multiparametric decision support system for the prediction of oral cancer reoccurrence
TRANSCRIPT

IEEE TRANSACTIONS ON INFORMATION TECHNOLOGY IN BIOMEDICINE, VOL. 16, NO. 6, NOVEMBER 2012 1127
Multiparametric Decision Support System for thePrediction of Oral Cancer Reoccurrence
Konstantinos P. Exarchos, Yorgos Goletsis, Member, IEEE, and Dimitrios I. Fotiadis, Senior Member, IEEE
Abstract—Oral squamous cell carcinoma (OSCC) constitutes thepredominant neoplasm of the head and neck region, featuring par-ticularly aggressive nature, associated with quite unfavorable prog-nosis. In this paper, we formulate a decision support system thatintegrates a multitude of heterogeneous data (clinical, imaging, andgenomic), thus, framing all manifestations of the disease. Our pri-mary aim is to identify the factors that dictate OSCC progressionand subsequently predict potential relapses (local or metastatic) ofthe disease. The discrimination potential of each source of data isinitially explored separately, and afterward the individual predic-tions are combined to yield a consensus decision achieving completediscrimination between patients with and without a disease relapse.
Index Terms—Classification, decision support system (DSS),gene expression, oral cancer, reoccurrence prediction.
I. INTRODUCTION
MALIGNANT neoplasms constitute the second cause ofdeath in the western world; oral cancer, in particular,
is the 8th most common neoplasm according to the worldwidecancer incidence ranking [1]. In the literature, several risk fac-tors have been associated with the induction and progressionof oral cancer; excessive smoking and especially when coupledwith alcohol consumption constitute predominant factors of oralsquamous cell carcinoma (OSCC) development. A significantcorrelation has also been identified between oral cancer and thesex of the patients, with men facing twice the risk of beingdiagnosed with OSCC [1]. Moreover, extensive sun exposurehas been proven a proliferating factor of OSCC development,particularly for neoplasms of the lip [1].
Besides the low quality of everyday life that is inherent toOSCC patients, a major issue is the high occurrence rate of lo-coregional relapses, which often leads to considerable inabilityand even death. Specifically, after successful treatment of thedisease, there is a state called remission where the patient isconsidered cancer free, and particles of the primary tumor massare no longer detectable. Patients in remission have a proba-bility of 25–48% to develop a disease relapse [2] owed to the
Manuscript received April 15, 2011; revised July 9, 2011; accepted August 6,2011. Date of publication August 18, 2011; date of current version November 16,2012. This work was supported by the European Commission NeoMark Project(FP7-ICT-2007-224483—ICT enabled prediction of cancer reoccurrence).
K. P. Exarchos and D. I. Fotiadis are with the Department of MaterialsScience and Engineering, Unit of Medical Technology and Intelligent Infor-mation Systems, University of Ioannina, Ioannina, GR 45110, Greece (e-mail:[email protected]; [email protected]).
Y. Goletsis is with the Department of Economics, University of Ioannina,Ioannina, GR 45110, Greece (e-mail: [email protected]).
Color versions of one or more of the figures in this paper are available onlineat http://ieeexplore.ieee.org.
Digital Object Identifier 10.1109/TITB.2011.2165076
deeply infiltrative nature of the disease. Due to the aggressivenature of OSCC and high rates of locoregional relapses, earlyidentification of a potential disease reoccurrence can prove verybeneficial for the prognosis of the patient [3] and the subsequentfine tuning of the followup treatment.
To this end, several studies have been conducted in the liter-ature aiming to predict a relapse after the disease has reachedremission; yet, the overall results remain unsatisfactory. Specif-ically, Roepman et al. [4], [5] have extracted a subset of genesable to identify lymph node metastasis from a primary head andneck squamous cell carcinoma. In a similar manner, Rickmanet al. [6] exploit gene expression information toward the predic-tion of future metastasis of a neoplasm originating in the regionof the head and neck. In the same context, Zhou et al., Watanabeet al., and Nagata et al. [7]–[9] have focused on carcinomas ofthe tongue, yielding a subset of genes able to predict nodalmetastasis. In [10], an artificial neural network (ANN) is em-ployed in order to predict the metastasis of primary esophagealcancer in the adjacent lymph nodes. In [11], a gene subset is ex-tracted aiming to predict lymph node metastasis from an initialoral squamous cell carcinoma.
In this paper, we propose an orchestrated approach in order tosystematically study and analyze the multifactorial basis of oralcancer evolvement. Three sources of data are employed, namelyclinical, imaging, and genomic in pursue of the most prominentOSCC proliferating factors. This multifaceted approach and thecomplementary analysis of the data are prone to capture all pos-sible manifestations of the disease. Subsequently, the identifiedfactors are utilized in a classification scheme in order to predicta potential disease reoccurrence and discriminate the patients interms of relapse probability. The timely and accurate identifica-tion of a disease relapse, can substantially determine the mostproper treatment, based on each patient’s specific prognosis.
II. MATERIALS AND METHODS
The flowchart of the proposed methodology is shown inFig. 1. Initially, all sources of data (clinical, imaging, and ge-nomic) are subject to certain basic preprocessing steps in orderto enhance the quality of the input data that are further fedto the next steps. Next, we either feed the input data directlyfor classification or we employ a feature selection algorithmto omit potentially redundant features. Besides the individ-ual clinical/imaging/genomic-based classifiers, we implementa consensus classifier that combines the aforementioned predic-tions in a complementary manner in order to procure a moreaccurate outcome.
1089-7771/$31.00 © 2012 IEEE

1128 IEEE TRANSACTIONS ON INFORMATION TECHNOLOGY IN BIOMEDICINE, VOL. 16, NO. 6, NOVEMBER 2012
Fig. 1. Flowchart of the proposed DSS.
Fig. 2. Clinical scenario.
A. Clinical Scenario
The clinical scenario adopted in this paper is depicted inFig. 2. Initially, a patient is diagnosed with OSCC through tra-ditional clinical procedures. The patient is then treated accord-ingly (surgery and chemoradiotherapy) in order to be relievedfrom the primary tumor. Consequently, the patient either reachescomplete remission or particles of the disease still remain intactand further treatment is advised. Only the former group of pa-tients meets the inclusion criteria of our study, and is subject tothe subsequent data collection process.
Specifically, at the baseline phase of the patient’s treatment,we collect a multitude of heterogeneous data that are prone to“frame” all possible aspects of the disease (see Fig. 3).
The patient is then monitored for a followup period of 18months, where a potential relapse is more likely to appear [2].Consequently, based on the disease outcome, patients are di-vided into relapsers and nonrelapsers. We employ a multileveldata integration approach that utilizes clinical, imaging, andgenomic data. The clinical data contain standard measurementsand laboratory markers from the patient’s medical record as wellas pathology and risk factor data referring to the organism as awhole. The imaging data contain image-extracted information
Fig. 3. Data collection process.
after the analysis of CT and MRI images of the primary tumormass and adjacent lymph nodes of the head and neck area. Asfor the genomic data, these include gene expression informationfrom the tumor specimen.
In this paper, 41 patients are considered, coming from twomajor clinical centers situated in Italy (University Hospitalof Parma, Parma) and Spain (MD Anderson Cancer Center,Madrid). Among the 41 patients, 13 patients have already devel-oped a relapse, whereas the remaining 28 are currently diseasefree.
B. Dataset Formulation
In the sections that follow, we describe the input variablescomprising each source of data as well as the preprocessingsteps employed to enhance the quality of input data.
1) Clinical Data: The specific features considered duringthis study from a clinical aspect are shown in Table I, and are inaccordance with current medical domain knowledge.
Certain features, however, contain missing values for a con-siderable number of patients. Specifically, features with morethan 90% of missing values (indicated with bold in Table I) arecompletely discarded from our analysis, whereas, the values offeatures with less percentage of missing values are imputed withmodes and means in the case of nominal and numerical features,respectively.
2) Imaging Data: As for the imaging data, 17 features (seeTable II) are extracted from CT and MRI images of the headand neck area.
For the case of imaging data, none of the features containmissing values; therefore, no imputation procedure is neededand the feature vector is used as it is.
3) Genomic Data: From the cancerous tissue specimen ofeach patient, we extract the expression of 45 015 genes. All mi-croarray experiments have been conducted with the same plat-form and feature extraction software in order to maintain solelybiological variability and avoid all other potentially contami-nating sources. The extracted gene expression files are initiallysubject to a series of basic preprocessing steps. Specifically, du-plicate and control features are discarded as well as genes with
EXARCHOS et al.: MULTIPARAMETRIC DECISION SUPPORT SYSTEM FOR THE PREDICTION OF ORAL CANCER REOCCURRENCE 1129
TABLE ICLINICAL FEATURES
TABLE IIIMAGING FEATURES
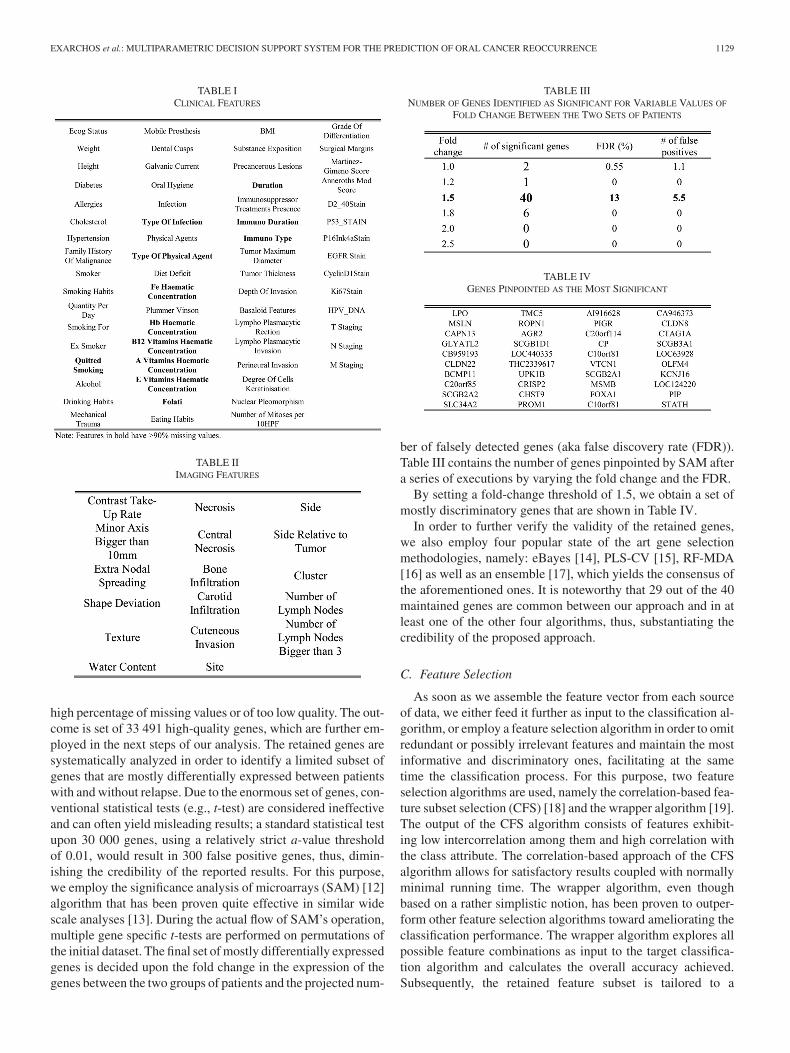
high percentage of missing values or of too low quality. The out-come is set of 33 491 high-quality genes, which are further em-ployed in the next steps of our analysis. The retained genes aresystematically analyzed in order to identify a limited subset ofgenes that are mostly differentially expressed between patientswith and without relapse. Due to the enormous set of genes, con-ventional statistical tests (e.g., t-test) are considered ineffectiveand can often yield misleading results; a standard statistical testupon 30 000 genes, using a relatively strict a-value thresholdof 0.01, would result in 300 false positive genes, thus, dimin-ishing the credibility of the reported results. For this purpose,we employ the significance analysis of microarrays (SAM) [12]algorithm that has been proven quite effective in similar widescale analyses [13]. During the actual flow of SAM’s operation,multiple gene specific t-tests are performed on permutations ofthe initial dataset. The final set of mostly differentially expressedgenes is decided upon the fold change in the expression of thegenes between the two groups of patients and the projected num-
TABLE IIINUMBER OF GENES IDENTIFIED AS SIGNIFICANT FOR VARIABLE VALUES OF
FOLD CHANGE BETWEEN THE TWO SETS OF PATIENTS
TABLE IVGENES PINPOINTED AS THE MOST SIGNIFICANT
ber of falsely detected genes (aka false discovery rate (FDR)).Table III contains the number of genes pinpointed by SAM aftera series of executions by varying the fold change and the FDR.
By setting a fold-change threshold of 1.5, we obtain a set ofmostly discriminatory genes that are shown in Table IV.
In order to further verify the validity of the retained genes,we also employ four popular state of the art gene selectionmethodologies, namely: eBayes [14], PLS-CV [15], RF-MDA[16] as well as an ensemble [17], which yields the consensus ofthe aforementioned ones. It is noteworthy that 29 out of the 40maintained genes are common between our approach and in atleast one of the other four algorithms, thus, substantiating thecredibility of the proposed approach.
C. Feature Selection
As soon as we assemble the feature vector from each sourceof data, we either feed it further as input to the classification al-gorithm, or employ a feature selection algorithm in order to omitredundant or possibly irrelevant features and maintain the mostinformative and discriminatory ones, facilitating at the sametime the classification process. For this purpose, two featureselection algorithms are used, namely the correlation-based fea-ture subset selection (CFS) [18] and the wrapper algorithm [19].The output of the CFS algorithm consists of features exhibit-ing low intercorrelation among them and high correlation withthe class attribute. The correlation-based approach of the CFSalgorithm allows for satisfactory results coupled with normallyminimal running time. The wrapper algorithm, even thoughbased on a rather simplistic notion, has been proven to outper-form other feature selection algorithms toward ameliorating theclassification performance. The wrapper algorithm explores allpossible feature combinations as input to the target classifica-tion algorithm and calculates the overall accuracy achieved.Subsequently, the retained feature subset is tailored to a

1130 IEEE TRANSACTIONS ON INFORMATION TECHNOLOGY IN BIOMEDICINE, VOL. 16, NO. 6, NOVEMBER 2012
specific classification algorithm and is chosen based on the high-est achieved performance. Another appealing characteristic ofthe wrapper algorithm is that it provides some protection againstoverfitting, owed to the internal cross-validation function usedfor accuracy estimation. It should be noted, however, that thewrapper algorithm is quite computationally intensive due to theexhaustive search that it performs across the feature space.
D. Class Imbalance
As noted previously, patients with and without disease relapseare unevenly distributed in the employed datasets. In the litera-ture, there have been proposed three sampling-based approachesin order to cope with class imbalance, namely: undersamplingthe majority class, oversampling the minority class, and a hybridapproach that combines the aforementioned ones. During under-sampling, some important patients from the majority class (i.e.,the nonrelapsers) might be omitted, thus, imposing bias on thereported results, with a subsequent reduction of the employeddataset. In the case of oversampling, patients from the minor-ity class (i.e., the relapsers) are replicated until both classesare equally distributed in the resulting dataset, which woulddeteriorate the generalization capability of the classifier. Thehybrid approach consequently suffers from both the aforemen-tioned approaches, nevertheless, to a lesser degree. In this pa-per, we employ the synthetic minority oversampling technique(SMOTE) [20] that neither discards potentially useful samplesnor merely replicates existing samples. Specifically, SMOTEidentifies a subset of the patients in the minority class, using ak-nearest neighbor approach and employs them to create newsamples by interpolating the existing ones.
E. Classification
After the datasets have been assembled, we examine theperformance of five popular classification algorithms towardthe discrimination between patients with and without diseaserelapse. Specifically, we employ Bayesian networks (BNs),ANNs, support vector machines (SVMs), decision trees (DTs),and random forests (RFs), which have all been used in a widerange of problems in the field of biomedical engineering. BNsalso known as belief networks are used to represent intuitivelydomain knowledge coupled with data-driven probabilistic de-pendences among variables of interest. ANNs are trained toexpress the output as a combination among the input variableswhereby the classification error is minimized. SVMs initiallymap the input vector into a feature space of higher dimensional-ity where the samples are linearly discriminated and identify thehyperplane that separates the instances of the classes with higherconfidence. DTs formulate a tree-structured classifier where thenodes correspond to the input variables and the leafs denote de-cision outcomes; by traversing the tree, given the features valuesof a new sample, we are able to conjecture about its class. RFshave emerged relatively recently and have been employed in avariety of classification problems. RFs use feature subsets toconstruct multiple DTs, each casting a vote on the predictedclass; the final outcome is the one collecting the majority ofindividual votes.
TABLE VCLINICAL-BASED CLASSIFICATION RESULTS WITHOUT PERFORMING
FEATURE SELECTION
TABLE VICLINICAL-BASED CLASSIFICATION RESULTS AFTER EMPLOYING THE CFS
ALGORITHM FOR FEATURE SELECTION
III. RESULTS AND DISCUSSION
For evaluation purposes, we employ in all cases the tenfoldcross-validation technique, which is widely used in order tomake the most out of limited datasets [21]. Specifically, eachdataset is split into ten stratified folds, whereby 9/10 are used fortraining and the remaining 1/10 is subsequently used for testing;this procedure is repeated in a round robin manner until allfolds are eventually used exactly once for testing. Afterward, theresults obtained from the ten testing sets are averaged in order toprocure the overall performance of the algorithm. The evaluationmetrics used to compare the employed classification schemesare: sensitivity, specificity, and accuracy; sensitivity is defined asthe fraction of correctly identified relapsing patients, specificitymeasures the proportion of disease-free patients predicted asnonrelapsing ones, and accuracy is the weighted average of thesensitivity and specificity denoting the overall correctness of themodel. Moreover, the receiver operating characteristic (ROC)analysis is also adopted to further assess the performance of theemployed classification algorithms [22].
As described in the previous section, several classificationschemes are employed for each source of data, varying in thepreprocessing process as well as the actual classification algo-rithm. Each feature vector is either employed as it is, or afteremploying the CFS and the wrapper algorithm for feature selec-tion; finally, five classifiers are employed in order to discriminatebetween patients with and without disease relapse.
A. Clinical-Based Classification
Table V shows the results obtained when we employ theclinical input vector without performing feature selection.
Next, we employ the CFS algorithm for feature selection thatdesignates the following features as most important: hyperten-sion, family history of malignance, infection, basaloid features,lymphoplasmacytic reaction, lymphovascular invasion, surgi-cal margins, and T-staging. The respective results are shown inTable VI.
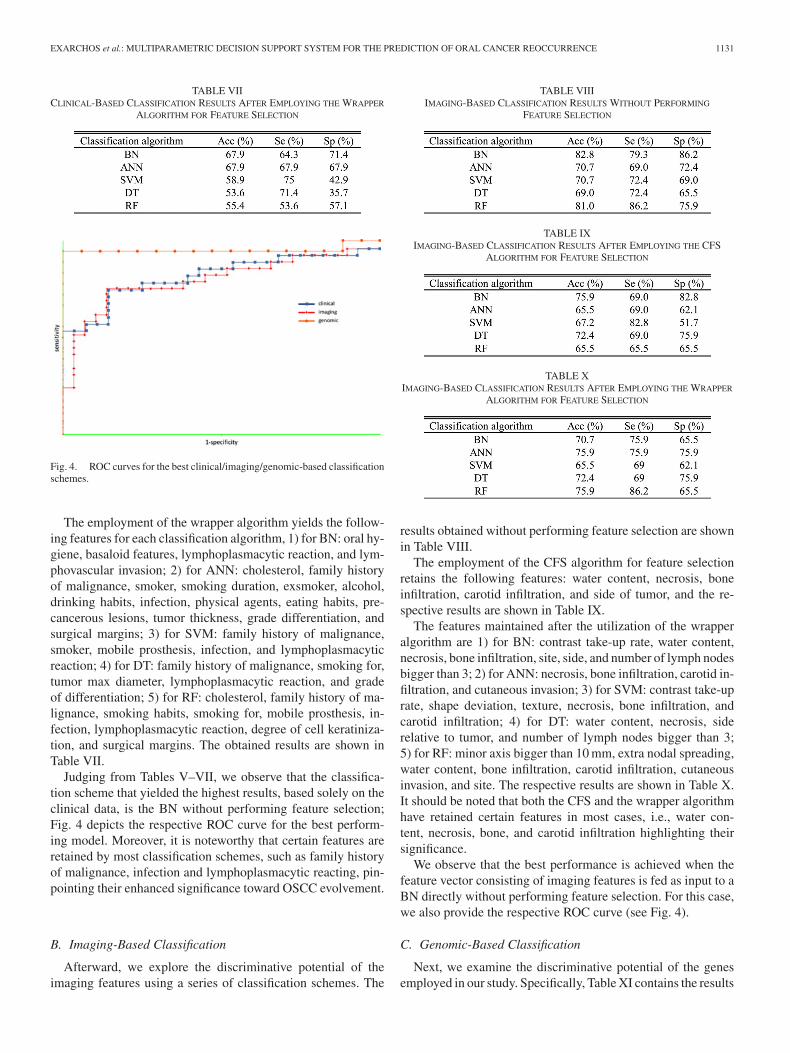
EXARCHOS et al.: MULTIPARAMETRIC DECISION SUPPORT SYSTEM FOR THE PREDICTION OF ORAL CANCER REOCCURRENCE 1131
TABLE VIICLINICAL-BASED CLASSIFICATION RESULTS AFTER EMPLOYING THE WRAPPER
ALGORITHM FOR FEATURE SELECTION
Fig. 4. ROC curves for the best clinical/imaging/genomic-based classificationschemes.
The employment of the wrapper algorithm yields the follow-ing features for each classification algorithm, 1) for BN: oral hy-giene, basaloid features, lymphoplasmacytic reaction, and lym-phovascular invasion; 2) for ANN: cholesterol, family historyof malignance, smoker, smoking duration, exsmoker, alcohol,drinking habits, infection, physical agents, eating habits, pre-cancerous lesions, tumor thickness, grade differentiation, andsurgical margins; 3) for SVM: family history of malignance,smoker, mobile prosthesis, infection, and lymphoplasmacyticreaction; 4) for DT: family history of malignance, smoking for,tumor max diameter, lymphoplasmacytic reaction, and gradeof differentiation; 5) for RF: cholesterol, family history of ma-lignance, smoking habits, smoking for, mobile prosthesis, in-fection, lymphoplasmacytic reaction, degree of cell keratiniza-tion, and surgical margins. The obtained results are shown inTable VII.
Judging from Tables V–VII, we observe that the classifica-tion scheme that yielded the highest results, based solely on theclinical data, is the BN without performing feature selection;Fig. 4 depicts the respective ROC curve for the best perform-ing model. Moreover, it is noteworthy that certain features areretained by most classification schemes, such as family historyof malignance, infection and lymphoplasmacytic reacting, pin-pointing their enhanced significance toward OSCC evolvement.
B. Imaging-Based Classification
Afterward, we explore the discriminative potential of theimaging features using a series of classification schemes. The
TABLE VIIIIMAGING-BASED CLASSIFICATION RESULTS WITHOUT PERFORMING
FEATURE SELECTION
TABLE IXIMAGING-BASED CLASSIFICATION RESULTS AFTER EMPLOYING THE CFS
ALGORITHM FOR FEATURE SELECTION
TABLE XIMAGING-BASED CLASSIFICATION RESULTS AFTER EMPLOYING THE WRAPPER
ALGORITHM FOR FEATURE SELECTION
results obtained without performing feature selection are shownin Table VIII.
The employment of the CFS algorithm for feature selectionretains the following features: water content, necrosis, boneinfiltration, carotid infiltration, and side of tumor, and the re-spective results are shown in Table IX.
The features maintained after the utilization of the wrapperalgorithm are 1) for BN: contrast take-up rate, water content,necrosis, bone infiltration, site, side, and number of lymph nodesbigger than 3; 2) for ANN: necrosis, bone infiltration, carotid in-filtration, and cutaneous invasion; 3) for SVM: contrast take-uprate, shape deviation, texture, necrosis, bone infiltration, andcarotid infiltration; 4) for DT: water content, necrosis, siderelative to tumor, and number of lymph nodes bigger than 3;5) for RF: minor axis bigger than 10 mm, extra nodal spreading,water content, bone infiltration, carotid infiltration, cutaneousinvasion, and site. The respective results are shown in Table X.It should be noted that both the CFS and the wrapper algorithmhave retained certain features in most cases, i.e., water con-tent, necrosis, bone, and carotid infiltration highlighting theirsignificance.
We observe that the best performance is achieved when thefeature vector consisting of imaging features is fed as input to aBN directly without performing feature selection. For this case,we also provide the respective ROC curve (see Fig. 4).
C. Genomic-Based Classification
Next, we examine the discriminative potential of the genesemployed in our study. Specifically, Table XI contains the results
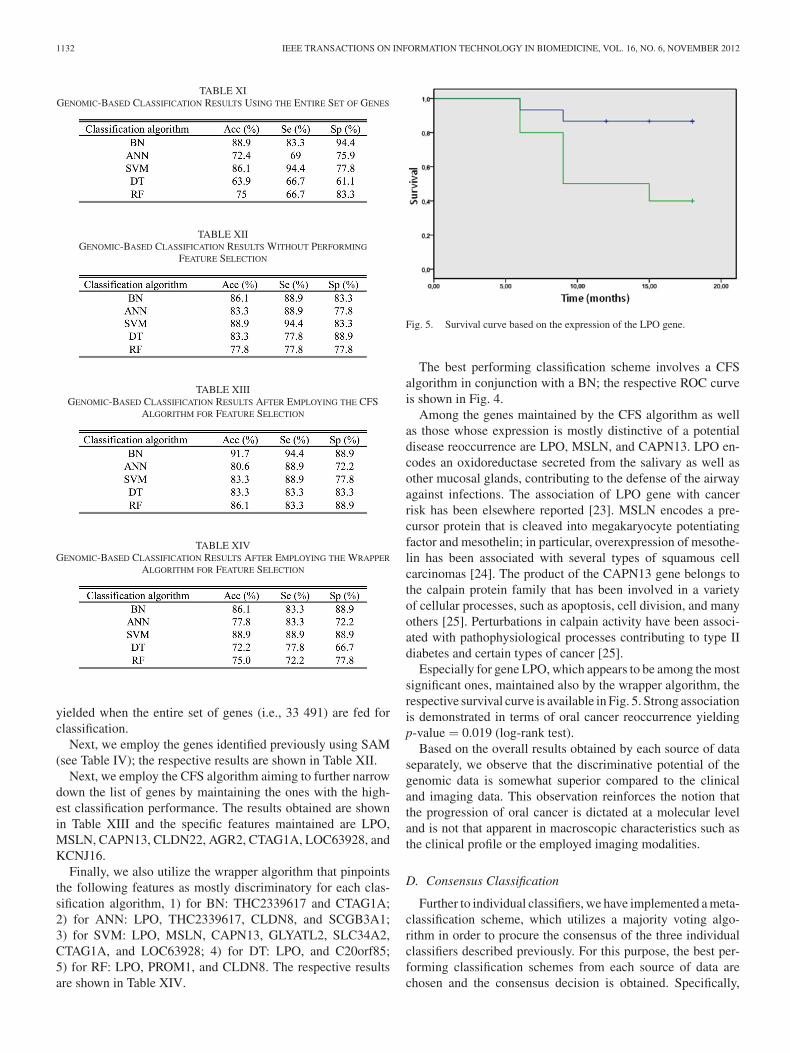
1132 IEEE TRANSACTIONS ON INFORMATION TECHNOLOGY IN BIOMEDICINE, VOL. 16, NO. 6, NOVEMBER 2012
TABLE XIGENOMIC-BASED CLASSIFICATION RESULTS USING THE ENTIRE SET OF GENES
TABLE XIIGENOMIC-BASED CLASSIFICATION RESULTS WITHOUT PERFORMING
FEATURE SELECTION
TABLE XIIIGENOMIC-BASED CLASSIFICATION RESULTS AFTER EMPLOYING THE CFS
ALGORITHM FOR FEATURE SELECTION
TABLE XIVGENOMIC-BASED CLASSIFICATION RESULTS AFTER EMPLOYING THE WRAPPER
ALGORITHM FOR FEATURE SELECTION
yielded when the entire set of genes (i.e., 33 491) are fed forclassification.
Next, we employ the genes identified previously using SAM(see Table IV); the respective results are shown in Table XII.
Next, we employ the CFS algorithm aiming to further narrowdown the list of genes by maintaining the ones with the high-est classification performance. The results obtained are shownin Table XIII and the specific features maintained are LPO,MSLN, CAPN13, CLDN22, AGR2, CTAG1A, LOC63928, andKCNJ16.
Finally, we also utilize the wrapper algorithm that pinpointsthe following features as mostly discriminatory for each clas-sification algorithm, 1) for BN: THC2339617 and CTAG1A;2) for ANN: LPO, THC2339617, CLDN8, and SCGB3A1;3) for SVM: LPO, MSLN, CAPN13, GLYATL2, SLC34A2,CTAG1A, and LOC63928; 4) for DT: LPO, and C20orf85;5) for RF: LPO, PROM1, and CLDN8. The respective resultsare shown in Table XIV.
Fig. 5. Survival curve based on the expression of the LPO gene.
The best performing classification scheme involves a CFSalgorithm in conjunction with a BN; the respective ROC curveis shown in Fig. 4.
Among the genes maintained by the CFS algorithm as wellas those whose expression is mostly distinctive of a potentialdisease reoccurrence are LPO, MSLN, and CAPN13. LPO en-codes an oxidoreductase secreted from the salivary as well asother mucosal glands, contributing to the defense of the airwayagainst infections. The association of LPO gene with cancerrisk has been elsewhere reported [23]. MSLN encodes a pre-cursor protein that is cleaved into megakaryocyte potentiatingfactor and mesothelin; in particular, overexpression of mesothe-lin has been associated with several types of squamous cellcarcinomas [24]. The product of the CAPN13 gene belongs tothe calpain protein family that has been involved in a varietyof cellular processes, such as apoptosis, cell division, and manyothers [25]. Perturbations in calpain activity have been associ-ated with pathophysiological processes contributing to type IIdiabetes and certain types of cancer [25].
Especially for gene LPO, which appears to be among the mostsignificant ones, maintained also by the wrapper algorithm, therespective survival curve is available in Fig. 5. Strong associationis demonstrated in terms of oral cancer reoccurrence yieldingp-value = 0.019 (log-rank test).
Based on the overall results obtained by each source of dataseparately, we observe that the discriminative potential of thegenomic data is somewhat superior compared to the clinicaland imaging data. This observation reinforces the notion thatthe progression of oral cancer is dictated at a molecular leveland is not that apparent in macroscopic characteristics such asthe clinical profile or the employed imaging modalities.
D. Consensus Classification
Further to individual classifiers, we have implemented a meta-classification scheme, which utilizes a majority voting algo-rithm in order to procure the consensus of the three individualclassifiers described previously. For this purpose, the best per-forming classification schemes from each source of data arechosen and the consensus decision is obtained. Specifically,
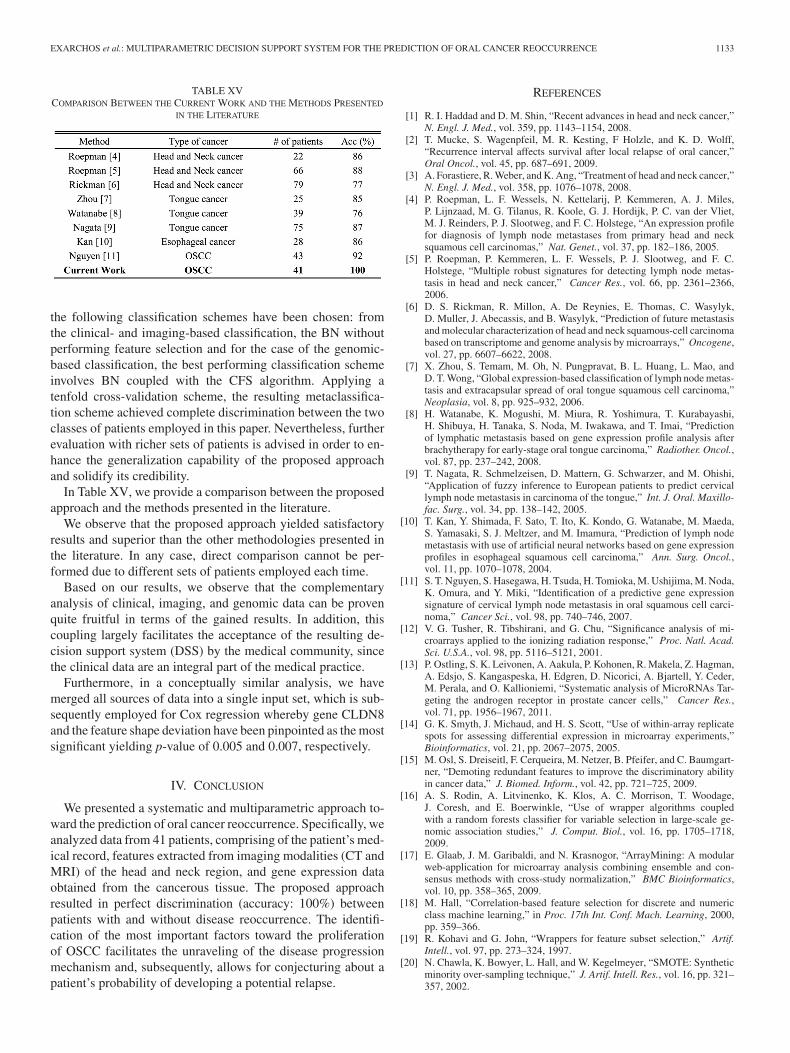
EXARCHOS et al.: MULTIPARAMETRIC DECISION SUPPORT SYSTEM FOR THE PREDICTION OF ORAL CANCER REOCCURRENCE 1133
TABLE XVCOMPARISON BETWEEN THE CURRENT WORK AND THE METHODS PRESENTED
IN THE LITERATURE
the following classification schemes have been chosen: fromthe clinical- and imaging-based classification, the BN withoutperforming feature selection and for the case of the genomic-based classification, the best performing classification schemeinvolves BN coupled with the CFS algorithm. Applying atenfold cross-validation scheme, the resulting metaclassifica-tion scheme achieved complete discrimination between the twoclasses of patients employed in this paper. Nevertheless, furtherevaluation with richer sets of patients is advised in order to en-hance the generalization capability of the proposed approachand solidify its credibility.
In Table XV, we provide a comparison between the proposedapproach and the methods presented in the literature.
We observe that the proposed approach yielded satisfactoryresults and superior than the other methodologies presented inthe literature. In any case, direct comparison cannot be per-formed due to different sets of patients employed each time.
Based on our results, we observe that the complementaryanalysis of clinical, imaging, and genomic data can be provenquite fruitful in terms of the gained results. In addition, thiscoupling largely facilitates the acceptance of the resulting de-cision support system (DSS) by the medical community, sincethe clinical data are an integral part of the medical practice.
Furthermore, in a conceptually similar analysis, we havemerged all sources of data into a single input set, which is sub-sequently employed for Cox regression whereby gene CLDN8and the feature shape deviation have been pinpointed as the mostsignificant yielding p-value of 0.005 and 0.007, respectively.
IV. CONCLUSION
We presented a systematic and multiparametric approach to-ward the prediction of oral cancer reoccurrence. Specifically, weanalyzed data from 41 patients, comprising of the patient’s med-ical record, features extracted from imaging modalities (CT andMRI) of the head and neck region, and gene expression dataobtained from the cancerous tissue. The proposed approachresulted in perfect discrimination (accuracy: 100%) betweenpatients with and without disease reoccurrence. The identifi-cation of the most important factors toward the proliferationof OSCC facilitates the unraveling of the disease progressionmechanism and, subsequently, allows for conjecturing about apatient’s probability of developing a potential relapse.
REFERENCES
[1] R. I. Haddad and D. M. Shin, “Recent advances in head and neck cancer,”N. Engl. J. Med., vol. 359, pp. 1143–1154, 2008.
[2] T. Mucke, S. Wagenpfeil, M. R. Kesting, F Holzle, and K. D. Wolff,“Recurrence interval affects survival after local relapse of oral cancer,”Oral Oncol., vol. 45, pp. 687–691, 2009.
[3] A. Forastiere, R. Weber, and K. Ang, “Treatment of head and neck cancer,”N. Engl. J. Med., vol. 358, pp. 1076–1078, 2008.
[4] P. Roepman, L. F. Wessels, N. Kettelarij, P. Kemmeren, A. J. Miles,P. Lijnzaad, M. G. Tilanus, R. Koole, G. J. Hordijk, P. C. van der Vliet,M. J. Reinders, P. J. Slootweg, and F. C. Holstege, “An expression profilefor diagnosis of lymph node metastases from primary head and necksquamous cell carcinomas,” Nat. Genet., vol. 37, pp. 182–186, 2005.
[5] P. Roepman, P. Kemmeren, L. F. Wessels, P. J. Slootweg, and F. C.Holstege, “Multiple robust signatures for detecting lymph node metas-tasis in head and neck cancer,” Cancer Res., vol. 66, pp. 2361–2366,2006.
[6] D. S. Rickman, R. Millon, A. De Reynies, E. Thomas, C. Wasylyk,D. Muller, J. Abecassis, and B. Wasylyk, “Prediction of future metastasisand molecular characterization of head and neck squamous-cell carcinomabased on transcriptome and genome analysis by microarrays,” Oncogene,vol. 27, pp. 6607–6622, 2008.
[7] X. Zhou, S. Temam, M. Oh, N. Pungpravat, B. L. Huang, L. Mao, andD. T. Wong, “Global expression-based classification of lymph node metas-tasis and extracapsular spread of oral tongue squamous cell carcinoma,”Neoplasia, vol. 8, pp. 925–932, 2006.
[8] H. Watanabe, K. Mogushi, M. Miura, R. Yoshimura, T. Kurabayashi,H. Shibuya, H. Tanaka, S. Noda, M. Iwakawa, and T. Imai, “Predictionof lymphatic metastasis based on gene expression profile analysis afterbrachytherapy for early-stage oral tongue carcinoma,” Radiother. Oncol.,vol. 87, pp. 237–242, 2008.
[9] T. Nagata, R. Schmelzeisen, D. Mattern, G. Schwarzer, and M. Ohishi,“Application of fuzzy inference to European patients to predict cervicallymph node metastasis in carcinoma of the tongue,” Int. J. Oral. Maxillo-fac. Surg., vol. 34, pp. 138–142, 2005.
[10] T. Kan, Y. Shimada, F. Sato, T. Ito, K. Kondo, G. Watanabe, M. Maeda,S. Yamasaki, S. J. Meltzer, and M. Imamura, “Prediction of lymph nodemetastasis with use of artificial neural networks based on gene expressionprofiles in esophageal squamous cell carcinoma,” Ann. Surg. Oncol.,vol. 11, pp. 1070–1078, 2004.
[11] S. T. Nguyen, S. Hasegawa, H. Tsuda, H. Tomioka, M. Ushijima, M. Noda,K. Omura, and Y. Miki, “Identification of a predictive gene expressionsignature of cervical lymph node metastasis in oral squamous cell carci-noma,” Cancer Sci., vol. 98, pp. 740–746, 2007.
[12] V. G. Tusher, R. Tibshirani, and G. Chu, “Significance analysis of mi-croarrays applied to the ionizing radiation response,” Proc. Natl. Acad.Sci. U.S.A., vol. 98, pp. 5116–5121, 2001.
[13] P. Ostling, S. K. Leivonen, A. Aakula, P. Kohonen, R. Makela, Z. Hagman,A. Edsjo, S. Kangaspeska, H. Edgren, D. Nicorici, A. Bjartell, Y. Ceder,M. Perala, and O. Kallioniemi, “Systematic analysis of MicroRNAs Tar-geting the androgen receptor in prostate cancer cells,” Cancer Res.,vol. 71, pp. 1956–1967, 2011.
[14] G. K. Smyth, J. Michaud, and H. S. Scott, “Use of within-array replicatespots for assessing differential expression in microarray experiments,”Bioinformatics, vol. 21, pp. 2067–2075, 2005.
[15] M. Osl, S. Dreiseitl, F. Cerqueira, M. Netzer, B. Pfeifer, and C. Baumgart-ner, “Demoting redundant features to improve the discriminatory abilityin cancer data,” J. Biomed. Inform., vol. 42, pp. 721–725, 2009.
[16] A. S. Rodin, A. Litvinenko, K. Klos, A. C. Morrison, T. Woodage,J. Coresh, and E. Boerwinkle, “Use of wrapper algorithms coupledwith a random forests classifier for variable selection in large-scale ge-nomic association studies,” J. Comput. Biol., vol. 16, pp. 1705–1718,2009.
[17] E. Glaab, J. M. Garibaldi, and N. Krasnogor, “ArrayMining: A modularweb-application for microarray analysis combining ensemble and con-sensus methods with cross-study normalization,” BMC Bioinformatics,vol. 10, pp. 358–365, 2009.
[18] M. Hall, “Correlation-based feature selection for discrete and numericclass machine learning,” in Proc. 17th Int. Conf. Mach. Learning, 2000,pp. 359–366.
[19] R. Kohavi and G. John, “Wrappers for feature subset selection,” Artif.Intell., vol. 97, pp. 273–324, 1997.
[20] N. Chawla, K. Bowyer, L. Hall, and W. Kegelmeyer, “SMOTE: Syntheticminority over-sampling technique,” J. Artif. Intell. Res., vol. 16, pp. 321–357, 2002.

1134 IEEE TRANSACTIONS ON INFORMATION TECHNOLOGY IN BIOMEDICINE, VOL. 16, NO. 6, NOVEMBER 2012
[21] P.-N. Tan, M. Steinbach, and V. Kumar, Introduction to Data Mining, 1sted. Reading, MA: Addison-Wesley, 2006.
[22] T. Fawcett, “An introduction to ROC analysis,” Pattern Recog. Lett.,vol. 27, pp. 861–874, 2006.
[23] P. Bhatti, P. A. Stewart, A. Hutchinson, N. Rothman, M. S. Linet, P. D.Inskip, and P. Rajaraman, “Lead exposure, polymorphisms in genes relatedto oxidative stress, and risk of adult brain tumors,” Cancer EpidemiolBiomarkers Prev., vol. 18, pp. 1841–1848, 2009.
[24] C. Y. Liu, M. C. Wu, F. Chen, M. Ter-Minassian, K. Asomaning, R. Zahi,Z. Wang, L. Su, R. S. Heist, M. H. Kulke, X. Lin, G. Liu, and D. C.Christiani, “A large-scale genetic association study of esophageal adeno-carcinoma risk,” Carcinogenesis, vol. 31, pp. 1259–1263, 2010.
[25] T. N. Dear and T. Boehm, “Identification and characterization of two novelcalpain large subunit genes,” Gene, vol. 274, pp. 245–252, 2001.
Konstantinos P. Exarchos was born in Ioannina,Greece, in 1983. He received the Diploma degreein computer science from the University of Ioannina,Ioannina, Greece, in 2006 and the Ph.D. degree fromthe Medical School of the University of Ioannina.His Ph.D. thesis is entitled “Processing and anal-ysis of biological data using intelligent computa-tional methods.” During his studies he has receivedseveral scholarships and fellowships from the StateScholarship Foundation as well as other national andinternational organizations.
Currently, he is with the Unit of Medical Technology and Intelligent Infor-mation Systems. His research interests include bioinformatics, computationalbiology, biomedical engineering and decision support systems in healthcare. Hehas worked in several R&D projects and has published more that 30 papers inscientific journals, conferences and books.
Yorgos Goletsis (M’03) received the Diploma degreein electrical engineering and the Ph.D. degree in op-erations research, both from the National TechnicalUniversity of Athens, Athens, Greece.
He is a Lecturer in the Department of Economics,University of Ioannina, Ioannina, Greece. His re-search interests include operations research, decisionsupport systems, multicriteria analysis, quantitativeanalysis, data mining, artificial intelligence, projectevaluation.
Dimitrios I. Fotiadis (M’01–SM’06) received theDiploma degree in chemical engineering from theNational Technical University of Athens, Athens,Greece, in 1985, and the Ph.D. degree in chemical en-gineering and materials Science from the Universityof Minnesota, Twin Cities, Minneapolis, MN, in1990.
He is currently a Professor of biomedical engi-neering in the Department of Materials Science andEngineering, the Director of the Unit of MedicalTechnology and Intelligent Information Systems,
Department of Materials Science and Engineering, University of Ioannina, Ioan-nina, Greece. He was a Visiting Researcher at the RWTH, Aachen, Germanyand the Massachusetts Institute of Technology, Boston, MA. He has publishedmore than 140 papers in scientific journals, 270 papers in peer-reviewed con-ference proceedings, and more than 25 chapters in books. He is the editor of16 books. His research interests include modeling of human tissues and organs,intelligent wearable devices for automated diagnosis and bioinformatics. Hiswork has received more than 1,500 citations.
Prof. Fotiadis is an Affiliated Member of FORTH, Biomedical ResearchInstitute. He is an Associate Editor in the IEEE TRANSACTIONS ON INFORMA-TION TECHNOLOGY IN BIOMEDICINE.