one-pot preparation of polymer–enzyme–metallic nanoparticle composite films for high-performance...
TRANSCRIPT

FULLPAPER
www.afm-journal.de
1784
One-Pot Preparation of Polymer–Enzyme–MetallicNanoparticle Composite Films for High-PerformanceBiosensing of Glucose and Galactose
By Yingchun Fu, Penghao Li, Qingji Xie,* Xiahong Xu, Lihong Lei, Chao Chen,
Can Zou, Wenfang Deng, and Shouzhuo Yao
[*] Prof. Q. Xie, Y. Fu, P. Li, L. Lei, C. Chen, C. Zou, W. Deng, Prof. S. YaoKey Laboratory of Chemical Biology and TraditionalChinese Medicine ResearchMinistry of Education of ChinaCollege of Chemistry and Chemical EngineeringHunan Normal UniversityChangsha, 410081 (P. R. China)E-mail: [email protected]
X. XuState Key Laboratory of Chemo/Biosensing and ChemometricsCollege of Chemistry and Chemical EngineeringHunan University, Changsha, 410082 (P. R. China)
DOI: 10.1002/adfm.200801576
New polymer–enzyme–metallic nanoparticle composite films with a high-
load and a high-activity of immobilized enzymes and obvious electrocatalysis/
nano-enhancement effects for biosensing of glucose and galactose are
designed and prepared by a one-pot chemical pre-synthesis/
electropolymerization (CPSE) protocol. Dopamine (DA) as a reductant and a
monomer, glucose oxidase (GOx) or galactose oxidase (GaOx) as the enzyme,
and HAuCl4 or H2PtCl6 as an oxidant to trigger DA polymerization and the
source of metallic nanoparticles, are mixed to yield polymeric
bionanocomposites (PBNCs), which are then anchored on the electrode by
electropolymerization of the remaining DA monomer. The prepared PBNC
material has good biocompatibility, a highly uniform dispersion of the
nanoparticles with a narrow size distribution, and high load/activity of the
immobilized enzymes, as verified by transmission/scanning electron
microscopy and electrochemical quartz crystal microbalance. The
thus-prepared enzyme electrodes show a largely improved amperometric
biosensing performance, e.g., a very high detection sensitivity
(99 or 129mA cm�2 mM�1 for glucose for Pt PBNCs on bare or platinized Au),
a sub-micromolar limit of detection for glucose, and an excellent durability,
in comparison with those based on conventional procedures. Also,
the PBNC-based enzyme electrodes work well in the second-generation
biosensing mode. The proposed one-pot CPSE protocol may be extended
to the preparation of many other functionalized PBNCs for wide
applications.
� 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
1. Introduction
The design of nanocomposites of polymer,nanoparticles, and more components withintegrated properties and synergetic effectsof the individuals, termed as polymericnanocomposites (PNCs), has attractedgreater and greater attention for theirobvious scientific and industrial interest invarious areas.[1–3] The incorporation ofnanoparticles can endow the PNCs withdiversified functions, which include catalysis(e.g., Pt nanoparticles, PtNPs),[4,5] electro-nics,[6,7] optics (e.g., quantum dots),[8–10] andmagnetics (e.g., magnetic particles).[11,12] Thepolymer, which includes conducting poly-mers,[13] virtually acts as a matrix to supportand disperse the nanoparticles,[4–6] and toachieve processibility for functionaliza-tion.[14] Entrapment or adsorption of bio-molecules in/on a conducting or non-conducting polymer has become one ofthe commonly used routes to immobilizebiomolecules for biosensing and bioelectro-nics,[15–18] and such PNCs have been usedfor highly efficient biosensing involvingDNA,[19] enzymes,[20–26] proteins (immuno-assay),[27] and so on. However, the researchto date has mainly focused on the post-immobilization of biomolecules on pre-
synthesized PNCs to yield polymeric bionanocomposites(PBNCs),[19–27] namely, by a multi-step route, which is primarilybecause the preparation of PNCs is often conducted underrigorous conditions, e.g., high acidity, high temperature, and/orutilization of organic solvents, under which the biomolecules ofinterest may suffer more severely from deactivation. To ourknowledge, the facile and efficient one-pot preparation of newPBNCs in an aqueous medium, i.e., simultaneous occurrence ofpolymer growth, formation and high dispersion of metallicnanoparticles, and highly effective entrapment of biomolecules inan aqueous solution that contains molecular-level-mixed pre-cursors, followed by co-electrodeposition of the PBNCs on anelectrode through electropolymerization of the remainingmonomers, has not been reported, and this new route is expectedto endow the prepared PBNC film with new properties forinteresting applications.
Adv. Funct. Mater. 2009, 19, 1784–1791

FULLPAPER
www.afm-journal.de
The neurotransmitter dopamine (DA) as a biogenic speciesplays an important physiological role as a chemical messenger inmammalians, whose extreme abnormality can result in someserious diseases such asParkinson’s disease.[28] DA is electroactiveandhasbeenelectrochemically studied innumerous reports,[29–31]
including its electropolymerization study in this laboratory.[32] DAat a micromolar-scale concentration was also used to chemicallyinduce the growth of Au nanoparticles (AuNPs) for visiblespectrophotometric assays for the detection of DA and oftyrosinase activity,[33] but the DA polymerization should not takeplace therein, since the water-phase polymerization of DA wasreported to occur only at a pH> 3.86 and a DA concentrationgreater than 0.2� 10�3
M.[32] In addition, inspired by thecomposition of adhesive proteins in mussels, the DA self-polymerization (air-driven chemical polymerization of DA) hasbeen used to prepare multifunctional biocompatible polydopa-mine (PDA) coatings on various surfaces for a variety of promisingapplications.[34] However, to our knowledge, DA has never actedsimultaneously as a reductant and amonomer to synthesize highlybiocompatible PBNCs, although some other reducing monomersthat are not biogenic have recently been used to prepareconventional PNCs,[13,35] e.g., a conducting polypyrrole–AuNPnanocomposite was prepared by a direct redox reaction of pyrrole(the reductant) and HAuCl4 (the oxidant).
[13]
Herein, we report on the preparation of polymer–enzyme–metallic nanoparticle films obtained by a one-pot chemical pre-synthesis/electropolymerization (CPSE) for high-performancebiosensing of glucose and galactose. DA is utilized as a reductantandamonomer, glucoseoxidase (GOx) orgalactose oxidase (GaOx)as a model enzyme to be entrapped, andHAuCl4 or H2PtCl6 as anoxidizing reagent to trigger DA polymerization and the source ofmetallic nanoparticles. For comparison, we also prepared a PDA–GOx film by conventional electropolymerization (CEP) in aphosphate buffer solution (PBS, 0.10 M KH2PO4–K2HPO4þ 0.10M K2SO4, pH 7.0) that contains DA and GOx, and a PDA–GOx–K3Fe(CN)6 composite filmby a one-pot chemical pre-oxidation andelectropolymerization of monomer (CPEM) in PBS (pH 7.0) thatcontains DA, GOx, and K3Fe(CN)6. The CPEM protocol hasenabled us to prepare poly(dithiol) filmswith a very high load and avery high activity of the immobilized enzyme.[36]We found that thebiosensors based on the PBNC films presented much betterperformance, in comparison with those based on the CEP andCPEMprotocols. Also, thePBNCfilm-based electrodes couldworkin a second-generation biosensing mode with high sensitivity,
Scheme 1. Schematic depiction of the construction and biosensing mechanism of the PDA–
GOx–PtNP PBNCs electrode.
broad linear detection range (LDR), low detec-tion potential, and high current output. Theone-pot CPSE protocol is highly recommendedas a new experimental platform for thepreparation of many other PBNCs with inter-esting properties and application potentials.
2. Results and Discussion
2.1. Fabrication and Characterization of the
PBNC Films
As illustrated in Scheme 1 (with GOx andH2PtCl6 as an example), DA and GOx were
Adv. Funct. Mater. 2009, 19, 1784–1791 � 2009 WILEY-VCH Verl
dissolved in a pH 7.0 PBS (I), and then H2PtCl6 was added tooxidize DA to yield Pt nanoparticles (PtNPs) and PDA (or DAoligomers) (for convenience, PDA for short below), as well asmany PDA–GOx–PtNP PBNCs (II). Some of the PBNCs werethen electrochemically codeposited during the oxidative poly-merization of excess DA in the solution, finally achieving a PDA–GOx–PtNP PBNC film on the electrode (III). The prepared PBNCelectrodes worked well in both first and second-generationbiosensing modes. In the presence of glucose and dissolved O2
(natural mediator of GOx, the first-generation biosensing mode),GOx oxidized the substrate glucose to gluconic acid, O2 oxidizedthe reduced GOx to the original GOx with simultaneousproduction of H2O2, and then the electro-oxidation of H2O2 toO2 catalyzed by PtNPs occurred at the Au electrode at suitablepotentials, which improved the detection sensitivity (H2O2-oxidation mode here) and speeded up the O2 cycle. In thepresence of glucose and an artificial mediator p-benzoquinone(BQ), the turnover of GOx was achieved by BQ instead of O2, andthe resultant quinol was electro-oxidized to BQ and the oxidationcurrent was taken as the analytical signal, thus completing theelectron shuttle between GOx and the electrode, and minimizingthe dependence of the biosensing signal on the dissolved O2 of alimited concentration here (the second-generation biosensor).The replacement of H2PtCl6 with HAuCl4 and of GOx with GaOxwas also tested.
The experimental conditions for the preparation of variousPBNCs/composites were optimized by varying the examinedcondition while others were fixed, and the optimized values wereselected so as to obtain the best detection sensitivities of theprepared enzyme electrodes, as listed in Table 1. It should benoted that the optimized concentration ratios of the reductant(DA) to the oxidant (K3Fe(CN)6, HAuCl4, or H2PtCl6) were alwayshigher than those for the corresponding saturated reactions, thusHAuCl4 or H2PtCl6 became fully exhausted in the solution, whichleaves excess DA for the subsequent electropolymerization toanchor the PBNCs onto the electrode surface.
Transmission electronic microscopy (TEM) pictures of theprepared PDA–GOx–AuNP and PDA–GOx–PtNP PBNCs werecollected. As shown in Figure 1 (Pictures 1 and 2), AuNPs of�4 nm diameter and PtNPs of �3 nm diameter on average wererather uniformly dispersed in the PBNCs prepared underoptimized conditions, respectively. The observed gray coloredsurrounding of the AuNPs or PtNPs is assumed to be the PDAand GOx. The diameter of the observed PBNCs ranged roughly
ag GmbH & Co. KGaA, Weinheim 1785

FULLPAPER
www.afm-journal.de
Table 1. The optimization of the experimental variables.
Experimental variable Testing range Optimized value
cDA [�10�3M] 10–50 30
cGOx [mg mL�1] 0.5–5.0 3.5
cDA : cK3FeðCNÞ6 [a] 30: 1–3: 1 10: 1
cDA : cHAuCl4 [a] 120: 1–6: 1 20: 1
cDA : cH2PtCl6 [a] 75: 1–3.3: 1 7.5: 1
pH 4.0–9.0 7.0
[a] cDA¼ 30� 10�3M.
Figure 1. The TEM pictures of PDA–GOx–AuNP (1) and PDA–GOx–PtNP
(2) nanocomposites as well as the SEM pictures of PDA–GOx (3), PDA–
GOx–K3Fe(CN)6 (4), PDA–GOx–AuNP (5), and PDA–GOx–PtNP (6) nano-
composites. The enzyme films were constructed under optimized con-
ditions, 30� 10�3M DA and 3.5mg mL�1 GOx were used, and 1.5� 10�3
M HAuCl4 or 4� 10�3M H2PtCl6 was used when needed. TEM scale
bar¼ 20 nm; SEM scale bar¼ 100 nm.
1786
from 50 to 200 nm. It should be noted that, by decreasing themolar concentration ratio of the reductant (DA) to the oxidant(metallic salt) (15: 1 to 3.75: 1), the size and dispersion density ofthe PtNPs were both increased, as shown in Figure S1, but thehighest biosensing sensitivity was obtained at a ratio of DA toH2PtCl6 of 7.5: 1, which indicates that the sizes andmorphologiesof the metallic nanoparticles and subsequently the conductingand catalytic nature of the PBNCs, can be facilely controlled, andit is possible to flexibly regulate the performance (e.g., detectionsensitivity) of the PBNC enzyme electrode. The nanoparticlesgenerally endow the PNCs with improved mechanical strength.[1]
Large numbers of uniformly dispersed AuNPs or PtNPs couldeffectively promote the mechanical strength of the PBNCs here,and the stable micro-surrounding for the entrapped enzymemolecules should be favorable to minimize their conformationalchanges and preserve their bioactivity during utilization andstorage of the relevant biosensors. However, the enzymemolecules that are mainly dispersed on the PNC surface or keptdistance from the PNCs may suffer more severely fromenvironmental perturbation, which gives rise to a more obviousloss of bioactivity during usage and storage of the relevantbiosensors.[20,22–25] Hence, the enzyme molecules entrapped inthe present PBNCs are expected to show a higher sensitivity andlonger storage ability, as discussed below.
Four Au-supported enzyme films, PDA–GOx, PDA–GOx–K3Fe(CN)6, PDA–GOx–AuNP, and PDA–GOx–PtNP, as well as aplatinized Au supported PDA–GOx–PtNP film were prepared bypotential scanning from �0.5 to 0.5 V for 35 cycles, and the filmthickness wasmonitored and presented as the frequency decreaseof an electrochemical quartz crystal microbalance (EQCM), asshown in Figure S2 (also see Tables 2 and S1 (GaOx case)). Thedynamic frequency decrease was observed only at potentialspositive of �0.1 V versus a saturated calomel electrode (SCE),which indicates the occurrence of DA electropolymerization onlyafter DA is oxidized to its quinone form. The EQCM responses tothe redox processes of DA and the relevant mechanism forelectropolymerization of DA have been discussed in detail in our
Table 2. The construction and performance of GOx-based electrodes as well
Pre-oxidant Film thickness [kHz] S [mA cmS2 mMS1] LDR [a] [�10S3
none 1.6 17 0.005–4.6
K3Fe(CN)6 2.2 43 0.002–2.6
HAuCl4 3.4 66 0.001–2.6
H2PtCl6 3.6 99 0.0005–4.5
[a] The values of r2 for LDR are all > 0.99.
� 2009 WILEY-VCH Verlag GmbH &
previous report.[32] The final frequency response and the massgain of the electrode after the potential-cycling treatmentfollowed the sequence of PDA/Au<PDA–GOx/Au<PDA–GOx–K3Fe(CN)6/Au<PDA–GOx–AuNP/Au<PDA–GOx–PtNP/Au<PDA–GOx–PtNP/platinized Au, and the net mass increasehere versus sole PDA growth should be a result of the effectiveintake of the corresponding species (GOx, and mass-enhancedcomposites or PBNCs) into the polymer film deposits. Scanningelectronic microscopy (SEM) pictures of the films were collected,as shown in Figure 1 (Pictures 3–6). In comparison with thecompact PDA–GOx film, the PDA–GOx–K3Fe(CN)6 compositefilm presented a more porous structure, being similar to ourprevious report.[36] The PDA–GOx–AuNP and PDA–GOx–PtNPPBNC films showed a porous and rugged surface, with someprotuberances of PBNC aggregations. It is interesting that the twoPBNC films showed ‘cemented’ surfaces, with some seeminglyfilose species linked through the gaps distributed on the film,which is presumed to arise from the interaction of the
as estimation of ESAi by the EQCM.
M] LOD [�10S6M] Df0e [kHz] QH2O2 [mC] ESAi [kU gS1]
2 0.50� 0.04 3.9� 0.1 17� 1
0.7 0.90� 0.06 11� 1 39� 2
0.4 1.2� 0.1 17� 1 45� 4
0.09 1.1� 0.1 25� 1 71� 5
Co. KGaA, Weinheim Adv. Funct. Mater. 2009, 19, 1784–1791

FULLPAPER
www.afm-journal.de
amidocyanogen of PDA and metallic nanoparticles dispersedaround the nanocomposites. A similar coordination effect hasbeen reported to be responsible for the formation of submi-crometer-scale, monodisperse, spherical colloids of organic–inorganic hybrid materials at room temperature.[37] In addition,atomic force microscopy images were also collected (Fig. S3),which agree well with the SEM results. The SEM images ofplatinized Au and PDA–GOx–PtNP/platinized Au are also shownin Figure S4. The porous but robust structure of the enzyme filmshere should be favorable for mass transfer and to prevent enzymeleakage during utilization and storage, thus improving thebiosensing performance.
2.2. The Performance of the PBNC Film-Based Enzyme
Electrodes
To test the electrocatalytic/nano-enhancement effects of thePBNCs films, the static currents at various potentials for severalenzyme electrodes were examined in stirred PBS, as shown inFigure 2. The static current response of PDA–GOx–K3Fe(CN)6/Au to 2.0� 10�3
M glucose started at �0.4 V and leveled off at0.7 V, but those at PDA–GOx–AuNP/Au and PDA–GOx–PtNP/Auelectrodes started at 0.3 and 0.2 V, and then leveled off at 0.7 and0.5 V, respectively. Also, the static current responses at the twoPBNC electrodes were obviously enlarged, which undoubtedlydemonstrates the effective electrocatalytic/nano-enhancementeffects of the PBNCs, especially in the Pt case. For convenience ofcomparison, all enzyme electrodes below were run at 0.7 V, exceptwhere otherwise stated. The amperometric glucose-biosensingresponses (panel a) and the calibration curves (panel b) of severalenzyme electrodes are shown in Figure 3, and the values ofsensitivity (S), LDR, and limit of detection (LOD, S/N¼ 3) arelisted in Table 2. The sensitivities of the PDA–GOx–AuNP/Au andPDA–GOx–PtNP/Au electrodes were 3.9 and 5.8 fold that of
Figure 2. The static current response of PDA–GOx–K3Fe(CN)6/Au (1),
PDA–GOx–AuNP/Au (2), and PDA–GOx–PtNP/Au (3) to 2� 10�3M
glucose in stirred pH 7.0 PBS at various potentials. The enzyme electrodes
were constructed under optimized conditions.
Figure 3. Chronoamperometric responses to successive additions of glu-
cose (a) and the calibration curves (b) on PDA–GOx/Au (1), PDA–GOx–
K3Fe(CN)6/Au (2), PDA–GOx–AuNP/Au (3), PDA–GOx–PtNP/Au (4), and
PDA–GOx–PtNP/platinized Au (5) at 0.7 V versus SCE in pH 7.0 PBS. The
enzyme electrodes were fabricated under optimized conditions. The lin-
early regressed lines are also shown.
Adv. Funct. Mater. 2009, 19, 1784–1791 � 2009 WILEY-VCH Verl
PDA–GOx/Au, and 1.5 and 2.3 fold that of PDA–GOx–K3Fe(CN)6/Au, respectively. In addition, the sensitivities of theproposed PBNC-based biosensors, especially for the PDA–GOx–PtNP/Au electrode, are notably higher than those for the reportedPNC-based biosensors (<31mA cm�2 mM
�1).[20–25] We also pre-synthesized PtNPs,[38] which were then mixed with GOx and DAto electrodeposit enzyme films on Au by a similar cyclicvoltammetry (pre-synthesized PtNP–PDA–GOx/Au). Alternately
ag GmbH & Co. KGaA, Weinheim 1787

FULLPAPER
www.afm-journal.de
1788
we synthesized PtNP–PDA composites by first reacting DA withH2PtCl6, and then adding GOx to interact with the composites,and the mixture was subjected to a similar cyclic voltammetry togrow enzyme films on Au ((PtNP–PDA)–GOx/Au). The (PtNP–PDA)–GOx/Au and pre-synthesized PtNP–PDA–GOx/Auenzyme electrodes so prepared showed a poorer performancethan the PDA–GOx–PtNP/Au electrode, as shown in Figure S5,which demonstrates the notable advantages of the proposed one-pot CPSE protocol. Also, when the PDA–GOx–PtNP PBNC wasimmobilized on platinized Au (PDA–GOx–PtNP/platinized Au),the sensitivity was largely promoted to 129mA cm�2 mM
�1, whichis amongst the best reported (Fig. 3 and Table S2), anddemonstrates that the PBNC is not substrate selective and maybe well fitted to other catalytic substrates (e.g., porous Pt andcarbon nanotubes) for further improvement. Again, we experi-mentally found that the enzyme electrode based on the direct co-electrodeposition (pre-oxidant-free, CEP protocol) of GOx andPDA on platinized Au gave a higher sensitivity than that preparedwith poly(o-aminophenol) instead of PDA (48mA cm�2 mM
�1 forPDA versus 33mA cm�2 mM
�1 for poly(o-aminophenol)), whilepoly(o-aminophenol) was widely used for enzyme immobiliza-tion.[39] The PBNC enzyme electrodes all showed a sub-micromolar LOD, and excellent anti-interferent ability againsturic acid, ascorbic acid, acetaminophenol, and glutathione, asshown in Figure S6. The storage stability of the prepared enzymeelectrodes were investigated, and the time periods for the enzymeelectrodes to retain 80% of their initial response were found to be8, 8, 12, and 10 weeks for PDA–GOx/Au, PDA–GOx–K3Fe(CN)6/Au, PDA–GOx–AuNP/Au, and PDA–GOx–PtNP/Au, respectively.The biosensor was used to detect glucose in five blood serumsamples, and the results are listed in Table S3. The biosensorresults agreed well with the reference ones that were obtained bythe GOx–peroxidasemethod in a local hospital, and good recoverywas also obtained.
Another oxidase, GaOx, was similarly examined, and thebiosensing sensitivities of PDA–GaOx–AuNP/Au and PDA–GaOx–PtNP/Au electrodes to galactose were 2.9 and 3.8 fold thatof PDA–GaOx/Au, as well as 1.9 and 2.5 fold that of PDA–GaOx–K3Fe(CN)6/Au, respectively, as listed in Table S1. All the dataundoubtedly demonstrate that the constructed PBNC enzymeelectrodes exhibit excellent biosensing performance.
2.3. Estimation of the Enzymatic Specific Activity (ESA) and
Brief Discussion on the Nano-Enhancement Effects of the
PBNC Films
To further investigate the enhancement of the sensitivity of thePBNC-based biosensors, the enzymatic load (expressed as thefrequency shift for the immobilized enzyme, (Df0e) and ESA(defined as the ratio of electrode-captured molar quantity of H2O2
in mmol in 60 s to the mass of enzyme in g) were quantitativelymeasured by EQCM, as given below.
One unit of GOx activity (U) is defined here as the enzymaticproduction of 1mmol of H2O2 in 60 s under our experimentalconditions. The ESA can be written as:[36,40–42]
ESA ¼ nH2O2
Dme
(1)
� 2009 WILEY-VCH Verlag GmbH &
where nH2O2(in mmol) is the molar quantity of H2O2 produced in
the 60 s enzymatic reaction, which may be amperometrically
quantified, and Dme (in g) is the mass of the enzyme. So the ESA
can be obtained after quantifying the nH2O2and the Dme, and the
Dme for the immobilized enzyme can be estimated by EQCM.The estimation of the ESA for the electrode-immobilized GOx
(ESAi) involved the following basic steps: 1) the mass of theimmobilized enzyme (Dme) could be quantified from the ‘dry’-frequency-shift change (Df0e) of the electrode films in thepresence (Dfg,p) and absence (Dfg,a) of GOx (Df0e¼Dfg,p�Dfg,a),according to the Sauerbrey equation;[43,44] 2) glucose was addedinto a stirred PBS to give a final concentration that saturated theenzymatic reaction at the enzyme electrode, and the subsequent60 s change in the oxidation charge (QH2O2
in mC) on the enzymeelectrode at 0.7 V was recorded and used to quantify theenzymatically generated H2O2 (nH2O2
). Therefore, the ESAi isexpressed as:
ESAi ¼ �2:264� 10�6QH2O2
f 20g
zFADf0e(2)
where z (2) is the number of electrons transferred during
oxidation of H2O2, F is the Faraday constant (96485.33C mol�1),
f0 g (9MHz) is the fundamental frequency of the piezoelectric
quartz crystal, and A (0.29 cm2) is the electrode surface area.As listed in Table 2, the enzymatic loads on the PDA–GOx–
AuNP/Au and PDA–GOx–PtNP/Au electrodes were 2.4 and 2.2fold that on the PDA–GOx/Au electrode, as well as 1.3 and 1.2 foldthat on the PDA–GOx–K3Fe(CN)6/Au electrode. The ESAi valuesfor the PBNCs were larger than those for PDA–GOx/Au andPDA–GOx–K3Fe(CN)6/Au, and also superior to those in otherreports.[36,40–42] In particular, the ESAi for the PDA–GOx–PtNP/Au electrode is very close to that of the native enzyme in solutionunder our experimental conditions (74� 4 kU g�1),[36] whichdemonstrates that the enzymatic activity of the immobilizedenzyme here was excellently retained, and the PtNPs in thePBNCs could effectively capture the enzymatically generatedH2O2 to avoid its leakage from the enzyme film. In terms of theabove results, the notable enhancement in the performance of thePBNC-based electrodes should be attributed to 1) the highbiocompatibility of the biogenic DA utilized, the in-situ solution-state entrapment of enzyme into the PBNCs,[36] the highadsorption capacity and biocompatibility of the AuNPs andPtNPs, and the promoted mechanical strength of the PBNCs bythe uniformly dispersed AuNPs or PtNPs, which could allimprove the load and activity of the immobilized enzyme; and 2)the pre-synthesis of PBNCs in solution which producesuniformly dispersed AuNPs or PtNPs with a small size andhigh abundance in a 3D manner, which are favorable for highlyefficient capture of the enzymatically generated H2O2 in thevicinity to effectively catalyze its oxidation.
2.4. The Second-Generation Biosensing Mode
The dissolved O2 acts as a natural mediator for GOx turnover, butit is of only �1.3� 10�3
M saturated concentration in water.[45]
Co. KGaA, Weinheim Adv. Funct. Mater. 2009, 19, 1784–1791
FULLPAPER
www.afm-journal.de
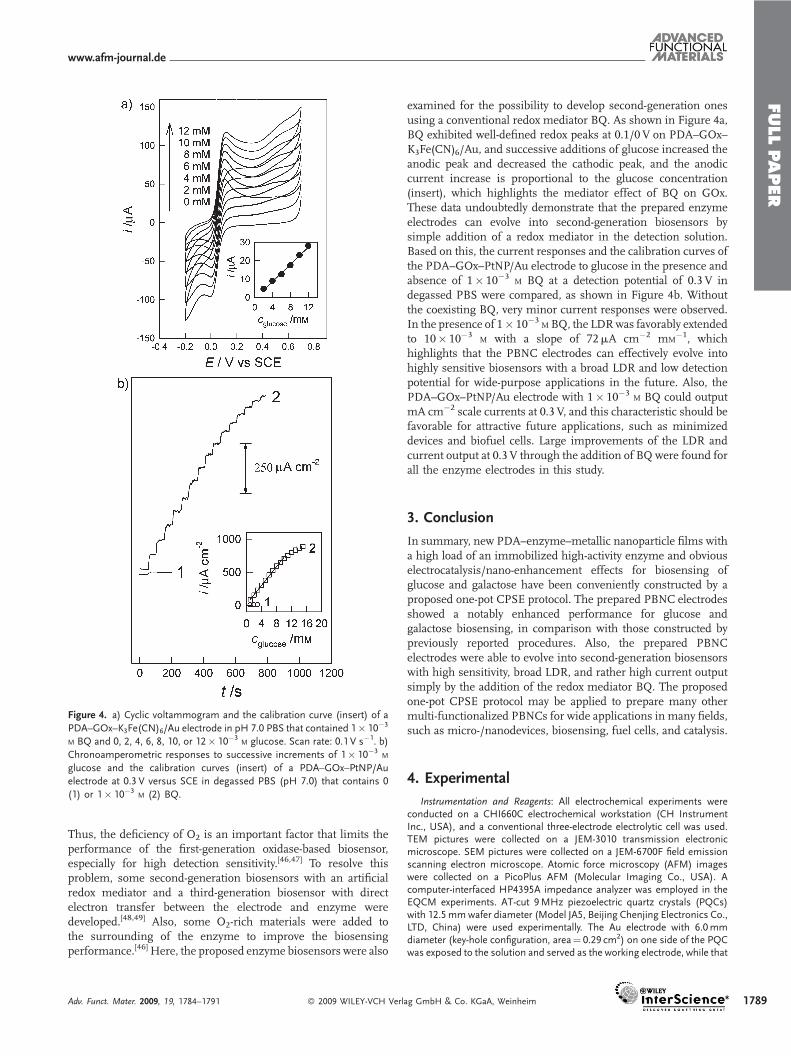
Figure 4. a) Cyclic voltammogram and the calibration curve (insert) of a
PDA–GOx–K3Fe(CN)6/Au electrode in pH 7.0 PBS that contained 1� 10�3
M BQ and 0, 2, 4, 6, 8, 10, or 12� 10�3M glucose. Scan rate: 0.1 V s�1. b)
Chronoamperometric responses to successive increments of 1� 10�3M
glucose and the calibration curves (insert) of a PDA–GOx–PtNP/Au
electrode at 0.3 V versus SCE in degassed PBS (pH 7.0) that contains 0
(1) or 1� 10�3M (2) BQ.
Thus, the deficiency of O2 is an important factor that limits theperformance of the first-generation oxidase-based biosensor,especially for high detection sensitivity.[46,47] To resolve thisproblem, some second-generation biosensors with an artificialredox mediator and a third-generation biosensor with directelectron transfer between the electrode and enzyme weredeveloped.[48,49] Also, some O2-rich materials were added tothe surrounding of the enzyme to improve the biosensingperformance.[46] Here, the proposed enzyme biosensors were also
Adv. Funct. Mater. 2009, 19, 1784–1791 � 2009 WILEY-VCH Verl
examined for the possibility to develop second-generation onesusing a conventional redox mediator BQ. As shown in Figure 4a,BQ exhibited well-defined redox peaks at 0.1/0V on PDA–GOx–K3Fe(CN)6/Au, and successive additions of glucose increased theanodic peak and decreased the cathodic peak, and the anodiccurrent increase is proportional to the glucose concentration(insert), which highlights the mediator effect of BQ on GOx.These data undoubtedly demonstrate that the prepared enzymeelectrodes can evolve into second-generation biosensors bysimple addition of a redox mediator in the detection solution.Based on this, the current responses and the calibration curves ofthe PDA–GOx–PtNP/Au electrode to glucose in the presence andabsence of 1� 10�3
M BQ at a detection potential of 0.3 V indegassed PBS were compared, as shown in Figure 4b. Withoutthe coexisting BQ, very minor current responses were observed.In the presence of 1� 10�3
M BQ, the LDRwas favorably extendedto 10� 10�3
M with a slope of 72mA cm�2 mM�1, which
highlights that the PBNC electrodes can effectively evolve intohighly sensitive biosensors with a broad LDR and low detectionpotential for wide-purpose applications in the future. Also, thePDA–GOx–PtNP/Au electrode with 1� 10�3
M BQ could outputmA cm�2 scale currents at 0.3 V, and this characteristic should befavorable for attractive future applications, such as minimizeddevices and biofuel cells. Large improvements of the LDR andcurrent output at 0.3 V through the addition of BQ were found forall the enzyme electrodes in this study.
3. Conclusion
In summary, new PDA–enzyme–metallic nanoparticle films witha high load of an immobilized high-activity enzyme and obviouselectrocatalysis/nano-enhancement effects for biosensing ofglucose and galactose have been conveniently constructed by aproposed one-pot CPSE protocol. The prepared PBNC electrodesshowed a notably enhanced performance for glucose andgalactose biosensing, in comparison with those constructed bypreviously reported procedures. Also, the prepared PBNCelectrodes were able to evolve into second-generation biosensorswith high sensitivity, broad LDR, and rather high current outputsimply by the addition of the redox mediator BQ. The proposedone-pot CPSE protocol may be applied to prepare many othermulti-functionalized PBNCs for wide applications in many fields,such as micro-/nanodevices, biosensing, fuel cells, and catalysis.
4. Experimental
Instrumentation and Reagents: All electrochemical experiments wereconducted on a CHI660C electrochemical workstation (CH InstrumentInc., USA), and a conventional three-electrode electrolytic cell was used.TEM pictures were collected on a JEM-3010 transmission electronicmicroscope. SEM pictures were collected on a JEM-6700F field emissionscanning electron microscope. Atomic force microscopy (AFM) imageswere collected on a PicoPlus AFM (Molecular Imaging Co., USA). Acomputer-interfaced HP4395A impedance analyzer was employed in theEQCM experiments. AT-cut 9MHz piezoelectric quartz crystals (PQCs)with 12.5mmwafer diameter (Model JA5, Beijing Chenjing Electronics Co.,LTD, China) were used experimentally. The Au electrode with 6.0mmdiameter (key-hole configuration, area¼ 0.29 cm2) on one side of the PQCwas exposed to the solution and served as the working electrode, while that
ag GmbH & Co. KGaA, Weinheim 1789

FULLPAPER
1790
on the other side faced the atmosphere. The reference electrode was a KClSCE, and a carbon rod served as the counter electrode. All potentialsreported in this paper are cited versus the SCE.
GOx (EC 1.1.3.4; type II from Aspergillus niger, �150 kU g�1) and GaOx(from Dactylium dendroides, 54 kU g�1) were purchased from Sigma. DAwas a Fluka product. A pH 7.0 PBS was used. All other chemicals were ofanalytical grade or better quality, and used as received. Milli-Q ultra-purewater (Millipore,�18 MV cm) was used throughout. All experiments wereperformed at room temperature at �20 8C.
General Procedures: To clean the Au electrode surface, one drop offreshly prepared concentrated H2SO4þH2O2 (3:1, v/v) was added for 15 s,followed by rinsing thoroughly with water and then drying with a stream ofpure nitrogen. The treatment was repeated three times. Prior toelectrochemical experiments, the Au electrode was subjected tocontinuous potential cycling (0–1.5 V, 30mV s�1) in 0.20 M aqueousHClO4, until the cyclic voltammogram became reproducible. The ‘dry’frequency of the clean bare Au electrode was recorded by the EQCM.
The GOx biosensors were prepared as follows. For CPSE, with the caseinvolving metallic salt H2PtCl6 as an example, to 0.10 M PBS (pH 7.0) thatcontained 30� 10�3
M DA (excess) and 3.5mg mL�1 GOx, 4� 10�3M
H2PtCl6 was slowly added. The mixture was stirred to allow a completereaction for 20min to form PDA–GOx–PtNP PBNCs, and then the co-electrodeposition of PDA–GOx–PtNP PBNC and PDA on a bare Auelectrode was performed by potential scanning from �0.5 to 0.5 V for 35cycles, simultaneously being monitored by the EQCM. The electrode wasthen thoroughly rinsed with Milli-Q water to remove physically adsorbedPDA and PDA–GOx–PtNP PBNCs, and the ‘dry’ frequency of the modifiedAu electrode was also recorded by the EQCM to compare with that on thebare electrode. Similar procedures were involved when themetallic salt wasHAuCl4 to obtain a PDA–GOx–AuNP PBNC film electrode, or when themetallic salt was replaced by oxidant K3Fe(CN)6 to obtain a PDA–GOx–K3Fe(CN)6 composites electrode. We also checked the modification of theprepared and isolated PBNCs on the electrode by directly dip-drying aPBNC/water mixture on the electrode surface, but the so-prepared enzymeelectrodes exhibited poor stability and reproducibility.
The platinization of Au electrodes was conducted at �0.3 V in 0.1 M
aqueous HCl that contained 2� 10�3M H2PtCl6 for 10min (EQCM
frequency decrease by�6 kHz,�33mg cm�2 load of Pt, calculated from theSauerbrey equation) [39]. The PDA–GOx–PtNP/platinized Au electrodewas similarly prepared.
For the CEP protocol, the potential scanning from �0.5 to 0.5 V for35 cycles in PBS that contained 30� 10�3
M DA and 3.5mgmL�1 GOx wasconducted to grow a �1.6 kHz film (PDA–GOx), with EQCM monitoring,and then the resultant electrode was rinsed thoroughly. The constructionconditions for the preparation of GaOx-based electrodes were similar tothose in the GOx case, but the concentration of GaOx was 80 U mL�1.
When not in use, the prepared enzyme electrodes were stored in pH 7.0PBS at 4 8C (refrigerator).
Amperometric measurements of the enzyme electrodes were carriedout under solution-stirred conditions, and the response current wasmarked with a change in value between the steady state current aftersubstrate addition and the initial background current without substrate.
Acknowledgements
The authors gratefully acknowledge the National Natural ScienceFoundation of China (20675029, 20335020, 90713018), the Foundationsof the Ministry of Education (MOE) of China and Hunan ProvincialEducation Department (05K009, 05A036), and the State Key Laboratory ofElectroanalytical Chemistry for financial support of this research.Supporting Information is available online at Wiley InterScience or fromthe author.Published online:
Received: November 22, 2008
Revised: December 17, 2008
Published online: April 14, 2009
� 2009 WILEY-VCH Verlag GmbH &
www.afm-journal.de
[1] R. Gangopadhyay, A. De, Chem. Mater. 2000, 12, 608.
[2] C. R. Martin, Chem. Mater. 1996, 8, 1739.
[3] R. A. Vaia, J. F. Maguire, Chem. Mater. 2007, 19, 2736.
[4] E. Granot, E. Katz, B. Basnar, I. Willner, Chem. Mater. 2005, 17, 4600.
[5] A. P. O’Mullane, S. E. Dale, J. V. Macpherson, P. R. Unwin, Chem. Commun.
2004, 1606.
[6] Z. M. Dang, Y. Q. Lin, H. P. Xu, C. Y. Shi, S. T. Li, J. B. Bai, Adv. Funct. Mater.
2008, 18, 1509.
[7] Y. Shen, Y. H. Lin, C. W. Nan, Adv. Funct. Mater. 2007, 17, 2405.
[8] X. D. Cao, C. M. Li, H. F. Bao, Q. L. Bao, H. Dong, Chem. Mater. 2007, 19,
3773.
[9] K. R. Choudhury, Y. Sahoo, P. N. Prasad, Adv. Mater. 2005, 17, 2877.
[10] M. J. Ventura, M. Gu, Adv. Mater. 2008, 20, 1329.
[11] X. Hong, J. Li, M. J. Wang, J. J. Xu, W. Guo, J. H. Li, Y. B. Bai, T. J. Li, Chem.
Mater. 2004, 16, 4022.
[12] M. Shokouhimehr, Y. Piao, J. Kim, Y. Jang, T. Hyeon, 2007, 119, 7169;
Angew. Chem. Int. Ed. 2007, 46, 7039.
[13] S. T. Selvan, J. P. Spatz, H. A. Klok, M. Moller, Adv. Mater. 1998, 10, 132.
[14] S. H. Stelzig, M. Klapper, K. Mullen, Adv. Mater. 2008, 20, 929.
[15] D. Chen, G. Wang, J. H. Li, J. Phys. Chem. C 2007, 111, 2351.
[16] S. Cosnier, Biosens. Bioelectron. 1999, 14, 443.
[17] Z. Q. Gao, G. Binyamin, H. H. Kim, S. C. Barton, Y. C. Zhang, A. Heller,
Angew. Chem. 2002, 114, 838; Angew. Chem. Int. Ed. 2002, 41, 810.
[18] R. A. Potyrailo, Angew. Chem. Int. Ed. 2006, 45, 702.
[19] J. P. Lellouche, G. Senthil, A. Joseph, L. Buzhansky, I. Bruce, E. R.
Bauminger, J. Schlesinger, J. Am. Chem. Soc. 2005, 127, 11 998.
[20] L. H. Xu, Y. H. Zhu, L. H. Tang, X. L. Yang, C. Z. Li, J. Appl. Polym. Sci. 2008,
109, 1802.
[21] Z. Liu, J. Wang, D. Xie, G. Chen, Small 2008, 4, 462.
[22] D. Foxx, E. E. Kalu, Electrochem. Commun. 2007, 9, 584.
[23] Y. H. Zhu, H. Y. Zhu, X. L. Yang, L. H. Xu, C. Z. Li, Electroanalysis 2007, 19,
698.
[24] Y. G. Liu, X. M. Feng, J. M. Shen, J. J. Zhu, W. H. Hou, J. Phys. Chem. B 2008,
112, 9237.
[25] J. Li, X. Q. Lin, Biosens. Bioelectron. 2007, 22, 2898.
[26] W. Zhao, S. X. Sun, J. J. Xu, H. Y. Chen, X. J. Cao, X. H. Guan, Anal. Chem.
2008, 80, 3769.
[27] W. Yan, X. J. Chen, X. H. Li, X. M. Feng, J. J. Zhu, J. Phys. Chem. B 2008, 112,
1275.
[28] A. Pezzella, M. d’Ischia, A. Napolitano, G. Misuraca, G. Prota, J. Med.
Chem. 1997, 40, 2211.
[29] S. Corona-Avendano, G. Alarcon-Angeles, M. T. Ramrez-Silva, G. Rosquete-
Pina, M. Romero-Romo, M. Palomar-Pardave, J. Electroanal. Chem. 2007,
609, 17.
[30] M. D. Hawley, S. V. Tatawawadi, S. Piekarski, R. N. Adarns, J. Am. Chem.
Soc. 1967, 89, 447.
[31] J. W. Mo, B. Ogorevc, Anal. Chem. 2001, 73, 1196.
[32] Y. L. Li, M. L. Liu, C. H. Xiang, Q. J. Xie, S. Z. Yao, Thin Solid Films 2006, 497,
270.
[33] R. Baron, M. Zayats, I. Willner, Anal. Chem. 2005, 77, 1566.
[34] H. Lee, S. M. Dellatore, W. M. Miller, P. B. Messersmith, Science 2007, 318,
426.
[35] J. M. Kinyanjui, D. W. Hatchett, J. A. Smith, M. Josowicz, Chem. Mater.
2004, 16, 3390.
[36] Y. C. Fu, C. Chen, Q. J. Xie, X. H. Xu, C. Zou, Q. M. Zhou, L. Tan, H. Tang, Y.
Y. Zhang, S. Z. Yao, Anal. Chem. 2008, 80, 5829.
[37] X. P. Sun, S. J. Dong, E. K. Wang, J. Am. Chem. Soc. 2005, 127, 13 102.
[38] R. Polsky, R. Gill, L. Kaganovsky, I. Willner, Anal. Chem. 2006, 78, 2268.
[39] Z. N. Zhang, H. Y. Liu, J. Q. Deng, Anal. Chem. 1996, 68, 1632.
[40] Y. Su, Q. Xie, C. Chen, Q. Zhang, M. Ma, S. Yao, Biotechnol. Prog. 2008, 24,
262.
[41] C. Y. Deng, M. R. Li, Q. J. Xie, M. L. Liu, Y. M. Tan, X. H. Xu, S. Z. Yao, Anal.
Chim. Acta 2006, 557, 85.
Co. KGaA, Weinheim Adv. Funct. Mater. 2009, 19, 1784–1791

FULLPA
www.afm-journal.de
[42] M. R. Li, C. Y. Deng, Q. J. Xie, Y. Yang, S. Z. Yao, Electrochim. Acta 2006, 51,
5478.
[43] D. A. Buttry, M. D. Ward, Chem. Rev. 1992, 92, 1355.
[44] G. Sauerbrey, Z. Phys. 1959, 155, 206.
[45] E. Wilhelm, R. Battino, R. J. Wilcock, Chem. Rev. 1977, 77, 219.
Adv. Funct. Mater. 2009, 19, 1784–1791 � 2009 WILEY-VCH Verl
[46] J. Wang, F. Lu, J. Am. Chem. Soc. 1998, 120, 1048.
[47] P. D. Hale, L. I. Boguslavsky, T. Inagaki, H. I. Karan, H. S. Lee, T. A.
Skotheim, Y. Okamoto, Anal. Chem. 1991, 63, 677.
[48] J. Wang, Chem. Rev. 2008, 108, 814.
[49] A. Heller, B. Feldman, Chem. Rev. 2008, 108, 2482.
P
ERag GmbH & Co. KGaA, Weinheim 1791