onderwijs 2014 oncology emergencies - … · disease is beyond the scope of this article. ......
TRANSCRIPT

Oncology emergencies
ICU Fellowship Training Radboudumc

Contents
• AKI
• Tumor lysis syndrome
• Hypercalcemia
• Renal toxicity of chemotherapy

AKI in cancer
• Overall 1-year risk 17.5% and 5-years risk 27%
• Highest 1-year risk in kidney cancer (44%), multiple myeloma (33%) and liver cancer (31.8%)
• Also high risk in acute lymphoma or leukemia
• With RRT 8-week mortality > 60%

Causes of AKI
• Prerenal - volume depletion, NSAID, hypercalcemia
• Renal - lymphomatous infiltration of the kidney, cast nephropathy (multiple myeloma), tumor lysis syndrome
• Postrenal - prostate, bladder, kidney, compression by abdominal/pelvic tumors

several studies, including three randomized controlled trials(RCTs) (30–37). One of these RCTs showed a survival ben-efit and improved renal function in the plasmapheresisgroup; however, patients in the plasmapheresis group re-ceived hemodialysis, whereas those patients in the controlgroup received peritoneal dialysis (31). The largest of theseRCTs randomized 104 patients with myeloma and AKI toconventional therapy with or without five to seven plasmaexchanges over 10 days and found that plasmapheresis didnot significantly reduce death, dialysis dependence, orGFR,30 ml/min per 1.73 m2 (32). Two limitations of thisstudy were that neither renal biopsy data nor quantitationof SFLC was provided. A recent systematic review byGupta et al. (38) concluded that plasmapheresis does notoffer a significant benefit over chemotherapy alone interms of survival, discontinuation of dialysis, or improve-ment in renal function. However, all of these studies were
performed before the introduction of proteasome inhibitors(e.g., bortezomib) and SFLC assays, and the role of plasma-pheresis in the setting of these important developmentsmay warrant additional investigation. In an analysis of 14patients with newly diagnosed or relapsed multiple mye-loma and cast nephropathy treated at the Mayo Clinic,treatment with bortezomib plus plasmapheresis resultedin normalization of serum creatinine levels in 43% of pa-tients, with another 43% of patients achieving .50% reduc-tion in serum creatinine or freedom from hemodialysiswithin 6 months of initiation (33). SFLCs were reducedby a mean of 74.6% on discontinuation of plasmapheresisand 96.5% by 3 months after treatment was initiated. Basedon these data, the work by Cagnoli et al. (34) concluded thatthere may be a role for plasmapheresis combined withbortezomib in reversing AKI secondary to cast nephropa-thy, but additional studies are needed.The recent development of high cutoff (HCO) dialyzers
used with extended hemodialysis sessions offers an alter-native approach to the efficient removal of SFLC (Figure 3)(39). In a study of 19 patients with biopsy-proven castnephropathy treated with conventional chemotherapy regi-mens and extended hemodialysis using the Gambro HCO1100 dialyzer (Gambro Dialysatoren GmbH, Hechingen,Germany), 13 patients experienced an early, sustained reduc-tion in SFLC (median585%) and became dialysis indepen-dent at a median of 27 days (39). The remaining six patientshad an interruption in chemotherapy, five of whom did notrecover renal function. Renal recovery was correlated withimproved survival. One small case control study has alsoshown improved rates of renal recovery and dialysis discon-tinuation with HCO hemodialysis (40). The promising ben-efits of HCO hemodialysis are being tested combined withbortezomib-based chemotherapy in the multicenter RCTEuropean Trial of Free Light Chain Removal by ExtendedHemodialysis in Cast Nephropathy.Monoclonal gammopathies in the absence of a myeloma
diagnosis are also an important cause of AKI, but adiscussion of the spectrum of paraprotein-mediated renaldisease is beyond the scope of this article. Additionalinformation can be found in recent reviews (41,42).Tumor Lysis Syndrome. Considered an oncologic emer-
gency, tumor lysis syndrome (TLS) is a common causeof AKI in the patient with malignancy. TLS occurs whentumor cells release their intracellular contents into the blood-stream, resulting in a constellation of metabolic disturbancesincluding hyperuricemia, hyperkalemia, hyperphosphatemia,and hypocalcemia. These electrolyte abnormalities also putpatients with TLS at risk for cardiac arrhythmias, seizures,and death. Although TLS can occur spontaneously, it ismost often seen as a consequence of treatment of malig-nancy by standard chemotherapy, radiation, corticoste-roids, immunotherapy, monoclonal antibodies, and othertargeted therapies. Malignancies with high tumor burden,rapid cell turnover, and increased sensitivity to chemother-apy (e.g., acute leukemias and high-grade lymphomas) areat highest risk for developing TLS; however, TLS has re-cently been associated with tumors that were previouslythought to be low risk, such as hepatocellular carcinoma,endometrial cancer, non–small cell lung cancer, colon car-cinoma, chronic myelogenous leukemia, and chronic lym-phocytic leukemia (43). The development of highly potent
Figure 1. | Lymphomatous infiltration of the kidney. (A) A 63-year-old man with a history of B cell chronic lymphocytic leukemia not ontherapy developed AKI with a serum creatinine of 3.2 mg/dl andleukocytosis of 87,000. Renal biopsy disclosed diffuse lymphoma-tous infiltration of the kidney characterized bymonomorphic cellularinfiltrates throughout the interstitium visible on hematoxylin andeosin stain. (B) The cellular infiltrate was positive for CD20 (brownstain), confirming the B cell identity of the infiltrate and consistentwith the patient’s B cell leukemia. (C) The patient was startedon fludarabine 1 rituxan therapy, and serum creatinine improved to1.8 mg/dl, where it has remained for 2 years. Fludar, fludarabine;ritux, rituxan. Images courtesy of Helmut Rennke.
1694 Clinical Journal of the American Society of Nephrology
B-cell CLL
Lymphomatous infiltration

Figure 2. | Cast nephropathy. Schematic diagram illustrating the pathophysiology of AKI in cast nephropathy. Free light chains filtered by theglomerulus are taken up by proximal tubular epithelial cells through the cubulin–megalin receptor complex and clathrin-dependent endo-cytosis, where they are metabolized in lysosomes. Excess free light chains overwhelm lysosomal capacity, leading to activation of redoxpathways, increasedNF-kB andmitogen-activated protein kinase expressions, and production of proinflammatory, profibrotic cytokines. Lightchains bind to Tamm–Horsfall protein in the lumen of the distal tubule, where they precipitate and form casts. CCL2, C–C motif chemokine 2;MAPK, mitogen-activated protein kinase; THP, Tamm–Horsfall protein. Modified from ref. 27, with permission.
Figure 3. | Comparison of sieving coefficients between high cutoff (HCO) and high-flux membranes. Clearance of higher molecular weightmolecules, including light chains, is greater with HCO dialyzers compared with conventional high-flux membranes. Reprinted from ref. 69,with permission.
Clin J Am Soc Nephrol 7: 1692–1700, October, 2012 AKI in the Cancer Patient, Lam and Humphreys 1695
Cast nephropathy

Treatment
• Volume expansion (urine alkalinization)
• Antimyeloma agents (bortezomib)
• Plasmapheresis?
• Extended high cut-off hemodialysis

Figure 2. | Cast nephropathy. Schematic diagram illustrating the pathophysiology of AKI in cast nephropathy. Free light chains filtered by theglomerulus are taken up by proximal tubular epithelial cells through the cubulin–megalin receptor complex and clathrin-dependent endo-cytosis, where they are metabolized in lysosomes. Excess free light chains overwhelm lysosomal capacity, leading to activation of redoxpathways, increasedNF-kB andmitogen-activated protein kinase expressions, and production of proinflammatory, profibrotic cytokines. Lightchains bind to Tamm–Horsfall protein in the lumen of the distal tubule, where they precipitate and form casts. CCL2, C–C motif chemokine 2;MAPK, mitogen-activated protein kinase; THP, Tamm–Horsfall protein. Modified from ref. 27, with permission.
Figure 3. | Comparison of sieving coefficients between high cutoff (HCO) and high-flux membranes. Clearance of higher molecular weightmolecules, including light chains, is greater with HCO dialyzers compared with conventional high-flux membranes. Reprinted from ref. 69,with permission.
Clin J Am Soc Nephrol 7: 1692–1700, October, 2012 AKI in the Cancer Patient, Lam and Humphreys 1695
Extended high cut-off hemodialysis

AKI after HCT• Myeloablative allogeneic (50%), nonmyeloablative
allogeneic (30-40%), myeloablative autologous (20%)
chemotherapeutic agents such as flavopiridol (a cyclin-dependent kinase inhibitor used to treat chronic lymphocyticleukemia) has also lead to a markedly increased risk fordeveloping TLS (44). The pathophysiology of AKI in TLSinvolves the formation of crystals comprised of uric acid,calcium phosphate, and/or xanthine, which can lead tointratubular obstruction and inflammation and a reductionin GFR. In addition, hyperuricemia can cause AKI throughcrystal-independent mechanisms, such as renal vasocon-striction, reduced renal blood flow, reactive oxygen species,and inflammation (43). The management of TLS is re-viewed elsewhere (19).
Postrenal Causes of AKIObstruction is an important cause of AKI and should
always be considered in the cancer patient. It ismost commonin cancers of the prostate, bladder, and kidney or secondary toextrinsic compression of the urinary outflow tract from bothprimary and metastatic abdominal or pelvic malignancies.Many renal cell carcinoma patients only have one kidney andtherefore, are susceptible to AKI from unilateral ureteralobstruction. Diagnosis of obstruction is usually establishedradiographically by the presence of hydronephrosis on eitherabdominal ultrasonography or computed tomography, butfalse-negative results can be seen in the setting of hypovolemia,early or partial obstruction, or obstruction caused by ret-roperitoneal fibrosis. Interventions to relieve the obstructioninclude placement of ureteral stents and percutaneous nephro-stomy tubes, which can lead to reversal of renal impairment.
AKI after HCTSince the 1960s, HCT, formerly known as bone marrow
transplantation, has been used to treat a number of ma-lignant and nonmalignant diseases. Although HCT offers apotential cure for several conditions that may be refractoryto chemotherapy, it is associated with a host of organ tox-icities, with AKI being one of the most common seriouscomplications of HCT (14). The incidence and timing ofAKI after HCT vary according to the type of transplant.The original report by Zager (45), which analyzed 272 pa-tients undergoing myeloablative HCT (89% allogeneic and11% autologous), found that 53% of patients developedAKI (defined as doubling of serum creatinine), with one-half of these patients requiring dialysis. This remarkablyhigh incidence of AKI in myeloablative allogeneic HCThas been confirmed in several more recent studies (46–49).In nonmyeloablative allogeneic HCT, which employs aless toxic conditioning regimen and has fewer complica-tions, the incidence of AKI is lower (29%–40.4%) (48,50,51).Myeloablative autologous HCT has the lowest incidenceof AKI (22%) (52,53), a difference that can be attributedto the lack of graft versus host disease (GVHD), the absenceof calcineurin inhibitors, and more rapid engraftment inthis population. Most cases of AKI occur within the first100 days after HCT, with an earlier onset in myeloablative(7–40 days) compared with nonmyeloablative regimens(22–60 days) (14). Overall mortality rates in patients withAKI range from 37% to 46% and are as high as 88% inpatients requiring dialysis (46,54). Importantly, AKI afterHCT predicts the subsequent development of CKD in bothmyeloablative and nonmyeloablative HCT (14).
The causes of AKI after HCT can be divided into thosecauses occurring early after HCT (within the first 30 days) andthose causes occurring later (.3–4 months) (Table 1). Duringthe peritransplant period, AKI is most commonly caused bysepsis, hypotension, and exposure to nephrotoxic agents(methotrexate, amphotericin B, acyclovir, aminoglycosides,angiotensin-converting enzyme inhibitors, intravenouscontrast, and calcineurin inhibitors), which predispose thepatient to acute tubular necrosis (45). In addition, the admin-istration of antibiotics and/or allopurinol can cause acuteinterstitial nephritis. TLS can occur as a result of the condi-tioning regimen, although the incidence of this occurrence islow in this population (45). Hepatic sinusoidal obstructionsyndrome presents early post-HCT with clinical and labora-tory features similar to hepatorenal syndrome. Late-onsetAKI after HCT has a more limited differential diagnosis,and it is usually attributed to thrombotic microangiopathyor calcineurin inhibitor toxicity (45).
Calcineurin Inhibitor ToxicityCalcineurin inhibitors (cyclosporine and tacrolimus) are
widely used for prevention of GVHD in patients undergo-ing allogeneic HCT. Both cyclosporine and tacrolimus, ineither oral or parenteral formulation, can acutely induce renalvasoconstriction and reduce GFR in a dose-dependent man-ner. Calcineurin inhibitor nephrotoxicity plays a more signif-icant role in the development of AKI in nonmyeloablativethan myeloablative HCT, where it has not been associatedwith AKI in multiple studies (55). In addition to their acutenephrotoxicity, calcineurin inhibitors can also cause progres-sive, irreversible CKD and are a risk factor for the devel-opment of HCT-associated thrombotic microangiopathy(55). Routine monitoring of serum creatinine and plasmadrug levels (cyclosporine5150–400 ng/ml; tacrolimus#15ng/ml) (56) is important in determining when appropriatedose reductions are necessary to help preserve renalfunction.
Table 1. Causes of AKI after hematopoietic cell transplant
Early onset (,30 days)SepsisHypotensionHypovolemia (vomiting and diarrhea)Nephrotoxic agentsAcyclovirAllopurinolAmphotericin BAngiotensin-converting enzyme inhibitorsAngiotensin receptor blockersCalcineurin inhibitorsContrast dyeMethotrexateNSAIDs
Tumor lysis syndromeHepatic sinusoidal obstruction syndrome
Late onset (.3 months)Thrombotic microangiopathyCalcineurin inhibitor toxicity
NSAIDS, nonsteroidal anti-inflammatory drugs.
1696 Clinical Journal of the American Society of Nephrology

Calcineurin inhibitor toxicity• Cyclosporine/tacrolimus - prevention of GVHD
• Renal vasoconstriction and dose-dependent GFR↓
• Especially in nonmyeloablative HCT
• Also progressive non-reversible CKD and risk-factor for thrombotic microangiopathy
• Drug levels essential (Cs 150-400 ng/ml, Ts ≤ 15 ng/ml

Vena occlusive disease liver• Hepatomegaly, fluid retention, weight gain and
icterus after high dose conditioning (cyclophosphamide, busulfan, TBI)
• Damage to sinusoidal endothelial cells in zone III resulting in thrombosis and portal HT
• Only after myeloablative HCT (15%)
• AKI in 50% ≈ hepatorenal syndrome - no structural abnormalities - mortality 80% with dialysis
Supportive therapy ± defibrotide (46% CR)

Thrombotic microangiopathy
• Resembles HUS and occurs 20 - 100 D after HCT
• Anemia, thrombocytopenia, AKI, LDH↑, schistocytes, often hypertension
• Often after TBI followed by GVHD/infection
• Supportive therapy - daclizumab substitution of CI - retuximab/defibrotide - plasmapheresis?
Hepatic Sinusoidal Obstruction Syndrome(Veno-Occlusive Disease)Hepatic sinusoidal obstruction syndrome (SOS), previ-
ously known as veno-occlusive disease, is the constellationof tender hepatomegaly, fluid retention, weight gain, andjaundice that occurs after the administration of high-doseconditioning regimens, including cyclophosphamide, bu-sulfan, and/or total body irradiation (57). The pathophys-iology of SOS involves damage to hepatic sinusoidalendothelial cells in zone 3 of the hepatic acinus, whichleads to sinusoidal thrombosis and obstruction and portalhypertension. Although SOS has historically been reportedin 5%–60% of patients, a recent review of studies per-formed between 1979 and 2007 found that the overallmean incidence of SOS was 13.7% (58). SOS occurs morecommonly after myeloablative allogeneic HCT than afterautologous HCT (59), and it is essentially nonexistent withnonmyeloablative regimens. Risk factors for developingSOS include older age, pre-existing liver disease, medications(methotrexate, itraconazole, sirolimus, and norethisterone),and certain conditioning agents (cyclophosphamide andbusulfan) (45,60).AKI develops in approximately 50% of patients with SOS
and is clinically indistinguishable from the hepatorenalsyndrome. Patients initially present with sodium retention,weight gain, peripheral edema, and ascites accompaniedby hepatic dysfunction and hyperbilirubinemia. AKI en-sues 10–16 days post-HCT, with approximately one-half ofpatients requiring dialysis (45). Patients typically have lowblood pressures and are frequently hyponatremic, andmost have persistently low fractional excretion of sodium.Urinalysis shows variable proteinuria and/or hematuria;the urine sediment is often bland but may reveal granularcasts in patients with tubular injury secondary to hypoten-sion or bilirubin toxicity. Renal biopsies and autopsies per-formed in patients with SOS have not shown evidence ofstructural kidney lesions, confirming the notion that AKI islikely hemodynamically mediated (54). Mortality is 37% inthose patients experiencing a doubling of serum creatinineand as high as 84% in those patients requiring dialysis (57).More than 70% patients with SOS will recover sponta-
neously with only supportive therapy, which consists ofmaintaining sodium and water balance, preserving renalblood flow, and treating symptomatic ascites with repeatedparacenteses (60). For patients with severe SOS, thereare no highly effective treatments, although the bestresults have been achieved with defibrotide, a single-stranded oligodeoxyribonucleotide with antithromboticand profibrinolytic properties that has a 46% complete re-sponse rate (61). Infusion of heparin and/or ursodeoxycholicacid administered immediately before induction ther-apy may also be moderately successful as preventivemeasures.
Thrombotic MicroangiopathyThrombotic microangiopathy (TMA) is a common cause
of late-onset AKI in patients who have undergone HCT.Previously known as bone marrow transplant nephropathyor radiation nephropathy, TMA after HCT resembles thehemolytic–uremic syndrome and usually occurs 20–99days post-transplant. The diagnosis of TMA can be chal-lenging, because characteristic features such as anemia,
thrombocytopenia, and renal insufficiency are commonlypresent in the HCT patient population for other reasons,and evidence of schistocytes or elevated serum lactate de-hydrogenase levels is also not entirely reliable (62). Hyper-tension is often present. Urinalysis can be normal or revealvariable proteinuria and/or hematuria, and cellular castsmay be seen on urine sediment. Renal biopsy is rarelyneeded to establish the diagnosis, except when the presen-tation is atypical. Typical histology includes mesangiolysis,basement membrane duplication, glomerular endothelialcell swelling, and tubular injury with interstitial fibrosis(Figure 4) (63).The pathogenesis of TMA after HCT is not well un-
derstood, but damage to renal endothelial cells likelyplays a central role. The conditioning regimen, particularlythe use of total body irradiation, is a primary cause of renalendothelial damage, with post-HCT factors such as GVHD,infections, and medications (such as the calcineurin inhib-itors) playing a later modulatory role (64) Strategies suchas partial shielding of the kidneys, hyperfractionation ofthe radiation dose, and slow radiation administration havebeen proposed to reduce radiation injury (45). However,these approaches run the risk of decreasing the effective-ness of tumor cell eradication. The management of HCT-associated TMA is otherwise largely supportive. Calcineurininhibitors are typically discontinued, although there is nosubstantial evidence that this discontinuation is necessary,especially in patients who require these medications forlife-threatening GVHD. Other oral agents that can beused for the prevention and treatment of GVHD includemycophenolate mofetil and corticosteroids (65). Substitu-tion of calcineurin inhibitors with daclizumab, a human-ized monoclonal antibody to the a-chain of the IL-2receptor, has been shown to improve TMA in patientswith both GVHD and TMA (66). Rituximab, a monoclonal
Figure 4. | Thrombotic microangiopathy after hematopoietic celltransplantation. (A) A 25-year-old woman was diagnosed with high-risk acute myelogenous leukemia, underwent induction chemother-apy, and experienced a slow hematologic recovery. She subsequentlyunderwent allogeneic double-cord blood hematopoietic cell trans-plantation; 6 months later, her serum creatinine rose from 1.2to 2.4 mg/dl in association with new hypertension, thrombocytopenia,and evidence of microangiopathic hemolysis. Kidney biopsy re-vealed diffuse and severe endothelial damage with double contoursand occlusion of capillary lumens (periodic acid–Schiff stain). (B)There is marked endothelial swelling (endotheliosis) with fragmentedredblood cells visible in capillary lumens and thedamagedmesangium(arrows, hematoxylin and eosin stain). (C) Electron microscopyof a single glomerular capillary loop reveals expansion of thesubendothelial space by electron lucent debris (asterisks), narrowingof the capillary lumen, and loss of endothelial fenestrae. Imagescourtesy of Helmut Rennke.
Clin J Am Soc Nephrol 7: 1692–1700, October, 2012 AKI in the Cancer Patient, Lam and Humphreys 1697

Tumor lysis syndrome• Clinical and laboratory sequelae resulting from
rapid release of intracellular contents of dying cancer cells
• Hyperkalemia, hyperphosphatemia, hypocalcemia, hyperuricemia, AKI and death
• After ALL, AML, Burkitt lymphoma and other rapidly growing tumors sensitive to chemotherapy

Laboratory TLS! Clinical TLS
Uric acid ≥ 0.48 mmol/l AKI (creatinine > 1.5 the upper
limit of normal for patient age and sex)
Potassium ≥ 6 mmol/l Cardiac arrhythmia
Phosphorus ≥ 1.49 mmol/l Seizure, tetany, or other symptom from hypocalcemia
Calcium ≤ 1.75 mmol/l
Cairo -Bishop classification
2 out of 4 laboratory criteria within 24 hrs within 3 D before and 7 D after chemotherapy

Uric acid

Uric acid• Impairs kidney function (crystal dependent and -
independent)
• Acid urine pH favors precipitation
• Multiple hemodynamic changes in renal vessels leading to ischemia
• Renal inflammatory injury

Phosphorus and calcium• Primary toxicity of phosphorus is the resulting
hypocalcemia
• Cardiac arrhythmias, seizures, tetany and death
• Calcium-phosphate crystals may precipitate in the renal parenchyma

IncidenceMalignancy! Incidence (%) Risk
Burkitt lymphoma B cell ALL Diffuse large-B cell lymphoma ALL AML (WBC > 75.000/mm3) AML (WBC < 25.000/mm3) Solid tumors
14.9 26.4
6 5.2 - 23
18 1
Unknown
High High
Intermediate Vary by WBC
High Low Low
Other risk factors: tumor burden (bulky lymphatic disease, LDH 2× normal, WBC > 25.000) baseline creatinine > 125 μmol/l (odds 10.7), baseline uric acid > 0.45 mmol/l)

Prophylaxis & treatment
• Volume expansion (3 L/day before chemotherapy)
• Allopurinol prophylaxis
• Rasburicase (urate oxidase) - 0.15 mg/kg (once)
• Dialysis/CRRT
Increase clearance of intracellular contents

Hypercalcemia
• 10 - 30% of all patients with cancer
• Usually with advanced disease and poor prognosis
• Nausea, vomiting, constipation, abdominal pain, anorexia, weight loss, bone pain, polyuria, fatigue, weakness, confusion and coma
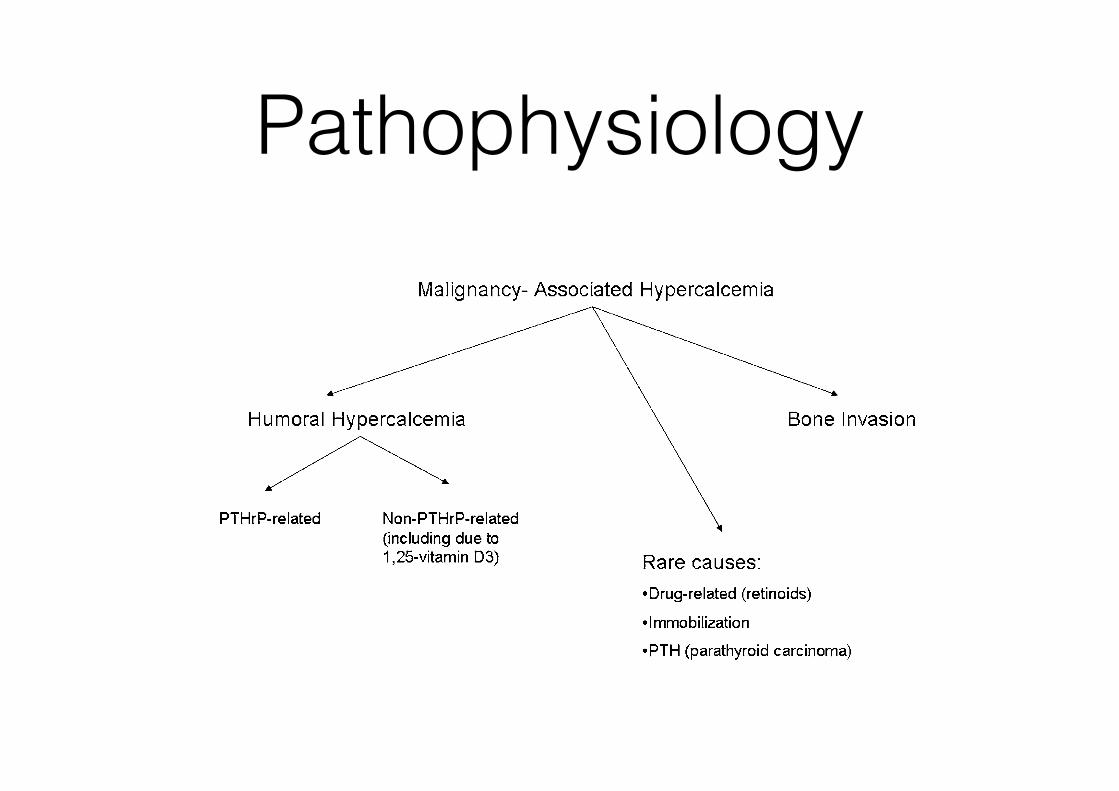
Pathophysiology
receptor activator of nuclear factor k B (RANK) ligand(RANKL) signaling pathway (10–12). RANK is a receptorexpressed on osteoclast precursor cells. The naturally oc-curring ligand, RANKL, is produced by osteoblasts anddrives proliferation and differentiation of the osteoclastsinto mature, multinucleated units. In addition, the osteo-blast cell produces osteoprotegerin (OPG), a decoy recep-tor that binds to and inactivates RANKL.The cellular regulation of RANK, RANKL, and OPG
expression is incompletely understood, but the osteoblast isthe focal point for the integration of endocrine and paracrinesignals that alter bone remodeling. For example, osteoblastcells express estrogen receptors that when occupied withligand can reduce RANKL and increase OPG (13). Directlyrelated to the hypercalcemia of malignancy, osteoblasts alsoexpress the cell surface receptor for parathyroid hormone(PTH) and parathyroid hormone–related hormone (PTHrP),PTH1R (14). Both PTH and PTHrP stimulate PTH1R, which,in turn, increases osteoblast activity and RANKL signalingto the osteoclast. OPG expression may decline as well. Insum, PTH/PTHrP signaling results in an increased boneturnover with a greater increase in bone resorption than for-mation, resulting in a net efflux of calcium and hypercalce-mia from the bone microenvironment.Tumors that commonly produce PTHrP include squa-
mous cell carcinomas of the lung, cervix, and esophagus;certain lymphomas; renal cell carcinoma; and adenocarci-noma of the breast, prostate, and ovary (15–17). In addi-tion, several case reports have documented PTHrPsecretion in other malignancies. Ectopic secretion of PTHsecretion is less common than that of PTHrP. The numberof cases in which tumor-related production of PTH hasbeen verified are few but include pulmonary, thyroid,ovarian, and pancreatic malignancies (18–20).Local osteolysis as the basis for hypercalcemia occurs
most frequently in widely metastatic disease, and the degreeof hypercalcemia correlates with the extent of tumor burden.Local osteolysis is most commonly seen in patients withmetastatic breast and lung cancers (21,22). Probably due to acommon pathophysiology, multiple myeloma, usually
extensive in nature, can present with significant areas of os-teolysis and hypercalcemia (23). Underlying the release ofcalcium from the bone microenvironment is increased oste-oclast activity, probably due to PTHrP and other factors thatcan increase resorption. As a result of the paracrine nature ofthis condition, circulating levels of PTHrP may be “normal”or only slightly above normal, in contrast to levels in patientswith humoral hypercalcemia due to PTHrP.Vitamin D has numerous physiologic actions, including
the enhancement of calcium and phosphate absorptionfrom the intestinal tract. Stored vitamin D (25-[OH]D) is1-hydroxylated in the kidney to the active compound 1,25-(OH)2D, which in turn acts via the vitamin D receptor. PTHactively drives the 1-hydroxylase step at the kidney, andstates of hyperparathyroidism, including ectopic PTH frommalignancies, are associated with elevated 1,25-(OH)2D lev-els. Of note, despite its action through the common PTH1R,PTHrP is a poor stimulus for 1a-hydroxylation comparedwith PTH, and levels of active vitamin D are often low ornormal in patients with humoral hypercalcemia of malig-nancy (24). Important for the understanding of hypercalce-mia, the 1-hydroxylase enzyme is expressed in tissuesoutside of the kidney, including macrophages and someneoplastic tissues. Patients with Hodgkin lymphoma andnon-Hodgkin lymphoma, as well as multiple myeloma,have been described as having vitamin D–mediated hyper-calcemia (25). In these individuals, 1,25-(OH)2D levels, alongwith calcium levels, are high while PTH is suppressed fromthe negative feedback to the parathyroid cells. Similarly,measurements of bone turnover markers are low in vitaminD–mediated hypercalcemia because the reduction in PTHresults in a diminution of osteoblast and osteoclast activity.
Laboratory InvestigationGiven that the malignancy is usually advanced by the
time hypercalcemia develops, a thorough history andphysical examination can often lead to the correct diagnosiswith limited laboratory evaluation. Measurement of intactPTH through an immunoradiometric or immunochemolu-minescent assay will confirm a PTH-independent process.
Figure 1. | Mechanism of malignancy-associated hypercalcemia. PTHrP, parathyroid hormone–related hormone.
Clin J Am Soc Nephrol 7: 1722–1729, October, 2012 Hypercalcemia of Malignancy, Rosner and Dalkin 1723

General therapy
In this case, the PTH levels will be suppressed (often,20 pg/ml) and should prompt further testing to distin-guish among the multiple possible causes in patients withmalignancy. This would include measurement of PTHrP,1,25-(OH)2D levels, serum and urine protein electrophoresis,assessment of serum free light chains, and possibly imagingstudies (such as a skeletal survey). Less common causes ofhypercalcemia in malignancy include drugs (e.g., retinoicacid [26]) and, rarely, parathyroid carcinomas producingexcessive PTH.
Therapy for Malignancy-Associated HypercalcemiaPatients presenting with acute and symptomatic hyper-
calcemia require prompt therapy that rapidly reduces theserum calcium level, restores the GFR, and leads to longer-term normalization of the serum calcium level. An algo-rithmic approach is shown in Figure 2.
Intravenous Fluid and DiureticsHypercalcemia leads to a decrease in the GFR through a
combination of the natriuretic effects of high serum levelsof calcium and renal vasoconstriction (27). In fact, polyuriais the most common renal manifestation of hypercalcemia,and it is thought that the impaired urinary concentratingability is due to activation of the calcium-sensing receptorin the thick ascending limb of the loop of Henle (28). Ac-tivation of the calcium-sensing receptor leads to decreasedresorption of sodium and chloride in the loop of Henle,resulting in decreased countercurrent multiplication anddecreased ability to concentrate the urine. In addition, ac-tivation of the calcium-sensing receptor in the collectingduct blunts the response of this segment to the actions ofarginine vasopressin (29). Thus, patients presenting withhypercalcemia are usually profoundly volume depleted.Furthermore, patients usually will present with poor oralintake and may have had nausea and vomiting that furtherworsen the volume depletion. The decrease in GFR leads toimpaired calcium clearance and, thus, restoration of extra-cellular fluid volume, and GFR is a key goal of therapy.Thus, initial therapy should begin with intravenous (IV)
volume expansion with 0.9% saline at 200–500 ml per hour.
Volume expansion will lead to an increase in GFR and anincrease in urine calcium excretion. The goal should be es-tablishing an adequate urine output (.75 ml per hour). Inmilder cases of hypercalcemia, this therapy can be sufficientto restore normal calcium levels (30). However, the effect ofrepleting the extracellular volume is transient, and other ther-apies are required. Careful monitoring of the patient’s vitalsigns and laboratory values must occur during the acutephase of volume repletion, along with vigilance for evidenceof volume overload. Hypernatremia may be seen during thisphase of therapy because of hypercalcemia-induced nephro-genic diabetes insipidus and may require changing from iso-tonic to hypotonic fluid therapy (31).A common practice is to add a loop diuretic to saline
therapy in an effort to increase urine calcium excretion(forced saline diuresis). LeGrand and colleagues reviewedthe evidence-base for this approach (32). By inhibiting thesodium-potassium-chloride (NKCC2) transporter in thethick ascending limb of the loop of Henle, a loop diureticdecreases the lumen positive charge that normally drivescalcium reabsorption; thus, a loop diuretic increases urinecalcium excretion and theoretically should be useful intherapy for hypercalcemia (33,34). However, LeGrandet al. could find only nine articles that documented theuse of furosemide in the treatment of hypercalcemia, thelast of which was published in 1983. These studiescomprised a total of 37 patients. The average dosage offurosemide was 1120 mg given over 24 hours, and serumcalcium normalized in 14 of 39 episodes of hypercalcemia;however, normalization was rapid (,12 hours) in onlytwo cases. Lower dosages of furosemide (40–60 mg/d)did not achieve normalization of the serum calcium.Most important, monitoring in these patients was inten-sive and included aggressive replacement of hourly urineoutput losses, and secondary electrolyte disorders, such ashypernatremia, hypophosphatemia, hypomagnesemia,and metabolic acidosis, were seen. Thus, the routine useof loop diuretics in the therapy for hypercalcemia cannotbe recommended, and their use should be restricted topatients who develop fluid overload while receiving ag-gressive volume resuscitation.
Figure 2. | Treatment algorithm for malignancy-associated hypercalcemia. IV, intravenous.
1724 Clinical Journal of the American Society of Nephrology

Therapy• Patients are usually profoundly dehydrated due to
nephrogenic DI and poor oral intake
• Therefore restoration of extracellular fluid volume and GFR is essential - volume expansion with NaCl 0.9% 250 - 500 mL/h (UP > 75 mL/h)
• Diuretics only indicated in case of volume overload
• Intravenous biphospohonates (decrease bone reabsorption) - pamidronate 60 - 90 mg over 2-3 hrs

Other therapies• Corticosteroids - malignancies with overproduction
of calcitriol ((non)-Hodgkin) - hydrocortison 200 - 300 mg/d for 3 - 5 days
• Denosumab (monoclonal AB against RANKL - osteoclast activator) ??

that regulate drug carrier proteins, which can impair drugexcretion and induce nephrotoxicity by increasing intracel-lular drug concentrations (13,15).Last, the renal handling of drugs is another risk factor for
the development of nephrotoxicity. The kidney is exposedto considerable drug concentrations based on the high renalblood flow rate—approximately 25% of cardiac output. Sig-nificant drug uptake occurs in the proximal tubular cellsthrough both apical uptake and basolateral transport (16,17).Trafficking of these agents through tubular cells explains, inpart, their nephrotoxicity. The high metabolic rate and hyp-oxic environment of loop of Henle and medullary collectingduct cells impart nephrotoxic drug risk (18,19). Metabolism
of drugs by several enzyme systems present in the kidneyfavors toxic metabolite and reactive oxygen species formation.Renal injury may occur, because drug byproducts cause harmthrough lipid peroxidation, protein damage, nucleic acidalkylation or oxidation, and DNA strand breaks (18–20).
Classification of Chemotherapy-Associated RenalLesionsThere are a number of ways that one can approach
classifying the kidney lesions caused by the various chemo-therapeutic agents. For example, one could categorize thechemotherapy-related kidney lesions based on the nephronsites primarily affected by the drug. Agents that injure therenal vasculature, glomerulus, proximal and distal tubularsegments, and collecting ducts are described (Table 2), recog-nizing that all nephrotoxic drugs cannot possibly be covered.
Renal Vasculature: Thrombotic MicroangiopathyChemotherapeutic agents, such as bevacizumab and
gemcitabine, can injure the renal vasculature and causethrombotic microangiopathy (TMA). TMA presents clinicallyas microangiopathic hemolytic anemia, thrombocytopenia,hypertension, and AKI with hematuria and proteinuria,although renal-limited TMA does occur.
BevacizumabRecognition that tumor growth was highly dependent on
pathologic angiogenesis induced by local production ofvascular endothelial growth factor (VEGF) paved the wayfor the development of drugs targeting this pathway (21).This pathway was a logical point of attack to supplementother tumor-directed therapies—and turned out to be a ben-eficial addition to the therapeutic armamentarium. Althoughthere are numerous drugs that target VEGF effects, the anti-VEGF antibody bevacizumab will be the focus of discussion,recognizing that there are differences among the agents.VEGF importantly regulates vasculogenesis and angio-
genesis during development and in disease through reg-ulation of vascular permeability, endothelial cell migration,proliferation, and survival (21). This regulation raises thepossibility that these drugs may be associated with ad-verse effects. In fact, this finding is the case, because anumber of adverse systemic end-organ effects have beendescribed, including kidney injury. This finding is not sur-prising, because VEGF is produced by renal visceral epi-thelial cells and binds to VEGF receptors located onglomerular endothelium and mesangium, as well as peri-tubular capillaries (21). Local VEGF production maintainsnormal functioning of all of these cells, including injuryrepair and cell turnover. Importantly, there is crosstalkbetween the glomerular endothelium and epithelium,maintaining the integrity of the filtration barrier.The most important renal effects described with anti-
angiogenesis therapy are new or worsened hypertensionand kidney-specific injury, including proteinuria and AKI.Importantly, the development of hypertension in patientspredicts a better tumor response to therapy (22), and itshould prompt clinicians to continue therapy and controlBP with antihypertensive agents rather than discontinuingantiangiogenic therapy. Animal experiments documented a
Table 1. Risk factors for chemotherapy-inducednephrotoxicity
Tumor-related kidney effectsdirect renal involvementmyeloma-related kidney injuryrenal infiltration (lymphoma and leukemia)urinary obstructionneoplasia-associated glomerulopathies
indirect renal involvementtrue volume depletion (N/V, diarrhea, andoverdiuresis)
effective volume depletion (cardiomyopathy,malignant ascites, and pleural effusions)
metabolic effects (hyperuricemia and hypercalcemia)Innate drug toxicityhigh-dose drug exposure and prolonged course oftherapy
insoluble drug or metabolite form crystals withinintratubular lumens
potent direct nephrotoxic effects of the drug or toxindrug combinations enhance nephrotoxicityNSAIDs, aminoglycosides, and radiocontrast
Patient factorsolder ageunderlying AKI or CKDimmune response genesincreased allergic reactions to drugs
pharmacogenetics favoring drug/toxin toxicitygene mutations in hepatic and renal CYP450enzyme systems
gene mutations in transport proteins and renaltransporters
Renal drug handlinghigh blood (and drug) delivery rate to the kidneysproximal tubular uptake of toxinsapical tubular uptake by endocytosisor another pathway
basolateral tubular transport through OATand OCT pathways
relatively hypoxic renal environmenthigh metabolic rate of tubular cells in the loop of Henleincreased drug/toxin concentration in renal medullaand interstitium
biotransformation of substances to ROS causingoxidative stress
N/V, nausea/vomiting; NSAIDs, nonsteroidal anti-inflammatory drugs; OAT, organic anion transporter; OCT,organic cation transporters; ROS, reactive oxygen species.
1714 Clinical Journal of the American Society of Nephrology
Renal toxicity of chemotherapy
two- to threefold increase in proteinuria in mice injectedwith a single dose of anti-VEGF antibody (23). Renal his-topathology revealed glomerular endothelial cell swelling,vacuolization, and detachment, as well as disruption of ep-ithelial cell slit diaphragms. Immunohistochemistry alsoshowed downregulation of nephrin, which was partiallyrestored with administration of recombinant VEGF. Therenal effects of bevacizumab therapy in six patients withvarious malignancies were described in detail (24). Renalfindings developed within 3–17 months of drug exposure:proteinuria occurred in all patients, with five patients hav-ing at least 1 g/d and two patients having nephrotic-levelproteinuria. Hypertension and AKI developed in 50% ofpatients, and all had TMA on kidney histology (Figure 1).Importantly, proteinuria, hypertension, and AKI generallyimproved on withdrawal of bevacizumab.Although a number of renal lesions have been described
with the antiangiogenesis drugs, the predominant histo-pathology is TMA. Other lesions described on kidneybiopsy include focal segmental glomerulosclerosis (FSGS),mebranoproliferative GN, glomerular endotheliosis,cryoglobulinemic GN, nonspecific immune complex GN,and acute interstitial nephritis (21,24,25).
GemcitabineGemcitabine is a cell cycle–specific pyrimidine antago-
nist that is an effective therapy for certain malignancies,primarily carcinomas of the lung, pancreas, bladder, andbreast. Unfortunately, as with other chemotherapeuticagents, it is complicated by kidney injury. Numerouscase reports and case series have documented AKI fromgemcitabine, primarily from TMA. In addition, hyperten-sion, microangiopathic hemolytic anemia, and ischemicskin lesion may be present. A recent case series of 29 pa-tients treated with gemcitabine described the various clin-ical renal manifestations (26). All patients developed AKI;TMA was seen in four patients who underwent kidneybiopsy. New or worsening hypertension occurred in 26of 29 patients, whereas edema (21/29) and congestiveheart failure (7/29) also complicated gemcitabine therapy.Classic systemic TMA occurred in all patients and wasmanifested by anemia, thrombocytopenia, and increasedlactate dehydrogenase levels. Suppressed haptoglobin lev-els (23/26) and schistocytes on peripheral smear (21/24)were also noted. Urinalysis revealed hematuria/proteinuria(27/29) and red blood cell casts (n58).Gemcitabine-associated TMA is relatively rare, with the
major risk factors being previous therapy with mitomycin-Cand total drug dose. Often, it is impossible to predict whowilldevelop this complication, although new or worsened hy-pertension may precede other clinical manifestations of TMA.Unfortunately, therapy is generally limited and mainly sup-portive, consisting of drug discontinuation, antihypertensivemedications, and dialysis when indicated. Plasmapheresis hasbeen used with minimal or no success. Renal outcomes arehighly variable. In the largest cases series (26), 19 patients hadfull or partial renal recovery, whereas 3 patients developedCKD and 7 patients had dialysis-requiring ESRD.
Glomerulus: PodocytopathyIFNIFN is a glycoprotein synthesized and released by
leukocytes, fibroblasts, T cells, and natural killer cells in
Table 2. Kidney injury associated with chemotherapeuticagents
Renal vasculaturehemodynamic AKI (capillary leak syndrome)IL-2, denileukin diftitox
thrombotic microangiopathyantiangiogenesis drugs (bevacizumab and tyrosinekinase inhibitors)
gemcitabine and cisplatinmitomycin C and IFN
Glomeruliminimal change diseaseIFNpamidronate
focal segmental glomerulosclerosisIFNpamidronatezoledronate (rare)
Tubulointerstitiumacute tubular necrosisplatinums, zoledronate, ifosfamide, and mithramycinpentostatin, imatinib, diaziquone, and pemetrexed
tubulopathiesFanconi syndromecisplatin, ifosfamide, and azacitadine,diaziquone, imatinib, and pemetrexed
salt wastingcisplatin and azacitadine
magnesium wastingcisplatin, cetuximab, and panitumumab
nephrogenic diabetes insipiduscisplatin, ifosfamide, and pemetrexed
syndrome of inappropriate antidiuresiscyclophosphamide and vincristine
acute interstitial nephritissorafenib and sunitinib
crystal nephropathymethotrexate
Figure 1. | A glomerulus exhibits mesangiolysis, endothelial de-nudation, red blood cell congestion, and glomerular basement mem-brane duplication in this example of thrombotic microangiopathy.(Jones methenamine silver stain; original magnification, 3600.) Cour-tesy of Glen S. Markowitz.
Clin J Am Soc Nephrol 7: 1713–1721, October, 2012 Nephrotoxicity of Chemotherapy, Perazella 1715
Renal vasculature - TMA bevacuzimap - gemcitabine
Glomerulus - FSGS interferon-α
response to pathogens, such as viruses, parasites, and bac-teria, as well as tumor cells. It is a protective defense thatallows communication between cells to eradicate infectionor malignant cells. In general, IFN-a and -b reduce viralreplication and protein synthesis in neighboring cells,whereas IFN-g activates macrophage and MHC expression(27,28). Based on these characteristics, exogenous IFN has anumber of therapeutic uses. The most commonly usedagent is IFN-a, which is used to treat hepatitis C and Bviruses and various malignancies. IFN-b is used to treatmultiple sclerosis, whereas IFN-g was studied as a treatmentfor chronic granulomatous disease but has been abandoned.Chronic IFN therapy is associated with clinical renal disease,
which seems to be associated primarily with podocyte injury(27,28). Based on published cases, minimal change diseasehas been described with both IFN-a (n56) and -b (n52). Thelesion developed anywhere from 5 days to 22 months aftertherapy (27). Nephrotic syndrome with urinary protein lev-els of 2.3–28 g/d occurred in all patients, whereas AKI com-plicated the course of two patients. In follow-up rangingfrom 53 days to 12 months, complete remission was notedin all patients, with discontinuation of IFN and steroid ther-apy in three patients (27,28).FSGS constitutes another form of podocytopathy that can
complicate IFN therapy. The lesion FSGS-not otherwisespecified has been described in 10 patients treated with IFNtherapy (IFN-a in 9 patients and -g in 1 patient) (27,28). Inthese cases, IFN therapy ranged from 19 days to 20 monthsand was associated with nephrotic syndrome in all 10 patients,with proteinuria of 6.3–42 g/d and AKI in 8 patients. Withfollow-up ranging from 1 to 16 months in 9 of 10 patients,complete or partial remission was noted in 4 of 9 patientswith discontinuation of IFN. Six patients received steroids,of which only two patients benefited with remission.Collapsing FSGS (Figure 2) also complicates IFN ther-
apy. In 14 patients with this lesion, IFN-a was the causa-tive agent in 9 patients, whereas IFN-b and -g therapy wasused in 3 and 2 patients, respectively (27,28). IFN exposureranged from 1 to 48 months; nephrotic syndrome was
present in 12 of 13 patients, with proteinuria rangingfrom 1.9 to 27 g/d. AKI occurred in 11 patients. Urinesediment was bland in nearly one-half of the patients,whereas red and white blood cells were seen in five andone patients, respectively. Remission was inconsistent andincomplete in most patients after IFN discontinuation.With follow-up in 10 patients, which ranged from 2 to54 months, complete remission was noted in 1 patientand partial remission was noted in 3 patients, whereassome improvement in proteinuria and kidney functionwas described in 5 patients. Of these 10 patients, 8 patientsreceived steroids, and 1 patient received cyclophospha-mide. Thus, although IFN discontinuation sometimeshelps, it is not always associated with remission. Steroidsseem unhelpful, especially in FSGS.The mechanism of podocyte injury with IFN is incom-
pletely understood. A number of putative mechanismshave been put forward (27). A direct IFN effect on thepodocyte is possible through receptor binding and activa-tion that promotes two potential injurious effects: (1) al-tered cellular proliferation and metabolism of thepododyte and (2) increased podocyte oxidative capacityand increased MHC class II antigen expression. IndirectIFN effects on the podocyte may also contribute to thedevelopment of FSGS. IFNs activate adaptive immunemechanisms that increase macrophage activation—an ex-ample is hemophagocytic syndrome, which is associatedwith collapsing FSGS. Viral diseases such as HIV and par-vovirus B19, which increase IFN production, are also as-sociated with collapsing FSGS. Finally, IFN may enhancesynthesis of pathogenic cytokines such as IL-6 and -13,which are permeability factors in FSGS and minimalchange disease.
Tubules: Acute Tubular InjuryCisplatinCisplatin is a platinum compound that is an effective
therapy for many carcinomas and sarcomas, as well aslymphomas. Its major adverse effect is nephrotoxicity,although ototoxicity also occurs. Both are dose-relatedtoxicities, causing apoptosis and necrosis of cells (29–32).Cisplatin injures multiple renal compartments, includingblood vessels, glomeruli, and most commonly, the tubules(29). Nephrotoxicity is generally reversible, but it can bepermanent. Tubular injury as manifested by AKI and tu-bular dysfunction syndromes will be described.Cisplatin’s mechanism of nephrotoxicity is related to its
drug characteristics, its renal handling, and the kidney re-sponse to the cisplatin molecule (29–32). Chloride at thecis-position of the molecule is one such factor that pro-motes kidney injury, whereas the pathway of excretionthrough the cell (in through organic anion transporter 1and out through efflux transporters) potentially increasesintracellular concentrations (Figure 3). After it is inside thetubular cell, a number of intracellular injury pathways oc-cur. These pathways include caspase activation, cyclin-dependent kinases, mitogen-activated protein kinaseactivation, and p53 signaling. In addition, cellular injuryalso develops from inflammation and oxidative stress,whereas vascular injury and decreased GFR with ischemicinjury also occur (29–32). These pathways of injury result
Figure 2. | A glomerulus exhibits global wrinkling and retraction ofthe glomerular basement membrane and diffuse swelling and hy-perplasia of overlying visceral epithelial cells in this example ofcollapsing focal segmental glomerulosclerosis. (Jones methenaminesilver stain; original magnification, 3600.) Courtesy of Glen S.Markowitz.
1716 Clinical Journal of the American Society of Nephrology
in renal tubular cell apoptosis and necrosis—resulting inclinical AKI and/or a tubulopathy.Tubular injury without AKI also complicates cisplatin
therapy and manifests as a number of tubulopathies, whichinclude isolated proximal tubulopathy (proteinuria andphosphate wasting) or full-blown Fanconi syndrome (2,29).Sodium wasting is another consequence of cisplatin therapy,which can be associated with hypovolemia, orthostasis,and prerenal AKI. In the distal nephron, cisplatin can im-pair reabsorption of magnesium, causing refractory hypo-magnesemia. Finally, water absorption in the collectingduct can be disturbed, resulting in a form of nephrogenicdiabetes insipidus.AKI is a dose-related complication of cisplatin—it can be a
functional decline in GFR or caused by TMA, but mostcommonly, it is a result of acute tubular injury/necrosis.Although AKI can recover, renal outcomes such as pro-
gressive CKD from chronic tubulointerstitial fibrosis andirreversible chronic tubulopathies may result (2,29).A focus of care for patients receiving cisplatin is pre-
vention of kidney injury (2,29). Forced diuresis with intra-venous normal saline or hypertonic saline is used tocounteract the toxic effect of chloride on the cis-positionof the molecule. The addition of mannitol to induce aforced diuresis is sometimes used, but evidence of benefitis lacking; in some cases, this approach may cause wors-ened kidney function (33). Amifostine is a glutathione an-alog taken up by normal cells that blunts cisplatin-inducedcellular injury. It is, however, complicated by nausea andvomiting. A number of other agents have been used toreduce nephrotoxicity, such as sodium thiosulfate,
nucleophilic sulfur thiols, neurotrophins, phosphonic acid,melanocortins, and free oxygen radical scavengers, but theirutility is unclear. In high-risk patients, other platinums suchas carboplatin and oxalaplatin are used based on their lessnephrotoxic profile compared with cisplatin. Two potentialexplanations for reduced nephrotoxicity exist. Neither ofthese molecules is transported by organic cation transporter2 (OCT-2), thereby reducing proximal tubular intracellularconcentrations (29–32). In addition, the chloride at cis-positionin cisplatin is replaced by carboxylate and cyclobutane in car-boplatin and oxalaplatin, respectively, which may further re-duce toxicity (29–32).Treatment of toxic renal manifestations is primarily
supportive (2,28). There are no effective therapies to reverseAKI or tubular dysfunction. Dialysis is reserved for ad-vanced AKI as manifested by uremia, metabolic disturban-ces, and hypervolemia. There is no role for dialytic removalof cisplatin. Maneuvers to correct hypovolemia from saltwasting (intravenous normal saline or oral sodium chloride)and address symptomatic hypomagnesemia with intrave-nous and/or oral magnesium are required. Fanconi syn-drome is notoriously difficult to treat.
IfosfamideIfosfamide is an alkylating agent used for certain cancers,
including sarcomas, testicular cancer, and some forms oflymphoma. Ifosfamide’s major adverse effect is kidney in-jury. Nephrotoxic manifestations include tubulopathiessuch as proximal tubular injury or Fanconi syndromeand nephrogenic diabetes insipidus; additionally, AKI is oftenreversible but can be permanent (2,34,35). Histopathology
Figure 3. | Cisplatin (Cis) nephrotoxicity is, in part, related to its uptake by proximal tubular cells. Cis enters cells through organic cationtransporters (OCTs), andwhen it accumulateswithin cells, it causes cell injury throughmultiplemechanisms. Apoptosis and necrosis of tubularcells result and cause clinical AKI and tubulopathy. CDKs, cyclin-dependent kinases; MAPK, mitogen-activated protein kinase; MRP, multidrug-resistant protein; NaDC, sodium dicarboxylate; OAT, organic anion transporter; P53, protein 53; Pgp, P glycoprotein; ROS, reactive oxygenspecies.
Clin J Am Soc Nephrol 7: 1713–1721, October, 2012 Nephrotoxicity of Chemotherapy, Perazella 1717
Tubulus - acute tubular injury cisplatin - ifosfamide
pemetrexed
Sodium loss - water loss - magnesium/phosphate loss

Tubules - magnesium wasting
including colorectal, head/neck, breast, and lung cancers.EGF overexpression present in these malignancies reducesapoptosis and enhances tumor cell growth. Cetuximab hasa 10-fold greater affinity for EGFR than natural ligand, makingit an effective targeted therapy in these cancers. However, oneof the drug’s adverse effects is hypomagnesemia (37–41).Cetuximab-induced hypomagnesemia results from a renal
leak of magnesium. Magnesium reabsorption in the distalconvoluted tubule is, in part, dependent on EGF binding itsreceptor on the basolateral membrane (37,38,41). Activationof the EGFR sets in motion intracellular signals that stimu-late the movement of the cation channel transient receptorpotential M6 into the apical membrane, which facilitates thereabsorption of magnesium from the urinary space into thecell (37,38,41). Cetuximab competitively inhibits EGF bind-ing to its receptor (Figure 5), thereby blunting the placementof transient receptor potential M6 into the apical membraneand causing renal magnesium wasting (37,38,41).The incidence of hypomagnesemia with cetuximab in
initial colorectal cancer trials was only 1.8%–5.8% (37,38).However, a higher incidence was noted when magnesiumlevels were measured more rigorously. More than one-halfof patients develop hypomagnesemia with cetuximab, andnearly 100% of patients have some decline in serum mag-nesium concentrations (37,38,41). In fact, a meta-analysisof randomized controlled trials of cetuximab versus othertherapies showed an odds ratio of 4.7 for all-grade hypo-magnesemia and 5.3 for grade 3/4 hypomagnesemia (42).In addition, hypokalemia and hypocalcemia occur withsevere hypomagnesemia. Risk factors for hypomagnesemiainclude duration of cetuximab therapy, older age, and base-line magnesium concentration. Panitumumab is also com-plicated by hypomagnesemia (36%) but of less severegrade, because only 3% developed grade 3/4 hypomag-nesemia (1,2).
Treatment of hypomagnesemia requires intravenousrepletion, because oral magnesium is ineffective and oftencomplicated by diarrhea. Along with magnesium sup-plementation, calcium and potassium repletion are alsocommonly required. In general, renal magnesium wastingimproves and eventually resolves approximately 4–6 weeksafter discontinuation of cetuximab.
Tubules: Crystal NephropathyMethotrexateThe dihydrofolate reductase inhibitor methotrexate is
widely used, especially in high dose, to treat malignanciessuch as high-grade lymphomas. Nephrotoxicity is a knowncomplication of high-dose therapy (1–12 g/m2) but rarelyoccurs with long-term conventional dosing (1,2). The in-cidence is highly variable depending on the patient’s riskfactors and the appropriate employment of preventivemeasures, such as intravenous fluids (1,2,43).Methotrexate and its major metabolite, 7-OH metho-
trexate, are filtered by the glomerulus and secreted intothe urinary space by proximal tubules. AKI is primarilythe result of acute tubular injury from precipitation ofmethotrexate/7-OH methotrexate in distal tubular lumens(2,43). However, tubular apoptosis/necrosis may also de-velop from oxygen radicals associated with decreasedadenosine deaminase activity (44). AKI incidence, whendefined as grade .2 nephrotoxicity, ranges from 1.8% to12% (2,43).Risk factors for nephrotoxicity include intravascular
volume depletion with sluggish urinary flow, acid urinepH, and underlying kidney disease (GFR,60 ml/min)(2,42). Based on these factors, prevention is focused onvolume repletion before/during drug infusion, appropri-ate drug dosing, and alkalinization of the urine (pH.7.1).
Figure 5. | Cetuximab (C) is an EGF receptor (EGFR) antibody that causes renal magnesium wasting by competing with EGF for its receptor.Normally, EGF binds its receptor (EGFR) and stimulates magnesium reabsorption in the distal convoluted cell. EGFR activation is associatedwith magnesium absorption through transient receptor potential M6 (TRPM6) in the apical membrane. NCC, sodium chloride cotransporter.
Clin J Am Soc Nephrol 7: 1713–1721, October, 2012 Nephrotoxicity of Chemotherapy, Perazella 1719
> 50% hypomagnesemia - but also hypokalemia

Outcome ICU admission Solid tumors
Mor
talit
y (%
)
0
15
30
45
60
All (N=717) Scheduled surgery (N=381) Emergency surgery (N=79) Medical (N=257)
ICU mortality Hospital mortality
Soares M. Crit Care Med 2010;38:9-15

Mortality prediction
critical care medicine score. They ob-served a hospital mortality rate of 42%.Over the last decade, advances in oncol-ogy and intensive care coupled with animproved selection of patients likely tobenefit from ICU management havetranslated into better survival rates.Nonetheless, most of the studies demon-strating improvement in patients’ out-comes were conducted in single centersand specialized hemato-oncologic ICUs.Very recently, Taccone et al evaluated 473patients with solid tumors and hemato-logic malignancies (15% of the patients)included in the SOAP study over a 2-wkperiod (3). They found a global hospitalmortality rate of 29% and the main out-come predictors were higher SAPS II
scores and the need for MV. However, asthat study was not specifically designed toevaluate patients with cancer, the lack ofinformation on cancer-related character-istics other than the group of malignan-cies (solid or hematologic) imposes limi-tations to the interpretation of its results.A new multicenter study in this popula-tion is of critical importance.
We found a relatively low global hos-pital mortality rate of 30%, but half of thepatients were admitted for postoperativecare after scheduled surgeries with anexpectedly lower mortality rate (11%). Al-though the outcomes may vary depend-ing on case-mix, hospital mortality rates(53%) described in medical and emer-gency surgical patients are similar to
those reported in more recent single-center studies on critically ill patientswith cancer (range ! 44%–63%) (4, 5,10, 11, 22–28). Adjusting for other co-variates including the type of admission,mortality was more dependent on the se-verity of organ failures at the time of ICUadmission, poor PS, need of MV, and ac-tive disease. Although these outcome pre-dictors have been reported in previousstudies (3, 4, 7, 11, 13, 24, 25, 27, 28), thepresent study confirms that the type ofmalignancy per se and the presence ofneutropenia do not mainly influence apatient’s short-term mortality. However,the relatively low number of patients withbone marrow transplant imposed limita-tions to evaluate appropriately their con-tribution to the patients’ outcomes.
The present study has many positivefeatures including a large sample size anda multicenter and prospective design. Byevaluating a contemporary cohort of pa-tients from institutions with differentcharacteristics, it provides a descriptionof the current ICU admission practicesand outcomes for these patients. Never-theless, the present study has also poten-tial limitations. Although almost thirtyICUs have participated, the study was car-ried out in a single country and somecaution is needed with the extrapolationof our results because possible selectionbiases concerning different standards ofcare cannot be excluded. On the otherhand, as patients from nonspecializedICUs were also evaluated, the findings ofthe present study may be more represen-tative of the practice in general hospitalsand therefore more suitable to generali-zation. Furthermore, the present studydoes not represent an audit of BrazilianICUs regarding the care provided to pa-tients with cancer. In general, the fre-quencies of EOL decisions were relativelylower than those reported in the litera-ture (6, 9, 10), but half of the patientswere admitted after a scheduled surgicalprocedure. If only medical and scheduledsurgical patients are considered, the fre-quency of EOL decisions is similar toother reports (6, 9, 10, 22, 25). However,as there is not yet a legal regulation onEOL care in Brazil (29), and only EOLdecisions shared by the ICU team, on-cologists, and patient’s relatives (which isgenerally the rule in Brazil in ICUs) wereconsidered, possible underestimationcannot be completely ruled out.
In conclusion, one in five ICU admis-sions in the participating centers was inpatients with malignancies. This large
Table 3. Multivariate analysis of predictors of hospital mortality in all patients admitted to the ICUs(n ! 717)
Variables Coefficients Odds Ratio (95% CI) p
Type of admissionScheduled surgical 1.00Emergency surgical 0.900 2.46 (1.28–4.73) .007Medical 1.733 5.66 (3.43–9.33) ".001
Hospital stay before ICU admission#Ln (days $ 0.5)%
0.165 1.18 (1.01–1.37) .033
SOFA on the 1st day of ICU (points) 0.224 1.25 (1.17–1.34) ".001Performance status
0–1 1.002–4 1.223 3.40 (2.19–5.26) ".001
Cancer statusControlled/remission 1.00Active-newly diagnosed 1.010 2.75 (1.19–6.32) .018Active-recurrence/progression 0.803 2.23 (0.96–5.20) .063
Mechanical ventilation 0.883 2.42 (1.51–3.87) ".001Constant &5.518
ICU, intensive care unit; SOFA, Sequential Organ Failure Assessment; CI, confidence interval.Area under receiver operating characteristic curve ! 0.88 (95% CI, 0.86–0.91); Hosmer-
Lemeshow goodness-of-fit ('2 ! 4.305; p ! .829).
Table 4. Multivariate analysis of predictors of hospital mortality in medical and emergency surgicalpatients (n ! 336)
Variables Coefficients Odds Ratio (95% CI) p
Type of admissionEmergency surgical 1.00Medical 0.825 2.28 (1.21–4.32) .011
Recent weight loss (10% 0.829 2.29 (1.15–4.55) .018SOFA on the 1st day of ICU (points) 0.196 1.22 (1.12–1.33) ".001Performance status
0–1 1.002–4 1.277 3.59 (2.08–6.18) ".001
Cancer statusControlled/remission 1.00Active-newly diagnosed 1.166 3.21 (1.33–7.76) .010Active-recurrence/progression 0.901 2.46 (1.01–6..01) .048
Mechanical ventilation 0.976 2.66 (1.50–4.71) .001Constant &4.544
ICU, intensive care unit; SOFA, Sequential Organ Failure Assessment; CI, confidence interval.Area under receiver operating characteristic curve ! 0.81 (95% CI, 0.77–0.86); Hosmer-
Lemeshow goodness-of-fit ('2 ! 8.707; p ! 0.368).
13Crit Care Med 2010 Vol. 38, No. 1
Soares M. Crit Care Med 2010;38:9-15