multiple sequence alignment dr alexei drummond department of computer science...
TRANSCRIPT

Multiple sequence alignment
Dr Alexei Drummond
Department of Computer Science
Semester 2, 2006

2
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Multiple alignment software
Really need approximation methods.
Four techniques
1. progressive global alignment of sequences starting with an alignment of the most similar sequences and then building a full alignment by adding more sequences
2. iterative methods that make an initial alignment of groups of sequences and then refine the alignment to achieve a better result (Barton-sternberg, Simulated annealing, stochastic hill climbing)
3. (alignments based on locally conserved patterns found in the same order in the sequences), and
4. use of probabilistic models of the indel and substitution process to do statistical inference of alignment. (“Statistical alignment”)
3
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
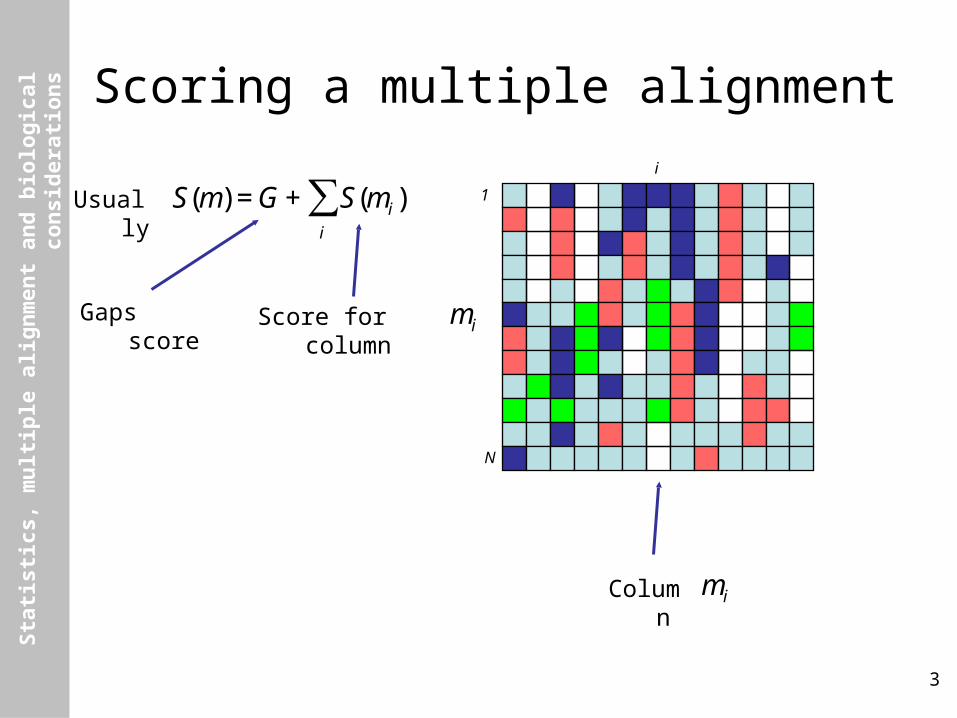
Scoring a multiple alignment
Usually
€
S(m) =G + S(mi)i
∑
Score for column
€
miGaps score
Column
€
mi
i
1
N
4
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
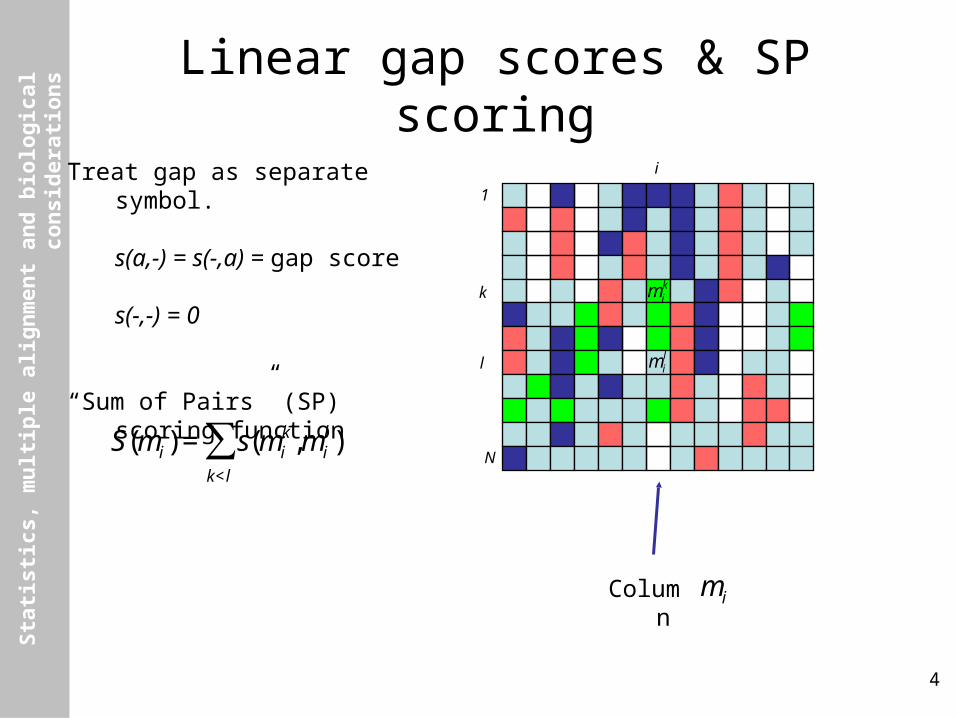
Linear gap scores & SP scoring
Treat gap as separate symbol.
s(a,-) = s(-,a) = gap score
s(-,-) = 0
“Sum of Pairs” (SP) scoring function
€
S(mi) = s(mik,mi
l )k<l
∑
Column
€
mi
k
l €
mik
€
mil
i
1
N
5
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
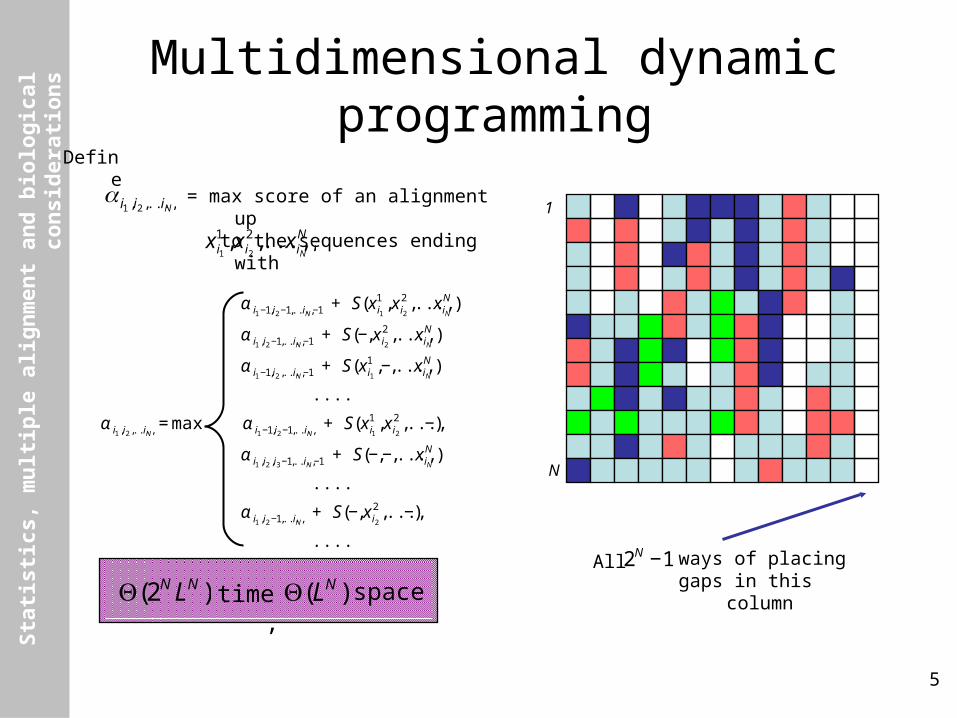
s Multidimensional dynamic programming
€
α i1 −1,i2 −1,...,iN −1 + S(x i11 ,x i2
2 ,...,x iNN )
α i1 ,i2 −1,...,iN −1 + S(−,x i22 ,...,x iN
N )
α i1 −1,i2 ,...,iN −1 + S(x i11 ,−,...,x iN
N )
....
α i1 ,i2 ,...,iN= max α i1 −1,i2 −1,...,iN
+ S(x i11 ,x i2
2 ,...,−)
α i1 ,i2 ,i3 −1,...,iN −1 + S(−,−,...,x iNN )
....
α i1 ,i2 −1,...,iN + S(−,x i2
2 ,...,−)
....
€
α i1 ,i2 ,...,iN
Define
= max score of an alignment up to the sequences ending with
€
x i11 ,x i2
2 ,...,x iNN ,
1
N
All ways of placinggaps in this column
€
2N −1
€
Θ(2N LN ) time,
€
Θ(LN ) space

6
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
MSA
Carrillo and Lipman (1988), Lipman, Altschul and Kececioglu (1989).
Can optimally align up to 5-7 protein sequences of up to 200 residues.
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
7
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
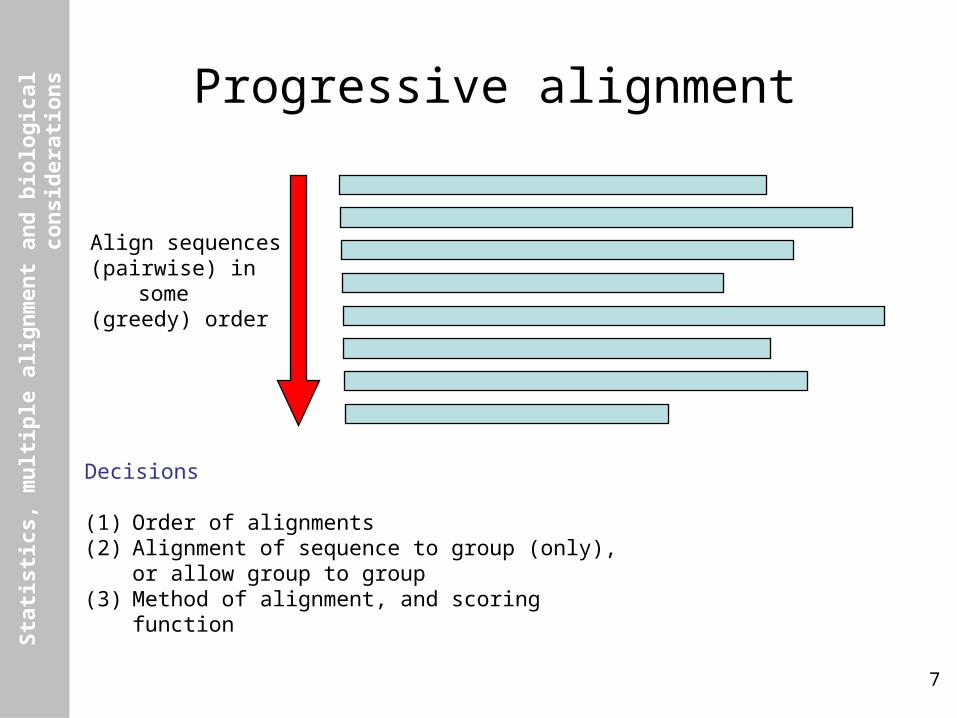
Progressive alignment
Align sequences(pairwise) in some(greedy) order
Decisions
(1) Order of alignments(2) Alignment of sequence to group (only), or allow group
to group(3) Method of alignment, and scoring function
8
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
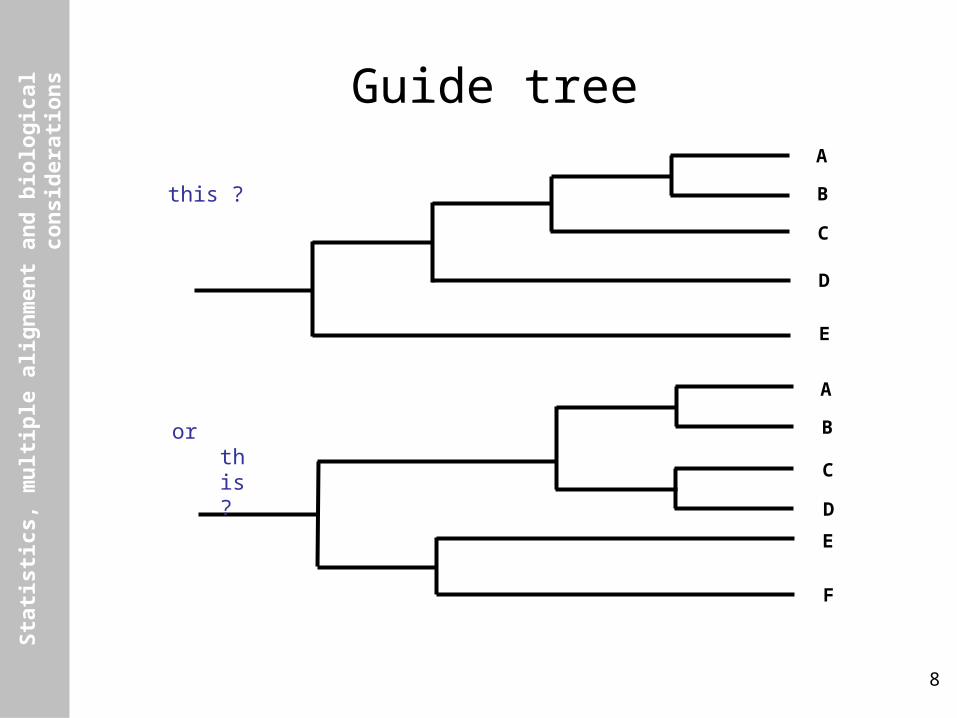
Guide treeA
B
C
D
E
A
B
C
D
F
this ?
or this ?
E

9
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
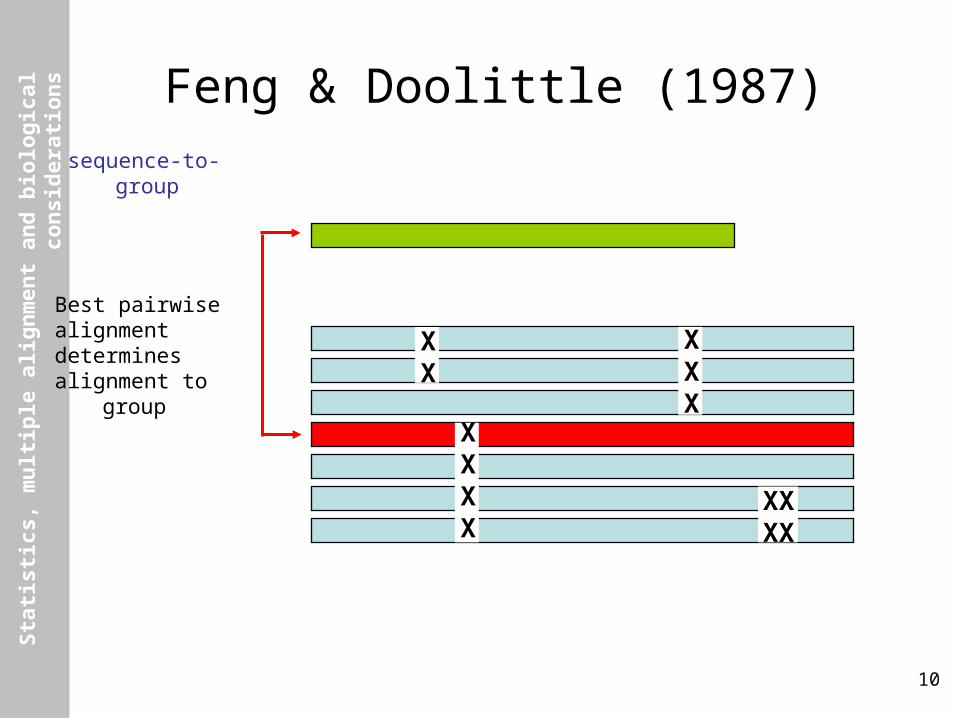
Feng & Doolittle (1987)
Overview
(1) Calculate diagonal matrix of N(N-1)/2 distances between all pairs of N sequences by standard pairwise alignment, converting raw alignment scores to approximate pairwise “distances”.
(2) Construct guide tree from the distance matrix by using appropriate clustering algorithm.
(3) Starting from first node added to the tree, align the child nodes (which may be two sequences, a sequence and an alignment, or two alignments). Repeat for all other nodes in the order that they were added to tree, until all sequences have been aligned.
10
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
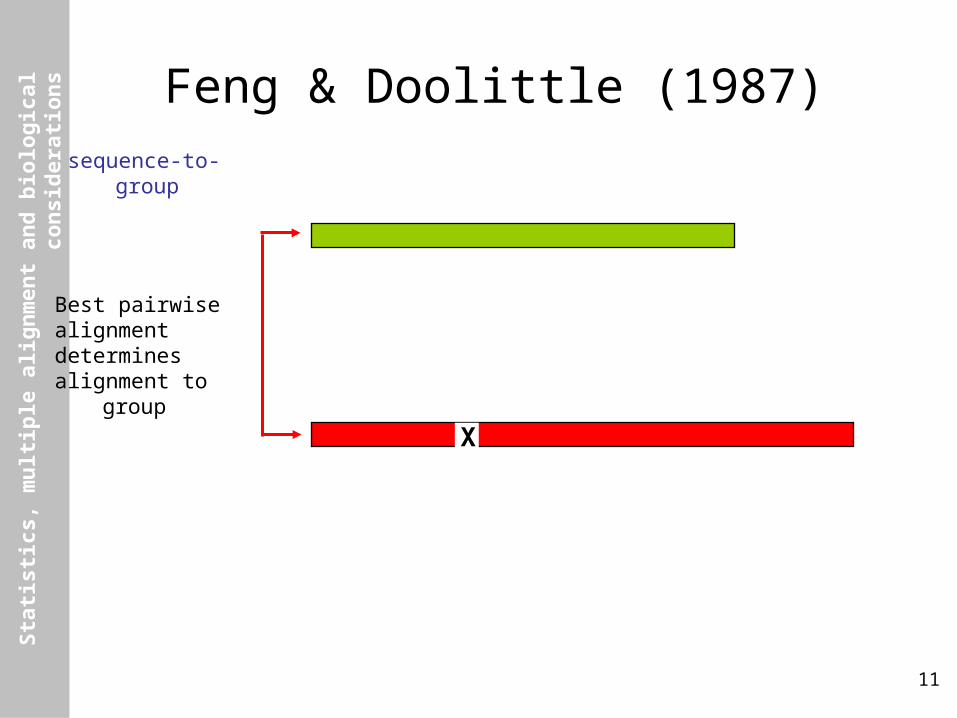
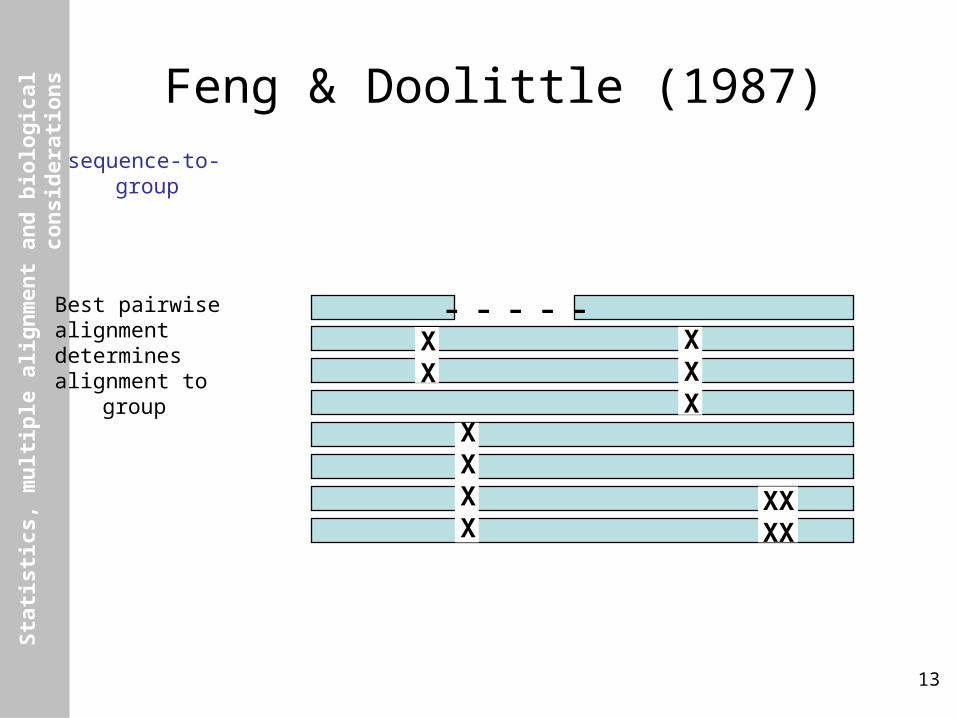
Feng & Doolittle (1987)sequence-to-group
Best pairwisealignmentdeterminesalignment to group
XX
XXX
XXXX
XXXX
11
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
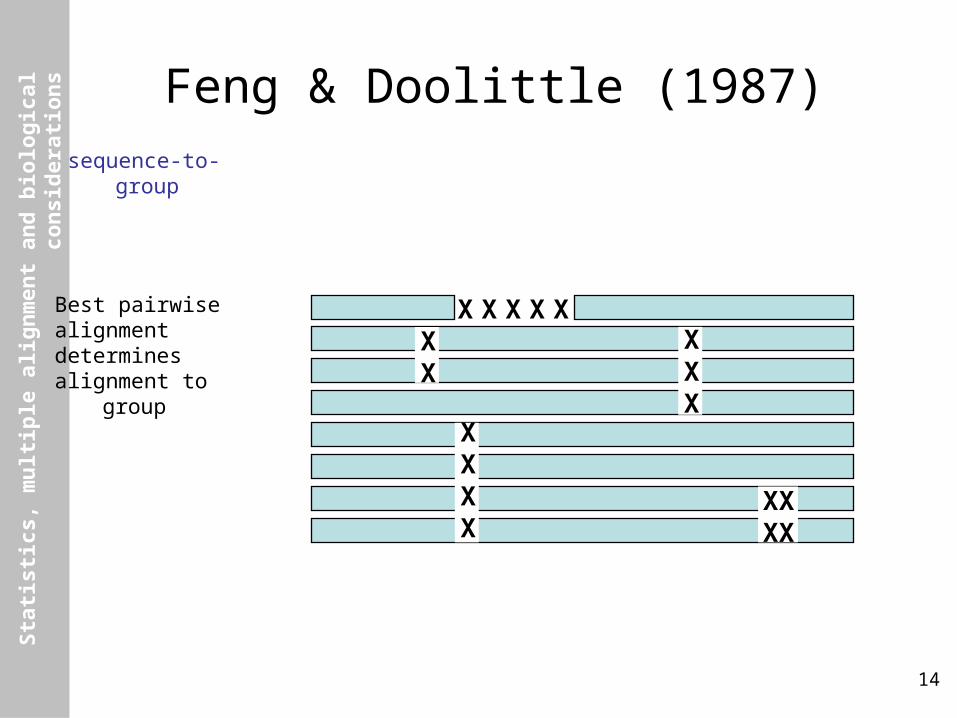
Feng & Doolittle (1987)sequence-to-group
Best pairwisealignmentdeterminesalignment to group
X
12
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Feng & Doolittle (1987)sequence-to-group
Best pairwisealignmentdeterminesalignment to group
X– – – – –
This column is encouraged because it has no cost
13
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Feng & Doolittle (1987)sequence-to-group
Best pairwisealignmentdeterminesalignment to group
XX
XXX
XXXX
XXXX
– – – – –
14
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Feng & Doolittle (1987)sequence-to-group
Best pairwisealignmentdeterminesalignment to group
XX
XXX
XXXX
XXXX
X X XX X
15
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
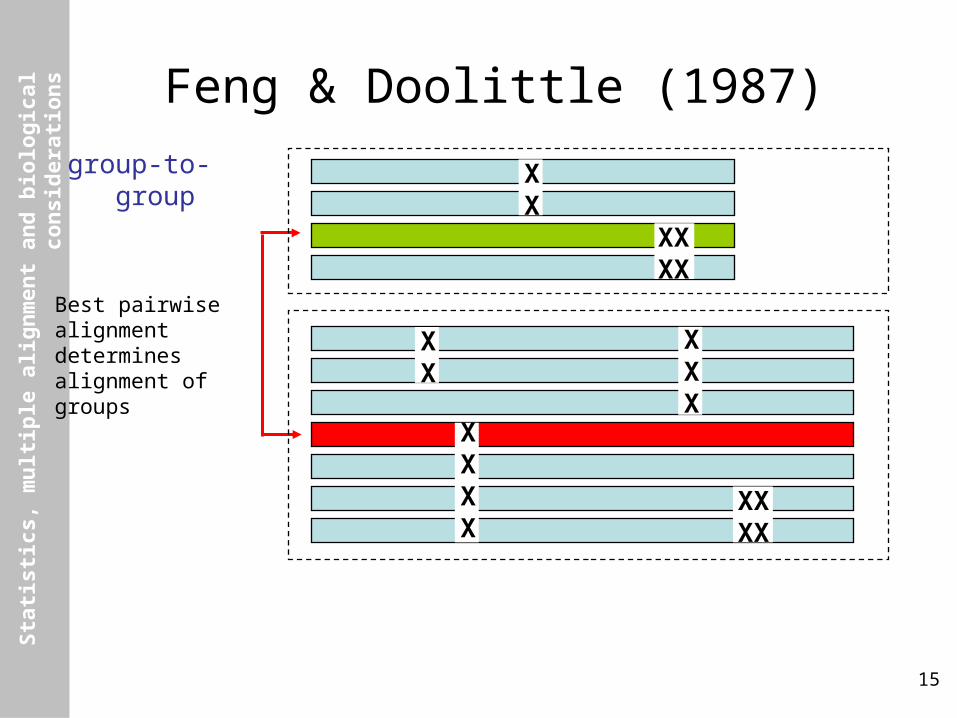
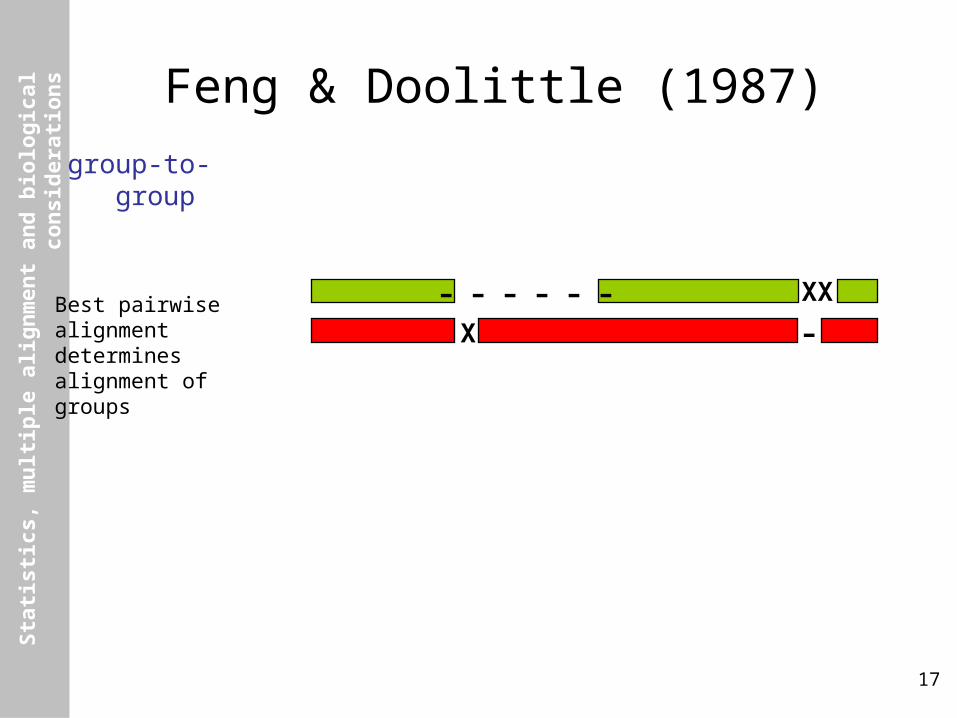
Feng & Doolittle (1987)
group-to-group
Best pairwisealignmentdeterminesalignment ofgroups
XX
XXX
XXXX
XXXX
XX
XXXX
16
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
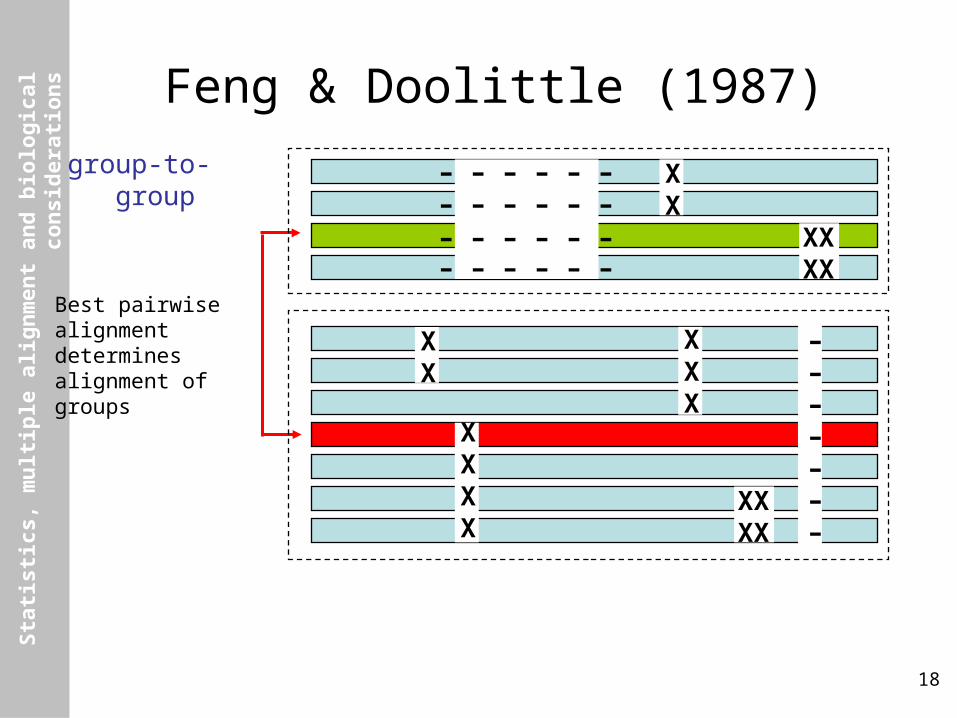
Feng & Doolittle (1987)
group-to-group
Best pairwisealignmentdeterminesalignment ofgroups
X
XX
17
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Feng & Doolittle (1987)
group-to-group
Best pairwisealignmentdeterminesalignment ofgroups
X
XX– – – – – –
–
18
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Feng & Doolittle (1987)
group-to-group
Best pairwisealignmentdeterminesalignment ofgroups
XX
XXX
XXXX
XXXX
XX
XXXX
– – – – – –– – – – – –– – – – – –– – – – – –
–––––––
19
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Feng & Doolittle (1987)
group-to-group
Best pairwisealignmentdeterminesalignment ofgroups
XX
XXX
XXXX
XXXX
XX
XXXX
–––––––
– – – – – –– – – – – –– – – – – –– – – – – –
20
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Feng & Doolittle (1987)
group-to-group
Best pairwisealignmentdeterminesalignment ofgroups
XX
XXX
XXXX
XXXX
XX
XXXX
XXXXXXX
XXXX
XXXX
XXXX
XXXX
XXXX
XXXX

21
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Feng & Doolittle (1987)
After alignment is completed gap symbols replaced by “X”.
“Once a gap, always a gap”.
Encourages gaps to occur in same columns in subsequent alignments.
Implemented by PILEUP (from GCG package).
22
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
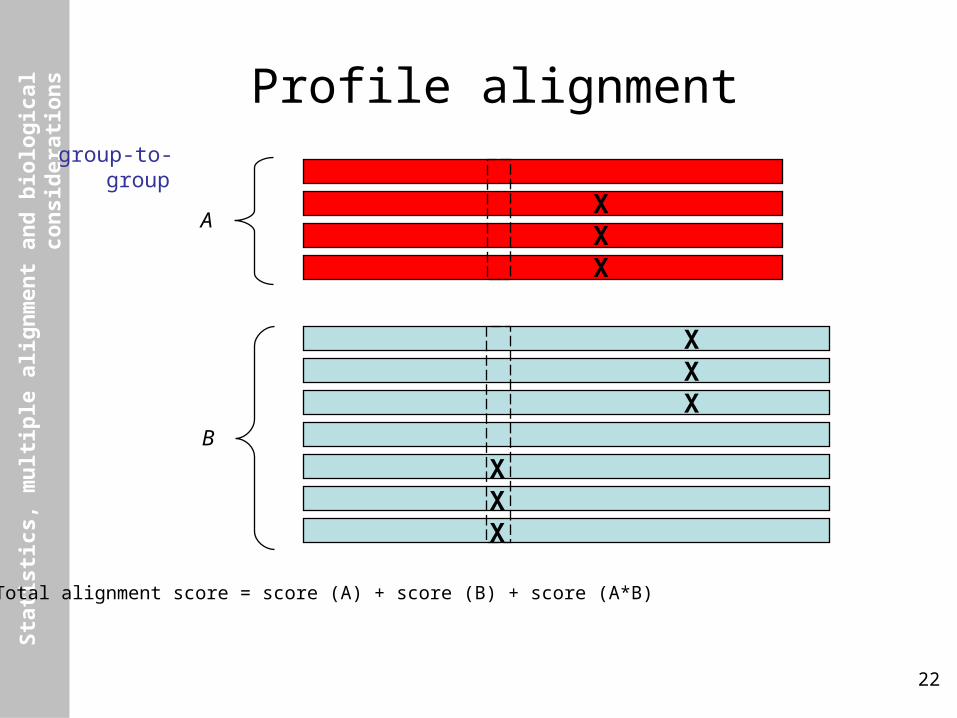
Profile alignmentgroup-to-group
A
B
Total alignment score = score (A) + score (B) + score (A*B)
XXX
XXX
XXX

23
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
CLUSTALW
Thompson, Higgins and Gibson (1994).
Widely used implementation of profile-based progressive multiple alignment.
Similar to Feng-Doolittle method, except for use of profile alignment methods.
Overview:
1. Calculate diagonal matrix of N(N-1)/2 distances between all pairs of N sequences by standard pairwise alignment, converting raw alignment scores to approximate pairwise “distances”.
2. Construct guide tree from distance matrix by using an appropriate neighbour-joining clustering algorithm.
3. Progressively align at nodes in order of decreasing similarity, using sequence-sequence, sequence-profile, and profile-profile alignment.
Plus many other heuristics.

24
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
CLUSTAL W heuristics
• Closely related sequences are aligned with hard matrices (BLOSUM80) and distant sequences are aligned with soft matrices (BLOSUM50).
• Hydrophobic residues (which are more likely to be buried) are given higher gap penalties than hydrophilic residues (which are more likely to be surface-accessible).
• Gap-open penalties are also decreased if the position is spanned by 5 or more consecutive hydrophilic residues.

25
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
CLUSTAL W heuristics
• Both gap-open penalties and gap-extend penalties are increased if there are no gaps in a column but gaps occur nearby in the alignment. This rule tries to force all gaps to occur in the same places in an alignment.
• In the progressive alignment stage, if the score of an alignment is low, the guide tree may be adjusted on the fly to defer the low scoring alignment until later in the progressive alignment phase when more profile information has been accumulated.

26
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Iterative refinement
i.e. “hill climbing”. Slightly change solution to improve score. Converge to local optimum.
e.g. Barton-Sternberg (1987) multiple alignment
(1) Find the two sequences with the highest pairwise similarity and align them using standard dynamic programming alignment.(2) Find sequence most similar to a profile of the alignment of the first two, and align it
to first two by profile-sequence alignment. Repeat until all sequences have been included in the multiple alignment.
(3) Remove sequence and realign it to a profile of the other aligned sequences by profile-sequence alignment. Repeat for sequences .
(3) Repeat the previous alignment step a fixed number of times, or until the alignment score converges.€
x1
€
x 2,...,xN
€
x 2,...,xN
27
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Clustal X
28
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Clustal X
29
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
CLUSTALX
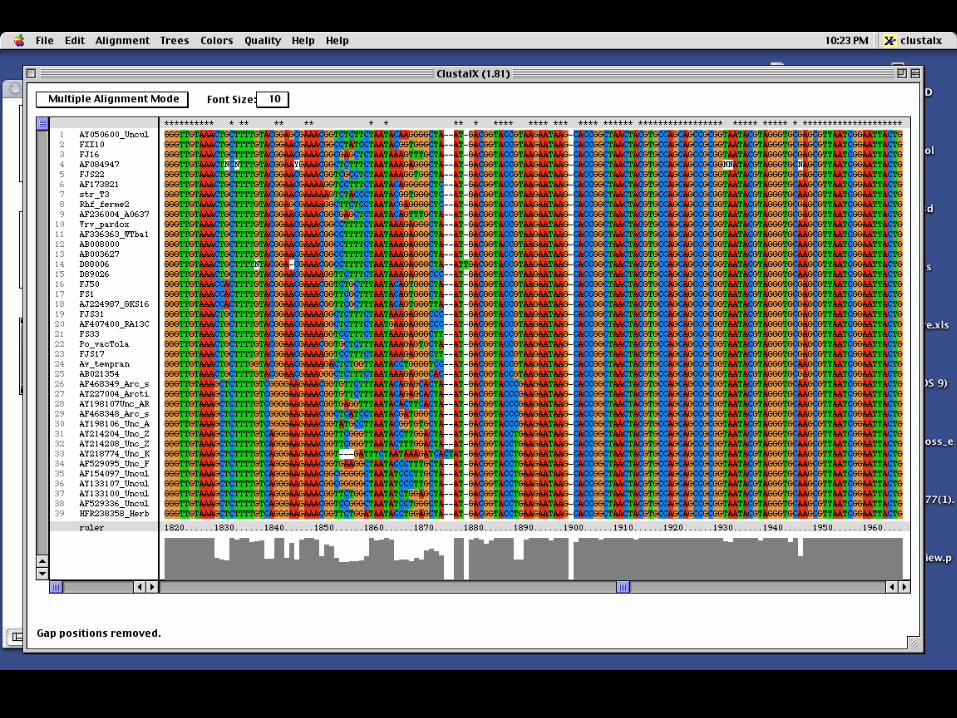
30
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
CLUSTALX

Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
C_aminophilum AGCT.YCGCA TGRAGCAGTG TGAAAA.... ............ACTCCGGT GGTACAGGAT C_colinum AGTA..GGCA TCTACAAGTT GGAAAA.... ............ACTGAGGT GGTATAGGAG C_lentocellum GGTATTCGCT TGATTATNAT AGTAAA.... ............GATTTATC GCCATAGGAT C_botulinum_D TTTA.TGGCA TCATACATAA AATAATCAAA ..........GGAGCAATCC GCTTTGAGAT C_novyi_A TTTA.CGGCA T....CGTAG AATAATCAAA ..........GGAGCAATCC GCTTTGAGAT C_gasigenes AGTT.TCGCA TGAAACA... GC.AATTAAA ..........GGAGAAATCC GCTATAAGAT C_aurantibutyricum A.NT.TCGCA TGGAGCA... AC.AATCAAA ..........GGAGCAAT.C ACTATAAGAT C_sp_C_quinii AGTT.T.GCA TGGGACA... GC.AATTAAA ..........GGAGCAATCC GCTATGAGAT C_perfringens AAGA.TGGCA T.CATCA... TTCAACCAAA ..........GGAGCAATCC GCTATGAGAT C_cadaveris TTTT.CTGCA TGGGAAA... GTC.ATGAAA ..........GGAGCAATCC GCTGTAAGAT C_cellulovorans ATTC.TCGCA TGAGAGA... .TGTATCAAA ..........GGAGCAATCC GCTATAAGAT C_K21 TTGR.TCGCA TGATCKAAAC ATCAAAGGAT ..TTTTCTTTGGAAAATTCC ACTTTGAGAT C_estertheticum TTGA.TCGCA TGATCTTAAC ATCAAAGGAA ..TTT..TTCGG..AATTTC ACTTTGAGAT C_botulinum_A AGAA.TCGCA TGATTTTCTT ATCAAAGATT ..T............ATT.. GCTTTGAGAT C_sporogenes AGAA.TCGCA TGATTTTCTT ATCAAAGATT ..T............ATT.. GCTTTGAGAT C_argentinense AAGG.TCGCA TGACTTTTAT ACCAAAGGAG ..T............AATCC GCTATGAGAT C_subterminale AAGG.TCGCA TGACTTTTAT ACCAAAGGAG ..T............AATCC GCTATGAGAT C_tetanomorphum TTTT.CCGCA TGAAAAACTA ATCAAAGGAG ..T............AAT.C GCTTTGAGAT C_pasteurianum AGTT.TCACA TGGAGCTTTA ATTAAAGGAG ..T............AATCC GCTTTGAGAT C_collagenovorans TTGA.TCGCA TGGTCGAAAT ATTAAAGGAG ..T............AATCC GCTTACAGAT C_histolyticum TTTA.ATGCA TGTTAGAAAG ATTAAAGGAG ..............CAATCC GCTTTGAGAT C_tyrobutyricum AGTT.TCACA TGGAATTTGG ATGAAAGGAG ..T............AATTC GCTTTGAGAT C_tetani GGTT.TCGCA TGAAACTTTA ACCAAAGGAG ..T............AATCT GCTTTGAGAT C_barkeri GACA.TCGCA TGGTGTT... .TTAATGAAA ............ACTCCGGT GCCATGAGAT C_thermocellum GGCA.TCGTC CTGTTAT... .CAAAGGAGA ............AATCCGGT ...ATGAGAT Pep_prevotii AGTC.TCGCA TGGNGTTATC ATCAAAGA.. ..............TTTATC GGTGTAAGAT C_innocuum ACGGAGCGCA TGCTCTGTAT ATTAAAGCGC CCTTCAAGGCGTGAAC.... ....ATGGAT S_ruminantium AGTTTCCGCA TGGGAGCTTG ATTAAAGATG GCCTCTACTTGTAAGCTATC GCTTTGCGAT

Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
C_aminophilum AGCT.YCGCA TGRAGCAGTG TGAAAA.... ............ACTCCGGT GGTACAGGAT C_colinum AGTA..GGCA TCTACAAGTT GGAAAA.... ............ACTGAGGT GGTATAGGAG C_lentocellum GGTATTCGCT TGATTATNAT AGTAAA.... ............GATTTATC GCCATAGGAT C_botulinum_D TTTA.TGGCA TCATACATAA AATAATCAAA ..........GGAGCAATCC GCTTTGAGAT C_novyi_A TTTA.CGGCA T....CGTAG AATAATCAAA ..........GGAGCAATCC GCTTTGAGAT C_gasigenes AGTT.TCGCA TGAAACA... GC.AATTAAA ..........GGAGAAATCC GCTATAAGAT C_aurantibutyricum A.NT.TCGCA TGGAGCA... AC.AATCAAA ..........GGAGCAAT.C ACTATAAGAT C_sp_C_quinii AGTT.T.GCA TGGGACA... GC.AATTAAA ..........GGAGCAATCC GCTATGAGAT C_perfringens AAGA.TGGCA T.CATCA... TTCAACCAAA ..........GGAGCAATCC GCTATGAGAT C_cadaveris TTTT.CTGCA TGGGAAA... GTC.ATGAAA ..........GGAGCAATCC GCTGTAAGAT C_cellulovorans ATTC.TCGCA TGAGAGA... .TGTATCAAA ..........GGAGCAATCC GCTATAAGAT C_K21 TTGR.TCGCA TGATCKAAAC ATCAAAGGAT ..TTTTCTTTGGAAAATTCC ACTTTGAGAT C_estertheticum TTGA.TCGCA TGATCTTAAC ATCAAAGGAA ..TTT..TTCGG..AATTTC ACTTTGAGAT C_botulinum_A AGAA.TCGCA TGATTTTCTT ATCAAAGATT ..T............ATT.. GCTTTGAGAT C_sporogenes AGAA.TCGCA TGATTTTCTT ATCAAAGATT ..T............ATT.. GCTTTGAGAT C_argentinense AAGG.TCGCA TGACTTTTAT ACCAAAGGAG ..T............AATCC GCTATGAGAT C_subterminale AAGG.TCGCA TGACTTTTAT ACCAAAGGAG ..T............AATCC GCTATGAGAT C_tetanomorphum TTTT.CCGCA TGAAAAACTA ATCAAAGGAG ..T............AAT.C GCTTTGAGAT C_pasteurianum AGTT.TCACA TGGAGCTTTA ATTAAAGGAG ..T............AATCC GCTTTGAGAT C_collagenovorans TTGA.TCGCA TGGTCGAAAT ATTAAAGGAG ..T............AATCC GCTTACAGAT C_histolyticum TTTA.ATGCA TGTTAGAAAG ATTAAAGGAG ..............CAATCC GCTTTGAGAT C_tyrobutyricum AGTT.TCACA TGGAATTTGG ATGAAAGGAG ..T............AATTC GCTTTGAGAT C_tetani GGTT.TCGCA TGAAACTTTA ACCAAAGGAG ..T............AATCT GCTTTGAGAT C_barkeri GACA.TCGCA TGGTGTT... .TTAATGAAA ............ACTCCGGT GCCATGAGAT C_thermocellum GGCA.TCGTC CTGTTAT... .CAAAGGAGA ............AATCCGGT ...ATGAGAT Pep_prevotii AGTC.TCGCA TGGNGTTATC ATCAAAGA.. ..............TTTATC GGTGTAAGAT C_innocuum ACGGAGCGCA TGCTCTGTAT ATTAAAGCGC CCTTCAAGGCGTGAAC.... ....ATGGAT S_ruminantium AGTTTCCGCA TGGGAGCTTG ATTAAAGATG GCCTCTACTTGTAAGCTATC GCTTTGCGAT
TCAAAGGAG
TCAAAGGAG

33
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Alignment - considerations
• The programs simply try to maximize the number of matches– The “best” alignment may not be the correct
biological one• Multiple alignments are done progressively
– Such alignments get progressively worse as you add sequences
– Mistakes that occur during alignment process are frozen in.
• Unless the sequences are very similar you will almost certainly have to correct manually

34
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Manual Alignment- software
Geneious 2.0- java application:– http://www.geneious.com/
CINEMA- Java applet available from:– http://www.biochem.ucl.ac.uk
Seqapp/Seqpup- Mac/PC/UNIX available from:– http://iubio.bio.indiana.edu
Se-Al for Macintosh, available from:– http://evolve.zoo.ox.ac.uk/Se-Al/Se-Al.html
BioEdit for PC, available from:– http://www.mbio.ncsu.edu/RNaseP/info/programs/
BIOEDIT/bioedit.html
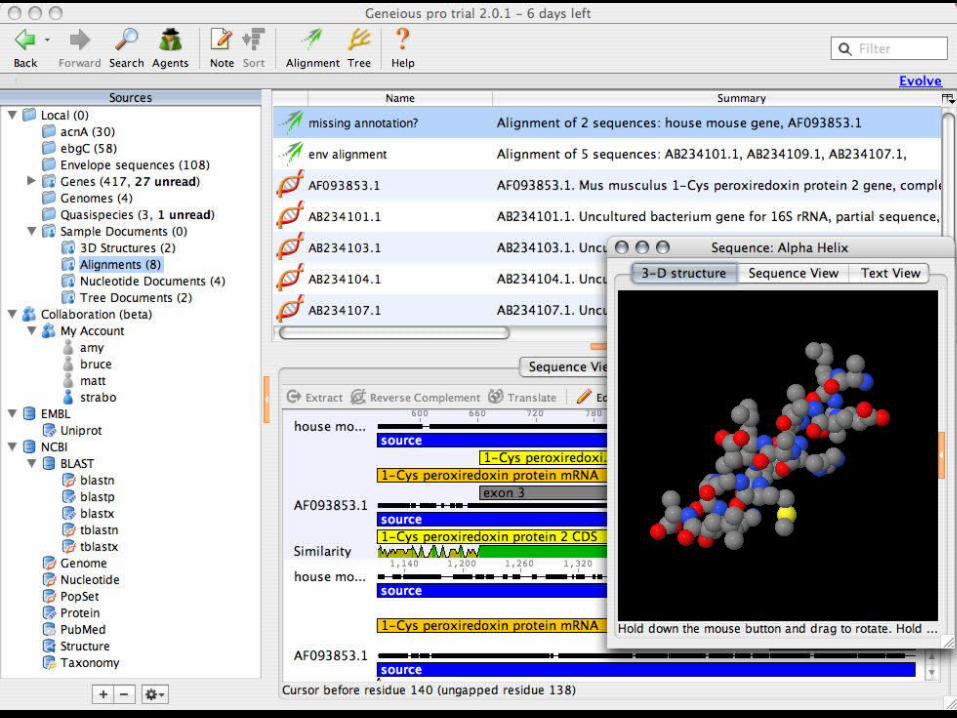
35
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
36
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s

37
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
38
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
sMACCLADE 4
39
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
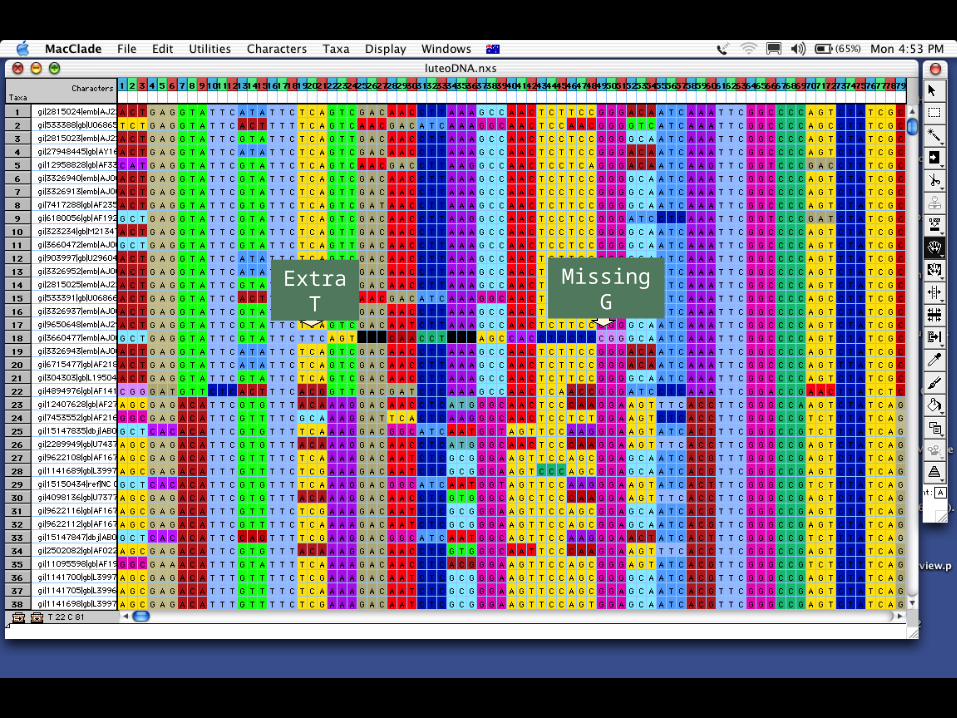
Extra T Missing G
40
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
41
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
42
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
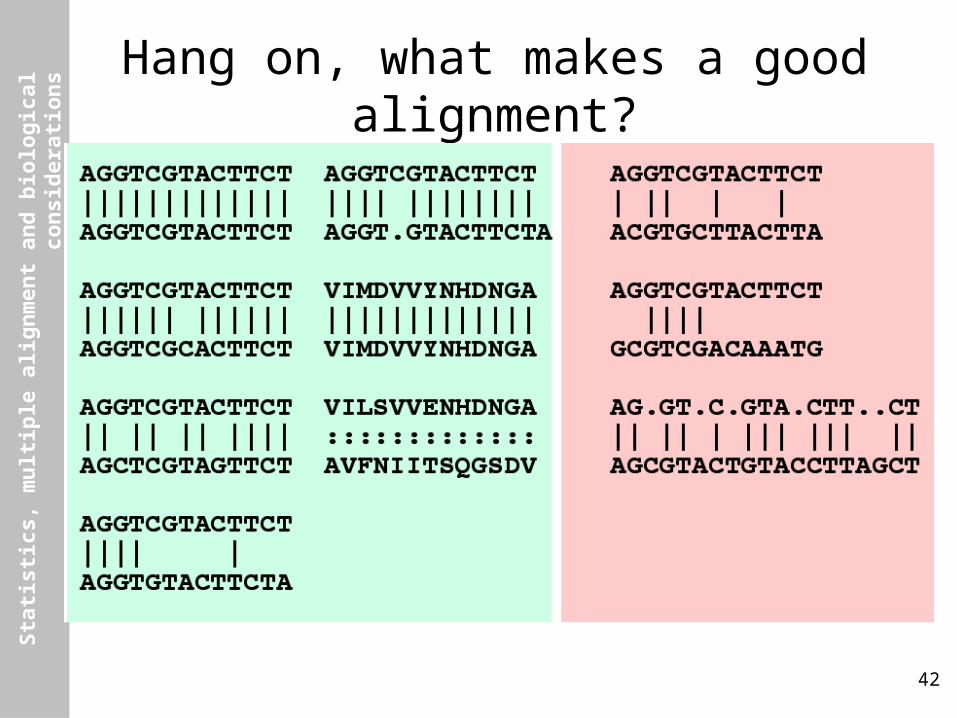
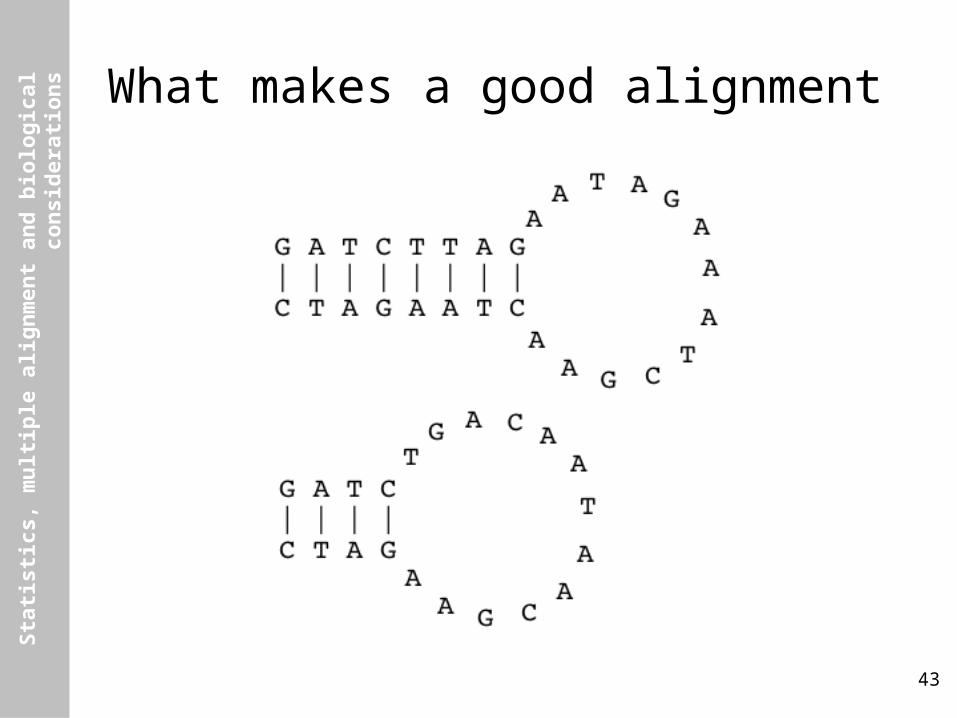
s Hang on, what makes a good alignment?
43
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
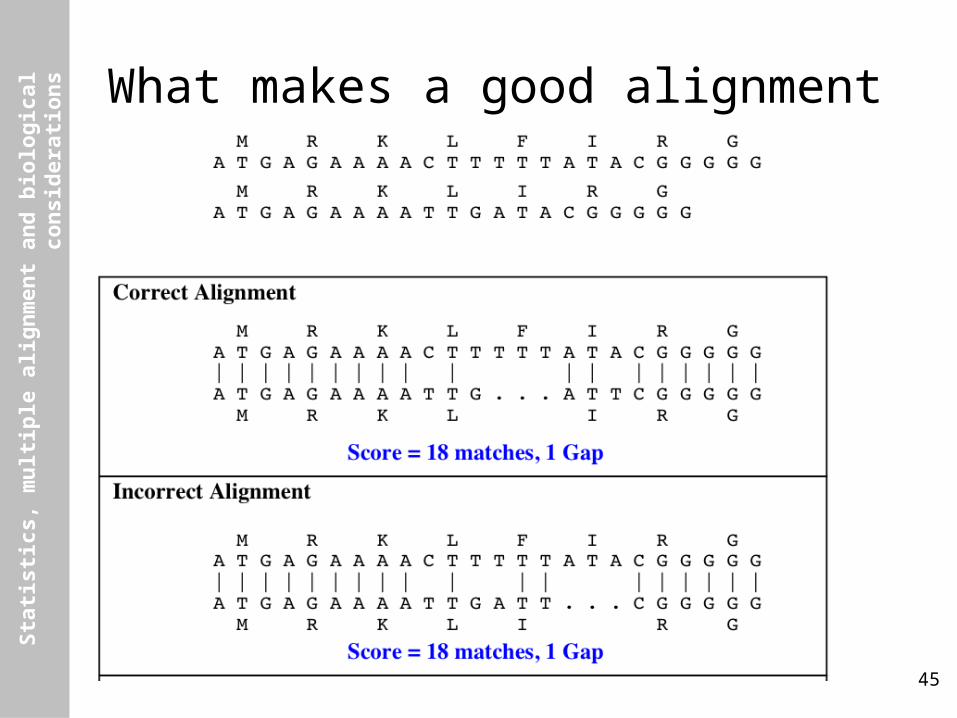
What makes a good alignment
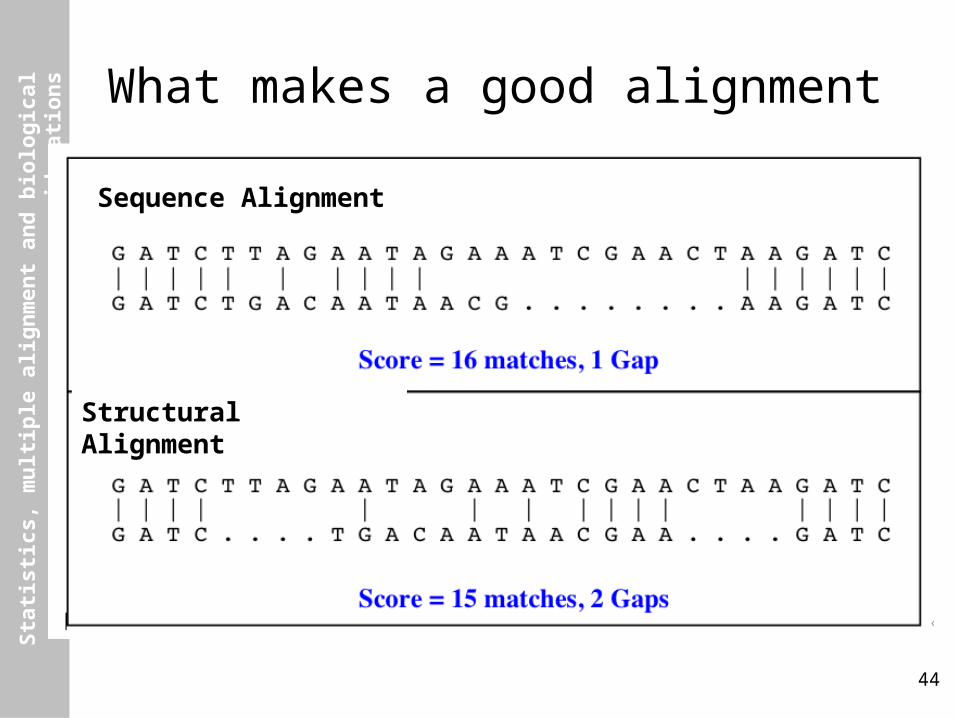
44
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
What makes a good alignment
Structural Alignment
Sequence Alignment
45
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
What makes a good alignment

46
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
I hate ad hoc algorithms and manual sequence alignment!
Is there an alternative?

47
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
An evolutionary hypothesis
AG
AAT AAC AC ACCG ACC
Insert CCInsert T
Delete G
G->C
G->A
Observations
Hypothesis/Model
T->C
Knowing the rates of different events (substitutions, insertions and deletions) provides a method of assessing the probability of these observations, given this hypothesis: Pr{D|T,Q}
T: the evolutionary tree
Q: parameters of the evolutionary process

48
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
Statistics: fitting versus modeling
• Statistical fitting of sequence variation– Count frequencies of changes in real data sets – Build empirical statistical descriptions of the data (Blosum62)– Compare observed frequencies to well defined null hypothesis
for testing (log-odds ratio and scores)– Use scores in ad hoc algorithms for search and alignment
(BLAST and ClustalX)
• Probabilistic models of sequence evolution– Describe a probabilistic model in terms of a process of
evolution, rates of substitution, insertion and deletion– Estimate parameters of the models and compare models using
model comparison (likelihood ratios, Bayes factors)– Use maximum likelihood and Bayesian inference to co-
estimate (uncertainty in) alignment and evolutionary history.
49
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
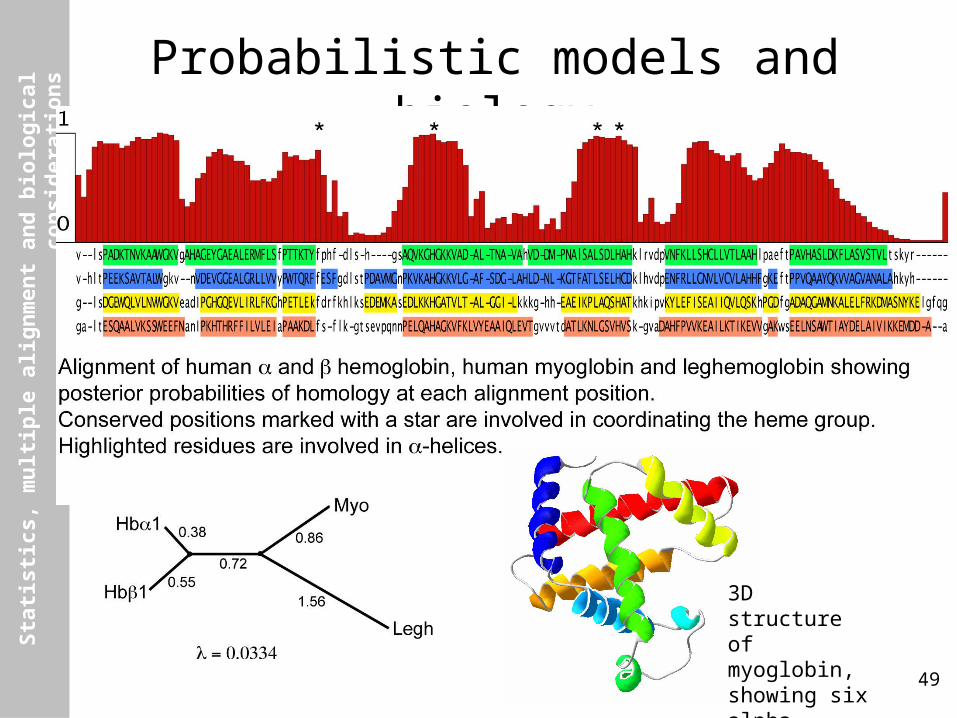
Probabilistic models and biology
3D structure of myoglobin, showing six alpha-helices.

50
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
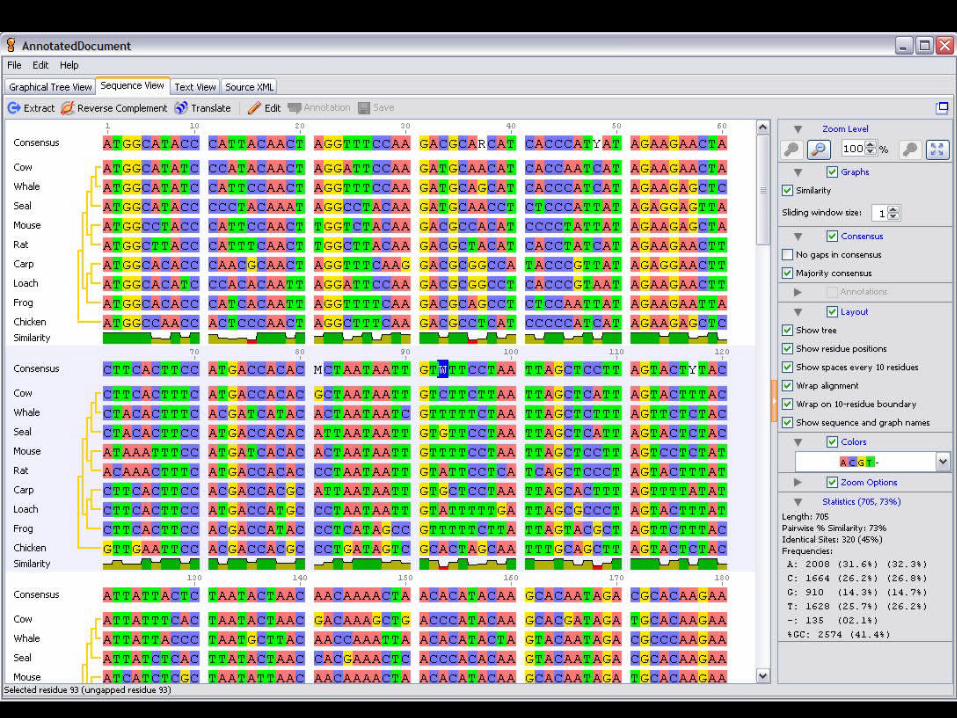
State of the art
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

51
Sta
tist
ics,
mu
ltip
le a
lign
men
t an
d b
iolo
gic
al c
on
sid
erat
ion
s
What does the future hold?
• No single “true” alignment– In most situations there are a set of alignments that
are consistent with the observations– Understanding this uncertainty is as important as
understanding the “best” alignment
• Explicit evolutionary model-based methods– Methods that co-estimate alignment and phylogeny
are beginning to appear– Co-estimation of protein structure and alignment
using evolutionary models may be on horizon
• Death of manual sequence alignment?