profile searches revised 07/11/06. overview introduction motif representation motif screening motif...
TRANSCRIPT

Profile Searches
Revised 07/11/06

Overview
• Introduction
• Motif representation
• Motif screening
• Motif Databases
• Exercise

Features characteristic for the whole family
Multiple sequence alignment
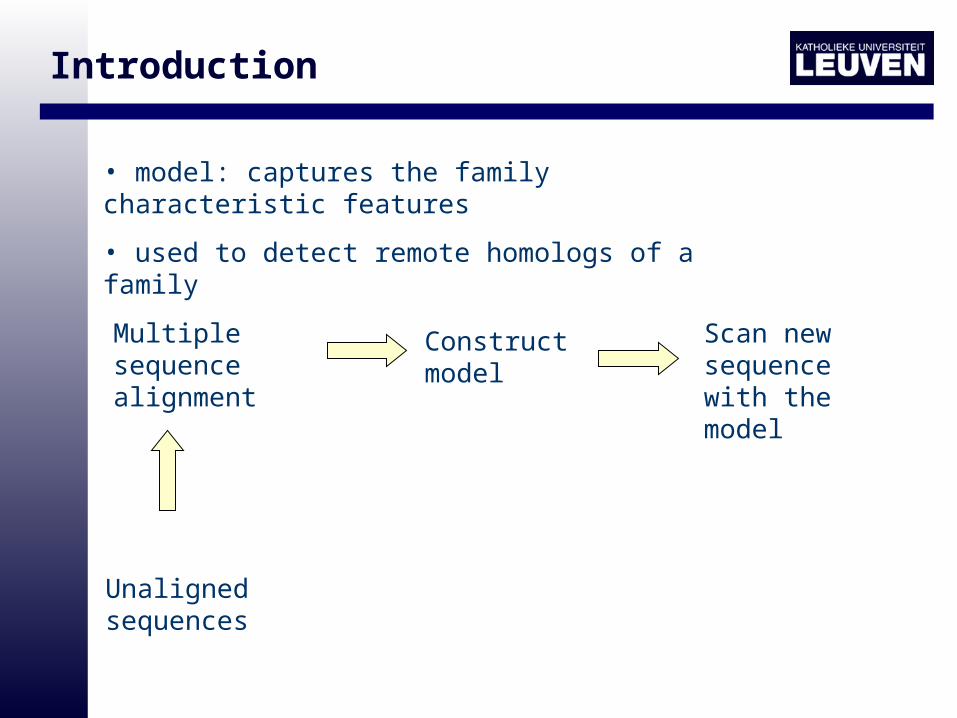
Introduction
How to represent the characteristic features?
Motif model: captures the family characteristic features
• regular expression, weight matrix, HMM profile
Introduction
Multiple sequence alignment
Construct model
Scan new sequence with the model
Unaligned sequences
• model: captures the family characteristic features
• used to detect remote homologs of a family

Overview
• Introduction
• Motif representation– String based representation
• Consensus
• Regular expression
– Probabilistic representation
• PSSM
• HMM
• Profile
• Motif screening
• Motif Databases
• Exercise
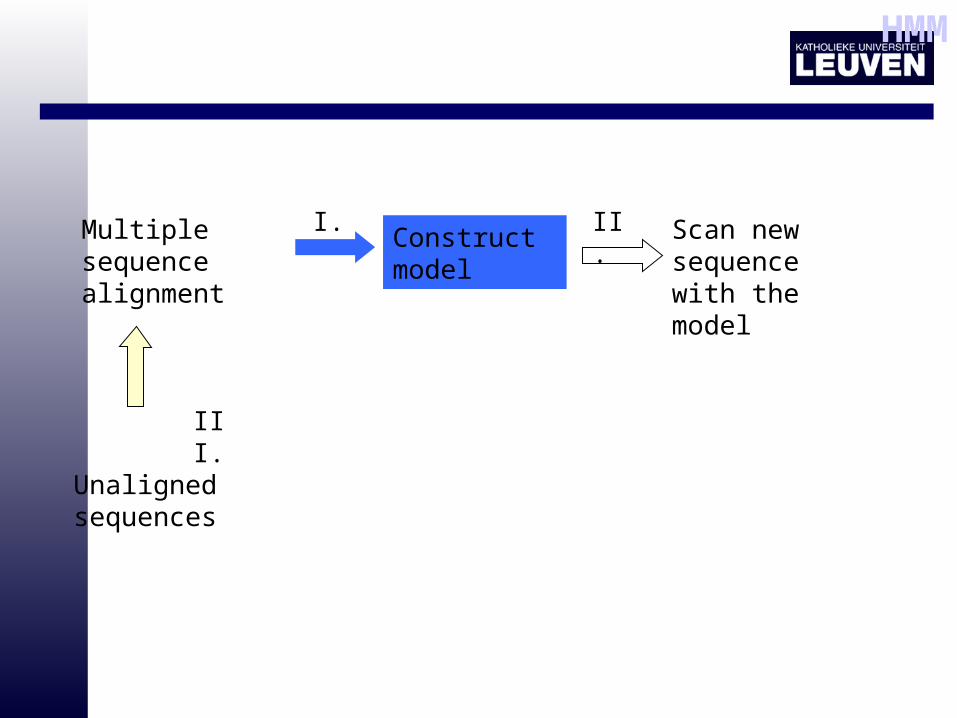
HMM
Multiple sequence alignment
Construct model
Scan new sequence with the model
I. II.
Unaligned sequences
III.

Consensus sequence:
– Reductionistic representation of a motif
– Most frequent instance is used as a representative
– Loss of information
Regular expression:
– More complex representation allowing motif degeneracy
String Based Representation
Symbol Meaning Origin of designation G G Guanine A A Adenine T T Thymine C C Cytosine R G or A puRine Y T or C pYrimidine M A or C aMino K G or T Keto S G or C Strong interaction (3 H bonds) W A or T Weak interaction (2 H bonds) H A or C or T not-G, H follows G in the alphabet B G or T or C not-A, B follows A V G or C or A not-T (not-U), V follows U D G or A or T not-C, D follows C N G or A or T or C aNy

CTTAATATTAACTTAATConsensus
CTTAAKRTTMAYTTAATRegular expression
String Based Representation
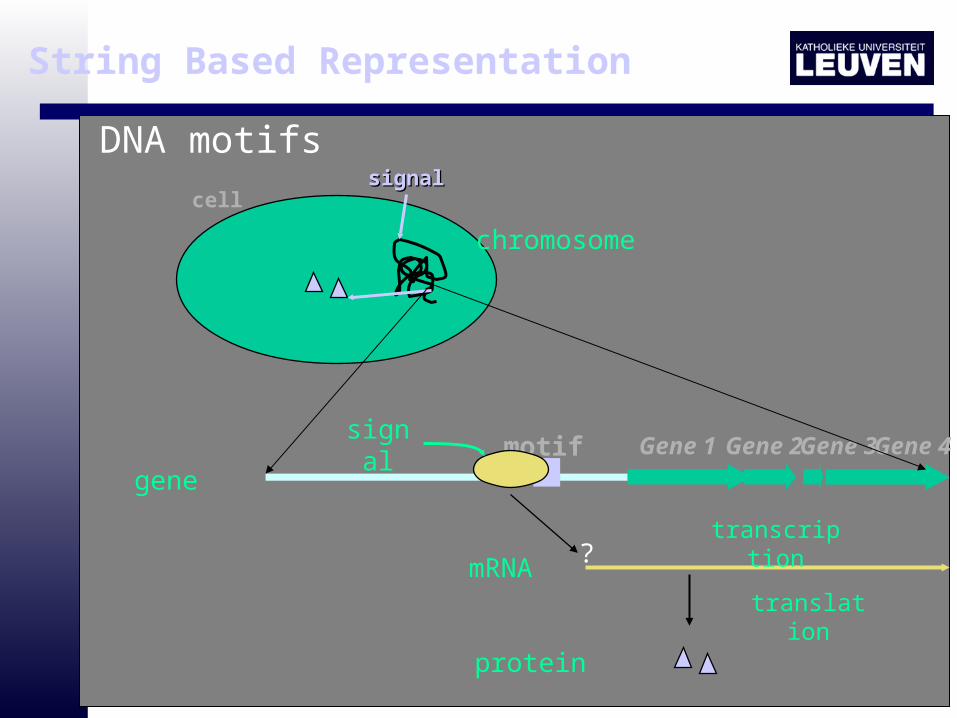
cellsignalsignal
motif Gene 1 Gene 2Gene 3Gene 4signal
?
translation
transcription
mRNA
protein
gene
chromosome
DNA motifs
String Based Representation

Sequences involved in enzymatic reactions (PROSITE)
String Based Representation

Overview
• Introduction
• Motif representation– String based representation
• Consensus
• Regular expression
– Probabilistic representation
• PSSM
• HMM
• Profile
• Motif screening
• Motif Databases
• Exercise

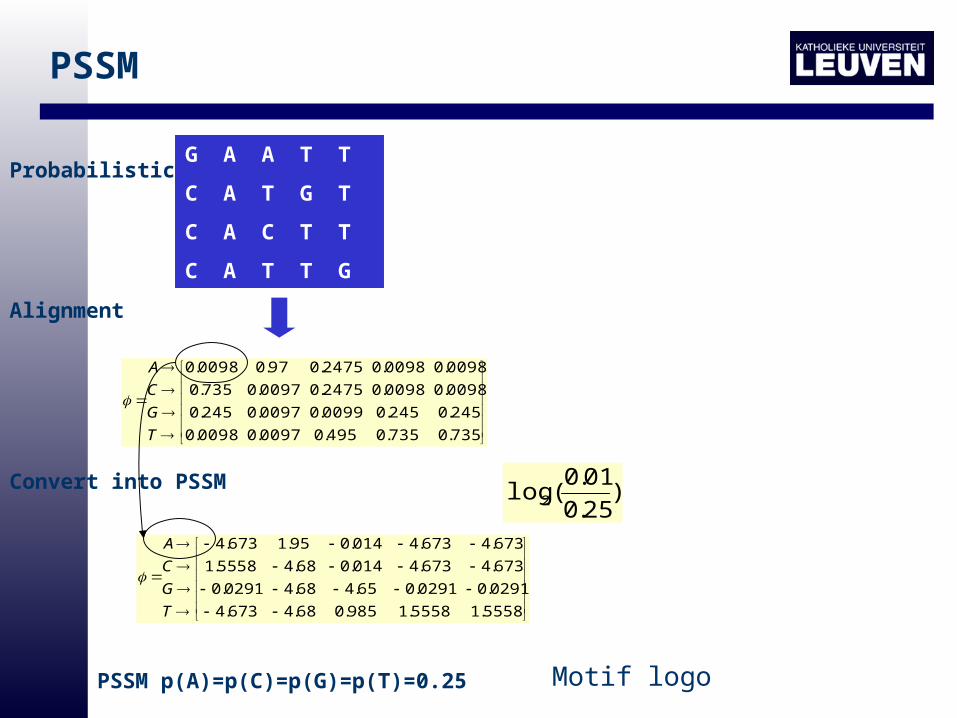
Probabilistic
PSSM
Frequency matrix
wTTT
wGGG
wCCC
wAAA
,...2,1,
,...2,1,
,...2,1,
,...2,1,
G A A T T
C A T G T
C A C T T
C A T T G
75.075.05.001.001.0
25.025.001.001.025.0
01.001.025.001.075.0
01.001.025.0101.0
T
G
C
A
75.075.05.00.00.0
25.025.00.00.025.0
0.00.025.00.075.0
0.00.025.010.0
T
G
C
A
Pseudo Counts
Frequency matrix
Alignment
735.0735.0495.00097.00098.0
245.0245.00099.00097.0245.0
0098.00098.02475.00097.0735.0
0098.00098.02475.097.00098.0
T
G
C
A
735.0735.0495.00097.00098.0
245.0245.00099.00097.0245.0
0098.00098.02475.00097.0735.0
0098.00098.02475.097.00098.0
T
G
C
A
5558.15558.1985.068.4673.4
0291.00291.065.468.40291.0
673.4673.4014.068.45558.1
673.4673.4014.095.1673.4
T
G
C
A
Probabilistic
PSSM
G A A T T
C A T G T
C A C T T
C A T T G
Convert into PSSM
Alignment
PSSM p(A)=p(C)=p(G)=p(T)=0.25
)25.0
01.0(log2
Motif logo
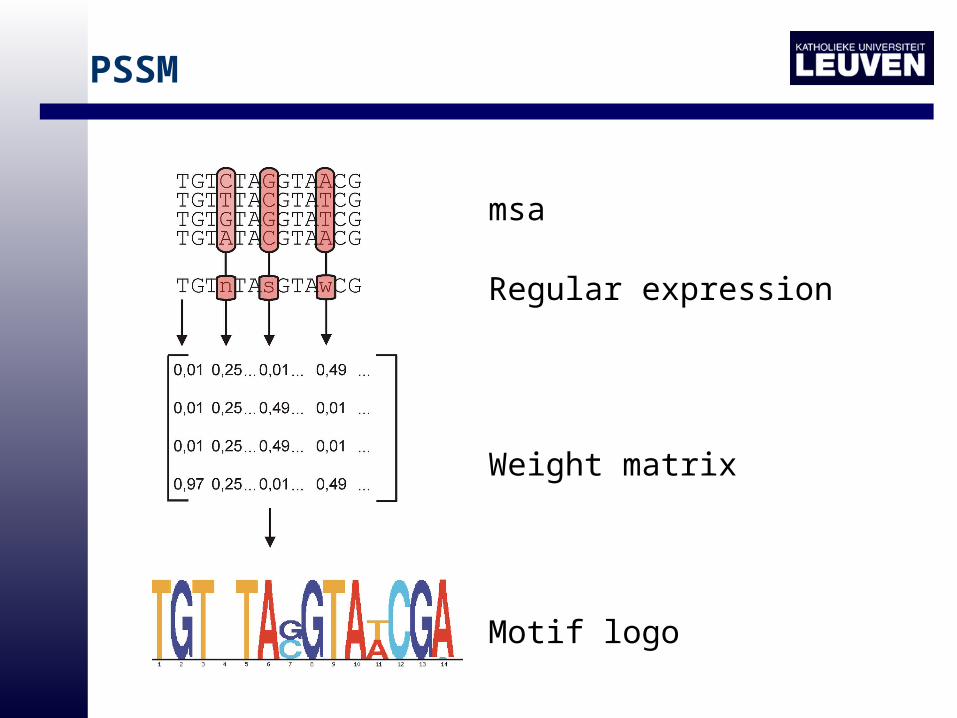
PSSM
msa
Regular expression
Weight matrix
Motif logo
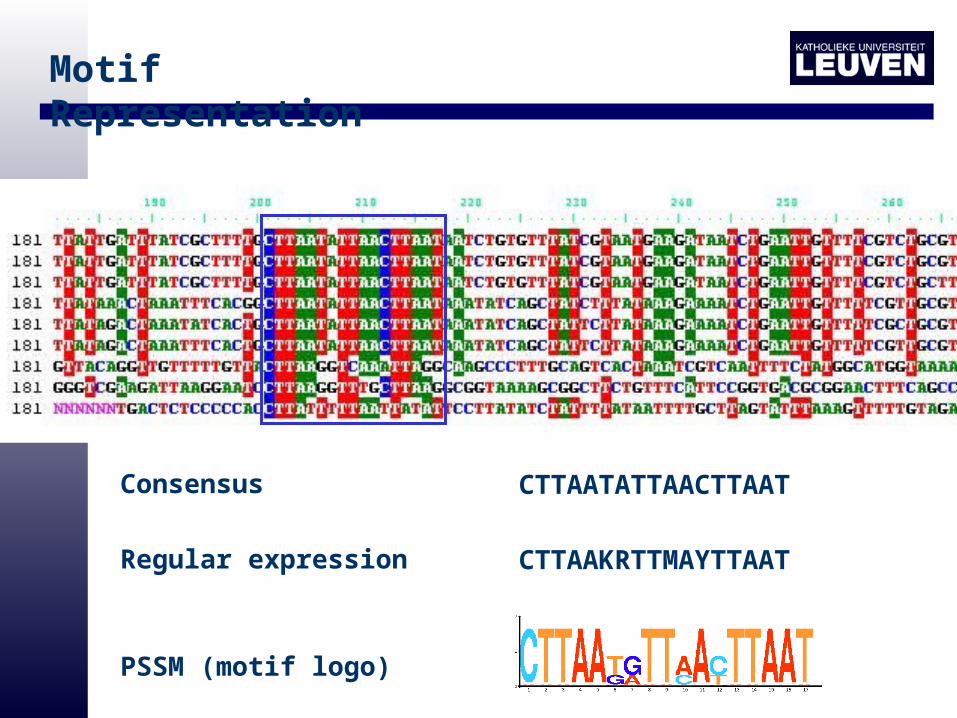
Motif Representation
CTTAATATTAACTTAATConsensus
CTTAAKRTTMAYTTAATRegular expression
PSSM (motif logo)
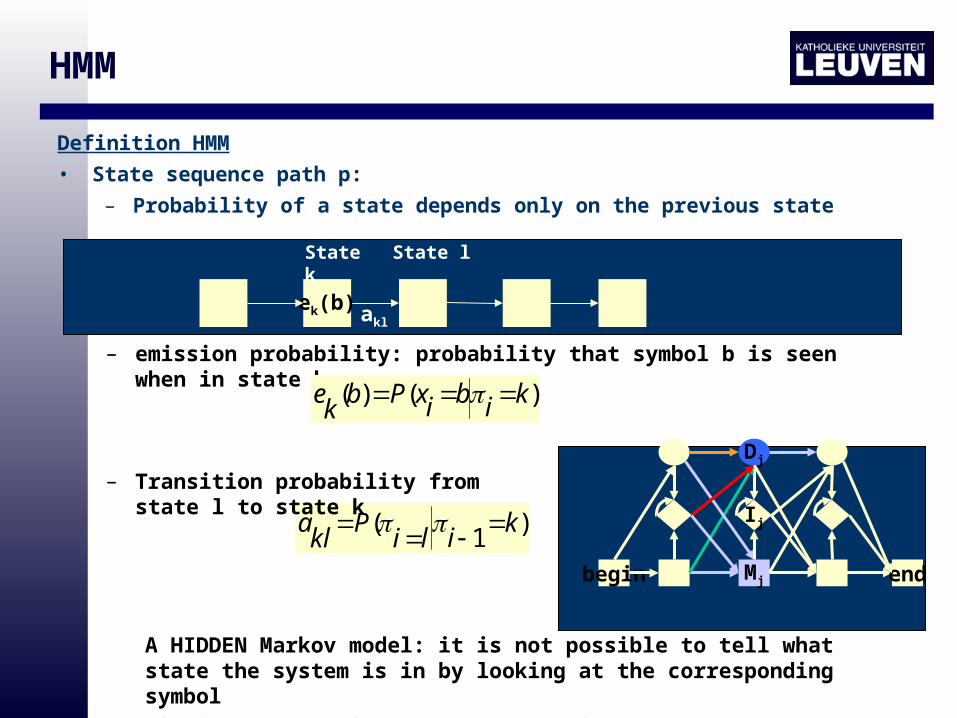
Definition HMM
• State sequence path p:
– Probability of a state depends only on the previous state
)1
( kili
Pkla
– Transition probability from state l to state k
– emission probability: probability that symbol b is seen when in state k
)()( kibixPbke
aklek(b)
State lState k
A HIDDEN Markov model: it is not possible to tell what state the system is in by looking at the corresponding symbol
Finding the possible paths = decoding
begin Mj
Ij
Dj
end
HMM

HMM
Probabilistic model that represents the alignment of the family
– Gapped multiple alignment
– Distinct states separated by transition probabilities (i.e. the probability of moving from one state to the next)
– The current stateis only dependent on the previous state (first order Markov process)
– The sequence of states followed in the model is called the path
– Each state has the probability of emitting a certain symbol of the alphabet (A,C,T,G for DNA) or one of the 20 amino acids for proteins: emission probability

• HMM can model any possible sequence
• It defines a probability distribution over the whole space of sequences
• Training a HMM: search for the parametrisation that makes this distribution peak around members of the family
• Parametrisation
– Determine model structure
• Length of alignment
• Number of insert states
– Determine the probability parameters
HMM
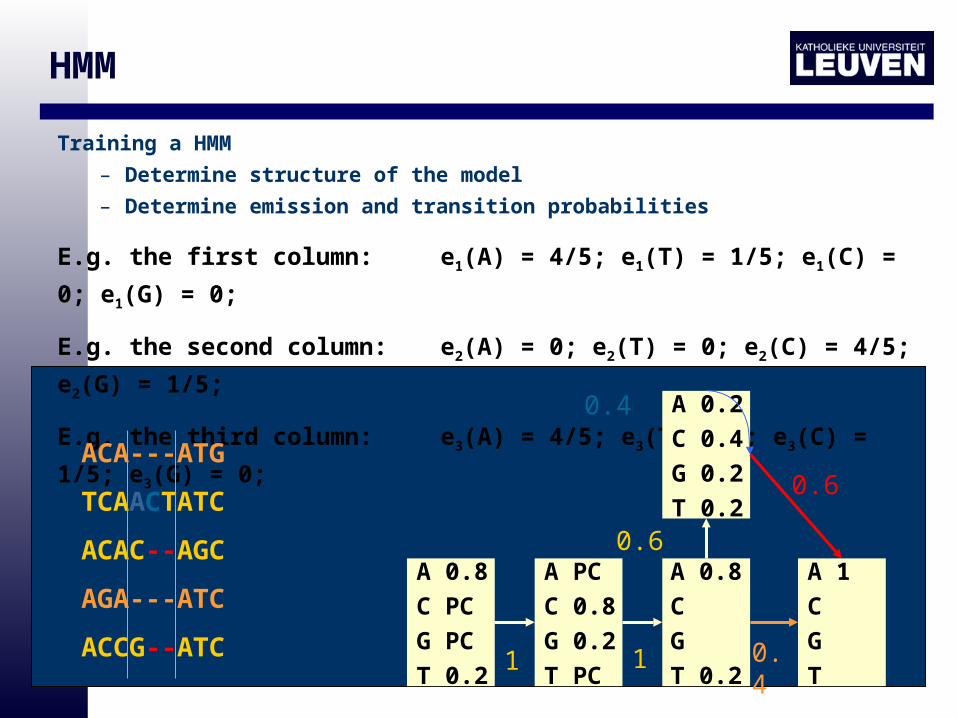
HMM
Training a HMM
– Determine structure of the model
– Determine emission and transition probabilities
E.g. the first column: e1(A) = 4/5; e1(T) = 1/5; e1(C) = 0; e1(G) = 0;
E.g. the second column: e2(A) = 0; e2(T) = 0; e2(C) = 4/5; e2(G) = 1/5;
E.g. the third column: e3(A) = 4/5; e3(T) = 0; e3(C) = 1/5; e3(G) = 0;
ACA---ATG
TCAACTATC
ACAC--AGC
AGA---ATC
ACCG--ATC
A 0.8
C PC
G PC
T 0.2
A PC
C 0.8
G 0.2
T PC
A 0.8
C
G
T 0.2
A 1
C
G
T 1 1 0.4
A 0.2
C 0.4
G 0.2
T 0.20.6
0.6
0.4

Profile representation
• Suppose I (amino acid b) is the ancestor
• What is the probability of observing a T (amino acid a) in the first column (position p) of the alignment
• This probability is reflected by the score M
M(p,a)= W(p,b) X Y(a,b)
M(1,I)= W(1,T) X Y(I,T)
M is dependent on
• The observed frequency of T in the first position of the alignment (W)
• The probability of mutating I => T (according to PAM) (Y)
I A …I ST TV AV AI LT VT VI AI V
b
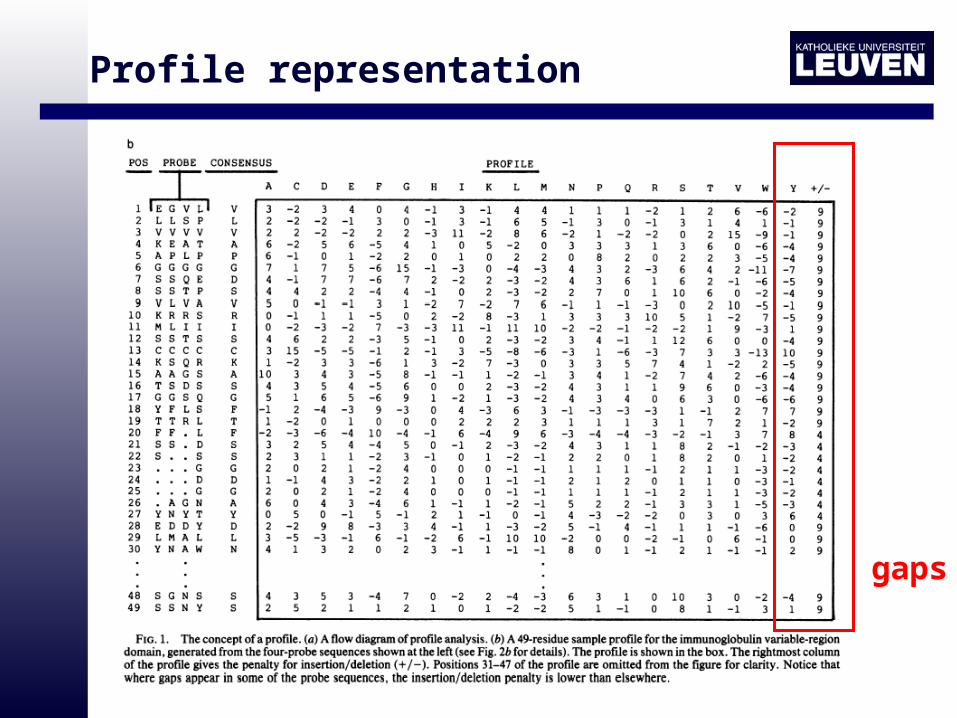
Profile representation
gaps

Overview
• Introduction
• Motif representation
• Motif screening
• Motif Databases
• Exercise
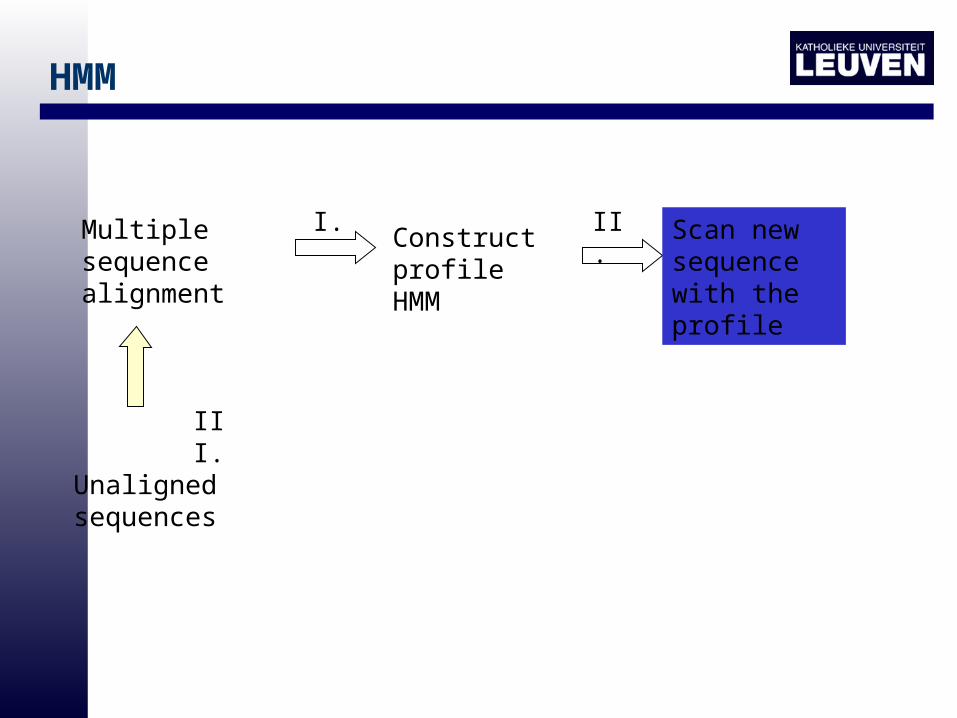
HMM
Multiple sequence alignment
Construct profile HMM
Scan new sequence with the profile
I. II.
Unaligned sequences
III.
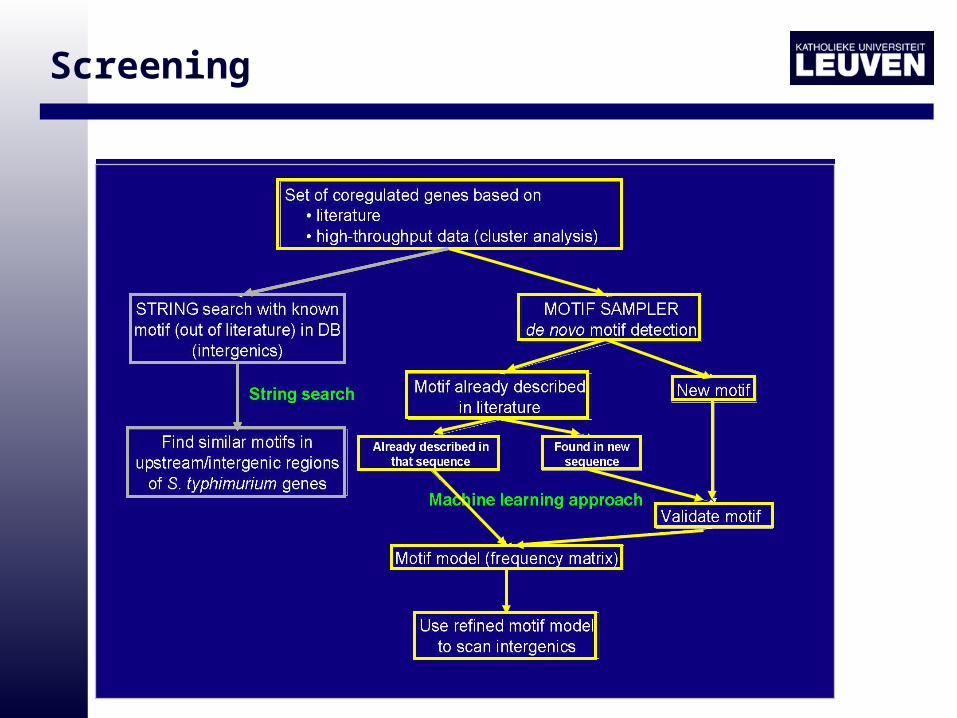
Screening

Screening
• The multiple alignment of the family is known (Clustal W)
• The motif to be detected is known but the multiple alignment does not yet exist– Motifs already described in literature
– Construct the multiple alignment, derive the model
• Neither the motif nor the multiple alignment exist– Probabilistic motif detection

• Obtained Motif Model used for genome wide screening (Motif Scanner)
• Identification of putative additional targets
• Use sliding window
• Attribute to each sequence within the sliding window a score
• Rank the hits based on their score and select the most promising candidates
Genome wide screening
Screening

Screening
• Distinct methods differ in the motif representation and the scoring system used
• Consensus Sequence or Regular expression (pattern match)
– Very conservative
– Do not allow mismatches
• PSSM / HMM: more complicated scoring schemes
– based on information content
– Log likelihood
– Less conservative
– Difficult choice of threshold score
– Tradeoff between sensitivity and selectivity

Screening
• FDR (1-Precision)
FP/(TP+FP)
• Precision
TP/(TP+FP)
• Specificity (related to the false positive fraction= 1-spec)
TN/(TN+FP)
• Sensitivity (true positive fraction = recall)
TP/(TP+FN)

Screening
• E- value: corresponds to the probability of finding a score equal or better than the one observed, by chance alone.

Screening with Regular Expression
Simple perl scripts

Screening with PSSM
i
i
x
ixW
i p
qscoreodds
1
2loglog
70,005,001,050,090,010,0
10,030,001,080,002,010,0
10,005,097,005,006,020,0
10,060,001,010,002,060,0
PSSM
Background frequency of each of the four nucleotides: 25,0
ixp
Sequentie Odds Log odds Opmerking
ATGCAT 720,8829 9,49362 consensus CTGCGT 120,1471 6,90866 Similar sequence CATGGC 0,0002 -11,99046 Different sequence
• Slide a window of length W over a sequence• Calculate for each subsequence within the window a log odds-score • The highest scoring positions correspond to the most likely locations of the motif
9.4 = log2(720)
(0.6*0.9*0.8*0.97*0.6*0.7)/(0.25^6)
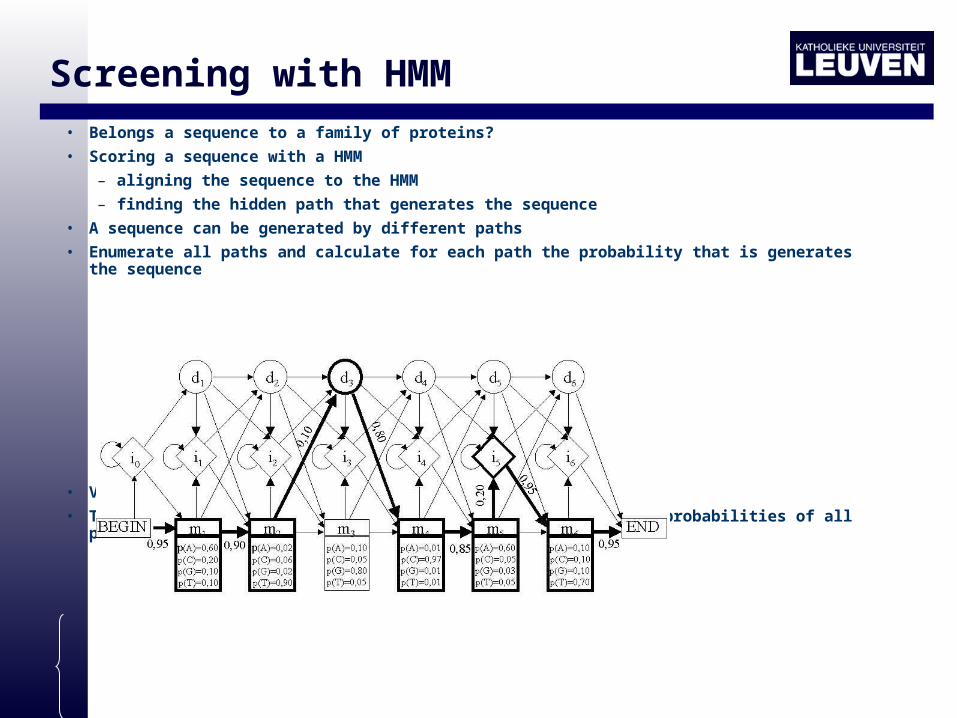
Screening with HMM
• Belongs a sequence to a family of proteins?
• Scoring a sequence with a HMM
– aligning the sequence to the HMM
– finding the hidden path that generates the sequence
• A sequence can be generated by different paths
• Enumerate all paths and calculate for each path the probability that is generates the sequence
• Viterbi Path: most likely path
• Total probability that sequence is generated by HMM = sum of probabilities of all possible paths
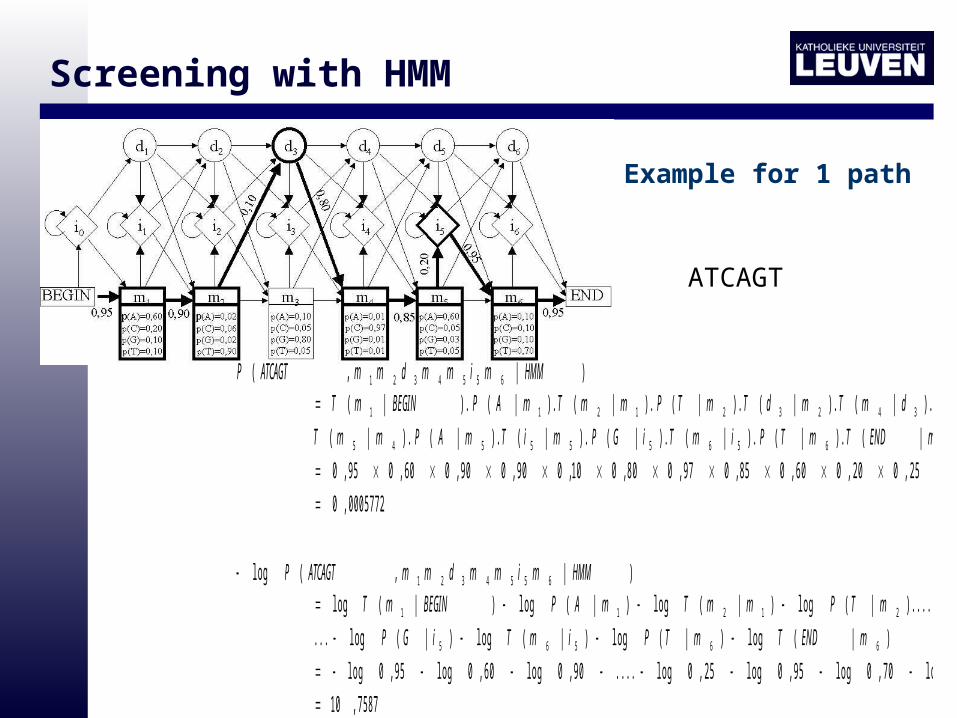
Screening with HMM
)|,( 6554321 HMMmimmdmmATCAGTP
0005772,0
95,070,095,025,020,060,085,097,080,010,090,090,060,095,0
)|().|().|().|().|().|().|(
).|().|().|().|().|().|().|(
6656555545
4342321211
mENDTmTPimTiGPmiTmAPmmT
mCPdmTmdTmTPmmTmAPBEGINmT
)|,(log 6554321 HMMmimmdmmATCAGTP
7587,10
95,0log70,0log95,0log25,0log....90,0log60,0log95,0log
)|(log)|(log)|(log)|(log...
)....|(log)|(log)|(log)|(log
66565
21211
mENDTmTPimTiGP
mTPmmTmAPBEGINmT
Example for 1 path
ATCAGT

Screening with HMM
• Calculate the probability of the sequence being generated by the HMM profile of a protein family versus a random model
= align the unknown sequence with the HMM
– The sequence can be generated by different paths
• Impossible to enumerate all possibilities
– What is the most probable path? (Viterbi, backtracking)
– What is the total probability? (Forward)
)(
)mod(2log
RandomdataP
eldataPBits score
A
T
A
-
-
T
ATT and
TTC

Screening with HMM
• Hidden Markov model because if we observe a sequence, the path of states that was followed by the Markov model to generate the observed sequence is unknown or hidden.
• This hidden path contains the information on how the observed sequence should be aligned with the profile.
• Usually a sequence can be generated in multiple ways by the Markov model and more hidden paths (corresponding to distinct alignments) are possible. Usually not all possible paths have an equal probability. Indeed some transitions are not very likely (low transition probability). Usually the path with the highest probability (highest score = most likely path) corresponds to the best alignment.
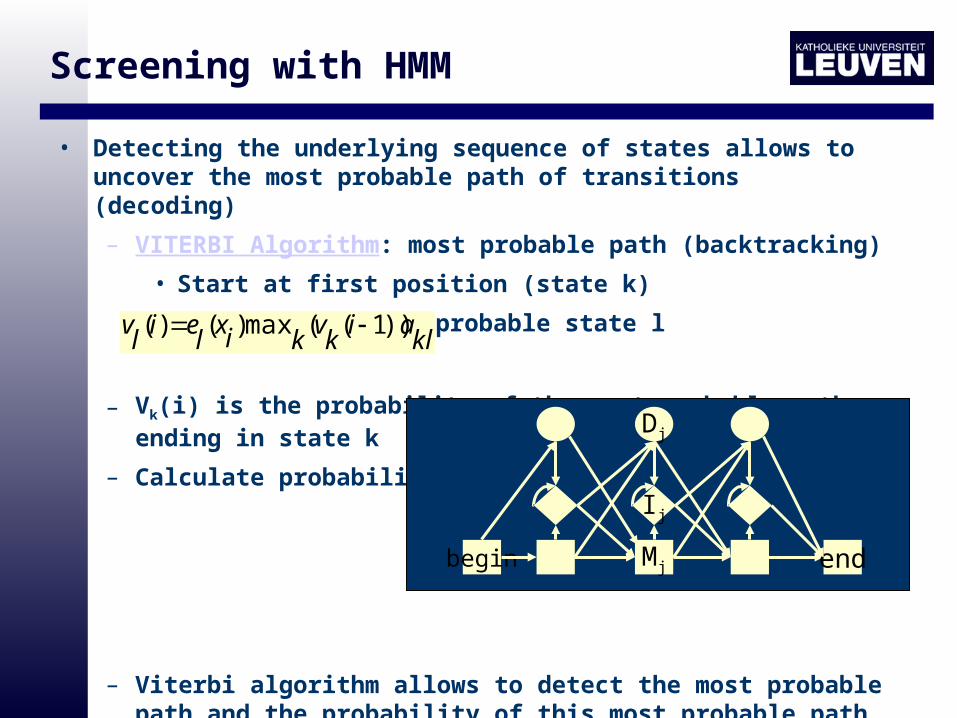
Screening with HMM
• Detecting the underlying sequence of states allows to uncover the most probable path of transitions (decoding)
– VITERBI Algorithm: most probable path (backtracking)
• Start at first position (state k)
• Move to next most probable state l
– Vk(i) is the probability of the most probable path ending in state k
– Calculate probability
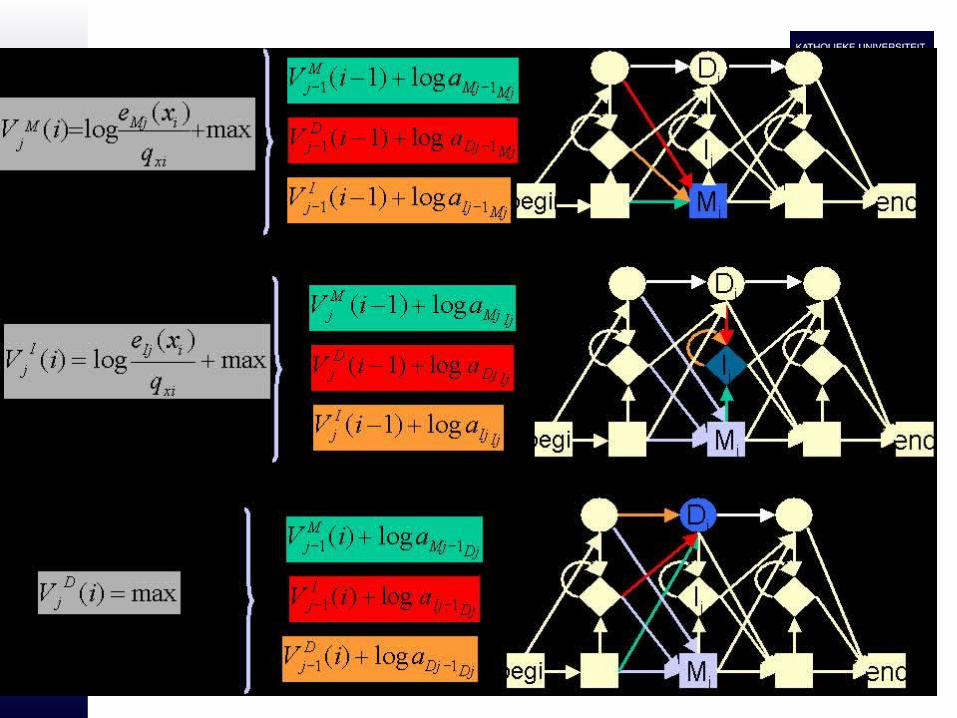
– Viterbi algorithm allows to detect the most probable path and the probability of this most probable path
klai
kv
kixlei
lv ))1((max)()(
begin Mj
Ij
Dj
end
begin Mj
Ij
Dj
end
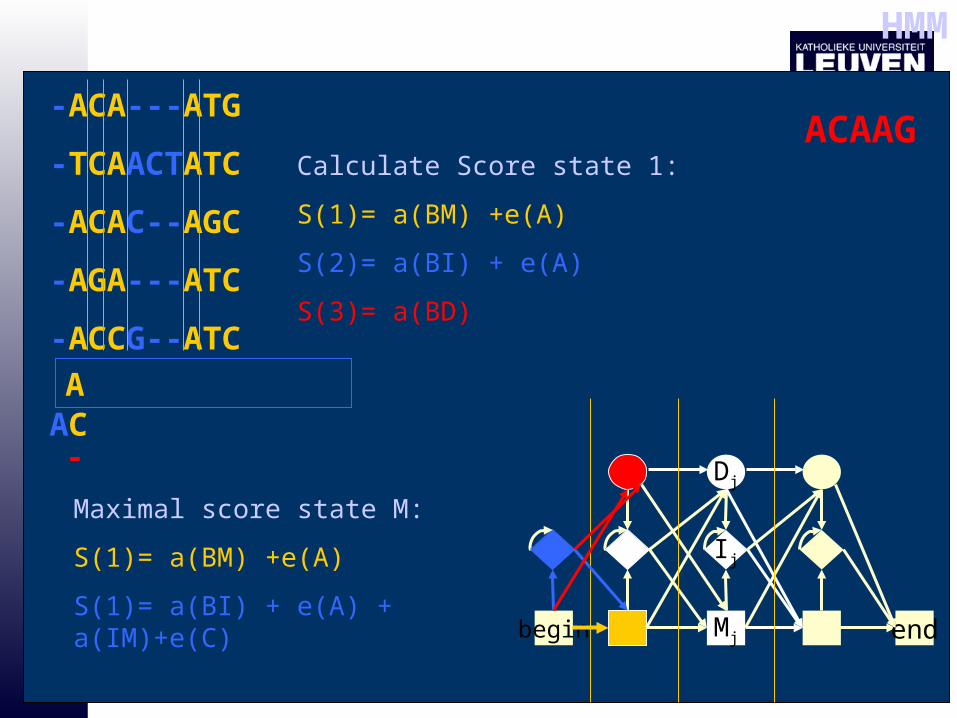
-ACA---ATG
-TCAACTATC
-ACAC--AGC
-AGA---ATC
-ACCG--ATC
AAC
Calculate Score state 1:
S(1)= a(BM) +e(A)
S(2)= a(BI) + e(A)
S(3)= a(BD)
-
Maximal score state M:
S(1)= a(BM) +e(A)
S(1)= a(BI) + e(A) + a(IM)+e(C)
HMM
ACAAG

Conclusion
• Distinct methods differ in the motif representation and the scoring system used
• Consensus Sequence or Regular expression (pattern match)
– Very conservative
– Do not allow mismatches
• PSSM / HMM: more complicated scoring schemes
– based on information content
– Log likelihood
– Less conservative
– Difficult choice of threshold score
– Tradeoff between sensitivity and selectivity

Overview
• Introduction
• Motif representation
• Motif screening
• Motif Databases– Prosite
– Blocks
– pFAM
• Exercise

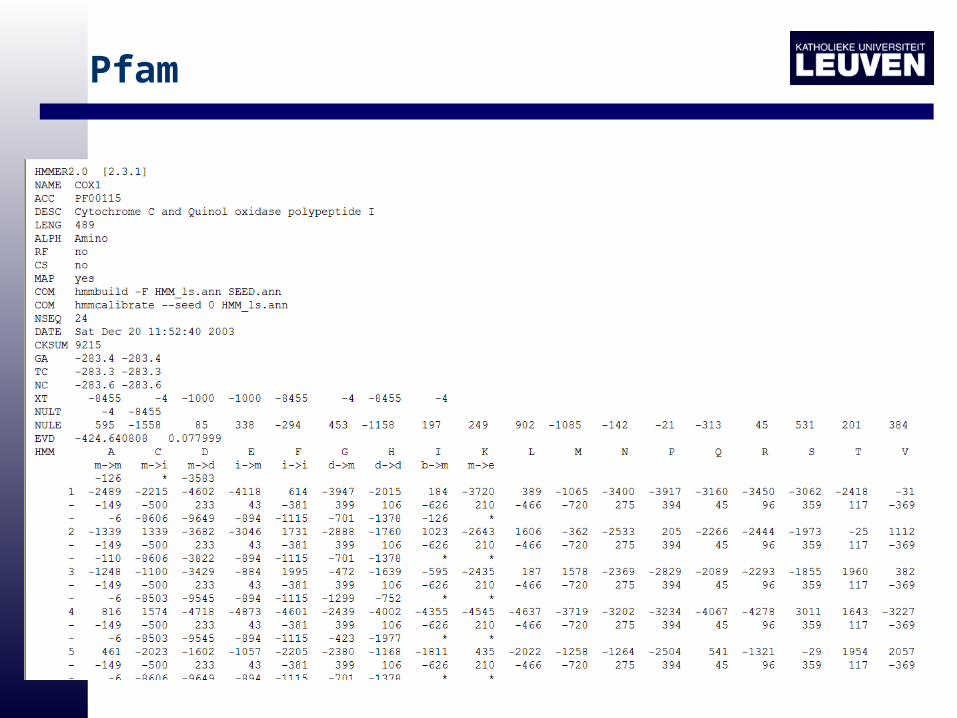
Pfam
• Pfam starts from a set of automatically generated domain alignments (generated by PsiBlast).
• From these alignments a HMM is calculated
• Subsequently all sequences in the SwissProt database of proteins are classified in protein families
– By scoring them with the representative HMMs
– Ranking sequences according to their score
– separate class members from the other sequences in the database based on a suitable threshold
• Pfam 7.0 is such a database that contains a total of 3360 families. Pfam contains multiple protein alignments and profile-HMMs of these families.
PSI-blastAutomatically generated
sequences
PfamB families
Construct seed HMM
PfamA
PRODOM alignment
Discover PRODOM alignments not covered by PfamA
Pfam

Pfam
• Full: alignment on which the Pfam HMM was based
• HMMs for global and fragment search
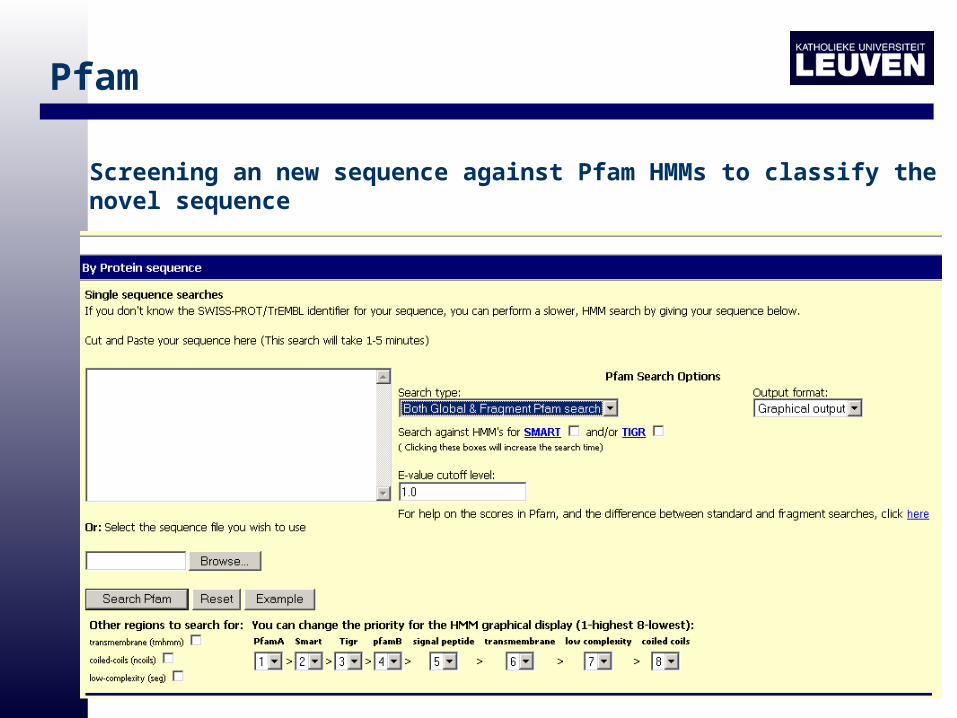
Pfam
Screening an new sequence against Pfam HMMs to classify the novel sequence

Pfam
• Each Pfam family: "trusted cutoff" and a "noise cutoff“
• TC1 is the lowest score for sequences included in the family
• NC1 is the highest score for sequences not included in the Full alignment
The probability that the sequence was generated by the HMM and the probability that the sequence was generated by a null model
E-value is the number of hits that would be expected to have a score equal or better than this by chance alone
•Raw score: bitscore
Scores in Pfam
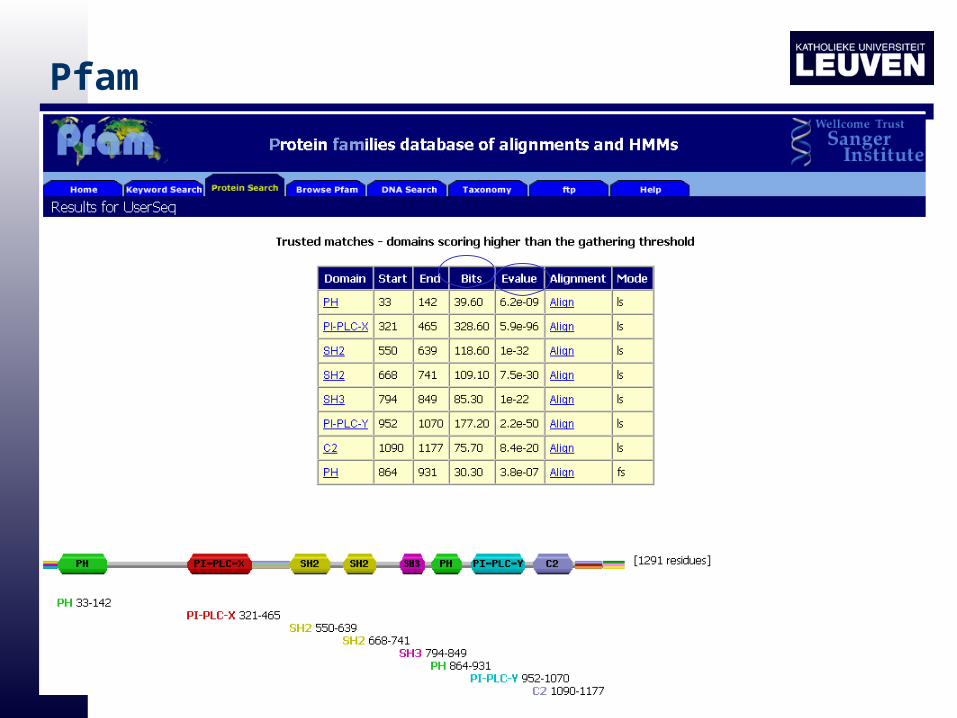
Pfam

PROSITE
Patterns (regular expressions) (ScanProsite)
– Shorter than Pfam
• Enzyme catalytic sites
• Prosthetic group attachment sites (heme, pyridoxal-phosphate, biotin, etc)
• Amino acids involved in binding a metal ion
• Cysteines involved in disulfide bonds
• Regions involved in binding a molecule (ADP/ATP, GDP/GTP, calcium, DNA, etc.) or another protein

PROSITE
• Profiles (Profile representation)
PROSITE
Aminael
renew

BLOCKS
• Database of ungapped alignments
• Motif models represented as PSSMs

Example sequence
>gi|1071819|pir||B54759 ba-type ubiquinol oxidase Paracoccus denitrificansMATFSNETTFLLGRLNWDAIPKEPIVWATFVVVAIGGIAALAALTKYRLWGWLWREWFTSVDHKKIGIMYIVLALIMFVRGFADAIMMRLQQVWAFGGSEGYLNSHHYDQIFTAHGVIMIFFVAMPFITGLMNYVVPLQIGARDVSFPFLNNFSFWMTVGGAVITMASLFLGEFAQTGWLAFPPLSGIGYSPWVGVDYYIWGLQVAGVGTTLSGINLLVTILKMRAPGMTMMRMPIFTWTSFCANILIVASFPVLTMTLILLTLDRYVGTNFFTNDLGGNPMMYINLIWIWGHPEVYILILPLFGVFSEVTSTFSGKRLFGYSSMVYATVCITVLSYLVWLHHFFTMGSGASVNSFFGITTMIISIPTGAKLFNWLFTMYRGRIRYELPMMWTIAFMLTFVIGGMTGVLLAVPPADFVLHNSLFLIAHFHNVIIGGVLFGLFAAINFWWPKAFGFKLDVFWGKVSFWFWVVGFWAAFMPLYILGLMGVTRRLRVFDDPDLRIWFAIAAFGAVLIACGIAAMFVQFGVSILRRDRPEYRDVSGDPWDGRTLEWATSSPPPAYNFAFNPISHGLDTWWEMKQQGATRPTGGYMPIHMPKNTGTGVILAALATVCGMALVWYVWWLAALSFLGIIAVSIAHTFNYNRDYYIPVSEIEATEDARTRQLAQGV
http://www.expasy.org/prosite/http://www.sanger.ac.uk/Software/Pfam/search.shtmlhttp://blocks.fhcrc.org/
Scan sequence with prosite, Pfam, Blocks

PSI-BLAST
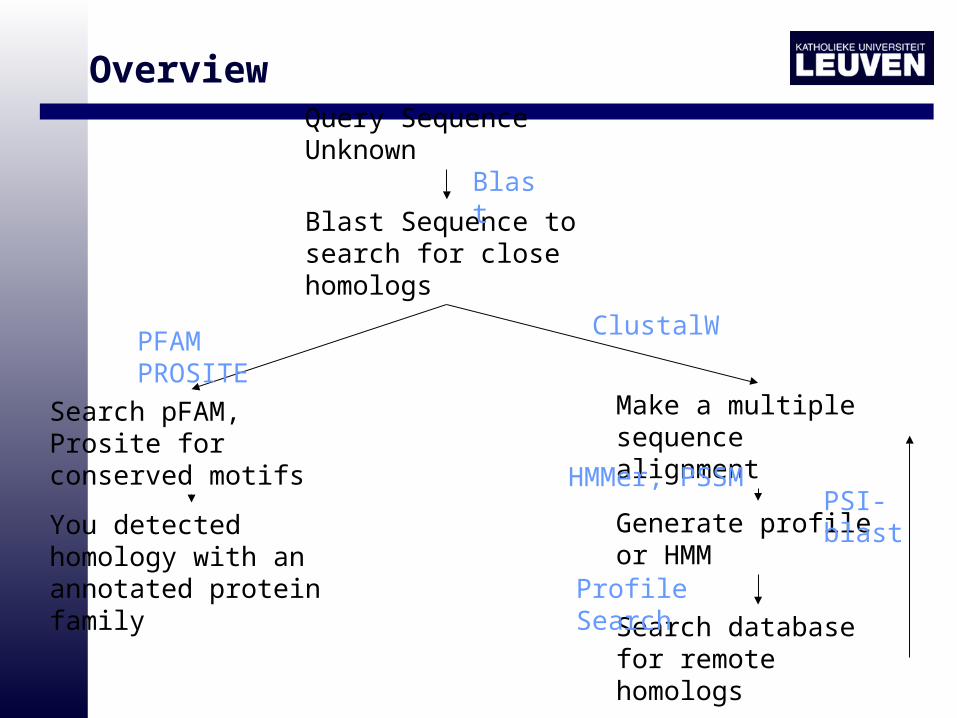
OverviewQuery Sequence Unknown
Blast Sequence to search for close homologs
Search pFAM, Prosite for conserved motifs
You detected homology with an annotated protein family
Make a multiple sequence alignment
Generate profile or HMM
Search database for remote homologs
Blast
ClustalWPFAM PROSITE
HMMer, PSSM
Profile Search
PSI-blast


• exercises

Bits score (log odd score Bayesian)
– Posterior: HMM model: is this a globin domain?
– Likelihood calculated: probability of the sequence being generated by the HMM model
– Prior probability:
• p(model)
– Bayes)()(),( DpDMpMDp
)( DMp
)( MDp )(Mp
)(
)()()(
Dp
MpMDpDMp
)()(),( MpMDpMDp
M
R
1 2 3 4