morbo di parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
TRANSCRIPT

Morbo di Parkinson idiopatico:aspetti clinici, diagnostici e terapeutici
F. Viallet, D. Gayraud, B. Bonnefoi, L. Renie, R. Aurenty
Il morbo di Parkinson idiopatico viene considerato come un’entità autonoma a causa dei suoi aspetticlinici e terapeutici peculiari che, tuttavia, lo distinguono solo in parte dalle altre sindromi parkinsoniane.Esso si caratterizza anche e soprattutto per il suo scenario eziologico e patogenetico ipotizzato,abbastanza specifico da conferirgli una storia naturale particolare e da delimitare il suo profilonosografico. Nell’ultimo decennio la progressione delle conoscenze, specialmente nel campo dellabiologia molecolare, ha permesso l’identificazione di molte forme genetiche, mentre la comparsa diipotesi fisiopatologiche feconde favoriva lo sviluppo, spettacolare nei suoi risultati terapeutici, dellaneurostimolazione del nucleo subtalamico, parallelamente all’introduzione del concetto farmacologico distimolazione dopaminergica continua. Dopo la consensus conference sul morbo di Parkinson,organizzata nel 2000 dalla Fédération française de neurologie, l’interesse che i neurologi in attività e ineurologi in formazione prestano alla gestione dei malati parkinsoniani non si è smentito: in effetti, l’iterclinico di osservazione e di valutazione resta ancora il principale determinante della qualità dei risultatiterapeutici e la base indispensabile di ogni ricerca epidemiologica e fisiopatologica.© 2010 Elsevier Masson SAS. Tutti i diritti riservati.
Parole chiave: Morbo di Parkinson idiopatico; Antiparkinsoniani; Tremori; Acinesia; Rigidità; L-dopa
Struttura dell’articolo
¶ Storia del morbo di Parkinson dall’origine ai nostri giorni 1An essay on the Shaking Palsy (1817) 1Evoluzione delle idee dopo il 1817 2Morbo di Parkinson nel XXI secolo 5
¶ Aspetti eziologici e patogenetici: ipotesi attuali 6Caratteristiche epidemiologiche 6Ipotesi eziologiche 7Meccanismi di morte dei neuroni dopaminergici 9Concezioni fisiopatologiche 10
¶ Aspetti clinici e diagnostici del morbo di Parkinson idiopatico 12Sintomi della malattia all’esordio 12Sintomi della malattia conclamata 13Sintomi della malattia avanzata 16Criteri di diagnosi clinica 17Ruolo degli accertamenti 18Diagnosi differenziale del morbo di Parkinson 20
¶ Aspetti terapeutici: gestione dei pazienti parkinsoniani 20Farmaci antiparkinsoniani 21Riabilitazione funzionale 22Trattamenti chirurgici 23Scale di valutazione utili 23Indicazioni terapeutiche 23
■ Storia del morbo di Parkinsondall’origine ai nostri giorni [1]
An essay on the Shaking Palsy (1817)Per definizione, il primo riferimento esplicito al morbo di
Parkinson è costituito dalla monografia del 1817 intitolata Anessay on the Shaking Palsy [2]. La descrizione principale postanell’apertura del lavoro caratterizza la malattia attraverso lacoesistenza apparente di due sintomi precisi, il tremore a riposoe la deambulazione festinante, che compare nel contesto di unariduzione della forza muscolare e in assenza di deficit intellet-tivo (Fig. 1). I dati precedenti probabilmente correlati a questamalattia sono riferiti quasi esclusivamente da Parkinson stessonel suo sviluppo, dedicato, da una parte, al tremore a riposo(tremor coactus) e, dall’altra, alla deambulazione festinante(scelotyrbe festinans). In realtà, le allusioni più antiche altremore a riposo sono state riscontrate nell’antico sistemamedico indù (detto Ayurveda che significa «scienza della vita» insanscrito) che risale all’anno 1000 a. C., sotto il nome diKampavata [3], molto prima di quelle degli scritti di Galeno (129-199). Secondo Parkinson, l’individualizzazione del tremore ariposo deve essere attribuita a De le Boë (1680), la descrizionedella deambulazione festinante risale a Gaubius (1758) e la suaconferma esplicita a Boissier de Sauvages (1768). Il contributocapitale di Parkinson resta dunque quello di avere affermato chela coesistenza di questi due sintomi con un deficit muscolarepoteva corrispondere a un’entità nosologica che egli ha chia-mato, in una sapiente abbreviazione, la «paralisi agitante». Lasua argomentazione si basa sulla descrizione clinica di sei casidi cui uno solo venne seguito per un lungo periodo, mentre glialtri cinque (di cui due incontrati in strada e uno osservato a
¶ I – 17-060-A-50
1Neurologia

distanza) hanno dato luogo solo a brevi presentazioni. Nonos-tante queste constatazioni quasi aneddotiche la storia naturaledella paralisi agitante è magistralmente descritta in alcunepagine, di cui questi estratti significativi illustrano le tappeevolutive:• «l’inizio insidioso di una sensazione di disagio con tremore,
il più delle volte localizzato a un arto superiore e che sidiffonde in alcuni mesi ad altre parti del corpo;
• la difficoltà a mantenere una postura eretta, soprattutto nelladeambulazione, associata a una grande difficoltà a compieredei movimenti precisi (scrittura);
• la comparsa di cadute per squilibrio alla deambulazione efestinazione incontrollata che conduce all’allettamento conipersalivazione, disturbi della deglutizione e incontinenzasfinterica».Evocando gli aspetti terapeutici di questa nuova malattia,
Parkinson formula una constatazione poco incoraggiante: «lamalattia è generalmente considerata come l’espressione di unariduzione irrimediabile dell’influsso nervoso derivantedall’invecchiamento».
Tuttavia, egli mitiga questo pessimismo proponendo al lettoreil caso di un paziente che soffriva di sintomi che richiamavanola paralisi agitante e che fu alleviato con l’applicazione divescicanti e la somministrazione di sali di mercurio a scopopurgativo, e questo risultato suggeriva di prendere in conside-razione «una certa influenza misteriosa del sistema simpatico».
Evoluzione delle idee dopo il 1817L’evoluzione delle idee si è costituita con grandi ondate
successive, di cui si può ritenere che lo scopo comune fu quellodi concorrere all’individuazione più precisa possibile delprocesso patologico e, quindi, della causa della malattia, inmodo da poterne definire la terapia. Storicamente, in effetti, losviluppo delle discipline neuroscientifiche si è svolto secondouna cronologia discontinua che ha particolarmente segnato
l’evoluzione dei concetti relativi al morbo di Parkinson. Sipossono così grossolanamente distinguere diversi periodi che sisono sovrapposti e completati successivamente e che hannocorrisposto allo sviluppo rispettivo della disciplina clinica eanatomopatologica in un primo tempo, seguito dalla comparsapiù recente degli approcci biochimico e farmacologico.
Approccio clinico [1, 4, 5]
Dopo Parkinson la paralisi agitante fu riconosciuta e citata inmolti trattati medici, senza contributi innovativi fino a Trous-seau e Charcot. Nella sua quindicesima lezione di clinica medica(1868) Trousseau presenta un’analisi clinica arricchita: gli sideve in particolare una descrizione esplicita della rigidità, unaspiegazione della deambulazione festinante («come se il suocentro di gravità fosse spostato in avanti, il malato deverincorrere se stesso») e il riscontro di un rallentamento progres-sivo nella prova di apertura-chiusura ripetuta della mano. Ilcontributo di Charcot è ancora meglio conosciuto nei suoiscritti in comune con Vulpian e nella sua quinta lezione sullemalattie del sistema nervoso (1872): gli si attribuisce, di solito,l’identificazione della rigidità muscolare e la denominazione di«morbo di Parkinson»; l’opera di Charcot è stata arricchita damolte illustrazioni di Richer che evidenziano i disturbi posturali.In seguito, furono pubblicati altri studi clinici esaustivi cheprecisavano la storia naturale della malattia (modalità diesordio, variabilità evolutiva e causa di decesso). L’epidemia diencefalite letargica ha avuto in Europa un impatto considere-vole a partire dagli anni Venti, poiché molti sopravvissutisvilupparono un parkinsonismo come sequela, rinforzandol’interesse per i lavori clinici con la trattazione della perdita deimovimenti associati (di Foerster), del rallentamento del movi-mento (di Cruchet) e delle cinesie paradosse (di Babinski). Inquesta rassegna cronologica deve essere sottolineato l’impor-tante contributo di Wilson (1925) in virtù della sua descrizioneesplicita dell’acinesia, riconosciuta, in seguito, come il sintomopiù specifico del morbo di Parkinson: osservata nella scrittura enei movimenti ripetitivi, l’acinesia comporta anche la difficoltàa iniziare il movimento evidenziata con un allungamento deitempi di reazione nei pazienti parkinsoniani, di cui Wilsonricordava la riduzione del bisogno o dell’impulso a fare deimovimenti: «così, la loro motivazione ad agire è alterata». Piùrecentemente, si è imposta la definizione clinica di PurdonMartin (1967) con due sintomi positivi, il tremore e la rigidità,e due sintomi negativi, l’acinesia e la perdita dei riflessiposturali. Le rassegne cliniche attuali riprendono in generequesta concezione dei sintomi motori aggiungendo gli altrisintomi non motori più recentemente documentati (disautono-mia, disturbi sensitivi e disturbi psichici).
Approccio anatomopatologico [1, 4-7]
Come prevedeva Parkinson, l’anatomia patologica ha ampia-mente contribuito a precisare il processo lesionale responsabiledella malattia che egli aveva descritto. Storicamente sembra cheil locus niger di Soemmering, o sostanza nera, sia stata la primastruttura sospettata da Brissaud nel 1895 a proposito diun’osservazione pubblicata da Blocq e Marinesco nel 1893 rela-tiva a un paziente portatore di un tubercoloma situato nellasostanza nera e affetto da un tremore parkinsoniano dell’emi-soma controlaterale; questi autori avevano avuto cura di notareche il fascio piramidale e il braccio connettivo da una parte edall’altra della lesione nigrica non contenevano alcuna fibra indegenerazione [4]. In realtà, è Tretiakoff (1919) che, nella suatesi, dimostra il ruolo determinante delle lesioni nigrichebasandosi sull’esame anatomico del cervello di nove casi dimorbo di Parkinson e di un caso di emiparkinsonismo: inquest’ultimo caso egli evidenziò il processo lesionale (depig-mentazione, perdita neuronale e gliosi) nella sostanza neracontrolaterale al lato clinicamente interessato, il che lo condussea chiamare in causa questa struttura nel controllo del tonomuscolare [5]. Tuttavia, altri autori, basandosi sull’esame dicervelli di pazienti portatori di lesioni vascolari diffuse dellostriato e del pallidum («stati cribrosi») che avevano presentatosintomi di tipo parkinsoniano, proponevano, all’epoca, un
Figura 1. Definizione della paralisi agitante [2].
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
2 Neurologia
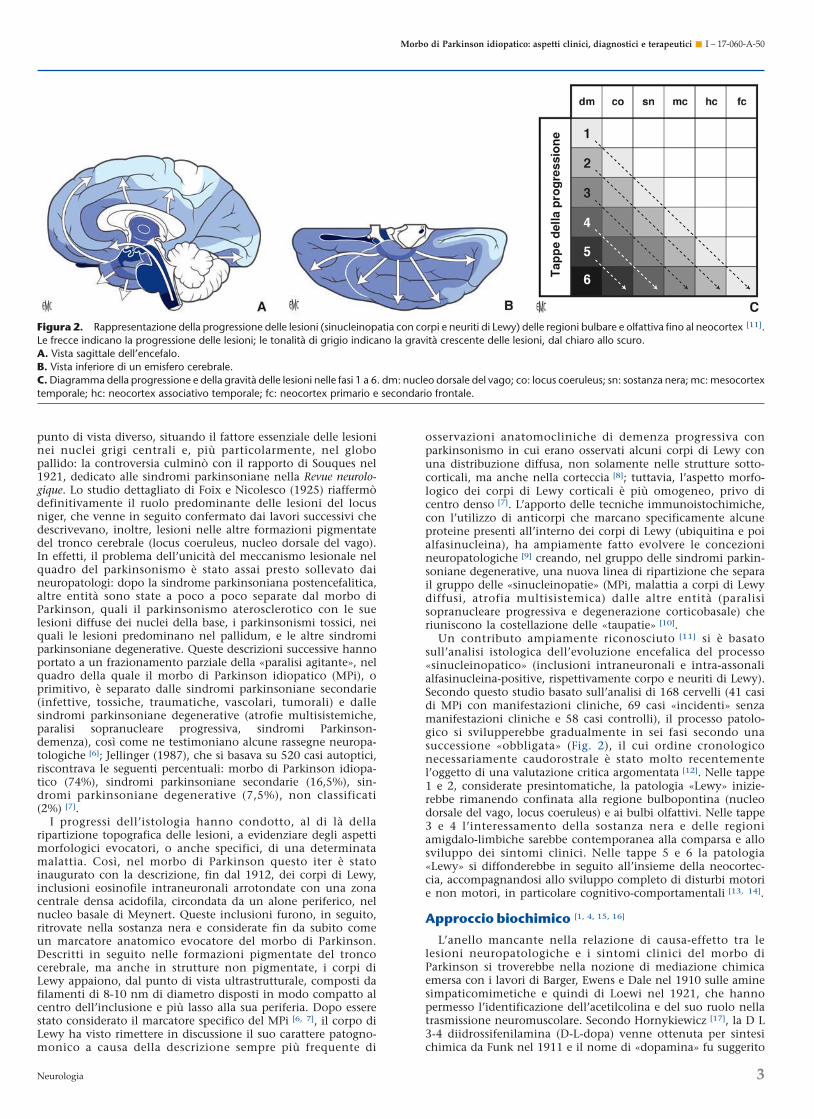
punto di vista diverso, situando il fattore essenziale delle lesioninei nuclei grigi centrali e, più particolarmente, nel globopallido: la controversia culminò con il rapporto di Souques nel1921, dedicato alle sindromi parkinsoniane nella Revue neurolo-gique. Lo studio dettagliato di Foix e Nicolesco (1925) riaffermòdefinitivamente il ruolo predominante delle lesioni del locusniger, che venne in seguito confermato dai lavori successivi chedescrivevano, inoltre, lesioni nelle altre formazioni pigmentatedel tronco cerebrale (locus coeruleus, nucleo dorsale del vago).In effetti, il problema dell’unicità del meccanismo lesionale nelquadro del parkinsonismo è stato assai presto sollevato daineuropatologi: dopo la sindrome parkinsoniana postencefalitica,altre entità sono state a poco a poco separate dal morbo diParkinson, quali il parkinsonismo aterosclerotico con le suelesioni diffuse dei nuclei della base, i parkinsonismi tossici, neiquali le lesioni predominano nel pallidum, e le altre sindromiparkinsoniane degenerative. Queste descrizioni successive hannoportato a un frazionamento parziale della «paralisi agitante», nelquadro della quale il morbo di Parkinson idiopatico (MPi), oprimitivo, è separato dalle sindromi parkinsoniane secondarie(infettive, tossiche, traumatiche, vascolari, tumorali) e dallesindromi parkinsoniane degenerative (atrofie multisistemiche,paralisi sopranucleare progressiva, sindromi Parkinson-demenza), così come ne testimoniano alcune rassegne neuropa-tologiche [6]; Jellinger (1987), che si basava su 520 casi autoptici,riscontrava le seguenti percentuali: morbo di Parkinson idiopa-tico (74%), sindromi parkinsoniane secondarie (16,5%), sin-dromi parkinsoniane degenerative (7,5%), non classificati(2%) [7].
I progressi dell’istologia hanno condotto, al di là dellaripartizione topografica delle lesioni, a evidenziare degli aspettimorfologici evocatori, o anche specifici, di una determinatamalattia. Così, nel morbo di Parkinson questo iter è statoinaugurato con la descrizione, fin dal 1912, dei corpi di Lewy,inclusioni eosinofile intraneuronali arrotondate con una zonacentrale densa acidofila, circondata da un alone periferico, nelnucleo basale di Meynert. Queste inclusioni furono, in seguito,ritrovate nella sostanza nera e considerate fin da subito comeun marcatore anatomico evocatore del morbo di Parkinson.Descritti in seguito nelle formazioni pigmentate del troncocerebrale, ma anche in strutture non pigmentate, i corpi diLewy appaiono, dal punto di vista ultrastrutturale, composti dafilamenti di 8-10 nm di diametro disposti in modo compatto alcentro dell’inclusione e più lasso alla sua periferia. Dopo esserestato considerato il marcatore specifico del MPi [6, 7], il corpo diLewy ha visto rimettere in discussione il suo carattere patogno-monico a causa della descrizione sempre più frequente di
osservazioni anatomocliniche di demenza progressiva conparkinsonismo in cui erano osservati alcuni corpi di Lewy conuna distribuzione diffusa, non solamente nelle strutture sotto-corticali, ma anche nella corteccia [8]; tuttavia, l’aspetto morfo-logico dei corpi di Lewy corticali è più omogeneo, privo dicentro denso [7]. L’apporto delle tecniche immunoistochimiche,con l’utilizzo di anticorpi che marcano specificamente alcuneproteine presenti all’interno dei corpi di Lewy (ubiquitina e poialfasinucleina), ha ampiamente fatto evolvere le concezionineuropatologiche [9] creando, nel gruppo delle sindromi parkin-soniane degenerative, una nuova linea di ripartizione che separail gruppo delle «sinucleinopatie» (MPi, malattia a corpi di Lewydiffusi, atrofia multisistemica) dalle altre entità (paralisisopranucleare progressiva e degenerazione corticobasale) cheriuniscono la costellazione delle «taupatie» [10].
Un contributo ampiamente riconosciuto [11] si è basatosull’analisi istologica dell’evoluzione encefalica del processo«sinucleinopatico» (inclusioni intraneuronali e intra-assonalialfasinucleina-positive, rispettivamente corpo e neuriti di Lewy).Secondo questo studio basato sull’analisi di 168 cervelli (41 casidi MPi con manifestazioni cliniche, 69 casi «incidenti» senzamanifestazioni cliniche e 58 casi controlli), il processo patolo-gico si svilupperebbe gradualmente in sei fasi secondo unasuccessione «obbligata» (Fig. 2), il cui ordine cronologiconecessariamente caudorostrale è stato molto recentementel’oggetto di una valutazione critica argomentata [12]. Nelle tappe1 e 2, considerate presintomatiche, la patologia «Lewy» inizie-rebbe rimanendo confinata alla regione bulbopontina (nucleodorsale del vago, locus coeruleus) e ai bulbi olfattivi. Nelle tappe3 e 4 l’interessamento della sostanza nera e delle regioniamigdalo-limbiche sarebbe contemporanea alla comparsa e allosviluppo dei sintomi clinici. Nelle tappe 5 e 6 la patologia«Lewy» si diffonderebbe in seguito all’insieme della neocortec-cia, accompagnandosi allo sviluppo completo di disturbi motorie non motori, in particolare cognitivo-comportamentali [13, 14].
Approccio biochimico [1, 4, 15, 16]
L’anello mancante nella relazione di causa-effetto tra lelesioni neuropatologiche e i sintomi clinici del morbo diParkinson si troverebbe nella nozione di mediazione chimicaemersa con i lavori di Barger, Ewens e Dale nel 1910 sulle aminesimpaticomimetiche e quindi di Loewi nel 1921, che hannopermesso l’identificazione dell’acetilcolina e del suo ruolo nellatrasmissione neuromuscolare. Secondo Hornykiewicz [17], la D L3-4 diidrossifenilamina (D-L-dopa) venne ottenuta per sintesichimica da Funk nel 1911 e il nome di «dopamina» fu suggerito
Figura 2. Rappresentazione della progressione delle lesioni (sinucleinopatia con corpi e neuriti di Lewy) delle regioni bulbare e olfattiva fino al neocortex [11].Le frecce indicano la progressione delle lesioni; le tonalità di grigio indicano la gravità crescente delle lesioni, dal chiaro allo scuro.A. Vista sagittale dell’encefalo.B. Vista inferiore di un emisfero cerebrale.C. Diagramma della progressione e della gravità delle lesioni nelle fasi 1 a 6. dm: nucleo dorsale del vago; co: locus coeruleus; sn: sostanza nera; mc: mesocortextemporale; hc: neocortex associativo temporale; fc: neocortex primario e secondario frontale.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
3Neurologia

da Dale nel 1952. Secondo Fahn [16], la scoperta della dopadecarbossilasi permise in seguito a Holtz e a Blaschko nel1939 di definire la dopa e la dopamina come metabolitiintermedi sulla via di biosintesi delle catecolamine (noradrena-lina e adrenalina), mentre la tirosina idrossilasi (che converte latirosina in dopa) è stata identificata solo nel 1964 da Nagatsu.Tuttavia, la presenza dei messaggeri chimici nel sistema nervosocentrale rimaneva ancora ipotetica. Inizialmente fu von Eulerche, nel 1946, dimostrò la presenza nel tessuto cerebrale di unasostanza che chiamò «simpatina», le cui proprietà erano similialla noradrenalina. Nello stesso periodo Raab e Gigee, nel 1951,descrissero sotto il nome di «encefalina», un’amina simpatico-mimetica ritenuta diversa dalla simpatina e riscontrata aconcentrazioni elevate nei gangli della base di cervelli umani [4,
16]: in effetti, secondo Fahn [16] Raab e Gigee sarebbero stati iprimi a dimostrare la presenza della dopamina nel cervello nellamisura in cui essi evidenziarono che, tra diverse sostanze testate,solo la L 3-4-idrossifenilalanina (L-dopa), isolata a partire daestratti di fava da Guggenheim nel 1913, poteva aumentare laconcentrazione cerebrale dell’«encefalina». Tuttavia, la lettera-tura classica attribuisce generalmente la scoperta della3-idrossitiramina, o dopamina, nel cervello umano a Carlssonnel 1958. Lavori successivi dimostrarono che la dopamina sitrovava concentrata all’80% nello striato e, grazie alla messa apunto delle tecniche di immunofluorescenza, identificarono lavia nigrostriata. La scoperta in pazienti parkinsoniani di unariduzione considerevole del contenuto di dopamina dello striatoe della sostanza nera a un esame del cervello postmortem, dauna parte, e di una diminuzione dell’escrezione urinaria didopamina dall’altra, confermò l’idea emergente di un ruolofondamentale della dopamina nella patogenesi del morbo diParkinson. A partire da questi dati di base, gli ultimi 20 annihanno visto uno sviluppo molto fecondo della patologiabiochimica, in particolare quella dedicata al morbo di Parkin-son [15]. I risultati sono stati ottenuti con differenti tecniche cheriflettono l’attività dei sistemi biochimici: tasso endogeno delneurotrasmettitore stesso, attività di enzimi di sintesi o didegradazione, tasso di prodotti del metabolismo del neurotras-mettitore e capacità di legame (densità) dei recettori con ligandispecifici. Così, i sistemi dopaminergici si sono rivelati colpitimolto selettivamente nel morbo di Parkinson, con un deficit didopamina che predomina all’interno della via nigrostriatale,dove è superiore all’80%, rispetto ai sistemi mesocorticolimbicoe ipotalamico, che sono colpiti in percentuali dell’ordine del50-60%. Anche gli altri sistemi biochimici sono affetti nelmorbo di Parkinson, ma in maniera meno costante e menomarcata: lo stesso vale per le vie noradrenergiche (che siproiettano dal locus coeruleus verso la neocorteccia e lacorteccia limbica), le vie serotoninergiche (proiezioni dal rafedel tronco cerebrale verso la corteccia da una parte e il midollospinale dall’altra), le vie colinergiche sotto-cortico-corticali(proiezione settoippocampica e sistema ascendente dal nucleobasale di Meynert e dal nucleo peduncolopontino, PPN, versola corteccia frontale e limbica) e anche i sistemi GABAergiciglutamatergici e peptidergici, le cui disfunzioni eventuali si sonorivelate molto più complesse da definire. Il confronto di questabiochimica cerebrale postmortem con i dati clinici ha suggeritol’idea che il morbo di Parkinson si caratterizzasse per un deficitdopaminergico puro progressivamente completato, nel corsodella sua evoluzione, dalla comparsa di lesioni, in parallelo o inserie, di altri sistemi di neurotrasmissione (Fig. 3) [15]. Il recentesviluppo della diagnostica per immagini cerebrale, realizzandoun approccio biochimico in vivo (tomografia a emissione dipositroni e a emissione monofotonica) ha permesso di iniziarea verificare questa ipotesi seguendo, in particolare, l’evoluzionedel deficit dopaminergico nel morbo di Parkinson e valutandoil suo tasso medio di progressione [18]. Accanto allo studio deineurotrasmettitori, l’approccio biochimico si è recentementededicato soprattutto a chiarire i meccanismi della scomparsa deineuroni dopaminergici: così, la nozione di un aumento delcontenuto totale di ferro della sostanza nera è stata confermatain vitro su materiale autoptico e in vivo con risonanza magne-tica (RM) ed ecografia; sono anche state elencate altre anomalie
del metabolismo ossidativo, quali l’aumento della perossida-zione dei lipidi e il deficit dell’attività del complesso I dellacatena respiratoria mitocondriale.
Approccio farmacologico [1, 4]
Si è dovuto attendere un mezzo secolo dopo Parkinsonperché emergessero dall’empirismo le prime terapie che racco-mandavano l’uso degli alcaloidi naturali della belladonna(scopolamina, iosciamina o canapa indiana) sulla base della loroattività simpaticolitica, e ciò fino agli anni Quaranta. L’identifi-cazione dell’acetilcolina come neuromediatore nel sistemanervoso centrale condusse poi rapidamente allo sviluppo disostanze anticolinergiche di sintesi, fin dal 1949 con il triesife-nidile, seguito da molte altre, con un meccanismo d’azione chesi basa sulla loro capacità di fissarsi sui recettori colinergici ditipo muscarinico che predominano nel sistema nervoso cen-trale. Nello stesso periodo, dopo alcuni tentativi di sezione delsistema corticospinale che miravano a interrompere il tremore,ma al prezzo di un’emiparesi, la neurochirurgia si indirizzava ainuclei della base per distruggervi il pallidum interno (GPi) e laregione dell’ansa lenticolare: utilizzando in seguito il metodostereotattico che permetteva di ridurre il trauma chirurgico e dimigliorare la precisione della localizzazione del bersaglio, laricerca dell’efficacia terapeutica sul tremore spostò progressiva-mente il bersaglio verso il nucleo ventrale intermedio (VIM) deltalamo.
La comparsa della dopaterapia alla fine degli anni Sessanta hafondamentalmente e durevolmente modificato il trattamentodel morbo di Parkinson, malgrado tappe preliminari difficili,con l’evoluzione delle idee in questo campo che si confondecon la storia della L-dopa. La levodopa è un aminoacidoaromatico neutro che costituisce un intermedio naturale nellavia di sintesi delle catecolamine a partire dalla L-tirosina diorigine alimentare (Fig. 4). Normalmente prodotta nei neuronidopaminergici grazie all’azione della tirosina-idrossilasi, lalevodopa è trasformata in dopamina sotto l’azione della dopadecarbossilasi. La dopamina è successivamente metabolizzatasotto le azioni congiunte della monoaminossidasi (MAO) e dellacatecol-O-metiltransferasi (COMT). Così, i primi risultati dellasomministrazione di D-L-dopa (miscela racemica) nel morbo diParkinson si rivelarono promettenti nella misura in cui fuosservato un beneficio clinico a piccole dosi (da 150 mg a200 mg) per via venosa. Tuttavia, studi ulteriori non poteronoconfermare queste impressioni iniziali, introducendo il dubbiosul loro carattere occasionale e transitorio; in questo contestomolto critico, l’interesse venne rilanciato dalla pubblicazione dinuovi risultati positivi ottenuti con la somministrazione orale di
Cx
ACH
NA
5HT
DA
VL GLU
GABA
SI
LC
NR
ATV
SN
DA
PALGABA
ME
PU
GABA
GABASPDYN
ACH
NC
Figura 3. Stato dei sistemi di trasmissione biochimica nel morbo diParkinson (modificato da Agid et al. [15]). I sistemi alterati sono rappre-sentati da un tratto discontinuo. ACH: acetilcolina; NC: nucleo caudato;Cx: cortex; DA: dopamina; DIN: dinorfina; GABA: acido gamma-aminobutirrico; GLU: glutamina; 5HT: serotonina; LC: locus coeruleus;ME: metionina-encefalina; NA: noradrenalina; PAL: pallidum; PU: puta-men; NR: nuclei del rafe; SI: sostanza innominata; SN: sostanza nera; SP:sostanza P; VL: nucleo ventrale laterale del talamo; ATV: area tegmentaleventrale.
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
4 Neurologia

D-L-dopa ad alte dosi (12 g/die e oltre), raggiungendo gradual-mente queste posologie per facilitare la tollerabilità digestiva. Inseguito, fu dimostrata l’evidenza dei benefici terapeuticiapportati dalla L-dopa (dal momento che la forma levogira si èrivelata più efficace della miscela racemica) [19]. Una tappaimportante venne quindi raggiunta grazie alla somministrazionecontemporanea alla L-dopa di inibitori periferici della dopadecarbossilasi (benzerazide, carbidopa) che, non oltrepassando labarriera ematoencefalica, permettevano di aumentare conside-revolmente la biodisponibilità della L-dopa e, quindi, didiminuirne dell’80% la dose utile, riducendo anche gli effetticollaterali gastrointestinali e cardiovascolari nella stessa propor-zione. Un altro aspetto importante della farmacologia dellaL-dopa comparve nel corso della somministrazione cronica: sitrattava dell’influenza dell’attraversamento della parete intesti-nale da una parte e della barriera ematoencefalica dall’altra sullabiodisponibilità della L-dopa (Fig. 4). In effetti, questi attraver-samenti hanno dimostrato di dipendere dai sistemi di trasportoattivi saturabili, altamente specifici per gli aminoacidi aromaticineutri: ne derivava che la biodisponibilità della L-dopa potevaessere ridotta attraverso un meccanismo di competizione omediante un apporto massivo di proteine di origine alimentareo mediante l’accumulo di 3-O-metildopa (3-OMD), prodottasotto l’azione della COMT epatica ed eritrocitaria. Le strategieproposte per far fronte a queste difficoltà hanno conosciutofortune diverse: in effetti, le diete povere di proteine si sonorivelate difficilmente applicabili e di efficacia farmacologicamodesta; viceversa, la recente introduzione di inibitori dellaCOMT (entacapone, tolcapone) ha dimostrato un’efficaciaindiscutibile sulla biodisponibilità della L-dopa. Una voltaliberata nello striato, la L-dopa è trasformata in dopamina e puòesercitare la sua azione biologica fissandosi sui recettori dopa-minergici. Una questione importante riguardava la localizza-zione dell’attività dopa-decarbossilasi e, parallelamente, ildeposito della L-dopa a livello dello striato. In effetti, benchéquesta attività dopa-decarbossilasi fosse riscontrata al 90% nelleterminazioni nigrostriatali dopaminergiche, altre terminazionimonoaminergiche, così come neuroni striatali intrinseci e anchedelle cellule di sostegno, potevano partecipare alla decarbossila-zione della L-dopa esogena. Un’altra questione riguardava lanatura dei recettori dopaminergici nello striato; sulla base dicriteri biochimici e farmacologici, sono state descritte due
famiglie di recettori della dopamina [20, 21]: si tratta di recettoriaccoppiati a una proteina G. Il tipo D1, a localizzazionepostsinaptica, è legato positivamente all’adenilato-ciclasi(secondo messaggero), mentre il tipo D2, a localizzazione pre- epostsinaptica, vi è legato negativamente. L’espressione completadell’attività biologica della dopamina richiede l’attivazionesimultanea e sinergica di questi due tipi di recettori striatali [22],d’altra parte situati su sottogruppi diversi di neuroni striatali [23].
Così, la L-dopa, trasformata in dopamina, si è rivelata moltoefficace sull’acinesia e sulla rigidità nel morbo di Parkinson:nella maggior parte dei pazienti, dopo un periodo più o menolungo di notevole efficacia che corrisponde alla fase detta di«luna di miele», è stato osservato un deterioramento dellarisposta terapeutica che conduce gradualmente alla fase detta di«declino motorio». I meccanismi di questo deterioramento sonostati e restano controversi: perdita continua delle terminazionidopaminergiche striatali che riduce la capacità di decarbossila-zione e/o di accumulo della L-dopa esogena, riduzione (orealizzazione di uno stato di scarsa affinità per desensibilizza-zione) dei recettori dopaminergici, principalmente di tipo D2.Questa emergenza progressiva dei problemi legati all’uso dellaL-dopa a lungo corso, trattandosi in particolare di fluttuazionidi efficacia e di movimenti anomali involontari attribuiti piùparticolarmente all’emivita breve della L-dopa, responsabile diuna stimolazione troppo «pulsatile» dei recettori striatali, haportato a rimettere in causa i principi della sua somministra-zione malgrado l’importante progresso farmacologico che essaha rappresentato. Per mantenere e prolungare l’efficacia tera-peutica della stimolazione dopaminergica, diverse soluzionialternative sono state e sono ancora attualmente oggetto distudi, alimentate dal concetto di «stimolazione dopaminergicacontinua»: l’introduzione di forme galeniche a liberazioneprolungata della L-dopa in una forma solubile (per via endove-nosa o duodenale) e, soprattutto, lo sviluppo di sostanzeagoniste della dopamina, che ha costituito un’altra tappafondamentale nell’approccio farmacologico del morbo diParkinson. Gli agonisti misti dei recettori D1 e D2, qualil’amantadina e l’apomorfina, furono i primi utilizzati, poicaddero nel dimenticatoio per alcuni anni prima di conoscerenuovamente un rinnovato interesse per ragioni diverse.L’amantadina si è vista riconoscere un’azione antagonistaglutamatergica che le conferisce una efficacia significativa suimovimenti anomali dopa-indotti [24]; l’apomorfina ha benefi-ciato dell’uso simultaneo del domperidone (che annulla i suoieffetti indesiderati digestivi), il che permette la sua somminis-trazione per via sottocutanea o in modo intermittente (coniniettore a penna) o in maniera continua (con minipompaprogrammabile). Gli altri agonisti della dopamina che sono statisuccessivamente sviluppati hanno in comune la proprietà dilegarsi più selettivamente ai recettori D2: si tratta della bromo-criptina, della lisuride e del pergolide da una parte e, dall’altra,del piribedil, del ropinirolo e del pramipexolo, questi ultimi nonderivati dalla segale cornuta. La loro emivita biologica, piùlunga di quella della L-dopa, ha condotto a proporre il loro usoin associazione con la L-dopa (permettendo di ridurre le dosiefficaci di quest’ultima) o in sostituzione della L-dopa (maspesso al prezzo dell’efficacia terapeutica, in particolare per gliagonisti più selettivi dei recettori D2).
Morbo di Parkinson nel XXI secoloLa constatazione attuale di un’evoluzione molto rapida delle
conoscenze sul morbo di Parkinson, con un impatto positivoconsiderevole sul comportamento dei pazienti e dei lorofamiliari a proposito delle modalità della gestione clinica eterapeutica, ha favorito la presa di coscienza della necessità diuna riflessione professionale approfondita su questo soggetto.L’iniziativa della Fédération française de neurologie per larealizzazione di una consensus conference nel 2000 ha avutoun’ampia partecipazione e ha portato alla pubblicazione deitesti studiati (rassegne bibliografiche commentate, prese diposizione argomentate da parte di esperti) e delle raccomanda-zioni formulate dalla giuria (testo breve e testo esteso) nellaRevue neurologique [25]. Il XXI secolo che inizia vede unosviluppo spettacolare degli indicatori biologici: si tratta, in
TH LAAAD DBH
COMT MAO COMT
COMT MAO
L-tirosina Levodopa Dopamina Noradrenalina
3-O-metildopa DOPAC 3-metossitiramina
HVA A
Stomaco Intestino tenue
LevodopaLAAAD
Vena porta
Circolazionesistemica Cervello
DA
LAAAD
COMT
LAAADDA
Fegato
DA3-OMD
1
2
BFigura 4. Farmacologia della levodopa.A. Vie metaboliche (biosintesi e degradazione).B. Trasporto intestinale ed ematico fino al cervello. COMT: catecol-O-metiltransferasi; DA: dopamina; DBH: dopamina betaidrossilasi; DOPAC:diidrossifenilacetato; LAAAD: decarbossilasi degli aminoacidi aromaticilevogiri; HVA: acido omovanillico; MAO: monoaminoossidasi; 3-OMD:3-O-metildopa; TH: tirosina idrossilasi; 1. Barriera intestinale; 2. Barrieraematoencefalica.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
5Neurologia

particolare, della genetica delle forme familiari, con quasi unadecina di geni identificati [26], e della diagnostica per immaginidella denervazione dopaminergica striatale nella tomografiamonofotonica [27]: questi dati alimentano alcuni progetti distudi longitudinali di coorte di casi presintomatici selezionatisulla base di indicatori clinici precoci (disturbi dell’olfatto,disturbi del comportamento motorio del sonno paradosso) egenetici nelle famiglie con casi indice, successivamente confer-mati con la diagnostica per immagini della denervazionedopaminergica, nell’attesa di misure terapeutiche mirate diprevenzione (o di neuroprotezione) future.
■ Aspetti eziologici e patogenetici:ipotesi attuali
Allo stato attuale delle conoscenze sul morbo di Parkinsonsembra necessario che ogni ipotesi eziologica che si vorrebbeglobale sia compatibile con i seguenti prerequisiti: lesioneselettiva di alcuni individui nel contesto di una popolazione, etàdi insorgenza relativamente tardiva ed evoluzione clinicaprogressiva che giunge dopo un periodo preclinico prolungatoe corrispondente a un processo di morte cellulare che colpiscein modo assolutamente preferenziale i neuroni dopaminergicinelle formazioni pigmentate del mesencefalo. Questi prerequisitifanno riferimento alle caratteristiche epidemiologiche del morbodi Parkinson (prevalenza e storia naturale del processo lesionalee della sua espressione clinica).
Caratteristiche epidemiologiche
Prevalenza
In una rassegna recente [28] le stime di prevalenza del morbodi Parkinson si sono rivelate molto variabili da uno studioall’altro (tra lo 0,1‰ e il 4‰). I fattori che possono influenzarei risultati sono, in particolare, la strategia di ricerca dei casi(inchieste porta a porta, triage iniziale telefonico, studi inambito istituzionale), i criteri di diagnosi utilizzati (la cuispecificità può variare inversamente alla loro sensibilità) e,infine, l’esaustività dell’indagine rispetto alla popolazionebersaglio. Il valore globale, e considerato fino a prova contrariacome un riferimento medio affidabile, è dell’1‰ con unrapporto fra i sessi di 1. Lo studio collaborativo Europarkinsonsi è sforzato di controllare nel modo migliore i fattori divariazione e di differenziare i casi di morbo di Parkinson daglialtri parkinsonismi [29]: i risultati confermano che la prevalenzadel morbo di Parkinson aumenta con l’età dopo i 50 anni [29],rappresentando complessivamente l’1,5% della popolazione dioltre 60 anni. L’analisi per decenni di età suggerisce unaumento di andamento esponenziale che passa dall’1,5‰ (tra i50 e i 59 anni) al 6‰ (tra i 60 e i 69 anni) e, quindi, all’1,5%(tra i 70 e i 79 anni) e al 3% (tra gli 80 e gli 89 anni), con unastima del 6% dopo i 90 anni [30].
Storia naturale
Evoluzione del processo patologico e della sua espressioneclinica (Fig. 5) [25, 31, 32]
La fase sintomatica corrisponde all’evoluzione progressivadell’handicap motorio descritta in cinque stadi di invaliditàcrescente da Hoehn e Yahr nel 1967 [33]. Questo lavoro pionie-ristico sulla storia naturale clinica del morbo di Parkinson(Tabella 1) [32, 33] conserva attualmente tutto il suo interessenella misura in cui è stato realizzato su una popolazione di672 pazienti affetti da morbo di Parkinson e seguiti dal 1949 al1964 prima dell’uso della L-dopa. L’età media di esordio diquesta fase sintomatica è stata stimata a 55 anni e la duratamedia degli stadi evolutivi è valutata pari a 3 anni per gli stadiI e II, a 1 anno per lo stadio III e a 2 anni per lo stadio IV,ovvero un totale di 9 anni per arrivare allo stadio V. Questirisultati sono stati confrontati in una revisione [32] con quelliottenuti da Martilla e Rinne nel 1977 in uno studio finlandese(Tabella 1).
La fase presintomatica [31] è una nozione che è emersarecentemente, tenuto conto dei progressi delle conoscenze sulprocesso di denervazione dopaminergica e sulla sua evolu-zione [25, 34-38]. Il suo inizio è mal definito nel tempo e corris-ponde al momento in cui compare un’accelerazione delprocesso fisiologico di perdita dei neuroni dopaminergici. Il suotermine (che corrisponde all’inizio della fase sintomatica) èanch’esso mal definito nel tempo, poiché l’inizio dell’espres-sione clinica dipende dalla percezione soggettiva di un disturbofunzionale da parte del paziente. È stata suggerita la presenza disintomi discreti (disturbi dell’olfatto, stipsi, modificazionidell’umore e disturbi del comportamento motorio del sonnoparadosso) come indicatori precoci del morbo di Parkinson [39-
44], ma la loro specificità resta insufficiente. La confermadell’esistenza di questa fase presintomatica è stata fornitamediante le indagini in tomografia a emissione di positroni(PET) che riscontra, in alcuni soggetti ancora asintomatici (la cuiulteriore evoluzione dimostrerà che essi sviluppano i segni dellamalattia), un deficit significativo della captazione striatale difluorodopa [45]: per estrapolazione a partire da valutazionilongitudinali, la PET ha permesso di valutare la durata media
Durata della vita (anni)
? Evento non determinato nel tempo e dinatura ignota, responsabile di un'accelerazionedel processo fisiologico di perdita dei neuronidopaminergici nel corso della vita
Pecentuale di neuronidopaminergici che sopravvivono nella sostanza nera
Zona stimata di comparsadei sintomi clinici
Fase pre-sintomatica
Fase sintomatica(stadi di Hoehn e Yahr)
100 %
90 %
80 %
70 %
60 %
50 %
40 %
30 %
20 %
10 %
0 %
0 10 20 30 40 50 60 70 80 90 100 110
7 anni 3 anni 3 anni 1,5 anni 2,5 anni
Età di esordio:55 anni
Evoluzione clinica senza trattamento(durate medie)
St I St II St III St IV St V
Figura 5. Storia naturale del morbo di Parkinson idiopatico(da Langston e Koller [31], Poewe e Wenning [32], Viallet in [25]).
Tabella 1.Stadi evolutivi della fase sintomatica del morbo di Parkinson: descrizione edurata media.
Stadio Descrizione della disabilità Durata media (anni)
A B
I Lesione monolaterale con disturbofunzionale minimo o nullo
3 2,9
II Lesione bilaterale o assiale senzaalterazione dell’equilibrio
3 2,6
III Comparsa di un’alterazionedell’equilibrio ai cambiamentidi direzione o alla prova della spinta(piedi uniti, occhi chiusi): esisteun disturbo funzionale certo,ma il proseguimento del lavoro restapossibile a seconda del tipo di impiego;l’autonomia è conservata
1 2
IV Sviluppo completo della malattiacon disabilità grave: la stazione erettae la deambulazione senza aiuto sonoancora possibili, ma con grandidifficoltà
2 2,2
V Senza supporto, il paziente rimaneconfinato alla sedia rotelle o al letto
- -
A: da Hoehn e Yahr [33]; B: da Martilla e Rinne, in [32].
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
6 Neurologia

della fase presintomatica a circa 7 anni [46], il che lascia,tuttavia, sospettare una grande variabilità interindividuale. Altrevalutazioni della durata della fase presintomatica sono stateeffettuate sulla base della ricerca di sintomi non specifici (ansia,superlavoro) che motivano un consumo di farmaci [47] o perestrapolazione a partire dalle lesioni neuropatologiche [34]
(Tabella 2) [34, 46, 47]. I dati più recenti riguardo agli indicatoriclinici di questo periodo presintomatico quali l’iposmia, lastipsi, la depressione e i disturbi del comportamento motoriodel sonno paradosso, considerando in particolare le ipotesipatogenetiche sulla progressione ascendente delle lesioni neltronco cerebrale [25] e sull’interessamento precoce del sistemaolfattivo [48], o anche dell’innervazione del tubo digerente [39,
49], dovrebbero apportare nuove indicazioni su questa tappaessenziale della storia naturale del morbo di Parkinson.
Specificità del processo lesionale
La comparsa della degenerazione dei neuroni dopaminergicinei pazienti non può essere datata con precisione, ma ilprocesso si estende su diversi decenni. I neuroni muoiono inmodo asincrono ma non aleatorio: in effetti, il processo èeterogeneo, con una perdita neuronale massiva (del 70-80%)nella sostanza nera compatta (SNpc o A9), intermedia (del40-50%) nelle regioni dell’area tegmentoventrale (A10) edell’area retro- e perirubrale (A8) e quasi nulla nella sostanzagrigia periacqueduttale. La distribuzione nel tempo mostra cheil processo inizia nella parte caudale e ventrolaterale della SNpce si estende poi progressivamente verso le regioni rostrale,mediale e dorsale del mesencefalo [35], in aree scarsamentemarcate dalla calbindina chiamate «nigrosomi» [50]. La mortalitàdifferenziale dei neuroni dopaminergici sembra essere correlataal loro contenuto di melanina (fattore di aggravamento), mainversamente correlata alla presenza di un ambiente astrocitariodenso (fattore di protezione).
Ipotesi eziologicheRuolo dell’invecchiamento del sistemadopaminergico
Questo invecchiamento è una realtà confermata dall’eviden-ziazione di una riduzione della dopamina striatale in funzionedell’età [37] e stimata al 5% di perdita neuronale per decen-nio [51]. Il suo ruolo nel morbo di Parkinson può tuttavia essereconsiderato soltanto marginale, come suggeriscono studianatomici [34] che mostrano una topografia delle lesioni dopa-minergiche (predominanza nella parte dorsale della sostanzanera) molto diversa da quella osservata nel morbo di Parkinson.Peraltro, uno studio anatomico suggerisce che la perditaneuronale può rimanere molto discreta anche in soggettinormali molto anziani [52].
Ruolo dei fattori ambientaliTra questi fattori, solo gli agenti tossici rappresentano una
pista ampiamente studiata sulla base di argomenti epidemiolo-gici confermati e di ipotesi biochimiche coerenti con i dati dimodelli sperimentali. L’ipotesi virale e/o immunitaria non sibasa attualmente su alcun argomento tangibile e il ruolo deitraumi cranici in senso lato resta controverso [51].
Intossicazione da 1-metil-4-fenil-1,2,3,6-tetraidropiridina(MPTP)
Sintetizzata fin dal 1947 e anche utilizzata in alcuni studinell’animale come agente antiparkinsoniano nel corso degli
anni Cinquanta, la MPTP aveva mostrato effetti particolarmentedisastrosi che avevano fatto abbandonare l’idea che questoprodotto potesse essere un agente terapeutico. Tuttavia, pocotempo più tardi, una molecola molto simile, la meperidina(MPPP), iniziò a essere sintetizzata clandestinamente conl’obiettivo di un uso illecito, poiché questo prodotto haproprietà narcotiche ed è relativamente facile da produrre. Ilprimo caso di parkinsonismo indotto fu osservato in un giovanestudente di 23 anni che aveva consumato MPPP per via endo-venosa come sostituto dell’eroina per 6 mesi nel 1976; inseguito a un’accelerazione accidentale della procedura di sintesila droga risultò contaminata da MPTP, che determinò lacomparsa rapida in questo soggetto giovane di una sindromeparkinsoniana grave, con decesso 2 anni più tardi e una perditaneuronale limitata alla sostanza nera evidente all’autopsia. Unaltro caso di un soggetto giovane e tossicodipendente che avevasintetizzato della MPPP contaminata da MPTP e l’aveva consu-mata per via nasale fu osservato nel 1980 a Vancouver: anchequesto soggetto divenne parkinsoniano e morì 2 anni più tardi.In queste due osservazioni iniziali il meccanismo dell’intossica-zione non era stato provato chiaramente, il che spiega perché,nell’estate del 1982, la fabbricazione e la vendita illecita dellaMPPP come sostituto sintetico dell’eroina si svilupparono sugrande scala nel nord della California, moltiplicando il rischiodi dosi contaminate dalla MPTP. In effetti, nei mesi successividiversi giovani tossicomani furono ricoverati in questa regioneper sindrome parkinsoniana grave, nella genesi della quale furapidamente incriminata la MPTP. In seguito a quest’ultimapubblicazione, che raggruppava sette osservazioni, la rivelazionedel caso di un chimico che lavorava nell’industria farmaceuticasulla MPTP utilizzata come intermedio chimico nella sintesi diprodotti analgesici e che aveva sviluppato un morbo di Parkin-son all’età di 38 anni suggerì l’ipotesi del ruolo della MPTPcome fattore ambientale del morbo di Parkinson [53]. Ilfollow-up ulteriore di un gruppo di 40 persone che soddisface-vano i criteri di un’esposizione certa al prodotto (uso delprodotto sospetto nel nord della California tra il gennaio el’agosto del 1982, sensazione di bruciore nel punto di iniezionee presenza di sintomi della serie parkinsoniana in modotransitorio nei giorni successivi all’iniezione) ha permesso diconfermare molte similitudini cliniche e farmacologiche con ilmorbo di Parkinson, con la metà del gruppo che aveva comin-ciato a presentare dei sintomi progressivi suggestivi dopo 2 annidi periodo asintomatico; in tre di essi, deceduti dopo aversviluppato un parkinsonismo grave, l’esame neuropatologico hariscontrato lesioni molto simili a quelle del morbo di Parkin-son [54], ma con le seguenti particolarità: assenza di corpi diLewy, lesione selettiva della sostanza nera che risparmia il locuscoeruleus e importante proliferazione microgliale con accumuloextracellulare di neuromelanina. Negli anni seguenti sono stateportate a termine tappe importanti nella conoscenza delmeccanismo di azione della MPTP grazie all’uso di modellianimali (roditori, primati). Così, è stato dimostrato che dopouna somministrazione sistemica la MPTP deve superare labarriera ematoencefalica per essere trasformata, sotto l’azionedella MAOB intracerebrale, in MPP+, che rappresenta la veraneurotossina. La MPP+ è, quindi, introdotta nei neuronidopaminergici grazie al sistema del reuptake selettivo delladopamina e si lega con la neuromelanina che la libera, inseguito, progressivamente: captata dai mitocondri, la MPP+eserciterà la sua azione tossica bloccando il complesso I dellacatena respiratoria e provocando la produzione di radicali liberi(nozione di «stress ossidativo»). Un altro modello animale, cheutilizza un insetticida inibitore del complesso I, il rotenone,somministrato in modo sistemico e il cui processo di reuptakenon è specifico del trasportatore della dopamina, è statosviluppato nei roditori e nei primati [55].
Pesticidi
Sull’esempio di quanto mostrato dalla MPTP, la ricerca di unatossina ambientale responsabile del morbo di Parkinson haprivilegiato l’analogia strutturale con la MPP+ (paraquat) o, piùrecentemente, l’analogia funzionale con l’azione della MPP+ sulcomplesso I mitocondriale (rotenone), dal momento che
Tabella 2.Durata stimata della fase presintomatica (da Gonera et al. [47], Fearnley eLees [34], Morrish et al. [46]).
Metodo di stima Durata
Indagini cliniche retrospettive 4-6 anni
Estrapolazione neuropatologica 4,7 anni
Estrapolazione sulle immagini PET 7 anni
PET: tomografia a emissione di positroni.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
7Neurologia
quest’ultima è confermata in modelli sperimentali [55]. Leindagini epidemiologiche si sono moltiplicate [51], fornendo avolte risultati contraddittori ma rilevando ripetutamenteun’aumentata prevalenza del morbo di Parkinson in regionifortemente industrializzate (industrie chimiche) o di agricolturaintensiva (pesticidi, erbicidi): si è trovato che il consumo diacqua di pozzo, che si suppone concentri i tossici contenutinelle acque di scorrimento, sia correlato a una prevalenza piùelevata di morbo di Parkinson senza che le analisi dell’acquapermettano di identificare alcuna sostanza tossica sospetta.
Genetica e morbo di ParkinsonL’incidenza familiare nel morbo di Parkinson è generalmente
stimata pari al 10%. L’approccio genetico si è basato su trecategorie di studi: studi epidemiologici per i piccoli aggregatifamiliari, studi del tasso di concordanza per i gemelli omozigotie dizigoti e, infine, studi di legame genetico per cosegregazionetra marker citogenetici e fenotipi parkinsoniani nelle grandifamiglie multigenerazionali dalla modalità di trasmissionedominante e in famiglie più limitate dalla modalità di trasmis-sione recessiva di un morbo di Parkinson a esordio precoce.
Epidemiologia degli aggregati familiariIn seguito al lavoro pionieristico di Mjönes nel 1949 erano
state sollevate numerose critiche a proposito dell’imprecisionedei dati clinici a causa delle frontiere mal definite del morbo diParkinson propriamente detto con i tremori isolati da una partee con i casi affetti da disturbi mentali dall’altra: altre criticheerano state formulate a proposito delle distorsioni di selezionedei casi-indice e dell’incertezza della diagnosi dei casi secondari,raramente esaminati. Alcuni studi epidemiologici controllati piùrecenti hanno tuttavia piuttosto confermato che i fenotipiclinici osservati negli aggregati familiari di morbo di Parkinsonnon erano significativamente diversi dal fenotipo clinico mediodel morbo di Parkinson sporadico. Inoltre, l’esplorazione in PETha riscontrato una riduzione della fissazione striatale di18-fluorodopa in soggetti asintomatici o che presentano untremore posturale isolato imparentati con malati parkinsoniani.Infine, il rischio di sviluppare un morbo di Parkinson è statotrovato più elevato nei soggetti imparentati con un pazienteparkinsoniano, con una predominanza di trasmissione verticalemonolaterale. Questi dati hanno permesso di concludere che ilfenotipo parkinsoniano potrebbe trasmettersi per segregazionedi geni dominanti con una penetranza incompleta.
Studi su gemelli
Un primo studio su 65 coppie di gemelli e una fratria diquadrigemini (19 monozigoti, 48 dizigoti), che evidenziava tassidi concordanza molto bassi identici nei monozigoti e neidizigoti, aveva contribuito a scartare l’ipotesi genetica nelmorbo di Parkinson; tuttavia, la possibilità di individuare formeprecliniche di denervazione dopaminergica striatale con glistudi PET e l’accresciuta precisione dei dati clinici riguardanti icasi-indice hanno permesso di riscontrare successivamente tassidi concordanza più elevati in queste coppie di gemelli, masenza un aumento significativo nei monozigoti. I risultati diquesti studi sono, in definitiva, considerati compatibili con uncontributo genetico nell’eziologia del morbo di Parkinson, maconfermano l’importanza dei fattori non genetici. Questaconstatazione è stata ancora rinforzata da un nuovo studio su161 coppie di gemelli (71 monozigoti, 90 dizigoti), il cui tassodi concordanza è più elevato nei monozigoti, considerando soloi casi esorditi prima dei 50 anni [56]: così, l’importanza deifattori genetici pare prevalere tanto più quanto più precoce èl’esordio del morbo di Parkinson.
Studi di legame genetico (Tabella 3) [26]
Trasmissione autosomica dominante. Le grandi famigliemultigenerazionali sono eccezionali e le loro similitudinifenotipiche, relativamente al fenotipo medio del morbo diParkinson sporadico o a quello degli aggregati familiari dimorbo di Parkinson, restano controverse. Esse hanno tuttaviafornito la possibilità di studi di legame genetico con markerscromosomici di dimensioni sempre più ridotte, beneficiando deiprogressi nella conoscenza del genoma umano. La grandefamiglia italoamericana originaria della città di Contursi nellaprovincia di Salerno nell’Italia meridionale [57] comporta592 membri identificati di cui 60 presentano un fenotipo ditipo parkinsoniano, ma con la particolarità di un’età media diesordio inferiore e di una durata di evoluzione ridotta rispettoa ciò che viene osservato nel morbo di Parkinson sporadico; sulpiano clinico, la triade classica (tremori, acinesia, rigidità) e lasensibilità alla L-dopa sono associate frequentemente a disturbimentali gravi; infine, i dati neuropatologici che riscontrano unadegenerazione dei neuroni della sostanza nera con corpi diLewy sono disponibili solo per due soggetti. È anche statadescritta un’altra famiglia grecoamericana nello stato delNebraska, con dati clinici e neuropatologici piuttosto simili a
Tabella 3.Geni implicati nelle forme familiari del morbo di Parkinson: «stato dei luoghi» nel 2008 (da Klein e Schlossmacher [26]).
Nomi Regionecromosomica
Gene/proteina Mutazioni (M) Numero di famiglie Modalità ditrasmissione
Età di esordio(anni)
Corpidi Lewy
Park1/4 4q21-q23 SNCA/
alfasinucleina
3 M puntiformi (A53T,A30P, E46K)
duplicazione, triplicazione
<10 AD 45 +
Park2 6q25.2-q27 Parkina >100 M puntiformi
delezione/moltiplicazionedi esoni
10%-20% dei MP giovanili AR <30 ±
Park3 2 p13 ? ? ? AD 60 +
Park5 4 p14 UCH-L1 Ile 93 Met 1 AD 50 +
Park6 1 p35 - p36 PINK 1 40 M puntiformi
Delezioni di esoni
1%-8% dei MP giovanili AR 40 ?
Park7 1 p36 DJ1 10 M puntiformi
Delezioni di esoni
1%-2% dei MP giovanili AR 33 ?
Park8 12 q12 LRRK2/
dardarina
>16 M puntiformi (la piùconosciuta G2019S)
1,5% dei MP giovanili oppureno (40% Maghreb)
AD 51 +
Park9 1 p36 ATP13A2/
ATPasi lisosomiale
3 M 2 famiglie MP giovanili(sindrome di Kufor-Rakeb)
AR ? ?
Park10 1p ELAV L4
RNF-11
Geni di suscettibilità ? ? ? ?
Park11 2q36-37 GIGYF2? 10 M puntiformi >10 AD ? ?
Park13 2p12 Omi/Htr A2 2 M puntiformi ? AD 57 ?
Park? 2q 22-q23 NR4A2/Nurr1 3 M ? AD 54 ?
MP: morbo di Parkinson; AD: autosomica dominante; AR: autosomica recessiva.
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
8 Neurologia

quelli del morbo di Parkinson sporadico. L’evidenziazione, nellafamiglia di Contursi, di un’associazione genetica con un markercromosomico situato sul cromosoma 4 nella regione 4 q21-q23 ha rappresentato un importante progresso nel contestodell’ipotesi genetica, seguito dall’identificazione di una muta-zione puntiforme (Ala53Thr) sul gene dell’alfasinucleina [58]:questa stessa mutazione è stata riscontrata in altre sei famigliedi origine greca, mentre un’altra mutazione (Ala30Pro) dellostesso gene era descritta in una famiglia tedesca. Più recente-mente, una terza mutazione puntiforme (E46K) è stata segnalatain una famiglia spagnola [59] con un fenotipo clinico didemenza a corpi di Lewy. Queste mutazioni sul gene dell’alfa-sinucleina sono raggruppate sotto l’etichetta di «Park1»(Tabella 3), così come altre anomalie che colpiscono questogene, come duplicazioni [60] o triplicazioni eterozigoti, questeultime corrispondenti a casi riferiti precedentemente sottol’etichetta Park4 [61], con una gravità della sinucleinopatia chesembra proporzionale al numero di copie del gene. Un’altramutazione (Ile93Met), che colpisce il metabolismo dell’ubiqui-tina [62] sul gene dell’idrolasi della parte C terminale dell’ubi-quitina (UCH-L1) è stata anch’essa catalogata sotto l’etichettaPark5; resta ancora un altro locus, il cui gene non è sempreidentificato, situato in 2p13 e catalogato Park3 [63]. Tuttavia,questi risultati riguardano solo un numero ridotto di casi dimorbo di Parkinson familiare, e queste diverse mutazioni nonsono state riscontrate nel morbo di Parkinson sporadico né neipiccoli aggregati familiari.
Negli ultimi 5 anni [64, 65] è emersa un’altra etichetta chesembra avere un’importanza quantitativa molto più grande, nonsolamente nelle forme familiari del morbo di Parkinson, maanche nelle forme sporadiche, con un fenotipo molto sovrap-ponibile a quello del morbo di Parkinson idiopatico: si tratta diPark8 che riguarda il gene della leucin-rich-repeat kinase di tipo2 (LRRK2), il quale comporta 51 esoni con numerose mutazionipuntiformi, di cui la più frequente in Europa e nel Maghreb(fino al 40% di prevalenza nelle forme familiari) è G2019S [66].Questa mutazione è stata riscontrata nell’1,5% dei casi-indicecon morbo di Parkinson a esordio tardivo, e l’espressione dellasinucleinopatia può variare in una stessa famiglia [26].
Infine, altre etichette (Park10, Park11, Park13) e le mutazionidel gene NR4A2 (Nurr1) restano mal determinate, con unapresumibile trasmissione autosomica dominante [26].
Trasmissione autosomica recessiva. Altre anomalie geneti-che che interessano la regione cromosomica 6q25.2-q27 sonostate riscontrate in modo nettamente più frequente in famigliepiù piccole nelle quali il morbo di Parkinson, trasmesso secondomodalità recessiva, comporta un’età di esordio molto precoce,con un’evoluzione forse più lenta. Queste anomalie sonomutazioni puntiformi o delezioni situate su differenti esoni delgene della parkina e corrispondono all’etichetta Park2. Esse sonostate osservate in alcune famiglie giapponesi [67], ma anche infamiglie europee e nordafricane [68], con una frequenza del 50%nelle forme recessive giovanili e del 15% nei casi isolati conesordio prima dell’età di 45 anni; delezioni sugli esoni del genedella parkina sono state osservate in alcune famiglie europeeche presentano un fenotipo di distonia dopasensible, ponendocosì nuovamente il problema già noto dei limiti tra morbo diParkinson giovanile e distonia dopasensible.
Le etichette Park6 [69] e Park7 [70] sono associate rispettiva-mente a mutazioni puntiformi sui geni PTEN-Induced kinase 1(PINK1) e DJ-1, e corrispondono anche a forme recessive dimorbo di Parkinson a esordio giovanile, con frequenze rispetti-vamente dell’1-8% e dell’1-2% [26]. L’etichetta Park9 corrispondea mutazioni del gene dell’ATP13A2 che codifica una ATPasilisosomiale: è stata descritta in due famiglie affette da parkinso-nismi atipici a esordio precoce con segni piramidali e demenzadi progressione rapida che corrisponde alla sindrome diKufor-Rakeb [71].
Interazione genetica-ambienteCome molte malattie croniche la cui prevalenza aumenta con
l’età, il morbo di Parkinson ha un’eziologia multifattoriale nellaquale i fattori genetici e ambientali contribuiscono in propor-zioni variabili a seconda degli individui. Quando l’età di esordio
è precoce, i fattori genetici sono considerati prevalenti, il che èstato confermato dall’aumento significativo del tasso di concor-danza nei monozigoti rispetto ai dizigoti, osservato in unostudio di gemelli nei casi con un’età di esordio inferiore a50 anni [56]; l’evidenziazione di anomalie Park2 in alcuni casisporadici di morbo di Parkinson illustra questo punto in modoancora più netto, nella misura in cui sono state riscontrate dellemutazioni nel 77% dei casi che esordiscono prima dei 20 annie nel 26% dei casi prima dei 30 anni, ma solo nel 3% dei casiche sono esorditi tra i 31 e i 45 anni [68]; lo stesso vale per leanomalie Park8 riscontrate in modo non eccezionale in alcunicasi sporadici di morbo di Parkinson [66]. Tuttavia, nellastragrande maggioranza dei casi l’età di esordio del morbo diParkinson è piuttosto tardiva (dopo i 60 anni); inoltre, lamaggior parte degli studi casi-controlli sull’esposizione a tossiciambientali o sul possibile intervento di fattori endogeni oesogeni suggerisce fortemente un’interazione genetica-ambiente.L’ipotesi di base di una tale interazione si basa sulla nozione dipolimorfismo dei geni di alcuni enzimi che intervengono nelmetabolismo di sostanze ambientali potenzialmente tossiche: glialleli associati a un fenotipo di attività metabolica detossificantelenta o incompleta esporrebbero a un rischio aumentato dimorbo di Parkinson. I primi lavori in questo campo si sonoincentrati sull’idrossilazione della debrisochina da parte delcitocromo P450 [72]: i risultati dei vari studi riguardanti inparticolare il polimorfismo del CYP2D6 sono variabili [73] e lemeta-analisi non permettono di concludere per un’associazioneformale tra la frequenza dell’allele B e un rischio aumentato dimorbo di Parkinson sporadico o familiare; ad oggi, lo studio didifferenti geni candidati a questi ruoli di fattore di suscettibilitào di protezione (geni 2D6 e 1A1 del citocromo P450, geni dellaN-acetiltransferasi 2, del sito trasportatore della dopamina edella glutatione-S-transferasi M1) resta negativo [74]. Infine, altristudi casi-controlli hanno suggerito una correlazione inversa trail consumo di tabacco [75], e più recentemente di caffè [76], e ilrischio di morbo di Parkinson: in assenza di una spiegazionemetabolica, questo risultato si è potuto globalmente assimilarealla personalità premorbosa dei futuri parkinsoniani, ai quali ildeficit asintomatico di dopamina potrebbe conferire una minoresuscettibilità a comportamenti di tipo additivo, senza tuttaviaignorare la notevole variabilità interindividuale in questocampo.
Meccanismi di morte dei neuronidopaminergici [51, 77]
Ruolo dello stress ossidativo (Fig. 6) [1]
Il metabolismo ossidativo della dopamina (azione della MAO,auto-ossidazione) e la funzione energetica della catena respira-toria mitocondriale sono suscettibili di produrre dei «radicaliliberi», agenti potenzialmente citotossici a causa della loroinstabilità elettrochimica (perossidazione dei lipidi di mem-brana, frammentazione dell’acido desossiribonucleico).
Radicali liberi citotossiciSi tratta dell’anione superossido (O2
-), del perossido diidrogeno (H2O2) e del radicale idrossile (OH•), quest’ultimoparticolarmente tossico essendo prodotto a partire dal perossidodi idrogeno in presenza di ione ferroso (Fe2+) mediante lareazione di Fenton.
Sistemi enzimatici di protezioneSi tratta della superossido dismutasi (SOD) che trasforma O2
-
in H2O2 e la cui azione deve essere coordinata con gli altrienzimi detossificanti (che neutralizzano la H2O2 in acqua), dauna parte la catalasi che si rivela poco efficace nel cervello e,dall’altra, la glutatione perossidasi la cui azione è fondamentalee che si trova strettamente localizzata nella glia astrocitaria.
Neuroni dopaminergici e stress ossidativoI neuroni dopaminergici della sostanza nera sono tanto più
suscettibili allo stress ossidativo quanto più sono ricchi dimitocondri (rischio aumentato di produzione di O-
2) e conten-gono neuromelanina (testimone dell’auto-ossidazione della
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
9Neurologia

dopamina), concentrazioni elevate di SOD (testimone dellapresenza importante di radicali liberi) e una grande quantità diferro; inoltre, questi neuroni sono tanto meno resistenti allostress ossidativo quanto più ridotto è il loro ambiente astrogliale(contenente la glutatione perossidasi).
Morbo di Parkinson e stress ossidativo
Indici di stress ossidativo sono stati osservati nella sostanzanera di pazienti parkinsoniani: aumento della perossidazione deilipidi e del tasso di ferro, diminuzione del glutatione ridotto edeficit specifico dell’attività del complesso I mitocondriale (dicui si sa, peraltro, che può essere inibito da tossici come laMPP+ o il rotenone).
Le lesioni dei neuroni dopaminergici prevalgono nelle regioninigrali più povere di ambiente astrogliale, ma anche più ricchedi neuromelanina e di mitocondri [78].
Ruolo del processo apoptotico [77]
Sono state osservate modificazioni caratteristiche di morte perapoptosi nella sostanza nera di pazienti affetti da morbo diParkinson. Il processo apoptotico sarebbe iniziato dall’attiva-zione, per mezzo di citochine come il tumor necrosis factor a, diuna via di segnalazione intracellulare che scinde la sfingomie-lina di membrana in ceramide. L’attivazione di questa viaconduce alla traslocazione del fattore nucleare Kappa B (NFKB)nel nucleo, dove stimola l’espressione di geni che conducono atermine alla «morte cellulare programmata» [77]. Le citochine,iniziatrici presunte del processo, sarebbero prodotte a partiredalla proliferazione microgliale la cui presenza è stata dimos-trata all’interno delle regioni nigrali in degenerazione [78]:tuttavia, il significato patogenetico dell’apoptosi nel morbo diParkinson rimane ancora controverso.
Concezioni fisiopatologicheIn maniera molto semplificata, la fisiopatologia del morbo di
Parkinson può essere riassunta attraverso la constatazione di unlegame di causalità tra da una parte la deplezione di dopaminadello striato (putamen in particolare e nucleo caudato in misuraminore) derivante dal processo di morte progressiva dei neuronidopaminergici e, dall’altra, la comparsa di manifestazionicliniche (oltre un livello di neuroni residui del 30% circa), la cui
triade classica (tremori, rigidità, acinesia) implica una descri-zione che privilegia aspetti esclusivamente motori.
La realtà è, in effetti, molto più complessa e le concezionifisiopatologiche attuali sul morbo di Parkinson devono prenderein considerazione non solamente la recente evoluzione delleconoscenze sull’organizzazione funzionale del sistema dei ganglidella base (troppo a lungo considerato come le «fondamentaoscure» del cervello), ma anche la multidimensionalità deicomportamenti (anche nel campo ristretto delle attività moto-rie), la cui analisi fenomenologica non può essere isolata dalcontesto della loro realizzazione.
Organizzazione funzionale dei gangli della baseIl circuito del sistema dei gangli della base è stato descritto a
partire dai dati morfologici e l’elettrofisiologia ha in seguitoconfermato la realtà delle vie anatomiche precisandone il ruolofunzionale. Così, il sistema comporta un ingresso principalerappresentato dalla proiezione corticostriatale glutamatergicaeccitatoria (Fig. 7) [79]. Questo ingresso corticostriatale èmodulato da un’influenza complessa legata alla dualità funzio-nale dell’afferenza dopaminergica nigrostriata, a cui corrispondeuna dualità della popolazione dei neuroni spinosi di dimensionimedie che rappresentano l’efferenza striatale GABAergicainibitoria. Un primo contingente di questi neuroni striataliefferenti, che contiene anche della sostanza P e che riceve uninflusso dopaminergico eccitatorio mediato dai recettori di tipoD1, inibisce direttamente le strutture di uscita del sistema chesono il GPi e la sostanza nera reticolare (SNr), mentre unsecondo contingente, contenente dell’encefalina e che riceve uninflusso dopaminergico inibitorio mediato da recettori di tipoD2, esercita un’azione indiretta su queste stesse strutture diuscita (GPi, SNr) passando attraverso l’inibizione dei neuroni
Feedback +
MAO-B
Monoamine
Mitocondri
Perossidazione
Melanina
Morte neuronale
OH-
OH-
GSH
H2O
R-CHO + NH3+
Superossidodismutasi
GS-SG
Alterazione dei lipidi di membranaBlocco del complesso I mitocondriale
Perdita dell'omeostasi calcicaDeficit energetico
Anionesuperossido
O2-
Radicaleidrossile
Aggiuntadi un e-
Fe3+
Fe2+
Glutationeperossidasi Perossido
di idrogeno
Catalasi
H2O2
Auto-ossidazione
Reazione di Fenton
Figura 6. Modellizzazione dello «stress ossidativo» nel morbo diParkinson (da Viallet [1]). GSH: glutatione ridotto; MAO: monoamino-ossidasi.
Corteccia cerebrale
SNc
(GLU) (GLU)
DA
Striato
<< Diretto >><< Indiretto >>
(GABA enc)
GPe(GABA sost P)
(GABA)
Tronco cerebraleMidollo spinale
PPNGPi/SNr
STNTal
(GABA)(GLU)
Uscita
Entrata
a
B
Figura 7. Diagramma schematico del circuito funzionale del sistema deinuclei della base (modificato da Alexander e Crutcher [79]). DA: dopa-mina; Enc: encefalina; GABA: acido gamma-aminobutirrico; GLU: glutam-mato; GPe: globus pallidus esterno; GPi: globus pallidus interno; PPN:nucleo peduncolo-pontino; SNc: sostanza nera compatta; SNr: sostanzanera reticolare; STN: nucleo subtalamico; Subst P: sostanza P; tal: talamo;in nero: legame inibitorio; in grigio: legame eccitatorio; a: insieme strio-pallido-nigrale («cuore» dei nuclei della base); b: sottoinsieme GPe-STN.
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
10 Neurologia

GABAergici del pallidum esterno (GPe) che inibiscono, a lorovolta, il nucleo subtalamico (NST), il quale esercita un influssofinale eccitatorio attraverso la sua proiezione glutamatergicasull’insieme GPi-SNr. Occorre inoltre considerare la proiezioneeccitatoria glutamatergica «iperdiretta» della corteccia sul NST, ilcui significato funzionale è stato sottolineato recentemente [80].L’uscita principale del sistema corrisponde ai neuroni GABAer-gici inibitori pallidotalamici e nigrotalamici. Così, il modello«classico» e globale di proiezione cortico-striato-pallidonigro-talamo-corticale [79] si caratterizza al suo livello striato-pallido-nigrale per il funzionamento in parallelo di due tipi di vie(Fig. 7): una via diretta (trans-striatale) che focalizza, attraversoil meccanismo di una disinibizione selettiva, il comandoselezionato («desiderato») all’ingresso corticostriatale e delle vieindiretta e iperdiretta (trans-sotto-talamiche) che contribuisconoa rinforzare il fenomeno di focalizzazione spaziotemporaleattenuando i segnali adiacenti («competitivi» e dunque «nondesiderati») a livello dei bersagli effettori talamocorticali e delmesencefalo [81]; questo insieme permetterebbe di focalizzare leinformazioni emesse a partire dall’entrata corticostriatale allamaniera dell’«inibizione laterale» descritta per i sistemi senso-riali e, su un tale sistema, la dopamina rinforzerebbe questafocalizzazione dell’informazione (Fig. 8) [81, 82]. Negli ultimi anniquesto modello «classico» è stato criticato [82], in particolare inseguito all’evidenziazione di una colocalizzazione possibile direcettori dopaminergici D1 e D2 sugli stessi neuroni striatali dauna parte e dall’altra di proiezioni dopaminergiche extrastriatali(verso il GPe, il GPi e il NST, così come SNr per liberazionedendritica). Peraltro, alcuni studi metabolici (attività dellasubunità 1 della citocromo ossidasi, ben correlata con il livellodi attività neuronale) hanno evidenziato l’assenza di riduzionedi questa attività nel GPe dopo una lesione dopaminergica (incontraddizione con le previsioni del modello classico): questorisultato è stato attribuito a un aumento dell’influenza eccitato-ria del NST (sul GPe), essa stessa conseguente a entrate eccita-torie aumentate a provenienza dal PPN e dal complesso centromediano-nucleo parafascicolare (Cm-Pf) del talamo [83]. Infine,al di là di queste osservazioni che mettono in luce alcuneinsufficienze del modello classico, la difficoltà più importanteda superare per avvicinarsi alla realtà fisiopatologica risiedenell’apprensione della dinamica spaziotemporale del tratta-mento dell’informazione nel sistema dei gangli della base.Questa apprensione della dinamica dovrebbe poter integrare icambiamenti di stato del sistema in funzione del contesto
generale e di eventi significativi, il che spinge a considerare piùprecisamente da una parte il ruolo chiave dell’innervazionedopaminergica nella regolazione «in linea» del sistema(Fig. 9) [82, 84] e nel suo adattamento con la motivazione el’apprendimento [84] e, dall’altra, la natura dell’informazioneveicolata sotto forma dei ritmi di scarica dei neuroni e degliinsiemi neuronali con l’aiuto di registrazioni in più sedisimultanee analizzando l’evoluzione delle loro relazioni di
Programma motorio«desiderato»
Programmi motori«competitivi»
(«non desiderati»)
Bersagli talamocorticalie mesencefalici
Eccitatore
Inibitore
Corteccia cerebrale
STN
GPi
Striato
Figura 8. Illustrazione della focalizzazione spaziale dell’informazioneall’interno dei nuclei della base (da Mink [81] e Viallet [82]). GPi: globuspallidus interno; STN: nucleo subtalamico.
Corteccia limbica
Contributo dei nuclei della base alla valutazionecosciente del contesto dell'azione
Motivazione
Corteccia prefrontale Corteccia parietale
DopaminaModulazione
Piano d’azione
Selezione delle strategiedi comportamenti adeguati
SMAProgrammazione
Corteccia motoria
Azione
Idea di azioneauto-iniziazione
Confronto con iparametri memorizzati
Intenzione Attenzione
Neocervelletto
Figura 9. Dall’intenzione all’azione: modulazione dopaminergica della gestione dell’informazione corticale (motoria, cognitiva e motivazionale) da parte deinuclei della base (da Nieoullon [84] e Viallet [82]). SMA: area motoria supplementare.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
11Neurologia

coerenza [85]. Un tale iter mira a favorire la sostituzione deimodelli congelati tipo «astuccio e frecce» a vantaggio dirappresentazioni «animate» più idonee a una modellizzazione altempo stesso dinamica e non lineare [86].
Gangli della base e controllo motorioSituato in una posizione del circuito funzionale tra la cortec-
cia associativa (dove sarebbe concepita l’idea di un’azione,motoria o mentale) e la corteccia frontale motoria (che neassicura l’esecuzione), il sistema dei gangli della base si vedelogicamente attribuire un ruolo di pianificazione e programma-zione. Prendendo in considerazione le dimensioni motivazionalie cognitive di qualsiasi azione, con la funzione delle strutturelimbiche la cui interfaccia con i gangli della base corrispondeallo striato ventrale (nucleo accumbens), e la posizione privile-giata dell’area motoria supplementare che rappresenta ilbersaglio corticale principale di questi stessi gangli della base, siè precisato il contenuto di questo ruolo, in particolare mettendoin risalto una forma di memoria detta «procedurale» (o senso-motoria in senso più restrittivo), intesa come la capacità diacquisire per apprendimento degli elementi di «savoir faire» (odegli automatismi) in vista di costituire dei repertori diazioni [87]: al di là di una visione rigida del concetto di pro-gramma motorio, tale processo di apprendimento, che si puòritenere si manifesti in modo continuo a gradi diversi nelleesperienze sensorimotorie della vita quotidiana, introduce unadimensione più dinamica con il concetto di «pianificazionemotoria» [88]. Così, la pianificazione motoria sarebbe iniziata daun’idea interna o da uno stimolo scatenante esterno, dopo ilconfronto con l’insieme del contesto sensorimotorio e unapercezione adeguata degli obiettivi dell’azione. Il piano motoriopropriamente detto corrisponde allora a una modellizzazioneglobale dell’azione motoria ottenuta dopo apprendimento,attraverso l’assemblaggio secondo un’organizzazione cronologicaprecisa di un certo numero di programmi motori elementarisuccessivamente eseguiti in modo simultaneo o sequenziale invista di realizzare precisamente questa azione motoria(Fig. 10) [1, 88]: il piano motorio è quindi distinto dai programmimotori che lo costituiscono e, secondo Marsden, i gangli dellabase sono incaricati dell’«esecuzione automatica dei pianimotori appresi» [88]. Una tale funzione è stata consideratainteressata precocemente e specificamente nel morbo di Parkin-son, poiché la disorganizzazione progressiva della struttura deipiani motori appresi si applica particolarmente bene a unsintomo come l’acinesia; in effetti, l’acinesia, sintomo cardinedel morbo di Parkinson e, inoltre, molto ben correlata con ildeficit di dopamina striatale, può presentarsi sotto aspettidiversi fenomenologici che si possono associare: aspetti psico-motori, aspetti motori. Le diverse modalità di espressionedell’acinesia parkinsoniana potrebbero rappresentare la tradu-zione comportamentale di deficit che coinvolgono le diverse fasidell’attività motoria (Fig. 11) [89]. Schematicamente, l’acinesia
psichica rifletterebbe il deficit motivazionale che può compor-tare esso stesso un disturbo dell’attenzione selettiva a stimoliesterni o interni e/o una perdita di interesse per la realizzazionedi alcuni movimenti, mentre l’acinesia motoria dipenderebbe daun deficit di regolazione istantanea della forza muscolare condegradazione dell’organizzazione dei piani motori che portagradualmente a un rallentamento del movimento (bradicinesia)e/o a una riduzione di ampiezza del movimento (ipocinesia).
■ Aspetti clinici e diagnosticidel morbo di Parkinson idiopatico
La storia naturale della fase sintomatica del morbo di Parkin-son, declinata inizialmente attraverso i cinque stadi evolutivi diHoehn e Yahr descritti prima della L-dopa, non può attualmenteconcepirsi senza tenere conto dell’influenza considerevole deitrattamenti dopaminergici: così, la pratica neurologica attualeporta a distinguere tre tappe evolutive principali nel corso dellequali le manifestazioni cliniche, l’iter diagnostico e il contestodi gestione sono differenti [25]. Queste tappe di un’evoluzioneche rimane progressiva e le cui frontiere conservano unadefinizione necessariamente sfocata, corrispondono innanzituttoal periodo di esordio, detto «ex novo» (che si prolunga insensi-bilmente nel periodo detto «di luna di miele»), quindi alperiodo di malattia conclamata (che si caratterizza per losviluppo progressivo di segni assiali da una parte e di sintomilegati al trattamento dall’altra) e, infine, al periodo di malattiaavanzata, detto di «declino» (che corrisponde a una perdita diautonomia e all’emergere di complicanze invalidanti).
Sintomi della malattia all’esordioI primi segni compaiono spesso in maniera insidiosa, manifes-
tandosi in modo intermittente, il che rende difficile la datazioneprecisa dell’inizio reale della fase sintomatica: il tremore resta ilmotivo di visita iniziale più frequente, ma le manifestazionidolorose (crampi muscolari, distonie) o ansiosodepressive nonsono eccezionali. Lo scopo dell’esame neurologico, a livello diquesta tappa, sarà quello di realizzare un inventario preciso deisegni motori e non motori riscontrabili all’esordio del morbo diParkinson. Durante tale periodo, che copre approssimativamentegli stadi I e II di Hoehn e Yahr, la diagnosi inizialmente sospettatasi trova progressivamente confermata quando la buona qualitàdella risposta dei sintomi al trattamento medico fa intravedereun’evoluzione relativamente controllata, che corrisponde alla«luna di miele» terapeutica.
Segni motori
Tremori
Si tratta di un tremore di «semi-riposo», che compare,quando è discreto e intermittente, solo in alcune posture dirilassamento muscolare parziale (mano appoggiata sulla coscia,
Piano
Motorio
Azione motoria
Contesto
Memorizzazione Apprendimento Azioneprevista
Azionerealizzata
Pianificazione - programma Esecuzione
Inizio
Svolgimento
Termine
SelezioneAssemblaggio
IdeaStimolo esterno
Programmi motori pronti
Indiziinterni
Indiziesterni
~Figura 10. Organizzazione dei piani motori (da Marsden [88] eViallet [1]).
(Deficit di energizzazione)
Acinesia
«Aspetti psicomotori» «Aspetti motori»
Acinesia psichica Bradicinesia Ipocinesia
( ampiezza)( velocità)
TR TM
Programmazione
AvvioIncitamento
Movimento
Iniziazione Esecuzione
( motivazione)
( attenzione selettiva)
Figura 11. Differenti modalità dell’acinesia parkinsoniana in funzionedelle fasi del movimento (da Viallet e Trouche [89]). TR: tempo di reazione;TM: tempo di movimento.
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
12 Neurologia

dita in lieve flessione) oppure dopo la sensibilizzazione (calcolomentale) o, ancora, durante la marcia. La contrazione muscolarevolontaria lo fa in genere sparire, anche se, in alcuni casi, puòesistere una componente «posturale» associata alla componentedi «semi-riposo»: analogamente, il tremore scompare nelrilassamento muscolare completo (segno detto «della poltronacoloniale») e nel sonno. Viceversa, il tremore è aggravato dallostress. La sua distribuzione topografica iniziale più tipica èmonolaterale (o quanto meno asimmetrica con predominanzanetta da un lato), localizzata alle estremità (il più delle voltemano, ma anche piede e regione periorale: labbra, mento,lingua); quando il tremore è più prossimale, riveste spesso uncarattere più ampio e invalidante, conservando frequentementeuna componente «posturale».
Lo studio elettrofisiologico conferma la frequenza «lenta», traquattro e sei cicli per secondo, del fenomeno oscillatorio(mediante registrazione accelerometrica dello spostamento delsegmento colpito) e della contrazione alternata dei muscoliagonista e antagonista (il cui segnale elettromiografico comportala successione ritmica di scariche di attività raggruppatealternate con fasi di silenzio elettrico).
Rigidità
Anch’essa asimmetrica all’esordio, predomina all’estremitàdove ha sede il tremore. Raramente isolata, predomina suigruppi muscolari flessori causando posture a volte distoniche,specialmente al piede. Corrisponde a un aumento del tonomuscolare e si manifesta con una resistenza accresciuta avvertitadall’esaminatore che effettua la mobilizzazione del segmentoaffetto: questa resistenza è, di regola, omogenea e continua,evocando quella di un tubo di piombo, salvo se la coesistenzadi un tremore a riposo la fa cedere a scatti, realizzando unfenomeno di «troclea». Quando la rigidità è discreta, la manovradi sensibilizzazione classica consiste nel chiedere al paziente,durante la mobilizzazione del suo polso, di sporgersi in avantiper afferrare un oggetto con l’altra mano oppure di eseguiremovimenti ampi con la spalla opposta: l’aumento della resis-tenza alla mobilizzazione del polso nel corso del movimentovolontario effettuato dal paziente corrisponde al segno diFroment, chiamato anche segno del «polso congelato» o «segnodel bancone». Come il tremore, la rigidità è aumentata dallostress e può scomparire durante il sonno: durante un esameprolungato, può variare leggermente in funzione dello stato dirilassamento generale del paziente.
Acinesia
Essa si valuta osservando tutti i movimenti del paziente, inparticolare degli arti (movimento di pronosupinazione alternatadelle mani, tamburellamento ritmico del tallone al suolo)ricercando le difficoltà a iniziare il movimento (acinesiapropriamente detta), il rallentamento e la riduzione di ampiezzadei gesti (bradicinesia, ipocinesia), così come la riduzione deimovimenti associati e automatici (mimica facciale, oscillazionedelle braccia durante la deambulazione). Questa acinesiacontrasta con la vivacità dei riflessi osteotendinei e con lacostanza di un riflesso nasopalpebrale inesauribile.
Disturbi della postura e della coordinazionepostura-movimento
Modificazioni posturali. Esse derivano essenzialmentedall’ipertonia che predomina sui gruppi muscolari flessori. Così,durante la stazione eretta la testa e il tronco sono inclinati inavanti, le spalle sono in anteposizione, gli avambracci insemiflessione e pronazione, i gomiti leggermente divaricati e leanche e le ginocchia leggermente flesse. Le reazioni posturalicorrettrici dopo un’alterazione possono essere ridotte o ritardate.
Disturbi della coordinazione postura-movimento. Sono, ingenere, poco marcati nel periodo di esordio, a eccezione dellascrittura, specialmente se il lato affetto corrisponde alla mano«dominante». La scrittura è allora tipicamente micrografica, e lamicrografia si accentua di pari passo al tracciato, ma spessorimane leggibile malgrado il suo restringimento; la malattia è, a
volte, rivelata da una contrattura dolorosa dei muscolidell’avambraccio durante la scrittura, simile a una distoniafunzionale. La parola può essere monotona con un’intensità piùdebole e un flusso irregolare, ma rimane in genere udibile;tuttavia, la gestualità del viso e degli arti superiori associata allacomunicazione si rivela spesso precocemente ridotta. La marciapuò essere contraddistinta da alcuni disturbi: esitazione allapartenza e a metà passo e riduzione moderata dell’ampiezza delpasso.
Segni non motori
Fenomeni sensitivi e dolorosi
Spesso legati all’ipertono muscolare localizzato, i fenomenisensitivi si esprimono sotto la forma di parestesie, di sensazionidi costrizione o di «tremore interno» e perfino di dolorimuscolari che presentano una modalità parossistica piuttostolocalizzata tipo crampi o una modalità più diffusa e continua.Interessando più spesso l’arto inferiore che l’arto superiore,questi fenomeni dolorosi sono responsabili di una presentazione«pseudo-reumatologica» della malattia all’esordio, che rende lasua diagnosi tanto più difficile da ipotizzare in quanto iltremore può essere assente. Un fenomeno tipo sindrome delle«gambe senza riposo» è, a volte, inaugurale e caratterizzato dallasua comparsa notturna. Un deficit della percezione olfattiva èstato riscontrato molto frequentemente all’inizio della malat-tia [90], ma esso rappresenta ancora raramente un segnale diallarme.
Disturbi psichici
I disturbi psichici osservati all’esordio della malattia silimitano essenzialmente all’ansia e alla depressione. Pocospecifica, l’ansia si manifesta a episodi, con attacchi di panico,ma anche in modo più permanente, associata spesso alladepressione. La frequenza media della depressione è statastimata pari al 50% in una meta-analisi [91]: una revisionesistematica recente degli studi di prevalenza fornisce unafrequenza media del 17% di disturbi depressivi maggiori, del13% di distimia e del 22% di depressione minore [92]. Nelcontesto dei segni motori iniziali della malattia (specialmentedell’acinesia) la depressione, che associa umore triste, disturbidell’appetito e del sonno, tensione nervosa e perdita di motiva-zione, si rivela difficile da separare dalla bradifrenia, checorrisponde all’espressione di disturbi cognitivi minori (rallen-tamento del pensiero e diminuzione dell’attenzione). Conside-rata, almeno in parte, come reattiva alla disabilità legata almorbo di Parkinson [91], la comparsa della depressione parecchianni prima dei segni motori è stata recentemente dimostrata instudi casi-controlli retrospettivi [43, 47], ma anche in uno studioprospettico che utilizzava la scala di depressione di Beck [90], chele conferisce anche lo stato di sintomo precoce e indipendentedallo stato motorio.
Sintomi della malattia conclamataQuesta fase evolutiva è caratterizzata dalla comparsa progres-
siva di segni che vengono detti «assiali» (instabilità posturale,disturbi più complessi della deambulazione e della voce),nonché di manifestazioni disautonomiche e di disturbi dellefunzioni esecutive, e l’insieme è meno controllato con la terapiamedica; inoltre, questo periodo vede svilupparsi sintomi nuovilegati al trattamento stesso che possono essere considerati effettisecondari indesiderati a causa del loro ruolo sempre piùinvadente, non solo nel campo motorio (fluttuazioni di effica-cia, movimenti anomali involontari), ma anche nei campineurovegetativo e psichico. Durante tale periodo il disturbofunzionale si accentua più chiaramente, interessando in misuravariabile a seconda dei pazienti la vita socioprofessionale efamiliare, il che ricopre approssimativamente gli stadi III e IV diHoehn e Yahr: in effetti, il livello di handicap diviene moltovariabile a causa dell’instabilità della risposta terapeutica nelcorso della giornata, che alternerà intervalli di buona mobilità(periodi on) con altri intervalli di mobilità ridotta (periodi off)o di stato motorio intermedio (periodi in-between).
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
13Neurologia

Sintomi specifici della malattia
Segni motori
I disturbi posturali tendono a svilupparsi, esagerando latendenza generale in semi-flessione nella stazione eretta efavorendo la comparsa di deformità articolari pseudo-reumatiche alla mano («mano da scrittore», «mano da fachiro»)o al piede (piede varo equino, dita ad «artiglio», estensionetonica dell’alluce); sono possibili patologie rachidee con cifosi oflessioni laterali in posizione eretta o seduta.
I disturbi della deambulazione e della parola diventano piùcomplessi, in particolare con la comparsa del fenomeno difreezing, chiamato ancora «blocco cinetico» (forse in riferimentoal fenomeno di slipping-clutch descritto nel 1956 da Denny-Brown) o «blocco motorio». Inizialmente descritto all’iniziodella deambulazione, il freezing corrisponde all’assenza di avviodel primo passo (piedi incollati al suolo) o all’abbozzo di questoavvio interrotto da una specie di calpestio (abasia trepidante): ilfreezing può comparire anche durante la marcia (superamento diuna porta, cambiamento di direzione o giravolta). La deambu-lazione in se stessa si caratterizza, dopo una partenza lenta, conuna riduzione netta della lunghezza del passo o della falcata:l’atteggiamento generale del paziente, il corpo inclinato insquilibrio assiale in avanti per accelerare il suo centro di gravitàe provocare un passo destinato a recuperare l’equilibrio, realizzail quadro tipico della deambulazione festinante dove il pazientedà l’impressione di «rincorrere il proprio centro di gravità».Come ciò che si osserva per la deambulazione, la produzionevocale, come la scrittura, possono essere perturbate dal feno-meno di freezing e dalla riduzione di ampiezza dell’attivitàmotoria prodotta (calo del volume vocale, micrografia) e, per laparola, da alterazioni della prosodia (riduzione dell’estensionedella gamma tonale a spese della gamma alta e accelerazioni delflusso, che corrispondono rispettivamente agli aspetti di parolamonotona e di tachifemia parossistica [93, 94].
Segni non motori
La disautonomia resta abitualmente molto discreta nel morbodi Parkinson, il che permette di differenziarlo dall’atrofiamultisistemica. Tuttavia, si possono osservare alcuni disturbispecifici del sistema nervoso autonomo in autentici casi dimorbo di Parkinson, che colpiscono, in particolare, le funzionidigestiva e cardiovascolare: così, i disturbi digestivi più frequentisono la scialorrea, che sarebbe il segnale precoce di unadifficoltà di deglutizione subclinica piuttosto che il risultato diuna produzione eccessiva di saliva, e la stipsi, che deriverebbesia da un rallentamento della peristalsi colica che da unadisfunzione dell’eliminazione rettale; i disturbi cardiovascolarisono dominati dall’ipotensione ortostatica e postprandiale chesi rivela raramente invalidante, tranne quando è aggravata daitrattamenti dopaminergici. Possono comparire altre manifesta-zioni disautonomiche (disturbi vescicosfinterici tipo urgenzeminzionali, ipersudorazione brutale) e divenire invalidanti inparticolare nei periodi off, mentre la disfunzione farmacologicaeventualmente sottostante resta ancora poco chiarita: questotipo di sintomo è spesso responsabile di un’alterazione dellaqualità del sonno notturno.
I disturbi psichici del morbo di Parkinson al di fuori dell’ansiae della depressione (che compaiono fin dall’esordio dellamalattia) e della demenza (che resta di comparsa tardiva, salvofar sospettare la diagnosi di malattia a corpi di Lewy) si limitanoalla nozione di disturbi cognitivi minori, dominati da dueaspetti: i disturbi del richiamo mnesico e i disturbi dellefunzioni esecutive [95-97]. Questi disturbi non interessano inmaniera significativa il comportamento del paziente e possonoessere evidenziati con l’aiuto di test neuropsicologici: così, il testdi Grober e Buschke mostra una normalizzazione con ilrichiamo indicizzato del deficit di rievocazione mnesica nelricordo libero [96]; molti altri test incentrati sulla valutazionedelle funzioni esecutive (generazione di concetti, cambiamentoo mantenimento di atteggiamenti mentali, risoluzione diproblemi) contribuiscono a mettere in risalto la sindromedisesecutiva sotto-cortico-frontale [96]. Descritta inizialmente da
Marin nel 1991, l’apatia rappresenta una sindrome comporta-mentale che deve essere isolata dalle manifestazioni clinichedella depressione [98], e la sua frequenza è stata stimata tra il20% e il 70% nel morbo di Parkinson, con criteri di identifica-zione e di valutazione variabili [99, 100] che restano ancoradiscussi [101].
Sintomi legati al trattamento [51, 102]
Segni motori
Fluttuazioni di efficacia. Definite come la ricomparsaintermittente dei sintomi del morbo di Parkinson, esse sisuddividono in fluttuazioni prevedibili e imprevedibili aseconda che appaiano o meno legate all’assunzione di farmaci.Per convenzione, poiché l’acinesia resta considerata il sintomopiù tipicamente dopa-sensibile, i vocaboli «fluttuazione» e«acinesia» sono spesso assimilati.
Le fluttuazioni prevedibili sono le prime a comparire epossono restare a lungo isolate: esse sono caratterizzate da unarelazione cronologica regolare con le assunzioni di farmaci. Èclassico distinguere l’acinesia di fine dose (wearing-off), chericompare prima dell’assunzione seguente, e l’acinesia notturna,con l’acinesia di risveglio o del primo mattino, che deriva dallamancata assunzione dopo la sera precedente. Queste fluttua-zioni prevedibili sarebbero complessivamente la conseguenzadella perdita progressiva della capacità di immagazzinamentodella L-dopa esogena da parte delle terminazioni dopaminergi-che striatali a causa della progressione della denervazionedopaminergica o, alternativamente, il risultato della progres-sione del deficit dopaminergico che renderebbe più elevata lasoglia di efficacia clinica della L-dopa. Questa perdita di effetto«tampone» (o questo innalzamento della soglia) determina lascomparsa della risposta farmacologica «a lungo termine»(dipendente dalla somma di somministrazioni successive) e unariduzione di durata della risposta «a breve termine» (stretta-mente legata a ogni assunzione).
Le fluttuazioni imprevedibili compaiono più tardivamente esi oppongono alle precedenti per la loro assenza di relazioni conle assunzioni di farmaci. Spesso situate all’inizio del pomeriggio(acinesia postprandiale) o alla fine del pomeriggio, tendono adassumere un orario fisso nella giornata malgrado gli adattamentidelle assunzioni farmacologiche (acinesia circadiana o nicteme-rale, acinesia resistente). L’acinesia paradossa, che compare dopouna assunzione, è stata considerata una variante ritardatadell’acinesia di fine dose, ma sembra avere un’esistenza realeassimilabile in parte a un ritardo di efficacia (delay-on). Infine,delle fluttuazioni improvvise (effetto on-off), di osservazione piùrara e di comparsa più tardiva, corrispondono a cambiamentirepentini dello stato motorio in alcuni minuti, o anche in pochisecondi. Queste fluttuazioni imprevedibili sono ancora pocospiegate: le ipotesi correnti chiamano in causa un’alterazionedello svuotamento gastrico oppure una competizione farmaco-dinamica con gli aminoacidi alimentari oppure, ancora, dellevariazioni dei livelli di affinità dei recettori dopaminergici.
Movimenti anomali involontari. Chiamati «discinesie», essipossono rivestire tutti gli aspetti clinici, andando da posturedistoniche prolungate fino a movimenti violenti pseudo-ballici,passando per tutta una gamma di movimenti coreiformi più omeno ripetitivi, che interessano tutte le parti del corpo, ma chepredominano piuttosto nel territorio dove è iniziato il morbo diParkinson. A seconda del livello della stimolazione dopaminer-gica, si distinguono le discinesie della porzione centrale delladose (che corrispondono a un livello sopraliminare) e lediscinesie di inizio e fine dose (che coincidono con un livellopiuttosto subliminare): le distonie di periodo off si osservanoquando l’attività dopaminergica è al minimo.
Le discinesie della porzione centrale della dose (o di «picco didose») possono comparire precocemente: sono spesso discrete eben tollerate secondo un modello relativamente stabile in unostesso paziente. Viceversa, le discinesie di inizio e fine dose (o«bifasiche»), che sono di comparsa più tardiva, sono molto piùviolente e mal tollerate, rivestendo l’aspetto di movimentialternativi ripetitivi o l’aspetto di posture distoniche dolorose.L’analisi cronologica delle discinesie su documento video, dopo
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
14 Neurologia
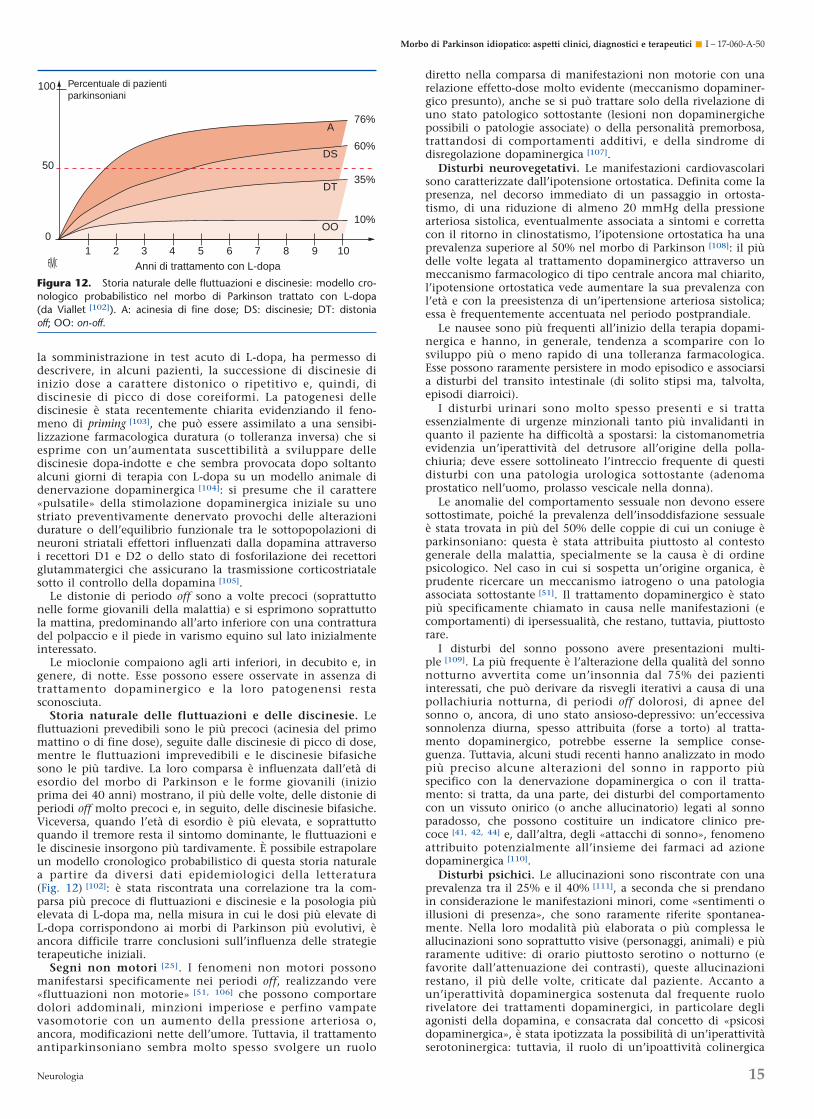
la somministrazione in test acuto di L-dopa, ha permesso didescrivere, in alcuni pazienti, la successione di discinesie diinizio dose a carattere distonico o ripetitivo e, quindi, didiscinesie di picco di dose coreiformi. La patogenesi dellediscinesie è stata recentemente chiarita evidenziando il feno-meno di priming [103], che può essere assimilato a una sensibi-lizzazione farmacologica duratura (o tolleranza inversa) che siesprime con un’aumentata suscettibilità a sviluppare dellediscinesie dopa-indotte e che sembra provocata dopo soltantoalcuni giorni di terapia con L-dopa su un modello animale didenervazione dopaminergica [104]: si presume che il carattere«pulsatile» della stimolazione dopaminergica iniziale su unostriato preventivamente denervato provochi delle alterazionidurature o dell’equilibrio funzionale tra le sottopopolazioni dineuroni striatali effettori influenzati dalla dopamina attraversoi recettori D1 e D2 o dello stato di fosforilazione dei recettoriglutammatergici che assicurano la trasmissione corticostriatalesotto il controllo della dopamina [105].
Le distonie di periodo off sono a volte precoci (soprattuttonelle forme giovanili della malattia) e si esprimono soprattuttola mattina, predominando all’arto inferiore con una contratturadel polpaccio e il piede in varismo equino sul lato inizialmenteinteressato.
Le mioclonie compaiono agli arti inferiori, in decubito e, ingenere, di notte. Esse possono essere osservate in assenza ditrattamento dopaminergico e la loro patogenensi restasconosciuta.
Storia naturale delle fluttuazioni e delle discinesie. Lefluttuazioni prevedibili sono le più precoci (acinesia del primomattino o di fine dose), seguite dalle discinesie di picco di dose,mentre le fluttuazioni imprevedibili e le discinesie bifasichesono le più tardive. La loro comparsa è influenzata dall’età diesordio del morbo di Parkinson e le forme giovanili (inizioprima dei 40 anni) mostrano, il più delle volte, delle distonie diperiodi off molto precoci e, in seguito, delle discinesie bifasiche.Viceversa, quando l’età di esordio è più elevata, e soprattuttoquando il tremore resta il sintomo dominante, le fluttuazioni ele discinesie insorgono più tardivamente. È possibile estrapolareun modello cronologico probabilistico di questa storia naturalea partire da diversi dati epidemiologici della letteratura(Fig. 12) [102]: è stata riscontrata una correlazione tra la com-parsa più precoce di fluttuazioni e discinesie e la posologia piùelevata di L-dopa ma, nella misura in cui le dosi più elevate diL-dopa corrispondono ai morbi di Parkinson più evolutivi, èancora difficile trarre conclusioni sull’influenza delle strategieterapeutiche iniziali.
Segni non motori [25]. I fenomeni non motori possonomanifestarsi specificamente nei periodi off, realizzando vere«fluttuazioni non motorie» [51, 106] che possono comportaredolori addominali, minzioni imperiose e perfino vampatevasomotorie con un aumento della pressione arteriosa o,ancora, modificazioni nette dell’umore. Tuttavia, il trattamentoantiparkinsoniano sembra molto spesso svolgere un ruolo
diretto nella comparsa di manifestazioni non motorie con unarelazione effetto-dose molto evidente (meccanismo dopaminer-gico presunto), anche se si può trattare solo della rivelazione diuno stato patologico sottostante (lesioni non dopaminergichepossibili o patologie associate) o della personalità premorbosa,trattandosi di comportamenti additivi, e della sindrome didisregolazione dopaminergica [107].
Disturbi neurovegetativi. Le manifestazioni cardiovascolarisono caratterizzate dall’ipotensione ortostatica. Definita come lapresenza, nel decorso immediato di un passaggio in ortosta-tismo, di una riduzione di almeno 20 mmHg della pressionearteriosa sistolica, eventualmente associata a sintomi e correttacon il ritorno in clinostatismo, l’ipotensione ortostatica ha unaprevalenza superiore al 50% nel morbo di Parkinson [108]: il piùdelle volte legata al trattamento dopaminergico attraverso unmeccanismo farmacologico di tipo centrale ancora mal chiarito,l’ipotensione ortostatica vede aumentare la sua prevalenza conl’età e con la preesistenza di un’ipertensione arteriosa sistolica;essa è frequentemente accentuata nel periodo postprandiale.
Le nausee sono più frequenti all’inizio della terapia dopami-nergica e hanno, in generale, tendenza a scomparire con losviluppo più o meno rapido di una tolleranza farmacologica.Esse possono raramente persistere in modo episodico e associarsia disturbi del transito intestinale (di solito stipsi ma, talvolta,episodi diarroici).
I disturbi urinari sono molto spesso presenti e si trattaessenzialmente di urgenze minzionali tanto più invalidanti inquanto il paziente ha difficoltà a spostarsi: la cistomanometriaevidenzia un’iperattività del detrusore all’origine della polla-chiuria; deve essere sottolineato l’intreccio frequente di questidisturbi con una patologia urologica sottostante (adenomaprostatico nell’uomo, prolasso vescicale nella donna).
Le anomalie del comportamento sessuale non devono esseresottostimate, poiché la prevalenza dell’insoddisfazione sessualeè stata trovata in più del 50% delle coppie di cui un coniuge èparkinsoniano: questa è stata attribuita piuttosto al contestogenerale della malattia, specialmente se la causa è di ordinepsicologico. Nel caso in cui si sospetta un’origine organica, èprudente ricercare un meccanismo iatrogeno o una patologiaassociata sottostante [51]. Il trattamento dopaminergico è statopiù specificamente chiamato in causa nelle manifestazioni (ecomportamenti) di ipersessualità, che restano, tuttavia, piuttostorare.
I disturbi del sonno possono avere presentazioni multi-ple [109]. La più frequente è l’alterazione della qualità del sonnonotturno avvertita come un’insonnia dal 75% dei pazientiinteressati, che può derivare da risvegli iterativi a causa di unapollachiuria notturna, di periodi off dolorosi, di apnee delsonno o, ancora, di uno stato ansioso-depressivo: un’eccessivasonnolenza diurna, spesso attribuita (forse a torto) al tratta-mento dopaminergico, potrebbe esserne la semplice conse-guenza. Tuttavia, alcuni studi recenti hanno analizzato in modopiù preciso alcune alterazioni del sonno in rapporto piùspecifico con la denervazione dopaminergica o con il tratta-mento: si tratta, da una parte, dei disturbi del comportamentocon un vissuto onirico (o anche allucinatorio) legati al sonnoparadosso, che possono costituire un indicatore clinico pre-coce [41, 42, 44] e, dall’altra, degli «attacchi di sonno», fenomenoattribuito potenzialmente all’insieme dei farmaci ad azionedopaminergica [110].
Disturbi psichici. Le allucinazioni sono riscontrate con unaprevalenza tra il 25% e il 40% [111], a seconda che si prendanoin considerazione le manifestazioni minori, come «sentimenti oillusioni di presenza», che sono raramente riferite spontanea-mente. Nella loro modalità più elaborata o più complessa leallucinazioni sono soprattutto visive (personaggi, animali) e piùraramente uditive: di orario piuttosto serotino o notturno (efavorite dall’attenuazione dei contrasti), queste allucinazionirestano, il più delle volte, criticate dal paziente. Accanto aun’iperattività dopaminergica sostenuta dal frequente ruolorivelatore dei trattamenti dopaminergici, in particolare degliagonisti della dopamina, e consacrata dal concetto di «psicosidopaminergica», è stata ipotizzata la possibilità di un’iperattivitàserotoninergica: tuttavia, il ruolo di un’ipoattività colinergica
Percentuale di pazientiparkinsoniani
100
50
0
76%
60%
35%
10%
1 2 3 4 5 6 7 8 9 10Anni di trattamento con L-dopa
OO
DS
DT
A
Figura 12. Storia naturale delle fluttuazioni e discinesie: modello cro-nologico probabilistico nel morbo di Parkinson trattato con L-dopa(da Viallet [102]). A: acinesia di fine dose; DS: discinesie; DT: distoniaoff; OO: on-off.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
15Neurologia

non può essere ignorato a causa del ruolo favorente dei farmacianticolinergici e, soprattutto, dell’esistenza di un deficitcolinergico sotto-cortico-frontale nella demenza a corpi di Lewy,affezione caratterizzata dalla precocità delle allucinazioni.
Gli episodi confusionali, che si manifestano in modo piùbrutale, sono talvolta associati a fenomeni allucinatori com-plessi, non criticati, e provocano disturbi gravi del comporta-mento [51]: spesso scatenati da un avvenimento intercorrente(trauma, infezione, intervento chirurgico), essi evolvonogeneralmente in modo regressivo, ma restano rivelatori di unprocesso di degenerazione cognitiva sottostante. L’età è unfattore predisponente, così come i farmaci anticolinergici eagonisti dopaminergici.
Sono stati attribuiti dei disturbi cognitivi in particolareall’effetto deleterio degli anticolinergici sulle capacità attentivee mnesiche: il concetto dell’emergenza progressiva, nel morbodi Parkinson, di lesioni non dopaminergiche e l’evidenziazionedel ruolo predittivo, nella comparsa di un processo di deterio-ramento delle prestazioni mentali, di un’età di esordio piùelevata e di una riduzione delle prestazioni in alcuni test, qualifluenza verbale e Stroop [112], suggeriscono la nozione di uncontinuum [113] tra i disturbi cognitivi sotto-cortico-frontalicaratterizzati da un deficit crescente delle funzioni esecutive [96]
e quella che si è convenuto chiamare la «demenza parkinso-niana», che deve essere differenziata dalla demenza a corpi diLewy e dal morbo di Alzheimer.
Sintomi della malattia avanzataDurante questo periodo detto «di declino», i disturbi legati
all’evoluzione della malattia stessa e le complicanze dovute altrattamento si confondono progressivamente e si accompa-gnano a un aumento della disabilità, caratterizzato dalla perditadi autonomia (fasi IV e V di Hoehn e Yahr). In effetti, le azionicombinate dell’età e dell’evoluzione della malattia, così comel’alterazione progressiva del rapporto efficacia/tollerabilità deifarmaci antiparkinsoniani che ne deriva, concorrono all’accen-tuazione dei segni «assiali» e della loro «doparesistenza» [114,
115]: questo processo porta concretamente alla riduzione delladurata dei periodi on, ma anche al deterioramento del punteg-gio di risposta alla L-dopa [116] ottenuto con la differenza tra ipunteggi motori in periodo off e in periodo on, i cui corrispon-denti livelli di disabilità si aggravano (Fig. 13) [116]. Il declinoriguarda al tempo stesso le attività motorie e cognitive, e le
funzioni neurovegetative la cui somma dei disturbi è responsa-bile di complicanze diverse (cadute con traumi, ab ingestis conpolmoniti, stati confusionali acuti), esse stesse fattori discompenso attraverso episodi che possono mettere in gioco laprognosi quoad vitam, tanto più che il paziente, divenuto piùanziano, si trova in uno stato generale più precario.
Declino motorio
Disturbi dell’equilibrio e della postura
Essi divengono prevalenti: l’instabilità alla stazione erettarichiede un sostegno più frequente, mentre quando ci si alza dauna sedia o da una poltrona si deve essere assistiti. Questedifficoltà traducono il deterioramento delle reazioni posturalicorrettive indispensabili al mantenimento della postura in piedistabile, così come la perdita di coordinamento tra postura emovimento. L’aggravamento delle deformità articolari delleestremità (mani e piedi) e, soprattutto, dell’asse vertebrale, chepuò presentare importanti angolazioni deformanti tipo campto-cormia [117], accentua ancora le difficoltà del controllo posturale.
Disturbi della deambulazione
Essi sono particolarmente invalidanti, associando i fenomenidi freezing e di festinazione con antepulsioni e retropulsioni chetestimoniano l’alterazione della coordinazione posturocinetica:quando la deambulazione resta possibile senza supporto, ilrischio di caduta diventa rilevante, a causa dell’insufficienzadelle reazioni di recupero.
Disturbi del linguaggio
Essi rendono la voce appena udibile a causa dell’ipofonia edei disturbi articolatori con accelerazione del flusso e riduzionedella gamma tonale [93, 94]: la comunicazione con i familiari e ilpersonale curante si riduce notevolmente, tanto più che lascrittura è divenuta da lungo tempo impraticabile.
Disturbi della deglutizione
Essi completano queste difficoltà [25] con un’alterazione deltempo orale (difetto di continenza labiale e di formazione delbolo) e, soprattutto, faringeo (ritardo nell’avviamento e insuffi-cienza del processo di propulsione, deficit di protezione dellevie aeree con rischio di ab ingestis immediato o primario e stasifaringea nelle vallecole e nei seni piriformi con il rischio di abingestis differito o secondario).
Declino cognitivoParallelamente ai disturbi motori tardivi, il declino cognitivo
colpisce sempre più la vita di relazione del paziente parkinso-niano riducendo le interazioni con i familiari e con le personeche li curano. La «demenza» parkinsoniana diventa menospecifica e più difficile da categorizzare, rappresentando unfattore che limita considerevolmente l’efficacia della gestioneterapeutica.
Declino neurovegetativoAggiungendosi alle alterazioni legate al declino motorio e
cognitivo, esso si manifesta sotto i seguenti aspetti: disturbidella regolazione pressoria responsabili di manifestazionisincopali, disturbi del controllo sfinterico urinario punteggiatida episodi infettivi, disturbi dell’evacuazione intestinale conrischio di episodi subocclusivi e dimagrimento a volte gravederivante da disturbi della deglutizione ma anche da unosquilibrio nutrizionale.
ComplicanzeLe cadute sono particolarmente frequenti negli stadi avanzati
della malattia e influenzano gravemente la prognosi a causadelle loro conseguenze traumatiche (fratture con ricovero eallettamento) e psicologiche (atteggiamento fobico del paziente,ma anche delle persone vicine, che peggiora ulteriormente ledifficoltà posturocinetiche). Il loro meccanismo è polimorfo,accidentale e favorito dall’instabilità posturale o provocato daun’ipotensione ortostatica o dai fenomeni di freezing e difestinazione alla deambulazione.
25
20
15
10
5
00 2 4 6 8 10 12 14 16
Decorso temporale della malattia (anni)
Punteggio di disabilità(scala di Webster)
Figura 13. Rappresentazione teorica dell’evoluzione, nel morbo diParkinson, dell’ampiezza della risposta motoria di «breve durata» (inchiaro) e di lunga durata (in scuro) alla L-dopa, misurata sulla scala diWebster. La linea obliqua ascendente illustra la progressione del punteg-gio senza trattamento e il rettangolo chiaro a sinistra fornisce la rispostamotoria (differenza tra il punteggio non trattato e quello trattato altrattamento iniziale, da Clissold et al. [116]).
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
16 Neurologia

Le complicanze viscerali, tipo broncopolmoniti ab ingestis,episodi subocclusivi e infezioni urinarie legate all’allettamento,rappresentano potenzialmente il pericolo più diretto per laprognosi vitale.
Criteri di diagnosi clinica [25]
Le manifestazioni cliniche, necessariamente presenti findall’inizio della fase detta sintomatica del morbo di Parkinson,rappresentano un’informazione facilmente disponibile, la cuiimprecisione inerente la soggettività del medico può essereattenuata ricorrendo a procedure standardizzate ed esaurienti, lacui attuazione deve restare la più semplice possibile: questeprocedure possono essere eventualmente integrate da registra-zioni video che permettono di raccogliere il parere di altriosservatori. Secondo una proposta [95], la condotta praticadell’esame neurologico di un paziente parkinsoniano potrebbeessere scomposta in quattro tempi e in 20 elementi. Il primotempo riguarda i dati dell’anamnesi (quattro elementi): età, datad’esordio della malattia, natura e localizzazione del sintomoiniziale ed eventuali anamnesi familiare di morbo di Parkinson.Il secondo tempo (tre elementi) è dedicato all’evidenziazionedella triade caratteristica (tremore, rigidità, acinesia) osservando,in particolare, la deambulazione e la scrittura. Il terzo tempo(quattro elementi) valuta la risposta al trattamento dopaminer-gico (la cui efficacia è espressa con il tasso di miglioramentopercepito) e rileva gli eventuali effetti collaterali (discinesie,fluttuazioni di efficacia, disturbi psichici). Il quarto e ultimotempo (nove elementi) consiste in un inventario sistematico deisegni «assiali» eventualmente presenti: anomalie della postura,instabilità posturale, disturbi della deambulazione, disartria,disfagia, rigidità nucale, anomalie oculomotorie, disturbicognitivi e disturbi sfinterici.
Nel contesto abituale dell’esame di un paziente parkinsoniano(sulla base dei soli sintomi clinici) l’esigenza di specificità nelladiagnosi del MPi imporrà, in un approccio di diagnosi differen-ziale, di escludere le altre sindromi parkinsoniane cosiddette«secondarie» e «atipiche», le cui particolarità cliniche, inparticolare nella fase iniziale della malattia, possono esseredifficili da discernere. In alcune casistiche autoptiche [118, 119] iltasso di conferma anatomopatologica della diagnosi clinica diMPi è stato riscontrato pari al solo 76%, il che corrisponde a un24% di errori per eccesso (falsi positivi), essenzialmente a spesedi sindromi parkinsoniane atipiche. Così, sulla base metodolo-gica del confronto anatomoclinico i criteri clinici di MPi sonostati sistematicamente analizzati allo scopo di stabilire i lororispettivi valori predittivi positivi (VPP) [120]. Un processo digerarchizzazione di questi criteri in funzione delle tappeevolutive del MPi ha portato, in seguito, a un approcciooperativo di tipo probabilistico della diagnosi di MPi [25, 121].
Analisi dei criteri cliniciConsiderando il tasso di errore per eccesso del 25% circa per
la diagnosi di MPi, un tale studio clinicopatologico è statocondotto su 100 casi consecutivi di MPi diagnosticati clinica-mente e ha ottenuto una conferma istologica della diagnosi diMPi solo in 76 di questi casi [118]. Conformemente a un iterclassico in materia di ragionamento medico, la procedurautilizzata dalla banca di cervelli della Società britannica delmorbo di Parkinson (UKPDSBB) per stabilire la diagnosi clinicadi MPi (Tabella 4) [120] comprendeva tre fasi: la prima tappaformulava la diagnosi iniziale di sindrome parkinsoniana, laseconda fase eseguiva la diagnosi differenziale scarttando lesindromi parkinsoniane diverse dal MPi e la terza fase confer-mava la diagnosi di MPi. Fra i criteri clinici così elencati, ilconfronto tra i 76 casi di MPi confermato e i 24 casi di diagnosidiversa dal MPi ha permesso di individuare i VPP più elevati peril tremore, l’asimmetria dei sintomi, la risposta netta al tratta-mento con L-dopa (miglioramento clinico superiore al 50% epresenza di fluttuazioni e discinesie) e, soprattutto, per l’asso-ciazione dei tre segni cardinali (tremori, rigidità, acinesia), conun esordio asimmetrico in assenza di sintomi atipici o diun’eziologia a favore di un’altra diagnosi (Tabella 5) [118]. Altristudi clinicopatologici hanno confermato i VPP particolarmente
elevati del tremore a riposo, dell’asimmetria dei sintomiall’esordio e della risposta marcata alla L-dopa [25]; una duratadell’evoluzione superiore a 5 anni è stata inoltre considerata uncriterio supplementare in favore di una diagnosi di MPi [119].
Tabella 4.Criteri diagnostici clinici del morbo di Parkinson idiopatico (MPi) secondola Società britannica del morbo di Parkinson (UKPDSBB, da Hughes etal. [120]).
Prima tappa:diagnosi diuna sindromeparkinsoniana
Bradicinesia + almeno uno dei tre seguentisintomi:
- rigidità
- tremore a riposo (4-6 Hz)
- instabilità posturale non atassica
Seconda tappa:esclusione di diagnosidiverse dal MPi
Assenza di sintomi atipici
- crisi oculogira
- remissione prolungata
- segni strettamente monolaterali dopo3 anni di evoluzione
- paralisi sopranucleare dello sguardo
- segni cerebellari
- disautonomia grave precoce
- demenza grave precoce con disturbidel linguaggio e della memoria e aprassia
- segno di Babinski
- risposta negativa a una posologia adeguatadi L-dopa
Assenza di eziologia in favore di una diagnosidiversa dal MPi
- pregressi ictus cerebrali con progressionea «scalini» della sindrome parkinsoniana,di traumi cranici ripetuti, di encefalite certa
- terapia con neurolettici al momentodell’inizio dei sintomi
- presenza di più di un altro caso nei parentiprossimi
- presenza di un tumore cerebrale odi idrocefalo comunicante sulla TC cerebrale
- esposizione alla MPTP
Terza tappa:criteri di diagnosipositiva di MPi(tre o piùsono necessari)
- esordio monolaterale
- presenza di tremore a riposo
- peggioramento progressivo
- asimmetria persistente con sintomo del latocolpito inizialmente che mostrauna maggiore sensibilità alla dopaterapia(miglioramento del 70-100%)
- movimenti anomali involontari sottodopaterapia di intensità grave
- risposta positiva alla dopaterapia per 5 annio più
- evoluzione clinica su 10 anni o più
MPTP: metilfeniltetraidropiridina.
Tabella 5.Valori predittivi positivi (VPP) dei principali criteri clinici del morbo diParkinson idiopatico (da Hughes et al. [118]).
Criteri clinici VPP (%)
Tremore presente associato 82
Tremore autosomico 91
Asimmetria dei sintomi 85
Sindrome akinetorigida dominante 57
Sindrome akinetorigida associata 82
Due su tre (tremori, rigidità, bradicinesia) 77
Tre su tre (tremori, rigidità, bradicinesia) 88
Esordio asimmetrico, assenza di sintomi atipici 90
Esordio asimmetrico, assenza di sintomi atipici e assenzadi altra eziologia
93
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
17Neurologia

Approccio operativo alla diagnosi
In un primo approccio globale è possibile considerare comecriteri clinici necessari alla diagnosi di MPi i tre segni cardinali(tremore a riposo, bradicinesia, rigidità), a cui si aggiungonol’asimmetria, l’assenza di segni atipici, l’assenza di altre eziologiee una risposta marcata alla L-dopa [25]. Tuttavia, l’utilizzo praticodi criteri diagnostici deve essere adattato alle tappe successivedell’evoluzione della malattia che pongono il neurologo insituazioni differenti.
Così, all’inizio della fase sintomatica (situazione detta «exnovo»), il criterio della risposta alla L-dopa non è disponibile.Basandosi sui criteri di VPP elevato (tremore a riposo distale,esordio asimmetrico), l’iter diagnostico si dedica a scartare lealtre sindromi parkinsoniane con un esame neurologico preciso(che verifica l’assenza di segni atipici) e un’anamnesi esaustiva(controllando l’assenza di eziologie che depongano in favore diuna sindrome parkinsoniana secondaria); solo in caso di dubbiodiagnostico sono necessari alcuni accertamenti.
Quando la malattia è conclamata, dopo un intervallo dialcuni anni nel corso del quale il trattamento dopaminergico èstato instaurato e stabilizzato, la risposta farmacologica divieneun criterio utilizzabile e pertinente il cui VPP per la diagnosi diMPi è elevato se il miglioramento clinico è superiore al 50%.L’obiettivo rimane incentrato sulla valutazione della risposta allaL-dopa e sulla ricerca di segni atipici per scartare le altresindromi parkinsoniane degenerative, al tempo stesso utiliz-zando l’andamento evolutivo per escludere le sindromi parkin-soniane secondarie.
Infine, negli stadi più avanzati dell’evoluzione la rispostafarmacologica si è confermata con la comparsa frequente didiscinesie e fluttuazioni di efficacia: tuttavia, lo sviluppoprogressivo di segni «assiali» pone nuove difficoltà diagnosticheche giustificano una valutazione clinica e neuropsicologicaattenta, destinata a scartare alcune forme prolungate di sin-dromi parkinsoniane atipiche (in particolare la demenza a corpidi Lewy diffusi).
In tutti i casi, al termine dell’evoluzione clinica del MPi,l’esame neuropatologico dell’encefalo, aiutato dal confronto conun esame neurologico attentamente documentato, apporta ilcriterio diagnostico di certezza attraverso l’evidenziazione dicorpi di Lewy molto predominanti nelle formazioni pigmentatedel tronco cerebrale e la prevalenza della denervazione dopami-nergica nella sostanza nera con una conseguente reazionemicrogliale in assenza di altre anomalie sistemiche specifiche diun’entità patologica riconosciuta diversa dal MPi. Le diverseproposte di criteri diagnostici di MPi pubblicate in letteratura,un esempio delle quali è illustrato nella Tabella 6 [121], siispirano a questo iter pragmatico che tiene conto, da una parte,della relativa variabilità del fenotipo clinico del MPi, che portaa preferire i criteri il cui VPP è più elevato, e, dall’altra,dell’evoluzione del livello di probabilità diagnostica nella storianaturale del MPi [25, 121].
Ruolo degli accertamenti [25]
Nella misura in cui la diagnosi del morbo di Parkinson sibasa essenzialmente su criteri clinici, gli accertamenti vedonoil loro ruolo limitato alle situazioni di dubbio diagnostico, oall’inizio della malattia (in presenza di segni atipici) o nel corsodell’evoluzione (in presenza di un peggioramento rapido o diuna degradazione della risposta alla L-dopa che fanno sospet-tare una diagnosi diversa dal MPi). Tuttavia, accanto al lorocontributo essenziale nella diagnosi differenziale del MPi,principalmente con le altre sindromi parkinsoniane degenera-tive (Parkinson «plus») ma anche con le sindromi parkinso-niane secondarie, gli accertamenti possono essere utili per unamigliore valutazione o una descrizione più precisa dei sintomiclinici e delle loro conseguenze, contribuendo inoltre allacomprensione dei loro meccanismi fisiopatologici in unobiettivo di «ricerca». Infine, alcuni accertamenti di realizza-zione molto recente e di uso ancora di nicchia, per ragioni dicosto eccessivo o di accessibilità insufficiente o perché il lororeale impatto nella gestione del MPi resta ancora poco definito,
possono essere considerati strumenti potenziali il cui campo diapplicazione ha dei limiti che devono essere precisati.
Esami utili alla diagnosi differenziale
LaboratorioResta tradizionale e consensuale escludere, nei pazienti in cui
una sindrome parkinsoniana esordisce prima dei 45 anni, unamalattia di Wilson attraverso il bilancio del rame (plasmatico eurinario) e il dosaggio della ceruloplasmina plasmatica: piùaneddoticamente, può essere giustificato, sempre in soggettigiovani, scartare le ipotesi di una neuroacantocitosi di presen-tazione atipica con la ricerca di emazie deformate al test diHam-Dacie o di una sindrome di Fahr con la ricerca di unipoparatiroidismo. Il campo di applicazione della genetica alladiagnosi differenziale, ma anche alla diagnosi delle formefamiliari ancora non differenziate di MPi, è attualmente inpieno sviluppo grazie all’evidenziazione di numerosi nuovigenotipi [26]: ogni ipotesi diagnostica, in particolare (ma nonesclusivamente) per le forme a inizio precoce, deve basarsi suun’indagine familiare esaustiva, dopo aver informato il pazientee con il suo consenso, con l’obiettivo di precisare la modalitàdi trasmissione, e su una descrizione più precisa possibile delfenotipo clinico in modo da orientare la ricerca delle mutazionifrequenti la cui diagnosi molecolare è accessibile.
Diagnostica per immagini morfologicaL’uso della TC cerebrale serve a escludere lesioni focali
(tumori, localizzazioni infettive) o più diffuse (traumatiche,vascolari, idrocefalo) il cui sviluppo eventualmente progressivopuò assumere un andamento pseudo-parkinsoniano con addi-rittura, in alcuni casi, una risposta alla L-dopa sufficientementenetta da essere ingannevole; tuttavia, le situazioni ben docu-mentate di questo tipo restano piuttosto rare [25].
La RM encefalica fornisce un contributo sempre più riconos-ciuto alla diagnosi dei Parkinson «plus» [122]: alcuni aspettisuggestivi sono stati, in effetti, descritti per le atrofie multisis-temiche di forma cerebellare (atrofia del ponte e del cervelletto
Tabella 6.Criteri di diagnosi di morbo di Parkinson idiopatico (MPi, da Gelb etal. [121]).
Criteri A Tremore a riposo
Rigidità
Bradicinesia
Esordio asimmetrico
Criteri B (in favoredi una diagnosi diversadal MPi)
Sintomi atipici all’esordio del MPi(3 anni): instabilità posturale marcata,fenomeno di freezing, allucinazioni non legateai farmaci, demenza
Paralisi sopranucleare dello sguardoo rallentamento dei movimenti saccadiciverticali
Disautonomia grave non legata ai farmaci
Evidenziazione di un’eziologia: lesionicerebrali focali che possono produrreuna disfunzione dopaminergica o usodi neurolettici negli 6 ultimi mesi
Criterio C Risposta farmacologica marcata e durevole
MPi possibile Presenza di almeno due dei quattro criteri A (dicui uno almeno è il tremore o la bradicinesia)
Assenza di criteri B
Presenza del criterio C a
MPi probabile Presenza di almeno tre dei quattro criteri A
Assenza dei criteri B
Presenza del criterio C a
MPi certo Ogni MPi possibile o probabile clinicamentecon conferma istopatologica della diagnosi
a Nel caso in cui la durata di evoluzione clinica del MPi sia inferiore a 3 anni, lapresenza del criterio C non è necessaria se la risposta alla terapia con L-dopa o conun agonista dopaminergico non è ancora stata documentata.
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
18 Neurologia

con segnale iperintenso lineare che delimita il putamen su unfondo ipointenso del putamen posterolaterale in sequenza T2),per la paralisi sopranucleare progressiva (atrofia a predominanzamesencefalica della regione tectale periacqueduttale con segnaleiperintenso in T2) e per la degenerazione corticobasale (atrofiacorticale frontoparietale asimmetrica a spese del lato controla-terale a quello dell’interessamento clinico iniziale).
Diagnostica per immagini funzionale
Lo sviluppo recente della tomoscintigrafia monofotonica(SPET) al 123I-Ioflupano (DaTSCAN®, tracciante specifico dei sitipresinaptici trasportatori di dopamina nello striato), dopol’autorizzazione all’immissione in commercio in Francia nel2004 [NdT: in Europa il 27 luglio 2000] nell’indicazione delladiagnosi differenziale tra tremore parkinsoniano e tremoreessenziale [123], richiede di precisare il ruolo di questa diagnos-tica per immagini funzionale dell’innervazione dopaminergicastriatale. Malgrado il suo costo ancora elevato, l’accessibilità diquesta indagine è stata considerevolmente migliorata, il checonduce a una riflessione sul suo buon uso. Due studi [124, 125]
hanno valutato il contributo di questo esame alla diagnosidifferenziale del tremore e confermano l’apporto diagnosticodella diagnostica per immagini della denervazione dopaminer-gica nel caso in cui i criteri diagnostici clinici siano incompletio dubbi (tremore isolato, assenza di progressione, rispostadubbia al trattamento antiparkinsoniano, tremore misto diriposo e di posizione), poiché tali condizioni non sono eccezio-nali, in particolare all’esordio del MPi. Si deve tuttavia precisareche, se la sensibilità e la specificità dell’analisi visiva per ilrilevamento della denervazione dopaminergica striatale caratte-ristica del MPi (riduzione del segnale a predominanza putami-nale dorsolaterale e più marcata sul versante controlaterale aquello dove sono presenti, o predominano, i sintomi clinici)sono molto elevate, esse non raggiungono il 100% [126]: èopportuno esigere una tecnica rigorosa e, a volte, ricorrere a unaquantificazione, il che non è alla portata di tutti i centri.Nonostante il costo elevato dei traccianti, alcune applicazionisono utili per la diagnosi differenziale tra MPi e atrofia multi-sistemica (in particolare la forma putaminale): si tratta dellascintigrafia cerebrale con 123I-iodobenzamide (IBZM, tracciantedei recettori dopaminergici D2 nello striato) [127] e dellascintigrafia miocardica con 123I-meta-iodobenzilguanidina(MIBG, tracciante dei recettori catecolaminergici cardiaci) [128].
Elettrofisiologia
L’elettroencefalogramma (EEG) può contribuire alla diagnosidifferenziale con una demenza a corpi di Lewy diffusi nellaquale spesso si osserva un rallentamento diffuso dell’attività difondo con onde lente frontotemporali. La polisonnografia puòcontribuire a caratterizzare i disturbi del sonno, specialmentequelli associati al sonno paradosso, evidenziando una perdita diatonia.
L’elettromiografia (specialmente perineale), abbinata all’uro-dinamica, conserva un’utilità confermata per la diagnosidifferenziale con le atrofie multisistemiche, caratterizzate daun’ipoattività del detrusore e da una denervazione marcata deglisfinteri uretrale e anale.
La caratterizzazione della disautonomia con la misurazione divariabilità dell’intervallo R-R su un elettrocardiogramma durantela manovra di Valsalva e la valutazione precisa dell’ipotensioneindotta con il passaggio in ortostatismo (attivo o inclinazionepassiva sul tavolo basculante) possono aiutare a differenziare leatrofie multisistemiche da autentici MPi con disautonomiamarcata [51].
L’elettro-oculografia permette, in modo non invasivo, dievidenziare anomalie di esecuzione dei movimenti saccadici, inparticolare verticali, così come alcuni errori perseverativi nel testdelle antisaccadi che caratterizzano la paralisi sopranucleareprogressiva.
Test neuropsicologici
La valutazione e la caratterizzazione dei disturbi cognitivi nelmorbo di Parkinson e nelle sindromi parkinsoniane si basano subatterie di test il cui scopo è quello di fare un inventario dellecapacità mnesiche e attentive, dell’interessamento eventuale difunzioni strumentali (prassia o anche linguaggio) e, soprattutto,dei disturbi delle funzioni esecutive. Una batteria che associa iseguenti test (scala di Mattis per una valutazione globale della«demenza», test di Grober e Buschke per la memoria immediatae il ricordo, test del Wisconsin, di Stroop, della fluenza verbalee di trail making per le funzioni esecutive e ricerca dei compor-tamenti di prensione, di utilizzo e di imitazione) è stataproposta in un iter di diagnosi differenziale [97], illustrato nellaTabella 7 [95, 97].
Esami di «ricerca»
Laboratorio
Si tratta, essenzialmente, del dosaggio della L-dopa plasmatica(con metodo di cromatografia liquida ad alta prestazione): talidosaggi si concepiscono solo in maniera ripetuta a intervalliprecisi in seguito all’assunzione di una dose di L-dopa per«disegnarne» la cinetica plasmatica rappresentativa del processodi assorbimento digestivo e, quindi, della sua eliminazioneprogressiva dall’ambiente ematico [129]. La concentrazionemassimale (Cmax) è molto variabile da un paziente all’altro(livelli plasmatici «efficaci» in media tra 1 000 e 3 000 ng/ml):anche la cinetica di assorbimento (tempo di salita fino al picco,Tmax) è molto variabile (in media tra i 60 e i 90 minuti); questidati si riferiscono a una forma galenica standard di levodopa,con un’emivita plasmatica molto breve (in media 120 min).
Diagnostica per immagini funzionale
La PET resta potenzialmente uno strumento fondamentale didiagnosi attraverso la marcatura in vivo, con L-dopa marcatacon 18-fluoro, della funzione del sistema dopaminergiconigrostriatale: questa applicazione, che è utile non solo all’iniziodella fase sintomatica, ma anche nella fase di progressione dellamalattia, rimane poco accessibile e di costo proibitivo con laPET, limitando il suo utilizzo a obiettivi di ricerca. La diagnos-tica per immagini funzionale cerebrale può anche essereutilizzata al di fuori degli scopi diagnostici per identificareanomalie di attivazione metabolica delle zone cerebrali, in
Tabella 7.Diversi profili dei test neuropsicologici: morbo di Parkinson idiopatico e sindromi affini (da Pillon et al. [97] e Agid [95]).
MP AMS DCB PSP MPD
Demenza Deterioramento globale - - - + +
Fluttuazioni - - - - -
Disturbi della memoria Consolidamanto - - - - -
Ricordo + + + ++ ++
Disturbi strumentali Linguaggio - - + ± ±
Prassia - - ++ ± ±
Disturbi delle funzioni esecutive Pianificazione + + + ++ ++
Comportamenti ± ± + ++ +
Allucinazioni - - - - ±
AMS:atrofiamultisistemica;DCB:degenerazionecorticobasale; PSP:paralisi sopranucleareprogressiva;MP:morbodiParkinson;MPD:morbodiParkinsoncondemenza;MH:malattiadiHuntington;MCL:malattia acorpidiLewy; -: assente;±:discreto:+:moderatoopresentenellamaggiorpartedei casi; ++: graveepresentenellamaggioranzadei casi.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
19Neurologia

particolare nel corso di compiti motori nel morbo di Parkinson,che si tratti di applicazioni in PET o i RM funzionale.
Diagnostica per immagini morfologica
Si tratta, qui, di applicazioni della risonanza magnetica mirateall’identificazione del contenuto molecolare nelle regionicerebrali: così, la RM ha contribuito a precisare la ridistribuzionedel ferro tra striato e sostanza nera nel MPi [130] e, con l’aiutodi tecniche evolute (T2 rho, 3 tesla, R2*, spin-lattice distributionimaging), permette di rivelare l’accumulo intranigrale diferro [131, 132] e alterazioni precoci del contenuto intracellulare.
L’ecografia della sostanza nera, con l’aiuto di sonde transcra-niche, rappresenta un approccio promettente e molto piùaccessibile dell’accumulo intranigrale di ferro nel morbo diParkinson fin dagli stadi precoci [133].
Elettrofisiologia
Registrazioni dei tremori (accelerometria con analisi dellospettro di frequenza, affiancata eventualmente all’elettro-miografia [EMG] di superficie). Esse possono contribuire aoggettivarne le caratteristiche cliniche e, in funzione dellevarianti tecniche utilizzate, possono apportare una forma divalutazione quantitativa: inoltre, questo tipo di analisi puòaiutare a riconoscere il tremore «mioclonico» dall’atrofiamultisistemica.
Studi dell’eccitabilità delle regioni corticali motorie(mediante stimolazione magnetica transcranica) e delle vieriflesse del tronco cerebrale. Partecipano a una migliorecomprensione dei fenomeni di inibizione laterale e di «focaliz-zazione» dei messaggi afferenti corticali, mentre l’analisi deiriflessi del tronco cerebrale (riflesso di ammiccamento, reazionidi soprassalto al rumore) permette di ritrovare un’alterazionedelle risposte nella paralisi sopranucleare progressiva, mentrequeste sono relativamente conservate nel MPi.
Studi della preparazione e dell’esecuzione del movimento.Questo approccio neurofisiologico dell’acinesia impiega proce-dure diverse: tempi di reazione, correlazioni elettrofisiologichedella preparazione motoria e misure della velocità e dellaprecisione dei movimenti. Le attività motorie più complesse(marcia, produzione vocale) necessitano di analisimultiparametriche.
Le misure del tempo di reazione (TR) dimostrano che esso è,di regola, allungato nel MPi e nelle sindromi parkinsoniane;tuttavia, il tempo centrale (differenza di durata tra TR di sceltae TR semplice, corrispondente al tempo supplementare necessa-rio alla selezione di una risposta) non è allungato nel MPirispetto alla norma [89]. Inoltre, la facilitazione della prepara-zione motoria con informazioni preliminari sulla risposta attesaresta più o meno conservata nel MPi a seconda che questeinformazioni siano fornite in modo esplicito o implicito.
Le correlazioni elettrofisiologiche della preparazione motoria(indici attentivi specifici come l’onda P300 dopo uno stimolosonoro, potenziali lenti e alterazioni dell’attività EEG nel verticeprecedenti l’avvio del movimento come la variazione contin-gente negativa, il Bereischaftspotential e la desincronizzazioneEEG legata all’evento) si rivelano anormali nel morbo diParkinson.
La misurazione della velocità di esecuzione del movimentoconferma il rallentamento di questo: le registrazioni EMGmostrano l’ampiezza insufficiente della scarica iniziale delmuscolo antagonista con un deficit di regolazione della forzamuscolare e una perdita degli aggiustamenti posturali anticipati.La precisione del movimento è conservata grazie a un usoaccresciuto dei riferimenti visivi, e la soppressione della visionedel movimento rivela una riduzione dell’ampiezza di questo.
L’analisi multiparametrica della postura e della deambula-zione rivela che, malgrado i cambiamenti evidenti della posturain piedi stabilizzata nel morbo di Parkinson, la posizione dellaproiezione verticale del centro di gravità resta poco modificatarispetto ai soggetti normali: all’inizio della deambulazione esisteuna netta riduzione delle forze propulsive che permettono diaccelerare il centro di gravità verso l’avanti nel morbo diParkinson, il che provoca una riduzione marcata della lunghezzadel passo; nella locomozione, la riduzione della lunghezza della
falcata e della velocità di progressione è confermata dal miglio-ramento di questi parametri con l’utilizzo accresciuto deiriferimenti visivi grazie a linee orizzontali poste sul terreno.Anche la rotazione assiale è molto alterata, il che penalizza icambiamenti di direzione [134].
Diagnosi differenziale del morbodi Parkinson
Nel quadro della gestione clinica del morbo di Parkinson l’iterdiagnostico rimane sotteso dalla problematica della sua diagnosidifferenziale [51] per tutta la sua evoluzione clinica a causadell’impatto sulla gestione terapeutica.
La Tabella 8 [135] presenta una lista non esaustiva dell’insiemedei parkinsonismi catalogati [51, 135] che possono essere classifi-cati, al di fuori del MPi, in due categorie principali: parkinso-nismi secondari di cui si può determinare il meccanismoeziologico e parkinsonismi «plus» che si caratterizzano piuttostoper dei sintomi atipici rispetto al quadro clinico del MPi e la cuicausa rimane in parte indeterminata.
Tra i parkinsonismi secondari, l’eziologia più frequente restaquella dei farmaci che possono essere implicati nella comparsadi una sindrome parkinsoniana la cui lista è presentata nellaTabella 9.
■ Aspetti terapeutici: gestionedei pazienti parkinsoniani
La gestione terapeutica nel morbo di Parkinson, che restaancora ampiamente dominata dai trattamenti dopaminergici i cuieffetti sintomatici restavano ineguagliati dalla loro introduzione30 anni or sono, si è tuttavia costantemente evoluta, nonsoltanto nella strategia della loro introduzione iniziale all’esordiodella malattia, ma anche nella gestione delle complicanze checompaiono nella fase conclamata: la progressiva comprensionedell’utilità delle scale di valutazione clinica e dell’apporto dellariabilitazione funzionale e, soprattutto, la messa a punto di nuoveprocedure chirurgiche, in particolare la stimolazione del NST i cuispettacolari effetti sintomatici sono stati confermati [136], influen-zano sempre più la pratica neurologica attuale. La consensusconference sul morbo di Parkinson [25] ha permesso di elaborarealcune raccomandazioni che sono state ampiamente diffuse e ilcui contenuto influenza necessariamente il testo di questoarticolo. Queste raccomandazioni sono state, in seguito, confer-mate con i criteri della «medicina basata sulle evidenze» [137].
“ Punti essenziali
Segni di allarme che devono far ipotizzare unparkinsonismo «plus» (da Tison, in [31]).• Instabilità posturale e cadute precoci• Disfagia, disartria precoce, sindrome pseudobulbare• Disautonomia precoce• Demenza iniziale o precoce• Segni piramidali• Segni cerebellari• Segni del corno anteriore• Segni corticali parietali asimmetrici: aprassia• Disturbi dell’oculomotricità verticale• Tremore «mioclonico»• Progressione rapida• Assenza di una risposta prolungata alla L-dopa• Discinesie dopa-indotte assenti o atipiche• Rapida perdita della deambulazione e della stazioneeretta• Antecollis esagerato e contratture distoniche fisse
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
20 Neurologia

Farmaci antiparkinsoniani [25, 51, 137]
Questi farmaci hanno, o si vedono riconoscere al momento,solo un effetto sintomatico. L’emergenza del concetto di«neuroprotezione» ha fatto intravedere la possibilità di rallen-tare la progressione della malattia, il che è stato smentito dairisultati negativi iniziali [138] e differiti [139] dello studioDATATOP, che ricercava eventuali effetti neuroprotettivi dellaselegilina e del tocoferolo: sono in corso altri studi su questopunto con farmaci della classe degli agonisti dopaminergici edegli inibitori della MAO-B.
La Tabella 10 presenta la lista dei farmaci antiparkinsonianiattualmente disponibili, con le loro forme galeniche e la loromodalità d’azione farmacologica presunta: ad oggi, la L-doparesta considerata come il trattamento di riferimento del morbodi Parkinson, anche se gli agonisti dopaminergici svolgono unruolo progressivamente crescente [25]. Altri farmaci hanno uneffetto sintomatico che passa attraverso il rallentamento dellametabolizzazione della dopamina (selegilina) o per la loroazione antagonista muscarinica (anticolinergici) o glutamater-gica (amantadina), mentre gli inibitori della COMT (entaca-pone, tolcapone) agiscono attraverso la L-dopa somministratasimultaneamente prolungando la sua durata d’azione, rendendocosì più continua la stimolazione dopaminergica.
Benché la L-dopa sia considerata «la pietra angolare dellagestione dei pazienti e il più efficace degli antiparkinsoniani» [25],la comparsa di complicanze motorie (fluttuazioni e discinesie) nelcorso della dopaterapia ha condotto a rimettere in questione lasua posizione dominante a vantaggio degli agonisti dopaminer-gici (in particolare all’esordio della malattia, quando il disturbofunzionale non richiede necessariamente un effetto sintomaticopotente) e anche a prendere in considerazione la sua tossicità suineuroni dopaminergici (a causa del suo metabolismo ossidativoattraverso la MAO). Tuttavia, la responsabilità della L-dopa nellacomparsa di queste complicanze motorie resta una questioneancora non risolta nella misura in cui l’ipotesi del suo ruolotossico «in vivo» è stata scartata sulla base di molte argomenta-zioni [25, 140, 141]; parallelamente, la fisiopatologia di questecomplicanze è stata rivisitata [104], con l’emergenza dell’ipotesipostsinaptica di una sensibilizzazione (o priming) nei recettoristriatali, più adatta alla spiegazione delle discinesie e checompleta il ruolo già riconosciuto della perdita di terminazionidopaminergiche nigrostriatali nel meccanismo delle fluttuazioni;il carattere «pulsatile» della dopaterapia diviene allora un fattoreessenziale di questa sensibilizzazione [142]. Sul piano pratico, ci sipuò anche interrogare sul fatto di sapere se queste complicanzemotorie sono legate direttamente alla data di introduzione dellaL-dopa e alla sua posologia o, prima di tutto, alla gravità iniziale
Tabella 8.Classificazione dei parkinsonismi (modificata da Jankovic [135]).
I - Idiopaticoo primitivo:morbodi Parkinson
II - Sintomaticoo secondario(eziologiadeterminata)
A - Infezioni virali o imparentate a queste
- encefalite letargica (postencefalitica)
- encefaliti virali, incluso HIV
- infezioni batteriche, fungine, parassitarie del SNC
- altre: prioni, panencefalite sclerosante subacuta,malattia di Whipple
B - Intossicazioni
- manganismo cronico
- monossido e solfuro di carbonio, cianuro, metanolo,idrocarburi, n-esano, solventi, disulfiram
- MPTP, BMAA (complesso SLA-demenza-Parkinsondi Guam)
- anossia/ipossia, encefalopatia dopo punturadi vespa
C - Farmaci
- neurolettici e affini
- reserpinici
- altri (alfametildopa, litio)
- calcioantagonisti
D - Altro
- tumori cerebrali
- trauma (encefalopatia dei pugili)
- vasculopatia (aterosclerosi, angiopatia amiloide,malattia di Binswanger)
- disturbi metabolici (degenerazione epatocerebraleacquisita, mielinolisi centrale pontina)
- idrocefalo a pressione normale
- calcificazione dei nuclei grigi (sindrome di Fahr)
III -Parkinsonismoplus (eziologianon o maldeterminata)
A - Malattie sporadiche
- paralisi sopranucleare progressiva
- atrofia multisistemica (forma putaminale, formacerebellare)
- degenerazione corticobasale
- demenza a corpi di Lewy diffusi
- malattia di Pick
- morbo di Alzheimer
- sclerosi laterale amiotrofica
- gliosi sottocorticale progressiva
- emiparkinson/emiatrofia
- sindrome di Rett
B - Malattie ereditarie
- malattia di Wilson
- malattia di Huntington
- malattia di Hallervorden-Spatz
- atassie spinocerebellari autosomiche dominanti(SCA3)
- neuroacantocitosi
- atrofia dentato-rubro-pallido-luisiana
- sindrome depressione ipoventilazione alveolare
- distonia dopasensible
- sindromi parkinsoniane familiari varie
- sindrome Parkinson-distonia legata all’X (Lubag)
- emocromatosi ereditaria
- encefalomiopatie mitocondriali
- malattie lisosomiali
HIV: virus dell’immunodeficienza umana; SNC: sistema nervoso centrale; MPTP:metilfeniltetraidropiridina; BMAA: beta-N-metilamino-L-alanina; SLA: sclerosilaterale amiotrofica; SCA3: spinocerebellar ataxia 3.
Tabella 9.Principali farmaci implicati nelle sindromi parkinsoniane iatrogene(da Montastruc et al. Fund Clin Pharmacol 1994;8:293-306.).
Antiadrenergici Reserpina, tetrabenazina
Neurolettici Fenotiazine, butirrofenoni, tioxanteni,dibutilpiridine, benzamidi, loxapina,risperidone, olanzapina, clozapina?
Antipertensivi Metildopa
Calcioantagonisti Flunarizina, cinarizina, diltiazem,verapamil
Antiaritmici Amiodarone
Antidepressivie timoregolatori
Fluoxetina?, fluvoxamina, amossapina, litio
Anticomiziali Valproato, fenitoina
Colinergici Betanecolo, piridostigmina, tacrina
Antinfettivi Cefazolidina, amfotericina B,trimetoprim-sulfametossazolo
Citostatici Vincristina + adriamicina,citosina-arabinosina
Altro Sulindac, trazodone, fenelzina,meperidina, dietilpropion, clorfeniramina,xilometazolina, codeina, difenidramina,procaina, ciclosporina
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
21Neurologia

della denervazione dopaminergica e alla sua evolutività, chedeterminerebbero esse stesse un’introduzione precoce e unaposologia elevata della L-dopa.
Riabilitazione funzionale [25]
Essa comporta diverse modalità a seconda che riguardil’attività fisica globale (motricità degli arti, postura, equilibrio eandatura) che fa intervenire il fisioterapista o l’attività dellasfera oro-faringo-laringea (parola, deglutizione) più specificata-mente gestita dal logopedista.
Riabilitazione motoria [143, 144]
Le tecniche sono empiriche e comportano dei programmi diesercizi adattati al sintomo motorio predominante e allo stadiodella malattia, eventualmente completati con la balneoterapia el’ergoterapia:• esercizi volti ad accrescere la coordinazione, la precisione e la
velocità del movimento;
• movimenti destinati a preservare il controllo posturale e lerotazioni assiali;
• lavoro sulla deambulazione utilizzando indizi visivi o sonori.
Logopedia [145]
Il metodo Lee Silverman Voice Therapy (LSVT) è basato su unarieducazione della prosodia (lavoro laringeo con mantenimentodi una vocale tenuta e lavoro sull’estensione della gammatonale) completata con una rieducazione respiratoria (lavorosull’inspirazione e sulla coordinazione pneumofonica).
Rieducazione della deglutizione [25]
Fondata sull’analisi dei disturbi della deglutizione precisandoil meccanismo dell’ab ingestis, comporta un tempo di consu-lenza alimentare (frazionamento del pasto, sapori accentuati,cibi semisolidi) e mira all’apprendimento di posture di «prote-zione» (posizione seduta e testa in antiflessione al momentodella deglutizione).
Tabella 10.Farmaci antiparkinsoniani disponibili in Italia.
Classe DCI Forme galeniche
(unità = mg)
Azione farmacologica
L-dopa associata a un inibitoredella decarbossilasi periferico
L-dopa + benserazide Standard: capsule 62,5, 125 e 250(rispettivamente: 50, 100 e 200 mg diL-dopa)
LP: capsula 125 (100 mg L-dopa, 25 mgbenserazide)
Dispersibile: compressa frazionabile 125(100 mg L-dopa)
Corregge il deficit di dopamina: captatadalle terminazioni dopaminergichemigrostriate residue (e dalla glia?),poi decarbossilata in dopamina cheagisce in seguito sui recettori (effettoD1 dominante)
L-dopa + carbidopa Standard: compresse divisibili 100 e 250(rispettivamente: 100, 250 mg L-dopa e 10,25 mg carbidopa) e LP: compresse divisibili100 e 200 (rispettivamente: 100, 200 mgL-dopa e 25, 50 mg carbidopa)
L-dopa/carbidopa associata aun inibitore della COMT
L-dopa + carbidopa + entacapone Compresse 50, 100, 150, 200(rispettivamente 50, 100, 150, 200 mgL-dopa, 12,5, 25, 37,5, 50 mg carbidopae 200 mg entacapone)
Agonisti dopaminergici Bromocriptina
(Bromocriptina)
Compressa 2,5 mg, capsule 5 e 10 mg
(farmaco generico: stessa preparazionegalenica)
Agiscono direttamente sui recettoridopaminergici
Azione D2 dominante, ma anche D1per il pergolide e, soprattutto,per l’apomorfina (profilo similealla L-dopa)
Lisuride Compresse divisibili da 0,2 e 0,5 mg(solubile, via sottocutanea)
Pergolide Compresse divisibili da 0,05, 0,25 e 1 mg
Piribedil Standard (compressa 20 mg), LP(compressa 50 mg)
Ropinirolo Standard: compresse da 0,25, 0,5, 1, 2,5 mg; LP 2, 4, 8 mg
Pramipexolo Compresse da 0,7 mg
Apomorfina Iniettore a penna 30 mg (via sottocutanea)
Anticolinergici Triesifenilide Compresse da 2 mg
Capsule LP da 5 mg
Antagonismo muscarinico
a livello centrale e
azione parasimpaticolitica
a livello periferico
Altri Compresse 2mg, 4 mg a rilascioprolungato, 5mg/1 ml soluzioneiniettabile, 2,3 mg/1 ml gocce
Inibitori della MAO-B Selegilina Compresse frazionabili 5 mg Blocco selettivo irreversibile MAO-B
(ritarda la metabolizzazionedella dopamina)
Inibitori della COMT Entacapone Compressa da 200 mg Blocco COMT periferico (prolungal’emivita plasmatica della L-dopa)e centrale per il tolcaponeTolcapone Compressa da 100 mg
Antiglutammatergici Amantadina Capsula da 100 mg Antagonismo glutammatergico(Rc NMDA)
DCI: denominazione comune internazionale; COMT: catecol-O-metiltransferasi; MAO: monoamino-ossidasi; LP: liberazione prolungata; NMDA: N-metil D-aspartato.
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
22 Neurologia

Trattamenti chirurgici [25, 146]
Solo l’approccio stereotattico permette di conciliare un’effica-cia significativa con un rischio minimo: esso si limita alleapplicazioni di neurochirurgia funzionale (lesione, stimolazione)su diversi bersagli (VIM, GPi, NST), mentre i trapianti di celluleembrionali restano allo stadio sperimentale.
Tecniche di neurochirurgia funzionaleLa tecnica lesionale classica con elettrocoagulazione presenta
il vantaggio di un atto operatorio singolo senza alcun impiantodi materiali, il che riduce il costo immediato dell’intervento; glisvantaggi risiedono nel suo carattere irreversibile e necessaria-mente monolaterale, con la difficoltà di definire l’estensionedella lesione utile.
La tecnica di neurostimolazione ad alta frequenza [147],nonostante il costo immediato del materiale impiantabile, ha ivantaggi della reversibilità, dell’adattabilità con la regolazionedei parametri di stimolazione e della buona tollerabilità diun’azione bilaterale.
Bersagli della neurochirurgia funzionaleIl VIM del talamo resta il bersaglio di elezione per un tremore
isolato e non controllato con i farmaci. Questo bersaglio si èanche rivelato efficace per ridurre le discinesie dopa-indotte.Uno studio recente, che confrontava gli effetti della lesione conquelli della stimolazione, ha rilevato una superiorità a favoredella stimolazione in virtù di una migliore efficacia e di effettiindesiderabili minori [148].
Il GPi, nella sua parte ventro-postero-laterale descrittainizialmente da Leksell e Svennilson, è stato riattualizzato eresta ancora molto utilizzato a causa degli effetti sintomaticianti-acinetici moderati, ma soprattutto di una riduzione moltomarcata delle discinesie dopa-indotte [25]. La stimolazione portachiaramente il vantaggio di una possibile bilateralizzazione [149],poiché la pallidotomia (lesionale) deve restare strettamentemonolaterale.
Il NST è stato aggredito più recentemente [150] ed è rapida-mente divenuto il bersaglio di elezione per una stimolazionebilaterale, con effetti spettacolari sull’insieme dei sintomi(tremori, rigidità, acinesia) che permettono di ridurre i farmaciantiparkinsoniani e di ottenere, per questo motivo, quasi unascomparsa delle fluttuazioni e delle discinesie [136] con laconferma del mantenimento di questi risultati terapeutici a5 anni [151].
Il PPN è divenuto molto recentemente un bersaglio, pertrattare più particolarmente i segni assiali (particolarmente ilfreezing della deambulazione), ma i risultati restano ancoraoggetto di controversie.
Altre tecnicheSolo l’allotrapianto striatale dei neuroni dopaminergici
embrionari resta eseguito in un quadro sperimentale, conrisultati sintomatici ancora molto inferiori a quelli dellastimolazione del NST, anche se sono state dimostrate la vitalitàe l’efficacia dopaminergica delle cellule trapiantate.
Scale di valutazione utiliEsse sono necessarie per il follow-up clinico obiettivo di una
patologia cronica ed evolutiva come il morbo di Parkinson [25].
Scala unificata di valutazione del morbodi Parkinson (Unified Parkinson’s Disease RatingScale [UPDRS])
Questa scala composita rappresenta attualmente lo standardinternazionale di valutazione clinica per l’insieme dei sintomidel morbo di Parkinson e dei problemi legati al suotrattamento [152].
Essa comporta 6 parti:• la parte I valuta i disturbi mentali con l’aiuto di quattro
elementi;• la parte II rappresenta la disabilità nelle attività quotidiane
secondo 13 elementi;
• la parte III descrive i sintomi motori al momento dell’esamesulla base di 27 elementi;
• la parte IV stabilisce le complicanze legate al trattamento;• le parti V e VI corrispondono all’incorporazione dello stadio
evolutivo secondo Hoehn e Yahr e del punteggio globale diautonomia nella vita quotidiana di Schwab ed England.La sua affidabilità globale è stata confermata [25], ma le parti
I, II e IV restano oggetto di critiche ed è in corso di elabora-zione una versione revisionata che prevede in particolare unosviluppo delle parti I e II per integrare più ampiamente i segninon motori, il che giustifica, nell’attesa, l’utilizzo di scalecomplementari.
Altre scale
Per i disturbi cognitivi, la scala di Mattis e la batteria rapidadi efficienza frontale (BREF) appaiono preferibili al Mini MentalState (MMS) di Folstein, mentre, per i disturbi timici, la scala didepressione di Montgomery e Asberg (MADRS) sembra la piùadeguata. Per completare la parte II dell’UPDRS, la qualità dellavita può essere valutata meglio con la PDQ-8 (versione riassuntadella PDQ-39) la cui affidabilità è stata validata. La parte IIIdell’UPDRS resta la più valutata e può, eventualmente, essereoggettivata con una registrazione video. Questa parte III sipresta particolarmente bene alla pratica di test farmacologici(alla L-dopa o all’apomorfina) permettendo di quantificare larisposta al trattamento con la differenza tra i punteggi ottenutiin periodo off e in periodo on. Infine, la parte IV può esserecompletata da un’autovalutazione a intervallo orario dei periodion e off per le fluttuazioni di efficacia e per la versione CAP-SIT [153] per le discinesie (distribuzione, intensità, tipologia).
Indicazioni terapeutiche
Gestione all’esordio della malattia [25, 51, 137]
Annuncio della diagnosi
Questa è una tappa fondamentale sul piano psicologico,poiché il morbo di Parkinson conserva l’immagine di unacondizione invalidante associata all’invecchiamento. È quindiconsigliabile, nel corso delle visite iniziali, mettere in risalto leprospettive positive che rappresentano l’efficacia sintomaticaattesa del trattamento dopaminergico, l’esistenza di forme aevoluzione lenta e il risparmio relativo delle funzioni mentali,aspetti che lasciano sperare, almeno durante i primi anni dettidi «luna di miele», la conservazione della qualità della vitasocioprofessionale e familiare. Occorre in seguito informare ilpaziente e le persone a lui vicine sulle diverse scelte terapeuti-che possibili.
Principi di base per la scelta del trattamento
Allo stato attuale delle conoscenze, non esiste alcun farmacodisponibile che si possa definire «neuroprotettore», vale a direin grado di rallentare o arrestare l’evoluzione progressiva delladenervazione dopaminergica [25].
Trattandosi del trattamento dopaminergico propriamentedetto, è opportuno ricordare il carattere puramente sintomaticodella sua azione. Questo trattamento è dunque iniziato soltantoquando il disturbo funzionale lo giustifica e la sua posologia èadattata progressivamente tenendo conto della tolleranza, inparticolare agli effetti collaterali digestivi e pressori, poichél’obiettivo è quello di ottenere la migliore efficacia possibile suisintomi motori con una posologia minima regolata in base alleesigenze. In effetti, questo trattamento sarà di lunga durata,tanto più quanto più giovane sarà il paziente, il che implica diprendere in considerazione una forma di compromesso strate-gico tra il beneficio sintomatico a breve termine, giustificatodalla necessità oggettiva di preservare la qualità della vitaprofessionale e familiare, e le potenziali complicanze deltrattamento a medio e a lungo termine più o meno prevedibili,dal momento che questo rischio è legato all’evolutività delladenervazione dopaminergica che resta, su scala individuale,un’incognita all’inizio della fase sintomatica del morbo diParkinson.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
23Neurologia

Strategie terapeutiche possibili
La L-dopa in monoterapia (L-dopa associata a un inibitoredella decarbossilasi) può essere motivata sulla base del suorapporto efficacia/tolleranza che rimane attualmente il piùelevato, in particolare nel periodo di esordio. Tuttavia, la suaemivita biologica breve le conferisce la qualifica di trattamento«pulsatile», con un rischio più elevato, e di insorgenza piùprecoce, di fluttuazioni di efficacia e di discinesie, rispetto aglialtri trattamenti dopaminergici [154]. L’ipotesi del suo effettoneurotossico, sospettata a partire da studi in vitro, sembraesclusa in vivo sulla base di molte argomentazioni indirette [25,
140] e dei risultati dello studio prospettico ELLDOPA [141].L’interesse di strategie complementari (L-dopa a liberazioneprolungata, associazioni con un inibitore della MAO-B e/o dellaCOMT), destinate a rendere la dopaterapia meno «pulsatile»,resta ancora da dimostrare [25].
Gli agonisti dopaminergici in monoterapia si sono vistiriconoscere recentemente un rapporto efficacia/tolleranzainiziale, se non analogo a quello della L-dopa, almeno a unlivello molto vicino, con un rischio potenziale a medio e alungo termine di complicanze motorie notevolmenteridotto [154]. Tuttavia, l’instaurazione del trattamento deverimanere molto progressiva, il che implica un’informazione delpaziente solida e continua, allo scopo di conservare la suaadesione agli obiettivi del trattamento. Inoltre, questo rapportoefficacia/tolleranza favorevole pare non mantenersi al di là dialcuni anni, il che richiede di associare la L-dopa come tratta-mento di supporto per mantenere l’effetto sintomatico: ineffetti, l’aumento della posologia degli agonisti dopaminergici,che sarebbe necessario per rimanere in monoterapia, si rivelafrequentemente mal tollerato, specialmente sul piano psichicoe nei soggetti anziani.
L’associazione precoce di L-dopa e di agonisti dopaminergici,dal momento che ogni farmaco è prescritto alla posologiaminima, si basa principalmente sul postulato di un effettofarmacologico sinergico: i rari studi dedicati alla valutazione diquesta strategia di compromesso pragmatico non consentono diconcludere definitivamente sui suoi eventuali vantaggi [25].
Raccomandazioni per il trattamento iniziale
Nel contesto attuale, in cui l’evoluzione del rapporto medico-paziente porta a prendere in considerazione in modo piùformale le nozioni di rischio terapeutico e di principio diprecauzione, la pubblicazione recente delle raccomandazionistabilite dalle istanze professionali può costituire un utileriferimento [25]: «l’età di esordio e l’importanza del disturbofunzionale sono i due fattori che guidano le scelte terapeutiche:• in assenza di rallentamento motorio (o di disagio legato al
tremore o all’ipertono), i trattamenti farmacologici non sonoindispensabili; i motivi dell’astensione terapeutica devonoessere forniti al paziente;
• quando il disturbo è minimo, possono essere utilizzatiagonisti dopaminergici, selegilina e anticolinergici, in fun-zione del sintomo che predomina e dell’età;
• quando esiste una ripercussione funzionale, l’età del pazientecondiziona il trattamento:C nel soggetto giovane è opportuno privilegiare gli agonisti
dopaminergici il più a lungo possibile; il ricorso alladopaterapia si giustifica in caso di intolleranza o di rispostaterapeutica insufficiente; il dosaggio di L-dopa deve restareil più basso possibile;
C nel soggetto anziano la L-dopa può essere utilizzata inprima scelta; la comparsa di un declino cognitivo deveportare a utilizzare le dosi minime efficaci».
Ruolo della riabilitazione funzionale [25]
All’esordio della malattia il disturbo funzionale rimane bassoe risponde, il più delle volte in modo evidente, al trattamentofarmacologico. Si può tuttavia proporre al paziente una gestionerieducativa, essenzialmente motoria, in modo pragmatico: essasi situa in un’azione di prevenzione volta a preservare lecapacità fisiche generali del paziente e ad attenuare eventualisintomi dolorosi.
Gestione nella fase evoluta
Trattamento farmacologico delle complicanzemotorie [25, 51, 102]
La gestione farmacologica delle fluttuazioni e delle discinesieindotte dal trattamento antiparkinsoniano è illustrata in modosinottico nella Tabella 11 [102].
Fluttuazioni prevedibili. Il principio di base è quello direndere il trattamento antiparkinsoniano meno «pulsatile».L’atteggiamento più semplice consiste nell’aumentare il numerodi assunzioni di L-dopa, se possibile riducendo la quantità aogni assunzione per non aumentare il dosaggio totale giorna-liero, il che corrisponde a un «frazionamento» delle somminis-trazioni. Può anche rivelarsi utile introdurre la L-dopa aliberazione prolungata (LP) realizzando, tenuto conto della suabiodisponibilità inferiore, un aumento della posologia quoti-diana dell’ordine del 30%: tuttavia, il mantenimento diun’assunzione di L-dopa standard o anche l’introduzione dellaforma dispersibile di L-dopa possono essere necessari come«dose-starter» in caso di acinesia del primo mattino persistente.L’aggiunta di agonisti dopaminergici o l’aumento della loroposologia se erano già prescritti possono costituire un’alterna-tiva; allo stesso modo, gli inibitori della COMT possonorappresentare il trattamento di elezione per l’acinesia di finedose in virtù del miglioramento che essi forniscono sullabiodisponibilità della L-dopa.
Fluttuazioni imprevedibili. I diversi approcci possibilimirano tutti a migliorare la biodisponibilità del trattamentoantiparkinsoniano: ottimizzare la cinetica di assorbimentointestinale della L-dopa (farmaci procinetici che accelerano losvuotamento gastrico, somministrazioni di L-dopa prima deipasti), ridurre la quantità degli aminoacidi alimentari (sposta-mento delle proteine del pasto di mezzogiorno verso quellodella sera), «cortocircuitare» la barriera intestinale (con l’utilizzodell’apomorfina sottocutanea), stimolare più fortemente irecettori dopaminergici (aumento della posologia di L-dopa,agonisti dopaminergici). In pratica, le misure che si basanosull’orario delle somministrazioni rispetto ai pasti, sullosvuotamento gastrico e sulla dieta proteica sono le prime aessere attuate; quando le fluttuazioni persistono, è giustificatointrodurre l’apomorfina in iniezioni sottocutanee unitarie(posologia tra i 3 e gli 8 mg, stabilita dopo un test farmacolo-gico valutando il rapporto efficacia/tolleranza) sotto coperturainiziale di domperidone a 60 mg/die. Nei casi più difficili, puòrivelarsi necessario sostituire del tutto o in parte la L-dopa conl’apomorfina in somministrazione continua sottocutanea conl’aiuto di una pompa portatile.
Discinesie del picco di dose. Quando divengono fastidiosea causa della loro intensità o di una componente dolorosa, laprima misura terapeutica è il «frazionamento» delle assunzioni,eventualmente associato a una riduzione della posologiagiornaliera di L-dopa, il che può essere facilitato, in caso dinecessità, dall’aggiunta misurata di agonisti dopaminergici.L’amantadina, con l’evidenziazione del suo ruolo antagonistaglutamatergico, si è rivelata spesso efficace per attenuare lediscinesie.
Discinesie di inizio dose e di fine dose. La loro gestioneresta spesso difficile e deludente. Una prima misura logicaconsiste nell’aumentare la posologia giornaliera di L-dopaaumentando il numero di assunzioni. Anche l’apomorfina el’amantadina possono essere utilizzate in questa indicazione.
Distonie del periodo off. La tossina botulinica è indicatanelle forme localizzate stabili. In altri casi, è giustificatoaumentare la posologia di L-dopa, tentare l’aggiunta di unaforma LP o, ancora, usare l’apomorfina.
Mioclonie. Raramente richiedono un trattamento. L’uso, inpratica, di amitriptilina a dosi moderate (25 mg) o di clonaze-pam la sera è una misura abituale.
Trattamento farmacologico dei disturbi non motori [25]
L’ipotensione ortostatica, spesso iatrogena nel morbo diParkinson, richiede, quando è poco tollerata, l’aggiunta di
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
24 Neurologia

fluoroidrocortisone, di simpaticomimetici e, in particolare, dimidodrina. Le misure complementari (calze elastiche e controllodegli apporti sodici) sono spesso utili.
I disturbi urinari tipo urgenze minzionali e pollachiurianotturna sono spesso attenuati con la semplice gestione deiperiodi off oppure corrispondono a una patologia associata. Inalcuni casi, l’uso prudente di ossibutinina e di amitriptilinapermette di attenuare l’iperattività del detrusore.
I disturbi sessuali tipo impotenza nell’uomo possono giustifi-care la prescrizione di sildenafil, dopo il controllo della funzio-nalità cardiaca e dopo aver verificato l’assenza di un’altraeziologia (effetti iatrogeni, stato depressivo, patologia endo-crina). In caso di ipersessualità fastidiosa può rivelarsi necessarioridurre il trattamento dopaminergico.
I disturbi digestivi, in particolare la stipsi, giustificano sempredelle misure igieniche (apporto di fibre, bevande sufficienti eattività fisica) eventualmente completate con blandi lassativi e,all’occorrenza, la sospensione dei farmaci anticolinergici. Lascialorrea fastidiosa per insufficienza di deglutizione spontaneapuò beneficiare del trattamento con l’iniezione di tossinabotulinica nelle ghiandole parotidi e sottomascellari sotto uncontrollo ecografico.
I disturbi ansioso-depressivi, quando non sono migliorati conil trattamento dopaminergico, possono essere trattati sintoma-ticamente con antidepressivi (triciclici o inibitori del reuptakedella serotonina) e con una gestione psicoterapeutica.
I disturbi cognitivi giustificano il fatto di evitare gli anticoli-nergici. Quando questi disturbi evolvono verso una demenza,può essere legittimo valutare prudentemente l’effetto di unanticolinesterasico ad azione centrale: in questa indicazione, larivastigmina dispone di un’autorizzazione all’immissione alcommercio [155].
La «psicosi» dopaminergica, la cui semeiologia può compor-tare un continuum tra alcune allucinazioni minime bencontrollate e degli accessi confusivo-deliranti acuti con gravidisturbi del comportamento, richiede sempre una valutazionemedica completa (alla ricerca di una patologia associata sottos-tante) e una riduzione dei trattamenti antiparkinsonianicominciando con gli anticolinergici, la selegilina, l’amantadina
e gli agonisti dopaminergici prima di ridurre, se necessario, laL-dopa stessa. Molto spesso, il peggioramento motorio richiededi riprendere la dopaterapia a un livello sufficiente, la cuitolleranza è resa possibile grazie alla prescrizione associata diclozapina alla dose minima efficace e sotto un controlloematologico [156].
I disturbi del sonno rispondono a meccanismi multipli,ognuno dei quali può richiedere una gestione specifica [25, 109],come illustrato nella Tabella 12 [25, 109].
Tabella 12.Tipologia e gestione specifica dei disturbi del sonno nel morbo diParkinson (da Pal et al. [109], Pollak e Tranchant in [25]).
Tipo di disturbi del sonno Trattamento
Frammentazione del sonno Aggiungere L-dopa LP o ICOMTla sera
Insonnia di addormentamento
- Ansia - depressione
- Sindrome delle gambe senzariposo
- Insonnia vera
Evitare selegilina la sera
- Triciclici la sera
- Agonisti DA a basse dosi la sera
- Tentativo con ciclopirroloni
Allucinazioni notturne Ridurre gli antiparkinsonianial bisogno, aggiungere clozapina(12,5-50 mg la sera)
Disturbi del comportamentodel sonno paradosso
Oggettivare con polisonnografiae aggiungere clonazepam a bassedosi la sera
Sonnolenza diurna Eliminare i disturbi del sonnonotturno
«Panico» del sonno Ridurre la dose di agonista DAo cambiare la molecola
Apnee del sonno Trattamento specialistico (ORL,CPAP) dopo polisonnografia
ICOMT: inibitore della catecol-O-metiltransferasi; DA: dopamina; ORL:otorinolaringoiatrico; CPAP: Continuous positive airways pressure; LP: liberazioneprolungata.
Tabella 11.Sinossi delle complicanze motorie legate al trattamento antiparkinsoniano e della loro gestione medica (da Viallet [102]).
Tipo Caratteristiche cliniche Fisiopatologia Gestione farmacologica
F prevedibili
A di fine dose
A del primo mattino
A notturna
F non motorie
Correlazione con le assunzionidi farmaci
Perdita dell’effetto tamponedelle terminazioni DA striatali
Innalzamento della soglia minima di effi-cacia della L-dopa
Trattamento meno «pulsatile»
- frazionamento delle assunzioni di L-dopa
- forme galeniche: LP, dispersibili
- agonisti dopaminergici
- ICOMT
F imprevedibili
A circadiana
A paradossa
A resistente
F improvvise
F non motorie
Non correlate alle assunzionidi farmaci
Ritardo nello svuotamento gastrico
Competizione con le proteine dei pasti
Riduzione della soglia di affinitàdei recettori DA
«Corto-circuitare» la barriera intestinale
Ridurre le proteine del pasto
Stimolare più fortemente i recettori DA
- apomorfina (penna, pompa)
- dietetica: modificazione della ripartizionedelle proteine
- aumento della posologia di L-dopa
- agonisti dopaminergici
- orario delle somministrazioni rispetto ai pasti
D di picco di dose Coreiformi, stabili, ben tollerate Stimolazione DA sopraliminare(attivazione preferenziale D1?)
Sensibilizzazione (priming) dei recettori(DA, glutammato)
Riduzione della posologia di L-dopa o semplicefrazionamento delle somministrazioni
Agonisti dopaminergici
Amantadina
D di inizio e fine dose Violente o distoniche, dolorose Stimolo DA subliminare(attivazione preferenziale D2?)
Aumento della posologia della L-dopacon frazionamento della somministrazioni
Apomorfina (penna, pompa)
Amantadina
Distonia periodo off Primo mattino, arto inferiore,dolorosa
Livello DA troppo basso Se localizzata, stabile: tossina botulinica
Altri casi: L-dopa LP, apomorfina
Mioclonie Decubito, arti inferiori, notturne Non conosciuta Metisergide, amitriptilina, vari
F: fluttuazioni; A: acinesia; D: discinesie; ICOMT: inibitore della catecol-O-metiltransferasi; LP: liberazione prolungata; DA: dopaminergico; D1, D2: recettori dopaminergici.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
25Neurologia

I dolori, al di fuori delle distonie dolorose dei periodi off,richiedono spesso una valutazione precisa alla ricerca di unapatologia associata, in particolare reumatica, che può beneficiaredi un trattamento locale antinfiammatorio.
Ruolo della riabilitazione funzionale [25, 51, 143, 144]
La riabilitazione motoria deve adattarsi allo stato motorio delpaziente con un lavoro attivo necessariamente limitato aiperiodi on, mentre i periodi off permettono di affrontare gli aiutialla mobilizzazione, le manovre di rotazione nel letto, ilcontrollo delle cadute e la prevenzione delle deformità articolaricon la gestione dei dolori muscolari. In pratica, dopo un tempodi ricondizionamento fisico generale la seduta di riabilitazioneattiva esercita la motricità degli arti e quindi il controllodell’equilibrio e della deambulazione, insistendo specificamentesulle rotazioni assiali di cui si devono mantenere le ampiezze.
Anche la riabilitazione della parola e della deglutizione deveadattarsi a questi vincoli, ma resta ancora ampiamente sottou-tilizzata se si considerano i bisogni dei pazienti.
In linea generale, la rieducazione funzionale rappresenta, nelmorbo di Parkinson, un elemento importante del trattamento,il cui costo economico si rivela quasi equivalente a quello deifarmaci [25]. A causa dell’approccio pragmatico che ha prevalsonella realizzazione di queste tecniche, le pratiche di riabilita-zione restano imperfettamente valutate nella loro efficacia:tuttavia, recenti puntualizzazioni con meta-analisi di studiprecedenti permettono di confermare una loro efficacia sullafunzione fisica e sulla qualità della vita, ma il livello di evidenzarimane insufficiente per l’efficacia sulla prevenzione dellecadute [143, 144]. Tenendo conto di questa situazione di fatto,appare auspicabile prevedere per l’avvenire una metodologia divalutazione di ogni campo specifico della motricità (deambula-zione, voce, deglutizione), con obiettivi precisi e quantificabiliin un quadro multidisciplinare [25].
Ruolo dei trattamenti chirurgici [25]
La neurostimolazione ad alta frequenza del NST, allo statoattuale delle conoscenze, appare la tecnica che presenta ilrapporto benefici/rischi più favorevole. A condizione di ungrande rigore nella realizzazione di reperi guidata dalla diagnos-tica per immagini e dalle registrazioni elettrofisiologiche, il NSTsembra, in effetti, essere il bersaglio che dà i migliori risultaticon un impianto bilaterale fin dall’inizio. La tecnica, messa apunto da Benabid nel 1993, la cui considerevole efficaciasintomatica è stata confermata [136, 151], si è diffusa progressiva-mente in Europa molto prima di raggiungere gli Stati Uniti,dove la pratica si sviluppa ampiamente dall’inizio degli anni2000. Il principale fattore che limita la sua utilizzazione resta ilcosto elevato del materiale impiantabile di stimolazione e dellagestione ospedaliera nel periodo perioperatorio [25]: tuttavia, aldi là dei costi economici diretti, occorre sottolineare anchel’importanza delle risorse mediche umane nella gestione e nelfollow-up, necessariamente pluridisciplinare, dei molti pazientiparkinsoniani candidati a questo trattamento.
Se si prende in considerazione l’insieme di questi fattori, ègiustificato proporre alcune raccomandazioni concernentil’indicazione di un tale trattamento neurochirurgico [25]: «Devetrattarsi di un MPi che evolve da più di 5 anni, con assenza dideficit cognitivi e/o di disturbi psichiatrici. La persistenza di unabuona sensibilità alla L-dopa è un criterio essenziale nellaselezione, con l’eccezione del tremore. L’età in sé non è uncriterio discriminante, contrariamente alla presenza di patologieassociate la cui frequenza e le cui ripercussioni aumentano conl’età: si fa riferimento, pertanto, a ogni stato patologico checontroindichi un atto chirurgico a scopo funzionale. La qualitàdel contesto sociofamiliare è un fattore di riuscita, poiché lagestione del paziente, qualunque sia il beneficio dell’intervento,rimarrà pesante. Infine, la decisione di proporre l’operazione alpaziente spetta all’equipe medico-chirurgica specializzata cheegli ha scelto, la quale gli deve garantire un’informazionecompleta».
Le altre tecniche chirurgiche, che restano attualmenteutilizzate, sono essenzialmente la stimolazione talamica a livello
del VIM e la stimolazione del GPi, mentre la pallidotomia vedele sue indicazioni regredire a causa della sua irreversibilità.
La stimolazione del VIM può essere unilaterale o bilaterale (aseconda dell’espressione del tremore): essa conserva indicazioninei tremori atipici e prevalenti del MPi quando questi sonoresistenti alla terapia medica e invalidanti, il più delle volte acausa della loro intensità, con una componente a riposoassociata a componenti posturali e di azione.
La stimolazione del GPi può essere anche monolaterale obilaterale e ritrova un certo interesse in Europa e negli StatiUniti.
■ Riferimenti bibliografici[1] Viallet F. Akinésie et parkinsonisme [mémoire HDR Neurosciences].
Université Aix-Marseille II; 1993 (218p).[2] Parkinson J. An essay on the shaking palsy. London: Whittingham and
Rowland; 1817.[3] Manyam BV. Paralysis agitans and LevoDOPA in ″Ayurveda″: ancient
Indian medical treatise. Mov Disord 1990;5:47-8.[4] Fahn S. The history of parkinsonism. Mov Disord 1989;4(suppl1):
S2-S10.[5] Pearce JMS. Aspects of the history of Parkinson’s disease. J Neurol
Neurosurg Psychiatry 1989(suppl special):6-10.[6] Gray F. Neuropathologie des syndromes parkinsoniens. Rev Neurol
1988;144:229-48.[7] Jellinger K. The pathology of Parkinsonism. In: Marsden CD, Fahn S,
editors. Movement disorders 2. London: Butterworth; 1987. p. 124-65.[8] Dickson DW, Davies P, Mayeux R. Lewy body disease:
neuropathological and biochemical studies of six patients. ActaNeuropathol (Berl) 1987;75:8-15.
[9] Schults CW. Lewy bodies. Proc Natl Acad Sci USA 2006;103:1661-8.[10] Dickson DW. Neurodegeneration: The molecular pathology of
dementia and movement disorders. Basel: ISN Neuropath Press; 2003(414p).
[11] Braak H, Del Tredici K, Rüb U, De Vos RA, Jansen Steur EN, Braak E.Staging of brain pathology related to sporadic Parkinson’s disease.Neurobiol Aging 2003;24:197-211.
[12] Burke RE, Dauer WT, Vonsattel JP. A critical evolution of the Braakstaging scheme for Parkinson’s disease. Ann Neurol 2008;64:485-91.
[13] Braak H, Bohl JR, Müller CM, Rüb U, De Vos RA, Del Tredici K.Stanley Fahn Lecture: The staging procedure for the inclusion bodypathology associated with sporadic Parkinson’s disease reconsidered.Mov Disord 2006;21:2042-51.
[14] Braak H, Del Tredici K. Invited article: nervous system pathology insporadic Parkinson’s disease. Neurology 2008;70:1916-25.
[15] Agid Y, Cervera P, Hirsch E, Javoy-Agid F, Lehricy S, Raisman R, et al.Biochemistry of Parkinson’s disease 28 years later: a critical review.Mov Disord 1989;4(suppl1):S126-S144.
[16] Fahn S. The history of dopamine and levodopa in the treatment ofParkinson’s disease. Mov Disord 2008;23(suppl3):S497-S508.
[17] Hornyckiewicz O. Dopamine miracle: from brain homogenate todopamine replacement. Mov Disord 2002;17:501-8.
[18] Vingerhoets FJG, Snow BJ, Lee CS, Schulzer M, Mak E, Calne DB.Longitudinal fluorodopa positron emission tomographic studies of theevolution of idiopathic parkinsonism. Ann Neurol 1994;36:759-64.
[19] Cotzias GC, Papavasiliou PS, Gellene R. Modification ofparkinsonism-chronic treatment with L-DOPA. N Engl J Med 1969;280:337-45.
[20] Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature1979;277:93-6.
[21] Stoof JC, Kebabian JW. Two dopamine receptors: biochemistry,physiology, and pharmacology. Life Sci 1984;35:2281-96.
[22] Robertson HA. Dopamine receptor interactions: some implications forthe treatment of Parkinson’s disease. Trends Neurosci 1992;15:201-6.
[23] Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ,et al. D1 and D2 dopamine receptor-regulated gene expression ofstriatonigral and striatopallidal neurons. Science 1990;250:1429-32.
[24] Chase TN, Oh JD, Blanchet PJ. Neostriatal mechanisms in Parkinson’sdisease. Neurology 1998;51:S30-S35.
[25] Conférence de consensus sur la maladie de Parkinson : textes desexperts, textes du groupe bibliographique et recommandations du jury(texte court et texte long). Rev Neurol 2000;156(suppl2b):1-294.
[26] Klein C, Schlossmacher MG. Parkinson’s disease, 10 years after itsgenetic revolution: multiple clues to a complex disorder. Neurology2007;69:2093-104.
.
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
26 Neurologia

[27] Tolosa E, Borght TV, Moreno E, DaTSCAN CUPS Study Group.Accuracy of DaTSCAN (123I- Ioflupane) SPECT in diagnosis ofpatients with clinically uncertain parkinsonism: 2-year follow-up of anopen-label study. Mov Disord 2007;22:2346-51.
[28] Zhang ZX, Roman GC. Worldwide occurrence of Parkinson’s disease:an updated review. Neuropidemiology 1993;12:195-208.
[29] De Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF,Baldereschi M, et al. Prevalence of Parkinson’s disease in Europe: Acollaborative study of population-based cohorts. Neurologic Diseasesin the Elderly Research Group. Neurology 2000;54(11suppl5):S21-S23.
[30] Tison F, Dartigues JF, Dubes L, Zuber M, Alperovitch A, Henry P.Prevalence of Parkinson’s disease in the elderly: a population study inGironde, France. Acta Neurol Scand 1994;90:110-5.
[31] Langston JW, Koller WC. Preclinical detection of Parkinson’s disease.The next frontier: presymptomatic detection. Introduction. Neurology1991;41(suppl2):5-7 (1991).
[32] Poewe WH, Wenning GK. The natural history of Parkinson’s disease.Ann Neurol 1998;44(3suppl1):S1-S9.
[33] Hoehn MM,Yahr MD. Parkinsonism: onset, progression and mortality.Neurology 1967;17:427-42.
[34] Fearnley JM, Lees A. Aging and Parkinson’s disease: substantia nigraregional selectivity. Brain 1991;114:2283-301.
[35] Hirsch EC, GraybielAM,AgidY. Melanized dopaminergic neurons aredifferentially susceptible to degeneration in Parkinson’s disease.Nature 1988;334:345-8.
[36] Kish SJ, Shannak K, Hornyckiewicz O. Uneven pattern of dopamineloss in the striatum of patients with idiopathic Parkinson’s disease. NEngl J Med 1988;318:876-80.
[37] Scherman D, Desnos C, Darchen F, Javoy-Agid F, Agid Y. Striataldopamine deficiency in Parkinson’s disease: role of aging. Ann Neurol1989;26:551-7.
[38] Viallet F, Witjas T. Neuroplasticité et maladie de Parkinson. Rev Neurol2002;158(suppl1):S42-S48.
[39] Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD,et al. Frequency of bowel movements and the future risk of Parkinson’sdisease. Neurology 2001;57:456-62.
[40] Doty RL, Deems DA, Stellar S. Olfactory dysfunction in parkinsonism:a general deficit unrelated to neurologic signs, disease stage, or diseaseduration. Neurology 1988;38:1237-44.
[41] Iranzo A, Molinuevo JL, Santamaria J, Serradell M, Marti MJ,Valldeoriola F, et al. Rapid-eye-movement sleep behaviour disorder asan early marker for a neurodegenerative disorder: a descriptive study.Lancet Neurol 2006;5:572-7.
[42] Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of aparkinsonian disorder in 38% of 29 older men initially diagnosed withidiopathic rapid eye movement sleep behaviour disorder. Neurology1996;46:388-93.
[43] Shiba M, Bower JH, Maraganore DM, McDonnell SK, Petterson BJ,Ahlskog JE, et al.Anxiety disorders and depressive disorders precedingParkinson’s disease: a case-control study. Mov Disord 2000;15:669-77.
[44] Stiasny-Kolster K, Dörr Y, Möller C, Höffken H, Behr TM, Oertel WH,et al. Combination of idiopathic REM sleep behavior disorder andolfactory dysfunction as possible indicator for alpha-synucleinopathydemonstrated by dopamine transporter FP-CIT SPECT. Brain 2005;128:126-37.
[45] Sawle GV, Wroe SJ, Lees AJ, Brooks DJ, Frackowiak RS. The identi-fication of presymptomatic parkinsonism: clinical and 18F. dopapositron emission tomography studies in an Irish kindred. Ann Neurol1997;32:609-17.
[46] Morrish PK, Rashki JS, Sawle GV, Brooks DJ. Measuring the rate ofprogression in Parkinson’s disease with 18F.dopa PET. J NeurolNeurosurg Psychiatry 1998;64:314-9.
[47] Gonera EG, Vant’hof M, Berger HJ, Van Weel C, Horstink MW.Symptoms and duration of the prodromal phase in Parkinson’s disease.Mov Disord 1997;12:871-6.
[48] Lerner A, Bagic A. Olfactory pathogenesis of idiopathic Parkinson’sdisease revisited. Mov Disord 2008;23:1076-84.
[49] Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alphasynucleinimmunorective inclusions in Meissner’s and Auerbach’s plexuses incases staged for Parkinsin’s disease-related brain pathology. NeurosciLett 2006;396:67-72.
[50] Damier P, Hirsch E, Agid Y, Graybiel A. The substantia nigra of thehuman brain I. Nigrosomes and the nigral matrix, a compartmentalorganization based on calbindin D28K immunohistochemistry. II.Patterns of loss of dopamine-containing neurons in Parkinson’sdisease. Brain 1999;122:1421-48.
[51] Rascol A. La maladie de Parkinson. Paris: Acanthe-Masson; 1998(246p).
[52] Kubis N, Faucheux B, Ransmayr G, Damier P, Duyckaerts C, Henin D,et al. Preservation of midbrain catecholaminergic neurons in very oldhuman subjects. Brain 2000;123:366-73.
[53] Langston JW, Ballard PA. Parkinson’s disease in a chemist workingwith 1-methyl-4-phényl-1,2,3,6,-tetrahydropyridine (MPTP). N EnglJ Med 1983;309:310.
[54] Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D.Evidence of active nerve cell degeneration in the substantia nigra ofhumans, years after 1-methyl-4-phenyl-1,2,3,6-tetrahydroxypyridineexposure. Ann Neurol 1999;46:598-605.
[55] Betarbet R, Sherer TB, McKenzie G, Garcia-Osuna M, PanovAV, et al.Chronic systemic pesticide exposure reproduces features ofParkinson’s disease. Nat Neurosci 2000;3:1301-6.
[56] Tanner CM, Ottman R, Goldman S, Ellenberg J, Chan P, Mayeux R,et al. Parkinson’s disease in twins: an etiologic study. JAMA 1999;281:341-6.
[57] Golbe LI, Di Iorio G, Sanges G, LazzariniAM. Clinical genetic analysisof Parkinson’s disease in the Contursi kindred. Ann Neurol 1996;40:767-75.
[58] Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, DehejiaA, DutraA,et al. Mutation in the alpha synuclein gene identified in families withParkinson’s disease. Science 1997;276:2045-7.
[59] Zarranz JJ,Alegre J, Gomez-Esteban JC, Lezcano E, Ros R,Ampuero I,et al. The new mutation, E46K, of alpha-synuclein causes Parkinsonand Lewy body dementia. Ann Neurol 2004;55:164-73.
[60] Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, et al.Causal relation between alpha-synuclein gene duplication and familialParkinson’s disease. Lancet 2004;364:1169-71.
[61] SingletonAB, Farrer M, Johnson J, SingletonA, Hague S, Kachergus J,et al. Alpha-synuclein locus triplication causes Parkinson’s disease.Science 2003;302:841.
[62] Leroy E, Boyer R, Auburger G. The ubiquitin pathway in Parkinson’sdisease (letter). Nature 1998;395:451-2.
[63] Gasser T, Müller-Myhsok B, Wszolek ZK, Oehlmann R, Calne B,Bonifati V, et al. A susceptibility locus for Parkinson’s disease maps tochromosome 2p13. Nat Genet 1998;18:262-5.
[64] Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A newlocus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 2002;51:296-301.
[65] Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al.Mutations in LRRK2 cause autosomal-dominant parkinsonism withpleiomorphic pathology. Neuron 2004;44:601-7.
[66] Lesage S, Janin S, Lohmann E, Leutenegger AL, Leclere L, Viallet F,et al. LRRK2 exon 41 mutations in sporadic Parkinson’s disease inEuropeans. Arch Neurol 2007;64:425-30.
[67] Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y,Minushima S, et al. Mutations in the Parkin gene cause autosomalrecessive juvenile Parkinsonism. Nature 1999;392:605-8.
[68] Lucking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T,et al. Association between early–onset Parkinson’s disease and muta-tions in the Parkin gene. N Engl J Med 2000;342:1560-7.
[69] Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K,Gispert S, et al. Hereditary early-onset Parkinson’s disease caused bymutations in PINK1. Science 2004;304:1158-60.
[70] Bonifati V, Rizzu P, Van Baren MJ, Schaap O, Breedveld GJ, Krieger E,et al. Mutations in the DJ-1 gene associated with autosomal recessiveearly-onset parkinsonism. Science 2003;299:256-9.
[71] Williams DR, HadeedA, al-DinAS,WreikatAL, LeesAJ. Kufor-Rakebdisease: autosomal recessive, levodopa-responsive parkinsonism withpyramidal degeneration supranuclear gaze palsy and dementia. MovDisord 2005;20:1264-71.
[72] Barbeau A, Roy M, Cloutier T, Plasser L, Paris S. Environmental andgenetic factors in the etiology of Parkinson’s disease. In: Yahr MD,Bergmann KJ, editors. Advances in Neurology (vol 45). New York:Raven Press; 1986. p. 299-306.
[73] Riedl AG, Watts PM, Jenner P, Marsden CD. P450 enzymes andParkinson’s disease: the story so far (review). Mov Disord 1998;13:212-20.
[74] Nicholl DJ, Bennett P, Hiller L, Bonifati V, Vanacore N, Fabbrini G,et al. A study of five candidate genes in Parkinson’s disease and relatedneurodegenerative disorders. Neurology 1999;53:1415-21.
[75] Jimenez-Jimenez FJ, Mateo D, Gimenez-Roldan S. Premorbidsmoking, alcohol consumption and coffee drinking habits inParkinson’s disease: a case-control study. Mov Disord 1992;7:339-44.
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
27Neurologia

[76] Ross GW, Abbott R, Petrovitch H, Morens DM, Grandinetti A,Tung KH, et al.Association of coffee and cafeine intake with the risk ofParkinson’s disease. JAMA 2000;283:2674-9.
[77] Ruberg M, France-Lanord V, Brugg B, Lambeng N, Michel PP,Anglade P, et al. La mort neuronale par apoptose dans la maladie deParkinson. Rev Neurol 1997;153:499-508.
[78] Hirsch EC, Hunot S, Damier P. Glial cell participation in thedegeneration of dopaminergic neurons in Parkinson’s disease. AdvNeurol 1999;80:9-18.
[79] Alexander GE, Crutcher MD. Functional architecture of basal gangliacircuits: neural substrates of parallel processing. Trends Neurosci 1990;13:266-71.
[80] Deniau JM. Le système des ganglions de la base : une revue critique desprincipes d’organisation fonctionnelle. Mouvements 2003;4:2-0.
[81] Mink JW. The basal ganglia: focused selection and inhibition ofcompeting motor programs. Prog Neurobiol 1996;50:381-425.
[82] Viallet F. Physiopathologie : organisation des ganglions de la base. In:Defebvre L, Verin M, editors. La maladie de Parkinson. Paris: Masson;2006. p. 31-43.
[83] Orieux G, François C, Feger J, Yelnik J, Vila M, Ruberg M, et al.Metabolic activity of excitatory parafascicular and pedunculopontineinputs to the subthalamic nucleus in a rat model of Parkinson’s disease.Neuroscience 2000;97:79-88.
[84] Nieoullon A. Dopamine and the regulation of cognition and attention.Prog Neurobiol 2002;67:53-83.
[85] Boraud T, Bezard E, Bioulac B, Gross CE. From single extracellularunit recording in experimental and human Parkinsonism to thedevelopment of a functional concept of the role played by the basalganglia in motor control. Prog Neurobiol 2002;66:265-83.
[86] Bar-Gad I, Bergman H. Stepping out of the box: information processingin the neural networks of the basal ganglia. Curr Opin Neurobiol 2001;11:689-95.
[87] Graybiel AM. Building action repertories: memory and learningfunctions of the basal ganglia. Curr Opin Neurobiol 1995;5:733-41.
[88] Marsden CD. The mysterious motor function of the basal ganglia: theRobert Wartenberg lecture. Neurology 1982;32:514-39.
[89] Viallet F, Trouche E. Troubles de l’initiation du mouvement chez leparkinsonien. JAMA 1991(suppl Ed. Fr):37-41.
[90] Montgomery Jr. EB, KollerWC, LaMantiaTJ, Newman MC, Swanson-Hyland E, Kaszni AW, et al. Early detection of probable idiopathicParkinson’s disease: Development of a diagnostic test battery. MovDisord 2000;15:467-73.
[91] Mayeux R, Stern Y, Rosen NJ, Leventhal J. Depression, intellectualimpairment and Parkinson’s disease. Neurology 1981;31:645-50.
[92] Reijnders JS, Ehrt U, Weber WE, Aarsland D, Leentjens AF. Asystematic review of prevalence studies of depression in Parkinson’sdisease. Mov Disord 2008;23:183-9.
[93] Gentil M, Pollak P, Perret J. La dysarthrie parkinsonienne. Rev Neurol1995;151:105-12.
[94] Viallet F, Meynadier Y, Lagrue B, Mignard P, Gantcheva R. Thereductions of tonal range and of average pitch during speech productionin ″off″ parkinsonians are restored by L-DOPA. Mov Disord 2000;15(suppl3):131.
[95] Agid Y. Une façon d’examiner un patient parkinsonien (mise au point).Rev Neurol 2000;156:17-22.
[96] Dubois B, Malapani C, Verin M, Rogelet P, Deweer B, Pillon B. Fonc-tions cognitives et noyaux gris centraux: le modèle de la maladie deParkinson. Rev Neurol 1994;150:763-70.
[97] Pillon B, Dubois B, Agid Y. Testing cognition may contribute to thediagnosis of movement disorders. Neurology 1996;46:329-34.
[98] Kirsch-Darrow L, Fernandez HH, Marsiske M, Okun M, Bowers D.Dissociating apathy and depression in Parkinson’s disease. Neurology2006;67:33-8.
[99] Dujardin K, Sockeel P, Devos D, Delliaux M, Krystkowiak P, DestéeA,et al. Characteristics of apathy. Mov Disord 2007;22:778-84.
[100] Pluck GC, Brown RG. Apathy in Parkinson’s disease. J NeurolNeurosurg Psychiatry 2002;73:636-42.
[101] Leentjens AF, Dujardin K, Marsh L, Martinez-Martin P, Richard IH,Starkstein S, et al. Apathy and anhedonia rating scales in Parkinson’sdisease: critique and recommendations. Mov Disord 2008;23:2004-14.
[102] Viallet F. Fluctuations et dyskinésies lors de la maladie de Parkinson.Neurologie 1998;6:21-4.
[103] Pearce RK, Banergi T, Jenner P, Marsden CD. De novo administrationof ropinirole and bromocriptine induces less dyskinesia than L-DOPAin the MPTP-treated marmoset. Mov Disord 1998;13:234-41.
[104] Damier P. Pathogénie des dyskinésies provoquées par le traitement dela maladie de Parkinson (revue générale). Mouvements 2000;2:6-17.
[105] Oh JD, Vaughan CL, Chase TN. Effect of dopamine denervation anddopamine agonist administration on serine phosphorylation of striatalNMDA receptor subunits. Brain Res 1999;821:433-42.
[106] Witjas T, Kaphan E,Azulay JP, Blin O, Ceccaldi M, Pouget J, et al. Nonmotor fluctuations in Parkinson’s disease: frequent and disabling.Neurology 2002;59:408-13.
[107] Giovannoni G, O’Sullivan JD, Turner K, Manson AJ, Lees AJ.Hedonistic homeostatic dysregulation in patients with Parkinson’sdisease on dopamine replacement therapies. J Neurol NeurosurgPsychiatry 2000;68:423-8.
[108] Senard JM, Rai S, Lapeyre-Mestre M, Brefel C, Rascol O, Rascol A,et al. Prevalence of orthostatic hypotension in Parkinson’s disease.J Neurol Neurosurg Psychiatry 1997;63:584-9.
[109] Pal PK, Calne DB, SamiiA, Fleming JAE.Areview of normal sleep andits disturbances in Parkinson’s disease. Parkinsonism Relat Disord1999;5:1-7.
[110] Ferreira J, Galitzky M, Montastruc JL, Rascol O. Sleep attacks andParkinson’s disease treatment. Lancet 2000;355:1333-4.
[111] Fénelon G, Mahieux F, Huon R, Ziegler M. Hallucinations inParkinson’s disease: prevalence, phenomenology and risk factors.Brain 2000;123:733-45.
[112] Mahieux F, Fénelon G, Flahault A, Manifacier MJ, Michelet D,Boller F. Neuropsychological factors predictive of dementia inParkinson’s disease. J Neurol Neurosurg Psychiatry 1998;64:178-83.
[113] Guillard A, Fénelon G, Mahieux F. Les altérations cognitives au coursde la maladie de Parkinson. Rev Neurol 1991;147:337-55.
[114] Bonnet AM, Loria Y, Saint-Hilaire MH, Lhermitte F, Agid Y. Doeslong-term aggravation of Parkinson’s disease result from nondopaminergic lesions? Neurology 1987;37:1539-42.
[115] Hely MA, Morris JG, Reid WG, Trafficante R. Sydney multicentrestudy of Parkinson’s disease: non L-dopa-responsive problemsdominate at 15 years. Mov Disord 2005;20:190-9.
[116] Clissold BG, McColl CD, Reardon KR, Shiff M, Kempster PA. Longi-tudinal study of the motor response to levodopa in Parkinson’s disease.Mov Disord 2006;21:2116-21.
[117] Viallet F, Gambarelli D, Alonzo B, Sedan R, Khalil R. Long-termfollow-up after stereotaxic basal ganglia surgery in Parkinsonism. MovDisord 1988;3:140-51.
[118] Hughes AJ, Daniel SE, Kifford L, Lees AJ. Accuracy of clinicaldiagnosis of idiopathic Parkinson’s disease: a clinico-pathologicalstudy of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181-4.
[119] Rajput AH, Rozdilsky B, Rajput A. Accuracy of clinical diagnosis inParkinsonism: a prospective study. Can J Neurol Sci 1991;18:275-8.
[120] Hughes AJ, Ben-Schlomo Y, Daniel SE, Lees AJ. What featuresimprove the accuracy of clinical diagnosis in Parkinson’s disease: aclinicopathologic study. Neurology 1992;42:1142-6.
[121] Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease.Arch Neurol 1999;56:33-9.
[122] SchragA, Good CD, Miszkiel K, Morris HR, Mathias CJ, LeesAJ, et al.Differentiation of atypical Parkinsonian syndromes with routine MRI.Neurology 2000;54:697-702.
[123] Benamer H, Patterson J. Grosset D and the 123I.-FP-CIT study group.Accurate differenciation of parkinsonism and essential tremor usingvisual assessment of 123I.-FP-CIT SPECT imaging. Mov Disord 2000;15:503-10.
[124] Catafau AM, Tolosa E, DaTSCAN CUPS study group. Impact ofdopamine transporter SPECT using 123I-Ioflupane on diagnosis andmanagement of patients with clinically uncertain Parkinsonian syndro-mes. Mov Disord 2004;19:1175-82.
[125] Jennings DL, Seibyl JP, Oakes D, Eberly S, Murphy J, Marek K. 123Ibeta-CIT and single-photon emission computed tomography imagingvs clinical evaluation in Parkinsonian syndrome: unmasking an earlydiagnosis. Arch Neurol 2004;61:1224-9.
[126] Thobois S, Rémy P. Controverse : Le SPECT a-t-il un intérêt dans lessyndromes parkinsoniens? Rev Neurol 2008;164(FMCHors série1):F17-F25.
[127] Vlaar AM, De Nijs T, Kessels AG, Vreeling FW, Winogrodzka A,Mess WH, et al. Diagnostic value of FP-CIT and IBZM SPECT scans in248 patients with parkinsonian syndromes. Eur Neurol 2008;59:258-66.
[128] Courbon F, Brefel-Courbon C, Thalamas C, Alibelli MJ, Berry I,Montastruc JL, et al. Cardiac MIBG scintigraphy is a sensitive tool fordetecting cardiac sympathetic denervation in Parkinson’s disease. MovDisord 2003;18:890-7.
[129] Simon N, Gantcheva R, Bruguerolle B,Viallet F.The effects of a normalprotein diet on levodopa plasma kinetics in advanced Parkinson’sdisease. Parkinsonism Relat Disord 2004;10:137-42.
I – 17-060-A-50 ¶Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici
28 Neurologia

[130] Ryvlin P, Broussolle E, Piollet H, Viallet F, Khalfallah Y, Chazot G.Magnetic resonance imaging evidence of decreased putaminal ironcontent in idiopathic Parkinson’s disease. Neurology 1995;52:583-8.
[131] Martin WRW, Wieler M, Gee M. Midbrain iron content in earlyParkinson’s disease: a potential biomarker of disease status. Neurology2008;70:1411-7.
[132] Michaeli S, Oz G, Sorce DJ, Garwood M, Ugurbil K, Majestic S, et al.Assessment of brain iron and neuronal integrity in patients withParkinson’s disease using novel MRI contrasts. Mov Disord 2007;22:334-40.
[133] Berg D. Ultrasound in the (premotor) diagnosis of Parkinson’s disease.Parkinsonism Relat Disord 2007;13(suppl3):S429-S433.
[134] Vaugoyeau M, Viallet F, Aurenty R, Assaiante C, Mesure S, Massion J.Axial rotation in Parkinson’s disease. J Neurol Neurosurg Psychiatry2006;77:815-21.
[135] Jankovic J. Parkinson-plus syndromes. Mov Disord 1989;4(suppl1):S95-S119.
[136] Limousin P, Krack P, Pollak P, Benazzouz A, Ardouin C, Hoffman D,et al. Electrical stimulation of the subthalamic nucleus in advancedParkinson’s disease. N Engl J Med 1998;339:1105-11.
[137] Goetz CG, Poewe W, Rascol O, Sampaio C. Evidence-based medicalreview update: pharmacological and surgical treatments of Parkinson’sdisease: 2001 to 2004. Mov Disord 2005;20:523-39.
[138] Parkinson Study Group. Effects of tocopherol and deprenyl on the pro-gression of disability in early Parkinson’s disease. N Engl J Med 1993;328:176-83.
[139] Parkinson Study Group. I. Impact of deprenyl and tocopherol treatmenton Parkinson’s disease in DATATOP subjects not requiring levodopa.II. Impact of deprenyl and tocopherol treatment on Parkinson’s diseasein DATATOP patients requiring levodopa. Ann Neurol 1996;39:26-45.
[140] Agid Y. Levodopa: is toxicity a myth? Neurology 1998;50:858-63.[141] Fahn S, Oakes D, Shoulson I. Levodopa and the progression of PD. N
Engl J Med 2004;351:2498-508.[142] Nutt JG. Continuous dopaminergic stimulation: Is it the answer to the
motor complications of levodopa? Mov Disord 2007;22:1-9.[143] Deane KH, Ellis-Hill C, Jones D, Whurr R, Ben-Shlomo Y,
Playford ED, et al. Systematic review of paramedical therapies forParkinson’s disease. Mov Disord 2002;17:984-91.
[144] Goodwin VA, Richards SH, Taylor RS, Taylor AH, Campbell JL. Theeffectiveness of exercise interventions for people with Parkinson’sdisease: a systematic review and meta-analysis. Mov Disord 2008;23:631-40.
[145] Ramig LO, Sapir S, Countryman S, Pawlas AA, O’Brien C, Hoehn M,et al. Intensive voice treatment (LSVT) for patients with Parkinson’sdisease: a 2-year follow-up. J Neurol Neurosurg Psychiatry 2001;71:493-8.
[146] Hallett M. Litvan I and the members of the task force on surgery forParkinson’s disease. Scientific position paper of the MovementDisorder Society: Evaluation of surgery for Parkinson’s disease. MovDisord 2000;15:436-8.
[147] Benabid AL, Pollak P, Louveau A, Henry S, De Rougemont J.Combined thalamotomy and stimulation in stereotactic surgery of theVIM thalamic nucleus for bilateral Parkinson’s disease. ApplNeurophysiol 1987;50:344-6.
[148] Schuurman PR, Bosch DA, Bossuyt PM. A comparison of continuousthalamic stimulation and thalamotomy for suppression of severetremor. N Engl J Med 2000;342:461-8.
[149] Kumar R, Lozano AM, Montgomery E, Lang AE. Pallidotomy anddeep brain stimulation of the pallidum and subthalamic nucleus inadvanced Parkinson’s disease. Mov Disord 1998;13:73-82.
[150] Pollak P, Benabid AL, Gross C, Gao DM, Laurent A, Benazzouz A,et al. Effets de la stimulation du noyau sous-thalamique dans la maladiede Parkinson. Rev Neurol 1993;149:175-6.
[151] Krack P, Batir A, Van Blercom N, Chabardes S, Fraix V, Ardouin C,et al. Five-year follow-up of bilateral stimulation of the subthalamicnucleus in advanced Parkinson’s disease. N Engl J Med 2003;349:1925-34.
[152] Fahn S, Members of the UPDRS development Committee. UnifiedParkinson’s disease rating scale. In: Fahn S, Marsden CD, Goldstein M,Calne DB, editors. Recent developments in Parkinson’s disease (VolII). New Jersey: MacMillan; 1987. p. 153-63.
[153] Defer G, Widner H, Marie RM, Remy P, Levivier M. Core assessmentprogram for surgical interventional therapies in Parkinson’s disease(CAPSIT-PD). Mov Disord 1999;14:572-84.
[154] Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE,for the 056 study group. . A five-year study of the incidence ofdyskinesia in patients with early Parkinson’s disease who were treatedwith ropinirole or levodopa. N Engl J Med 2000;342:1484-91.
[155] Emre M, Aarsland D, Albanese A, Byrne EJ, Deuschl G, De Deyn PP,et al. Rivastigmine for dementia associated with Parkinson’s disease. NEngl J Med 2004;351:2509-18.
[156] French Clozapine Parkinson Study group. Clozapine in drug-inducedpsychosis in Parkinson’s disease. Lancet 1999;353:2041-2.
F. Viallet, Praticien hospitalier, habilitation à diriger les recherches, chef de service ([email protected]).D. Gayraud, Praticien hospitalier.B. Bonnefoi, Praticien hospitalier, docteur ès sciences.L. Renie, Praticien hospitalier.Service de neurologie, Centre hospitalier du Pays d’Aix, avenue des Tamaris, 13616 Aix en Provence, France.
R. Aurenty, Ingénieur de recherche CNRS, docteur ès sciences.Association France Parkinson, 4, avenue du Colonel-Bonnet, 75016 Paris, France.
Ogni riferimento a questo articolo deve portare la menzione: Viallet F., Gayraud D., Bonnefoi B., Renie L., Aurenty R. Morbo di Parkinson idiopatico: aspetticlinici, diagnostici e terapeutici. EMC (Elsevier Masson SAS, Paris), Neurologia, 17-060-A-50, 2010.
Disponibile su www.em-consulte.com/it
Algoritmidecisionali
Iconografiasupplementare
Video-animazioni
Documentilegali
Informazioniper il paziente
Informazionisupplementari
Autovalutazione Casoclinico
Morbo di Parkinson idiopatico: aspetti clinici, diagnostici e terapeutici ¶ I – 17-060-A-50
29Neurologia