrófilos [7].La hidrólisis de las pirimidinonas alquiladas se llevó a cabo
en medio ácido a 120°C y permitió obtener los aminoácidoscorrespondientes con buenos rendimientos y, lo más impor-tante, sin racemización. La tabla 2 presenta los resultadosobtenidos para los cuatro diferentes aminoácidos que fue-ron sintetizados con esta metodología [6a].
21
22
ESQ
UEMA4. Síntesis de la pirimidinona enantioméricam
ente pura.
23
Tabla 1ALQUILACIÓN DE LA PIRIMIDINONA
ENANTIOMÉRICAMENTE PURA
[a]28D p. f.E (c = 1, 1N HCl) °C ds (%) ee (%)
CH3I +39.5 121-122 >95 77n-C4H9I +26.7 80-81 95 75n-C6H13I +31.2 70-71 95 80PhCH2Br −64.0 173-174 >95 80
##
N
N
O
Bz
LDA / THF
-78 °C
N
N
O-Li+
Bz
RX N
N
O
Bz
R
##
FIGURA 4. Estructura de rayos X de la 1-benzoíl-2-t -butil-3-metil-pirimidin-4-ona,mostrando la orientación axial del grupo t -butilo.

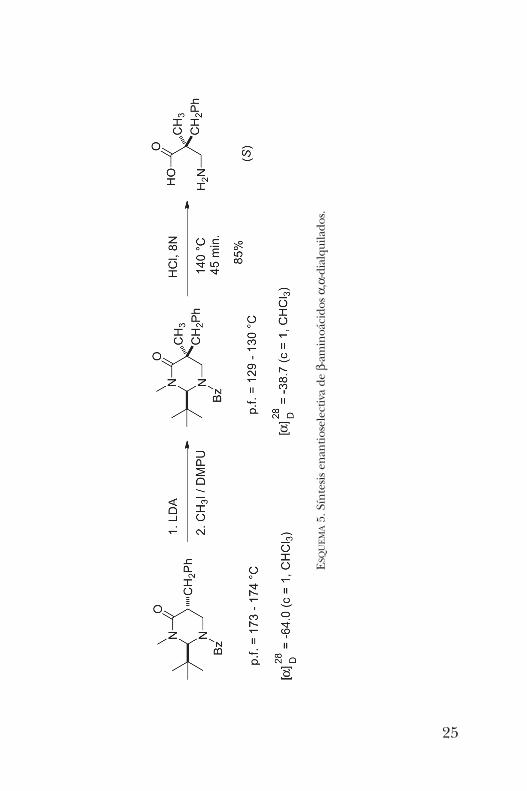
Esta misma pirimidinona resultó ser un sustrato útil en lasíntesis de b-aminoácidos dialquilados enantioméricamentepuros [8]. Como ejemplo, en el esquema 5 se presenta ladialquilación de la pirimidinona previamente alquilada conbromuro de bencilo. Este sustrato se hizo reaccionar con LDApara la formación del enolato y posteriormente se adicionóyoduro de metilo como segundo electrófilo. Nuevamente,este proceso ocurrió de manera altamente diastereoselectivapor la cara opuesta al grupo t -butilo, obteniéndose el com-puesto dialquilado correspondiente. La hidrólisis en medioácido condujo al aminoácido enantioméricamente purocon buen rendimiento.Otra aplicación de estas pirimidinonas fue en la síntesis
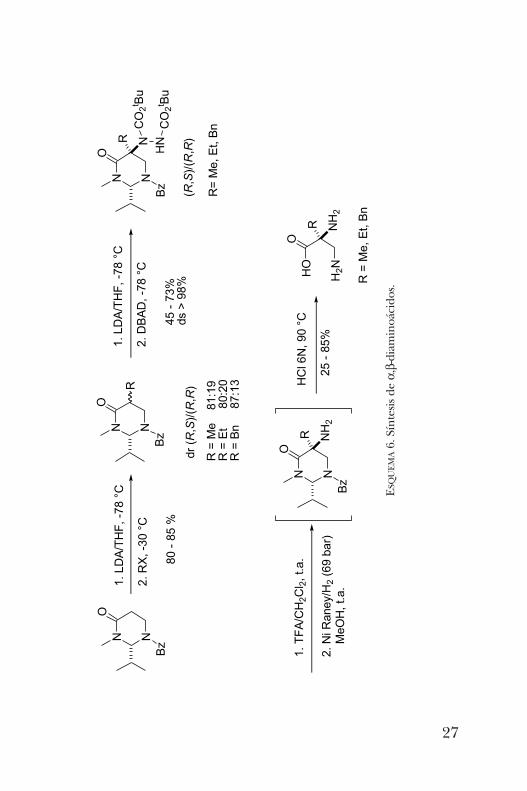
de a,b-diaminoácidos. En este nuevo ejemplo, se empleó lapirimidinona sustituida con un grupo iso -propilo en lugarde t -butilo. La primera alquilación con diferentes electrófilosprocedió con buen rendimiento pero diastereoselectividadmoderada. Sin embargo, el segundo proceso de alquilacióncon azodicarboxilato de di-t -butilo produjo los productos
24
Tabla 2HIDRÓLISIS DE LAS PIRIMIDINONAS ALQUILADAS
Y OBTENCIÓN DE LOS b-AMINOÁCIDOS CORRESPONDIENTES
p. f. RendimientoRX [a]28D °C (%)
CH3I −11.8 185-186 80n-C4H9I +5.3 170-171 80n-C6H13I +6.6 219-220 80PhCH2Br +11.3 225-226 85
##
25
ESQ
UEMA5. Síntesis enantioselectiva de b-aminoácidos a,a-dialquilados.

disustituidos con diastereoselectividad mayor al 98%. Final-mente, la hidrólisis en medio ácido condujo a los a,b-dia-minoácidos de interés (esquema 6) [9].Otro aminoácido sintetizado en nuestro grupo de investi-
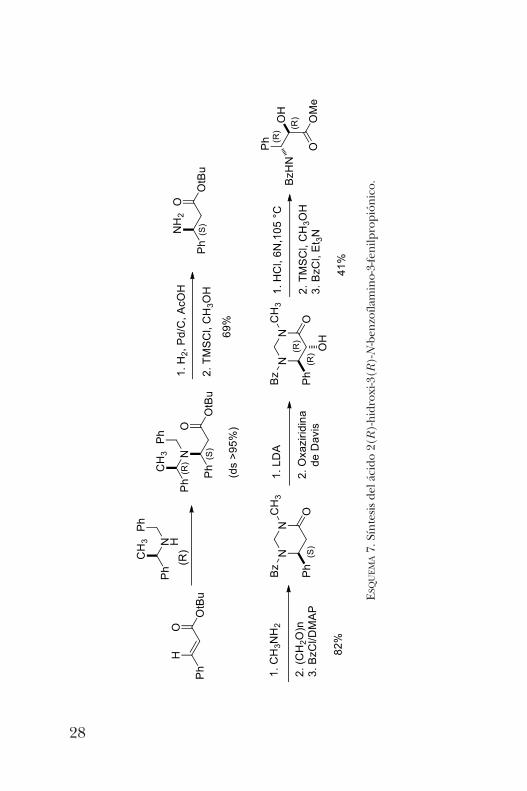
gación es la cadena lateral del Taxol, un importante antican-cerígeno que se aísla de la corteza del árbol Taxus brevifolia [10].Esta ruta de síntesis parte de la adición 1, 4 de la (R)-a-feni-letilamina N-bencilada al cinamato de t -butilo, que procedecon alta diastereoselectividad. Posteriormente se lleva a cabola hidrogenólisis para remover los grupos bencilo y feniletilo.Con este sustrato se sintetizó una pirimidinona que fue hidro-xilada con excelente diastereoselectividad, empleando laoxaziridina de Davis. Finalmente, la hidrólisis permitió aislarel b-aminoácido de interés (esquema 7).Otro aminoácido de gran importancia biológica es la
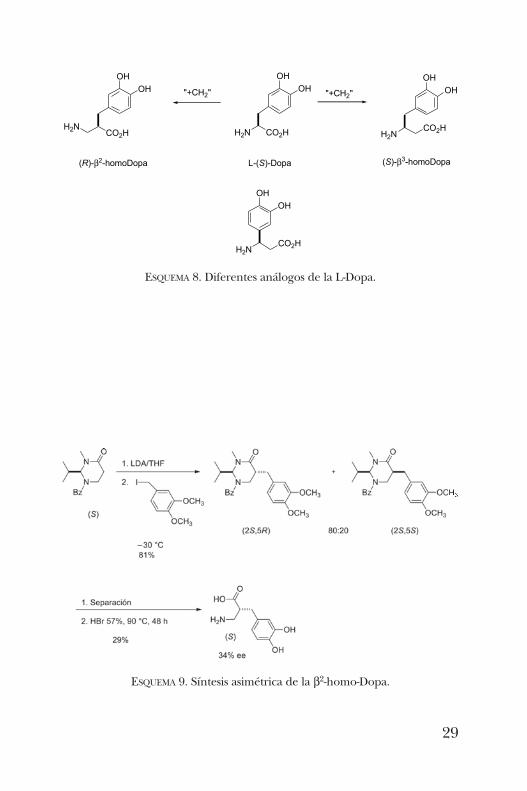
L-Dopa, cuya aplicación es en el tratamiento contra el mal deParkinson. Este aminoácido se obtiene de la planta Vicia fabade manera natural. En 1998, Steglich y colaboradores [11]aislaron un derivado de este aminoácido que es el respon-sable del color azul-morado del hongo Cortinarius violaceus.Cabe mencionar que dependiendo de dónde se introduzcael grupo metileno tendremos b2- o b3-aminoácidos, como semuestra en el esquema 8. La ubicación de la cadena lateralindicará el descriptor que se ha de utilizar.En una primera aproximación, se llevó a cabo la síntesis asi-
métrica de la b2-homo-Dopa empleando la pirimidinonapreviamente reportada [12]. Aunque la alquilación conyoduro de veratrilo procedió con buena selectividad y losdiastereómeros se lograron separar por cromatografía encolumna, la hidrólisis en medio ácido requirió condicionesdrásticas. Es así que se logró obtener el producto de interéscon un rendimiento modesto y un exceso del 34%, debidoa racemización parcial durante la hidrólisis (esquema 9).
26
27
##
N N
O
Bz
1. L
DA/
THF,
-78
°C
2. R
X, -3
0 °C
80 -
85 %
N N
O
Bz
R
dr (R
,S)/(R
,R)
R =
Me
81
:19
R =
Et
8
0:20
R =
Bn
8
7:13
1. L
DA/
THF,
-78
°C
2. D
BAD
, -78
°C
45 -
73%
ds >
98%
N N
O
Bz
R NH
N
CO
2t Bu
CO
2t Bu
(R,S
)/(R
,R)
R=
Me,
Et,
Bn
1. T
FA/C
H2C
l 2, t.
a.
2. N
i Ran
ey/H
2 (6
9 ba
r)
MeO
H, t
.a.
N N
O
Bz
R NH
2
HC
l 6N
, 90
°C
25
- 85
%H
O
H2N
O
R NH
2
R =
Me,
Et,
Bn
ESQ
UEMA6. Síntesis de a,b-diaminoácidos.
28
!
!
!
ESQ
UEMA7. Síntesis del ácido 2(R)-hidroxi-3(R)-N-benzoílamino-3-fenilpropiónico.
29
##
H2N CO2H
OHOH"+CH2" "+CH2"
H2N
OHOH
CO2HCO2H
OHOH
H2N
(R)-!2-homoDopa L-(S)-Dopa (S)-!3-homoDopa
H2NCO2H
OHOH
ESQUEMA 8. Diferentes análogos de la L-Dopa.
ESQUEMA 9. Síntesis asimétrica de la b2-homo-Dopa.

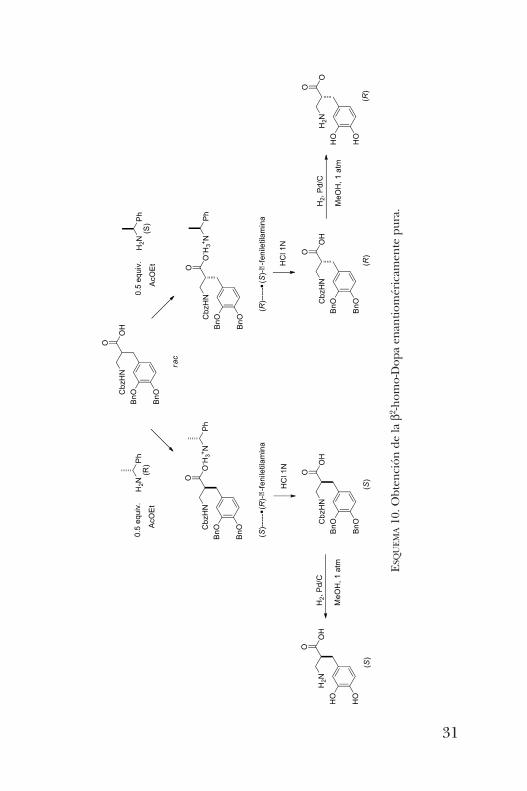
Es así que se optó por una metodología alternativa. Eléster metílico del ácido b-aminopropiónico protegido fuealquilado con yoduro de 3,4-dibenciloxibencilo y el pro-ducto se saponificó con hidróxido de sodio en metanol y agua.Se tomaron dos muestras del derivado carboxílico de lab2-homo-Dopa y se trataron por separado con medio equiva-lente de (R) y (S)-a-feniletilamina, respectivamente. En cadacaso, la sal precipitada se recristalizó hasta obtener valoresconstantes de rotación óptica y punto de fusión. Cuando seempleó (R)-a-feniletilamina, la b2-homo-Dopa recuperadacorrespondió al enantiómero (S) con 96% de exceso enan-tiomérico, mientras que la (S)-a-feniletilamina condujo a la(R)-b2-homo-Dopa con 94% ee (esquema 10) [12].Una vez obtenidos los dos enantiómeros de la b2-homo-
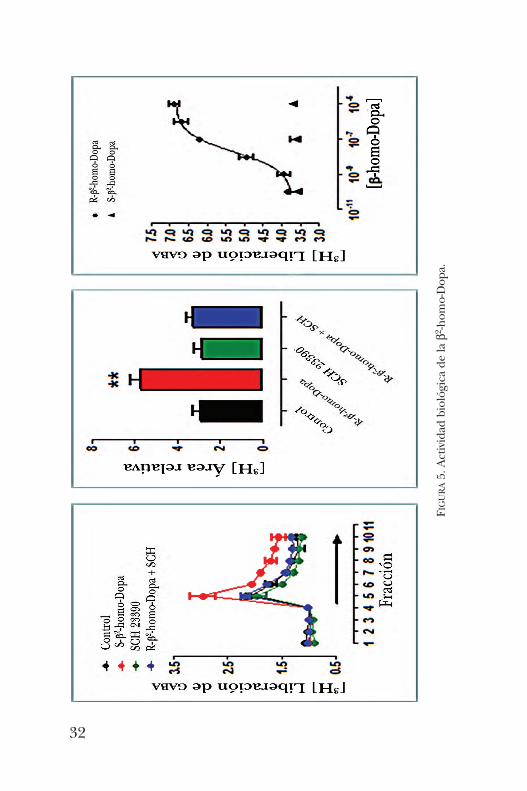
Dopa se iniciaron las pruebas para analizar su posible activi-dad biológica. Hasta el momento, el enantiómero de confi-guración (R) ha mostrado tener actividad sobre los recep-tores que promueven la liberación de GABA (figura 5) [13].Después de desarrollar todas estas estrategias para la sín-
tesis de b-aminoácidos el paso siguiente era analizar sus posi-bles aplicaciones. Estudios de Seebach [14] y Gellman [15]muestran que la presencia de b-aminoácidos modifica drás-ticamente la actividad e incrementa la estabilidad frente ala hidrólisis de varios péptidos naturales con actividad bio-lógica. Asimismo, la unión de estos b-péptidos con ciertosgrupos funcionales en la cadena peptídica puede inducir a laformación de hélices y conformaciones más estables.Un trabajo de nuestro grupo publicado en 2008 [16]
muestra la síntesis en fase sólida de 34 a,b-tetrapéptidos.El esquema 11 muestra la metodología general de este pro-ceso empleando la resina de Wang.El Método de Espectrometría de Masas de Ionización por
Electrospray (ESI-MS, por sus siglas en inglés) ha demostradoser una herramienta muy útil en el análisis de complejos su -pramoleculares formados en solución y transportados a la
30
31
!
ESQ
UEMA10. O
btención de la b2 -hom
o-Dopa enantioméricam
ente pura.
32
FIGURA5. Actividad biológica de la b2 -hom
o-Dopa.

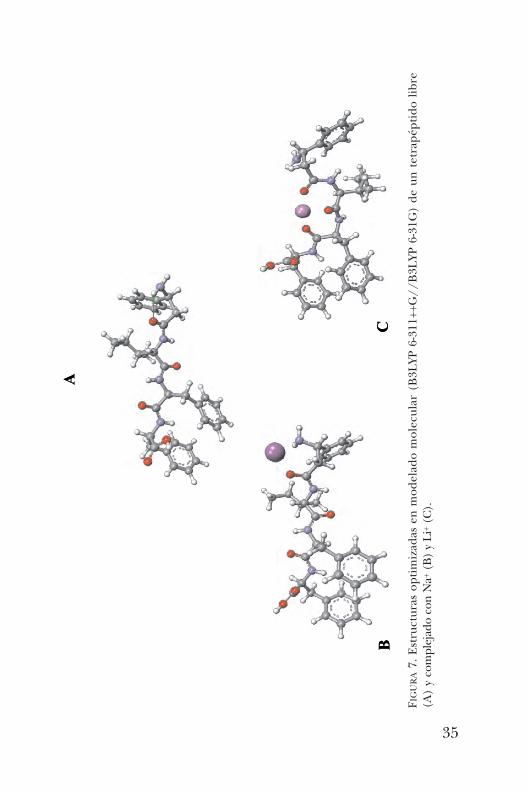
fase gaseosa. En particular, la ionización por electrospray eslo suficientemente suave para permitir la transferencia decomplejos no-covalentes de solución al estado gaseoso sinromper dichas interacciones. Con base en la intensidad delos complejos en el espectro resultante, es posible estimarlas afinidades relativas de los diferentes huéspedes metáli-cos y los péptidos. Es así que se encontró que los a,b-pép-tidos presentaron la siguiente escala de afinidad con los meta-les analizados (figura 6).El modelado molecular del tetrapéptido libre A mostró una
conformación de menor energía lineal, de la cual resulta claroque los posibles centros de coordinación con metales seríanlos grupos carbonilo y el grupo amino terminal (figura 7).
33
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
ESQUEMA 11. Metodología general para la síntesis de péptidos en fase sólida.
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
FIGURA 6. Afinidad relativa de a,b-tetrapéptidos frente a diferentes iones metá-licos, observada por ESI-MS [16].

En presencia de los metales sodio y litio los resultados arro-jaron cambios conformacionales. En el caso del sodio B, elmodelado mostró una estructura del péptido con el sodiocoordinándose de preferencia al amino terminal y un car-bonilo en el extremo del péptido. Mientras que el Litio, porser más pequeño, permite el plegamiento del péptido for-mando una estructura cerrada C colocando al metal en elcentro coordinado a los grupos carbonilo.En años más recientes, se desarrolló una nueva ruta de
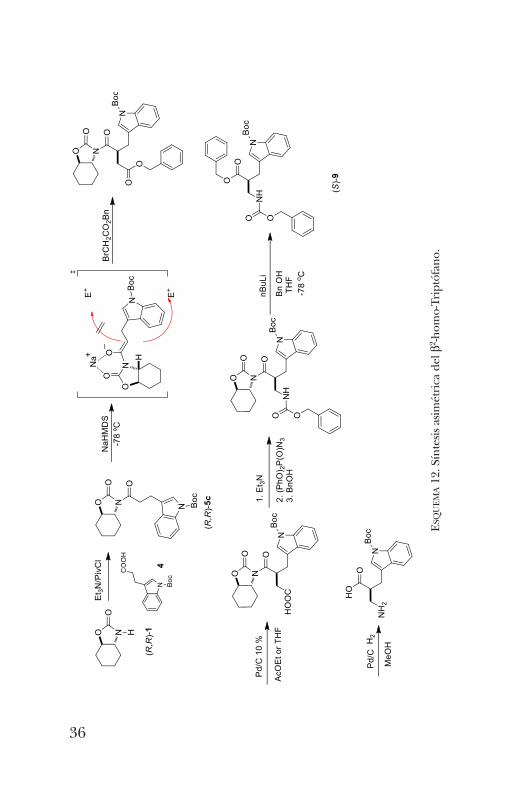
síntesis para la obtención de b-aminoácidos empleando unaoxazolidinona preparada a partir del óxido de ciclohexenousando como auxiliar quiral a la a-feniletilamina [17]. Este sus-trato permitió la obtención del b2-homo-Triptófano. Primera-mente, se llevó a cabo una N-acilación con el ácido 3-(1-t-butoxicarbonil)-1H-indol-3-il)propanóico. El producto obte-nido fue alquilado de manera altamente diastereoselectivacon bromoacetato de bencilo, seguido de una hidrogenacióny una reacción de Curtius que permitió obtener el amino -ácido deseado (esquema 12) [18].El b2-homo-Triptófano mostró actividad biológica intere-
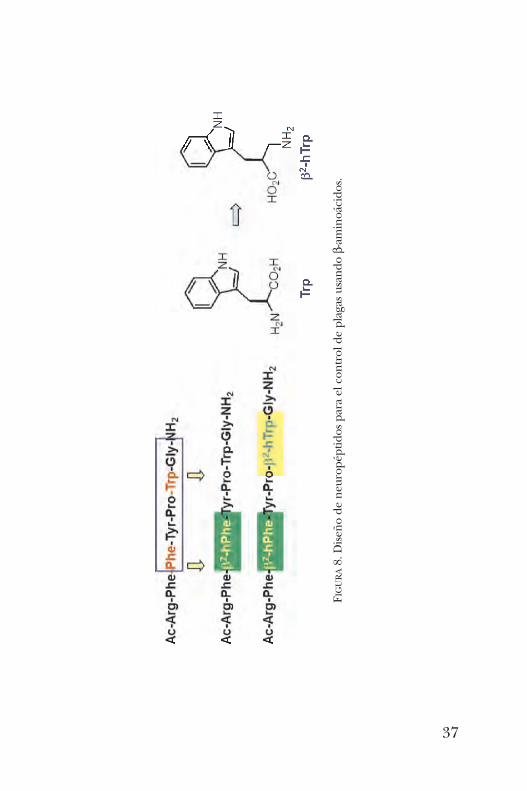
sante al sustituir su análogo a en un neuropéptido quemodifica el sistema de diuresis de los insectos conduciendoa su muerte por deshidratación, siendo de utilidad en elcontrol de plagas (figura 8) [19].Finalmente, el a-aminoácido no-proteinogénico tert -Leu-
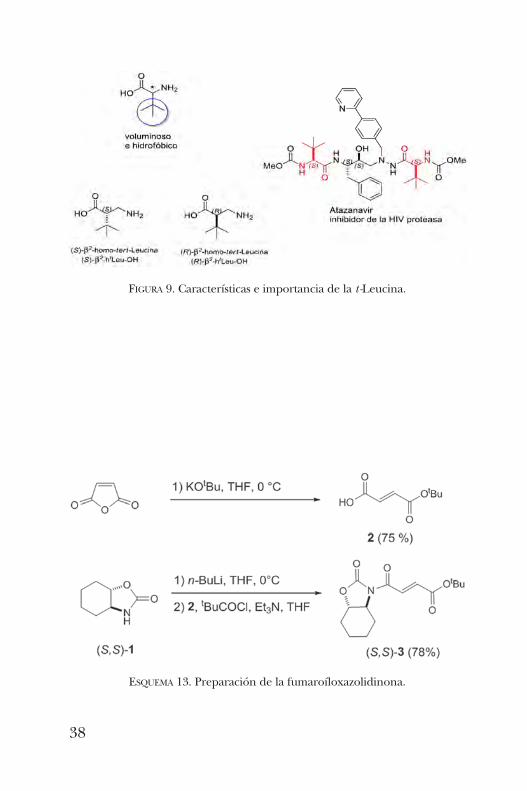
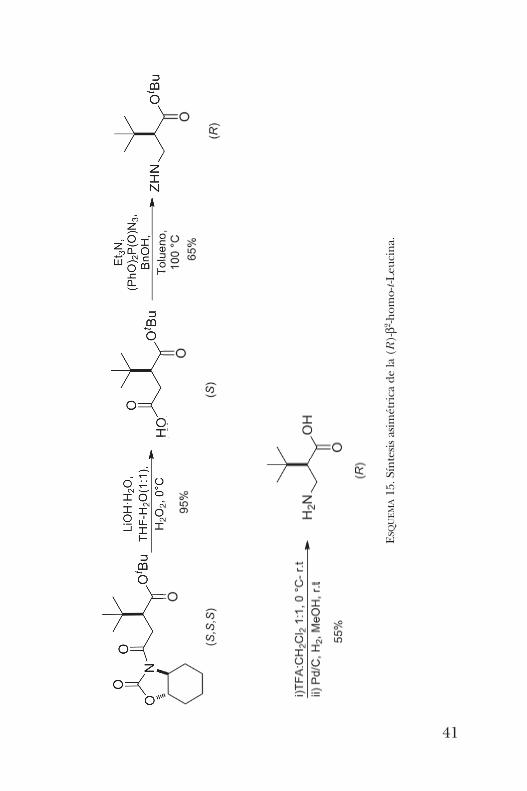
cina (figura 9) ha sido usado como bloque de construcciónpara la síntesis de fármacos y auxiliares quirales, ligantes encatálisis asimétrica y como un organocatalizador quiral muyefectivo. Es por estas razones que resultaba de interés teneracceso a los dos enantiómeros de sus análogos b.La síntesis de la b2-homo-t -Leucina se llevó a cabo como
sigue [20]. Primeramente, se realizó la isomerización del anhí-drido maleico con t -BuOK y activado a través de la formacióndel anhídrido mixto con cloruro de pivaloílo. Posteriormente,se hizo reaccionar con el amiduro litiado de la oxazolidinona
34
35
##
A
B
C
##
##
##
##
##
##
##
##
##
##
##
##
A
##
##
##
##
##
##
##
##
B
##
##
C
##
##
##
FIGURA7. Estructuras optimizadas en modelado molecular (B3LYP 6-311++G//B3LYP 6-31G
) de un tetrapéptido libre
(A) y complejado con Na+(B) y Li+(C).
36
!
ESQ
UEMA12. Síntesis asimétrica del b2 -hom
o-Triptófano.
37
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
FIGURA8. Diseño de neuropéptidos para el control de plagas usando b-aminoácidos.
38
ESQUEMA 13. Preparación de la fumaroíloxazolidinona.
FIGURA 9. Características e importancia de la t -Leucina.

obteniéndose así fumaroíloxazolidinona, como se muestra enel esquema 13. Posteriormente, la adición del radical t -BuHgCl a la
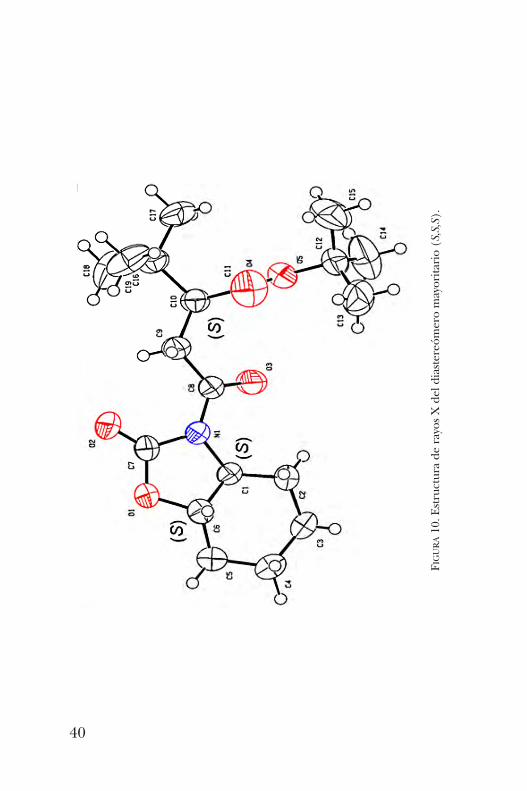
N-fumaroílhexahidrobenzo-2-oxazolidinona condujo al pro-ducto esperado en un rendimiento del 70%. La reacciónprocedió con completa regioselectividad para dar el productode adición en la posición b con una relación de diastereó-meros 9:1 en favor del isómero (S,S,S) (esquema 14), comose dedujo por RMN 1H y más tarde se confirmó por difrac-ción de rayos X (figura 10).Finalmente, se realizó la remoción del auxiliar quiral y un
reordenamiento de Curtius, seguido de la hidrólisis del ésterde t -butilo para dar lugar a la obtención de la (R)-b2-homo-t -Leucina como se muestra en el esquema 15. Este breve resumen de los avances, en cuanto a la síntesis
y aplicación de a- y b-aminoácidos, permite ver que ese meti-leno de diferencia permite modificar y diseñar nuevasmo léculas con propiedades diferentes a las de partida.Muchos de estos análogos presentan una mayor resistencia ala hidrólisis enzimática, que se traducirá en fármacos deefecto más duradero o en nuevos pesticidas que permitanel control de plagas transmitidas por insectos.
39
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
##
#
o
##
#
##
#
l
##
#
##
#
o
##
#
##
#
##
#
##
#
##
#
m
##
#
m
##
#
t
##
#
o
##
#
.
##
#
!
ESQUEMA 14. Adición del radical t -BuHgCl a la N -fumaroílhexahidrobenzo-2-oxazolidinona.
40
FIGURA10. Estructura de rayos X del diastereómero mayoritario (S,S,S).
41
ESQ
UEMA15. Síntesis asimétrica de la (R)-b2 -hom
o-t-Leucina.

NOTAS Y REFERENCIAS BIBLIOGRÁFICAS
[1] a) Juaristi E. Introducción a la Estereoquímica y al Análisis Conforma-cional, SEP: México, 1989; Minal: México, 1998; El Colegio Nacio-nal: México, 2007.
[2] a) Fassihi A. R. Int. J. Pharm., 1993, 92, 1. b) Waldeck, B. Chirality,1993, 5, 330.
[3] Morrison J. D., Mosher H. S. Asymmetric Organic Reactions, Ameri-can Chemical Society: Washington, D. C., 1976.
[4] Seebach D., Juaristi E., Miller D., Schickli C. y Weber T. Helv. Chim.Acta, 1987, 70, 237.
[5] Juaristi E. y García-Barradas O. Tetrahedron, 1995, 51, 3423.[6] a) Juaristi E. y Quintana D. Tetrahedron: Asymmetry, 1992, 3, 723. b) Jua-
risti E. Chiral Reagents for Organic Synthesis, Paquette, L. A., Ed., Wiley:Nueva York, 2003.
[7] Juaristi E., Quintana D., Balderas M. y García-Pérez E. Tetrahedron:Asymmetry, 1996, 7, 2233.
[8] Juaristi E., Balderas M. y Ramírez-Quirós Y. Tetrahedron: Asymmetry,1998, 9, 3881.
[9] Castellanos E., Reyes-Rangel G. y Juaristi E. Helv. Chim. Acta, 2004,87, 1016.
[10] Escalante J. y Juaristi E. Tetrahedron Lett., 1995, 36, 4397.[11] a) von Nussbaum F., Spiteller P., Rueth M., Steglich W., Wanner G.,
Gamblin B., Stievano L. y Wagner F. E. Angew. Chem., Int. Ed., 1998,37, 3292. b) Spiteller P., Rueth M., von Nussbaum F. y Steglich W.Angew. Chem., Int. Ed., 2000, 39, 2754.
[12] Ávila-Ortiz C. G., Reyes-Rangel G. y Juaristi E. Tetrahedron, 2005, 61,8372.
[13] Florán B., Caballero I., Paz F., Reyes-Rangel G. y Juaristi E. Resulta-dos no publicados.
[14] Seebach D., Beck A. K. y Bierbaum D. J. Chem. Biodiv., 2004, 1, 1111.[15] Gelman M. A. y Gellman S. H. en Second Edition of Enantioselective
Synthesis of b-Amino Acids; Juaristi E. y Soloshonok V. A., eds., Wiley-VCH: Nueva York, 2005.
[16] Bandala Y., Aviña J., Rivero I. y Juaristi E. J. Phys. Org. Chem.,2008, 21, 349.
[17] Anaya de Parrodi C., Juaristi E., Quintero L. y Clara-Sosa A. Tetra-hedron: Asymmetry, 1997, 8, 1075.
42

[18] Reyes G., Jiménez E., Olivares J. L. y Juaristi E. Tetrahedron: Asym-metry, 2008, 19, 2839.
[19] Zubrzak P., Williams H., Coast G., Isaac R., Reyes-Rangel G., Jua-risti E., Zabrocki J. y Nachman R. Biopolymers: Peptide Science, 2007,88, 76.
[20] Escudero-Casao M. y Juaristi E. Helv. Chim. Acta, 2012, 95, 1714.
43


ESTUDIO DE LA RELACIÓN ESTRUCTURA-FUNCIÓN DE LAS PROTEÍNAS
GABRIEL DEL RÍODepartamento de Bioquímica y Biología Estructural
Instituto de Fisiología Celular Universidad Nacional Autónoma de México
INTRODUCCIÓN
Las proteínas son moléculas fascinantes que han capturadonuestra atención por mucho tiempo. La digestión de la carne,causada por secreciones gástricas, o la conversión de almi-dones en azúcares se han conocido desde principios delsiglo XVII, pero no fue hasta 1833 que fueron descubiertas porAnselme Payen y más tarde fueron referidas como fermentospor Louis Pasteur al estudiar la fermentación alcohólica.Además de estas transformaciones químicas, las proteínasparticipan en muy diversas funciones fundamentales paralos seres vivos; por ejemplo, en el movimiento muscular, lacomunicación de las células, crecimiento y diferenciacióncelular entre muchas otras. ¿Cómo logran generar tan diver-sos fenómenos las proteínas? Para abordar esta pregunta,los científicos comúnmente purifican a las proteínas y lesmiden diversas características estructurales, tales como lasecuencia de aminoácidos o su estructura tridimensional.
45

Una vez conociendo estas dos características de las proteínas,el fenotipo y su estructura, el problema que enfrentan loscientíficos es cómo relacionarlas. ¿Cómo explicar el fenotipoa partir de la estructura de una proteína? ¿Cómo explicar laestructura a partir del fenotipo generado por una proteína?A este problema se le conoce comúnmente como el proble-ma de la relación estructura-función de las proteínas, perodado el contexto matemático de este trabajo, nos referiremosa este problema como el de la relación estructura-fenotipode las proteínas, para mayor claridad. ¿Por qué este problemaes tan importante y ha mantenido el interés de los científi-cos por más de un siglo sin lograr una solución todavía? Laimportancia de este problema tiene dos aspectos: uno rela-cionado a entender las bases del mecanismo de acción deestas moléculas, y el otro está relacionado con la aplicaciónde este conocimiento para resolver diversos problemas bio-tecnológicos, tales como la producción de cerveza, pan, vino,medicamentos, entre muchos otros. Hoy en día se reconoceque para abordar este problema se requiere de la participa-ción de físicos, matemáticos, biólogos y químicos. Para explo-rar y comunicar los avances en este problema tanto en Méxicocomo en Alemania, el Club Humboldt de México organizóuna reunión de expertos en este campo para que compar-tieran sus conocimientos en El Colegio Nacional. En el pre-sente trabajo, resumiré los avances que hemos tenido en laUniversidad Nacional Autónoma de México abordando esteproblema con formalismos matemáticos y métodos numéricos.Como material de apoyo para el lector, al final de cada sec-ción se mencionarán artículos científicos relacionados concada tema, publicados por mi grupo de trabajo.
46

EXPLORANDO LA NATURALEZA DE LA RELACIÓN
ESTRUCTURA-FENOTIPO DE LAS PROTEÍNAS
Para abordar el problema de la relación estructura-feno-tipo en las proteínas, en mi grupo estamos desarrollandodiferentes formalismos matemáticos. Por una parte, nosinteresa conocer qué tipo de relación es la que mantienenla estructura y el fenotipo en el caso de las proteínas. Enmatemáticas se reconoce que existen diversas clases de rela-ciones: reflexiva, simétrica, antisimétrica, transitiva y de equi-valencia. Una relación en matemáticas se define como laasociación que existe entre los elementos de un grupo (alque se le refiere como dominio) y los elementos de otro con-junto (conocido como codominio). Por ejemplo, el conjuntodominio carros puede contener los siguientes elementos{VW Sedán, BMW Coupé, Nissan Tsuru, Ford Lobo} los cua-les pueden estar asociados a los elementos del conjuntocodominio características_de_carros {caro, accesible en precio,grande, pequeño}; podemos establecer que los elementosVW Sedán y Nissan Tsuru en el conjunto dominio están aso-ciados con los elementos pequeño y accesible en precio delconjunto codominio. Podemos diferir en estas asociaciones,pero el punto en este ejemplo es que existen relaciones entrelos elementos de estos dos conjuntos. Existe una clase derelación llamada función en matemáticas, en la cual sóloexiste una relación para un elemento del conjunto codominiocon los elementos del conjunto dominio ; considerando estadefinición, se puede reconocer que la relación entre el con-junto carros y sus atributos no es una función. Las funcio-nes son una clase de relación que puede ser convenientepara representar la relación entre la estructura y el fenotipode las proteínas, ya que las funciones permiten usar la mismaequivalencia para diferentes formas del mismo objeto. Porejemplo, digamos que la estructura de una proteína la repre-
47

sentamos por x y su fenotipo como y ; si estos dos objetosestán relacionados por una función equivalente, entoncespodemos definir que x = y y, por ende, f(x) = f(y); lo con-trario no es necesariamente cierto, es decir, podemos defi-nir que f(x) = f(y) y eso no necesariamente implica que x = y.Aplicado a proteínas esto implica que, si reconocemos quela estructura de una proteína cambia (por ejemplo, la estruc-tura tridimensional de una proteína puede cambiar porefecto de la temperatura, unión a un cofactor, etcétera) y queel fenotipo de una proteína también cambia (por ejemplo,expresar la misma proteína en diferentes contextos puederesultar en fenotipos diferentes), el uso de funciones mate-máticas para describir la relación que guardan la estructuray el fenotipo resulta conveniente.Si vamos a usar funciones matemáticas para estudiar la
relación estructura-fenotipo en las proteínas, tenemos quedefinir qué tipo de función existe entre estos objetos. Existentres tipos de funciones: inyectivas, sobreyectivas y biyectivas.Para explicar estas funciones, definamos que el conjuntodominio está constituido por las estructuras de una proteína(conjunto S) y el conjunto codominio por los fenotipos (con-junto P). Las funciones inyectivas son aquellas en las que noexiste necesariamente una relación entre cada elemento delos conjuntos S y P, pero para satisfacer la condición de fun-ción, los elementos en el conjunto P sólo se relacionan conun elemento en S y viceversa. Un ejemplo para este tipo defunciones lo constituyen aquellas proteínas para las cualesse conoce su estructura pero no un fenotipo; alternativa-mente, pueden haberse descrito diversos fenotipos paraalguna proteína cuya estructura aún se desconoce. De esteejemplo, podríamos concluir que la relación estructura-fenotipo de las proteínas es una función inyectiva; sin embar-go, podemos reconocer que este tipo de relaciones sóloocurren como consecuencia del carácter temporal de los
48

descubrimientos científicos y no necesariamente sea unacaracterística de esta relación. Por otra parte, las sobre-yec-ciones ocurren cuando más de dos elementos del conjuntodominio establecen una relación con el mismo elemento delconjunto codominio. Las sobre-yecciones son consistentescon la evolución molecular en la que se proponen que pro-teínas similares (varios elementos del conjunto S) puedenpresentar fenotipos similares (un elemento del conjunto P).En este caso, podemos reconocer que fenotipos similaresno implican fenotipos idénticos y consecuentemente, pode-mos concluir que las sobreyecciones no representan la natu-raleza de esta relación. Finalmente, las biyecciones ocurrencuando cada elemento del conjunto dominio tiene una rela-ción con un elemento diferente del conjunto codominio.Es decir, para una estructura de proteína existe uno y sóloun fenotipo. Nosotros proponemos que este tipo de funciónsatisface las características conocidas de esta relación y almismo tiempo presenta un formalismo conveniente paraexplorar el problema de la relación estructura-fenotipo delas proteínas.En las siguientes secciones describiré las herramientas
computacionales que hemos desarrollado en mi grupopara explorar esta relación en las proteínas. Para ahondar en el tema descrito en esta sección, se reco-
mienda al lector consultar la siguiente publicación y lasreferencias bibliográficas en ésta:Ambriz-Rivas M., Pastor N., del Río G. Relating Protein
Structure and Function Through a Bijection and Its Impli-cations on Protein Structure Prediction. En: Protein Structure.InTech-Open Access Publisher, 2012.Esta publicación está disponible gratis en la web en la
siguiente dirección:http://cdn.intechopen.com/pdfs/32430/InTech-Relating_
protein_structure_and_function_through_a_bijection_and_its_implications_on_protein_structure_prediction.pdf.
49

LOS ELEMENTOS MÁS TRANSITADOSEN LA ESTRUCTURA DE PROTEÍNAS
Para identificar el fenotipo asociado a una estructura deproteína, propusimos usar los elementos de la proteína crí-ticos para el fenotipo de interés; dichos elementos se cono-cen como residuos críticos de aminoácido. En ese sentido,cada fenotipo asociado a una proteína es el resultado de lainteracción de ésta con otras moléculas y los residuos críticosmedian estas interacciones. El mecanismo mediante el cuallos residuos críticos determinan la interacción de las proteí -nas depende de la forma en la que las proteínas inter actúancon otras moléculas. Se han descrito tres mecanismos parala interacción de las proteínas con otras moléculas: llave-cerradura, ajuste inducido y selección de confórmeros. En elmecanismo llave-cerradura, la velocidad de la inter accióndepende de la velocidad de difusión de la molécula inter -actuante al sitio activo en la proteína; estos eventos ocurrenen lapsos de tiempo en el orden de 10-9 segundos. Alterna-tivamente, la velocidad de interacción en el mecanismo deajuste inducido depende de la velocidad en la que ocurrencambios de conformación en la proteína al unirse a otra mo -lécula; estos eventos ocurren en lapsos de tiempo de segun-dos a microsegundos. Finalmente, en el mecanismo de selec-ción de confórmeros, se postula que las proteínas existenen conjuntos de conformaciones (por ejemplo, conjuntosdiferentes de estructuras tridimensionales de una proteína)y que sólo algunos de estos conjuntos participan en la unióncon otras moléculas. Cuando ocurre la interacción, hay uncambio en el equilibrio de los conjuntos de conformacio-nes, lo cual ocurre en rangos de tiempo que van de segundosa horas. En todos estos mecanismos, se requieren cambios enla conformación de las proteínas para que ocurra una inter -acción entre la proteína y otra molécula (note que en el casodel mecanismo de llave-cerradura, los cambios de confor-
50

mación son los relacionados al plegamiento de la proteína).De esta forma, nuestra hipótesis es que los residuos que de -terminan estos cambios de conformación en las proteínasdeben ser críticos para el fenotipo asociado a una proteína.Esta hipótesis la evaluamos en cinco proteínas de orígenesdiversos: beta-lactamasa de bacterias, lisozima de pollo, pro-teasa del virus de inmunodeficiencia humano tipo 1, unaribonucleasa bacteriana (barnasa) y la proteína que estácodificada por el gen V del bacteriófago f1. Se escogió esteconjunto de proteínas, porque estas son las únicas reporta-das de las cuales se conoce tanto la estructura tridimensionalcomo cada uno de sus residuos críticos. La estructura tridi-mensional de estas proteínas se representó como una redde contactos entre sus aminoácidos y a esta red se le aplica-ron diversos métodos para identificar los residuos críticos. Eneste ejercicio aprendimos que los residuos que se encuen-tran en medio de las inter acciones (tanto directas como indi-rectas) de todos los residuos correlacionan significativamen-te con los residuos críticos. Estos resultados son consistentescon nuestra hipótesis, ya que los residuos que están en mediode todas las fuerzas de interacción en una proteína (tambiénconocidos como los residuos más transitados) determinan lasconformaciones accesibles a las proteínas.Adicionalmente, estudiamos si los residuos de aminoácido
involucrados directamente en la unión con otras moléculasestaban correlacionados con los residuos más transitados conmayor frecuencia en conformaciones de proteínas, interac-tuando con otras moléculas que en conformaciones en don-de no ocurre esta interacción. Para este experimento, usa-mos proteínas para las cuales su estructura tridimensionalse haya descrito tanto en presencia como en ausencia deuna molécula interactuante. De este conjunto de proteínasdeterminamos sus residuos más transitados y evaluamos enqué estructuras se encontraba la mayor proporción de resi-duos críticos relacionados con los transitados. En este expe-
51

rimento aprendimos que, efectivamente, las conformacionesde las proteínas interactuando con otras moléculas contienenuna mayor proporción de residuos más transitados que corre-lacionan con residuos críticos para la inter acción con otrasmoléculas. En otras palabras, fuimos capaces de identificar laestructura asociada a un fenotipo conociendo los elemen-tos críticos del fenotipo (es decir, residuos críticos).En resumen, la relación entre la estructura de una pro-
teína y un fenotipo puede ser evaluada a través de los resi-duos más transitados. Para leer más sobre este tema, el lector puede consultar
las siguientes publicaciones y las citas bibliográficas referi-das en éstas:Thibert B., Bredesen D. E., del Río G. Improved predic-
tion of critical residues for protein function based on net-work and phylogenetic analyses. En: BMC Bioinformatics,2005, 6 : 213. Esta publicación está disponible en la siguiente dirección
en internet sin costo:http://www.biomedcentral.com/1471-2105/6/213.Cusack M. P., Thibert B., Bredesen D. E., del Río G. Effi-
cient identification of critical residues based only on proteinstructure by network analysis. PLOS ONE. 2007, 2(5): e421. Esta publicación está disponible en la siguiente dirección
en internet sin costo:http://www.plosone.org/article/info%3Adoi%2F10.137
1%2Fjournal.pone.0000421.Montiel Molina H. M., Millán-Pacheco C., Pastor N., del Río G.
Computer-based screening of functional conformers of pro-teins. PLOS Comput. Biol., 2008, 4(2): e1000009. Esta publicación está disponible en la siguiente dirección
en internet sin costo:http://www.ploscompbiol.org/article/info%3Adoi%2F
10.1371%2Fjournal.pcbi.1000009.
52

CONCLUSIONES Y PERSPECTIVAS
Hemos aprendido que la estructura de las proteínas y losfenotipos puede relacionarse a través de una biyección usandolos residuos más transitados como criterio. Aunque estosresultados nos han ayudado a entender mejor la naturalezade esta relación, aún no estamos cerca de poder predecirde forma eficiente la estructura de una proteína a partir deun fenotipo y viceversa. Estas limitaciones se basan en quela estructura tridimensional para muchas proteínas es aúndesconocida (sola o en complejo con otra molécula) y/oque para muchas proteínas no se han identificado aún losresiduos críticos. Para resolver estas limitaciones, actual-mente estamos desarrollando:a) Un modelo matemático que facilite el modelado de la
relación estructura-fenotipo y la predicción de la estructuratridimensional de las proteínas.b) Una metodología experimental para identificar de for-
ma exhaustiva y efectiva cada uno de los residuos críticos delas proteínas.Esperamos que en el futuro cercano reuniones, como la
organizada por el Club Humboldt, ayuden a promover elnuevo conocimiento en el estudio de la relación estructura-fenotipo de las proteínas y con ello se promueva la fase deldescubrimiento y aplicación de estos nuevos conceptosy métodos.El doctor Gabriel del Río Guerra trabaja en el departa-
mento de bioquímica y biología estructural en el Institutode Fisiología Celular en la Universidad Nacional Autónoma deMéxico. Para mayor información sobre su trabajo, visite lasiguiente dirección en internet:http://www.ifc.unam.mx/investigadores/gabriel-delrio.
53


LA INFLUENCIA DE LOS CAMPOSMAGNÉTICOS EN LA CRISTALIZACIÓN Y ESTRUCTURA TRIDIMENSIONAL
DE LAS PROTEÍNAS
ABEL MORENO
EUGENIO DE LA MORA
NURIA SÁNCHEZ-PUIGDepartamento de Química de Biomacromoléculas
Instituto de QuímicaUniversidad Nacional Autónoma de México
RESUMEN
En este trabajo, hemos cuantificado y comparado los efec-tos de los campos electromagnéticos sobre la nucleación y elproceso de crecimiento cristalino de proteínas. En esta inves-tigación se estudió el efecto de un campo magnético intensoen el crecimiento de cristales y en la estructura tridimen-sional de proteínas modelo y proteínas nuevas. Esto se llevóa cabo a través de difracción de rayos X.
Palabras clave: Lisozima, Taumatina, campo magnético, cre-cimiento de cristales en geles, agarosa, hidrogeles de alcoholpolivinílico.
55

INTRODUCCIÓN
Para el estudio de rayos X, y la subsecuente determina-ción estructural de proteínas, se requiere preferentementede cristales de adecuado tamaño; sin embargo, esto repre-senta un problema difícil de superar a pesar de que se hayandesarrollado nuevos métodos para obtener cristales, enpequeñas gotas, en tiempos más cortos e inclusive usandodispensadores automáticos a través de robots. En la actuali-dad, se han hecho muchos esfuerzos para implementar pro-cedimientos que faciliten el procesamiento de datos em -pleando computadoras personales; aun cuando se han enfo-cado algunos esfuerzos en entender los aspectos cinéticosde la cristalización de proteínas o el estudio de los paráme-tros que afectan la conducta de cristalización. Sólo muypocas investigaciones han llevado a cabo estudios sistemáti-cos para entender cómo estas variables físicas y químicas in -fluyen de forma positiva en el rendimiento en términos detamaño cristalino y la calidad cristalina.En la última década, mucha de la investigación que se ha
hecho se ha enfocado en investigar el crecimiento de cris-tales de macromoléculas biológicas o complejos macromo-leculares para incrementar la calidad cristalina, con el obje-to de tener una cristalografía de alta resolución (figura 1).Particularmente, las proteínas modelo como la Lisozima (Mr
14.3 kDa) y la Taumatina (Mr 22.2 kDa), entre otras, se hanestudiado de forma extensiva, pero las investigaciones sólose han enfocado en entender todos los aspectos relacionadoscon el crecimiento de los cristales. El entender el proceso decrecimiento cristalino requiere no solamente obtener cris-tales, sino que va desde conocer acerca de la sobreexpresión,la purificación, la cristalización, el polimorfismo cristalinohasta el análisis de la estructura tridimensional (estimación dela calidad cristalina, la colecta de datos, la investigación sobre
56

57
contactos cristalinos). Actualmente, las investigaciones bio-lógicas requieren no sólo de cristales de alta calidad para uncorrecto análisis de la estructura vía rayos X, sino también ladifracción de neutrones de cristales biológicos que requierede un tamaño considerable para tener una difracción ade-cuada. Existen también métodos de alta eficiencia disponi-bles para obtener cristales empleando robots o dispensadoresautomáticos.Sin embargo, a pesar de todos estos adelantos, existen aún
muchas proteínas que no se han cristalizado o, en el mejorde los casos, se obtienen cristales con una difracción muypobre. Generalmente, las proteínas que no cristalizan pormétodos convencionales se descartan, y pocos esfuerzos se
FIGURA 1. Las macromoléculas biológicas (líneas en azul) y sus combinacionesproducen diferentes complejos macromoleculares importantes en la investiga-ción biológica y son el centro de investigaciones estructurales actuales. Los lípidosno son macromoléculas biológicas, pero participan en la formación de las mem-branas celulares al unirse con las proteínas.

hacen para obtener información adicional acerca de su con-ducta de cristalización o para encontrar nuevas formaso métodos para incrementar su calidad cristalina.La mejoría de esta calidad cristalina puede obtenerse redu-
ciendo la convección natural y el flujo de oscilaciones, lo queresulta en una homogeneidad química [1]. Estas vías paraincrementar la calidad cristalina son: 1) crecer cristales encondiciones de microgravedad, 2) crecer cristales en tuboscapilares y 3) crecer cristales en geles.Otro procedimiento efectivo para crecer cristales de alta
calidad cristalina, que ha atraído la atención de algunoscientíficos, es la aplicación de un campo magnético en el pro-ceso de crecimiento cristalino. Cualquier molécula suscep-tible de ser influida por un campo magnético debe tener unaalta conductividad eléctrica.Es bien conocido que las proteínas que tienen una baja
conductividad eléctrica incitan a preguntarse si son o no afec-tadas por un campo magnético. A nivel molecular, las pro-teínas que poseen alfa hélices en su estructura secundaria,presentan una anisotropía de la susceptibilidad diamagné-tica, debido a su alineamiento axial del enlace peptídico. Estaanisotropía de la susceptibilidad diamagnética resulta en laorientación de un núcleo cristalino de proteína, crecido enpresencia de un campo magnético intenso [2]. Adicional-mente, si combinamos el crecimiento de cristales en gelesbajo la influencia de campos magnéticos, resulta un incre-mento en la formación de un cristal, ya que las unidades decrecimiento se ubican en el cristal. La razón es que estos pro-cesos de transporte en geles proceden de una difusión con-trolada; sin embargo, existen reportes contradictorios quemuestran que los campos magnéticos no ejercen efectosobre el crecimiento de cristales de proteínas, en particularcuando estos experimentos son llevados a cabo en solucióny cuando se usan campos magnéticos débiles o no muyintensos. Estas observaciones con los casos de estudios mos-
58

trados en este artículo, requieren investigaciones más pro-fundas para investigar qué parámetros son los decisivos paraincrementar esta calidad cristalina.Los trabajos pioneros sobre la influencia de campos mag-
néticos en la cristalización de proteínas, fueron hechos porSazaki y col., [3], Ataka y col., [4]. Ellos encontraron un ali-neamiento importante de los cristales de proteínas conrespecto al campo magnético aplicado; además los cristalesfueron más grandes y hubo una nucleación controlada.También existen reportes sobre el uso de campos magnéti-cos alternos y se ha observado un incremento en la actividadcatalítica de una enzima si los cristales crecen en estas con-diciones bajo la influencia de campos magnéticos intensos [5].El primer reporte que combina el crecimiento de cristales
en geles con campo magnético fue llevado a cabo por Kinos-hita y col., [6] su objetivo fue incrementar la calidad cristalinay facilitar la colecta de cristales para el diseño de fármacosbasados en la estructura de las macromoléculas biológicas.Lübbert y col., [7] analizaron por medio de curvas de topo-grafía de rayos X y midieron las diferencias en el mosaicis-mo entre los cristales crecidos en presencia y ausencia decampos magnéticos de 2.4T, así como midieron el efectode los campos magnéticos en geles y sin geles.Hemos enfocado nuestro interés en entender el papel que
juegan los campos magnéticos en el proceso de cristaliza-ción de proteínas modelo; sin embargo, en este trabajo enparticular hemos cuantificado y comparado estos efectos decampos electromagnéticos sobre la nucleación hasta el pro-ceso de crecimiento de diferentes proteínas. La influenciade campos magnéticos intensos sobre el crecimiento crista-lino y sobre la estructura 3D en proteínas modelo y en nue-vas proteínas, ha sido comparada empleando la cristalogra-fía por rayos X.
59

PROCEDIMIENTOS EXPERIMENTALES
Cristalización en geles bajo la influenciade un campo magnético
La cristalización de proteínas, empleando el método“batch”, en presencia de campos magnéticos intensos, sebasó en el método propuesto por Surade y col., [8]. Lascondiciones “batch” consisten en mezclar volúmenes igualesde proteína, agente precipitante y la solución gelificada (estegel puede ser agarosa [9] o alcohol polivinílico, PVA por sussiglas en inglés [10]). La solución gelificada (casi un fluidoviscoso) se introduce en un capilar (en un diámetro entre0.3-0.5 mm) y los extremos del capilar se sellan con silicóno plastilina. El tubo capilar que contiene la muestra dentrodel gel (proteína/agente precipitante) se pone 10 minutosa 10°C y se introduce en el magneto de resonancia de11.75 Tesla (aproximadamente de 500 MHz), éste se aplicade 24 a 48 horas, o por el tiempo en el que aparezca la nuclea -ción de la proteína que se estudia. El campo magnético esaquel que puede obtenerse dentro de un magneto clásico deResonancia Magnética Nuclear (RMN, por sus siglas en inglés).Las figuras 2 y 3 muestran este efecto cuando se aplica al cre-cimiento cristalino un campo magnético intenso con dos nue-vas proteínas, una llamada SdsA [11] y un módulo de cris-talización denominado 2TEL-Lys [12]. Ambas se orientana lo largo de su eje C y puede notarse una regularidad tantoen el tamaño de los cristales obtenidos, como en la orienta-ción respectiva. Puede observarse que cuando los cristalescrecen en geles de PVA suelen ser un poco más grandes quelos crecidos en agarosa, además el hábito de los mismos esmás euhédrico.
60

Análisis por difracción de rayos X
La caracterización de la calidad cristalina de dos proteí-nas modelo, como la lisozima (de la clara de huevo) y la tau-matina (del fruto de la pasión) se llevó a cabo por medio dela difracción de rayos X empleando radiación sincrotrón.Todos los cristales analizados fueron aproximadamente delmismo tamaño y se empleó el mismo procedimiento. Losexperimentos de difracción se llevaron a cabo en la líneaID14-4 del sincrotrón europeo (ESRF) ubicado en Grenoble(Francia). Para cada proteína se tomaron 180 grados y lacolecta se hizo con un detector de rayos X marca ADSC Q315.
RESULTADOS Y DISCUSIÓN
Publicaciones recientes han mostrado que el uso combi-nado de crecimiento de cristales en geles junto con camposmagnéticos intensos, incrementa la calidad cristalina. Esta me -jora en la calidad cristalina, se observa como un mejor límitede resolución, así como una mejor estadística en los datoscristalográficos —reducción de datos y afinamiento [7, 8]—.
61
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
FIGURA 2. Cristales de SdsA en batch crecidos en: a) solución (controles) y b) cre-cidos en geles de PVA en presencia de un campo magnético intenso de 11.74 Tesla(aproximadamente 500 MHz).
62
!
FIGURA 3. Cristales de la proteína 2TEL-Lis crecidos en: a) batch en presencia deun campo magnético intenso de 500 MHz, b) cristales crecidos por la técnicasde contra-difusión en geles de PVA y c) cristales crecidos por la técnicas de con-tradifusión en geles de PVA en presencia de un campo magnético intenso de500 MHz. En ausencia de campo magnético no se obtuvieron cristales en batch.La escala está representada por una barra de 100 micrones en cada caso.

La combinación lógica de ambos efectos en el crecimientode cristales de proteínas cuando usamos geles y camposmagnéticos, resulta en un claro efecto global en el incre-mento de la calidad cristalina, como puede juzgarse de lacalidad de los datos colectados de rayos X. Desde el puntode vista cualitativo, los cristales de SdsA crecidos en geles dePVA y empleando un campo magnético de 500 MHz, duranteel proceso de crecimiento cristalino produjo cristales conun hábito bien definido, los cristales tuvieron tamaños máshomogéneos y una orientación preferencial, comparadoscon los controles que no tenían campo aplicado (figura 2).En el caso de una proteína rica en alfa-hélices denominada2TEL-Lys, el campo magnético tuvo un efecto mucho másmarcado, en condiciones “batch” los cristales crecieron per-fectamente alineados con el vector del campo magnético, queen ausencia del mismo fueron agujas delgadas o diáboloscon cabezas formadas de muchos policristales (figura 3). Estemismo efecto, en donde el campo regula el tamaño y orientaperfectamente los cristales, fue reportado previamente paraproteínas modelo como la lisozima y la taumatina [8]. Elanálisis de rayos X mostró una mejora en todos los datosy parámetros cristalográficos cuando los cristales crecieronen geles y bajo la influencia de campos magnéticos intensos(tabla 1). En algunos casos la resolución no se modificó, nila mosaicidad, pero sí se observó una mejora en el factor Bde Wilson, donde éste tuvo valores mucho menores y loscristales crecieron en ambiente difusivo (geles) y en pre-sencia de un campo magnético intenso. Inclusive cuando secrecieron cristales en solución, sólo con la presencia de cam-pos magnéticos, los cristales fueron de mejor calidad quelos controles.
63
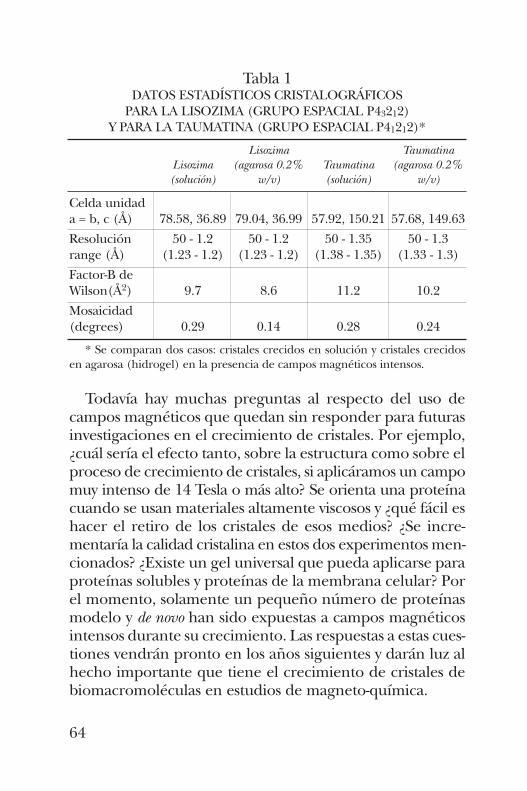
Todavía hay muchas preguntas al respecto del uso decampos magnéticos que quedan sin responder para futurasinvestigaciones en el crecimiento de cristales. Por ejemplo,¿cuál sería el efecto tanto, sobre la estructura como sobre elproceso de crecimiento de cristales, si aplicáramos un campomuy intenso de 14 Tesla o más alto? Se orienta una proteínacuando se usan materiales altamente viscosos y ¿qué fácil eshacer el retiro de los cristales de esos medios? ¿Se incre-mentaría la calidad cristalina en estos dos experimentos men-cionados? ¿Existe un gel universal que pueda aplicarse paraproteínas solubles y proteínas de la membrana celular? Porel momento, solamente un pequeño número de proteínasmodelo y de novo han sido expuestas a campos magnéticosintensos durante su crecimiento. Las respuestas a estas cues-tiones vendrán pronto en los años siguientes y darán luz alhecho importante que tiene el crecimiento de cristales debiomacromoléculas en estudios de magneto-química.
64
Tabla 1DATOS ESTADÍSTICOS CRISTALOGRÁFICOS PARA LA LISOZIMA (GRUPO ESPACIAL P43212)
Y PARA LA TAUMATINA (GRUPO ESPACIAL P41212)*
Lisozima TaumatinaLisozima (agarosa 0.2% Taumatina (agarosa 0.2% (solución) w/v) (solución) w/v)
Celda unidad a = b, c (Å) 78.58, 36.89 79.04, 36.99 57.92, 150.21 57.68, 149.63
Resolución 50 - 1.2 50 - 1.2 50 - 1.35 50 - 1.3range (Å) (1.23 - 1.2) (1.23 - 1.2) (1.38 - 1.35) (1.33 - 1.3)
Factor-B de Wilson(Å2) 9.7 8.6 11.2 10.2
Mosaicidad (degrees) 0.29 0.14 0.28 0.24
* Se comparan dos casos: cristales crecidos en solución y cristales crecidosen agarosa (hidrogel) en la presencia de campos magnéticos intensos.

4. CONCLUSIONES Y PERSPECTIVAS
Los experimentos para estudiar la calidad cristalina, ya seaen solución o en geles, deben implicar el trabajo con pro-teínas de alta pureza (mayor a 99%) y siempre debe trabajar-se con la misma muestra. De nuestros resultados, los mejorescristales se obtuvieron cuando el transporte convectivo seminimizó (para ello se empleó el crecimiento de cristales engeles y en medios capilares, aplicando campos magnéticosintensos). Es claro que las impurezas macromoleculares fue-ron removidas previamente. Finalmente, las futuras investiga-ciones en este campo deben enfocarse con proteínas modeloy de novo, que tengan grupos espaciales polares como losP21 y C2. Estos grupos espaciales mostrarán interesantesresultados relacionados con la orientación cristalográficay nos darán luz en la relación que existe entre la estructura-función.
AGRADECIMIENTOS
Uno de los autores (A. M.) agradece al Conacyt el pro-yecto número 175924 por la financiación para llevar a caboesta investigación. N. S. P., agradece el apoyo del Conacytcon el proyecto número 167359. E. de la Mora agradece a laDGAPA-UNAM por el apoyo de la beca postdoctoral.
REFERENCIAS
[1] Moreno A., Quiroz-García B., Yokaichiya F., Stojanoff V. y Rudolph P.Crystal Research and Technology, 2007, 42, 231-236.
[2] Astier J. P., Veesler S. y Boistelle R. Acta Crystallogr. D: Biol. Crystallogr.,1998, 54, 703-706.
[3] Sazaki G., Yoshida E., Komatsu H., Nakada T., Miyashita S. y Wata-nabe K. Journal of Crystal Growth, 1997, 173, 231-234.
65

[4] Ataka M., Katoh E. y Wakayama N. I. Journal of Crystal Growth, 1997,173, 592-596.
[5] Sakai Y., Oishi A. y Takahashi F. Biotechnol. Bioeng., 1999, 62, 363-367.[6] Kinoshita T., Ataka M., Warizaya M., Neya M. y Fujii T. Acta Crysta-
llogr. D: Biol. Crystallogr., 2003, 59, 1333-1335.[7] Lubbert D., Meents A. y Weckert E. Acta Crystallogr. D: Biol. Crysta-
llogr., 2004, 60, 987-998.[8] Surade S., Ochi T., Nietlispach D., Chirgadze D. y Moreno A. Crystal
Growth & Design, 2009, 10, 691-699.[9] Sugiyama S., Maruyama M., Sazaki G., Hirose M., Adachi H., Taka-
no K., Murakami S., Inoue T., Mori Y. y Matsumura H. J. Am.Chem. Soc., 2012, 134, 5786-5789.
[10] Pietras Z., Lin H. T., Surade S., Luisi B., Slattery O., Pos K. M.y Moreno A. Journal of Applied Crystallography, 2010, 43, 58-63.
[11] Hagelueken G., Adams T. M., Wiehlmann L., Widow U., Kolmar H.,Tummler B., Heinz D. W. y Schubert W. D. Proc. Natl. Acad. Sci.USA, 2006, 103, 7631-7636.
[12] Nauli S., Farr S., Lee Y. J., Kim H. Y., Faham S. y Bowie J. U. ProteinSci., 2007, 16, 2542-2551.
66

FUENTES DE RAYOS X COMPACTAS Y BRILLANTES PARA ESTUDIAR LA DISTRIBUCIÓN ATÓMICA
EN LAS PROTEÍNAS
XIM BOKHIMI
Instituto de FísicaUniversidad Nacional Autónoma de México
RESUMEN
Describimos las nuevas fuentes de rayos X compactas y bri-llantes para el estudio de la distribución atómica de las pro-teínas. Su brillantez es mayor que la de los tubos selladosde rayos X, y similar a la de los sincrotrones de las primeray segunda generaciones. Describimos también el desarrollode las nuevas fuentes de rayos X basadas en la aceleración deelectrones en el campo de la estela generada al interactuarun intenso pulso de láser con un gas de helio. Estas fuentesproducen pulsos de rayos X con brillantez comparable conla de los sincrotrones de tercera generación. En un futuro cer-cano, estas fuentes de rayos X reemplazarán a los sincrotronespara estudiar la distribución atómica de proteínas, lo quepermitirá que un mayor número de científicos realicen estetipo de análisis, porque estas nuevas fuentes de rayos X ten-drán un costo bajo y consumirán menos energía que losactuales sincrotrones.
67

INTRODUCCIÓN
El conocimiento de la distribución atómica y molecularen cualquier sustancia es básico para entender sus propie-dades macroscópicas. Cuando los átomos interactúan entre sí,formando arreglos ordenados con simetría de traslación, sudistribución puede obtenerse directamente de experimentosde difracción de rayos X o de neutrones. Este arreglo de áto-mos forma cristales con dimensiones desde nanómetros hastametros, dependiendo del sistema y su entorno.Cuando el material de interés es orgánico (por ejemplo,
formado por proteínas), y se analiza por difracción de rayos X,hay que prestar atención a la densidad de energía del hazde radiación incidente y el tiempo de irradiación de la mues-tra para evitar dañar el arreglo atómico durante el análisis.En un experimento de difracción de rayos X en proteínas
es necesario usar una fuente de rayos X que proporcione unadensidad de fotones alta sobre la muestra. Esto permite obte-ner un patrón de difracción con suficiente intensidad en untiempo relativamente corto, de manera que la informaciónobtenida sobre la distribución atómica es confiable. Para analizar el arreglo atómico en proteínas con difrac-
ción de rayos X en un laboratorio estándar, el difractómetro derayos X usado normalmente tiene un tubo sellado de rayos Xque consume entre 0.15 y 2 kW [1]. La brillantez de estostubos es de entre 107 y 108 fotones/s/mrad2/mm2/0.1% ab–0.1% (ab significa un ancho de banda de 10-3 la energíade los rayos X empleados). Por ejemplo, si la energía de losrayos X es de 8 KeV, un ancho de banda de 0.1% es iguala 8 × 10-3 KeV = 8 eV.La brillantez del tubo sellado de rayos X es demasiado
baja si el interés del experimento de difracción es determi-nar la estructura atómica de una proteína. Por consiguiente,llevar a cabo esta clase de experimentos en un laboratorio
68
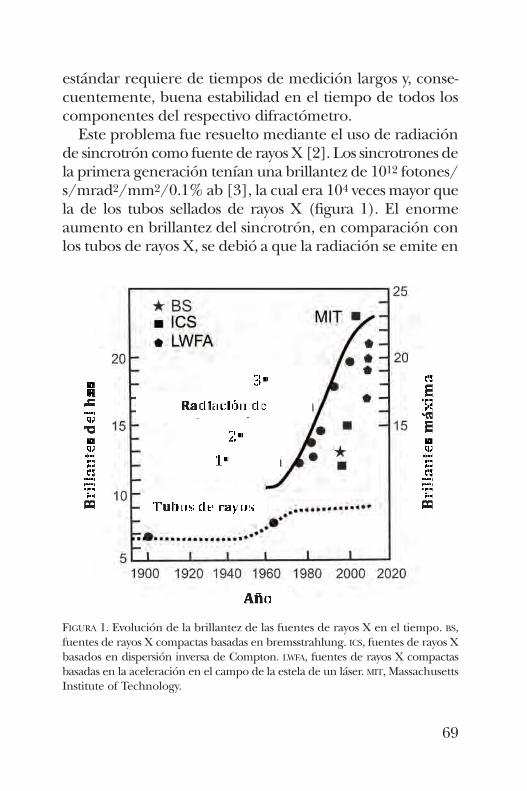
estándar requiere de tiempos de medición largos y, conse-cuentemente, buena estabilidad en el tiempo de todos loscomponentes del respectivo difractómetro.Este problema fue resuelto mediante el uso de radiación
de sincrotrón como fuente de rayos X [2]. Los sincrotrones dela primera generación tenían una brillantez de 1012 fotones/s/mrad2/mm2/0.1% ab [3], la cual era 104 veces mayor quela de los tubos sellados de rayos X (figura 1). El enormeaumento en brillantez del sincrotrón, en comparación conlos tubos de rayos X, se debió a que la radiación se emite en
69
##
!"#$$%&'()*+($*,%)*
!
!"#$$%&'()*+,-#+%!
!
!"#!
!!!!
!!!!!!!
!
"#$%#&%'(!$)!' !
./0,*!$)!+#1,*!! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
##
*!
!!
!
!!
!!!
!
! !!
! ! !! !
FIGURA 1. Evolución de la brillantez de las fuentes de rayos X en el tiempo. BS,fuentes de rayos X compactas basadas en bremsstrahlung. ICS, fuentes de rayos Xbasados en dispersión inversa de Compton. LWFA, fuentes de rayos X compactasbasadas en la aceleración en el campo de la estela de un láser. MIT, MassachusettsInstitute of Technology.

un ángulo sólido pequeño. Esto es importante si el haz se usapara estudios de difracción de cristales de proteínas pequeños.Hoy en día los sincrotrones estándar de la tercera genera-ción tienen una brillantez de 1018 a 1023 fotones/s/mrad2/mm2/0.1% ab [4].Es interesante mencionar que los flujos totales de rayos X
(fotones/s) de un tubo sellado de rayos X y de un sincrotrónson similares; la gran diferencia entre ellos es la divergenciadel haz, la cual está relacionada con la coherencia espacialde los fotones. Los láseres de electrones libres que emitenrayos X (XFEL, por su siglas en inglés, X-ray Free Electron Laser)también producen coherencia temporal de los paquetesde electrones que producen los rayos X. Esto ge nera flujos derayos X que son 108 veces más brillantes que los producidosen los sincrotrones. Actualmente sólo hay dos fuentes de estetipo, habiendo sido la primera la Fuente Lineal de Luz Cohe-rente (Linear Coherent Light Source) en Stanford, Estados Uni -dos [5]. El grupo de investigación asociado a ese centro yaha reportado el uso de esta fuente de rayos X para analizarla distribución atómica de una proteína [6]. La alta brillantez del sincrotrón permite obtener patrones
de difracción de cristales de proteínas en poco tiempo; sinembargo, la intensidad del haz de rayos X puede dañar ladistribución atómica de la proteína [7]. Una ventaja impor-tante de los sincrotrones es la capacidad de sintonizar (selec-cionar) la longitud de onda de los rayos X usados en losexperimentos de difracción, a diferencia de los tubos selladosde rayos X que tienen una longitud de onda constante.Aunque los sincrotrones tienen gran brillantez, su número
en el mundo es pequeño, en comparación con el número decientíficos y estudiantes que podrían trabajar sobre la difrac-ción de cristales de proteínas. Además, el uso de estas fuen-tes de rayos X implica tiempos de espera largos, lo cual esinadecuado cuando el problema científico es el aislamiento
70

de una proteína nueva, lo que requiere muchos experimen-tos de difracción.El desarrollo de nuevas fuentes de rayos X sugiere que en
el futuro, las grandes fuentes de rayos X de sincrotrones seránsustituidas por otras, compactas y brillantes [8-10], que pue-dan instalarse en áreas menores a 10 × 10 m2 y que consu-man mucha menos energía que los sincrotrones actuales.Esto significa que estarán disponibles en cualquier laborato-rio estándar, en cualquier hospital o en cualquier fábrica. Actualmente, la brillantez de algunas de estas nuevas fuen-
tes compactas de rayos X es incluso mejor que la obtenidaen la generación actual de sincrotrones (tercera generación).Las nuevas fuentes compactas de rayos X no sólo ocupanespacios pequeños, sino que también consumen muchamenos energía que los sincrotrones actuales.En los párrafos siguientes describiremos tres de estas nue-
vas fuentes compactas de rayos X, con un énfasis especialsobre aquellas basadas en la aceleración de electrones en elcampo de la estela generada por la interacción de pulsos deláser, intensos y temporalmente cortos, con un gas de helio.Esta selección se hizo porque su desarrollo sugiere que seránlas fuentes compactas de rayos X con alta brillantez en elfuturo cercano.
FUENTES COMPACTAS BRILLANTES DE RAYOS X
Hay más de tres conceptos físicos básicos diferentes parala construcción de fuentes de rayos X compactas y brillan-tes, pero en el presente documento se presentarán sólo tresde ellas. 1) Sincrotrón compacto con ánodo que genera radiación
de bremsstrahlung (BS, por sus siglas en inglés, Bremsstrah-lung Scattering). En esta fuente de rayos X, brillante y com-
71

pacta, los electrones se aceleran a energías menores a 10 MeV,lo que permite almacenarlos en un anillo con un diámetromenor a 1 m. Para producir los rayos X se introduce unánodo (alambre metálico) transversal a la trayectoria de loselectrones, en el anillo de almacenamiento. Cada vez que unelectrón interactúa con este ánodo se produce radiación X debremsstrahlung (radiación por desaceleración) y los rayos Xcaracterísticos del ánodo [11]. 2) Sincrotrón compacto basado en la dispersión inversa
de Compton (CS, por sus siglas en inglés, Compton Scattering).En esta fuente de rayos X, compacta y brillante, los electro-nes son acelerados a energías de algunas decenas de MeV,lo que permite almacenarlos en un anillo del orden de 1 m dediámetro. Los rayos X se generan al interactuar estos elec-trones con un pulso de láser intenso [12]. 3) Aceleración de electrones mediante un pulso de láser
(LWFA, por sus siglas en inglés, Laser-Wakefield Acceleration).En esta fuente de rayos X los electrones se generan y sonacelerados a velocidades relativistas al interactuar un pulsointenso y temporalmente corto, de láser con gas de helio con-tenido en un capilar. Los electrones relativistas se generanen pulsos temporalmente cortos, los cuales al pasar por unarreglo periódico de imanes (ondulador) generan un pulsode rayos X intenso y temporalmente corto [13].A continuación describimos en detalle cada una de estas
fuentes de rayos X compactas y brillantes.
Sincrotrón compacto con ánodo que genera radiación de bremsstrahlung (BS)
Se pueden producir fuentes de rayos X compactas y bri-llantes cambiando la geometría de un tubo sellado de rayos Xconvencional. En estos tubos, se producen electrones al calen-tar un filamento metálico, los cuales son acelerados en un
72

campo eléctrico a energías de algunos KeV antes de que inter -actúen con un ánodo metálico. En las fuentes compactas derayos X, descritas en la presente sección, los electrones seaceleran con radiofrecuencia para producir pulsos de elec-trones con energía de algunos MeV y duración de unospocos microsegundos, que se almacenan en un anillo conun diámetro menor a 1 m.En la trayectoria de los electrones relativistas almacena-
dos en el anillo se inserta un ánodo perpendicular tanto en latrayectoria de los electrones como en el diámetro del anillo.Este ánodo es, típicamente, un alambre de Mo o de W, conun diámetro entre 10 y 20 µm. Dado que la velocidad de loselectrones es relativista, los electrones en un pulso pasan unnúmero considerable de veces en la región donde se encuen-tra el ánodo. Se estima que para un ánodo con un diámetrode 10 µm, cada electrón en el pulso interactúa 200 veces conel ánodo [11]. Esta interacción genera bremsstrahlung y losrayos X característicos del ánodo; la energía más alta de los fo -tones del bremsstrahlung es igual a la energía de los elec-trones incidentes, es decir, algunos MeV. La brillantez deesta fuente de rayos X depende del diámetro del alambreque funciona como ánodo; existe un diámetro para el cualla brillantez es máxima.Como se mencionó previamente, la brillantez de los sin-
crotrones es mayor que la de los tubos de rayos X porque elhaz de rayos X producido en un sincrotrón tiene una diver-gencia más estrecha; sin embargo, la emisión de rayos X totalpor unidad de tiempo es la misma para ambas fuentes derayos X. Es importante hacer notar que el ángulo sólido enque emiten los rayos X está directamente relacionado con laenergía de los electrones que producen la radiación, a mayorenergía, menor ángulo sólido. En un tubo de rayos X sellado,la energía de los electrones empleados para generar losrayos X es menor a 100 KeV.
73

Cuando se desacelera un paquete de electrones conenergía E mayor que la energía asociada al electrón enreposo, mec = 0.510998 MeV (me es la masa del electrónen reposo), seproduce un haz de rayos X con un ángulo dedivergencia determinado por E [14], según la relación<sen2q> = (0.510998/E)2. El sincrotrón compacto que estamos describiendo ya está
disponible comercialmente [8]. Su nombre comercial esMIRRORCLE RAY CV4L, el cual es fabricado por la compañíaPhoton Production Laboratory Ltd. [15] con un precio de alre-dedor de 1.5 millones de dólares americanos.La línea del haz de esta fuente de rayos X compacta se
diseña de acuerdo con la aplicación. Por ejemplo, para laespectroscopía de fluorescencia de absorción extendida (EXAFS,por sus siglas en inglés, Extended X-ray Absorption FluorescenceSpectroscopy), la difracción de rayos X y la microtomografíacomputarizada de rayos X. Esta fuente compacta de rayos X consume alrededor de
50 kVA y tiene dos líneas de haz: una para la radiación debremsstrahlung y otra para la emisión de rayos X caracte-rísticas del ánodo.La brillantez del bremsstrahlung es de 1013 fotones/s/
mrad2/mm2/0.1% ab, que es comparable a la brillantez delos sincrotrones de la primera generación (figura 1); la dis-tribución angular del haz es de 125 mrad, esto debido a quela energía de los electrones acelerados es de 4.5 MeV. Esto pro-duce una densidad de fotones de bremsstrahlung de 4 × 109fotones/s/mrad2/0.1% ab y una fluorescencia de rayos Xcaracterística del ánodo de 9 × 1010 fotones/s/mrad2/0.1% ab.En la misma sala del laboratorio donde se encuentre esta
fuente compacta se puede conectar el haz a cualquier difrac-tómetro de rayos X (de monocristales o de polvo) para llevara cabo experimentos de difracción sobre proteínas, porquetodos los componentes de esta fuente de rayos X caben en unespacio de 120 cm de altura, 45 cm de ancho y 45 cm de fondo.
74

La compañía que produce esta fuente de rayos X compactay brillante vende también el difractómetro para la difracciónde rayos X de proteínas. La fuente de rayos X también sepuede conectar a cualquier difractómetro que se use para elmismo fin, incluyendo los empleados con tubos de rayos Xconvencionales. Por supuesto, el difractómetro ideal para rea-lizar difracción sobre muestras de proteínas con esta fuentede rayos X es el usado con sincrotrones convencionales o simi-lares a éste, porque tienen la óptica que genera poca abe-rración y el dispositivo para seleccionar una longitud deonda determinada.
Sincrotrón compacto basado en la dispersión inversa de Compton (CS)
Se pueden producir fuentes de rayos X de sincrotrón com-pactas y brillantes. Para ello, se sustituye el ondulador mag-nético convencional con un pulso de láser intenso en dondelos fotones se mueven en dirección opuesta a la direcciónde los electrones relativistas acelerados. Los onduladores soncomponentes clave de los sincrotrones de tercera genera-ción [3], porque en él se desaceleran los electrones paragenerar los rayos X.En un sincrotrón convencional, un ondulador es un arre-
glo periódico de imanes permanentes; el período espaciales de algunos centímetros (típicamente 30-40 cm). La lon-gitud de onda del haz de rayos X emitido es proporcionala este período, en tanto que su intensidad la determina elnúmero de períodos presentes en el arreglo de imanes. Unvalor típico de este número es cinco, lo que da una longitudtotal del ondulador de entre 1.5 y 2.0 m.Si se sustituye al ondulador magnético con un pulso de
láser intenso que se mueve en dirección opuesta al pulsode electrones relativistas, éstos experimentan los camposeléctricos y magnéticos oscilantes en el pulso de láser [12],
75

de modo semejante a como experimentan el campo mag-nético cambiante de un ondulador magnético.Para un ondulador de láser con longitud de onda de 1 µm,
la interacción electrón-láser es equivalente a la interacción delpulso de electrones con un ondulador magnético de 25 milperíodos. Como la longitud de repetición equivalente deeste ondulador es pequeña (1 µm en comparación con los30 cm de un ondulador magnético), para generar rayos Xde una longitud de onda dada, la energía de los electronesrequerida es menor que en un sincrotrón convencional [16].Por ejemplo, si la longitud de onda del láser es de 1 µm, paraproducir rayos X con una longitud de onda de 0.1 nm, la ener-gía de los electrones debe ser de 25 MeV; lo que conllevaa un diámetro del anillo de almacenamiento de sólo 1.5 m.En contraste, en un sincrotrón actual, dichos rayos X se pro-ducen por electrones de 6 GeV, con un correspondiente ani-llo de almacenamiento de 300 m de diámetro. En la litera-tura, la interacción electrón-láser descrita en este párrafo seinterpreta como una dispersión inversa de Compton [12].El uso de pulsos de láser como onduladores fue desarro-
llado a finales de los años noventa [12]. Actualmente, la com-pañía Lyncean Technologies, Inc. [9] vende ya un sincrotróncompacto del tipo descrito en el párrafo anterior. Este sin-crotrón compacto produce un flujo promedio de 1012 foto-nes/s en un círculo de 300 µm de diámetro. La brillantezde la fuente es del orden de 1012 fotones/s/mrad2/mm2/0.1% ab. Esta compañía ofrece un sistema de difracción com-pleto para estudiar la cristalografía de proteínas, el cual, porejemplo, fue usado para determinar la estructura cristalinadel sistema de proteínas H de Mycobacterium tuberculosis [17].Cuando la fuente compacta de rayos X que describimos
en la presente sección se emplea para analizar proteínas,produce 4.0 × 1011 fotones/s en un círculo de 30 µm de diá-metro, lo que permite analizar cristales de sólo 10 µm de
76

diámetro. Como esta fuente compacta es sintonizable, tam-bién es posible realizar difracción anómala de longitudes deonda múltiples sobre un solo cristal de proteína, para deter-minar su estructura cristalina tridimensional [18].El Instituto Tecnológico de Massachusetts ha desarrollado
una nueva fuente de rayos X compacta y brillante basada enla dispersión inversa de Compton, pero usando un aceleradorlineal. La brillantez promedio de la fuente es de 1015 fotones/s/mrad2/mm2/0.1% ab para rayos X de 12 KeV y emite picospulsados de rayos X de 1 ps con una brillantez de hasta 1023fotones/s/mrad2/mm2/0.1% ab, con 108 fotones por pulso.La brillantez promedio de esta fuente compacta compitecon la brillantez de algunos de los sincrotrones actuales detercera generación; además los aventaja en el modo pulsadopor el hecho de generar un pulso más corto en el tiempo [19].
Aceleración de electrones mediante un pulso de láser (LWFA)
En todas las fuentes de rayos X, éstos se producen desa-celerando electrones relativistas. Las dimensiones de lafuente están determinadas principalmente por el sistemaque suministra dichos electrones. Por ejemplo, en los tubossellados de rayos X, el cañon que proporciona los electronesrelativistas tiene dimensiones de unos pocos centímetros [20].Cuando se generan rayos X en un sincrotrón, se aceleran
electrones con una fuente de radiofrecuencia de gran escalaque genera un voltaje máximo de aceleración de 100 MV/m.Luego los electrones acelerados se almacenan en un anillocon un diámetro que depende de la energía de los electrones.Por ejemplo, en la Instalación Europea de Radiación de Sin-crotrón (European Synchrotron Radiation Facility) se aceleranelectrones a 6 GeV; por tanto, sólo el anillo de almacena-miento es de 300 m de diámetro [21]. Estos elementos delos sincrotrones determinan sus dimensiones.
77

Recientemente se ha demostrado que se pueden acelerarelectrones a energías de 1 GeV en capilares de unos cuantosmm de longitud [22]. Esto sucede al interactuar un pulso deláser intenso (de más de 50 TW) y corto en el tiempo (pocosfs) con gas de helio en un capilar. Esta interacción genera unplasma que suministra electrones y forma una estela ondu-lante detrás del pulso de láser. Los electrones del plasma sonatrapados en una depresión que se forma justo detrás delpulso de láser y son acelerados por el gradiente de potencialgenerado en dicha estela. Estos electrones acelerados siguenal pulso de láser que se mueve con una velocidad de grupocercana a la velocidad de la luz. Como resultado de todas estasinteracciones se generan pulsos de electrones intensos conduración de algunos fs. Finalmente, este pulso de electronespasa por un ondulador y genera pulsos de rayos X intensoscon una longitud temporal de algunos fs.Las dimensiones de estas fuentes de rayos X compactas
y brillantes están determinadas por las dimensiones del láserutilizado para producir los pulsos de láser que interactúancon el gas de helio. Por ejemplo, hoy en día los láseres queproducen los pulsos más intensos (1 PW), que son adecuadospara construir la fuente de rayos X descrita, requieren unárea de 4.2 × 6 m2 [23]. Esto significa que las dimensiones deestas fuentes de rayos X son significativamente más peque-ñas que las de un sincrotrón convencional. Conforme avancela tecnología de los láseres, se reducirá su tamaño y, en con-secuencia, las dimensiones respectivas de las fuentes derayos X descritas en esta sección.Como los electrones son generados y acelerados en el
campo de la estela producida por el pulso de láser cuandointeractúa con el gas de helio, el proceso se llama acelera-ción en el campo de la estela de láser (LWFA, por sus siglas eninglés, Laser-Wakefield Acceleration). Este fenómeno fue pro-puesto desde finales de los años setenta [24], pero los expe-rimentos para observarlo se realizaron hasta que se pudieron
78

ge nerar pulsos de láser muy intensos y cortos en el tiempo [25].Vale la pena mencionar que el pulso de láser para producirel campo de estela se puede sustituir con un pulso de elec-trones [26] o un pulso de protones [27].Se han reportado hasta ahora tres formas diferentes de
producir rayos X con los electrones acelerados en el campode una estela de láser.En la primera, las oscilaciones transversales que sufren los
electrones acelerados en el campo de la estela generan pul-sos de rayos X que siguen al pulso de láser [22]. Los rayos Xemitidos son pulsos cortos (de algunos fs) con brillantezmáxima del orden de 1022 fotones/s/mrad2/mm2/0.1% ab;esta brillantez es mayor que la de los sincrotrones actuales detercera generación (figura 1). Este hecho debe ser tomadocuidadosamente en cuenta por los grupos interesados enconstruir sincrotrones nuevos de esta generación. En el segundo caso, los electrones acelerados pasan por
un ondulador magnético para generar rayos X [28]. En estecaso la brillantez obtenida es, hasta ahora, de 1.3 × 1017 foto-nes/s/mrad2/mm2/0.1% ab, con un ancho temporal delpulso de rayos X de 10 fs. La ventaja de este método paragenerar rayos X es que la energía de los rayos X es fácil-mente sintonizable a través de la configuración del ondulador.Vale la pena mencionar que en este caso la brillantez essimilar a la obtenida en sincrotrones de tercera generación.En el tercer caso, los electrones acelerados interactúan
con un pulso de láser intenso que se mueve en direcciónopuesta a la de los electrones [29, 30], lo cual produce dis-persión de Compton inversa y genera rayos X de modosimilar al descrito arriba en la sección 2.2. En este caso, losrayos X se emiten con una brillantez máxima de entre1019 fotones/s/mrad2/mm2/0.1% ab [30] y 1021 fotones/s/mrad2/mm2/0.1% ab [29] y longitudes temporales del ordende 10 fs. Estos valores dependen del arreglo experimentaly cambiarán en el futuro a través de su mejoramiento.
79

En todas estas formas para producir rayos X, a partir depulsos de electrones acelerados en el campo de una estelade láser, los rayos X son pulsos de duración de unos pocos fsy dispersos en ángulos del orden de mrad [28-30]. Esto ge -nera densidades espaciales altas de fotones de rayos X útilespara hacer difracción sobre cristales pequeños de proteínas,en tiempos de medición menores a los usados en sincrotro-nes de tercera generación. Como los pulsos de rayos X tie-nen una dimensión temporal de sólo algunos pocos fs, pue-den usarse para estudiar procesos dinámicos en la materia.Es interesante resaltar que la duración de los pulsos de las
fuentes de rayos X, descritas en esta sección es menor que laduración de los pulsos generados en los sincrotrones decuarta generación existentes hasta ahora para producirrayos X [5], y que la duración de los pulsos de rayos X obte-nidos en los sincrotrones de tercera generación, la cual esdel orden de los ps.En la actualidad, ninguno de los grupos que han desarro-
llado las fuentes de rayos X compactas y brillantes, reporta-dos en la presente sección, han realizado experimentos dedifracción de rayos X en proteínas pero, con seguridad, loharán pronto.Es interesante agregar un comentario que se hizo en el
Centro de Noticias del Laboratorio de Berkeley en diciem-bre de 2009:
Los aceleradores están lejos de lograr las energías más altas a lasque aspiran sus constructores, pero su tamaño y costo puedenlimitar la clase de instalaciones que pueden apoyar las agenciasde financiamiento. En el futuro, se requerirán nuevos tipos demáquinas para hacer mayores progresos. Tal vez la más pro-metedora es el acelerador de estela de láser.
Actualmente los láseres más poderosos generan pulsos conuna potencia de 1 PW y con una duración de unos pocos fs.
80

Estos láseres probablemente serán usados tanto en los sin-crotrones de cuarta generación como en las fuentes derayos X basadas en la aceleración en el campo de una estelade láser. Por lo tanto, ambas fuentes producirán pulsos derayos X con duraciones similares, pero sus costos serán total-mente diferentes.
CONCLUSIONES
Hay nuevas posibilidades para que un número mayor degrupos de científicos alrededor del mundo determinen la dis-tribución atómica de proteínas. Esto se debe a que han apare-cido en el mercado fuentes de rayos X compactas con una bri-llantez similar a la generada en los sincrotrones de la primerageneración. En un futuro próximo, estas fuentes de rayos Xserán las nuevas para realizar difracción de rayos X de pro-teínas, con el atractivo de que la energía de los rayos X essintonizable.El futuro para llevar a cabo la difracción de rayos X en
proteínas es aún mucho más prometedor. Actualmente, seencuentran en desarrollo fuentes de rayos X compactasque tienen una brillantez similar o mayor que la de los sin-crotrones actuales de tercera generación, pero con pulsos derayos X temporalmente más cortos. Estas nuevas fuentesde rayos X están basadas en la aceleración de electrones enel campo de la estela generada por la interacción de pulsosde láser, intensos y cortos en el tiempo, con un gas de helio.El gran atractivo de estas fuentes de rayos X compactas esque la aceleración de los electrones y la producción de losrayos X se puede realizar en un espacio menor a 10 m × 10 my con bajo consumo de energía. Esto significa que muchoslaboratorios en el mundo tendrán la posibilidad de integrarestas fuentes de rayos X a sus instalaciones de difracción.
81

REFERENCIAS
[1] Fultz B. y Howe J., Transmission Electron Microscopy and Diffractometryof Materials, Chapter 1, Springer-Verlag, Berlín, 2013.
[2] Duong T., Park K., Kim T., Kang S. W., Hahn M. J., Hwang H. Y.y Kim K. K., Acta Cryst. D, 2014, 70, 615-626.
[3] Winick H., Brown G., Halbach K. y Harris J., Physics Today, 1981, 54,50-63.
[4] “Interim Report of the Scientific and Technical Issues (XFEL-STI)Working Group on a European XFEL Facility in Hamburg”, Deuts-ches Elektronen-Synchrotron Notkestrasse, 2005, 85, D-22607Hamburgo, Alemania. Recuperado de la URL: http://xfel.desy.de.
[5] Emma P. et al., Nature Photonics, 2010, 4, 641-647.[6] Chapman H. N. et al., Nature, 2011, 470, 73-77.[7] Shimizu N., Hirata K., Hasegawa K., Ueno G., y Yamamotoa M.,
J. Synchrotron Rad., 2007, 14, 4-10.[8] MIRRORCLE-CV4, Photon Production Laboratory, Ltd. Shiga Prefec-
ture techno-factory #7, Nojihigashi 7-3-46, Kusatsu, SHIGA, 525-0058, Japón. Correo electrónico: [email protected].
[9] Lyncean Technologies Inc., 370 Portage Avenue, Palo Alto, CA94306, EUA. Recuperado de la URL: http://www.linceantech.com.
[10] Nakajima K., Nature Physics, 2008, 4, 92-93.[11] Yamada H., Jpn. J. Appl. Phys., 1996, 35, L182-L185.[12] Huang Z. y Ruth R. D., Phys. Rev. Lett., 1998, 80, 976-979.[13] Kneip S., Nagel S. R., Bellei C., Bourgeois N., Dangor A. E., Gopal A.,
Heathcote R., Mangles S. P. D., Marques J. R., Maksimchuk A., Nil-son P. M., Ta Phuoc K., Reed S., Tzoufras M., Tsung F. S., Willin-gale L., Mori W. B., Rousse A., Krushelnick K. y Najmudin Z., Phys.Rev. Lett., 2008, 100, 105006.
[14] Schwinger J., On the Classical Radiation of Accelerated Electrons,Phys. Rev., 1949, 75, 1912-1925.
[15] Hasegawa D., Yamada H., Toyosugi N., Noh Y. D., Yamada T., Mori-ta M., Mantey E., y Masaoka S., AIP Conference Proceedings, 2007, 879,26-29.
[16] Motz H., Thon W. y Whitehurst R. N., J. Appl. Phys., 1953, 24, 826-833.[17] Abendroth J., McCormick M. S., Edwards T. E., Staker B., Loewen R.,
Gifford M., Rifkin J., Mayer C., Guo W., Zhang Y., Myler P., KelleyA., Analau E., Hewitt S. N., Napuli A. J., Kuhn P., Ruth R. D. y StewartL. J. J. Struct. Funct. Genom., 2010, 11, 91-100.
[18] Hendrickson W. A., Science, 1991, 254, 51-58.
82

[19] Schoenlein R. W., Chattopadhyay S., Chong H. H. W., Glover T. E.,Heimann P. A., Shank C. V., Zholents A. A. y Zolotorev M. S., Scien-ce, 2000, 287, 2237-2240.
[20] Kamiya Y., J. Phys. Soc. J., 1959, 14, 1327-1333.[21] http://www.esrf.eu.[22] Kneip S., McGuffey C., Martins J. L., Martins S. F., Bellei C., Chvy-
kov V., Dollar F., Fonseca R., Huntington C., Kalintchenko G.,Maksimchuk A., Mangles S. P. D., Matsuoka T., Nagel S. R., PalmerC. A. J., Schreiber J., Ta Phuoc K., Thomas A. G. R., Yanovsky V.,Silva L. O., Krushelnick K. y Najmudin Z., Nature Physics, 2010, 6,980-983.
[23] Alpha XS 1 PW brochure, Thales Optronique S. A. S. 2, AvenueGay-Lussac–CS 90502 78995 Élancourt Cedex, Francia. Recupe-rado de la URL:https://www.thalesgroup.com/en/petawatt-multi-petawatt-systems.
[24] Tajima T. y Dawson J. M., Phys. Rev. Lett., 1979, 34, 267-270.[25] Kasouleas T., Nature, 2004, 431, 515-516.[26] Conde M. E., Doran D. S., Gai W., Konecny R., Liu W., Power J. G.,
Antipov S., Jing C., Wisniewski E. y YusofZ., Proceedings ofIPAC2013, 2013, pp. 1111-1113, Shanghai, China.
[27] Muggli P., Caldwell A., Reimann O., Oz E., Tarkeshian R., Bracco C.,Gschwendtner E., Pardons A., Lotov K., Pukhov A., Wing M.,Mandry S. y J. Vieira, Proceedings of IPAC2013, 2013, pp. 1179-1181,Shanghai, China.
[28] Fuchs M., Weingartner R., Popp A., Major Z., Becker S., Osterhoff J.,Cortrie I., Zeitler B., Hörlein R., Tsakiris G. D., Schramm U., Row-lands-Rees T. P., Hooker S. M., Habs D., Krausz F., Karsch S. y Grü-ner F., Nature Phys., 2009, 5, 826-829.
[29] Phuoc K. T., Corde S., Thaury C., Malka V., Tafzi A., Goddet J. P.,Shah R. C., Sebban S. y Rousse A., Nature Photonics, 2012, 6, 308-311.
[30] Chen S., Powers N. D., Ghebregziabher I., Maharjan C. M., Liu C.,Golovin G., Banerjee S., Zhang J., Cunningham N., Moorti A.,Clarke S., Pozzi S. y Umstadter D. P., Phys. Rev. Lett., 2013, 110,155003.
83


REDES DE RESIDUOS PROTEÍNICOSESTUDIO DE UN CASO DE GRÁFICAS
GRANDES Y SU ENTROPÍA
JOSÉ ANTONIO DE LA PEÑA
Instituto de Matemáticas Universidad Nacional Autónoma de México
Centro de Investigación en Matemáticas Guanajuato, México
INTRODUCCIÓN
La Teoría de las gráficas nació en 1736 cuando LeonhardEuler publicó “Solutio problematis ad geometriam situs pertinentis”(“La solución de un problema relativo a la teoría de la posi-ción”, Euler, 1736), un artículo sobre los famosos siete puen-tes de Königsberg. Esta historia está bien documentada y am -pliamente disponible en cualquier libro de texto sobre teoríade gráficas o de redes. No obstante, la palabra gráfica apare-ció por primera vez en el contexto de las ciencias naturales en1878, cuando el matemático inglés James J. Sylvester escribióun artículo titulado “Chemistry and Algebra” (“Químicay Álgebra”), el cual fue publicado en Nature (Sylvester, 1877-1878) y en el que escribió que Cada invariante y covariante, porconsiguiente, se torna expresable mediante una gráfica precisamenteidéntica a un diagrama de Kekulé o quimicográfico. El primer librosobre teoría de gráficas fue escrito por Dénes König y publi-cado en 1936. Otro libro, por Frank Harary, publicado en
85

1969, fue considerado el libro de texto definitivo sobre eltema y permitió a los matemáticos, químicos, ingenieros elec-tricistas y electrónicos y científicos sociales comunicarseentre sí. En nuestros días hay pocas áreas de las matemáticas,ciencias de la computación, física o ciencias naturales en elsiglo XXI, si es que las hay, en las que no estén implicadas lasgráficas y las redes directa o indirectamente. De hecho, lossistemas complejos van de los sistemas moleculares y bioló-gicos hasta los ecológicos, sociales y tecnológicos.
Alcance y contenido de este artículo. Luego de una introduc-ción corta sobre terminología y algunos resultados esencia-les de la teoría espectral de gráficas, presentamos, siguiendotrabajo reciente (Estrada de la Peña, 2013) el concepto deentropía de caminos. En la sección 2 presentaremos demostra-ciones completas de los Teoremas 1 y 2 que expresan larelación entre regularidad de caminos y la entropía máximade las gráficas. Son dos las razones para presentar los resulta-dos de Estrada de la Peña (2014) en detalle: por una parte,introducimos un tema de interés para el autor y, lo que esmás importante, los resultados ilustran el uso de variasherramientas para estimar sumas de la forma
que aparece en varias áreas de las matemáticas y la física, rela-cionadas en particular con la entropía de los sistemas. Entreotras herramientas, usamos:
• La desigualdad entre media aritmética y media geométrica• La desigualdad de Chebysheff• La desigualdad de Borwein-Girhensohn• La desigualdad de la suma de logaritmos
86

87
Las secciones 3 y 4 son de carácter expositivo; la primerapresenta conceptos de la teoría de gráficas y la segundarepasa algunos hechos de la bioquímica sobre las proteínasy la estructura de las gráficas asociadas a ellas. En la sección 3repasamos varias propiedades de las gráficas, con énfasis par-ticular sobre las gráficas grandes:
• Propiedades de mundo pequeño• Crecimiento libre de escala• Raleza de aristas
Estas propiedades se consideran en la sección 4 para elcaso particular de algunas redes del mundo real, tales comolas proteínas y las redes de interacción proteína-proteína.La red de residuos proteínicos, asociada a una proteína, es unagráfica cuyos nodos son los aminoácidos constituyentesy ciertas aristas de conexión definidas por una distancia decorte de alrededor de 7.0 Å. Las interacciones proteína-proteínaocurren cuando dos o más proteínas se enlazan, a me nudopara llevar a cabo su función biológica. Muchos de los pro-cesos moleculares más importantes de la célula, tales como lareplicación del ADN, se llevan a cabo en grandes máquinasmoleculares constituidas por un gran número de proteínasorganizadas por sus interacciones proteína-proteína. En lasección 5 demostramos el Teorema 3, que muestra que nues-tro concepto de entropía de caminos de una gráfica se com-porta de manera similar a la entropía física de Gibbs, a saber:que la entropía de las proteínas disminuye conforme éstasse doblan. Este último resultado no se publicará en ningúnotro lado.
El artículo está basado en una conferencia dictada duranteel simposio Humboldt organizado por El Colegio Nacionalen noviembre de 2013.

1. EL LENGUAJE DE LAS GRÁFICAS Y LAS REDES
Lo primero que se necesita aclarar es que los términos grá-ficas y redes se usan como sinónimos en la literatura. Reser-varemos el término gráfica para el concepto matemático abs-tracto, referido a formaciones pequeñas y artificiales de nodosy aristas. Reservaremos el término red para las gráficas querepresentan objetos del mundo real en los que los nodos re -presentan a entidades del sistema y las aristas representana las relaciones entre ellas.
Se recomienda al lector que para los conceptos básicos dela teoría de gráficas consulte el libro introductorio de Harary(1967). Para los conceptos algebraicos relacionados con lateoría espectral de gráficas referimos al lector al libro deCvetkovic, Doob y Sachs (1980). Hay muchos libros excelen-tes de publicación reciente sobre la teoría algebraica de grá-ficas, entre otros, Biggs (1993), Godsil (1993) y Estrada (2012).
1.1 Sea G una gráfica con vértices {1, …, n}, suponemos quela gráfica es simple, es decir, que no hay aristas múltiples olazos. La matriz A(G) = (aij) de n × n definida como aij = 1si hay una arista entre i y j, y 0 en otro caso, se llama la matrizde adyacencia de G.
Denotamos por jG(x) (o simplemente j(x) cuando no hayposibilidad de confusión) al polinomio característico de la grá-fica G, definido como:
un polinomio mónico de grado n cuyas raíces son los eigen-valores l1, …, ln de la matriz de adyacencia A(G). ComoA(G) es simétrica, los eigenvalores l1, …, ln son númerosreales. Supondremos que l1 ≥ l2 ≥ … ≥ ln. Llamamos aleigenvalor máximo l1 el radio espectral, que posee muchaspropiedades importantes, entre otras…
88

• Existe un eigenvector u1, que corresponde a l1, es decir,A(G)u1 = l1u1, tal que todos los elementos u1( j) > 0,para j = 1, …, n.
• dmin = min {Snj=1aij:i = 1, …, n} l1 max {Sn
j=1aij:i = 1, …,n} = dmax,
donde los valores extremos se toman sobre los gradosdi = Sn
j =1aij .
1.2 Hay muchas identidades importantes que relacionana los eigenvalores l1, …, ln con las propiedades estructura-les de la gráfica G. Mencionamos las siguientes:
• Sni =1li = 0, ya que tr (A(G)) = # lazos = 0
• Sni =1l
2i = 2m, donde m cuenta el número de aristas en G
• Sni =1l
3i = 6t, donde t cuenta el número de triángulos en G
Dados nodos p, q en G, definimos a a(m)pq como el número
de caminos entre p y q de longitud m, es decir, A(G)m = (a(m)ij ).
Sea uj el eigenvector correspondiente a lj, es decir,A(G)uj = ljuj para j = 1, …, n. Entonces…
• a(m)pq = Sn
j =1uj(p)uj(q)lmj
1.3 Decimos que una gráfica G es m-camino-regular (resp.camino-regular) si a(m)
pq es independiente de los nodos p, qseleccionados (resp. para todos los nodos p, q y todos losenteros m ≥ 1).
El primer resultado se demostró recientemente. La demos-tración ilustra varios conceptos; en particular, se terminarásólo después de introducir la definición y las propiedadesde la entropía de caminos en la siguiente sección.
≥
89

Teorema 1 (Estrada de la Peña, 2014): sea A la matriz deadyacencias de una gráfica conexa G. Entonces las siguien-tes condiciones son equivalentes:
(a) G es camino-regular.(b) Ak tiene una diagonal constante para números natu-
rales 0 k n – 1.(c) e A tiene diagonal constante.(d) e bA tiene diagonal constante, para b > 0.
Demostración (con excepción de (d) implica (a)): (a) cla-ramente implica a (b). Para (b) implica (a), sea…
el polinomio característico de la gráfica G. Por el teorema deCayley-Hamilton, p(A) = 0. Si Ak tiene diagonal constantepara números naturales 0 k m y n – 1 m, entonces…
tiene diagonal constante.Claramente, (a) implica (d), que es equivalente a (c). Demos-
traremos que (d) implica (b). Seguimos las técnicas utilizadaspara el Teorema 2.1 en [6]. Para 1 i n, consideramos
≥ ≥
≥ ≥ ≥
≥≥
90
FIGURA 1. Una gráfica que es 0- y 2-camino-regular, pero no es 1- ni 3-camino-regular.

una función analítica real. Como serie de potencias
aprovechando que G no tiene lazos y a(2)ii = di es el grado del
vértice i. Considérese la función analítica
y el límite , que es independiente delvértice, lo que muestra que G es regular. Repitiendo el argu-mento obtenemos sucesivamente que a(k)
ii es independientedel vértice i para k = 3, 4,
2. SOBRE EL CONCEPTO DE ENTROPÍA
2.1 Es obvio que la matriz e bA cuenta todos los caminosde un nodo a otro de la gráfica, de modo que los caminos máscortos tienen mayor peso que los más largos. Así
representa la suma de los caminos de cualquier longitud,en la que los caminos de longitud k están penalizados por elrecíproco del factorial de su longitud. La función de particiónpara esta gráfica se define entonces como .
91

Comenzamos por definir la probabilidad de seleccionaral azar un camino cerrado que comienza (y termina) en elnodo i de todos los caminos cerrados de la gráfica. Esto es,
Ahora, usando la fórmula de Shannon, definimos la entro-pía de camino basada en los nodos de la gráfica como sigue:
Nuestro primer resultado relaciona a la entropía de cami-no de los vértices con el concepto de camino-regularidad.
Lema 1: una gráfica con n nodos tiene entropía máximamax SV(G, b) = lnn si y sólo si es 0-camino-regular.
Demostración: la máxima entropía de camino de un vértice ise alcanza cuando (e A)ii es constante, en cuyo caso a todos losvértices les toca la misma probabilidad pi y SV(G, b) = lnn.Por definición, (e A)ii es constante si y sólo si la gráfica es 0-camino-regular.
En el siguiente resultado demostramos que los cuadradosde los elementos del eigenvector principal de la matriz deadyacencias representan una clase de probabilidad de cami-no cuando la temperatura del sistema tiende a cero.
Proposición 2 : sea n1(i ) el i -ésimo elemento del eigenvec-tor principal de la matriz de adyacencias. Entonces, n 2
1(i ) esla probabilidad de seleccionar al azar un camino cerrado
92
.
.
n

que comienza (y termina) en el nodo i, en el límite de tem-peratura cero de la gráfica, es decir, cuando
Demostración: Sea y Entonces,
y , tal que .
2.2 Hemos calculado la entropía de camino de los vérticespara todas las gráficas conexas de hasta ocho nodos (~12,000gráficas). De ahí, encontramos las gráficas con la mínimaentropía de camino de los vértices para un número dado denodos. Por ejemplo, para n = 8 las gráficas de mínima entro-pía de camino de los vértices consisten de un clique de cinconodos y tres nodos colgantes conectados en diferentes posi-ciones del clique (véase la figura 2). Se obtienen resultadossimilares para n = 7, donde las gráficas que minimizan a SV(G)tienen un clique de cinco nodos y dos nodos colgantes.
La característica distintiva principal de estas gráficas esque consisten de dos tipos de nodos según el grado delocali zación de una partícula que se difunde a través de lagráfica entera siguiendo un camino cuasi aleatorio. Unos pocosnodos exhiben valores muy altos de Gpp, en tanto que los de -más exhiben valores muy bajos de dicho parámetro, es decir,
! !".
! !".
93
FIGURA 2. Las gráficas con n = 8 que tienen las entropías de camino mínimas.
A B C
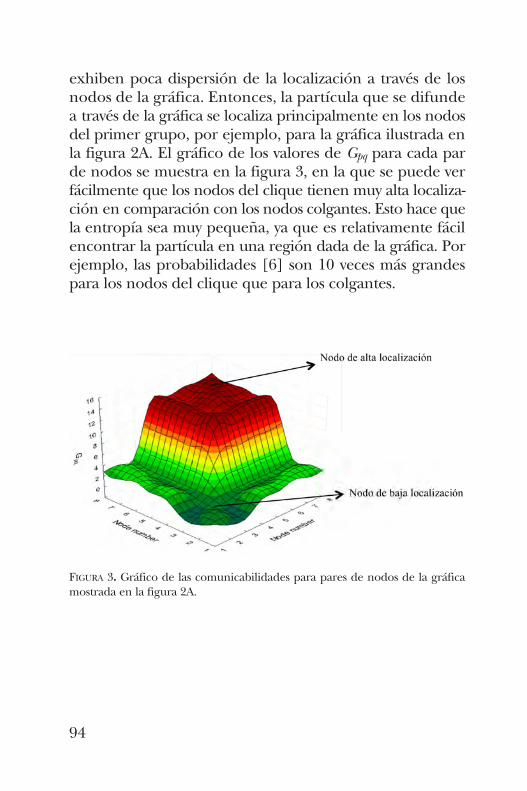
exhiben poca dispersión de la localización a través de losnodos de la gráfica. Entonces, la partícula que se difundea través de la gráfica se localiza principalmente en los nodosdel primer grupo, por ejemplo, para la gráfica ilustrada enla figura 2A. El gráfico de los valores de Gpq para cada parde nodos se muestra en la figura 3, en la que se puede verfácilmente que los nodos del clique tienen muy alta localiza-ción en comparación con los nodos colgantes. Esto hace quela entropía sea muy pequeña, ya que es relativamente fácilencontrar la partícula en una región dada de la gráfica. Porejemplo, las probabilidades [6] son 10 veces más grandespara los nodos del clique que para los colgantes.
94
FIGURA 3. Gráfico de las comunicabilidades para pares de nodos de la gráficamostrada en la figura 2A.
Teorema 2 (Estrada de la Peña, 2014): sea A la matriz deadyacencias de una gráfica G. Entonces se da una de lassiguientes condiciones:
(a) G es camino-regular. Entonces SV(G, b) = lnn, paratoda b > 0;
95
FIGURA 4. Cambio en la entropía de camino de los vértices con respecto alaumento de la temperatura recíproca para (izquierda) tres gráficas no regularesy (derecha) tres gráficas cúbicas (gráficas regulares de grado 3). Las estructurasde las seis gráficas se ilustran a continuación. Los gráficos de las gráficas noregulares son: curva continua roja (gráfica A), curva punteada verde (gráfica B)y curva punteada azul (gráfica C). Para las gráficas regulares, los gráficos son:curva continua azul (gráfica D), curva punteada verde (gráfica E) y curva pun-teada roja (gráfica F).

(b) G es una gráfica regular pero no es camino-regular.Entonces SV(G, b) < lnn, para toda b > 0. Además…
;
(c) Existe una e> 0 tal que SV(G, b) < lnn – e, para toda b > 0.
2.3 La entropía de camino y la desigualdad de Borwein-Girgensohn.
Sean l1 ≥ l2 ≥ … ≥ ln los eigenvalores de A con Sni =1li ≥ 0.
Para el vector de los elementos diagonales y = (y1, …, yn) dee bA, para alguna b > 0, definimos un vector z = lny = (lny1,…, lnyn) de números reales. Tenemos:
con donde la desigualdad es una aplicación directa del teoremade Hadamard para la matriz positiva definida e A.
El resultado notable de Borwein y Girgensohn [7] afirma que
donde cn = 2, si n = 2, 3, 4 y cn = e(1 – 1n ), para n ≥ 5.
Demostración del Teorema 2: sabemos que SV(G, b) lnnpara toda b > 0.
Observando que Z(b) = tr(e bA) y que la entropía de losvértices es
≥
96
,

la desigualdad de Borwein-Girgensohn arroja:
Más aún, la desigualdad entre media aritmética y mediageométrica (MA-MG) da:
Distinguimos dos situaciones con b > 0: (1) Sn
i =1zi2|b = 0, es decir, yi(b) = 1 para i = 1, …, n. Entonces
y por consiguiente:
.
En particular, para cualquier g > 0, la desigualdad MA-MG da:
lo que implica que todas las e g li tienen el mismo valor, esdecir, todas las g li tienen el mismo valor. Como tr(A) = 0entonces:
li = 0,
97
.

para i = 1, …, n. Entonces la gráfica G es discreta y
SV(G, g) = lnn
para cualquier g > 0.(2) Sn
i =1zi2 > 0. Entonces existe una función diferenciable
cn dn(b) tal que:
Como Z ≥ n, existe una función diferenciable en que satis-face 0 < en(b) dn(b) tal que:
Para toda M > 0, y usando la compacidad del intervalo,[0, M], existe una e(M) > 0 tal que en(b) ≥ e(M) para b [0, M].Además, recordando de [6] que:
donde u1, al igual que antes, es el eigenvector (de Perron)de A que corresponde al eigenvalor máximo. Este límite es< lnn excepto cuando hay un valor común u1(i) = c1, i = 1,…, n. Esta última propiedad implica que G es una gráficaregular. Consideramos estos casos por separado.
(3) Supongamos que G no es una gráfica regular. Enton-ces SV(G, ) < lnn. Por tanto existe una e > 0 tal quepara b [0, ], tenemos:
≥
!
≥
! !".!!
98
.
,

es decir, SV(G, b) lnn – e. (4) Supongamos que G es una gráfica regular. Podemos
suponer que G no es camino-regular. Entonces, según elanálisis de [11], el valor máximo SV(G, b) = lnn no se alcanzapara ninguna b . Más aún:
2.4 La parte (d) implica (a) en la demostración del Teo-rema 1:
De hecho, afirmamos que (a) y (d) son equivalentes a (e) SV(G, 1) = lnn.(d) Implica (e): sea y el valor constante de los elementos
de e A. Entonces Z(1) = ny y
(e) Implica (a): se sigue del Teorema 2.
3. GRÁFICAS GRANDES
Revisamos algunos índices de gráficas cuyos valores sonconocidos para muchas redes de la vida real.
3.1 Sea G una gráfica con matriz de adyacencias A y eigen-valores ln ln – 1 … l1. La energía de G se define como:
≥
≥≥≥
99
.
.
,

La varianza de los eigenvalores es:
donde am = media aritmética.Proposición: (a)
(b)
donde es la densidad de G.
Corolario: (a) (b)
Se dice que una red G es rala si d(G) << 1. Por ejemplo,si d(G(i)) k para alguna constante k y todos los vértices i,entonces:
Por consiguiente, si todas las restricciones G(i) son ralas,entonces G es rala.
3.2 De la desigualdad entre la media geométrica y lamedia aritmética obtenemos:
Proposición: Sea m0 la multiplicidad de 0 como eigenvalorde A. Entonces:
Demostración: Si A es invertible, entonces:
≥
100

Supongamos que m0 = 1. Como tenemos, para los poli-nomios característicos,
obtenemos un nodo j tal que cG (j)(0) 0. Como la energíadecrece para las subgráficas, entonces:
El resultado se sigue por inducción.3.3 La matriz de transición de la gráfica G es P = (pij), donde
es la probabilidad de que un camino que pasa por i
continuará hacia j. La matriz P es estocástica y como tal tienea 1 como un solo eigenvalor con un eigenvector p positivoen todos lados, es decir, pP = p. Al vector p se le llama vectorestacionario y sus elementos
miden la probabilidad de que una trayectoria arbitrariapase por i.
3.4 Para vértices p, q de la gráfica G, la distancia d(p, q) esla longitud mínima de los caminos entre p y q. El diámetrode G se define como:
y es una invariante global simple de la gráfica G.Un descriptor importante de la estructura de una gráfica
es la longitud de trayectoria media definida como:
!
101

Para un árbol seleccionado al azar, la longitud de trayec-toria media será cercana a la de un árbol generado aleato-riamente, que es asintótica a , donde K 0.5682 … Para n suficientemente grande, este número es menor que
. El invariante
se conoce como índice de Wiener. Se sabe que:
.
Si la longitud de trayectoria media es relativamente peque-ña en comparación con el tamaño de la red, se dice que lared tiene la propiedad de mundo pequeño, según se ha popula-rizado en muchos contextos diferentes. En general, se aceptaque una red satisface la propiedad de mundo pequeño sil(G) lnn.
Proposición: sea G una gráfica con n vértices. Supongamosque G1, …, Gm son subgráficas inducidas que cubren a G, esdecir, cada vértice i de G pertenece a alguna Gj. Supongamosque estas subgráficas satisfacen una condición de mundopequeño de la forma l(Gi) pi lnni , donde ni es el númerode vértices de Gi y 0 < pi . Si
entonces G satisface la condición de mundo pequeño l(G) lnn.
!
≥
≥
≥
102
.

Demostración: Obsérvese que, para i = 1, …, m, tenemos
Para p, q G tendremos
Por lo tanto
y
El resultado sigue.3.5 El índice de Estrada de una gráfica G con matriz de
adyacencia A es
Supongamos que G tiene n vértices y m aristas. Se demos-tró en [15] que
!
103
.
.
.
.
.
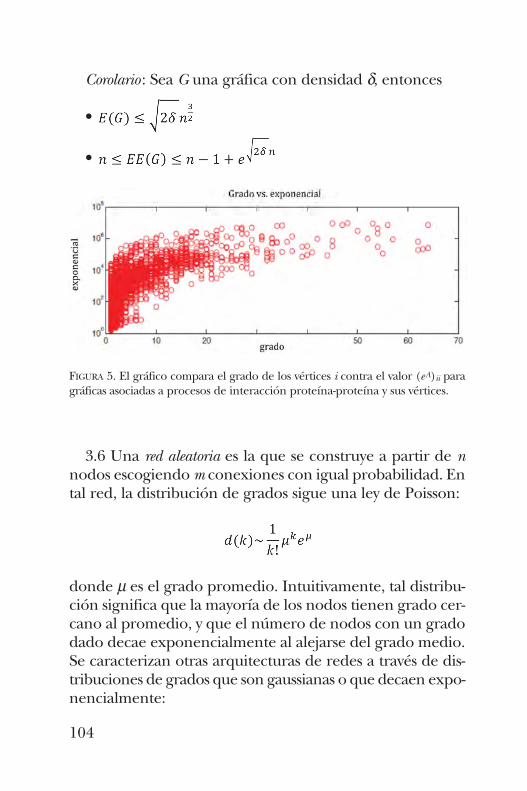
Corolario : Sea G una gráfica con densidad d, entonces
•
•
3.6 Una red aleatoria es la que se construye a partir de nnodos escogiendo m conexiones con igual probabilidad. Ental red, la distribución de grados sigue una ley de Poisson:
donde m es el grado promedio. Intuitivamente, tal distribu-ción significa que la mayoría de los nodos tienen grado cer-cano al promedio, y que el número de nodos con un gradodado decae exponencialmente al alejarse del grado medio.Se caracterizan otras arquitecturas de redes a través de dis-tribuciones de grados que son gaussianas o que decaen expo-nencialmente:
104
FIGURA 5. El gráfico compara el grado de los vértices i contra el valor (eA)ii paragráficas asociadas a procesos de interacción proteína-proteína y sus vértices.

o, tal vez más comúnmente, que siguen una ley potencial:
donde 0 < a < 2. Una de las características más importan-tes de las distribuciones de grados con ley potencial es que,a diferencia de las distribuciones de Poisson, gaussianasy exponenciales, se caracterizan por una alta variabilidad enel número de conexiones, teniendo un número pequeño denodos muchas conexiones y un número mucho mayorde nodos con pocas conexiones.
Considérese la distribución de grados d(k) de la gráfica G, esdecir, d(k) es el número de vértices i con di = k. Se dice quela distribución de grados es de tipo libre de escala si el númerode vértices con k vecinos es
para alguna g tal que 0 < g < 1. 3.7 Sea G una gráfica y A = A(G) su matriz de adyacencias.
Considérese el sistema de eigenvalores y eigenvectores de A,correspondiendo el eigenvector uj a lj, es decir, Auj = ljujpara j = 1, …, n. Entonces:
•
•
Recordemos que . Utilizando el hecho deque l1 es un eigenvalor máximo único, obtenemos
105
.

Es decir, para temperaturas grandes b podemos suponer
para números r(b) y vector a constantes.
4. EJEMPLOS DE GRÁFICAS MOLECULARES GRANDES
4.1 Redes de residuos proteínicos. Para entender la estruc-tura de las gráficas que representan a las moléculas de proteí -nas conviene revisar los hechos fundamentales de la estructuramolecular de las proteínas.
La estructura primaria de una proteína se refiere a lasecuencia lineal de aminoácidos en la cadena polipeptídica.El término residuos de aminoácidos se usa al hablar de proteínas,porque cuando se forma un enlace peptídico, se pierde unamolécula de agua, de modo que las proteínas están com-puestas por residuos de aminoácidos. Por ejemplo, la insulina
106
##
F
Cada uno de estos aminoácidos tiene un diseño fundamental compuesto
d
Por ejemplo, la i
Una cadena tiene 31 aminoácidos, y la otra tiene
2 La secuencia de una proteína es única para esa proteína, y define la estructura
y función de la misma. Sabemos que hay más de 10,000 proteínas en el cuerpo humano, que
e Además de la
e
Glicina Tirosina Cisteína naicilG
##
na nairosiT
##
naíetsCi
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
(
##
(
##
(
##
(
##
##
##
##
##
##
FIGURA 6. Hay 20 aminoácidos diferentes de los que se componen esencialmentetodas las proteínas en la Tierra. Cada uno de estos aminoácidos tiene un diseñofundamental compuesto de un carbono central (llamado también el carbonoalfa) ligado a un hidrógeno, un grupo carboxilo, un grupo amino y una cadenalateral o R-grupo único.
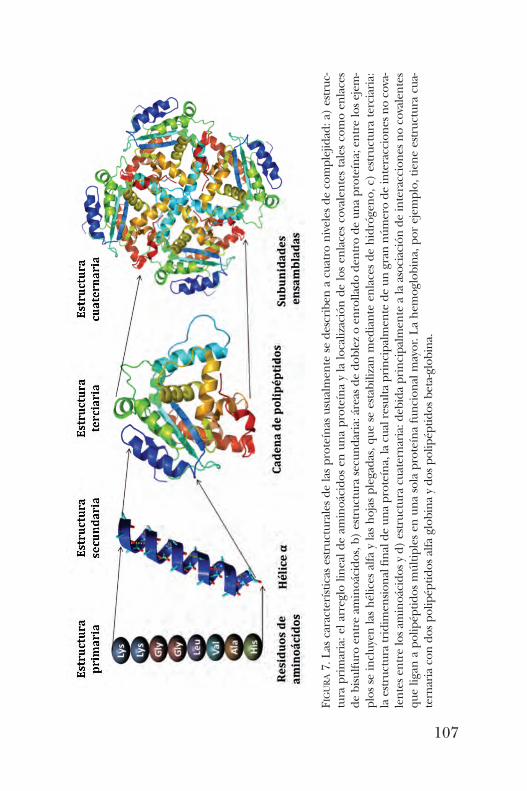
107
FIGU
RA
7. L
as c
arac
terísticas
estru
ctur
ales
de
las pr
oteí
nas us
ualm
ente
se
desc
ribe
n a
cuat
ro n
ivel
es d
e co
mpl
ejid
ad: a
) es
truc
-tu
ra p
rim
aria
: el a
rreg
lo li
neal
de
amin
oácido
s en
una
pro
teín
a y la
loca
lizac
ión
de lo
s en
lace
s co
vale
ntes
tal
es c
omo
enla
ces
de b
isul
furo
ent
re a
min
oácido
s, b)
estru
ctur
a se
cund
aria
: áre
as d
e do
blez
o e
nrol
lado
den
tro
de u
na p
rote
ína; e
ntre
los ej
em-
plos
se
incluy
en la
s hé
lices
alfa
y la
s ho
jas pl
egad
as, q
ue se
esta
biliz
an m
edia
nte
enla
ces de
hid
róge
no, c
) es
truc
tura
ter
ciar
ia:
la e
stru
ctur
a trid
imen
sion
al fi
nal d
e un
a pr
oteí
na, l
a cu
al res
ulta
princ
ipal
men
te d
e un
gra
n nú
mer
o de
inte
racc
ione
s no
cova
-le
ntes
ent
re lo
s am
inoá
cido
s y d)
estru
ctur
a cu
ater
naria: d
ebid
a pr
incipa
lmen
te a
la a
sociac
ión
de in
tera
ccio
nes no
cov
alen
tes
que
ligan
a p
olip
éptid
os m
últip
les en
una
sol
a pr
oteí
na fun
cion
al m
ayor
. La
hem
oglo
bina
, por
eje
mpl
o, tie
ne e
stru
ctur
a cu
a-te
rnar
ia con
dos
pol
ipép
tidos
alfa
glo
bina
y d
os p
olip
éptid
os b
eta-gl
obin
a.
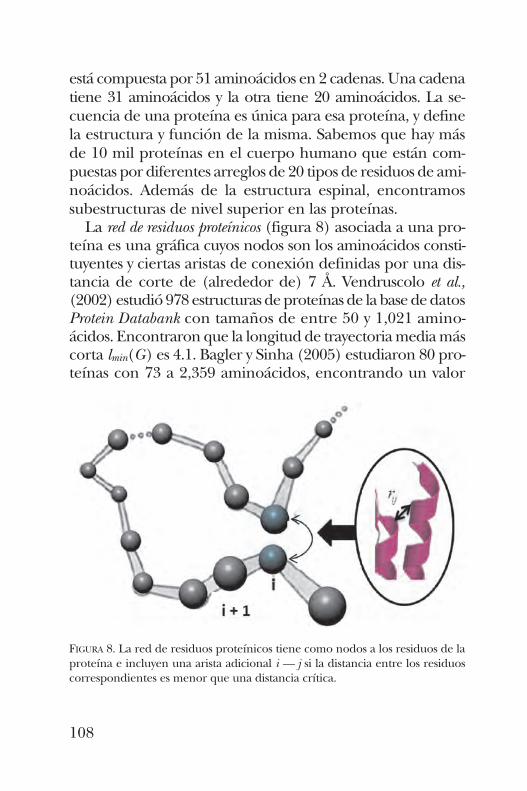
está compuesta por 51 aminoácidos en 2 cadenas. Una cadenatiene 31 aminoácidos y la otra tiene 20 aminoácidos. La se -cuencia de una proteína es única para esa proteína, y definela estructura y función de la misma. Sabemos que hay másde 10 mil proteínas en el cuerpo humano que están com-puestas por diferentes arreglos de 20 tipos de residuos de ami-noácidos. Además de la estructura espinal, encontramossubestructuras de nivel superior en las proteínas.
La red de residuos proteínicos (figura 8) asociada a una pro-teína es una gráfica cuyos nodos son los aminoácidos consti-tuyentes y ciertas aristas de conexión definidas por una dis-tancia de corte de (alrededor de) 7 Å. Vendruscolo et al.,(2002) estudió 978 estructuras de proteínas de la base de datosProtein Databank con tamaños de entre 50 y 1,021 amino-ácidos. Encontraron que la longitud de trayectoria media máscorta lmin(G) es 4.1. Bagler y Sinha (2005) estudiaron 80 pro-teínas con 73 a 2,359 aminoácidos, encontrando un valor
108
##
Encontraron que la
l
Estos comentarios se pueden usar para ver que estas redes de residuos
p
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
FIGURA 8. La red de residuos proteínicos tiene como nodos a los residuos de laproteína e incluyen una arista adicional i — j si la distancia entre los residuoscorrespondientes es menor que una distancia crítica.

promedio para la longitud de trayectoria l(G) = 6.88. Estoscomentarios se pueden usar para ver que estas redes de resi-duos proteínicos tienen la propiedad de mundo pequeño.
4.2 Las interacciones proteína-proteína ocurren cuando doso más proteínas se enlazan, a menudo para llevar a cabo sufunción biológica. Muchos de los procesos moleculares másimportantes en las células, tales como la replicación del ADN,se llevan a cabo mediante grandes máquinas molecularesconstituidas por un gran número de proteínas organizadaspor sus interacciones proteína-proteína.
Una red de interacciones proteína-proteína (RIP) de un procesobiológico (u organismo) es una gráfica construida como sigue:
• Los nodos son todas las proteínas que interactúan en elproceso.
• Dos nodos están conectados por una arista si se esperaque las dos proteínas correspondientes interactúen conun grado de confianza alto o medio.
La red de interacciones proteína-proteína (RIP) de la leva-dura, Saccharomyces cerevisiae, fue compilada por Bu et al. Losdatos originales fueron obtenidos por von Mering et al., valo-rando un total de 80,000 interacciones entre 5,400 proteí-nas reportadas previamente y asignando a cada interacciónun nivel de confianza. Bu et al., se centraron en 11,855 inter -acciones entre 2,617 proteínas con niveles alto y medio deconfianza, para reducir la interferencia de positivos falsos.
Se observan las siguientes características en las redes deinteracciones proteína-proteína (RIP):
• Las redes son ralas, con densidades
en muchos ejemplos, .
109
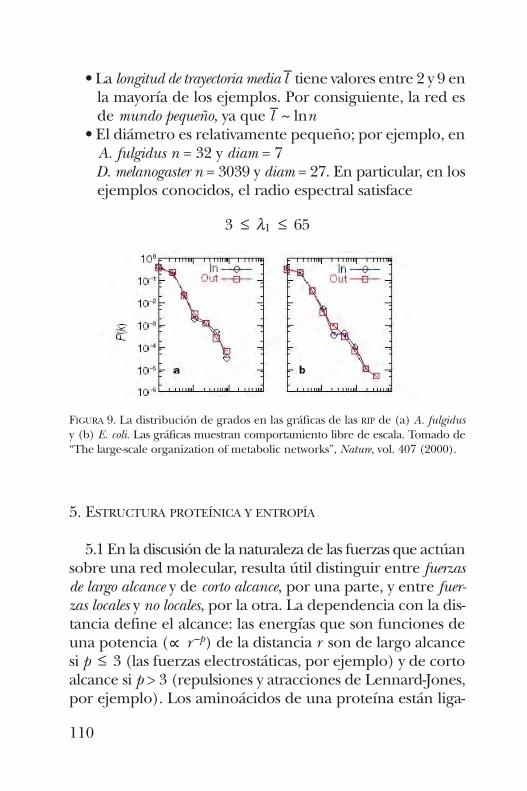
• La longitud de trayectoria media l– tiene valores entre 2 y 9 enla mayoría de los ejemplos. Por consiguiente, la red esde mundo pequeño, ya que l– ~ lnn
• El diámetro es relativamente pequeño; por ejemplo, enA. fulgidus n = 32 y diam = 7D. melanogaster n = 3039 y diam = 27. En particular, en losejemplos conocidos, el radio espectral satisface
3 l1 65
5. ESTRUCTURA PROTEÍNICA Y ENTROPÍA
5.1 En la discusión de la naturaleza de las fuerzas que actúansobre una red molecular, resulta útil distinguir entre fuerzasde largo alcance y de corto alcance, por una parte, y entre fuer-zas locales y no locales, por la otra. La dependencia con la dis-tancia define el alcance: las energías que son funciones deuna potencia ( r –p) de la distancia r son de largo alcancesi p 3 (las fuerzas electrostáticas, por ejemplo) y de cortoalcance si p > 3 (repulsiones y atracciones de Lennard-Jones,por ejemplo). Los aminoácidos de una proteína están liga-
≥≥
##
F
Tomado de T
La dependencia con la distancia define el alcance: las energías que son
f ! ! !
El polipéptido se dobla en tres dimensiones mediante interacciones que no forman
e Estas interacciones, que pueden ser alteradas fácilmente por pH extremo, temperatura,
p
• Interacciones hidrofóbicas (< 10 kcal/mol)
P
≥
110
##
F
Tomado de T
La dependencia con la distancia define el alcance: las energías que son
f ! !
El polipéptido se dobla en tres dimensiones mediante interacciones que no forman
e Estas interacciones, que pueden ser alteradas fácilmente por pH extremo, temperatura,
p
• Interacciones hidrofóbicas (< 10 kcal/mol)
P
FIGURA 9. La distribución de grados en las gráficas de las RIP de (a) A. fulgidusy (b) E. coli. Las gráficas muestran comportamiento libre de escala. Tomado de“The large-scale organization of metabolic networks”, Nature, vol. 407 (2000).

dos por interacciones de enlace covalente. El polipéptido sedobla en tres dimensiones mediante interacciones que no for-man enlaces. Estas interacciones, que pueden ser alteradasfácilmente por pH extremo, temperatura, presión y desna-turalizantes, son:
• Interacciones electrostáticas (5 kcal/mol)• Interacciones de enlace de hidrógeno (3-7 kcal/mol)• Interacciones Van Der Waals (1 kcal/mol)• Interacciones hidrofóbicas (< 10 kcal/mol)
Para las cadenas poliméricas, tales como las proteínas, laposición del segmento en la cadena también es importante,además del alcance de la fuerza.
Las interacciones locales son aquellas que ocurren entresegmentos de la cadena que son vecinos conectados (i, i + 1)o vecinos cercanos en la secuencia. No locales se dice de lasinteracciones entre residuos que están significativamente sepa-radas en la secuencia. Las interacciones locales pueden sur-gir de fuerzas de largo o de corto alcance, al igual que lasinteracciones no locales (figura 10).
El acomodamiento de la proteína a partir de una cadenade péptidos extendida libremente implica una variedad defuerzas físicas, algunas de las cuales favorecen a la cadenade péptidos sin plegar y otras que favorecen a la proteína biendoblada. Al evaluar las contribuciones entrópicas a la energíalibre de Gibbs con el propósito de determinar la espontaneidad,debemos ver no sólo el sistema, sino también su entorno. Escierto que la entropía (de Gibbs) de la proteína decrece al ple-garse porque la cadena se acomoda de modo más ordenado.Sin embargo, hay otras consideraciones entrópicas en elentorno de la proteína. En una proteína desdoblada, todaslas porciones hidrofóbicas del polipéptido están expuestasal entorno acuoso, y las moléculas de agua se ordenan en
111
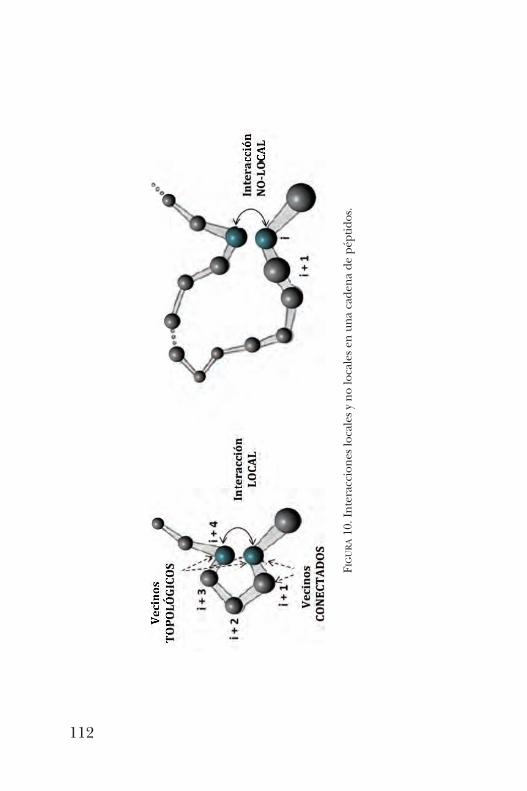
112
FIGU
RA
10. I
nter
accion
es lo
cale
s y no
loca
les en
una
cad
ena
de p
éptid
os.

torno a los residuos hidrofóbicos en estructuras ordenadasllamadas esferas de hidratación. Conforme se pliega una pro-teína, estos residuos hidrofóbicos inicialmente expuestosal entorno acuoso quedan sepultados en el interior de laproteína, escondidos del contacto con moléculas de agua,y la entropía éstas aumenta conforme la necesidad de esfe-ras de hidratación disminuye, superando en efecto la dismi-nución de la entropía para la proteína sola. En otras palabras,en tanto que la entropía del sistema (la proteína) decrece, laentropía del entorno aumenta en mayor grado, lo que llevaa un aumento total en la entropía para el universo (véanselas referencias sobre bioquímica).
5.2 El siguiente resultado del presente artículo queda muybien ilustrado por el proceso de formación de una red de inter -acciones proteína-proteína, donde una red dada G espontá-neamente tiene dos nodos (marcados como n – 1 y n) queinteractúan de modo que aparece el enlace n – 1 – n y seforma una nueva red . Según el Teorema, bajo hipótesis!G
113
##
! ! !
! ! Según el Teorema, bajo hipótesis muy
g
Queda por explicar cómo entra este hecho en el cuadro
c
F
La
t
! !
! ! !
! !! !
!
$$ �
! ! !
!
#############$%&#
$#
FIGURA 11. La gráfica de la RIP de Archeoglobus fulgidus. La interacción posible delas proteínas correspondientes a los nodos n - 1 y n determinan la existenciade la arista entre estos nodos. La transición de la gráfica sin la arista a la gráficacon la arista implica una disminución en la entropía.

114
muy generales, la entropía de gráfica del sistema disminuye,resultado que es compatible con la disminución en la entro-pía de Gibbs. Queda por explicar cómo entra este hecho enel cuadro completo del universo. Teorema 3 : sea A = (aij) la matriz de adyacencia de la grá-
fica G (resp. A = (aij) la matriz de adyacencia de G ). Ambasgráficas tienen n vértices. Supongamos que aij = aij para (i, j)(n – 1, n) y, además, an – 1, n = 0 en G y an – 1, n = 1 en G . Supon-gamos que una de las siguientes condiciones se cumple paralos elementos diagonales yi de la matriz eA:
(a) , donde es la energía de G ;
(b) , donde , para una matriz
ortonormal V y D = diag(l1, …, ln) es la matriz diagonal for-mada con los eigenvalores de G.
Entonces SV(G , 1) < SV(G, 1).Demostración : obsérvese que A = A + B con B = En – 1, n +
En, n – 1, donde Eij denota a la matriz elemental con 1 en elelemento ij y 0 en todos los demás.
Dividimos la demostración en varios pasos. Suponemosque n ≥ 3.
(1) Cálculo de eB = .
En efecto, el polinomio mínimo de B es mT = T(T 2 – 1)con raíces 0, 1, –1. En consecuencia, hay un polinomio degrado dos que satisface
!G!G!G!!G
!G!G
!G!G

115
que da el sistema b0 = 1 y
cuya solución implica el resultadodeseado.
(2) Aplicamos la poderosa identidad de Lie
Sea V una matriz ortonormal tal que A = VDVtr, dondeD = diag (l1, …, ln). Por lo tanto eA = VeDVtr, donde eD = diag(el1, …, eln). Entonces
Definamos 0 < k 1 tal que
Por lo tanto
(3) Definimos yi = (eA)ii e y i = (eA)ii, para i = 1, …, n.Obsérvese que y i ≥ yi, para i = 1, …, n. Más precisamente, dela identidad de Lie,
≥
!G!G
!G

por lo que tenemos
y
Entonces
Podemos suponer, sin pérdida de generalidad, que
yn – 1 + yn = 2rZ
para alguna 0 < r 1/2. La desigualdad de la suma delogaritmos da
y
(4) La entropía de los vértices es
≥
116

(5) Los valores k, r, b2 que aparecen arriba son constan-tes y Z = Sni=1 eli ≥ n.
(a) Si entonces podemos escoger . Porlo tanto
(b) Si entonces podemos escoger k 2r.Por lo tanto
como se desea.
REFERENCIAS
Libros de matemáticas
[1] Harary F. Graph Theory. Addison Wesley, 1967. [2] Cvetkovic D., Doob M. y Sachs H. Spectra of Graphs. Theory and Appli-
cations. Academic Press. Nueva York, San Francisco, 1980. [3] Biggs N. Algebraic Graph Theory. Cambridge University Press, Cam-
bridge, 1993.[4] Godsil C. D. Algebraic Combinatorics. Chapman and Hall, 1993. [5] Estrada E., The Structure of Complex Networks. Theory and Applications,
Oxford University Press, UK, 2011.
Artículos de matemáticas
[6] Benzi M. A note on walk entropies in graphs, Linear Algebra Appl.,2014, 445, 395-399.
≥
117

[7] Borwein J. y Girgensohn R., A class of exponential inequalities (Pre-print). http://docserver.carma.newcastle.edu.au/148/2/01_174-Borwein-Girgensohn.pdf.
[8] Dehmer, M. y Mowshowitz, A. A history of graph entropy measures.Inform. Sci., 2011, 181, 57-78.
[9] Deng H., Radenkovic S. y Gutman I., The Estrada index. Applica-tions of Graph Spectra, Math. Inst., Belgrade, 2009, 123-140.
[10] Estrada E., de la Peña J. A. y Hatano N., Walk entropies in graphs,Linear Algebra Appl., 2014, 443, 235-244.
[12] Estrada E. y Rodríguez-Velázquez J. A., Subgraph centrality in com-plex networks, Phys. Rev. E., 2005, 71, 671-696.
[13] Estrada E., Hatano N. y Benzi M., The physics of communicabilityin complex networks, Phys. Rep., 2012, 514, 89-119.
[14] Estrada E. y de la Peña J. A. Maximum walk-entropy implies walkregularity. Por aparecer en Linear Algebra Appl.
[15] Gutman I., de la Peña J. A. y Rada J., Estimating the Estrada index,Linear Algebra Appl., 2007, 427, 70-76.
[16] Estrada E. Graph and network theory in physics. A short intro-duction. arXiv: 1302.4378.
Bioquímica
[17] Dill K. Dominant forces in protein folding. Biochemistry, 1990,29(31).
[18] Kollman P. et al. Calculating Structures and Free Energies of Com-plex Molecules: Combining Molecular Mechanics and ContinuumModels. Acc. Chem. Res., 2000, 33, 889-897.
[19] Science technologies. Essential Biochemistry. Wiley, 2004.
Bioinformática
[20] Aittokallio T. y Schwikowski B. Graph-based methods for analyzingnetworks in cell biology. Briefings Bioinformatics, 2006, 7, 243-255.
[21] Barabási A. L. y Oltvai Z. N. Network biology: understanding thecell’s functional organization. Nature Reviews Genetics, 2004, 5, 101-113.
[22] Patil A., Kinoshita K. y Nakamura H. Hub Promiscuity in Protein-Protein Interaction Networks. International Journal of MolecularSciences, 2010, 11, 1930-51.
[23] Barabási A. L. y Albert R. Emergence of scaling in random net-works. Science, 1999, 286, 509-512.
118
´

[24] Vallabhajosyula R. R., Chakravarti D., Lutfeali S., Ray A. y Raval A.Identifying Hubs in Protein Interaction Networks. PLOS ONE, 2009,4, e5344.
[25] Vendruscolo M., Dokholyan N. V., Paci E. y Karplus M.. Small-worldview of the amino acids that play a key role in protein folding.Phys. Rev. E., 2002, 65:061910.
[26] Bagler G. y Sinha S. Network properties of protein structures.Physica A: Statistical Mechanics and its Applications, 2005.
119


ENERGÉTICA DE LA TRANSICIÓN PLEGAMIENTO/DESPLEGAMIENTO
DE PROTEÍNAS:UNA HERRAMIENTA PARA APRENDER SOBRE
LA CONFORMACIÓN DE PROTEÍNAS EN UN SISTEMA BIOLÓGICO
ALICIA ORTEGA
JOEL MEDINA
Departamento de Bioquímica Facultad de Medicina
Universidad Nacional Autónoma de Mé[email protected]
Life is the existence form of protein structures, and this existence form consists essentially of the constant
self-renewal of the chemical components of these structures.1
FRIEDRICH ENGELS
(Científico social), 1894
Durante muchos años, científicos de todas las áreas del cono-cimiento han intuido y tratado de interpretar a la estructurade las proteínas y su función asociada. Uno de los retos actua-les más importantes en la biología, especialmente en bio-química, consiste en entender las reglas que determinan cómoun polipéptido naciente adquiere una estructura tridimen-
121
1 “La vida es la forma de existir de la estructura de proteínas, y esta forma deexistir consiste esencialmente en una constante autorrenovación de los com-ponentes químicos que constituyen a estas estructuras”.

sional en un tiempo biológico (~milisegundos) y con unafunción fisiológica definida. El rápido progreso en la secuen-ciación de genomas, incluido el genoma humano con sus~25 mil genes que codifican para la síntesis de proteínas, elplegamiento y la modificación postraduccional, han gene-rado una enorme avalancha de datos. Con el aumento y dis-ponibilidad de información de las secuencias de proteínas,que constituye la primera traducción del código genético,y con la información sobre el plegamiento de proteínas,que constituye el segundo mensaje codificado en el ADN,han hecho que el estudio de la estructura de proteínas quehabía sido tradicionalmente visto como un problema físico-químico, en la actualidad sea un paradigma explorado porlas herramientas de la teoría matemática para una extrac-ción más eficiente de la información, que será determinantepara entender el comportamiento de las proteínas. El para-digma del plegamiento de proteínas en una célula viva esun proyecto hoy en día de un esfuerzo multidisciplinario.
INTRODUCCIÓN
La conformación nativa de una proteína, definida comola estructura con un estado de mínima energía libre, se logramediante un plegado específico de la cadena polipeptídica.La estructura resultante tendrá la actividad específica másalta para una función definida. Las cadenas polipeptídi-cas que se sintetizan en los ribosomas y que dan origen a lasproteínas, van desde 50 aminoácidos para la proteína más pe -queña hasta una cadena polipeptídica de 34,354 aminoácidos,que corresponde a la proteína más grande que se haya des-cubierto hasta hoy, y que está codificada en el genoma delos vertebrados (la Titina, una proteína del tejido muscular).Todas las proteínas pasan por un proceso para encontrar su
122

123
propia estructura tridimensional, fenómeno que puede suce-der espontáneamente o facilitado por otras proteínas (Cha-peronas) u otras enzimas. La síntesis de proteínas y su plegadoocurren en milisegundos y en pocas ocasiones se requierenpocos segundos. Este proceso de síntesis y plegamiento deproteínas ocurre miles de veces en cada una de las célulasde las cerca de 40 × 1013 células que constituyen por ejem-plo, el cuerpo de un humano adulto.
Se han utilizado enfoques termodinámicos y cinéticospara caracterizar la energética del plegamiento de proteínas.Desde los experimentos famosos de Anfinsen en la décadade los sesenta se ha creído y, generalmente aceptado, que elplegamiento y la estructura nativa de las proteínas están gober-nados y determinados por la secuencia de aminoácidos y lanaturaleza del solvente en el que se encuentran [1]. La supo-sición de que una cadena polipeptídica transite de manera
FIGURA 1. El Dogma Central, del ADN a las proteínas funcionales. Lo que se sabesobre el plegamiento de proteínas a partir de los experimentos in vitro y pre-dicciones matemáticas, podría no ser cierto para las proteínas in vivo en susentornos biológicos. Actualmente se desconoce si las reglas que dirigen el meca-nismo de plegado de las proteínas in vitro son válidas para entender el mecanismode plegado dentro de la célula viva.

aleatoria en todas las posibles configuraciones hasta encon-trarse en el estado de mínima energía libre, no considera elfactor tiempo para un proceso biológico. Un ejemplo clásicodado para esta hipótesis es el de una proteína de 100 ami-noácidos, en donde cada enlace peptídico puede tener tresposibles conformaciones. Por lo que esta proteína podría exis-tir en 3100 = 5 × 1047 conformaciones diferentes y asumiendoque cada conformación se puede muestrear tan rápido como1 × 1013/segundo, tomaría 1 × 1027 años para pasar por todaslas conformaciones posibles. Teniendo en cuenta el ejem-plo anterior y siguiendo la hipótesis termodinámica, resultadifícil visualizar la complejidad de plegado, por ejemplo dela proteína Titina con sus 34,354 aminoácidos (334,354). LaTitina es una proteína presente en grandes cantidades en elmúsculo estriado (cardiaco y esquelético), en donde se sinte-tiza en pocos segundos, y se pliega y despliega durante cadaciclo de la contracción/relajación muscular que sucede enun rango de milisegundos, por lo que la Titina, no llegaríaa su conformación nativa de manera aleatoria. Contrario a loque ocurre en la mayoría de las proteínas, el proceso de ple-gado/desplegado de la Titina es esencial para su función(resorte molecular), siendo esta proteína responsable de laelasticidad pasiva del músculo.
Levinthal, señaló (1969) la inconsistencia de la hipótesistermodinámica del plegamiento de proteínas en relacióncon los tiempos biológicos y sugirió la existencia de rutasespecíficas para el plegamiento [2]. La hipótesis de la ciné-tica de plegamiento está basada en un mecanismo jerárquico,en donde la estructura secundaria de la proteína o las inter -acciones hidrofóbicas son las fuerzas iniciales que impulsan elproceso de propagación del plegamiento; sin embargo, estoaún no está definido. El concepto de rutas de plegamiento esfundamental en la hipótesis cinética del plegamiento. Estudiosexperimentales dirigidos a encontrar intermediarios con-
124

formacionales específicos para entender las reglas de ple-gamiento, han generado varias hipótesis complementariasrelacionadas con la iniciación-propagación y la conclusióndel proceso.
Más recientemente, basados en el concepto metafórico delpaisaje epigenético, propuesto por Waddington en la década delos cincuenta, que simula en caída libre a un cigoto totipo-tente en una cascada de diferenciación celular que en elcaso del mamífero genera casi 220 diferentes tipos de célu-las, propone que estos cambios en la diferenciación celularestán gobernados por estados energéticos favorables. Estemodelo sugiere que cada linaje celular sigue su propia tra-yectoria en un proceso en principio, irreversible. El conceptode paisaje epigenético dio origen al concepto del paisajeenergético. Bryngelson en 1987, propuso el uso de la teoría delos vidrios de espín, utilizando el concepto del paisaje ener-gético para estudiar el plegamiento de proteínas [3]. Este mo -delo es una herramienta matemática basada en mecánicaestadística utilizado para ayudar a entender el comportamien-to energético de un sistema molecular desordenado en buscade la conformación de menor energía libre [4].
En el caso de las proteínas, el plegamiento sucede a tra-vés de un número indefinido de estados intermedios parallegar a la conformación nativa. Los ejemplos de proteínascon paisajes energéticos sinuosos, con muchos mínimos deenergía libre, pasan por estructuras intermedias metaestables(figura 2A). Es importante destacar que los paisajes ener-géticos sinuosos son el resultado del estudio de secuenciaspolipeptídicas elegidas aleatoriamente. Por el contrario, lasecuencia de las proteínas biológicas está lejos de ser alea-toria y es más bien el resultado de una optimización evolu-tiva, en donde el proceso de plegamiento sucede con unmínimo de intermediarios metaestables, por lo que el pro-ceso de plegamiento se desarrolla con un mínimo de frus-
125

tración y la trayectoria es más suave, más parecida a la formade un embudo, el embudo de plegamiento [5] (figura 2B).
Sin embargo, proteínas biológicas de gran tamaño conestructuras globulares y fibrilares pueden tener un paisajeenergético tan complejo como se ha observado que ocurre enlas proteínas no biológicas, lo que implicaría la coexistenciade conformaciones metaestables significativamente pobladasen equilibrio. Estos intermediarios alcanzan mínimos de ener-gía libre que para continuar su trayectoria cuesta abajo, desdeel punto de vista energético, tienen que superar barreras ener-géticas en busca de la conformación del estado nativo. Con-formaciones metaestables pueden ser detectadas por métodosbioquímicos convencionales y pueden tener actividades aso-ciadas a la función de la proteína nativa o tener diferentesactividades.
El modelo cinético-termodinámico de una transición deltipo , en donde la población de proteínas está sólo enlos estados Desplegados y Nativos, no considera característicascinéticas, tales como a los intermediarios transitorios y tram-pas cinéticas, por lo que es necesario considerar dos aspectosque pueden complicar aún más el estudio de la energéticade plegamiento así como la hipótesis del paisaje energé-
D! N
126
##
E
(Fig. 2A). Es importante destacar que los paisajes energéticos sinuosos son el
r
desarrolla con un mínimo
d más parecida a la forma de un embudo, e
FIGURA 2. Representación esquemática de paisajes energéticos de proteínasA) con varios intermediarios metaestables y B) con dos poblaciones; desple-gadas (D) y nativas (N).

tico para las proteínas biológicas in vivo: 1) la formación dedisulfuros y 2) el plegamiento dependiente de Chaperonas.
1) La formación de enlaces entre cisteínas (disulfuros).Esta reacción se lleva a cabo de manera enzimática (disul-furo isomerasas) o no enzimática (potencial redox, DE°). Losdisulfuros son elementos clave en la estructura tridimensionalde muchos polipéptidos y proteínas. Tales enlaces covalentesestabilizan la estructura nativa de manera significativa, redu-ciendo la entropía del estado desplegado y estabilizando laestructura del estado nativo. El término de plegamiento oxi-dativo, que describe a este proceso, implica que bajo las con-diciones redox del medio se pueda llevar a cabo el plegamientohasta los estados de mínima energía. Por ejemplo, en un am -biente reducido (–230 mV), como sucede dentro de unacélula eucariota, las proteínas con cisteínas reactivas adquie-ren su estructura nativa formando disulfuros, que son ener-géticamente desfavorables en estas condiciones. Cuando sereducen los disulfuros de la proteína, la estructura nativase recupera mediante la formación de los disulfuros nativos(regeneración de disulfuros) invirtiendo energía de reacciónde enzimas específicas. Las proteínas en las células eucario-tas son lineales y algunas células procariotas sintetizan pro-teínas cíclicas, que son estructuras muy estables. La forma-ción de disulfuros da origen a ciclos que pueden darse enuna misma cadena polipeptídica o conectando a dos cade-nas polipéptidicas, dando origen a la formación de nudos.Alrededor de un 10% de las proteínas celulares están impli-cadas en la formación de disulfuros, dando origen a estruc-turas de interés para la topología [6].
A los polipéptidos con disulfuros que forman nudos, seles conoce genéricamente como Knottinas, que son proteínaspequeñas ricas en disulfuros y que muestran una estabili-dad estructural excepcional, tanto térmica como biológica,parecida a la de las proteínas cíclicas en las células proca-
127
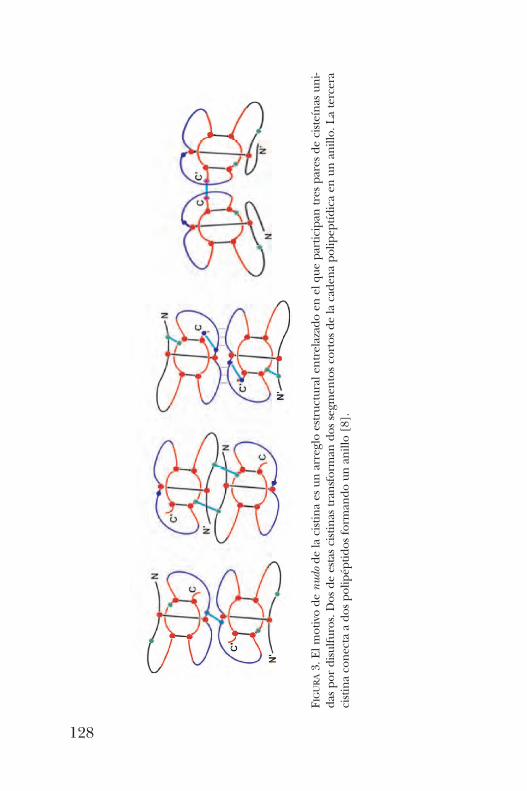
128
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
FIG
URA
3. E
l mot
ivo
de nud
ode
la c
istin
a es
un
arre
glo
estr
uctu
ral e
ntre
laza
do e
n el
que
par
ticip
an tr
es p
ares
de
cisteí
nas un
i-da
s po
r di
sulfu
ros.
Dos
de
esta
s ci
stin
as tr
ansfor
man
dos
seg
men
tos co
rtos
de
la c
aden
a po
lipep
tídic
a en
un
anill
o. L
a te
rcer
aci
stin
a co
nect
a a
dos po
lipép
tidos
form
ando
un
anill
o [8
].

riotas. Las Knottinas han sido estudiadas sistemáticamentecon la teoría combinatoria y optimización [7] y constituyenun conjunto considerado una biblioteca combinatoria en laNaturaleza [8] (figura 3).
2) El plegamiento de proteínas-dependientes de Chape-ronas. Las proteínas denominadas Chaperonas, tambiénconocidas como proteínas de choque térmico, proporcionanmicroambientes para asistir al plegado de otras proteínas,facilitan el plegado de ~80% de las proteínas celulares [9].La disfunción o ausencia de Chaperonas resultaría en unatrampa cinética en el proceso de plegamiento. Las Chape-ronas por sí mismas tienen funciones y una estructura pareci-das a un embudo estructural de plegamiento, que se alimentadel polipéptido naciente del ribosoma y libera a la proteínaen su forma nativa (figura 4). Las Chaperonas también seunen a proteínas que se han plegado incorrectamente, dán-doles una segunda oportunidad para llegar a la estructuranativa. En general, la función clave de las Chaperonas es elreconocimiento de superficies hidrofóbicas.
Las estructuras metaestables intermedias en el proceso deplegamiento (estructuras parcialmente plegadas) son par-ticularmente vulnerables a la agregación, muy probablementea través de interacciones moleculares específicas entre sussuperficies hidrofóbicas. Estas conformaciones tienden a agre-garse de manera dependiente de la concentración de pro-teína, por lo que son más propensas a la agregación en com-paración a las configuraciones desplegadas, en donde lascadenas laterales hidrofóbicas están en una configuraciónaleatoria.
Se ha demostrado en varios estados patológicos relacio-nados con la agregación de proteínas, tales como en la ami-loidosis, el Alzheimer y otras patologías neurodegenerativas,que la formación de los cuerpos de inclusión (agregados dela proteína) hallados en las células [10] son determinantes
129
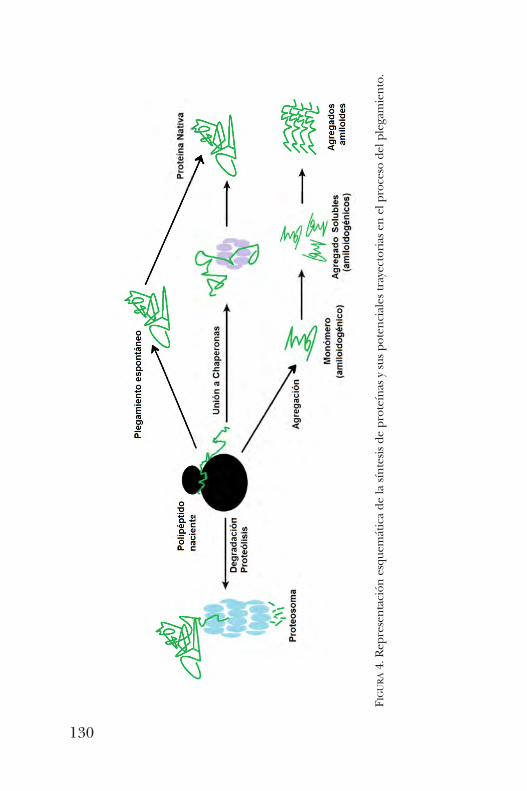
130
FIG
URA
4. R
epre
sent
ació
n es
quem
átic
a de
la sín
tesis de
pro
teín
as y sus
pot
enci
ales
tray
ecto
rias
en
el p
roce
so d
el p
lega
mie
nto.

para el desarrollo de la enfermedad. Una disponibilidadreducida de las Chaperonas, ya sea por una síntesis muyrápida de proteínas, la formación de intermediarios meta-estables con vidas medias más largas y/o la presencia de Cha-peronas defectuosas, resulta en una inminente agregaciónde proteínas y con ello la disfunción celular [11].
Las condiciones experimentales en las que se estudia elplegamiento de proteínas consiste, generalmente, en utilizarpreparaciones puras de proteínas pequeñas, colocadas ensoluciones acuosas, habitualmente diluidas, puestas a tempe-raturas bajas, en un entorno de pH amortiguado en dondecuidadosamente se considera el punto isoeléctrico (pI) de laproteína a estudiar para evitar la agregación.
Por el contrario, el interior de una célula es un sistema ter-modinámico abierto, con un entorno dinámico altamentepoblado y con una concentración de proteína de ~100 mg/ml= 3mM en un volumen celular de entre 10-2 a 10-4 µm3 parauna célula promedio de mamífero; en donde las proteínassolubles coexisten con una red bien definida formada deproteínas estructurales que forman el citoesqueleto [12].Esto implicaría que las estructuras y funciones asociadas a lasproteínas estudiadas en un sistema acuoso, no necesaria-mente son las mismas que si fueran estudiadas en un am -biente coloidal. Asimismo, cambios en el potencial redox (DE°),fluctuaciones transitorias de pH cercano al pI de muchasproteínas e incluso fluctuaciones de temperatura y presiónestán sucediendo continuamente dentro de una célula, loque influiría en la energética del plegamiento. La estructuranativa y los paisajes energéticos del plegamiento determi-nados bajo condiciones experimentales pueden no coinci-dir con el fenómeno in vivo.
Por otro lado, la misma estructura de la proteína nativaen diferentes conformaciones puede tener varias actividadesque podrían ajustar con modelos de importancia fisiológica.
131

Por ejemplo, en enzimología, el paradigma de la especifici-dad de sustrato (la llave y la cerradura) tiene sus excepcionesy reconoce la importancia biológica de la llamada promis-cuidad catalítica [13]. La promiscuidad catalítica, en contrastecon la especificidad de sustrato, es un fenómeno poco cono-cido asociado a la estructura de la proteína, y por lo tantoderiva de la energética del plegamiento/desplegamiento delas mismas. La promiscuidad catalítica pudiese ser la naturalezacomún de la función de las proteínas más que una excepción.
LA DESNATURALIZACIÓN (DESPLEGAMIENTO) TÉRMICA
DE LAS PROTEÍNAS COMO HERRAMIENTA PARA ESTUDIAR
LA ENERGÉTICA DEL PLEGAMIENTO/DESPLEGAMIENTO
DE PROTEÍNAS Y SUS CONSECUENCIAS FUNCIONALES
Wu (1931) fue el primero en describir las relaciones fun-damentales entre el estado nativo y el desnaturalizado de unaproteína. A diferencia de la estructura nativa, en donde mu -chas variables todavía deben ser consideradas para revelarlas reglas del mecanismo de plegado, la desnaturalizaciónde proteínas es un mecanismo que ofrece la posibilidad deinvestigar la energética del plegamiento de proteínas con unenfoque experimental directo si, y sólo si la desnaturalizaciónresultante es un proceso reversible del tipo . Aunquela desnaturalización no es el proceso inverso del plegamiento,puede ser útil para entender los eventos tempranos de ple-gamiento de proteínas. Es importante destacar que la des-naturalización térmica de la mayoría de las proteínas grandes,multidominio y con múltiples subunidades, es irreversible enlas condiciones experimentales en las que han sido estudiadas.
La Calorimetría Diferencial de Barrido (CDB) es usada parainvestigar la estabilidad de proteínas y, en combinación conotros métodos biofísicos y bioquímicos ha sido muy útil para
N ! D
132

vincular la termodinámica del proceso a la estructura y lafunción de las proteínas. Para que una proteína se desplieguenecesita que se rompan las fuerzas que la estabilizan. La CDB
mide el cambio en la capacidad calorífica (DCp) de la pro-teína durante la desnaturalización (ruptura de su conforma-ción tridimensional) en función del cambio de temperaturaa una tasa constante. Los cambios en la DCp se deben, prin-cipalmente, a cambios en la hidratación de las cadenas late-rales de los aminoácidos que estaban ocultos en el estadonativo y que se exponen al disolvente en el estado desnatu-ralizado. De la resta de la contribución de la Cp del estadodesnaturalizado y el estado nativo, se obtiene la absorción decalor causada por cambios conformacionales (exceso en lacapacidad calorífica, exCp). La integral de los cambios enexCp a presión constante es igual a la entalpía.
La entalpía calorimétrica total obtenida de la desnatura-lización completa de la proteína está dada por:
En donde Cp se obtiene experimentalmente.Se han utilizado modelos cinéticos para modelar la des-
naturalización de proteínas. Los parámetros termodinámi-cos se obtienen directamente del ajuste de las curvas del exCp,de acuerdo con modelos previamente descritos [14, 15, 16].La temperatura de transición (temperatura de fusión, Tm)se define como la temperatura media de la entalpía y corres-ponde a la temperatura en donde hay una distribución del50% de las poblaciones nativas y desnaturalizadas. En tér-minos simples, cuando la proteína alcanza la Tm, la energíalibre de Gibbs, DG = 0.
Considerando que…
""
!!"#$ ! !"#$! ! !"!"#$%&
!!!"!#!$%
C
!
!
e
133
.

DG = DH-TDS
…y DH se obtiene experimentalmente, DS se calcula a par-tir de la información obtenida experimentalmente,
Al calentar una proteína con un incremento de tempe-ratura constante, hay ruptura de los puentes de hidrógenosin afectar los enlaces covalentes, por lo que la estructura tri-dimensional se pierde progresivamente en función del incre-mento de temperatura. Cada proteína con una estructurao estado conformacional estable, tiene parámetros termodi-námicos definidos y únicos para las condiciones experimen-tales en las que está contenida. Los parámetros termodiná-micos obtenidos a partir de la desnaturalización térmica deproteínas en condiciones experimentales específicas, consti-tuyen una huella dactilar que puede ser utilizada para suidentificación, y por lo tanto puede ser útil para examinar lasbarreras energéticas del estado nativo (figura 5).
Por ejemplo, la proteína de membrana ATPasa de calcio(SERCA), es un polipéptido de 994 aminoácidos, cuya fun-ción es transportar calcio en contra de un gradiente de con-centración al retículo endoplásmico durante la relajación dela célula muscular. SERCA ha sido cristalizada en dos confor-maciones determinadas por la unión de calcio [17]. Aunqueel calcio se une a unos cuantos aminoácidos en el dominiotransmembranal de la proteína, la unión del calcio induceuna reorganización completa de su estructura, este cambioes necesario para completar su ciclo catalítico. El perfil dedesnaturalización de estas dos conformaciones (estructuraprimaria conservada) sigue una cinética diferente con pará-metros termodinámicos resultantes diferentes [14] (figura 6).
""
!!
C
!
!! ! !!!"
e
134
.

La información obtenida de los perfiles de desnaturaliza-ción (energética del desplegamiento) de la SERCA procedentede músculos en diferentes condiciones fisiológicas, tales comola fatiga [18, 19] o la adaptación al ejercicio [20] revelan unaenergética de desplegamiento diferente, lo que indica quedurante estos procesos, la SERCA adquiere otra conformaciónestable capaz de mantener la función (figura 7).
De la misma manera que otras conformaciones establesde la SERCA ocurren en condiciones fisiológicas de estrésmecánico y metabólico, como en la fatiga, en algunas distro-fias musculares, la SERCA modifica su estructura en una formadiferente a la que ocurre en la fatiga [18]. En condicionespatológicas como en la mayoría de las distrofias musculares, quese caracterizan por una disminución en la fuerza de con-tracción, la energética de desplegamiento de la SERCA es sig-
135
##
A
##
##
##
##
##
##
##
s
##
n
##
c
##
a
##
e
##
o
##
o
##
##
p
##
g
##
i
##
g
##
)
##
!
FIGURA 5. Representación esquemática de la desnaturalización térmica de dosconformaciones estables de la SERCA (enzima de origen muscular, ATPasa decalcio). Las imágenes iniciales en cada línea fueron obtenidas por cristalogra-fía de la SERCA en presencia y en ausencia de calcio [17]. Las imágenes consecu-tivas son representaciones esquemáticas de la pérdida de estructura en funcióndel incremento en la temperatura.
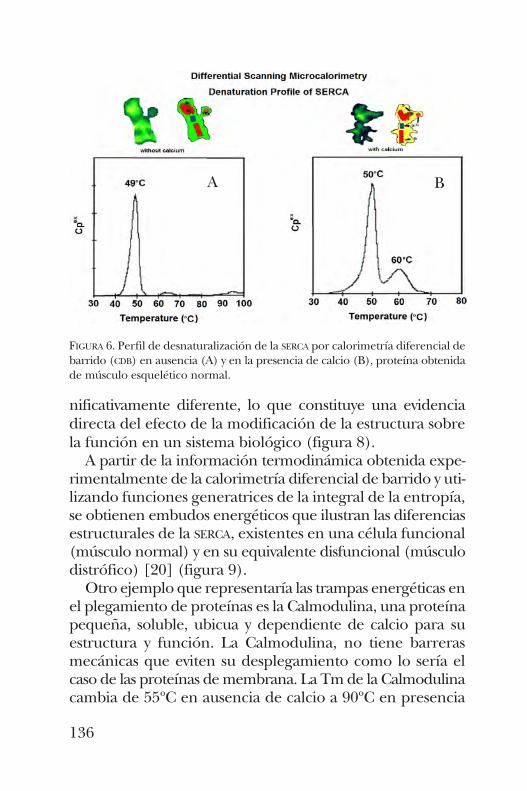
nificativamente diferente, lo que constituye una evidenciadirecta del efecto de la modificación de la estructura sobrela función en un sistema biológico (figura 8).
A partir de la información termodinámica obtenida expe-rimentalmente de la calorimetría diferencial de barrido y uti-lizando funciones generatrices de la integral de la entropía,se obtienen embudos energéticos que ilustran las diferenciasestructurales de la SERCA, existentes en una célula funcional(músculo normal) y en su equivalente disfuncional (músculodistrófico) [20] (figura 9).
Otro ejemplo que representaría las trampas energéticas enel plegamiento de proteínas es la Calmodulina, una proteínapequeña, soluble, ubicua y dependiente de calcio para suestructura y función. La Calmodulina, no tiene barrerasmecánicas que eviten su desplegamiento como lo sería elcaso de las proteínas de membrana. La Tm de la Calmodulinacambia de 55ºC en ausencia de calcio a 90ºC en presencia
136
##
por calorimetría diferencial de barrido (CDB)
e
FIGURA 6. Perfil de desnaturalización de la SERCA por calorimetría diferencial debarrido (CDB) en ausencia (A) y en la presencia de calcio (B), proteína obtenidade músculo esquelético normal.
A B
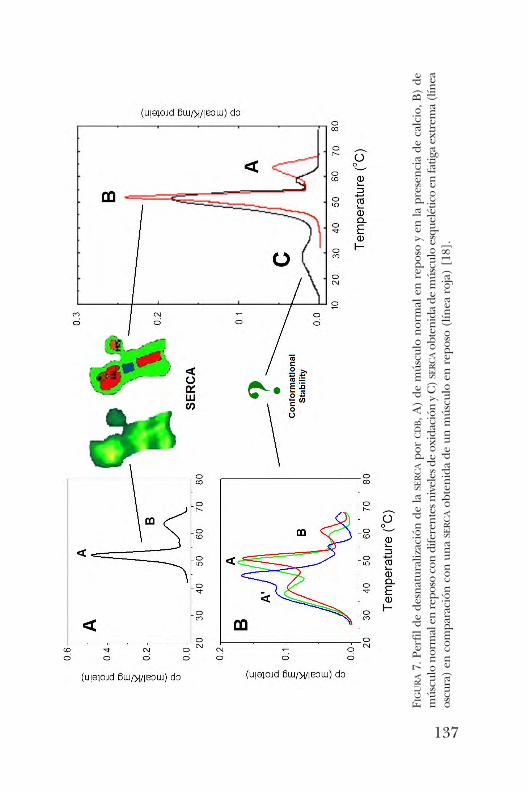
137
$$
FIG
URA
7. P
erfil
de
desn
atur
alizac
ión
de la
SERCA
por
CD
B, A
) de
mús
culo
nor
mal
en
repo
so y
en
la p
rese
ncia
de
calc
io, B
) de
mús
culo
nor
mal
en
repo
so con
dife
rent
es n
ivel
es d
e ox
idac
ión
y C)
SERCAob
teni
da d
e m
úscu
lo esq
uelé
tico
en fa
tiga ex
trem
a (lín
eaos
cura
)en
com
para
ción
con
una
SERCA
obte
nida
de
un m
úscu
lo e
n re
poso
(lín
ea roj
a) [
18].
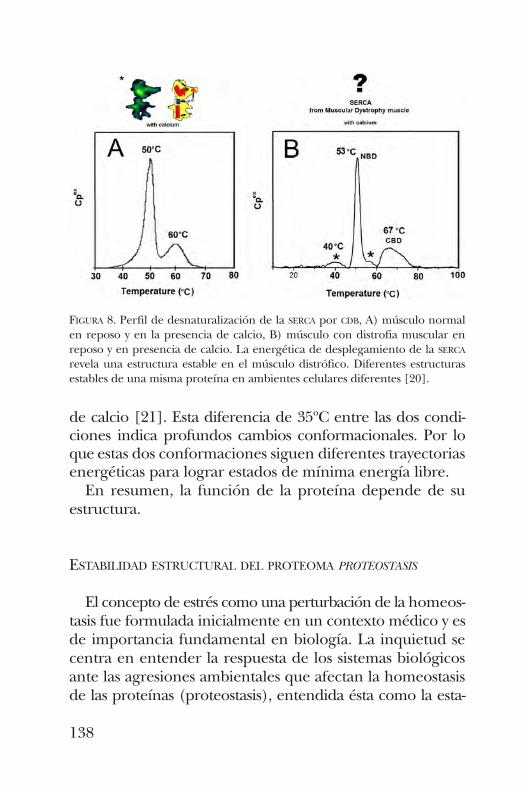
de calcio [21]. Esta diferencia de 35ºC entre las dos condi-ciones indica profundos cambios conformacionales. Por loque estas dos conformaciones siguen diferentes trayectoriasenergéticas para lograr estados de mínima energía libre.
En resumen, la función de la proteína depende de suestructura.
ESTABILIDAD ESTRUCTURAL DEL PROTEOMA PROTEOSTASIS
El concepto de estrés como una perturbación de la homeos-tasis fue formulada inicialmente en un contexto médico y esde importancia fundamental en biología. La inquietud secentra en entender la respuesta de los sistemas biológicosante las agresiones ambientales que afectan la homeostasisde las proteínas (proteostasis), entendida ésta como la esta-
138
$$
(A) Músculo normal en reposo y en
l
funciones generatrices de la integral de la entropía, se
o
FIGURA 8. Perfil de desnaturalización de la SERCA por CDB, A) músculo normalen reposo y en la presencia de calcio, B) músculo con distrofia muscular enreposo y en presencia de calcio. La energética de desplegamiento de la SERCA
revela una estructura estable en el músculo distrófico. Diferentes estructurasestables de una misma proteína en ambientes celulares diferentes [20].
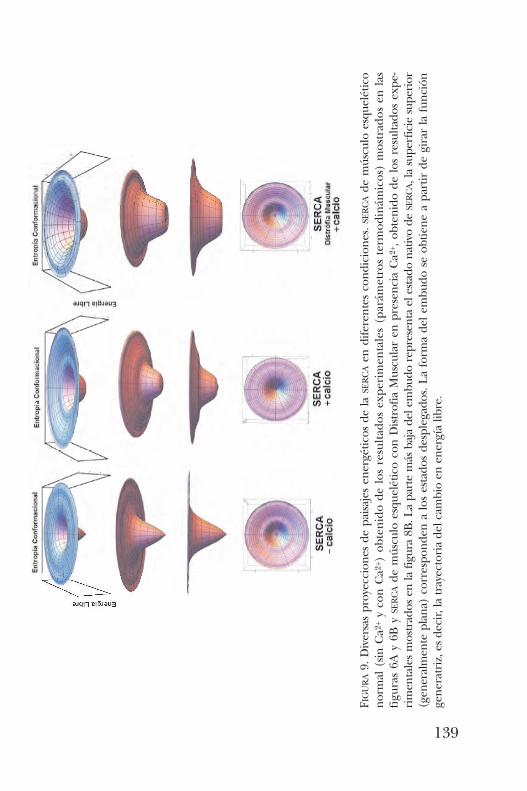
139
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
FIG
URA
9. D
iver
sas pr
oyec
cion
es d
e pa
isaj
es e
nerg
étic
os d
e la
SERCA
en d
ifere
ntes
con
dici
ones
. SERCA
de m
úscu
lo e
sque
létic
ono
rmal
(sin
Ca2
+y
con C
a2+ )
obt
enid
o de
los
res
ulta
dos
expe
rim
enta
les
(par
ámet
ros
term
odin
ámic
os)
mos
trad
os e
n las
figu
ras
6A y
6B y
SERCA
de m
úscu
lo e
sque
létic
o co
n D
istr
ofia
Mus
cula
r en
pre
senc
ia C
a2+ , o
bten
ido
de los
res
ulta
dos
expe
-rim
enta
les m
ostr
ados
en
la fi
gura
8B. L
a pa
rte
más
baj
a de
l em
budo
rep
rese
nta
el e
stad
o na
tivo
de S
ERCA, l
a su
perf
icie
sup
erio
r(g
ener
alm
ente
pla
na)
corr
espo
nden
a los
estad
os d
espl
egad
os. L
a fo
rma
del em
budo
se
obtie
ne a
par
tir d
e gi
rar
la fun
ción
gene
ratr
iz, e
s de
cir,
la tr
ayec
toria
del c
ambi
o en
ene
rgía
libr
e.

bilidad del proteoma. Factores físicos que constituyen unestímulo estresante son: el estrés térmico (calor y frío), el pHy las fluctuaciones en el DE°, cambios en la osmolaridad y elestrés mecánico.
El choque térmico por calor es, sin duda, el factor estresantedel que mejor se entienden las consecuencias físico-químicassobre el proteoma. La exposición a altas temperaturas puedecambiar críticamente el equilibrio conformacional de las pro-teínas a estados parcialmente desplegados. Como resultado,el peligro de la agregación de proteínas aumenta abrupta-mente, explicando la necesidad de una rápida regulaciónpor parte de las Chaperonas (proteínas de choque térmico).La agregación de las proteínas no necesariamente resultaen una estructura térmicamente más estable, como se ha vis-to ocurre con las proteínas amiloide que incrementan su Tmhasta en 40°C, dependiendo del tiempo de incubación y con-centración de proteína que da origen al agregado [22].Muchos de los agregados de proteína que ocurren en bio-logía, por ser más estables se dificulta su degradación, ade-más de que la formación de agregados limita la función dela proteína nativa interfiriendo con la función de otros sis-temas, siendo éste el origen de patologías más complejas.
Independientemente del estímulo estresante que se apli-que, las células parecen estar mejor equipadas para respon-der al estrés agudo que al estrés crónico. Cada vez más sereconoce al estrés crónico como el origen de la enfermedady es, en cierto modo, la causa que afecta la proteostasis en elproceso de envejecimiento.
REFERENCIAS
[1] Anfinsen C. B. The formation and stabilization of protein structure.Biochem J., 1972, 128(4): 737-749.
140

[2] Levinthal C. How to fold graciously. En: Mossbauer Spectroscopy in Bio-logical Systems, Proceedings of a Meeting Held at Allerton House, Monti-cello, Illinois, eds. J. T. P. DeBrunner, E. Munck, 1969, pp. 22-24.
[3] Bryngelson J. D. y Wolynes P. G. Spin glasses and the statistical mecha-nics of protein folding. Proc. Natl. Acad. Sci. USA, 1987, 84, 7524-7528.
[4] Bryngelson J. D., Onuchic J. N., Socci N. D. y Wolynes P. G. Funnels,pathways, and the energy landscape of protein folding: a synthesis.Proteins, 1995, 21(3): 167-95.
[5] Leopold P. E., Montal M. y Onuchic J. N. Protein folding funnels:A kinetic approach to the sequence-structure relationship. Proc.Natl. Acad. Sci. USA, 1992, 89, 8721-8725.
[6] Sikora M. y Cieplak M. Formation of Cystine Slipknots in DimericProteins PLOS ONE, 2013, 8(3).
[7] Glotzbach B., Reinwarth M., Weber N., Fabritz S., Tomaszowski M.Fittler H., Christmann A., Avrutina O. y Kolmar H. CombinatorialOptimization of Cystine-Knot Peptides towards High-Affinity Inhi-bitors of Human Matriptase-1. PLOS ONE, 2013 DOI: 10.1371.
[8] Gelly J. C., Gracy J., Kaas Q., Le-Nguyen D., Heitz A. et al. The KNOTTIN
website and database: a new information system dedicated to theknottin scaffold. Nucleic Acids Res., 2004 32 : D156-159.
[9] Fink A. L. Chaperone-Mediated Protein Folding. Physiological Reviews,1999, 79(2).
[10] Muñoz V. Protein Folding, Misfolding and Aggregation: Classical Themesand Novel Approaches. Ed. Royal Society of Chemistry, 2008.
[11] Selkoe D. J. Folding proteins in fatal ways. Nature, 2003, 426, 18-25. [12] John Ellis R. Macromolecular crowding: obvious but underappre-
ciated. Trends in Biochemical Sciences 2001, 26, No. 10.[13] Honaker M. T., Acchione M., Sumida J. P. y Atkins W. M. Ensem-
ble Perspective for Catalytic Promiscuity, calorimetric analysisof the active site conformational landscape of a detoxificationenzyme. The J. of Biol. Chem., 2011, 286(49), 42770-42776.
[14] Lepock J. R., Rodahl A. M., Zhang C., Heynen M. L., Waters B. yCheng K. H. Thermal denaturation of the Ca2+-ATPase of sarco-plasmic reticulum reveals two thermodynamically independent-domains. Biochemistry, 1990, 29 : 681-689.
[15] Ortega A. y Lepock J. R.: Use of thermal analysis to distinguishmagnesium and calcium stimulated ATPase activity in isolatedtransverse tubules from skeletal muscle. Biochem. Biophys. A., 1995,1233 : 7-13.
141

[16] Johnson C. M. Differential scanning calorimetry as a tool for pro-tein folding and stability. Archives of Biochemistry and Biophysics,2013, 531, 100-109.
[17] Toyoshima C., Nakasako M., Nomura H. y Ogawa H. Crystal struc-ture of the calcium pump of sarcoplasmic reticulum at 2.6 Å reso-lution. Nature, 2000, 405 : 647-655.
[18] Ortega A., Álvarez R., Pérez F., Jiménez A. M., Gutiérrez J. A. y Váz-quez P. Redox regulation of vicinal dithiol groups of the SERCA1participates in muscle fatigue. J. Muscle Res. Cell Motil., 2004, 25597-608.
[18] Álvarez R., Vázquez P., Pérez F., Jiménez A., Tirado A., Irles C.,Gon zález-Serratos H., Ortega A., Regulation of fast skeletalmuscle activity by SERCA1 vicinal-cysteines. J. Muscle Res. Cell Motil.,2009, 30 5-16.
[19] Becker V., González-Serratos H., Álvarez R., Bäermann M., Irles C.y Ortega A. Effect of endurance exercise on the Ca2+ pumps fromtransverse tubule and sarcoplasmic reticulum of rabbit skeletalmuscle. J. Appl. Physiol., 2004, 97 : 467-474.
[20] Solares-Pérez A., Álvarez R., Crosbiee R. H., Vega-Moreno J., Medina-Monares J., Estrada F., Ortega A. y Coral-Vazquez R. Altered calciumpump and secondary deficiency of sarcoglycan and microspan insarcoplasmic reticulum membranes isolated from sarcoglycanknockout mice. Cell Calcium, 2010, 48 28-36.
[21] Brzeska H., Venyaminov S. V., Grabarek Z. y Drabikowski, W. 1983.Comparative studies on thermostability of calmodulin, skeletalmuscle troponin C and their tryptic fragments. FEBSLett., 153: 169-173.
[22] Powers E. T., Morimoto R. I., Dillin A., Kelly J. W. y Balch W. E.Biological and Chemical Approaches to Diseases of ProteostasisDeficiency. Annu. Rev. Biochem., 2009, 78 : 959-91.
[23] Morel B., Varela L. y Conejero-Lara F. The Thermodynamic Stabi-lity of Amyloid Fibrils Studied by Differential Scanning Calori-metry. Phys. Chem. B, 2010, 114, 4010-4019.
142

TRANSICIONES ESTRUCTURALES DEL TIPODESORDEN-AL-ORDEN EN PROTEÍNAS
TRANSFERIDORAS DE LÍPIDOS Y SU ASOCIACIÓN A LA FUNCIÓN
JAIME MAS OLIVA
Instituto de Fisiología CelularUniversidad Nacional Autónoma de México
PLEGAMIENTO DE PROTEÍNAS
Hoy en día se ha establecido que el plegamiento de proteí-nas está directamente relacionado con los mecanismos de lastransiciones reversibles del desorden-al-orden, por el cual unacadena polipeptídica desplegada se pliega en una estructuranativa funcional específica [1, 2]. Aunque las cadenas de poli-péptidos desplegados, para adquirir un estado nativo, requie-ren la intervención de las interacciones débiles, cuando secolocan en un medio acuoso, la cadena comienza a plegar-se, impulsada por interacciones hidrofóbicas, convirtiéndoserápidamente en un glóbulo fundido. La estabilización del gló-bulo fundido se logra, principalmente, a través de la distribu-ción de los residuos hidrofóbicos fuera de la matriz del agua.De esta manera, los residuos polares, contenidos en la pro-teína, desarrollan enlaces de hidrógeno con la red de agua,y tanto a-hélices como láminas b-plegadas se pueden formar.Se ha calculado que las uniones podrían ser del orden que
143
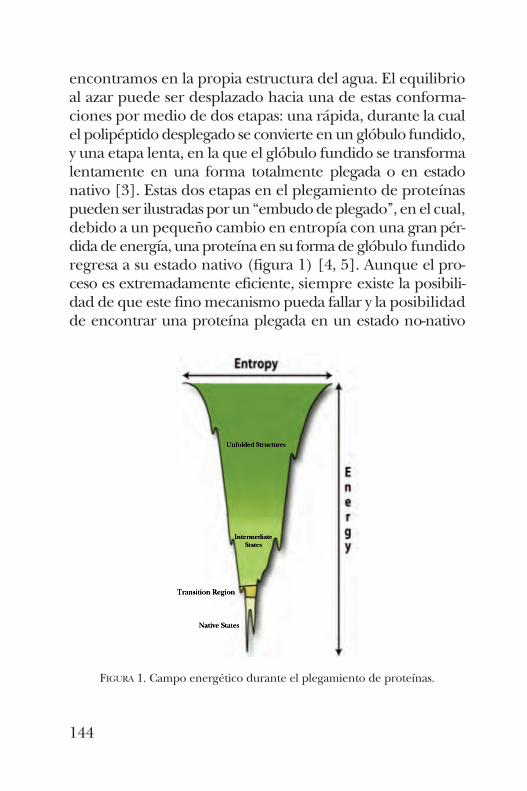
encontramos en la propia estructura del agua. El equilibrioal azar puede ser desplazado hacia una de estas conforma-ciones por medio de dos etapas: una rápida, durante la cualel polipéptidodesplegado se convierte en un glóbulo fundido,y una etapa lenta, en la que el glóbulo fundido se transformalentamente en una forma totalmente plegada o en estadonativo [3]. Estas dos etapas en el plegamiento de proteínaspueden ser ilustradas por un “embudo de plegado’’, en el cual,debido a un pequeño cambio en entropía con una granpér-dida de energía, una proteína en su forma de glóbulo fundidoregresa a su estado nativo (figura 1) [4, 5]. Aunque el pro-ceso es extremadamente eficiente, siempre existe la posibili-dad de que este fino mecanismo pueda fallar y la posibilidadde encontrar una proteína plegada en un estado no-nativo
144
FIGURA 1. Campo energético durante el plegamiento de proteínas.

145
que pueda ser factible [6]. Las proteínas que siguen esta víapueden presentar conformaciones transitoriamente estables,promoviendo su interacción con otras moléculas y facilitar elhecho de que se puedan formar oligómeros amorfos y ter-minar en un estado de agregación, la que no surge de unestado de estructura al azar, sino a partir de una serie deintermediarios que, basados en el tipo de estructura secun-daria adquirida durante el plegado, pueden o no parecerseal estado nativo (figura 2) [5, 7]. Es bien sabido ahora quelas secuencias primarias de polipéptidos se convierten en elfactor clave en este proceso, mientras que el entorno querodea a la proteína es un factor importante para explicarel proceso de plegado [8].
Por otro lado, las proteínas no plegadas de forma nativa,careciendo de la presencia de estructuras secundarias y ter-
FIGURA 2. Campo energético en la agregación de proteínas.

ciarias permanentes, se han reconocido, por lo menos, enausencia de otras proteínas, para presentar la tendencia a or -ganizarse en estructuras amiloidogénicas, teniendo en cuentaque el estado nativo se encuentra en el punto más bajo del“embudo de plegado’’. Esto indica que, en condiciones fisio-lógicas, esta región es la configuración termodinámicamentemás estable de la cadena de polipéptidos. Para las proteínas,cuyo estado funcional consiste en un pliegue globular apre-tado, un paso clave en la formación de fibrillas se desarrollay por lo tanto permanece protegido contra la agregación [9].A este respecto, se ha propuesto que las estructuras más tran-sitorias así formadas en las proteínas, son determinantesclave en la formación de fibrillas de amiloide que se encuen-tran [10]. Por lo tanto, muchas de las formas conocidas deenfermedades amiloides, asociadas con mutaciones genéti-cas que reducen la estabilidad de la proteína y promuevenel desarrollo [10], están ambas relacionadas con el trastornode las transiciones conformacionales. La primera evidenciaexperimental sobre una transición específica-trastorno-al-orden se presentó hace más de treinta años con la descrip-ción del mecanismo para la conversión del tripsinógenoa tripsina [11]. Este mecanismo se caracteriza por la elimi-nación enzimática de un hexapéptido de la región N-ter-minal del tripsinógeno con el fin de formar la tripsina. Estecambio básico promueve la transición de un estado desorde-nado del tripsinógeno a un estado ordenado en tripsina[12], dado que se sabe que varios aminoácidos que formanuna proteína favorecen fuertemente un estado desordenado,donde la influencia de las condiciones externas o ambienta-les es clave en la definición entre ambos. En la actualidadesta nueva visión del plegado está comenzando a estudiarsemás a fondo en cadenas de polipéptidos específicos con-tenidos en proteínas o proteínas completas que carecen deestructuras terciarias definidas que, se sabe, tienen la capa-
146

cidad de experimentar transiciones del desorden-al-ordentras la formación de uniones específicas [13] o múltiplesparejas [14]. Es precisamente esta capacidad la que permiteque el concepto de trastornos en la proteína se propongacomo una característica importante en la capacidad de lasproteínas para presentar regiones con propiedades de cam-bio estructural local [15-17].
Desde un punto de vista evolutivo, parece que el desordenintrínseco en proteínas podría haber sido la fuerza impul-sora detrás de muchos de los procesos de adaptación que seencuentran en las proteínas [6, 18]. Teniendo en cuenta queel número de proteínas que presentan trastornos en regionesdirectamente relacionadas con la función, y por lo tanto conla enfermedad, está aumentando cada vez más, existe actual-mente un gran interés de generar bancos de datos accesiblespara la mejora del manejo de la información. Por lo tanto,la base de datos de proteínas desordenadas (DisProt) fuecreada y liberada en agosto de 2006 por el grupo de Dunker[19], con muy buenos resultados en la actualidad [20].
Desde entonces, otros sistemas para el estudio del desor-den en proteínas han sido liberados, como el analizador decambio de proteínas que tiene como objetivo identificary predecir regiones desordenadas en proteínas [21], así comoalgoritmos para la predicción y evaluación de la agregación delos llamados “puntos calientes’’ (AGGRESCAN) [22]. De acuerdocon el grupo de Dunker, y como se predijo por PONDR® [23],un gran porcentaje de todas las proteínas implicadas conalgún tipo de enfermedad se han identificado como direc-tamente relacionadas con las regiones desordenadas en lasproteínas. Desde un punto de vista general, las regiones desor-denadas en las proteínas se han dividido en dos clases: enprimer lugar, en la que las proteínas retienen un porcentajebajo de la estructura secundaria, junto con estructuras tercia-rias inestables durante un estado de glóbulo fundido conocido
147

como colapsado, y en segundo, la clase extendida en la que lasproteínas, con una columna vertebral muy extendida, se ase-mejan a una conformación hoja-b [24, 25]. En general, lasproteínas que contienen regiones desordenadas se han reco-nocido como presentes en varias enfermedades humanas,incluyendo las cardiovasculares, el cáncer, las degenerativasy la diabetes. De forma interesante, ya que enmuchos de estoscasos la función de señalización celular está involucrada, existeuna fuerte posibilidad de que las transiciones estructurales lo -cales en las proteínas pudieran desempeñar un papel normalen la célula, pudieran distorsionarse y, por lo tanto, suprimiro transformar el lenguaje normal (proteína-proteína) enunoaberrante. Por lo tanto, las propiedades básicas de un meca-nismo que podríamos llamar “interruptor molecular” debenbasarse en el equilibrio entre una alta especificidad y las afi-nidades débiles, acompañadas de una gran disminución de laentropía conformacional. Este fenómeno se basa, principal-mente, en el hecho de que después de la unión, las transicio-nes del desorden-al-orden pueden superar las restriccionesestéricas y así permitir interacciones de superficie, importan-tes en un complejo proteína-proteína, en comparación conaquellas interacciones que pudieran obtenerse de proteínasrígidas. A pesar de la extraordinaria importancia de este tipode transición, seguimos careciendo de estudios biofísicosdetallados que podrían demostrar la estrecha relación entreeste tipo de organización de desorden-al-orden, así como lafunción de las proteínas.
APOA1 EN LA FORMACIÓN DE LAS PARTÍCULAS DE HDL
La apolipoproteína A1 (ApoA1) ha sido estudiada enmodelos en estado libre y asociada a membranas, tomandoen cuenta la importancia que implica conocer los procesos
148

que dan lugar a las HDL nacientes, así como los mecanismosprecisos que apoyan este fenómeno en relación con el pro-ceso del transporte reverso del colesterol. La cadena de poli-péptido de 243 aminoácidos de la ApoA1 se organiza enbloques característicos de 22 y 11 residuos, los cuales se pre-dicen como segmentos anfipáticos helicoidales. Las hélicesque forman la ApoA1 han sido clasificadas como sigue:(1-45 aa) G*, (44-65, 66-87, 121-142, 143-164, 165-186,187-208 aa) A1, (88-98, 99-120, 209-219, 220-241) Y. Lashélices clasificadas como tipo G corresponden a las hélicesanfipáticas que forman el interior de las proteínas globu-lares, razón por la cual los aminoácidos que contienen corres-ponden a un carácter de tipo hidrofóbico. Las hélices anfi-páticas de tipo A1 presentan la característica de tener ungrupo de aminoácidos cargados positivamente en la inter-fase hidrófoba/ hidrófila, conteniendo residuos negativosen el centro de la cara polar. Por otra parte, las hélices detipo Y presentan la característica de tener aminoácidos carga-dos positivamente, separados por aminoácidos negativos [26].Actualmente, existen dos estructuras cristalinas de la ApoA1libre de lípidos en dos diferentes conformaciones. En laestructura cristalina, obtenida por (Borhani et al., 1997)(D1-43) [27], el segmento N-terminalestá truncado. Esta estruc-tura es única en la presentación de una conformación similara la que sería en presencia de lípidos. Además, la estructuraApoA1 (1-243) obtenida por (Ajees et al., 2006) [28], presen-ta dos dominios formados por cuatro a-hélices, dos en elN-terminal y dos en el C-terminal. Además, las técnicas espec-troscópicas han demostrado que la ApoA1, libre de lípidosen solución, presenta una disposición tridimensional endos dominios similares a la observada en la estructura crista-lina, pero menos organizada [29]. En nuestro laboratorio,hemos analizado tres péptidos diseñados de acuerdo con lasecuencia descrita para el ApoA1 nativa y su estructura cris-
149

talina [27], denominados DRV (D9-D24) y KLL (K45-P63) situa-dos dentro de sus segmentos helicoidales N-terminal y VLE
(V221-K239), situado en su segmento C-terminal. Varios estudios han evaluado la propensión de unión a lí -
pidos de cada una de las hélices que componen la ApoA1,tomando nota de que el dominio N-terminal determina laestructura abierta o cerrada de la proteína modulada porla presencia de colesterol, mediante la interacción con elcasete de unión a ATP (ABC) A1 (ABCA1) dando lugar a unaHDL naciente. Asimismo, debido a su alta hidrofobicidad, eldominio C-terminal de la ApoA1 facilita el anclaje a membra-nas lipídicas [30, 31]. Estos estudios se basan en el modelo,ampliamente aceptado, para una HDL discoidal que corres-ponde a un disco hecho de una bicapa lipídica rodeada pordos hélices de ApoA1 con su eje largo y perpendicular a lascadenas acilo de los fosfolípidos [32]. Estas propiedades sepueden observar fácilmente en un perfil de hidrofobicidadde ApoA1 obtenido con el uso del algoritmo de EMBOSS Pe -pinfo, empleando una ventana de nueve aminoácidos y la es -cala deKyte J. y DoolittleR. F. (1982) (figura 3) [33] . Mientrasque su perfil negativo es característico de las proteínas demembrana en el extremo N y las regiones C-terminales de laproteína (10-17, 213-229), un perfil positivo indica los hidró-fobos. Por otro lado, el uso del servidor HCA, que predicebloques hidrófobos en función de la estructura secundariade la cadena polipeptídica, muestra tres segmentos altamen-te hidrofóbicos (aa 13-22, 45-49, 216-232) (figura 3a, Clustershidrófobos) [34]. La distribución en la hélice de las cargasnegativas, aminoácidos positivos o neutros, generan los dife-rentes tipos de hélices presentes en la ApoA1. Este cambioen la distribución de aminoácidos es importante en la com-prensión de la forma en que las proteínas se asocian con loslípidos [26]. Por ejemplo, los segmentos correspondientes
150
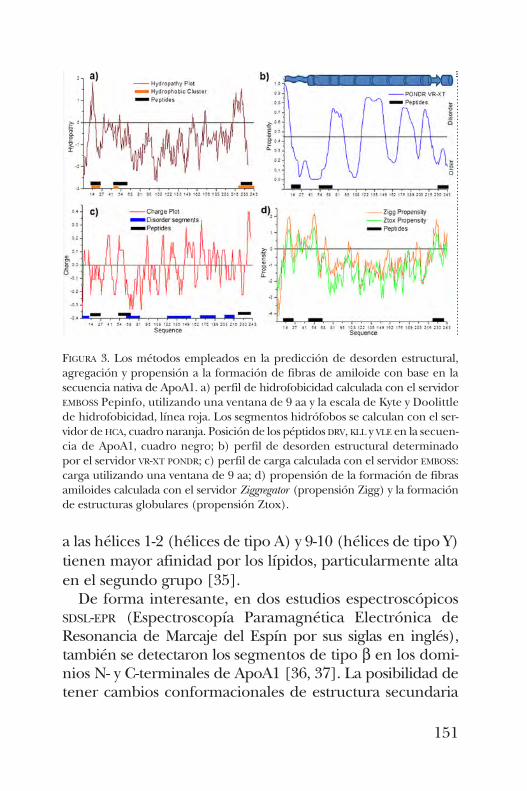
a las hélices 1-2 (hélices de tipo A) y 9-10 (hélices de tipo Y)tienen mayor afinidad por los lípidos, particularmente altaen el segundo grupo [35].
De forma interesante, en dos estudios espectroscópicosSDSL-EPR (Espectroscopía Paramagnética Electrónica deResonancia de Marcaje del Espín por sus siglas en inglés),también se detectaron los segmentos de tipo b en los domi-nios N- y C-terminales de ApoA1 [36, 37]. La posibilidad detener cambios conformacionales de estructura secundaria
151
FIGURA 3. Los métodos empleados en la predicción de desorden estructural,agregación y propensión a la formación de fibras de amiloide con base en lasecuencia nativa de ApoA1. a) perfil de hidrofobicidad calculada con el servidorEMBOSS Pepinfo, utilizando una ventana de 9 aa y la escala de Kyte y Doolittlede hidrofobicidad, línea roja. Los segmentos hidrófobos se calculan con el ser-vidor de HCA, cuadro naranja. Posición de los péptidos DRV, KLL y VLE en la secuen-cia de ApoA1, cuadro negro; b) perfil de desorden estructural determinadopor el servidor VR-XT PONDR; c) perfil de carga calculada con el servidor EMBOSS:carga utilizando una ventana de 9 aa; d) propensión de la formación de fibrasamiloides calculada con el servidor Ziggregator (propensión Zigg) y la formaciónde estructuras globulares (propensión Ztox).

han sido también observados en otras proteínas. Por ejem-plo, se ha demostrado que la unidad HA2 fusogénica de lahemaglutinina del virus de la influenza puede presentarestos tipos de transiciones conformacionales. El segmentoHA2 corresponde a un segmento que contiene 36 amino -ácidos y que presenta la capacidad para llevar a cabo transi-ciones de una estructura al azar a una a-hélice. La presenciade estos cambios conformacionales de estructura secunda-ria en ApoA1, en la presencia de lípidos, podría servir comoun mecanismo para disminuir la barrera de energía en suinteracción con estas moléculas, como paso crítico en elflujo de colesterol y ensamblaje de las partículas lipoprotéi-cas de alta densidad (HDL) [37, 38].
DESORDEN INTRÍNSECO EN APOA1
ApoA1 se considera dentro del grupo de proteínas noestructuradas de forma nativa [39]. Recientemente, este tipode proteína ha tomado una gran importancia y ha dado lugara la expresión unfoldomics: un grupo altamente disfuncionalde proteínas que se ha asociado con un número de condi-ciones, tales como la amiloidosis, el cáncer, la diabetes, lasenfermedades neurodegenerativas y otros. Los sitios alterados,contenidos en muchas proteínas desordenadas, han demos-trado ser altamente susceptibles a la proteolisis. En la ApoA1libre de lípidos, específicamente el segmento N-terminaly mediante diversas técnicas como RMN y EPR, se ha observadoque la movilidad presenta una gran variabilidad en su estruc-tura secundaria [30, 36, 40-43]. Usando el servidor VR-XT
PONDR con la secuencia nativa de la ApoA1 se estimó un altoporcentaje de desorden en cinco segmentos de ApoA1 (fi -gura 3b). El primer segmento (aa 1-10) corresponde a un sitioque podría servir como un sensor de lípidos cuando la ApoA1
152

se encuentra en una partícula discoidal [30,44]. El segundositio (aa 69-89) tiene una distribución de carga particular-mente negativa en comparación con los otros sitios no estruc-turados (figura 3c). El sitio también incluye una transiciónentre un tipo de hélice A de un tipo Y que, se ha postulado,podría ser un factor de desestabilización en la continuidadde la estructura secundaria de una a-hélice. Wu Z. et al., [44]han propuesto un modelo de HDL discoidal donde el tercermás largo y desordenado segmento (aa 116-150) presentauna región que podría ser considerada como bisagra. Elmismo modelo incluye un rizo (159 a 180) que correspondeal cuarto segmento desordenado (aa 172-194). Además, estesitio es adyacente al segmento postulado como el que inter -actúa con la enzima LCAT (aa 159-170). El quinto segmentodesordenado está situado cerca de una región de transiciónde un tipo de hélice A a un tipo Y. Aunque el último sitiopresenta una estructura a-hélice en la estructura cristalina deApoA1, por EPR este segmento muestra una estructura b quepodría servir comoun mecanismo para facilitar la interaccióncon los lípidos. Por otro lado, se han encontrado in vivoconcentraciones muy bajas de las fibrillas, del tipo amiloide,formadas por un segmento de 10 kD N-terminal de ApoA1nativo, [45]. Estas estructuras están constituidas por estructu -ras-b transversales que, a su vez, producen filamentos-b orien-tados de forma perpendicular con respecto al eje largo de lafibra, lo que resulta en el aumento de su capacidad de difu-sión. Posteriormente, protofibrillas se asocian lateralmenteo giran juntas para formar fibras de mayor diámetro [46].Mediante el análisis de la secuencia de la ApoA1 nativa, em -pleando el servidor Ziggregator [47], se ha observado que estaproteína presenta varios sitios con propensión a formar fibri-llas del tipo amiloide (A15-D20, F57 y W50-S224-L230) y que,interesantemente, se incluyen en la secuencia de los péptidosDRV, KLL (figura 3d, Zigg propensión). Este servidor utiliza un
153

algoritmo que considera los patrones de hidrofobicidad, losaminoácidos polares y el contenido aromático observado enlas cadenas polipeptídicas de proteínas amiloidogénicas. Lossegmentos con tendencia a la agregación presentan bloquesaltamente hidrofóbicos compuestos por seis o siete amino-ácidos, flanqueados por las cargas negativas y/o positivas. Porotro lado, Ziggregator calcula la tendencia a formar estruc-turas globulares, que se han observado como paso interme-diario en la formación de fibras amiloidogénicas. También,mediante experimentación in vitro se ha observado que estasestructuras globulares, formadas por péptidos amiloides,están implicadas en un proceso de citotoxicidad celular, debi-do a su contribución en la formación de poros en la mem-brana [48]. Los patrones para formar estructuras amiloidesen ApoA1 calculados por Ziggregator, muestran el mismopatrón observado durante la formación de fibras de amiloide(figura 3d, propensión Ztox).
Interesantemente, dentro de los mutantes existentes deApoA1, existen cuatro isoformas asociadas con formas sisté-micas de amiloidosis hereditaria. Las mutaciones correspon-den a Gly26-Arg, Leu60-Arg, Trp50-Arg y una deleción/inser-ción del segmento (Leu60-Phe71)-(Val-Val-Thr). Teniendo encuenta la estructura de la ApoA1 libre de lípidos propuestapor Ajees (Ajees et al., 2006), estas mutaciones proporcionan,generalmente, cargas positivas a la interfase hidrófoba for-mada entre los dos pares de hélices del segmento N-terminal(aa 1-188). El cambio en el dieléctrico, creado por la muta-ción de la interfase hidrofóbica de la ApoA1 libre de lípidos,probablemente impide la formación de un grupo dea-hélicesen la estructura N-terminal. Esto también impide la forma-ción de parches hidrófilos situados en diferentes zonas de laproteína (figura 4) [49]. Estos parches hidrófilos se ha pos-tulado que interactúan con ABCA1 para la transferencia defosfolípidos y colesterol. Una consecuencia de este obstáculo
154

podría ser que la formación de una HDL discoidal de tamañoadecuado y necesario para interactuar con la enzima LCAT,como se observa en estas mutaciones, no se logra adecua-damente [31, 50]. Esta interacción es crucial en la transiciónde las formas discoidales a las esféricas de HDL [51]. Recien -temente, en nuestro grupo de trabajo hemos demostradoque varios péptidos a-helicoidales derivados de los dominiosN- y C-terminales de la ApoA1 son capaces de activar la en -zima Lecitin-Colesterol Aciltransferasa (LCAT) [52].
Las formas discoidales de HDL, formadas por una de lasvarias isoformas de la ApoA1 amiloidogénica, se catabolizanrápidamente y no se convierten en HDL esféricas [50]. Enesta etapa, es interesante mencionar que la vía metabólicapor la cual estas isoformas de la ApoA1 causan la deposiciónde fibrillas del tipo amiloide, todavía no se ha aclarado. Sinembargo, cualquiera de estas mutaciones que se han encon-trado en la hélice uno y la hélice G* podrían estar generandoregiones susceptibles a la proteólisis entre las hélices dosy tres (segundo sitio desordenado de la ApoA1 con baja afi-nidad a lípidos y con un marcado carácter negativo (figura 3ay 3b). En todos los casos, las fibras amiloides se generan conpolipéptidos a partir de los primeros 83-94 aminoácidos en elextremo N-terminal de la ApoA1. Las mutaciones en los ami-noácidos 26, 50 y 60 también generan cambios de carga, carac-terística que favorece la formación de hojas b-extendidas [53].Estos péptidos liberados, independientemente de la muta-ción de origen, siempre tienen la misma carga neta, lo queindica la conservación del perfil hidrófilo. Del mismo modo,el valor de mH y la hidrofobicidad total promedio disminu-yen en las mutaciones con respecto a las secuencias nativas.
Es notable cómo varios datos teóricos y experimentales,relacionados con los segmentos N-terminal y C-terminal dela ApoA1, indican que ambos tienen una alta propensióna la agregación y sólo el primero se encuentra en las placas
155
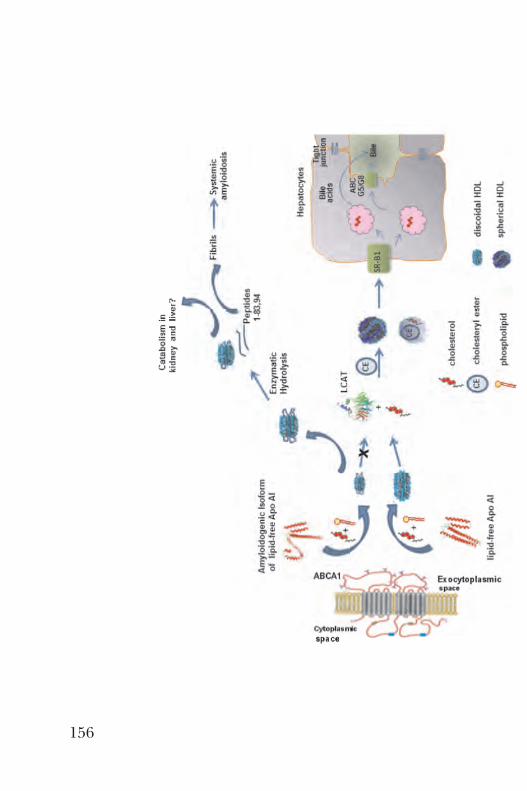
156
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##

157
FIGU
RA
4. E
lrec
epto
r de
ABCA1
prom
ueve
la tr
ansfer
encia de
fosfol
ípid
osa un
a fo
rmapo
bre
en lí
pido
sde
la A
poA1
nativ
a, e
lco
mpo
nent
e pr
incipa
lde
las
HDL. E
lmec
anism
o po
r el
cua
lse
prod
uce
este
proc
eso
nose
ent
iend
e co
mpl
etam
ente
; pro
ba-
blem
ente
ABCA1
tran
sloc
afo
sfol
ípid
os y
cole
ster
olde
la m
embr
ana
plas
mát
ica
a la
s H
DL
Apo
A1
para
pro
ducir un
a pa
rtíc
ula
disc
oida
l. Estas
par
tícul
asdi
scoi
dale
sse tran
sfor
man
en
las H
DLes
férica
sen
circ
ulac
ión
por la
acc
ión
dela
enzim
a LCAT
. Las
HDL
esfé
rica
sse
aso
cian
con
el rec
epto
rSR
-B1
en la
mem
bran
a pl
asm
ática
de lo
s he
pato
cito
sy tran
sfie
ren
cole
ster
ollib
re y e
ster
i-fic
ado
al h
ígad
o pa
ra su
excr
eció
n e
nla
bili
sco
mo
cole
ster
ol li
bre
(vía
ABCG
5/G
8) o
a su
pos
terior
con
vers
ión
asa
les bi
lia-
res. E
nel
cas
o de
la fo
rmac
ión
dela
s iso
form
asam
iloid
ogén
icas
dela
Apo
A1,
una
inte
racc
ión
adec
uada
con
ABCA1
está
impe
-di
day,
por lo
tant
ola
form
ació
n de
HDLdi
scoi
dale
sse
con
vier
te e
nun
pro
ceso
defic
ient
e, con
losco
nsig
uien
tespr
oble
mas
enel
rec
onoc
imie
nto
dela L
CAT
y ta
mbi
énSR
-B1.
Se co
noce
que
las H
DLan
orm
ales
seca
tabo
lizan
rápi
dam
ente
libe
rand
odi
fere
ntes
pépt
idos
, los
que
,a su
vez, g
ener
anpé
ptid
os for
mad
ores
de
fibra
sam
iloid
esco
n la
con
secu
ente
pos
ibili
dad
de ini
ciar
una
amilo
idos
issistém
ica.

amiloidogénicas aisladas de individuos con amiloidosis fami -liar o Alzheimer.
Los segmentos de la ApoA1 que presentan una tendenciaa mantenerse en un estado desordenado, junto con su capa-cidad para unirse a los sitios de baja afinidad para lípidos,podría ser la clave para la comprensión de la formación delas partículas de HDL. Debido a estas características, pareceque la ApoA1 tiene la capacidad de modular su estructurasecundaria con base en la presencia de interfases hidrófo-bas/hidró filas, las que a su vez pueden activar o inhibir lafunción de las proteínas que regulan el metabolismo de HDL.
LA PROTEÍNA TRANSFERIDORA DE ÉSTERES
DE COLESTEROL (CETP)
La proteína CETP está constituida por 476 residuos, presentaun peso molecular de 66 kDa, los residuos Asp88, Asp240,Asp341 y Asn396 que están glicosilados, contienen cinco cis-teínas libres y un alto contenido de residuos hidrofóbicos encomparación con otras proteínas plasmáticas (aproximada-mente 44%). La estructura tridimensional de la CETP, con unaresolución de 2,2 Å, se dio a conocer a principios de 2007.En términos generales, el cristal refleja una estructura alar-gada semejante a un boomerang, con dimensiones de 135 ×30 × 35 Å y un plegamiento similar a la proteína BPI (proteínaque incrementa la permeabilidad bacteriana). La descrip-ción estructural de CETP puede centrarse en cuatro dominios:un barril en cada lado de la proteína, llamados barriles N- y C,una conexión de hoja-b central entre los dos barriles y unaextensión C-terminal llamada hélice-X que está ausente en laproteína BPI.
La estructura cristalográfica revela un túnel de 60 Å delargo. De acuerdo con este modelo de estructura tridimen-
158

sional, la proteína CETP puede acomodar a dos moléculasde éster de colesterol en el interior y una molécula de fos-folípido asociada a cada puerta del túnel, orientadas de talma nera que las cadenas de ácido graso se ubican hacia elinterior del túnel y los grupos de cabeza polar hacia la inter-fase con el agua. Las puertas del túnel son lo suficientementegrandes para permitir que los lípidos ingresen, donde una deellas está protegida por la hélice-X en la región del barril-Ny dos estructuras movibles (W1 yW2) en la región del barril-C (fi gura 5). Del mismo modo, estudios tanto de mutagéne-sis como estructurales sugieren que los triglicéridos, comolos ésteres de colesterol (lípidos neutros), pueden moversea lo largo del túnel a través de la región central con dimen-siones de 10 × 5 Å [54].
Sin embargo, en nuestro laboratorio hemos encontradoque una posibilidad alternativa para facilitar el transportede lípidos, entre las partículas de lipoproteínas, puede sermediante la formación de estructuras micelares asociadas ala región C-terminal de la CETP, cuando esta región se estruc-tura en forma a-helicoidal [55, 56].
159#
#
P
!"
!"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
##
"
"
FIGURA 5. Estructura cristalina de CETP (modificada de [53]). El rectángulo con-tiene el carboxilo-terminal de la proteína, segmento en el cual se llevan a cabolas transiciones de estructura secundaria en presencia de lípidos específicos.

Puesto que el dominioC-terminal de la CETP mantiene unaalta flexibilidad intramolecular, con un factor-B alto asociadoa la interacción con lípidos, esta propiedad debe estar direc-tamente asociada con el tipo y concentración de los lípidosespecíficos que permiten o previenen de cambios en la estruc-tura secundaria [56]. Por ejemplo, se ha descrito que cuandola LPA en su CMC se coloca en contacto con la proteína b2-microglobulina, promueve la formación y extensión de lasfibrillas de amiloide. Sin embargo, se encontró que el empleode una amplia gama de concentraciones, por encima de laCMC, LPA y en menor medida la lisofosfatidilcolina, promueveun cambio hacia estructuras a-helicoidales y, por lo tanto,previene la formación de fibrillas del tipo amiloide cuandoel del péptido hélice-Z (mutación D470N) se deriva la C-ter-minal del dominio de la CETP que es estudiada [57]. Por lotanto, el control de los cambios conformacionales en sitiosespecíficos de las proteínas, dadas por lípidos específicos,podría ser tomado como la base para el diseño de molécu-las similares a lípidos que, a su vez, pudieran mantener o esta-bilizar el estado nativo de proteínas propensas a la agrega-ción [56]. Este tipo de organización molecular llevada a cabopor el segmen to C-terminal de la CETP, siguiendo transicionesde es tructura secundaria del tipo desorden-al-orden y siendocontroladas por la presencia de lípidos específicos, en nuestraopinión, puede entenderse como ejemplo de un interruptormolecular [57, 58]. Esto permitiría que la función de la CETP
pudiera realizarse bajo condiciones metabólicas específicasy mantener su actividad de transferencia asociada a un cam-bio de entropía mínima relacionada con la formación deestructuras de tipo micelar en el segmento C-terminal de laproteína, proceso que reducirá drásticamente la barrera deenergía para la transferencia de lípidos a través de unmedio acuoso.
160

Durante muchos años, la función de las proteínas ha resi-dido en el hecho de que estructuras bien ordenadas, en sumayoría fundamentadas en bloques tridimensionales rígidos,pueden ser fundamentales en la comprensión de la forma enque las proteínas funcionan. Sin embargo, hoy en día esteconcepto ha sido superado por el reconocimiento realizadopor organismos filogenéticamente avanzados, donde la fun-ción se desarrolla a través de la función, en un número impor-tante de proteínas intrínsecamente desordenadas y donde elconcepto de desorden estructural se concibe como una formade estructura secundaria de proteínas [59]. Sin lugar a dudas,podemos decir que en el futuro cercano muchas enferme-dades, con orígenes poco conocidos, no sólo encontraránuna explicación molecular en la forma en que las proteínasimportantes adquieren su plegamiento final, sino tambiénen la manera en que las regiones intrínsecamente desorde-nadas de las mismas son moduladas.
AGRADECIMIENTOS
Las investigaciones que se describen en el presente capí-tulo han sido realizadas por el grupo de JMO y apoyadaspor el Consejo Nacional de Ciencia y Tecnología de Méxi -co (Conacyt) y la Universidad Nacional Autónoma de México(DGAPA-UNAM).
REFERENCIAS
[1] Rose G. D., Fleming P. J., Banavar J. R. y Maritan A. A backbone-based theory of protein folding. Proc. Natl. Acad. Sci. USA, 2006,103, 16623-16633.
[2] Eaton W. A., Muñoz V., Hagen S. J., Jas G. S., Lapidus L. J. y Henry E. R.Fast kinetics and mechanisms in protein folding. Annu. Rev. Biophys.Biomol. Struct., 2000, 29, 327-359.
161

[3] Huang K. Lectures on statistical physics and protein folding. WorldScientific, New Jersey, 2005.
[4] Dobson C. M. Protein folding and misfolding. Nature, 2003, 426,884-890.
[5] Gsponer J. y Vendruscolo M. Theoretical approaches to proteinaggregation. Protein Pept. Lett., 2006, 13, 287-293.
[6] Dobson C. M. Protein misfolding, evolution and disease. TrendsBiochem. Sci., 1999, 24, 329-332.
[7] Eisenberg D., Nelson R., Sawaya M. R., Balbirnie M., Sambashivan S.,Ivanova M. I., Madsen A. O. y Riekel C. The structural biology ofprotein aggregation diseases: fundamental questions and someanswers. Acc. Chem. Res., 2006, 39, 568-575.
[8] Fink A. L. Protein aggregation: folding aggregates, inclusion bodiesand amyloid. Fold. Des., 1998, 3, R9-R23.
[9] Dobson C. M. Protein chemistry: in the footsteps of alchemists.Science, 2004, 304, 1259-1262.
[10] Ohnishi S. y Takano K. Amyloid fibrils from the viewpoint of pro-tein folding. Cell. Mol. Life Sci., 2004, 61, 511-524.
[11] Bode W. y Huber R. Induction of the bovine trypsinogentrypsintransition by peptides sequentially similar to the N-terminus oftrypsin. FEBS Lett., 1976, 68, 231-236.
[12] Huber R. y Bode W. Structural basis of the activation and actionof trypsin. Acc. Chem. Res., 1978, 11, 114-122.
[13] Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci.,2002, 27, 527-533.
[14] James C. y Tawfik D. S. Conformational diversity and protein evo-lution a 60-years-old hypothesis revisited. Trends Biochem. Sci., 2003,28, 361-368.
[15] Bustos D. M. e Iglesias A. A. Intrinsic disorder is a key characteris-tic in partners that bind 14-3-3 proteins. Proteins, 2006, 63, 35-42.
[16] Kriwacki R. W., Hengst L., Tennant L., Reed S. I. y Wright P. E.Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity.Proc. Natl. Acad. Sci. USA, 1996, 93, 11504-11509.
[17] Dalal S. y Regan L. Understanding the sequence determinants ofconformational switching using protein design. Protein Sci., 2000,9, 1651-1659.
[18] Dunker A. K., Garner E., Guilliot S., Romero P., Albrecht K., Hart J.,Obradovic Z., Kissinger C. y Villafranca J. E. Protein disorderand the evolution of molecular recognition: theory, predictions andobservations. Pac. Symp. Biocomput., 1998, 473-484.
162

[19] Sickmeier M., Hamilton J. A., LeGall T., Vacic V., Cortese M. S.,Tatos A., Szabo B., Tompa P., Chen J., Uversky V. N. y Obradovic Z.,Dunker A. K. DisProt: the database of disordered proteins. NucleicAcids Res., 2007, 35, D786-D793.
[20] Cortese M. S., Uversky V. N. y Dunker A. K. Intrinsic disorder inscaffold proteins: getting more from less. Prog. Biophys. Mol. Biol.,2008, 98, 85-106.
[21] Su C. T., Chen C. Y. y Hsu C. M. iPDA: integrated protein disorderanalyzer. Nucleic Acids Res., 2007, 35, W465-W472.
[22] Conchillo-Sole O., de Groot N. S., Aviles F. X., Vendrell J., Daura X.y Ventura S. AGGRESCAN a server for the prediction and evaluationof ‘‘hot spots’’ of aggregation in polypeptides. BMC Bioinformatics,2007, 8, 65.
[23] Romero P. Z., Obradovic C. y Dunker A. K. Intelligent data analysisfor protein disorder prediction. Artif. Intell. Rev., 2001, 14, 447-484.
[24] Dunker A. K., Lawson J. D., Brown C. J., Williams R. M., Romero P.,Oh J. S., Oldfield C. J., Campen A. M., Ratliff C. M., Hipps K. W.,Ausio J., Nissen M. S., Reeves R., Kang C., Kissinger C. R., BaileyR. W., Griswold M. D., Chiu W., Garner E. C. y Obradovic Z. J.Intrinsically disordered protein. J. Mol. Graph. Model., 2001, 19, 26-59.
[25] Uversky V. N. What does it mean to be natively unfolded? Eur. J.Biochem., 2002, 269, 2-12.
[26] Segrest J. P., Jones M. K., De Loof H., Brouillette C. G., Venkata -chalapathi Y. V. y Anantharamaiah G. M. The amphipathic helixin the exchangeable apolipoproteins: a review of secondary struc-ture and function. J. Lipid Res., 1992, 33, 141-166.
[27] Borhani D. W., Rogers D. P., Engler J. A. y Brouillette C. G. Crystalstructure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc. Natl. Acad. Sci. USA, 1997, 94, 12291-12296.
[28] Ajees A. A., Anantharamaiah G. M., Mishra V. K., Hussain M. M.y Murthy H. M. Crystal structure of human apolipoprotein A-I:insights into its protective effect against cardiovascular diseases.Proc. Natl. Acad. Sci. USA, 2006, 103, 2126-2131.
[29] Tanaka M., Koyama M. y Dhanasekaran P., Nguyen D., Nickel M.,Lund-Katz S., Saito H., Phillips M. C. Influence of tertiary struc-ture domain properties on the functionality of apolipoprotein A-I. Biochemistry, 2008, 47, 2172-2180.
[30] Kono M., Okumura Y., Tanaka M., Nguyen D., Dhanasekaran P.,Lund-Katz S., Phillips M. C. y Saito H. Conformational flexibility
163

of the N-terminal domain of apolipoprotein A-I bound to spheri-cal lipid particles. Biochemistry, 2008, 47, 11340-11347.
[31] Fang Y., Gursky O. y Atkinson D. Lipid-binding studies of human apo-lipoprotein A-I and its terminally truncated mutants. Biochemistry,2003, 42, 13260-13268.
[32] Garda H. A. Structure-function relationships in human apolipo-protein A-I: role of a central helix pair. Future Lipidol., 2007, 2, 95-104.
[33] Kyte J. y Doolittle R. F. A simple method for displaying the hydro-pathic character of a protein. J. Mol. Biol., 1982, 157, 105-132.
[34] Callebaut I., Labesse G., Durand P., Poupon A., Canard L., Chomi -lier J., Henrissat B. y Mornon J. P. Deciphering protein sequenceinformation through hydrophobic cluster analysis (HCA): currentstatus and perspectives. Cell Mol, Life Sci, 1997, 53, 621-645.
[35] Mishra V. K., Palgunachari M. N., Datta G., Phillips M. C., Lund-Katz S., Adeyeye S. O., Segrest J. P. y Anantharamaiah G. M. Studiesof synthetic peptides of human apolipoprotein A-I containing tan-dem amphipathic alpha-helixes. Biochemistry, 1998, 37, 10313-10324.
[36] Lagerstedt J. O., Budamagunta M. S., Oda M. N. y Voss J. C. Electronparamagnetic resonance spectroscopy of site-directed spin labelsreveals the structural heterogeneity in the N-terminal domain ofapoA-I in solution. J. Biol. Chem., 2007, 282, 9143-9149.
[37] Oda M. N., Forte T. M., Ryan R. O. y Voss J. C. The C-terminaldomain of apolipoprotein A-I contains a lipid-sensitive conforma-tional trigger. Nat. Struct. Biol. 2003, 10, 455-460.
[38] Tamm L. K. Hypothesis: spring-loaded boomerang mechanism ofinfluenza hemagglutinin-mediated membrane fusion. Biochim.Biophys. Acta, 2003, 1614, 14-23.
[39] Uversky V. N., Gillespie J. R. y Fink A. L. Why are “natively unfol-ded” proteins unstructured under physiologic conditions?Proteins, 2000, 41, 415-427.
[40] Okon M., Frank P. G., Marcel Y. L. y Cushley R. J. Heteronuclear NMR
studies of human serum apolipoprotein A-I. Part I. Secondarystructure in lipid-mimetic solution. FEBS Lett., 2002, 517, 139-143.
[41] Okon M., Frank P. G., Marcel Y. L. y Cushley R. J. Secondary struc-ture of human apolipoprotein A-I (1-186) in lipid-mimetic solu-tion, FEBS Lett., 2001, 487, 390-396.
[42] Wang G., Sparrow J. T. y Cushley R. J. The helix-hinge-helix struc-tural motif in human apolipoprotein A-I determined by NMR spec-troscopy. Biochemistry, 1997, 36, 13657-13666.
[43] Wang G., Treleaven W. D. y Cushley R. J. Conformation of humanserum apolipoprotein A-I (166-185) in the presence of sodium
164

dodecyl sulfate or dodecylphosphocholine by 1H-NMR and CD.Evidence for specific peptide-SDS interactions. Biochim. Biophys.Acta, 1996, 1301, 174-184.
[44] Wu Z., Wagner M. A., Zheng L., Parks J. S., Shy J. M. 3rd., Smith J. D.,Gogonea V. y Hazen S. L. The refined structure of nascent HDL
reveals a key functional domain for particle maturation and dys-function. Nat. Struct. Mol. Biol., 2007, 14, 861-868.
[45] Schmidt H. H., Haas R. E., Remaley A., Genschel J., Strassburg C. P.,Buttner C. y Manns M. P. In vivo kinetics as a sensitive method fortesting physiologically intact human recombinant apolipoproteinA-I: comparison of three different expression system. Clin. Chim.Acta, 1997, 268, 41-60.
[46] DuBay K. F., Pawar A. P., Chiti F., Zurdo J., Dobson C. M. y Ven -druscolo M. Prediction of the absolute aggregation rates of amy-loidogenic polypeptide chains. J. Mol. Biol., 2004, 341, 1317-1326.
[47] Tartaglia G. G., Pawar A. P., Campioni S., Dobson C. M., Chiti F.y Vendruscolo M. Prediction of aggregation-prone regions in struc-tured proteins. J. Mol. Biol., 2008, 380, 425-436.
[48] Lashuel H. A. y Lansbury P. T. Jr. Are amyloid diseases caused byprotein aggregates that mimic bacterial pore-forming toxins? Q. Rev.Biophys., 2006, 39, 167-201.
[49] Oram J. F. ATP-binding cassette transporter A1 and cholesterol traf-ficking. Curr. Opin. Lipidol., 2002, 13, 373-381.
[50] Genschel J., Haas R., Pröpsting M. J. y Schmidt H. H. J. Hypothesis.Apolipoprotein A-I induced amyloidosis. FEBS Letters, 1998, 430,145-149.
[51] Calabresi L. y Franceschini G. Lecithin:cholesterol acyltransferase,high-density lipoproteins, and atheroprotection in humans.Trends Cardiovasc. Med., 2010, 20, 50-53.
[52] Aguilar-Espinosa S. L., Mendoza-Espinosa P., Delgado-Coello B.y Mas-Oliva J. Lecithin cholesterol acyltransferase (LCAT) activity inthe presence of Apo-AI- derived exposed to disorder-to-order confor-mational transitions. Biochem. Biophys. Res. Comm., 2013, 441, 469-475.
[53] Andreola A., Bellotti V., Giorgetti S., Mangione P., Obici L., StoppiniM., Torres J., Monzani E., Merlini G. y Sunde M. Conforma tionalswitching and fibrillogenesis in the amyloidogenic fragment ofapolipoprotein A-1. J. Biol. Chem., 2003, 278, 2444-2451.
[54] Qiu X., Mistry A., Ammirati M. J., Chrunyk B. A., Clark R. W., Cong Y.,Culp J. S. et al. Crystal structure of cholesteryl ester transfer pro-tein reveals a long tunnel and four bound lipid molecules. Nat.Struct. Mol. Biol., 2007; 14, 106-113.
165

[55] García-González V. y Mas-Oliva J. Structural Arrangement thatSupports Lipid Transfer in the cholesteryl-ester transfer protein(CETP). USA-Mexico Workshop in Biological Chemistry: Multi disciplinaryApproaches to Protein Folding, Ciudad de México, México, 25-27 demarzo de 2009.
[56] García-González V., Gutiérrez-Quintanar N., Mendoza Espinosa P.,Brocos P., Piñeiro Á. y Mas-Oliva J. Key Struc tural Arrangementsat the C -terminus Domain of CETP Suggest a Potential Mechanismfor Lipid-transfer Activity. J. Struct. Biol., 2014. En prensa.
[57] García-González V. y Mas-Oliva J. Amyloid fibril formation of pep-tides derived from the C -terminus of CETP modulated by lipids.Biochem. Biophys. Res. Comm., 2013, 434, 54-59.
[58] Mendoza-Espinosa P., García-González V., Moreno A., Castillo R.y Mas-Oliva J. Disorder-to-order conformational transitions in pro-tein structure and its relationship to disease. Mol. Cell Biochem.,2009, 330, 105-120.
[59] Mas Oliva J. y García González V. El Concepto de Enfermedad Aso -ciado a la Conformación de Proteínas. Programa Universitario deInvestigación en Salud, UNAM/El Manual Moderno. 2012.
166

PROTEÓMICA DE LA TIOLACIÓNDE LA MODIFICACIÓN DE TIOLES EN LA PROTEÍNA
A LA DISFUNCIÓN CELULAR
PÁVEL VÁZQUEZDepartamento de Farmacología y Toxicología
Facultad de QuímicaUniversidad Autónoma del Estado de México
Departamento de BioquímicaFacultad de Medicina
Universidad Nacional Autónoma de Mé[email protected]@hotmail.com
In Thiology, only God understands thiols.1CARLOS GITLER
La proteómica tiene como objeto de estudio un grupo demacromoléculas biológicas muy específicas y dinámicas,que corresponden en general al “proteoma”, (la palabraproteoma deriva del conjunto de prote ínas expresadas enun organismo —bajo determinadas condiciones por su infor-mación genética o genoma— siendo éste, la dotación com-pleta de genes). El proteoma está comprendido por dichasmacromoléculas o biopolímeros, llamados proteínas, for-mados por largas cadenas de aminoácidos ensambladosuno a uno y con una determinada secuencia por la maqui-naria celular —considerando; que el ribosoma es el encar-
167
1 “En Tiología, sólo Dios entiende a los tioles”.

gado de transcribir y decodificar la información almacenadapor los ácidos nucleicos—. Una vez sintetizados los biopolí-meros de aminoácidos, estos pueden organizarse estructu-ralmente en distintos niveles; que van desde la estructuraprimaria, hasta la cuaternaria, tomando en cuenta que cadanivel estructural de la proteína es esencial para la funciónfinal de la macromolécula. Algunas de las cadenas laterales de los residuos de amino-
ácidos que constituyen la proteína, pueden sufrir alteracioneso modificaciones químicas, conocidas como: modificacionespostraduccionales, llevadas a cabo en la proteína ya sintetizadamediante mecanismos enzimáticos o no enzimáticos; alte-rando las propiedades químicas, físicas, la estabilidad, la acti-vidad y finalmente la función de la proteína. Por ejemplo,si se consideraran a las proteínas como palabras, entoncespodría decirse que las modificaciones postraduccionales equi-valdrían al uso de signos diacríticos como: el uso de acentoso diéresis en diversas lenguas, dando un valor a las letrasy, por consiguiente, modificando el significado de la palabra.El propósito de las modificaciones postraduccionales es
incrementar la diversidad de grupos funcionales, más allá delas simples cadenas laterales de los aminoácidos. Es decir, lavariedad de estructura y función de las proteínas aumenta,ya que éstas adquieren una nueva química, nuevos patronesde reconocimiento, regulan el encendido y apagado de suactividad, además de controlar el tiempo de vida y su loca-lización en la célula.Las modificaciones postraduccionales no sólo incluyen la
ruptura de enlaces peptídicos y la unión de otras proteínas,sino también la adición covalente de pequeños grupos fun-cionales como: la fosforilación, la metilación, nitración, ace-tilación, carbonilación, entre otras (véase tabla 1), tomandoen cuenta que una de las principales características de lascadenas laterales de los aminoácidos —donde se lleva a cabo
168

169
la unión o modificación— es la presencia de grupos comoalcohol (-OH) o los grupos amino (-NH2). Sin embargo, mu -chas de las proteínas encargadas de regular determinadosprocesos en las células contienen una cantidad significativadel aminoácido cisteína, cuyos grupos tiol o sulfhidrilo (-SH)desempeñan un papel muy importante en gran parte de lasfunciones celulares. Por las características químicas quepresenta el azufre en el grupo tiol, presente en la cisteína,el aminoácido adquiere una determinada reactividad fren-te a una gran variedad de moléculas, incluyendo reaccionesentre cisteínas adyacentes o vecinales. Se denomina tiolacióna todas aquellas modificaciones postraduccionales que ocu-rren específicamente sobre el grupo SH de este aminoácido. Una de las características principales de todas las modifi-
caciones postraduccionales es la trasferencia de electronesdurante la formación de los enlaces entre la proteína y lamolécula modificadora, ya sea en la adición de un sencillo
Tabla 1ALGUNAS MODIFICACIONES POSTRADUCCIONALES CON SUS RESPECTIVOS RESIDUOS DE AMINOÁCIDO,
DONDE SE LLEVA A CABO LA REACCIÓN
Modificación Aminoácido modificado
Fosforilación Ser, Thr y TyrHidroxilación Lys y ProAcetilación Lys y SerMetilación Lys, His y ArgSulfatación TyrCarboxilación GluSelenización SerCitrulinación ArgUbiquitinación LysNitración TyrGlicosilación VariedCarbonilación Cys, LysTiolación Cys

grupo funcional o una macromolécula. Esta transferenciade electrones —conocida como reacciones redox u óxido/reducción—, se basa en la pérdida o ganancia de electrones,donde cualquier especie química que acepte dichas partícu-las se verá reducida; en tanto que cualquier agente químicoque ceda o done sus electrones se oxida.
Este tipo de reacciones redox juegan un papel importanteen muchos procesos celulares como: en la regulación de laexpresión de genes, la respiración celular, la proliferación celu-lar, la muerte celular programada o apoptosis, la respuestacelular a distintos niveles de oxígeno, radicales libres y otrosoxidantes, etcétera. Es decir, las modificaciones sufridas porla transferencia de electrones, entre la proteína y una espe-cie donadora o aceptora, modula alostéricamente la fun-ción de la biomolécula.Las propiedades químicas que presenta el átomo de azu-
fre en cuanto a sus números de oxidación, su electronega-
170#
#
donde se lleva a cabo la reacción.
de electrones, donde cualquier especie química que acepte dichas
p
F
reacciones redox juegan un papel importante en muchos procesos celulares,
c
modula
a
L
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
FIGURA 1. Las reacciones redox involucran una transferencia de electrones. La pér-dida de electrones ocasiona una oxidación, en tanto que la ganancia implica unareducción.

tividad, multivalencia, etcétera, le permiten, en general, alaminoácido cisteína desempeñar diferentes funciones en lasproteínas, como por ejemplo: participar en reacciones decatálisis redox, reacciones de hidrólisis, procesos de quelacióno unión de una gran variedad de metales y metaloides y, nomenos importante, la formación de enlaces disulfuro o puen-tes de azufre. De esta manera se considera al azufre, y en par-ticular a la cisteína, como el “camaleón redox”, debido a sucapacidad de adaptación y de reacción frente a una granvariedad de especies químicas bajo diferentes circunstan-cias, además de las propiedades antes mencionadas. Otra característica importante de la cisteína es que puede
ser afectada por diversas modificaciones postraduccionales,como por ejemplo: la sulfonación, la unión de un gruposulfénico, sulfínico o sulfónico, la nitrosilación, etcétera—de las cuales muchas pueden ser reversibles o no—, locual puede modificar a la proteína definiendo su funcióno marcándola para su posible degradación por el proteosoma.Sin embargo, la habilidad del azufre para tener distintosestados de oxidación (–2, 0, +2, +4, +6), le permiten a éste,actuar como un sensor bajo determinados estados redox enla célula; detectando, uniendo y estabilizando, desde radica-les libres hasta distintas especies reactivas. Es decir, cuandose ve alterado el ambiente redox en la célula, el proteomatiólico es el responsable de adaptarla para hacer frente a lanaturaleza altamente reactiva, tanto de las Especies Reactivasde Oxígeno (ERO), Especies Reactivas de Nitrógeno (ERN)y las Especies Reactivas de Carbono (ERC) utilizando a estaspequeñas moléculas como potentes moléculas señalizadoraspara intentar controlar la homeostasis celular; sin embargo,muchas proteínas tiólicas, al ser modificadas estructural y fun-cionalmente por ataques nucleofílicos sobre sus grupos tiol,son altamente dañadas y, como consecuencia, pueden cau-sar un daño irreversible a la célula. El proteoma tiólico es
171
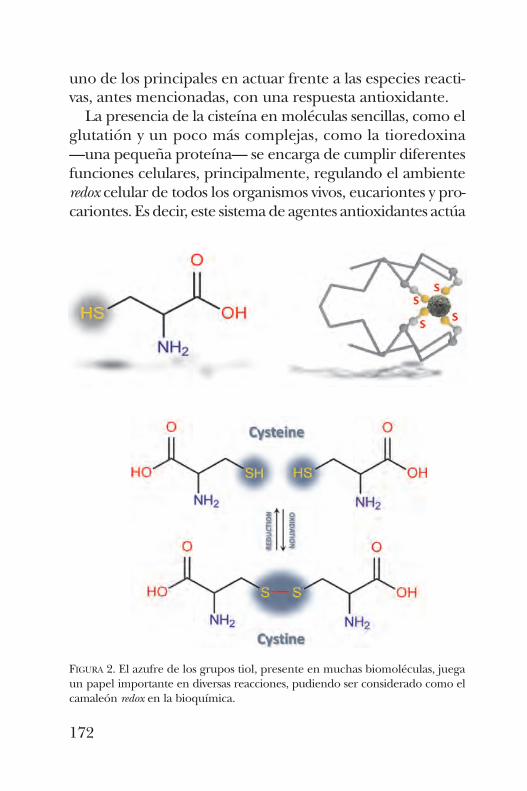
uno de los principales en actuar frente a las especies reacti-vas, antes mencionadas, con una respuesta antioxidante.La presencia de la cisteína en moléculas sencillas, como el
glutatión y un poco más complejas, como la tioredoxina—una pequeña proteína— se encarga de cumplir diferentesfunciones celulares, principalmente, regulando el ambienteredox celular de todos los organismos vivos, eucariontes y pro-cariontes. Es decir, este sistema de agentes antioxidantes actúa
172
FIGURA 2. El azufre de los grupos tiol, presente en muchas biomoléculas, juegaun papel importante en diversas reacciones, pudiendo ser considerado como elcamaleón redox en la bioquímica.
$$
y no menos importante, la formación de enlaces
d
debido a su capacidad de adaptación y de reacción frente a una gran
v
diversas reacciones, pudiendo ser considerado como el camaleón
r
nitrosilación, etc., -de las cuales muchas pueden ser reversibles
o no- lo cual puede modificar a la proteína definiendo su función o marcándola para su posible
d
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$

como amortiguador frente a las ERO, ERN, ERC y otras especiesreactivas, manteniendo un balance frente al estrés oxidativo.En este sistema, tanto el glutatión como la tioredoxina actúancomo donadores de electrones, mediante la formación y rup-tura de enlaces disulfuro en sus grupos tiol, siendo los gruposquímicos reguladores más comunes involucrados en estetipo de reacciones redox.
Muchas proteínas, principalmente aquellas encargadasde regular procesos específicos, ya sea en vías de señaliza-ción o en rutas metabólicas, contienen una cantidad signi-ficativa de cisteínas, no sólo en su secuencia de aminoácidos,sino también en su estructura tridimensional. Sin embargo,una característica notable es la cercanía que presentan dichascisteínas dentro de una misma proteína, las cuales cumplencon el siguiente motivo:
173
$$
ser modificadas estructural y funcionalmente por ataques nucleofílicos
s
Siendo, los grupos químicos reguladores más comunes involucrados en
e
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
FIGURA 3. El sistema antioxidante, comprendido por glutatión y tioredoxina,juega un papel importante en la homeostasis celular, involucrando sus grupos SHdurante las reacciones redox.

Cys – (X)n – Cys
donde X es cualquier residuo de aminoácido y n es el númerode aminoácidos presentes entre ambas cisteínas; que va de dosa seis residuos. Esta característica le permite a los grupos SH,la formación de enlaces disulfuro de manera reversible, de -pendiendo del ambiente redox al que se vean sometidos. Estasproteínas, conocidas como Proteínas Tiol Vecinal (PTV), pue -den alternar de un estado oxidado (mediante la formaciónde puentes de azufre o disulfuros, tanto intramolecularescomo intermoleculares) a un estado reducido, con una con-figuración ditiólica. Estos grupos tiol vecinal pueden formarenlaces covalentes reversibles actuando como nanosesoreso nanoswitches.
Así como existe una proteómica para cada una de las modi-ficaciones postraduccionales ya antes mencionadas, comola proteómica de la fosforilación —encargada del estudio de
174
$$
Tiol Vecinal (PTV), pueden
a
$$
a
$$
e
$$
t
$$
s
$$
ó
$$
q
$$
n
$$
p
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
m
$$
a
$$
o
$$
$$
a
$$
e
$$
d
$$
u
$$
t
$$
!
FIGURA 4. Modelo de una proteína tiol vecinal, donde el estado redox de sus gru-pos SH juega un papel importante tanto en su estructura como en su función.

todas aquellas proteínas en las que se lleva a cabo la unióndel grupo fosfato PO4
3– existe la proteómica de la tiolación,la cual centra especial atención en el estudio de todas aque-llas proteínas reguladoras; cuya característica principal es lapresencia de grupos tiol vecinal, ya que el estado redox de losresiduos de cisteína está involucrado; tanto en la estructuracomo en la función de muchas enzimas, receptores y canales.Aproximadamente, un 5% del proteoma corresponde a pro-teínas en cuya estructura tridimensional se encuentran estosresiduos. La proteómica de la tiolación nos indica que dichas prote-
ínas requieren de una alta especificidad para el control desus funciones y el papel que juegan en la célula. Debidoa que la presencia de estos residuos de cisteína habla de unagran precisión en su control, no en el resto del proteoma;donde fácilmente pueden ser modificadas postraduccional-mente mediante fosforilaciones o reguladas por otros gru-pos químicos. Es decir, a menor cantidad mayor especifici-dad; como ejemplo: el caso de la tioredoxina, una proteínaque basa su actividad catalítica únicamente en la presenciade dos grupos tiol en su estructura.Las técnicas utilizadas para el estudio del proteoma tiólico
se basan en técnicas de separación, como la cromatografíade afinidad. Este proceso se lleva a cabo haciendo eluir pro-teínas a través de una columna de sefarosa acomplejada conun arsenical, como lo es el óxido de fenilarsina —mejorconocido por sus siglas en inglés como PAO (Phenil ArsineOxide)—. Este compuesto reacciona selectivamente con lasproteínas tiol vecinal, quedando unidas a la columna en suinterior formando ditioarsina; sin embargo, las proteínasmonotiólicas, o aquellas proteínas no tiol vecinal, eluyenlibremente. Posterior a esto, mediante el uso de un agentereductor como 2-b-mercaptoetanol o el ditiotreitol (DTT),se lleva a cabo la reducción del complejo ditioarsina, permi-tiendo ahora la elución de las PTV.
175

Para el marcaje de las PTV se utiliza el compuesto N-iodo-acetil-3-[125I]iodotirosina [I125]IAIT, un reactivo altamente tiolespecífico, el cual se une y marca de manera selectiva losgrupos SH de las proteínas, con la característica de no verseafectado por la presencia de reactivos como el DTT o el 2-b -mercaptoetanol en concentraciones de hasta 10mM. La reac-ción de [I125]IAIT con tioles vecinales se puede ver bloqueadapor el uso de arsenicales, marcando a las proteínas conarsenitos AsO2 o PAO, permitiendo un método sensible paradistinguir reacciones con monotioles y tioles vecinales. El uso de estas técnicas de separación y marcaje ha per-
mitido suponer que, aproximadamente, un 5% del proteomacelular corresponde a proteínas en cuya estructura se encuen-tran grupos tiol vecinal. Esto puede demostrarse con elhecho de que el marcaje con [I125]IAIT se ve reducido pro-gresivamente con la adición de PAO, ya sea en células intactas
176
$$
L
de sefarosa acomplejada con un arsenical; como lo es el
ó
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
FIGURA 5. Cromatografía de afinidad. Columna de sefarosa-PAO, a través de la cual
se hacen eluir las proteínas, quedando unidas las PTV. Posteriormente se haceneluir con un agente reductor.

o extractos celulares. Como se muestra en la figura 6B, lasPTV reaccionan selectivamente con arsenicales trivalentes,como el PAO, formando ditioarsinas, las cuales no son mar-cadas por [I125]IAIT.
La presencia de los grupos tiol vecinal permite regular, demanera reversible, la actividad de las proteínas en respuestaa determinados ambientes redox, es decir, la regulación deuna proteína reguladora o PTV se lleva a cabo específica-mente en sus grupos SH regulando, a su vez, determinadasfunciones celulares; como en algunas vías de señalización,rutas metabólicas y en general algunos procesos fisiológicos.Uno de los procesos más representativos, donde se ve invo-lucrado este tipo de regulación, por PTV, es la contracción/relajación del músculo esquelético. El músculo, como principal reservorio de proteínas, lleva
a cabo el proceso de contracción principalmente por dosproteínas estructurales, como lo son la actina y la miosina. Sinembargo, algunas proteínas reguladoras encargadas de con-
177
$$
E
la
r
$$
$$
$$
a
$$
n
$$
[
$$
I
$$
u
$$
e
$$
s
$$
m
$$
$$
$$
$$
a
$$
$$
$$
$$
$$
$$
$$
$$
n
$$
e
$$
e
$$
u
$$
s
$$
!
FIGURA 6. A) Estructura química de la molécula [I125]IAIT; B) efecto de la adi-ción de PAO en linfoblastos con proteínas tiólicas marcadas con IAIT.

trolar la cinética del ion calcio Ca2+, durante la contraccióny la relajación, son la ATPasa de Calcio del Retículo Sarco-plásmico (SERCA1) y el Receptor de Rianodina (RyR1), lascuales son PTV; dependiendo únicamente de la SERCA1, elproceso de relajación y del RyR1, el proceso de contracción.
La SERCA1 y el RyR1 PTV, como se muestra en la figura 8,pueden experimentar una regulación redox tiol específica,modificando su estructura y por consiguiente su función.El modelo representado en la figura 8A permite apreciarque cuando la SERCA1 y RyR1 tienen sus grupos Tiol en unestado reducido, o (SH)2, permiten la regulación del flujo
178
$$
E
$$
e
$$
$$
T
$$
$$
e
$$
$$
$$
$$
1
$$
$$
$$
$$
$$
$$
$$
$$
s
$$
S
$$
$$
$$
$$
r
$$
a
$$
ó
$$
d
$$
d
$$
!
FIGURA 7. El músculo esquelético corresponde, aproximadamente, al 75% delpeso total en los vertebrados adultos, además de ser el principal representantede la conversión de la energía química en energía mecánica.
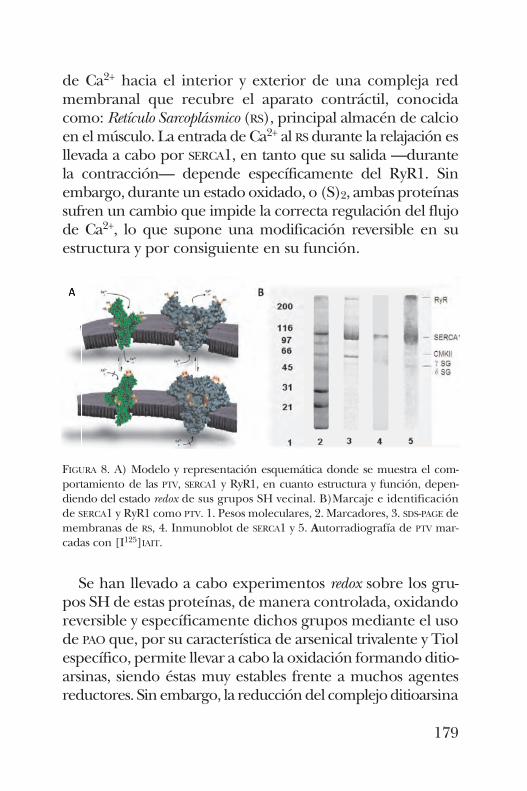
de Ca2+ hacia el interior y exterior de una compleja redmembranal que recubre el aparato contráctil, conocidacomo: Retículo Sarcoplásmico (RS), principal almacén de calcioen el músculo. La entrada de Ca2+ al RS durante la relajación esllevada a cabo por SERCA1, en tanto que su salida —durantela contracción— depende específicamente del RyR1. Sinembargo, durante un estado oxidado, o (S)2, ambas proteínassufren un cambio que impide la correcta regulación del flujode Ca2+, lo que supone una modificación reversible en suestructura y por consiguiente en su función.
Se han llevado a cabo experimentos redox sobre los gru-pos SH de estas proteínas, de manera controlada, oxidandoreversible y específicamente dichos grupos mediante el usode PAO que, por su característica de arsenical trivalente y Tiolespecífico, permite llevar a cabo la oxidación formando ditio-arsinas, siendo éstas muy estables frente a muchos agentesreductores. Sin embargo, la reducción del complejo ditioarsina
179
$$
(B
-
e
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
l
$$
e
$$
$$
d
$$
a
$$
-
$$
(
$$
a
$$
e
$$
o
$$
a
$$
$$
a
$$
n
$$
!
FIGURA 8. A) Modelo y representación esquemática donde se muestra el com-portamiento de las PTV, SERCA1 y RyR1, en cuanto estructura y función, depen-diendo del estado redox de sus grupos SH vecinal. B)Marcaje e identificaciónde SERCA1 y RyR1 como PTV. 1. Pesos moleculares, 2. Marcadores, 3. SDS-PAGE demembranas de RS, 4. Inmunoblot de SERCA1 y 5. Autorradiografía de PTV mar-cadas con [I125]IAIT.

se puede realizar con un agente altamente reductor como loes el 2,3-Dimercaptopropanol, mejor conocido como: BritishAnti-Lewisite (BAL) el cual, por sus propiedades como agentereductor y quelante, —debido a sus grupos SH— permite re -mover el arsenical manteniéndolo estable, restableciendo elfuncionamiento de la proteína y, por ende, el funciona-miento de la célula muscular. Esto ha permitido proponer unmecanismo que explique el fenómeno de fatiga muscular;un fenómeno fisiológico reversible que experimenta princi-palmente el músculo esquelético, donde las células muscu-lares, después de ser sometidas a una estimulación intensay continua, comienzan a disminuir su fuerza de contracción,recuperándose después de un tiempo (figura 9). Duranteuna estimulación muscular prolongada el tejido comienzaa disminuir su fuerza de contracción hasta sufrir el fenómenode fatiga; sin embargo, al oxidar los SH vecinal con el arse-nical, el fenómeno se presenta con mucho más rapidez. Alrevertir el efecto, con el uso de BAL, reduciendo dichos gru-pos tiol mediante la eliminación de PAO, el músculo resta-blece su fuerza de contracción. Experimentos como los mostrados en la figura 10 expo-
nen el efecto que tienen los compuestos oxidantes y reduc-tores directamente sobre las proteínas SERCA1 y RyR1. Enla figura 10A, se muestra la actividad de la ATPasa durante lacaptura de Ca2+, reflejando el poder oxidante del PAO conuna disminución de la actividad de SERCA1 ( ), con respectoa su control ( ). En la figura 10B, vesículas de RS, cargadascon Ca2+, son estimuladas con cafeína —un alcaloide queinduce la apertura del canal de rianodina o RyR1— permi-tiendo la salida del ion, el cual forma un complejo coloridocon el reactivo arzenaso III. Cuando el canal es oxidado conPAO se inhibe la salida de Ca2+, efecto revertido al agregar BAL. La idea de utilizar a los arsenicales como el PAO, a modo
de herramienta, para estudiar el estado redox en las proteínas
$$
!
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
!
$$
$$
$$
$$
$$
$$
$$
$$
"
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
"
$$
$$
$$
$$
$$
$$
$$
$$
$$
180

reguladoras o PTV se debe a que durante la Guerra Mundial,los alemanes conocían el efecto farmacológico de estos com-puestos, utilizados como agentes vesicantes, causando pro-blemas respiratorios y principalmente debilidad muscular.Sin embargo, los ingleses diseñaron el antídoto BAL contralos efectos causados por estos arsenicales, principalmente con-tra el compuesto 2-cloroetenildicloroarsina, mejor conocidocomo Lewisita. Esto ha permitido estudiar el efecto que tienela oxidación en las proteínas, principalmente las PTV, encar-gadas de regular procesos importantes en la célula, permi-tiendo abrir un campo de estudio en otras enfermedadescomo, el cáncer, las distrofias, etcétera.
181
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
FIGURA 9. Efecto del agente oxidante PAO y un agente reductor BAL sobre la fun-ción contráctil del músculo esquelético con respecto al tiempo. Se observa unadisminución en la fuerza de contracción en presencia de PAO; sin embargo, laremoción del mismo por el BAL restablece la fuerza de contracción. Al lado, unmodelo del efecto de los dos agentes químicos sobre las PTV.
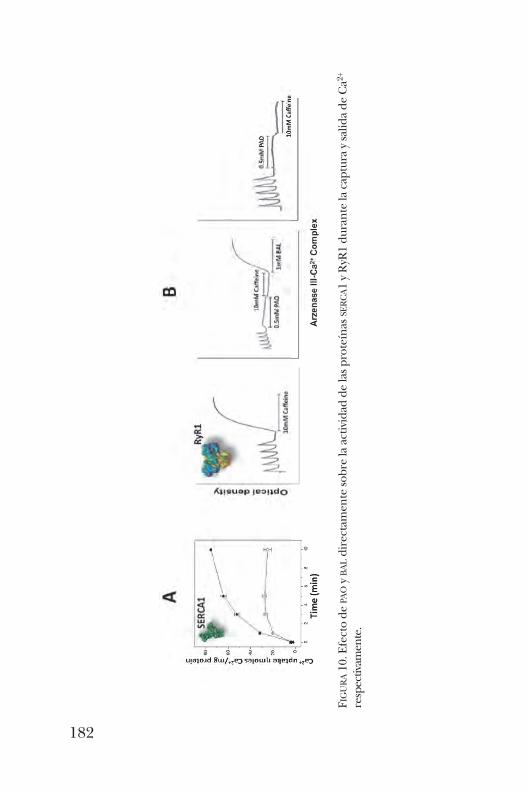
182
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
##
FIGURA10. Efecto de PAOy BALdirectam
ente sobre la actividad de las proteínas SERCA1y RyR1 durante la captura y salida de Ca2+
respectivam
ente.

REFERENCIAS
Álvarez R., Vázquez P., Pérez F., Jiménez A., Tirado A., Irles C. et al. Regu-lation of fast skeletal muscle activity by SERCA1 vicinal-cysteines.J. Muscle Res. and Cell Motil., 2009, 30(1): 5-16.
Antelmann H. y Helmann J. D. Thiol-based redox switches and generegulation. Antioxid. Redox Signal., 2011, 14(6): 1049-63.
Carmel-Harel O. y Storz G. Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and saccha-romyces cerevisiae responses to oxidative stress. Annu. Rev. Micro-biol., 2000, 54: 439-61.
Gitler C., Mogyoros M. y Kalef E. Labeling of protein vicinal dithiols: roleof protein-S2 to protein-(SH)2 conversion in metabolic regulationand oxidative stress. Methods Enzymol., 1994, 233: 403-15.
Gitler C., Zarmi B. y Kalef E. General method to identify and enrich vici-nal thiol proteins present in intact cells in the oxidized, disulfidestate. Anal. Biochem., 1997, 252(1): 48-55.
Gordon J. J. y Quastel J. H. Effects of organic arsenicals on enzyme sys-tems. Biochem. J., 1948, 42(3): 337-350.
Halliwell B. Oxidative stress and cancer: have we moved forward? Bio-chem. J., 2007, 401(1): 1-11.
Jacob C., Giles G. I., Giles N. M. y Sies H. Sulfur and selenium: the roleof oxidation state in protein structure and function. Chem. Int. Ed.Engl., 2003, 42(39): 4742-58.
Lu J. y Holmgren A. The thioredoxin antioxidant system. Free Radic. Biol.Med., 2013, S0891-5849: 00380-8.
Ortega A., Álvarez R., Pérez F., Jiménez A., Gutiérrez J. y Vázquez P.Redox regulation of vicinal thiol groups of the SERCA1 participatesin muscle fatigue. J. Muscle Res. Cell Motil., 2004, 25: 597-598.
Paget M. S. y Buttner M. J. Thiol-based regulatory switches. Annu. Rev.Genet., 2003, 37: 91-121.
Prabakaran S., Lippens G., Steen H. y Gunawardena J. Post-translationalmodification: nature’s escape from genetic imprisonment and thebasis for dynamic information encoding. Rev. Syst. Biol. Med.,2012, 4(6): 565-83.
Reid M. B. Nitric oxide, reactive oxygen species, and skeletal musclecontraction. Med. Sci. Sports Exerc., 2001, 33(3): 371-376.
Winterbourn C. C. Radical scavenging by thiols: Implications for redoxsignaling and antioxidant. En: Gitler C. y Danon A. eds. CellularImplications of Redox Signalling. Imperial College Press; 2003, 175-185.
183


EL CAMBIO TOPOLÓGICO DE LA MEMBRANACAUSADO POR LAS PROTEÍNAS
TRANSMEMBRANALES
SAMUEL GITLERDepartamento de Matemáticas
Centro de Investigación y Estudios AvanzadosMiembro de El Colegio [email protected]
INTRODUCCIÓN
La doctora Alicia Ortega me pidió hablar sobre la topologíade las proteínas de la membrana. Para ello, hablaré sobre laclasificación de las superficies orientables bidimensionales,el resultado más importante de la topología algebraica en elsiglo XIX obtenido por B. Riemann, uno de los más grandesmatemáticos de la historia.No usaré funciones en esta exposición, sino lo que se llama
geometría de hule o topología visual. Uno de los topólogos másfamosos, J. Minor, dijo (cuando se le pidió que hiciera estaclasificación a partir de principios básicos y que escribieralas funciones que definen las transformaciones) que esa cla-sificación requeriría en verdad de una demostración mate-mática larga y difícil. Dicho esto, permítaseme comenzar con algunas defini-
ciones en 0, 1 y 2 dimensiones.
185

Un 0-disco es un punto.Un 1-disco abierto es un intervalo
que no contiene los puntos extremos a y b.
Un 1-disco cerrado D1 es un intervalo…
en el que los extremos sí pertenecen a D1.
Un 2-disco abierto D2 es…
usando geometría analítica:
x2 + y2 < 1.
Definición 1. Una n-variedad, n-dimensional cerrada yconexa, Mn para n = 1 y 2, es un conjunto Mn que puede sercubierto por n-discos abiertos, de manera tal que cada parde puntos x y y están contenidos en discos Dnx, Dny, cuyaintersección puede ser cubierta por discos abiertos.Definición 2. Una variedad n-dimensional Mn con frontera
es un conjunto que puede ser cubierto por discos abiertosy discos semiabiertos Dn.
186
$$
! !
!
$$
! !
!
##
D
E
Para
e
Uno de los topólogos más famosos, J. Minor, dijo, cuando se le pidió que hiciera esta
c
U

Ejemplo:
La frontera es x2 + y2 = 1 y los puntos de la frontera estáncubiertos por discos semiabiertos.
Pasemos ahora a la geometría de hule. Supongamos quese nos da una mesa y la cubrimos con un mantel completa-mente elástico, es decir, podemos extenderlo o contraerlosiempre que no lo rompamos.
EJEMPLOS DEM1
A y B son (topológicamente) equivalentes porque pode -mos deformar uno en cualquier otro. Si los llenamos obte-nemos ejemplos de M2.De modo que si dibujamos variedades con frontera y
cada punto del interior es parte de M o –M y las fronterasson equivalentes, entonces las variedades con fronteraserán equivalentes.¿Cuándo obtenemos una M2 con frontera? Supongamos
que cortamos un hueco en la figura
187
$$
! !
y !
$$
!! !
Supongamos que cortamos un hueco en la figura
$$
!! !
Supongamos que cortamos un hueco en la figura
A) B)

entonces obtenemos otra frontera en el interior de la varie-dad, disjunta de la frontera exterior, y que no puede ser equi-valente a la que no tiene huecos. Podemos seguir haciendo huecos:
que son la frontera de un 3-disco. Continuando con esteproceso de agregar huecos, obtenemos una familia M2i coni huecos y, entonces, ésta es la clasificación de las variedadesconexas con frontera.
Clasificación de las 2-variedades con frontera
¿Cómo obtenemos variedades sin deformar? Ahora esta-mos en el 3-espacio:
Continuamos hasta reducir:
188
$$
Continuando con este proceso de agregar huecos, obtenemos
u !
Ahora estamos en el 3-espacio
$$
Continuando con este proceso de agregar huecos, obtenemos
u !
Ahora estamos en el 3-espacio
$$
Continuando con este proceso de agregar huecos, obtenemos
u !
Ahora estamos en el 3-espacio
$$
n

a un punto, obteniendo la frontera de una pelota, la 2-esferax2 + y2 + z2 = 1, que es la frontera de un 3-disco. ¿Cómo pro-ducimos una 2-variedad cerrada no equivalente mediantela adición de un hueco?Consideremos ahora las variedades cerradas de género 2,
que significa que la variedad se forma añadiendo 2 asas aldisco. Así, la clasificación de las variedades conexas M2 noequivalentes es la siguiente:
Le anexamos un asa
T1 no es equivalente a T0.La clasificación de variedades cerradas, donde el género 12
significa que la variedad cerrada tiene 12 asas o la variedadtiene género 12.
Esfera
Variedades conexas no equivalentes.
189
$$
n
$$
Sin cambiar la topología de
l
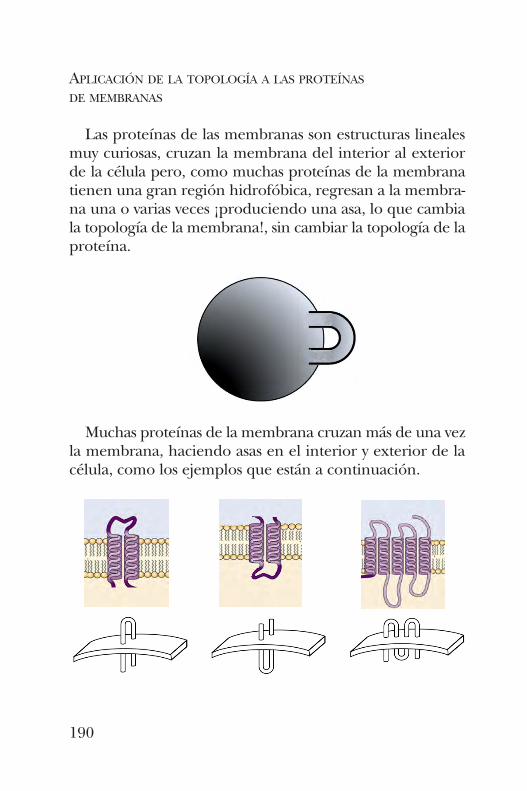
APLICACIÓN DE LA TOPOLOGÍA A LAS PROTEÍNAS
DE MEMBRANAS
Las proteínas de las membranas son estructuras linealesmuy curiosas, cruzan la membrana del interior al exteriorde la célula pero, como muchas proteínas de la membranatienen una gran región hidrofóbica, regresan a la membra-na una o varias veces ¡produciendo una asa, lo que cambiala topología de la membrana!, sin cambiar la topología de laproteína.
Muchas proteínas de la membrana cruzan más de una vezla membrana, haciendo asas en el interior y exterior de lacélula, como los ejemplos que están a continuación.
190
$$
Sin cambiar la topología de
l
$
$
Sin cambiar la topología de
l

La parte hidrofóbica de la proteína lleva a esta estructuralineal de regreso a la membrana, creando una o varias asas,cambiando la topología de la membrana en cada cruce,pero sin cambiar la topología de la proteína. Aquí entramos en territorio desconocido y surgen las
siguientes preguntas:1) El número de cambios en el género de la membrana
¿es característico de cada proteína?2) ¿Es característico de ciertas familias de proteínas? ¿Qué
propiedades de las proteínas de la membrana las hacenpertenecer a una familia dada?
191


ESTUDIANDO LA SÍNTESIS DE FOSFATIDILCOLINA EN BACTERIAS:
DE LÍPIDOS DE MEMBRANA A PROTEÍNAS DE MEMBRANA
CHRISTIAN SOHLENKAMPROSA LIDIA SOLÍS OVIEDO
OTTO GEIGERCentro de Ciencias Genómicas
Universidad Nacional Autónoma de México Cuernavaca, México
PRÓLOGO
Durante el Kolleg Proteins, at a crossroad between mathe-matics, physics, chemistry and biolog” que se celebró del6 al 8 de noviembre de 2013 en El Colegio Nacional, en laciudad de México, se presentó y se discutió una ampliagama de enfoques que se usan para el estudio de proteínas.A pesar de los avances tecnológicos recientes, el estudio deproteínas integrales de membrana todavía significa un granreto. Mi presentación en este Kolleg pretendía ejemplificaralgunos de los problemas típicos que con frecuencia ocurrenen el estudio de proteínas integrales de membrana y cómologramos obtener cierta caracterización estructural y fun-cional de la proteína integral de membrana fosfatidilcolinasintasa (PCS) utilizando técnicas de biología molecular.
193

Para mí, este Kolleg abrió una oportunidad especial de en -contrarme con matemáticos, físicos y químicos que estudiandiferentes aspectos de las proteínas. Este encuentro permitióel intercambio de ideas y opiniones de una manera interdisci-plinaria. Vale la pena mencionar dos ejemplos: 1) tuve discu-siones largas con el doctor Samuel Gitler, un matemático muydistinguido, miembro de El Colegio Nacional, sobre los dis-tintos significados del término topología en matemáticas y bio-química. 2) El Kolleg también me ofreció la oportunidad deconocer al ganador del premio Nobel, el doctor RobertHuber, discutir problemas científicos y conocer su razona-miento científico.
SÍNTESIS DE FOSFATIDILCOLINA EN BACTERIAS
Las membranas biológicas están mayoritariamente com-puestas por lípidos de membrana anfifílica y proteínas mem-branales. Cada tipo de membrana presenta una composi-ción específica y contiene una gran variedad de lípidos. Enla mayoría de los casos se trata de los fosfolípidos fosfatidi-letanolamina, fosfatidilglicerol, fosfatidilinositol y fosfatidil-colina (PC). La PC es el fosfolípido mayoritario en membranaseucariotas que tiene una plétora de diferentes funciones.Durante muchos años, el dogma era que solamente bacteriaspatógenas, simbiontes y algunas bacterias fotosintéticas acu-mulan PC en sus membranas; sin embargo, un análisis dedatos genómicos muestra que la presencia de PC no se res-tringe a estos grupos. A partir de datos genómicos se puedeestimar que aproximadamente 15% de las bacterias secuen-ciadas tienen la capacidad de formar PC (Sohlenkamp et al.,2003; Geiger et al., 2013). En mi presentación les platicaré lahistoria de cómo salimos buscando una nueva ruta de síntesis
194

de PC en bacterias y llegamos, finalmente, a estudiar la rela-ción estructura-función y la topología de la proteína inte-gral de membrana fosfatidilcolina sintasa (PCS).
DESCUBRIENDO LA RUTA DE FOSFATIDILCOLINA SINTASA
Por muchos años, la única ruta de síntesis de la PC en bac-terias que se conocía era la N-metilación de fosfatidiletano-lamina (PmtA). En esta ruta, PC se forma por tres metilacionesconsecutivas de PE. Hace aproximadamente 15 años, el grupode investigación del doctor Otto Geiger, ubicado en la Tech-nische Universität Berlin, Alemania, había empezado a estu-diar la síntesis de PC utilizando como organismo modelo laa-proteobacteria S. meliloti, la cual causa la formación de nódu-los en las raíces de plantas leguminosas. Se observó que mutan-tes deficientes en la ruta de PmtA todavía formaban PC enla presencia de colina libre en el medio de cultivo, lo queindicaba que existía una segunda ruta de biosíntesis de PCen este organismo. Esta ruta se conoció posteriormentecomo ruta de fosfatidilcolina sintasa(PCS) (de Rudder et al.,1997). La PCS utiliza citidina difosfato-diacilglicerol (CDP-DAG)y colina como sustratos para formar PC y citidina monofos-fato (CMP) (de Rudder et al., 1999). Con el propósito de aislarcepas mutantes de S. meliloti, deficientes en este ruta biosin-tética, se desarrolló un método basado en la autorradiografíade colonias (Raetz, 1975). Se plaqueó una población de cepasmutantes químicas de S. meliloti en un medio de cultivo sólido,luego se tomó una réplica del patrón de colonias con unpapel filtro y las bacterias adheridas al filtro se incubaroncon [14C] colina. En las condiciones experimentales selec-cionadas, la [14C] colina se incorporaba preferencialmenteen PC. Los fosfolípidos y las proteínas de las bacterias adhe-ridas al filtro se precipitaron con ácido tricloroacético (TCA)
195

y las colonias se tiñeron con azul de Coomassie. Comparandolos patrones de colonias que incorporaron radioactividady colonias teñidas con azul de Coomassie, se aislaron 15 mu -tantes deficientes en la actividad de PCS, dentro de una pobla -ción de 26 mil mutantes analizados (Sohlenkamp et al., 2000).Se seleccionó una de las cepas mutantes obtenidas y se movi-lizó un banco de cósmidos de ADN genómico de S. meliloti a lacepa mutante. Se repitió el método de autorradiografía decolonias, descrito anteriormente, pero esta vez buscando colo-nias que incorporaban [14C] colina en PC. De las cepas mu -tantes complementadas se aislaron los cósmidos, se sub-clonaron sus insertos y se volvió a repetir la autorradiografíade colonias. De esta manera, se identificó un fragmento decuatro genes que complementaba la cepa mutante causandola formación de PC. Uno de estos genes codificaba para unaproteína, altamente hidrofóbica de 241 aminoácidos, la cualpresentaba una versión modificada de un motivo descrito enmiembros de la superfamilia de proteínas de las CDP-alcoholfosfotransferasas (DGX2ARX12GX3DX3D para PCS versusDGX2ARX8PX3GX3DX3D para otros miembros de esta super-familia) (Sohlenkamp et al., 2000). La gran mayoría de las pro-teínas que pertenecen a esta superfamilia está involucrada enla síntesis de fosfolípidos. Algunos ejemplos son la fosfatidili-nositol sintasa, la fosfatidilserina sintasa, la fosfatidilglicerolfosfato sintasa y la cardiolipina sintasa del tipo eucarionte. Lospocos miembros de esta superfamilia que no están relacio-nados con la síntesis de fosfolípidos están involucrados en lasíntesis de solutos compatibles derivados de inositol en variasbacterias hipertermófilos (Rodrigues et al., 2007).
196

IDENTIFICACIÓN DE AMINOÁCIDOS ESENCIALES PARA
LA ACTIVIDAD EN VIVO DE LA FOSFATIDILCOLINA SINTASA
A pesar de la gran importancia que tienen los miembrosde esta superfamilia de proteínas en la síntesis de fosfolípi-dos, aún no se tienen datos estructurales sobre este hecho.En el estudio de la enzima CPT1 de levadura (responsable delúltimo paso en la síntesis de PC a través de la ruta de CDP-colinao ruta de Kennedy en eucariotas) se encontró que varios delos residuos de aminoácidos del motivo conservado DGX2
ARX8PX3GX3DX3D son importantes para la actividad de laenzima. Nosotros sospechábamos que también los residuosde aminoácidos fueran del motivo conservado, podrían serimportantes para la actividad enzimática. Por esto, queríamosestudiar la PCS de S. meliloti con un enfoque por mutagénesismás amplio. Para esto, se alinearon las secuencias de amino-ácidos de PCS de diferentes bacterias, cuya actividad había sidodemostrada, incluyendo a las secuencias de S. meliloti, Agro-bacterium tumefaciens, Rhizobium leguminosarum, Brucella abor-tus, Mesorhizobium loti, Pseudomonas aeruginosa, Legionella pneu-mophila, y Borrelia burgdorferi (Comerci et al., 2006; Conoveret al., 2008; Martínez-Morales et al., 2003; Wang et al., 2004;Wessel et al., 2006). Asimismo se identificaron los residuosde aminoácidos conservados y se seleccionaron 55 para sercambiados por mutagénesis sitio dirigido. Todos los residuosconservados fueron cambiados por alanina, pero en casode que el residuo conservado ya fuera una alanina, se cam-bió por glicina. La actividad de PCS de las enzimas mutadasse evaluó expresándolas en E. coli, que no forma PC, pero síes el precursor CDP-DAG y además puede incorporar colinadel medio de cultivo a la célula. Las cepas mutantes se clasi-ficaron en tres grupos, según la actividad de PCS que presen-taban: (I) actividad similar a la PCS silvestre (>80% de activi-dad comparada con la PCS silvestre), (II) actividad de PCS
197

reducida (entre 20% y 80% de actividad comparada con laPCS silvestre), (III) actividad perdida (<20% de actividad com-parada con la PCS silvestre). Nueve de las cepas mutantes sitio-dirigido carecían por completo de actividad de PCS o solamentemostraron una actividad residual y se clasificaron en el grupo III.Además de los residuos que forman parte del motivo con-servado (DGX2ARX12GX3DX3D) se identificaron cuatro resi-duos de aminoácidos esenciales (Solís-Oviedo et al., 2012).
TOPOLOGÍA DE LA FOSFATIDILCOLINA SINTASA DE S. MELILOTI
Para poder mapear los residuos de aminoácidos esencialesen la estructura secundaria, era necesario conocer la topolo-gía de la proteína PCS. Predicciones de la estructura secunda-ria arrojaron entre cuatro y ocho hélices transmembranalespredichas. Con el propósito de determinar la topología, expe-rimentalmente, construimos 23 fusiones traduccionales; entreversiones truncadas de la PCS de S. meliloti y las enzimas repor-teras b-galactosidasa (lacZ) y fosfatasa alkalina (phoA). Lasenzimas lacZ solamente muestran actividad enzimática cuandoestán ubicadas dentro del citoplasma, mientras que las phoAsolamente muestran actividad cuando están ubicadas dentrodel periplasma (Manoil, 1991). Las proteínas de fusión seexpresaron en una cepa de E. coli que carecía de actividadesde phoA y lacZ. Ensayando las actividades enzimáticas de lasproteínas de fusión en esta cepa de E. coli pudimos determi-nar las posiciones aproximadas de siete hélices transmem-branales (Nies et al., 1998; Aguilera et al., 2004). Desafortu-nadamente, este enfoque no permitió la determinación dela topología del N-terminus de la proteína, lo cual queríadecir que aún no sabíamos si el N-terminus estaba orientadohacia el citoplasma o hacia el periplasma. Basado en esterazonamiento se seleccionó el mé todo de accesibilidad de cis-
198

teínas sustituidas (Substituted Cysteine Accessibility Method, SCAM)como enfoque complementario (Bogdanov et al., 2005;Bogdanov et al., 2010), el cual permite localizar residuos decisteína específicos dentro de la proteína de interés. La ver-sión silvestre de la PCS de S. meliloti no presenta residuos decisteína. Se construyeron tres cepas mutantes sitio-dirigidos,una presentando un residuo de cisteína, ubicado en el N-ter-minus, (serina 18 cambiada por cisteína-S18C), una en unresiduo de cisteína expuesto al periplasma (S155C) y una enun residuo de cisteína en el citoplasma (V68C). Cuando lastres versiones mutadas de la PCS se expresaron en E. coli,mos-traron actividad de PCS en el mismo nivel que la proteína sil-vestre de PCS indicando que su topología no se había cam-biado por la mutación introducida. En los ensayos se ocupa un reactivo especial (biotina aco-
plada a maleimida por un linker -MPB) que reacciona espe-cíficamente con grupos sulfhidrilo y que no puede pasar lamembrana interna. En las condiciones experimentales selec-cionadas MPB solamente reaccionaría con residuos de cis -teína expuestos al lado periplasmático de la membrana, perono con residuos de cisteína ubicados dentro del citoplasma.La PCS que lleva la mutación S155C reacciona con el agen-te MPB, lo que confirma que S155C está expuesta al peri-plasma. En el caso de la PCS que lleva la mutación V68C, lacisteína solamente reacciona con MPB cuando la muestra essonicada en presencia de MPB, confirmando que V68C estáexpuesta al citoplasma. La PCS que lleva la mutación S18Cse comporta igual que la PCS que lleva la mutación en V68C.Esto implica que el N-terminus de la PCS está orientado haciael citoplasma. Los datos obtenidos del método SCAM, juntocon los datos obtenidos de las fusiones traduccionales, per-mitieron la construcción de un modelo topológico de la PCSde S. meliloti , la cual presenta ocho hélices transmembrana-les, con el N- y el C-terminus ubicados dentro del citoplasma.
199

Los residuos de aminoácidos identificados como esencialespara la actividad enzimática, forman parte de las hélicestransmembranales uno y dos, del asa citoplasmática, conec-tando las hélices dos y tres.
CONCLUSIONES
La PCS de S. meliloti es una proteína integral de membrana,lo que dificultó su purificación para estudios estructurales.Empezamos la caracterización de la proteína PCS usandovarios métodos de biología molecular como mutagénesissitio-dirigido, fusiones traduccionales, entre la PCS y enzimasreporteros, y el método de accesibilidad de cisteínas susti-tuidas. La combinación de estos tres enfoques con un mode-laje computacional nos permitió obtener un modelo tridi-mensional de la PCS de S. meliloti.
REFERENCIAS
Aguilera S., Aguilar M. E., Chávez M. P., López-Meza J. E., Pedraza-Reyes M., Campos-García J. y Cervantes C. Essential residues in thechromate transporter ChrA of Pseudomonas aeruginosa. FEMS Micro-biol. Lett., 2004, 232, 107-112.
Bogdanov M., Zhang W., Xie J. y Dowhan W. Transmembrane proteintopology mapping by the substituted cysteine accessibility method(SCAMTM): application to lipid-specific membrane protein topoge-nesis. Methods, 2005, 36, 148-171.
Bogdanov M., Heacock P., Guan Z. y Dowhan W. Plasticity of lipid-pro-tein interactions in the function and topogenesis of the membraneprotein lactose permease from Escherichia coli. Proc. Natl. Acad. Sci.USA, 2010, 107, 15057-15062.
Comerci D. J., Altabe S., de Mendoza D. y Ugalde R. A., Brucella abortussynthesizes phosphatidylcholine from choline provided by thehost. J. Bacteriol., 2006, 188, 1929-1934.
200

Conover G. M., Martínez-Morales F., Heidtman M. I., Luo Z. Q., Tang M.,Chen C., Geiger O. y Isberg R. R. Phosphatidylcholine synthesis isrequired for optimal function of Legionella pneumophila virulencedeterminants. Cell. Microbiol., 2008, 10, 514-528.
de Rudder K. E., Thomas-Oates J. E. y Geiger O. Rhizobium melilotimutants deficient in phospholipid N-methyltransferase still con-tain phosphatidylcholine. J. Bacteriol., 1997, 179, 6921-6928.
de Rudder K. E., Sohlenkamp C. y Geiger O. Plant-exuded choline isused for rhizobial membrane lipid biosynthesis by phosphatidyl-choline synthase. J. Biol. Chem., 1999, 274, 20011-20016.
Geiger O., López-Lara I. M. y Sohlenkamp C. Phosphatidylcholine biosyn -thesis and function in bacteria. BBA-Molecular and Cell Biology ofLipids, 2013, 1831, 503-513.
Manoil C. Analysis of membrane protein topology using alkaline phos -phatase and beta-galactosidase gene fusions. Methods Cell. Biol.,1991, 34, 61-75.
Martínez-Morales F., Schobert M., López-Lara I. M. y Geiger O. Pathwaysfor phosphatidylcholine biosynthesis in bacteria. Microbiology, 2003,149, 3461-3471.
Nies D. H., Koch S., Wachi S., Peitzsch N. y Saier M. H. Jr. CHR, a novelfamily of prokaryotic proton motive force-driven transporters pro-bably containing chromate/sulfate antiporters. J. Bacteriol., 1998,180, 5799-5802.
Raetz C. R. H. Isolation of Escherichia coli mutants defective in enzymesof membrane lipid biosynthesis. Proc. Natl. Acad. Sci. USA, 1975,72, 3461-3471.
Rodrigues M. V., Borges N., Henriques M., Lamosa P., Ventura R., Fer-nandes C., Empadinhas N., Maycock C., da Costa M. S. y Santos H.Bifunctional CTP:Inositol-1-phosphate cytidylyltransferase/CDP-inositol:Inositol-1-phosphate transferase, the key enzyme for di-myo -inositol-phosphate synthesis in several (Hyper)thermophiles.J. Bacteriol., 2007, 189, 5405-5412.
Sohlenkamp C., de Rudder K. E., Röhrs V. V., López-Lara I. M. y GeigerO. Cloning and characterization of the gene for phosphatidylcho-line synthase. J. Biol. Chem., 2000, 275, 27500.
Sohlenkamp C., López-Lara I. M. y Geiger O. Biosynthesis of phospha-tidylcholine in bacteria. Prog. Lipid Res., 2003, 42, 115-162.
Solís-Oviedo R. L., Martínez-Morales F., Geiger O. y Sohlenkamp C. Func-tional and topological analysis of phosphatidylcholine synthase
201

from Sinorhizobium meliloti. BBA-Molecular and Cell Biology of Lipids,2012, 1821, 573-581.
Wang X. G., Scagliotti J. P. y Hu L. T. Phospholipid synthesis in Borreliaburgdorferi : BB0249 and BB0721 encode functional phosphatidyl-choline synthase and phosphatidylglycerolphosphate synthaseproteins. Microbiology, 2004, 150, 391-397.
Wessel M., Klüsener S., Gödeke J., Fritz C., Hacker S. y Narberhaus F.Virulence of Agrobacterium tumefaciens requires phosphatidylcholinein the bacterial membrane. Mol. Microbiol., 2006, 62, 906-915.
Williams J. G. y McMaster C. R. Scanning alanine mutagenesis of theCDP-alcohol phosphotransferase motif of Saccharomyces cerevisiae cho-linephosphotransferase. J. Biol. Chem., 1998, 273, 13482-13487.
202

AGRUPACIONES PROTEICASCUANDO LAS PROTEÍNAS FORMAN
UN EQUIPO PARA TRABAJAR
AURA MATILDE JIMÉNEZ-GARDUÑODepartamento de Bioquímica
Facultad de MedicinaUniversidad Nacional Autónoma de México
Desde el principio de la vida en la Tierra se ha demostradoque las unidades individuales no logran lo mismo solas queagrupadas con otras entidades de la misma o de diferentenaturaleza. Desde unidades muy pequeñas como los áto-mos, hasta unidades más grandes como los organismos, lasuma de las fuerzas individuales es uno de los factores másimportantes para alcanzar metas importantes y trascendentes.Este fenómeno se observa incluso en procesos a largo plazocomo la evolución y la especiación. Así pues, para el enten-dimiento de muchas funciones a nivel celular, debemos teneren mente que la vida siempre ha trabajado en agrupacioneso mó dulos, con el fin de avanzar al siguiente nivel de orga-nización. Uno de los más claros ejemplos son las biomo -léculas. Las proteínas son los módulos de los aminoácidos,así como los ácidos nucleicos son los módulos de los nucleó -tidos. Ambos ejemplos constituyen, sin duda, los blancos deinvestigación más estudiados en las ciencias biomédicas y nopuede ser negada su trascendencia en el proceso evolutivo.
203

Sin embargo, los módulos de lípidos y de carbohidratos tam-bién juegan un papel importante en muchos procesos celu-lares y han sido relegados por muchos investigadores. Eneste texto trataremos de enfatizar la importancia de los módu-los de lípidos representados por la membrana plasmáticadentro de la organización y función celular.Empezaremos por definir una agrupación como el con-
junto de objetos que se unen y que comparten propiedadesdiscretas y medibles. En un sentido más aplicado a la célula,un módulo funcional se define como: una entidad discretacuya función se puede separar de otros módulos. Esta separa-ción depende de un aislamiento químico que se basa, ya seaen la localización espacial o por la especificidad química [1].Puesto que la membrana plasmática es un organelo espa-cialmente aislado de otros, provee un módulo óptimo parasu aislamiento y estudio.Antes de analizar los métodos existentes para el estudio de
las agrupaciones, la primera pregunta que emerge es: ¿cuáles la importancia de las agrupaciones en los procesos celu-lares? Debemos tener en cuenta que el simple aglutinamientode moléculas no sería suficientemente específico para explicarsu significado biológico, es por eso que se sabe que las agru-paciones tienen un propósito. Hasta ahora, tres funciones ge -nerales se han propuesto, por lo menos, para agrupacionesproteicas de la membrana plasmática. En neuronas, por ejem-plo, el rastreo de proteínas involucradas en la liberación delas vesículas sinápticas en la membrana presináptica ha de -mostrado que el agrupamiento del complejo SNARE (Receptorde la Proteína de Unión Soluble NSF) tiene como propósito elmantenimiento y el reciclaje del complejo como una entidadúnica para evitar la difusión de sus componentes a lo largode la membrana [2]. Reciclar el complejo, como una unidadsupramolecular, toma sentido cuando tenemos en cuenta lavelocidad que requiere la comunicación sináptica, así como
204

el ahorro de energía que se genera. Una segunda funciónpropuesta para las agrupaciones proteicas es la que se rela-ciona con canales y transportadores de la membrana plas-mática. El transporte de la mayor parte de las moléculas,a través de la membrana plasmática, se lleva a cabo por pro-teínas transmembranales que permiten el flujo de moléculasa través de su poro (canales) o por la translocación de molécu -las a lo largo de la membrana, con una fuente de energía(transportadores). El agrupamiento de este tipo de proteínasen puntos específicos de la membrana permite la elevaciónde la concentración de la molécula transportada en el lugarrequerido para ejercer la función correspondiente. Un ejem-plo típico es el de la familia de los canales de calcio. La mo -dulación fina de las concentraciones de calcio intracelulares trascendental para muchas funciones celulares, inclu-yendo la muerte celular. La concentración de calcio depen-derá de cuántos canales permitan el flujo de los iones fueradel retículo endoplásmico o la mitocondria. En el caso delreceptor de IP3 (receptor de inositol 1.4.5-trifosfato, un canalde calcio dependiente de ligando), localizado en la mem-brana del retículo endoplásmico, se lleva a cabo una agru-pación dinámica en la presencia de IP3 o a partir de altas con-centraciones de Ca2+. Dichos estímulos inducen el recluta-miento reversible de hasta cuatro canales que permiten elflujo de calcio por períodos más largos, alcanzando mayoresconcentraciones [3]. Otra función determinada de las agru-paciones es, justamente la opuesta a la previamente descrita,es decir, cuando la agrupación interfiere con el cambio con-formacional impidiendo la activación de las proteínas. Estoha sido descrito para los canales KV2.1. En estas proteínasse ha descrito que las corrientes que se emiten por el KV2.1,cuando están agrupadas, son mucho más bajas comparadascon un canal único abierto [4].
205

Estos tres tipos de funciones son sólo algunos de la grangama de posibilidades existentes y que, seguramente con elmejoramiento de la tecnología la lista se irá ampliando. Enlas siguientes secciones discutiremos por qué la membranaplasmática es un blanco excelente para el análisis de lasagrupaciones proteicas y cómo se aborda su estudio.
Desde los años ochenta, la formación de fases en las mem-branas artificiales de mezclas lipídicas ya se conocía. Losesteroles constituían una parte fundamental en la forma-ción de fases que se denominaron líquido-ordenadas (Lo)y que podía coexistir con la fase líquido-desordenadas (Ld )(figura 1). Más tarde, los primeros experimentos en mem-branas biológicas se realizaron en células epiteliales y se des-cubrió una localización polarizada de los lípidos. La asocia-ción entre una función determinada, dependiente de unacomposición lipídica en un compartimento membranal,abrió el panorama para revisar el modelo existente de lamembrana y proponer un nuevo sistema de organizaciónde este organelo. La primera propuesta se realizó a fina-les de los años noventa cuando el concepto de las “balsas lipí-dicas” (lipid rafts en inglés) fue introducido. “Balsa lipídica”
206
$$
entre una función determinada dependiente de una composición lipídica en un
c
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
o
$$
D
$$
$$
a
$$
e
$$
a
$$
a
$$
u
$$
a
$$
$$
r
$$
l
$$
q
$$
a
$$
l
$$
!
FIGURA 1. Modelo de las fases líquido-ordenadas (Lo, enriquecidas en colesterol)y líquido desordenadas que coexisten en membranas artificiales.

se refiere a la fase Lo enriquecida en colesterol y esfingolípi-dos que regula funciones proteicas. Con esta propuesta elenfoque y estudio de la membrana plasmática se modificódrásticamente [5]. Sin embargo, existían limitaciones téc-nicas, principalmente en el área de la óptica, que generabanel debate sobre su existencia en las células vivas, puesto quetoda la información se basaba principalmente en experi-mentos de membranas aisladas. El aislamiento de los domi-nios ricos en colesterol y esfingolípidos por métodos bio-químicos se realiza a partir de una teoría que propone quelas “balsas lipídicas” son insolubles a detergentes no iónicosa 4°C y que pueden ser separadas por diferencia de densi-dades del resto de la membrana solubilizada. El materialque se obtiene por este método se conoce como “Membra-nas Resistentes a Detergentes” (MRD) y por la modificación dela temperatura y la homogenización celular existe polémicasobre si representan fidedigna y confiablemente las “balsaslipídicas” de la célula viva. Recientemente, gracias a la intro-ducción de la microscopía de luz de súper-resolución (SRLM,por sus siglas en inglés) [6], el análisis de las “balsas lipídicas”en la célula viva fue posible y se han podido observar balsasquiescentes de 10 a 30nm. SRLM incluye técnicas como STED(depleción por emisión estimulada), PALM (microscopía deluz fotoactivada)/STORM (microscopía de reconstrucciónóptica estocástica) y SIM (microscopía de iluminación estruc-turada). La confirmación, por estos métodos ópticos, de quelas proteínas halladas en las MRD se encontraban en domi-nios observados en células vivas, apoyó el modelo de lasbalsas y sus diferentes abordajes de estudio. En las últimasdécadas, el modelo de la membrana compartamentalizada,que inició con el modelo de las “balsas lipídicas”, se extendióa un modelo de mesoescala, donde el citoesqueleto sub-membranal se introdujo como un nuevo participante queforma “bardas” a lo largo de toda la membrana. Así, no sólo
207

los lípidos, sino también las proteínas moldean y organizanlas diferentes regiones y dominios de la membrana plasmá-tica para alcanzar una regulación de alta especificidad.Con el fin de ilustrar cómo las balsas afectan o regulan el
agrupamiento de las proteínas en un proceso específico, semencionará el ejemplo de la sinapsis inmunológica, uno delos primeros modelos de estudio para las balsas lipídicas.Para responder a los agentes externos, nuestro cuerpo cuentacon el sistema inmunológico, el cual es capaz de reconocerepítopes extraños (dominios inmunogénicos de una pro-teína). Las células que realizan esto son el linfocito T y laCélula Presentadora de Antígenos (APC). El epítope es pre-sentado en la membrana de la APC, rodeado por un grancomplejo proteico que también debe ser reconocido juntocon el epítope por la célula T, para que el proceso sea exi-toso. Todo el complejo se agrupa en un punto específico dela membrana gracias a la coalescencia de varias balsas lipí-dicas, tanto en la APC como en el linfocito T, que reúnen a lasproteínas necesarias de una manera eficiente y específica.Las membranas de ambas células se ubican muy cercanaspara la interacción proteica correspondiente, a lo cual se ledenomina sinapsis inmunológica (figura 2). Diversos experi-mentos que utilizan compuestos para manipular las concen-traciones de colesterol y esfingolípidos en la membrana deestas células han demostrado que ambos juegan un papelimportante, no sólo afectando la localización de las proteí-nas, sino también su conformación y función [7].Como se mencionó previamente, cuando se habló de
agrupaciones proteicas en la membrana plasmática, los cana-les iónicos constituyen proteínas protagonistas, puesto queel transporte de iones que ellos regulan participa en unagran cantidad de procesos celulares. En esta última sección,nos concentraremos en los canales de voltaje dependientesde potasio para ilustrar cómo los diferentes compartimentos
208
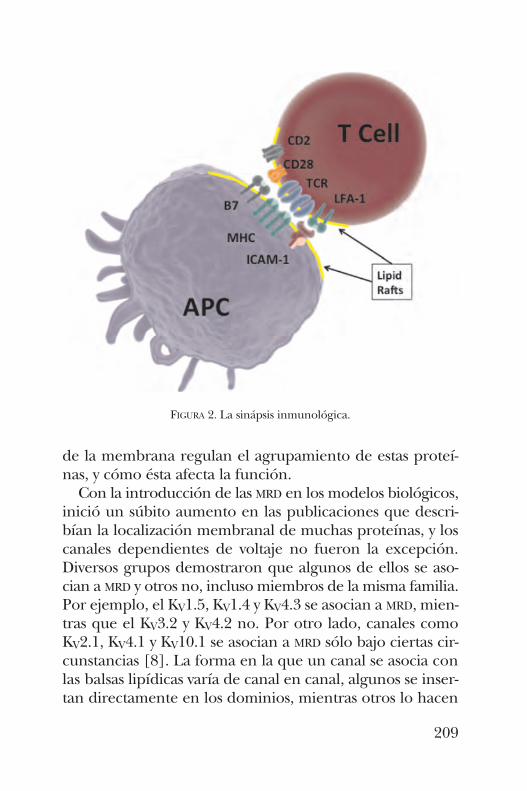
de la membrana regulan el agrupamiento de estas proteí-nas, y cómo ésta afecta la función.Con la introducción de las MRD en los modelos biológicos,
inició un súbito aumento en las publicaciones que descri-bían la localización membranal de muchas proteínas, y loscanales dependientes de voltaje no fueron la excepción.Diversos grupos demostraron que algunos de ellos se aso-cian a MRD y otros no, incluso miembros de la misma familia.Por ejemplo, el KV1.5, KV1.4 y KV4.3 se asocian a MRD, mien-tras que el KV3.2 y KV4.2 no. Por otro lado, canales comoKV2.1, KV4.1 y KV10.1 se asocian a MRD sólo bajo ciertas cir-cunstancias [8]. La forma en la que un canal se asocia conlas balsas lipídicas varía de canal en canal, algunos se inser-tan directamente en los dominios, mientras otros lo hacen
209
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
a
$$
r
$$
,
$$
p
$$
p
$$
q
$$
s
$$
e
$$
c
$$
r
$$
e
$$
m
$$
e
$$
i
$$
i
$$
!
FIGURA 2. La sinápsis inmunológica.

indirectamente a través de interacciones proteicas o lipídicas.La existencia de diferentes compuestos y enzimas que modu-lan las concentraciones de lípidos en la membrana puederomper la estabilidad de los dominios para analizar el efectoen la conformación, función y distribución de las proteínas.En el caso de KV2.1, la formación de agrupaciones es muyclara cuando se observa con el microscopio confocal en con-diciones control, pero después de la extracción de colesterollas agrupaciones ya no pueden ser identificadas y los canalesse difunden por toda la membrana [9]. Finalmente, el ejem-plo del canal KV10.1 demuestra cómo la localización diná-mica del canal constituye una regulación muy específica queejerce la misma membrana plasmática. El caso de KV10.1 esespecialmente interesante porque además de función de con-ductor de iones, también ha sido asociado con un potencialoncogénico cuando se expresa en células no neuronales.Muchos tumores humanos fuera del sistema nervioso cen-tral expresan KV10.1, no así sus contrapartes sanas. A pesar deque todavía existen muchas incógnitas en lo que conciernea su regulación, expresión y vía de promoción de la proli-feración, es una proteína atractiva para ser blanco en el diag-nóstico y tratamiento de procesos proliferativos. La locali-zación de KV10.1 en la membrana plasmática, en neuronascontrol, presenta una distribución bipartita, encontrándose60% dentro de las balsas lipídicas y 40% fuera de ellas. Estadistribución se relaciona con una cantidad específica decorrientes cuando se estudia en sistemas heterólogos (célu-las HEK-293 transfectadas). Cuando la estructura de la mem-brana se afecta con la extracción de colesterol, aproxima-damente 80 a 90% de los canales se localizan fuera de unabalsa lipídica y las corrientes se duplican [8]. En este caso,la balsa lipídica podría proponerse como un regulador nega-tivo de los canales (figura 3). Este hecho se torna interesantesi lo combinamos con su potencial oncogénico, puesto que
210
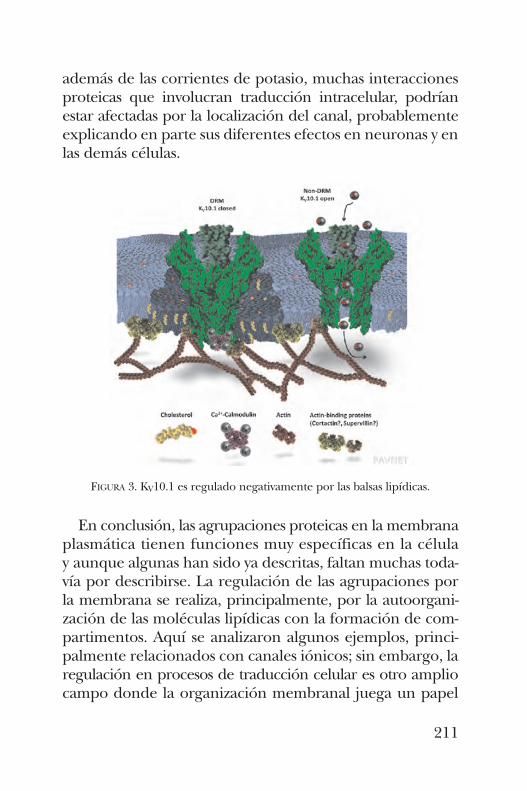
además de las corrientes de potasio, muchas interaccionesproteicas que involucran traducción intracelular, podríanestar afectadas por la localización del canal, probablementeexplicando en parte sus diferentes efectos en neuronas y enlas demás células.
En conclusión, las agrupaciones proteicas en la membranaplasmática tienen funciones muy específicas en la célulay aunque algunas han sido ya descritas, faltan muchas toda-vía por describirse. La regulación de las agrupaciones porla membrana se realiza, principalmente, por la autoorgani-zación de las moléculas lipídicas con la formación de com-partimentos. Aquí se analizaron algunos ejemplos, princi-palmente relacionados con canales iónicos; sin embargo, laregulación en procesos de traducción celular es otro ampliocampo donde la organización membranal juega un papel
211
$$
$$
$$
$$
$$
$$
$$
.
$$
o
$$
.
$$
R
$$
s
$$
o
$$
$$
4
$$
2
$$
,
$$
a
$$
a
$$
a
$$
5
$$
D
$$
,
$$
e
$$
!
FIGURA 3. KV10.1 es regulado negativamente por las balsas lipídicas.

importante. La preservación de las agrupaciones, en losorganismos evolutivamente más antiguos como las bacteriashasta los más complejos, apoya el hecho de que la organi-zación modular juega un papel muy importante en la pre-servación de la vida. Este concepto debe, sin duda, mante-nerse siempre en cuenta cuando se exploren nuevas fun-ciones de proteínas y complejos moleculares.
REFERENCIAS
[1] Hartwell L. H. et al. From molecular to modular cell biology. Nature,1999, 402(6761 Suppl): p. C47-52.
[2] Willig K. I. et al. STEDmicroscopy reveals that synaptotagmin remainsclustered after synaptic vesicle exocytosis. Nature, 2006, 440(7086):935-9.
[3] Rahman T. Dynamic clustering of IP3 receptors by IP3. Biochem. Soc.Trans., 2012, 40(2): 325-30.
[4] O’Connell K. M., Loftus R. y Tamkun M. M. Localization-depen-dent activity of the Kv2.1 delayed-rectifier K+ channel. Proc. Natl.Acad. Sci. USA, 2010, 107(27): 12351-6.
[5] Lingwood D. y Simons K. Lipid rafts as a membrane-organizingprinciple. Science, 2010, 327(5961): 46-50.
[6] Lang T. y Rizzoli S. O. Membrane protein clusters at nanoscale reso-lution: more than pretty pictures. Physiology (Bethesda), 2010,25(2): 116-24.
[7] Irles C., Arias M. J., Guzmán B. J. y Ortega A. Plasma membrane sub-domain partitioning of Lck in primary human T lymphocytes.Can. J. Physiol. Pharmacol., 2010, 88(4): 487-96.
[8] Jiménez-Garduño A. M., Mitkovski M., Alexopoulos I. K., Sánchez A.,Stühmer W., Pardo L. A., Ortega A. Kv10.1 K+-channel plasmamembrane discrete domain partitioning and its functional corre-lation in neurons. BBA Biomembranes, 2013, 1838 : 921-31.
[9] O’Connell K. M. y Tamkun M. M. Targeting of voltage-gated potassiumchannel isoforms to distinct cell surface microdomains. J. Cell Sci.,2005, 118(Pt 10): 2155-66.
212

BIOFÍSICA DE CANALES IÓNICOS SENSIBLESAL VOLTAJE: MODULACIÓN DE CANALES
DE CALCIO POR PROTEÍNAS G
DAVID E. GARCÍAKARINA BERMEOHÉCTOR CASTRO
Departamento de Fisiología Facultad de Medicina
Universidad Nacional Autónoma de Mé[email protected]
INTRODUCCIÓN
Esta revisión, primeramente, hace un bosquejo sobre el ori-gen de las corrientes iónicas sensibles al voltaje mediante lateoría del movimiento de carga y describe la aplicación delmétodo de patch clamp en la comprensión de los procesossubyacentes a la regulación de los canales iónicos. Posterior-mente, expone un ejemplo notable de un modelo de regu-lación de los canales de calcio mediado por proteínas G.Hodgkin y Huxley abrieron la era de la biofísica, en las cien-
cias biomédicas, al establecer las bases para la comprensiónde la excitabilidad y el desarrollo del método de patch clamp(Hodgkin y Huxley, 1952). Después de Hodgkin y Huxley,hubo un estudio exhaustivo de la corriente generada por laapertura y el cierre de los canales para corroborar la presen-cia de moléculas polares en la bicapa de las membranas celu-
213

lares. Esto fue posible mediante el uso de un cable axial parafijar el voltaje a lo largo de la fibra en el músculo (Schneidery Chandler, 1973) y en el axón gigante de calamar (Arms-trong y Bezanilla, 1973), antes de la invención del métodode patch clamp.Durante el estudio de la generación del potencial de
acción en el nervio, Hodgkin y Huxley (1952) predijeron,por primera vez, la existencia de moléculas cargadas cuyomovimiento dipolar las distribuye u orienta, en respuestaa cambios en el campo eléctrico. Se observó que el movi-miento de carga se enmascara casi completamente por lascorrientes iónicas y capacitivas que son, al menos, una ordende magnitud mayor y que únicamente pueden ser detectadasbajo condiciones particulares de experimentación, que con-sisten en bañar constantemente a las células con solucionespropiamente diseñadas para eliminar componentes iónicos,tales como corrientes de sodio, potasio y calcio.Hoy en día es bien aceptado que el movimiento de carga
se correlaciona con la apertura y el cierre de los canales ióni-cos de sodio y potasio (Almers y Armstrong, 1980; Armstrong,1975; Armstrong y Bezanilla, 1974; Bezanilla et al., 1982),que son responsables de los cambios de la permeabilidaddurante un potencial de acción. Los aminoácidos con cargapositiva, que constituyen el sensor de voltaje de los canalesiónicos, se encuentran en el cuarto segmento (S4) de cadadominio de la proteína. Se ha observado que las mutacio-nes de S4 alteran el movimiento de carga y la dependenciade voltaje de los canales (Perozo et al., 1994; Stühmer et al.,1989). Como consecuencia del movimiento del sensor delvoltaje se produce una corriente llamada movimiento de carga(figura 1). Puesto que el movimiento de estas moléculas car-gadas es el primer paso en la traducción de cambios de vol-taje, el movimiento de carga precede a la apertura y sigue alcierre de los canales iónicos.
214

215
MOVIMIENTO DE CARGA
Existe una variedad de funciones fisiológicas reguladas porcambios en el voltaje de membrana a través de diferentes pro-teínas, como los receptores acoplados a proteínas G y aquellasproteínas que están implicadas en mecanismos de transportede membrana (incluidos los canales iónicos, transportadoresy bombas) (Ben-Chaim et al., 2006; Hodgkin y Huxley, 1952).Los eventos moleculares, que enlazan los cambios en el vol-taje de membrana y los cambios estructurales de estas pro-teínas, parecen comenzar con el desplazamiento o reorien-
FIGURA 1. Modelo que resume el origen del movimiento de carga. De acuerdocon el modelo clásico de compuerta (Horn, 2004; Starace y Bezanilla, 2004),una porción estrecha de S4 se encuentra en la compuerta del poro donde elcampo eléctrico de la membrana (área que rodea el sensor de voltaje) ejerce suefecto. La despolarización de la membrana (DV) ejerce una fuerza electrostáticasobre las cargas positivas de S4, que están dentro o cerca del campo eléctricode la membrana y, por lo tanto, pueden detectar cambios en el voltaje (el lla-mado movimiento de carga). Esto produce el movimiento de S4, resultando enuna transferencia de carga a través de la compuerta del poro (modificada deRebolledo-Antúnez et al., 2009).

tación de algunos aminoácidos capaces de detectar el voltajede membrana. Este sensor de voltaje es la maquinaria nece-saria para traducir el potencial de membrana a respuestastales como la conducción del potencial de acción, la libera-ción de neurotransmisores, la contracción muscular, la secre-ción de las células endocrinas y la sensibilidad de unión deagonistas (Almers, 1978). Por lo tanto, la facilidad o dificul-tad del desplazamiento del sensor de voltaje puede regularestas funciones fisiológicas. Desde la invención de la técnica patch clamp por Neher
y Sakmann (Hamill et al., 1981), las corrientes iónicas macros-cópicas pueden ser registradas en neuronas de un diámetropequeño, tales como las neuronas corticales de mamíferos(Edwards et al., 1989), e incluso en terminales sinápticas (Stuarty Sakmann, 1994), con el fin de determinar la sensibilidad alvoltaje de muchos fenómenos fisiológicos. Además, es posi-ble registrar corrientes unitarias de un canal sensible al vol-taje en un pequeño parche en la membrana plasmática. Lascorrientes macroscópicas y unitarias permiten comprenderlos procesos que tienen lugar cuando los canales ya estánabiertos (Hamill y Sakmann, 1981; Sakmann et al., 1980).Cuando se aplica un pulso de voltaje despolarizante (pulso
de prueba) a una célula bañada con la solución que eliminalos componentes iónicos, el registro se compone de unacorriente capacitiva, una pequeña corriente de fuga y el mo -vimiento de carga. Sin embargo, la señal del movimiento decarga no puede ser observada debido al lento componentecapacitivo lineal. Este componente no cambia con el tiemponi con el voltaje y se puede medir desde el potencial de mem-brana en reposo. Aprovechando las propiedades lineales delas corrientes capacitivas y de fuga, el movimiento de cargapuede ser aislado por medio de un protocolo de substracciónaplicado antes o después del pulso de prueba. La figura 2A
216
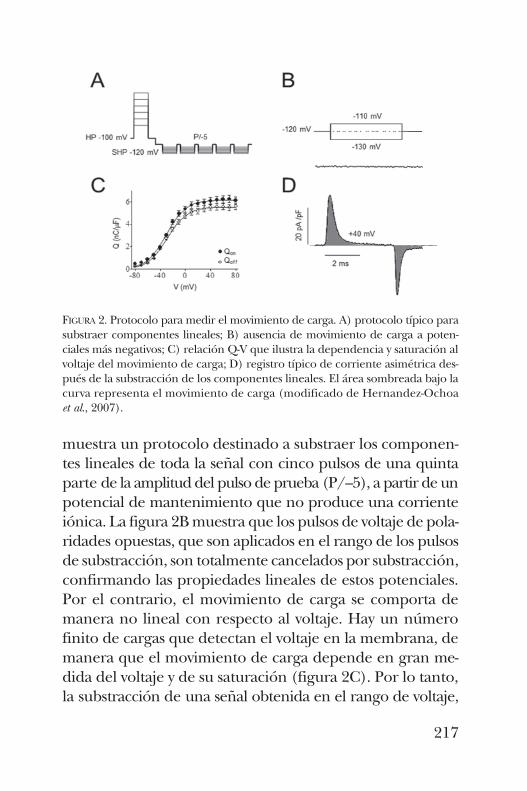
muestra un protocolo destinado a substraer los componen-tes lineales de toda la señal con cinco pulsos de una quintaparte de la amplitud del pulso de prueba (P/–5), a partir de unpotencial de mantenimiento que no produce una corrienteiónica. La figura 2B muestra que los pulsos de voltaje de pola-ridades opuestas, que son aplicados en el rango de los pulsosde substracción, son totalmente cancelados por substracción,confirmando las propiedades lineales de estos potenciales.Por el contrario, el movimiento de carga se comporta demanera no lineal con respecto al voltaje. Hay un númerofinito de cargas que detectan el voltaje en la membrana, demanera que el movimiento de carga depende en gran me -dida del voltaje y de su saturación (figura 2C). Por lo tanto,la substracción de una señal obtenida en el rango de voltaje,
217
FIGURA 2. Protocolo para medir el movimiento de carga. A) protocolo típico parasubstraer componentes lineales; B) ausencia de movimiento de carga a poten-ciales más negativos; C) relación Q-V que ilustra la dependencia y saturación alvoltaje del movimiento de carga; D) registro típico de corriente asimétrica des-pués de la substracción de los componentes lineales. El área sombreada bajo lacurva representa el movimiento de carga (modificado de Hernandez-Ochoaet al., 2007).

donde solamente las corrientes capacitivas y de fuga estánpresentes, nos permite obtener un registro del movimientode carga (figura 2D).
ORIGEN DE LAS CORRIENTES IÓNICAS SENSIBLES A VOLTAJE
Como se dijo antes, el movimiento de carga es de al menosun orden de magnitud menor que las corrientes iónicas ycapacitivas; además, la magnitud de éste depende del númerode sensores de voltaje en movimiento en la membrana (Arms-trong y Bezanilla, 1997; Noceti et al., 1996). Por lo tanto, el sis-tema ideal para estudiar el movimiento de carga es una célulacon una capacitancia pequeña pero con una alta densidadde canales. Como modelos de estudio se han utilizado exito-samente sistemas de expresión, como los oocitos de Xenopus(Bezanilla et al., 1991; Conti y Stühmer, 1989; Neely et al.,1994) o células HEK (Starace et al., 1997), para expresar unsolo tipo de canal.La expresión heteróloga de canales ha dado lugar a avances
significativos en el conocimiento molecular y funcional delmovimiento de carga. La expresión de canales en oocitosde Xenopus ha permitido establecer: 1) que el movimiento decarga es la consecuencia de los cambios conformacionalesde ciertas proteínas (Villalba-Galea et al., 2009), donde cadatransición estructural implica un quantum de carga (Contiy Stühmer, 1989); 2) que el sensor de voltaje de los canaleses de cuatro segmentos (Logothetis et al., 1992; Perozo et al.,1994), y 3) cada transición del estado cerrado al abierto tieneuna única constante de velocidad (Schoppa y Sigworth, 1998). Por otra parte, registrar el movimiento de carga en células
nativas tiene la ventaja de estudiar los fenómenos relacio-nados en condiciones inalteradas. Sin embargo, el estudiodel movimiento de carga en células nativas se complica por
218

la presencia de diferentes tipos de canales, lo que hace difí-cil saber la contribución de cada tipo de canal en el registrode movimiento de carga total (Chameau et al., 1995). En elcaso de la preparación clásica de los axones de calamar, elestudio del movimiento de carga no representa un proble-ma, debido a la presencia de sólo dos poblaciones de cana-les a muy altas densidades, sodio y potasio, que podrían serseparadas fácilmente por su cinética y cambios en la tem-peratura (Bezanilla, 1985; Gilly y Armstrong, 1980). En con-traste, en mamíferos se encontró que en el movimiento decarga de células cardiacas ventriculares, participan pobla-ciones de canales de sodio y calcio, las cuales no podrían serseparadas (Bean y Ríos, 1989).En un reporte anterior mostramos cómo aislar e identificar
los canales que están contribuyendo al movimiento de cargatotal, en neuronas del ganglio cervical superior (SCG), me -diante dos enfoques diferentes. El primer enfoque analiza lacinética del movimiento de carga donde la hipótesis es quesi los canales de sodio (NaV), calcio, (CaV) y potasio (KV(A)),con diferentes constantes de tiempo, están contribuyendo almovimiento de carga, cada componente debe observarsepor separado en el tiempo. Este enfoque es esencialmenteuna separación cinética del movimiento de carga y clásica-mente se ha utilizado el músculo esquelético para su estu-dio (Adrián y Peres, 1979; Francini et al., 2001; Huang,1982; Huang, 1988). Un análisis adicional compara la inmovilización de la carga
con la inactivación rápida sensible a voltaje de corrientes ióni-cas, tales como INa o IK(A) (Armstrong y Bezanilla, 1977; Beza-nilla, 2000). La inmovilización dependiente de voltaje o detiempo conduce a diferencias en la carga entre la despola-rización (Qon) o la repolarización (Qoff). Para probar esto,la dependencia de voltaje de la inmovilización de la cargaes comparada con la dependencia de voltaje de la inactiva-
219
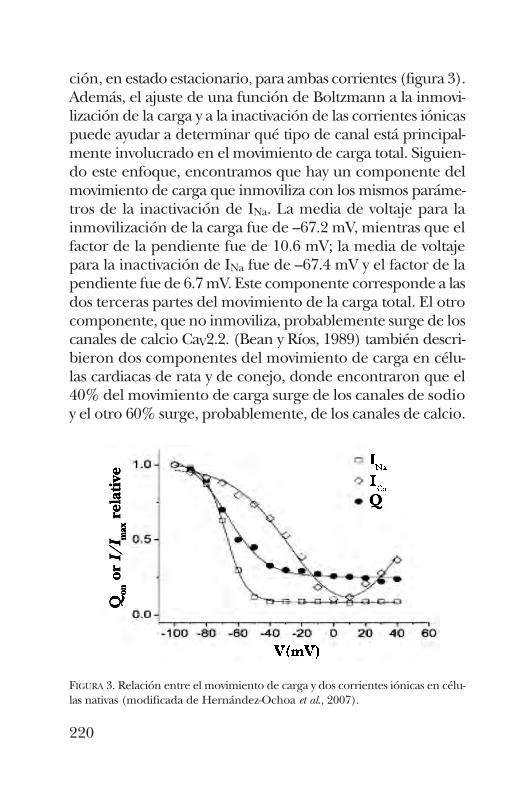
ción, en estado estacionario, para ambas corrientes (figura 3).Además, el ajuste de una función de Boltzmann a la inmovi-lización de la carga y a la inactivación de las corrientes iónicaspuede ayudar a determinar qué tipo de canal está principal-mente involucrado en el movimiento de carga total. Siguien -do este enfoque, encontramos que hay un componente delmo vimiento de carga que inmoviliza con los mismos paráme-tros de la inactivación de INa. La media de voltaje para lainmovilización de la carga fue de –67.2 mV, mientras que elfactor de la pendiente fue de 10.6 mV; la media de voltajepara la inactivación de INa fue de –67.4 mV y el factor de lapendiente fue de 6.7 mV. Este componente corresponde a lasdos terceras partes del movimiento de la carga total. El otrocomponente, que no inmoviliza, probablemente surge de loscanales de calcio CaV2.2. (Bean y Ríos, 1989) también descri-bieron dos componentes del movimiento de carga en célu-las cardiacas de rata y de conejo, donde encontraron que el40% del movimiento de carga surge de los canales de sodioy el otro 60% surge, probablemente, de los canales de calcio.
220
FIGURA 3. Relación entre el movimiento de carga y dos corrientes iónicas en célu-las nativas (modificada de Hernández-Ochoa et al., 2007).

El segundo enfoque utiliza un análisis de fluctuacionesno estacionarias de las corrientes. Este análisis se puede utili-zar para estimar el número de canales en el cuerpo celular(Álvarez et al., 2002; Sigworth, 1980) y comparar la cargamáxima transferida durante el movimiento de carga (Qmax)con la carga transferida mediante un tipo de canal. Para estefin, las corrientes se generan a través de una serie de 80 a 200pulsos de voltaje idénticos y consecutivos. El curso temporalde la varianza (s2) y la media de las corrientes se calcula.Dado que los cambios en s2 surgen no sólo de la compuertaestocástica de los canales, sino también del ruido de fondotérmico, el curso temporal de s2 en el potencial de manteni-miento se resta. Después de la corrección de s2 se puedemontar la siguiente ecuación:
s2 = i(I ) – (I )2/N
Donde I es la media de la corriente, i es la corriente unita-ria y N es el número de canales. Esta teoría se basa en tressupuestos generales acerca de los canales dependientes devoltaje: 1) los canales se componen de una población; 2) la po -blación de canales tiene un número fijo de compuertas decanal independiente y 3) se asume que los canales que exis-ten se encuentran en un estado conductor o de no conducción.Cuando la probabilidad en el estado de conducción es la
máxima, la teoría predice una relación parabólica entre s2
y la media de la corriente que se puede utilizar para estimarel número de canales y la amplitud de la corriente unitaria.En neuronas SCG de 40 pF se encontraron 3.8 × 104 de los ca -nales de calcio en el cuerpo celular. Considerando que 12 car-gas elementales (e0) equivalen al movimiento de carga de uncanal (Noceti et al., 1996) y que Qmax es igual a 6.2 nC/cm2
o, traducido en cargas elementales, 390 e0 serían 32 cana-les/µm2 por neurona. Por lo tanto, el movimiento de carga
221

derivada de los canales de calcio comprende, aproximada-mente, un tercio del movimiento de carga total. Este valorse correlaciona con la primera aproximación en la que dostercios del movimiento de carga total corresponden a cana-les NaV, mientras que otro tercio a los canales CaV.
REGULACIÓN DE CANALES DE CALCIO POR PROTEÍNAS G
Existen varios estados cerrados antes de la apertura de uncanal y hasta la fecha, los procesos que subyacen a estosestados están parcialmente entendidos. Es por esto, que elmovimiento de carga y la regulación extrínseca por proteí-nas G, que ocurre durante los estados cerrados, deben serestudiados. Los canales de calcio, de potasio y de sodio sensibles al vol-
taje son modulados por neurotransmisores (Hille, 1994; Ikeday Dunlap, 1999). Ellos ejercen su efecto mediante la activaciónde los receptores acoplados a proteínas G (GPCRs). La activa-ción de GPCRs por neurotransmisores puede activar varias víasde señalización que tienen efectos pleiotrópicos sobre las neu-ronas (Hille, 1994; Hille et al., 1995). La molécula efectora dela regulación de los canales iónicos ha sido identificada y es lasubunidad bg de las proteínas G (Gbg) (Herlitze et al., 1996;Ikeda, 1996). La unión deGbg al canal induce cambios carac-terísticos como los siguientes: disminuye la amplitud de lacorriente (Dunlap y Fischbach, 1981), altera la dependenciaal voltaje del canal (Bean, 1989) y enlentece la cinética de acti-vación de la corriente (Marchetti et al., 1986); de manera queel efecto es reversible o sensible y puede ser separado de loscanales por una fuerte despolarización (Elmslie et al., 1990;Grassi y Lux, 1989). Sin embargo, todavía es controvertidosi el dímeroGbgdisminuye el número de canales disponiblespor un cambio en la conductancia unitaria o por la modifi-cación de la estructura del canal.
222

Las corrientes iónicas pueden abolirse mediante la susti-tución de los iones permeables, en ambos lados de la mem-brana, por compuestos que no pasan a través de los canalespresentes en la membrana. Algunas sustituciones típicas sonel tetraetilamonio (TEA), N-metilglucamina (HMG) y algunoscationes orgánicos, tales como aspartato o glutamato y cesio.Además de sustituir los iones permeables, es común el usode bloqueadores, tales como la tetrodotoxina (TTX), nifedi-pina y el cadmio. Se debe considerar el efecto que tienenalgunos iones o bloqueadores sobre el movimiento del sen-sor de voltaje, ya que daría lugar a una mala interpretación delos resultados. Por ejemplo, hemos diseñado una solucióninterna para estudiar el movimiento de carga en las neuro-nas SCG después de varios ensayos y errores experimentales; seha diseñado con cesio como el ion no permeable en la solu-ción interna. Esta solución mejora el sellado e incrementa laduración del experimento mediante el bloqueo de las corrien-tes iónicas sin alterar la cinética o la dependencia de voltajedel movimiento de carga. Adicionalmente, TTX, cadmio y lan-tano se añaden a la solución externa para bloquear corrien-tes iónicas residuales (Hernández-Ochoa et al., 2007).Se ha propuesto que la regulación de la proteína G se lleve
a cabo en los estados cerrados del canal que transitan haciasu apertura (Bean, 1989; Boland y Bean, 1993). El registro enconfiguración de célula completa ha sido la técnica más fre-cuente utilizada para estudiar el efecto de las proteínas Gsobre los canales después de que se han abierto; sin embargo,los canales están ya regulados antes de que el canal se abra.Por lo tanto, el efecto de las proteínas G durante los estadosde transición de los canales, todavía no está claro. Dado quela transición entre los estados cerrados implica el movimien-to del sensor de voltaje, la regulación de los canales iónicos enestos estados se ha entendido mejor a través de registros delmovimiento de carga. La hipótesis de que los canales iónicosson regulados durante los estados cerrados es fuertemente
223

apoyada por el hallazgo de que la activación de las proteí-nas G reduce el movimiento de carga (Hernández-Ochoaet al., 2007). La diálisis de GTPgS, un activador no hidroli-zable de las proteínas G, tiene tres efectos principales en ladependencia de voltaje de la carga total en las neuronas SCG:1) una disminución del 34% en Qmax, 2) un corrimientohacia potenciales más positivos de la dependencia de voltajede 10 mV, y 3) un aumento del 63% en la pendiente. Estasobservaciones sugieren que la activación no selectiva de laproteína G modifica tanto la dependencia de voltaje comoel número de canales disponibles presentes en las neuronasSCG. Además, la activación de una vía de señalización especí-fica, mediante la aplicación de neurotransmisores, tales comola noradrenalina (NA) o la angiotensina II, también reduceel movimiento de carga. Los cambios en el movimiento de carga, bajo condiciones
de modulación, sugieren que el sensor de voltaje de los cana-les se altera en tales condiciones. Sin embargo, ¿cómo puedenmodificar las proteínas G la actividad del sensor de voltaje?Se han propuesto diferentes mecanismos para la modula-ción de las propiedades de los sensores de voltaje medianteel estudio del movimiento de carga.La inmovilización de carga está relacionada con la inac-
tivación de los canales, un proceso intrínseco de regulaciónde algunos canales iónicos sensibles al voltaje (Armstrongy Bezanilla, 1977; Armstrong et al., 1973; Bezanilla y Arms-trong, 1977; Bezanilla et al., 1991; Nonner, 1980); sin em -bargo, la inactivación parece no ser el único proceso en elcual se inmoviliza carga. Recientemente se ha demostradoque la modificación del entorno lipídico induce la inmovi-lización de carga en los canales de potasio. La eliminaciónenzimática de grupos fosfato de esfingomielina disminuyeel movimiento de carga en un 90% sin cambios en la depen-dencia de voltaje (Xu et al., 2008); además, diversas toxinas
224

alteran el movimiento de la carga (Sheets et al., 1999) restrin-giendo los cambios conformacionales entre los estados detransición (Catterall et al., 2007; Sokolov et al., 2008). Estemecanismo se denomina “atrapamiento” o “captura” que, entérminos moleculares, se puede interpretar como la reten-ción del sensor de voltaje en posición activa o de reposo.Las toxinas pueden inhibir la activación, la desactivación o lainactivación de canales iónicos, ya que atrapan el canal enalguna de sus tres conformaciones: abierto, cerrado o inactivo.Otro proceso que modula los canales iónicos, principal-
mente los canales de potasio, es la fosforilación (Kaczmarek,1988; Levitan, 1985), que reduce el movimiento de cargay modifica la dependencia de voltajes potenciales positivos(Augustine y Bezanilla, 1990). La modulación de los cana-les de potasio está mediada por interacciones electrostáticasentre grupos del sensor de voltaje y grupos fosfato (Perozoy Bezanilla, 1990).Otras moléculas que afectan al movimiento de carga son
las subunidades accesorias de los canales iónicos y los com-plejos de rutenio. Mientras que las subunidades accesoriasde los canales de calcio mejoran el acoplamiento entre laexcitación y la apertura (Lacinova y Klugbauer, 2004), loscomplejos de rutenio desacoplan el movimiento del sensorde voltaje en la transición de apertura (Jara-Oseguera et al.,2011). Todos estos posibles cambios: inmovilización, capturao atrapamiento, selección, acoplamiento o desacoplamiento,son ejemplos de cómo los canales iónicos pueden ser modu-lados por medio de cambios en las propiedades del movi-miento de carga. Es así como el registro del movimiento decarga es una herramienta para entender cómo se modulanlos canales iónicos. Actualmente se desconoce el mecanismo que explica la
inhibición del movimiento de carga por las proteínas G enneuronas SCG; sin embargo, se sabe que existe una altera-
225

ción de la dependencia de voltaje del movimiento de cargay una reducción de la carga efectiva (Rebolledo-Antúnez et al.,2009). Esta reducción de la carga efectiva, que se transfieredurante la activación del canal con un cambio de potencial(figura 4), apoya la idea de que las proteínas G ejercen unamodulación alostérica del movimiento de carga, condiciónen la que los sensores de voltaje responden diferencialmentea los cambios del potencial de membrana (Herlitze et al.,2001; Monod et al., 1965).
226
FIGURA 4. Modelo alostérico para explicar la modulación del movimiento de cargapor proteínas G. Este modelo postula que la modulación alostérica del movi-miento de carga, por la subunidad Gbg, es mediada por una redistribución delcampo eléctrico local que rodea al sensor de voltaje (mostrado como un cambioen el patrón de color) (modificado de Rebolledo-Antúnez et al., 2009).

LOS CANALES DE CALCIO SENSIBLES AL VOLTAJE SON REGULADOS
POR LA SUBUNIDAD b DE LAS PROTEÍNAS G
Las subunidades Gbg de las proteínas G heterotriméricasson importantes reguladores de los canales iónicos sensi-bles al voltaje (Agler et al., 2005; Currie, 2010) y juegan unpapel crítico en el acoplamiento de los GPCRs con sus diver-sos efectores, incluyendo las isoformas g de la fosfolipasa C y dela fosfatidilinositol 3-cinasa (Clapham y Neer, 1997; Tedfordy Zamponi, 2006). Aunque Gbg puede activar directamentemuchos de estos efectores a través de interacciones proteí-na-proteína in vitro, no queda claro cómoGbg coordina espa-cial y temporalmente la activación de un buen número deefectores in vivo. Una vía en común por la que afectan la fun-ción celular es a través de la inhibición sensible al voltaje delos canales Cav2.2 (Marchetti et al., 1986; Bean, 1989; Ikeday Schofield, 1989; Elmslie et al., 1990; Hille, 1994). Los canalesCav2.2, a menudo traducen, una señal eléctrica en una señalquímica en las células excitables y su regulación puede modu-lar la comunicación intercelular. La regulación del canal decalcio, producida por neurotransmisores, se realiza a travésde la activación de GPCRs (Rosenthal et al., 1988), un ejemplonotable es la NA; sin embargo, no se sabe qué moléculas deter-minan la especificidad de un neurotransmisor en la regula-ción sensible al voltaje. Como las subunidades Gb mediandirectamente la inhibición sensible al voltaje de los canalesCav2.2 (Herlitze et al., 1996; Ikeda, 1996), una posibilidades que las subunidades Gbmedien esta especificidad. Ante-riormente se ha mostrado que las corrientes Cav2.2 en neu-ronas SCG de rata presentan un enlentecimiento cinéticoy una facilitación sensible al voltaje después de la sobreexpre-sión deGb1 oGb2, similar a la producida por NA (García et al.,1998), lo que sugiere que la inhibición inducida por NANA esprincipalmente a través de Gb1 y/o subunidades Gb2. Sin
227

embargo, no se ha examinado si algunas subunidades Gbllevan la información de un neurotransmisor específico queinduce una regulación sensible al voltaje y un enlentecimien-to cinético. Se ha encontrado que los canales Cav2.2 soncaracterísticamente modulados por cada isoforma de la sub -unidad Gb y que los cambios en la velocidad de activaciónde la corriente, y los cambios de la población willing-reluctantproducidos por NA, son imitados por la sobreexpresión de laisoforma Gb2. Estos resultados contribuyen a comprenderel mecanismo por el cual las subunidades Gbmedian espe-cíficamente la inhibición de la corriente Cav2.2 producida porun neurotransmisor (Hernández-Castellanos et al., 2014).Varios neurotransmisores y hormonas, que actúan a través
de receptores acoplados a proteínas G provocando una regu-lación sensible al voltaje de los canales Cav2.2, tienen pro-fundos efectos en la función celular y en el organismo. Seha planteado la hipótesis de que las interacciones proteína-proteína definen la especificidad en la traducción de señal;sin embargo, se desconoce cómo las interacciones molecu-lares, en una cascada de señalización intracelular, determinanla especificidad de la regulación sensible al voltaje producidapor un neurotransmisor específico. Se ha sugerido la exis-tencia de regiones efectoras específicas de las subunidadesGb,de las proteínas G, que son responsables de la regulaciónsensible al voltaje. Estos estudios contribuyen a comprenderlos mecanismos que permiten respuestas homeostáticas pre-cisas a partir de la activación de una variedad de receptoresde membrana.Con el fin de evaluar las capacidades de los diferentes
subtipos Gb para modular los canales de calcio, se ha explo-rado el enlentecimiento cinético típico de la corriente CaV2.2producido por NA en las neuronas que sobreexpresan dife-rentes subunidades Gb. La figura 5A muestra trazos decorriente, superpuestos y obtenidos con un protocolo de dos
228

pulsos separados por aplicación de una fuerte despolariza-ción (+80 mV), llamada prepulso (PP). En la ausencia de so -breexpresión de Gb en neuronas SCG, es decir, en condiciónde control, la corriente de calcio fue inhibida por la aplica-ción de NA y después de la aplicación de PP se observó la des -inhibición típica de la corriente (figura 5Aa). La figura 5Ab-fmuestra ejemplos de las diferentes cinéticas producidas pordiferentes subunidades Gb, siendo Gb2 la que produjo la
229
FIGURA 5. Las subunidades Gb producen diferentes cinéticas sobre la activaciónde Cav2.2. A) trazos de corriente superpuestos fueron generados con un par depulsos despolarizantes a –8 mV (P1, P2), a partir de un potencial de manteni-miento de –80 mV, separados por un prepulso (PP) de +80 mV durante 50 ms(protocolo de doble pulso, sobre los trazos de corriente en Aa). Las corrientesde Ba2+ se registraron en ausencia (trazos inferiores, Pre-NA) y en presencia (tra-zos superiores, NA) de 10 µM de NA para neuronas control (Aa) y neuronas quesobreexpresaron Gb1g3 (Ab), Gb2g3 (Ac), Gb3g3 (Ad), Gb4g3 (Ae) y Gb5g3 (Af);B) gráfica de cinéticas de activación de corrientes de Ba2+. Promedio de los pri-meros 10 ms de los trazos de corriente (P1) para neurona control ( ), célulasque sobreexpresaron Gb5g3 ( ), y Gb2g3 (D) (media ± SEM, n = 4). Los datos seajustaron a una función de decaimiento exponencial; las líneas continuas indi-can los mejores valores de ajuste.

mayor oclusión del efecto producido por NA, mientras queGb5 no tuvo ningún efecto significativo (figura 5Ac-f). La cons -tante de tiempo de activación obtenida en condiciones con-trol (figura 5B) y la obtenida con la sobreexpresión de Gb5fueron muy similares (1.56 ± 0.47 vs 1.85 ± 0.15 ms, respecti-vamente), mientras que la constante de tiempo de activaciónde Gb2 fue mucho más lenta de manera significativa (2.76 ±0.25 ms). Hasta la fecha no queda claro si la activación de un GPCR
específico por un neurotransmisor libera una cierta combi-nación de subunidades Gbg para estimular una vía de seña-lización en particular (García et al., 1998; Ruiz-Velasco y Ikeda,2000; Krumins y Gilman, 2006). A fin de abordar este tema,se ha investigado qué subunidad imita el enlentecimientocinético de los canales de calcio producido por NA. Se hancomparado las constantes de activación de Gb1-Gb4 con lasobtenidas mediante la aplicación de NA. La figura 6A ilustrael enlentecimiento cinético producido por Gb2, comparadocon el obtenido en presencia de 10 mM de NA. Como se pue-de observar, la sobreexpresión de subunidades Gb2 imita elenlentecimiento cinético observado en los canales de calciopor NA. El ajuste monoexponencial del curso temporal dela activación dio valores t de activación para el control: 1.56± 0.47, 3.22 ± NA: 0.4 y G�2: 2.76 ± 0.25 (figura 6B). Esteresultado muestra el mimetismo y la oclusión de las respues-tas de NA por la sobreexpresión de Gb2.El modelo propuesto por Bean (Bean, 1989) sigue siendo
el mejor modelo para explicar cómo los neurotransmisoresregulan los canales de calcio. Este modelo plantea que haydos estados funcionales de los canales de calcio: los canaleswilling no regulados y los canales reluctant regulados por pro-teínas G, dicha regulación es liberada en respuesta a unafuerte despolarización. La principal característica de esta regu-lación es el enlentecimiento cinético (Hille, 1994; Dunlap,
230
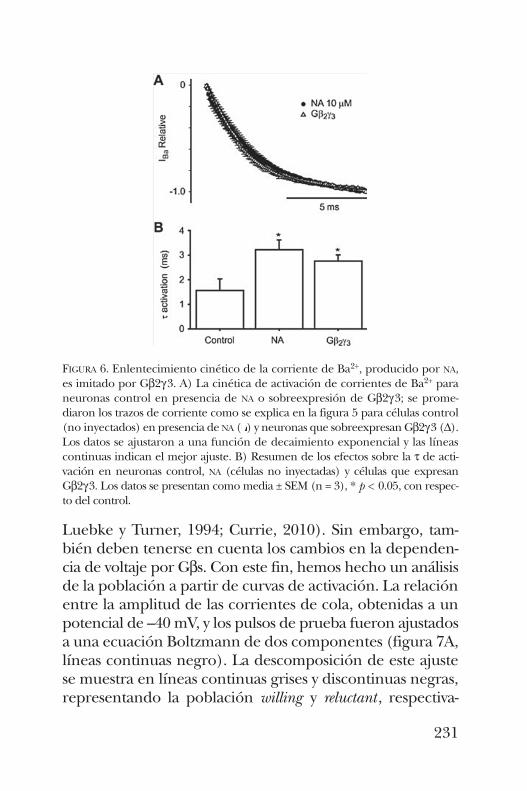
Luebke y Turner, 1994; Currie, 2010). Sin embargo, tam-bién deben tenerse en cuenta los cambios en la dependen-cia de voltaje por Gbs. Con este fin, hemos hecho un análisisde la población a partir de curvas de activación. La relaciónentre la amplitud de las corrientes de cola, obtenidas a unpotencial de –40 mV, y los pulsos de prueba fueron ajustadosa una ecuación Boltzmann de dos componentes (figura 7A,líneas continuas negro). La descomposición de este ajustese muestra en líneas continuas grises y discontinuas negras,representando la población willing y reluctant , respectiva-
231
FIGURA 6. Enlentecimiento cinético de la corriente de Ba2+, producido por NA,es imitado por Gb2g3. A) La cinética de activación de corrientes de Ba2+ paraneuronas control en presencia de NA o sobreexpresión de Gb2g3; se prome-diaron los trazos de corriente como se explica en la figura 5 para células control(no inyectados) en presencia de NA ( ) y neuronas que sobreexpresan Gb2g3 (D).Los datos se ajustaron a una función de decaimiento exponencial y las líneascontinuas indican el mejor ajuste. B) Resumen de los efectos sobre la t de acti-vación en neuronas control, NA (células no inyectadas) y células que expresanGb2g3. Los datos se presentan como media ± SEM (n = 3), * p < 0.05, con respec-to del control.

mente (figura 7). El análisis de la población de las colas decorriente se muestra en el pulso 1 (P1) y en el pulso 2 (P2)para las neuronas en condiciones control, durante la apli-cación de NA y en las neuronas que sobreexpresan Gb2. Laaplicación de 10 µM de NA aumentó la población reluctantconcomitantemente y disminuyó la población willing en elprimer pulso. Después del prepulso para liberar la regula-ción sensible al voltaje, el componente willing aumentó y lafracción reluctant disminuyó en el segundo pulso. La inhibi-ción constitutiva sensible al voltaje por las proteínas G y suliberación por fuertes despolarizaciones en neuronas delSCG están bien documentadas (Elmslie et al., 1990; Garcíaet al., 1998). Notablemente, la sobreexpresión de Gb2 imitael efecto de NA en el pulso 2 (figura 7B). Estos resultados su -gieren que cada isoforma de Gb ejerce efectos diferencialessobre las poblaciones del canal que se pueden reflejar, no sólocomo diferencias en las constantes de asociación, sino tam-bién en la unión sensible al voltaje.Miles de botones de las terminales presinápticas conver-
gen en las dendritas celulares y el soma, liberando neuro-transmisores para provocar una variedad de respuestas deseñalización intracelular. Notablemente, las interaccionespro teína-proteína y la especificidad son fenómenos estre-chamente relacionados (Pawson y Nash, 2000). Como resul-tado, las acciones sobre efectores se conservan en gran me -dida para que las respuestas homeostáticas estén coordinadasoportuna y espacialmente. Sin embargo, no queda claro cómoun neurotransmisor promueve la activación de una ciertacombinación de subunidades Gbg para generar una respuestaparticular por una vía de señalización. Existen varios repor-tes que están de acuerdo en que NA produce principalmenteuna inhibición sensible al voltaje de los canales Cav2.2 (Hille,1994). Aquí encontramos que la sobreexpresión de Gb2 imitalas acciones biofísicas producidas por NA, como el enlente-
232
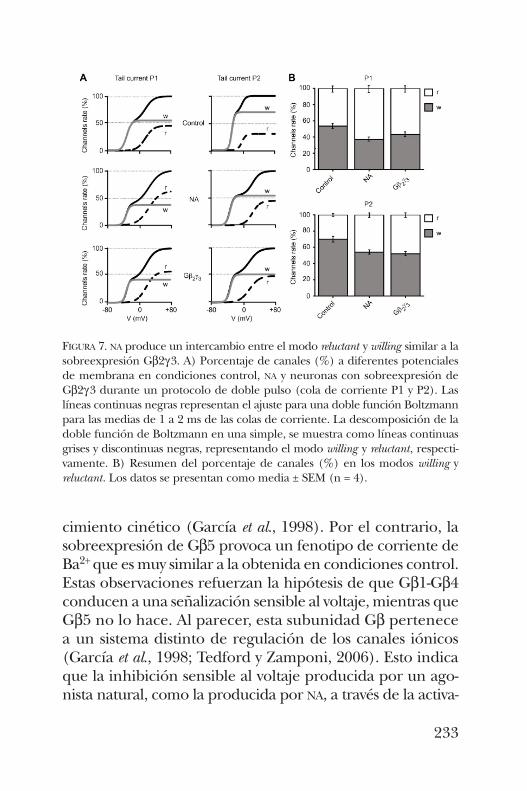
cimiento cinético (García et al., 1998). Por el contrario, lasobreexpresión de Gb5 provoca un fenotipo de corriente deBa2+ que es muy similar a la obtenida en condiciones control.Estas observaciones refuerzan la hipótesis de que Gb1-Gb4conducen a una señalización sensible al voltaje, mientras queGb5 no lo hace. Al parecer, esta subunidad Gb pertenecea un sistema distinto de regulación de los canales iónicos(García et al., 1998; Tedford y Zamponi, 2006). Esto indicaque la inhibición sensible al voltaje producida por un ago-nista natural, como la producida por NA, a través de la activa-
233
FIGURA 7. NA produce un intercambio entre el modo reluctant y willing similar a lasobreexpresión Gb2g3. A) Porcentaje de canales (%) a diferentes potencialesde membrana en condiciones control, NA y neuronas con sobreexpresión deGb2g3 durante un protocolo de doble pulso (cola de corriente P1 y P2). Laslíneas continuas negras representan el ajuste para una doble función Boltzmannpara las medias de 1 a 2 ms de las colas de corriente. La descomposición de ladoble función de Boltzmann en una simple, se muestra como líneas continuasgrises y discontinuas negras, representando el modo willing y reluctant, respecti-vamente. B) Resumen del porcentaje de canales (%) en los modos willing yreluctant. Los datos se presentan como media ± SEM (n = 4).

ción de GPCRs en neuronas del SCG de rata, puede ser mediadapor una subunidad Gb en particular. Ciertamente, también seespera que una serie de Gbs, en lugar de una en particular,podría ser responsable de dicha regulación. Anteriormente,nosotros reportamos por primera vez que la sobreexpresiónde la subunidad Gb2 inhibe la corriente Cav2.2, aumenta elíndice de facilitación y ocluye parcialmente el efecto de NA;además, Gbs no es capaz de reproducir la regulación sensibleal voltaje de los canales Cav2.2 (Herlitze et al., 1996). En gene-ral, la sobreexpresión de Gb1-Gb4 produce efectos cualitati-vamente similares; en contraste, Gb5 sólo efectos débiles (Ruiz-Velasco e Ikeda, 2000). Esta observación se correlaciona biencon el porcentaje de identidad entre las diferentes Gbs (Yanet al., 1996; Clapham y Neer, 1997). De acuerdo con nues-tra hipótesis actual, cada isoforma de la subunidad Gb pro-voca una inhibición diferencial sensible al voltaje sobre lascorrientes de calcio, caracterizada por el enlentecimientocinético, aumento del índice de facilitación y la oclusión deagonista, excepto para Gb5. También abre la posibilidadde que la regulación sensible al voltaje, producida por dife-rentes neurotransmisores, pueda estar mediada por diferen-tes combinaciones de subunidades Gb que llevan la especifi-cidad de la modulación sensible al voltaje para cada tipo decanal iónico, enzima o transportador. Por lo tanto, los futu-ros experimentos deben dirigirse a estudiar otros GPCRs conuna amplia variedad de neurotransmisores. Además, debe-rían ser consideradas las diferentes constantes de disociaciónde las isoformas de las subunidades Gb de los GPCRs. En con-clusión, una posibilidad interesante para explicar la variedadde funciones vitales del organismo, a partir de un númerolimitado de elementos que participan en la señalización ycómo éstas conservan su especificidad a partir de la regula-ción sensible al voltaje, también, es la variedad de posiblesprogramas de subunidades Gb. Así, la especificidad de la
234

señalización intracelular para una amplia gama de neuro-transmisores y hormonas es un mecanismo fundamental,cuyas respuestas adecuadas a cada condición dependen delestado computacional neuronal de los eventos espacio-tem-porales que mantienen el equilibrio homeostático del orga-nismo (Hille, 1994).
AGRADECIMIENTOS
Este trabajo fue apoyado por una beca de UNAM-DGAPA-PAPIIT IN215813 y The Alexander von Humboldt Stiftung,Germany, to DEG.
REFERENCIAS
Adrián R. H. y Peres A. Charge movement and membrane capacity infrog muscle. J. Physiol., 1979, 289 : 83-97.
Agler H. L., Evans J., Tay L. H., Anderson M. J., Colecraft, H. M. y Yue, D.T. G protein-gated inhibitory module of N-type (ca(v)2.2) ca2+channels. Neuron., 2005, 46(6): 891-904.
Almers W. Gating currents and charge movements in excitable membranes.Rev. Physiol. Biochem. Pharmacol., 1978, 82 : 96-190.
Almers W. y Armstrong C. M. Survival of K+ permeability and gatingcurrents in squid axons perfused with K+-free media. J. Gen. Physiol.,1980, 75 : 61-78.
Álvarez O., González C. y Latorre R. Counting channels: a tutorial guideon ion channel fluctuation analysis. Adv Physiol Educ., 2002, 26 :327-341.
Armstrong C. M. Currents associated with the ionic gating structures innerve membrane. Ann. N. Y. Acad. Sci., 1975, 264 : 265-77.
Armstrong C. M. y Bezanilla F. Currents related to movement of thegating particles of the sodium channels. Nature, 1973, 242 : 459-61.
Armstrong C. M. y Bezanilla F. Charge movement associated with theopening and closing of the activation gates of the Na channels.J. Gen. Physiol., 1974, 63 : 533-52.
235

Armstrong C. M. y Bezanilla F. Inactivation of the sodium channel.II. Gating current experiments. J. Gen. Physiol., 1977, 70 : 567-590.
Armstrong C. M., Bezanilla F. y Rojas E. Destruction of sodium con-ductance inactivation in squid axons perfused with pronase. J. Gen.Physiol., 1973, 62 : 375-391.
Augustine C. K. y Bezanilla F. Phosphorylation modulates potassiumconductance and gating current of perfused giant axons of squid.J. Gen. Physiol., 1990, 95 : 245-271.
Bean B. P. Neurotransmitter inhibition of neuronal calcium currents bychanges in channel voltage dependence. Nature, 1989, 340: 153-156.
Bean B. P. y Ríos E. Nonlinear charge movement in mammalian cardiacventricular cells. Components from Na and Ca channel gating.J. Gen. Physiol., 1989, 94 : 65-93.
Ben-Chaim Y., Chanda B., Dascal N., Bezanilla F., Parnas I. y Parnas H.Movement of ‘gating charge’ is coupled to ligand binding in aG-protein-coupled receptor. Nature, 2006, 444 : 106-9.
Bezanilla F. Gating of sodium and potassium channels. J. Membr. Biol.,1985, 88 : 97-111.
Bezanilla F. The voltage sensor in voltage-dependent ion channels.Physiol. Rev., 2000, 80 : 555-592.
Bezanilla F. y Armstrong C. M. Inactivation of the sodium channel.I. Sodium current experiments. J. Gen. Physiol., 1977, 70 : 549-566.
Bezanilla F., Perozo E., Papazian D. M. y Stefani E. Molecular basis ofgating charge immobilization in Shaker potassium channels.Science, 1991, 254 : 679-683.
Bezanilla F., White M. M. y Taylor R. E. Gating currents associated withpotassium channel activation. Nature, 1982, 296 : 657-9.
Boland L.M. y Bean B. P. Modulation of N-type calcium channels in bull-frog sympathetic neurons by luteinizing hormone-releasing hor-mone: kinetics and voltage dependence. J. Neurosci., 1993, 13: 516-533.
Catterall W. A., Cestele S., Yarov-Yarovoy V., Yu F. H., Konoki K. yScheuer T. Voltage-gated ion channels and gating modifier toxins.Toxicon., 2007, 49 : 124-141.
Chameau P., Bournaud R. y Shimahara T. Asymmetric intramembranecharge movement in mouse hippocampal pyramidal cells. Neurosci.Lett., 1995, 201 : 159-162.
Clapham D. E. y Neer E. J. G protein beta gamma subunits. Annu RevPharmacol. Toxicol., 1997, 37 : 167-203.
236

Conti F. y Stühmer W. Quantal charge redistributions accompanyingthe structural transitions of sodium channels. Eur. Biophys. J., 1989,17 : 53-9.
Currie K. P. G-protein modulation of CaV2 voltage-gated calciumchannels. Channels (Austin), 2010, 4(6): 497-509.
Dunlap K. y Fischbach G. D. Neurotransmitters decrease the calciumconductance activated by depolarization of embryonic chick sen-sory neurones. J. Physiol., 1981, 317 : 519-35.
Dunlap K., Luebke J. I. y Turner T. J. Identification of calcium channelsthat control neurosecretion. Science, 1994, 266(5186): 828-31.
Edwards F. A., Konnerth A., Sakmann B. y Takahashi T. A thin slice pre-paration for patch clamp recordings from neurones of the mam-malian central nervous system. Pflügers Arch., 1989, 414 : 600-612.
Elmslie K. S., Zhou W. y Jones S. W. LHRH and GTP-gamma-S modify cal-cium current activation in bullfrog sympathetic neurons. Neuron.,1990, 5 : 75-80.
Francini F., Bencini C., Piperio C. y Squecco R. Separation of charge move-ment components in mammalian skeletal muscle fibres. J. Physiol.,2001, 537 : 45-56.
García D. E., Li B., García-Ferreiro R. E., Hernández-Ochoa E. O., YanK., Gautam N., Catterall W. A., Mackie K. y Hille B. G-proteinbeta-subunit specificity in the fast membrane-delimited inhibitionof Ca2+ channels. J. Neurosci., 1998, 18(22): 9163-70.
Gilly W. F. y Armstrong C. M. Gating current and potassium channels inthe giant axon of the squid. Biophys. J., 1980, 29 : 485-492.
Grassi F. y Lux H. D. Voltage-dependent GABA-induced modulation ofcalcium currents in chick sensory neurons. Neurosci. Lett., 1989,105 : 113-9.
Hamill O. P. y Sakmann B. Multiple conductance states of single acetyl-choline receptor channels in embryonic muscle cells. Nature, 1981,294 : 462-464.
Hamill O. P., Marty A., Neher E., Sakmann B. y Sigworth F. J. Improvedpatch-clamp techniques for high-resolution current recording fromcells and cell-free membrane patches. Pflügers Arch., 1981, 391:85-100.
Herlitze S., García D. E., Mackie K., Hille B., Scheuer T. y Catterall W. A.Modulation of Ca2+ channels by G-protein beta gamma subunits.Nature, 1996, 380 : 258-262.
Herlitze S., Zhong H., Scheuer T. y Catterall W. A. Allosteric modula-tion of Ca2+ channels by G proteins, voltage-dependent facilita-tion, protein kinase C, and Ca(v)beta subunits. Proc. Natl. Acad.Sci. USA. 2001, 98 : 4699-704.
237

Hernández-Castellanos J. M., Vivas O., Garduño J., De la Cruz L., Are-nas I., Elías-Viñas D., Mackie K. y García D. E. Gb2 mimics activationkinetic slowing of CaV2.2 channels by noradrenaline in rat sympathe-tic neurons. Biochem. Biophys. Res. Commun., 2014, 445(1): 250-4.
Hernández-Ochoa E. O., García-Ferreiro R. E. y García D. E. G proteinactivation inhibits gating charge movement in rat sympatheticneurons. Am. J. Physiol. Cell Physiol., 2007, 292 : C2226-2238.
Hille B. Modulation of ion-channel function by G-protein-coupledreceptors. Trends Neurosci., 1994, 17 : 531-536.
Hille B., Beech D. J., Bernheim L., Mathie A., Shapiro M. S. y Wollmuth,L. P. Multiple G-protein-coupled pathways inhibit N-type Ca chan-nels of neurons. Life Sci., 1995, 56 : 989-92.
Hodgkin A. L. y Huxley A. F. A quantitative description of membranecurrent and its application to conduction and excitation in nerve.J. Physiol., 1952, 117 : 500-544.
Horn R. How S4 segments move charge. Let me count the ways. J. Gen.Physiol., 2004, 123 : 1-4.
Huang C. L. Pharmacological separation of charge movement compo-nents in frog skeletal muscle. J. Physiol., 1982, 324 : 375-387.
Huang C. L. Intramembrane charge movements in skeletal muscle.Physiol. Rev., 1988, 68 : 1197-1147.
Ikeda S. R. Voltage-dependent modulation of N-type calcium channelsby G-protein beta gamma subunits. Nature, 1996, 380 : 255-258.
Ikeda S. R. y Dunlap K. Voltage-dependent modulation of N-type cal-cium channels: role of G protein subunits. Adv. Second MessengerPhosphoprotein Res., 1999, 33 : 131-51.
Ikeda S. R. y Schofield G. G. Somatostatin cyclic octapeptide analogswhich preferentially bind to SOMa receptors block a calcium currentin rat superior cervical ganglion neurons. Neurosci. Lett., 1989,96(3): 283-8.
Jara-Oseguera A., Ishida I. G., Rangel-Yescas G. E., Espinosa-Jalapa N.,Pérez-Guzmán J. A., Elías-Vinas D., Le Lagadec R., Rosenbaum T.e Islas L. D. Uncoupling charge movement from channel openingin voltage-gated potassium channels by ruthenium complexes.J. Biol. Chem., 2011, 286 : 16414-16425.
Kaczmarek L. K. The regulation of neuronal calcium and potassiumchannels by protein phosphorylation. Adv. Second Messenger Phosp-hoprotein Res., 1988, 22 : 113-138.
Krumins A. M. y Gilman A. G. Targeted knockdown of G protein subu-nits selectively prevents receptor-mediated modulation of effectors
238

and reveals complex changes in non-targeted signaling proteins.J. Biol. Chem., 2006, 281(15): 10250-62.
Lacinova L. y Klugbauer N. Modulation of gating currents of the Ca(V)3.1calcium channel by alpha 2 delta 2 and gamma 5 subunits. Arch.Biochem. Biophys., 2004, 425 : 207-213.
Levitan I. B. Phosphorylation of ion channels. J. Membr. Biol., 1985, 87 :177-190.
Logothetis D. E., Movahedi S., Satler C., Lindpaintner K. y Nadal-Ginard B. Incremental reductions of positive charge within the S4region of a voltage-gated K+ channel result in correspondingdecreases in gating charge. Neuron., 1992, 8 : 531-40.
Marchetti C., Carbone E. y Lux H. D. Effects of dopamine and nora-drenaline on Ca channels of cultured sensory and sympatheticneurons of chick. Pflügers Arch., 1986, 406 : 104-11.
Monod J., Wyman J., Changeux J. P. On the nature of allosteric transi-tions: a plausible model. J. Mol. Biol., 1965, 12 : 88-118.
Neely A., Olcese R., Wei X., Birnbaumer L. y Stefani E. Ca(2+)-dependentinactivation of a cloned cardiac Ca2+ channel alpha 1 subunit (alpha1C) expressed in Xenopus oocytes. Biophys. J., 1994, 66 : 1895-903.
Noceti F., Baldelli P., Wei X., Qin N., Toro L., Birnbaumer L. y Stefani E.Effective gating charges per channel in voltage-dependent K+ andCa2+ channels. J. Gen. Physiol., 1996, 108: 143-155.
Nonner W. Relations between the inactivation of sodium channels and theimmobilization of gating charge in frog myelinated nerve. J. Physiol.,1980, 299 : 573-603.
Pawson T. y Nash P. Protein-protein interactions define specificity insignal transduction. Genes Dev., 2000, 14(9): 1027-47.
Perozo E. y Bezanilla, F. Phosphorylation affects voltage gating of thedelayed rectifier K+ channel by electrostatic interactions. Neuron.,1990, 5 : 685-690.
Perozo E., Santacruz-Toloza L., Stefani E., Bezanilla F. y Papazian D. M.S4 mutations alter gating currents of Shaker K channels. Biophys. J.,1994, 66 : 345-54.
Rebolledo-Antúnez S., Farías J. M., Arenas I. y García D. E. Gating char-ges per channel of Ca(V)2.2 channels are modified byG protein acti-vation in rat sympathetic neurons. Arch. Biochem. Biophys., 2009,486 : 51-57.
Rosenthal W., Hescheler J., Trautwein W. y Schultz G. Control of voltage-dependent Ca2+ channels by G protein-coupled receptors. FASEB J.,1988, 2(12): 2784-90.
239

Ruiz-Velasco V. e Ikeda S. R. Multiple G-protein betagamma combina-tions produce voltage-dependent inhibition of N-type calciumchannels in rat superior cervical ganglion neurons. J. Neurosci.,2000, 20(6): 2183-91.
Sakmann B., Patlak J. y Neher E. Single acetylcholine-activated channelsshow burst-kinetics in presence of desensitizing concentrations ofagonist. Nature, 1980, 286 : 71-73.
Schneider M. F. y Chandler W. K. Voltage dependent charge movementof skeletal muscle: a possible step in excitation-contraction cou-pling. Nature, 1973, 242 : 244-6.
Schoppa N. E. y Sigworth F. J. Activation of shaker potassium channels.I. Characterization of voltage-dependent transitions. J. Gen. Physiol.,1998, 111 : 271-94.
Sheets M. F. y Hanck D. A. Gating of skeletal and cardiac muscle sodiumchannels in mammalian cells. J. Physiol., 1999, 514(Pt2): 425-436.
Sheets M. F., Kyle J. W., Kallen R. G. y Hanck D. A. The Na channel vol-tage sensor associated with inactivation is localized to the externalcharged residues of domain IV, S4. Biophys. J., 1999, 77 : 747-757.
Sigworth F. J. The variance of sodium current fluctuations at the nodeof Ranvier. J. Physiol., 1980, 307 : 97-129.
Sokolov S., Kraus R. L., Scheuer T. y Catterall W. A. Inhibition of sodiumchannel gating by trapping the domain II voltage sensor with pro-toxin II. Mol. Pharmacol., 2008, 73 : 1020-1028.
Starace D. M. y Bezanilla F. A proton pore in a potassium channel vol-tage sensor reveals a focused electric field. Nature, 2004, 427: 548-53.
Starace D. M., Stefani E. y Bezanilla F. Voltage-dependent proton trans-port by the voltage sensor of the Shaker K+ channel. Neuron., 1997,19 : 1319-27.
Stuart G. J. y Sakmann B. Active propagation of somatic action potentialsinto neocortical pyramidal cell dendrites. Nature, 1994, 367 : 69-72.
Stühmer W., Conti F., Suzuki H., Wang X. D., Noda M., Yahagi N., KuboH. y Numa S. Structural parts involved in activation and inactiva-tion of the sodium channel. Nature, 1989, 339 : 597-603.
Tedford H. W. y Zamponi G. W. Direct G -protein modulation of Cav2calcium channels. Pharmacol. Rev., 2006, 58(4): 837-62.
Villalba-Galea C. A., Sandtner W., Dimitrov D., Mutoh H., Knöpfel T.y Bezanilla F. Charge movement of a voltage-sensitive fluorescentprotein. Biophys. J., 2009, 96 : L19-21.
240

Xu Y., Ramu Y. y Lu Z. Removal of phospho-head groups of membranelipids immobilizes voltage sensors of K+ channels. Nature, 2008,451 : 826-829.
Yan K., Kalyanaraman V. y Gautam N. Differential ability to form the Gprotein betagamma complex among members of the beta andgamma subunit families. J. Biol. Chem., 1996, 271(12): 7141-6.
241


MARCADORES EPIGENÉTICOS
PABLO PADILLA LONGORIAYURIRIA CORTÉS POZA
Instituto de Investigaciones en Matemáticas Aplicadas y en Sistemas, IIMAS
Universidad Nacional Autónoma de México
All we’ve got are metaphors, and they’re never exactly right.1
JENNIFER EGAN
Sometimes a cigar is just a cigar.2
SIGMUND FREUD
RESUMEN
En muchas ocasiones las metáforas son herramientas podero-sas para ayudar a entender ciertos fenómenos naturales ycrear nuevos significados. Frecuentemente dan lugar a analo-gías más precisas o a modelos matemáticos. El concepto depaisaje epigenético fue propuesto originalmente por C. Wad -dington como una metáfora de los procesos evolutivos y deldesarrollo en biología. En años recientes, esta propuesta hasido estructurada en el contexto de las redes de regulacióngenética mediante una metodología sistemática. En estetrabajo buscamos presentar tal metodología en dos ejemplos
243
1 “Sólo tenemos metáforas y nunca son del todo correctas”.2 “A veces un puro es sólo un puro”.

específicos provenientes de la biología del desarrollo, ambosrelacionados con el estudio de la determinación del destinocelular durante los primeros estadios del desarrollo de unorganismo o de la formación de órganos.El primer ejemplo se centra en la diferenciación celular
durante las divisiones iniciales del embrión de ratón, mientrasque la segunda se refiere a la dinámica espacio-temporaldurante la formación de órganos en el meristemo floral deArabidopsis thaliana.
INTRODUCCIÓN
Probablemente la metáfora de una canica que se mueveen un paisaje fue usada en física, donde de hecho puedeconsiderarse no como una metáfora, sino como un sistemamecánico real. Sin embargo, esta analogía ha sido muy fruc-tífera en la descripción de sistemas dinámicos en donde elpaisaje está asociado con las funciones de energía para pro-cesos conservativos. El paisaje (figura 1) está relacionado conalgún tipo de potencial.
244
$$
visto desde el Valle de Chalco cerca de la Ciudad de México. (
los estados conformacionales estables de una proteína.
E
Waddington.
FIGURA 1. Paisaje: el Iztaccíhuatl visto desde el Valle de Chalco cerca de la Ciudadde México. (De Wikipedia: Iztaccíhuatl).

245
Este enfoque ha sido usado en otras áreas, incluyendo labiología del desarrollo, por ejemplo en el estudio del ple-gamiento de proteínas y otros procesos biológicos, en dondelos mínimos locales del paisaje se identifican con los estadosconformacionales estables de una proteína. En este caso, elpaisaje energético ha sido incluso propuesto para tratar deentender la eficiencia con la que ocurre el proceso y porqué es mucho más rápido de lo que pudiera esperarse. (Dillet al., 1997).Un paisaje epigenético, concepto originalmente propuesto
por Waddingon (figura 2), permite visualizar la historia deldesarrollo de una célula en un embrión u otros procesos bio-lógicos. Conforme la canica rueda tiene varias posibilidadesde a dónde moverse (de forma similar, el embrión en desa-rrollo puede ser influenciado por varios factores genéticosy ambientales) y cuando llega al fondo de un valle el procesode determinación celular habrá sido completado y un tipocelular habrá sido “escogido”, por así decirlo. Goldberg ofreceuna descripción completa, así como una perspectiva actualsobre este tema (Goldberg, 2007).
FIGURA 2. Paisaje epigenético. (Del libro The Strategy of the Genes de Waddington).

Hasta cierto punto es sorprendente que aun cuando lametáfora de Waddington era de carácter más bien abstracto,jugó un papel fundamental en los esfuerzos para describiry en tender diferentes procesos. Esto se debe probablementeal hecho de que, como ocurre con otras metáforas, hay másque la simple analogía. En efecto, recientemente algunasmetodologías detalladas han sido propuestas con el objetode construir paisajes epigenéticos en algunos fenómenosbiológicos que van desde la dinámica de las células madreshasta el proceso de iniciación del cáncer, entre otros.En este artículo presentamos dos ejemplos que surgen en
la biología del desarrollo, en los cuales se puede construir unpaisaje epigenético y esto resulta ser útil en la prediccióny comprensión de los procesos subyacentes. El primer ejem-plo está relacionado con la pérdida de potencialidad en losestadios tempranos de desarrollo de las primeras divisiones deun embrión de ratón. En este caso, la construcción de un pai-saje epigenético está asociada a la identificación de marcadoresepigenéticos. Estos marcadores nos permiten distinguir entrelas células que formarán parte del trofectodermo, (TE) queconstituirán la placenta y cuáles serán parte de la MasaCelular Interna, (ICM, por sus siglas en inglés).El segundo ejemplo se refiere a la determinación del des-
tino celular durante el proceso inmediatamente previo a laformación de órganos florales en Arabidopsis thaliana.Aquí, la construcción del paisaje epigenético se vuelve una
herramienta útil para entender la dinámica espacio-temporal.De hecho, a partir de esta construcción es posible realizaralgunas predicciones experimentales que han sido corrobo-radas; más aún, se puede formular una hipótesis relativa depor qué en la flor Lacandonia schismatica el orden especialde dos verticilos (anillos) florales, el de estambres y el de car-pelos, está invertido en relación con las otras 250 mil espe-cies de plantas con flores o angiospermas.
246

De acuerdo con lo anterior, el resto del trabajo está organi-zado en dos partes: en la primera, se presenta la construc-ción del paisaje epigenético para el proceso de pérdida depotencialidad en un embrión de ratón y, en la segunda, sedescribe el paisaje epigenético para la determinación del des-tino celular en el proceso de floración en Arabidopsis thaliana.
Marcadores epigenéticos
El proceso en el cual una sola célula, el embrión, da ori-gen a un organismo complejo aún permanece, básicamente,sin entenderse. A través de una serie de divisiones, el cigotollega a diferenciarse en células de muchos tipos. Conforme eldesarrollo avanza, el nivel de especialización aumenta. Enotras palabras, las “elecciones” que la célula puede hacer selimitan y se llega así a un destino de diferenciación. Este pro-ceso está caracterizado por la pérdida de potencialidad: deser totipotencial (el embrión tiene la posibilidad de conver-tirse en una célula de cualquier tipo) llega a la diferenciacióntotal. Entender cómo esto sucede no sólo es interesante, sinoesencial para el uso eficiente de las células madre como unaalternativa terapéutica realista.A pesar de que durante algún tiempo se pensó que las
células resultantes de las primeras divisiones del cigoto eranidénticas, una gran cantidad de trabajo ha sido dedicadoa identificar las tendencias en la pérdida de potencialidad apartir de etapas muy tempranas en el desarrollo. De hecho, unpaso muy importante ocurre cuando las células en el cigotose diferencian en una masa celular interna y en células de laplacenta (trofectodermo). En un trabajo reciente (Burtonet al., 2013), se muestra que no sólo patrones de activacióngenética, sino también factores epigenéticos (por ejemplo,modificadores de cromatina) pueden ser utilizados como mar-cadores para diferentes destinos celulares.
247

La preimplantación del embrión de ratón resulta serun modelo ideal para estudiar los cambios en totipotenciay di ferenciación in vivo. Un conjunto bastante grande dege nes, mo dificadores de cromatina, factores de transcripcióny otros modificadores epigenéticos fueron analizados duran-te las primeras divisiones del embrión. Inicialmente se tratóde determinar si diferentes estados de la célula y grados depotencialidad podrían ser definidos con base en estos com -ponentes epigenéticos.Para ello, los autores realizaron un análisis de cúmulos jerár-
quicos no supervisados de todos los blastómeros y todos losgenes (figura 3). Este análisis mostró que las células se acumu-lan, principalmente, de acuerdo con su estado de desarrollo.Posteriormente se realizó un procesamiento estadístico con -
vencional (análisis de componentes principales). Este proce-dimiento permite seleccionar, o más bien combinar los fac-tores que mejor expliquen la variabilidad de los datos. Comopuede verse en la figura 4, en ella se agrupan en cúmulos depuntos que son similares de acuerdo con esta combinaciónde factores.A partir de este análisis se puede observar que los datos
se agrupan de acuerdo con las diferentes etapas de división.Basados en estos resultados, es posible identificar los cen-tros de diferentes cúmulos, como los fondos de los valles delpaisaje, y utilizar esta información para completar los resul-tados (figura 4).
248
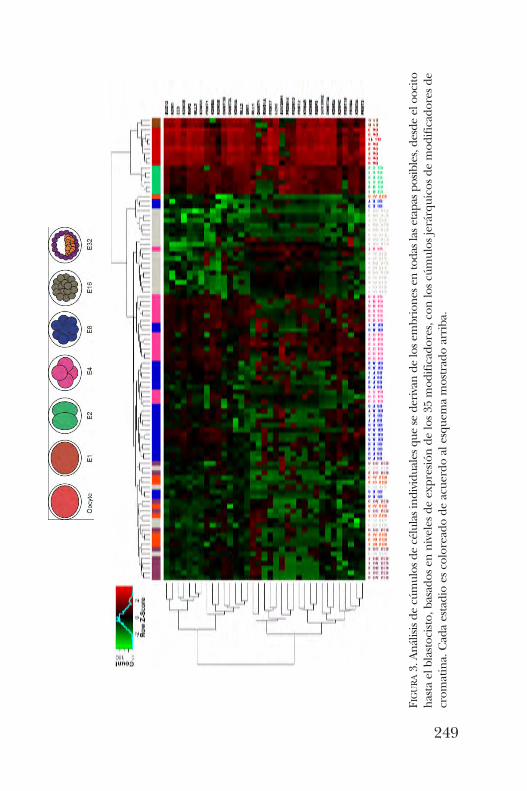
249
##
FIGURA3. Análisis de cúm
ulos de células individuales que se derivan de los embriones en todas las etapas posibles, desde el oocito
hasta el blastocisto, basados en niveles de expresión de los 35 modificadores, con los cúmulos jerárquicos de modificadores de
crom
atina. Cada estadio es coloreado de acuerdo al esquema mostrado arriba.
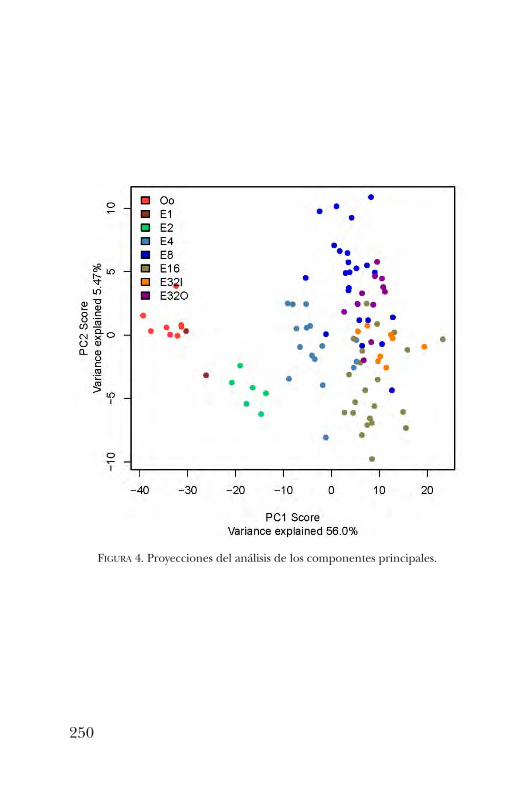
250
$$
estadio es coloreado de acuerdo al esquema mostrado arriba.
FIGURA 4. Proyecciones del análisis de los componentes principales.
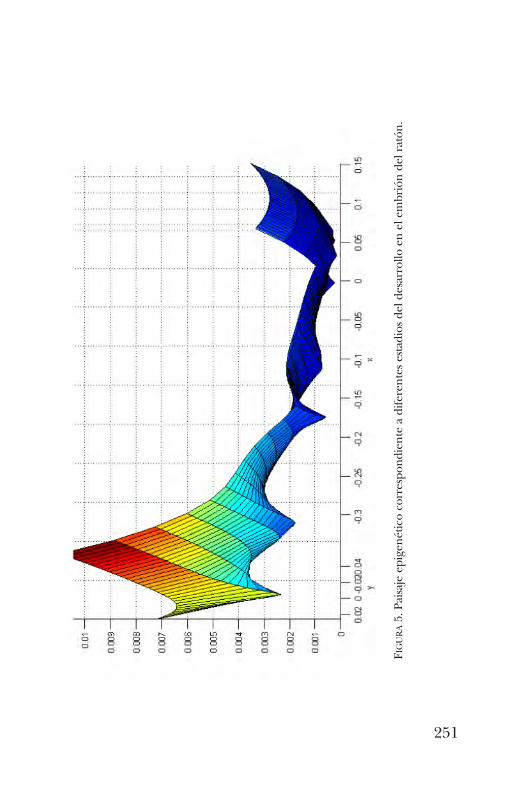
251
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
FIGURA5. Paisaje epigenético correspondiente a diferentes estadios del desarrollo en el embrión del ratón.

DETERMINACIÓN DEL DESTINO CELULAR EN LA FLORACIÓN
DE ARABIDOPSIS THALIANA
1. Introducción
Los órganos florales de todas las especies de Angiospermas(aproximadamente 250 mil) están organizados en cuatrocírculos concéntricos, llamados verticilos.
La única excepción conocida en esta configuración es laobservada en la flor Lacandonia schismática, donde la posi-ción de sus estambres y carpelos está invertida.Trabajamos con la flor de la planta Arabidopsis thaliana, la
primera flor cuyo genoma completo fue secuenciado y queha sido estudiada extensamente.
252
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
g
$$
c
$$
$$
$$
e
$$
d
$$
$$
$$
$$
d
$$
$$
$$
$$
$$
$$
$$
!
FIGURA 1. Verticilos.

2. El modelo
El modelo consiste de un sistema de ecuaciones de reac-ción-difusión cuya dinámica está gobernada por un campopotencial. Este potencial es el paisaje epigenético de la for-mación de los órganos florales, que modela la forma en quediferentes fuerzas ambientales y genéticas afectan la dife-renciación celular. Para definirlo, usamos la construcciónde la Red Regulatoria Genética (RRG) de la flor que estamosestudiando, utilizando información experimental.
253
$$
A
$$
s
$$
$$
$$
a
$$
F
$$
$$
$$
m
$$
$$
$$
$$
t
$$
o
$$
$$
$$
$$
á
$$
e
$$
$$
$$
n
$$
á
$$
a
$$
$$
$$
e
$$
!
FIGURA 2. Lacandonia schismática.
$$
A
$$
s
$$
$$
$$
a
$$
F
$$
$$
$$
m
$$
$$
$$
$$
t
$$
o
$$
$$
$$
$$
á
$$
e
$$
$$
$$
n
$$
á
$$
a
$$
$$
$$
e
$$
!
FIGURA 3. Arabidopsis thaliana.

Al resolver este sistema de ecuaciones de reacción difu-sión, con técnicas analíticas y numéricas, observamos que elmodelo reproduce correctamente la configuración espacialde la formación de los órganos florales. Para validar nuestromodelo repetimos el procedimiento con los mutantes homeó-ticos de la flor, los cuales tienen, cada uno, diferentes redesde regulación genética y, por lo tanto, una distinta configu-ración espacial e incluso órganos faltantes. Los resultadosobtenidos corresponden con los experimentos.El flujo de datos de nuestro modelo es el siguiente:
2a. Sistema dinámico discreto
En este sistema utilizamos una red reguladora booleanaque consiste en 13 nodos (cada uno correspondiente a ungen específico), cuyo estado (0 o 1) se actualiza de acuerdocon las reglas obtenidas experimentalmente y, además, corres-ponde a la interacción entre los genes (figura 5).Cada punto fijo de la red, correspondiente a un órgano
floral (sépalos, pétalos, estambres y carpelos), tiene 13 com-ponentes (uno por cada gen) y es tratado como un vector enun espacio de 13 dimensiones. Del número total de posiblescondiciones iniciales, contamos el número de éstas, que vana dar a cada uno de estos puntos de equilibrio y obtenemos152, 160, 3744 y 3608 para sépalos, pétalos, estambres y car-pelos, respectivamente.
254
FIGURA 4. Flujo de datos.

2b. Reducción de coordenadas
La dimensionalidad del sistema se reduce utilizando ladescomposición en valores singulares, para lo que encontra-mos una base que genera el plano y después proyectamoscada uno de los cuatro vectores en este plano. Así obtene-mos lo siguiente:
(uS, vS) = (–1.7878, 0.1504); (uP, nP) = (–2.5841, –0.2055)(uT, vT) = (–2.8277, –0.1695); (uC, vC) = (–2.3844, 0.2067)
2c. Paisaje epigenético
El paisaje epigenético es un campo potencial F(u, v) concuatro cuencas, cada una centrada en uno de los puntos fijosy cuyos tamaños (aS, aP, aE, aC) están dados por el númerorecíproco de condiciones iniciales que se dan en cada unade las cuencas (figura 6):
F(u, v) = min{aS((u –uS)2 + (v – vS)2), aP((u – uP)2 + (v – vP)2),aE((u – uE)2 + (v – vE)2), aC((u – uC)2 + (v – vC)2)}
255
##
número de estas que van a dar a cada uno de estos puntos de equilibrio
y obtenemos 152, 160, 3744 y 3608 para sépalos, pétalos, estambres y carpelos respectivamente.
2
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
##
#*#
)
) ) ) !#
' )
FIGURA 5. Sistema dinámico discreto.

2d. Ecuaciones de reacción-difusión
A continuación, interpolamos el sistema dinámico discretopara obtener uno continuo, cuya dinámica está dada por elsiguiente sistema de ecuaciones diferenciales parciales dereacción-difusión, gobernadas por el campo potencial F(u, v):
ut = d1Du + f(u, v)vt = d2Dv + g(u, v)
donde (f, g) = F (definido localmente) y d1, d2 son cons-tantes de difusión.Las variables u y v representan una combinación lineal
de los estados de activación de los genes, lo cual resulta al ele-gir un sistema de ejes coordenados en el plano bidimensio-nal ajustado.
3. Mutantes homeóticos y el modelo ABC
Los genes homeóticos son responsables del desarrollo deestructuras específicas en plantas y animales, las mutaciones
D
256
$$
! !!!!!!
! !!
! ! !
! ! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
$$
! !!!!!!
! !!
! ! !
! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !
FIGURA 6. Potencial con cuatro cuencas.

en estos genes pueden causar que un órgano sea reemplazadopor otro. Al estudiar estas mutaciones, Cohen y Meyerowitz(1991) formularon el modelo ABC para el desarrollo floral.De acuerdo con este modelo, la identidad de los órganos flo-rales está determinada por tres clases de genes: A, B y C. Estasclases codifican los factores transcripcionales que, combina-dos, provocan la especialización de tejidos en regiones espe-cíficas durante el desarrollo.
Una serie de mutantes de la flor Arabidopsis thaliana hasido caracterizada; ésta sirven como material experimentalen estudios de laboratorio. En particular, nosotros trabajamoscon los mutantes Apetala (Apt1) y Agamus (Ag).
4. Resultados
Las soluciones al Sistema de reacción-difusión visitan lascuatro cuencas de atracción en el orden correcto (figura 8).En A1, B1 y C1 (flor silvestre, mutantes Apt1 y Ag respectiva-
257
FIGURA 7. Modelo ABC.
##
(
!
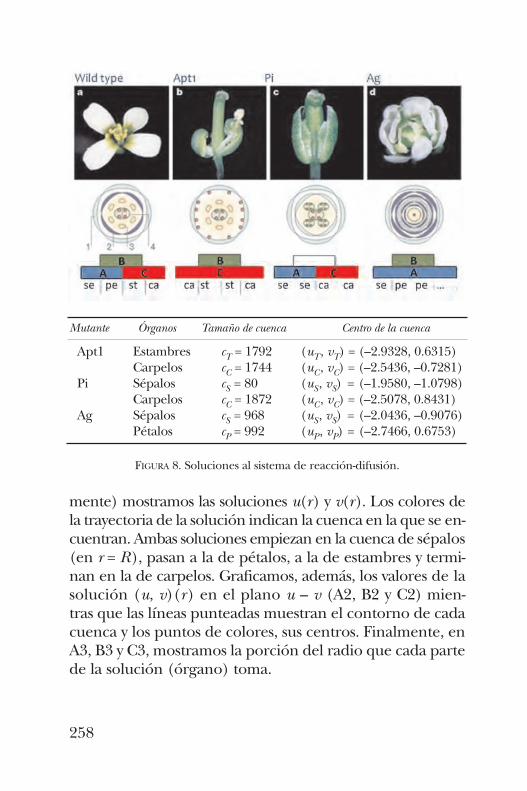
mente) mostramos las soluciones u(r) y v(r). Los colores dela trayectoria de la solución indican la cuenca en la que se en -cuentran. Ambas soluciones empiezan en la cuenca de sépalos(en r = R), pasan a la de pétalos, a la de estambres y termi-nan en la de carpelos. Graficamos, además, los valores de lasolución (u, v)(r) en el plano u – v (A2, B2 y C2) mien-tras que las líneas punteadas muestran el contorno de cadacuenca y los puntos de colores, sus centros. Finalmente, enA3, B3 y C3, mostramos la porción del radio que cada partede la solución (órgano) toma.
258
##
!
##
##
##
##
n
##
##
##
##
u
##
##
##
##
a
##
A
##
1
##
!
##
!
##
!
##
u
##
r
##
##
##
##
p
##
o
##
y
##
u
##
!
Mutante Órganos Tamaño de cuenca Centro de la cuenca
Apt1 Estambres cT = 1792 (uT, vT) = (–2.9328, 0.6315)Carpelos cC = 1744 (uC, vC) = (–2.5436, –0.7281)
Pi Sépalos cS = 80 (uS, vS) = (–1.9580, –1.0798)Carpelos cC = 1872 (uC, vC) = (–2.5078, 0.8431)
Ag Sépalos cS = 968 (uS, vS) = (–2.0436, –0.9076)Pétalos cP = 992 (uP, vP) = (–2.7466, 0.6753)
FIGURA 8. Soluciones al sistema de reacción-difusión.
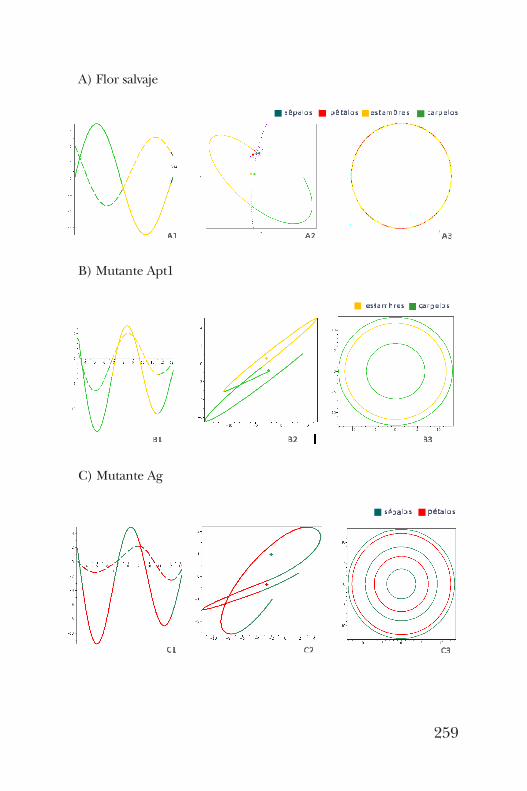
A) Flor salvaje
B) Mutante Apt1
C) Mutante Ag
259
$$
A) Flor salvaje
B) Apt 1 mutantB
B) Apt1 mutant
!!!!!!!"#$%&'"!!!!!!!!!$#(%&'"!!!!!!)"(%*+,)"!!!!!!!!!-%,$)&'"
%"$ %!$ %#$
or sl) FA
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
ejvaajlaor s
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
!!!!!!! !!!!!!!! !!!!! !!!!!!!!!
$ $ $
$$
B) Mutante Apt1
!!!!!!!!!!!)"(%*+,)"!!!!!!!-%,$)&'"
&"$ &!$ &#$
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
autB) M
$$
!!!!!!!!!!! !!!!!!!
$ $ $
1pt Aenta
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
!!!!!!!!!!! !!!!!!!
$ $ $
$$
B) Mutante AG
%"$ %!$ %&$
!!!!!!!"#$%&'"!!!!!!!!$#(%&'" autB) M
$$
$ $ $
!!!!!!! !!!!!!!! G Aenta
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!
$$
$ $ $
!!!!!!! !!!!!!!!

5. Conclusiones
En este trabajo logramos modelar correctamente el pro-ceso de determinación del destino celular en los órganosflorales de la planta Arabidopsis thaliana. Al resolver las ecua-ciones recuperamos la configuración espacial de los órga-nos florales que aparecen en la flor analizada. El modelo,además, reproduce los resultados experimentales observadosen mutantes homeóticos y tiene la gran cualidad de que estábasado en datos experimentales.
REFERENCIAS
Álvarez-Buylla E., Benítez M., Chaos Á., Cortés-Poza Y., Espinosa-Soto C.,Escalera G. y Padilla-Longoria. Floral Morphogenesis StochasticExplorations of a Gene Network’s Epigenetic Landscape, Plos ONE,2008, 3, Issue 11.
Álvarez-Buylla E. R., Benítez M., Chaos Á., Cortés Y., Escalera-Santos G.,Espinosa C. y Padilla P.. Variational Problems Arising in Biology,Centre de Recherches Mathématiques, CRM Proceedings and LectureNotes, 2008, 44.
Álvarez-Buylla E. R., Chaos Á., Cortés-Poza Y., Espinosa-Soto C. y Padilla-Longoria P. De genes a patrones: una propuesta metodológica, pp.473-486. En: García-Colín Scherer L., Dagdug L., Miramontes P.,Roja A. (coord.). La Física Biológica en México: Temas Selectos. ElColegio Nacional, México, 2006.
Burton et al. Single-Cell Profiling of Epigenetic Modifiers IdentifiesPRDM14 as an Inducer of Cell Fate in the Mammalian Embryo. CellReports, 2013. Recuperado de la URL: http://dx.doi.org/10.1016/j.celrep.2013.09.044
Dill K. A. y Chan H. S. From Levinthal to pathways to funnels. Nat.Struct. Biol., 1997, 4 : 10-19.
Goldberg A. D., Allis C. D. y Bernstein E. Epigenetics: A landscape takesshape. Cell, 2007, 128 : 635-638.
Waddington C. H. The Strategy of the Genes. Allen and Unwin, 1957.
260

¿CÓMO ES UNA PROTEÍNA CAPAZ DE PROMOVER LA PROLIFERACIÓN,
LA DIFERENCIACIÓN Y LA MUERTE CELULAR?
SUSANA CASTROInstituto de Fisiología Celular
Universidad Nacional Autónoma de Mé[email protected]
Una pregunta intrigante para los estudiosos de la vida esentender cómo a partir de una sola célula, el huevo reciénfecundado, se genera la gran diversidad de células que com-ponen nuestro cuerpo. Un mamífero tiene alrededor de 200tipos diferentes de células, cada una con formas y funcionesdiferentes y definidas. El proceso por el cual una célula seconvierte en un tipo particular de célula se llama diferenciacióny la disciplina que estudia los mecanismos por los cuales seforma un nuevo individuo, a partir del huevo fertilizado,se llama biología del desarrollo. Desde las observaciones deAristóteles sabemos que el huevo fecundado se divide paragenerar los millones de células que conforman el organismoadulto, pero cómo se regula, qué tipo celular se debe dividir,cuántas veces y cuándo, sigue aún sin entenderse del todo.Si se observa el desarrollo temprano de un humano, por ejem-plo, se nota desde el segundo día de desarrollo que unas cé -lulas se dividen antes que otras, y para el quinto día de desa-rrollo ya se observan dos tipos diferentes de células (figura 1):las de alrededor (llamadas trofoectodermo), que darán lugar
261

262
FIGURA1.Evolución de la brillantez de las fuentes de rayos X en el tiem
po. B
S, fuentes de rayos X com
pactas basadas en
brem
sstrahlung. ICS, fuentes de rayos X basados en dispersión inversa de Com
pton. LW
FA, fuentes de rayos X com
pactas
basadas en la aceleración en el cam
po de la estela de un láser.

263
a estructuras extraembrionarias como la placenta y unaspocas células que quedan acomodadas en un pequeño grupoen el interior (llamadas masa celular interna), que darán lugaral embrión como tal. Estas últimas células se pueden aislardel embrión (llamado blastocisto en esta etapa) y mantenerseproliferando en una caja de cultivo en el laboratorio, puestienen la capacidad de diferenciarse hacia todos los tiposcelulares del embrión, es decir que son pluripotenciales, asícomo de renovarse a sí mismas. Estas dos características defi-nen a las células troncales (mejor conocidas en los mediosde comunicación y publicidad como células madre). Si bieninvestigadores en todos los continentes están buscando lamanera de inducir la diferenciación de estas células troncalesembrionarias hacia un tipo celular particular, aún hoy (año2014) no se ha podido desarrollar un protocolo que genereuna población 100% pura de un tipo particular de céluladiferenciada.Otro proceso celular esencial para el desarrollo embrio-
nario es la muerte celular programada. Desde etapas tan tem-pranas como el blastocisto, así como durante la formación decada órgano, ocurre la muerte celular como parte del desa-rrollo del embrión. Un ejemplo clásico ocurre durante la for-mación de las extremidades, en el que se mueren las célulasque se encuentran entre los dígitos para que se puedan indi-vidualizar. Si esta muerte celular no ocurre (por defectos gené-ticos o exposición a fármacos) los dedos se quedan pegadosy no se forman bien las extremidades. Como la eliminaciónde las células destinadas a morir sucede siempre en la mismaetapa de desarrollo y en una región precisa, se entendió quela muerte de esas poblaciones celulares está programada den-tro del desarrollo embrionario, es decir, que hay genes queinstruyen a esas células a morir. Lo interesante ahora es enten-der cómo se regula y cuándo se deben utilizar esos genes.El concepto de la muerte celular programada fue contra-

intuitivo, ya que es difícil imaginarse la selección evolutivade genes que instruyan a las células a matarse a sí mismas.Resulta interesante que la misma maquinaria genética seutiliza en el organismo adulto para eliminar células no desea-das, ya sea para mantener el tamaño de los órganos (debehaber un balance entre la proliferación y la muerte celular),para renovar ciertos tejidos o para eliminar células infectadaso dañadas. Este mecanismo de muerte celular se llama apop-tosis. Cuando una célula se empieza a morir por apoptosiscambia las propiedades de su superficie, de manera que célu-las vecinas (en el embrión) o especializadas (macrófagos si yase formó el sistema inmune) reconocen que la célula se estámuriendo y la engullen para eliminarla.Es muy importante regular cuándo y qué células se deben
morir, pues cuando ocurren mutaciones en los genes demuerte celular o de su regulación, surgen enfermedadescomo el cáncer, en el que las células se dejan de morir, o dege-nerativas, en las que las células se mueren fuera de contexto.Por eso, entender las bases moleculares de la regulación dela muerte celular tiene un gran impacto en la medicina. Eneste contexto resulta interesante conocer que además de laapoptosis hay otros mecanismos de muerte celular que ocu-rren tanto en el desarrollo embrionario como en el orga-nismo maduro. Cuando se eliminan estructuras completas en el desarrollo
embrionario, como ocurre en la metamorfosis, son tantas lascélulas que desaparecen en ese momento que ellas mismasayudan a su eliminación al degradar parte de sí mismas antesde ser engullidas por células vecinas. Este proceso catabólicose llama autofagia.Durante la autofagia, dentro de la célula se forman unas
vesículas especializadas (llamadas autofagosomas) que engu-llen el material citoplásmico a degradar, que bien pueden serproteínas, ribosomas e incluso organelos. Todas las células
264

eucariotas, incluídos organismos unicelulares, tienen unasestructuras membranales a partir de las cuales se forman losautofagosomas. Una vez que se encerró el material a degra-dar dentro de los autofagosomas, estos se fusionan con liso-somas, que son organelos que contienen enzimas hidrolíticasque rompen las macromoléculas, como lípidos, proteínas,etcétera, en sus componentes unitarios. El destino del mate-rial degradado varía según el estímulo inductor, el tipo celu-lar, etcétera. La maquinaria molecular que permite la forma-ción de los autofagosomas está conservada en la evolución,a tal grado que experimentalmente se pueden sustituir genesde una especie por otra, por ejemplo, se pueden comple-mentar genes mutantes de levadura con genes humanos. Todas las células eucariotas tienen un nivel basal de auto-
fagia y en respuesta a diversos estímulos aumenta la cantidadde autofagosomas y se elige el material específico a degradar.El estímulo más común para aumentar la autofagia es la faltade nutrientes, que induce la degradación de proteínas y otroscomponentes no esenciales para reciclar los elementos quelas constituyen, como aminoácidos para sintetizar proteínasesenciales. Esto les permite resistir más tiempo en la esperade que lleguen los nutrientes nuevamente. Sin embargo, nosiempre es bueno para el organismo. Utilizar la autofagiapara resistir períodos de hambruna favorece el crecimientode tumores, ya que las células que quedan en el centro deltumor no reciben nutrientes de la sangre hasta que se for-man nuevos vasos sanguíneos que alimentan el tumor. Estollevaría a considerar inhibir la autofagia para evitar el esta-blecimiento del tumor, pero no es tan sencillo, pues otrasobservaciones han mostrado lo contrario: que cuando laautofagia no funciona correctamente en un organismo, sur-gen tumores. Entender este papel aparentemente contra-dictorio de la autofagia es una área activa de investigación.
265

Otros estímulos de la autofagia son la presencia de micro-organismos invasores o compuestos tóxicos: las células delsistema inmune eliminan a algunos microorganismos porautofagia y las células cancerosas utilizan a la autofagia paraeliminar las drogas de la quimioterapia. En estas ocasiones elmaterial engullido no se utiliza para sintetizar nuevas pro-teínas sino que probablemente es secretado. Fisiológicamente,la autofagia limpia a la célula de organelos dañados, como lasmitocondrias, y así evita que las células mueran por apoptosis.También ayuda a mantener la integridad del genoma por unmecanismo que se desconoce. Con los ejemplos aquí men-cionados se puede concluir que en la gran mayoría de susfunciones, la autofagia ayuda a mantener a las células salu-dables. Sin embargo, en otras ocasiones promueve la muertecelular, aunque tampoco se sabe el mecanismo por el cual con-tribuye a la muerte. Parece razonable suponer que se debea que elimina componentes esenciales o bien, a que degradaexcesivamente componentes celulares llevando a un colapsometabólico. En nuestro laboratorio investigamos el meca-nismo molecular que lleva a que la autofagia favorezca lamuerte celular en lugar de la sobrevivencia.El papel de la autofagia como parte de un mecanismo de
muerte celular programada se demostró al estudiar el desa-rrollo de la mosca de la fruta. Durante la metamorfosis dela larva, para convertirse en mosca, se desintegra la glándulasalival, entre otros órganos. Si se compara el desarrollo demoscas mutantes en los genes apoptóticos con el desarrollode moscas normales, se observa que la glándula salival sesigue eliminando aunque la apoptosis no se pueda realizar(eso es que la muerte de estas células no es apoptótica), mien-tras que durante el desarrollo de moscas mutantes en losgenes de la maquinaria central de la autofagia ya no desa-parece la glándula salival (eso es, que se necesita la autofagiapara eliminar estas células). Si bien durante el desarrollo
266

embrionario de mamíferos también se eliminan estructurascompletas —como los conductos de Wolf o de Müller—y las células que se mueren presentan morfologías que ase-mejan células con autofagia, aún no se han hecho experi-mentos en los que se inhiba la autofagia para demostrarque esa muerte celular programada efectivamente dependede la autofagia. Este tipo de demostración es importante, yaque la función de la autofagia que más se ha observado, esla de favorecer la sobrevivencia de las células, como se men-cionó anteriormente. En organismos más complejos, como los vertebrados, se
ha adaptado la maquinaria central de la autofagia para con-tribuir en otros procesos de desarrollo, mediante la adqui-sición de proteínas accesorias que modulan tanto el materiala degradar como el destino del contenido de los autofagoso-mas; estas proteínas accesorias son específicas para cada tipocelular o proceso particular. Un ejemplo de ello se observaen el desarrollo del sistema nervioso del ratón, el cual no seforma correctamente cuando ocurre una mutación en ungen que sólo se encuentra en vertebrados (llamado AMBRA)y que regula la autofagia. En estos casos, los embriones tienenel cerebro expuesto (exoencefalia) y una espina bífida, queson malformaciones bastante frecuentes en humanos. Igualque la muerte celular programada, los efectos celulares dela autofagia tienen un impacto en la fisiología del organismotambién en el adulto, de tal suerte que la pérdida de su regu-lación promueve el surgimiento de enfermedades como elcáncer, la neurodegeneración o enfermedades metabólicas.Por otro lado, diversos estímulos que aumentan la autofagiaaumentan la longevidad, por lo que se ha propuesto que sucorrecto funcionamiento ayuda a prevenir cambios asocia-dos al envejecimiento. En nuestra investigación estudiamoscómo la autofagia regula el destino celular, tanto en el desa-rrollo embrionario como en el envejecimiento.
267

Resulta interesante que durante el desarrollo embrionariono siempre se eliminan los restos de las células después desu parcial degradación. Por ejemplo, durante la formacióndel cristalino del ojo se forman unas células muy especiali-zadas que se rellenan de una proteína llamada cristalina,que permite el paso de la luz sin perturbaciones. Sin embargo,los otros componentes de las células como las mitocondriasy demás organelos, el material genético, etcétera, sí impi-den el paso de la luz, por lo que deben ser eliminados. Eneste caso, las células digieren parte de sí mismas de maneramuy controlada, garantizando que las células vecinas no lasidentifiquen como moribundas para que no las eliminen.Este es un ejemplo tanto de muerte como de autofagia par-cial, que muestra cómo las células utilizan los mismos pro-cesos fundamentales con variantes adicionales para realizarprocesos especializados.Una ventaja de que existan mecanismos alternos de muerte
celular es que al conocer sus componentes moleculares sepuede proponer el desarrollo de nuevos fármacos que per-mitan activarlos para matar células cancerosas, o al contrario,evitarlos para contender con enfermedades degenerativas.En esta búsqueda, estudiando diversos modelos de muertecelular, descubrimos que una proteína llamada NR4A1 esesencial para que las células se mueran por un mecanismodiferente a la apoptosis y que depende de la autofagia. Demanera interesante, otros grupos de investigación encontra-ron que la misma proteína es esencial para que ciertos tiposcelulares mueran por apoptosis, mientras que en otras con-diciones es necesaria para evitar que se mueran las células.También se descubrió que participa en el proceso de dife-renciación neuronal. De ahí entonces que surja la pregunta:¿cómo puede una misma proteína promover que una célulaprolifere, muera por apoptosis, autofagia o incluso diferencie?
268

La proteína NR4A1 es el producto de un gen que perte-nece a una familia de genes cuya función es la de censar lapresencia de hormonas tiroideas. A este tipo de proteínasque perciben señales extracelulares que instruyen a las célu-las para que respondan de cierta manera se les llama receptores.Normalmente se encuentran en la membrana plasmática,viendo hacia el espacio extracelular, y trasmiten la informa-ción de la señal hacia el interior de la célula, generalmenteinstruyendo la expresión de un subconjunto de genes. Lasproteínas como NR4A1 tienen la particularidad de estar loca-lizadas en el interior de la célula, de entrar al núcleo donde seencuentra en material genético y cambiar ellas mismas laexpresión de ciertos genes, por lo que este tipo de receptoresse llaman receptores nucleares. Los receptores nucleares son proteínas que están forma-
das por tres regiones estructurales o dominios, que les per-miten interactuar con diferentes moléculas para realizar susfunciones. Con uno de esos dominios los receptores interac-cionan con otras proteínas (llamado dominio de transactiva-ción), en el caso de NR4A1 esa interacción cambia la funciónde las proteínas con las que interactúa; con otro dominio seunen a una secuencia particular del ADNpara regular la expre-sión de los genes que contengan esta secuencia; con un tercerdominio interactúan con su ligando u hormona, cuya pre-sencia detectan. De entre los receptores nucleares NR4A1 esparticular, ya que tiene la habilidad de modificar la funciónde otras proteínas, así como de inducir o reprimir la expre-sión de genes blancos. ¿Cómo se determina qué funciónen particular va a realizar?, y ¿qué le pasa a la célula cuan-do ejerce cada una de esas funciones? Ese es precisamentenuestro tema de estudio.La proteína NR4A1 es un gen que se expresa sólo en
respuesta a ciertos estímulos y no parece detectar algunahormona. Durante el desarrollo del sistema inmune, por
269

ejemplo, se expresa para eliminar a células T que producenauto-anticuerpos. En el adulto se expresa en respuesta alejercicio, a tener una dieta baja en calorías o cuando haydaño al ADN. Acorde a los estímulos inductores, la función deNR4A1, en el organismo completo, se refleja en el controldel metabolismo, ya que cambios en su función favorecen eldesarrollo de obesidad e intolerancia a la insulina, así comoen promover la reparación del ADN dañado, con lo que man-tiene la estabilidad del genoma. Interesantemente, esas mis-mas funciones fisiológicas tiene la autofagia y esos mismosestímulos aumentan la autofagia. A nivel celular, la actividadde NR4A1 causa que las células proliferen, diferencien o semueran, igual que sucede con la autofagia. Conjuntandoestas observaciones con nuestros propios descubrimientos, enlos que observamos que en diversos modelos de muerte celu-lar por autofagia es necesaria la función de NR4A1, propo-nemos que esta proteína regule la maquinaria molecular dela autofagia para dirigir el destino de la célula, es decir, regu-lando la autofagia influye sobre la proliferación, diferencia-ción o muerte celular. ¿Cómo puede NR4A1 promover la pro-liferación, diferenciación o muerte celular? Nos imaginamosque deben variar sus propiedades fisicoquímicas para podervariar las moléculas con las que interactúa en cada caso, esdecir, que debe tener diferentes modificaciones químicas des-pués de haber sido sintetizada; dicho de otra forma, que essusceptible a recibir diferentes modificaciones post-traduc-cionales que cambiarán su función particular. El tipo de modificación más frecuente es la fosforilación,
es decir, la unión covalente de un grupo fosfato a un ami-noácido particular, ya sea tirosina, treonina o serina. Cuandouna proteína es fosforilada, en general, cambia su actividady recupera su estado original al ser eliminado el fosfato. Enel caso de NR4A1, la fosforilación en una serina que seencuentra en el dominio de unión al ADN evita que participe
270

en la muerte de células T durante el desarrollo del sistemainmune, ya que no puede activar la expresión de genes demuerte apoptótica. En cambio, nosotros descubrimos quees necesario que se fosforile en otro aminoácido fuera deldominio de unión al ADN (en este caso una treonina) parainducir la muerte dependiente de autofagia. Otro tipo de modificaciones post-traduccionales no siem-
pre son reversibles, lo que aporta un nivel mayor de regu-lación al propiciar un cambio estable en la actividad. Tal esel caso de la SUMOilación o unión covalente de una proteínapequeña (o péptido) llamada SUMO, a alguna lisina que seencuentre en la proteína blanco, dentro de un contextoparticular de aminoácidos. Como diría elocuentemente eldoctor Luis Fernando Lara: “Respetando la gramática”. El pép-tido SUMO puede ser despegado de la proteína blanco de -jando a la proteína con una conformación estructural dife-rente a la que tenía antes de ser SUMOilada, aunque tambiénla puede dejar en su estado original, es decir, una misma pro-teína puede tener tres características diferentes con un posibleimpacto en su función: antes de ser SUMOilada, SUMOiladay después de ser desSUMOilada.Cuando una proteína es modificada por SUMOilación
puede cambiar no sólo su actividad sino la velocidad a lacual se degrada su localización dentro de la célula, o puedeadquirir o perder la habilidad de interactuar con otrasmoléculas. En nuestros estudios de muerte celular autofá-gica, observamos que NR4A1 cambia de localización intra-celular, se degrada más lentamente e interactúa con ciertasproteínas; por lo tanto, propusimos que NR4A1 podríainducir la muerte autofágica, en particular, sólo cuando estéSUMOilada. Para probar esta hipótesis comparamos la secuen-cia de aminoácidos de NR4A1 con otras proteínas que separecen a ella y pertenecen a la misma familia filogenética, lla-madas NR4A2 y NR4A3, y buscamos sitios potenciales de
271

SUMOilación, como un lingüista buscaría las raíces etimoló-gicas de palabras semejantes. Resulta que las tres proteínasconservan los mismos aminoácidos en el contexto de secuen-cia conocido para ser unidas a SUMO, aumentando la proba-bilidad de que nuestras predicciones fueran ciertas. Investi-gadores de otros países pensaron lo mismo y publicaron queefectivamente NR4A3 se SUMOila en uno de esos aminoácidosque predijimos, aumentando nuestra confianza en nuestrahipótesis. Nos dimos a la tarea, entonces, de demostrar expe-rimentalmente que NR4A1 se SUMOila en los tres sitios quepredijimos, para lo cual sustituimos tales lisinas por argini-nas, un aminoácido al cual no se puede unir SUMO. Susti-tuímos una lisina a la vez, por parejas y las tres lisinas simul-táneamente. Encontramos que efectivamente NR4A1 seSUMOila en esos sitios y que cuando eso sucede disminuye sucapacidad para activar la expresión de sus genes blancos,para lo cual se sale del núcleo de las células. Lo más intere-sante fue descubrir que efectivamente, sólo cuando NR4A1se SUMOila puede inducir la muerte celular autofágica. Losexperimentos que hicimos para demostrar esto consistieronen eliminar la expresión del gen NR4A1 de las células, parapoder sustituirlo por el gen mutante con las diferentes sus-tituciones de lisinas. En ausencia de NR4A1 las células ya nose mueren por autofagia, cuando reintroducimos el gen sil-vestre (normal) las células sí se mueren. En cambio, si intro-ducimos el gen con argininas en lugar de lisinas, las célulasya no se mueren por autofagia. En conclusión, una misma proteína puede tener diferen-
tes funciones, dependiendo de las modificaciones químicasque se le hagan, que aquí ejemplificamos con NR4A1. El men-saje que quisiera transmitir es que, si bien con la secuencia-ción del genoma humano se generó una especie de decepciónpor descubrir que el número de genes que tenemos noresultó ser abrumadoramente mayor que el de otros orga-
272

nismos notablemente menos complejos, la variabilidad defunciones que pueden tener las proteínas de nuestro cuerposí son abrumadoramente mayores; no sólo por la gran varie-dad de modificaciones post-traduccionales que pueden reci-bir, como la fosforilación, SUMOilación, acetilación, metilación,glucosilación, lipidación, etcétera, sino por la gran variabi-lidad de proteínas que se pueden producir a partir de unmismo gen alterando la combinación de ARN así como losmensajeros que se pueden generar, entre otros mecanismos.Dado que una misma proteína puede tener diversas fun-
ciones es un reto contar con herramientas que permitanmodular cada una de sus actividades de manera específica.En el instituto Max Planck de Genética Molecular, en Ber-lín, el doctor Zoltán Konthur (quien actualmente trabajaen el Instituto Max Planck de Coloides e interfases) desa-rrolló un método para obtener moléculas pequeñas queinteractúan fuertemente y de manera muy específica con laproteína de interés. Estas dos características, en principio,permitirían obtener moléculas que interfirieran con unafunción particular, al competir y ganar la interacción de laproteína de interés con otra proteína o ADN. Por ejemplo,podríamos obtener una molécula pequeña que se una congran avidez al dominio de unión con otras proteínas (de trans-activación), de tal manera que no permita que otras molécu-las se unan a ese mismo dominio, sin alterar la habilidad deinteractuar con el ADN u otras moléculas en otros dominios.Este tipo de moléculas se llaman aptámeros. En experienciadel doctor Konthur, si la naturaleza de los aptámeros es deácidos nucléicos (ADN o ARN), no son capaces de ganar la com-petencia de la unión entre dos proteínas. En cambio, si losaptámeros fueran también de proteína, posiblemente sí sepodría impedir la interacción entre dos proteínas que natu-ralmente se unan. De entre las proteínas, los anticuerpossuelen tener afinidades muy altas y específicas con las regiones
273

de las proteínas que reconocen. Teniendo esta informaciónen consideración, el doctor Konthur desarrolló un métodopara aislar fragmentos de anticuerpos (justo la parte que reco-noce a la proteína blanco, conocida como región variable)que se unan específicamente a una proteína de interés. Porotro lado, el doctor Jörn Glöcker, también del Instituto MaxPlanck de Genética Molecular en Berlín, razonó que losaptámeros de ácidos nucléicos posiblemente sí competiríancon la interacción de la proteína con el ADN, especialmentelos de ARNporque suelen formar uniones más estables. El doc-tor Glöcker optimizó un método para aislar, en unos cuantosdías, aptámeros de ADN y de ARN que se unen a la proteínade interés.Gracias al apoyo recibido por parte de la Fundación Ale-
xander von Humboldt, tuve la oportunidad de hacer unaestancia de investigación en el laboratorio del doctor Kont-hur y aplicar su método para obtener proteínas pequeñasque se unieran a NR4A1 con la intención de impedir lainteracción de NR4A1 con otras proteínas, sin que perdierasu habilidad de inducir la expresión de genes blancos. Simul-táneamente, en el laboratorio del doctor Glöcker buscamosaptámeros de ARN y de ADN que se unieran a NR4A1, espe-rando encontrar alguno que, al pegarse al dominio de unióndel ADN de NR4A1, compitiera con la unión de NR4A1 a susgenes blancos, impidiendo así que induzca su expresión. Afortunadamente, encontramos varios aptámeros y actual-
mente estamos investigando si funcionan como esperamos.En caso de que resulte, podríamos utilizarlos para estudiarel mecanismo por el cual NR4A1 induce no sólo la muerteautofágica, sino cómo le hace para favorecer la estabilidaddel genoma, que es importante para evitar el surgimiento depadecimientos como el cáncer, o modular su efecto en elmetabolismo, y para controlar la obesidad y resistencia a lainsulina, enfermedades de preocupante incidencia en México.
274

LECTURAS SUGERIDAS PARA AMPLIAR LA INFORMACIÓN
Bouzas-Rodríguez J., Zarraga-Granados G., Sánchez-Carbente M. del R.,Rodríguez-Valentín R., Gracida X., Anell-Rendon D., Covarrubias L.y Castro-Obregón S. The nuclear receptor NR4A1 induces a formof cell death dependent on autophagy in mammalian cells. PLOSONE, 2012, 7(10): e46422. doi: 10.1371/journal.pone. 0046422.
Castro-Obregón S. The Discovery of Lysosomes and Autophagy. NatureEducation, 2010, 3(9): 49. Recuperado de la URL:http://www.nature.com/scitable/topicpage/the-discovery-of-lysosomes-and-autophagy-14199828
Fuchs Y. y Steller H. Programmed cell death in animal development anddisease. Cell, 2011, 147(4): 742-58. doi: 10.1016/j.cell.2011.10.033.
Lo A. S., Zhu Q. y Marasco W. A. Intracellular antibodies (intrabodies)and their therapeutic potential. Handb. Exp. Pharmacol., 2008,181 : 343-73.
Dahm R. Dying to see. Scientific American, 2004, 13 : 19.Feng Y., He D., Yao Z. y Klionsky D. J. The machinery of macroautophagy.
Cell Research, 2014, 24: 24-41. doi: 10.1038/cr.2013. 168.
275


LA SIMPLICIDAD DEL CEREBRO Y LA PERCEPCIÓN MUSICAL
¿TIENEN ALGO QUE VER CON LAS PROTEÍNAS?
HÉCTOR RASGADO-FLORESDepartamento de Fisiología y Biofísica
Universidad de Medicina y Ciencia Rosalind FranklinNorth Chicago, EUA
RESUMEN
El simposio Humboldt Kolleg sobre la intersección entre labioquímica y biofísica de las proteínas, las matemáticas y lashumanidades constituye una visita hacia el futuro. Conformeavanza la comprensión de nuestro entorno y de lo que somos,las barreras artificiales que separan las disciplinas del cono-cimiento se van desvaneciendo. Somos y estamos inmersosen un universo tan complejo que para tratar de entenderloy entendernos requerimos una visión multidimensionale interdisciplinaria.El diálogo establecido en esta reunión comenzó con una
discusión sobre las relaciones entre dos lenguajes aparente-mente muy distintos: el de las proteínas y el de los hombres,la lingüística. Seguidamente se discutió cómo el lenguajede las matemáticas sirve como herramienta para explicar quelas relaciones proteína-proteína establecen las bases de la bio-química y la fisiología. Finalmente, con esta presentación se
277

completa el ciclo al considerar la ma nera en que un lenguajehumano, el de la música, constituye el agente más poderosoque conocemos (aparte de las drogas psicotrópicas), capazde modificar nuestras emociones. La respuesta está en en -tender el cómo y el por qué el lenguaje musical afecta tanintensamente la bioquímica y fisiología cerebral. En otraspalabras, se trata de descubrir cómo es que oír música afectalas proteínas pertinentes en nuestro cerebro.
INTRODUCCIÓN
La música tiene un alto valor en las sociedades humanas.Confucio (500 a. C.) razonó que la música posee una fuerzamoral capaz de generar buena voluntad y armonía entrefamilias y comunidades. Platón (428 a. C.) afirma en suRepública que la música puede hacer ciudadanos mejoreso peores. En la Grecia clásica, el adjetivo “persona musical”se reservaba a individuos altamente educados y bondadosos,y G. F. Handel (1685-1759) afirmó que su música no teníael propósito de entretener, sino más bien el de hacer mejoresciudadanos.
Funciones del cerebro
Nosotros y nuestro universo somos tan complejos y mis-teriosos que, para sobrevivir, el propósito más importantede nuestro cerebro es encontrar un sentido comprensible,tanto de nosotros como de nuestro entorno. Para cumplircon este objetivo, nuestro cerebro —a través de los sentidos—escoge de entre la miríada cambiante de piezas de infor-mación que inciden en nosotros, analiza sólo la esencia quees pertinente y se las arregla para hacerla parecer “estable”y “significativa”. Con esta información, crea una imagen de
278

nosotros y de nuestro entorno, la cual es artificial y limitada,pero suficientemente compleja para permitir que nos adap-temos, crezcamos y nos reproduzcamos.
Las ondas de presión llegan a la membrana timpánica y cau-san que vibre a la misma frecuencia de las ondas sonoras. Eltímpano, por medio de la cadena osicular, transmite y ampli-fica la vibración hacia el fluido coclear. Este fluido vibra a lamisma frecuencia que el sonido y produce deflexiones enla membrana basilar. Esta membrana está sintonizada parasufrir una deflexión máxima dependiente de la frecuenciadel sonido. Las frecuencias bajas son detectadas máxima-mente en el ápice y las frecuencias altas en la porción basal.Conforme oscila la membrana basilar, las estereocilias o célu-las ciliadas en el órgano de Corti se sujetan a esfuerzos cor-tantes. La deflexión de las estereocilias resulta en la gene-ración de un potencial que puede producir potenciales deacción. Las corrientes iónicas en el órgano de Corti generanlos potenciales microfónicos cocleares. Estos potenciales
279
FIGURA 1. La imagen ilustra un ejemplo de una de las funciones de nuestro cere-bro: la audición. Este proceso comienza con la generación de ondas sonoras,que son ondas alternantes de presión del aire. Las ondas sonoras están deter-minadas por su amplitud (volumen), frecuencia (tono) y fase. El oído humanopuede detectar sonidos entre 20 y 18,000 Hz con una sensibilidad óptima alsonido de 1,000 a 3,000 Hz. Es interesante notar que este rango óptimo de fre-cuencias corresponde a las frecuencias que usamos cuando hablamos.

tienen una frecuencia idéntica a la frecuencia del sonido.Hay una organización tonotópica del órgano de Corti. Estaorganización se mantiene a través de las trayectorias aferen-tes, así como en la corteza auditiva (temporal). Una vez quese activa la corteza auditiva, tiene lugar la percepción dellenguaje y de la música en la corteza. Conforme estas expe-riencias se graban en nuestros cerebros, las percepcionespueden suscitarse aún en ausencia de los estímulos que lasoriginaron. Por ejemplo, podemos recordar palabras, ora-ciones y libros completos en ausencia de sonidos y podemoscomunicarnos susurrando palabras. En este caso, no hay soni-dos asociados con las palabras y nuestro cerebro aún es capazde reconocer los conceptos que se suscitan. Un ejemplo máselaborado es el hecho de que se puede provocar la percep-ción de música como resultado de recordar una pieza musi-cal o leer una partitura de música en ausencia de sonidos. Másaún, las respuestas emocionales y estéticas asociadas conescuchar música pueden ser evocadas en ausencia de seña-les auditivas, mediante la activación directa de actividad neu-ronal asociada con la percepción musical.Es interesante notar que la mayoría de las fibras nerviosas
que conectan al órgano de Corti con el cerebro no trans-miten información al cerebro, sino que llevan informacióndel cerebro al órgano de Corti. En el órgano de Corti hayunas 3,500 células ciliares interiores que están inervadas porneuronas aferentes (que transmiten información al cerebro)y 20,000 células ciliares exteriores que están principalmen-te inervadas por neuronas eferentes (que transmiten infor-mación desde el cerebro) (Livingston, 1990). Tal arreglo des-taca la capacidad de nuestro cerebro para modular y filtrarla información que recibimos. Un ejemplo excelente es unexperimento realizado por el doctor Izquierdo, quien regis-tró los potenciales evocados de la corteza auditiva de ungato al escuchar un clic. Cuando se le mostraba un ratón al
280

gato, los potenciales evocados desaparecían aunque los clicscontinuaran sin interrupción. Esta capacidad de nuestrocerebro de filtrar información innecesaria, de hacer que lainformación que nos rodea parezca “estable” para facilitarnuestra supervivencia, puede llamarse “simplicidad”.
Simplicidad del cerebro
Nuestro cerebro está urgido de darle sentido al universopara ayudarnos a sobrevivir, adaptarnos, crecer y reprodu-cirnos. Para lograr este objetivo, el cerebro filtra la informa-ción total que nos rodea y selecciona la mínima informaciónnecesaria para hacer que parezca simple y estable. Cuatroejemplos ilustran este principio: 1) Reconstrucción aural. Nuestro cerebro está tan necesitado
de darle sentido a su entorno que pacientes profundamentesordos desarrollan ilusiones aurales; “oyen” música que leses muy familiar, como canciones de cuna o marchas (Sacks,2007). Se puede interpretar esto como algo que su giere quesu sistema auditivo está tan desesperado por recibir infor-mación para interpretar su entorno, que incluso en ausenciade tal información recurre a “fabricarla”. Estas ilusiones desa-parecen si dichos pacientes reciben implantes cocleares. Elhecho de que dichos implantes realmente funcionen paraayudar a los pacientes a oír, también constituye otro ejem-plo de la necesidad desesperada que nuestro cerebro tienede darle sentido a su entorno. Los implantes cocleares sondispositivos electrónicos implantados quirúrgicamente queconsisten de un sensor de sonido que se implanta cerca de laoreja del paciente. La activación de este sensor por el sonidoes seguida por la transmisión de esta información a un recep-tor implantado dentro del cráneo del paciente. Dependiendode la frecuencia del sonido recibido, el sensor produce
281

señales eléctricas que viajan a la cóclea a través de alambresy activan a la cóclea en el área apropiada para dicha fre-cuencia. Un arreglo de hasta 24 electrodos (que sustituyena las 16 mil delicadas células ciliares que se usan en la audi-ción normal) se insertan en la cóclea, que envía los impulsosa los nervios de la rampa timpánica y de ahí directamente alcerebro a través del sistema nervioso auditivo. Más de 200 milpersonas en todo el mundo han recibido implantes cocleares.Estos implantes pueden restaurar audición suficiente parapermitir la comprensión del habla sin asistencia en un fondosilencioso, pero la audición que ofrece es muy limitada para laapreciación musical o para la comprensión del habla enambientes ruidosos. El hecho de que 24 electrodos sean sufi-cientes para rescatar la capacidad de oír el habla apunta a lanecesidad desesperada de nuestro cerebro por “oír” su entor-no, y la extrema importancia del habla y de la audición delhabla para que nuestro cerebro se comunique y nos permitasobrevivir.2) Reconstrucción visual. El Departamento de Energía de
los Estados Unidos ha desarrollado una prótesis de retinamicroelectrónica implantable que restaura la visión útil a per-sonas cegadas por enfermedades de la retina, tales como reti-nitis pigmentosa y la degeneración macular relacionada conla edad. El implante consiste en una cámara miniatura loca-lizada en anteojos usados por el paciente. Las cámaras regis-tran imágenes del entorno y las transmiten de modo inalám-brico a una retina artificial que consiste en una película consensores implantada en la retina de los pacientes. A la fechase han implantado retinas artificiales con 16 y 60 electrodos.Vale la pena notar que con este número tan limitado de elec-trodos los pacientes pueden reconocer caras. Esta hazañaapoya nuevamente la necesidad desesperada que tiene nues-tro cerebro por darle sentido a la información que recibepara ayudarnos a comunicarnos y sobrevivir.
282

3) Fabricación de orden aparente. Un corolario del principiode simplicidad en nuestros cerebros es el hecho de que, conel propósito de darle sentido a nuestro entorno, nuestroscerebros pueden fabricar un orden aparente escogiendoselectivamente de entre la información que se les presentasólo la que es pertinente para hacernos creer que nuestro en -torno es estable y que tiene sentido. La doctora Diana Deutch(1995) ha desarrollado varios experimentos para demostrareste principio. Tres de estos ejemplos son la ilusión de laoctava, la ilusión de la escala y el reconocimiento de melodía(véanse a continuación). Además, ya sea conscientementeo no, las ilusiones musicales han sido efectivamente usadaspor los compositores (véase a continuación).i) Ilusión de la octava. Al escuchar con audífonos estereofó-
nicos, en el oído derecho, un patrón de notas de una octava(por ejemplo, sol alto sol bajo) mientras se escucha simul-táneamente, en el otro oído, el patrón invertido (es decir,sol bajo sol alto) produce en la gran mayoría de los oyen-tes un patrón simplificado en la que un oído escucha sol bajo
silencio sol alto, en tanto que el otro escucha silencio sol alto silencio. Así, el sujeto percibe un sol alto y unobajo que se alternan de un oído al otro. Sin importar la posi-ción de los audífonos, los individuos diestros escuchan elpatrón opuesto al que escuchan los zurdos.ii) Ilusión de la escala. Si se le presentan simultáneamente
patrones de notas a cada oído y ninguno contiene una escala,y si una combinación de notas alternadas presentadas a cadaoído constituye una escala ascendente o descendente (diató-nica o cromática), el cerebro del sujeto reacomoda la percep-ción de las notas de modo que lleva a “escuchar” en efectouna escala en cada oído. En otras palabras, el cerebro sim-plifica la información recibida para hacerla parecer mássimple y más ordenada.
!
!
! ! !!
283

iii) Reconocimiento de melodías. Si una melodía muy cono-cida (por ejemplo, Mary Had a Little Lamb— María tenía uncorderito) le es presentada a un oyente con la secuencia denotas correcta pero con la posición de las notas revuelta, esdecir, se tocan las notas al azar en escalas bajas, medianaso altas, algunos oyentes con formación musical pueden reco-nocer la melodía pero la mayoría de los no músicos no podránreconocerla. No obstante, si a estos oyentes se les expone a lamelodía interpretada con las posiciones correctas de las notas,una nueva exposición a la melodía revuelta será reconocidapor la mayoría de los oyentes. iv) Ilusiones musicales. Federico Chopin (1810-1849) no era
fisiólogo ni psicólogo, pero entendía sobre la percepción delsonido en nuestro cerebro. Escribió 24 estudios en todas lastonalidades, mayores y menores, con el propósito de formarpianistas. Todas son piezas musicales magníficamente tra-bajadas. El Estudio no. 13, op. 25 en la bemol tiene la caracterís-tica notable de que hace uso de la capacidad de nuestro cere-bro de simplificar su insumo de información para hacerloparecer más simple. Esta pieza tiene 2,278 notas pero, cuan-do se interpreta, el oyente sólo escucha realmente unas 380notas. La figura 2 muestra los primeros tres compases deesta composición. Las notas encerradas en círculos son lasprincipales que escucha el oyente. La pieza suena agradable,
284
FIGURA 2. Primeros tres compases del Estudio no. 13, op. 25 en la bemol de FedericoChopin.

gratificante y melodiosa porque todas las notas escritas tra-bajan juntas para producir ese efecto.Otro ejemplo de una ilusión musical es el último movi-
miento de la Sexta Sinfonía de Piotr Ilich Chaikovski (1840-1893), su última composición. Muchos eruditos argumentanque él sabía que iba a morir, porque en realidad se suicidó.En todo caso, escribió esta sinfonía como una despedida dela vida justo antes de morir. El último movimiento es extre-madamente triste y expresivo. Comienza con una melodíasimple y evocadora, interpretada por los violines. La observa-ción de la partitura (figura 3) ilustra que la percepción de talmelodía es, de hecho, una ilusión musical. La melodía quenuestros cerebros perciben no es tocada por los primeros nipor los segundos violines; de hecho es una combinación delas notas que ambos grupos de violines tocan. Ninguna de lasmelodías que tocan los primeros o los segundos violines esparticularmente memorable. Sin embargo, la combinaciónapropiada de notas de ambas melodías produce la muybella y memorable melodía que cualquier oyente realmentepercibe. Las notas que constituyen esta melodía están ence-rradas en círculos rojos en la partitura (figura 3). El hechode que en realidad escuchemos una melodía que resulta dela combinación de las notas tocadas por los dos grupos
285
FIGURA 3. Fragmento de la partitura de la Sexta Sinfonía de Piotr Ilich Chaikovski.

de violines ilustra la capacidad y la necesidad que nuestroscerebros tienen de simplificar y “embellecer” la informaciónque incide sobre nosotros todo el tiempo. Chaikovski en -tendía esto y creó una ilusión musical muy efectiva.
LA MÚSICA, LA FÍSICA, LAS MATEMÁTICAS Y LAS EMOCIONES
Pitágoras (570-495 a. C.) descubrió que pulsar una cuerdala hace vibrar entera, así como en mitades, tercios y así suce-sivamente. La vibración más baja (fundamental) se generapor la vibración de la cuerda entera y transmite el tono delsonido; las demás vibraciones son sus armónicas. Los mate-máticos llaman a una sucesión de números como 1, 2, 3, 4,5, 6…una sucesión armónica. En la música, las notas de fre-cuencias, que son múltiplos enteros de la fundamental, sellaman armónicas. Pitágoras estableció las relaciones mate-máticas sobre cómo se pueden conseguir tonos (notas) dife-rentes variando la longitud, grosor y tensión de la cuerda.Pitágoras también descubrió que al dividir la longitud de lacuerda en ciertas proporciones se producen tonos diferentes.Por ejemplo, si la vibración fundamental o principal alcan-zada por la cuerda era un do, dividir la cuerda en ½ produ-cía una octava (el do siguiente); 1/3 producía una quinta(sol); ¼ producía una octava más alta (do) y, lo que era muyimportante, si la cuerda se dividía en fracciones de 1/8 a 1/16se producía la escala diatónica (do-re-mi-fa-sol-la-si-do).¿Por qué algunos intervalos musicales suenan agradables
en tanto que otros suenan desagradables? Pitágoras tam-bién descubrió que pulsar dos cuerdas simultáneamente pro-ducía dos notas (intervalos musicales) que en algunos casostenían un sonido agradable, en otros moderadamente desa-gradable e inclusive en algunos casos el sonido era muydesagradable. Pitágoras derivó las relaciones matemáticas
286

de esos intervalos y encontró que cuando la razón de losmismos era simple, el sonido era agradable; pero cuando larelación era complicada, el sonido era desagradable. Encontróademás que había una relación directa entre la belleza delin tervalo musical y la simplicidad de la razón entre los dossonidos. Esto se ilustra en la figura 5. La figura muestratodos los intervalos que se pueden obtener de tocar las 12notas de una escala cromática en un instrumento musicalcomo el piano. Por ejemplo, si la primera nota es un Do, las12 notas serían Do-Do#-Re-Re#-Mi-Fa-Fa#-Sol-Sol#-La-La#-Si.Si el primer Do se toca junto con la nota inmediata superior(Do#), constituiría una segunda menor. La razón de las fre-cuencias de estas notas es 256:243, un número más biencomplicado para entender y escuchar; el sonido resulta muy
287
FIGURA 4. En la imagen se muestran las consecuencias físicas de pulsar una cuerdacuya frecuencia fundamental es un do muy bajo con frecuencia de 33 Hz. Lafigura muestra las armónicas 8 a 16 de esta frecuencia fundamental y las fre-cuencias en Hz de esas armónicas. La figura ilustra que 8 de las armónicas dela nota fundamental constituyen, de hecho, la escala diatónica (del do4 en elcentro de un piano al siguiente do, do5). Estas armónicas son el producto de mul-tiplicar la frecuencia fundamental (es decir, 33 Hz) por los enteros de 8 a 16.Estrictamente estas notas pueden ser armónicas sólo si las razones de sus fre-cuencias son exactamente números enteros. En la vida real, por razones mecá-nicas y físicas no todas estas notas corresponden a múltiplos enteros de la fun-damental, especialmente para la armónica 13 (la).

desagradable. Sin embargo, si el intervalo fuera de una quinta,es decir, un do y un sol, la razón, 3:2, sería muy simple y elsonido resultaría muy agradable al oído. Así, ¡Pitágoras logróel notable avance de relacionar los números con la aprecia-ción de la belleza!, y más aún, la relación entre la compleji-dad de los intervalos de sonido y lo desagradable del sonido:apoya nuevamente la idea de que nuestros cerebros buscanla simplicidad para darle sentido a nuestro entorno. Las rela-ciones matemáticas o las ondas sonoras complicadas sonrechazadas por nuestros cerebros como algo que genera unasensación des agradable. Esto se ilustra de modo más patenteen la figura 6. La figura 6A ilustra las frecuencias armónicasde sonido que se obtienen cuando un tono puro se mantieneconstante (línea roja horizontal), en tanto que se cambia lafrecuencia de otro tono comenzando en la misma frecuenciadel tono estacionario y elevándola hasta llegar una octava másarriba (línea roja inclinada). Tanto la abscisa como la orde-nada son frecuencias de sonido. Las razones de las frecuencias
288
$$
Si el primer Do se toca
j La razón de las
f
u
simple y el sonido resultaría muy agradable al oído. Así,
¡Pitágoras logró el notable avance de relacionar los números
con la apreciación de la belleza! Más aún, la relación entre la
complejidad de los intervalos de sonido y lo desagradable del
sonido apoya nuevamente la idea de que nuestros cerebros
buscan la simplicidad para darle sentido a nuestro entorno.
Las relaciones matemáticas o las ondas sonoras complicadas
son rechazadas por nuestros cerebros como algo que genera
una sensación desagradable. Esto se ilustra de modo más
p La Figura 6A ilustra las frecuencias
a
Tanto la abscisa como la ordenada son frecuencias de sonido.
L
La Figura 6B muestra las ondas sonoras que en efecto
s Las
l Claramente, ya sea que
s
!"#$%&'()*!
!+(,-#!
.#/0*#*!1%23#4(!5%#*&!1%23#4(!5(6*&!7%&8%&(!5%#*&!7%&8%&(!5(6*&!93(&$(!93(&$(!(35%#$(4(!:3;#$(!1%<$(!5%#*&!1%<$(!5(6*&!1=>$;5(!5%#*&!1=>$;5(!5(6*&!?8$('(!!
@A@!BCDABEF!GAH!FBABI!H@ADE!EAF!IBGAC@B!FAB!@BHAH@!BIA@D!@DAG!BEFA@BH!BA@!
FIGURA 5. Intervalos obtenidos de tocar las 12 notas de la escala cromática.

de sonido en los puntos indicados con letras son: a=1:1;b=15:16; c=4:5; d=2:3; e=20:31; f=30:59; g=1:2. La figura 6Bmuestra las ondas sonoras que, en efecto, se producen cuan-do se tocan simultáneamente las dos notas de los intervalos(de la a a la g). Las letras a a la g corresponden a las fre-cuencias en los trazos de la derecha. Claramente, ya sea quese vean las razones de las frecuencias de sonido generadascuando los dos sonidos se producen simultáneamente (figu-ra 6A) o las ondas sonoras que de hecho se generan condichos sonidos (figura 6B), la conclusión es la misma: a nues-tros cerebros les resulta más agradable encontrarse con razo-nes armónicas simples, cuyas ondas sonoras se repiten másrápido en el tiempo (por ejemplo, a, c, d y g) que cuando losdos sonidos simultáneos producen una razón armónica com-pleja y cuyas frecuencias sonoras tardan más tiempo en repe-tirse (por ejemplo, b, e y f ).Por lo tanto, hay una correlación estricta y directa entre
qué tan rápido se repite la frecuencia del sonido y qué tanagradable suena el intervalo. Entonces, ¿la interpretaciónemocional de la música es un proceso aprendido o intuitivo?
289
FIGURA 6. Figuras armónicas de sonido (modificada de Taylor, 1992).
$$
Encontraron una tribu que vive en las montañas Mandara de
C Realizaron varias
p
La Figura 7 exhibe algunos de los resultados. La Figura 7A muestra que cuando
l
Figura 6. Figura 6B
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
P
$$
$$
$$
$$
$$
r
$$
a
$$
p
$$
$$
o
$$
c
$$
z
$$
v
$$
o
$$
r
$$
d
$$
d
$$
d
$$
r
$$
!
A B
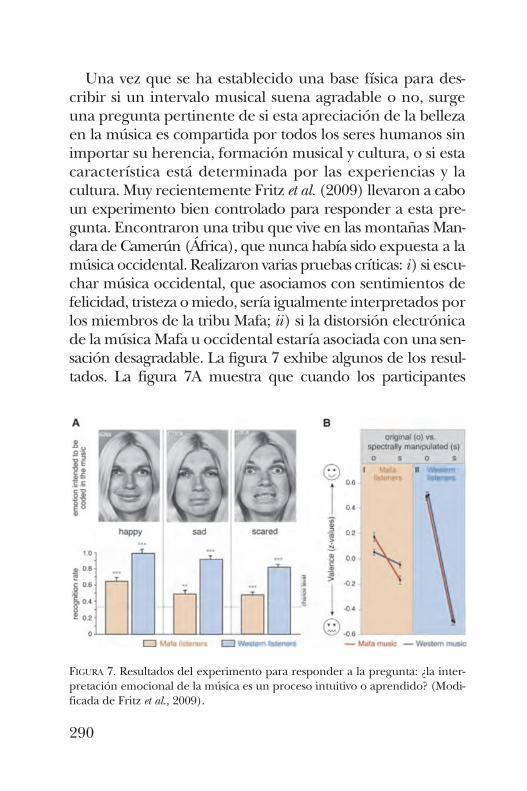
Una vez que se ha establecido una base física para des-cribir si un intervalo musical suena agradable o no, surgeuna pregunta pertinente de si esta apreciación de la bellezaen la música es compartida por todos los seres humanos sinimportar su herencia, formación musical y cultura, o si estacaracterística está determinada por las experiencias y lacultura. Muy recientemente Fritz et al. (2009) llevaron a caboun experimento bien controlado para responder a esta pre-gunta. Encontraron una tribu que vive en las montañas Man-dara de Camerún (África), que nunca había sido expuesta a lamúsica occidental. Realizaron varias pruebas críticas: i) si escu-char música occidental, que asociamos con sentimientos defelicidad, tristeza o miedo, sería igualmente interpretados porlos miembros de la tribu Mafa; ii) si la distorsión electrónicade la música Mafa u occidental estaría asociada con una sen-sación desagradable. La figura 7 exhibe algunos de los resul-tados. La figura 7A muestra que cuando los participantes
290
$$
La figura muestra que tanto los sujetos Mafa (rectángulos rojos) como los
o
Estos valores fueron significativamente
m Es interesante
n La Figura 7B
i
Nuevamente las respuestas de los
o
No obstante, esta componente se puede
r
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
$$
FIGURA 7. Resultados del experimento para responder a la pregunta: ¿la inter-pretación emocional de la música es un proceso intuitivo o aprendido? (Modi-ficada de Fritz et al., 2009).

(Mafa y occidentales) escuchaban la música tenían quedecidir cuál de las tres caras (que ilustran expresiones emo-cionales) correspondía mejor al estímulo musical percibido.La figura muestra que tanto los sujetos Mafa (rectángulosrojos) como los de Occidente (rectángulos azules) tuvieroninterpretaciones similares del valor emocional de la músicaque escucharon (***p < 0.001, **p < 0.05). Estos valoresfueron significativamente mayores que el nivel de azar (1/3)representado por la línea horizontal punteada. Es interesantenotar que los sujetos occidentales tuvieron respuestas másfuertes que los Mafa. La figura 7B ilustra que tanto los suje-tos Mafa como los occidentales interpretaron como desa-gradable el escuchar la música distorsionada, fuera Mafau occidental. Nuevamente, las respuestas de los oyentes occi-dentales fueron más fuertes que las de los Mafa.Estos resultados demuestran que hay un fuerte compo-
nente innato que determina nuestra respuesta emocional alescuchar música. No obstante, dicho componente se puedereforzar significativamente mediante la formación cultural.
CONCLUSIONES
1) La función principal de nuestro cerebro es la de ayu-darnos a sobrevivir y por esta razón manipulamos la inter-pretación de nuestro entorno para hacerlo parecer “estable”,que “tiene sentido”, es “ordenado” y “bello”. Esta funciónpuede llamarse el Principio de simplicidad.2) La apreciación de lo agradable, por parte de nuestros
cerebros, está correlacionada con la simplicidad de los estímu-los con los que nos encontramos.3) Las respuestas emocionales al escuchar música tienen
componentes tanto innatos (naturaleza) como culturales(crianza).
291

AGRADECIMIENTOS
El autor está en deuda con el doctor Ernest Sukowskipor su lectura crítica del manuscrito, con la doctora AliciaOrtega por apoyar y alentar la escritura del mismo y conTania Gitler por su apoyo en la traducción inicial delmanuscrito.
REFERENCIAS
Deutsch D. Musical Illusions and Paradoxes. Philomel Records. La Jolla,California, 1995.
Fritz T., Jentschke S., Gosselin N., Sammler D., Peretz I. y Turner R.Universal Recognition of Three Basic Emotions in Music. CurrentBiology, 2009, 19, 573-576, Issue 7, 2009.
Sacks O. Musicophilia. Vintage Press, 2007.Taylor C. Exploring Music. Institute of Physics Publishing. Bristol & Phi-
ladelphia, 1992.
292

Proteínas. En la intersección entre las matemáticas, la física, laquímica y la biología se terminó de imprimir y encuader-nar el 30 de enero de 2015 en HEMES IMPRESORES,Cerrada Tonantzin 6, Col. Tlaxpana, Deleg. MiguelHidalgo, C. P. 11370, México, D. F. En su composiciónse usó tipo New Baskerville 12:14, 10:12, 9:11 puntos. Laedición consta de 1000 ejemplares. Coordina ción edi-torial: María Elena Ávila Urbina. Corrección: AlejandraRuiz Díaz. Cuidado de la edición: Mario Medina Márquez.
