panels for quality assurance and quality - who · cv coefficient of variation ... qa quality...
TRANSCRIPT

Technical Guidance Series (TGS)
for WHO Prequalification – Diagnostic Assessment
Panels for quality
assurance and quality
control of in vitro
diagnostic medical
devices
TGS–6
Draft for comment 22 May 2017

© World Health Organization 2017 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; email: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for non-commercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; email: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by the World Health Organization to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either expressed or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use. Contact: Irena Prat, EMP Prequalification Team Diagnostics WHO – – 20 Avenue Appia – – 1211 Geneva 27 Switzerland

WHO Prequalification – Diagnostic Assessment: Technical Guidance Series
The WHO Prequalification Programme is coordinated through the Department of
Essential Medicines and Health Products. WHO prequalification of in vitro diagnostic
medical devices (IVDs) is intended to promote and facilitate access to safe, appropriate
and affordable IVDs of good quality in an equitable manner. The focus is on IVDs for
priority diseases and their suitability for use in resource-limited settings. The WHO
Prequalification Programme undertakes a comprehensive assessment of individual IVDs
through a standardized procedure that is aligned with international best regulatory
practice. It also undertakes post-qualification activities for IVDs to ensure ongoing
compliance with prequalification requirements.
Products that are prequalified by WHO are eligible for procurement by United Nations
agencies. The products are then commonly purchased for use in low- and middle-income
countries.
IVDs prequalified by WHO are expected to be accurate, reliable and be able to perform as
intended for the lifetime of the IVD under conditions likely to be experienced by a typical
user in resource-limited settings. The countries where WHO-prequalified IVDs are
procured often have minimal regulatory requirements, and the use of IVDs in these
countries presents specific challenges. For instance, IVDs are often used by health care
workers without extensive training in laboratory techniques, in harsh environmental
conditions, without extensive pre- and post-test quality assurance (QA) capacity, and for
patients with a disease profile different to those encountered in high-income countries.
Therefore, the requirements of the WHO Prequalification Programme may be different to
the requirements of high-income countries, or of the regulatory authority in the country of
manufacture.
The Technical Guidance Series was developed following a consultation – held on 10–
13 March 2015 in Geneva, Switzerland – which was attended by experts from national
regulatory authorities, national reference laboratories and WHO prequalification dossier
reviewers and inspectors. The guidance series is a result of the efforts of this and other
international working groups.
This guidance is intended for manufacturers interested in WHO prequalification of their
IVD. It applies in principle to all IVDs that are eligible for WHO prequalification for use in
WHO Member States. It should be read in conjunction with relevant international and
national standards and guidance.
The TGS guidance documents are freely available on the WHO website.
WHO
Prequalification
– Diagnostic
Assessment
Procurement of
prequalified
IVDs
Prequalification
requirements
About the
Technical
Guidance
Series
Audience and
scope

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
4 Draft for comment 22 May 2017
Contents
Acknowledgements .................................................................................................................... 6
1 Abbreviations and definitions ................................................................................... 7
1.1 Abbreviations .......................................................................................................................7
1.2 Definitions ............................................................................................................................7
2 Introduction ........................................................................................................... 12
2.1 Key concept ...................................................................................................................... 12
2.2 Rationale for the use of QA and QC panels ...................................................................... 12
2.3 Purpose of this document ................................................................................................ 13
2.4 Limitations of this guidance .............................................................................................. 13
3 WHO prequalification requirements ....................................................................... 15
3.1 Manufacturer responsibility ............................................................................................. 15
3.2 Standards .......................................................................................................................... 15
3.3 Suitability for use in Member States ................................................................................ 16
4 Basic principles for developing panels ..................................................................... 17
4.1 Core principle .................................................................................................................... 17
4.2 Quantitation is essential ................................................................................................... 17
4.3 Verification and validation ................................................................................................ 18
4.4 Use of surrogate specimens in panels .............................................................................. 18
5 Use of panels .......................................................................................................... 19
5.1 Release-to-sale panels ...................................................................................................... 19
5.2 In-process panels .............................................................................................................. 19
5.3 Stability panels .................................................................................................................. 19
5.4 Reproducibility during evaluations ................................................................................... 20
5.5 Analytical sensitivity and range: quantitative assays ....................................................... 21
5.6 Control materials provided to users ................................................................................. 21

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
5 Draft for comment 22 May 2016
6 Selection of specimens ........................................................................................... 22
6.1 General comments ........................................................................................................... 22
6.2 In-house panels ................................................................................................................. 23
6.3 Certified reference materials ............................................................................................ 29
6.4 International conventional calibration materials ............................................................. 29
6.5 Measurement variation .................................................................................................... 30
6.6 External regulatory panels ................................................................................................ 30
7 Relationship between panel members and claims ................................................... 33
7.1 Process control charts ...................................................................................................... 34
8 Maintenance of panels ........................................................................................... 35
8.1 Storage .............................................................................................................................. 35
8.2 Replacement ..................................................................................................................... 35
9 References.............................................................................................................. 38
10 Annex 1 – Examples ................................................................................................ 41
10.1 Example 1: Correlation of a QA / QC panel member with a critical specimen ................. 41
10.2 Example 2: Correlation under stress conditions ............................................................... 44
10.3 Example 3: Validation of a release-to-sale panel for anti-HCV ........................................ 47
10.4 Example 4: A release panel for a flow cytometer for the enumeration of CD4 T-cells .... 49
10.5 Example 5: Nucleic acid testing ........................................................................................ 51
10.6 Example 6: Use of imposed specimens as QA / QC panel members ................................ 53

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
6 Draft for comment 22 May 2017
Acknowledgements
The document Panels for quality assurance and quality control of in vitro diagnostic
medical devices was developed as part of the Bill & Melinda Gates Foundation Umbrella
Grant and the UNITAID grant for “Increased access to appropriate, quality-assured
diagnostics, medical devices and medicines for prevention, initiation and treatment of
HIV/AIDS, TB and malaria”. The draft was prepared in collaboration with Dr J Duncan,
London, United Kingdom; Ms K Richards, WHO Geneva, Switzerland and Ms R Meurant,
WHO Geneva, Switzerland and with input and expertise Dr S Hojvat, Maryland, United
States of America; Dr E Cowan, Maryland, United States of America and Dr D Milic, WHO
Geneva, Switzerland. This document was produced under the coordination and
supervision of Kim Richards and Josée Hansen WHO/HIS/EMP, Geneva, Switzerland.
The draft guidance has been posted on the WHO website for public consultation on 22
May 2017.
1

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
7 Draft for comment 22 May 2016
1 Abbreviations and definitions 2
1.1 Abbreviations 3
CE Conformité Européenne (European Conformity)
CV Coefficient of variation
CLSI Clinical and Laboratory Standards Institute
CRM Certified Reference Material
EIA Enzyme-linked immunoassay
GHTF Global Harmonization Task Force
HBsAg Hepatitis B surface antigen
HBV Hepatitis B virus
HCV Hepatitis C virus
HIV Human immunodeficiency virus
IFU Instructions for Use
IS International Standard
ISO International Organization for Standardization
IU International Unit
IVD In vitro diagnostic or in vitro diagnostic device
NAT Nucleic Acid Test, Nucleic Acid Testing
OD Optical density
PEI Paul Ehrlich Institute
QA Quality assurance
QC Quality control
QMS Quality management system
RDT Rapid diagnostic test
R&D Research and development
SI International System of Units/Système International d’Unités
1.2 Definitions 4
The definitions given below apply to the terms used in this document. They may have 5
different meanings in other contexts. 6

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
8 Draft for comment 22 May 2017
Acceptance criteria: A defined set of conditions that must be met to establish the performance of 7 a system. 8
Source: (1) 9 10 Numerical limits, ranges, or other suitable measures for acceptance of the results 11
of analytical procedures. 12 Source: (2) 13 14 Batch/lot: A defined amount of material that is uniform in its properties and has been 15
produced in one process or series of processes. 16
Note: The material can be either starting material, intermediate material or 17 finished product. 18
Source: (1), definition 3.5 19
Component: Part of a finished, packaged and labelled in vitro diagnostic device (IVD). 20
Note: Typical kit components include antibody solutions, buffer solutions, 21 calibrators or control materials. 22
Source: (1), definition 3.12 23
Constituent: Raw materials used to make a component. 24
Source: (1), definition 3.57 25
Control material: A substance, material or article intended by its manufacturer to be used to 26 verify the performance characteristics of a medical IVD. 27
Source: (3), definition 3.4 and (1), definition 3.13 28
Certified reference material (CRM): Reference material, accompanied by a certificate, one or 29 more of whose property values are certified by a procedure that establishes 30 metrological traceability to an accurate realization of the unit in which the 31 property values are expressed, and for which each certified value is accompanied 32 by an uncertainty at a stated level of confidence. 33
Source: (4), definition 3.8 34
Design input: The physical and performance requirements of an IVD that are used as a basis for 35 IVD design. 36
Source: (5), definition f 37
Diagnostic sensitivity: The proportion of patients with a well-defined clinical disorder (or 38 condition of interest) whose test values are positive or exceed a defined decision 39 limit (i.e. a positive result and identification of the patients who have a disease). 40
Note 1: The clinical disorder must be defined by criteria independent of the test 41 under consideration. 42
Note 2: The term "diagnostic sensitivity" (Europe) is equivalent to "clinical 43 sensitivity" (United States) 44
Source: [http://htd.clsi.org] 45

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
9 Draft for comment 22 May 2016
Evidence: Information that can be proved true, based on facts obtained through 46 observation, measurement, test or other means. 47
Source: Modified from (6), definition 3.8.3 48
Instructions for use (IFU): Information supplied by the manufacturer to enable the safe and proper 49 use of an IVD. 50
Note: Includes the directions supplied by the manufacturer for the use, 51 maintenance, troubleshooting and disposal of an IVD, as well as warnings and 52 precautions. 53
Source: (1), definition 3.30 54 WHO comment: In the United States, the acronym IFU occasionally stands for 55 “indications for use”, and the acronym IU stands for “intended use” or 56 “indications for use”. The International Organization for Standardization 57 (ISO) definition and requirements (1) for IFU cover the intended use and the 58 precise method of use. 59
International conventional calibrator: A calibrator whose value of a quantity is not metrologically 60 traceable to the International System of Units (SI), but is assigned by 61 international agreement. 62
Note: The quantity is defined with respect to the intended clinical application. 63
Source: (4) definition 3.11 64
In vitro diagnostic medical device: A medical device, whether used alone or in combination, 65 intended by the manufacturer for the in vitro examination of specimens derived 66 from the human body, solely or principally to provide information for diagnostic, 67 monitoring or compatibility purposes. 68
Note 1: IVDs include reagents, calibrators, control materials, specimen 69 receptacles, software and related instruments or apparatus or other articles; 70 they are used, for example, for diagnosis or to aid diagnosis, screening, 71 monitoring, predisposition, prognosis, prediction and determination of 72 physiological status. 73
Note 2: In some jurisdictions, certain IVDs may be covered by other regulations. 74
Source: (7) 75
International unit: An arbitrary unit assigned to a WHO International Standard by the WHO Expert 76 Committee of Biological Standardisation. 77
Source: (4), Section 4.2.6 78
IVD reagent: Chemical, biological or immunological components, solutions or preparations 79 intended by the manufacturer to be used as an IVD. 80
Source: (1), definition 3.28 81 WHO comment: This document uses the terms “IVD” and “IVD” reagent 82 interchangeably. 83

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
10 Draft for comment 22 May 2017
Life cycle: All phases in the life of a medical device, from the initial conception to final 84 decommissioning and disposal. 85
Source: (8), definition 2.7 86
Metrological traceability: Property of the result of a measurement or the value of a standard 87 whereby it can be related to stated references, usually national or international 88 standards, through an unbroken chain of comparisons, all of which have stated 89 uncertainties. 90
Note 1: Each comparison is affected by a (reference) measurement procedure 91 defined in a calibration transfer protocol. 92
Source: (4) 93
Performance claim: Specification of a performance characteristic of an IVD as documented in the 94 information supplied by the manufacturer. 95
Note 1: The specification can be based on prospective performance studies, 96 available performance data or studies published in the scientific literature. 97
WHO comment: “Information supplied by the manufacturer” includes but is not 98 limited to: statements in the IFU, in the dossier supplied to WHO and /or other 99 regulatory authorities, in advertising, on the internet. 100
Referred to simply as “claim” or “claimed” in this guide. 101
Source: (1), definition 3.51 102
103
Quality assurance (QA): Part of the quality management focused on providing confidence that 104 quality requirements will be fulfilled. 105
106 Source: (6), definition 3.3.6 107 108 Quality control (QC): Part of quality management focused on fulfilling quality requirements. 109 110 Source: (6), definition 3.3.7 111 112 Risk management: The systematic application of management policies, procedures and practices 113
to the tasks of analysing, evaluating, controlling and monitoring risk. 114
Source: (8) 115
Statistical process control: Activities focused on the use of statistical techniques to reduce 116 variation, increase knowledge about the process and steer the process in the 117 desired way. 118
Source: (9), definition 2.1.8 119
Trueness of measurement: Closeness of agreement between the average values obtained from a 120 large series of results of measurements and a true value. 121
Source: (4), definition 3.33 122

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
11 Draft for comment 22 May 2016
Validation: Confirmation by examination and provision of objective evidence that the 123 particular requirements for a specific intended use can be consistently fulfilled. 124
Source: (5), definition z; (6), definition 3.8.13. 125
Verification: Confirmation by examination and provision of objective evidence that specified 126 requirements have been fulfilled. 127
Source: (5), definition aa; (6), definition 3.8.12. 128
129

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
12 Draft for comment 22 May 2017
2 Introduction 130
2.1 Key concept 131
A “panel” is a collection of well-characterized materials and specimens that is used to 132
monitor aspects of the function of an IVD or its components. Probably the most 133
important use of a panel is to verify, before a lot of an IVD can be released-to-sale, that 134
the lot will consistently meet all its quality critical metrics until the end of its assigned 135
shelf life (not just at the time of its release-to-sale, also referred to as batch release). 136
Panels are also used for in-process control, during reproducibility and stability studies and 137
for some aspects of design validation. The same materials might be used for each of these 138
purposes, but would be assigned different acceptance criteria for the different functions. 139
The manufacturer should be able to justify their rationale for assigning specific 140
acceptance limits when panel samples are being tested at any point throughout the 141
product lifecycle. The rationale can be documented as part of the risk management 142
process; alternatively, it can be included in the design control documentation when 143
statistical techniques are used as part of the process for establishing performance 144
characteristics. 145
2.2 Rationale for the use of QA and QC panels 146
There is a regulatory requirement for CE-marked products for the detection of infectious 147
diseases listed in Annex II of the In Vitro Diagnostic Medical Devices Directive 98/79/EC 148
(IVDD) that, “The manufacturer’s release testing criteria shall ensure that every batch 149
consistently identifies the relevant antigens, epitopes, and antibodies” (10: Section 3.4.1). 150
It is expected that this will be shown for all of the relevant specimen types claimed for the 151
IVD (e.g. serum, whole blood and urine), even for rest-of-world products that fall into the 152
high-risk categories C and D of the Global Harmonization Task Force (GHTF) classification 153
(10). However, subject to documented risk evaluation by the manufacturer, this 154
requirement could be relaxed to testing of only the most searching specimen type(s). It is 155
also a regulatory requirement that the manufacturer provides objective, scientifically 156
sound evidence to support all claims made regarding the performance of an IVD (e.g. 157
stability, reproducibility and sensitivity). It is not reasonable to verify all performance 158

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
13 Draft for comment 22 May 2016
metrics for every lot manufactured, but use of a well-designed lot release panel will – 159
subject to appropriate risk assessment and validation work – provide a high degree of 160
assurance that each lot of the IVD will consistently perform as claimed for its assigned life. 161
In addition to testing of the IVD at release, conventional practice is to evaluate 162
intermediates in the of manufacture (referred to as “in-process testing”). Use of carefully 163
chosen panels will provide evidence that the intermediates meet their specifications and 164
that the manufacturing process is in statistical control (9). In-process panels are also used 165
to provide evidence that a lot is homogeneous in performance metrics from the 166
beginning to the end of each production process. 167
Panels are also needed for stability and reproducibility studies used during IVD 168
development, and for aspects of verification at the end of the product’s assigned shelf 169
life. Results over time – as dictated by the developer’s quality management system (QMS) 170
– must be shown to be within predetermined and validated specifications. 171
2.3 Purpose of this document 172
The purpose of this document is to provide IVD manufacturers with guidance on possible 173
approaches to preparing validated panels for QA and QC; for example, choosing the 174
materials, assigning meaningful criteria to them, storing them and replacing them when 175
necessary. It describes the expectations for WHO prequalification in terms of the QA and 176
QC information to be provided in dossiers submitted according to PQDx_018 (11: Section 177
6.2.1). It also provides guidance on information that might be requested during QMS 178
inspections according to PQDx_014 (12: Section 7.2.2), following requirements in 179
ISO 13485:2016 (13: Clauses 7.3.4 and 8.2.6). 180
2.4 Limitations of this guidance 181
This document should not be taken as a prescriptive checklist of what must be 182
performed, but as a guide on how to improve processes and generate the evidence 183
needed to ensure a comprehensive, systematic procedure with an appropriate risk 184
management plan. As explained in Section 3.3, the expectations of the WHO 185
prequalification might be more stringent than the requirements of the users and 186

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
14 Draft for comment 22 May 2017
regulatory authority in the country of manufacture. Wherever possible the guidance 187
attempts to explain the reasons for these additional expectations. Other approaches to 188
accommodating these further expectations can be provided in dossiers submitted for 189
WHO prequalification, if supported by rigorous risk assessment or other evidence. 190
The examples included in this document apply to the principles outlined here only. 191
Manufacturers must perform their own product-specific risk assessment for each of their 192
IVDs. The risk evaluation must be related to the specific product, in its specific format 193
(e.g. antigen sandwich, next-generation sequence methodology, etc.), and to the specific 194
target analyte and the specific, claimed intended use and users. 195
Depending on the particular categorization of the product, additional requirements may 196
apply in particular jurisdictions. Such regulatory and legal requirements are specific for 197
each regulatory authority; they are beyond the scope of this document but should be 198
documented in the design input requirements and their effects should be evaluated by 199
risk analyses. 200
201

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
15 Draft for comment 22 May 2016
3 WHO prequalification requirements 202
WHO requires the following details for prequalification: 203
…any in-process and final product testing ... 204
… an overview of verification, validation and quality control activities for all 205
stages of design and manufacture (including purchased components, 206
in-process products, and finished products). 207
Provide the batch release criteria … 208
Source: (11: Section 6.2.1) 209
The extent of the information provided in the dossier will vary. For in-process, the control 210
points and test methods would probably be noted on process flow diagrams, with a link 211
to the risk assessments that describe the necessity for that control. For stability or for 212
reproducibility work, and especially for release-to-sale (i.e. batch release), a full 213
description of the panel would be expected, including the reason for the inclusion of each 214
panel member, its characterization, the criteria assigned and the validation of the test 215
method. If required during any on-site QMS inspection (12), the full information about 216
each QA and QC control point and test method should be available in the design history 217
file. 218
The information provided must demonstrate the link to the predetermined user 219
requirements and to product development. 220
3.1 Manufacturer responsibility 221
It is a manufacturer’s responsibility to ensure that all claims made regarding the 222
performance of the IVD are supported by evidence that is objective and scientifically 223
sound. 224
3.2 Standards 225
WHO recommends that manufacturers be familiar with the standards and guidance 226
documents listed in the Standards applicable to the WHO prequalification of in-vitro 227
diagnostics (14), and take them into account when planning, assessing risk and 228
developing QA and QC procedures. 229

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
16 Draft for comment 22 May 2017
3.3 Suitability for use in Member States 230
Information on the use and value of panels for QA and QC in the dossiers of a product 231
submitted to WHO must reflect the expected environmental conditions and the normal 232
usage conditions and methods encountered by the users in WHO Member States. The 233
environmental conditions might differ from, and be more extreme than, those in the 234
country of manufacture; for example, more extreme temperature and humidity, and 235
different contaminating microorganisms. Each of these factors must be considered not 236
only in the design input documentation (in the risk management section) but also in 237
validating the IVD and in developing QA procedures to verify adequate performance 238
through the life cycle of the IVD. 239

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
17 Draft for comment 22 May 2016
4 Basic principles for developing panels 240
4.1 Core principle 241
The core principle underlying any testing is that the results of the test are relevant to the 242
investigation; that is, the test methods must be validated (15). For development of QA 243
and QC panels, the materials and panel members chosen must be applicable to the task in 244
hand, and their utility must be validated and documented. A panel member should be 245
chosen for the purpose of showing stability (or sensitivity, reproducibility, etc.) of some 246
aspect of an IVD, so that if the IVD becomes unstable (or insensitive, irreproducible, etc.) 247
in that respect, the test result from that panel member will reflect the potential for 248
generation of an incorrect result, with resulting incorrect patient management. Often, the 249
data provided in WHO prequalification submissions do not adequately support the 250
conclusions drawn because the panel members have not been properly characterized and 251
validated for their stated use. Examples of appropriate (and inappropriate) choice of 252
specimens are given in Annex 1 of this document, and in the guides and sample dossiers 253
available online (16). 254
4.2 Quantitation is essential 255
Panels are used in quantitative, semiquantitative and qualitative processes. It is important 256
to be able to show whether a parameter is different from what is expected and, if so, by 257
how much. That change can then be related to the predetermined limits within which the 258
IVD will function, as shown by the panel validation work (see Section 0 of this document). 259
Finding a panel member to be merely positive or negative is often uninformative. Many 260
IVDs are not intended to produce quantitative results; however, for certain situations 261
(e.g. release-to-sale, stability work, robustness studies and process control) it is essential 262
to be able to assign some form of quantitative values to the results from panel members. 263
The methods for doing this should be determined through research and development 264
(R&D). 265
For most rapid diagnostic tests (RDTs), the intensity of the colour of the signal from the 266
IVD can be compared with a calibration scale. Degradation of signal intensity may be a 267
sign that the IVD is degenerating. It is sometimes argued that the relative intensity of the 268

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
18 Draft for comment 22 May 2017
signal from a qualitative IVD antibody test is not related to the antibody content of the 269
specimen. Although that is true when comparing different specimens it is not true for the 270
relative signal generated from an IVD that has degenerated or been manufactured 271
incorrectly (nor for dilutions of a single specimen); in such cases, the signal changes and 272
that change can be used to monitor potential changes in the device. 273
Some IVDs based on a nucleic acid test (NAT) cannot be forced to give a quantitative 274
signal, and some qualitative IVDs need to be assigned a sensitivity (with confidence limits) 275
relative to an international conventional calibrator. In such cases, the parameter that can 276
most easily be used to monitor stability, and verification at release, is the detection limit 277
of each claimed analyte, which is almost always required to be stated either in the IFU or 278
in regulatory submissions. The detection limit (or any other threshold value) is found by 279
probit or logistic analysis of replicates of dilutions of the target analytes in each claimed 280
specimen type, but it is beyond the scope of this document to discuss the use of these 281
techniques in panels. For an introductory text see (17: Section 6.2.6) and (18: Appendix C). 282
The data should be analysed statistically rather than by eye, and a statistical plan should 283
be developed before starting the experiments. 284
4.3 Verification and validation 285
The following discussion often uses the terms “verification” (or “verify”) and “validation” 286
(or “validate”). These terms are defined in Section 1.1; however, to reiterate, verification 287
means providing proof of meeting predetermined specifications, whereas validation 288
means providing proof of consistently meeting predetermined needs, one of which is 289
always that the validated factor is robust and reliable. 290
4.4 Use of surrogate specimens in panels 291
Frequently, IVD lots must be verified as being able to detect particular specimens that are 292
often available in only limited quantities, because of either rarity or restriction in supply 293
from an authority. In such cases, surrogate panel members must be found that, when 294
tested in the assay of interest, mimic the rare specimens but are more readily available 295
(see Section 6 and Example 1 in Annex 1). 296

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
19 Draft for comment 22 May 2016
5 Use of panels 297
5.1 Release-to-sale panels 298
As noted in Section 2.2, for many years it has been accepted as best practice that the 299
release criteria for IVD will ensure that the device consistently performs as claimed over 300
the assigned shelf life of the lot. It is not necessary to verify every aspect of performance 301
at release-to-sale with every specimen type claimed (provided that R&D analytical matrix 302
studies have shown performance equivalence). Nevertheless, the documentation of 303
claims that are not verified at release must include a stringent risk assessment and 304
evidence of validation during the development of the device. 305
5.2 In-process panels 306
QA panels used to verify individual process stages (including testing of incoming 307
materials) and the performance of manufactured intermediates will be varied, and will 308
require critical evaluation and validation. Materials used in these in-process panels might 309
be similar to those used in other panels, particularly the release-to-sale panel, but the 310
criteria assigned will usually be different. For example, the in-process criteria assigned to 311
a panel member used for release of a coated microplate to the next stage of manufacture 312
might be wider than the criteria assigned to the same panel member when used at 313
release-to-sale, because some variability in the coating is accommodated by adjustment 314
of a conjugate. 315
5.3 Stability panels 316
Stability of each critical aspect of the IVD must be evaluated, subject to risk assessment 317
where the necessity to evaluate is determined. Some of the performance claims that are 318
not verified lot by lot at release-to-sale will probably be validated as part of the stability 319
programme. Hence, the QA panels used for stability studies, although similar to those 320
used for releasing lots to sale, are likely to be more comprehensive. 321
It is good practice to establish an ongoing stability monitoring programme where 322
necessary (19: Section 4.1), to verify that lots released-to-sale maintain their claimed 323
performance at the end of their assigned shelf lives. This verification is achieved using 324

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
20 Draft for comment 22 May 2017
IVDs retained by the manufacturer and evaluated at some predetermined time at or after 325
their expiration date, employing a panel designed specifically for the purpose (20). The 326
panel will probably be the same as that used for release-to-sale but it is likely to include 327
additional material to monitor some of the quality metrics validated for stability and 328
therefore not verified at release (e.g. in-use stability, open-pack stability and specificity). 329
The criteria might be different from those of the same panel members in the release-to-330
sale panel. For example, if there is a known and allowable degeneration of signal over the 331
life of the device the requirement at release will be higher than that at end of life so that 332
the device will continue to meet the claim over its assigned life. 333
The manufacturer can extend a product’s shelf-life claim by using IVD lots kept under 334
ongoing stability conditions and evaluated at appropriate intervals. The panel used for 335
this, as with the panel used for validation, will probably be more complex in its 336
composition than the release-to-sale panel. 337
5.4 Reproducibility during evaluations 338
The QA panel used for repeatability and reproducibility studies need not be complex – 339
unless a risk assessment shows otherwise. It is generally sufficient to use dilutions of 340
selected specimens or calibrants with concentrations near decision points (e.g. cut-off 341
values or clinically important concentrations) in each of the claimed specimen types. A 342
significant aspect of precision studies that is frequently absent from submissions to WHO 343
prequalification is the relationship between the repeatability and reproducibility as 344
measured by the developers of the assay and by intended users in their own 345
environment. If there is an important difference between the findings of the developers 346
and of the intended users, the assay needs further development in some respect. Best 347
practice is to give the users involved in external performance evaluation / clinical 348
validation a reproducibility panel that is identical to the one used by the R&D department 349
during development work, and then to ensure that the panel is tested, along with the 350
specimens, each time the assay is performed. The reproducibility data collected during 351
external performance evaluation should cover the variability between the testing sites, 352
the testers and the lots of IVD used. Comparison of these data with similar in-house data, 353

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
21 Draft for comment 22 May 2016
and the development of an experimental design that will enable the data to be collected 354
in the first place, requires statistical advice that is beyond the scope of this guide. 355
5.5 Analytical sensitivity and range: quantitative assays 356
Panel members used in development work for verifying the limits of detection, 357
quantitation and sensitivity at clinically important thresholds and range of quantitative 358
assays are likely to be a subset of the materials eventually used in the release-to-sale 359
panel. For most quantitative assays, certified reference materials or international 360
conventional calibrators will be available. In the worst case, only a manufacturer’s 361
calibrator will be available – ISO 17511 (4: Section 4.2.2g ) provides information about 362
assigning values to such material. These panels are of fundamental importance in 363
verifying the performance of an IVD. 364
5.6 Control materials provided to users 365
Control materials provided to users are intended to assure users that IVD performance is 366
consistent with its intended use and the manufacturer’s claims – a function related to 367
that of QA / QC panels. Such materials are sometimes called “run controls”, and they 368
differ from materials provided as part of external QA programmes. These controls are not 369
part of the manufacturer’s own QA system; nevertheless, the controls chosen and the 370
criteria they must meet are similar to those of the manufacturer’s internal QA panels. 371
Controls may be supplied along with the IVD as individual components or as a line on a 372
membrane (e.g. in the case of an RDT), or they may be available to purchase separately; 373
however, in all cases, ISO 15198 (3) is applicable. The claims given for run controls in IFU 374
need careful validation, and it must be shown that these controls do in fact provide 375
evidence that the IVD has or will function as claimed. Submissions to WHO 376
prequalification rarely document that the control materials meet the relevant claim in the 377
IFU; for example, for serological assays, most run controls merely show addition of a 378
reagent, or flow, but not that the IVD would meet its claimed quality critical performance. 379

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
22 Draft for comment 22 May 2017
6 Selection of specimens 380
6.1 General comments 381
Some IVD require use of fresh specimens; for example, assays for CD4, some NIVDD 382
AT IVDs and some assays requiring capillary whole blood. If stable surrogate specimens 383
cannot be generated and validated then all the required panels must be generated and 384
evaluated before each use. This will require use of a predicate test method, validated for 385
both the purpose and the specific specimen type to be used. The variance of the test 386
method must be proven not to conceal variance in the IVD being verified. This 387
requirement is usually studied as gauge repeatability and reproducibility (R&R), but the 388
process and methodology applies to any measuring system, not just to gauges. Statistical 389
analysis of gauge R&R is well documented (21). 390
Inactivation 6.1.1391
Many specimens used in QA panels are reactive for infectious organisms and must 392
therefore be handled with appropriate caution. All control materials should undergo 393
universal screening; for example, for human immunodeficiency virus (HIV), hepatitis B 394
virus (HBV), hepatitis C virus (HCV), human T-lymphotrophic virus (HTLV- I/II) and syphilis. 395
They should also be shown to be negative for transmissible (i.e. infectious) agents, unless 396
a particular analyte is essential for demonstrating a performance characteristic of an IVD 397
or suitable negative specimens are not available. 398
It is often convenient to inactivate the organisms specific for the assay so that the 399
specimens can be handled with lower risk; for control materials provided to users (see 400
Section 5.6), inactivation is generally essential. Thermal inactivation is commonly used for 401
HIV-positive specimens but it is often not realized that the routine conditions for such 402
inactivation (≥56 °C for ~30–60 minutes) do not adequately inactivate virus dried down in 403
blood fractions, as might be found round stoppers or seals (22). Even the conditions 404
commonly used for HBV inactivation (≥65 °C for ~16–20 hours) might not completely 405
inactivate dried specimens containing HIV. Where thermal inactivation is used, it must be 406
performed and documented correctly, using properly calibrated thermal measurements 407
and taking care to ensure that no dried specimen resides on the tops of containers. 408

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
23 Draft for comment 22 May 2016
Chemical inactivation with tri(n-butyl)phosphate–detergent mixtures has been shown to 409
efficiently inactivate enveloped viruses (23, 24), and for some purposes chemical 410
inactivation might be more appropriate than thermal inactivation. 411
Whatever method of inactivation is used, it is important to verify that the treatment does 412
not affect the analyte of interest. For antibodies there is rarely a direct problem; 413
however, heat-treated serum or plasma are well known to cause false reactivity in some 414
assays. For antigen and nucleic acid detection, the risk of affecting the analyte by 415
inactivation is relatively high. Care must be taken to show that the measurement is not 416
affected by the treatment. It must at least be shown that signal and end-point titre from 417
the specimen are not affected, and that both newly manufactured and aged IVD product 418
detect the treated specimen in exactly the same way as they detect the untreated 419
specimen. If the treated specimen is to be used as a control material for more than one 420
IVD, the proof of validity must be documented for each IVD. 421
Dilution 6.1.2422
Most specimens used in QA panels will probably be dilutions of stronger specimens. In 423
such cases, it is important to document that the diluent is appropriate. The diluent must 424
not interfere in the assay in any way and must give a negative signal. In addition, if the 425
diluent is a specimen negative for the analyte of interest, it must be shown not to contain 426
any related antibodies, antigens, polynucleotides or inhibitors, even if it appears not to 427
interact with the IVD under test. When the QA / QC process requires a titration curve 428
(e.g. to monitor sensitivity relative to an international conventional calibrator), the result 429
from the diluent must always be included in the documented measurements. A titration 430
curve in the absence of documentation of the signal from the diluent is invalid. 431
6.2 In-house panels 432
Characterization 6.2.1433
Panel members must be well characterized. Annex 1 of this document provides some 434
examples of characterization for release and stability panels. Normally, specimens are 435
chosen that are non-reactive for infectious agents other than the agent of specific 436
interest, but sometimes this is not possible. If the reagent is reactive for an agent other 437

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
24 Draft for comment 22 May 2017
than that of interest, then it must be proven and documented that the other agents will 438
not affect the primary assay. Where specimens are to be used in routine panels, the 439
process documentation must contain warnings alerting operating staff to the presence of 440
other infectious agents. 441
Verification or validation 6.2.2442
It is incumbent on the manufacturer to develop appropriate QA and QC panels. Risk 443
assessment of each key performance and functionality aspect, and experience with 444
development of the IVD, should indicate which features must be verified at release-to-445
sale, during stability work, in the process of manufacture and for end-of-life security, and 446
which aspects can be validated as routinely fit for purpose and only rarely need to be 447
verified. Efficient development work will minimize the need for verification by providing 448
comprehensive documentary evidence of validation for as many aspects as possible. 449
However, according to regulation or current practice, some functionalities must always be 450
verified at release-to-sale, at least to some extent. The correct, consistent functionality of 451
each critical constituent (enzyme, primer, antibody, antigen, other active biological 452
substances such as protein A and streptavidin, and agents to suppress or monitor false 453
reactivity or immune complex disruption etc.) is normally expected to be verified at 454
release-to-sale. However, the ability to function with each specimen type claimed should 455
be determined unless other strong evidence is available. 456
Sensitivity 6.2.3457
As noted previously, although verification panels for QA at release-to-sale might contain a 458
restricted set of material, the panels used to validate key metrics must be comprehensive 459
and provide the evidence required to allow documentation of the validity. The example 460
that follows illustrates what might be expected for validation of epitope functionality 461
safer. In antibody detection assays, recombinant fusion proteins are frequently used. For 462
example, for syphilis antibody detection, the fusion protein might comprise epitopes from 463
the three treponemal proteins (TpN15, TpN17 and TpN47); for anti-HIV-1 it might 464
comprise epitopes of gp41 and gp120; and for anti-HCV it might comprise epitopes of NS3 465
and core. It must be validated that each of those epitopes will function consistently to the 466

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
25 Draft for comment 22 May 2016
end of the assigned life of the IVD. The development panel will therefore prove that for all 467
lots of the recombinant, in all lots of the complete device, each epitope functions as 468
expected and remains consistently functional throughout stability studies. Testing one lot 469
of recombinant, even in several lots of IVD, is not sufficient to validate the system, 470
because it is known that different purification runs can subtly modify the epitopic 471
efficiency, particularly that of fusion proteins and the various epitopes within them. The 472
development panel used in these studies will therefore specifically and independently 473
monitor the epitopes involved, and show that the activities remain consistent and stable. 474
The R&D department faces the difficult challenge of selecting specimens to provide such 475
proof. Usually, specimens can be found that react strongly by Western blot (or another 476
method) with just one of the epitopes required, and that by dilution can be made 477
essentially specific for that epitope, at least so far as a development panel is concerned 478
( see also Section 6.2.5). Once the panel members have been identified (usually 479
throughout the R&D phase of an IVD life cycle) they can be used each time the 480
recombinant is prepared and used. The results, collected and analysed over time, can 481
either support validation of only one of those specimens in the release panel, or show the 482
need for all of them as part of the in-process panels. 483
Prozones 6.2.4484
Prozones, also known as “hook effects” occur when dilution series of specimens with a 485
high concentration of analyte give a maximum signal stronger than the signal from the 486
undiluted specimen. The following relates to assays for which the immunological 487
response profile for a target analyte is not clearly established or in which the analyte is 488
heterogeneous; for example, assays for antibodies or for antigens occurring in multiple 489
forms, such as hepatitis B surface antigen (HBsAg) and histamine rich protein 2 (HRP2) for 490
malaria. Assays for well-characterized, homogeneous analytes (e.g. small metabolites and 491
some hormones) can usually be verified not to prozone with a single artificial specimen 492
containing well above the highest concentration of the analyte found in a clinical setting. 493
The possibility of a prozone depends largely on the format of the assay. If the format 494
chosen is known to prozone, it is imperative to show that the balance of reagents will 495
make prozones rare and prozoning to negative even more rare. Prozones are commonly 496

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
26 Draft for comment 22 May 2017
associated with particle agglutination assays, but can also occur with immunometric 497
assays and with lateral flow devices. Two-step assays – that is, add specimen to capture 498
system, separate (wash), add label, wash – are not susceptible to prozones. However, the 499
normal form of lateral flow device – that is, add specimen to conjugate pad, cause 500
specimen and conjugate together to flow over capture line – will always have the 501
potential to prozone with sufficiently strong specimens. Prozones are very sensitive to the 502
exact concentrations of reagent in the IVD; hence, studies of lot-to-lot variation are 503
necessary. 504
Selection of specimens for the panel to validate consistent lot-to-lot absence of prozone 505
is not difficult, but there are two issues that need to be taken into account. 506
The first issue, as noted, is that of the number of specimens to be chosen. Because 507
prozones are relatively rare, a reasonable number of specimens must be evaluated. The 508
number would need to be obtained from knowledge of the concentration range of 509
analyte likely to be found and a stringent risk analysis – at least 10 specimens would be a 510
suitable minimum. 511
The second issue is the selection of the specimens. It is necessary to choose specimens 512
that have a strong signal and a very high titre in an assay format that cannot prozone. 513
Once the specimens and their numbers have been selected it is a simple matter to test all 514
of them on several lots of the device, to show that there is no increase in signal after 515
dilution of the specimens in a diluent validated not to interfere with the test. Any increase 516
whatever in signal strength for any of the specimens implies a potential prozone and 517
needs further analysis. 518
Rare specimens 6.2.5519
Sometimes it is necessary to validate detection of critical specimens (e.g. seroconversion, 520
rare subtypes or antigen or nucleic acid in immune complexes); however, the genuine 521
specimens might be too valuable to use in lot release-to-sale panels or in stability panels. 522
In such cases, the reactivity or the classes of antibody and epitopic specificity are unlikely 523
to be found in commonly available specimens. Instead, it is necessary to validate 524
surrogate specimens. For immune complex and seroconversion work, near-525

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
27 Draft for comment 22 May 2016
seroconversion specimens can sometimes be shown to mimic the critical individual 526
seroconversion specimens claimed. Such specimens might be available in existing 527
seroconversion series where the later members are not of particular interest in 528
themselves. Immune complexes at later stages of an infection can be important – for 529
example, in HBsAg testing (25) – in that case, specimens can be prepared by dilution of 530
analyte into individual specimens to cover an appropriate range of antibodies (at least ad 531
and ay related, in the HBsAg testing example). For rare subtypes, it might be necessary to 532
prepare surrogates synthetically either biochemically or by evaluating numbers of 533
available specimens until one that mimics the target is found. Further information is given 534
in Example 1 of Annex 1. 535
For instance, a specimen with a rare marker might be tested once with a device and the 536
result recorded. Such a single evaluation does not provide evidence of consistency and it 537
gives no indication of the extent of any lot-to-lot variability that might be present. 538
Therefore, a properly constituted panel is required that can be used on a sufficient 539
number of occasions to prove – using lots that are as varied as possible, and lots that are 540
at the end of their assigned lives – that any variability is within a predetermined 541
acceptable range. 542
Specificity 6.2.6543
Specificity of IVD is largely controlled by the additives in wash solutions and diluents (e.g. 544
detergents, chaotropic agents, cell constituents and masking proteins); monitoring the 545
functioning of such additives is an important role of QA / QC panels. Specificity must be 546
monitored during stability work both for components and for complete devices. 547
It is usual to collect false reactive specimens and interfering specimen types (26) during 548
the R&D phase of device design, and then to monitor and control lot-to-lot variation by 549
using these specimens in release-to-sale panels. When an unusual type of false reactive or 550
interfering substance is identified post-market, either submitted as a complaint or 551
identified in published literature, the specimen (if available in sufficient volume) should 552
be added to release-to-sale panels, and monitored during the statistical process control 553
to ensure that lots do not show excessive reactivity with that specimen. 554

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
28 Draft for comment 22 May 2017
Specimen types 6.2.7555
The claims to usability of different specimen types are usually validated (and 556
documented) in the R&D phase of the IVD life cycle, and verification at lot release is 557
restricted to one or two specific specimen types. This is a particular issue for whole blood 558
specimens with some flow IVD where there is variability between lots and between 559
devices in clearance of the red cell debris from the results window. In general, the whole 560
blood to be tested must be fresh unless otherwise validated, and a sufficient number of 561
individual specimens must be used to verify that the device will operate as expected. 562
Associated materials 6.2.8563
Some materials used in an IVD are not directly regarded as functionality related; for 564
example, antimicrobials, stabilizers for enzymes, agents to quench false reactants and 565
desiccants. Each of these materials must be validated in some way, and a panel of 566
materials, or a chemical method, prepared with which to document acceptable 567
performance. The antimicrobials used in an IVD submitted to WHO prequalification need 568
special consideration, both in terms of the material used (which must be capable of 569
protecting the device against a range of aggressive microorganisms) and stability under 570
harsh conditions. The panels of microorganisms used to validate antimicrobial efficacy 571
can be derived from various pharmacopoeia – for example (27) – and should be used to 572
show efficacy at the end of the assigned shelf life of the IVD. 573
Acceptance criteria 6.2.9574
Acceptance criteria are specific indicators or measures employed in assessing the ability 575
of a component, structure or system to perform its intended function. The manufacturer 576
should be able to justify the rationale for assigning specific acceptance limits when panel 577
samples are being tested at any point throughout the product lifecycle. The rationale can 578
be documented either as part of the risk management process, or as part of the design 579
control documentation when statistical techniques are used as part of the process for 580
establishing performance characteristics. 581

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
29 Draft for comment 22 May 2016
Conclusion 6.2.10582
Selection of specimens for preparation of appropriate panels to verify or support 583
validation of claims for all manufactured lots requires an in-depth knowledge of the 584
particular analyte, the mechanism of the particular assay and the clinical intent of the 585
IVD. Each claim (which will originate from a requirement in the input documentation) and 586
each statement in any documentation of an IVD needs careful consideration as to 587
whether the claim or statement is to be validated or verified for lot release. This 588
consideration needs start early in R&D so that appropriate specimens and procedures can 589
be identified and proper validation performed. 590
6.3 Certified reference materials 591
Certified reference materials (CRM) are available for many IVDs intended to measure 592
analytes that are homogeneous at a molecular level. CRM can frequently be traced to 593
calibrants with an accurate concentration and uncertainty in SI units. Whenever they are 594
available, materials traceable to SI units must be used to validate and verify quantitative 595
IVD. For this type of analyte the production of panels is usually a matter of diluting the 596
pure material into each specimen type required and proving commutability (4). CRM not 597
traceable to SI units used in biological assays are usually international conventional 598
calibrators, as discussed in the sections below. 599
6.4 International conventional calibration materials 600
Many standards from WHO and the Paul Ehrlich Institute (PEI) fall within the category of 601
international calibration materials. Examples include the 3rd International Standard for 602
HBsAg (28), and the 1st International Reference Panel for HBV genotypes for HBsAg-603
based assays (29). Such standards are used for calibration of quantitative assays and for 604
evaluation of analytical sensitivity of qualitative assays. These materials are in too limited 605
supply to use routinely in panels, so it is usual for to prepare manufacturer`s working 606
standards from more readily available specimens, calibrated against the reference 607
materials by methods described in ISO 17511 (4). 608
The 2nd HBsAg WHO international standard is serotype adw2, genotype A, while the 3rd 609
international standard is a mixture of serotypes ayw1 and adw2. Relative reactivity of 610

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
30 Draft for comment 22 May 2017
these standards in HBsAg assays from different manufacturers is different, and so when 611
verifying claims related to this type of standard, it is important to define the version used. 612
The variation can be a result of molecular heterogeneity, which is likely to be detected 613
differently by different antibodies. The same applies for IVDs intended to detect 614
antibodies – if reference materials are changed it is likely that the apparent sensitivity of 615
devices from different manufacturers will also change. 616
6.5 Measurement variation 617
It is expected that any quantitative result will be accompanied by an uncertainty 618
statement (4); therefore, any sensitivity claimed in IFU or assigned to panel members 619
should always be associated with the uncertainty. The uncertainty should take into 620
consideration variation both within and between lots. 621
6.6 External regulatory panels 622
Further details on external regulatory panels are given in Example 6 of Annex 1. Some 623
national agencies and some regulatory authorities provide panels of specimens that the 624
IVD must detect correctly before it can be released-to-sale. These panels cannot control 625
all aspects and all claims about the IVD from all the manufacturers that they regulate 626
because of the variety of reagents in those IVDs and the different specimen types 627
involved. A properly designed regulatory panel might be able to verify some key metrics, 628
but not all that a manufacturer has claimed. For example, some HIV-1 antibody detection 629
systems include only gp41, whereas others include a variety of epitopes from gp41, 630
gp120, gp160 and p24. It is unlikely that a panel provided by a regulator could monitor 631
each of these epitopes in a critical way so as to ensure that the claims related to each 632
could be verified. 633
In the example of the previous paragraph, depending on how the regulator designed and 634
measured the balance of antibodies in a changed panel member, as assay that did not 635
detect anti-p24 could, after a change to a panel member that resulted in reduced anit-636
gp41 but augmented anti-p24, suddenly appear insensitive although it had not changes at 637
all and would continue to perform as claimed. An IVD that detected anti-p24 might 638
appear more sensitive or remain that same on the changed panel member. 639

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
31 Draft for comment 22 May 2016
A different problem might be encountered if correct detection of a regulatory panel were 640
used as the manufacturer’s sole criterion for release-to-sale. Some large national 641
regulatory panels – intended to set a baseline of performance, although each 642
manufacturer’s IVD is unique – set an accuracy requirement of, for example, correct 643
detection of nine out of 10 specimens. Such a criterion is perfectly acceptable if used 644
appropriately; however, if it were used as a sole criterion, a manufacturer could release a 645
lot with an acknowledged 10% false detection rate. 646
Manufacturers must not rely exclusively on external panels to prove compliance but must 647
control products according to their claims while also meeting and using regulatory 648
requirements intelligently. Despite these cautionary comments, from a manufacturer’s 649
point of view, regulatory panels might be useful in maintaining lot-to-lot consistency. 650
For certain high-risk IVD in GHTF class D (10), the European Commission Common 651
Technical Specifications (30: Section 3.4.2) states that, “The manufacturer’s batch release 652
testing for screening assays shall include at least 100 specimens negative for the relevant 653
analyte”, but gives no further comment on choice of specimens or the required result. 654
Presumably, the intent is to detect specificity problems but the (two-sided) 95% 655
confidence interval around zero false reactives in 100 specimens is 0–3.6% false reactive. 656
For the manufacturer of an IVD claiming a typical 0.05% or fewer false reactives, these 657
100 specimens superficially add little value, particularly if the 100 specimens have been 658
pre-selected as negative on that IVD and then stored. Certainly, the results with these 100 659
specimens could not be used as the sole criterion for verification of specificity at release. 660
Again, for a manufacturer, the importance of these types of panels is as part of a 661
performance consistency monitoring programme. 662
For assays with a quantitative output, the 100 specimens might give information if the 663
background were monitored and corrected for minor changes in blank readings. A change 664
in apparent signal for the negative specimens or an increased skewness towards the cut-665
off value might signal that specificity problems might be found with larger numbers of 666
specimens. However, experience also shows that false reactions are likely to be from 667
sporadic high signals, unrelated to the background as a whole but related to impurities in 668
the system, changes in conformations of proteins (leading to new possibility of incorrect 669

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
32 Draft for comment 22 May 2017
epitopic reactions) or changes in the population or individuals tested, such as a 670
vaccination programme (e.g. vaccination against influenza in the autumn). 671

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
33 Draft for comment 22 May 2016
7 Relationship between panel members and claims 672
The particular panel members must fail their criteria if the IVD will not attain the relevant 673
claims at the end of its labelled life. For example, if there is a known change in activity or 674
a drift in signal, the release panel criteria must be set so that each lot will still meet claims 675
at the end of life, despite the drift. Similarly, the end-of-life panel must show that the 676
device has, or has not, met its claims. The signals from the panel members must therefore 677
be correlated with the signals from the critical specimens, providing information on how 678
signals from both the panel member and the critical specimens will change over the life of 679
the IVD. For panel members controlling sensitivity and specificity, the routine R&D work 680
on stability and robustness should have shown the correlation and the change of signal 681
that can be tolerated for as wide a range of conditions as possible; for example, different 682
constituent purification or syntheses, different balances of constituents and various forms 683
of stress (e.g. temperature and humidity) of routine devices. This gives assurance of 684
correlation under conditions that might occur in manufacturing; it also emphasizes the 685
necessity of preparing and validating potential panel members throughout the R&D phase 686
of the IVD life cycle, when these parameters are all accessible. For some components that 687
appear completely stable under routine conditions, extremely stressful conditions must 688
be used to force change of signal in the critical specimens and the panel members to 689
obtain points for the correlation studies (see Examples 1 and 2 in Annex 1). Correlation is 690
the key: the signals from the panel member and the related critical specimen or antigen 691
must follow each other. It is not necessary that the signals be the same, just that the 692
correlation be understood and applied. For example, in many microplate enzyme 693
immunoassays (EIAs) the positive control material is manufactured with an optical density 694
(OD) of around 1.5–2.0, and the criterion in the IFU for a successful assay is >0.8 OD. 695
Thus, the device could have lost more than 50% of its activity and still be declared as 696
efficacious. These same EIA seroconversion series are often claimed have a specimen to 697
cut-off ratio of 1:1. Even if the control material is a dilution of a specimen from a late 698
stage of the infection and bears no relationship to seroconversion, it is unlikely that those 699
seroconversion claims would be upheld if the device had lost more than 50% of its activity 700
on any kind of specimen. It is irrelevant that the device is normally stable and does not 701

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
34 Draft for comment 22 May 2017
lose activity – if the control material is claimed to function at 0.8 OD, then so must the 702
IVD, otherwise the method of validation of the control material would have failed. The 703
same principle applies to all QA / QC panel members. 704
7.1 Process control charts 705
As noted in Section 4.2, data from panel members should be made quantitative. Best 706
practice is to plot successive values lot by lot for both release-to-sale and in-process data, 707
generating “process control” charts. There is a large body of literature on how to design 708
and interpret charts to monitor incipient or actual changes in the process, or in the state 709
of the product at release-to-sale, as shown in (31). Such charts for release-to-sale data are 710
expected to be available for inspection (12), given that this is a method of verifying lot-to-711
lot consistency in manufacture, assuming the QA panel used is valid. 712
713

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
35 Draft for comment 22 May 2016
8 Maintenance of panels 714
8.1 Storage 715
Fully characterized and validated specimens are valuable; therefore, appropriate storage 716
is wise. To minimize the possibility of degradation, the stock from which the specimens 717
are to be prepared by dilution is usually split between two locations, and stored frozen at 718
a temperature below its eutectic point (usually between –20 °C and –40 °C), so that liquid 719
water does not occur. Larger stocks of prepared panel members are also usually stored at 720
these temperatures, as are the working size aliquots. Storage of some of the working 721
aliquots at ≤ –70 °C acts as insurance in case of doubt over the stability of those stored at 722
–40 °C. It is unlikely that material stored at both –40 °C and –70 °C would degrade at the 723
same rate; hence, if differences are found, instability could be suspected. Although it is 724
probably impractical to undertake full stability studies on frozen stored specimens, the 725
number of freeze–thaw cycles permissible and the time allowable for storage unfrozen 726
must be documented unless specimens are thawed, used within a short space of time and 727
then discarded. 728
The volume in the stored working aliquots needs to be determined from the rate of use, 729
the stability as a liquid and the validated number of freeze–thaw cycles permissible. 730
Storing in too small a volume in too large a vial should be avoided to minimize any effects 731
from potential freeze drying. 732
The temperature in freezers used for storing panel members must be monitored, ideally 733
electronically, and the temperature record must be retained. Normally, there is an alarm 734
system to alert staff remotely if a freezer shows signs of failure. 735
8.2 Replacement 736
Panel members are key in monitoring and verifying the performance of the IVD and its 737
components; thus, any change must be controlled rigorously. The pre-launch risk 738
evaluation of an IVD should consider the situation that the parent stock of a panel 739
member might not satisfy requirements for the expected commercial life of the product. 740
Factors to be taken into consideration are the likelihood of being able to obtain an 741
equivalent specimen and the extent of characterization already available for the existing 742

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
36 Draft for comment 22 May 2017
specimen. When fresh specimens must be used in QA panels, the risk assessment must 743
also evaluate the validation of the acceptance criteria. As pointed out previously, a 744
thorough, documented understanding of the role of each panel member and of the 745
characteristics required to achieve the role is vital and is an essential part of the 746
development effort of the IVD. This documentation and standard operating procedure 747
(SOP) to control and direct replacement of panel members should be in place from the 748
time the finalized manufacturing documentation of the IVD is prepared (i.e. before 749
verification and validation of the device). 750
Replacement from an existing stock or with well-defined analytes 8.2.1751
Replacement of working vials of panel members is simple if the parent material (e.g. 752
serum pack or manufacturer’s working calibrator (4)) is available. Replacement is simply a 753
matter of dilution into a validated diluent (see Section 6.1.2) and assignment of new 754
criteria to the diluted material. To be sure of conformity, it is best to verify that the signal 755
from the newly made panel member is the same as that from a stock previous version, by 756
assaying the new and previous members side by side in sufficient replication and on 757
sufficient lots of the IVD (or component if it is an in-process control), taking into account 758
the issues discussed in the next paragraph. If there are slight differences, a new criterion 759
can usually be applied by proportion of the difference. The new panel members must be 760
prepared under the usual change control system, and it must be possible to trace the 761
change from the identification number of the new working stocks and the records of use 762
of the panels. 763
Changes by comparison are known to lead to drift. It is therefore important to reserve 764
some of the working panel members validated during device development and stored at 765
≤ –70°C for use in the comparisons when panel members are changed, and not merely to 766
compare with the previous version of that panel member. 767
Replacement from a new stock 8.2.2768
Paragraph 8.2.1 could apply to replacement of panels of analytes with a well-defined 769
molecular structure (e.g. polynucleotides and some viral proteins), although the parent 770
stock might be exhausted. Even for relatively well-defined entities such as HBV it is clear 771

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
37 Draft for comment 22 May 2016
that the serotype (for HBsAg testing) or genotype (for NAT) must be taken into 772
consideration for replacement panel members, the precise level of identity required must 773
be defined (e.g. ad or ay, ayw or ayr, and ayw1 or ayw3) and the replacement must be 774
shown to correlate with the original in several distinct lots of the IVD. 775
Replacement of panel members from a new parent material is more difficult for 776
heterogeneous analytes. In antibody testing, the spectrum of antibodies (e.g. exact 777
epitopes detected, exact conformation of those epitopes, affinities and composition in Ig 778
classes) varies between specimens, making replacement of like for like tedious. Ideally, in 779
this situation a complete re-validation of the new specimen should be performed, 780
showing correlation with claimed critical specimens for different lots and for accelerated 781
stability. Subject to stringent and documented risk assessment related to product claims 782
and consistency, this might not be necessary; however, it does require detailed 783
consideration of exactly what is required, particularly for specimens intended to control 784
critical epitopes such as in seroconversion to HIV, HCV NS3 or syphilis TpN15. 785

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
38 Draft for comment 22 May 2017
9 References 786
1 ISO 18113–1:2009 In vitro diagnostic medical IVDs – information supplied by the 787 manufacturer (labelling) – Part 1: Terms, definitions and general requirements. Geneva: 788 International Organization for Standardization (ISO); 2009. 789
2 ICH. ICH Harmonised tripartite guideline for elemental impurities Q3D: Current Step 4 790 version. Rockville, MD: International Conference on Harmonisation of Technical 791 Requirements for Registration of Pharmaceuticals for Human Use (ICH); 2014 792 (https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3D/793 Q3D_Step_4.pdf, accessed April 2017). 794
3 ISO 15198:2004 Clinical laboratory medicine – in vitro diagnostic medical IVDs – validation 795 of user quality control procedures by the manufacturer. Geneva: International 796 Organization for Standardization (ISO); 2004. 797
4 ISO 17511:2003. In vitro diagnostic medical IVDs – measurement of quantities in biological 798 samples – metrological traceability of values assigned to calibrators and control materials. 799 Geneva: International Organization for Standardization (ISO); 2003. 800
5 HHS U. Code of Federal Regulations Title 21. Sec. 820.3 Definitions. United States 801 Department of Health and Human Services (US HHS); 2016 802 (https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=820.3&Sea803 rchTerm=definitions, accessed April 2017). 804
6 ISO 9000:2015. Quality management systems – fundamentals and vocabulary. Geneva: 805 International Organization for Standardization (ISO); 2015. 806
7 GHTF Steering Committee. GHTF/SG1/N071:2012: Definition of the terms ‘medical device’ 807 and ‘in vitro diagnostic medical device’. Global Harmonization Task Force (GHTF); 2012. 808
8 ISO 14971:2007: Medical IVDs – application of risk management to medical IVDs. Geneva: 809 International Organization for Standardization (ISO); 2007. 810
9 ISO 3534–2:2006 Statistics — vocabulary and symbols — Part 2: Applied statistics. Geneva: 811 International Organization for Standardization (ISO); 2006. 812
10 GHTF Steering Committee. GHTF/SG1/N045: Principles of in vitro diagnostic (IVD) medical 813 devices classification. Global Harmonization Task Force (GHTF); 2008. 814
11 WHO Prequalification of In Vitro Diagnostics Programme. PQDx_018: Instructions for 815 compilation of a product dossier. Geneva: World Health Organization (WHO); 2014 816 (http://www.who.int/diagnostics_laboratory/evaluations/141015_pqdx_018_dossier_instr817 uctions_v4.pdf, accessed April 2017). 818
12 WHO Prequalification of In Vitro Diagnostics Programme. PQDx_014: Information for 819 manufacturers on the manufacturing site(s) inspection (assessment of the quality 820 management system. Geneva: World Health Organization (WHO); 2014 821 (http://www.who.int/diagnostics_laboratory/evaluations/140717_pqdx_014_info_manufa822 cturers_pq_inspections_final.pdf, accessed April 2017). 823
13 ISO 13485:2016: Medical IVDs – quality management systems – requirements for 824 regulatory purposes. Geneva: International Organization for Standardization (ISO); 2016. 825

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
39 Draft for comment 22 May 2016
14 WHO. Technical Guidance Series (TGS): TGS1 – Standards applicable to the WHO 826 prequalification of in vitro diagnostics Geneva: World Health Organization (WHO); 2016 827 (http://www.who.int/diagnostics_laboratory/guidance/technical_guidance_series/en/, 828 accessed April 2017). 829
15 WHO. Technical Guidance Series (TGS): TGS4 – Test method validation for WHO 830 prequalification of in vitro diagnostic devices (draft). Geneva: World Health Organization 831 (WHO); 2016 832 (http://www.who.int/diagnostics_laboratory/guidance/technical_guidance_series/en/, 833 accessed April 2017). 834
16 WHO. In vitro diagnostics and laboratory technology – Guidance and training. Geneva: 835 World Health Organization (WHO); 2017 836 (http://www.who.int/diagnostics_laboratory/guidance/en/, accessed April 2017). 837
17 Magnusson B, Örnemark U, (ed). Eurachem Guide: The fitness for purpose of analytical 838 methods: A laboratory guide to method validation and related topics (2nd edition). 839 Eurachem. 2014 (https://www.eurachem.org/index.php/publications/guides/mv, accessed 840 April 2017). 841
18 CLSI. Evaluation of detection capability for clinical laboratory measurement procedures. 842 CLSI document EP17-A2 (2nd edition). Wayne, PA, Clinical and Laboratory Standards 843 Institute (CLSI). 2012. 844
19 ISO 23640:2011 In vitro diagnostic medical devices – evaluation of stability of in vitro 845 diagnostic reagents. Geneva: International Organization for Standardization (ISO); 2011. 846
20 WHO. Technical Guidance Series (TGS): TGS2 – Establishing stability of an in vitro diagnostic 847 for WHO prequalification (draft). Geneva: World Health Organization (WHO); 2015 848 (http://www.who.int/diagnostics_laboratory/guidance/technical_guidance_series/en/, 849 accessed April 2017). 850
21 Burdick RK, Borror CM, Montgomery DC. Design and analysis of gauge R&R studies: making 851 decisions with confidence intervals in random and mixed ANOVA models. SIAM. 2005. 852
22 Tersmette M, de Goede RE, Over J, de Jonge E, Radema H, Lucas CJ et al. Thermal 853 inactivation of human immunodeficiency virus in lyophilised blood products evaluated by 854 ID50 titrations. Vox Sang. 1986;51(3):239–243 855 (https://www.ncbi.nlm.nih.gov/pubmed/3643679, accessed April 2017). 856
23 Horowitz B, Prince AM, Horowitz MS, Watklevicz C. Viral safety of solvent/detergent 857 treated blood products. Dev Biol Stand. 1993:237–237. 858
24 Piet MP, Chin S, Prince AM, Brotman B, Cundell AM, Horowitz B. The use of tri(n-859 butyl)phosphate detergent mixtures to inactivate hepatitis viruses and human 860 immunodeficiency virus in plasma and plasma's subsequent fractionation. Transfusion. 861 1990;30(7):591–598 (https://www.ncbi.nlm.nih.gov/pubmed/2402772, accessed April 862 2017). 863
25 Brunetto MR. Chance and necessity of simultaneous HBsAg and anti-HBs detection in the 864 serum of chronic HBsAg carriers. J Hepatol. 2014;60(3):473–475. 865

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
40 Draft for comment 22 May 2017
26 CLSI. Interference testing in clinical chemistry. CLSI document EP07-A2. Wayne, PA, Clinical 866 and Laboratory Standards Institute (CLSI). 2005. 867
27 USP-NF. US Pharmacopeial Convention. Rockville, MD: United States Pharmacopeia and 868 National Formulary (USP-NF); 2008 (http://www.usp.org/usp-nf, accessed April 2017). 869
28 NIBSC. 3rd International Standard for HBsAg (HBV genotype B4, HBsAg subtypes 870 ayw1/adw2) – NIBSC code: 12/226. Potters Bar, Hertfordshire: National Institute for 871 Biological Standards and Control (NIBSC). 872
29 Chudy M, Scheiblauer H, Hanschmann K-M, Kress J, Nick S, Wend U et al. Performance of 873 hepatitis B surface antigen tests with the first WHO international hepatitis B virus genotype 874 reference panel. J Clin Virol. 2013;58(1):47–53. 875
30 GHTF Steering Committee. GHTF/SG1/N068:2012: Essential principles of safety and 876 performance of medical devices. Global Harmonization Task Force (GHTF); 2012. 877
31 CLSI. Statistical quality control for quantitative measurement procedures principles and 878 definitions; approved guideline. CLSI document C24-A3 (3rd edition). Wayne, PA, Clinical 879 and Laboratory Standards Institute (CLSI). 2006. 880
881

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
41 Draft for comment 22 May 2016
10 Annex 1 – Examples 882
The anecdotal examples that follow are not comprehensive but are intended to be 883
illustrative. Manufacturers and their R&D groups know their own assays better than 884
anyone else, so they are in the best position to develop and validate their panels. The 885
suggestions in the examples are based on selection of the panels for confirmation of 886
claims but the same principles apply to all method validations; that is, assurance that the 887
test method is providing a meaningful result (e.g. if it is a stability test, that the output 888
really does prove stability relative to claims and not just stability for a particular, possibly 889
irrelevant, spectrum of reactivity). 890
It is regarded as good practice, subject to the evaluation of residual risk before launch of 891
the product, to verify the validated claims on occasion over the life cycle of the IVD. This 892
will be achieved using the QA panels and test methods initially devised. For some 893
attributes this is a requirement (e.g. whole device stability, ISO 14971 (1) and ISO 23640 894
(2)) but for others it is more flexible (e.g. specificity, by active review of data from users). 895
10.1 Example 1: Correlation of a QA / QC panel member with a critical specimen 896
Background 10.1.1897
Critical specimens are likely to be rare and expensive, and our unlikely to be available 898
over the commercial life of an IVD. It might be possible to use critical specimens at all 899
stages of device characterization and at release-to-sale, but in view of the expense and 900
restricted availability of such specimens, validation and documentation of the methods 901
involved in their replacement becomes crucial. Therefore, the critical specimens 902
themselves are not particularly suited for inclusion in QA / QC panels and substitutes 903
must be found. Dilutions of random specimens from late stages of the infection are 904
unlikely to substitute for seroconversion, because the class, the affinity and even the 905
epitopes involved are likely to be different. Similarly, dilutions of specimens from one 906
stage of an infection or specimen type may well not monitor the behaviour of the IVD for 907
a different stage or specimen type. For NAT, the presence of different amounts of analyte 908
in organisms in immune complexes or in perinatal whole blood could present problems 909
hence, throughout the development work, R&D should be searching for readily available 910

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
42 Draft for comment 22 May 2017
specimens that mimic the rare, critical ones, and proving that the specimens chosen do 911
indeed monitor the expected characteristic. Obtaining the correct panel is not an 912
insignificant task, the main thing is that throughout the development of the device, the 913
potential panel member and the critical specimen type should correlate, and should have 914
been tested together whenever possible. Frequently, several specimens must be used 915
and tested every time the critical specimen is tested – this allows the correlation to be 916
obtained and non-correlating specimens to be rejected. Once the correlation is 917
understood, it is possible to establish the criteria for the signal generated by the IVD for 918
that panel member and hence to have evidence of meeting the corresponding claim. 919
The following example is drawn from the development of an EIA for antibodies to 920
Treponema pallidum when it was known (from the input requirements) that a particular 921
specimen in a particular regulator’s collection had to be detected in order to allow that 922
IVD to be sold in various jurisdictions. The cut-off of the EIA had been fixed at 0.15 OD 923
above the mean value of the control negatives and the regulator’s specimen gave a signal 924
of ~ 0.3 OD above the mean value negatives on routine lots. The specimen was only 925
available to IVD manufacturers in limited quantities but it was in the regulator’s own 926
panel (along with other specimens that presented no challenges). After risk evaluation, it 927
was decided that it was necessary to supplement the QA release-to-sale panel with a 928
mimic of this specimen to ensure consistent compliance with the regulator’s requirement 929
because no other specimen in the panel monitored behaviour with this type of specimen. 930
Development of a valid panel member 10.1.2931
R&D characterization of the specimen showed it to have predominantly IgM antibodies to 932
TpN17 (found by in-house Western blots against a panel of recombinant proteins made 933
for the assay’s development). A search through the in-house collection of specimens 934
found only a limited number that had this characteristic. To be sure that the chosen 935
specimen would mimic the regulator’s specimen, it was tested alongside the regulator’s 936
specimen when the IVD was in development, with several different experimental batches 937
of the recombinant fusion protein and with the lots also severely stressed by accelerated 938
stability-like studies. That led to a Passing & Bablok regression (3), as shown in Fig. A1. 939
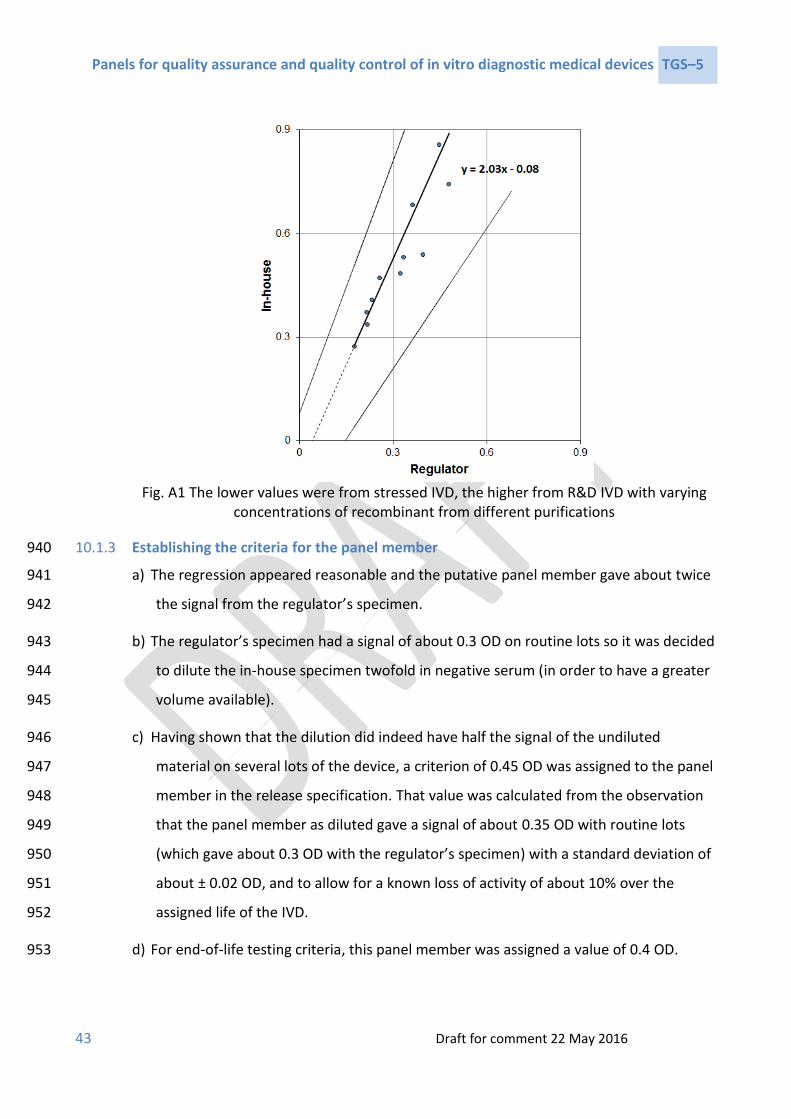
Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
43 Draft for comment 22 May 2016
Fig. A1 The lower values were from stressed IVD, the higher from R&D IVD with varying
concentrations of recombinant from different purifications
Establishing the criteria for the panel member 10.1.3940
a) The regression appeared reasonable and the putative panel member gave about twice 941
the signal from the regulator’s specimen. 942
b) The regulator’s specimen had a signal of about 0.3 OD on routine lots so it was decided 943
to dilute the in-house specimen twofold in negative serum (in order to have a greater 944
volume available). 945
c) Having shown that the dilution did indeed have half the signal of the undiluted 946
material on several lots of the device, a criterion of 0.45 OD was assigned to the panel 947
member in the release specification. That value was calculated from the observation 948
that the panel member as diluted gave a signal of about 0.35 OD with routine lots 949
(which gave about 0.3 OD with the regulator’s specimen) with a standard deviation of 950
about ± 0.02 OD, and to allow for a known loss of activity of about 10% over the 951
assigned life of the IVD. 952
d) For end-of-life testing criteria, this panel member was assigned a value of 0.4 OD. 953

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
44 Draft for comment 22 May 2017
Comment 10.1.4954
This was an exceptional case – it will not always be possible to perform such complete 955
regression studies – but it serves to illustrate the principle. For qualitative IVD or IVD with 956
discrete steps of signal (e.g. when read from a scale of colour with integral values), 957
regression is much more difficult to show. However, important factors are the proof that 958
the panel member varies with the critical specimens it is to monitor, and that the 959
criterion assigned will ensure the claims are met at end of life. 960
10.2 Example 2: Correlation under stress conditions 961
Background 10.2.1962
In Example 1, correlation between a rare, critical specimen and potential panel member 963
was proven using data obtained from both accelerated stability-like stress and changes in 964
recombinant preparation and concentration. Sometimes, routine accelerated stability 965
conditions are insufficient to effect change in the IVD, and it appears to be completely 966
stable. It is still necessary to show that the panel member and specimen will correlate as 967
the IVD is stressed, thus helping to ensure that unforeseen issues in manufacture are 968
likely to be detected (see Section 0). 969
This example is of a stable RDT for anti-HIV based on lateral flow and with a recombinant 970
polymeric gp41 as the constituent for HIV-1 detection. The RDT provided a qualitative 971
result but an in-house step-wise graduated colour chart was developed for QA / QC 972
purposes to quantify line strength. The chart was validated both by serial dilution of 973
specimens and by changes to the concentration balances of recombinants used. 974
Independent readers were in agreement with the assigned classification of results of test 975
devices about 90% of the time, and were never more than one grade-step in 976
disagreement, over the whole range of gradation. On this basis it was concluded the 977
colour chart was valid for routine use, with both ratios and differences of grade being 978
meaningful. 979
Detection of critical specimens in a number of commercial seroconversion series was 980
claimed. Between different recombinant purification lots, the appropriate panel members 981
were shown to correlate well with both IgG-first and IgM-first seroconversions (as 982

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
45 Draft for comment 22 May 2016
reflected by class-specific Western blots, relative activity on second generation (IgG only) 983
and third generation (IgG and IgM) commercial assays, and by protein A-based and 984
protein L-based research assay). There appeared to be no loss of activity either with panel 985
members or the critical seroconversion specimens over the claimed life of the assay (24 986
months claimed, 27 months stability data, 4–40 °C, humid or dry storage). Risk 987
assessment found that because activity of the gp41 component of the assay depended 988
critically on the conformation of the recombinant (4, 5), further correlation to include 989
stress conditions would give more assurance that the panel members could detect subtle 990
changes in the recombinant that might not be detected in R&D conditions, but might 991
affect the manufacturing, perhaps in the long term. 992
Extended correlation work 10.2.2993
Knowledge of the IVD suggested that stressful conditions could include storing the IVD 994
out of its protective pouch for various lengths of time under humid conditions at elevated 995
temperatures or by freezing and thawing it several times, again out of its protective 996
pouch. Trial experiments with the putative panel members showed that both these sets 997
of conditions caused loss of sensitivity: the more the stress, the more the loss. Neither of 998
these conditions were likely to occur either with users or in manufacture; nevertheless, 999
they were judged to give additional, different and useful information on the state of the 1000
IVD beyond simply using different lots and concentrations of the recombinant. 1001
It was not practical to test the critical specimens more than once under each condition 1002
because of rarity value, but the panel members could be tested several times at each 1003
condition. The work was done with three lots of the IVD made to final documentation on 1004
routine equipment. Lot A was at the end of its assigned life, whereas Lot B and Lot C were 1005
recently manufactured but from different lots of critical constituents. 1006
Some of the correlation data for the QA panel member (QA 123) and critical 1007
seroconversion specimens 1 and 2 is shown in Fig. A2. The work was done under 1008
isochronous conditions (6: Section 4.3.2), with RDT being taken from routine storage and 1009
placed under stress at different times, starting at 10 hours before testing, then 7 hours 1010
and so on for the data shown, in order that all specimens could be tested in random order 1011

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
46 Draft for comment 22 May 2017
at about the same time. Each number represents the result from an individual test of an 1012
RDT kept under the stated conditions before use. The result was read by two readers 1013
independently and the score relative to the colour scale was recorded for each. 1014
”Correlation” data
Fig. A2. “Correlation” data
A value such as 3.5 indicates that the two independent readers gave different values, but
these were never more than 1 grade different (e.g. 3 and 4 gives 3.5, whereas 1 and 0
gives 0.5).
Comments 10.2.31015
Although the analysis of these data are not statistically rigorous, the general trends can 1016
easily be seen; for example, the panel member was inactivated at about the same rate as 1017
this pair of critical specimens (which happened to be IgG-first seroconversions). Data from 1018
IgM-first seroconversions and the relevant QA / QC panel member and from the 1019
corresponding freeze–thaw experiments were equivalent to these results. 1020

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
47 Draft for comment 22 May 2016
This study was approved by the risk assessment group as sufficient to show satisfactory 1021
relationships between the QA panel members and the critical specimens, when taken 1022
together with the similar data obtained with different lots of the recombinant, as stated 1023
above. Approval to manufacture and release the IVD was given in relation to these panel 1024
members, which were considered to have a low risk of failing to detect non-compliant 1025
product. 1026
The panel member QA 123 was assigned a release-to-sales criterion of ≥3 but ≤5 on the 1027
basis of this and other evidence, and of ≥3 for end-of-life verification testing (for which 1028
the IVD is kept at a constant 40 °C for more than its assigned life). 1029
10.3 Example 3: Validation of a release-to-sale panel for anti-HCV 1030
This example applies both to RDTs with line intensity related to a graduated scale and to 1031
EIAs. 1032
Background 10.3.11033
As noted previously (Section 5.1), a release-to-sale panel must be validated to provide 1034
evidence that the tested lot will meet all the claims for the device for at least the entirety 1035
of its labelled shelf life. The claims will be derived from user input requirements and will 1036
generally include specificity, sensitivity to seroconversion, sensitivity to genotypes, 1037
capability to work on whole blood (for RDTs) and on serum and various plasma types, a 1038
functional internal control and so on. A review of the input documentation and the risk 1039
analyses is important, and is likely to produce a number of other attributes that will be 1040
claimed for the IVD but are not directly performance related (e.g. microbiological stability 1041
and operating temperature ranges). R&D should have developed the device with these 1042
and the other input requirements in mind. During the development, R&D will probably 1043
have noticed that at least NS3 and core antigens from HCV are required, and that some 1044
lots of antigen (especially NS3 1) detect some seroconversion series later than other lots. 1045
They will probably have noticed also that some lots of antigen are less specific than 1046
1 NS3 (proteases and RNA helicase) is a known to be a difficult antigen to use in product development; it seems to include both conformational and linear epitopes, and the exact conditions of recombinant culture and purification are critical to functionality – see, for example, European patent EP 1 471 074 (7) and Mondelli et al. (1994) (8).

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
48 Draft for comment 22 May 2017
others, especially NS5, if that is included in the IVD. Depending on the format of the assay 1047
there might be separate components to detect IgG and IgM. Whole blood is known to 1048
cause flow problems in many IVD, and there should be an input requirement related to 1049
invalid rates; the risk analysis should have reviewed this and R&D should have developed 1050
the device to overcome the problem. A competent design input risk assessment should 1051
have identified a range of other factors related to safe usage and other factors that will 1052
bring “added value” to the users of the device and so bring commercial advantage. 1053
Development of a valid panel 10.3.21054
For release-to-sale panels, not all claims associated with the IVD need to be addressed, 1055
depending on the risk assessment (e.g. microbiological stability, which can be validated 1056
during R&D from antibiotic efficacy and stability studies). For HCV, genotype detection 1057
can probably be validated in R&D and not verified at release. Risk evaluation must occur 1058
continuously in R&D to minimize the amount of work at release by proven and 1059
documented validation of as many factors as possible. 1060
Characteristics of IVD that are known from R&D studies to vary between lots must be 1061
verified at release (e.g. sensitivity, specificity or invalid result rate), and lot variance must 1062
be shown to be within acceptable limits. Also, it is generally accepted that concentrations 1063
and functions of critical constituents (e.g. antigens, antibodies, and biologicals such as 1064
protein A and streptavidin) must be verified at release. The number of specimens to be 1065
tested can be minimized by making appropriate choices, so that each specimen can 1066
monitor the condition of more than one thing (e.g. anti-NS3 first seroconversion and the 1067
NS3 antigen in the system). 1068
Once the claims to be verified have been decided, the specimens used to monitor them 1069
can be selected and evaluated as in Example 1 of Annex 1. A comparison of the published 1070
commercial line-assay results for critical specimens with those from [dilutions of] 1071
potential panel members might help in deciding on the initial choices for an anti-HCV IVD. 1072
For specificity monitoring, it is usual to include known falsely reactive specimens found 1073
during R&D; some falsely reactive specimens are available from the commercial suppliers 1074
for inclusion in QA / QC panels. Methods for monitoring invalid result rates must be 1075

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
49 Draft for comment 22 May 2016
devised and validated on a device-by-device basis because fresh specimens must normally 1076
be used for these studies, particularly if there is a whole blood claim. 1077
Suggested anti-HCV QA / QC panel members with these considerations in mind are given 1078
in the TGS2 guidance document for establishing stability of in vitro diagnostics assays and 1079
components (9). 1080
Establishing the criteria for panel members 10.3.31081
The particular panel members must fail their criteria if the device will not attain any of 1082
the claims at the end of its labelled life. The signals from the panel members must 1083
therefore be correlated with the signals from the critical specimens, as noted in Example 1084
1. For panel members controlling sensitivity and specificity, the routine development, 1085
stability and robustness work should have shown the correlation and the loss (or gain) of 1086
signal that can be tolerated; it should also have shown that the test method is 1087
satisfactorily less variable than the variability between lots of the device itself (see gauge 1088
R&R in Section 6.1). 1089
10.4 Example 4: A release panel for a flow cytometer for the enumeration of 1090
CD4 T-cells 1091
Background 10.4.11092
The user input requirements for a quantitative assay generally include: 1093
a specified limit of detection (LoD), with or without a specified limit of 1094
quantitation (LoQ); 1095
a measure of accuracy at clinically relevant thresholds; 1096
(possibly) a specified number of calibrator points, and hence provision of a 1097
calibrator solution) and 1098
(probably) a specified linear range or, more scientifically, a range with a specified 1099
accuracy and precision. 1100
For a CD4+ T-cell enumerating assay, there will almost certainly be a specificity 1101
requirement for CD4+ T-cells but not for other cells that might carry the CD4 determinant 1102
(e.g. monocytes). The design input risk analysis should have identified other well-known 1103

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
50 Draft for comment 22 May 2017
CD4-related problems, including potential analyte-specific interference (e.g. tuberculosis 1104
with its effects on CD4-bearing monocytes and, for some RDTs, malaria with its effects on 1105
red blood cell haemolysis). A competent design input risk assessment should have 1106
identified a range of other factors related to safe usage, and others that will bring “added 1107
value” to the users of the device and so bring commercial advantage. Many of these 1108
factors might lead to claims that require verification at release in addition to evaluation 1109
during device characterization; see, for example, the WHO sample product dosser for CD4 1110
IVD (10). 1111
Development of a valid panel 10.4.21112
For release-to-sale panels, not all claims associated with the IVD need to be addressed, 1113
depending on the risk assessment. For example, monoclonal antibodies from a reputable 1114
source are unlikely to change in avidity. However, device specificity could conceivably 1115
change from lot to lot, depending on the precise purity of the monoclonal antibodies; 1116
also, there could easily be variation between lots in the exact proportions of the 1117
monoclonal antibodies used in manufacture, which could lead to differences in ratios of 1118
cell counts and effects from analyte-specific interference. As noted previously, risk 1119
evaluation must occur continuously in R&D to minimize the amount of work at release, by 1120
proven and documented validation of as much as possible; in particular, any 1121
characteristics of the IVD that are known by R&D studies to be variable between lots must 1122
be verified at release. 1123
Selection of QA / QC panels for an analysis such as CD4+ T-cell enumeration presents 1124
problems because the analyte cannot easily be stored – even short periods of time at 2–1125
8 °C can affect the apparent cell count. It might be possible to design the device so that 1126
blood could be stored, or some form of stabilized or artificial “cells” might be developed. 1127
In each of those cases, the relationship between the behaviour of real blood and the 1128
substitute must be proven rigorously throughout device development. As in the HCV 1129
example, it must be proven and documented that there is satisfactory correlation 1130
between the real specimens and the substitutes. 1131

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
51 Draft for comment 22 May 2016
Where fresh blood is used at release-to-sale, the relationship between the device being 1132
tested and the device used as the calibration method must be validated for all lots of the 1133
tested device. A previous batch of the device is unlikely to adequately meet the 1134
requirements as a validated test method because it could lead to a drift in device 1135
performance. 1136
Establishing the criteria for panel members 10.4.31137
The general comments from the previous examples are applicable; however, for the 1138
quantitative aspects of testing the criteria must be established directly from the 1139
numerical claims (e.g. LoQ: 20 CD4+ T-cells/µL, accuracy: ±25 at 350 CD4+ T-cells/µL). Of 1140
course, the criteria must be set so that the claims will be met at end of life, so the criteria 1141
might well be different from the claim. The criteria for any substitutes for fresh blood 1142
must be shown to correlate with the same attributes in the blood in the same way as the 1143
substitutes for critical specimens in Section 10.3.3. 1144
Criteria must be established and documented during R&D for panel members monitoring 1145
claims that are not related directly to quantitation of the analyte (e.g. specificity on 1146
specimens from patients with tuberculosis and invalidity rate on specimens from patients 1147
with malaria). Similarly, criteria must be established and documented for panel members 1148
monitoring appropriate reactivity of the critical, potentially variable, components of the 1149
device. 1150
10.5 Example 5: Nucleic acid testing 1151
Background 10.5.11152
There is a wide variety of NAT methodology, qualitative and quantitative, based on DNA 1153
and RNA, and with intended use ranging from blood donor screening to infant diagnosis. 1154
The main issues relating to the assay concern specimen collection and preparation, 1155
inhibitory substances varying by specimen, stability of the enzyme systems involved and 1156
contamination during manufacture or in use. Occasionally, sub-genotypes present 1157
problems with some systems. Claimed specimen types usually include whole blood, often 1158
in the form of dried blood spots, in addition to buccal fluid, various types of respiratory 1159
samples, cerebrospinal fluid, stool, serum and plasma. Control materials, including 1160

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
52 Draft for comment 22 May 2017
calibrators for quantitative assays, are normally supplied with the IVD. As for all devices, 1161
each of the issues found during design input and process risk management processes 1162
must be dealt with and the methodology developed to minimize any effects. The nature 1163
and type of specimens to control all the hazards must be defined during the R&D phase of 1164
the work. 1165
Development of a valid panel 10.5.21166
As usual, each of the claims that cannot be validated during R&D for all lots over the 1167
commercial life of the device must be verified lot by lot at release-to-sale. In particular, 1168
the panels must demonstrate maintenance of sensitivity or LoD, or accuracy and precision 1169
for all claimed specimen types and all claimed genotypes if the latter cannot be validated 1170
in R&D. There are international conventional calibrators against which in-house QA / QC 1171
panel members can be standardized for most organisms of concern to WHO1 (see also 1172
Section 6.4), and the derived secondary standards should be enough to monitor most 1173
claims for both qualitative and quantitative assays. ISO 17511 (14) describes the types of 1174
calibrators and methods for tracing secondary, in-house, standards to internationally 1175
accepted calibrators that are expensive and in restricted supply. For genotype claims, it 1176
might be necessary to refer to collections such as the 1st WHO International Reference 1177
Panel for HBV Genotypes for NAT-Based Assays (15), or the 1st International Reference 1178
Panel HIV-1 RNA Genotypes (16). However, it is likely that the manufacturer will be more 1179
aware of the difficult subtypes from in-house and user testing than might be revealed by 1180
these panels. In that case, critical specimens will probably need to be included in the 1181
panels, at least for in-house validation of attributes such as stability and inhibition. 1182
Whenever in-house panels are made, the correlation under varying conditions with the 1183
internationally accepted materials must be demonstrated, as for any substitute specimen; 1184
proof of ISO 17511 (14) metrological traceability alone is not sufficient. 1185
1 For example, the 3rd HIV 1 International Standard (11), the 4th International Standard for Hepatitis C Virus for Nucleic Acid Amplification Techniques (12) and the Third International Standard for HBsAg (13).

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
53 Draft for comment 22 May 2016
Proof of functionality of extraction, especially from dried blood spots, and removal of 1186
inhibitory substances will almost certainly need to be verified lot by lot. This might 1187
require specimens in the panel or simply chemical analysis of the reagents. 1188
Establishing the criteria for panel members 10.5.31189
Refer to previous examples. 1190
10.6 Example 6: Use of imposed specimens as QA / QC panel members 1191
Background 10.6.11192
Some regulatory authorities require that specimens supplied by them (with criteria also 1193
supplied by them) be detected appropriately before each lot of an IVD can be released-to-1194
sale. Regulators cannot know the claims of every IVD within their purview, so such 1195
specimens cannot act as a valid release-to-sale panel. They are a requirement and, as 1196
such, will be in the design input documentation, but they should not be used as a sole 1197
basis for the manufacturer to verify release-to-sale. 1198
Reasons for not using imposed panels for QA / QC as the main panel members 10.6.21199
See also Section 6.6. 1200
a) A regulator’s panel might not control all components. The following are actual 1201
examples. A regulator provided a panel for HCV release-to-sale that contained 1202
virtually no NS3 antibodies, so the lot could still be released even though the NS3 1203
component of a device could be lacking. Another regulator changed an HIV release 1204
panel member from one that contained virtually no anti-p24 to one that contained 1205
high levels of anti-p24, both dilutions of positive specimens. The signals on some 1206
EIAs appeared to be equivalent, but not those on others, and the specimen did not 1207
correlate with seroconversion sensitivity – in fact it contributed little and certainly 1208
did not monitor critical aspects of devices. 1209
b) A panel member might be changed by the regulator, as in point (a), so that 1210
although still being detected appropriately as “positive”, it could allow a change in 1211
assay sensitivity to occur without being detected; alternatively, the device 1212
manufacturer could change his manufacturing to meet the new imposed panel 1213

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
54 Draft for comment 22 May 2017
member acceptance criteria with no control over the influence of the 1214
manufacturing change on specificity or sensitivity to meet his established claims. 1215
c) The requirements on the panel might be such that the device is not controlled 1216
within its claims. For example, if 19 of 20 panel members were required to be 1217
detected as negative, then a lot with about 5% false reactives could be released 1218
despite a claim of a much better specificity. 1219
d) If a lot meets the regulator’s requirements but the manufacturer decides the lot 1220
would not meet its claims and so does not release it, the manufacturer would be 1221
expected to document why the lot was not released as part of its QMS, to 1222
establish an auditable trail. 1223
e) Even international calibrators are not necessarily valid for release of a device 1224
against a sensitivity claim. When the first international HBsAg standard was 1225
replaced by the second, this changed the apparent sensitivity of a number of 1226
assays (17). Claims must always be cited against particular versions of 1227
international conventional calibrators. 1228
f) A manufacturer is responsible for confirming and maintaining his claims over the 1229
commercial life cycle of the IVD, despite external regulatory changes. 1230
Appropriate uses of regulators’ panels by manufacturers 10.6.31231
It might be possible to validate the regulator’s panel as in Section 10.3.3, and use it 1232
appropriately. However, the correlation work would be the same as the manufacturer 1233
choosing the specimens and there would be no security against the regulator changing 1234
the panel without notice. 1235
Using the regulator’s panel alongside the manufacturer’s own panel to monitor changes 1236
by routine trend analysis might occasionally be informative – usually about changes by 1237
the regulator. However, panel testing is done independently of the manufacturer by a 1238
regulatory authority. For example, for NAT devices, the panels are designed to test for a 1239
lot’s ability to detect a minimally acceptable number of copies/mL, defined by a 1240
regulatory authority. Any adverse comments on the composition of a regulator’s panel 1241
should always be discussed with that authority. 1242

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
55 Draft for comment 22 May 2016
1243

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
56 Draft for comment 22 May 2017
1 ISO 14971:2007: Medical IVDs – application of risk management to medical IVDs. Geneva: 1244 International Organization for Standardization (ISO); 2007. 1245
2 ISO 23640:2011 In vitro diagnostic medical devices – evaluation of stability of in vitro 1246 diagnostic reagents. Geneva: International Organization for Standardization (ISO); 2011. 1247
3 Passing H, Bablok W. A new biometrical procedure for testing the equality of 1248 measurements from two different analytical methods. Application of linear regression 1249 procedures for method comparison studies in clinical chemistry, Part I. J Clin Chem Clin 1250 Biochem. 1983;21(11):709–720. 1251
4 Pinter A, Honnen WJ, Tilley SA, Bona C, Zaghouani H, Gorny MK et al. Oligomeric structure 1252 of gp41, the transmembrane protein of human immunodeficiency virus type 1. J Virol. 1253 1989;63(6):2674–2679 (https://www.ncbi.nlm.nih.gov/pubmed/2786089, accessed April 1254 2017). 1255
5 Yuan W, Li X, Kasterka M, Gorny MK, Zolla-Pazner S, Sodroski J. Oligomer-specific 1256 conformations of the human immunodeficiency virus (HIV-1) gp41 envelope glycoprotein 1257 ectodomain recognized by human monoclonal antibodies. AIDS Res Hum Retroviruses. 1258 2009;25(3):319–328 (https://www.ncbi.nlm.nih.gov/pubmed/19292593, accessed April 1259 2017). 1260
6 CLSI. Evaluation of stability of in vitro diagnostic reagents; approved guideline. CLSI 1261 document EP25-A. Wayne, PA, Clinical and Laboratory Standards Institute (CLSI). 2009. 1262
7 Innogenetics N.V. Patent EP1471074: Methods for improving the conformation of proteins 1263 by means of reducing agents. 2004 1264 (https://register.epo.org/application?number=EP04103238&tab=main, accessed April 1265 2017). 1266
8 Mondelli MU, Cerino A, Boender P, Oudshoorn P, Middeldorp J, Fipaldini C et al. 1267 Significance of the immune response to a major, conformational B-cell epitope on the 1268 hepatitis C virus NS3 region defined by a human monoclonal antibody. J Virol. 1269 1994;68(8):4829–4836 (https://www.ncbi.nlm.nih.gov/pubmed/7518528, accessed April 1270 2017). 1271
9 WHO. Technical Guidance Series (TGS): TGS2 – Establishing stability of an in vitro diagnostic 1272 for WHO prequalification (draft). Geneva: World Health Organization (WHO); 2015 1273 (http://www.who.int/diagnostics_laboratory/guidance/technical_guidance_series/en/, 1274 accessed April 2017). 1275
10 WHO Prequalification of In Vitro Diagnostics Programme. Sample product dossier for WHO 1276 prequalification. Geneva: World Health Organization (WHO); 2014 1277 (http://www.who.int/diagnostics_laboratory/guidance/160613_simu_poc_cd4_dossier_w1278 eb.pdf?ua=1, accessed April 2017). 1279
11 NIBSC. 3rd International Reference Panel for HIV-1 RNA Genotypes – NIBSC code: 10/152. 1280 Potters Bar, Hertfordshire: National Institute for Biological Standards and Control 1281 (NIBSC);(http://www.nibsc.org/documents/ifu/10-152.pdf, accessed April 2017). 1282
12 NIBSC. 4th International Standard for Hepatitis C Virus for Nucleic Acid Amplification 1283 Techniques – NIBSC code: 06/102. Potters Bar, Hertfordshire: National Institute for 1284

Panels for quality assurance and quality control of in vitro diagnostic medical devices TGS–5
57 Draft for comment 22 May 2016
Biological Standards and Control (NIBSC);(http://www.nibsc.org/documents/ifu/06-1285 102.pdf, accessed April 2017). 1286
13 NIBSC. 3rd International Standard for HBsAg (HBV genotype B4, HBsAg subtypes 1287 ayw1/adw2) – NIBSC code: 12/226. Potters Bar, Hertfordshire: National Institute for 1288 Biological Standards and Control (NIBSC). 1289
14 ISO 17511:2003. In vitro diagnostic medical IVDs – measurement of quantities in biological 1290 samples – metrological traceability of values assigned to calibrators and control materials. 1291 Geneva: International Organization for Standardization (ISO); 2003. 1292
15 WHO. WHO 1st International Reference Panel: Hepatitis B virus genotype panel for NAT-1293 based assays – PEI code 5086/08. 1294
16 NIBSC. 1st International Reference Panel for HIV-1 RNA Genotypes – NIBSC code: 01/466 1295 Potters Bar, Hertfordshire: National Institute for Biological Standards and Control 1296 (NIBSC);(http://www.who.int/bloodproducts/publications/IFU_03-1961.pdf, accessed April 1297 2017). 1298
17 Ferguson M, Health A, Lelie N, Nubling M, Nick S, Gerlich W et al. WHO Working Group on 1299 Hepatitis and HIV Diagnostic Kits: report of a collaborative study to 1) assess the suitability 1300 of candidate replacement international standard for HBsAG and a reference panel for 1301 HBsAG and 2) to calibrate the candidate standard in IU. 1302 2003;(http://apps.who.int/iris/handle/10665/68581, accessed April 2017). 1303
1304