matrix metalloproteinase inhibitors: past lessons and future prospects in breast cancer
TRANSCRIPT

The Breast (2001) 10, 368–378
# 2001 Harcourt Publishers Ltd
doi:10.1054/brst.2000.0254, available online at http://www.idealibrary.com on
REVIEW
Matrix metalloproteinase inhibitors: past lessons and future prospects in breast
cancer
R. M. Glasspool and C. J. Twelves
Cancer Research Campaign Department of Medical Oncology, Alexander Stone Building,Glasgow G61 3BD, UK
SUMMARY . The matrix metalloproteinases (MMPs) play a central role in invasion and metastasis. However,despite striking activity in preclinical models, the clinical development of the matrix metalloproteinase inhibitors(MMPIs) has been difficult. The results of important phase III trials are now emerging and it is therefore opportuneto review the current state of the MMPIs. In this article the evidence for the role of MMPs in the progression ofbreast cancer, the development of the MMPIs and the recent phase III results are discussed. Despite the problemsencountered it is hoped that the MMPIs may yet provide another mechanism for the long-term control ofmicrometastatic disease. Furthermore, important lessons can be learnt from their development that are relevant tothe development of other biological agents. # 2001 Harcourt Publishers Ltd
INTRODUCTION
During the past few years there has been an extra-ordinary range of new developments in the systemictreatment of breast cancer. New cytotoxics especially thetaxanes, have already become established as therapy foradvanced disease and, studies suggest that as adjuvanttreatment, they appear to significantly improve survivalwhen added to standard treatment regimens of adria-mycin and cyclophosphamide. Likewise, with endocrinetherapy the aromatase inhibitors are clearly superior tothe progestogens or aminoglutethamide. Herceptin, ahumanized monoclonal antibody directed at the extra-cellular domain of the HER-2 receptor, is alreadylicensed for the treatment of advanced breast cancerboth as a single agent and in combination withchemotherapy. Nevertheless, there remains a need forother new therapies as breast cancer remains a majorcause of death in women.
Address correspondence to: Dr C. Twelves, Cancer ResearchCampaign Department of Medical Oncology, Alexander StoneBuilding, Garscube Estate, Switchback Road, Bearsden,Glasgow G61 3BD, UK. Tel.: 0141 211 1712; Fax.: 211 1869;E-mail: [email protected]: 4 September 2000Accepted: 6 September 2000Published online: 19 April 2001
368
As our understanding of the molecular biology ofcancer increases, there is an appreciation of the potentialbenefits to be gained by controlling a cancer, rather thanattempting to eradicate all malignant cells. In the future,cancer may become a chronic disease, like diabetesmellitus, with which people live for many years.Inhibitors of matrix metalloproteinases proteins(MMPs) represent one of the potentially importantdevelopments in this area. With results of importanttrials now emerging, it is opportune to look at thecurrent state of MMP inhibitors (MMPIs). In preclinicalmodels the MMPIs are well tolerated and have clearantitumour activity. In clinical trials, however, theirdevelopment has been less straightforward. There areimportant lessons to be learnt from experience with theMMPIs that are relevant to the development of otherbiological agents in the future.
THE MATRIX METALLOPROTEINASES
The MMPs are a family of secreted and transmembraneenzymes capable of digesting most components of theextracellular matrix. They were first identified in relationto the role of a collagenase responsible for theabsorption of tadpole tails more than 30 years ago.1
More than 20 MMPs have been described to date and

Table 1 The matrix metalloproteinases and their substrates
Group MMP (name) Substrates
Collagenases 1 (Interstitial) Fibrillary collagens I,II,III,VI,IX,Proteoglycans
8 (Neutrophil) Collagens I,II,III
13 (Collagenase 3) Collagens I,II,III18 (Xenopus)
Gelatinases 2 (Gelatinase-A) Gelatin, I,II,III, ElastinCollagen IV,V,VII,X, Fibronectin.
9 (Gelatinase B) Gelatin I, VCollagen IV,V.
Stromelysins 3 (Stromelysin-1) Proteoglycans, Collagens II,IV,XI,Fibronectin, Laminin, Procollagenase Gelatin I,III,IV,V,Collagen III,IV,V,IX.Gelatin I,III,IV,V, Proteoglycan, Fibronectin,
7 (Matrilysin) Procollagenase, TNF alpha precursor, Collagen IV.Gelatin I,III,IV,V, Collagen III,IV,V,
10 (Stromelysin 2) Fibronectin, Procollagenase.Casein, Serine protease inhibitors
11 (Stromelysin 3)
MT-MMPS 14 (MT1-MMP) Activates Progelatinase-A15 (MT2-MMP) Activates Progelatinase-A16 (MT3-MMP) Activates Progelatinase-A17 (MT4-MMP)
21 (MT5-MMP)
Others 12 (metalloelastase) Elastin, non fibrillar collagen, Progelatinase A.19 Rheumatoid arthritis-associated Undefined20 enamelysin Amelogenenin23 Undefined24 Undefined
Matrix metalloproteinase inhibitors 369
others are likely to emerge (Table 1). Soluble MMPs areproduced as inactive pro-enzymes and activated in acascade by cleavage of a 10 kDa amino terminaldomain. Once activated, they are individually regulatedby endogenous inhibitors such as a-2-macroglobulinand, more specifically, by a family of four tissueinhibitors of metalloproteinases (TIMPs). This complexbalance of activation and breakdown means that MMPoverexpression in itself does not necessarily implyincreased enzymatic activity.
The MMPs derive their name from a common, highlyconserved active site containing a zinc atom. They canbe classified into groups according to their sequencehomology. The membrane type or MT-MMPs form adistinct group that share the same active site but alsocontain a transmembrane domain. MT1-MMP has beenidentified on the cell membrane of tumour cells. It is ableto activate pro-gelatinase A (MMP2) produced bystromal cells adjacent to tumours. In this way, MT1-MMP can increase gelatinase A activity at the advan-cing edge of the tumour where it is relatively shieldedfrom non-specific inhibitors.2
In order to metastasize, cells must first escape fromthe primary tumour, enter into the lymphatic or bloodcirculation and survive to be transported through the
circulatory system. They need then to arrest in distantorgans, extravasate and continue to grow to formsecondary tumours in the new environment with theformation of new blood vessels (Fig. 1). In order to dothis, tumour cells must breach the extracellular matrixand basement membrane barriers. Since MMPs arecapable of degrading most of the components of theextracellular matrix, it was postulated that they might beinvolved in the early stages of metastasis.
Increased levels of MMPs have been found in arange of different solid tumours including breastcancer.3 Originally it was assumed that MMPs wereproduced by tumour cells. The concept that MMPs alsooriginated from the stroma came from observations withstromelysin 3 (MMP11) in breast cancer.4 In situhybridization localized most MMPs to the stroma ofbreast tumours.5 Stromelysin 3, for example, is ex-pressed on stromal fibroblasts and is detected moreoften in invasive ductal rather than lobular carcinomas.6
Similarly, collagenase 3 (MMP-13) is expressed bystromal cells and its transcription appears to becontrolled by diffusable factors released by the adjacentepithelial tumour cells.7 MT1-MMP, which is detectedin most breast cancers,8 differs in that it is localized onthe tumour cells.

Fig. 1 The role of MMPs in invasion and metastasis.
Fig. 2 Expression and activation of MMPs involves interaction between tumour and stromal cells.
370 The Breast
In general, there is a correlation between tumourprogression and MMP expression. Expression of gela-tinase A (MMP2) is common in breast cancer, but theratio of activated to total gelatinase A increases inmetastatic disease.3,9 The expression of MT1-MMPcorrelates with the presence of lymph node metastasesand other markers of advanced disease, supporting itskey role in tumour progression through activation ofprogelatinase A.10 Similarly, progelatinase B (MMP9) ispresent in DCIS and T1 invasive tumours but active
MMP-9 has been detected only in larger tumours.11
Membrane vesicles shed from the surface of breastcancer cells in vitro carry large amounts of MMP-9,emphasizing again the interaction between tumour andstroma (Fig. 2).12
The association between MMP activity and tumouraggressiveness supports a functional role for MMPs inmetastasis.6,9,13,14 This is reinforced by reports of MMPsas being prognostic factors. In 110 primary breastcancers moderate or strong staining for stromelysin-3

Matrix metalloproteinase inhibitors 371
(ST3) independently predicted for a shorter disease-freeand overall survival.6 Ahmad et al. (1998) confirmedthat in women with node positive, invasive ductalcarcinoma the level of ST3 staining was a strong,independent prognostic factor predicting disease-freesurvival (P=0.005).15 In a series of 177 primary breastcancers, MMP2 immunostaining was seen in 84%.Again, the intensity of staining correlated with survival;56% of patients who had strongly MMP2 positive werealive at 10 years compared to 88% with negativetumours. This effect was independent of age, tumourgrade, receptor status or stage of disease.14 Others,however, have found an apparently paradoxical rela-tionship between TIMP expression and a poor prog-nosis.16–18 For example, levels of TIMP-1 are higher inbreast carcinomas than fibroadenomas and in thecancers high TIMP-1 concentrations predicted a pooroutcome.19,20 Raised circulating TIMP-1 levels havealso been reported in 19 patients with advanced breastcancer.21 Interestingly, TIMP-1 acts as a growth factorfor one breast cancer cell line,22 whereas transfection ofanother human breast cancer xenograft with TIMP-4inhibits growth in vivo.23 Clearly, absolute levels ofMMPs or their inhibitors are a crude measure of overallenzyme activity. There is a complex interplay betweenactivators and inhibitors of each enzyme and alsobetween the tumour cells and their surroundingstroma.24
More recently, it has been appreciated that the role ofMMPs in metastasis goes beyond degradation of thebasement membrane and extracellular matrix allowingintravasation and extravasation of tumour cells. Indeed,degradation of the extracellular matrix is a critical stepin angiogenesis allowing endothelial cells to migratefrom pre-existing vessels into the tumour mass. Cham-bers and Matrisiam25 have reviewed evidence for theinvolvement of MMPs in angiogenesis and regulation oftumour growth. MMP-2 expression in human breastcancers correlates with vascular endothelial growthfactor (VEGF) expression.26 Synthetic inhibitors ofMMPs can inhibit angiogenesis both in vitro and inanimal models.27–30 In addition to contributing toproteolysis, MMP-2 may also modulate cell attachmentby lysing cell surface components that mediate attach-ment to the extracellular matrix thus facilitating cellmigration and invasion. Inhibition of MMP-2 in humanmelanoma cells resulted in enhanced cellular adhesion.31
Recent data suggest that the MMPs may have an evenmore fundamental role, actually initiating or promotingthe growth of breast tumours. The tumourigenicity ofhuman breast cancer cells is increased by injecting themalong with fibroblasts transfected with stromelysin-3.32
In transgenic mice, expression of Matrilysin (MMP 7)
enhances the tumourigenicity of the neu oncogene.33
Similarly, Sternlicht et al. (1999) showed an increase inhyperplastic, dysplastic and malignant lesions in trans-genic mice overproducing stromelysin-1(MMP3).34
These data suggest that MMPs may act as tumourpromoters and raise the possibility of using MMPIs aspreventative treatment in high-risk women.
DEVELOPMENT OF MATRIX
METALLOPROTEINASES INHIBITORS
The central role of MMPs in tumour invasion andmetastasis makes them an attractive target for drugdevelopment. Agents directed at MMPs might beexpected to have a broad range of activity. In addition,MMPIs are likely to be cytostatic rather than cytotoxicso there is expectation that they may be better toleratedthan current cytotoxics.
The naturally occurring MMP inhibitors TIMP-1 andTIMP-2 are an obvious choice for potential therapeuticagents. However, their relatively high molecular weightand poor oral bioavailability make them less attractivefor clinical use. The possibility that TIMPs have othereffects on cellular function also limits their appeal astherapeutic agents. Identification of the active domainsof the TIMPs may in the future lead to compounds withclinical potential. To date, however, the majority ofattention has focused on the clinical development ofsynthetic inhibitors. These include: batimastat (BB-94)and marimastat (BB-2516), both developed by BritishBiotech; CGS27023A (MMI 270B) from Novartis;AG3340 from Agouron; BAY12-9566 from Bayer; andBMS-275291 from Bristol Myers Squibb (Fig. 3). Thesecompounds have in the main been developed though theapplication of structure-based design rather thanthrough conventional screening technology. They actas competitive inhibitors, binding reversibly at the activesite of the MMPs.35
Many of the problems encountered during thedevelopment of the MMPIs are common to otherbiological modulators and relevant to development ofother novel anticancer agents that are emerging into theclinic. As with conventional cytotoxics, the first stage inclinical development in MMPIs is to establish a safe andpotentially effective dose for further evaluation. Withconventional cytotoxics this is relatively straightforwardin a Phase I trial. Dose escalation in sequential patientcohorts will usually identify a maximum tolerated dose(MTD). Experience with conventional cytotoxics hasshown that clinically important activity is most likely tobe achieved at doses close to the MTD. In this respect,antiproliferative treatment toxicities such as myelo
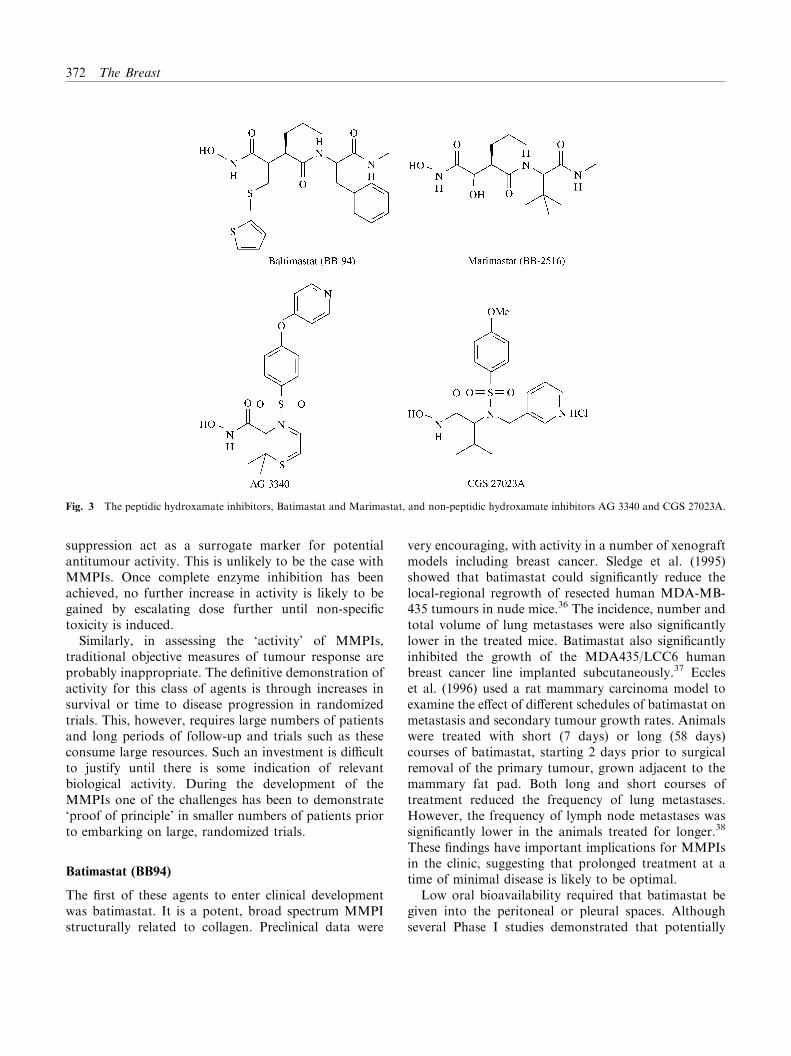
Fig. 3 The peptidic hydroxamate inhibitors, Batimastat and Marimastat, and non-peptidic hydroxamate inhibitors AG 3340 and CGS 27023A.
372 The Breast
suppression act as a surrogate marker for potentialantitumour activity. This is unlikely to be the case withMMPIs. Once complete enzyme inhibition has beenachieved, no further increase in activity is likely to begained by escalating dose further until non-specifictoxicity is induced.
Similarly, in assessing the ‘activity’ of MMPIs,traditional objective measures of tumour response areprobably inappropriate. The definitive demonstration ofactivity for this class of agents is through increases insurvival or time to disease progression in randomizedtrials. This, however, requires large numbers of patientsand long periods of follow-up and trials such as theseconsume large resources. Such an investment is difficultto justify until there is some indication of relevantbiological activity. During the development of theMMPIs one of the challenges has been to demonstrate‘proof of principle’ in smaller numbers of patients priorto embarking on large, randomized trials.
Batimastat (BB94)
The first of these agents to enter clinical developmentwas batimastat. It is a potent, broad spectrum MMPIstructurally related to collagen. Preclinical data were
very encouraging, with activity in a number of xenograftmodels including breast cancer. Sledge et al. (1995)showed that batimastat could significantly reduce thelocal-regional regrowth of resected human MDA-MB-435 tumours in nude mice.36 The incidence, number andtotal volume of lung metastases were also significantlylower in the treated mice. Batimastat also significantlyinhibited the growth of the MDA435/LCC6 humanbreast cancer line implanted subcutaneously.37 Eccleset al. (1996) used a rat mammary carcinoma model toexamine the effect of different schedules of batimastat onmetastasis and secondary tumour growth rates. Animalswere treated with short (7 days) or long (58 days)courses of batimastat, starting 2 days prior to surgicalremoval of the primary tumour, grown adjacent to themammary fat pad. Both long and short courses oftreatment reduced the frequency of lung metastases.However, the frequency of lymph node metastases wassignificantly lower in the animals treated for longer.38
These findings have important implications for MMPIsin the clinic, suggesting that prolonged treatment at atime of minimal disease is likely to be optimal.
Low oral bioavailability required that batimastat begiven into the peritoneal or pleural spaces. Althoughseveral Phase I studies demonstrated that potentially

Matrix metalloproteinase inhibitors 373
therapeutic systemic levels of batimastat could beachieved in this manner, problems with pain at the siteof administration halted further clinical developmentand British Biotech then subsequently focused theirattention on the oral compound, marimastat.
Marimastat (BB-2516)
Marimastat is also a broad spectrum MMPI that wasactive in preclinical studies, reducing both tumourgrowth and metastasis.39,40 In contrast to conventionalanticancer agents, initial human studies with marimastatwere conducted in healthy volunteers. These confirmedthat marimastat was almost completely absorbed afteroral administration with a high and predictable bio-availability, and a half life of 8–10 h that justified twicedaily dosing in later clinical trials.41
Subsequently, a dose-escalation Phase I trial set out toidentify the MTD and toxicities of oral marimastat inpatients with advanced lung cancer.42 Pharmacokineticswere measured, as were plasma levels of MMP2 andMMP9. Earlier studies had suggested that plasma MMPlevels may be elevated in patients with breast and coloncancer43 and it was hoped that changes in MMP levelsmight provide a marker of a biologically effective dose.Patients were treated with between 25 and 100mg ofmarimastat given twice daily (BID). The lower doseswere well tolerated over an 8-week period but 100mgtwice daily was associated with a characteristic inflam-matory polyarthritis that appeared within 3 weeks ofstarting treatment and increased in severity if the patientcontinued marimastat. This was consistent with animalstudies, which showed fibrosis and inflammation aroundweight-bearing joints. Marimastat levels were severalfold higher at the lowest dose level of 25mg/m2 thanthose required to achieve MMP inhibition in vitro. Theexception was MMP3 where peak levels remained belowthe IC50 at all doses. Unfortunately, no consistentchanges were seen in the plasma levels or activity ofMMP2 and 9 so this could not be used as a surrogatemarker of efficacy.
A series of Phase I/II trials were then undertakenin an attempt to identify a safe and effective doseof marimastat. These studies were built around thehypothesis that tumour markers may serve as asurrogate end-point for activity in identifying theoptimal dose of marimastat. Six non-randomized trialswere undertaken in patients with colorectal, ovarian,prostate and pancreatic cancers, tumours that are oftenassociated with elevated tumour markers. The keyeligibility criterion for these studies was that patientsshould have a 25% or greater rise in marker levelsduring the 4 weeks preceding study entry. Patients then
received marimastat at a range of different doses over a4-week period initially, at which point tumour markerswere measured again. It was hoped that an alteration inthe rate of increase of tumour markers might act as anindicator of activity of marimastat. A ‘biological effect’was defined as a fall, or no rise, in markers during thetreatment period; ‘partial biological effect’ was definedas a rise of less than 25%.
In all, 415 patients were recruited and treated at 11different dose levels of marimastat, starting at 25mgBID with initial escalation to 75mg BID which provedintolerable.44 The dose was then de-escalated as low as2mg daily in some patient cohorts. The key finding wasthat the rate of rise of tumour markers appeared slowerwhen patients were receiving marimastat than it hadbeen during the preceding 4 weeks. When the dose levelswere compared, the effect on tumour makers appearedgreater at the high dose levels. There are, however,difficulties in interpreting this trial. Firstly, fullyassessable patients represented only half of thoseinitially recruited to the study. Secondly, the hetero-geneity of the patient populations and wide range ofdose levels used are not ideal. Finally, the use of tumourmarkers as a surrogate end-point in this setting isunproven. Although carefully defined changes in mar-kers can be used as an alternative to alterations intumour dimensions in assessing patients with ovariancancer,45 neither the use of tumour markers in thissetting, nor the scheme of a 4-week screening periodfollowed by 4 weeks of observation on treatment, hasbeen validated. Indeed, the natural history of tumourmarkers in this situation is unclear. Gore et al.46
highlighted the slowing down in rate of increase intumour markers that may be seen in patients who arenot on any active treatment. Moreover, it is possiblethat any fall in tumour markers may be the result ofchanges in the shedding of tumour markers rather thanalterations in tumour proliferation.
On the basis of these data, marimastat given at dosesof between 10 and 25 mg twice daily were identified asappropriate for further randomized studies. The firsttrial to be completed compared three doses of marima-stat alone with gemcitabine in 400 patients withadvanced pancreatic cancer.47,48 The primary end-pointwas survival and the trial was designed to detect at leasta 16% reduction in mortality of patients receiving 10mgor 25mg twice daily marimastat. The study did not meetthis primary end-point with no significant difference insurvival between patients treated with gemcitabine orthe three doses of marimastat. However, a secondaryanalysis using Cox’s Proportional Hazards model gavemore encouraging results. In this analysis both the25mg marimastat dose level and gemcitabine appeared

374 The Breast
superior to either the 10mg and 5mg groups takentogether. This suggests that marimistat 25mg/day maybe equivalent to gemcitabine and superior to lower dosesof marimastat.
The results of a Phase III, placebo controlled, study ofmarimastat in patients with inoperable gastric cancerwere recently presented at the American Society ofClinical Oncology meeting in New Orleans. Onehundred and eighty five patients were randomized toreceive 10mg BID of marimastat for up to 18 monthswith 184 receiving placebo. The data were analysed atthe pre-defined clinical cut off when 85% mortality wasfirst recorded in one of the arms. The primary end-pointwas survival and at the pre-defined clinical cut off,median survival was 167 days for marimastat and 135days in the placebo arm. This difference was notstatistically significant (P=0.07) although there was astatistically significant benefit in progression free survi-val (P=0.015). With longer follow-up the difference insurvival in favour of marimastat just reached signifi-cance (P=0.046). Subset analysis suggested that pa-tients without distant metastases and those who hadreceived prior chemotherapy may benefit from marima-stat. As expected, musculoskeletal side-effects were morecommon in the marimastat patients, 18 of whomstopped treatment because of toxicity compared withtwo of those randomized to placebo.
These marimastat trials are intriguing but difficult tointerpret. In each case the primary analysis was negativebut additional analyses suggested that as a single agentmarimastat may have significant activity. Neverthelessthe negative intention-to-treat analysis must be viewedas the definitive result, especially since there are dangersin making multiple analyses of subgroups. Theseadditional analyses did generate apparently significantresults but should only be used to generate newhypotheses to be tested definitively in prospective trials.In women with metastatic breast cancer who haveresponding or stable disease after initial chemotherapy,a Phase III trial is underway in which patients arerandomized to marimastat or placebo. It is likely,however, that cytostatic agents such as the MMPIs willultimately be given either in combination with che-motherapy or to treat patients with minimal disease. Ina Phase I trial that is relevant to breast cancer givingmarimastat (10mg BID) with doxorubicin and cyclo-phosphamide was feasible and well tolerated.49
AG3340 (Prinomastat)
Like marimastat, AG3340 is a hydroxamic acidderivative but it was designed from a knowledge of theX-ray crystal structure of human MMPIs. AG3340
differs from marimastat in being a selective inhibitor ofMMP2, MMP9, MMP13 and MMP3 with little activityagainst MMP1. Although patterns of expression varybetween differing tumour types, MMP2, MMP9,MMP13 and MMP3 have consistently been associatedwith tumour progression. By contrast, inhibition ofMMP1 is thought to contribute to joint toxicity.AG3340 is a potent inhibitor of tumour growth in vivoagainst several human tumour xenografts including theMDA-MB-435 breast cancer model.50 There was alsosynergistic activity between AG3340 and both paclitaxeland carboplatin in gastric and non-small cell lung cancerlines, respectively.51 Pharmacodynamic studies in nudemice bearing human colon tumours showed thatantitumour efficacy was associated with maintenanceof minimum effective plasma concentrations of AG3340and not with total daily dose, exposure or peak plasmaconcentrations.52
Like marimastat, AG3340 also causes time and dose-dependent musculoskeletal pain. Doses below 25mgBID were well tolerated with few joint complaints andmedian plasma concentrations were achieved thatremained above the Ki of MMP-152 and were equivalentto those that produced maximal tumour inhibition inmice. In the current Phase III trials patients withunresectable non-small cell lung cancer or metastatichormone refractory prostate cancer are receiving stan-dard first-line chemotherapy with or without AG3340.Two doses of AG3340 are being given to address thequestion of whether therapeutic benefit can be achievedwithout toxicity. The lower dose of AG3340 achievesplasma trough levels above the Ki values for theinhibition of MMP-2 but should not cause musculoske-letal toxicity; the higher dose would be expected toinhibit a wider range of MMPs with a moderately highincidence of joint effects.53
CGS27023A (MMI 270B)
CGS27023A is a broad spectrum MMPI, also said toshow additive or synergistic cytotoxicity with cisplatinin preclinical models. A single agent, Phase I doseescalation study reported a widespread but self-limitingmaculopapular rash and mild to moderate joint pains.54
At doses of 300mg bd and below, CGS27023A was welltolerated and rapidly absorbed with plasma levels morethan 10 fold greater than the IC50 for the target MMPs.There was no evidence of additive toxicity or apharmacokinetic interaction when CGS27023A wasgiven in combination with 5-fluouracil/folinic acid;55
once again, arthritis was seen at the highest dose level. Atrial of carboplatin and paclitaxel in combination withCGS27023A has also been undertaken. The therapeutic

Matrix metalloproteinase inhibitors 375
potential of CGS27023A remains unclear as no PhaseIII studies are underway at present.
BAY12-9566 (Tanomastat)
Bay 12-9566 is structurally different from other MMPIs,being a non-peptide butanoic acid analogue. It isanother potent and specific inhibitor of MMP2,MMP9 and MMP3 with little activity against collage-nase I. Acitivity has been reported preclinically in theB16 melanoma and Lewis lung carcinoma tumourmodels, with inhibition of primary growth, number ofmetastases and secondary growth.56 Similar data havealso been described for the human colon carcinomaHCT116 in nude mice.57
Several dose-finding trials with BAY12-9566 havebeen reported and reviewed by Lathia et al.58 In all, 90patients received between 100 and 1600mg BAY12-9655. In contrast to other MMPIs, there was a strikinglack of musculoskeletal toxicity, although mild tomoderate thrombocytopenia and changes in liverbiochemistry tests were reported. BAY12-9566 wasbeing evaluated through Phase II and III trials in smallcell and non-small cell lung cancer, ovarian andpancreatic cancer. However, in September 1999 anIndependent Data Safety Monitoring Board recom-mended that the small cell lung cancer trial be stoppedas BAY12-9566 was performing worse than placebo. Onthe basis of this Bayer has halted all other trials withBAY12-9566. Not surprisingly, this news was describedby their Head of Worldwide Drug Development as‘disappointing’.59
DISCUSSION
The development of the MMPIs has proved at least aschallenging as that of a new cytotoxic. Like many newtherapies, MMPIs appear to have gone through a cycleof initial overoptimism followed by rebound pessimismbefore hopefully finding their correct place. Preclinicalresults with MMPIs were exciting but the controversysurrounding initial clinical trials with batimastat andmarimastat spilled over into the media. Bayer thenhalted development of BAY12-9566 and Novartis havesuspended work on CGS27023A. Recent results are alittle more encouraging, with marimistat potentiallyhaving significant activity in gastric cancer. Howeverthis study incorporated six different survival analysesand the primary end-point did not show benefit formarimastat. Certainly marimastat cannot be consideredstandard therapy in routine treatment. Nor should it bethe control arm in future trials. Rather these additional
analyses have pointed the way for future trials. ThusAG3340 and marimastat remain in clinical develop-ment, with other MMPIs at earlier stages of develop-ment, so this group of agents is still under investigationalthough the future of AG3340 is uncertain.
One clear lesson is that classical drug developmentparadigms are impractical for biological modifiers.Rather than escalating doses to toxicity in patients withadvanced disease, the aim of early MMPI trials shouldbe to demonstrate the safety of a biologically activedose. This requires either a reliable surrogate marker ofactivity or identification of a ‘target’ dose derived bymodelling preclinical pharmacokinetic and pharmaco-dynamic data. The lack of cytotoxic side-effects foragents such as the MMPIs could be turned to advantageby doing Phase I trials earlier in the history of a patient’sdisease so that chronic toxicities are more likely to berecognized. Having demonstrated safety, classical re-sponse criteria are not appropriate in defining theactivity of MMPIs. Unless surrogate markers forbiological activity can be validated, we may need toadopt new trial designs. For example it has beenproposed that a novel agent be considered ‘active’ if itdelays disease progression for substantially longer thanthe previous standard treatment. Alternatively, Phase IItrials may be omitted altogether in favour of Phase IIItrials incorporating rules for early stopping in the eventof unexpected adverse effects.
With the benefit of hindsight, the marimastat Phase IIprogramme added little and it is the Phase III trials thatwill determine its fate. The current Phase III trials ofMMPIs have time to progression and survival as theirend-points but their designs differ. The most ambitiousare a straightforward comparison of the MMPI andplacebo as initial treatment. More often the samerandomization is made at the point when patients haveresponded to initial chemotherapy. Other studies look atthe addition of an MMPI to standard chemotherapy.In the Agouron study of non-small-cell lung cancerprinomastat is administered with chemotherapy but canbe continued after progression on chemotherapy. Theoptimal trial design for these agents is unclear and islikely to differ according to the tumour type and stage aswell as the effectiveness of current standard treatments.
The current trials have left important questions suchas the timing and duration of MMPI treatment still tobe answered. Efficacy is greatest in animal studies whentreatment is started early in the history of metastases.Some Phase III trials have looked at the use of MMPIsas ‘consolidation’ treatment in patients who haveresponded to standard treatment. Arguably, these trialswould best be conducted in the adjuvant setting but thisrequires prolonged follow-up and large numbers of

376 The Breast
patients. The optimal duration of therapy is also unclearand difficult to define if not dictated by cumulativetoxicity. Since MMPs are involved at so many stages oftumour evolution, should treatment continue indefi-nitely? This is a potentially expensive option andcontrasts with our current practice of stopping cytotoxicor endocrine therapy once disease progresses. Resistanceto MMPIs may not develop in the same way as it does tocytotoxics, and continued MMPI treatment might slowthe rate of progression or limit damage to surroundingnormal tissue.
Should MMPIs be given with, rather than following,conventional therapy? If so, are there advantages incombining MMPIs with specific chemotherapy regi-mens? AG3340 is synergistic with paclitaxel in pre-clinical models60 and this may be especially relevant forstudies in breast cancer. Options for combinationtreatment are not limited to cytotoxics. The bispho-sphonate ibandronate inhibits the formation of bonemetastases by MDA-231 cells in nude mice. A similareffect can be achieved by transfecting MDA-231 withTIMP-2, but there is marked synergism when ibandro-nate treatment is combined with TIMP-2 transfection.61
Finally, we do not know whether selective inhibitorsprovide any advantages over broad spectrum inhibitors,or whether certain MMPs and their inhibitors are ofgreater importance in different cancers. There is no testto identify patients most likely to benefit from MMPIsso that they can be targeted more effectively. Althoughthe MMPIs lack the classical toxicities of chemotherapy,they do have side-effects. Marimastat may prove tootoxic to be administered at effective doses for prolongedperiods of time. If the MMPIs do have modest butsignificant effects on disease progression, better toler-ated compounds will need to be identified.
CONCLUSIONS
The MMPIs are an exciting class of drugs with strikingactivity in preclinical models. They can be administeredwith tolerable side-effects, at doses that produce plasmalevels above those required for inhibition of MMPsin vitro and in animal models. However, their develop-ment demonstrates, the problems of extrapolating fromencouraging results in animal models, which arebiologically artificial, to an expectation of clinicalbenefit. The potential cross talk and redundancy withinsignal transduction pathways may be too great to makeinhibition of a single target effective in vivo. Phase IIItrials have not yet convincingly demonstrated the benefitof MMPIs in patients with advanced cancer. They have
not, however, been adequately tested in patients withlow volume disease so their future remains unclear.
There is good evidence that the MMPs play animportant role in the progression of breast cancer. Theconcept of novel, non-cytotoxic treatment with MMPIsshould appeal to clinicians treating women with breastcancer familiar with endocrine therapy and morerecently Herceptin. Similarly, targeting the stroma isalso a familiar concept in the management of breastcancer where the use of bisphosphonates is widespread.Likewise, the role of adjuvant therapy is best establishedin breast cancer and preclinical data suggest thatMMPIs may be most effective given early in this setting.Despite the difficulties encountered in their develop-ment, hopes remain that MMPIs may yet provideanother much needed mechanism through which thelong-term control of macrometastatic disease can beimproved.
References
1. Gross J, Lapiere C M. Collagenolytic activity in amphibian tissues:a tissue culture assay. Proc Natl Acad Sci USA 1962; 48:1014–1022.
2. Sato H, Takino T, Okada Y, et al. A matrix metalloproteinaseexpressed on the surface of invasive tumour cells. Nature 1994;370: 61–65.
3. Brown P D, Bloxidge R E, Anderson J E, Howell A. Expression ofactivated gelatinase in human invasive breast cancer. Clin ExplMetastasis 1993; 11: 183–189.
4. Basset P, Bellocq J P, Wolf C, et al. A novel metalloproteinasegene specifically expressed in stromal cells of breast carcinomas.Nature 1990; 348: 699–704.
5. Heppner K J, Matrisian L M, Jensen R A, Rodgers W H.Expression of most matrix metalloproteinase family members inbreast cancer represents a tumour-induced host response. Am JPathol 1996; 149: 273–282.
6. Chenard M P, OSiorian L, Shering S, et al. Stromelysin 3association with prognosis in breast cancer. Int J Cancer 1996; 69:448–451.
7. Uria J A, Stahle-Backdahl M, Seiki M, Fueyo A, Lopez-Otein C.Regulation of collagenase-3 expression in human breastcarcinomas is mediated by stromal-epithelial cell interactions.Cancer Res 1997; 57: 4882–4888.
8. Polette M, Nawrocki B, Billes C, et al. MT-MMP expression andlocalization in human lung and breast cancers. Virchows Archiv1996; 428: 29–35.
9. Davies B, Miles D W, Happerfield L, et al. Activity of type IVcollagenases in benign and malignant breast disease. Br J Cancer1993; 67: 1126–1131.
10. Ueno H, Nakamura H, Inoue M, et al. Expression and tissuelocalization of membrane-types 1, 2 and 3 matrixmetalloproteinases in human invasive breast carcinomas. CancerRes 1997; 57: 2055–2060.
11. Rha S Y, Kim J H, Roh J K, et al. Sequential production andactivation of matrix-metalloproteinase-9 (MMP-9) with breastcancer progression. Breast Cancer Res Treat 1997; 43: 175–181.
12. Dolo V, Ginestra A, Cassara D, et al. Selective localization ofmatrix metalloproteinase 9, beta integrins, and human lymphocyteantigen class I molecules on membrane vesicles shed by 8701-BCbreast carcinoma cells. Cancer Res 1998; 58: 4486–4474.

Matrix metalloproteinase inhibitors 377
13. Daidone M G, Silvestrini R, D’Errico, AD, et al. Lamininreceptors, collagenase IV and prognosis in node-negative breastcancers. Int J Cancer 1991; 48: 529–532.
14. Talvensaari-Mattila A, Paakko P, Hoyhtya M, et al. Matrixmetalloproteinase-2 immunoreactive protein - a marker ofaggresiveness in breast carcinoma. Cancer 1998; 83: 1153–1162.
15. Ahmad A, Hanby A, Dublin E, et al. Stromelysin 3: anindependent prognostic factor for relapse-free survival in node-positive breast cancer and demonstration of novel breastcarcinoma cell expression. Am J Pathol 1998; 152(3): 721–728.
16. Azzam H S, Arand G, Lippman M E, Thompson E W.Association of MMP-2 activation potential with metastaticprogression in human breast cancer cell lines independent ofMMP-2 production. J Natl Cancer Inst 1993; 85: 1758–1764.
17. Ree A H, Florenes V A, Berg J P, Malandsmo G M, Nesland J M,Fodstad O. High levels of messenger RNAs for tissue inhibitor ofmatrix metalloproteinase (TIMP-1 and TIMP-2) in primary breastcarcinomas are associated with development of distant metastases.Clin Cancer Res 1997; 3: 1623–1628.
18. Visscher D W, Hoyhtya M, Ottosen S K, et al. Enhancedexpression of tissue inhibitor of metalloproteinase-2 (TIMP-2) inthe stroma of breast carcinomas correlates with tumourrecurrence. Int J Cancer; 1994; 59: 339–344.
19. Yoshiji H, Gomez D E, Thorgeirsson U P. Enhanced RNAexpression of tissue inhibior of metalloproteinases-1 (TIMP-1) inhuman breast cancer. Int J Cancer 1996; 69: 131–134.
20. McCarthy K, Maguire T, McGreal G, McDermott E,O’Higgins N, Duffy M J. High levels of tissue inhibitor ofmetalloproteinase-1 predict poor outcome in patients with breastcancer. Int J Cancer 1999; 84: 44–48.
21. Holten-Andersen M N, Murphy G, Nielsen H J, et al.Quantitation of TIMP-1 in plasma of healthy blood donors andpatients with advanced cancer. Br J Cancer 1999; 80: 494–503.
22. Luparello C, Avanzato G, Carella C, Pucci-Minafra I. Tissueinhibitor of metalloprotease (TIMP)-1 and proliferative behaviourof clonal breast cancer cells. Breast Cancer Res Treat 1999; 54:235–244.
23. Wang M, Liu Y E, Green J, et al. Inhibition of tumor growth andmetastasis of human breast cancer cells transfected with tissueinhibitor of metalloproteinase 4. Oncogene 1997; 14: 2767–2774.
24. Noel A C, Polette M, Lewalle J M, et al. Coordinate enhancementof gelatinase A mRNA and activity levels in human fibroblasts inresponse to breast adenocarcinoma cells. Int J Cancer 1994; 56:331–336.
25. Chambers A, Matrisian L. Changing views of the role of matrixmetalloproteinases in metastasis. J Natl Cancer Inst 1997; 89(17):p 1260–1270.
26. Kurizaki T, Toi M, Tominaga T. Relationship between matrixmetalloproteinase expression and tumor angiogenesis in humanbreast carcinoma. Oncol Rep 1998; 5: 673–677.
27. Fisher C, Gilbertson-Beadling S, Powers E A, et al. Interstitialcollagenase is required for angiogenesis in vitro. Dev Biol 1994;162: 499–510.
28. Taraboleti G, Garofalo A, Belotti D, et al. Inhibition ofangiogenesis and murine haemangioma growth by batimastat, asynthetic inhibitor of matrix metalloproteinases. J Natl CancerInst 1997; 87: 293–298.
29. Manthey C, Zhou Z, Haslow K, et al. Inhibition of endothelial cellsprout formation and extracellular matrix invasion by a smallmolecule MMP inhibitor. Clin Cancer Res 1999; 5 (SS): 51.
30. Thorgeirsson U P, Yoshiji H, Sinha C C, Gomez D E. Breastcancer; tumor neovasculature and the effect of tissue inhibitor ofmetalloproteinases-1 (TIMP-1) on angiogenesis. In Vivo 1996; 10:137–144.
31. Ray J, Stetler-Stevenson W. Gelatinase A activity directlymodulates melanoma cell adhesion and spreading. EMBO J 1995;14: 908–917.
32. Masson R, Lefebvre O, Noel A, et al. In Vivo evidence that thestromelysin-3 metalloproteinase contributes in a paracrine mannerto epithelial cell malignancy. J Cell Biol 1998; 140(6): 1535–1541.
33. Rudolph-Owen L A, Chan R, Muller W J, Matrisian L M. Thematrix metalloproteinase matrilysin influences early-stagemammary tumorigenesis. Cancer Res 1998; 58: 5500–5506.
34. Sternlicht M D, Lochter A, Sympson C J, et al. The stromalproteinase MMP3/Stromelysin-1 promotes mammarycarcinogenesis. Cell 1999; 98: 137–146.
35. Brown P D. Matrix metalloproteinase inhibitors. Breast CancerRes Treat 1998; 52: 125–136.
36. Sledge G, Qulali M, Goulet R Bone E, Fifes R. Effect of matrixmetalloproteinase inhibitor batimastat on breast cancer regrowthand metastasis in athymic mice. J Natl Cancer Res 1995; 87:1546–1550.
37. Low J A, Johnson M D, Bone E A, Dickson R B. The matrixmetalloproteinase inhibitor batimastat (BB-94) retards humanbreast cancer solid tumor growth but not ascites formation in nudemice. Clin Cancer Res 1996; 2: 1207–1214.
38. Eccles S A, Box G M, Court W J, Bone E A, Thomas W,Brown P D. Control of lymphatic and haematogenous metastasesof a rat mammary carcinoma by the matrix metalloproteinasebatimastat (BB-94). Cancer Res 1996; 56: 2815–2822.
39. Beckett R P, Davidson A H, Drummond A H, Huxley P,Whittaker M. Recent advances in matrix metalloproteinaseinhibitor research. Drug Discovery Today 1996; 1: 16–26.
40. Drummond A H, Beckett P, Bone E A, et al. BB-2516, an orallybioavailable metalloproteinase inhibitor with effcacy in animalcancer models. Proc Am Assoc Cancer Res 1995; 36: 100 (abstr595).
41. Millar A, Brown R, Moore J, et al. Results of single and repeatdose studies of the oral metalloproteinase inhibitor marimastat inhealthy male volunteers. Br J Clin Pharm 1998; 45: 21–26.
42. Wajtowicz-Praga S, Torri J Johnson M, et al. Phse I ofmarimastat, a novel matrix metalloproteinase inhibitor,administered orally to patients with advanced lung cancer. J ClinOncol 1998; 16: 2150–2156.
43. Zucker S, Lysik R, Zarragi H, et al. Plasma assay ofmetalloproteinases (MMPs) and MMP-inhibitor complexes incancer. Potential use in predicting metastasis and monitoringtreatment. Ann NY Acad Sci 1994; 732: 248–262.
44. Nemunaitis J, Poole C, Primrose J, et al. Combined analysis ofstudies of the effects of the metalloproteinase inhibitor marimastaton serum tumour markers in advanced cancer: selection of abiologically active and tolerable dose for longer term studies.Clin Cancer Res 1998; 4: 1101–1109.
45. Rustin G J, Nelstrop A E, Bentzen S M, Bond S J, McClean P.Selection of active drugs for ovarian cancer based on CA-125 andstandard response rates in Phase II trials. J Clin Oncol 2000; 18(8):1733–1739.
46. Gore M, A Hern R, Stankiewicz M, Slevin M. Tumour markerlevels during marimastat therapy. Lancet 1996; 348: 2631.
47. Rosemrgy A, Buckels J, Charnley R. A randomised studycomparing marimastat to gemcitabine as first line therapy inpatients with non-resectable pancreatic cancer. Proc Am Soc ClinOncol 1999; 18: 261a (abstr 1005).
48. British Biotech plc. Results of marimastat study 128: Pancreaticcancer monotherapy trial. Realease date, 2/5/99.Http:www.britbio.co.uk/news/128.txt
49. Gradishar W, Sparano J, Cobleigh M, et al. A Phase I study ofmarimastat in combination with Doxorubicin andCyclophosphamide in patients with metastatic breast cancer. ProcAm Soc Clin Oncol 1998; 17: 144a (abstr 548).
50. Shalinsky D R, Brekken J, Sou H, et al. Broad antitumor andantiangiogenic activities of AG3340, a potent and selective MMPinhibitor undergoing advanced oncology clinical trials. Ann N YAcad Sci 1999; 878: 236–270.
51. Shalinsky D R, Brekken J, Zou H, et al. Marked antiangiogenicand antitumor efficacy of AG3340 in chemoresistant human non-small cell lung cancer tumours: Single ages and combinationchemotherapy studies. Clin Cancer Res 1999; 5(7): 1905–1917.
52. Pithavala Y, Shalinsky D, Wilding G, Hande K, Dixon M, CollierM. Comparison of preclinical efficacy and associated plasma

378 The Breast
concentration of AG3340, a matrix metalloprotease (MMP)inhibitor, with plasma concentrations achieved clinically. Proc AmSoc Clin Oncol 1999; 18: 223a (abst 860).
53. Collier M, Shepherd F, Ahmann F, et al, and the Lung andProstate Cancer Study Groups. A novel approach to studying theefficacy of AG3340. A selective inhibitor of matrixmetalloproteases (MMPs). Proc Am Soc Clin Oncol 1999; 18: 482a(abstr 18l61).
54. Levitt N, Eskens F, Propper D, et al. A phase Onepharmacokinetic study of CGS27023A, a matrix metalloproteinaseinhibitor. Proc Am Soc Clin Oncol 1998; 17: 213a (abstr 823).
55. Eatock M, Cassidy J, Johnson J, et al. A Phase 1 study of thematrix metalloproteinase inhibitor MMI270 (previously termedCGS270523A) with FU and folinic acid. Proc Am Soc Clin Oncol1999; 18: 209a (abstr 803).
56. Hibner B, Bull C, Flynn C, et al. Activity of the matrixmetalloproteinase inhibitor BAY 12-9566 against murinesubcutaneous and metastatic in vivo models. Ann Oncol 1998; 9(S2): 283.
57. Flynn C, Bull C, Matherne C, Eberwein D, Gibson N, Hibner B.Anti-invasive and anti-metastatic activity of the novel MMPInhibitor BAY 12-9566 in subcutaneous and orthotopic models usingthe human colon carcinoma, HCT 116. Ann Oncol 1998; 9(S2): 284.
58. Lathia C, Seymour L, Grochow L, et al. BAY 12-9566, a selective,non-peptidic biphenyl inhibitor of matrix metalloproteases(MMPs): summary of phase I clinical and pharmacokinetic (PK)results. Clin Cancer Res 1999; 5 (SS): 9.
59. Bayer Corporation. Bayer Halts Clinical Trials Evaluating MMPI.Release date, 24/09/99. Http:www.bayerus.com/news/1999/09.99ud.html
60. Shalinsky D R, Brekken J, Zou H, et al. marked antiangiogenicand antitumor efficacy of AG3340 in chemoresistant humannon-small cell lung cancer tumours: single agent and combinationchemotherapy studies. Clin Cancer Res 1999; 5(7): 1905–1917.
61. Yoneda T, Sasaki A, Dunstan C, et al. Inhibition of osteolyticbone metastasis of breast cancer by combined treatment with thebisphosphonate ibandronate and tissue inhibitor of the matrixmetalloproteinase-2. J Clin Invest 1997; 99: 2509–2517.