microfabricated bioreactor chips for immobilised enzyme assays
TRANSCRIPT

Analytica Chimica Acta 486 (2003) 149–157
Microfabricated bioreactor chips for immobilised enzyme assays
Shahanara Banu, Gillian M. Greenway, Tom McCreedy∗, Rachael ShaddickDepartment of Chemistry, University of Hull, Cottingham Road, Hull HU6 7RX, UK
Received 5 December 2002; received in revised form 10 April 2003; accepted 23 April 2003
Abstract
A microfabricated device is reported that has been designed to permit the in situ packing of a section of channel withenzyme immobilised onto controlled pore glass (CPG). It is fabricated from glass and polydimethylsiloxane and to preventdead volumes, has dedicated channels for packing the reactor. The device has the advantage of being simple in design,the flow through enzyme reactor channel being simply a widened section of the analyte channel. The system is suitablefor both hydrodynamic and electro-osmotic pumping, and is designed such that when the packing is exhausted it can berepacked. Controlled pore glass provides a reproducible none swelling, high porosity medium onto which the enzyme couldbe immobilised. The large particle size meant that it was vital to optimise the immobilisation procedure in order to achieveacceptable enzyme activity. The microfabricated device was developed with two enzymes of different molecular masses;alkaline phosphatase and xanthine oxidase. The pore size of the CPG was found to be very important for xanthine oxidase,where the 697 Å pore size (120–200 mesh) CPG was found to give the highest activity (18–20% activity retained afterimmobilisation). The microfabricated device was used for the assay ofp-nitrophenyl phosphate and hypoxanthine withspectrophotometric detection at 405 and 470 nm, respectively. The limits of detection were 5 and 8�M, respectively.© 2003 Elsevier Science B.V. All rights reserved.
Keywords: Bioreactor; Controlled pore glass; Microfabricated device; Xanthine oxidase; Alkaline phosphatase
1. Introduction
The miniaturisation of analytical systems, i.e. micrototal analytical systems (�TAS) has been growing inimportance for a number of years, and is now acceptedas an alternative to some conventional analytical ap-proaches. There are a number of advantages with suchsystems including a reduction in reagent consumption,the incorporation of separation and detection onto asingle planar device, and low cost production. En-zymes can be used in these devices to provide se-lectivity in detection, either as the free enzyme[1,2]
∗ Corresponding author. Tel.:+44-1482-466407;fax: +44-1482-466410.E-mail address: [email protected] (T. McCreedy).
or immobilised onto a surface or support[3–7]. Im-mobilised enzymes are frequently used in preferenceto the free enzyme because they can be reused formany assays but microfabricated devices pose particu-lar challenges when such supports are used, primarilyhow to incorporate the enzyme into the device, giventhe minute dimensions of the channels.
To employ immobilised enzymes in�TAS, it is nec-essary to develop a method to immobilise the enzymein a spatially resolved location within the device. Sur-face modification is a possible way to achieve this, butdifficulties can be encountered in precisely controllingwhere the surface reaction occurs and ensuring thereis sufficient surface area to give a measurable substrateconversion. Kasai et al. were able to precisely controlsurface immobilisation to produce separate spatially
0003-2670/03/$ – see front matter © 2003 Elsevier Science B.V. All rights reserved.doi:10.1016/S0003-2670(03)00503-8

150 S. Banu et al. / Analytica Chimica Acta 486 (2003) 149–157
resolved spots of enzyme[3], but it is more difficultwithin a closed system. Mao et al. have immobilisedalkaline phosphatase in a glass and PDMS microre-actor, but the spatial control was limited[4]. An al-ternative approach was to fabricate porous polymermonoliths within channels of a microchip and immo-bilise the enzyme onto the support[5]. Microporoussilica structures[6] have also been used as a support,but if hydrodynamic pumping is utilised the backpres-sure in forcing liquids through such structures canmake the system hard to operate. Packing the chan-nel with enzyme, pre-immobilised onto a support, is areproducible and attractive alternative, but since typi-cal channels are 200�m or less in diameter the prac-tical challenges can be significant. One approach hasinvolved the use of enzymes immobilised onto mag-netic beads. By incorporating a magnet into the chip,precise spatial resolution was possible[7]. Oleschuket al. packed octatdecylsilane coated silica beads in achip by trapping the beads between two weir struc-tures incorporated in the design of the chip[8]. Thisworked well for electrokinetic pumping, but would beless effective for hydrodynamic pumping as the flow-ing solution would just pass over the top of the beadbed.
Another challenge in using solid supports is theneed to get sufficient enzyme activity into the reactor.The enzyme can be immobilised in a number of ways,but perhaps covalent immobilisation offers the great-est advantages, by increasing the stability of the en-zyme and preventing it from leaching into solution. Inconventional bioreactors, there are numerous supportsavailable for enzyme immobilisation including nylon,porous alumina, Sepharose, and controlled pore glass(CPG)[9]. Polymer supports based on polysaccharideare very popular because they contain hydroxyl groupsfor binding and form hydrogen bonds thus giving ahydrophilic environment, but they tend to swell andhave particle sizes that are too large for micro reactorwork. CPG has the advantage of being chemical sta-ble, resistant to swelling, and allows relatively simpleimmobilisation procedures. CPG particles are howeverrelatively large (120–200 mesh) and the efficiency ofthe immobilisation procedure is often quite low[9].
In the work presented here, we demonstrate a novelimmobilised enzyme�TAS, created by packing aspecially fabricated channel section with CPG, ontowhich an enzyme was previously immobilised. The
use of dedicated channels for packing (keeping theanalytical flow network separate) overcomes the in-crease in dead volume while allowing easy movementof the CPG. By simply creating a widened sectionin the analyte channel a flowthrough immobilisedenzyme bed is created that is suitable for both hydro-dynamic and electro-osmotic pumping. The CPG canbe removed from the system when the immobilisedenzyme is exhausted and replaced. The system isdemonstrated using two enzymes of different molecu-lar mass; alkaline phosphatase and xanthine oxidase.The immobilisation procedure is optimised for theenzymes to ensure sufficient activity within the reac-tor and the reactions catalysed by the enzymes aremonitored by on-line spectrophotometry.
2. Experimental details
2.1. Reagents
Alkaline phosphatase, tetrazolium (XTT) sodiumsalt, hypoxanthine, sodium nitrite (99.5%), and CPGwere obtained from Sigma (Poole, UK). Ethanol(95%), sodium bicarbonate (AR grade) and sodiumdihydrogenorthophosphate dihydrate (AR grade)were obtained from Fisher Scientific, di-sodium hy-drogen orthophosphate (AR grade) from Fisons and(p-aminophenyl)trimethoxysilane (90%), from ABCR(UK). Hydrochloric acid (35.4%) and xanthine ox-idase (from Buttermilk) were obtained from PhilipHarris (UK) and Biochemika, respectively.
2.2. Device fabrication
A microfabricated device had to be developed thatallowed relatively large particles to be introduced intothe device without increasing the channel size in theanalytical flow network. The channels must be sealedprior to packing in order to prevent the packing mate-rial fouling the seal between layers. The actual chan-nel where the immobilised reagent was incorporatedhad to be deepened slightly to permit the CPG toform a regular packing without the presence of voids.The channel from reservoir B was also widened to in-troduce the CPG. The device was fabricated from acomposite of glass and polydimethylsiloxane (PDMS).The glass base plate was etched using a previously

S. Banu et al. / Analytica Chimica Acta 486 (2003) 149–157 151
Fig. 1. The chip design, showing the channel configuration. Thefour circles represent the location of the reservoirs. X is wherethe channel depth changes. The enzyme reactor where the CPGis packed is represented by the portion of thick black line alongthe channel A–D.
reported method[10] to produce a network of inter-connected channels as shown inFig. 1. A top plate ofPDMS was prepared with reservoirs (each 2 mm di-ameter) moulded into the required locations. The de-vice was closed by the application of pressure. Themicrochip reactor was packed with immobilised en-zyme after creating the sealed channel manifold.
The channel from reservoir C to D was 150�mwide and 50�m deep. The channel from reservoir Bto the enzyme reactor was 200�m wide, 80�m deep,and the enzyme reactor section (where the CPG waspacked) was 350�m wide and 80�m deep. The in situenzyme reactor was created by introducing etchant so-lution (1% HF/NH4F) into the closed channel fromreservoirs B up to point X (seeFig. 1). The etchant so-lution was left in the channel for approximately 10 minand then it was pumped out via reservoir C. As theetchant was slowly advanced along the channel frompoint X the tapered geometry was created with thetapering occurring in both the width and depth. Theprocess was optimised experimentally to achieve thebest packing conditions. A similar design of reactorhas been reported for use as a gas phase catalyst reac-tor [11].
On-chip absorbance detection was performed byaligning a light source with an optical fibre via reser-
voir D of the microchip. The reservoir provided anon-chip detection cell of volume 88�l. The opticalfibre detecting light passing through the detectioncell was 1 mm in diameter (2 mm with cladding). Ab-sorbance measurements were made using an OceanOptics S2000 fibre optic spectrometer and spectracollected using Ocean Optics OOIBase32 software.
2.3. Immobilisation of alkaline phosphatase
Alkaline phosphatase was immobilised covalentlyonto CPG according to the modified procedure ofPerininiger and Danielsson[12]: 0.5 g of CPG wascleaned with boiling 5% nitric acid for 30 min. Thesupport was washed on a Büchner funnel until thewashings attained a neutral pH and the beads were thendried in an oven at 95◦C. The dried glass beads weretreated with a 5% (3-aminopropyl)trimethoxysilanesolution prepared in anhydrous toluene for 4 h at 37◦Cin a water bath. The support was filtered and washedwith toluene and acetone before being dried at 115◦Cin an oven for >16 h. Once cooled the beads wereactivated for enzyme immobilisation at room temper-ature by treatment with a 2.5% aqueous gluteralde-hyde solution prepared in 0.1 M, pH 7.0 phosphatebuffer. On addition the beads were maintained at re-duced pressure for 30 min followed by 30 min at at-mospheric pressure. The activated support was filteredand washed well with distilled water and phosphatebuffer to remove any remaining unbound gluteralde-hyde before being reacted immediately with enzymesolution at<4◦C for 24 h. The enzyme solution wasprepared by adding 10 mg (170 units) alkaline phos-phatase to 3 ml of pH 7.0, 0.1 M phosphate buffer. Theactivity of the immobilised enzyme was monitoredoff-line using a modified version of a published assaymethod, in which the solution containing the beadswas stirred prior to measuring the absorbance[13].
2.4. Immobilisation of xanthine oxidase
Xanthine oxidase was covalently immobilised ontoCPG by modification and optimisation of the pre-viously published method[14,15] using a diazo-coupling method. 0.15 g of 697 Å (120–200 mesh)CPG was cleaned with 5% HNO3 by gently boilingfor 30 min. The CPG was then washed with plenty ofwater until the pH of the filtrate reached neutral and it

152 S. Banu et al. / Analytica Chimica Acta 486 (2003) 149–157
was dried in an oven at 95◦C. The cleaned CPG wasthen silanised with 5% (p-aminophenyl)trimethoxysilane in 95% ethanol with degassing for 15 min fol-lowed by another 30 min at normal pressure. The solidwas washed with a few millilitres of 95% ethanol andcured by heating to 110◦C for 25 min. After curingthe silanised CPG was treated with 2% NaNO2 in 2 Mhydrochloric acid at 0–4◦C for 30 min. The reactionresulted in the formation of a diazonium derivative ofsilica which was then added to a 2 ml aliquot of xan-thine oxidase (1.5 units ml−1) in 50 mM phosphatebuffer at pH 7.3 containing 10 mM EDTA for enzymecoupling. The CPG was then mechanically shakenfor 24 h. After 24 h the immobilised xanthine oxi-dase CPG was filtered and washed with the couplingbuffer. As mentioned previously, the activity of theimmobilised enzyme was monitored off-line usinga modified version of a published assay method, inwhich the solution containing the beads was stirredprior to measuring the absorbance[13].
2.5. Packing the chip
The enzyme immobilised on the CPG was dispersedwithin the buffer solution, this was pipetted into theentrance of the reactor cavity through reservoir B anda negative pressure was applied to the exit of the cavityvia reservoir C. The path B to C was used to avoid thepacking process compromising the analytical channel.When buffer solution was pumped through the chan-nel section B to C however, beads moved down thechannel to form a regular packing structure in the widesection of the channel. The beads travelled throughthe reactor until reaching the narrow analytical chan-nel (between C and D) where key stoning of the beadsled to their retention within the reactor. Any beadsthat did not get trapped were removed through reser-voir B or C without blocking the analytical channel.It was observed that when the device to be packedwith immobilised alkaline phosphatase was subjectedto sonication during the packing process, the qualityand density of the packing was improved. The soni-cation did not cause any detectable effect on the en-zyme activity, provided that the duration of the soni-cation was sufficiently short to avoid heating effects.The CPG could easily be removed by unplugging thereservoirs and reversing the process and this meantthat if the immobilised enzyme was denatured the
chip could be repacked with new reagent. The chipcould be repacked as many times as required with-out the system collapsing. If the channel was visiblypacked correctly, without gaps or air bubbles the re-sults obtained from the reactor were consistent. Some-times the packing failed, leaving visible channels andgaps and the beads then needed to be removed andrepacked.
2.6. On-chip assay of p-nitrophenyl phosphate
The immobilised alkaline phosphatase was usedto monitor the hydrolysis ofp-nitrophenyl phosphateto p-nitrophenol, which could be measured as an in-crease in absorbance at 405 nm. Calibration solutionsof p-nitrophenyl phosphate were prepared in pH 9.8,1.0 M diethanolamine buffer at concentrations of 0.5,0.4, 0.3, 0.2 and 0.1 mM. The buffer contained thealkaline phosphatase activators magnesium and zincat 1.25 × 10−5 and 1.25 × 10−4 M concentrations,respectively. The blank solution consisted of thepH 9.8, 1.0 M diethanolamine buffer. The stock andstandard solutions were protected from the light dueto the light sensitive nature of the substrate. Refer-ence measurements were made against pH 9.8, 1.0 Mdiethanolamine buffer. Calibration solutions werepumped at 50�l min−1 via a syringe pump in contin-uous flow mode from reservoir A through the reactorpacked with immobilised alkaline phosphatase. Ab-sorbance measurements were made in duplicate at405 nm.
2.7. On-chip assay of hypoxanthine
The assay method was adapted from the work ofUkeda et al.[16]. Calibration solutions of hypox-anthine were prepared in pH 9.4, 50 mM carbonatebuffer at concentrations of 12.25, 17.5, 25, 50, and100�M. The blank solution, which was used for ref-erence measurements, consisted of 50 mM carbonatebuffer and 50�M of reagent XTT salt (tetrazolium).The calibration solutions were pumped continuouslyat 20�l min−1 via a syringe pump in continuous flowmode from reservoir A through the reactor packedwith immobilised xanthine oxidase. Absorbance mea-surements were made in triplicate at 470 nm. Thereaction being monitored (seeFig. 2) was the reduc-tion of XTT (tetrazolium salt) by superoxide anion

S. Banu et al. / Analytica Chimica Acta 486 (2003) 149–157 153
Fig. 2. Reaction scheme for the xanthine oxidase reaction.
radicals generated from a xanthine oxidase catalysedreaction where hypoxanthine was the substrate[16].
3. Results and discussion
3.1. Selection of immobilisation technique
Several different techniques were screened forimmobilising the enzymes within the channels ofmicrofabricated devices, taking into account the previ-ous work described inSection 1 [3–7]. Both covalentbonding and physical entrapment were investigatedfor in situ immobilisation of the enzyme directlyonto the walls of the channels (using both glass andPDMS devices). Enzymes were successfully immo-bilised in the channel network, but when assays werecarried out, the sensitivity was very low. This waseither because there was insufficient enzyme activity,or perhaps more likely in these miniaturised systems,insufficient mixing of enzyme and substrate coupledwith a low surface area.
Using a pre-immobilised support offers greater re-producibility but then the challenge is in packing thechannels. Small silica beads were investigated (50�m)onto which the enzyme was covalently attached butthese particles were not porous and again insufficientsensitivity was achieved. Sol–gel particles with physi-cally entrapped enzyme was also investigated, this hadhigh enzyme loadings but, swelling effects, the ten-
dency to dissolve at higher pH and diffusion problemsprevented the use of sol–gel in our present system. Itwas finally concluded that controlled pore glass wasthe best support, having good mechanical stability anda high surface area, both for enzyme immobilisation,and for allowing the substrate to come into contactwith the enzyme. The problem with using controlledpore glass was two-fold. Firstly the enzyme activityper unit volume of CPG is low and secondly the par-ticle size is relatively high (typically 120–200 mesh,i.e. 80–120�m).
3.2. Enzyme immobilisation procedures
Procedures for the immobilisation of enzymes tocontrolled pore glass are well documented[17]. Oftenhowever general “recipes” are used and the enzymeactivity per g of support is very low. In this partic-ular application in which only a few mg (packed)of CPG are used, efficient immobilisation was vital.The selection of the type of covalent immobilisationutilised is highly dependent on the particular enzyme.Both the configuration and the size of the enzymeare important. Supports with a small pore size give alarge total surface area, but are only suitable for smallenzymes that can easily enter the pores of the CPG.In this work two different enzymes were selectedalkaline phosphatase with a relatively low molecularweight (140,000) and xanthine oxidase, which wasmuch larger (275,000). In carrying out the optimisa-tion of the immobilisation the initial activity of thefree enzyme was determined. The immobilisation pro-cedure was then carried out with a known amount ofenzyme. The activity of the enzyme left in the filtratecould then be used to determine how much enzymehad been immobilised. The activity of the immo-bilised enzyme was then also measured to identify anyloss of activity in enzyme due to the immobilisationprocess.
It was found that the alkaline phosphatase could berelatively easily immobilised on CPG with a mediumpore size of 239 Å using a straightforward gluteralde-hyde cross linking method based on the proceduredescribed by Perininiger and Danielsson[12]. Themethod was modified however with silianisation beingperformed using a 5% (v/v) silane solution for 4 h at37◦C followed by a 16 h period of curing at 115◦C.When assayed the alkaline phosphatase was found to
154 S. Banu et al. / Analytica Chimica Acta 486 (2003) 149–157
be successfully immobilised onto controlled pore glasswith an activity of 2 units g−1 support.
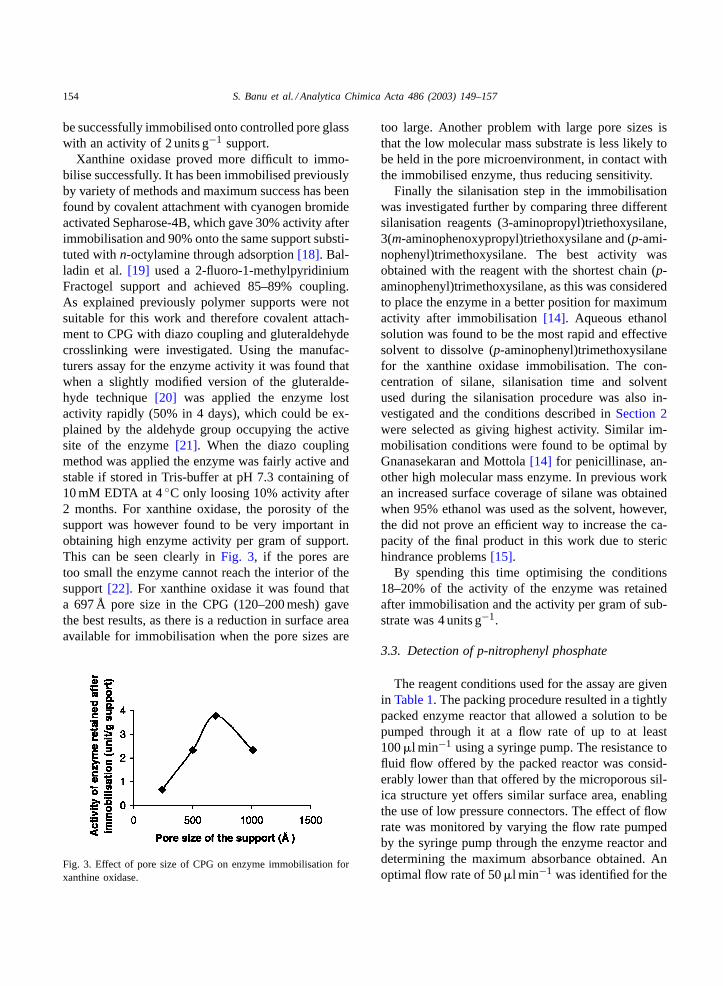
Xanthine oxidase proved more difficult to immo-bilise successfully. It has been immobilised previouslyby variety of methods and maximum success has beenfound by covalent attachment with cyanogen bromideactivated Sepharose-4B, which gave 30% activity afterimmobilisation and 90% onto the same support substi-tuted withn-octylamine through adsorption[18]. Bal-ladin et al.[19] used a 2-fluoro-1-methylpyridiniumFractogel support and achieved 85–89% coupling.As explained previously polymer supports were notsuitable for this work and therefore covalent attach-ment to CPG with diazo coupling and gluteraldehydecrosslinking were investigated. Using the manufac-turers assay for the enzyme activity it was found thatwhen a slightly modified version of the gluteralde-hyde technique[20] was applied the enzyme lostactivity rapidly (50% in 4 days), which could be ex-plained by the aldehyde group occupying the activesite of the enzyme[21]. When the diazo couplingmethod was applied the enzyme was fairly active andstable if stored in Tris-buffer at pH 7.3 containing of10 mM EDTA at 4◦C only loosing 10% activity after2 months. For xanthine oxidase, the porosity of thesupport was however found to be very important inobtaining high enzyme activity per gram of support.This can be seen clearly inFig. 3, if the pores aretoo small the enzyme cannot reach the interior of thesupport[22]. For xanthine oxidase it was found thata 697 Å pore size in the CPG (120–200 mesh) gavethe best results, as there is a reduction in surface areaavailable for immobilisation when the pore sizes are
Fig. 3. Effect of pore size of CPG on enzyme immobilisation forxanthine oxidase.
too large. Another problem with large pore sizes isthat the low molecular mass substrate is less likely tobe held in the pore microenvironment, in contact withthe immobilised enzyme, thus reducing sensitivity.
Finally the silanisation step in the immobilisationwas investigated further by comparing three differentsilanisation reagents (3-aminopropyl)triethoxysilane,3(m-aminophenoxypropyl)triethoxysilane and (p-ami-nophenyl)trimethoxysilane. The best activity wasobtained with the reagent with the shortest chain (p-aminophenyl)trimethoxysilane, as this was consideredto place the enzyme in a better position for maximumactivity after immobilisation[14]. Aqueous ethanolsolution was found to be the most rapid and effectivesolvent to dissolve (p-aminophenyl)trimethoxysilanefor the xanthine oxidase immobilisation. The con-centration of silane, silanisation time and solventused during the silanisation procedure was also in-vestigated and the conditions described inSection 2were selected as giving highest activity. Similar im-mobilisation conditions were found to be optimal byGnanasekaran and Mottola[14] for penicillinase, an-other high molecular mass enzyme. In previous workan increased surface coverage of silane was obtainedwhen 95% ethanol was used as the solvent, however,the did not prove an efficient way to increase the ca-pacity of the final product in this work due to sterichindrance problems[15].
By spending this time optimising the conditions18–20% of the activity of the enzyme was retainedafter immobilisation and the activity per gram of sub-strate was 4 units g−1.
3.3. Detection of p-nitrophenyl phosphate
The reagent conditions used for the assay are givenin Table 1. The packing procedure resulted in a tightlypacked enzyme reactor that allowed a solution to bepumped through it at a flow rate of up to at least100�l min−1 using a syringe pump. The resistance tofluid flow offered by the packed reactor was consid-erably lower than that offered by the microporous sil-ica structure yet offers similar surface area, enablingthe use of low pressure connectors. The effect of flowrate was monitored by varying the flow rate pumpedby the syringe pump through the enzyme reactor anddetermining the maximum absorbance obtained. Anoptimal flow rate of 50�l min−1 was identified for the
S. Banu et al. / Analytica Chimica Acta 486 (2003) 149–157 155
Table 1Reaction conditions for the determination ofp-nitrophenyl phosphate
pH Buffer Enzyme activators Reaction temperature Flow rate
9.4 1.0 M diethanolamine Mg2+ 1.25 × 10−5 M Room temperature 50�l min−1
Zn2+ 1.25 × 10−5 M
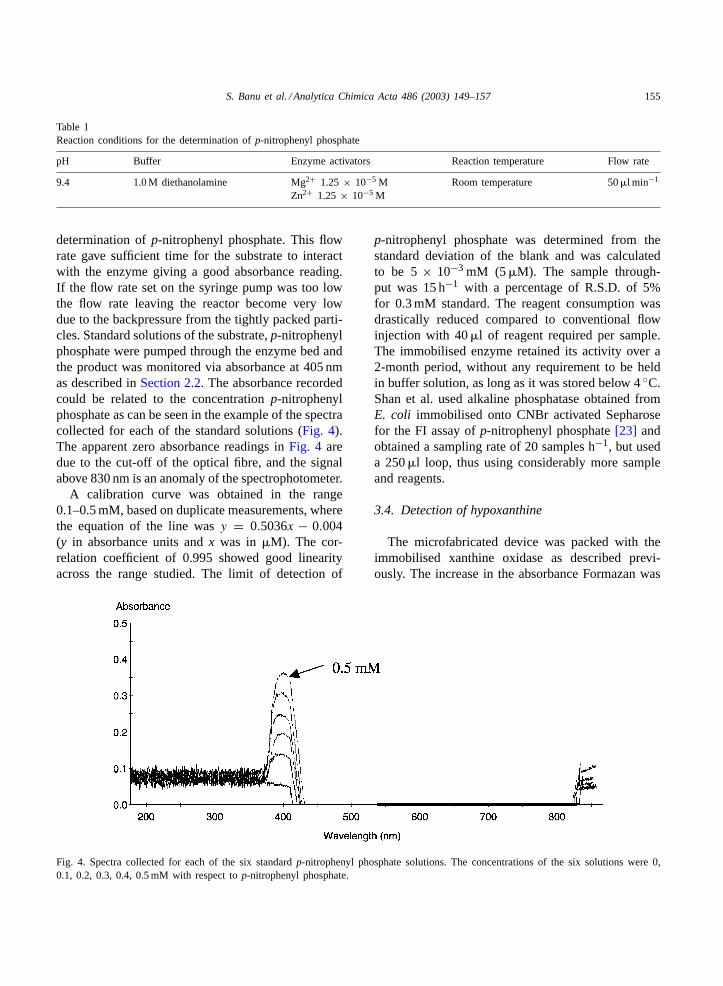
determination ofp-nitrophenyl phosphate. This flowrate gave sufficient time for the substrate to interactwith the enzyme giving a good absorbance reading.If the flow rate set on the syringe pump was too lowthe flow rate leaving the reactor become very lowdue to the backpressure from the tightly packed parti-cles. Standard solutions of the substrate,p-nitrophenylphosphate were pumped through the enzyme bed andthe product was monitored via absorbance at 405 nmas described inSection 2.2. The absorbance recordedcould be related to the concentrationp-nitrophenylphosphate as can be seen in the example of the spectracollected for each of the standard solutions (Fig. 4).The apparent zero absorbance readings inFig. 4 aredue to the cut-off of the optical fibre, and the signalabove 830 nm is an anomaly of the spectrophotometer.
A calibration curve was obtained in the range0.1–0.5 mM, based on duplicate measurements, wherethe equation of the line wasy = 0.5036x − 0.004(y in absorbance units andx was in �M). The cor-relation coefficient of 0.995 showed good linearityacross the range studied. The limit of detection of
Fig. 4. Spectra collected for each of the six standardp-nitrophenyl phosphate solutions. The concentrations of the six solutions were 0,0.1, 0.2, 0.3, 0.4, 0.5 mM with respect top-nitrophenyl phosphate.
p-nitrophenyl phosphate was determined from thestandard deviation of the blank and was calculatedto be 5× 10−3 mM (5�M). The sample through-put was 15 h−1 with a percentage of R.S.D. of 5%for 0.3 mM standard. The reagent consumption wasdrastically reduced compared to conventional flowinjection with 40�l of reagent required per sample.The immobilised enzyme retained its activity over a2-month period, without any requirement to be heldin buffer solution, as long as it was stored below 4◦C.Shan et al. used alkaline phosphatase obtained fromE. coli immobilised onto CNBr activated Sepharosefor the FI assay ofp-nitrophenyl phosphate[23] andobtained a sampling rate of 20 samples h−1, but useda 250�l loop, thus using considerably more sampleand reagents.
3.4. Detection of hypoxanthine
The microfabricated device was packed with theimmobilised xanthine oxidase as described previ-ously. The increase in the absorbance Formazan was

156 S. Banu et al. / Analytica Chimica Acta 486 (2003) 149–157
Fig. 5. The effect of flow rate on the determination of hypoxanthine.
monitored according to the scheme inFig. 2. Thismethod was adapted from the flow injection methoddescribed by Ukeda et al.[16] with the same opti-mised reagent conditions being used.Fig. 5shows theeffect of changing the flow rate through the systemusing the same method as described previously forp-nitrophenyl phosphate. As described previously, ifthe flow rate was too fast there is insufficient time forthe enzyme reaction to occur, if however the flow rateis too slow the sensitivity of the method decreasesdue to the back pressure in the enzyme reactor. Theconditions for the assay are given inTable 2.
A linear calibration was obtained (y = 0.0015x −0.0075, y in absorbance units andx in �M) with acorrelation coefficient of 0.9984 for standards acrossthe range 12–100�M (n = 15). The limit of detectionwas 8�M and the percentage of R.S.D. for 25�M so-lution was 4.4% (n = 3). The light was collected forsix minutes to obtain a stable absorbance reading, al-though it was found that after 4 min both absorbanceand intensity were in steady state, and this gave a sam-ple throughput of six samples per hour with a flowrate of 20�l min−1. The immobilised xanthine oxi-dase had to be stored under wet conditions (50 mMphosphate buffer, pH 7.3 with 10 mM EDTA), below4◦C to retain its activity. If the immobilised enzymewas packed into the micro reactor system, and usedfor measurements (whilst storing it a 4◦C in betweenmeasurements) it could still be successfully used up to2 months after packing. The response did not greatly
Table 2Reaction conditions for the determination of hypoxanthine
pH Buffer XTTconcentration
Reactiontemperature
Flow rate
9.4 50 mMcarbonatebuffer
50�M Roomtemperature
20�l min−1
decrease if the enzyme was not used for measure-ments, for example, the response did not change fora 50�M solution for 1 month when the chip was notused, but the signal then decreased by 43% when itwas used continuously for 40 h. The results obtainedare comparable with those obtained by Balladin et al.who determined hypoxanthine concentrations in fishsamples using flow injection analysis[19]. In theirwork xanthine oxidase was immobilised on a Fracto-gel support (85–89% activity) and they obtained limitsof detection of 4�M, but these results can not be di-rectly compared due to differences in procedures, forexample, they carried out the reaction at 35◦C.
4. Conclusion
This work has demonstrated a novel method forproducing a microfabricated bio reactor for analytical(or synthetic) applications. A flowthrough, high sur-face area packed reactor has been produced that canbe used successfully with hydrodynamic flow. The de-vice allows the use of well documented immobilisedenzyme reagents, but is also suitable for other supportreagents.
It is also designed such that when the packing isexhausted it can be repacked with relative ease. Workis now continuing in optimising specific enzymes sys-tems for applications where expensive enzymes andsmall samples make the utilisation of micro reactorsparticularly appropriate.
References
[1] A.G. Hadd, S.C. Jacobson, J.M. Ramsey, Anal. Chem. 71(1999) 5206–5212.
[2] J. Wang, M.P. Chatrathi, A. Ibanez, Analyst 126 (2001) 1203–1206.
[3] S. Kasai, Y. Hirano, N. Motochi, H. Shiku, M. Nishizawa,T. Matsue, Anal. Chim. Acta 458 (2002) 263–270.
[4] H.B. Mao, T.L. Yang, P.S. Cremer, Anal. Chem. 74 (2002)379–385.
[5] D.S. Peterson, T. Rohr, F. Svec, J.M.J. Frechet, Anal. Chem.74 (2002) 4081–4088.
[6] N.G. Wilson, T. McCreedy, G.M. Greenway, Analyst 125(2000) 237–239.
[7] J.W. Choi, K.W. Oh, J.H. Thomas, W.R. Heineman, H.B.Halsall, J.H. Nevin, A.J. Helmicki, H.T. Henderson, C.H.Ahn, Lab Chip 2 (2002) 27–30.

S. Banu et al. / Analytica Chimica Acta 486 (2003) 149–157 157
[8] R.D. Oleschuk, L.L. Shultz-Lockyear, Y. Ning, D.J. Harrison,Anal. Chem. 72 (2000) 585–590.
[9] G.F. Bickerstaff, Immobilisation of Enzymes and Cells(Methods in Biotechnology), Humana Press, Totowa, NJ,1997.
[10] T. McCreedy, Anal. Chim. Acta 427 (2001) 39–43.[11] B.R.M. Al-Gailani, T. McCreedy, Chem. Commun. (2003)
120–121.[12] C. Perininiger, B. Danielsson, Analyst 121 (1996) 1717–1720.[13] www.worthington-biochem.com/manual, Worthington Bio-
chemical Corp., Lakewood, NJ, 2002.[14] R. Gnanasekaran, H.A. Mottola, Anal. Chem. 57 (1985)
1005–1009.[15] M.A. Marshall, H.A. Mottola, Anal. Chem. 55 (1983) 2089–
2093.
[16] H. Ukeda, S. Maeda, M. Sawamura, J. Flow Injection Anal.15 (1998) 39–46.
[17] H.H. Weetal, Appl. Biochem. Biotechnol. 41 (1993) 157–188.[18] J. Tramper, F. Müller, H.C. Van Der Plas, Biotechnol. Bioeng.
20 (1978) 1507–1522.[19] D.A. Balladin, D. Na rinesingh, V.A. Stoute, T.T. Ngo, Appl.
Biochem. Biotechnol. 62 (1997) 317–328.[20] S. Lam, G. Malikin, Analytical Applications of Immobilised
Enzyme Reactors, Blackie, London, 1994, pp. 133–134.[21] W.W. Westerfeld, D.A. Richert, R.J. Bloom, J. Biol. Chem.
234 (1959) 1889–1896.[22] S. Lam, G. Malikin, Analytical Applications of Immobilised
Enzyme Reactors, Blackie, London, 1994, pp. 12–13.[23] Y. Shan, I.D. McKelvie, B.T. Hart, Anal. Chem. 65 (1993)
3053–3060.