production of biodiesel from jatropha curcas l. oil
TRANSCRIPT

P
HC
a
ARRAA
KJBPT
1
vitrmeo1imSdocoNMaMM
3
2f
0d
Computers and Chemical Engineering 33 (2009) 1091–1096
Contents lists available at ScienceDirect
Computers and Chemical Engineering
journa l homepage: www.e lsev ier .com/ locate /compchemeng
roduction of biodiesel from Jatropha curcas L. oil�
oufang Lu, Yingying Liu, Hui Zhou, Ying Yang, Mingyan Chen, Bin Liang ∗
ollege of Chemical Engineering, Sichuan University, Chengdu 610065, PR China
r t i c l e i n f o
rticle history:eceived 11 February 2008eceived in revised form 5 August 2008ccepted 25 September 2008vailable online 7 October 2008
a b s t r a c t
A two-step process consisting of pre-esterification and transesterification was developed to producebiodiesel from crude Jatropha curcas L. oil. The free fatty acids (FFAs) in the oil were converted to methylesters in the pre-esterification step using sulfuric acid or solid acid prepared by calcining metatitanic acidas catalysts. The acid value of oil was reduced from the initial 14 mg-KOH/g-oil to below 1.0 mg-KOH/g-oilin 2 h under the conditions of 12 wt% methanol, 1 wt% H2SO4 in oil at 70 ◦C. The conversion of FFAs washigher than 97% at 90 ◦C in 2 h using 4 wt% solid acid and a molar ratio of methanol to FFAs of 20:1. Phos-
eywords:atropha curcas L. oiliodiesel
pholipid compounds were eliminated during pre-esterification and a separate degumming operation wasunnecessary. The yield of biodiesel by transesterification was higher than 98% in 20 min using 1.3% KOH
io of
Japp
tl(I&1
t
pCst
re-esterificationransesterification
as catalyst and a molar rat
. Introduction
Biodiesel is an alternative fuel produced from renewableegetable oils, animal fats or recycled cooking oils by transester-fication reaction. Biodiesel has drawn significant attention dueo increasing environmental concern and diminishing petroleumeserves (Ma & Hanna, 1999). Presently, biodiesel is produced com-ercially in Europe and USA to reduce air pollution and the net
mission of greenhouse gas. Surplus edible oils, such as rapeseedil and soybean oil, are used as raw materials for biodiesel (Körbltz,999; Wood, 2005). However, using edible oils to produce biodiesels not encouraged in China because China imports more than 400
illion tons of edible oils annually to satisfy its consumption needs.ome Chinese biodiesel producers use recycled waste oils to pro-uce biodiesel, but the scale is limited. Although the use of wasteils can lower the feedstock cost significantly, complicated pro-edures are needed to remove the impurities, resulting in highperating costs (Al-Widyan & Al-Shyoukh, 2002; van Kasteren &isworo, 2007; Zhang, Dubé, McLean, & Kates, 2003a; Zhang, Dubé,
cLean, & Kates, 2003b). Non-edible oils like Jatropha curcas L. oilre attractive (Foidl, Foidl, Sanchez, Mittelbach, & Hackel, 1996;ohibbe Azam, Waris, & Nahar, 2005; Sarin, Sharma, Sinharay, &alhotra, 2007; Tiwari, Kumar, & Raheman, 2007; Wood, 2005).
� Supported by the Key Grant Project of Chinese Ministry of Education (No.07023).∗ Corresponding author at: College of Chemical Engineering, Sichuan University,
4 South Section 1, Yihuan Road, Chengdu 610065, PR China. Tel.: +86 28 85460556;ax: +86 28 85460557.
E-mail address: [email protected] (B. Liang).
h
(isPrurvTl
098-1354/$ – see front matter © 2008 Elsevier Ltd. All rights reserved.oi:10.1016/j.compchemeng.2008.09.012
methanol to oil 6:1 at 64 ◦C.© 2008 Elsevier Ltd. All rights reserved.
. curcas L. trees can grow in arid, semiarid and wastelands. It hashigh-seed yield and high oil content (Wood, 2005). In China, itslantation area is being expanded quickly along the Yangzi River asromoted by an environment protection act.
In conventional processes, biodiesel is manufactured by theransesterification of oils with methanol in the presence of cata-ysts, such as alkalis (KOH, NaOH) or their corresponding alkoxidesFreedman, Pryde, & Mounts, 1984; Holser & Harry-O’Kuru, 2006;kwuagwu, Ononogbu, & Njoku, 2000; Jitputti et al., 2006; Leung
Guo, 2006; Ma & Hanna, 1999; Siler-Marinkovic & Tomasevic,998):
riglycerides + methanol → biodiesel + glycerol
The process design and operation parameters vary with theroperties of the feedstock oils and the desired biodiesel quality.ommercial biodiesel processes using rapeseed oil (in Europe) andoybean oil (in the USA) have been well researched, and therebyhe properties of their biodiesel products have also been compre-ensively investigated.
However, J. curcas L. oil with high content of free fatty acidsFFAs) cannot be directly used in an alkali catalyzed transester-fication process because FFAs react with alkali catalyst to formoaps, resulting in serious emulsification and separation problems.re-esterification catalyzed by homogeneous acids, such as sulfu-ic acid, phosphorous acid, or sulfonic acid, is a conventional and
seful method to reduce the content of FFAs, which can turn theaw oils transesterificable by an alkali catalyst and convert FFAs toaluable fatty acid methyl esters (FAME) (Ghadge & Raheman, 2005;iwari et al., 2007). Compared with conventional liquid acid cata-ysts, solid acid catalyst is more environmentally friendly (López,

1 ical En
SNFo(fa
bFc
p
soof
ects
2
2
SgIlmptJppicf
w(pa
Crf
2
Tctswmei
wtn
2p
tIt
twtd
abmtwsTlaat
2
spws
2
vG8mlmdloco
3
3
f
F
092 H. Lu et al. / Computers and Chem
uwannakarn, Bruce, & Goodwin, 2007; Mbaraka & Shanks, 2005;arasimharao et al., 2007). The effect of methanol to oil ratios onFA conversion at the reaction temperature of 50 ◦C, a reaction timef 1 h, and a H2SO4 to oil ratio of 1% (w/w) has been investigatedBerchmans & Hirata, 2008). The optimum methanol to oil ratio wasound to be 60% (w/w) for an FFA concentration less than 1%, andn acid value (AV) of 2 mg-KOH/g-oil.
We propose a two-step method to convert raw J. curcas L. oil intoiodiesel. A pre-esterification operation was applied to eliminateFAs by reacting the oil with methanol in the presence of an acidatalyst. The process can be simply described as
re-esterificatin → purification → transesterification
→ phase separation
Raw oil was firstly reacted with methanol, followed by phaseeparation to remove acidic water and gum impurities. The purifiedil was further reacted with methanol in Section 2.4 in the presencef an alkali catalyst. Finally, the biodiesel product was separatedrom the glycerol by-product by phase separation.
In this work, factors influencing the pre-esterification and trans-sterification were systematically investigated, and the solid acidatalyst SO4
2−/TiO2 prepared by calcining metatitanic acid wasested and characterized. The properties of the biodiesel were mea-ured.
. Experimental
.1. Materials
The J. curcas L. seeds were collected from the Panzhihua area,ichuan Province, southwest China. Jatropha oil was obtained byrinding the seeds. The oil was filtrated to remove solid impurities.n order to investigate the effects of FFAs, water and phospho-ipids on the reaction kinetics, we used simulated oils obtained by
ixing refined oil with different impurities. The refined oil wasrepared by processing the crude Jatropha oil with NaOH solu-ion and active earth to remove FFA and moisture. The refinedatropha oil contained FFAs < 0.2 mg-KOH/g-oil, water < 0.1% andhospholipids < 0.04%. Oil samples with different FFAs, water andhospholipids contents were prepared by adding oleic acid, deion-
zed water and soluble phospholipids into the refined Jatorphaurcas L. oil in order to quantitatively investigate the effects of theseactors on the pre-esterification step.
The solid acid catalyst used in the pre-esterification reactionas prepared by calcining metatitanic acid. The metatitanic acid
the molar ratio of S/Ti is 0.1) was obtained from a commercial TiO2igment process of the Titanium Industry Company, Panzhihua Ironnd Steel Groups.
Phospholipids were purchased from Beijing Huaqing Meihengo. Ltd. (China), in which the content of acetone insoluble mate-ial > 98%. Other chemical reagents were of analytical grade withouturther purification.
.2. Pre-esterification process catalyzed by sulfuric acid
Pre-esterification was conducted in a 250 ml three-neck flask.he flask was equipped with a mechanical agitator and a refluxondenser, and heated with a water bath to control the reactionemperature. In the experiments, flasks loaded with Jatropha oil
amples were firstly heated to the designated temperature. Thisas followed by the addition of the methanol and sulfuric acidixture before turning on the agitator, marking the start of thesterification reaction. The esterification products were separatedn a tap funnel to obtain the upper oil layer, which was then washed
oos
gineering 33 (2009) 1091–1096
ith water several times until the pH of washing water was closeo 7.0. The resultant pre-esterified oil was dried by anhydrous mag-esium sulfate before subsequent transesterification.
.3. Preparation of solid acid catalyst and its application inre-esterification process
The metatitanic acid was a semifinished product collected fromhe hydrolysis section of the commercial TiO2 pigment process.t was a hydrolysis product containing adsorbed sulfuric acid anditanium sulfates without further purification.
The metatitanic acid sample was dried at 110 ◦C in air for 5 h, andhen crushed and sieved. The particles with a size under 125 �mere further calcined to prepare the solid acid catalyst. In order
o investigate the effect of calcination on the catalytic properties,ifferent calcination temperatures and times were tested.
Pre-esterification using the solid acid catalyst took place insiden autoclave. Both the reactants and the catalyst were added at theeginning, and the reactor was rapidly heated at 7 ◦C/min under aechanical agitation of 1500 rpm. After the completing the reac-
ion, the reactor was quenched to stop the reaction. The slurryas filtered under vacuum and the liquid phases were allowed to
ettle in a tap funnel to separate the acidic water and oil phase.he acidic water and methanol are the components in the upperayer. The oil phase was obtained at the lower layer and was keptt 110 ◦C for 90 min in an oven to evaporate the residual moisturend methanol. The treated oil was then used as the feedstock forransesterification.
.4. Transesterification
Transesterification experiments were carried out under atmo-pheric pressure using KOH as the catalyst. The reactor and therocess were similar to those at pre-esterification. The productsere FAME and glycerol. FAME was washed with water to remove
oap and catalyst before drying.
.5. Analytical method
The moisture, acid value, glycerol content and saponificationalue were determined following the National Standards of PRC:B/T 5528-1995, GB/T 5530-1998, GB/T 13216.6-91 and GB9104.2-8, respectively. The content of phospholipids was determined byolybdenum blue colorimetry. The specific surface area of cata-
yst was determined by the BET method. The sulfur content waseasured by elemental sulfur analysis. The acid concentration was
etermined by titration. The composition of fatty acid was ana-yzed by a Shimadzu GCMS-QP2010 after methylation. The qualitiesf biodiesel including density, kinematic viscosity, flash point andold filter plug point (CFPP) were measured following the Standardsf PRC: GB/T 2540, GB/T 265, GB/T 261 and SH/T 0248, respectively.
. Results and discussion
.1. Homogeneous pre-esterification reaction
In the presence of an acid catalyst, FFAs react with methanol toorm FAME. The reaction can be represented as
FAs + methanol = FAME + water
Under conditions favorable to esterification, the reaction ratef triglycerides with methanol was much slower compared to thatf FFAs. The conversion of the pre-esterification reaction was mea-ured by comparison of the acid values before and after the reaction.
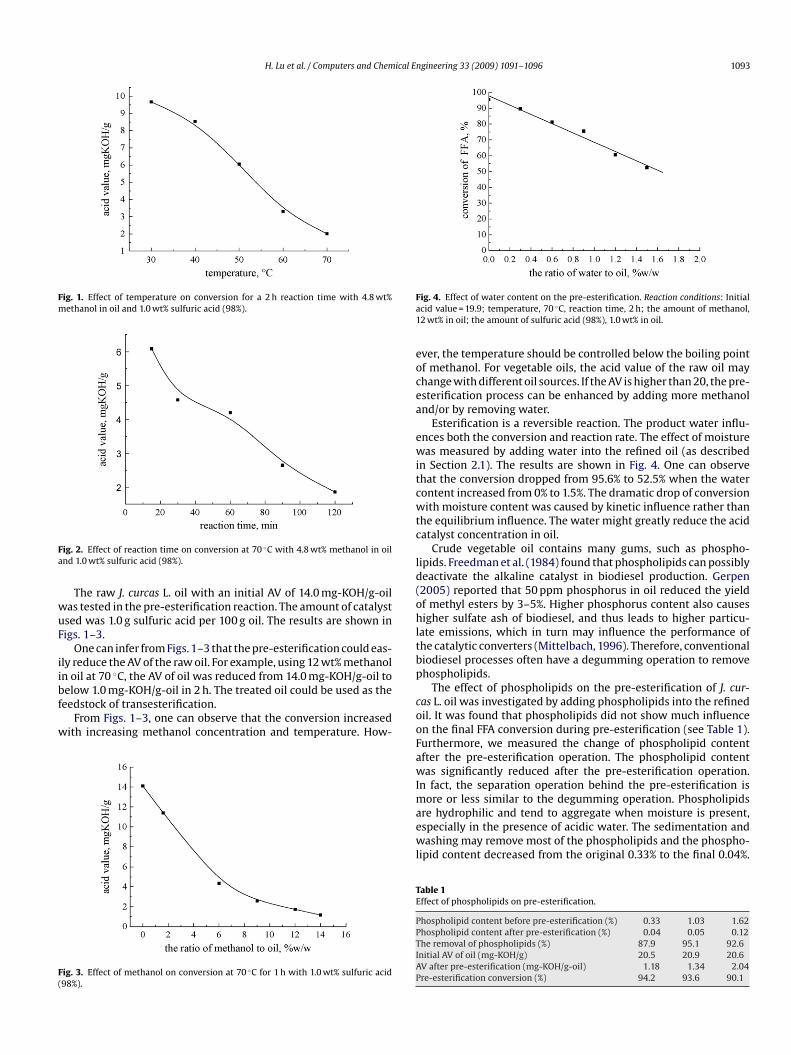
H. Lu et al. / Computers and Chemical Engineering 33 (2009) 1091–1096 1093
Fig. 1. Effect of temperature on conversion for a 2 h reaction time with 4.8 wt%methanol in oil and 1.0 wt% sulfuric acid (98%).
Fa
wuF
iibf
w
F(
Fa1
eocea
ewitcwtc
ld(ohltbp
ig. 2. Effect of reaction time on conversion at 70 ◦C with 4.8 wt% methanol in oilnd 1.0 wt% sulfuric acid (98%).
The raw J. curcas L. oil with an initial AV of 14.0 mg-KOH/g-oilas tested in the pre-esterification reaction. The amount of catalystsed was 1.0 g sulfuric acid per 100 g oil. The results are shown inigs. 1–3.
One can infer from Figs. 1–3 that the pre-esterification could eas-ly reduce the AV of the raw oil. For example, using 12 wt% methanoln oil at 70 ◦C, the AV of oil was reduced from 14.0 mg-KOH/g-oil to
elow 1.0 mg-KOH/g-oil in 2 h. The treated oil could be used as theeedstock of transesterification.From Figs. 1–3, one can observe that the conversion increasedith increasing methanol concentration and temperature. How-
ig. 3. Effect of methanol on conversion at 70 ◦C for 1 h with 1.0 wt% sulfuric acid98%).
cooFawImaewl
TE
PPTIAP
ig. 4. Effect of water content on the pre-esterification. Reaction conditions: Initialcid value = 19.9; temperature, 70 ◦C, reaction time, 2 h; the amount of methanol,2 wt% in oil; the amount of sulfuric acid (98%), 1.0 wt% in oil.
ver, the temperature should be controlled below the boiling pointf methanol. For vegetable oils, the acid value of the raw oil mayhange with different oil sources. If the AV is higher than 20, the pre-sterification process can be enhanced by adding more methanolnd/or by removing water.
Esterification is a reversible reaction. The product water influ-nces both the conversion and reaction rate. The effect of moistureas measured by adding water into the refined oil (as described
n Section 2.1). The results are shown in Fig. 4. One can observehat the conversion dropped from 95.6% to 52.5% when the waterontent increased from 0% to 1.5%. The dramatic drop of conversionith moisture content was caused by kinetic influence rather than
he equilibrium influence. The water might greatly reduce the acidatalyst concentration in oil.
Crude vegetable oil contains many gums, such as phospho-ipids. Freedman et al. (1984) found that phospholipids can possiblyeactivate the alkaline catalyst in biodiesel production. Gerpen2005) reported that 50 ppm phosphorus in oil reduced the yieldf methyl esters by 3–5%. Higher phosphorus content also causesigher sulfate ash of biodiesel, and thus leads to higher particu-
ate emissions, which in turn may influence the performance ofhe catalytic converters (Mittelbach, 1996). Therefore, conventionaliodiesel processes often have a degumming operation to removehospholipids.
The effect of phospholipids on the pre-esterification of J. cur-as L. oil was investigated by adding phospholipids into the refinedil. It was found that phospholipids did not show much influencen the final FFA conversion during pre-esterification (see Table 1).urthermore, we measured the change of phospholipid contentfter the pre-esterification operation. The phospholipid contentas significantly reduced after the pre-esterification operation.
n fact, the separation operation behind the pre-esterification isore or less similar to the degumming operation. Phospholipids
re hydrophilic and tend to aggregate when moisture is present,specially in the presence of acidic water. The sedimentation andashing may remove most of the phospholipids and the phospho-
ipid content decreased from the original 0.33% to the final 0.04%.
able 1ffect of phospholipids on pre-esterification.
hospholipid content before pre-esterification (%) 0.33 1.03 1.62hospholipid content after pre-esterification (%) 0.04 0.05 0.12he removal of phospholipids (%) 87.9 95.1 92.6nitial AV of oil (mg-KOH/g) 20.5 20.9 20.6V after pre-esterification (mg-KOH/g-oil) 1.18 1.34 2.04re-esterification conversion (%) 94.2 93.6 90.1
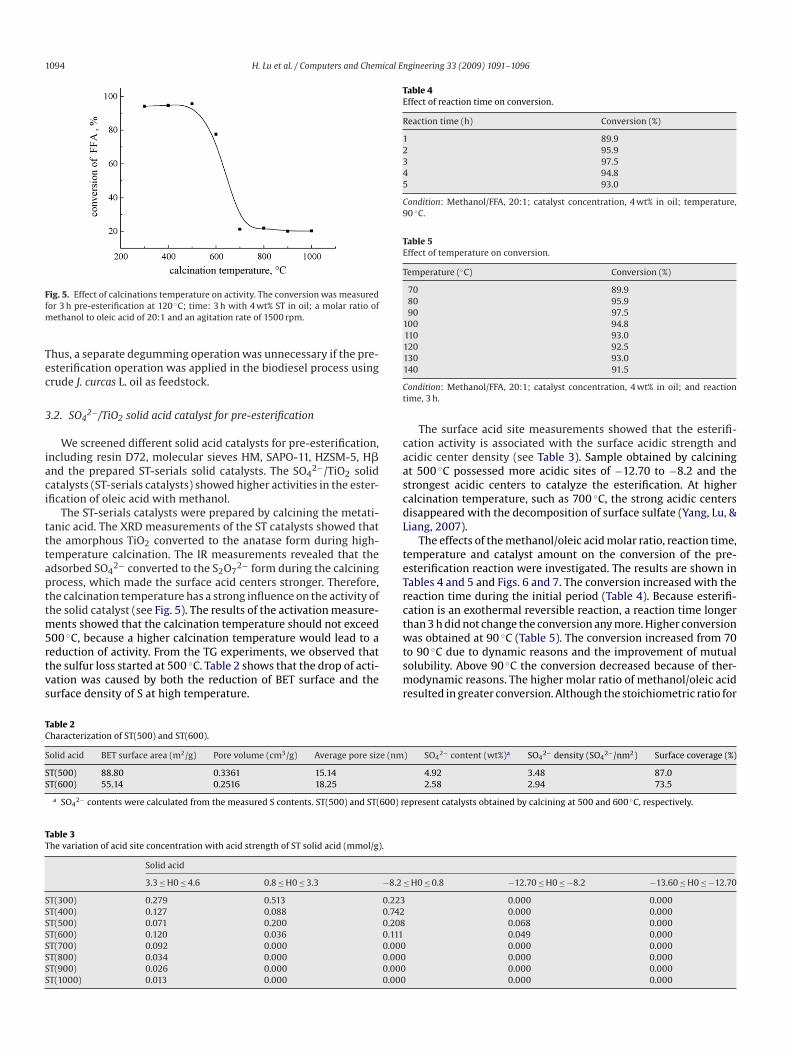
1094 H. Lu et al. / Computers and Chemical Engineering 33 (2009) 1091–1096
Ffm
Tec
3
iaci
tttapttm5rtvs
Table 4Effect of reaction time on conversion.
Reaction time (h) Conversion (%)
1 89.92 95.93 97.54 94.85 93.0
Condition: Methanol/FFA, 20:1; catalyst concentration, 4 wt% in oil; temperature,90 ◦C.
Table 5Effect of temperature on conversion.
Temperature (◦C) Conversion (%)
70 89.980 95.990 97.5
100 94.8110 93.0120 92.5130 93.0
Ct
caascdL
teTrctwas obtained at 90 ◦C (Table 5). The conversion increased from 70
TC
S
SS
TT
SSSSSSSS
ig. 5. Effect of calcinations temperature on activity. The conversion was measuredor 3 h pre-esterification at 120 ◦C; time: 3 h with 4 wt% ST in oil; a molar ratio of
ethanol to oleic acid of 20:1 and an agitation rate of 1500 rpm.
hus, a separate degumming operation was unnecessary if the pre-sterification operation was applied in the biodiesel process usingrude J. curcas L. oil as feedstock.
.2. SO42−/TiO2 solid acid catalyst for pre-esterification
We screened different solid acid catalysts for pre-esterification,ncluding resin D72, molecular sieves HM, SAPO-11, HZSM-5, H�nd the prepared ST-serials solid catalysts. The SO4
2−/TiO2 solidatalysts (ST-serials catalysts) showed higher activities in the ester-fication of oleic acid with methanol.
The ST-serials catalysts were prepared by calcining the metati-anic acid. The XRD measurements of the ST catalysts showed thathe amorphous TiO2 converted to the anatase form during high-emperature calcination. The IR measurements revealed that thedsorbed SO4
2− converted to the S2O72− form during the calcining
rocess, which made the surface acid centers stronger. Therefore,he calcination temperature has a strong influence on the activity ofhe solid catalyst (see Fig. 5). The results of the activation measure-
ents showed that the calcination temperature should not exceed00 ◦C, because a higher calcination temperature would lead to a
eduction of activity. From the TG experiments, we observed thathe sulfur loss started at 500 ◦C. Table 2 shows that the drop of acti-ation was caused by both the reduction of BET surface and theurface density of S at high temperature.tsmr
able 2haracterization of ST(500) and ST(600).
olid acid BET surface area (m2/g) Pore volume (cm3/g) Average pore size (nm
T(500) 88.80 0.3361 15.14T(600) 55.14 0.2516 18.25
a SO42− contents were calculated from the measured S contents. ST(500) and ST(600) r
able 3he variation of acid site concentration with acid strength of ST solid acid (mmol/g).
Solid acid
3.3 ≤ H0 ≤ 4.6 0.8 ≤ H0 ≤ 3.3 −8.2
T(300) 0.279 0.513 0.223T(400) 0.127 0.088 0.742T(500) 0.071 0.200 0.208T(600) 0.120 0.036 0.111T(700) 0.092 0.000 0.000T(800) 0.034 0.000 0.000T(900) 0.026 0.000 0.000T(1000) 0.013 0.000 0.000
140 91.5
ondition: Methanol/FFA, 20:1; catalyst concentration, 4 wt% in oil; and reactionime, 3 h.
The surface acid site measurements showed that the esterifi-ation activity is associated with the surface acidic strength andcidic center density (see Table 3). Sample obtained by calciningt 500 ◦C possessed more acidic sites of −12.70 to −8.2 and thetrongest acidic centers to catalyze the esterification. At higheralcination temperature, such as 700 ◦C, the strong acidic centersisappeared with the decomposition of surface sulfate (Yang, Lu, &iang, 2007).
The effects of the methanol/oleic acid molar ratio, reaction time,emperature and catalyst amount on the conversion of the pre-sterification reaction were investigated. The results are shown inables 4 and 5 and Figs. 6 and 7. The conversion increased with theeaction time during the initial period (Table 4). Because esterifi-ation is an exothermal reversible reaction, a reaction time longerhan 3 h did not change the conversion any more. Higher conversion
o 90 ◦C due to dynamic reasons and the improvement of mutualolubility. Above 90 ◦C the conversion decreased because of ther-odynamic reasons. The higher molar ratio of methanol/oleic acid
esulted in greater conversion. Although the stoichiometric ratio for
) SO42− content (wt%)a SO4
2− density (SO42−/nm2) Surface coverage (%)
4.92 3.48 87.02.58 2.94 73.5
epresent catalysts obtained by calcining at 500 and 600 ◦C, respectively.
≤ H0 ≤ 0.8 −12.70 ≤ H0 ≤ −8.2 −13.60 ≤ H0 ≤ −12.70
0.000 0.0000.000 0.0000.068 0.0000.049 0.0000.000 0.0000.000 0.0000.000 0.0000.000 0.000

H. Lu et al. / Computers and Chemical Engineering 33 (2009) 1091–1096 1095
Fc
trawcomc
3
ttt
t
ocmsipamoes
t
Fm
Fr
co(tott
sm1bwrttaw(
osarmh
ig. 6. Effect of the ratio of methanol to FFA on conversion at 90 ◦C for 3 h with 4 wt%atalyst in oil.
he esterification is 1:1, the excessive methanol could increase theate and promote the completion of the reaction (Fig. 6). When themount of catalyst was lower than 2%, the conversion increasedith the amount of catalyst. When the amount was over 4%, the
onversion did not increase further (Fig. 7). The optimal conditionsf the pre-esterification reaction for the J. curcas L. oil are: 20:1olar ratio of methanol/FFA, 4% solid catalyst, 90 ◦C and 2 h. The
onversion of FFAs reached 97%.
.3. Transesterification process
In pre-esterification, FFA was converted to methyl ester, whileriglyceride was further converted to methyl ester in the followingransesterification in the presence of KOH. The reaction during theransesterification can be represented as
riglyceride (oil) + methanol = FAME + glycerol
The inter-solubility of FAME–methanol–glycerol, J. curcas L.il–FAME–methanol, J. curcas L. oil–glycerol–methanol, and J. cur-as L. oil–FAME–glycerol between 298.15 and 333.15 K has beeneasured by Zhou, Lu, & Liang (2006a). Methanol is completely
oluble in both FAME and glycerol, but insoluble in oil. With increas-ng mass fraction of FAME, the solubility of methanol in oil-FAMEhase increases. As a result, the transesterification reaction showsn induction period because the reaction is carried out in theethanol phase. When the content of FAME increases to 70%, the
il–methanol–FAME mixture becomes a homogeneous phase. Glyc-rol has a low solubility in both oil and FAME and is thus easilyeparated from the final product biodiesel.
The yield of methyl ester and glycerol can be used to representhe progress of the transesterification reaction. For the high FFA
ig. 7. Effect of the amount of catalyst on conversion at 90 ◦C for 3 h with aethanol/FFA ratio of 20:1.
a
3
FptpdctnpflbKrcFcs
ig. 8. Relationship between the yield of products and acid value of raw oil for aeaction time of 1 h at 64 ◦C with a methanol to oil molar ratio of 6:1.
ontent oil, such as the crude J. curcas L. oil, we found that the yieldf methyl ester is often lower than the yield of glycerol (Fig. 8)Liu, Lu, Liang, & Chen, 2007). The loss of methyl ester is due tohe product loss during the separation process and the washingperation. The presence of FFAs results in emulsification that makeshe separation of ester difficult. Therefore, it is important to lowerhe AV below 1.5 mg-KOH/g-oil during the pre-esterification.
Higher molar ratios of methanol to oil lead to greater conver-ion for a given reaction time. Many researchers indicated that aolar ratio of 6:1 was the best for numerous oils (Freedman et al.,
984; Holser & Harry-O’Kuru, 2006; Leung & Guo, 2006). Reactionetween J. curcas L. oil and methanol was found to be similar. KOHas a possible catalyst for this system. Temperature influenced the
eaction rate and higher yield was obtained at a higher tempera-ure between 35 and 65 ◦C. The conversion increased with reactionime. The first 15 min is the fastest period of the reaction, in whichconversion of 90% is possible. The optimal conditions were: 64 ◦Cith 1.3% KOH as catalyst and a molar ratio of methanol to oil at 6:1
Zhou, Lu, Tang, & Liang, 2006b).Furthermore, the kinetics of the transesterification reaction
f J. curcas L. oil and methanol was investigated. The rate con-tant for the transesterification of J. curcas L. oil is 0.6628, 0.8045nd 0.9474 L/(min mol) at 32, 41 and 51 ◦C, respectively. Theseesults showed that the reaction follows a pseudo-second-orderechanism, while the reaction system can be described as pseudo-
omogeneous. The activation energy Ea was 15. 46 kJ/mol (Zhou etl., 2006b).
.4. Quality of biodiesel from J. curcas L. oil
The properties of crude J. curcas L. oils vary with their origins.atty acid contents in the crude J. curcas L. oil affect the biodieselroduction process and the biodiesel fuel properties. Table 6 listshe fatty acid compositions of two J. curcas L. oil samples. They wererepared in the two-step process under the optimized conditionsescribed above. The results indicated that the two-step processould convert the J. curcas L. oils to diesel. After esterification andransesterification, the AV and kinematic viscosity decreased sig-ificantly and the flash point was much higher than those of theetro-diesel (Table 7). The AV, density, viscosity, free glycerin, andash point meet the biodiesel standards of ASTM D6751. AV foriodiesel is primarily an indicator of FFA and AV higher than 0.8 mg-OH/g have been associated with fuel system deposits causing
educed life of fuel pumps and filters. Higher viscosity fuels canause poor fuel combustion that leads to deposit formation. TheAME’s flash point is much higher than that of petro-diesel whichan result in improved fire safety. The free glycerin number mea-ures the amount of by-product glycerin presenting in the biodiesel.
1096 H. Lu et al. / Computers and Chemical Engineering 33 (2009) 1091–1096
Table 6Fatty acid content in the refined oil.
Composition
C14:0 C16:1 C16:0 C18:2 C18:1 C18:0 C20:0 C22:0 C26:0 Content of unsaturated
Sample 1 (wt%) 0.13 1.18 18.97 38.36 35.28 5.60 0.13 – 0.37 74.82Sample 2 (wt%) 0.04 0.79 13.98 38.38 40.16 6.45 0.17 0.03 – 79.33
Table 7Quality of biodiesel from Jatropha curcas L. oil.
Property AV (mg-KOH/g-oil) (20 ◦C) Density (g/ml) Kinematic viscosity (mm2/s) (40 ◦C) Content of glycerol (%) Flash point (◦C) CFPP (◦C)
Sample 1, oil 10.1 0.9160 36.80 – – –Sample 1, FAMA 0.29 0.8810 5.13 0.02 164 2SSA
IiatmosbcccJ
4
btt1ticcbFaoomp
A
(
R
A
B
F
F
G
G
H
I
J
K
L
L
L
M
M
M
M
N
S
S
T
v
W
Y
Z
Z
ample 2, oil 2.80 0.9176 33.49ample 2, FAMA 0.18 0.8764 4.06STM D6751 <0.8 0.82–0.90 1.9–6.0
f the number is too high, storage tank, fuel system and engine foul-ng can occur. The cold flow properties of biodiesel and petro-dieselre extremely important because they can start to freeze or gel ashe temperature gets colder. The cold filter plug point (CFPP) is a
easure of low-temperature operability. The CFPP of the J. curcas L.il biodiesel product is nearly as high as 0# petro-diesel, whereasome animal fats biodiesel and plant oils biodiesel such as palm oiliodiesel have much higher CFPP (12 ◦C) that restricts their appli-ation at lower temperature. Therefore, the J. curcas L. oil biodieselan be directly used like 0# petro-diesel. The diversity of fatty acidontents only resulted in a small difference in the properties of theatropha biodiesel.
. Conclusion
A two-step process was developed to prepare J. curcas L. oiliodiesel. FFAs in the raw oil were converted to methyl esters inhe pre-esterfication catalyzed by sulfuric acid or solid acid beforeransesterification. The acid value of oil lowered from the initial4.0 mg-KOH/g-oil to below 1.0 mg-KOH/g-oil in 2 h at 70 ◦C underhe condition of 12 wt% methanol in oil, 1 wt% H2SO4 in oil. Watern oil could reduce the reaction rate and phospholipids compoundsould be eliminated in the pre-esterifiction process. The solid acidatalyst SO4
2−/TiO2 (ST) for the pre-esterification was preparedy calcining metatitanic acid. It was found that the conversion ofFAs was higher than 97% at 90 ◦C for 2 h using 4 wt% ST-serial cat-lysts with a molar ratio of methanol to FFAs of 20:1. The yieldf biodiesel by transesterification was higher than 98% in 20 minf reaction time using 1.3% KOH as catalyst, and a molar ratio ofethanol to oil of 6:1 at 64 ◦C. The transesterification reaction is of
seudo-second-order with an activation energy Ea of 15. 46 kJ/mol.
cknowledgment
We thank the key grant project of Chinese Ministry of EducationNo. 307023) for financial support.
eferences
l-Widyan, M. I., & Al-Shyoukh, A. O. (2002). Experimental evaluation of the transes-terification of waste palm oil into biodiesel. Bioresource Technology, 85, 253–256.
erchmans, H. J., & Hirata, S. (2008). Biodiesel production from crude Jatropha curcasL. seed oil with a high content of free fatty acids. Bioresource Technology, 99,1716–1721.
oidl, N., Foidl, G., Sanchez, M., Mittelbach, M., & Hackel, S. (1996). Jatropha curcaslL. as a source for the production of biofuel in Nicaragua. Bioresource Technology,58, 77–82.
reedman, B., Pryde, E. H., & Mounts, T. L. (1984). Variables affecting the yields of fattyesters from transesterified vegetable oils. Journal of the American Oil Chemists’Society, 61, 1638–1643.
Z
Z
– – –0.02 166 0
≤0.02 ≥130 –
erpen, J. V. (2005). Biodiesel processing and production. Fuel Processing Technology,86, 1097–1107.
hadge, S. V., & Raheman, H. (2005). Biodiesel production from mahua (Madhucaindica) oil having high free fatty acids. Biomass Bioenergy, 28, 601–605.
olser, R. A., & Harry-O’Kuru, R. (2006). Transesterified milkweed (Asclepias) seedoil as a biodiesel fuel. Fuel, 85, 2106–2110.
kwuagwu, O. E., Ononogbu, I. C., & Njoku, O. U. (2000). Production of biodiesel usingrubber [Hevea brasiliensis (Kunth. Muell.)] seed oil. Industrial Crops and Producst,12, 57–62.
itputti, J., Kitiyanan, B., Rangsunvigit, P., Bunyakiat, K., Attanatho, L., & Jenvanitpan-jakul, P. (2006). Transesterification of crude palm kernel oil and crude coconutoil by different solid catalysts. Chemical Engineering Journal, 116, 61–66.
örbltz, W. (1999). Biodiesel production in Europe and North America, an encour-aging prospect. Renewable Energy, 16, 1078–1083.
eung, D. Y. C., & Guo, Y. (2006). Transesterification of neat and used frying oil:Optimization for biodiesel production. Fuel Processing Technology, 87, 883–890.
iu, Y. Y., Lu, H. F., Liang, B., & Chen, P. (2007). Pre-esterification of Jatropha curcas L.seed oil for biodiesel production. China Oils & Fats, 32, 43–46 (In Chinese).
ópez, D. E., Suwannakarn, K., Bruce, D. A., & Goodwin, J. G., Jr. (2007). Esterificationand transesterification on tungstated zirconia: Effect of calcination temperature.Journal of Catalysis, 247, 43–50.
a, F. R., & Hanna, M. A. (1999). Biodiesel production: A review. Bioresource Technol-ogy, 70, 1–15.
baraka, I. K., & Shanks, B. H. (2005). Design of multifunctionalized mesoporoussilicas for esterification of fatty acid. Journal of Catalysis, 229, 365–373.
ittelbach, M. (1996). Diesel fuel derived from vegetable oils. VI: Specifications andquality control of biodiesel. Bioresource Technology, 56, 7–11.
ohibbe Azam, M., Waris, A., & Nahar, N. M. (2005). Prospects and potential of fattyacid methyl esters of some non-traditional seed oils for use as biodiesel in India.Biomass Bioenergy, 29, 293–302.
arasimharao, K., Brown, D. R., Lee, A. F., Newman, A. D., Siril, P. F., Tavener, S. J., & Wil-son, K. (2007). Structure–activity relations in Cs-doped heteropolyacid catalystsfor biodiesel production. Journal of Catalysis, 248, 226–234.
arin, R., Sharma, M., Sinharay, S., & Malhotra, R. K. (2007). Jatropha–palm biodieselblends: An optimum mix for Asia. Fuel, 86, 1365–1371.
iler-Marinkovic, S., & Tomasevic, A. (1998). Transesterification of sunflower oil insitu. Fuel, 77, 1389–1391.
iwari, A. K., Kumar, A., & Raheman, H. (2007). Biodiesel production from jatrophaoil (Jatropha curcas) with high free fatty acids: An optimized process. BiomassBioenergy, 31, 569–575.
an Kasteren, J. M. N., & Nisworo, A. P. (2007). A process model to estimate the costof industrial scale biodiesel production from waste cooking oil by supercriticaltransesterification. Resources Conservation and Recycling, 50, 442–458.
ood, P. (2005). Out of Africa: Could Jatropha vegetable oil be Europe’s biodieselfeedstock? Refocus, (July/August), 40–44.
ang, Y., Lu, H. F., & Liang, B. (2007). Influence of the calcination conditions of sul-phated titania on the activity in the esterification of high acid value oil. ChemicalReaction Engineering and Technology, 23, 13–18 (In Chinese).
hang, Y., Dubé, M. A., McLean, D. D., & Kates, M. (2003a). Biodiesel production fromwaste cooking oil. 1. Process design and technological assessment. BioresourceTechnology, 89, 1–16.
hang, Y., Dubé, M. A., McLean, D. D., & Kates, M. (2003b). Biodiesel production fromwaste cooking oil. 2. Economic assessment and sensitivity analysis. BioresourceTechnology, 90, 229–240.
hou, H., Lu, H. F., & Liang, B. (2006). Solubility of multicomponent systems inthe biodiesel production by transesterification of Jatropha curcas L. oil withmethanol. Journal of Chemical & Engineering Data, 51, 1130–1135.
hou, H., Lu, H. F., Tang, S. W., & Liang, B. (2006). Study on the trans-esterificationreaction of bio-diesel with Jatropha curcas L. oil. Applied Chemical Industry, 35(4),284–287 (In Chinese).