patologias de colon
TRANSCRIPT

UNIVERSIDAD LIBRE VIII SEMESTRE MEDICINA

Pedro Plaza R.

La poliposis adenomatosa familiar (PAF) es un trastorno hereditario autosómico dominante en el que se desarrollan múltiples pólipos premalignos en el colon debido a la mutación del gen APC (adenomatous polyposis coli), situado en el brazo largo del cromosoma 5.

• Edad de aparición de pólipos 16 años.
• Inicio de los síntomas 29 años.
• 40 años en adelante aparición de CA colorrectal.
• Prevalencia en todo el mundo de la PAF es 1/24.000 habitantes.

La asociación de PAF con tumoraciones extracolónicas (tumores óseos o tejidos blandos) constituye el síndrome de Gardner, descrito en 1952.

• tumores óseos. • tumores desmoides.
• quistes epidermoides.
• Pilomatrixomas. • quistes sebáceos.
• Leiomiomas. • anomalías dentarias.
• hipertrofia congénita del epitelio pigmentario de la retina.
• Pólipos gastroduodenales y de intestino delgado.
• predisposición especial para el cáncer periampular y de tiroides.

En el momento actual, la exéresis profiláctica del colon es el único tratamiento eficaz para prevenir la degeneración maligna de uno o más de los adenomas del colon.

LUIS CARLOS PINTO USTATE
Libro SCHWARTZ PRINCIPIO DE CIRUGIA TOMO 2 PAG:1086

Es un trastorno autosómico dominante en el que el paciente desarrolla cientos de pólipos en el colon y el recto.
Estas lesiones pueden degenerarse en adenoma y finalmente en carcinoma.




Son relativamente frecuente de modo que un 2% de los niños menores de 10 años lo presenta.
Tiene una prevalencia de 1/50.000 habitantes y una incidencia de 1/100.000 nacidos vivos

Hemorragias de vía digestiva
Anemia
Dolor abdominal
Obstrucción

Quirúrgico y de pende en parte del grado de afección rectal:
Si no esta afectando el recto:
Colectomia abdominal total con anastomosis iliorectal.
Si el recto esta diseminado por pólipos
Prontocolectomia total

CÉSAR GUSTAVO POLO GÓMEZ
“SINDROME DE LYNCH”

El 80% de los cánceres colorrectales son de aparición esporádica.
El 10% son familiares.
5-10% tienen carácter hereditario.
40-45 Años
Incidencia para tumores sincrónicos 18%
Incidencia para tumores metacronicos 24%

LYNCH
TIPO I
LYNCH
TIPO II

ENDOMETRIO
68%
GASTRICO
28%
OTROS
LYNCH TIPO II

PROTEINAS REPARADORAS: RERMMR
MMR (mist.match.repair):
(hMSH2, hMSH3, hMSH6, hMLH1, hMLH3, hPMS1, hPMS2)
MICROSATELITES
NO CODIFICANTES
CODIFICANTES
rTGF-ßII rIGF-II E2F4 BAX

Secuencia Adenoma-Carcinoma
Epitelio
normal
Epitelio
hiperproliferativo
Adenoma
precoz
Adenoma
tardio
Adenocarcinoma
Mutación
de APC
5q
Superexpresión
de COX-2
Mutación
de K-ras
12p
LOH 17q Mutación
de p53
LOH 18q
DCC Hipometilación
de DNA
Otras
alteraciones
Metástasis
Adenoma
intermed.
~ 7 - 8 años ~ 57 años ~ 65 años

Secuencia Adenoma Carcinoma
predisposición hereditaria: Síndrome de Lynch
Epitelio
normal
Epitelio
hiperproliferativo
Adenoma
precoz
Adenoma
tardio
Adenocarcinoma
Otras
alteraciones
Metástasis
Adenoma
intermed.
~ 2 - 3 años ~ 40 años ~ 43 años
MUTACIONES MSH2 MLH1

IMAGENES

1) Tres o más familiares consanguíneos, uno de ellos de primer grado en relación a los otros con cáncer colorrectal, endometrio, ovario, transicional de vías urinarias, intestino delgado, histológicamente verificado.
2) Compromiso de dos o más generaciones.
3) Afectación de un paciente antes de los 50 años.
Criterios de Amsterdam (1999).
CLINICO

Se recomienda la colonoscopia a los 20-25 años o 10 años antes, de la edad mas temprana diagnosticada en la familia.
Ultrasonido transvaginal o biopsia endometrial por aspiración de forma anual después de los 25 – 35 años de edad.

ANGEL A. PITRE

es una entidad rara autosómica dominante, caracterizada por lesiones hiperpigmentadas en boca, manos y pies; con presencia de pólipos gastrointestinales que ocasionan cuadros de anemia aguda o crónica, obstrucción intestinal y dolor abdominal.

En Estados Unidos se ha calculado una incidencia de 1 por cada 120.000 a 200.000 nacimientos y la prevalencia se ha estimado en 1 por cada 8.300 a 29.000 nacimientos.
Es igual en hombres y en mujeres, en todas las razas, y la edad promedio de diagnóstico es a los 23 años.

En Colombia no hay estadística sobre incidencia ni prevalencia y la literatura colombiana es escasa y casi siempre menciona este síndrome únicamente como factor de riesgo en artículos que tratan generalmente sobre cáncer y poliposis o complicaciones abdominales.

Hasta el momento se han descrito más de 145 mutaciones relacionadas con este síndrome, la mayoría de las cuales son pequeñas deleciones, inserciones o sustituciones simples de bases, en el gen Serina treonina kinasa (STK11) localizado en la región telomérica del brazo corto del cromosoma 19 en la banda 13,3 (19p13.3).

Este gen expresa una proteína (serina treonina kinasa) de 433 aminoácidos que se encuentra en el núcleo y en el citoplasma y cuya función no es completamente conocida pero que aparentemente está envuelta en el detenimiento del ciclo celular en G1

Esta proteína tiene que ver además en el desarrollo de la arquitectura celular, manteniendo la polaridad celular y su mutación conlleva a una pérdida de la polaridad y una tendencia al prolapso epitelial que resulta finalmente en la formación de pólipos.

el gen se asocia físicamente con el gen p53 regulando específicamente la vía de apoptosis así como la relación existente con el factor de crecimiento endotelial vascular VEGF

La mutación del gen además codifica una proteína truncada, una proteína con plegamiento anormal, o una proteína con estructura alterada, una proteína sin sentido con dominios catalíticos incompletos que tiene como consecuencia la disminución de la actividad kinasa de la misma la cual es importante en su efecto como gen supresor tumoral .

La clínica en estos pacientes es variable desde paciente asintomático con pigmentaciones de melanina mucocutáneas hasta emergencias abdominales y cáncer.
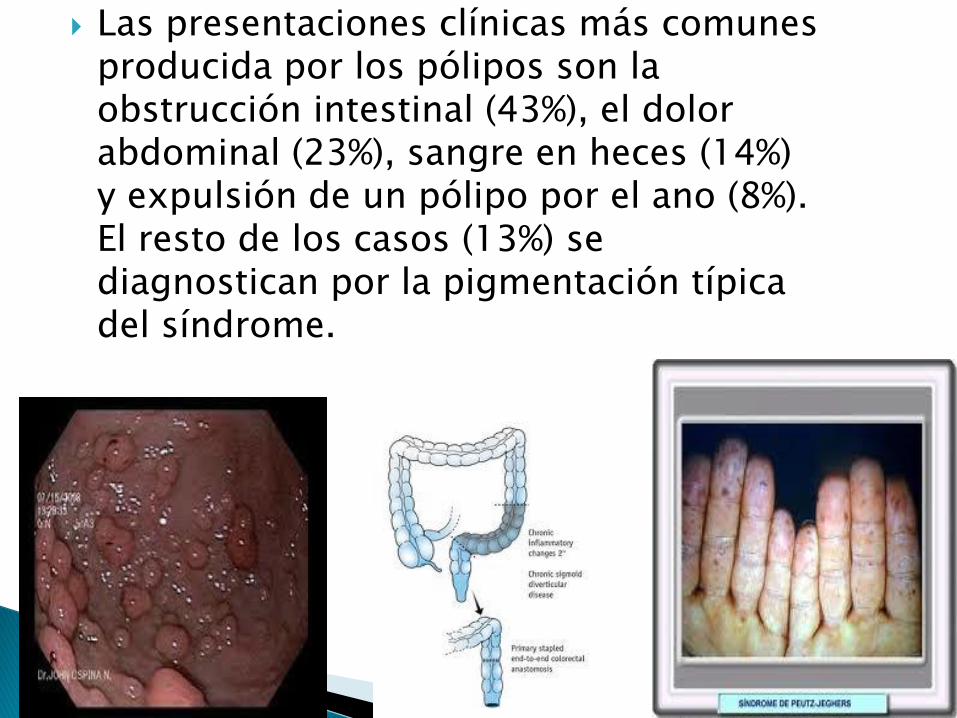
Las presentaciones clínicas más comunes producida por los pólipos son la obstrucción intestinal (43%), el dolor abdominal (23%), sangre en heces (14%) y expulsión de un pólipo por el ano (8%). El resto de los casos (13%) se diagnostican por la pigmentación típica del síndrome.

Clásicamente los pacientes se caracterizan por las máculas melanocíticas café oscuro o café azuladas de 1 a 5 mm de diámetro bien definidas, localizadas agrupadas en la región alrededor de los orificios corporales: boca, ojos, narinas y el ano, y es el compromiso labial-oral (típicamente cruzando el bermellón) el más frecuente de todos en un 94%; en un 66% se encuentra compromiso de las palmas de las manos y las plantas de los pies.

La otra característica clínica predominante del síndrome es el resultado de la poliposis gastrointestinal, la cual se manifiesta a una edad temprana. Una tercera parte de los pacientes presentan síntomas en la primera década de la vida y hasta un 50-60% antes de la segunda década

Esófago, estómago, intestino delgado, colon, recto, páncreas y vesícula biliar son los tumores gastrointestinales más comunes.

el seno es el más común y el riesgo de cáncer es similar al encontrado en las formas hereditarias asociadas a las mutaciones de los genes BRCA-1 y BRCA-2.
Otros tumores extragastrointestinales asociados al síndrome de Peutz-Jeghers son de pulmón, cuello uterino, ovario y testículo, y se presentan en estos tres últimos órganos tumores que no son muy frecuentes en la población como son el adenocarcinoma bien diferenciado de cuello uterino

El diagnóstico se hace en pacientes con pólipos hamartomatosos por medio de la endoscopia y con al menos dos de las siguientes características clínicas presentes:
• Depósitos labiales de melanina.
• Historia familiar del síndrome.
• Pólipos del intestino delgado.

Otra manera de detectar el SPJ es la evaluación genética. La identificación de la mutación del SPJ en el gen STK11 es muy exacta: la tasa de detección es de más del 90%.

Histológicamente los pólipos son hamartomas verdaderos caracterizados por el sobrecrecimiento desordenado de células nativas del órgano del que provienen, incluidas células de los tres tipos germinales en los pólipos del intestino delgado y de un solo tipo germinal en los pólipos del colon y del estómago , con características propias que incluyen su estructura frondosa, “arboriforme”, epitelio de recubrimiento específi co del segmento intestinal en el que se encuentra el pólipo y un corazón central consistente en la proliferación de bandas de músculo liso de la muscularis mucosae que perforan la lámina propia y que se ramifica en cada pliegue del pólipo.

Las endoscopías altas y bajas y la enteroscopía intraoperatoria con polipectomía respectiva, constituye el tratamiento de elección, mejorando cualitativamente el pronóstico en estos pacientes.
Sin embargo, si los pólipos son muy grandes o hay riesgo de que se conviertan en cancerosos, puede requerirse cirugía.

Giardello y Tomlinson recomiendan polipectomía para los pólipos gástricos y del colon que sean mayores de un cm. Igualmente, se recomienda cirugía para pólipos del intestino delgado que sean sintomáticos, mayores de 1 a 1,5 cms y pólipos de crecimiento rápido.

Algunos autores sugieren que la técnica de “clean sweep” (enterotomía y polipectomías) puede ser llevada a cabo con éxito en cirugía y aparentemente logra disminuir la necesidad de múltiples resecciones de intestino delgado.

Dai y colaboradores sugieren clasificar los pacientes de acuerdo al número de pólipos por segmento, recomendando tratamiento endoscópico (enteroscopia, gastroscopia y colonoscopia) para los que tengan menos de 50 pólipos y tratamiento quirúrgico con “clean sweep” para los pacientes que tengan más de 50 pólipos.

El estudio y el seguimiento de los pacientes con diagnóstico de síndrome de Peutz-Jeghers se inician con endoscopia digestiva alta y una colonoscopia
Se le debe solicitar un estudio baritado de tránsito intestinal para valorar los pólipos del intestino delgado, o en su defecto un estudio con cápsula de endoscopia, o enteroscopia, una ecografía abdominal total con énfasis en páncreas

STEPHANIE POLO RAMOS

• El Síndrome de Cowden o síndrome de hamartomas
múltiples.
• Es una genodermatosis que se hereda de forma
autosómica dominante.
• Se caracterizada por la presencia de múltiples
hamartomas cutáneos, fibromas orales y queratosis
acras benignas.
• Afecta a múltiples órganos (mama, tiroides, estómago
o colon), pudiéndose presentar en estos órganos
neoplasias malignas.

Afecta por igual a ambos sexos y se manifiesta en la segunda o tercera década de la vida.
Su etiología es desconocida.
Se relaciona con la presencia de alteraciones en el gen llamado PTEN en el brazo largo del cromosoma 10 (10q23.31, 10q22.3) o MMAC1 gen supresor tumoral.

La presencia de lesiones mucocutáneas típicas en 80% de los pacientes.
Triquilemomas o tricolemomas, se manifiestan como pápulas faciales, de color piel, parecidas a verrugas (boca, nariz o pabellones auriculares.)

Fibromas orales como pápulas lisas rosado-blanquecinas en las mucosas de la cavidad bucal (empedrado.)
Vitíligo, manchas café con leche, melanosis o pápulas queratósicas en partes acras.
La presencia de hamartomas o enfermedad fibroquística, típicas en zona tiroidea, mamaria y a cualquier nivel del aparato reproductor femenino.
Pueden presentar xerostomía y paladar ojival.




CLINICO:
1/ Criterios patognomónicos Son todas las lesiones mucocutáneas de cualquier tipo. (triquilemomas faciales, queratosis, lesiones papilomatosas y mucosas). 2/Criterios mayores Carcinoma de mama, carcinoma de tiroides, macrocefalia, hamartomas múltiples en cerebelo o enfermedad de l´Hermitte Duclos, y carcinoma endometrial.
3/ criterios menores Otras enfermedades tiroideas, retraso mental, hamartomas y tumores gastrointestinales, enfermedad fibroquística mamaria, lipomas, fibromas, y malformaciones o tumores genitourinarios.

Para el diagnóstico se requieren una de las 4 posibilidades siguientes:
A- Presencia exclusiva de lesiones mucocutáneas aisladas: 6 pápulas de las cuales tres correspondan a triquilemomas, o queratosis acral o un mínimo de seis lesiones de queratosis palmo-plantar
B- Presencia de dos criterios mayores, uno de los cuales debe ser obligatoriamente macrocefalia o enfermedad de l´Hermitte Duclos.
C- Presencia de 1 criterio mayor y tres menores.
D- Presencia de cuatro criterios menores .

Esclerosis Tuberosa.
Algunos tipos de Síndrome de Endocrinopatía Múltiple.
Síndrome de Byars-Jurkiewicz.
Síndrome de Gardner.
Proteinosis Lipoidea.
Hiperplasia Multifocal del Epitelio (lesiones orales).
Granulomatosis Orofacial

No existe tratamiento curativo para la enfermedad, aunque si se consiguen controlar las lesiones mucocutáneas con cirugía.
Los triquilemomas faciales responden al tratamiento con láser.


Es un raro desorden hereditario caracterizado por la asociación de pólipos adenomatosos en la capa mucosa del tracto digestivo con tumores del sistema nervioso central (glioma-poliposis).
Su etiolopatologia es desconocida.
Se transmite por un gen autosómico recesivo.
Algunos autores creen que es una variante de la poliposis familar adenomatosa.

En las neoplasias colorrectales hay dos patrones de herencia caracterizados molecularmente:
La poliposis adenomatosa familiar (gen APC)
El cáncer colorrectal no hereditario o síndrome de Lynch. (Genes hMLH1,hPMS1 y hPMS2)

Tipo 1, múltiples pólipos colónicos (20 a 100) con transformación maligna.
Tipo2, menos de 10 pólipos de más de 3 cm de diámetro con patrón de herencia incierto.
Tipo 3, similar a la poliposis adenomatosa familiar, con manifestaciones de carcinoma colorrectal antes de los 30 años de edad.

Los síntomas asociados a la formación de pólipos incluyen:
Diarrea
Rectorragia
Fatiga
Dolor abdominal
Pérdida de peso

Los individuos afectados pueden también experimentar síntomas neurológicos, dependiendo de la localización y tamaño del tumor cerebral.
Los pacientes con síndrome de Turcot tienen un riesgo sustancialmente elevado de desarrollar tarde o temprano un cáncer de colon u otros cánceres (del tiroides, adrenales y/o abdominales).

Historia del paciente, una detallada evaluación clínica y algunas pruebas especiales.
Se aconseja un examen sigmoidoscópico regular hasta la edad de 35 o 40 años.
La radiología también permite detectar la presencia de pólipos intestinales, y también la presencia de algún tumor cerebral.

Enfocado para paliar los síntomas de cada individuo.
Eliminación Qx de los pólipos del recto y del colon (proctocolectomía)
Si se realiza una ileoproctostomia, los pólipos rectales pueden regresar.

El rápido desarrollo de nuevos pólipos puede aconsejar una ileostomía o incluso una anastomosis ileoanal.
Examinar neurológicamente a intervalos regulares para detectar cuanto antes la presencia de un tumor cerebral.
El tratamiento de este dependerá de su tipo, localización y tamaño y consistirá cirugía acompañado de radio y/o quimioterapia.

Poliposis adenomatosa familiar
Síndrome de Gardner
Síndrome de Peutz-Jeghers (Poliposis intestinal de tipo II)
Síndrome de Cronkhite-Canada
Poliposis familiar juvenil

Diana Patricia Prado

Nombres alternativos: Gen MMAC1 o TEP1
Localización: 10q22-23
Función: Codifica la proteína PTEN
Estructura cristalográfica de PTEN humanos. La N-terminal de dominio
fosfatasa de color azul, mientras que el C-terminal de dominio C2 es de
color rojo.


Este termino fue propuesto por Cohen en 1990 para unificar las diferentes denominaciones que recibió este síndrome: Riley-Smith (1960), Bannayan-Zonana (1979), Ruvalcaba-Myhre (1980)¹
Es una enfermedad hereditaria rara de transmisión Autosómica Dominante, caracterizada por mutaciones germinales en el PTEN en el 60% de los casos, y la mayoría se halla en los exones 6 y 9.
1. Ruvalcaba R, Myhre S, Smith D. Soto’s syndrome with intestinal polyposis
and pigmentary changes of the genitalia. Clin Gent. 1980;18:413-6.

Dado el considerable solapamiento entre el gen del SBRR y la enfermedad de Cowden y demostrado que en estos pacientes el gen PTEN estaba delecionado en el cromosoma 10, Arch y cols. sugirieron que el SBRR y la enfermedad de Cowden eran anomalías alélicas.

Los síntomas de esta enfermedad varían mucho de un caso a otro.
Generalmente los pacientes tienen un peso y talla anormalmente elevados al nacer, retrasándose posteriormente el crecimiento hasta alcanzar la normalidad.

Cutis Marmorata
Lentiginosis genital
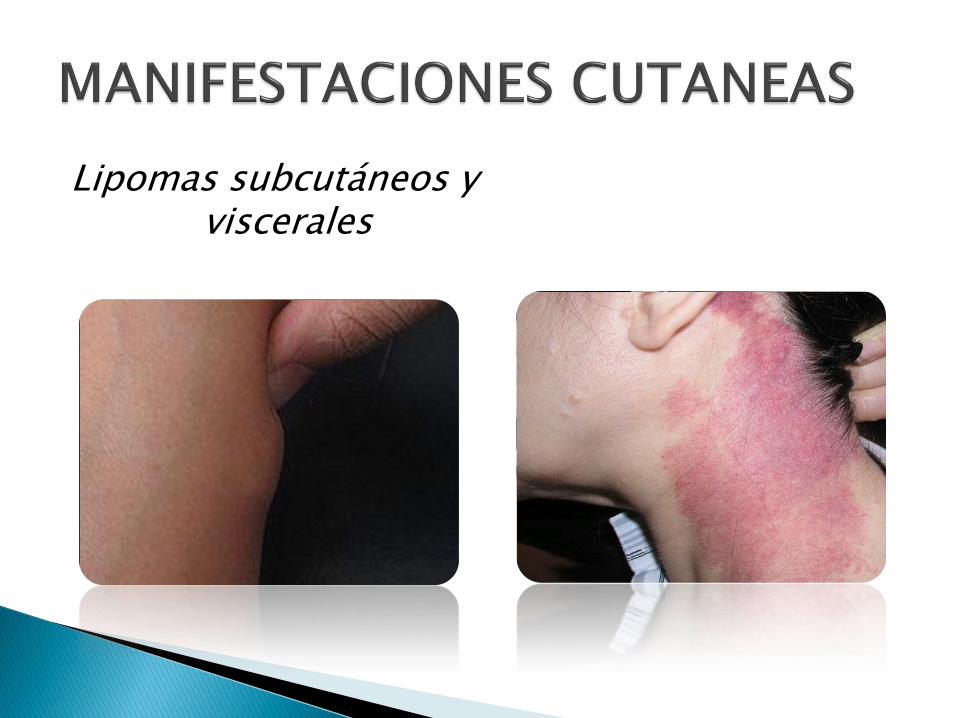
Lipomas subcutáneos y viscerales
Angiomas

Acantosis nigricans

Macrocefalia
Retardo mental
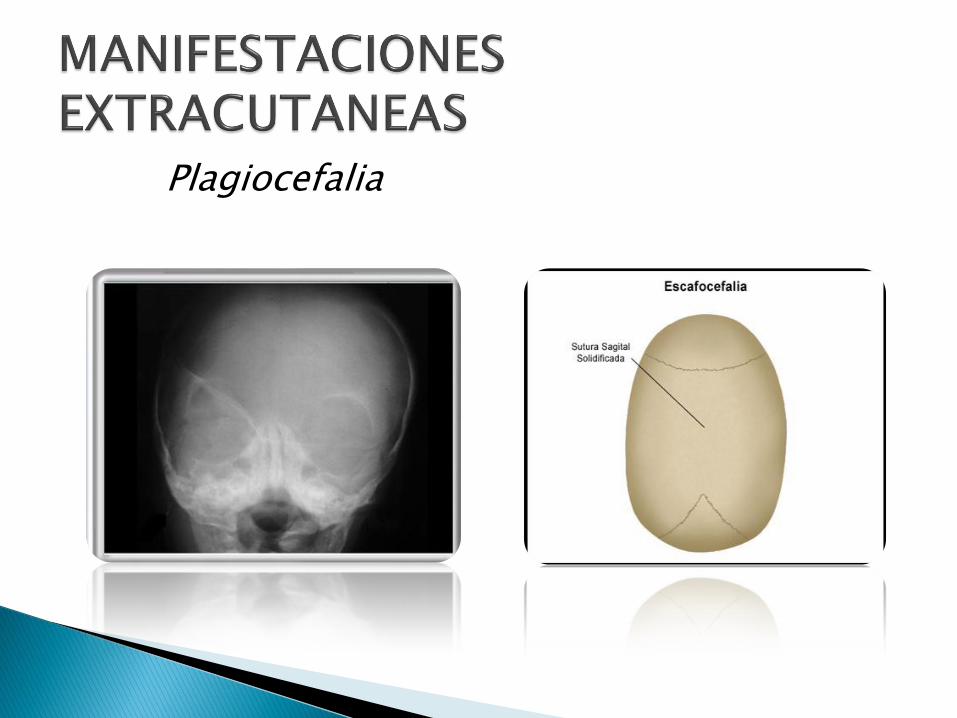
Plagiocefalia
Escafocefalia

Hipotonía
Pólipos

Estrabismo
Exotropia
Pseudopapiledema

El riesgo de cáncer en estos pacientes es muy elevado
