malignant tumors of bones
TRANSCRIPT

Malignant
Tumors of bones
Done by :
عبدالجليل خالد محمد عبدالستار
• Supervised by :
Dr : Ghazi AL-Ariki
الجمهورية اليمنية وزارة التعليم العالي
جامعة تعز كلية الطب و العلوم الصحية
قسم العظام–طب بشري
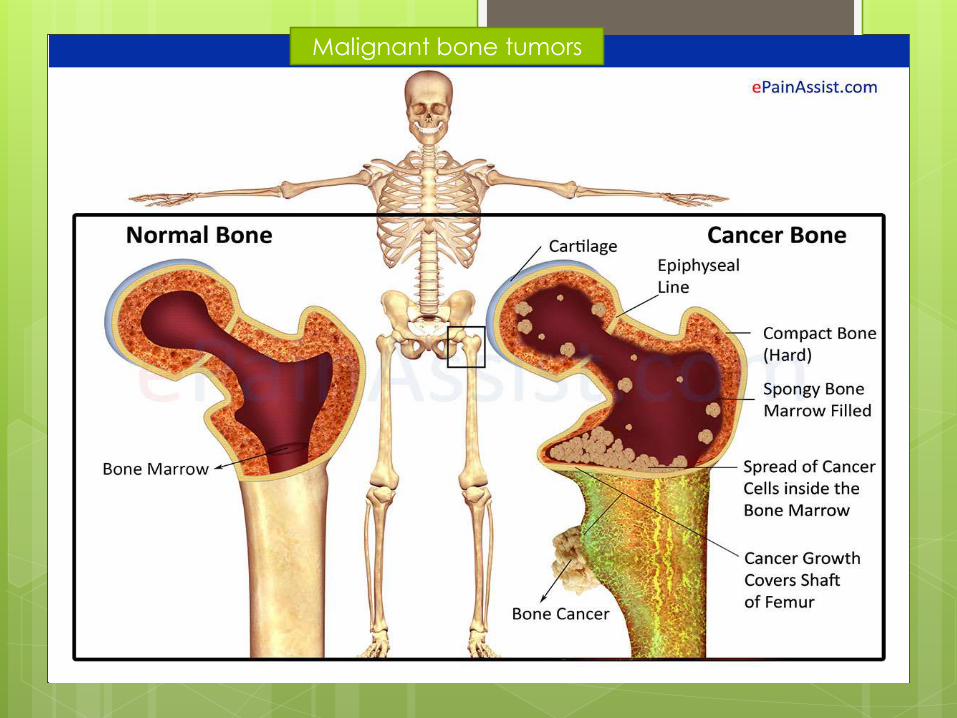
Malignant bone tumors

OUTLINE Introduction
What is a bone tumor ?
Epidemiology ( Incidence )
Causes & Risk factors .
Classification of Bone tumors
Diagnosis of bone tumors
Staging of Bone tumors
Malignant Bone Tumors
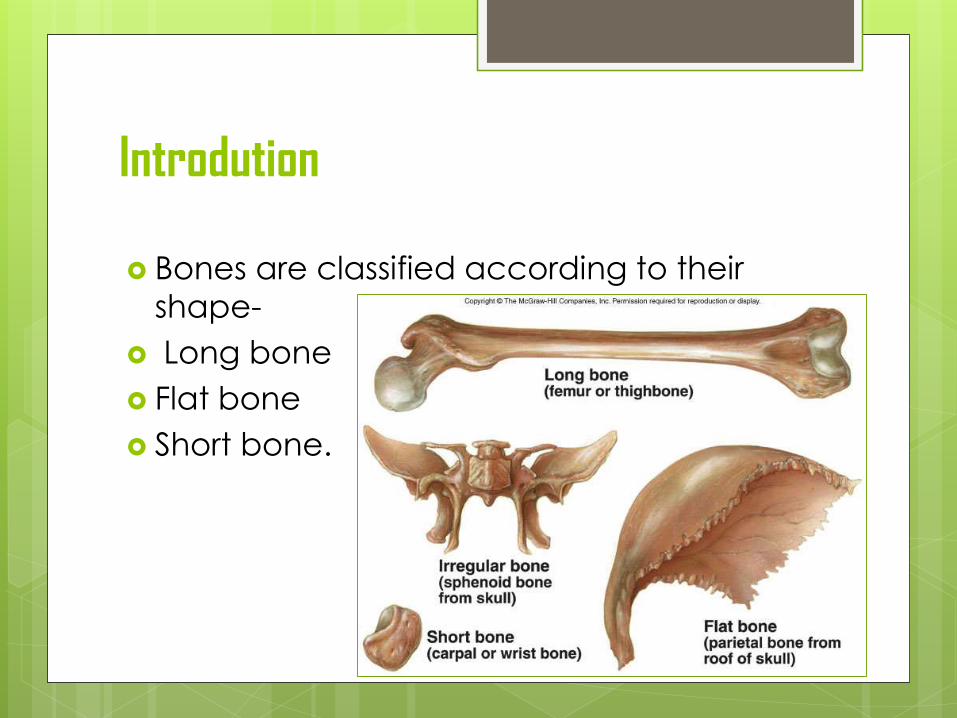
Introdution
Bones are classified according to their
shape-
Long bone
Flat bone
Short bone.

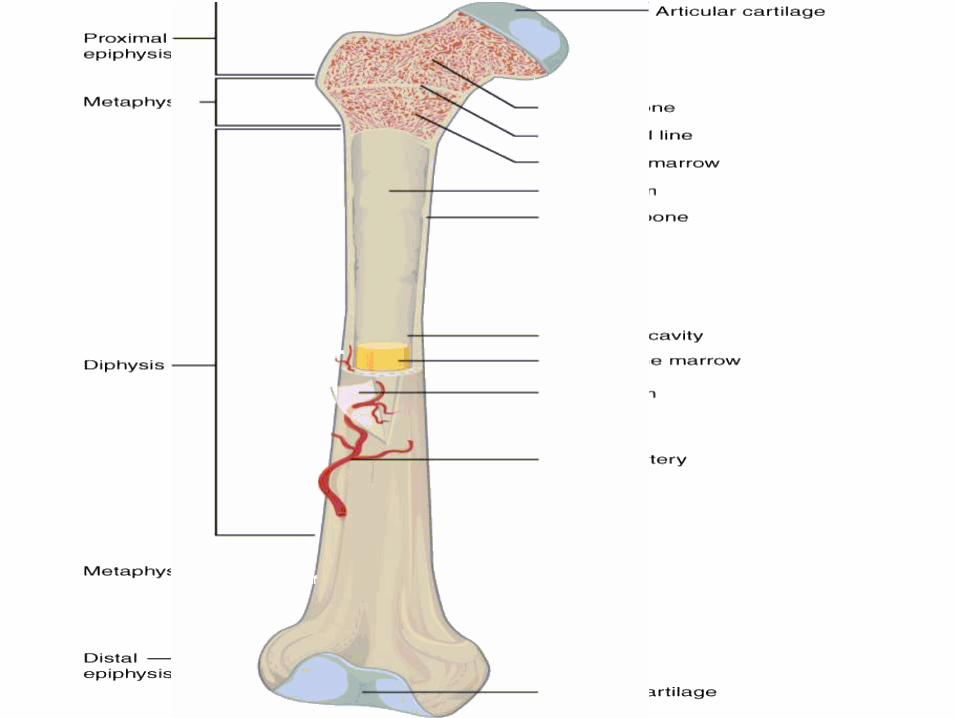
Long bone anatomy•Long bone consists of :
–Diaphysis: long shaft of bone–Epiphysis: ends of bone
–Epiphyseal plate: growth plate–Metaphysis: b/w epiphysis and diaphysis
–Articular cartilage: covers epiphysis–Periosteum: bone covering (pain sensitive)–Sharpey’s fibers: periosteum attaches to
underlying bone–Medullary cavity: Hollow chamber in bone
- red marrow produces blood cells- yellow marrow is adipose.
–Endosteum: thin layer lining the medullary cavity
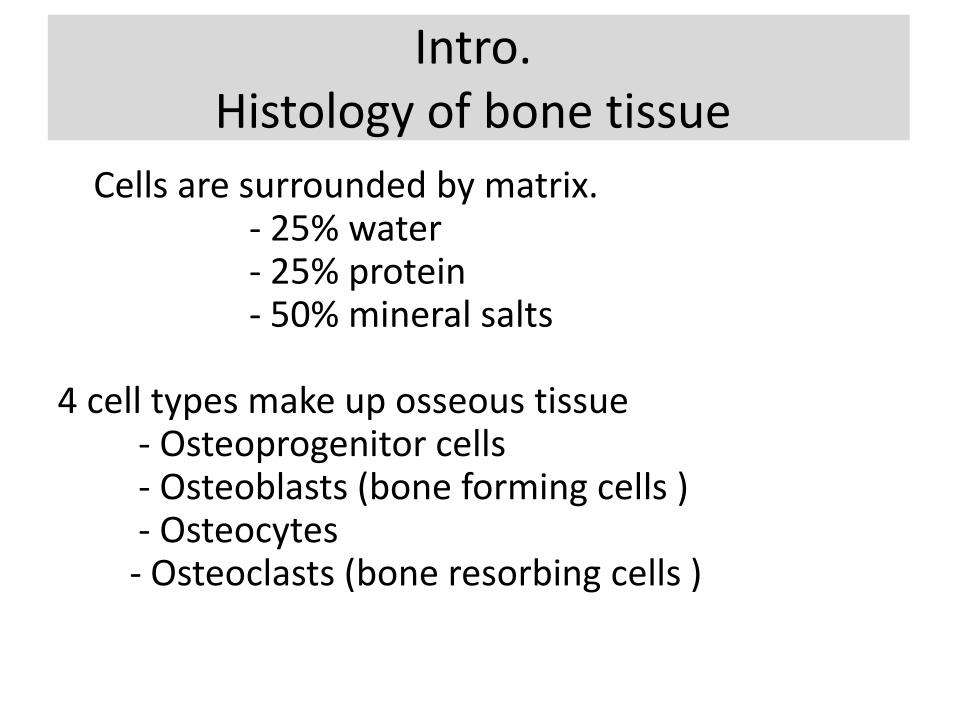
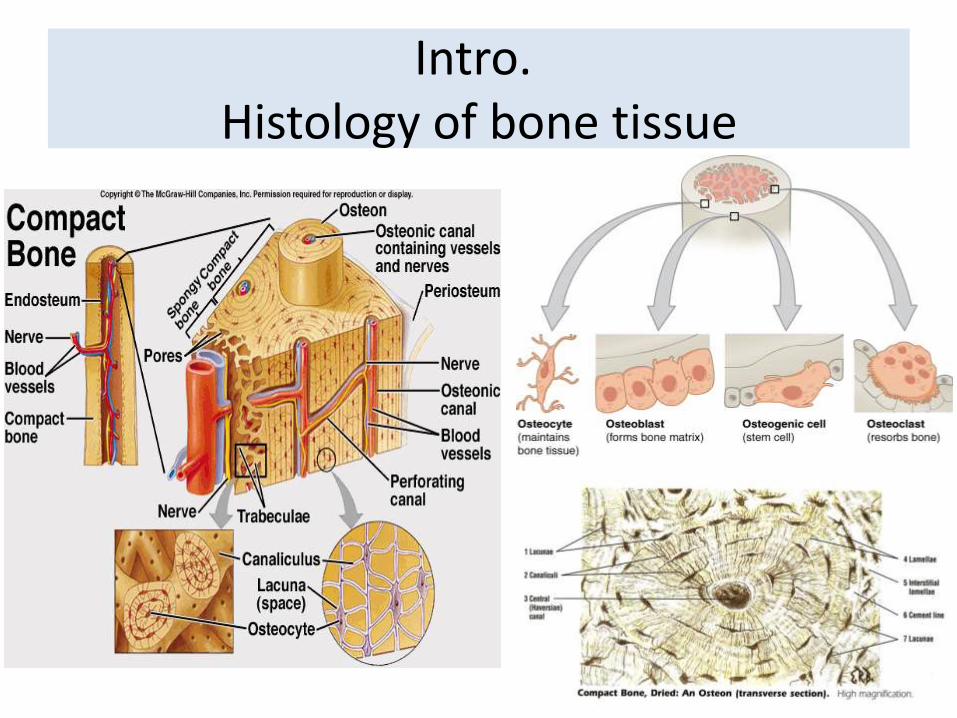
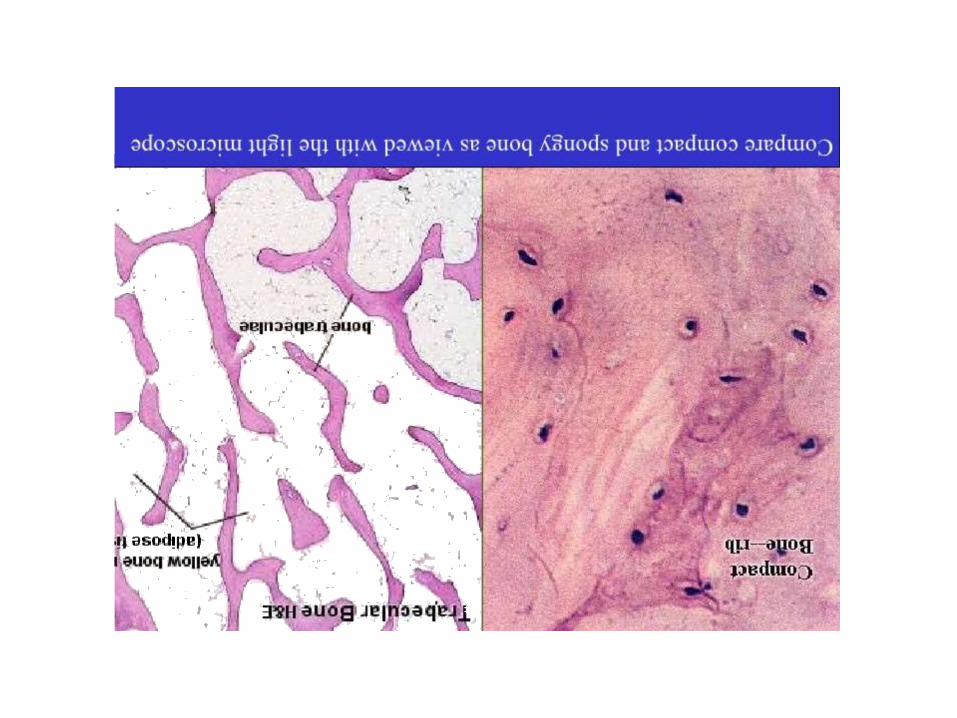
Intro. Histology of bone tissue
Cells are surrounded by matrix.- 25% water- 25% protein- 50% mineral salts
4 cell types make up osseous tissue- Osteoprogenitor cells- Osteoblasts (bone forming cells )- Osteocytes- Osteoclasts (bone resorbing cells )
Intro. Histology of bone tissue

Osteoprogeniter cells &Osteoblast
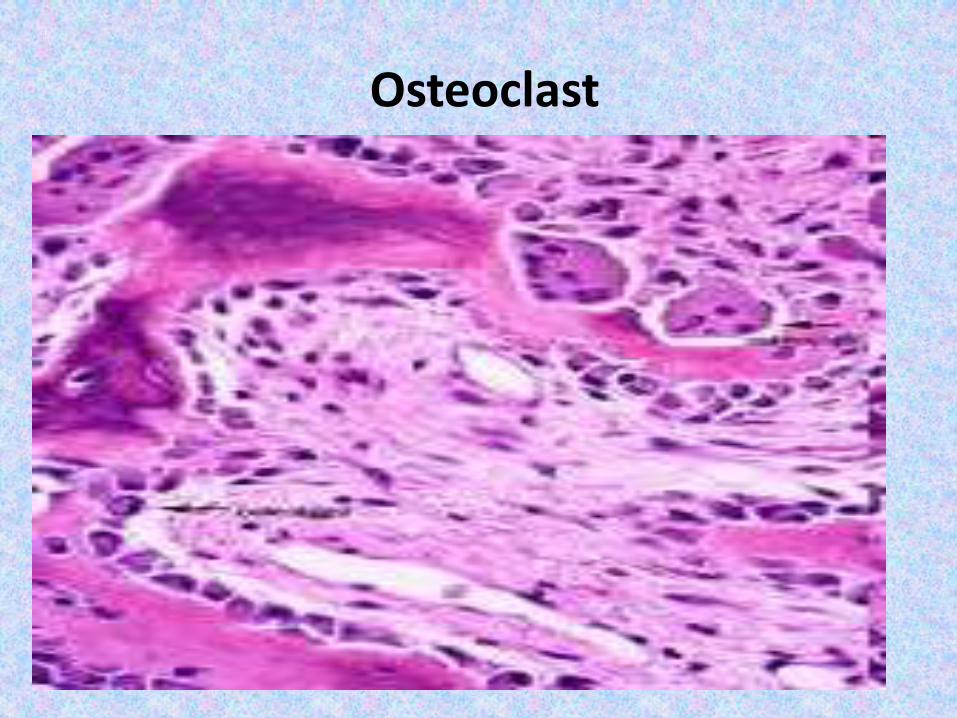
Osteoclast
Introduction A tumor : is a lump or mass of tissue that forms when cells
divide uncontrollably. A growing tumor may replace healthy
tissue with abnormal tissue .
A bone tumor, is a neoplastic growth of tissue in bone.
Abnormal growths found in the bone can be either benign
or malignant.
It may weaken the bone, causing it to break (fracture).
Most bone tumors are noncancerous (benign). Some are
cancerous (malignant).
Benign tumors are usually not life-threatening.
Malignant tumors can spread cancer cells throughout the body (metastasize). This happens via the blood or lymphatic
system
By far the commonest malignant lesions in bones
are metastatic tumors which are not strictly speaking
‘bone, tumors, i.e. not of mesenchymal origin .
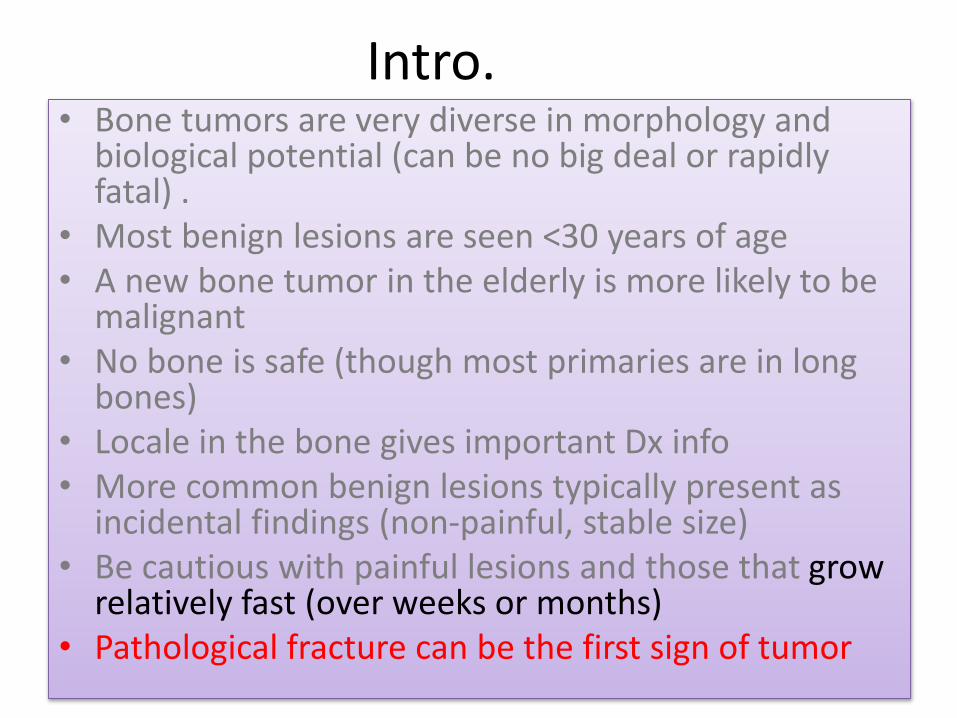
Intro.• Bone tumors are very diverse in morphology and
biological potential (can be no big deal or rapidly fatal) .
• Most benign lesions are seen <30 years of age• A new bone tumor in the elderly is more likely to be
malignant• No bone is safe (though most primaries are in long
bones)• Locale in the bone gives important Dx info• More common benign lesions typically present as
incidental findings (non-painful, stable size)• Be cautious with painful lesions and those that grow
relatively fast (over weeks or months)• Pathological fracture can be the first sign of tumor

Epidemiology & incidince
Primary bone tumors are relatively uncommon .
the incidence of benign bone tumors is higher than
the incidence of primary malignant tumors .
bone sarcomas account for 0.2% of all
malignancies diagnosed in the United States , and
the adjusted incidence rate for all bone and joint
malignancies is 0.9 per 100,000 persons per year,
while the 5-year overall survival rate is 67.9% .
The age specific incidence rates of bone sarcomas
typically show a bimodal distribution, with a first peak
occurring in the second decade, and a second peak
occurring in patients older than sixty years of age.

Epidemiology & incidence children are frequently affected .
The most frequently diagnosed histologic subtypes
were :
In adults, over 40% of primary bone cancers are
chondrosarcomas. This is followed by
osteosarcomas (28%), chordomas (10%), Ewing
tumors (8%), and malignant fibrous
histiocytoma/fibrosarcomas (4%). The remainder
of cases are several rare types of bone cancers.
In children and teenagers (those younger than 20
years), osteosarcoma (56%) and Ewing
tumors (34%) are much more common than
chondrosarcoma (6%).

Epidemiology & incidence

Causes & Risk factors
The cause of bone tumors is unknown.
Most people with bone cancers do not
have any apparent risk factors.
1-Genetic disorders A very small number of bone cancers
(especially osteosarcomas) appear to be
hereditary and are caused by defects
(mutations) in certain genes.
e.g. Li-Fraumeni syndrome osteosarcoma
- Rothmund-Thomson syndrome.
- Familial retinoblastoma syndrome .

Causes & Risk factors
2) multiple osteochondromas .
3) paget disease (pre-cacerous
lesion) .
4) Radiation
5) Bone marrow transplantation .
6) Injuries .

Classification of bone tumors
Bone tumors may be classified
as “primary tumors", which originate in bone
or from bone-derived cells and tissues,
and “secondary tumors" which originate in
other sites and spread ( metastatic ) .
Most classifications of bone tumours are based
on the recognition of the dominant cell tissue
(histopathology ) in the various lesions .
the current WHO classification (2002)includes a
total of 45 main bone tumor types .

WHO CLASSIFICATION OF
BONE TUMOURS
Bone-forming tumors (osteogenic )
Cartilage forming tumors (chondrogenic )
Giant-cell tumor
Marrow tumors
Vascular tumors
Other connective tissue tumors
Other tumors
Secondary malignant tumors of bone

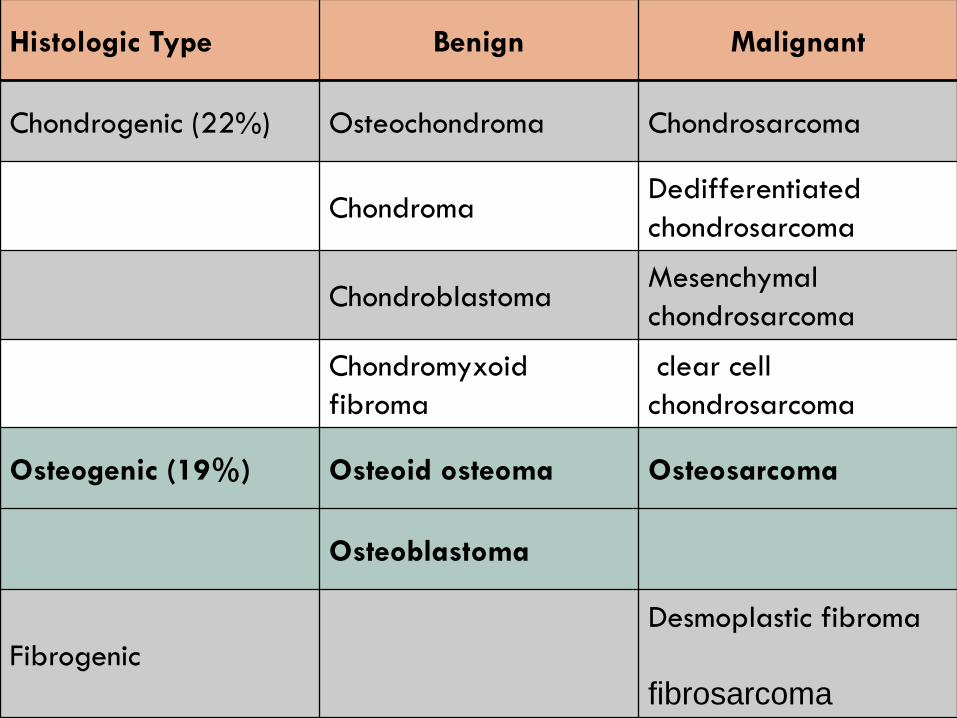
Histologic Type Benign Malignant
Chondrogenic (22%) Osteochondroma Chondrosarcoma
ChondromaDedifferentiated
chondrosarcoma
ChondroblastomaMesenchymal
chondrosarcoma
Chondromyxoid
fibroma
clear cell
chondrosarcoma
Osteogenic (19%) Osteoid osteoma Osteosarcoma
Osteoblastoma
Fibrogenic
Desmoplastic fibroma
fibrosarcoma
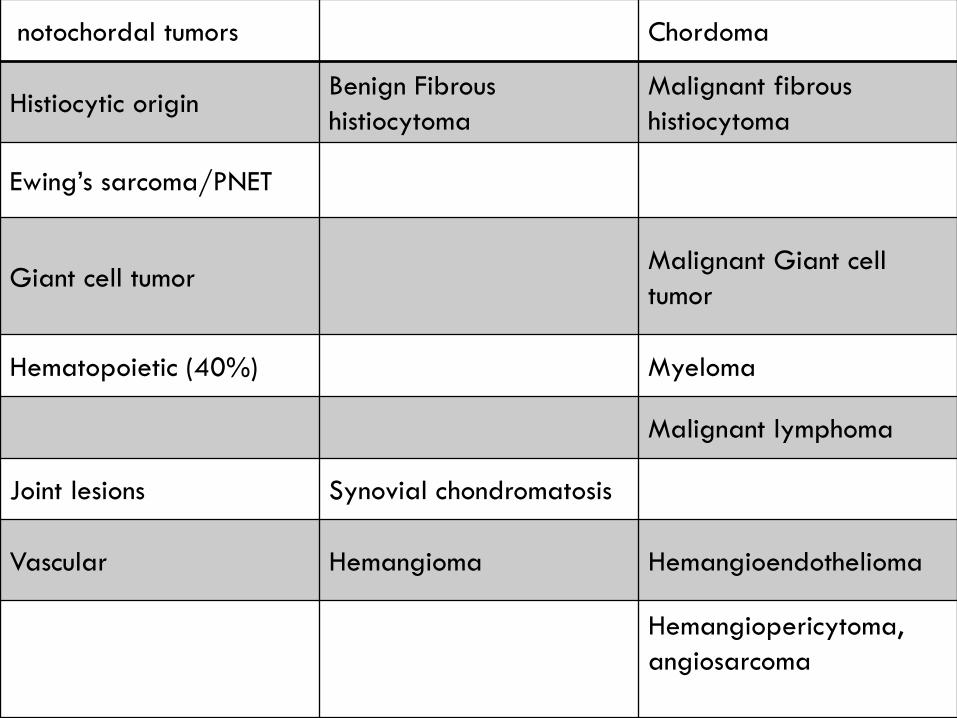
notochordal tumors Chordoma
Histiocytic originBenign Fibrous
histiocytoma
Malignant fibrous
histiocytoma
Ewing’s sarcoma/PNET
Giant cell tumorMalignant Giant cell
tumor
Hematopoietic (40%) Myeloma
Malignant lymphoma
Joint lesions Synovial chondromatosis
Vascular Hemangioma Hemangioendothelioma
Hemangiopericytoma,
angiosarcoma
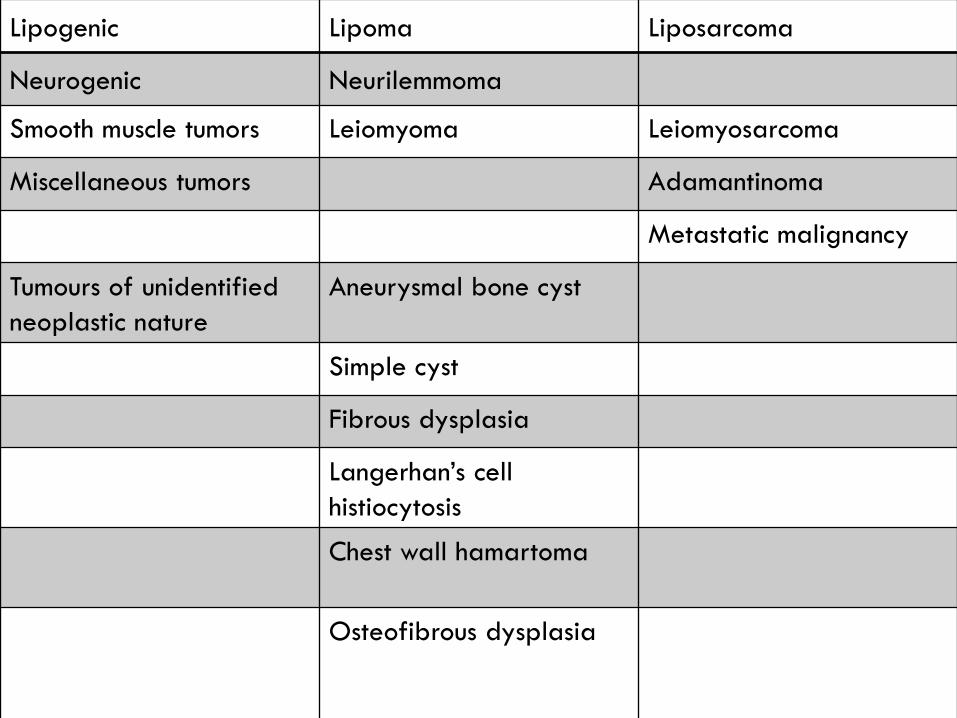
Lipogenic Lipoma Liposarcoma
Neurogenic Neurilemmoma
Smooth muscle tumors Leiomyoma Leiomyosarcoma
Miscellaneous tumors Adamantinoma
Metastatic malignancy
Tumours of unidentified
neoplastic nature
Aneurysmal bone cyst
Simple cyst
Fibrous dysplasia
Langerhan’s cell
histiocytosis
Chest wall hamartoma
Osteofibrous dysplasia
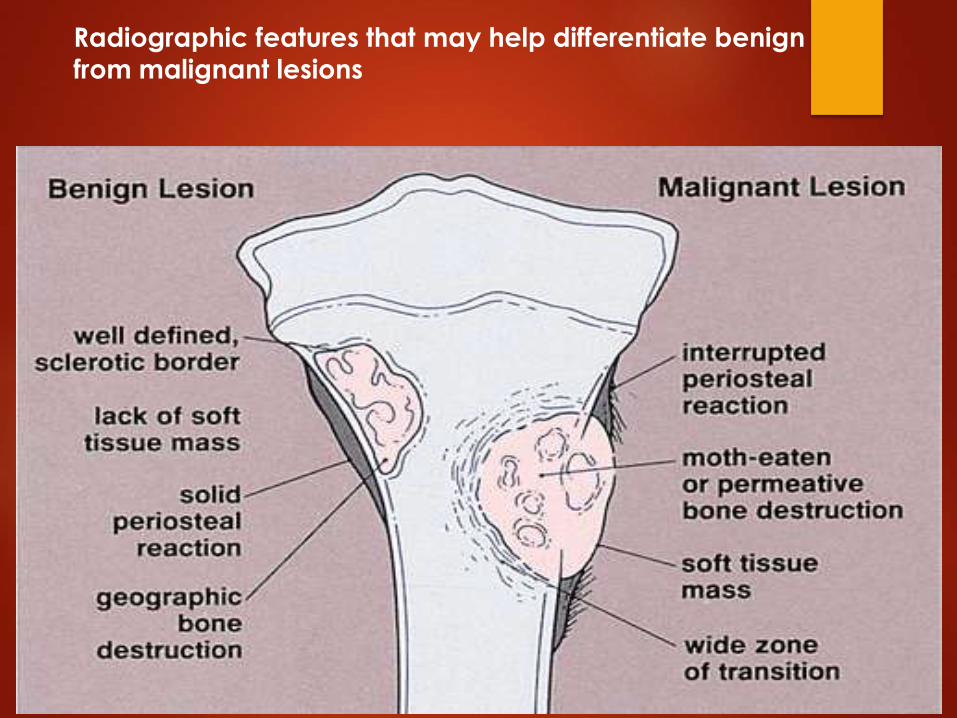
Radiographic features that may help differentiate benign from malignant lesions
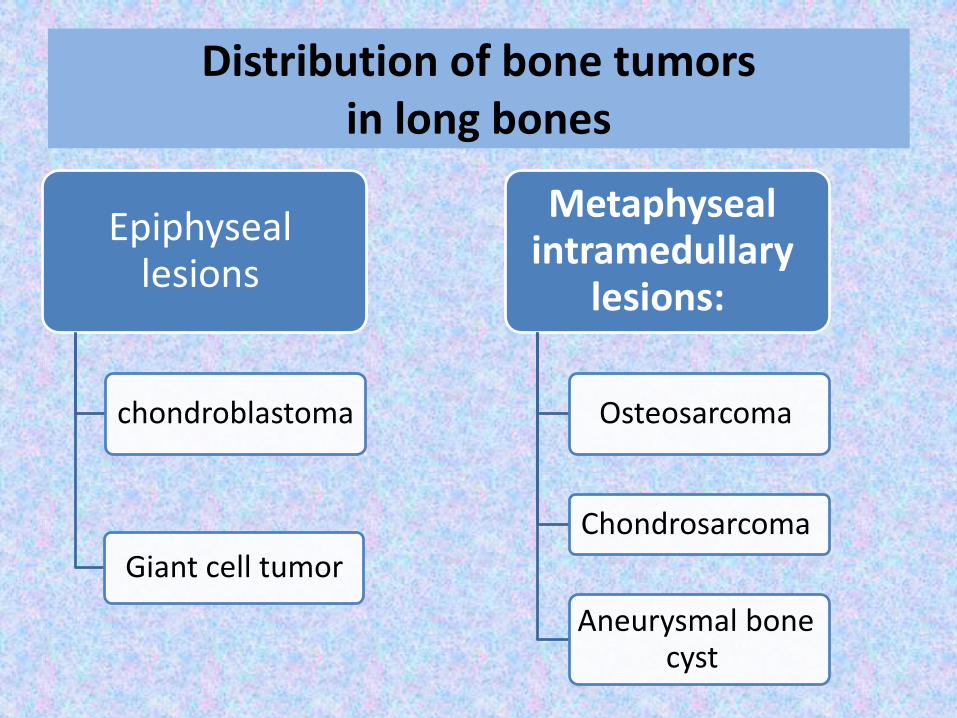
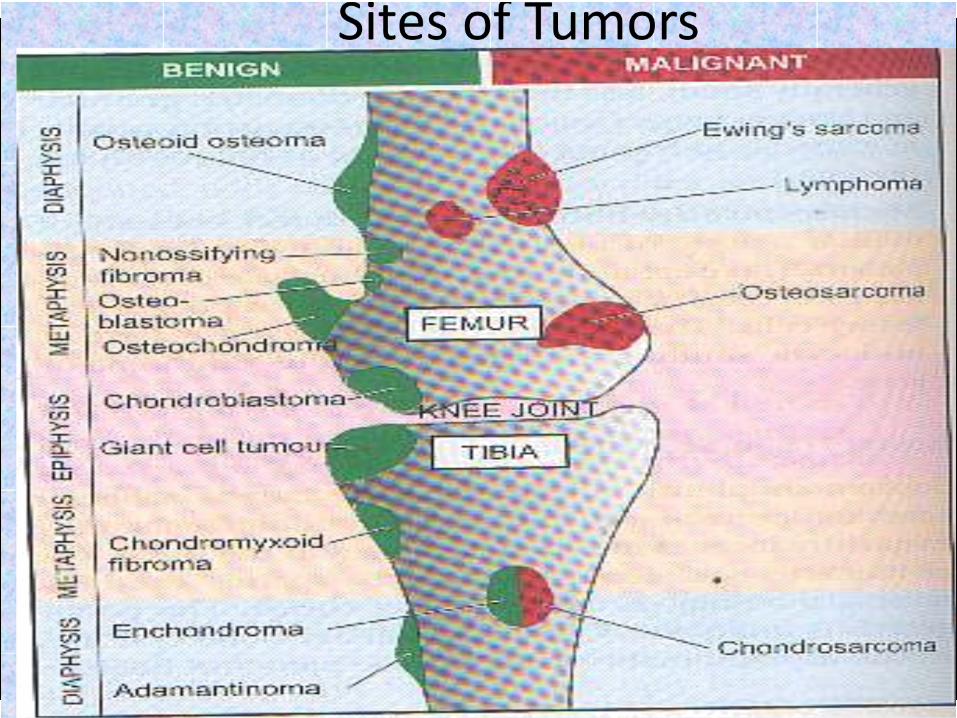
Distribution of bone tumors in long bones
Epiphyseal lesions
chondroblastoma
Giant cell tumor
Metaphysealintramedullary
lesions:
Osteosarcoma
Chondrosarcoma
Aneurysmal bone cyst
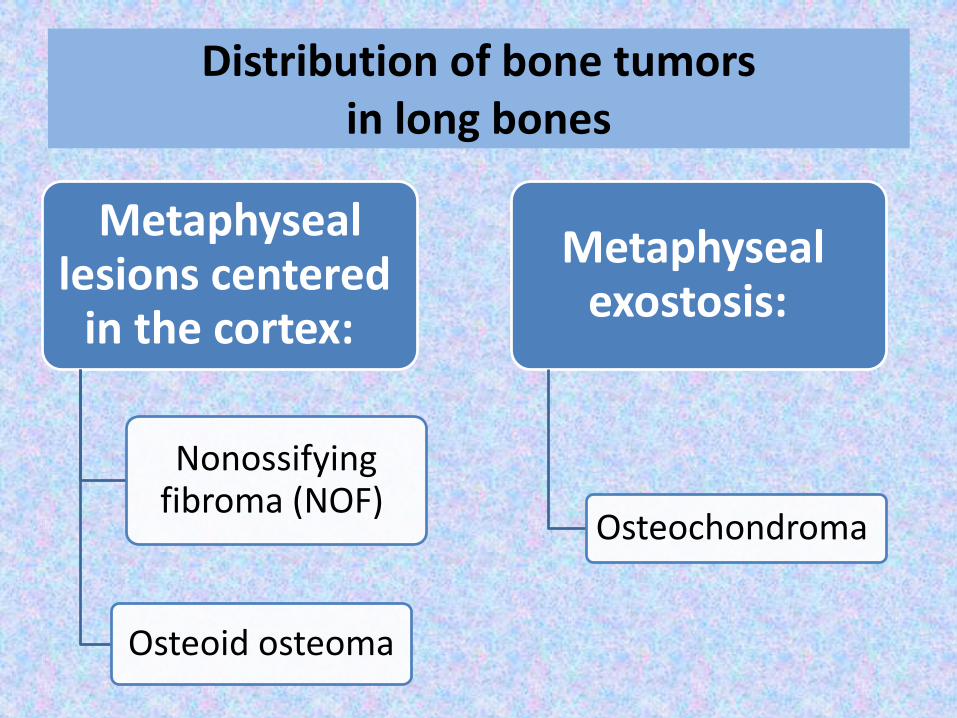
Distribution of bone tumors in long bones
Metaphyseallesions centered
in the cortex:
Nonossifying fibroma (NOF)
Osteoid osteoma
Metaphysealexostosis:
Osteochondroma
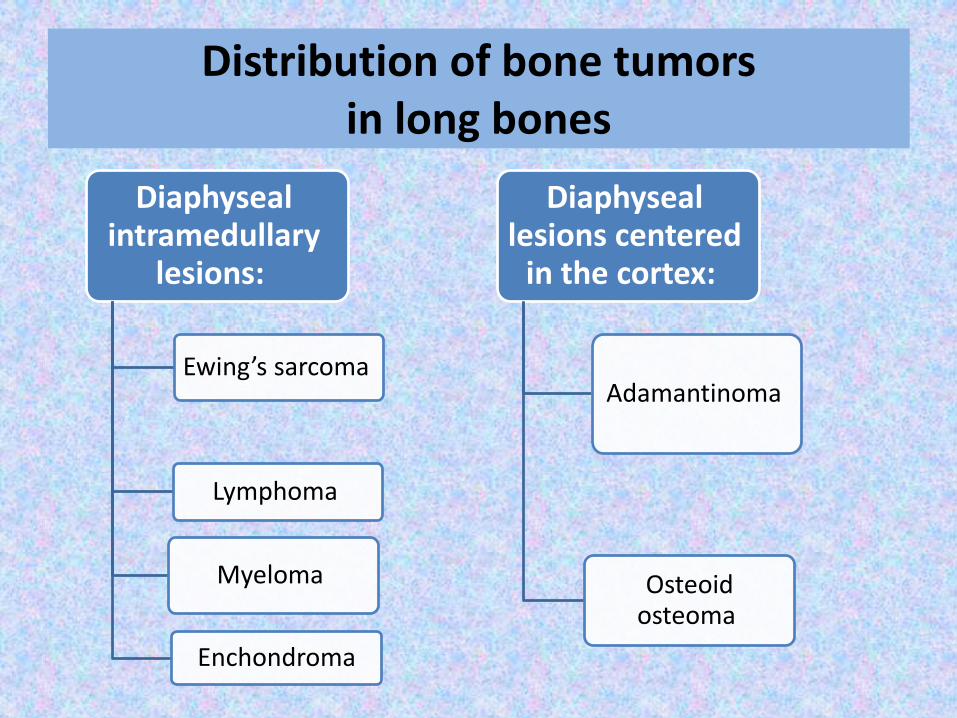
Distribution of bone tumors in long bones
Diaphysealintramedullary
lesions:
Ewing’s sarcoma
Lymphoma
Myeloma
Enchondroma
Diaphyseallesions centered
in the cortex:
Adamantinoma
Osteoid osteoma
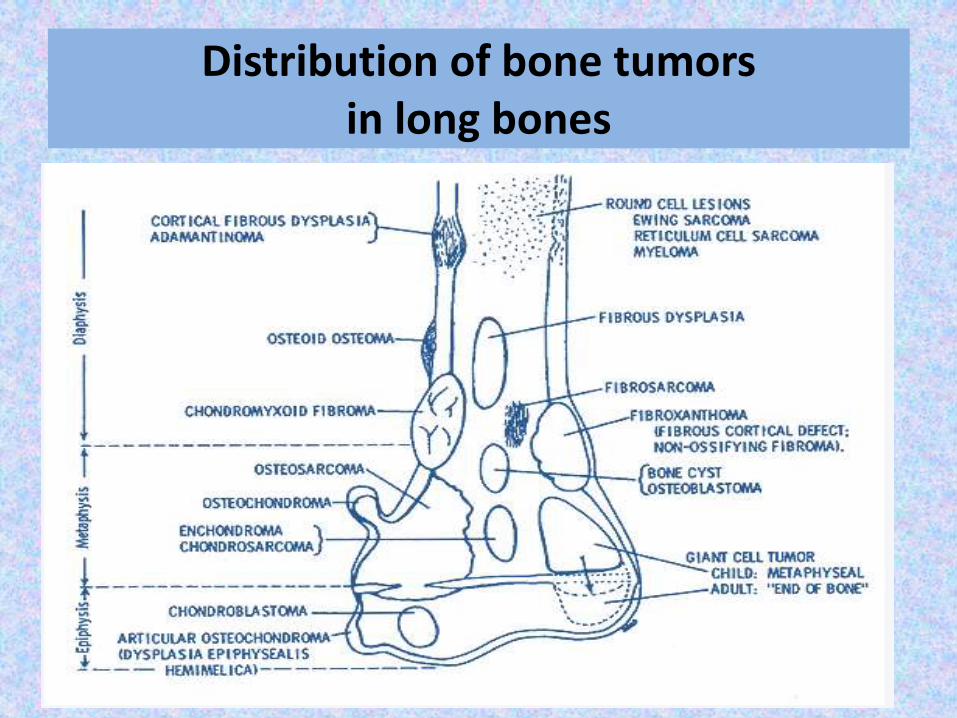
Distribution of bone tumors in long bones

Diagnosis of bone tumors
Signs and Symptoms of Bone Cancer
• - Pain in the bone.
• - Tenderness and swelling near the area
• which is affected.
• - The affected bone may break (pathologic #)
“may be the first and only clinical sign.”
• - Tiredness.
• - Weight loss which is unintentional.

Diagnosis of bone tumors
Imaging tests to detect bone cancer :
- X-rays >>> Most useful of all imaging
techniques.
>> There might be obvious
abnormality of the bone:
1 * Cotrical thickening
2 * Discrete lump
3 * Cyst
4 * Ill-defined destruction
- CT AND MRI

Diagnosis of bone tumors
- Biopsy There are three ways:
1.Needle biopsy: Must be performed by
experienced personal.
2. Open biopsy: most reliable way of
obtaining a representative sample.
3. Excisional biopsy: for benign tumors.


Staging of bone tumors
Staging is the process of finding out how
far the cancer may have spread.
This is very important because the type of
treatment and the outlook for recovery
(prognosis) depend on the stage of the
cancer.

Staging of bone tumors
In treating tumors we are facing two
conflicting principles:
1. Lesion must be removed widely to
ensure it doesn’t recur.
2. Damage must be kept minimal.
Staging of bone tumors
The balance between the 2 conflicting objectives depends on knowing:
1. How the tumor behaves (Aggressiveness)
2. How far it has spread.
The answers to these two questions are embodied in the staging system of Enneking.
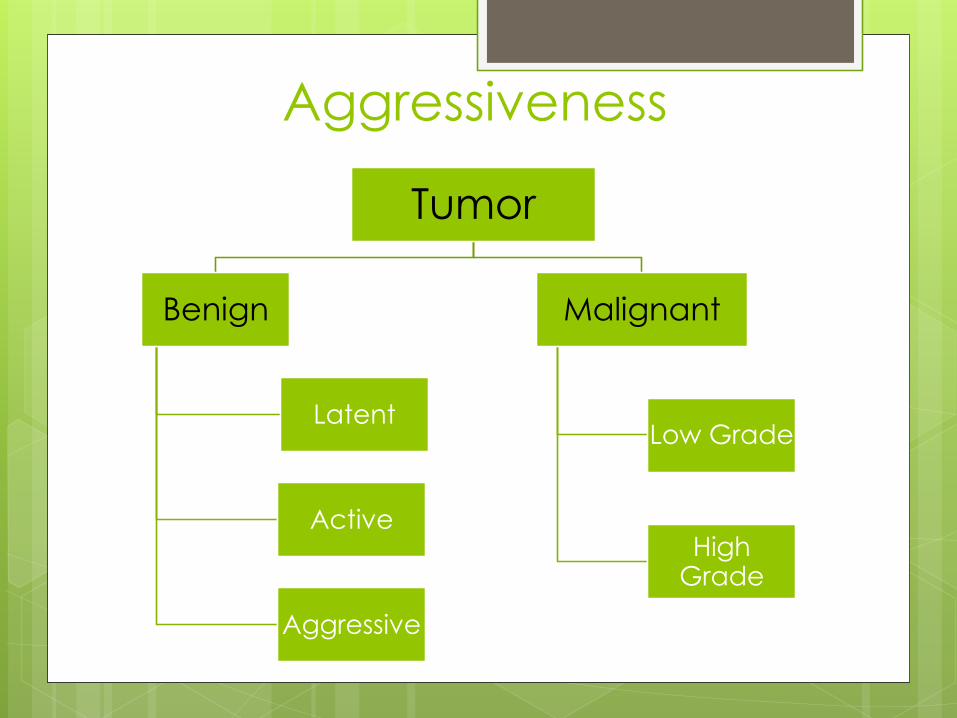
Tumor
Benign
Latent
Active
Aggressive
Malignant
Low Grade
High Grade
Aggressiveness

Enneking Staging system of
bone tumors
Benign Tumors
Latent Well defined margin. Grows
slowly and then stops.
Remains static/heals
spontaneously E.g Osteoid
osteoma
Active Progressive growth limited
by natural barriers.
Not self limiting. Tendency to
recur E.g Aneurysmal Bone
cyst
Aggressive Growth not limited by
natural Barriers E.g Gaint cell
tumor

Enneking Staging system of
bone tumors
Malignant Tumors
Low Grade Moderatly aggressive and
takes a long time to
metastasize
High Grade Very aggressive and
metastasize early

Spread
Assuming that there is no metastases, the
local extent of the tumor is the most
important factor in deciding how much
tissue to be removed.
Spread
Intracompartmental
Extracompartmental

Spread
Lesions that are confined to an enclosed
space (e.g Bone cavity, joint cavity or
muscle group within its fascial envelope)
are called Intracompartmental.
Lesions that extend into interfascial or
extrafascial with no natural barrier to
proximal or distal spread are called
Extracompartmental. (E.g pelvis, axilla)

Surgical stage
Staging the tumor is an important step
towards selecting the best operation
suited to the patient.
Bone sarcomas are divided as follows:
1. Stage 1: All low grade sarcomas
2. Stage 2: Histologically high grade lesions
3. Stage 3: Sarcomas which have
metastasized.
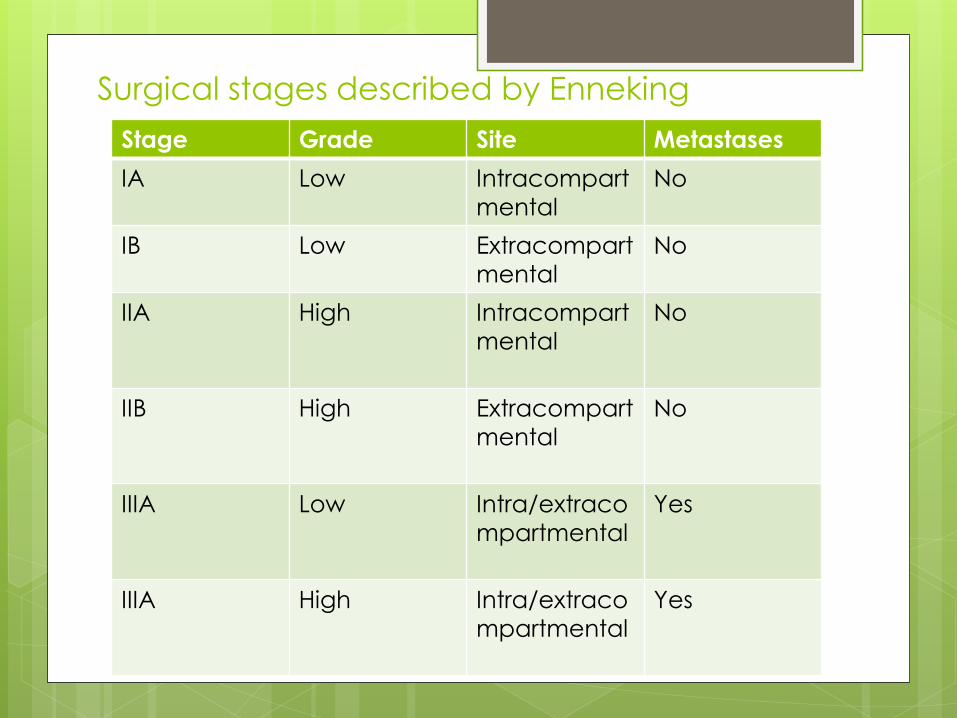
Surgical stages described by Enneking
Stage Grade Site Metastases
IA Low Intracompart
mental
No
IB Low Extracompart
mental
No
IIA High Intracompart
mental
No
IIB High Extracompart
mental
No
IIIA Low Intra/extraco
mpartmental
Yes
IIIA High Intra/extraco
mpartmental
Yes

Primary malignant bone
tumors
Bone cancers are differentiated into types depending on the cell type in which the cancer started. Given below are some of the common types of bone cancer:
1) osteosarcoma
2) chondrosarcoma
3) Ewing’s sarcoma
Multiple myeloma
Metastatic bone diseases
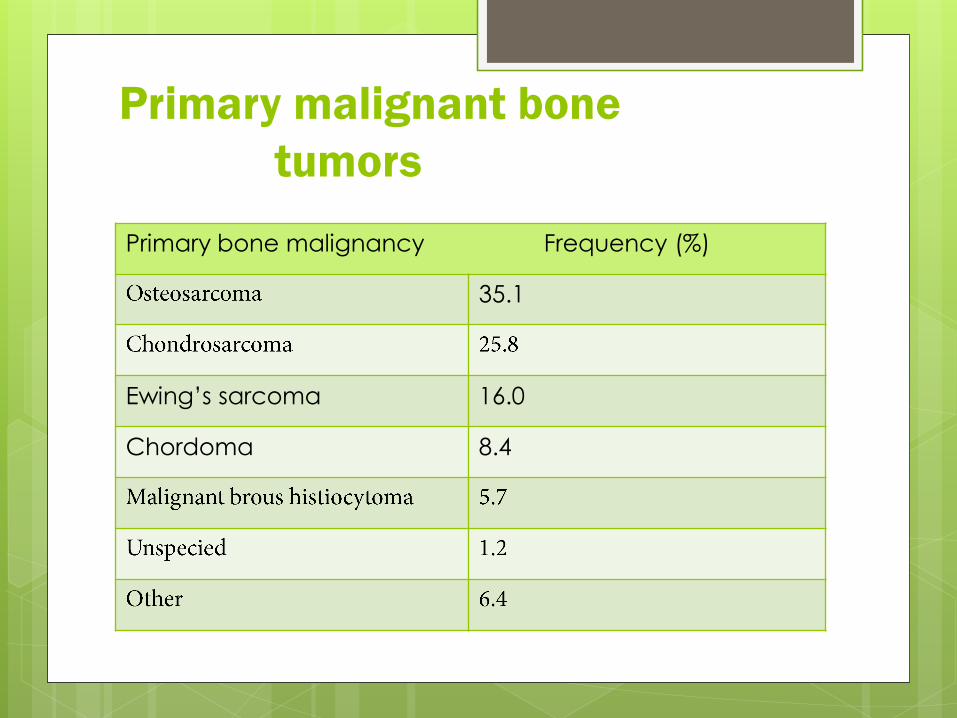
Primary malignant bone
tumors
Primary bone malignancy Frequency (%)
35.1
16.0Ewing’s sarcoma
8.4Chordoma
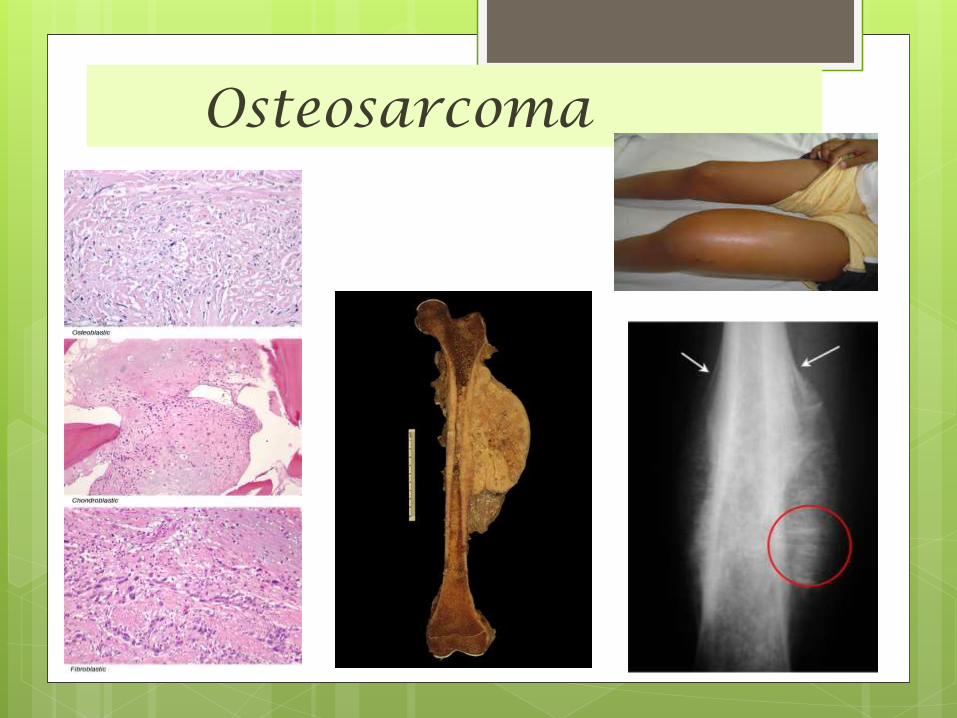
Osteosarcoma

Osteosarcoma Also called Osteogenic Sarcoma .
Is the most common primary bone cancer.
Osteosarcomas is a mesenchymally derived
malignant tumor that produces
osteoid and/or bone.
This cancer starts in the
bone cells.
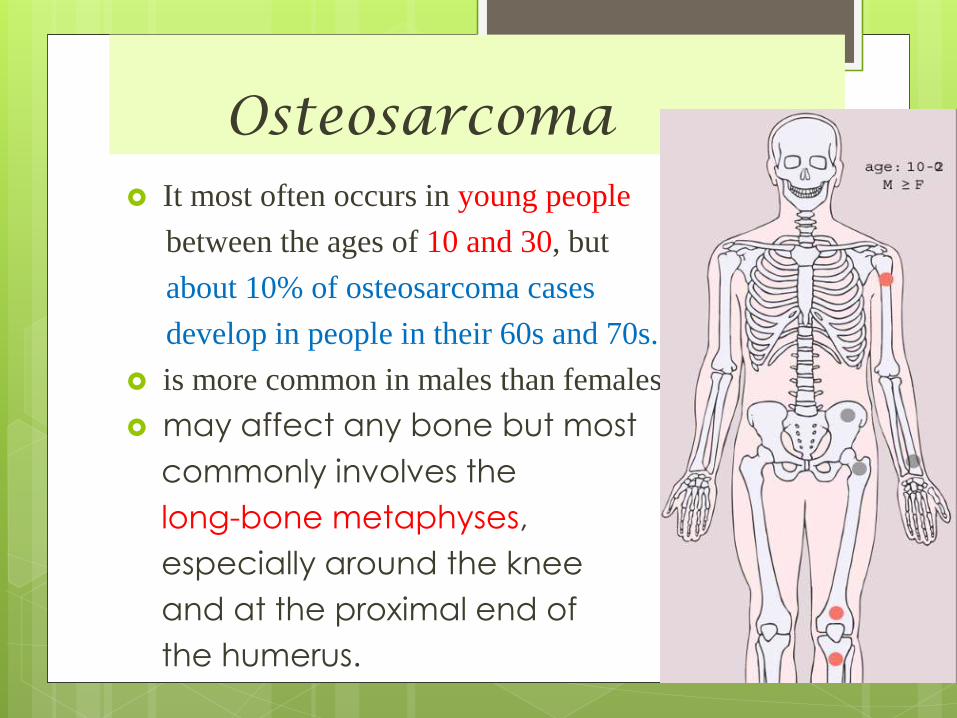
Osteosarcoma It most often occurs in young people
between the ages of 10 and 30, but
about 10% of osteosarcoma cases
develop in people in their 60s and 70s.
is more common in males than females.
may affect any bone but most
commonly involves the
long-bone metaphyses,
especially around the knee
and at the proximal end of
the humerus.

Osteosarcoma
They can be:
INTRAMEDULLARY OS
INTRACORTICAL OS
JUXTACORTICAL OS
In its classic (intramedullary) form,
osteosarcoma is a highly malignant tumour
arising within the bone and spreading rapidly
outwards to the periosteum and surrounding
soft tissues .
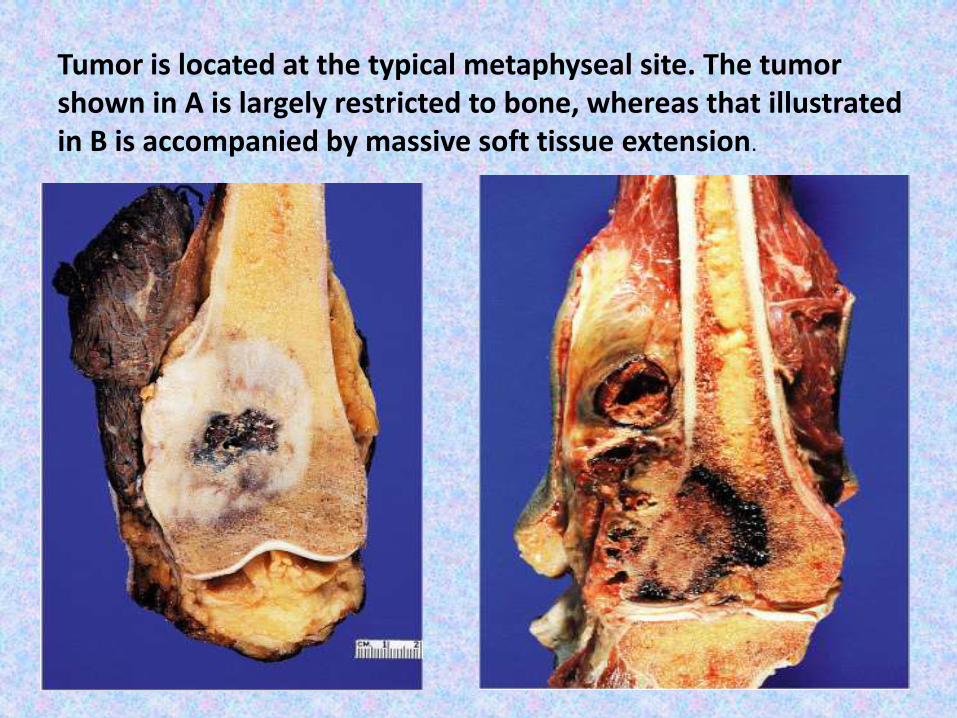
Tumor is located at the typical metaphyseal site. The tumor shown in A is largely restricted to bone, whereas that illustrated in B is accompanied by massive soft tissue extension.

Osteosarcoma invasive, variable histology; frequent
metastases without treatment
painful, poorly defined swelling
Pathological fracture is rare.
x-ray shows
characteristic periosteal elevation and
spicule formation representing tumour
extension into periosteum .
The endosteal margin is poorly defined.
sunburst’ effect and Codman’s triangle
.
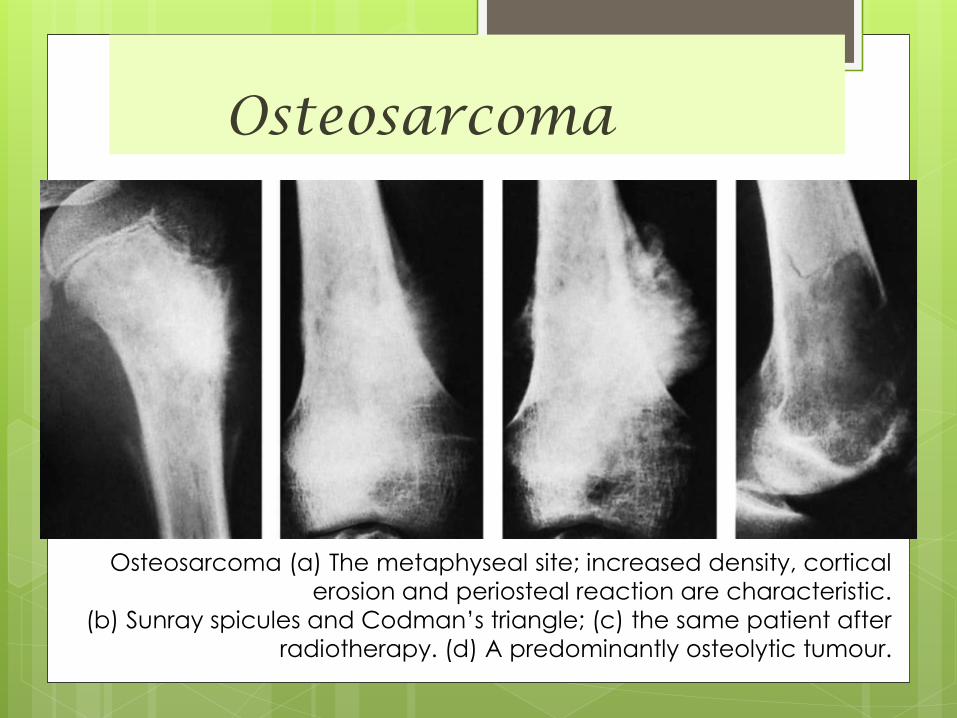
Osteosarcoma
Osteosarcoma (a) The metaphyseal site; increased density, cortical
erosion and periosteal reaction are characteristic.
(b) Sunray spicules and Codman’s triangle; (c) the same patient after
radiotherapy. (d) A predominantly osteolytic tumour.
Osteosarcoma
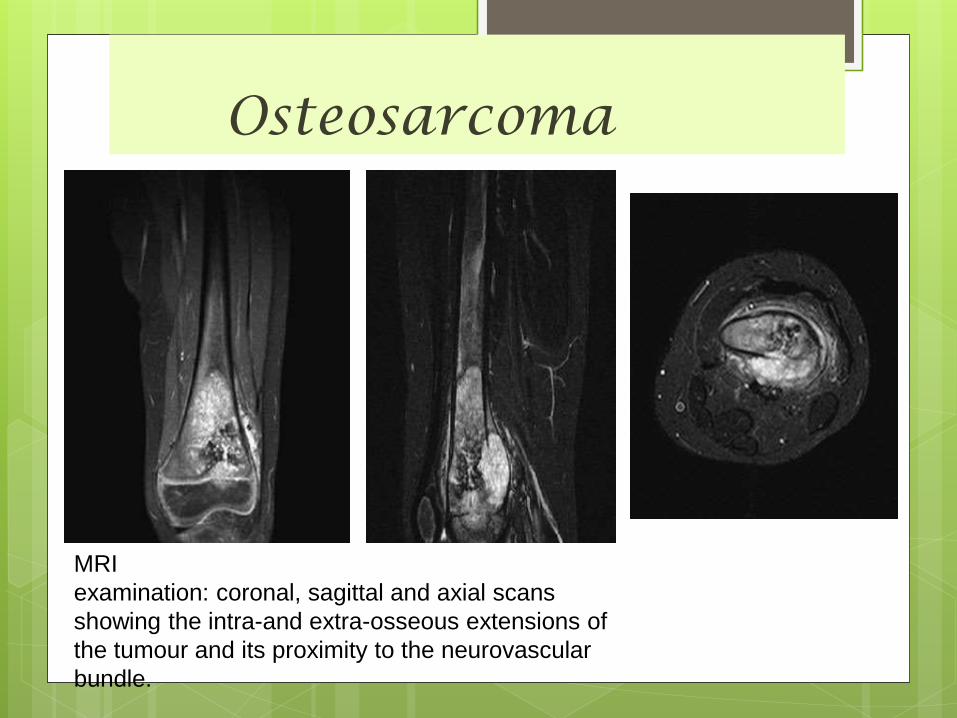
Osteosarcoma
MRI
examination: coronal, sagittal and axial scans
showing the intra-and extra-osseous extensions of
the tumour and its proximity to the neurovascular
bundle.
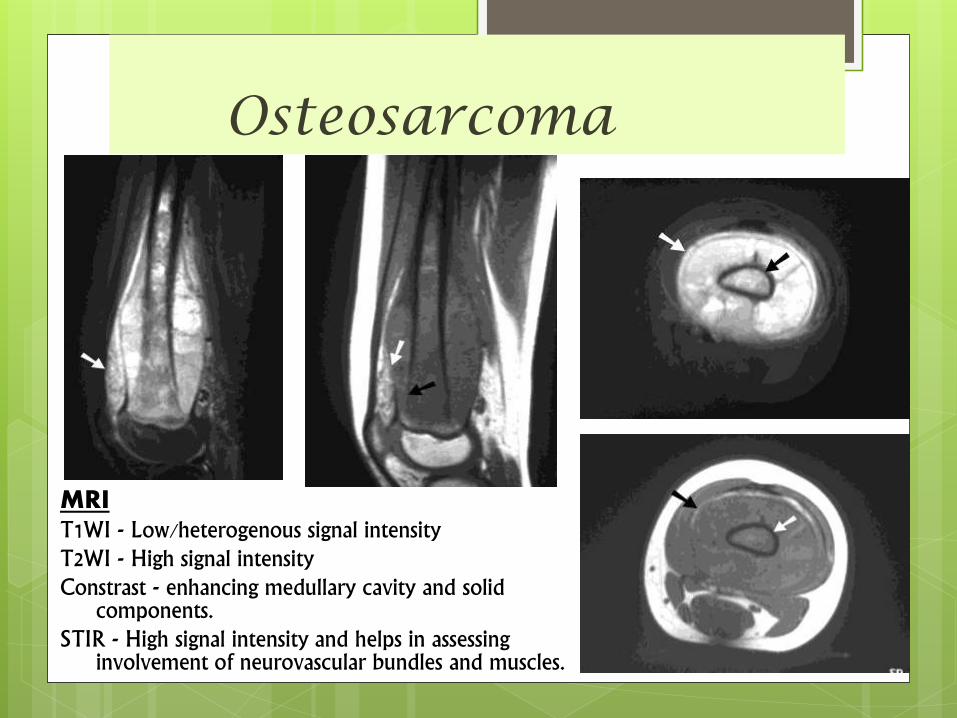
Osteosarcoma
MRIT1WI - Low/heterogenous signal intensityT2WI - High signal intensityConstrast - enhancing medullary cavity and solid
components.STIR - High signal intensity and helps in assessing
involvement of neurovascular bundles and muscles.
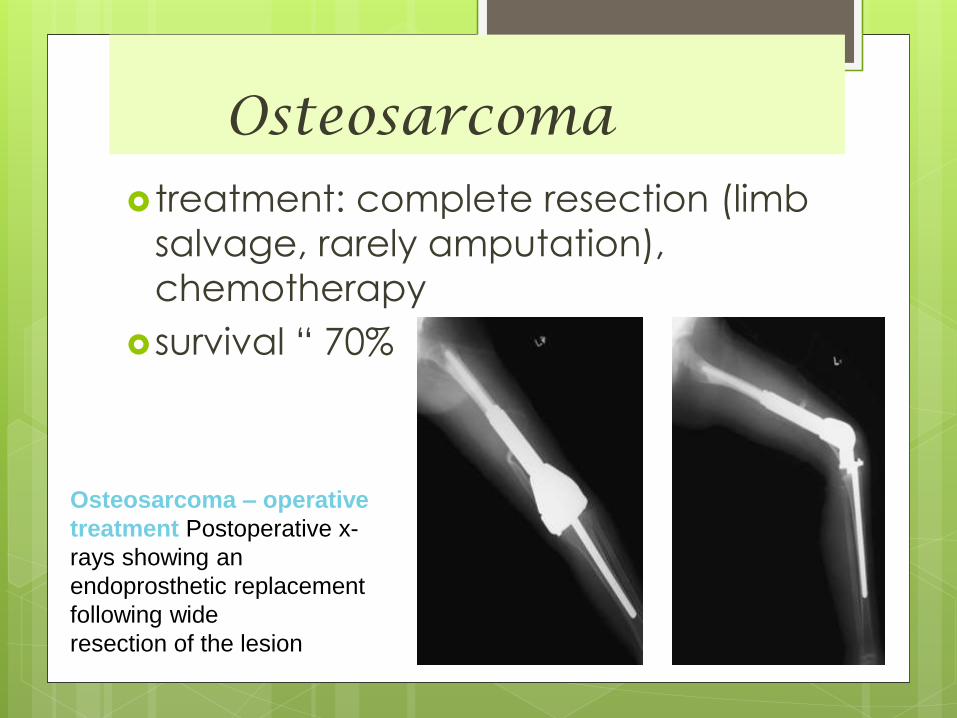
Osteosarcoma treatment: complete resection (limb
salvage, rarely amputation),
chemotherapy
survival “ 70%
Osteosarcoma – operative
treatment Postoperative x-
rays showing an
endoprosthetic replacement
following wide
resection of the lesion

Variants Of Osteosarcoma
1) Juxtacortical (parosteal)
low-grade sarcoma situated on the
surface of one of the tubular bones,
usually at the distal femoral or proximal
tibial metaphysis.
Slightly older age group.
X-ray shows a dense bony mass on
the surface of the bone or encircling it;
the cortex is not eroded and usually a
thin gap remains between cortex and
tumour
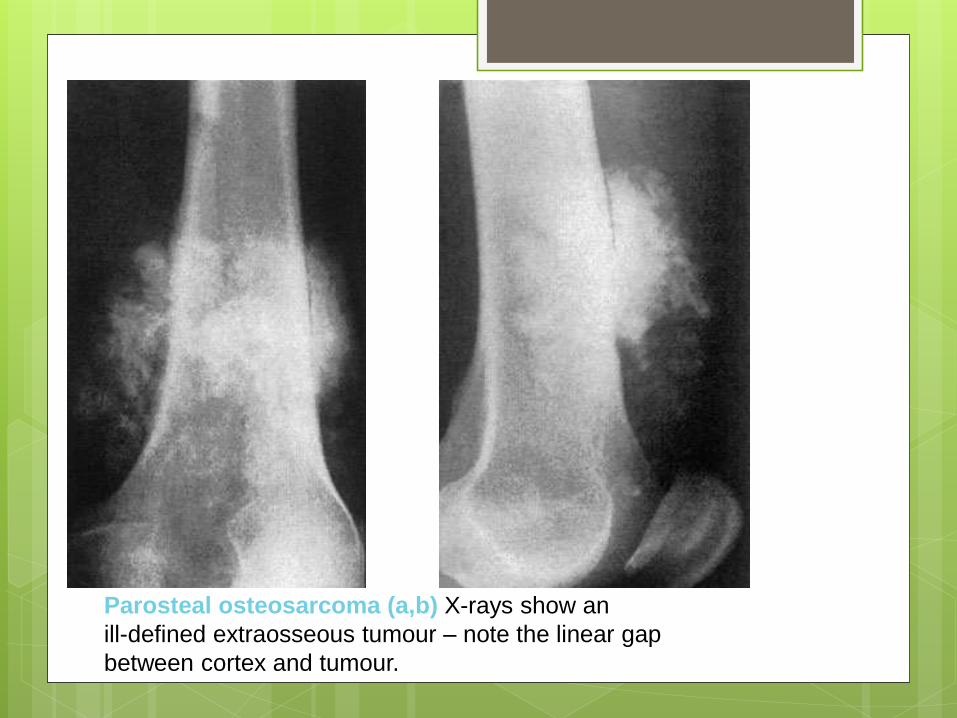
Parosteal osteosarcoma (a,b) X-rays show an
ill-defined extraosseous tumour – note the linear gap
between cortex and tumour.
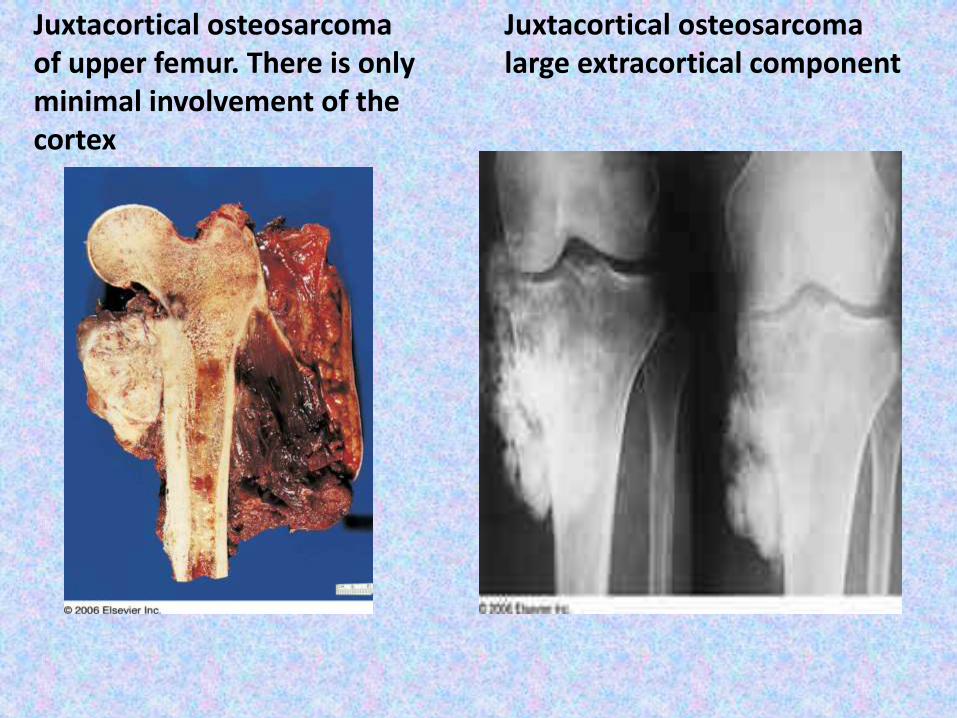
Juxtacortical osteosarcomaof upper femur. There is only minimal involvement of the cortex
Juxtacortical osteosarcomalarge extracortical component
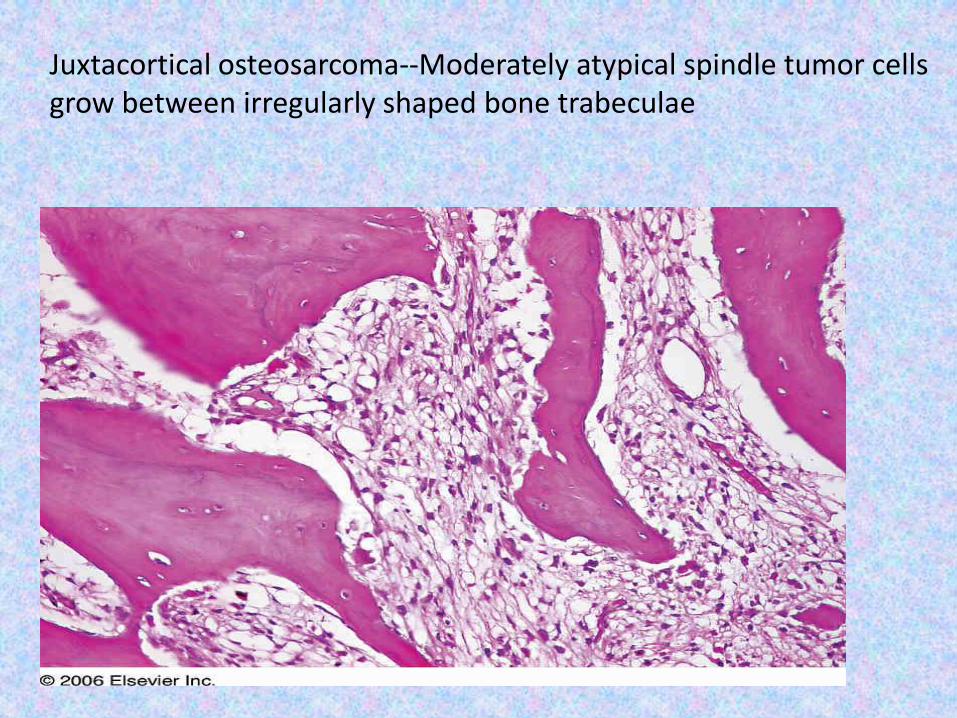
Juxtacortical osteosarcoma--Moderately atypical spindle tumor cells grow between irregularly shaped bone trabeculae

Variants Of Osteosarcoma
1) Juxtacortical (parosteal)
Treatment
- For a low-grade parosteal osteosarcoma,
wide excision without adjuvant therapy is
sufficient to ensure a recurrence rate
below 10 per cent.
- Dedifferentiated parosteal osteosarcoma
should be treated in the same way as
intramedullary sarcoma.

Variants Of Osteosarcoma
2) PERIOSTEAL OSTEOSARCOMA
It is more like an intramedullary
osteosarcoma, but situated on the
surface of the bone
Upper tibial shaft or femur.
X-ray shows a superficial defect of the
cortex, but
CT and MRI may reveal a larger soft-
tissue mass
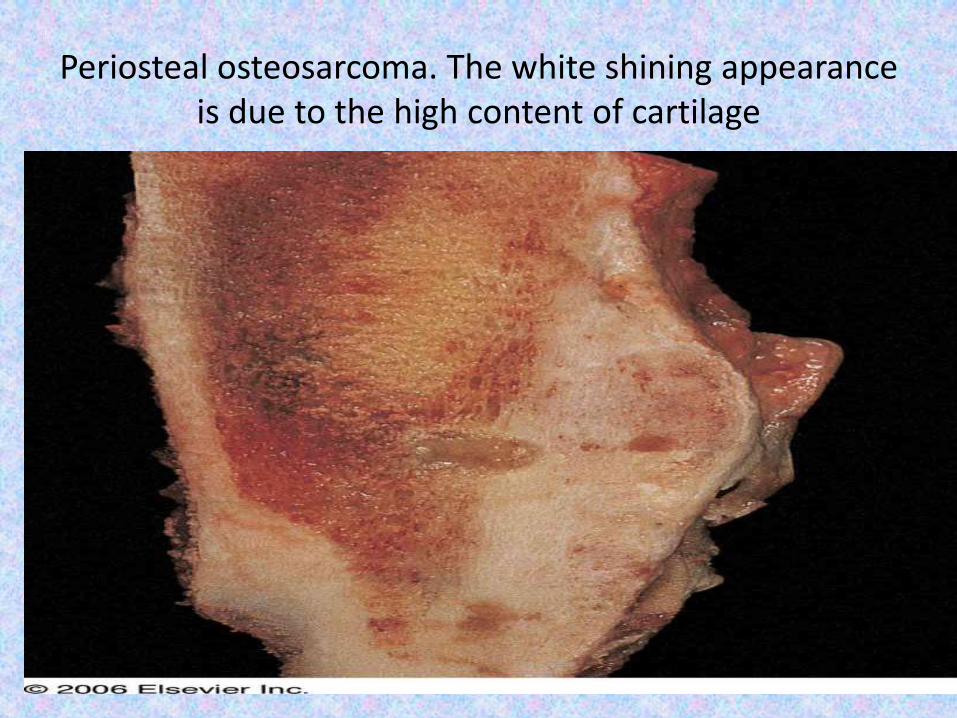
Periosteal osteosarcoma. The white shining appearance is due to the high content of cartilage
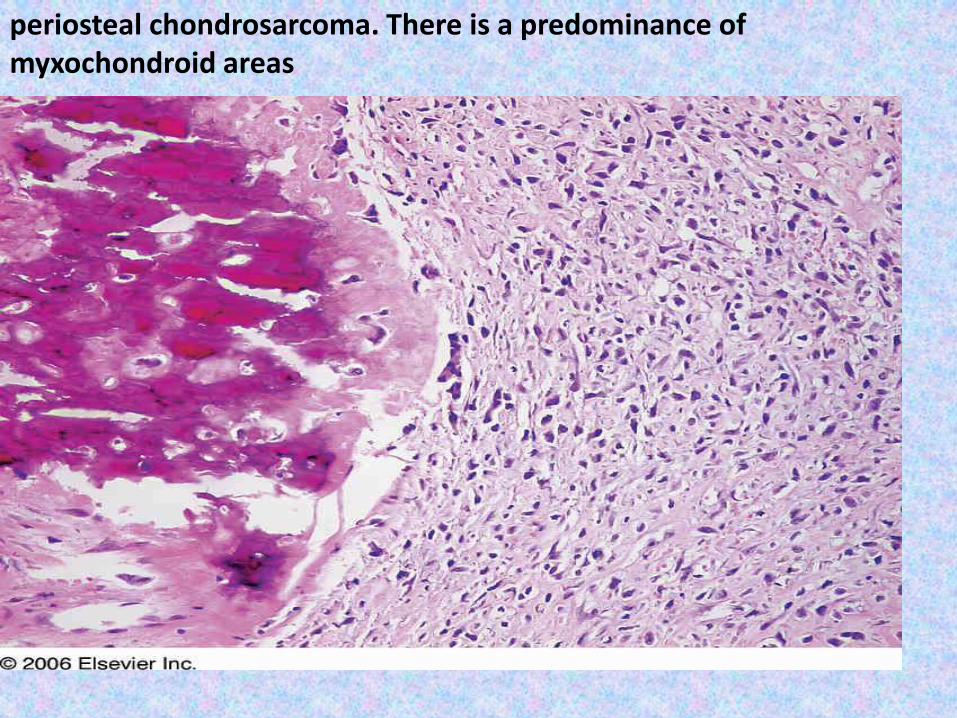
periosteal chondrosarcoma. There is a predominance of myxochondroid areas
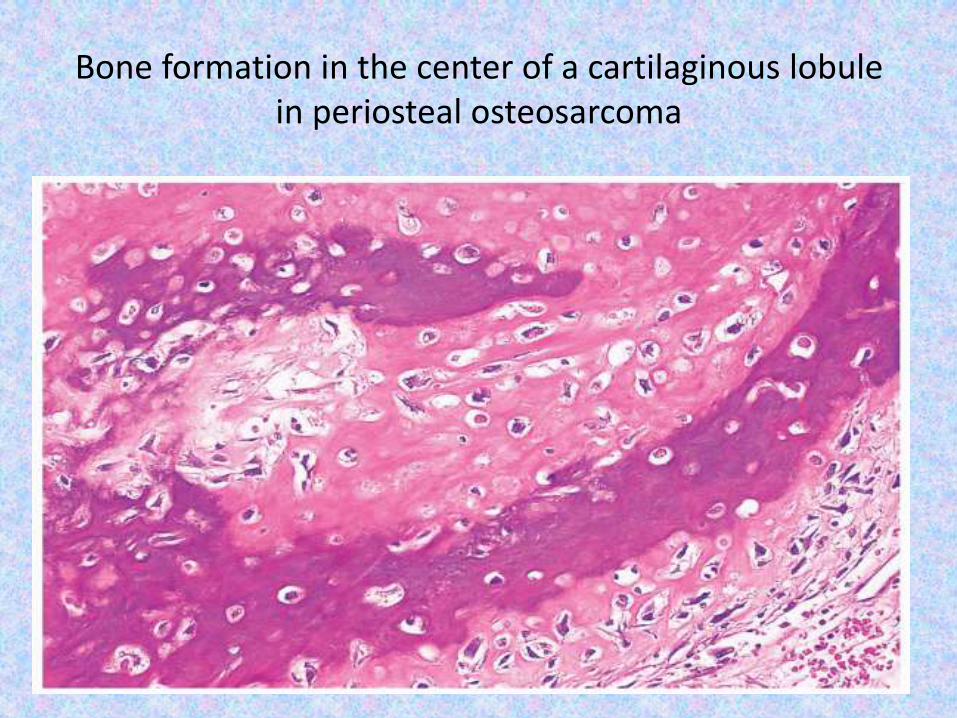
Bone formation in the center of a cartilaginous lobule in periosteal osteosarcoma

Variants Of Osteosarcoma
3) Osteosarcoma in Paget’s disease.
Osteosarcoma are of the polyostotic type
Pelvis, humerus, femur tibia & skull.
Large number of osteoclasts alternating
with atypical osteoblast.
X-ray shows the usual features of Paget’s
disease, but with areas of bone destruction
and soft-tissue invasion.
This is a high-grade tumour most patients have pulmonary metastases by the time the tumour is
diagnosed
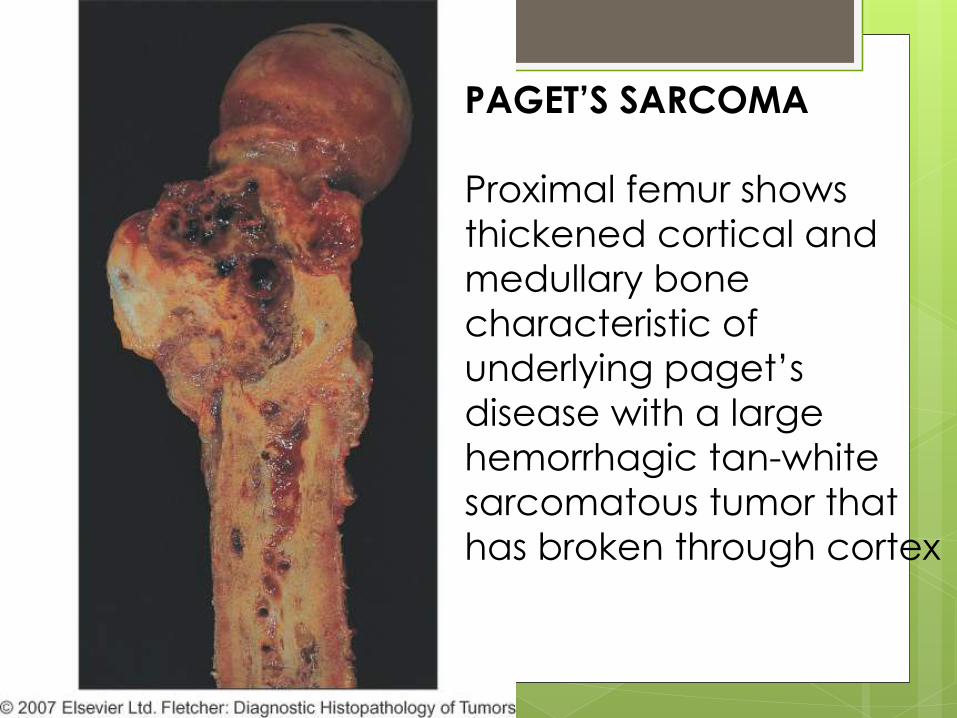
PAGET’S SARCOMA
Proximal femur shows
thickened cortical and
medullary bone
characteristic of
underlying paget’s
disease with a large
hemorrhagic tan-white
sarcomatous tumor that
has broken through cortex
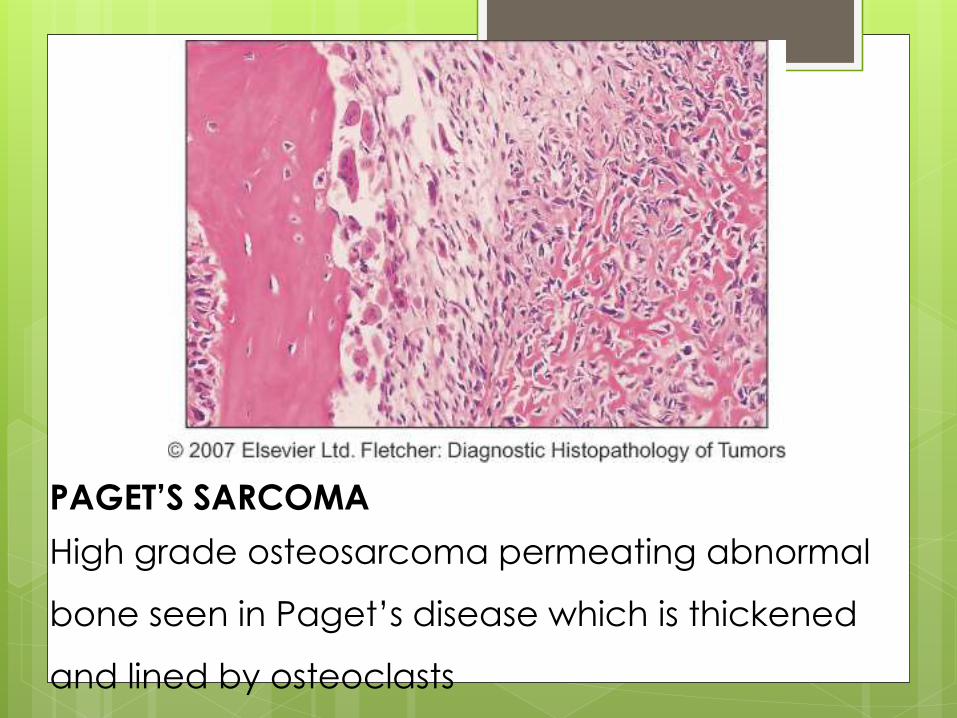
PAGET’S SARCOMA
High grade osteosarcoma permeating abnormal
bone seen in Paget’s disease which is thickened
and lined by osteoclasts

Variants Of Osteosarcoma
3) Osteosarcoma in Paget’s disease.
Treatment
Even with radical resection or amputation
and chemotherapy the 5-year survival
rate is low.
If the lesion is definitely
extracompartmental, palliative treatment
by radiotherapy may be preferable;
- chemotherapy is usually difficult because
of the patient’s age
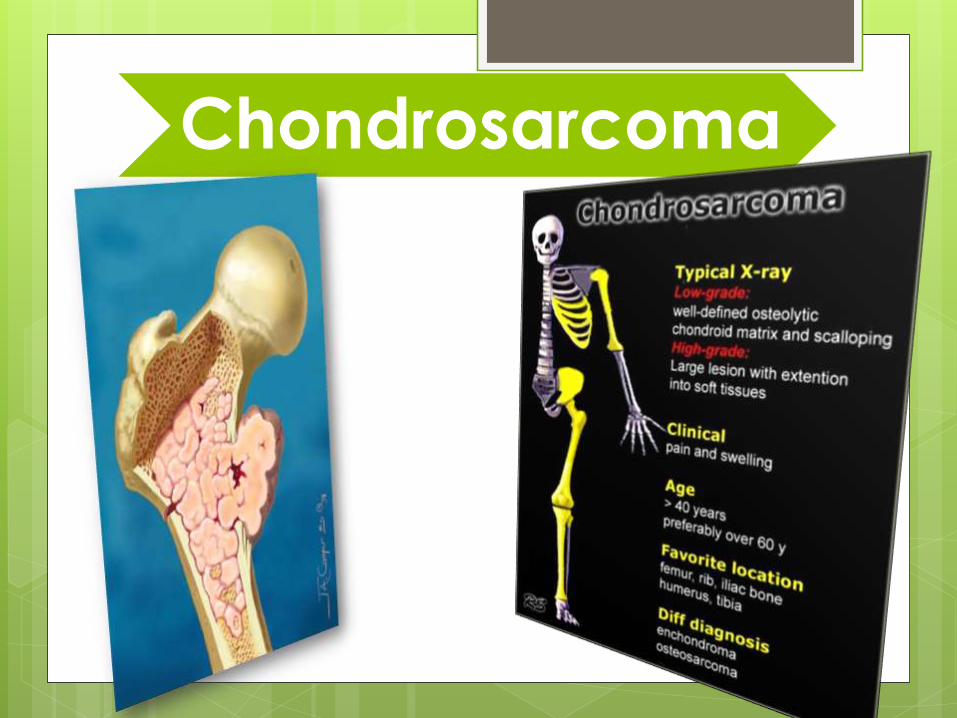
Chondrosarcoma

Chondrosarcoma Chondrosarcoma is malignant cartilaginous tumor and the second
most common malignant tumors originating in bone.
Arise de novo or from pre-existing benign cartilagenous tumor
i.e. enchondroma or osteochondroma
accounts for about 20% of bone tumors and is diagnosed in
approximately 600 patients each year in the United States.
The highest incidence is in the fourth and fifth decades.
men are affected more often than women.
These tumours are slow-growing and are usually present for many
months before being discovered .
Patients may complain of a dull ache or a gradually enlarging
lump. Medullary lesions may present as a pathological fracture.

Chondrosarcoma
Most common bones involved are
metaphysis of long tubular bones, mostly in
the lower limbs (45%),
pelvis (25%),
ribs (8%),
spine (7%),
scapula (5%),
sternum (2%)other bones
Despite the relatively frequent occurrence of benign cartilage tumours in the small bones of the
hands and feet, malignant lesions are rare at these sites

Chondrosarcoma Patients with multiple hereditary exostoses, Ollier's disease
and Maffucci's syndrome have a higher risk for malignant
transformation of a cartilaginous lesion.
DIAGNOSTIC FEATURES OF SECONDARY MALIGANT CHANGE
OF A BENING CARTILAGE TUMOR INDUCES :- Evidence of soft tissue calcification or
a soft tissue mass.
- Endo-osteal erosion and permeative
features in an enchondroma including
areas of lysis.
- Growth in a previously stable exostosis
- Expansion of the cartilage cap of an exoxtosis
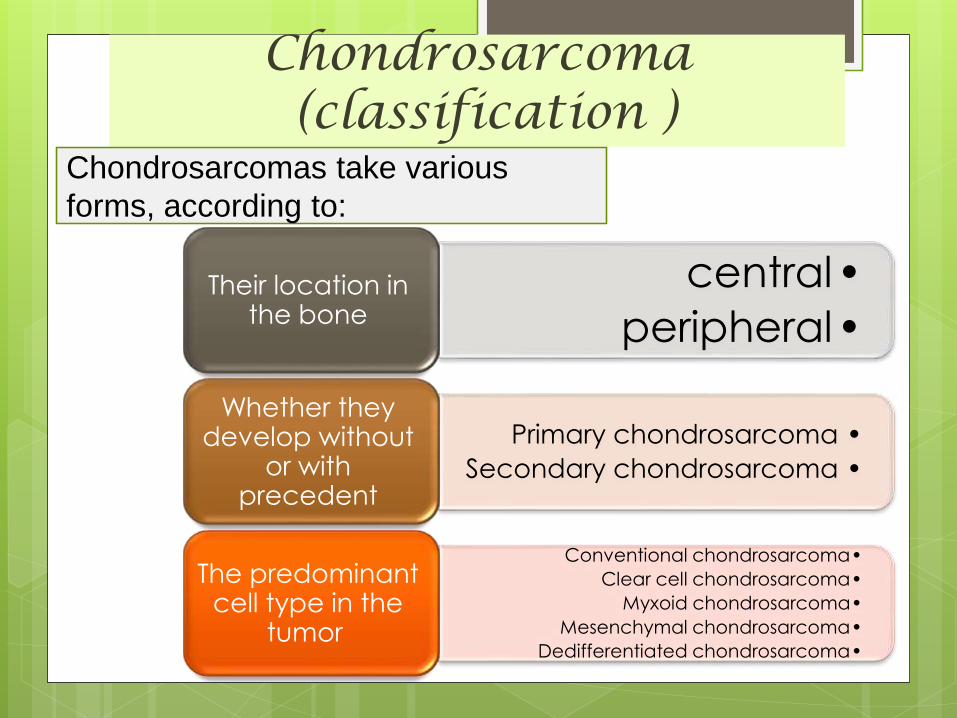
Chondrosarcoma(classification )
•central
•peripheralTheir location in
the bone
•Primary chondrosarcoma
•Secondary chondrosarcoma
Whether they develop without
or with precedent
•Conventional chondrosarcoma
•Clear cell chondrosarcoma
•Myxoid chondrosarcoma
•Mesenchymal chondrosarcoma
•Dedifferentiated chondrosarcoma
The predominant cell type in the
tumor
Chondrosarcomas take various
forms, according to:

Chondrosarcoma(classification )
Central chondrosarcoma
Also called
The tumor develops in the
medullary cavity of either tubular
or flat bones .
lobulated., translucent, bluish
whitish cartilaginous mass within
medullary cavity
accounting for more than 90 %
of chondrosarcomas
Most commonly at the proximal
end of the femur or in the
Innominate bone of the pelvis.
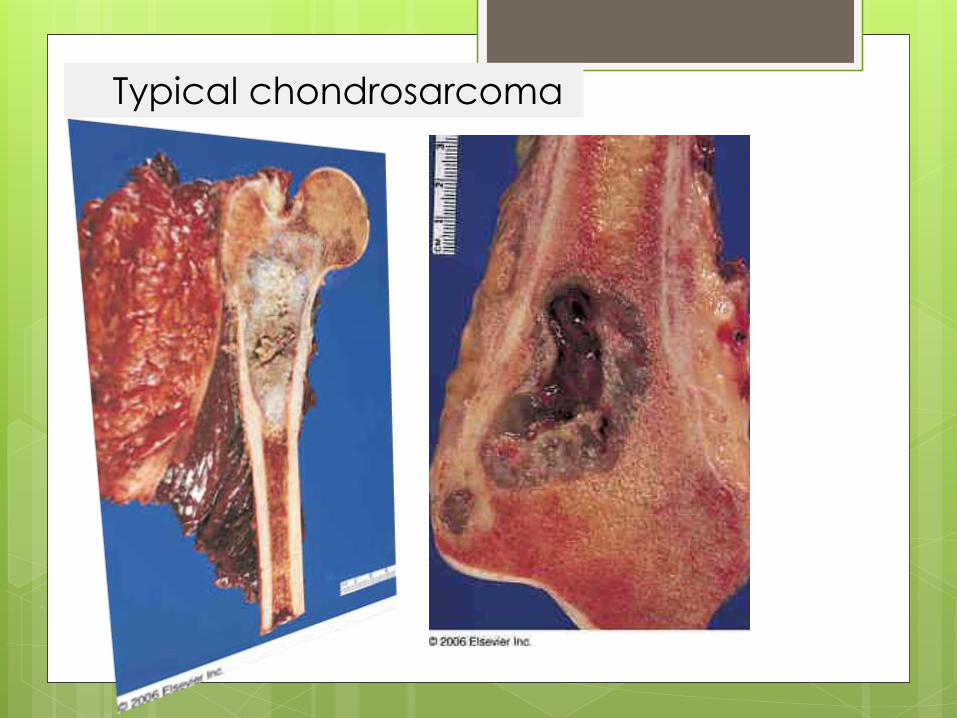
Typical chondrosarcoma

central Chondrosarcoma
large mass at the time of diagnosis, usually over 4 cm in diameter in 50% of cases.
Osteolytic lesion (50%)
intralesional calcification(s): 60-78% (rings and arcs calcification or popcorn calcification)
endosteal scalloping
affects more than two thirds of the cortical thickness (c.f. less than 2/3 in enchondromas)
moth eaten appearance or permeativeappearance in higher grade tumours
cortical remodelling, thickening and periosteal reaction are also useful in distinguishing between an enchondroma and low grade chondrosarcoma
Radiographic appearance
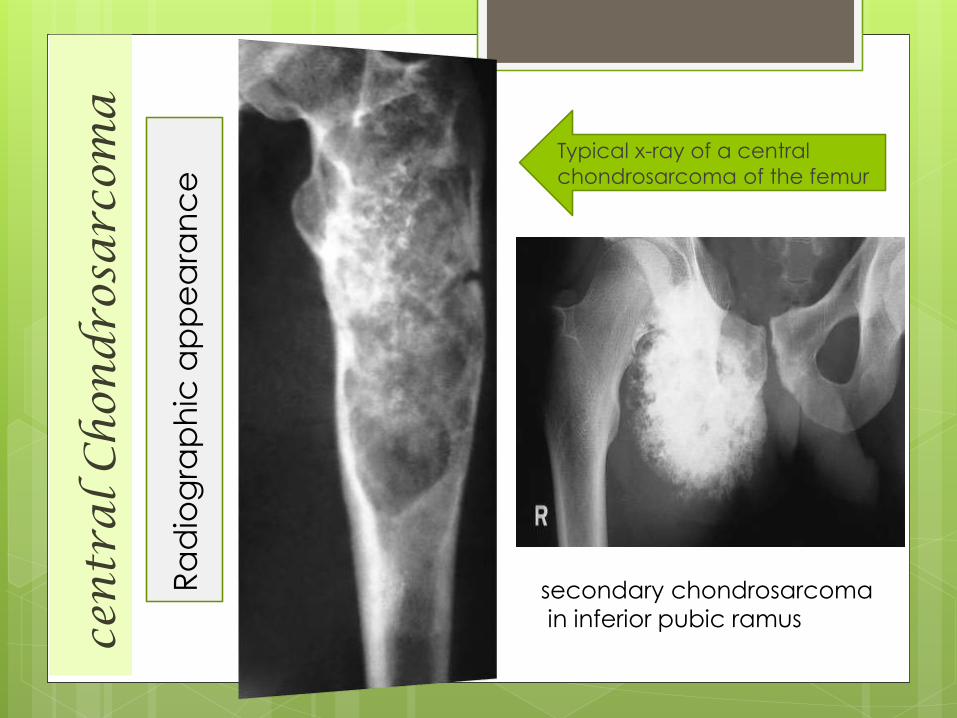
cen
tra
l C
hon
dro
sarc
oma
Ra
dio
gra
ph
ic a
pp
ea
ran
ce
Typical x-ray of a central
chondrosarcoma of the femur
secondary chondrosarcoma
in inferior pubic ramus
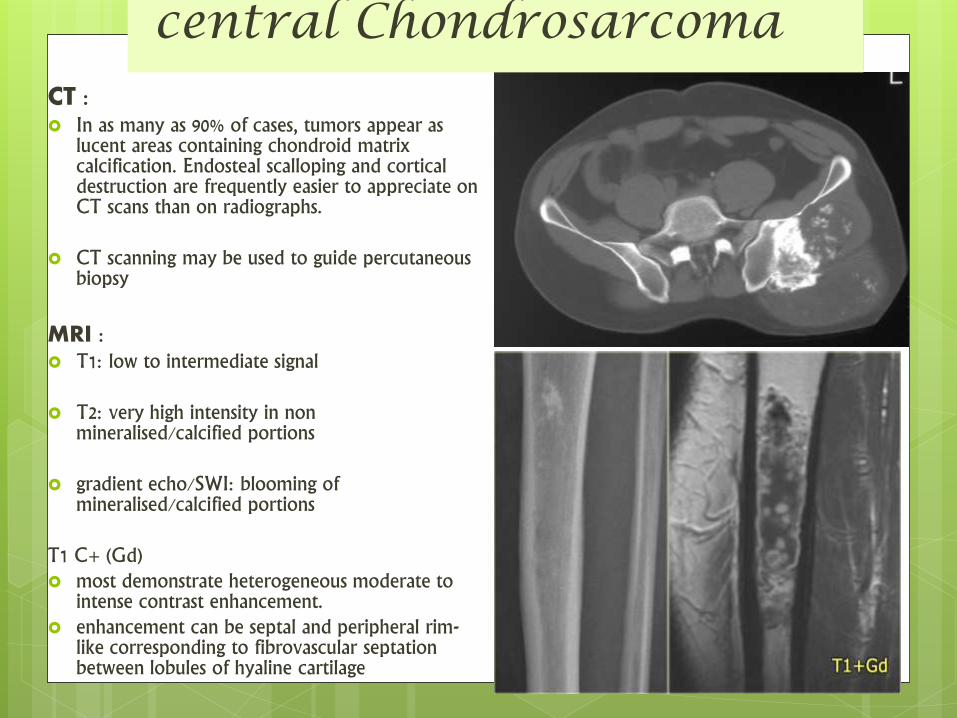
CT : In as many as 90% of cases, tumors appear as
lucent areas containing chondroid matrix calcification. Endosteal scalloping and cortical destruction are frequently easier to appreciate on CT scans than on radiographs.
CT scanning may be used to guide percutaneous biopsy
MRI : T1: low to intermediate signal
T2: very high intensity in non mineralised/calcified portions
gradient echo/SWI: blooming of mineralised/calcified portions
T1 C+ (Gd) most demonstrate heterogeneous moderate to
intense contrast enhancement. enhancement can be septal and peripheral rim-
like corresponding to fibrovascular septationbetween lobules of hyaline cartilage
central Chondrosarcoma
central Chondrosarcoma
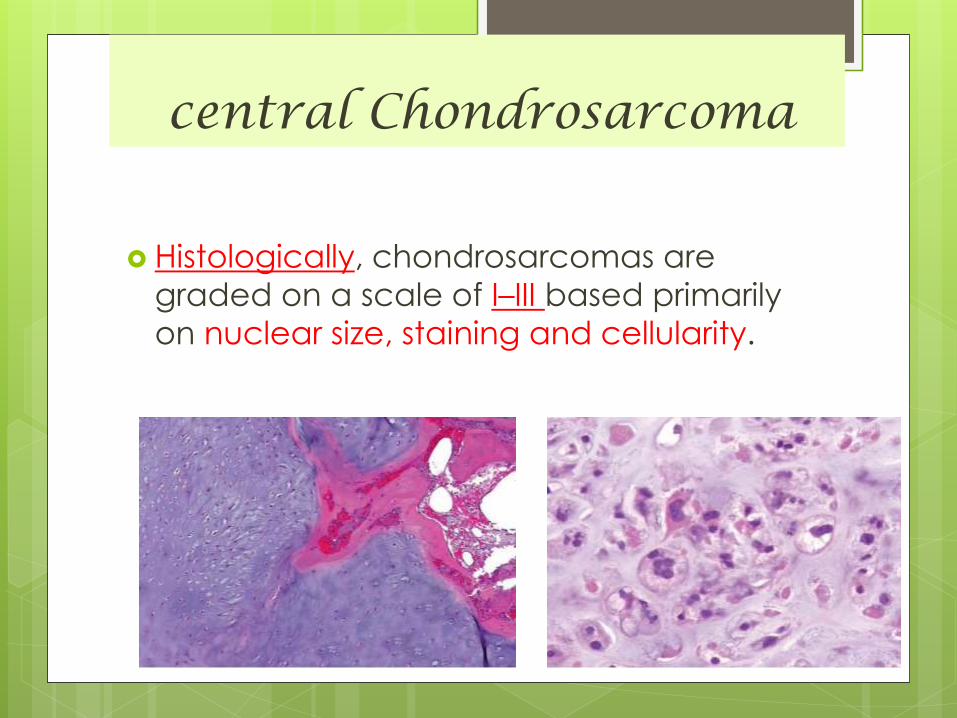
Histologically, chondrosarcomas are
graded on a scale of I–III based primarily
on nuclear size, staining and cellularity.

central Chondrosarcoma
Histologically, chondrosarcomas are
graded on a scale of I–III based primarily
on nuclear size, staining and cellularity.
Wide range of differentiation and graded
into:
- well differentiated
- moderately differentiated
- poorly differentiated

central Chondrosarcoma

Well-differentiated chondrosarcoma. The
tumor has a distinctly lobulated quality
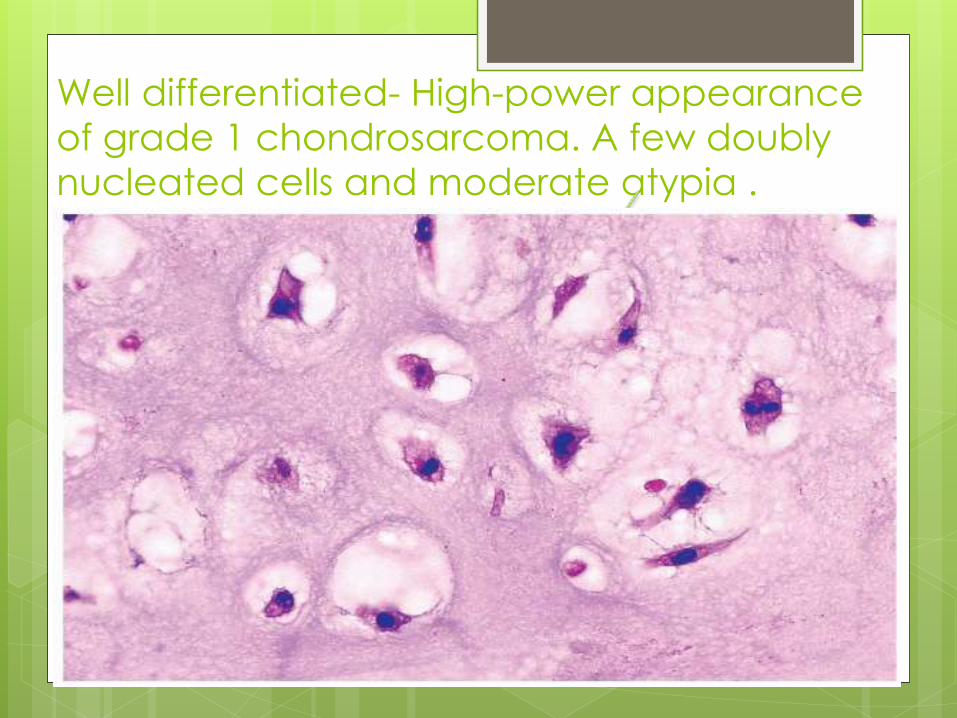
Well differentiated- High-power appearance
of grade 1 chondrosarcoma. A few doubly
nucleated cells and moderate atypia .

Moderately differentiated -High-power appearance of grade 2
chondrosarcoma with necrosis . The nuclei
are crowded and hyperchromatic
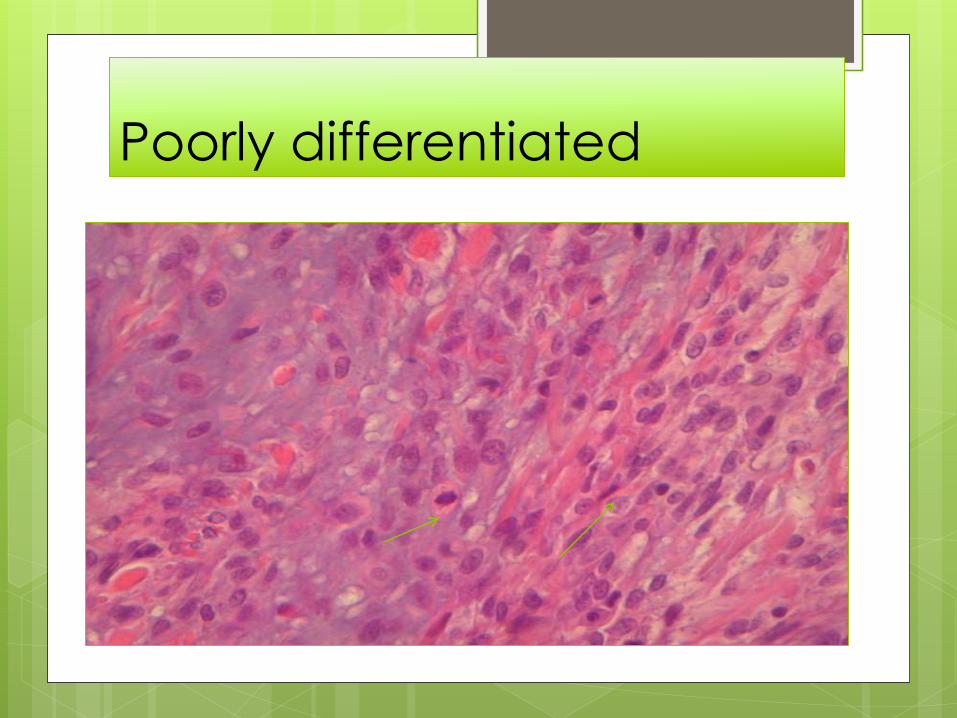
Poorly differentiated

Chondrosarcoma(classification )
Peripheral chondrosarcoma
Tumor is present on the surface of bone.
May arise de-novo or from cartilaginous cap of
preexisting osteochondroma .
X-rays show :
the bony exostosis, often surmounted by clouds of
patchy calcification
in the otherwise unseen lobulated cartilage cap.
MRI is the best means of showing the size and internal
features of the cartilage cap.
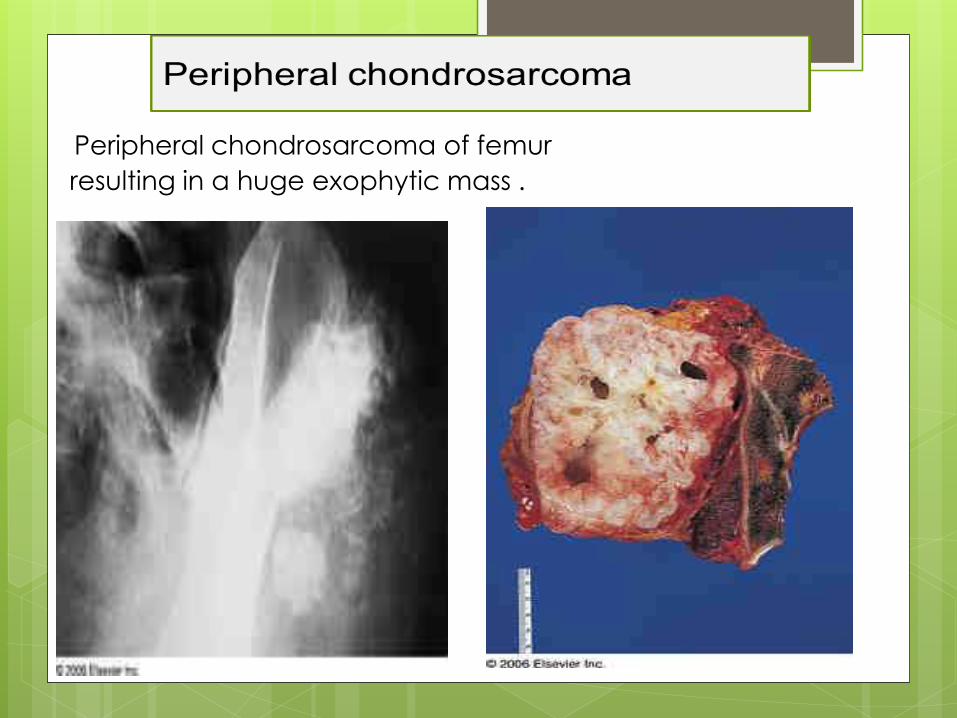
Peripheral chondrosarcoma of femur
resulting in a huge exophytic mass .

Chondrosarcoma(classification )
Juxtacortical (periosteal ) chondrosarcoma
the lesion appears as an excrescence on the surface of one
of the tubular bones – usually the femur.
It arises from the outermost layers of the cortex, deep to the
periosteum .
Cartilaginous lobular pattern with areas of:
spotty calcification
endochondral ossification
Closely related to periosteal osteosarcoma .
Chondrosarcoma(classification )
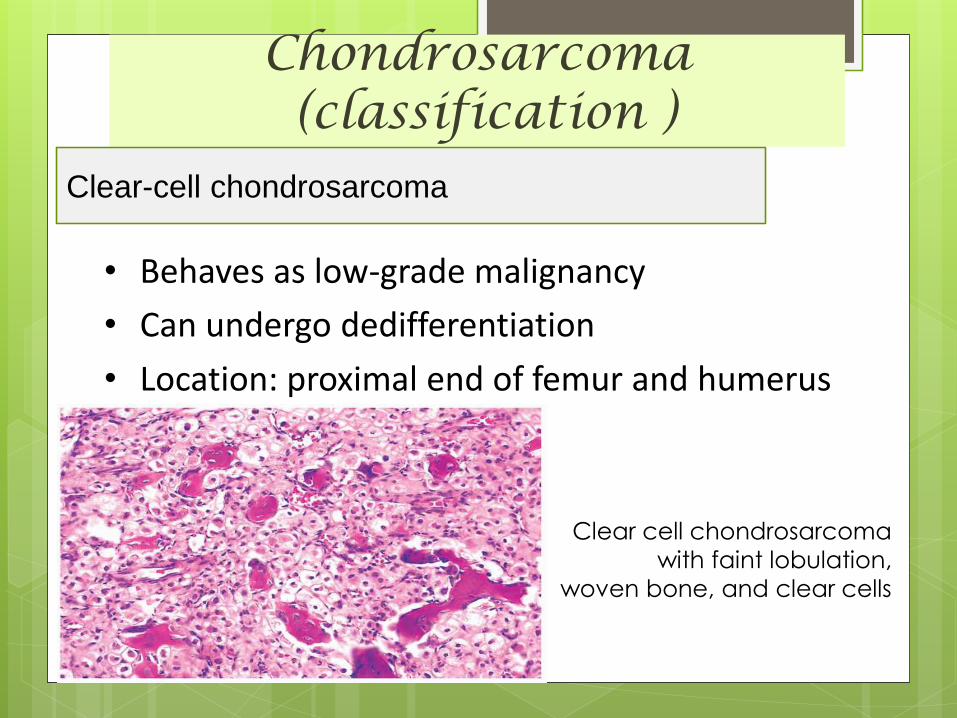
Clear-cell chondrosarcoma
• Behaves as low-grade malignancy
• Can undergo dedifferentiation
• Location: proximal end of femur and humerus
Clear cell chondrosarcoma
with faint lobulation,
woven bone, and clear cells

Chondrosarcoma(classification )
Mesenchymal chondrosarcoma
This is an equally controversial entity.
It tends to occur in younger individuals .
and in about 50 per cent of cases the tumor lies in the
soft tissues outside an adjacent bone.
The x-ray appearances are similar to those of the
common types of chondrosarcoma
but the clinical behavior of the tumor is usually more
aggressive.
Histology shows a mixture of mesenchymal cells and
chondroid tissue

Chondrosarcoma(classification )
Dedifferentiated chondrosarcoma:
Worst prognosis.
Age/sex: sixth decade/ M:F =1:1.
Bones involved: pelvis, femur.
Poorly differentiated sarcomatouscomponent at periphery of otherwise typical low-grade chondrosarcoma
usually central type
can be peripheral

Chondrosarcoma(treatment)
• These tumors are not generally sensitive to chemotherapy and radiotherapy.
• Surgery is the only reliable treatment with wide excision and prosthetic replacement, for these tumors.
• CHONDROSARCOMA IN SITU:
can be treated with intra lesional excision
coupled with adjuvants like phenol or
cryotherapy instead of wide excision .

Chondrosarcoma(prognosis)
• Most of pulmonary metastasis or local reccurences occur with in first five years of presentation.
• Survival rates at the end of 10 years were 89% for grade 1 ,
53% for grade2
and 38% for grade3 chondrosarcomas .
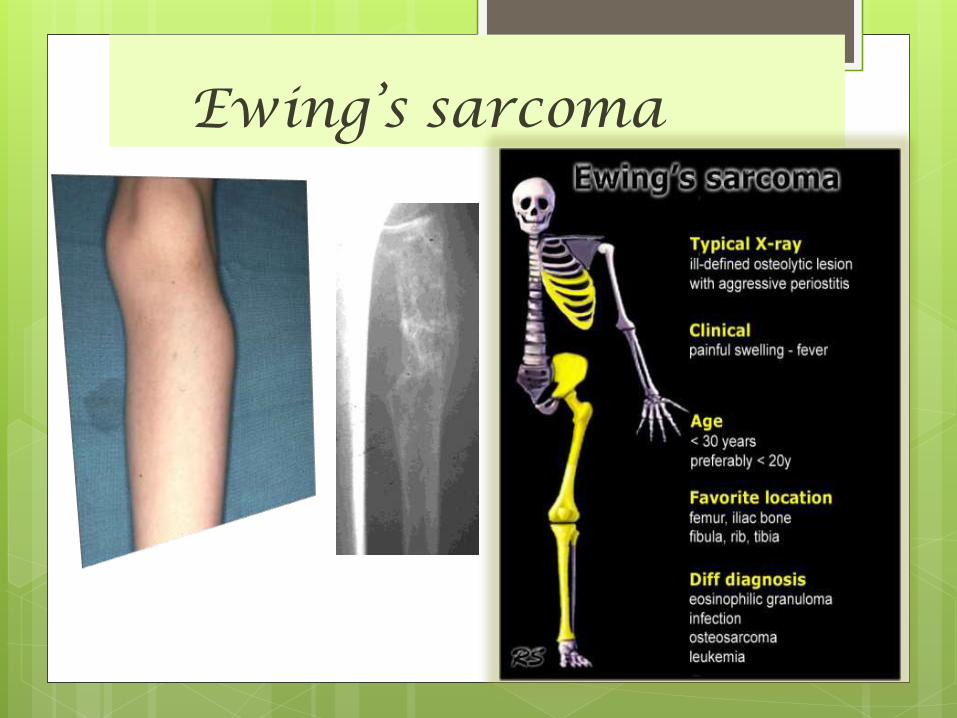
Ewing’s sarcoma

Ewing’s sarcoma Ewing’s sarcoma (ES) was first described by James Ewing
in 1921 as
a "diffuse endothelioma of bone"
He observed that this highly aggressive bone
cancer was remarkably sensitive to radiation
therapy.
Ewing sarcoma, a highly malignant primary bone tumor
that is derived from red bone marrow second most
common primary bone tumour of childhood.
Ewings sarcoma is thought to be of either neuro-
ectodermal or stem cell origin
All Ewings sarcoma’s are considered high grade malignancy

Ewing’s sarcoma
9% of primary bone sarcomas
4th most common primary malignancy of bone but
2nd most common below 30yrs.
Age Group – 95% patients age between 5 to 30 yrs
- of these most range between 5 to 15 yrs
Sex - slight male predilection – 60%
More common in Whites (95%)
No known predisposing factor
Chromosomal translocation – t(11;22)(q24;q12)
leading to fusion of EWS and FLI gene.
also t(21;22) and t(7;22)
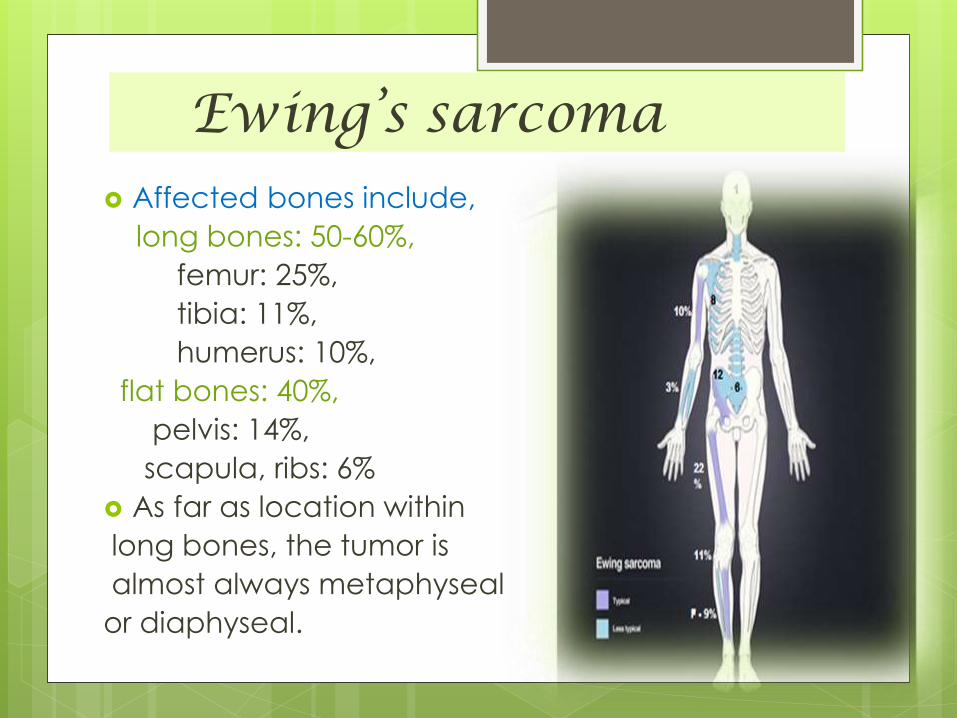
Ewing’s sarcoma Affected bones include,
long bones: 50-60%,
femur: 25%,
tibia: 11%,
humerus: 10%,
flat bones: 40%,
pelvis: 14%,
scapula, ribs: 6%
As far as location within
long bones, the tumor is
almost always metaphyseal
or diaphyseal.

Ewing’s sarcoma
The patient presents with pain
– often throbbing in character –
and swelling. May also have systemic symptoms:
Fever
Anemia
Weight loss
Elevated WBC & ESR,LDH
Pathological fracture .
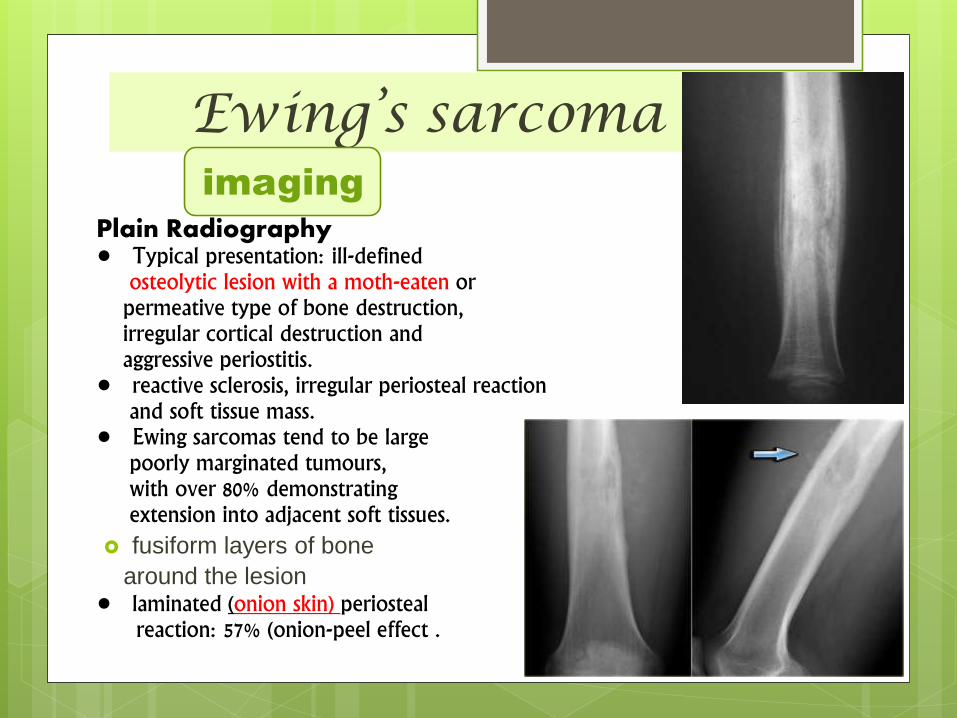
Ewing’s sarcoma
Plain Radiography• Typical presentation: ill-defined
osteolytic lesion with a moth-eaten orpermeative type of bone destruction,irregular cortical destruction andaggressive periostitis.
• reactive sclerosis, irregular periosteal reaction and soft tissue mass.
• Ewing sarcomas tend to be largepoorly marginated tumours,with over 80% demonstratingextension into adjacent soft tissues.
fusiform layers of bone
around the lesion • laminated (onion skin) periosteal
reaction: 57% (onion-peel effect .
imaging
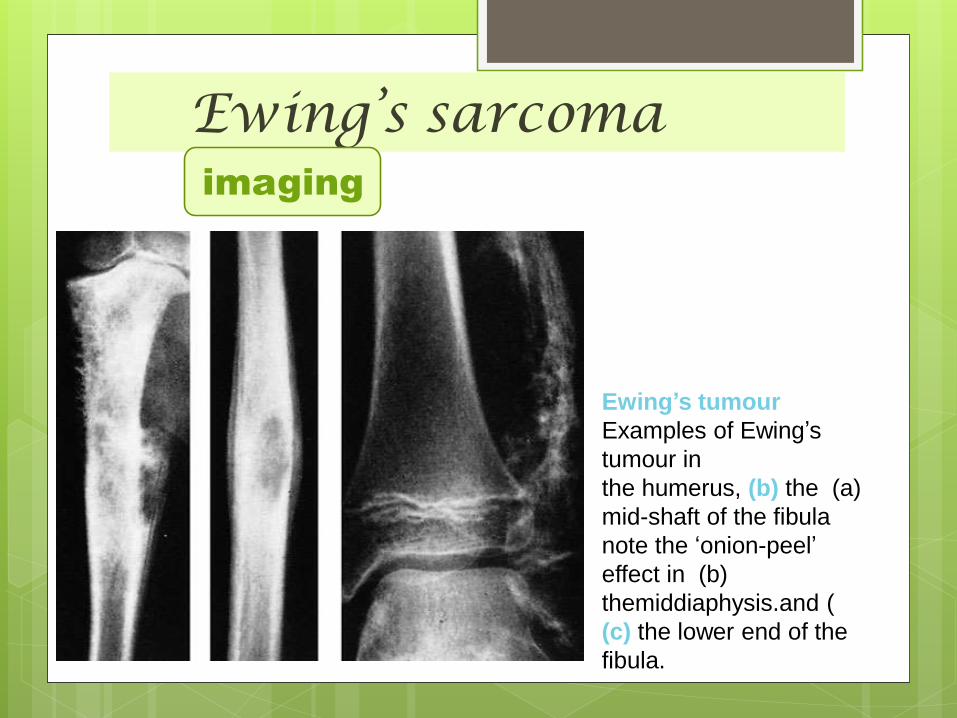
Ewing’s sarcomaimaging
Ewing’s tumour
Examples of Ewing’s
tumour in
(a)the humerus, (b) the
mid-shaft of the fibula
note the ‘onion-peel’
(b)effect in
themiddiaphysis.and (
(c) the lower end of the
fibula.
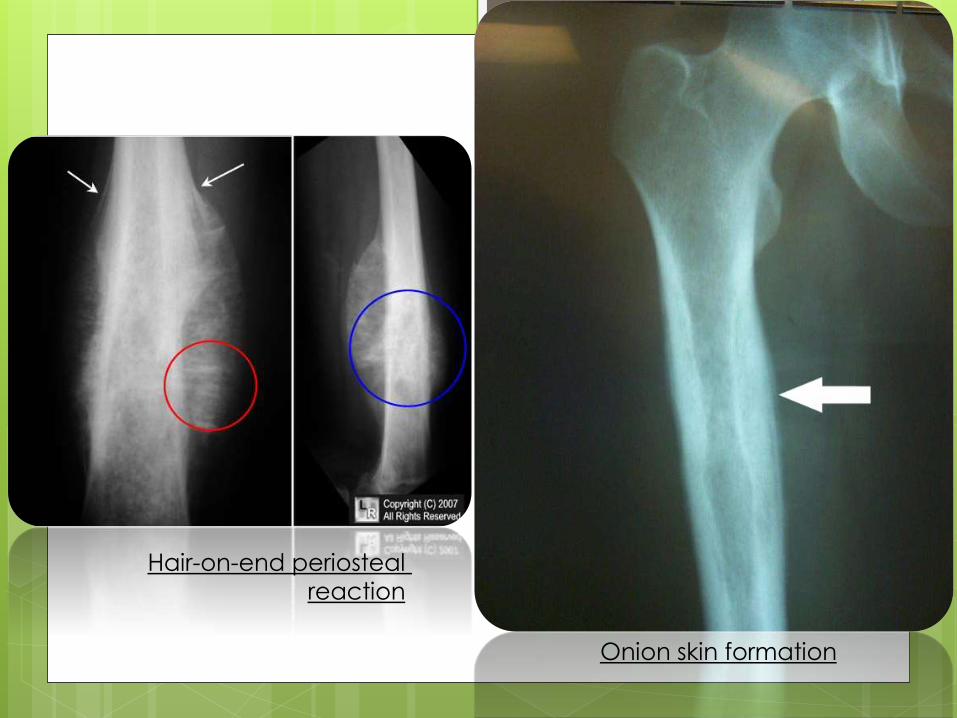
Hair-on-end periosteal
reaction
Onion skin formation
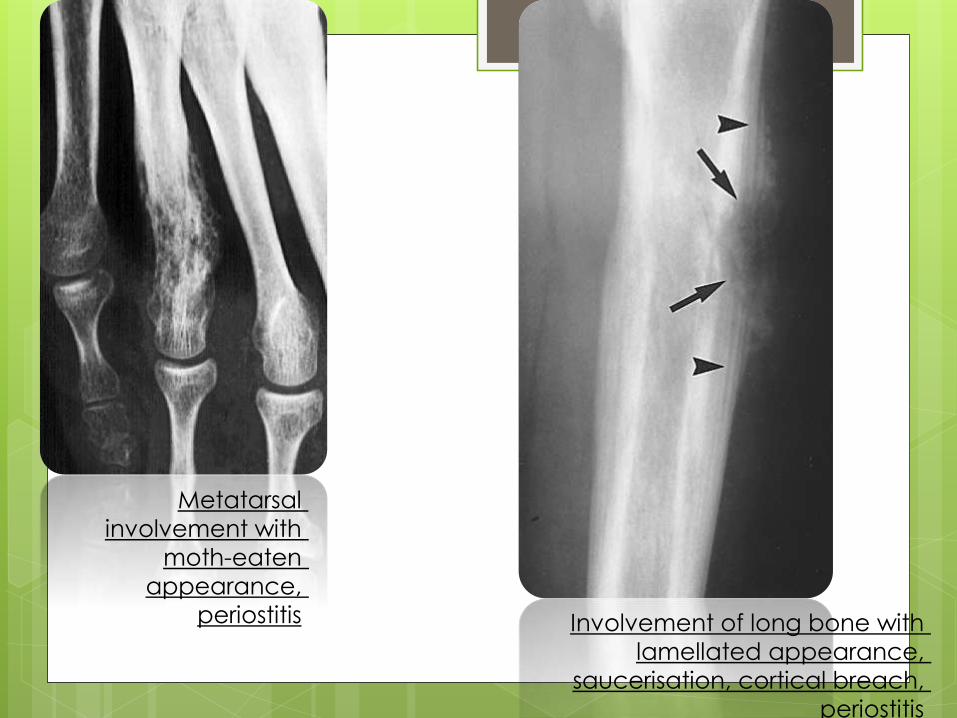
Metatarsal
involvement with
moth-eaten
appearance,
periostitis Involvement of long bone with
lamellated appearance,
saucerisation, cortical breach,
periostitis
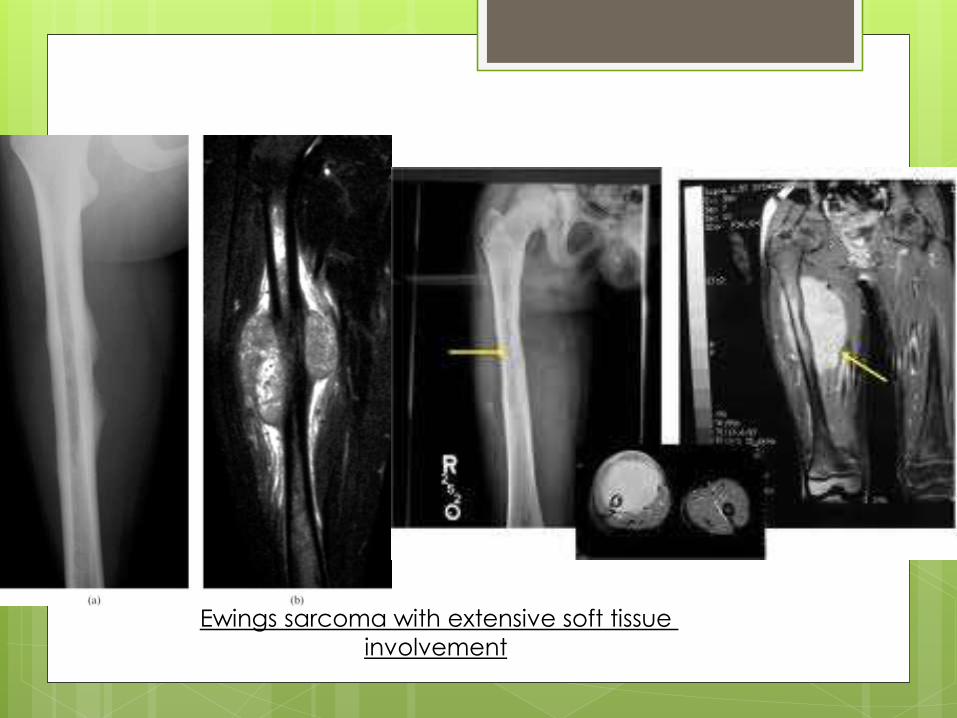
Ewings sarcoma with extensive soft tissue
involvement
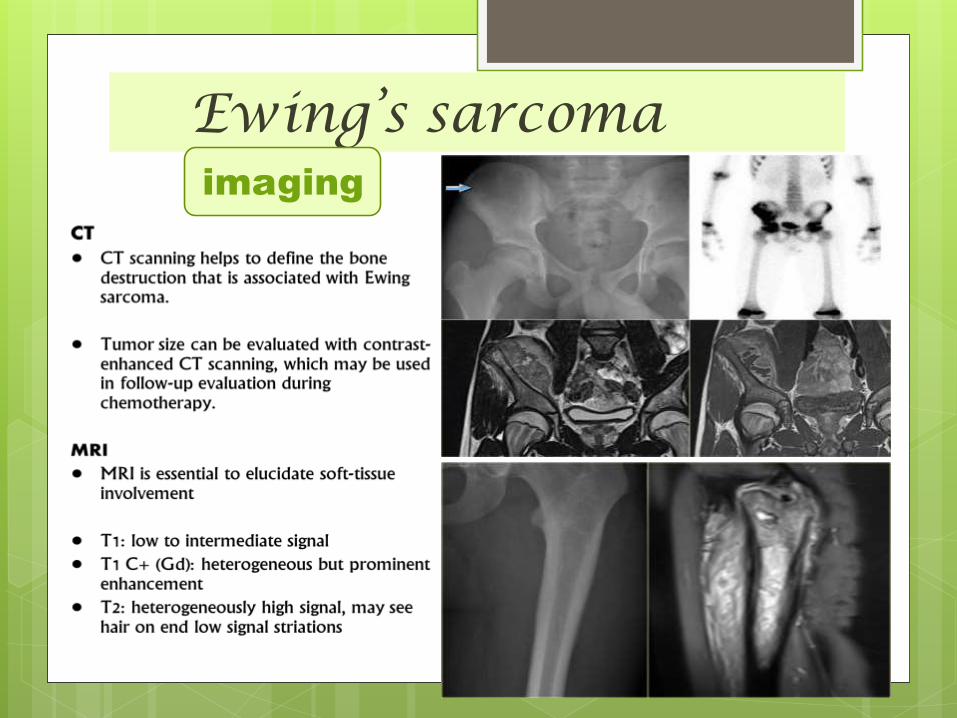
Ewing’s sarcomaimaging

Ewing’s sarcoma
• Greyish-white to pink in color
• soft, friable
• Often of semi-liquid consistency
macroscopy
• sheets of small dark polyhedral cells with no regular arrangement and no ground substance are seen.
microscopy
pathology
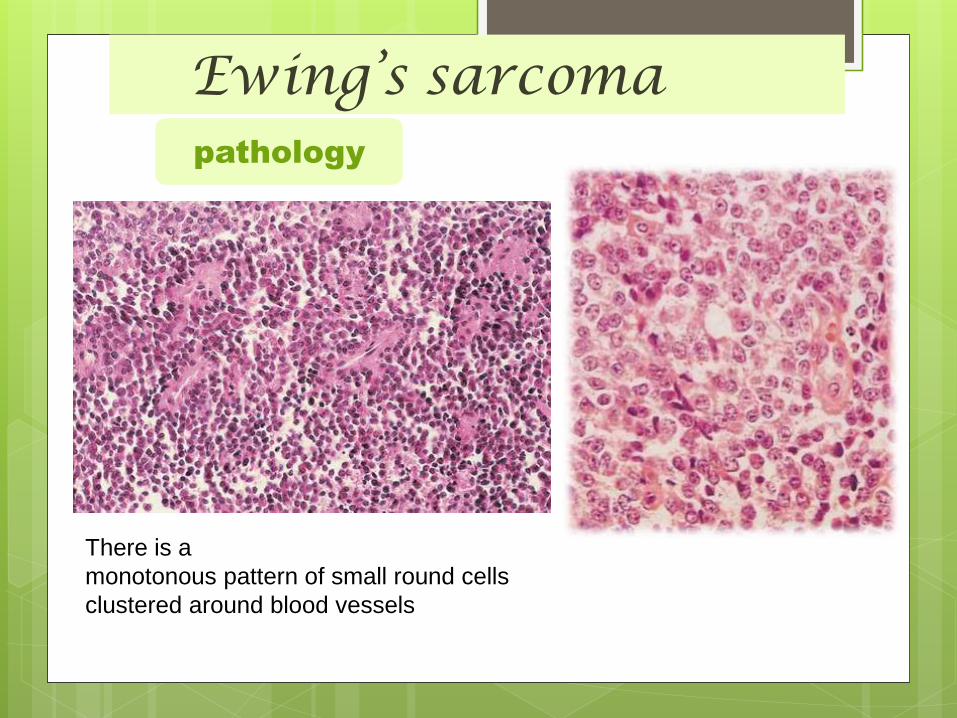
Ewing’s sarcomapathology
There is a
monotonous pattern of small round cells
clustered around blood vessels

Differential diagnosis
Osteosarcoma
Lymphoma
Leukemia
Osteomyelitis
PNET

Ewing’s sarcomatreatment
best results are achieved by a combination
of :
- a course of preoperative neo-adjuvant
chemotherapy;
- then wide excision if the tumor is in a
favourable site,
- or radiotherapy followed by local
excision if it is less accessible;
- and then a further course of
chemotherapy for 1 year.

Ewing’s sarcomaprognosis
The prognosis is always poor , esp. if
Proximal segment involvement
Sacral involvement
Patients above 18 yrs
Increased LDH and ESR
Size greater than 8cm
The prognosis has improved dramatically since the introduction of multi-agent chemotherapy
– from an erstwhile 10 per cent survival rate to the current 70 per cent for patients with nonmetastatic
Ewing’s sarcoma.
NON-HODGKIN’S LYMPHOMA(RETICULUM-CELL
SARCOMA )

NON-HODGKIN’S LYMPHOMA(RETICULUM-CELL SARCOMA)
Rare malignant condition that accounts for less than
5% of all primary bone tumors .
Like Ewing’s sarcoma, this is a round-cell tumor of
the reticuloendothelial system.
It occurs in the second to seventh decades, with a
peak age of occurrence from 45 to 75 years. M>F
It has also been called reticulum cell sarcoma,
malignant lymphoma of the bone, and more
recently osteolymphoma.
Distinguishing primary bone lymphoma from other
bone tumors is important because the former has a
better response to therapy and a better prognosis.
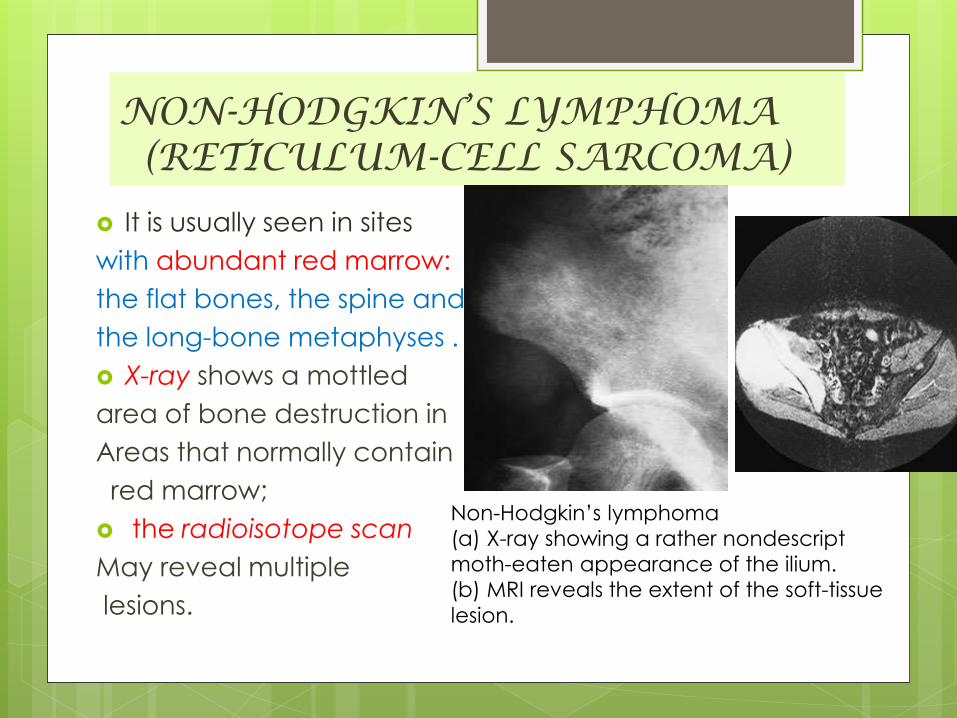
NON-HODGKIN’S LYMPHOMA(RETICULUM-CELL SARCOMA)
It is usually seen in sites
with abundant red marrow:
the flat bones, the spine and
the long-bone metaphyses .
X-ray shows a mottled
area of bone destruction in
Areas that normally contain
red marrow;
the radioisotope scan
May reveal multiple
lesions.
Non-Hodgkin’s lymphoma
(a) X-ray showing a rather nondescript
moth-eaten appearance of the ilium.
(b) MRI reveals the extent of the soft-tissue
lesion.
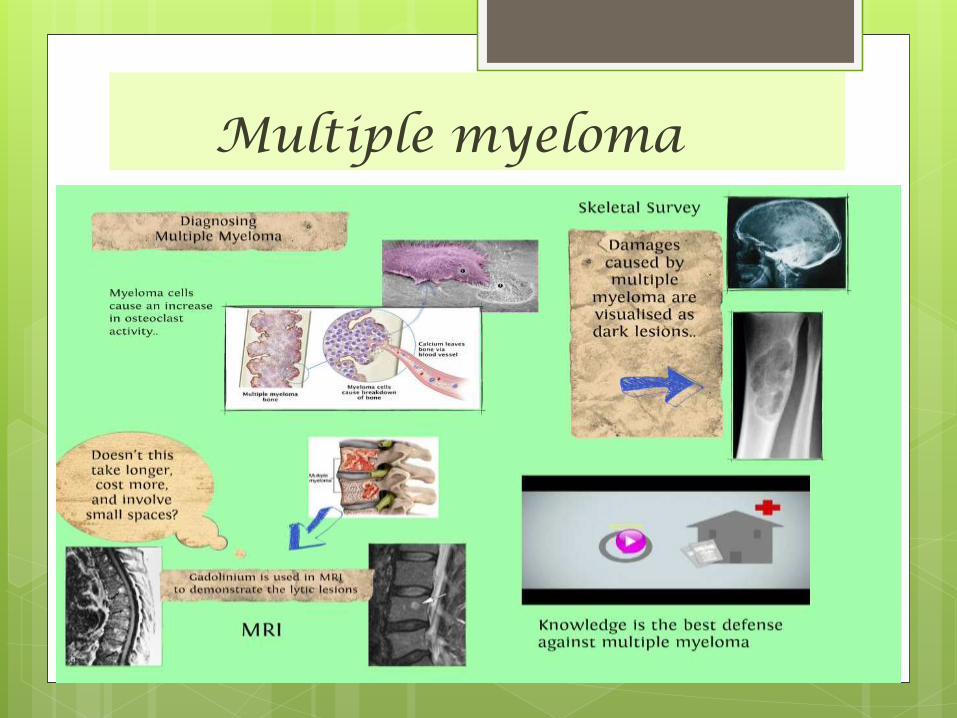
Multiple myeloma

Multiple myeloma Multiple myeloma is the most common primary malignant
neoplasm of the skeletal system. The disease is a malignancy of plasma cells “plasma cell myeloma,”
IT is a malignant B-cell lymphoproliferative disorder of the marrow, with plasma cells predominating, resulting in the overproduction of monoclonal immunoglobulins .
The effects on bone are due to marrow cell proliferation and increased osteoclastic activity, resulting in osteoporosis and the appearance of discrete lytic lesions throughout the skeleton.
Remember that myeloma is one of the commonest causes of osteoporosis and vertebral compression fracture in men over the age of 45 years.
Average age is 50-70 yrs
Much more common in men than women .
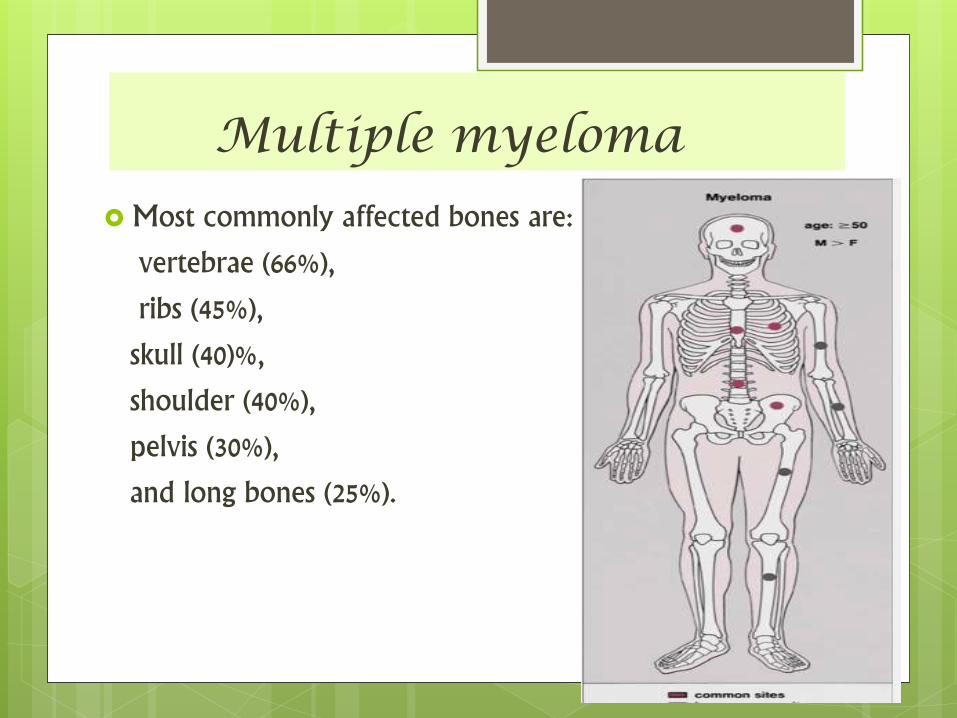
Multiple myeloma
Most commonly affected bones are:
vertebrae (66%),
ribs (45%),
skull (40)%,
shoulder (40%),
pelvis (30%),
and long bones (25%).
Multiple myeloma Associated features of the marrow-cell disorder are
- plasma protein abnormalities, and Bence-Jones protein in 40-60% of pts .
- increased blood viscosity and anaemia.
- Bone resorption leads to hypercalcaemia in 1/3 of cases.
Late secondary features are
- due to renal dysfunction and spinal cord or root compression
caused by vertebral collapse.
presents with :
weakness, backache, bone pain or a pathological fracture.
Hypercalcaemia may cause symptoms such as
thirst, polyuria and abdominal pain .

Multiple myeloma
Mu
ltip
le m
yel
oma
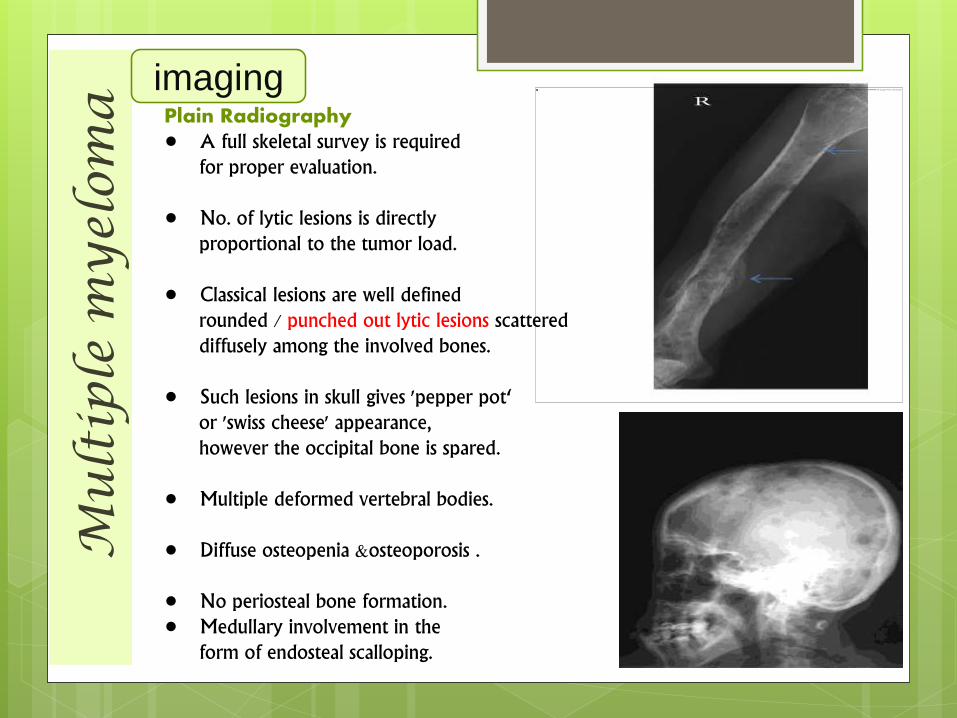
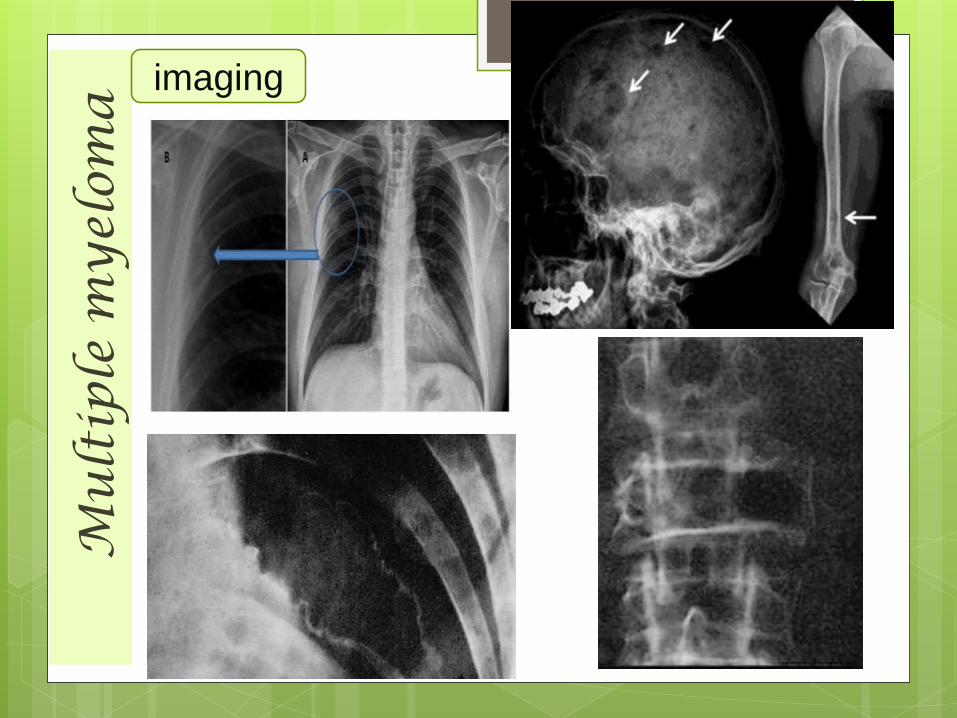
Plain Radiography• A full skeletal survey is required
for proper evaluation.
• No. of lytic lesions is directlyproportional to the tumor load.
• Classical lesions are well definedrounded / punched out lytic lesions scattereddiffusely among the involved bones.
• Such lesions in skull gives 'pepper pot‘or 'swiss cheese' appearance,however the occipital bone is spared.
• Multiple deformed vertebral bodies.
• Diffuse osteopenia &osteoporosis .
• No periosteal bone formation.• Medullary involvement in the
form of endosteal scalloping.
imaging
Mu
ltip
le m
yel
oma
imaging

Mu
ltip
le m
yel
oma
imaging
CT Computed tomography (CT) scanning readily
depicts osseous involvement in myeloma.
CT allowed a more accurate evaluation of areas at risk of fracture.
Tool of choice utilised in image guided spinal or pelvic bone biopsy.
MRI Most sensitive imaging modality at detecting
diffuse and focal multiple myeloma in the spine, as well as the extra-axial skeleton
Mainly bone marrow based lesions.
T1WI - Low signal intensity
T2WI and STIR - High signal intensity.
Show enhancement on contrast enhanced images.

Lab findings Anemia, leukopenia, thrombocytopenia
ALb, reversed A:G ratio
serum creat, uric acid, urea
Abnormal coagulation
Serum Ca
Proteinuria and cast
ESR
LOW NORMAL ALKALINE PHOSPHATASE
Red cells show rouleaux formation
BENCE-JONES PROTEIN in urine in 30%
Serum electrophoresis

Mu
ltip
le m
yel
oma
Major criteria: A biopsy-proven plasmacytoma.
A bone marrow sample
showing 30% plasma cells.
Elevated monoclonal Ig levels
in the blood or urine
Minor criteria: A bone marrow 10%-30% plasma cells.
Minor monoclonal Ig levels in blood or urine
Imaging --- holes in bones due to tumor growth
Antibody levels (not produced by the cancer cells) in the blood are abnormally low.
Diagnosis

Multiple myeloma
Durie-Salmon system :
clinical stage of disease (stage I, II, or III) is based
levels of M protein,
number of lytic bone lesions,
hemoglobin values and serum calcium levels.
Stages are further divided (A/B) according to renal function
International Staging
System (ISS)
new, simpler, more
cost-effective :
beta 2-micro
globulin (β2-M) and
albumin
staging

Multiple myeloma Stage Durie-Salmon Criteria ISS Criteria
I • Hb >10 g/dL
• S. Ca++≤12 mg/dL
• x-ray, normal bone stru.
or solitary bone plasmacytoma
• Low M-component production rate —
( IgG <5 g/dL; IgA <3 g/dL)
• Bence Jones protein <4 g/24 h
β2-M < 3.5 mg/dL and
albumin ≥3.5 g/dL
II* Neither stage I nor stage III Neither stage I nor stage III
III •Hb <8.5 g/dL
•S. Ca++
>12 mg/dL
•Advanced lytic bone lesions
•High M-component production rate —
(IgG>7 g/dL; IgA>5 g/dL)
•Bence Jones protein >12 g/24 h
β2-M ≥ 5.5 mg/dL
*Stage II = β2-M <3.5 or β2-M 3.5 – 5.5 mg/dL, and albumin <3.5 g/dLsub classification A) S. Creat.<2.0 mg/dl &
B) S. Creat.>2.0 mg/dl

Mu
ltip
le m
yel
oma
Management
Supporative :- The immediate need is for pain
control and, if necessary,
treatment of pathological
fractures.
- correction of fluid balance
and hypercalcaemia.
- antibiotic prophylaxis is important as there is a
higher than usual risk of infection and wound
breakdown.

Mu
ltip
le m
yel
oma
Management
Radiation :- Myeloma is radiosensitive
- Eventually looses its susceptibility
- Relieves pain
- Can be used for control of local disease (Solitary
plasmacytomas )
- Total body irradiation not advised .
Surgical options :
- Compression of intra-spinal nerves >>>
laminectomy, removal of myelomatoustissue
and post-op irradiation
- In cases with instability spinal fusion

Mu
ltip
le m
yel
oma
Management
Systemic anti-myeloma treatment :- alkylating cytotoxic agents (e.g. melphalan) with
Prednisone (has stem cell toxicity ) .
If stem cell transplantation is NOT planned :
- Melphalan/prednisone/thalidomide (MPT)
- Bortezomib/melphalan/prednisone (VMP)
- Thalidomide/dexamethasone(thaldex)
- levalidomide/low dose dexa (Revlodex)
If stem cell harvest is planned
- Bortezomib/thalidomide/dexamethasone (VTD)
- VCD -VELCADE/Cyclophosphamide/Dexa
- VRD - VELCADE/Revlimid/Dexa

Mu
ltip
le m
yel
oma
Management
Newer chemotherapy drugs for MM:
- Pomalidomide
- Next-generation proteasome
inhibitors- carfilzomib, NPI-0052
- Doxil-pegylated liposomal doxorubicin
- Histone deacetylase (HDAC) inhibitors
ex. vorinostat, panobinostat
- Monoclonal antibodies-elotuzumab

Mu
ltip
le m
yel
oma
Management
Maintenance therapy :
Alpha Interferon
Prednisone
Melphalan
Velcade
Revlimid
Dexamethasone
The prognosis in established cases is poor, with
a median survival of between 2 and 5 years.
Prognosis

Rajkumar et al. Mayo Clin Proc. 2002;77:814

Management of Complications
• UREMIA: rehydratation, diuretics,steroids,antibiotics if renal infection is suspected, hemodialysis if these measures fail.
• HYPERCALCEMIA: rehydratation, steroids, bisphosphonates, diuretics.
• PARAPLEGIA: decompressive laminectomy, radiotherapy, chemotherapy.
• BONE LESIONS: if painful and localised, chemo or local radio-therapy, analgetics, biphosphonates.
• SEVERE ANEMIA: transfusions, erytropoetin
• HYPERVISCOSITY SYNDROME: plasmapheresis, correction of hypercalcemia.
• BLEEDING: platelet concentrates, fresh frozen plasma
• INFECTIONS: antibiotic treatment


Adamantinoma Rare primary malignant bony tumor, only approximately 200 cases have been
reported.
The tumor occurs almost exclusively in the long bones; tumors in the tibia account for more than 80% of cases. The diaphyseal region is the area most commonly affected. but is occasionally found in other long bones.
Typically presents in the 2nd to 3rd decades as a locally aggressive mass 3-15 cm in diameter .
Adamantinoma is a low-grade tumor which metastasizes late
It may present as a solitary focus or multicentric lucencies or slightly expansileosteolytic lesion
May extend into the marrow cavity.
Lesions tend to have an eccentric epicenter and a lack of periosteal reaction.
usually no periosteal reaction is noted in the surrounding bone
Long-standing tumors produce marked cortical thickening and spool-shaped bulges of the outer cortex in an eggshell fashion.
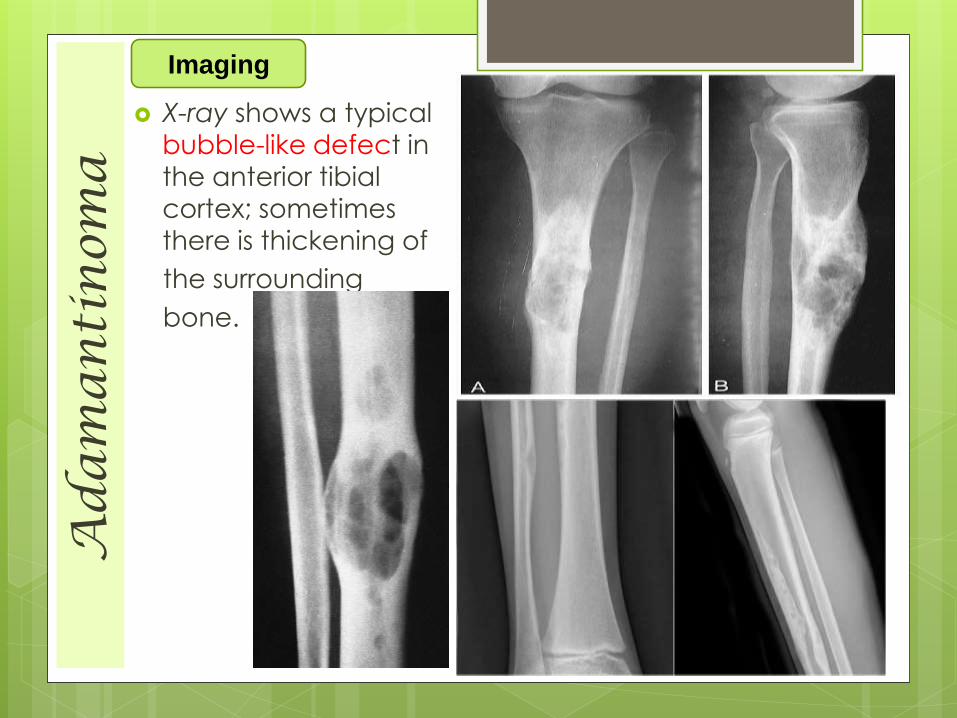
Ad
am
an
tin
oma
X-ray shows a typical
bubble-like defect in the anterior tibial
cortex; sometimes
there is thickening of
the surrounding
bone.
Imaging
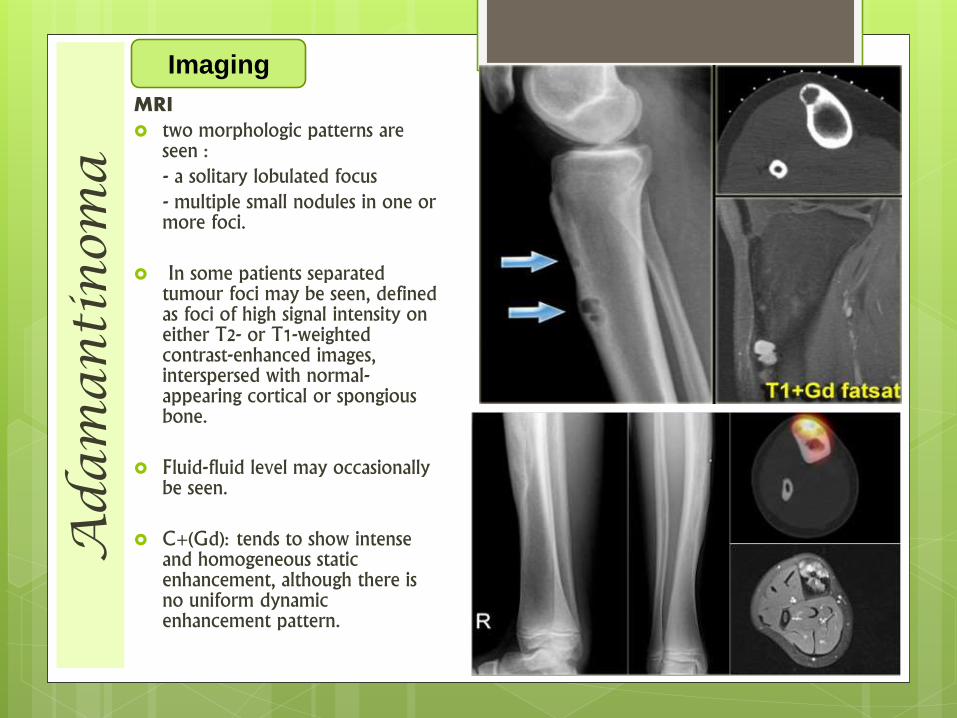
Ad
am
an
tin
oma
MRI two morphologic patterns are
seen :- a solitary lobulated focus - multiple small nodules in one or more foci.
In some patients separated tumour foci may be seen, defined as foci of high signal intensity on either T2- or T1-weighted contrast-enhanced images, interspersed with normal-appearing cortical or spongiousbone.
Fluid-fluid level may occasionally be seen.
C+(Gd): tends to show intense and homogeneous static enhancement, although there is no uniform dynamic enhancement pattern.
Imaging

Adamantinoma
If the diagnosis is made reasonably early,
wide local excision with a substantial margin of
normal bone is adequate .
- the gap is filled with a vascularized graft or a suitable
endoprosthesis, or managed by distraction
osteogenesis .
If there has been more than one recurrence, or if
the tumor extends into the surrounding soft
tissues, radical resection or amputation is
advisable.
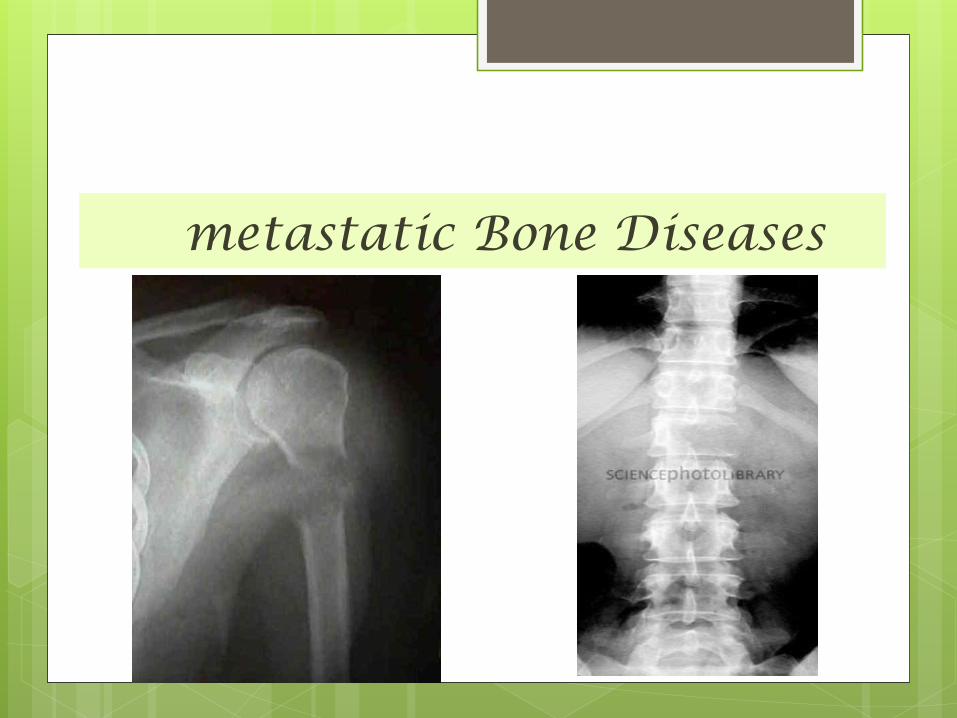
metastatic Bone Diseases

metastatic Bone Diseases The skeleton is one of the commonest sites of secondary
cancer .
in patients over 50 years bone metastases are seen more
frequently than all primary malignant
bone tumours together. (Approximately 70% of all malignant
tumors are metastatic in origin. )
Metastases are usually osteolytic, and pathological
fractures are common .
Bone resorption is due either to the direct action of tumor
cells or to tumor-derived factors that stimulate osteoclastic
activity.
Osteoblastic lesions are uncommon; they usually occur in prostatic carcinoma.

metastatic Bone Diseases
Sites :
- The commonest sites for bone metastases
are the vertebrae, pelvis, the proximal half
of the femur and the humerus.
Sources :- The commonest source is carcinoma of the breast; next
in frequency are carcinomas of the prostate, kidney,
lung, thyroid, bladder and gastrointestinal tract.
- In about 10 per cent of cases no primary tumor
is found.

metastatic Bone Diseases
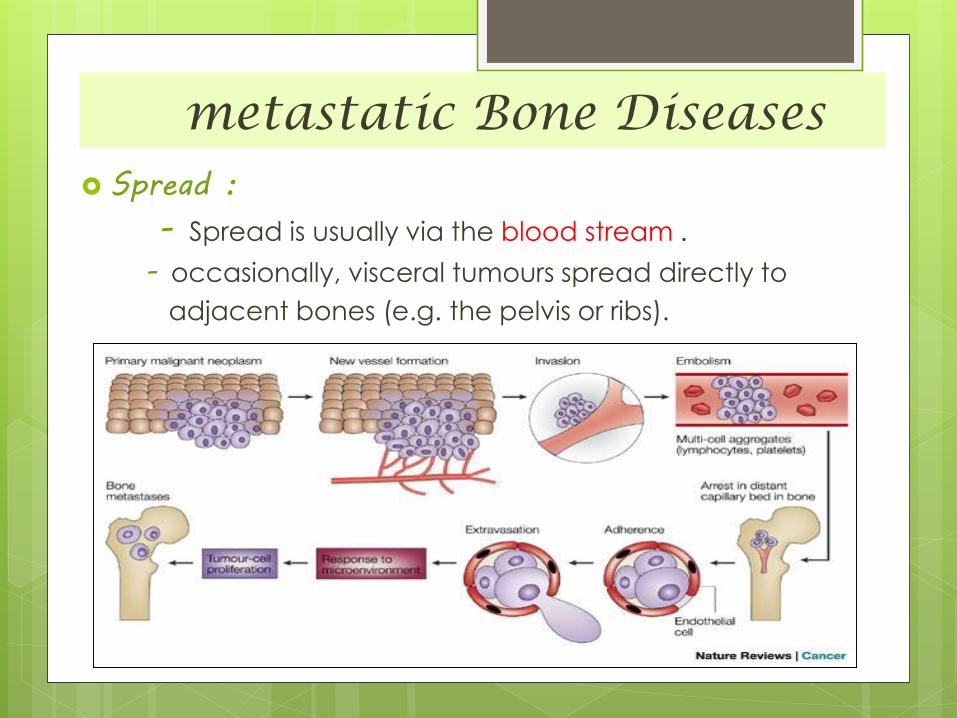
metastatic Bone Diseases Spread :
- Spread is usually via the blood stream .
- occasionally, visceral tumours spread directly to
adjacent bones (e.g. the pelvis or ribs).

metastatic Bone Diseases types :
- osteolytic metastases- sclerotic/osteoblastic metastases- mixed lytic and sclerotic metastases
Osteoblastic metastases Osteolytic metastases Mixed metastases
prostate carcinoma (most common) RCC Lung carcinoma (25%)breast carcinoma (may be mixed) Thyroid carcinoma Breast carcimoma (15%)transitional cell carcinoma (TCC) Pheochromocytoma carcinoma of cervixcarcinoid Wilms tumor testicular tumorsmedulloblastoma Ewings sarcoma Prostatic carcinoma (15%)neuroblastoma Carcinomas of GITmucinous adenocarcinoma of GIT Melanomalymphoma HCC
SCC of skin
Uterine carcinoma

metastatic Bone Diseases Imaging :
• little or no soft tissue mass associated with them• Usually no periosteal reaction• May appear as moth-eaten, permeative or geographic lesions• Indistinct zones of transition, no sclerotic margins and may be
sharply circumscribed or have indistinct borders• Lesions distal to elbows and knees - 50% are from lung and breast• Diffuse skeletal sclerosis or multiple round, well-circumscribed
sclerotic lesions - Prostate & Breast• Expansile and lytic (soap-bubbly) - RCC• Cookie-bite lesions of the cortices of long bones - Lung• Bone scans are extremely sensitive but not very specific• 10-40% of lesions will not be visible on plain film but will be positive
on bone scans• CT or MRI can be used to show findings in patients with negative
conventional radiographs and positive bone scans
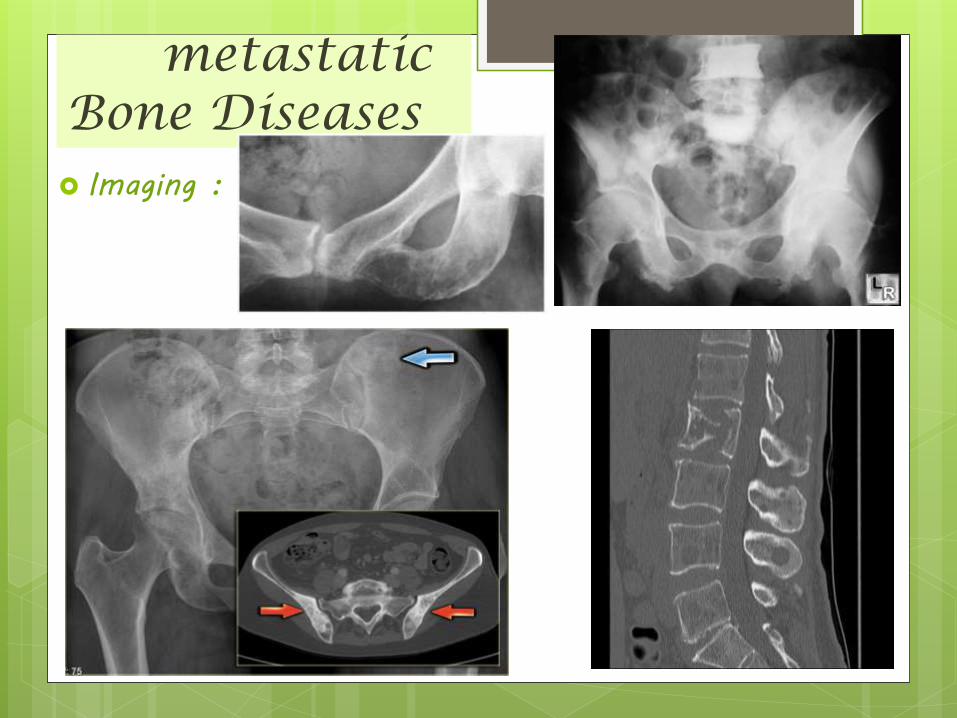
metastatic Bone Diseases Imaging :
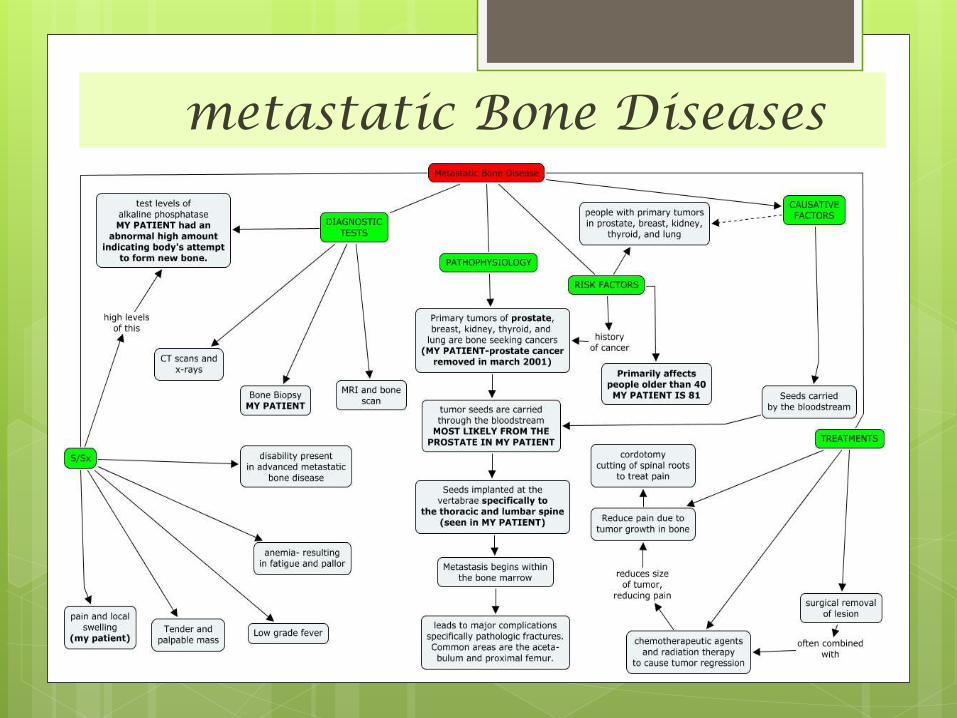
metastatic Bone Diseases

metastatic Bone Diseases treatment:
- By the time a patient has developed secondary deposits the
prognosis for survival is poor.
- Occasionally, radical treatment (combined chemotherapy,
radiotherapy and surgery) targeted at a solitary secondary
deposit and the parent primary lesion may be rewarding and even
apparently curative.
- but in the great majority of cases, and certainly in those with
multiple secondaries, treatment is entirely symptomatic.
Palliative care

metastatic Bone Diseases prognosis:
- Bauer (1995) has suggested useful criteria for assessing prognosis .
In his series of patients, survivorship at 1 year was as follows:
• of patients with 4 or 5 of Bauer’s criteria 50 per cent were alive
of patients with 2 or 3 criteria 25 per cent were alive
of patients with only 1 or none of the criteria,
the majority survived for less than 6 months and none were alive at 1 year.
BAUER’S POSITIVE CRITERIA FOR SURVIVAL
• A solitary metastasis
• No pathological fracture
• No visceral metastases
• Renal or breast primary
• No lung cancer

المراجع
Diagnostic Imaging -Imaging of Bone Tumors and
Tumor-Like Lesions_Techniques and Applications ,Editors: A. L. Baert, Leuven &M. Knauth, G ttingen
1sted.
Apley’s System of Orthopaedics and Fractures 9th ed
part 1 chapter 9 .
American cancer society
www.cancer.org/bonecancer.

المراجع http://www.slideshare.net/ARUNHALDIA/osteogenic-bone-
tumors.
http://www.slideshare.net/narmadaptiwari/bone-tumours-by-dr-narmada-prasad-tiwari
http://www.slideshare.net/kalhamadani/tumors-orthopedic
http://www.slideshare.net/ARUNHALDIA/osteogenic-bone-tumors?qid=1617d0c6-734b-445a-8762-d880bcebfb0b&v=default&b=&from_search=1
http://www.slideshare.net/upenderus/chondrosarcoma?qid=d5f04678-4690-4c04-aed2-e230aa79c0a1&v=default&b=&from_search=1
http://www.slideshare.net/vandana_rt/ewings-sarcoma-dr-vandana
http://www.slideshare.net/PruthvirajNistane/osteosarcoma-1
http://www.slideshare.net/xanthoid/an-approach-to-malignant-bone-tumors?qid=7476aa52-5476-460f-8119-eefe656419bc&v=qf1&b=&from_search=1

:عمل محمد عبدالستار عبدالجليل حمرة