method optimization for quantitative analysis of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine...
TRANSCRIPT

A
eficLs©
K
1
(mhec[Hs(caoa
0d
Talanta 70 (2006) 455–459
Method optimization for quantitative analysis ofoctahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine(HMX) by liquid chromatography-electrospray
ionization mass spectrometry
Xiaoping Pan ∗, Kang Tian, Lindsey E. Jones, George P. CobbThe Institute of Environmental and Human Health (TIEHH), Department of Environmental Toxicology, Texas Tech University,
Lubbock, TX 79409-1163, USA
Received 2 February 2006; received in revised form 5 March 2006; accepted 5 March 2006Available online 18 April 2006
bstract
A simple, sensitive LC-ESI-MS method was optimized for quantitative analysis of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) innvironmental samples. Under negative ionization mode, HMX can form adduct ions with various organic acids and salts, including acetic acid,ormic acid, propionic acid, ammonium nitrate, ammonium chloride, sodium nitrite, and sodium nitrate. Acetic acid was chosen as additive and the
−
on, [M + CH3COO] with m/z = 355 was used for selective ion monitoring (SIM) in this study. Good sensitivity was achieved with low acetic acidoncentration in the mobile phase and relatively low capillary temperature. The method detection limit was 0.78 pg for HMX in standard solution.inearity (R2 > 0.9998) was obtained at low concentrations (0.5–50 �g/L). This method has been used to determine HMX concentrations in wateramples and lizard egg samples from an animal exposure study.2006 Elsevier B.V. All rights reserved.
ation
AfaHmRcwCsamL
eywords: Explosive; HMX; Liquid chromatography (LC); Electrospray ioniz
. Introduction
Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX)Fig. 1) is one of the most important explosives used in variousilitary operations [1–4]. Limited animal studies indicate
epatic and central nervous system toxicities following HMXxposure [1]. However, HMX toxicity studies are limitedompared to two other important explosives: RDX and TNT5–7], especially in wildlife species such as reptiles. To aid inMX toxicity studies, it is necessary to develop selective and
ensitive analytical methods. In general, gas chromatographyGC) for nitramine explosive analysis, including HMX, isonsidered to be impractical due to their low vapor pressures
nd thermal liabilities [8]. Nevertheless, successful GC analysisf nitramine explosives using deactivated injection-port linersnd short, wide-bore capillary columns has been reported [8].∗ Corresponding author. Tel.: +1 806 885 4567.E-mail address: [email protected] (X. Pan).
TfFaptt
039-9140/$ – see front matter © 2006 Elsevier B.V. All rights reserved.oi:10.1016/j.talanta.2006.03.005
(ESI); Mass spectrometry (MS); Adduct ion; Water; Egg
dditionally, good GC separation of RDX and its derivativesrom a DB-5 column was obtained using low injector temper-ture and fast carrier gas flow in our laboratory [9]. However,MX failed to elute in the same system, probably due to itsuch lower vapor pressure than RDX (HMX: 10−14 Torr versusDX: 10−9 Torr at 20 ◦C) [8]. As a good alternative, liquidhromatography coupled with different detectors has beenidely employed for nitramine explosive analysis [10–13].ompared with common analytical detection techniques, mass
pectrometry detection is advantageous for its good sensitivitynd selectivity. The formation of HMX adduct ions was com-only observed in many studies [14–17]. However, quantitativeC-ESI-MS analysis of HMX in real world samples is rare.he aim of this study was to optimize a LC-ESI-MS method
or the trace quantification of HMX in environmental samples.ormation of HMX adduct ions with a variety of organic acids
nd salts were investigated, and critical LC-ESI-MS operationarameters such as additive concentration and heated capillaryemperature were also optimized. This method has been appliedo determine HMX concentrations in lizard egg extracts. Lizards
456 X. Pan et al. / Talanta 70 (2006) 455–459
aeivobs
2
2
cS(tws
apa5dsaST
2
oaipn
Table 1Some important optimized LC-ESI-MS operation parameters
LC conditions MS conditions
Mobile phase A Methanol Mode NegativeMobile phase B 0.5 mM aqueous
acetic acidIon spray voltage(kV)
3.5
A:B 60:40 (v/v) Sheath gas flow rate(L/h)
44.0
Flow rate 0.5 mL/min Aux/sweep gas flowrate (L/h)
53.1
Injection 25 �L Capillary voltage(V)
−6.3
Capillarytemperature (◦C)
140.0
Multipole 1 offset(V)
1.7
Lens voltage (V) 25.6Multipole 2 offset(V)
7.0
mipa
soucaathur
2
oLepswutEalc(
Fig. 1. Chemical structure of HMX (molecular weight = 296).
re among one of the most important bioindicator species forcological risk assessment [18]. Eggs are generally depositedn their habitats, and developing embryos are susceptible toarious environmental contaminants including HMX. Studyf contaminant residue in egg is thus an important index foriomonitoring of potential adverse effects of contaminants onome wildlife populations [19].
. Experimental
.1. Chemicals and instruments
Standard HMX (CAS No. 2691-41-0; 99.0% pure) at aoncentration of 1000 mg/L in acetonitrile was obtained fromupelco (Bellefonte, PA, USA). Methanol and glacial acetic acidboth HPLC grade), C18 cartridges (1 g), and PTFE syringe fil-er (0.2 �m) were from Fisher (Pittsburg, PA, USA). Ultra-pureater (>18 M�) obtained from a Barnstead NANOpure infinity
ystem (Dubuque, IA, USA) was used for all aqueous solutions.For instruments, the LC part was a Finnigan system including
vacuum membrane degasser, a gradient pump and an autosam-ler (San Jose, CA, USA). Chromatographic separation waschieved using a Supelco RP C18 column (4.6 mm × 250 mm,-�m packing) (Bellefonte, PA, USA). MS analyses were con-ucted using a Thermo-Finnigan LCQ advantage ion trap masspectrometer. Helium was used as the damping gas for ion trapnd nitrogen was the sheath and auxiliary gas for ion source.ome important LC-MS operation conditions are shown inable 1.
.2. LC-ESI-MS method optimization
To achieve better sensitivity, the LC-ESI-MS method wasptimized. First, capacities of different organic acids and salts
s additives in helping the formation of HMX adduct ion werenvestigated. Additives studies included acetic acid, formic acid,ropionic acid, ammonium nitrate, ammonium chloride, sodiumitrite, and sodium nitrate. Additives were dissolved in theltu2
Multiple RF Amp(Vp-p, sp)
500.0
obile phase and delivered at 0.5 mL/min, and HMX wasnjected continuously post-column at 5 �L/min using a syringeump. These two streams mixed together at a “T” coupling unitnd infused into the mass spectrometer interface.
Based on HMX–acetate adduct ion, operation parametersuch as ion spray voltage, sheath gas, and sweep gas wereptimized using the “tune” function and was done by contin-ous after-column injection of HMX. Acetic acids at differentoncentrations in the mobile phase and heated capillary temper-tures were optimized by changing the mobile phase manuallynd changing the heated capillary temperatures manually. Forhese two critical parameters, acetic acid concentration andeated capillary temperature, HMX was injected into LC col-mn via autosampler and the signal responses as peak areas wereecorded.
.3. Method application
In order to further test the developed method, lizard eggsbtained from an ongoing HMX exposure study were analyzed.izards were dosed using HMX contaminated cricket daily, andggs were collected during the experiment period. Eggs sam-les were extracted using a protocol reported previously withome modifications [20]. Briefly, egg samples (∼0.2 g each)ere homogenized and dehydrated with 4–5 g of dried Na2SO4sing a small mortar and pestle. The soil sample–Na2SO4 mix-ure was then extracted using a Dionex Accelerated Solventxtractor (Model 200, Salt Lake City, UT, USA) using 100%cetonitrile as extraction solvent. Extraction program is as fol-ows: preheat 5 min, heat 5 min, and static extraction 5 min atonstant temperature (100 ◦C) and pressure (1500 psi). Extracts15–20 mL/sample) were then purged from cells into glass col-
ection vials using nitrogen gas. The extract volume was reducedo ∼1 mL using a vortex evaporator for the following cleanupsing a C18 cartridge. C18 cartridge was conditioned using× 3 mL acetonitrile, sample extract (∼1 mL) was then loaded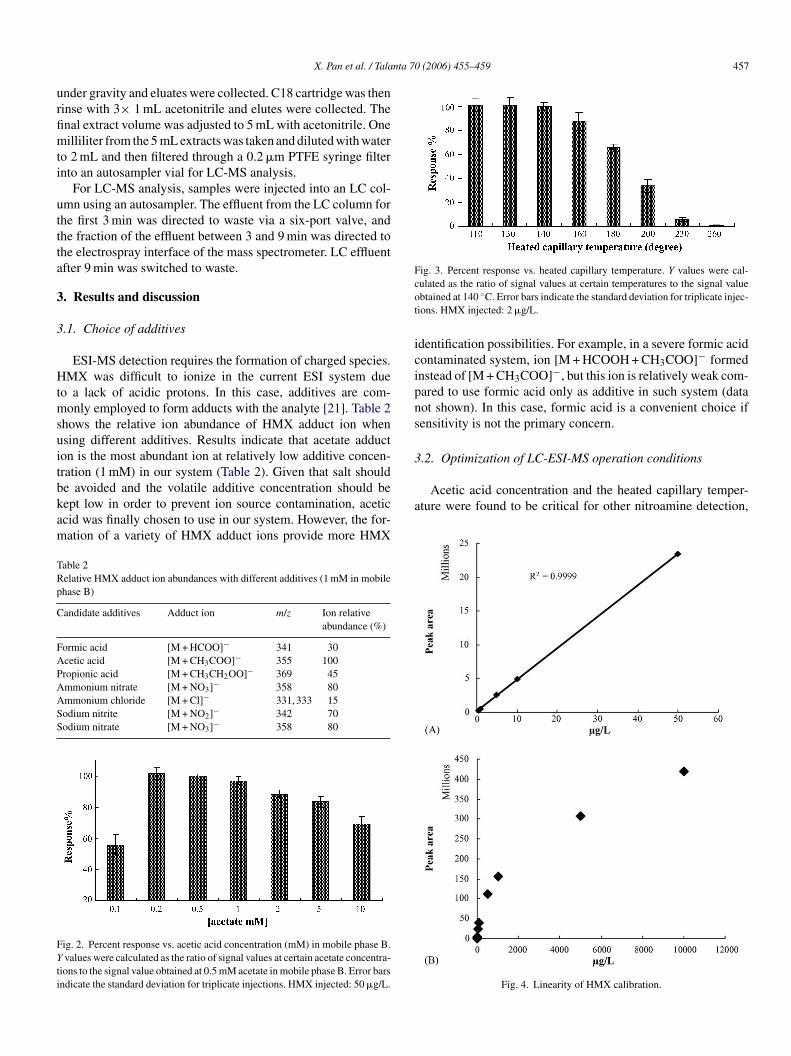
nta 70 (2006) 455–459 457
urfimti
uttta
3
3
Htmsuitbkam
TRp
C
FAPAASS
FYti
Fig. 3. Percent response vs. heated capillary temperature. Y values were cal-cot
icipns
3.2. Optimization of LC-ESI-MS operation conditions
X. Pan et al. / Tala
nder gravity and eluates were collected. C18 cartridge was theninse with 3× 1 mL acetonitrile and elutes were collected. Thenal extract volume was adjusted to 5 mL with acetonitrile. Oneilliliter from the 5 mL extracts was taken and diluted with water
o 2 mL and then filtered through a 0.2 �m PTFE syringe filternto an autosampler vial for LC-MS analysis.
For LC-MS analysis, samples were injected into an LC col-mn using an autosampler. The effluent from the LC column forhe first 3 min was directed to waste via a six-port valve, andhe fraction of the effluent between 3 and 9 min was directed tohe electrospray interface of the mass spectrometer. LC effluentfter 9 min was switched to waste.
. Results and discussion
.1. Choice of additives
ESI-MS detection requires the formation of charged species.MX was difficult to ionize in the current ESI system due
o a lack of acidic protons. In this case, additives are com-only employed to form adducts with the analyte [21]. Table 2
hows the relative ion abundance of HMX adduct ion whensing different additives. Results indicate that acetate adducton is the most abundant ion at relatively low additive concen-ration (1 mM) in our system (Table 2). Given that salt should
e avoided and the volatile additive concentration should beept low in order to prevent ion source contamination, aceticcid was finally chosen to use in our system. However, the for-ation of a variety of HMX adduct ions provide more HMXable 2elative HMX adduct ion abundances with different additives (1 mM in mobilehase B)
andidate additives Adduct ion m/z Ion relativeabundance (%)
ormic acid [M + HCOO]− 341 30cetic acid [M + CH3COO]− 355 100ropionic acid [M + CH3CH2OO]− 369 45mmonium nitrate [M + NO3]− 358 80mmonium chloride [M + Cl]− 331, 333 15odium nitrite [M + NO2]− 342 70odium nitrate [M + NO3]− 358 80
ig. 2. Percent response vs. acetic acid concentration (mM) in mobile phase B.values were calculated as the ratio of signal values at certain acetate concentra-
ions to the signal value obtained at 0.5 mM acetate in mobile phase B. Error barsndicate the standard deviation for triplicate injections. HMX injected: 50 �g/L.
a
ulated as the ratio of signal values at certain temperatures to the signal valuebtained at 140 ◦C. Error bars indicate the standard deviation for triplicate injec-ions. HMX injected: 2 �g/L.
dentification possibilities. For example, in a severe formic acidontaminated system, ion [M + HCOOH + CH3COO]− formednstead of [M + CH3COO]−, but this ion is relatively weak com-ared to use formic acid only as additive in such system (dataot shown). In this case, formic acid is a convenient choice ifensitivity is not the primary concern.
Acetic acid concentration and the heated capillary temper-ture were found to be critical for other nitroamine detection,
Fig. 4. Linearity of HMX calibration.
458 X. Pan et al. / Talanta 70 (2006) 455–459
HMX
ssptAihcapuautT
3
5ssG2up(pttwam
3
a
(teeH
4
ecaoaptdt
A
&eTQa
R
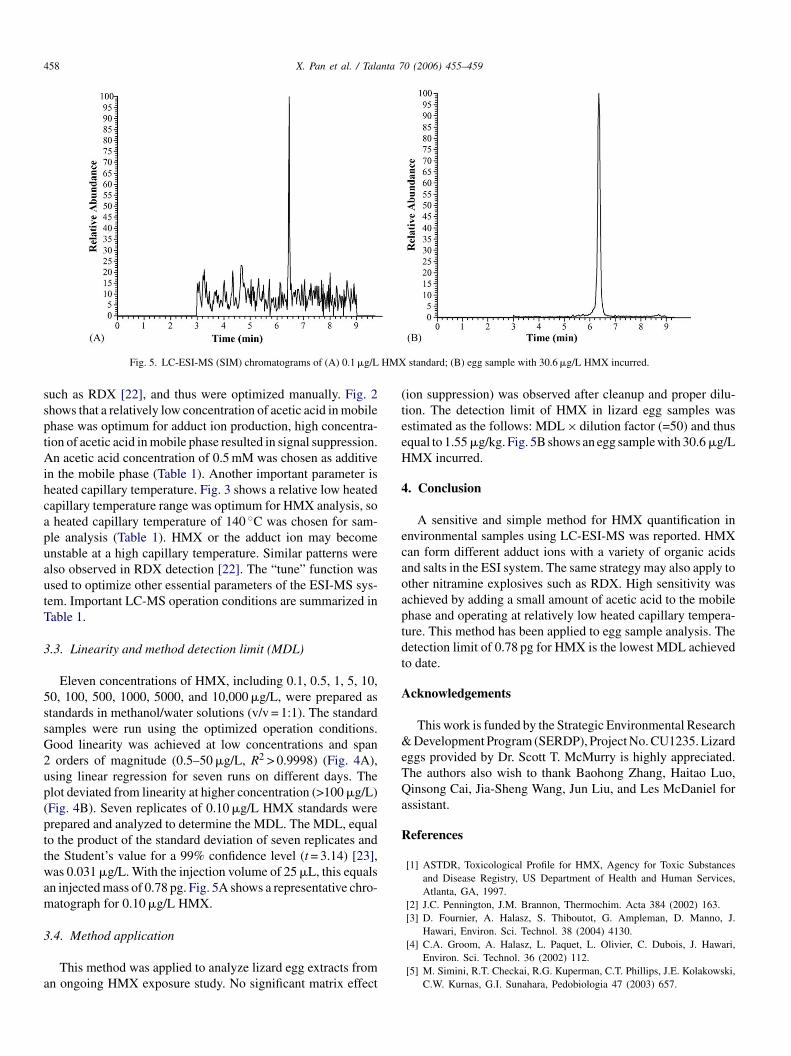
Fig. 5. LC-ESI-MS (SIM) chromatograms of (A) 0.1 �g/L
uch as RDX [22], and thus were optimized manually. Fig. 2hows that a relatively low concentration of acetic acid in mobilehase was optimum for adduct ion production, high concentra-ion of acetic acid in mobile phase resulted in signal suppression.n acetic acid concentration of 0.5 mM was chosen as additive
n the mobile phase (Table 1). Another important parameter iseated capillary temperature. Fig. 3 shows a relative low heatedapillary temperature range was optimum for HMX analysis, soheated capillary temperature of 140 ◦C was chosen for sam-
le analysis (Table 1). HMX or the adduct ion may becomenstable at a high capillary temperature. Similar patterns werelso observed in RDX detection [22]. The “tune” function wassed to optimize other essential parameters of the ESI-MS sys-em. Important LC-MS operation conditions are summarized inable 1.
.3. Linearity and method detection limit (MDL)
Eleven concentrations of HMX, including 0.1, 0.5, 1, 5, 10,0, 100, 500, 1000, 5000, and 10,000 �g/L, were prepared astandards in methanol/water solutions (v/v = 1:1). The standardamples were run using the optimized operation conditions.ood linearity was achieved at low concentrations and spanorders of magnitude (0.5–50 �g/L, R2 > 0.9998) (Fig. 4A),
sing linear regression for seven runs on different days. Thelot deviated from linearity at higher concentration (>100 �g/L)Fig. 4B). Seven replicates of 0.10 �g/L HMX standards wererepared and analyzed to determine the MDL. The MDL, equalo the product of the standard deviation of seven replicates andhe Student’s value for a 99% confidence level (t = 3.14) [23],as 0.031 �g/L. With the injection volume of 25 �L, this equals
n injected mass of 0.78 pg. Fig. 5A shows a representative chro-atograph for 0.10 �g/L HMX.
.4. Method application
This method was applied to analyze lizard egg extracts fromn ongoing HMX exposure study. No significant matrix effect
standard; (B) egg sample with 30.6 �g/L HMX incurred.
ion suppression) was observed after cleanup and proper dilu-ion. The detection limit of HMX in lizard egg samples wasstimated as the follows: MDL × dilution factor (=50) and thusqual to 1.55 �g/kg. Fig. 5B shows an egg sample with 30.6 �g/LMX incurred.
. Conclusion
A sensitive and simple method for HMX quantification innvironmental samples using LC-ESI-MS was reported. HMXan form different adduct ions with a variety of organic acidsnd salts in the ESI system. The same strategy may also apply tother nitramine explosives such as RDX. High sensitivity waschieved by adding a small amount of acetic acid to the mobilehase and operating at relatively low heated capillary tempera-ure. This method has been applied to egg sample analysis. Theetection limit of 0.78 pg for HMX is the lowest MDL achievedo date.
cknowledgements
This work is funded by the Strategic Environmental ResearchDevelopment Program (SERDP), Project No. CU1235. Lizard
ggs provided by Dr. Scott T. McMurry is highly appreciated.he authors also wish to thank Baohong Zhang, Haitao Luo,insong Cai, Jia-Sheng Wang, Jun Liu, and Les McDaniel for
ssistant.
eferences
[1] ASTDR, Toxicological Profile for HMX, Agency for Toxic Substancesand Disease Registry, US Department of Health and Human Services,Atlanta, GA, 1997.
[2] J.C. Pennington, J.M. Brannon, Thermochim. Acta 384 (2002) 163.[3] D. Fournier, A. Halasz, S. Thiboutot, G. Ampleman, D. Manno, J.
Hawari, Environ. Sci. Technol. 38 (2004) 4130.[4] C.A. Groom, A. Halasz, L. Paquet, L. Olivier, C. Dubois, J. Hawari,
Environ. Sci. Technol. 36 (2002) 112.[5] M. Simini, R.T. Checkai, R.G. Kuperman, C.T. Phillips, J.E. Kolakowski,
C.W. Kurnas, G.I. Sunahara, Pedobiologia 47 (2003) 657.

nta 7
[
[
[
[
[
[[
[
[
[
[
X. Pan et al. / Tala
[6] G. Rosen, G.R. Lotufo, Environ. Toxicol. Chem. 24 (2005) 2887.[7] S. Rocheleau, R.G. Kuperman, M. Martel, L. Paquet, G. Bardai, S.
Wong, M. Sarrazin, S. Dodard, P. Gong, J. Hawari, R.T. Checkai, G.I.Sunahara, Chemosphere 62 (2006) 545.
[8] M.E. Walsh, T.A. Ranney, Determination of Nitroaromatic, Nitramine,and Nitrate Ester Explosives in Water Using SPE and GC-ECD, Compar-ison with HPLC, US Army Corps of Engineers, Cold Regions Research& Engineering Laboratory, 1998.
[9] X.P. Pan, B.H. Zhang, G.P. Gobb, Talanta 67 (2005) 816.10] K. Bratin, P.T. Kissinger, B.C. Briner, C.S. Bruntlett, Anal. Chim. Acta
130 (1981) 295.11] D.H. Fine, W.C. Yu, E.U. Goff, E.C. Bender, D.J. Reutter, J. Forensic
Sci. 29 (1984) 732.12] T.F. Jenkins, P.H. Miyares, K.F. Myers, E.F. McCormick, A.B. Strong,
Anal. Chim. Acta 189 (1994) 69.13] T.M. Chow, M.R. Wilcoxon, M.D. Piwoni, N.R. Adrian, J. Chromatogr.
Sci. 42 (2004) 470.
[[
[
0 (2006) 455–459 459
14] D.W. Berberich, R.A. Yost, D.D. Fetterolf, J. Forensic Sci. 33 (1988)946.
15] B. Casetta, F. Garofolo, Org. Mass Spectr. 29 (1994) 517.16] J. Yinon, J.E. McClellan, R.A. Yost, Rapid Commun. Mass Spectrom.
11 (1997) 1961.17] E. Holmgren, H. Carlsson, P. Goede, C. Crescenzi, J. Chromatogr. A
1099 (2005) 127.18] C.D. Klaassen, Toxicology: The Basic Science of Poisons, sixth ed.,
2001, pp. 144–145.19] C.B. Pepper, T.R. Rainwater, S.G. Platt, J.A. Dever, T.A. Anderson, S.T.
McMurry, J. Wildl. Dis. 40 (2004) 493.20] B.H. Zhang, X.P. Pan, G.P. Cobb, T.A. Anderson, J. Chromatogr. B 824
(2005) 277.21] X. Zhao, J. Yinon, J. Chromatogr. A 977 (2002) 59.22] X.P. Pan, B.H. Zhang, S.B. Cox, T.A. Anderson, G.P. Cobb, J. Chro-
matogr. A 1107 (2006) 2.23] SW846 Test Methods SW846, US EPA, Washington, DC, 2000.