polymer blends are physical mixtures of two or more...
TRANSCRIPT


Polymer blends are physical mixtures of two or more structurally
M e r e n t polymers with no covalent bonds between them. The compon-
ents in polyblends adhere together through van der Wads forces, &pole
interactions or hydrogen bonding. Some level of thermodynamic
compatibility between the components is necessary to prevent phase
separation during processing and use. The other types of mixed
polymer pairs are graft copolymers, block copolymers and interpene-
trating networks. Polymer blends are the most attractive of the 'mixed
polymers' in view of the ease of designing and producing the blends
compared to other mixed polymer types',2. The relationship between
blend, IPN, block and graft copolymers can be understood from
Fig. 1.1.
For the processor and the end user, the blending technology
permits tailoring of a polymer compound to their spec& application
requirements often at a lower cost than a new material and over a
short development period.
Polymers to be combined in blends are generally selected to
complement each other in one or more of the following properties: cost,
processabdity, mechanical properties, chemical resistance, weather-
abdity, flammabhty resistance, thermal performance and a variety of
other proper tie^'.^. Blends are typically viewed as cost saving devices,
whereby an expensive polymer may be combined with a less costly
polymer to provide adequate performance at a signficantly reduced
price to the consumer. The versatihty of matching the pricelperfor-
mance requirements of spec&c application allows for a myriad of

Fig. 1.1 Schematic d i a g r a m o f s o m e s i m p l e two polymer combinations (Source Ref.5)
(a) a polymer blend; (b) a graft copolymer; (c) a block copolymer; (d) a semi - IPN; (e) an IPN (f) a cross - linked copolymer. The solid line represents polymer I, dotted line represents polymer 11, Enlarged intersections represent cross - link sites.

M e r e n t products from combination of the miscible polymers. The blend
can offer a set of properties that are not possible with either of the
polymers comprising the blend. By simply varying the concentration of
the constituents of a miscible blend, an innumerable variety of mate-
rials, each with a unique set of properties can be obtained.
Polymer alloys are the synergestic polymer combinations with
real property advantages derived from high level of thermodynamic
compatibility and greater intermolecular attractive forces between the
constituents. Alloys form practically a single phase system with unique
glass transition temperature.
1.2 COMMERCIAL IMPORTANCE OF POLYMER
BLENDS
The combination of two or more commercially available polymers
through blending represents an inexpensive route to produce new class
of materials. For example, in 1986, engineering polymer blends and
alloys represented 300 million pounds of commercial sales in the USA'.
It is estimated that by the end of 1996, US consumption of engineering
alloys and blends wdl reach approximately 700 million pounds.
Sivaram"rojected that 25% of the current world wide consumption of
high performance polymer is composed of blendslalloys which could
grow as high as 50% by the end of this decade.

1.3 BLENDING TECHNIQUES
Blenhng of polymers are carried out by a variety of means
including melt blenhng, solution blendmg, and latex or dispersion
blenhng, partial block or graft copolymerisation and synthesis of
interpenetrating net works (IPNs)~'~.
1.3.1 Melt blending
Mixing of polymer melt is the most common industrial method of
preparing polymer blends. Melt mixing avoids problems of contami-
nation, solvent or water removal etc. The disadvantages are the high
energy demands of mixing, high viscosity of the polymer melts and the
possibility of macromolecular changes like degradation, crosslinking
and chemical decomposition at high temperatures and stresses. Also,
the rheology of molten polymer systems is very complex and it is very
=cult to forecast the structure of the material from a knowledge of
the mixing conhtions.
Melt mixing of rubbers with plastics in an open-roll mill in air a t
elevated temperature induces oxidative degradation. Two techniques
which can be applied to mix a c u l t systems using a two-roll mill are
fugitive plasticisation and sequencing4. The former is useful when one
of the components has a much higher glass transition or melting
temperature than the other. The material can be softened by the
adhtion of a small amount of volatile solvent. This softened component
is then added to the fluxed second component. As mixing proceeds, the
solvent escapes. The other technique, sequencing, is similar in purpose
4

but involves heat instead of solvent. The step includes fluxing the high
Tg material first using a high roll temperature followed by addition of
second component slowly to the first with the temperature of the rolls
reduced accordingly. In addition, the roll gap must be adjusted
continuously to accommodate the increasing volume of the blend.
A widely used mixing device in the industry is the Banbury
mixer. Similar to the two-roll mill, this device has two counter rotating
rolls. In addition, the rotors are enclosed which produces more areas of
high shear and also allows the melt to be forced against the rolls by a
ram. Mixing in a Banbury is impressively rapid and efficient. But i t is
critical to have exactly the right volume of melt. Also it is possible to
blanket the Brabender with inert gas. Extruders can be used for
blending purpose. An extruder is almost a must if enough blend is to be
made for injection mouldmg.
Another useful mixing device is Wni-Max mixer. The melt is
mixed by torsional flow between two heated plates, but mixing across
flow lines can also be accomplished. This is done by periohcally chan-
ging the gap of the plates. Very small amount of polymers can be mixed
using Mini-Max mixer.
1.3.2 Latex blending
Mixing of low viscous latices is another method of obtaining
polymer blends without any organic solvent resulting to a heterogenity
of the order of micrometers. The limitation of this method is that the
components should be free from impurities and should be miscible. 5

Blenhng of latices of BR with SBR or NR is an example of latex
blending1.
1.3.3 Solution blending
Casting of a blend from a common solvent is the simplest mixing
method available and is widely practiced for coatings because i t allows
rapid and easy mixing of the components. This method causes neither
degradative colour changes nor premature crosslinking reactions. For
the preparation of solid polymer blends, the solvent is removed by
evaporation or precipitation of the polymeric components which usually
leads to phase separation and poorly controlled morphology in the
product4.
1.3.4 Mechanochemical blending
Under certain conditions, the mechanical working of a mixture of
polymers can lead to interpolymerisation leading to block copolymer
formation or graft copolymer formation. In such cases, mixing is
carried out in closed device in the absence of air. The property -
composition relationships of these blends differ from those of simple
mdl mixture. Synthesis of NR-SBR blend is an example of mechano-
chemical blenhng, where mechanical breaking of a primary bond in
the polymer chain backbone leads to the formation of free radicals4.
1.3.5 Freeze drying
With freeze drying, a solution of the two polymers is quenched
down to a very low temperature and the solvent is frozen. Freeze 6

drying has some advantages over solution casting which may be
critical for blending work. Ideally the polymers wdl have little chance
to phase segregate, but will collect randomly in regions throughout the
frozen solvent. Thus the state of Mute solution is somewhat preserved.
The freezing occurs rapidly, if the solution is single phase. Solvent is
removed by sublimation4.
1.3.6. Partial block and graft polymerisation
Partial block and graft copolymerisation is carried out in such a
way that its products are mainly homopolymers, but sufficient block or
graft copolymer is produced to ensure good adhesion between otherwise
incompatible components. Products of this process are suitable for
further mixing (in the latex or melt form) with the same or M e r e n t
homopolymers or copolymers. eg. Styrene butadiene block polymers1.
1.4 COMMERCIAL POLYMER BLENDS
1.4.1 Two phase polymer blends
Many miscible or partially miscible polymer blends have been
investigated recently because of their commercial importance. Blends of
PVC and nitnle rubber based barrier polymers are the examples where
elastomeric modification yields desired toughness by the incorporation
of an immiscible polymer phase. Other examples of commercial polymer
blends where miscibility is not achieved include bis phenol A poly-
carbonate - ABS', poly propylenetethylene propylene rubber1', chlorin-
ated polyethylene - PVC" and poly (methyl methacrylate) - PVC". The
list5of two component blends are given in Table I. 1. 7

Table 1.1
Commercial blend of immiscible components
System Property Advant,ages Applications PVC-ABS Better Processability and Mass-t,ransit interiors,
toughness than PVC, better appliance housings fire ret,ardancy than ABS
PVC-acrylic Imr~act-modified, similar Mass-transit interiors. to PVC-ABS appliance housings
PC-ABS Better toughness and heat Appliance and business distortion temperature than machine housings, ABS, t)et,t,er processability automotive components and lowcr cost t.han PC
PSF-ABS Similar to PC-ABS, composit,ion Plumbina fixtures. can be elect,roplated, lower cost food-service trays than polysulfone (FSF)
PC-PI? Bettor flow and energy Automotive applications absorpt,ion than PC
PC-PET Bett.er chemical resistance Tubing, auto bumpers, and processabilit,y and business machine housings . lower cost than PC
PC-PBT Better solvent resistance Tubing, auto bumpers, and processabilit,y t,han PC business machine housings
PET-PMMA Lower cost t,han PMMA, lower Electrical and warp and shrink than PET electronic applications
PC-SMA Impact.-modified, better Automot,ive applications, toughness and ductility than SMA, better retent,ion of properties upon ageing a t high temoerat,ure and lower cost than PC
PP-EPDM Better impact and Wire and cable insulation, toughness t,han PP auto- bumpers, hose and
gaskets PE-ethylene Bet,ter chemical resistance, Film c:opolymers impact,, st,rength and
toughness than PE Nylon-ethylene Better t,oughness Transport cont,ainers, copolymers sport,s equipments.
Source - Ref. 16

1.4.1.1 Interpenetrat ing polymer n e t work (IPN)
Interpenetrating polymer networks, (IPNs) are a class of mate-
rials formed by interlocking the net work of two or more polymers
synthesised in the presence of each other, aiming a t the enhanced
compatibility of thermodynamically incompatible net works. They are
synthesised by swelling a crosslinked polymer (I) with a second
monomer (11) together with crosslinhng and activating agents and by
polymerising monomer 01) in situ. IPN exhibits high degree of compa-
tibility. If one polymer is elastomeric and other is plastic the combina-
tion tends to behave synergestically, resulting in either reinforced
rubber or impact - resistant plastics depending upon which phase
predominates13.
Styrene butadiene rubber-polystyrene (SBR-PS) IPNs are
relatively incompatible, showing distinct phase separation14, even-
though both are non polar polymers. Density measurements of PU-PS
simultaneous 1 ~ N ' b a d e at a temperature between the glass transition
temperatures (Tgs) of the components showed a density 3% higher
than expected for intermediate IPN composition. This was attributed to
partial mixing or interpenetration of chains of the rubbery and glassy
polymer components. Examples of IPNs are listed in Table 1.2

Table : I. 2
Examples of IPN's
Source - Ref. 13,15
Component, I
Polyurethane Polyurethane Polyurethane Poly (ethyl acrylate) Poly (ethyl acrylate) Polybutadiene
1.4.1.2 Rubber - rubber polyblends
Component I1
Polyester Polyacrylat,e epoxy Poly (styrene - co - methyl methacrylate) Poly (methyl methacrylate) Polystyrene
Blends of two or more less incompatible rubbers are
commonly used in the automobile tyre industry inorder to improve
processabllity. EPDM blended with SBR has shown improvements in
ozone and chemical resistance with better compression set properties'7.
The use of EPDM in polybutahene or natural rubber resists the
formation of crack by ozone attack1'. Blends of natural rubber and
polybutadiene have shown various advantages, including heat stability,
improved elasticity and abrasion resistancelg.
Recently, blends of natural rubber (NR) and ethylene vinyl
acetate copolymer (EVA) have gained importancez0. These materials
combine the excellent ageing and flex crack resistance of EVA and the
good mechanical properties of NR. DSC and DMTA results showed
that the blend components are incompatible in the crosslinked and
uncrosslinked states. The adhtion of NR to EVA decreases the
10

crystallinity of the samples as supported by DSC and X-ray anal-
ysis30-23. Table 1.3 gives the examples of rubber - rubber blends.
Table : 1.3
Example for rubber - rubber blend
I Natural rubber (NR) 1 Polybut,adiene (PB) I Component I
Nalural rubber (NR)
Nitrile rubber (NBR)
Natural rubber (NR)
Component I1
Poly (ethylene-co-vinyl acetate) (EVA)
Poly (ethylene -co-vinyl acetate) (EVA)
Ethylene propylene diene monomer (EPDM)
Natural rubber (NR) Styrene but,adiene rubber (SBR) I
1.4.1.3 Rubber - plastic polyblends ,/' l2 $ \ ', 6; :',".&, ' .r
Among polymer blends, the rubber lends which
are commonly known as thermoplastic elastomers are the most familiar
ones. The improvement of impact strength of polystyrene by the
incorporation of a low modulus rubber phase is dramatic. Similarly,
the brittle PVC after blending with elastomer has improved toughness.
For example PVC - MBS blends have good impact strength and optical
clarity. The incorporation of a random copolymer of butahene and
acrylonitrile to PVC enhances the toughness2~ppreciably. In ABS -
PVC blends, ABS offers improved heat distortion temperature and
processability, whereas PVC offers flame-retardant properties. The
Chloroprene (CR)
Butyl rubber (IIR)
Styrene butadiene copoly
Nat,ural rubber (NR) .-.'
Source - Ref. 5,17,19,20. I /' i
{

blend can have higher notched impact strength than either of the
components. Applications include power tool handles, sanitary ware,
communication relays, electrical terminal blocks and electronic
housingsz5. Sen et a12' developed a novel series of cable sheathing
compounds by blending PVC and functionahsed EPDM. EPDM has
been functionahsed by grafting dtbutyl maleate @BM) using dicumyl
peroxide as initiator. Fire retardant low smoke (FRLS) compounds
made from PVC-functionahsed polyolefin blends possess the special
characteristics of low smoke, low acid generation, increased fire-
retardance and improved volume resistivity which are requirements to
the cable industry. Examples of rubber - plastic polyblends are given in
Table 1.4.
Table : 1.4
Examples of rubbecplas t ic polyblends
Source - Ref. 3,25,26
Component I
Acrylonitrile butadiene st,yrene copolymer (ABS)
Nit,rilc rubber (NBR)
Poly (ct,hylme - co - vinyl acetate) (EVA)
Natural rubber (NR)
1.4.1.4 Plastic -plas t ic poly blends
Component I1
Poly (vinyl chloride) (PVC)
Poly (vinyl chloride) (PVC)
Poly (vinyl chloride) (PVC)
Poly (vinyl chloride) (PVC)
Blends of thermoplastics also play an increasingly impor-
tant role in industry. Polyethylene/polystyrene (PEPS) blend has been
the most thoroughly investigated of all polymer blend systems27.
12

Huarng et al." reported on the immiscible behaviour of PVC-
SAN blends. They reported that most of the compositions of PVC-SAN
blends have two distinct glass transition temperatures. Blends of high
molecular weight PS and PMMA exhibit two phase morphology and
20,30 have been shown to be incompatible by m e r e n t techniques .
Commercially important plastic - plastic blends are listed in Table 1.5.
Table : 1.5
Examples for plastic-plastic blends
Source - Ref. 3,5,27,30.
Component I
Polyethylene (PE)
Poly (vinyl chloride) (PVC)
Poly (vinyl chloride) (PVC)
Poly (methyl methacrylate) (PMMA)
Poly (met,hyl methacrylat,e) (PMMA)
Polystyrene (PS)
Poly (vinylidene fluoride) (PVFz)
1.4.1.5 Block copolymers
Component I1
Polystyrene (PS)
Poly (met,hyl methacrylate) (PMIMA)
Poly (styrene - co - acrylonitrile) (SAW
Poly (styrene - co - acrylonitrile) (SAN)
Poly (ethyl methacrylate) (PEMA)
Polp (methyl methacrylat,e) (PMMA)
Poly (methyl methacrylate) (PMMA)
In block copolymers two phase behaviour is the key factor
resulting to elastomeric properties at normal use temperatures and
thermoplastic characteristics at temperatures suitable for conventional
thermoplastic fabrication. To achieve this, the continuous phase must
be amorphous with a Tg below the normal use temperature whereas
13

the dispersed phase must have a Tg or Tm above the normal use
temperature range. The dispersed phase physically restricts the soft-
block chain ends to a specific boundary and therefore present a situa-
tion similar to cross-linlung. The dispersed phase is also a reinforcing
material. " Examples of block copolymers include styrene - isoprene-
styrene (SIS), styrene-butadiene-styrene (SBS) etc.
1.4.2 One phase polymer blends
Compatibility is the fundamental property deciding the
practical u th ty of a polymer blend. In polymer blends, the property
(P) depends on average properties of the constituents and can be
described by the following equation,
where P is the property of the blend, PI and Pa the properties of the
isolated components and C1 and C2 the respective concentrations of the
constituents. I is an interaction parameter which can be positive, zero
or negative as shown in Fig. 1.2. When I is positive the property is
synergestic, when I is zero the property is additive (one phase blend)
and when I is negative the property is non synergestic (two phase
blend).
Miscible blends have only one phase and are rnorpholog-uxxlly the
simplest case. Commercially important examples of this subclass
include PPO-PS and PVC-nitrile rubber. The glass transition temper-
ature (Tg) is the primary thermal transition for these blends, and it
14

COMPOSITION
Fig 1.2. Variation of property with composition for a binary polymer blend (Source Ref.5)

varies monotonically with composition following FOX^' equation and
Gordon Taylor equation 33, which were originally written to describe
the composition dependence of Tg of copolymers. On the property
composition diagram (Fig. 1.2) the Tg usually falls below the tie-line
connecting the Tg's of the pure components in accordance with these
equations, although values above the tie line have been reported in
some noncommercial systems involving very strong intercomponent
hydrogen bonds34.
The glass transition temperature dependence on composition in
this subclass has considerable commercial significance because it
largely determines the heat distortion temperature (HDT) or the
maximum use temperature of the blend.
Exposure to high energy rahation causes various damage to
certain polymers and limits their use in applications where radiation
stenlisation is required. Incorporation of phenyl units into the stru-
cture, for example by copolymerisation provides protection of the
rahation- sensitive units. This mechanism apparently acts over a short
range and is not operative in phase-separated materials such as graft
35,36 copolymers or immiscible blends . There is growing evidence that
such protection can occur in miscible blends where mixing is at the
segmental level. For example, PMMA undergoes chain scission and
therefore loss of mechanical properties upon exposure to y -radiation.
The studles of Nguyen and ~ a u s c i ? ~ show that the extent of chain
scission of PMMA is greatly reduced in miscible blends with SAN
copolymer.

As observed in Tables 1.1 and 1.6, i t is possible to obtain a
particular improvement in properties by forming either a miscible or
immiscible blend. For example, the heat distortion temperature (HDT)
of ABS can be enhanced by mixing ABS with a miscible styrene maleic
anhydride component".
The material properties required for engineering applications
are high heat distortion temperature (HDT), toughness, solvent resist-
ance, low shrinkage upon moulding, low cost, and ease of moulding
which usually means a highly shear-dependent melt viscosity a t
moderate temperature combined with good melt thermal stability. No
material, blend or homopolymer, meets all these criteria. High melting
polyesters and polyamides meet most of the requirements. But these
materials are often =cult to mould because of low melt viscosities.
Amorphous materials with toughness and high Tg such as poly-
sulphone,polycarbonate and poly (para phenylene oxide) (PPO) possess
low mould shrinkage. But these materials are difficult to mould
because their melt viscosities are high and shear independent. This is
especially true in the case of PPO, which can be used commercially
only in blended form with PS as a flow aid. Glassy polymers, both
blends and homopolymers, are prone to crack upon exposure to
solvents and also more expensive than high volume polyesters and
polyamides. Miscible and immiscible blends of these two classes of
materials are a logical way to meet the growing needs for high perfor-
mance materials and the polycarbonate blends with poly (ethylene
terephtalate) (PET) and poly (butylene terephthalate) (PBT) are
commercial examples of this approach.
17
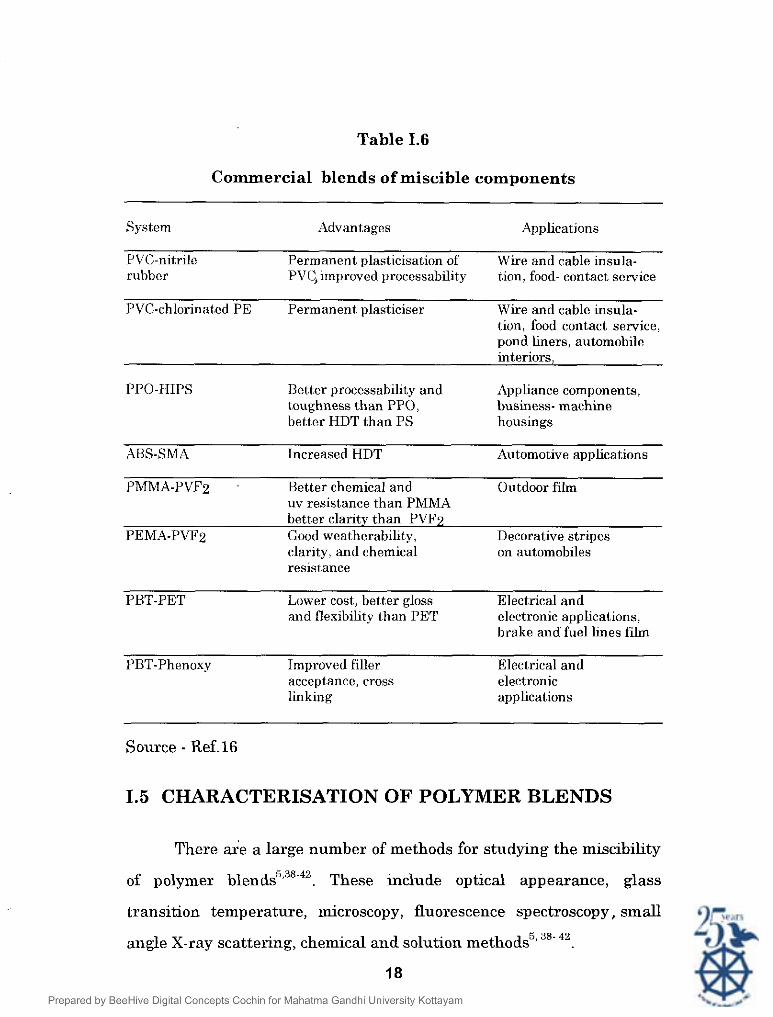
Table 1.6
Commercial blends of miscible components
System Advantages Applications
PVC-nitrilc Permanent plasticisation of Wire and cable insula- rubber PVC,~mprovcd processability t,ion, food- contact service
PVC-chlorinat,ed PE Permanent plasticiser Wire and cable insula- tion, food contact service, pond liners, automobile interiors,
PPO-HIPS Better processabihty and Appliance components, toughness than PPO, business- machine better HDT than PS housings
ABS-SMA Increased HDT Automotive applications
PMMA-PVFZ Better chemical and Outdoor film uv resistance than PMMA better clarity t,han PVFZ
PEMA-PVF2 Good weatherability, Decorative stripes clarity, and chemical on automobiles resistance
PBT-PET Lower cost, better gloss Electrical and and flexibility than PET electronic applications,
brake and fuel lines film
PBT-Phenoxy Improved filler Electrical and acceptance, cross electronic linking applications
Source - Ref.16
1.5 CHARACTERISATION OF POLYMER BLENDS
There are a large number of methods for studying the miscibility
538-42 of polymer blends , These include optical appearance, glass
transition temperature, microscopy, fluorescence spectroscopy, small
angle X-ray scattering, chemical and solution methods 5. 38- 42
18

1.5.1 Optical clarity
The study of optical properties of blends is important because
the knowledge of the factors leading to turbidity of a blend leads to an
understanding of factors which cause scattering. By knowing these
factors one can design blends of superior appearance.If the blend is
homogeneous it will have a refractive index in between those of the
individual polymers. If the system is heterogeneous with one polymer
present as a dispersed phase embedded in a second polymer it scatters
light according to the size of the hspersed particle5.
1.5.2 Glass transition temperature (Tg)
Determination of Tg is the most commonly used method for
studying the miscibility of polyblends. The Tg of the blend is usually
compared to that of inhvidual constituents. For polymer blends
exhibiting miscibility over the entire composition range, three genera-
Lised curves (Fig. 1.3) are possible: a linear relationship and the
minimum and maximum deviations from linearity. Examples of a linear
variation of Tg versus composition include blends of nitrocellulose and
poly (methyl r n e t h a ~ r ~ l a t e ) ~ ~ .
Examples of minimum variation from linearity are quite
common. One example is (poly hydroxy ether) of bisphenol Alpoly (E
capr~lactone)~~. Many of these systems obey the Fox equation3',
(eqn.I.2) and the Gordon Taylor equationJ3 (eqn 1.3)

Fig. I. 3. Variation of Tg with composition (Source Ref.41)

Tgblend = [Wa Tga + k (I-Wa) Tgbll [Wa + k (1-Wa)] 0.3)
In these equations Tga and Tgb represent the glass transitions of the
undiluted polymer components a and b, Wa and Wb are the weight
fractions of a and b, and k is the ratio of the thermal expansion
coefficient between the rubber and the glass states of the component
polymers, which is given by
The Kelley - Bueche equation45 is similar to the Gordon - Taylor
equation except that the volume fraction $i is used instead of the
weight fraction.
As ( a , - a,) has been proposed to be constant for all polymers46,
k = 1.0 and the Gordon - Taylor and Kelley - Bueche equations reduce
to the linear form
Tgblend = Wa Tga + Wb Tgb
%blend = $ a Tga + $ b Tgb
The above equations are useful for miscible polymer blends.
Tg of any polymer is affected by its previous history, the
experimental time scale and other factors that affect its intra and
intermolecular forces. Owing to the sensitivity of Tg to the disruption
of the local structure that results from mixing two M e r e n t polymers,
the existence of the single and sharp or single and broad, or double 21

and shifted or double and non shifted Tg of a polyblend reveals the
particular macroscopic characteristics of the blend. Broadening of the
transition occurs in the case of blends showing borderline miscibility.
Two separate transitions appear in case of total immiscibility. This
method is valuable only when a quasi-binary polyblend contains
polymers whose Tg values differ by more than 20°C.
A very large variety of physical measurements have been
proposed for the determination of glass transition temperatures. The
most commonly used techniques are
1. Calorimetric determination of heat capacities as a function of temperature Qifferential scanning calorimetry-DSC)
2. Dynamic mechanical (low strain) measurements of complex modulus as a function of temperature
3. Dielectric relaxation spectroscopy
4. Thermo optical technique.
DSC technique has successfully demonstrated polymer - polymer
47 miscibility for many systems'"". Schneier used DSC to study the
effects of mixing conditions on the compatibihty of PMMA and poly
(vinyl acetate) (PVA).ln DSC, the glass transition is distinguished by a
discontinuity in the specific heat (CP) Vstemperature curve.
Mechanical methods are the more frequently cited techniques for
the determination of the transition behaviour of polymer blends. The
elastic and viscoelastic properties of polymers derived by subjecting
polymers to small amplitude cyclic deformation can also yield important
information concerning transitions occurring on the molecular scale. 22

Data obtained over a broad temperature range can be used to ascertain
the molecular response of a polymer in blends with other polymers. In
a highly phase-separated polymer blend, the transitional behaviour of
the individual components will be unchanged. Likewise, in a miscible
blend, a single and unique transition corresponding to the glass
transition will appear. The glass transition temperature is obtained
from the plot of log. of loss modulus (GI) or mechanical loss (tans) vs.
temperature.
The electrical properties of polymers are analogous to mechanical
properties. The &electric loss factor @")and the dissipation factor
(tan6) are commonly used to ascertain polymeric transitions. The
experimental advantage of obtaining transition data from electrical
measurements over dynamic mechanical testing is in the ease of
changing frequency. The major disadvantage is the difficulty in
determining the transitions of non polar polymers. Generally non polar
polymers require slight mod&cation. Both mechanical and dielectric
methods are successfully used for establishing the miscibility of various
' 41 polymer systems.
1.5.3 Spectroscopy
Among the spectroscopic methods, NMR, IR and FTIR are the
most commonly used techniques for the determination of miscibihty,
nature of interaction and phase separation and influence of tacticity on
miscibility of polymer blends 48,49,50 . Kwei and CO-workersso used NMR
to study the influence of tacticity of PMMA on its miscibility with poly
(styrene-co-vinyl phenol). 23

Nuclear magnetic resonance experiments are possible on solid
polymers and polymer melts. Elmqvist and vans son^^ showed that
broad line NMR is a sensitive tool for the detection of small amounts of
a soft phase embedded in a hard matrix. The resonance of proton in the
soft phase is relatively sharp compared with the resonance band of the
matrix protons. The intensity of the band due to the soft segment is
accordingly very high.
Infrared spectroscopy has most often been used in the analysis
of polymer mixtures. Specific interactions in the systems poly (acrylic
acid)-poly (ethylene imine) and poly (methacrylic acid)-poly (ethylene
imine) were demonstrated by infrared spectroscopy by Zezin et a15'.
The system PMMA - poly (vinylidene fluoride) exhibits speclfic
interaction involving the carbonyl group, accordmg to infrared spectro-
scopy performed by Coleman and Co- workersG2. FTIR technique gives
evidence of the existence of speclfic interactions and elucidate the
nature of such interactions. Frequency s w t s and band broadening for
blends have been ascribed to intermolecular chemical interactions and
to changes in polymer chain conformations. Coleman and CO-workers4"
used FTIR technique to determine the lower critical solution behaviour
of PVCIEVA and EVAJchlorinated polyethylene.
Infrared and ultraviolet spectroscopy s tuhes on PSlPPO blend by
Wellinghoft and CO-workers" provided evidence for the conclusion
that, PPO is loosely packed in the glassy state and the addition of PS
reduces the free volumes. They further reported that the chains of the
two components interpenetrate significantly. The high extent of

miscibility is associated with the strong interaction between the phenyl
group of the PS and the phenylene group of PPO.
Ultraviolet emission spectroscopy has been suggested as a tool
for quantifying the degree of miscibility of polymeric components54. To
employ this technique the components of the blend must contain
chromophoric structures active in the uv region.
1.5.4 Scattering techniques
The commonly used scattering methods include X-ray scattering,
neutron scattering, light scattering, pulse induced critical scattering
and the cloud point method. Electron scattering and Rayleigh-Brillowin
scattering are also used in the characterisation of chain conformation, 55.56 local order and the morphology . All scattering experiments are
based on the existence of variation in the homogenity of the scattering
medium.
1.5.5 Inverse gas chromatography
Inverse gas chromatography (IGC) has been applied successfully
to the description of polymer blend miscibility in the liquid state. It
has also been used in studying the concentration dependence of the
glass transition temperature of polymer blend57. Glass transition
temperature, crystallinity, adsorption isotherms, heats of adsorption,
surface area and interfacial energy can be obtained from IGC~'. su5'
applied IGC to the study of thermodynamic interactions in poly (vinyl
chloride) (PVC) plasticised by di-n-octyl phthalate @ OP).

1.5.6 Fluorescence spectroscopy
Two fluorescence methods have been used for the study of
polymer blends. They are excimer fluorescence and non radiative
energy transfer techniques.
1.5.6.1 Excimer fluorescence
Excimer fluorescence is an effective and sensitive morphological
tool for the study of miscibility of an aromatic vinyl polymer with a non
fluorescent host polymer. A convenient analysis of the degree of mixing
at the molecular level is by the measurement of photostationary
excimer to monomer fluorescence intensity ratio Ie/Im. Tao, and
rank'" used excimer fluroscence technique to study miscible blends of
poly (2-vinyl naphthalene) (PZVN) and poly (cyclohexyl methacrylate).
Using fluorescence technique, Monnerie and Co-workersG0 have
determined the boundaries (binodal and spinodal curves) of the phase
hagram of anthracene-labeled polystyrene/poly (vinyl methyl ether)
blends.
1.5.6.2 Non radiative energy transfer fluorescence spectroscopy
Non rahative energy transfer (NRET) can be used to probe
polymer miscibihty and phase separation. In NRET technique, the
amount of energy transferred from a donor to an acceptor group is
assessed. Because the transfer is non radiative, the donor and acceptor

must be very close. A large amount of energy transfer imply miscibility.
Teyssie and CO-workers" used NRET technique successfully to deter-
mine the miscibility and phase separation in PVCPMMA blends. The
ratio of the fluorescence emission intensities of the donor and acceptor
(INIIA) (naphthalene and anthracene were used to label PVC and
PMMA respectively) is related to the efficiency of energy transfer. This
ratio is a measure of the degree of miscibility of two polymers. The
advantage of these methods is that the proximity of groups a t the
several Angstrom level can be unambiguously assessed.
1.5.7 Microscopy
Microscopy is used to find out both the presence and conne-
ctivities of the phases of a polyblend. Scanning electron microscope,
phase contrast microscope and transmission electron microscope are
extensively used for studying the morphology of polymer blends. For
the d e t d e d characterisation of the phase morphology in blends,
microscopy is unmatched by any other techniques. Scanning electron
microscopy (SEM) offers the simplest procedurefi? Photomicrographs of
fracture surfaces often give information about the extent of adhesion
between phases. Several polymer- polymer systems which are reported
to be miscible by less sensitive techniques have been shown to contain
domains by using the electron microscope. Matsuo et al.'"ound micro
level heterogenity (400AO) in the system of PVC-NBR containing 40%
acrylonitrile, by microscopy although only one glass transition was
observed in this blend.

Transmission electron microscopy64 ('!?EM) has been attempted
for the purpose of defining the scale of mixing. One example of an early
application of TEM to blends was the investigation of structures
produced by the spinodal decomposition of a PMMAISAN blend65.
Morphology of blends is the organisation of components i n
supermolecular scale. It indicates the form, size and orientation of
blend's crystallites, structure of groups of molecules of the components
and other boundaries, degree of crystallinity and the spatial arran-
gement of blend component phases. The major factors that govern
morphology of the blend system are 1) viscosity of the components 2)
ratio of the components 3) processing conditions such as shear rate
and temperature and (4) presence of additives. Additionally the
niorphology is also dependent on the thermodynamic properties of the
components and the mixing process.
Makarewiez and ~ i l k e s " carried out extensive stuhes on
morphology associated with the liquid-induced crystallisation of poly
(ethylene terephthalate) (PET) blend with poly (tetramethylene
terephthalate) (PTMT), atactic polystyrene (APS) and poly carbonate of
bis phenol A. SEM was used to study the morphology. They observed
that melt mixed blends of PET and PC showed no large scale phase
separation prior to liquid-induced crystahsation. This was accounted
for by copolymer formation due to interchange reaction occurring
during sample fabrication. The morphology of both pure PET and
PETIPC blends after liquid-induced crystallisation appears identical.

Danesi and porterfi7 have reported that composition, processing
history and difference in melt viscosity influence the morphology.
Similar melt viscosity values lead to uniform chstribution of minor
component in the major one with a very fine morphology. If the minor
component has lower viscosity than the major, the minor phase will be
finely and uniformly chspersed as domains oriented in the extrusion
direction. The minor component gets coarsely dispersed in essentially
spherical domains if its viscosity is higher than that of the major
component. SEM and phase contrast microscopy have been successfully
used in these studies by Danesi and
Morphology of different polymers has been studied by many 68.68 groups using a number of techniques .
1.5.8 Melting point depression
In polymer - polymer blends in which one component is
crystalhne, melting point depression can be used to determine the
miscibility of the system. Examples include isotactic po lys tyrene-~~070
and poly (vinylidene fluoride) - poly (methyl methacrylatejl,. Nishi and
wang7' calculated interaction parameter from melting point depression.
1.5.9 Viscosity studies
The basis for using dilute solution viscosity as a parameter for
compatibility determination of polymer blends lies in the fact that
while in solution the repulsive interaction may cause shrinkage of the
polymer coils resulting in a viscosity of the polymer mixture that is

lower than the value calculated from viscosities of the pure components
on the assumption of the adhtivity law. On the other hand attractive
interation increases the viscosity of the system.
Kulshreshtha et alT2 applied the viscosity method to the
PVCIABS polyblend system to study the compatibility. They found
that the plot of absolute viscosity versus composition deviates from
linearity accordmg to the degree of compatibility. chee7' also proved
viscometry as a simple and reliable tool for identifying the compati-
bllity of PVCJPMMA, PMMAfPiBMA and PVCffiBMA blends.
1.5.10 Mechanical properties
Utlimate mechanical properties such as tensile strength, tough-
ness, abrasion resistance, elongation, fatigue resistance and environ-
mental stress crack are of considerable interest for end application of
blends74. Usually compatible polymer blends show synergism in
mechanical properties while in the case of miscible blends an adhtive
value is obtained between the component polymers5.
Modulus of rigidity is roughly intermediate between the two
components and it depends fairly linearly on the ratios of the two3'.
When the two polymers exist in separate phases the relationship
between the composition and modulus is not nearly as simple. When
one of the components is present in larger amount it should form the
continuous matrix phase and should play the primary role in deter-
mining the modulus. A steep transition between the two components is
expected in the region where both components are present in equal
30

amounts. On the other hand, the dispersed phase may be considered as
a filler and effect of such content on the modulus of the filled matrix
can be calculated.
Strong exothermic interactions between components of miscible
blends leads to denser packing and loss of free volume. In such cases
mechanical properties are expected to be higher than prehcted by
simple additivity rule. Experimental results in some systems generally
conform to this expectationT5. Poor interfacial adhesion results in
inferior ultimate properties compared to the expected average property
of the constituents in totally incompatible blends. In a polyblend where
a moderate concentration of tiny rubber particles are hs-n a ,*.::, 4 L!li>
glassy plastic matrix, tremendous improvement i y d m P H 3 t r e &
// .< .
.1 results.
1.5.11 Rheological properties
The two or more polymers used for malung
difference in molecular structure. This may result in a different
rheological behaviour for the blendsT6.
Usachev et al.I6 proposed that the flow of polymer blends must
be regarded as the combined flow of M e r i n g viscosity. Parasiewicz et
al.I7 found that polymers of similar chemical structure exhibit
rheological behaviour deducible from adhtivity considerations and
blends with polymers having different chemical structure generally
have lower viscosity than the individual component of the blend.
Polymer blends with a second component, which is rubbery with high
31

molecular weight, can provide improvements in melt strengthm. This
improves processability for blowing and thermoforming.
The use of solution or melt rheology to judge interactions in
blends is largely empirical in nature. The expectation in rheology is
that, polymers with strong favourable or unfavourable interactions will
show viscosity Vs. concentration response that are largely n ~ n - l i n e a r ~ ~ .
A typical expression 64 to indcate the blend viscosity is
This is capable of fitting quite complex blend viscosity behaviour. In
this equation q, is the blend viscosity, q, and q, are the viscosities of
the components, and B1 and B2 are interaction terms. W, and W, are
the weight fractions of components.
1.5.12 Empirical approach at predicting compatibility:
solubility parameter approach
According to the solubility parameter approach at predicting
compatibihty, two polymers mix well if the difference in the pure
component solubihty parameter is s m d , typically 1.7-2.0'~. For
polymer molecules, the solubility parameter is best calculated using
tables of molar attraction coefficients, E, by using the equation

where E is summed over the structural units of the polymer, M the
'mer' molecular weight and e is the density7" Tables of calculated
values of 6 have been publisheds0.
Although the solub~lity parameter approach is not rigorous, it
allows a useful first approximation to polymer solubility. The difFerent
temperature coefficient for the pure component solubility parameter
has even been suggested as the reason for high temperature phase
separations1.
sanchezsz compared the predictions of polymer-polymer compati-
bility from the equation of state theories and the solubility parameter
method.
1.5.13 Interaction parameter and critical interaction
parameter
The miscibhty of a polymer blend system can be predicted by
calculating the interaction parameter and critical interaction para- 5 meter . The interaction parameter XAB, can be written in terms of
solubility parameter as
where Vr is the reference volume, R the gas constant, 6 the solubility
parameter and T the temperature in the absolute scale.
The critical interaction parameter can be calculated using the
equation
33

where XI and X2 are the degrees of polymerisation of polymer 1 and 2
respectively. Miscibility can occur over the entire cayosition range only 83
if (X ~B)er >XAB.
1.5.14 Enthalpy of mixing of polymer-polymer blends
The heat of mixing is an approximate measure of free energy of
84.85 mixing and thus may indicate the degree of compatibility. schneirsG
suggested that the following equation may be deduced for the heat of
mixing for two component polymer blends.
where XI, p and M are the weight fraction of polymer, polymer density
and the monomer unit molecular weight respectively and 6 is the
solubihty parameter of a polymer. Singh et a1.8"redicted the compati-
bility of poly (methyl methacrylate) / poly (vinyl acwtate) (PMMAIPVA)
and poly (methyl methacrylate/polystyrene (PMMAIPS) blend systems
using the above equation.
1.6 THERMODYNAMICS AND PHASE SEPARATION
OF POLYMER BLENDS
The structure and stability of polymer blends depend primarily
on the miscibility of polymers used in making the blend. The stability

of any binary system requires that the Gibb's free energy of mixing
AGm be negative. AGm is given by
A Gm - - AHm - TA Sm
In this equation A Gm = change in free energy of mixing
AHm = change in enthalpy of mixing
A s m = change in entropy of mixing
T - - absolute temperature
A negative AGm is necessary, although not a sufficient condition for
the stability of the mixture. Thermodynamic stability for a one phase
system exists when
where is the compositional variable. Graphically, the above
conditions are shown in Figure 1.4. The restriction of eq.0.14) requires
that the free energy-composition diagram be concave downward. For a
system that is miscible over the entire composition range a t one
temperahre, a curve similar to Fig. 1.4a would be followed. A partially
miscible system would include some compositions that are incompa-
tible. Several interesting features of the phase diagram can be derived
from Figure 1.4 b. Between the composition C and the pure component
1 and composition D to pure component 2, the behaviour of AGm with
composition obeys eq. 0.14). However, phase separation of two phases
denoted by A and B would lower the free energy from G' to G? The
binary compositions between A and C, D and B exist in a metastable
35

COMPOSITION
Fig. 1.4 Free energy versus blend composition for (a) miscible, and (b) partly miscible polymer blends (Source Ref.78)

state; stable to small perturbations in the system but unstable to large
disturbances. The boundary between the stable region and the
metastable region (point A and B) is termed the binodal. Compositions
between C and D do not satisfy the restriction of eq. 0.14) and are
unstable. The boundary between the unstable and metastable state
(points C and D) is called the spinodal.
The locus of the spinodal is in principle easy to calculate. The
spinodal boundary is simply the inflection points of the free energy-
composition hagram given as
The binodal is much more difficult to calculate since the
boundary does not necessarily coincide with any critical points on the
free energy - composition diagram. The locus of the binodal is found by
setting the chemical potentials of both homopolymers equal in the two
co-existing phases. Graphically this means that the binodal is found by
drawing a common tangent between the two concave section of the free
energy hagram (dashed line in Figure 1.4 b)78.
As the temperature is changed, so does the free energy plot.
Upon an increase in temperature, the binary system can phase
separate. The temperature a t which phase separation first appears is
called the lower critical solution temperature (LCST). Analogously,
phase separation upon a decrease in temperature is inhcative of the
existence of an upper critical solution temperature (UCST). The
position of the critical solution temepratures can be calculated by 78
37

Figure 1.5 illustrates the phase diagram for binary polymer-
polymer blends. The solid line is the binodal and the dashed line
indicates the spinodal. Figure 1.5 (a) shows UCST behaviour. This is
very common in polymer solution thermodynamics, where the critical
point at infinite molar mass represents the well-known Flory, 9 ,
condltions8? The existence of a polymer-polymer UCST has been
predicted but convincing evidence has not been found. Based on the
appearance of two mechanical loss peaks below O°C, Koningsveld et
al.87 reported that blends of SBR and natural rubber exhibit UCST
behaviour.
Figure 1.5 (b), showing LCST behaviour, is the normal phase
hagram for high molar mass homoploymer blends where specific
interactions are present. Many systems showing LCST behaviour have
been reported in literature". Examples are PSIpoly (vinyl methyl
ether)", poly (methyl methacry1ate)lpoly (styrene-co a ~ r ~ l o n i t i r i l e ) ~ ~
and poly (capro1actone)l poly (styrene-co a~r~lonitrile)"'. Figure 1.5 (c)
shows a phase diagram which exhibits a UCST and a LCST above it.
This behaviour is common in polymer solutions where the LCST is
usually above the boding point of solvent. It has been suggested that
polystyrenelpoly (2-chlorostyrene) blends might be in this categoryg2.
There are experimental =culties in determining such phase hagrams
since the UCST may be below the glass transition temperature (Tg)
and the LCST may lie above the decomposition temperature. It is also
38

Fig. 1.5. Schematic phase diagrams for polymer polymer blends:- (Source Ref.88)
--spinodd lines - binodal lines (a) a phase diagram of the UCST type : (b) a phase hagram of the LCST type :
(c) a phase diagram in which both an UCST and LCST occur, (d) an 'hourglass' phase hagram; and
(e) a phase diagram in which the UCST occurs above the LCST

possible for the UCST to merge with the LCST, producing an 'hour
glass' type of phase hagram as shown in Bg.I.5(d) or the UCST can lie
above the LCST, producing a 'closed loop' phase diagram as shown in
Figure 1.5(e). The 'hour glass' phase diagram (FigureI.5(d)) has no
temperature region for which a single phase exists over the entire
compdtion range. This represents the phase behaviour of compatible
blends. For homopolymers, the equation of state theory would predict
that UCST should always occur at or below an LCST. But such
restrictions may not apply to copolymer blends.
1.6.1 Kinetics of phase separation
The phase separation in miscible polymer systems are generally
brought about by variation in temperature, pressure andlor compo-
sition of the mixture. In polymer blends, the mixture can subsist inside
the spinodal below Tg and remain in a one phase system.
The lunetics of phase separation are different inside the spinodal
as opposed to the metastable region. Between the spinodal and binodal,
the system is stable to small compositional fluctuations whereas inside
the spinodal, any change in composition results in a change in free
energy, favouring phase separation. Two different mechanisms of phase
separation have been porposed to explain the lunetics in the two
regions.
Inside the spinodal, there is no thermodynamic barrier to phase
separation and the process should be spontaneous until separation is
complete. The phase separation requires diffusion against the concen-
40

tration grahent ie, a negative diffusion coefficient or so called "uphill"
hffusion. (Fig.1.6) Phase separation by this process is called spinodal
93,94 decompo&ion and has been treated theoretically by Cahn . Since
the spinodal decomposition mechanism is spontaneous and continuous,
the morphology is characterised by interconnected phases.
In the metastable region, the Wusion coefficient becomes
positive and phase separation is by nucleation and growth. A nucleus
is formed from a large fluctuation in composition and once established,
grows by normal Musion processes. The work required to form a
nuclueusdepends on the metastability of the system, vanishing to zero
a t the spinoda19? In the nucleation and growth mechanism, composition
of the growing phases remains constant. Schematic representation of
the two mechanisms are given in Egure 1.6. A good review on the
kinetics of phase separation is given by Kwei and wangY5.
1.6.2 Polymer mixture theories
The various theories of polymer mixture are arranged chronolo-
gically in this section. Many reviews on polymer solution thermodyna- 96-90 mics have been published . The first attempt to describe polymer
solution thermodynamics was made by Flory R5,99.100 and Huggins 101.102
(F-H theory). The expression derived by Flory and Huggins contained
a combinatorid entropy of mixing term due to the length and volume
of the polymer in solution. A van Laar type enthalpy of mixing was
also added to obtain the free energy of mixing. Although the F-H
theory was useful as a first approximation, it was unable to predict the
details of polymer solution behaviour.
41

Nucleation and growth
Nucleation Growth \ Equilibrium
\
M
$ 2 ~ --
Spinodal phase separation / Fluctuation Growth of Saturation and continued
fluctuation separation (Phase hardening)
$M = Composition of mixture
41 13 = Equilibrium composition of phase
Fig. 1.6. Modes of phase separation in miscible blends (Source Ref. 88)

Extension of F-H theory to polymer-polymer system was done by
scottIo3 and omp pa'^! The expression for the free energy of mixing
two polymers derived by Scott was103
RTV A G ~ = ----(v, /X,)lnV,+V,/X,lnV, +x,,V,V, (1.17) V1t
where V is total mixture volume,VR the reference volume which equals
the molar 'mer' volume,V, and V2 are the volume fractions, X, and X,
are the polymer chain length and x,, is the polymer- polymer intera-
ction parameter. The terms containing X, are entropic and as X,
increases, these terms go to zero. In other words, the entropy of mixing
of polymers goes to zero at high molecular weights. Given the above
situation, AG, can only be negative if the enthalpic term is negative,
ie. negative x,, Thus the interaction parameter reflects the strength of
interpolymeric contacts.
Calculations based on Flory-Huggins theory can account for the
existence of an UCST, but not for the LCST. The extension of F-H
theory to include free volume was done by Prigogine'06 and lor^''^,
~ c ~ a s t e r " applied the Prigogine-Flory theory to polymer-polymer
systems. Although the equations derived by McMaster were very
complex, several important conclusions were reached. The existence of
a polymer-polymer LCST was predicted. Moreover, it was found that
the thermal expansion coefficient was the most important parameter
for polymer blend systems. Small merences in the thermal expansion
coefficient of the pure components was the cause for LCST behaviour.

Patterson and ~ o b a r d ' ~ offered a simpler form of the Prigogine-
Flory Theory as applied to polymer-polymer systems. In their treat-
ment, they concluded that the primary cause of LCST behaviour was
not unfavourable free volume effects, as McMaster" implied, but
instead on the favourable polymer-polymer interaction at low tempera-
tures which decreases with temperature. However, the predictions of
McMaster and Patterson are similar.
1.6.3 Lattice fluid theory
Sanchez 82,108-110 formulated a new theory which has been charac-
terised as a lattice fluid theory and M e r s from the corresponhng state
theories of Prigogine and Flory.
Several conclusions of the phase behaviour of blends could be
reached using lattice fluid theory. In all cases, LCST behaviour was
prehcted. An increase in molecular weight of one of the components
serves to decrease the LCST and increase the UCST. The pressure
dependence of the CST's predicts a larger change in the LCST than the
UCST. Finally, very few polymer pairs would be expected to be
miscible. The approach used by Sanchez is a useful tool for under-
standmg the mechanism of polymer-polymer phase behaviour.
1.6.4 Phase behaviour of ternary polymer blends
Su and ~ r i e d " ' applied the Flory-Huggins theory to polymer
blends of three monodisperse homopolymers. The free energy of mixing

AGm of three monohpserse homopolymers may be expressed in terms
of volume fractions (c$~) as
G = ( A G ~ 1 k ~ ) (VU 1 V) 0.18)
= (4, +(& /m2)ln42+(43 /m3)ln$3
+X12 $1 $2 +X23 4 2 43 +x3143 41 (1.19)
where Vu is the volume per lattice site, V is the mixture volume, mi is
chain length and xij is the Flory interaction parameter between
segments of polymers i and j. The equation of the spinodal may be
obtained from the relation
2 Jsp = G,, G,, -(GZ3) = 0
where Gij = (6 G 1 6$i 6$j) ,p
From equations 1 and 2, the equation of the spiondal for a
ternary solution becomesLL',
ml$l +m,$, +m,43
-2[m,m2 ( X I + ~ a ) $ l 4 2 f m 2 m 3 ( ~ 2 + ~ 3 ) 4 2 4 3 +m3m1 (XS + ~ 1 ) 4 1 4 3 ]
+4m,m,m3 (xIxz + X ~ X , +~3~1)41$243 = 0 0.22)
where X, = (xij + xik - xjk) 12 0.23)
The critical points must satisfy the additional condition112
G,, SJSP I 64, -G,, s JSP 164, = o 0.24)
If desirable, blends of one or more random copolymers may be
included in the development by adopting an appropriate expression for
45

113-116 Xi, . For example x,, between two copolymers where copolymer 1
has segzment fractions fA and f, of comonomers A and B respectively,
and copolymer 2 has corresponding segment fractions fc and fD may be
given as
where xkl is the segmental interaction parameter between comonomers
k andl .
1.6.5 Experimental determination of phase separation
in polymer blends
Wideline and pulsed NMR", FTIR 117-119 , light scattering120
uv-visible absorption spectroscopy and fluorescence spectroscopy61 have
been successfully used to the study of phase separation in polymer
blends. In wide Line NMR, the temperature variation of the line width
and in pulsed NMR, the relaxation time can be used for characterising
the composition of the phases. The relative signal contributions give
information concerning the amounts of the phases. In FTIR, phase
separation is studied by monitoring the frequency of vibration of the
interacting groups as a function of temperature. A non linear relation-
ship is observed in this case. The temperature at which the relative
strength of the interactions appears very weak is the LCST. The light
scattering invariant can be quantitatively used to describe the early
stages of phase separation.

In uv-visible absorption spectroscopy'" appearance of an
adhtional peak at higher wave length region inhcates the onset of
phase separation. In excimer fluorescence spectroscopy, the LCST is
indicated by the point where the plot of Ie/Im Vs temperature shows a
change in slope.
1.7 PVC BASED BINARY BLENDS
Poly (vinyl chloride) represents one of the most rigorously
investigated components of polymer blends. PVC has been found to be
miscible with a number of structurally M e r e n t polymers and
copolymers. The capability of weak specific interaction is possible with
PVC. The a-hydrogen of PVC is capable of hydrogen bonding,
particulady with polymers which have electron donor groups (amides,
c a r b ~ n ~ l ) ' ~ ~ .
For the miscible blend of PVC and poly (E-caprolactone) charge-
transfer interactions between the pendant chloride and ester oxygen
has been proposed1".
1.7.1 PVCINBR blends
Blends of PVC and butadiene/acrylonitnle copolymers WBR)
historically represent the initial observation that miscibility with
polymer mixtures is possible124. In the technical literature, this blend
has been described as miscible, partially miscible and even hetero-
geneous based on m e r e n t experimental techniques.Generally dynamic
mechanical results inhcate miscibility with some broadening of glass
47

transition temperature. 'Using microscopic techniques separate phase
resolution was possible69. ~mbler '" observed that under certain poly-
merisation conditions, compositional variation in butadiene acrylo-
nitrile copolymers would result in nonhomogeneous materials. The
range of acrylonitrile content sufficient to yield miscibility in PVC
appears to be quite large and equal to 23-45%'"".
1.7.2 PVCIEVA blends
Blends of PVC with ethylene-vinyl acetate copolymers (EVA)
have been widely studied 48, 121-133 . Miscibility appears optimum a t
vinyl acetate contents of 62-70%''~. Marcincin et d."* studied EVA
(45%VA)/PVC and chlorinated EVAPVC blends. Definite phase
separation was observed with the EVAPVC blends. But chlorinated
EVAIPVC blends has single Tg.
Feldman and R U S U ' ~ ~ reported that PVCIEVA-45 blends are
miscible based on the studies of mechanical and &electric loss data.
The composition dependence of the tensile strength and ultimate
elongation for these blends exhibited the characteristics of mechanical
compatibility. EVA with vinyl acetate content 45% has been experi-
mentally observed to have limited miscibility1".
hbscibihty has also been inferred from diffusion data of gas
molecules, which can be used as probes to assess the level of molecular
131.13" mixing . Permeabhty results show that phase inversion occurs in
the case of PVCIEVA-45 blends at higher compositions of EVA. The two
polymers are assumed to be largely incompatible. PVC and EVA form
48

separate phases and may be molecularly dissolved only to a very small
extent. Small amounts of EVA polymer (addition of 5 5%) could be
dissolved in the PVC enclosed in voids or adsorbed on certain surfaces
of the PVC grains. Monteiro and ~haumaturgo'" used viscometry
studies to chatacterise the miscibihty behaviour of PVClEVA blends
with various vinyl acetate content and reported that PVClEVA
mixtures from EVAs with 45 to 70% vinyl acetate content may be
considered miscible.
Nuclear magnetic resonance (NMR)~' data on blends based on a
copolymer of 45% vinyl acetate content indicated partial miscibility
and the extent of miscibility was very much dependent on sample
preparation conditions.
Terpolymers of ethylene vinyl acetatelsulphur dioxide have been
shown to exhibit miscibihty with PVC over the entire composition
range by Hickman and Ikeda '". Sulphur dioxide incorporation allowed
the utihzation of much higher concentrations of ethylene in the
terpolymer than in EVA copolymer while still maintaining miscibility
with PVC. Similar results were noted by Robeson and McGrath13%th
terpolymers of ethylene vinyl acetatelcarbon monoxide and ethylene
ethyl acrylatetcarbon monoxide. With ethylene ethyl acrylate copoly-
mers, it was noted that no copolymer composition exhibited miscibility
with PVC. However, as low as 5 wt% carbon monoxide in the
terpolymer yielded miscible blends with PVC. As with the ethylene
vinyl acetate/S02 terpolymers, a broad range of terpolymer compo-
sitions was observed to be miscible with PVC. These results were

believed to be due to the specific interaction of the carbonyl group of
the terpolymer with the a-hydrogen of poly (vinyl chloride). The
interaction was classified as a weak 'acid-base' type where PVC
represented the proton donor and the terpolymer carbonyl represents
the proton acceptor. This interaction, although weak, allowed a large
variation in the composition of the terpolymer with retention of
miscibility with PVC.
1.7.3 PVCIPCL blends
Poly (E-caprolactone) (PCL) was reported by Koleske and
~ u n d b e r ~ ' " to be miscible with PVC over the entire composition range.
In fact, the Tg-composition data were used to determine the Tg of
amorphous PCL by extrapolation of the amorphous blend data to 100%
PCL. Crystallization kinetics of PCL in PCL-PVC blends were reported 123 . by ~obeson'". Olabisi investigated the PCL-PVC blends using
solvent probes by inverse gas chromatography technique. The
experimental data allowed for estimation of the interaction parameter
for PCL and PVC which prehcted miscibihty based on its negative
value. Khambatta et al . I3 ' studied the morphology of these blends
using small angle X-ray and light scattering.
1.7.4 PVCIPMMA blends
The effect of tacticity of poly (methyl methacrylate) (PMMA) on
its miscibility with PVC was studied by Schurer et al.'"". From their
s tuhes, it was concluded that the isotactic (i-) PhiIMA and PVC formed
an immiscible system with phase separation between two phases, and
50

two different Tgs were found over the entire composition range. One
phase was rich in PVC and the other in i-PMMA. On the contrary, the
Tg of syndiotactic (s) PMMA and PVC blends increased regularly with
composition upto 60 wt% s-PMMA. At higher s-PMMA contents, this
Tg did not change any more but was accompanied by a second Tg above
120°C indicating the value of pure s-PMMA. These results indicated
that a miscible system was formed up to 60wt% s-PMMA. Beyond
this concentration, a higher PMMA blend showed a separate phase
representing the excess pure s-PMMA.
The e ~ p l a n a t i o n ' ~ ~ of the variation comes from structural
Merences. Isotactic PMMA has a helical conformation while synho-
tactic PMMA possesses a planar structure. The helical structure makes
the ester groups less accessible to intermolecular interactions. Since the
microstructure of commercial atactic PMMA is much more syndiotactic
than isotactic, it is understandable that blends of an a-PMMA with
PVC give nearby the same DSC and dynamic mechanical results as
blends of S-PMMA do, and the blend should be considered a miscible
one.
1.7.5 PVCIPU blends
Blends of polyurethane and poly (vinyl chloride) (PUIPVC) have
been studiedI4'. PUPVC polymer blends offer increased flexibility,
abrasion resistance, tensile strength, impact strength, fire retardance14'
and acoustic damping1":'. They can be used as foams, elastomers,
coatings, adhesives and plastics. It was reported that PCL based
polyurethanes were miscible with PVC at all compositions over a broad, 51

temperature range144. The miscibility is most likely due to the hydrogen
bonding between the ester group in PCL and the a- H (H-C-C1) in PVC.
On a broader basis, there is evidence to suggest that polyether based
urethanes are more miscible with PVC than similar polyester based
polyurethanes143. This suggestion is based on thermal analysis which
yielded a single Tg.
1.7.6 PVCICPE blends
Chlorinated polyethylene (CPE) is structurally similar to PVC
with the only difference in chlorine content. The compatibility is
dependent upon the chlorine content and the distribution of the
chlorine atoms on the polyethylene back bone. Polymers containing
less than 25% C1 are incompatible with PVC. Those with 24-40% C1 are
the best impact modifiers having practical miscibility'45. CPE having
42 wt% C1 was found to be miscible with PVC with a lower critical
solution temperature b e h a v i ~ u r ' ~ ~ .
Xu et ~111~' stuhed binary blends of PVC, CPE, high density
polyethylene (HDPE) and low density poly ethylene (LDPE). They
concluded that CPE increased the impact strength of PVC.
1.7.7. PVC/ a - methyl styrene based polymer blends
Several high Tg polymers based on a-methyl styrene exhibit
miscibdity with P V C . ~ ~ Shur and ~ a n b ~ ' ~ ' reported on blends of PVC
with ABS. Using a series of experimental methods for characterisation

of these blends, they concluded that the styrene-acrylonitrile matrix of
ABS was miscible with PVC. However, other investigators have
reported two phase behaviour of styrene acrylonitile and PvCI4'.
Studies of Huarng et al." have shown that PVClSAN blends are
partially miscible depenhng on composition.
1.8 COMPATIBILISATION OF IMMISCIBLE BINARY
POLYMER BLENDS
Most pairs of high molecular weight polymers are incompa- tible150.152 . They have high interfacial tension and poor adhesion
between the phases. As a consequence of this they often exhibit poor
mechanical properties. This problem can be alleviated by the addition
or the in situ formation of a compatiblliser.
The use of block or graft copolymers as compatibilisers in binary
polymer blends has been well studied 5.153-157 . The choice of a block or
graft copolymer as compatibiliser is based on the miscibility or
reactivity of its segments with atleast one of the blend components. A
properly chosen block or graft copolymer preferentially locates at the
interface between the two immiscible phases. As pointed out by ~ a u l ' ,
this type of surface activity should reduce the interfacial energy
between the phases, permit a finer dispersion during mixing, provide a
measure of stability against gross segregation and result in improved
interfacial adhesion.

1.8.1 Addition of block copolymers for compatibilisation
Several s tuhes have been reported on the compatibilising action
of block copolymers in heterogeneous polymer systems. Molau and Co-
worker 155-157 demonstrated the ability of block copolymers to emulsify
polymer dispersions in solutions.
Inoue et a1.15%eported on the mechanism of domain formation
on a ternary system consisting of PS/poly(styrene- b- isoprene)/
polyisoprene. The domain structure was investigated by light and
electron microscopies using an osmium tetroxide fixation technique.
They concluded that when the molecular weight of the homopolymer is
much higher than that of the corresponding arm of the copolymer, the
block copolymer can no longer act as an emulsifier. Riess and Co-
workers 159,160 found that block copolymers are more effective than graft
copolymers i n increasing the compatibility of polystyrenelpoly (methyl
methacrylate) (PSIPMMA) and PSI polyisoprene blends. In these
studies the compatibility was monitored by the degree of optical trans-
parency of thin f lms cast from various solutions. These authors also
reported that the best compatibihsing action is obtained with a block
copolymer whose composition is 50:50 and whose molecular weight is
higher than those of the homopolymers. They found that &blocks were
more efficient than triblocks.
Gailard et al.'" have examined the surface activity of copoly-
mers by studying the interfacial tension reduction in demixed polymer
solutions. Addition of poly (styrene-b-butahene) to PS/polybutahene/
styrene ternary system showed first a characteristic decrease in inter-
54

facial tension followed by a levelling off. Several additional studies in
the area of compatibilisation of binary blends by the addition of
162-164 copolymers have also been reported . For example, the studies of
Coumans et al.'" and Paul and CO-workersIG3 deal with the emulsi-
fication of heterogeneous polyethylene (PE)/PS blends by the addition of
block copolymers.
Teyssie and CO-workers'" observed a significant reduction in
the dispersed phase size and an increase in interfacial adhesion as a
result of melt 'blending PE and PS with as little as 2 wt.% of poly
(butadiene-b-styrene). The copolymer also stabllised the system against
coalescence. Moreover Teyssie and CO- worker^"^ clearly demonstrated
that the copolymer is uniformly adsorbed a t the interface between the
two polymers.
LeiblerIG6 and Noolandi and Hong 167, 168 have proposed statistical
thermodynamic theories concerning the emulsifying effect of copoly-
mers. The theory of Leibler holds for nearly compatible systems,
whereas the theories of Noolandi and Hong 167.168 apply to the case of
highly incompatible systems, for concentrations below the critical
micelle concentration (CMC). Leiblerlti6 developed a mean field
formahsm to study the interfacial properties of mixtures of two
polymers, A and B, with an AB copolymer. ~ o o l a n d i ' ~ ~ reported that
both copolymer concentration and molecular weight are equally
important in reducing the interfacial tension. The locahsation of the
copolymer at the interface and the separation of the blocks into

corresponding homopolymer phases lead to various phenomena such as
the lowering of the interaction energy between the two immiscible
homopolymers, the broadening of the interface between the
homopolymers, the reduction in entropy of the system, a decrease in the
energy of interaction of the two blocks with each other, and a large
decrease in the interaction energy of the oriented blocks with
homopolymers. The sum of all these contributions should be considered
to determine the effect of copolymers on the surface tension between
the two phases'".
169 . Thomas and Prud' homme investigated quantitatively the
effect of molecular weight, composition and concentration of &block
copolymer of PS and PMlMA on the morphology of PSPMMA binary
blends. A sharp decrease in dispersed phase dimension was observed
with the addition of a few percent of block copolymer having equal
segment mass (50150 PSIPMMA), followed by a levelling off as the
copolymer content was increased above the critical micelle concen-
tration. For concentrations below the critical value, the particle size
reduction is linear with copolymer volume fraction. The experimental
results were in agreement with the prehctions of Noolanh and
on^'".
Chen et a1.I7O examined the compatibilising effect of block copoly-
mers on various binary systems like PEIPS, PEI Nylon-6, PS/Nylon-6
and polystyrenelpoly (ethylene terephthalate).

1.8.2 Addition of graft copolymers for compatibilisation
Adhtion of graft copolymer was reported as a means of impro-
ving the properties of high impact polystyrene (PS), poly (acrylonitrile-
171,172 co- butahene-co-styrene) (ABS) and PSPE blends . The decrease of
particle size of the hspersed phase upon the addition of graft copolymer
was substantiated by optical and electron microscopies. Chen et al.'73
studied the effect of added graft copolymers, on compatibilisation of
nylons with polyethylenes and polystyrene. They found that the
tendency of the melts to coalesce is decreased by the addition of maleic
anhydride graft polypropylene. (MA-g-PP)
Heino and ~ e ~ ~ a l a ' ~ ~ stuhed the compatibilising effect of maleic
anhydride grafted polypropylene, (PP-g-MA) and a reactive ethylene
based terpolymer on blends of polypropylene and an aromatic polyester-
type thermotropic main-chain liquid crystalline polymer. It was found
that the PP-g-MA compatibfiser h d not improve the impact strength
of PPLCP polymer blends. But clear enhancement in tensile strength
and elastic modulus was found.
1.8.3 Reactive compatibilisation
1.8.3.1 In situ formed copolymers
Recently the in situ formed compatibihsation in polyblends has
attracted great attention as an alternative to replace the conventional
block or graft copolymers. Ide and ~ a s e g a w a ' ~ ~ studied the use of
maleic anhydride modified isotactic polypropylene (ipp) in iPP-nylon 6
blends. During the melt mixing process, the anhydride groups react
57

with the amino end groups of nylon to yield a graft copolymer. HIPS
and ABS'"%~~ the classical examples of systems compatibilised by
block or graft copolymers formed through free radical reactions in situ.
Saleem and ~ a k e r ' " compared the compatibilising action of
in sit;u formed copolymer of polystyrene having oxazoline reactive
groups (OPS), polyethylene with carboxylic acid groups (CPE) and a
preblended graft copolymer of OPS-g-CPE in PSPE blends. The
preblended graft copolymer OPS-g-CPE imparts compatibility to PS-PE
blend but not effectively. They suggested that the addition of OPS and
CPE during melt mixing of PS and PE forms OPS-g-CPE polymer at
the interface and that these ingradients act as in situ reactive
compatibilisers and thus improve physical properties.
The in situ compatibilised polymer blends involving the reactive
glycidyl methacrylate (GMA) monomer have become important because
of the versatile application in many blenhng systems1"*.
Maa and changl"' used styrene-glycidyl methacrylate (SG) as in
situ compatibhser for the blends of poly (ethylene terephthalate) (PET)
and polystyrene (PS). The copolymer contains reactive epoxy functional
groups that are able to react with PET end groups (-OH and - COOH)
under melt conditions to form SG graft PET copolymer. The presence of
small amount of phosphonium catalyst (200PPM) accelerates the graft
reaction and results in a better compatiblised blend. The compatibllised
PETPS blend has a smaller phase domain and higher viscosity than
that of the correspondmg noncompatibllised blend. Mechanical

properties of the com&ibilised blends are superior to the correspon&ng
non compatibilised blend.
Nando and co- worker^.'^' studied the effect of the ethylene -
methacrylate copolymer as a chemical compatiblliser in the 50:50 blend
of low density polyethylene (LDPE) and poly (&methyl siloxane)
(PDMS). Ethylene - methacrylate (EMA) reacted with PDMS rubber
during melt mixing at 180°C to form EMA -grafted PDMS rubber (EMA
-g- PDMS) in situ which acted as a compatibihser in the LDPE -
PDMS rubber blend. The optimum amount of the compatibiliser ( E m )
was found t o be 6 wt% based on the results of dynamic mechanical
analysis, adhesion studles and phase morphology. Dynamic mechanical
analysis showed a single glass transition (Tg). X-ray diffraction
stu&es exhibited a remarkable increase in the degree of crystallinity.
The phase morphology showed a drastic reduction in the size of the
hspersed phase at the optimum concentration of E m .
1.8.3.2. Added reactive copolymer
Reactive copolymers of the type A-C may also compatibilise the
immiscible polymer pair A and B provided that C is capable of forming
a chemical reaction with B'". Although the non-reactive segment of
the copolymer has often a f e r e n t chemical and structural identity from
component A, i t may be capable of having miscibdity by specfic
interactions. ~ x a m ~ l e s ' ~ ' of added reactive copolymers as compati-
biliser include poly (styrene-co-acrylonitrile) (SAN)/maleic anhydride
for the system ABSIpolyamide 6 @A6) and poly ethylene-co-propylene

elastomer (EPM)/maleic anhydnde for the system PPIpoly amide 6.
(PA61
1.8.4 Homopolymers as compatibilisers
From the practical view point the ternary systems offer the possi-
bility of extending the list of miscible or mechanically compatible
blends as practised in utilising scrap or recycled plastic material.
Kwei and CO-workers182 conducted the first systematic study of
a miscible blend involving poly(methy1 methacrylate) (PMMA)lpoly
(ethyl methacrylate) (PEMA) compatibilised with poly (vinylidene
fluoride) (F'VDF2).
Paul and CO-workersJs3 reported on the compatibilisation of
polaarbonate (PC)/SAN using aliphatic polyesters. Ternary blends
comprising polycarbonate, styrene acrylonitrile copolymer and a
polyester of either poly (1,4-butylene adipate) (PBA), poly(l,4 cyclo-
hexane &methyl succinate) (PCDS) or poly (E - caprolactone) (PCL),
were found to be miscible based on the presence of a single glass
transition temperature at many compositions. For all systems, the
addition of 1% by weight of polyester resulted in a miscible blend for
SANlPC ratios of 111 and 311 and a region of immiscibility was
generally observed for PC rich composition with low polyester content.
A thermodynamic analysis was attempted in which the melting point
depression of the PCL in the miscible region of the ternary and in the
miscible binary solutions with PC and SAN, respectively was used to
evaluate the binary interaction parameters associated with the heat of
60

mixing and to predict the locus of ternary compositions which mark the
boundary between miscible and multiphase behaviour.
Paul and CO-workersla4 further made an attempt to predict the
boundary between miscible and immiscible compositions using the
binary interaction parameter obtained from PCL melting point
depression in ternary blends comprising of bisphenol - A polycarbonate
(PC), the polyhydroxy ether of bisphenol - A phenoxy and poly
(E-caprolactone) CPCL). In this case the interaction parameters were
used to calculate the locus of composition for which the heat of mixing
is zero. The locus was found to agree well with the observed boundary
between miscible and multiphase behaviour in the ternary. It is
concluded that the phase behaviour of ternary blend is largely
determined by the same enthalpic considerations known to govern the
phase behaviour of binary blends.
The phase equilibrium behaviour of a more complex system, poly
(vinyl chloride)(PVC)/styrene acrylonitrile copolymer (SAN)/ poly
(methyl methacrylate) (PMhL4) has been studied by Huarng, Min and
~ h i t e " ~ . They made a ternary phase diagram for the PVCISANIPMMA
system by both turbidimetry and DSC measurements. Miscibility was
investigated using a combination of turbidity, scanning electron micro-
scopy [SEMI and Merential scanning calorimetry experiments. PMMA
was found to be a good choice to add to PVCISAN blends to induce
miscibility.
White and Co-workers 28 have reported the miscibility behaviour
of binary and ternary blends of poly (vinyl chloride) (PVC), polycapro- 61

lactone (PCL) and styrene acrylonitrile copolymer using differential
scanning calorimetry (DSC) and turbidity techniques. PCLlPVC and
PCLISAN are largely miscible systems while PVCISAN is immiscible.
The ternary system shows considerable miscibihty. The blends are
characterised by polarised light microscopy and wide angle X-ray
diffraction. The former measurements characterise the structure of the
spherulites. Adchtion of PVC, SAN or PVCISAN causes the spherulites
observed in PCL to grow in size and become coarse. Melting point
depression measurements were used to calculate the Flory interaction
parameters x for PCLIPVC and PCLISAN blends.
The phase behaviour and morphology of a ternary blend
consisting of polystyrene (PS), bisphenol A polycarbonate PC) and
tetramethyl bisphenol A polycarbonate (TMPC) were investigated by
thermal and mechanical analysis and transmission electron micro-
s ~ o p ~ ' ~ ~ . It is shown that the TMPC, which is miscible with each of the
other components of the blend, does not solubllise the two immiscible
polymers, PS and PC. Two glass transition temperatures are observed
for most of the blend compositions. Based on the predictions of
~ o m ~ a " ~ , Zeman and ~atterson'" and Su and ~r ied" ' , Landry et al!"
developed a simple schematic hagram (Fig. 1.7) to represent the
various morphologies.

Structure type Miscibhty Examples
Figure 1.7. Basic phase structure types that may occur for a blend of three
high-molecular weight polymers,

In Figure 1.7, structure type I represents the simple case where
all the binary blends are immiscible ( x i j > 0). Three phases will thus
be present each consisting of the pure component. Many ternary
blends show this type of morphology. Structure type 2 and 3 occur
when two of the binary blends are either miscible ( ~ i j '0) or partially
miscible ( ~ i j ~ o ) , and the third pair is immiscible (~ij>o). Thus
depending on the relative x values and the symmetry of the system (a
symmetry system is one in which the ~ i j values of two or three of the
pairs are similar to each other in magnitude and sign.) the ternary
blend may separate iC to either two phases (type 3) or three phases
(type 2). The glass transition temperature will be shifted relative to
those of the pure components. Single phase compositions may also
occur. When AB and BC are miscible, but CA is marginally miscible or
immiscible (type 3) phase separation is predicted11' to occur in a
manner that preserves the most energetically favourable pair
interactions. Also, symmetry xAB = xBC favours miscibility. In some
cases component B may act as a compatibihser for component A and C.
Su and ~ r i e d ' " prehcted that the compatibiliser would be effective
only when the base blend is marginally immiscible (xCA>o). The
ternary blend of poly (ethyl methacrylate) (PEMA), poly (methyl
methacrylate) (PML\L4) and poly (vinylidene fluoride) (PVDF) s tuhed
by Kwei et a ~ . ' * ~ is an example of a symmetric system of type 3 where
PVDF acts as a compatibiliser for PEMA and PMMA to produce a
single-phase ternary blend over a wide range of composition. Two -
phases are observed at low PVDF concentrations.

Finally structure type 4 occurs when all thlee pairs of polymers
are miscible. If all three x i values are similar in magnitude (symme-
tric) then a single phase wdl be obtained. The ternary blend (PS/PC/
TMPC)'*"~~IS into structure type 3. In this case single phases are
obtained only at very high TMPC content. It has been found that the
relative molecular weights of the polymers have a considerable effect
on the phase diagram of the ternary blend.
Kalfoglou and CO-workers1*' tried to compatibihse the immi-
scible blends of chlorinated polyethylene (CPE) and poly (vinyl chloride)
(PVC) using epoxidised natural rubber (ENR). Based on the single glass
transition data the miscibility behaviour was studied. The ternary
blends were found to have good mechanical properties even at low
amounts of the compatibiliser.
Ternary blends were developed by melt mixing up to 30% poly
(butylene terephthalate) (PBT) with poly carbonate (PC) and phenoxy
in an attempt to improve the miscibhty of the PCIphenoxy binary
blend'". Although most of the blends with a PBT content higher than
10% appeared as transparent, two Tgs appeared at all the blend
compositions. These Tgs correspond to PC-rich and phenoxy rich
phases.
191 Young and Lee used poly (E-caprolactone) (PCL) to compati-
bilize polycarbonate (PC)/styrene acrylonitrile copolymer (SAN). The
improvement of compatibility was confirmed by mechanical properties
and morphology. Tensile strength showed maximum while impact
strength and elongation at break increased with increasing PCL 65

content in the blend. The domain size decreased with increasing PCL
content. Addition of PCL to the PCISAN blends increased the light
transmittance of the blends due to improved compatibility. At higher
concentrations of PCL light transmission decreased because of crysta-
llisation of PCL and PC in the blend.
Perrin and ~ r u d ' h o m m e ' ~ investigated the miscibility behaviour
of ternary poly(viny1 chloride)/poly(n-propyl methacrylate)/ poly
(n-amyl methacrylate) (PVCIPPMAIPAMA) and poly(viny1 chloride)/
poly (n-butyl methacry1ate)lpoly (n-amyl methacrylate) (PVCPBMAI
PAMA) blends by merent ia l scanning calorimetry. In both systems, a
binary mixture of the two polymethacrylates is totally immiscible
(PPMA with PAMA),(and PBMA with PAMA). For PVCPPMAPAMA
blends containing less than 70% PVC, the immiscible phase consists of
two coexisting binary PVCIPPMA and PVCPAMA phases. For an equal
amount of the two polymethacrylates in the ternary blend, the PVCI
PPMA phase contains 65% of the total weight of PVC and the whole
quantity of PPMA. The total amount of PAMA mixes with the remai-
ning 35% PVC to form the PVCPAMA phase. In contrast, the miscibi-
lity zone is predominant in the ternary P V C P B W A M A system,
since blends containing 30% or more of PVC exhibit a single glass
transition temperature. In the immiscible zone, the PVC is distributed
equally between PBMA and PAMA, which is in contrast to the 65-35%
distribution found in the previous system.
Another interesting work on miscible ternary blend is due to
Pomposo et a l . I g 3 They used poly (p-vinyl phenol) (PVPh) to compatibi-

lise immiscible blends of poly (methyl methacry1ate)lpoly (ethyl metha-
crylate). The assessment of miscibility was based mainly on the
presence of a single glass transition temperature. In this ternary blend,
PVPh is miscible with PMMA and PEMA. More than 60 weight
percent PVPh was required to cause miscibility between PMlMA and
PEMA. Based on the glass transition temperature data, a ternary phase
diagram was constructed. The miscibihty behaviour of the ternary
PMMAIPEWVPh blends as well as of the P W E M A I poly
(vinylidene fluoride) (PIT2) and PMMAPEMAlpoly(styrene-co- acrylo-
nitrile) (SAN) systems were rationahsed using the spinodal condition
and the Flory- .Huggins theory.
Machado and ~ee'"%sed PMMA to compatibilise four immis-
cible blend systems, styrene-maleic anhydridelstyrene-acrylonitrile
( S W SAN), styrene-maleic anhydridelacrylonitrile-butahene-styrene
(SMAI ABS), poly (vinylidene fluoride)/styrene acrylonitrile (PVF2/SAN)
and PWzl ABS. PMMA is miscible with each blend component. In
every case, the adhtion of PMMA led to the improvement of properties
such as tensile strength, tensile elongation and notched impact
strength. Further more, the addition of PMMA resulted in finer, more
uniform dispersion of the primary blend components.
Carty and whiteIg5 found that incorporation of polypropylene
into the ABSIPVC system has signficant effects on flammabihty and
smoke density. Recently Valenza and ~cierno'"' s tuhed ternary blends
of poly propylenelnylon-12lfunctionalized polypropylene. The effects of
addition of modified polypropylenes (one func t ionbed with maleic

anhydride and the other with acrylic acid) to blends of nylon 12 and
polypropylene were studied by considering morphological, thermal,
rheological and mechanical properties. The thermal property data
indicated that both the moddied polypropylenes influence the
crystallisation behaviour of the PP causing different shdts of the peaks
and lowering the crystallisation enthalpy. The differences between the
two modi£ied PPs also correspond to difference in the morphology. In
tensile measurements, yielding phenomenon was observed.

AIM AND SCOPE OF THE INVESTIGATION
The purpose of the study is to prepare novel class of miscible
ternary blends of PVC, EVA AND SAN (PVCIEVAISAN) by
compatibilising the immiscible pair EVMAN using PVC as a
compatibiliser. In this study, we selected PVC owing to its inherent
flame retardancy, chemical resistance, versatility, low cost and
capability to interact with other polymers. But without modification
processabAty, heat stability and impact strength of PVC are poor.
However conventional low molecular weight additives like plasticisers,
lubricants, stabilisers and other additives are usually used to attain
these properties. But these additives have an adverse effect on flame
retardant properties. Therefore a novel attempt has been made to
increase the processability, impact strength, thermal stability and
smoke characteristics of PVC by blending i t with EVA. Interestingly,
EVA copolymer is reported to have good smoke supprasant characte-
ristics. For applications like wire and cable insulation, the blend
formulation should have good mechnical properties. In view of this,
PVCBVA blend can be added with the SAN copolymers which possess
excellent tensile,tear and wear properties. Thus by preparing ternary
blends of PVC, EVA and SAN (PVCIEVAISAN) one can combine the
flame retardant properties of PVC, excellent low smoke characteristic of
EVA and superior tensile properties of SAN. Since PVC is miscible with
EVA and partially miscible with SAN, in these ternary blends, PVC
acts as an interfacial agent for the immiscible SANIEVA blend.

For the manlfestation of superior properties, miscibility of the
homopolymers is a requirement. Phase behaviour of the ternary blend
depends on the phase behaviour of binaries. Therefore it is important
to investigate the miscibhty characterstics and phase behaviour of both
binary and ternary blends. In view of this, various techniques have
been used to investigate the miscibility, phase separation and phase
behaviour of binary and ternary blends of PVC, EVA and SAN.
Infrared spectroscopy, DSC, TG, density, viscosity, microscopy (SEM
and optical) and flow characteristics have been used to analyse the
miscibhty. Heat of mixing and interaction parameters are also
calculated. Attempts have been made to correlate miscibility with
various other properties such as thermal stability, mechanical
properties and rheology. Study of blend morphology is important
because it relates the properties of the blend to the manner in which it
is processed. Therefore optical and scanning electron microscopic
techniques have been used t o study the morphology of the various
polymer blends. Optical microscopic studies of blends after phase
separation can clearly explain the mechanism of phase separation.
The LCST values have been used as a quantitative guide to the degree
of miscibhty in the binary and ternary blends.
Solution rheology stuhes provide a simple method to study the
flow behaviour of polymers and blends in solution which can be related
to solution processing of polymer blends. Solution rheology studies
can also give information about miscibility behaviour of polymer
blends. Therefore, the solution rheology of the blends has been
analysed.
70

Since these blends find potential applications in wire and cable
industry, various thermal and flammability characteristics such as
static thermal stability, smoke density, limiting oxygen index and
amount of volatile gas evolution have been assessed. It is expected that
by compounding this novel ternary polymer base matrix with various
additives such as acetyl tributyl citrate @lasticiser),DBTDL (stabiliser),
hydrated alumina (filler), ferrocene (pigment as well as smoke
supprasant) and stearic acid (lubricant) in controlled amount can give
rise to novel ternary blend formulations which can replace many of the
existing flame retardant polymeric compounds.

References
Gesner, B.D in Encylopeha of Polymer Science and Technology
vol. 10 Mark, H.F. Gaylord, N.G. and Bikales.Ni (eds) New York
Wiley Interscience (1969)
Keskkula, H; Polymer Moch6cation of Rubber and Plastics, New
york, Wiley Interscience (1970)
Bruins, P.F., Polymer Blends and Composites, New York Wiley
Interscience (1970)
Shaw,M.T., Polymer Blends and Mixtures, Series E, No.89
Walsh, D.J., Higgins J.S. and Maconnactie, A.(ed) Martinus
Nijhoff (pub.) Dordrecht (1985) Ch.4
Paul, D.R. and Newman,S., 'Polymer Blends' eds. Vol.1 and I1
Acad. Press, New York (1978)
Sivaram, S., IPI Transcations, vol5 No 1&3. 16 (1990)
Angove, S.N., Rubber J., 149 (3), 37 (1967)
Shundo, M., Imoto, M and Minoura, Y., J.Appl Polym. Sci., 10.
939 (1966)
Deanin, R.D, Geoffroy, R.R., Am. Chem. Soc. Div .Org. Coat.
Plast.Chem. Pap., 37(1), 257 (1977)
Morris, H,L. J.Elast. Plast., 6, 119 (1974)
Bier, G., Kunststoffe, 55, 694, 1965)
Alsup, R.G.., Tech. Pap. Reg. Tech. Conf. Soc. Plast.. Eng Ohio
Sect., 66 (1976)
Sperling, L.H. and mhalalus, E.N., J.App1. Polym Sci., 17, 3811
(1973)

Donatelli, A.A., Thomas, D.A. and Sperling, L.H., in "Recent
Avances in polymer Blends, Grafts, and Block," (Sperling
L.H.eds.) Plenum, New York (1974)
&m,S.C., Klempner, D., Frisch, K.C., Frisch, H.L., Macromole-
cules, 9, 263 (1976)
Mark H.F., Bikales,N.M, Overberger, C.G., Menges, G. and
Kroscchwitz, J.I. Encyclopeha of Polym. Sci. Eng. Vo1.12, Wiley - Interscience Pub. New York (1988).
Sutton, M.S., Rubber world, 149 (5) 62, (1964)
O'Mahoney, J.F., Rubber Age, 102, 47,(1970)
Corish, P.J., and Powell B.D.W., Rubber Chem.Technol., 47.48 1
(1974)
Koshy, A.T., Kuriakose, B., Thomas, S., and Varghese, S.,
Polymer, 34, 3428 (1993)
Koshy, A.T., Kuriakose, B., and Thomas, S., Indlan J . Nat.
Rubber Res.,3 (2), 87, (1990)
Koshy, A.T. Kurikose, B.and Thomas, S., Polym. Degr. Stab., 36,
137 (1992)
Koshy, A.T., Kuriakose, B., Thomas, S. and Varghese, S.,
Kausch. Gummi Kunststoffe, 45, 852, (1992)
Matsuo, M., Japan Plastics, 2, 6 (1968)
Grundmeier, M., Binsack, R., Vernaleken, H.R.U.S. Patent
4,056,504 assigned to Bayer (1977)
Sen. A.K., Mukherjee, B., Bhattacharya, A.S., Sanghi, L.K.,
De,P.P., and Bhowmick, A.K., J.App1. Polym.Sci.,43,1673 (1991)

Min,K., White, J.L. and Feller, J.F., J.App1. Polym.Sci., 29,2117
(1984)
Huarng, J.C., Min, K. and White,J.L., Polym Eng. Sci., 28 ,1590
(1988)
Brown, H.R., Macromolecules 2 2 , 2859, (1989)
Hong, S.D. and Burns, C.M., J.App1. Polym, Sci 15, 1995, (1971)
Allport, D.C. Block Copolymers, Allport, D.C. and Janes, W.H.
(Eds) Applied Science Pub. London (1973)
Fox, T.G., Bull. Am. Phys. Soc., [2] 2 , 123 (1956)
Gordon, M., and Taylor, J.S., J.App1. Chem., 2, 493 (1952)
Ting, S.P., Pearce, E.M. and Kwei, T.K., J. Polym. Sci. Polym.
Lett. Ed.,18, 201 (1980)
Nguyen, T.Q., and ~ a u h , H.H., J Appl Polym. Sci., 29, 455
(1984)
Joseph,E.A., Paul, D.R. and Barlow, J.W., J.App1. Polym. Sci.,
27,4807 (1982)
Hall, W.J., Kruse, R.L., Mendelson, R.A.and Trementozzi, Q.A.,
ACS Symp. Ser., 229 , 4 9 (1983)
Polymer Blends and Alloys - Guide book to commercial products,
Technomic. Pub. Co. Inc. Pennsylvania (1988)
Stoelting, J., Karasz, F.E., and MacKnight, W.J. Polym. Eng Sci.,
10, 133 (1970)
Buchanan, D.R., J . Polym. Sci., Part, A-2, 9, 645 (1971)
Olabisi, O., Robeson, L.M., and Shaw, M.T., Polymer- Polymer,
Miscibihty. Eds. Academic Press, New York (1979)

Walsh, D.J., Higgins, J.S., Doube, C.P. and Mckeown, J.G.,
Polymer,22,168 (1981)
Kargin, V.A., J . Polym.Sci. Part C4,1601 (1963)
Robeson, L.M. and Furtek, A.B., Am-Chem. Soc. Div. Org. Coat.
Plast. Chem. Pap., 37 (1) 136 (1977)
Kelley, F.N., Bueche, F.J., Polym. Sci., 50, 549, (1961)
Tobolsky, A.V., "Structure and Properties of Polymers", P.85,
Wiley, New York. (1960)
Schneier, B.J., Appl. Polym. Sci., 18, 1999 (1974)
Elmqvist, C. and Svanson, S.E., Eur. Polym. J., 11 789 (1975)
Coleman, M.M., Moskala, E.J., Painter P.C., Walsh, D.J., and
Rostami, S., Polymer, 24, 1410, (1983)
Jong. L., Pearce, E.M., and Kwei , T.K., Polymer, 34 (l), 48,
(1993).
Zezin A.B., Rogacheva, V.B., Komarov, V.S., Razvodovslui and
Ye. F., Polym. Sci., USSR (Engl. Transl.) 17, 3032 (1975)
Coleman, M.M., Zarian, J . , Varnell D.F., and Painter P.C., J .
Polym. Sci. Polym. Lett. Ed. 15, 745 (1977)
Wellinghoft, S.T., Koenig, J.L., and Baer, Jr., E., J . Polym. Sci.
Polym. Phys. Ed., 15, 1913, (1977)
Morawetz, H. and Amrani, F., Macromolecules, 11, 281 (1978)
Fischer, E.W., Wendorff, J.H., Dettenmaier, M.,Lieser, G., and
Voigt - Martin, I., J. Macromol. Sc.Phys., 12, 41 (1976)
Patterson, G.D., J. Macromol. Sci. Phys., 12 61 (1976)

Braun, J.M. and Guillet, J.E., in "Progress in Gas Chroma-
tography" (Purnell, J.H. ed.) Wiley Interscience, New York.
107 (1976)
Su, C.S., Patterson, D. and Schreiber, H.P., J.Appl. Polym. Sci.,
20, 1025 (1976)
Tao, W.C. and Frank, C.W., Macromolecules, 23 3275, (1990).
Halary, J.L., Ubrich, J.M., Nunzi, J.M., Monnerie, L. and Stein
R.S., Polymer, 25, 956 (1984)
Granville, M.H. Kyuda, K., Jerome, R., Teyssie, Ph. and De
Schryver, F.C., Macromolecules, 23, 1202 (1990).
White, J.R. and Thomas, E.L., Rub. Chem. Technol., 57, 457,
(1984)
Matsuo, M., Nozalu, C. and Jyo, Y., Polym. Eng. Sci., 9,197
(1969)
Shaw, M.T., Polymer Blends and Mixtures, Series, E No.89
Walsh, D.J., Higgins, J.S. and Maconnachie, A,, (ed) Martinus
Nijhoff (pub.) Dordrecht, (1985) ch.3
McMaster, L.P., Adv. Chem. Ser., 142,43 (1975)
Makarewiez, P.J. and Wilkes G.L., J. Appl. Polym. Sci., 23, 1619
(1979)
Danesi, S. and Porter, R.S., Polymer, 19, 448 (1978)
Baer, M., J . Appl. Polym. Sci., 16, 1109 (1972)
Mastsuo, M., Polym. Eng. Sci., 9 206, 1969
Wenig, W., Karasz, F.E. and MacKnight, W.J., J . Appl. Phys.,
46,4194 (1975)
Nishi T., and Wang, T.T., Macromolecules, 8, 909 (1975)

Kulshreshtha, A.K., Singh, B.P., and Sharma, V.N., Eur. Polym.
J., 2, 191 (1988)
Chee, K.K., Eur. Polym. J., 20,423 (1990)
Robeson, L.M., in'Polymer Compatibility and Incompatibility
Principles and Practices, Ed., Karel Solc. Harwood Acad. Pub.
New York 177 (1982)
Yee, A.F., Polym. Eng. Sci., 17, 213 (1977)
Usachev, S.V., Zahharov, M.D., Kuleznev, V.N., and Vetoshkin,
A.B., Intl. Polym. Sci. Technol., 7 T 148, (1980).
Parasiewicz, N. and Gaczynslu, R., Intl. Polym. Sci. Technol., 6,
Tl95, (1979)
Ryan, C.L Jr., Ph.D. Dissertation, University of Massachusetts
(1979)
Billmeyer, F.W., Textbook of Polymer Science, Wiley - Inter
science New York. 1971.
Hoy, K.L, J.Paint Tech.42,76 (1970)
Caspar, R., and Morbitzer, L., Angew, Makrom. Chem., 58/59, 1,
859, (1977)
Sanchez, I.C., in Polymer Blends, Vol.1 Paul D.R ed. Academic
Press, New York.(1978)
Singh, Y.P and Singh, R.P, Eur. Polym. J, 19, 6, 529 (1983)
Krause, S.J.,J. Macromol. Sci., C7.251 (1972)
Flory.P.J., Principles of Polymer Chemistry, Cornell University
Press, New York (1953)
Schneir, B.O., J.Appl. Polym.Sci.,l7, 3175 (1973)

87. Koningsveld, R., Kleintjens, L.A, and Schoeffeleers, H.M., Pure.
Appl. Chem, 39, l(1974)
88. MacKnight, W.J. and Karasz F.E., Polymer Blends in
Comprehensive Polymer Science Vo1.7 Ch.4, Allen, G and
Bevington J.C. (eds.) Pergamon Press (1989)
89. Bank, M.,Leffingwell, J., and Thies, C., J.Polym. Sci. A-2
10,1097 (1972)
90. Mc Master,L.P., Adv.Chem.Series, 142. 43 (1975).
91. Mc Master,L.P., Macromolecules, 6, 760 (1973)
92. Ryan, C.L., ph.D thesis, University of Massachusetts (1980)
93. Cahn, J.W., J.Chem. Phys., 42 (I), 93 (1965)
94. Cahn, J.W., Acta Metal., 9, 795 (1961)
95. Kwei, T.K., and Wang, T.T., in Polymer Blends, Paul D.R.,(Ed.)
Academic Press, New York, (1978)
96. Patterson, D. Rubber Chem. Technol., 40, 1.(1967)
97. Patterson,D., Macromolecules, 2 (6), 672 (1969)
98. Patterson D. and Robard,A., Macromolecule, 11 (4), 690 (1978).
99. Flory P.J.,J.Chem. Phys., 9 660 (1941)
100. Flory P.J., J.Chem. Phys., 10 ,51 (1942)
101. Huggins, M.L., J.Chem. Phys., 9,440 (1941).
102. Huggins, M.L.,J.Phys.Chem., 46,151 (1942).
103. Scott.R.L.,J.Chem. Phys., 17,279 (1949).
104. Tompa, H., Trans. Farad.Soc., 45,1142 (1949)
105. Krause,S., J.Macrom.Sci. Revs. Macrom.Chem.,C/7(2),251 (1972).
106. Prigogine, I., The molecular Theory of solutions, Interscience
New York. (1957)

Flory, P.J. Disc.Farad Soc., 49, 7 (1970)
Sanchez, I.C. and Lacombe, R.H.J. Phys. Chem., 80,2352,(1976).
Sanchez,I .C.,J.Polym. Sci. Polym. Lett., Ed. 15,71 (1977).
Sanchez, I.C.,Lacombe, R.H., Nature, (London) 252, 381 (1974).
Su, A.C. and Fried, J.R., Polym, Eng. Sci., 27, 1657 (1987).
Zeman, L., and Patterson, D., Macromolecules, 7,320 (1972).
Krause, S.,Smith, A.L., and Duden, M.G., J.Phys. Chem., 43
2144 (1965).
Roe, R.J.and Zin, W.C., Macromolecules, 13, 1221 (1980).
Kambour, R.P., Bender, J.J., and Boppi R.C., Macromolecules,
16, 753 (1983).
Ten Brinke, G., Karasz, F.E. and MacKnight, W.J., Macromole-
cules, 16, 1827 (1983).
Elmqvist, C., and Svanson, S.E., Colloid Polym. Sci.,253,327
(1975)
Kwei, T.K., Nishi, T. and Roberts, R.F., Macromolecules, 7, 667
(1974).
Nishi, T., Wang, T.T., and Kwei, T.K., Macromolecules, 8, 227
(1975).
Snyder, H.L., Meakin, P. and Reich, S., Macromolecules, 16, 757
(1983).
Eastmond, G.C. and Kotomin S.V., Polymer, 35,4, 882 (1994).
Lieberman, E.P., Off. Dig. Fed. Soc. Paint. Techno1.,34,30, (1962)
Olabisi.0, Macromolecules, 9, 316 (1975)
Emmett, R.A., Ind. Eng. Chem., 36, 730 (1944)
Ambler, MR., J. Polym. Sci. Polym. Chem. Ed., 11, 1505 (1973)

Zakrzewslu, G.A., Polymer,l4, 348 (1973)
Hammer, C.F., Macromolecules, 4, 69 (1971)
Marcincin, K., Romanov, A., and Pollack, V., J . Appl. Polym .
Sci., 16 2239 (1972)
Feldman, D. and Rusu, M.,Eur. Polym. J., 10, 41 (1974)
Elmqvist, C. and Svanson, S.E., Eur. Polym.J., 12, 559, (1976)
Shur, Y. J. and Ranby, B.G.,J. Appl. Polym. Sci., 19, 1337 (1975)
Ranby,B.G., J. Polym. Sci Polym. Symp., 51,89 (1975)
Monteiro, E.C., and Thaumaturgo, C., Polymer Bulletin, 30, 697
(1993)
Hickman J.J., and Ikeda, R..M., J . Polym. Sci. Polym. Phys. Ed.,
11, 1713 (1973)
Robeson, L.M. and McGrath, J.E., Polym. Eng. Sci., 17, 300
(1977).
Koleske, J.V. and Lundberg, R.D., J.Polym Sci. Part A-2 7., 795
(1969)
Robeson, L.M., J.Appl.Polym, Sci.,l7, 3607 (1973)
Khambatta, F.B., Warner, F., Russell,T. and Stein, R.S.,
J.Polym.sci.Po1ym.Phys. Ed., 14, 1391 (1976).
Schurer, J.W., deBoer, A. and Challa, G. Polymer, 16, 201 (1975)
Huarng, J.C., Chang, D.C. and Deanin, R.D., Advances in
Polymer Technology, 12, (I), 81 (1993).
Omoto, M., Klempner, D.and Frisch, K.C., Advances in Polymer
Blends and Alloys Technology vo1.2, Kohudic, M.A. and
Finlayson, K.(eds) Technomic, USA (1989).
Kalfoglou, N.K., J.Appl. Polym.Sci., 26 (3), 823 (1981)

Hoursten, D.J. and Hughes, I.D.,J.Appl. Polym.Sci., 26 (lo),
3467 (1981)
Ball, G.L. and Salyer, I.O., J.Acoust. Soc. Amer., 39, 663 (1966)
Nass, L., Encyclopedia of PVC vol. 2 Marcel Dekker, New York,
(1977)
Doube, C.P. and Wash, D.J., Polymer, 20, 1115 (1978).
Xu,X,. Zang, L., and Li, H., Polym. Eng.Sci., 27, 398 (1987).
Shur, Y.J., and Ranby, B.G.,J.Appl. Polym, Sci., 20, 3121 (1976).
Shaw, M.T., J. Appl. Polym. Sci., 20, 3121 (1976).
Paul D.R., and Barlow, J.W.,J Macromol. Sci. Rev. Macromol.
Chem., 1980 C18 (1) 109.
Utracki, L.A., Polymer Alloys and Blends, Hanser, New York
(1990).
Shaw, M.T., Polym Eng. Sci., 22, 115, (1982)
Paul, D.R., and Barlow, J.W., Am. Chem. Soc. Adv.Chem. Ser.,
176,315 (1979).
Gaylord, N.G., Am. Chem. Soc. Adv. Chem. Ser., 142, 76 (1975).
Molau, G.E., J Polym. Sci., A3,1267 (1965)
Molau, G.E., J. Polym. Sci., A3,4235 (1965).
Molau, G.E.,and Wittbrodt, W.M. Macromolecules, 1 260 (1968).
Inoue, T., Soen., T., Hashimoto, T. and Kawai, H., Macromole-
cules, 3,87, (1970).
Periard, J., and Riess, G., Colloid Polym. Sci., 253, 362, (1975).
Riess, G., and Jolivet, Y. Am.Chem. Soc. Adv. Chem. Ser., 142,
243 (1975).

Gailard, P., Ossenbach Sauter, M. and Riess, G., Makromol.
Chem. Rapid. Commun, 1, 771 (1980)
Coumans, W.J., Heikens, D. and Sjoerdsma, S.D., Polymer, 21,
103 (1980).
Lindsey, C.R., Paul, D.R., and Barlow, J.W., J.App1. Polym. Sci.,
26, l(1981).
Fayt, R., Jerome, R. and Teyssie, Ph., Makromol, Chem., 187,
837 (1986).
Fayt,R., Jerome, R., and Teyssie, Ph. J . Polym. Sci. Polym. Lett.
Edn.,24 25, 1986.
Leibler, L., Macromolecules, 15, 1283 (1982)
Noolan&.,J. and Hong,K.M., Macromolecules, 17, 1531 (1984)
Noolandi.,J. and Hong,K.M., Macromolecules, 15, 482 (1982)
Thomas, S. and Prud'homme, R.E., Polymer, 33,20, 4260 (1992).
Chen, C.C. and White, J .L., Polym. Eng. Sci., 33,923 (1993).
Turley, S.G., Appl. Polym. Symp., 7,237 (1968).
Locke, C.E. and Paul, D.R.J, Appl. Polym. Sci., 13, 2791 (1973).
Chen, C.C., Fontan, E., Min,K. and White, T.L. Polym. Eng. Sci.,
28, (2), 69 (1988).
Heino, M.T. and Seppala, J.V., J. Appl. Polym. Sci., 48, 1677
(1993).
Ide, F. and Hasegawa, A., J.App1. Polym. Sci.,18,963 (1974).
Manson J.A.and Sperling,L. H., Polymer Blends and Composites,
Plenum Press, New York (1976).
Saleem, M., andBaker, W.E., J.App1. Polym. Sci.,39,655 (1990).

Chang, F.C. and Hwu, Y.C. Polym. Eng. Sci., 31, 1509 (1991).
Maa, C.T. and Chang, F.C., J.Appl. Polym.Sci., 49, 913 (1993).
Santra, R.N., Samantaray, B.K., Bhowmick, A.K., and Nando,
G.B., J.Appl. Polym. Sci., 49,1145 (1993).
Xanthos, M., Polym, Eng. Sci., 28, 1392 (1988)
Kwei, T.K., Frisch, H.L., Radigan, W. and Vogel, S. Macromole-
cules, 10, 157 (1977).
Shah, V.S., Keitz.J.D., Paul, D.R. and Barlow, J.W., J . Appl.
Polym.Sci., 32,3863 (1986.)
Christiansen, H.H. Paul, D.R. and Barlow, J.W, J. Appl. Polym.
Sci., 34, 537 (1987).
Huarng, J.C. m n , K. and White J.L., Polym. Eng. Sci., 28,
1085/(988).
Landry, C.J.T., Yang, H. and Machell, J.S., Polymer, 32 44
(1991).
Tompa, H., Polymer solutions, Butterworths London (1956).
Zeman, I. and Patterson, D., Macromolecules, 5, 513 (1972)
Koklas, S.N., Sotiropolou, D.D., Kallitsis, J.K., and Kalfoglou,
N.K., Polymer, 32, 66 (1991).
Remiro, P.M. and Nazabal, J., J.Appl. Polym. Sci., 42, 1475
(1991).
Young W.Y. and Lee, D.S., Polym. Bulletin, 26, 701 (1991).
Perrin, P. and Prud homme, R.E., Acta Polymer, 44, 307 (1993).

193 Pomposo, J.A., Calahorra, E., Eguiazabal I. and Cortazar, M.
Macromolecules, 26, 2104 (1993).
194 Machado, J.M. and Lee, C.S., Polym. Eng. Sci. 34, 59 (1994).
195 Carty, P. and White, S., Polym. Deg. and Stab., 44, 93 (1994)
196 Valenza, A. and Acierno, D., Eur. Polym. J., 30,1121 (1994).