malformatii
TRANSCRIPT

UNIVERSITATEA “OVIDIUS” CONSTANŢAFACULTATEA DE MEDICINĂ GENERALĂ
DISCIPLINA ANATOMIE
MALFORMAŢII CONGENITALE ŞI VARIANTE ANATOMICE ALE
APARATULUI GENITAL
CONF. UNIV. DR. CHIRCOR LIDIA
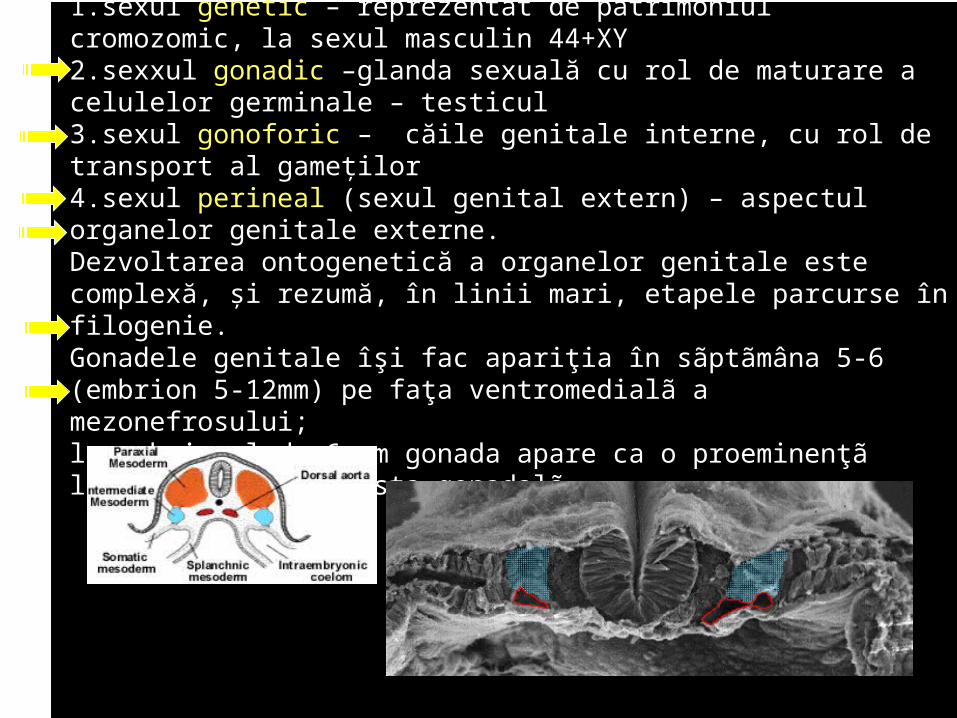
1.sexul genetic – reprezentat de patrimoniul cromozomic, la sexul masculin 44+XY2.sexxul gonadic –glanda sexuală cu rol de maturare a celulelor germinale – testicul3.sexul gonoforic – căile genitale interne, cu rol de transport al gameţilor 4.sexul perineal (sexul genital extern) – aspectul organelor genitale externe.Dezvoltarea ontogenetică a organelor genitale este complexă, şi rezumă, în linii mari, etapele parcurse în filogenie.Gonadele genitale îşi fac apariţia în sãptãmâna 5-6 (embrion 5-12mm) pe faţa ventromedialã a mezonefrosului; la embrionul de 6 mm gonada apare ca o proeminenţã longitudinalã, creasta gonadalã.

Etapa nediferenţiată

Gonadele – Etapa nediferenţiată –pe faţa antero-medială a mezonefrosului apare o eminenţă genital, progonadă alcãtuitã din epiteliu germinativ. Epiteliul celomic al progonadei proliferează, dă naştere cordoanelor sexuale, care sunt colonizate în sãptãmâna a 6-a de gonocite, celulele germinale provenite din peretele sacului vitelin. Există 2 tipuri de cordoane sexuale:
cordoane corticale – scurte şi groase; cordoane medulare – lungi şi subţiri Cromozomul Y transformã medulara gonadei în testicul şi cordoanele sexuale primitive în tubi seminiferi. Gonada, prin secreţia de hormoni, determinã diferenţierea canalelor genitale şi a organelor genitale externe.

Cãile genitale / Etapa nediferenţiatã
Mezodermul intermediar este segmentat în unităţi, tubuli mezonefrotici, căror porţiuni laterale se deschid în ductul Wolff. În afara canalului mezonefrotic care este
complet în sãptãmâna a 4-a, lateral de acesta, la embrionul de 10mm (sãptãmâna a 6-a ) se diferenţiazã canalele paramezonefrotice (Muller). La extremitatea cranialã rãmân deschise, în rest se alungesc, încrucişeazã ventral canalul mezonefrotic, se aşeazã medial de acesta iau contact cu sinusul uro-genital printr-o uşoară îngroşare, tuberculul sinusal

Organele genitale externe / Etapa nediferenţiatã În aceastã etapã sunt aceleaşi pentru ambele sexe. La finele sãptãmânii a 4-a sub inserţia cordonului ombilical, mezodermul formeazã o ridicãturã conicã, tuberculul genital. Buzele şanţului uretral în continuare cu cele ale deschiderii sinusului, alcãtuiesc plicile urogenitale.. Lateral de plicile urogenitale îşi fac apariţia tuberculii labioscrotali. În săptămâna a 8-a de dezvoltare, organele genitale externe sunt identice morfologic, la ambele sexe

Etapa de diferenţiere

Diferenţierea testiculului începe în sãptãmâna a 7-a i.u.Testis-determining factor (TDF) cunoscut şi ca proteina SRY (Sex-determining Region Y) este o proteina ADN codificata de gena SRY, responsabila de initierea diferentierii sexuale masculine.Epiteliul superficial germinativ este separat de blastemul central prin apariţia albugineei care la începutul lunii a 3-a i.u. este complet diferenţiatã. În medulara testiculului se diferenţiazã cordoane celulare aşezate radiar, viitori tubi seminiferi, în care se deosebesc celulele interstiţiale care secretã:
testoteron ce induce diferenţierea organelor genitale externe şi dezvoltarea canalului mezonefrotic
factorul MIS cu efect supresor asupra canalului paramezonefrotic.Tubii seminiferi rãmân solizi pânã la pubertate. Din blastemul central se organizeazã o reţea de cordoane anastomozate între ele schiţând rete testis din care pleacã muguri ce reprezintã viitorii tubi drepţi. Aceştia fac joncţiunea cu tubii seminiferi în luna a 3-a fetalã.
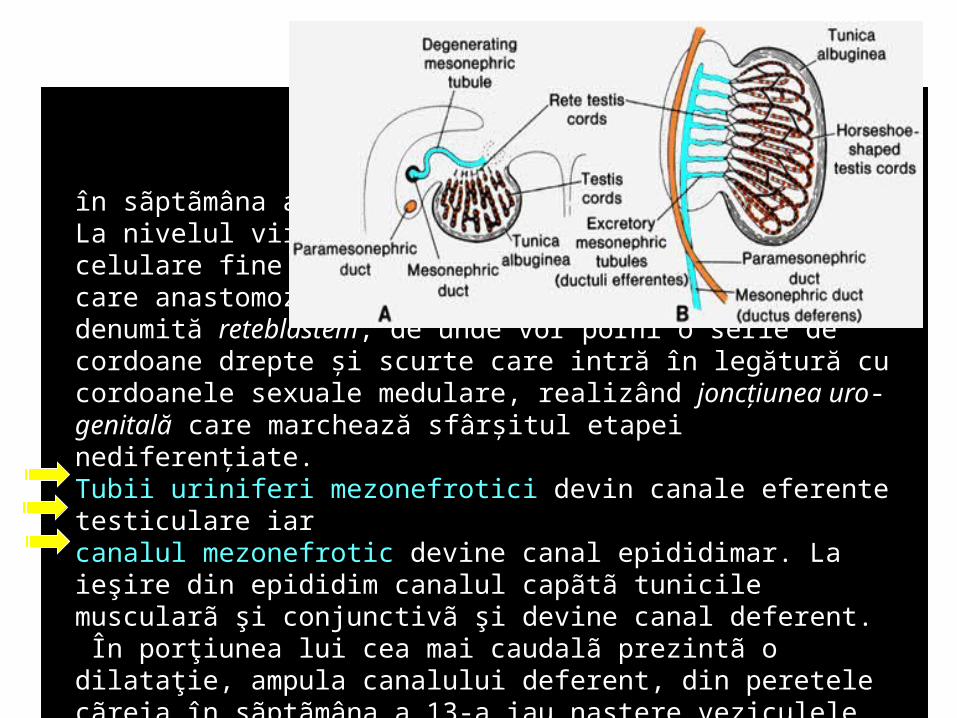
în sãptãmâna a 4-a. La nivelul viitoarei gonade apar o serie de conducte celulare fine din epiteliul canalelor mezonefrotice, care anastomozându-se între ele alcătuiesc o reţea, denumită reteblastem, de unde vor porni o serie de cordoane drepte şi scurte care intră în legătură cu cordoanele sexuale medulare, realizând joncţiunea uro-genitală care marchează sfârşitul etapei nediferenţiate.Tubii uriniferi mezonefrotici devin canale eferente testiculare iar canalul mezonefrotic devine canal epididimar. La ieşire din epididim canalul capãtã tunicile muscularã şi conjunctivã şi devine canal deferent. În porţiunea lui cea mai caudalã prezintã o dilataţie, ampula canalului deferent, din peretele cãreia în sãptãmâna a 13-a iau naştere veziculele seminale. În luna a 7-a glanda seminalã are formã definitivã, creşte lent pânã la pubertate când începe o creştere explozivã.
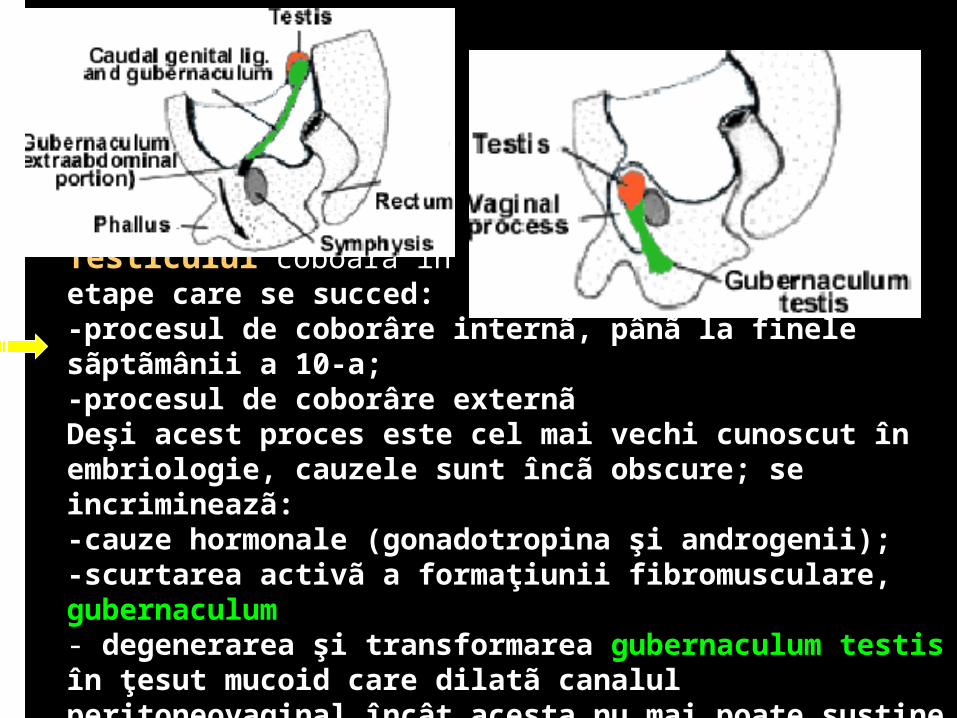
Testiculul coboarã în bursa scrotalã în douã etape care se succed: -procesul de coborâre internã, pânã la finele sãptãmânii a 10-a;-procesul de coborâre externãDeşi acest proces este cel mai vechi cunoscut în embriologie, cauzele sunt încã obscure; se incrimineazã: -cauze hormonale (gonadotropina şi androgenii);-scurtarea activã a formaţiunii fibromusculare, gubernaculum- degenerarea şi transformarea gubernaculum testis în ţesut mucoid care dilatã canalul peritoneovaginal încât acesta nu mai poate susţine testiculul care cade în interiorul sãu ajutat fiind şi de presiunea intraabdominalã.
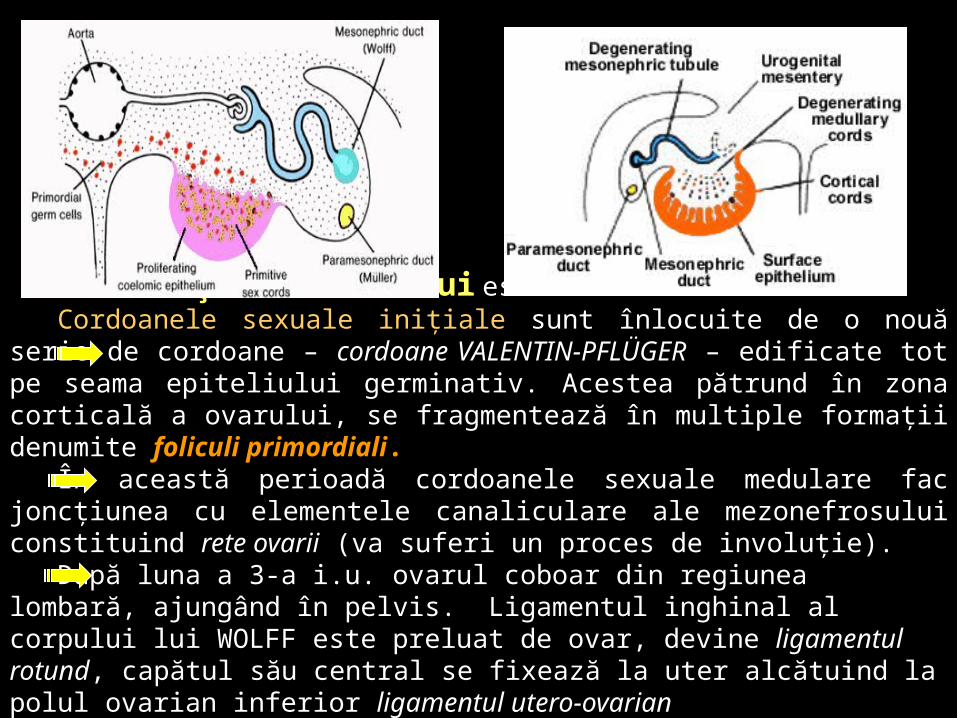
Diferenţierea ovarului este mai tardivă. Cordoanele sexuale iniţiale sunt înlocuite de o nouă serie de cordoane –
cordoane VALENTIN-PFLÜGER – edificate tot pe seama epiteliului germinativ. Acestea pătrund în zona corticală a ovarului, se fragmentează în multiple formaţii denumite foliculi primordiali.
În această perioadă cordoanele sexuale medulare fac joncţiunea cu elementele canaliculare ale mezonefrosului constituind rete ovarii (va suferi un proces de involuţie).
După luna a 3-a i.u. ovarul coboar din regiunea lombară, ajungând în pelvis. Ligamentul inghinal al corpului lui WOLFF este preluat de ovar, devine
ligamentul rotund, capătul său central se fixează la uter alcătuind la polul ovarian inferior ligamentul utero-ovarian
.
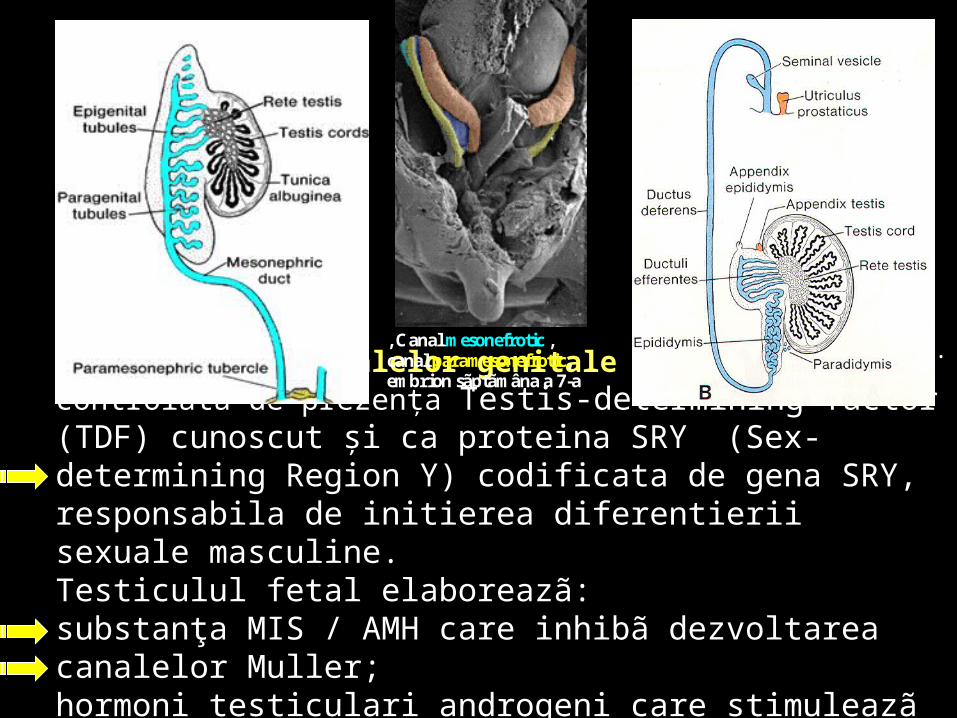
Diferenţierea canalelor genitale la bărbat este controlată de prezenţa Testis-determining factor (TDF) cunoscut şi ca proteina SRY (Sex-determining Region Y) codificata de gena SRY, responsabila de initierea diferentierii sexuale masculine.Testiculul fetal elaboreazã:substanţa MIS / AMH care inhibã dezvoltarea canalelor Muller; hormoni testiculari androgeni care stimuleazã creşterea şi diferenţierea canalului mezonefrotic Wolff .
, Canal mesonefrotic , canal paramesonefrotic, embrion sãptãmâna a 7-a

.
Structuri în etapaindiferentã
Structuri definitive masculine
GonadaCortexMedulara
Ligamente genitale
Tubi colectorimezonefroticiGrup cranialGrup caudal
Canal mezonefrotic
TesticulTubi seminiferiRete testis
MezorchimLig.testicularGubernaculum testis (segm. caudal)Gubernaculum testis (ca intreg)
Canale eferenteCanale aberante cranialeParadidimCanale aberante caudaleApendice epididimarCanal epididimarCanal deferent, vezicule seminaleCanal ejaculatorUreter, pelvis,calice, tubi colectori
.
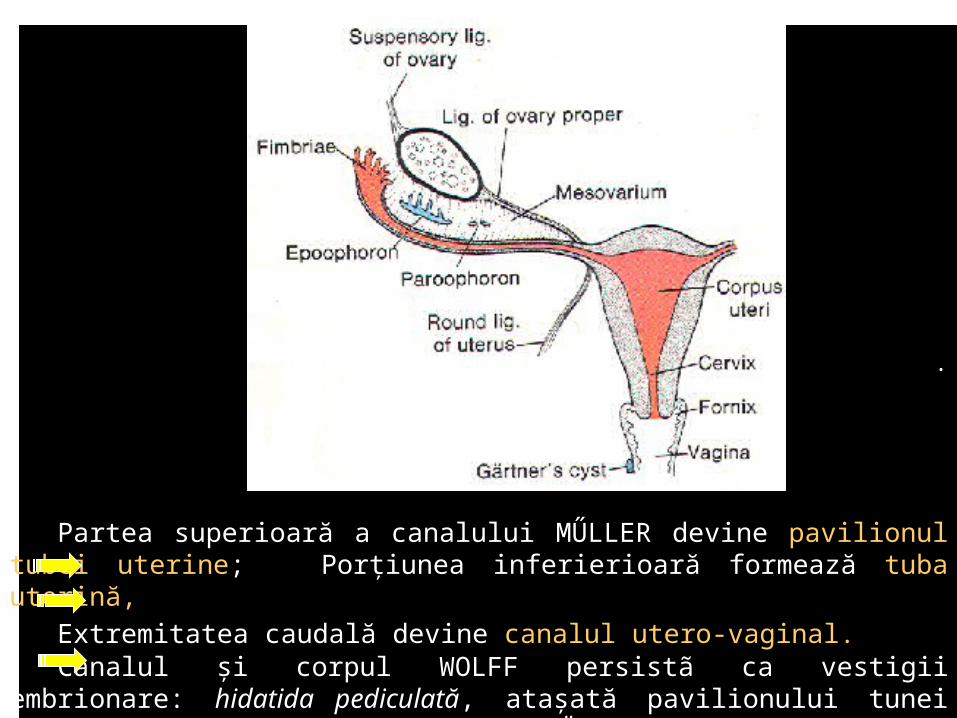
Partea superioară a canalului MŰLLER devine pavilionul tubei uterine; Porţiunea inferierioară formează tuba uterină,
Extremitatea caudală devine canalul utero-vaginal. Canalul şi corpul WOLFF persistã ca vestigii embrionare: hidatida pediculată,
ataşată pavilionului tunei uterine, epooforul, parooforul, ductul GÄRTNER.

.
A = la 9 sãptãmâni B=la finele lunii a 3-a C = la naştereDiferenţierea gonoforului feminin se face sub influenţa lipsei de
hormoni masculini. Diferenţierea gonoforului feminin se caracterizează prin:Persistenţa canalelor MÜLLERRegresia canalelor WOLFF. Uneori poate persista septul despărţitor: malformaţii uterine. Vagina se formează în luna a treia de viaţă embrionară.

.
În timp ce se formează canalul utero-vaginal ţesutul endodermal al tuberculului sinusal proliferează formând 20% din partea inferioară a vaginei. Extremitatea inferioară este obstruată printr-o placă vaginală (viitor himen) care se tunelizează prin descuamare centrală.
Peretele fibromuscular vaginal are originea în mezodermul canalului utero-vaginal.
Glandele genitale accesorii se dezvoltă prin înmugurire, din uretră (glande parauretrale SKENE) şi sinusul uro-genital definitiv (glande vestibulare mari BARTHOLIN).

.
Structuri în etapa indiferentã Structuri definitive feminine
GonadaCortexMedulara
Ligamente genitale
Tubi colectori mezonefroticiGrup cranialGrup caudalCanal mezonefrotic
Canal paramezonefrotic
OvarCortex (foliculi ovarieni)Medulara primaraRete ovariiMezovariumLigamentul suspensor al ovaruluiLig. propriu al ovaruluiLig. rotund al uteruluiLig.lateral al uterului
Epoofor.Canale aberanteParooforApendice veziculosCanalul epooforuluiCanalul lui GartnerUreter, pelvis,calice, tubi colectoriHidatida (Morgagni)Tuba uterinaUterVagina( porţiunea superioarã)

Diferenţierea organelor genitale externe în jurul membranei cloacale
Făt în vârstă de 56 zile
• Cloaca se divide în 2 regiuni:– anorectalã – urogenitalã
• se separă– membrana
urogenitala– membrana analã.
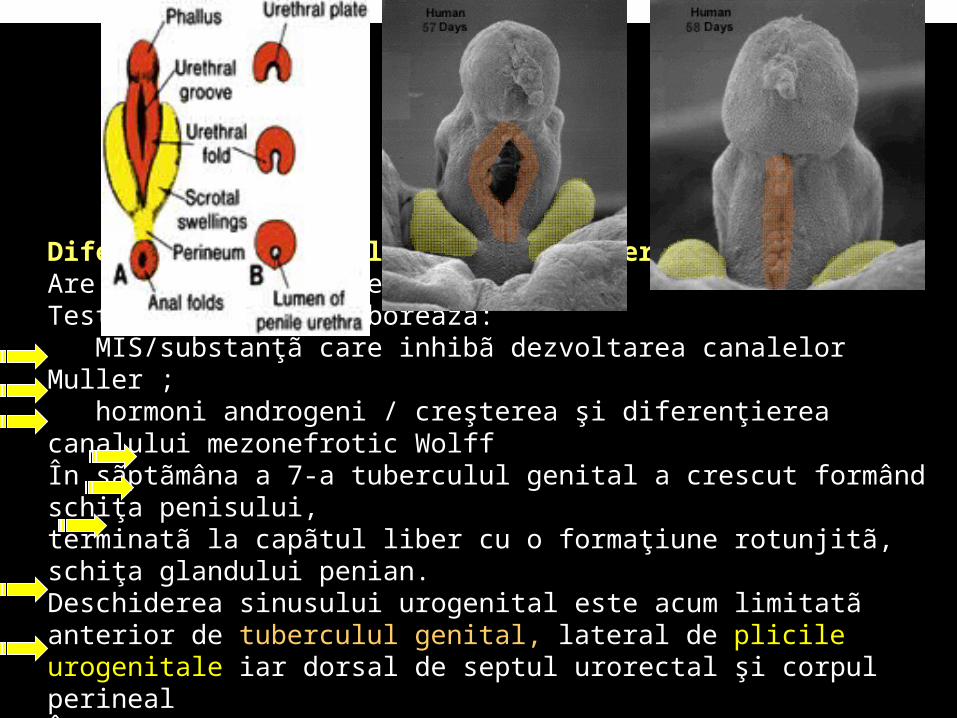
Diferenţierea organelor genitale externeAre loc sub influenţe hormonaleTesticulul fetal elaboreazã:
MIS/substanţã care inhibã dezvoltarea canalelor Muller ; hormoni androgeni / creşterea şi diferenţierea canalului mezonefrotic
Wolff În sãptãmâna a 7-a tuberculul genital a crescut formând schiţa penisului, terminatã la capãtul liber cu o formaţiune rotunjitã, schiţa glandului penian. Deschiderea sinusului urogenital este acum limitatã anterior de tuberculul genital, lateral de plicile urogenitale iar dorsal de septul urorectal şi corpul perinealÎn decursul sãptãmânii a 9-a hormonii androgeni induc creşterea penisului, formarea uretrei peniene, fuzionarea tuberculilor scrotali, dezvoltarea prostatei şi glandelor seminale.
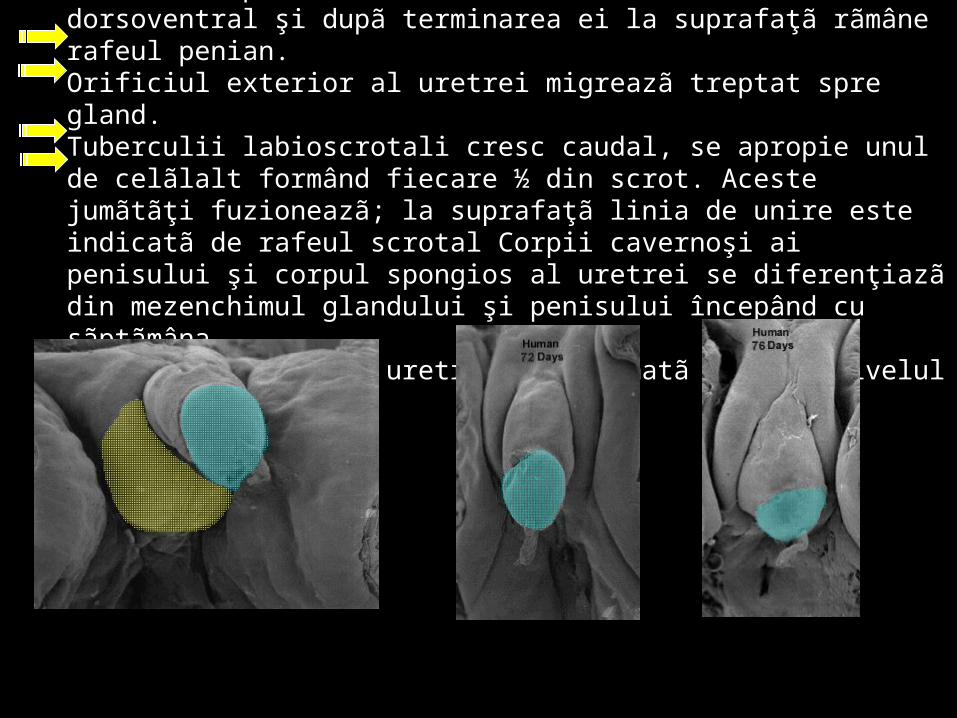
\Plicile urogenitale fuzioneazã formând uretra spongioasã. Fuzionarea plicilor uretrale are loc în sens dorsoventral şi dupã terminarea ei la suprafaţã rãmâne rafeul penian. Orificiul exterior al uretrei migreazã treptat spre gland. Tuberculii labioscrotali cresc caudal, se apropie unul de celãlalt formând fiecare ½ din scrot. Aceste jumãtãţi fuzioneazã; la suprafaţã linia de unire este indicatã de rafeul scrotal Corpii cavernoşi ai penisului şi corpul spongios al uretrei se diferenţiazã din mezenchimul glandului şi penisului începând cu sãptãmâna. În sãptãmâna a 14-a uretra este formatã pânã la nivelul glandului.

.
Structuri în etapaindiferentã
Structuri definitivemasculine
Canalparamezonefrotic
Sinus urogenital
Tubercul sinusalTubercul genital
Plici urogenitaleTuberculi labioscrotali
Apendice testicularUtriculaVezica urinaraUretra (fara fosanaviculara)ProstataGlande bulbouretraleColicul seminalPenisGland penianCorp cavernos penisCorp spongios aluretreiFaţa uretralã penisScrotRafeu scrotal

Prostata se dezvoltã din muguri ai epiteliului ce învelesc porţiunea falicã a sinusului urogenital, cranial şi caudal de locul de intrare a conductului genital ce va deveni uretra prostaticã.Canalul paramezonefrotic Műller degenereazã, Canalul mezonefrotic Wolff devine canal deferent, se dezvoltã dinspre testicul spre regiunea prostatei.
Dezvoltarea glandelor sexuale accesorii
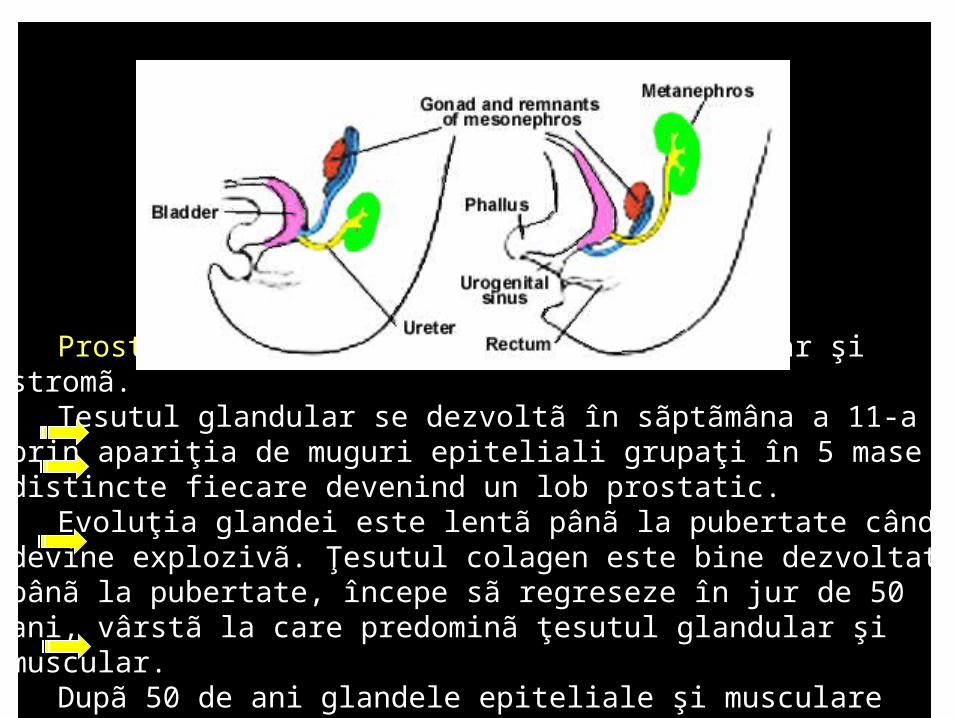
Prostata este alcãtuitã din ţesut glandular şi stromã. Ţesutul glandular se dezvoltã în sãptãmâna a 11-a prin apariţia de muguri
epiteliali grupaţi în 5 mase distincte fiecare devenind un lob prostatic. Evoluţia glandei este lentã pânã la pubertate când devine explozivã.
Ţesutul colagen este bine dezvoltat pânã la pubertate, începe sã regreseze în jur de 50 ani, vârstã la care predominã ţesutul glandular şi muscular.
Dupã 50 de ani glandele epiteliale şi musculare încep sã regreseze în timp ce sporeşte colagenul şi ţesutul sclerotesticular.

Glandele seminale se dezvoltã printr-o evaginare a peretelui ampulelor canalului deferent în sãptãmâna 13. În luna a 7-a au formã definitivã. Ele cresc lent pânã la pubertate, dupã care ritmul se accelereazã.
Glandele bulbouretrale la bãrbat apar în însãptãmâna a 9-a sub forma unor muguri solizi ai epiteliului endodermal care cãptuşeşte sinusul urogenital la nivelul uretrei cavernoase. În luna a 4-a epiteliul lor devine glandular.
Glandele uretrale apar în luna a 3-a la nivelul uretrei cavernoase prin acelaşi proces de înmugurire al epiteliului uretral, în special pe faţa lui dorsalã.

.
La femeie tuberculul genital are dezvoltare redusă şi dă naştere clitorisului. Din marginile şanţului uro-genital se dezvoltă cutele genitale care se transformă în labii mici. Lateral de pliurile uro-genitale se dezvoltă protuberanţele labio-scrotale care în lipsa androgenilor vor deveni labii mari.
Sinusul uro-genital definitiv va da naştere vestibulului vaginal.

.
Structuri în etapa indiferentã Structuri definitive feminineSinus urogenital
Tubercul sinusalTubercul genital
Plici urogenitaleTuberculi labioscrotali
Proeminenţa medialãcranialã
Vezica urinaraUretra,vestibul vaginalGlande uretrale şiparauretraleGlande mari vestibulareHimenClitorisGland clitoridianCorp cavernos clitoridianBulb vestibularLabii miciLabii mariComisura posterioarãMons Pubis

Malformatii congenitale
.
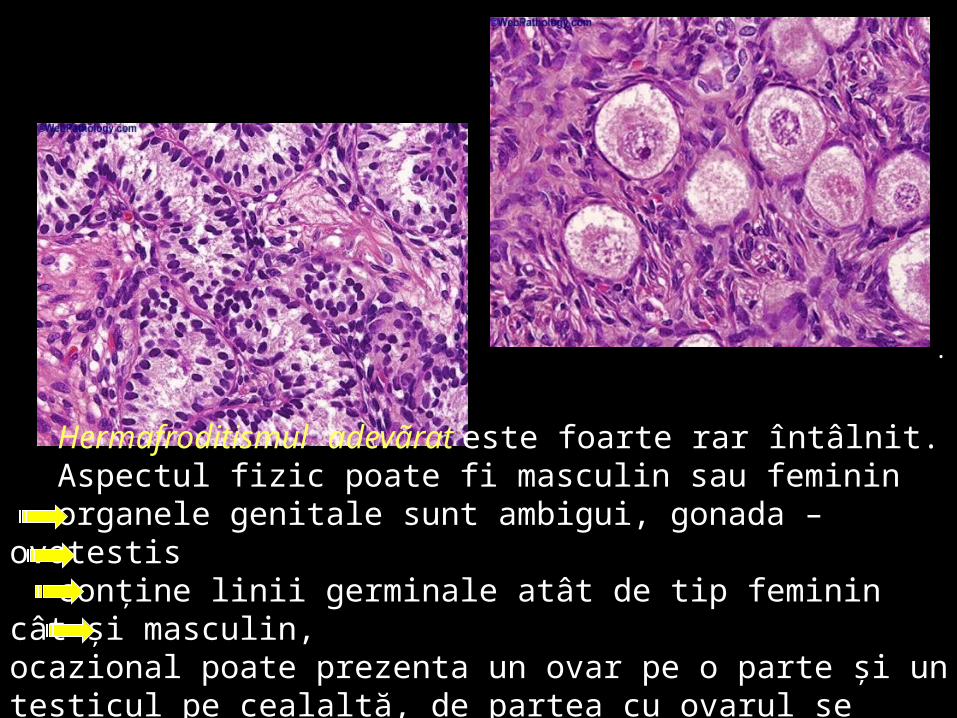
Hermafroditismul adevãrat este foarte rar întâlnit. Aspectul fizic poate fi masculin sau feminin organele genitale sunt ambigui, gonada – ovotestis conţine linii germinale atât de tip feminin cât şi masculin,
ocazional poate prezenta un ovar pe o parte şi un testicul pe cealaltă, de partea cu ovarul se poate dezvolta un salpinx şi un singur corn uterin.
.
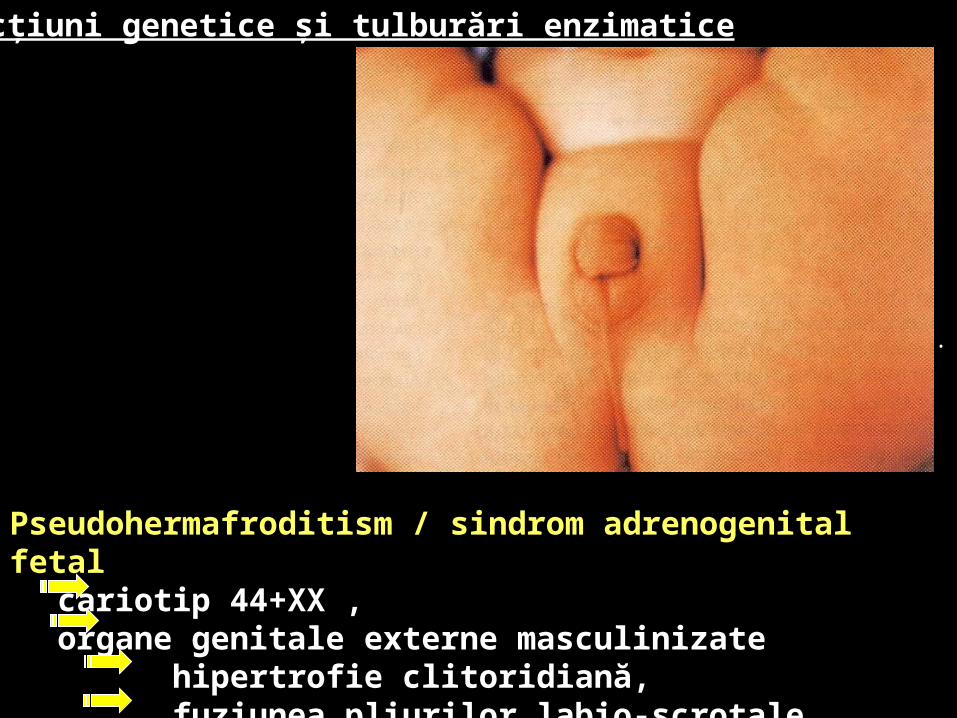
Pseudohermafroditism / sindrom adrenogenital fetal cariotip 44+XX , organe genitale externe masculinizate hipertrofie clitoridiană, fuziunea pliurilor labio-scrotale.
I. Afecţiuni genetice şi tulburări enzimatice

Pseudohermafroditism / sindrom adrenogenital congenital 1/16000 nn, cariotip 44+XX , deficit 21-hidroxilaza organe genitale externe masculinizate
clitoris peniform
labii mari de aspect pseudoscrotal

.
Pseudohermafroditism / Sindromul de feminizare testicularã: 44 +XY, organe genitale de tip feminin.hormoni androgeni cu nivel normal anomalii ale receptorilor din ţesuturile ţintã pentru androgeni

.
1.Afecţiuni genetice şi tulburări enzimatice: disgenezia gonadală (sindrom TURNER) asociază malformaţii multiple, cele genitale fiind generate de insuficienţe gonadale, deficitul de 17 alfa-hidroxilază (amenoree primară, absenţa caracterelor sexuale secundare, fenotip feminin, hipertensiune, hipokaliemie).
Anomalii în dezvoltarea organelor genitale feminine
.
II. Malformaţii congenitale ale gonadei masculine
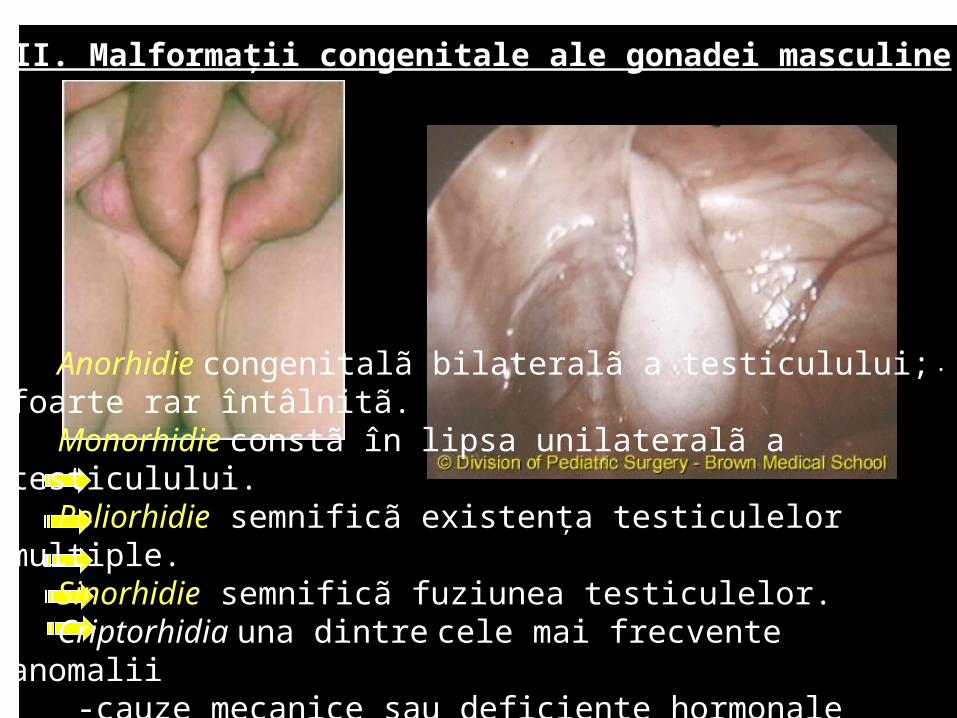
Anorhidie congenitalã bilateralã a testiculului; foarte rar întâlnitã.
Monorhidie constã în lipsa unilateralã a testiculului. Poliorhidie semnificã existenţa testiculelor multiple.Sinorhidie semnificã fuziunea testiculelor. Criptorhidia una dintre cele mai frecvente anomalii -cauze mecanice sau deficienţe hormonale -consecinţa opririi în coborâre a gonadei pe traiectul normal

Criptorhidie• unilateral • bilateral 20-40% .
• 30% dintre prematuri • 4% dintre nn la termen

Ectopic testes
• Testicul Ectopic – Tumori testiculare 20-40%; – Sterilitate
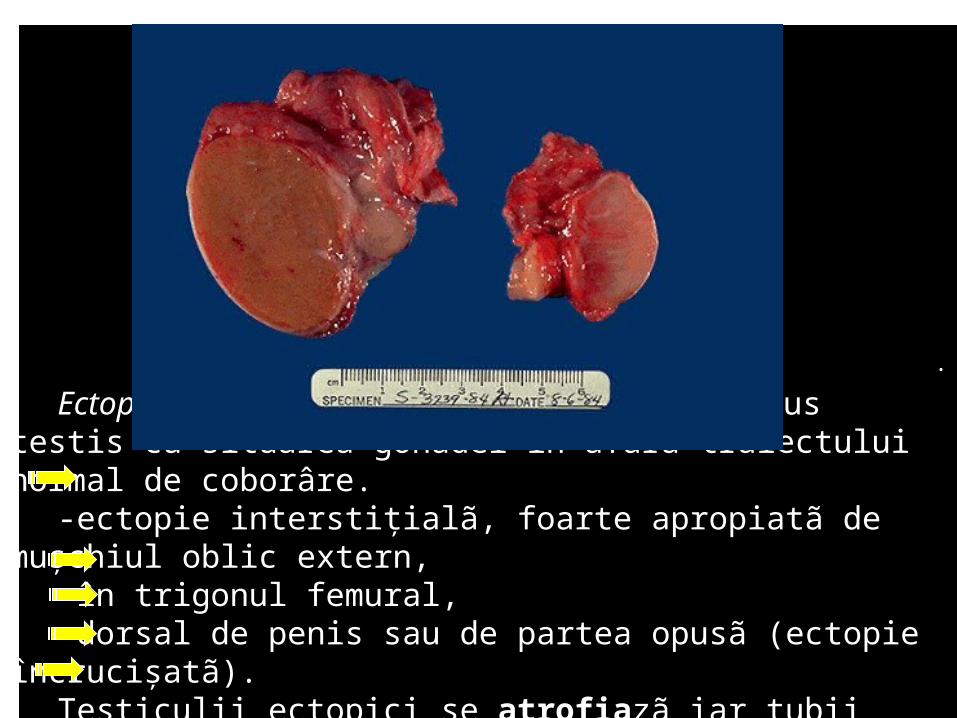
.
Ectopia testicularã: anomalii în descensus testis cu situarea gonadei în afara traiectului normal de coborâre.
-ectopie interstiţialã, foarte apropiatã de muşchiul oblic extern, -în trigonul femural, -dorsal de penis sau de partea opusã (ectopie încrucişatã). Testiculii ectopici se atrofiazã iar tubii seminiferi suferã un proces
de sclerozã şi degenerescenţã.

• Hernia Inghinală
hernia inghinala Incompleta
Hernia inghinala Completa
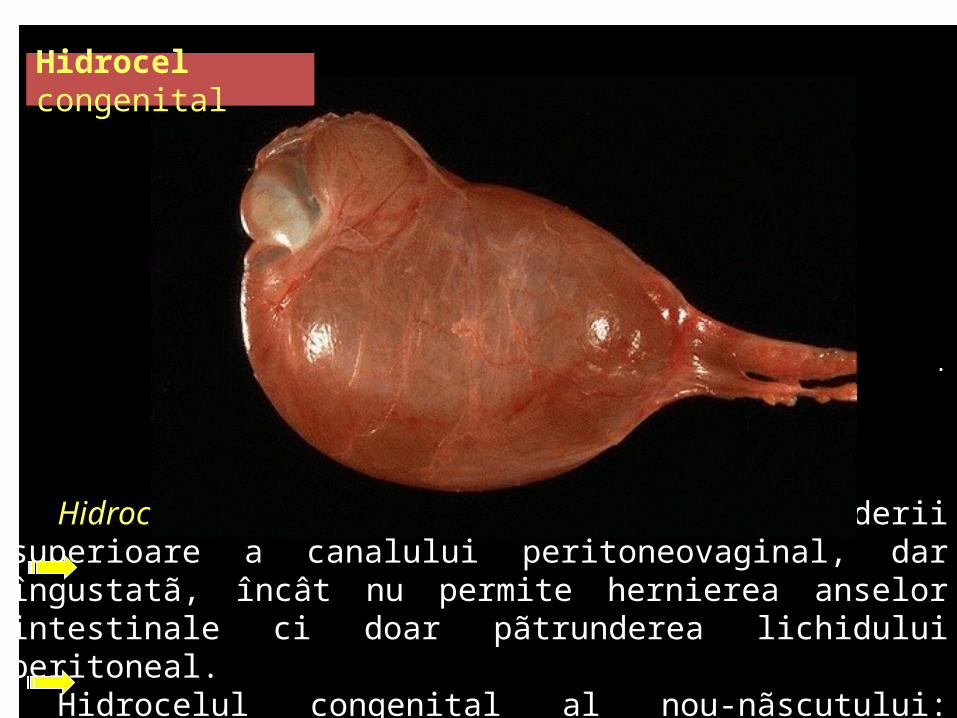
.
Hidrocel congenital prin persistenţa deschiderii superioare a canalului peritoneovaginal, dar îngustatã, încât nu permite hernierea anselor intestinale ci doar pãtrunderea lichidului peritoneal.
Hidrocelul congenital al nou-nãscutului: vindecare spontanã.
Hidrocel congenital

.
Coborârea incompletã a testiculului sau lipsa ligamentului scrotal cresc mobilitatea testicului putând determina torsiunea sa urmatã de infarctul hemoragic al testiculului prin suprimarea drenajului venos.
Torsiunea testiculului
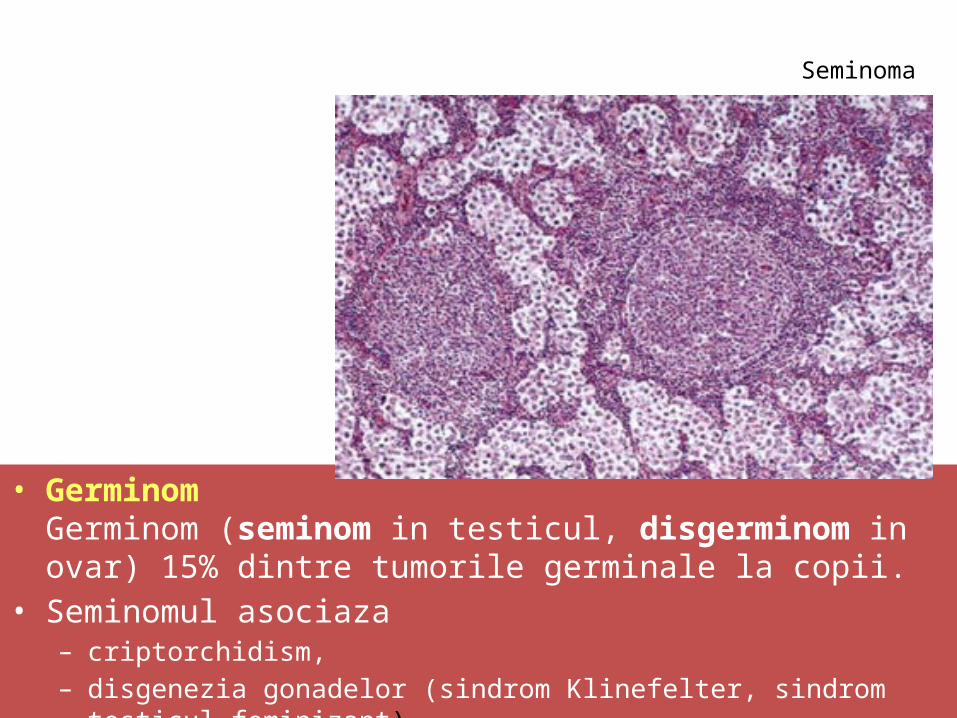
• GerminomGerminom (seminom in testicul, disgerminom in ovar) 15% dintre tumorile germinale la copii.
• Seminomul asociaza – criptorchidism, – disgenezia gonadelor (sindrom Klinefelter, sindrom testicul feminizant).
Seminoma

• carcinom Embrionar 1/3 dintre tumorile germinale testiculare
Embryonic carcinoma Embryonic carcinoma

• Teratom combinatie de tesuturi derivate din ecto-, mezo- si endoderm).– teratom matur ( cistic),– teratom imatur (solid)– teratom malign degenerat.
• Complicatii– Torsiune ovar si tuba
uterina
Teratom Ovarian

III. Malformaţii congenitale ale organelor genitale externe masculine
Hipospadias Semnificã deschiderea uretrei pe faţa ventralã a glandului sau penisului, în cazuri mai severe prin mai multe deschideri
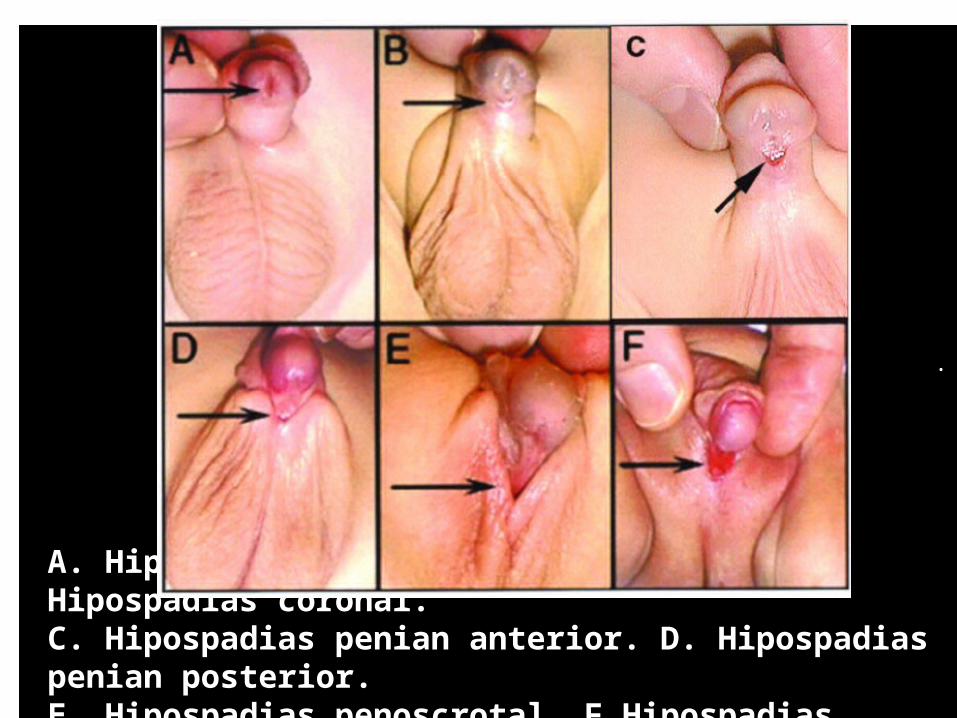
.
A. Hipospadias la nivelul glandului. B. Hipospadias coronal. C. Hipospadias penian anterior. D. Hipospadias penian posterior.E. Hipospadias penoscrotal. F.Hipospadias scrotal
.
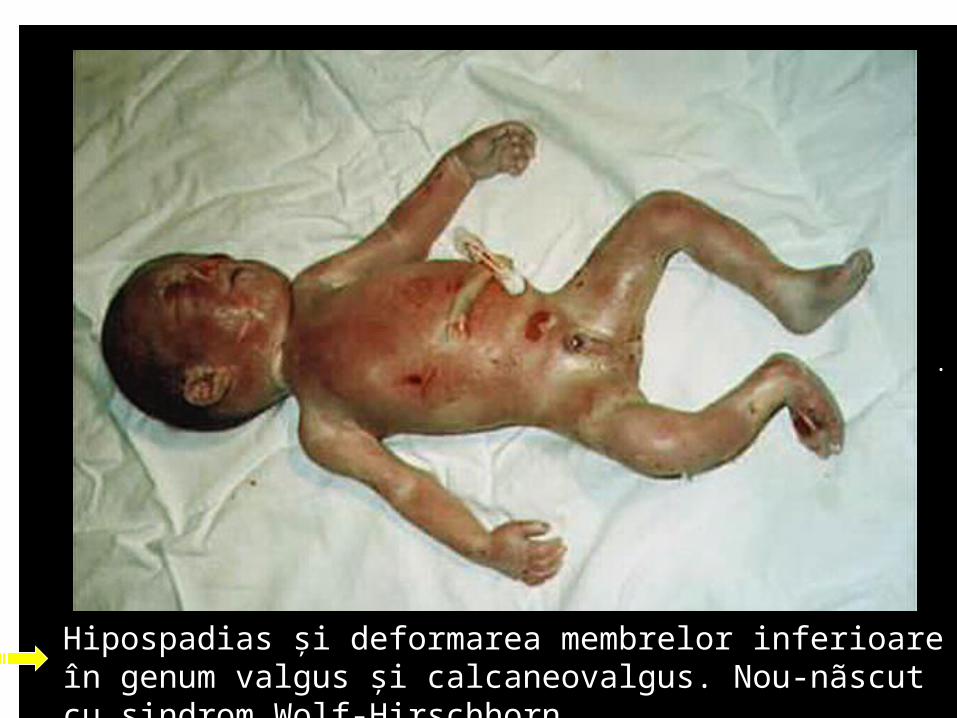
Hipospadias şi deformarea membrelor inferioare în genum valgus şi calcaneovalgus. Nou-nãscut cu sindrom Wolf-Hirschhorn.
.
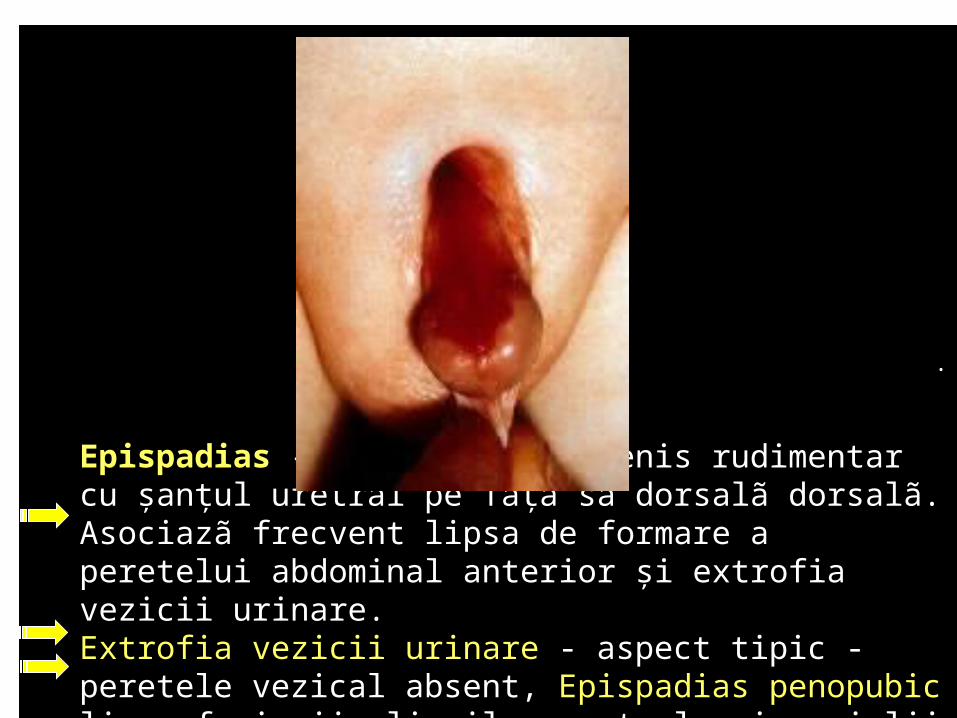
Epispadias - prezenţa unui penis rudimentar cu şanţul uretral pe faţa sa dorsalã dorsalã. Asociazã frecvent lipsa de formare a peretelui abdominal anterior şi extrofia vezicii urinare. Extrofia vezicii urinare - aspect tipic -peretele vezical absent, Epispadias penopubic lipsa fuziunii pliurilor uretrale şi a pielii la nivelul glandului penian

.
Agenezia sau absenţa penisului prin lipsa de dezvoltare a tuberculului genital. Uretra se deschide în perineu lângã anus.
Penis bifid - prin lipsa de fuziune a celor douã jumãtãţi ale tuberculului genital, frecvent asociat cu extrofia vezicalã.
Micropenis - asociat de obicei cu hipopituitarism. Penisul are dimensiuni atât de reduse încât este mascat de grãsimea supraspubicã.
Penis retroscrotal sau transpoziţia penisului şi scrotului. Penisul este posterior de scrot prin lipsa de fuziune a tuberculilor labioscrotali saui formarea lor anterior de tuberculul genital.
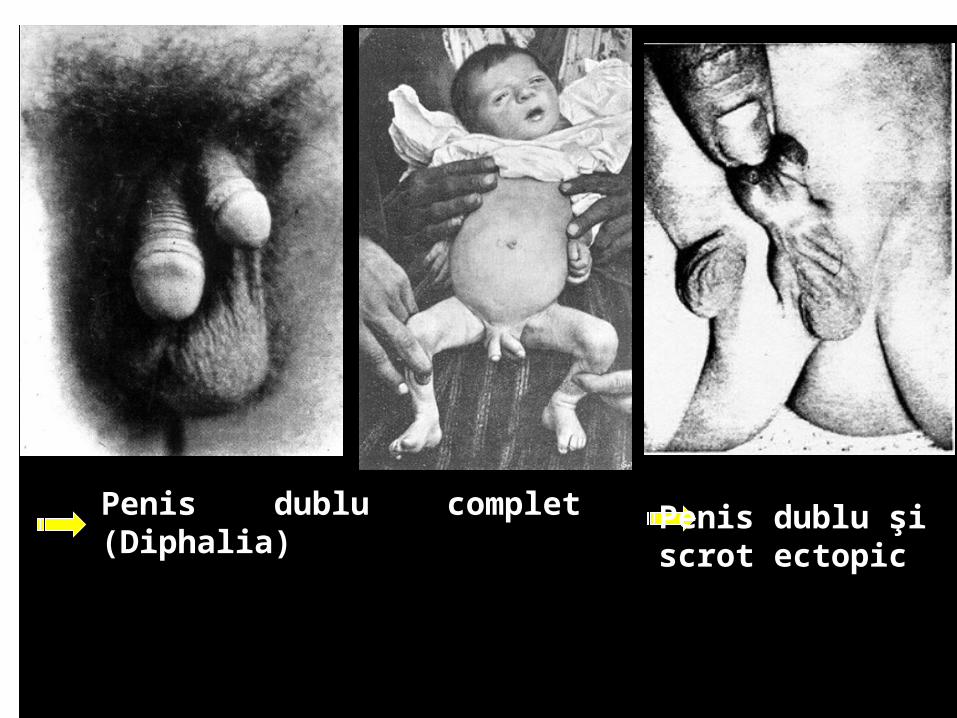
Diphalia - Penis dublu - prin dezvoltarea a doi tuberculi genitali; însoţit frecvent de anomalii ale cãilor urinare şi anus imperforat.La
.
Penis dublu complet (Diphalia) Penis dublu şi scrot ectopic

.
Malformaţii utero-vaginale – apar la 0,16% dintre femei; cauze: fuziunea anormală a canalelor paramezonefrotice, dezvoltarea incompletă a unui duct paramezonefrotic, insuficienţa de dezvoltare a unei părţi a unui duct paramezonefrotic, absenţa tunelizării sau tunelizarea incompletă a plăcii vaginale.

.
Uter unicorn imagine Rezonanţã Electromagneticã
Uter unicorn Laparoscopie
Uter unicorn
Uter unicorn Radiografie cu substanţã de contrast
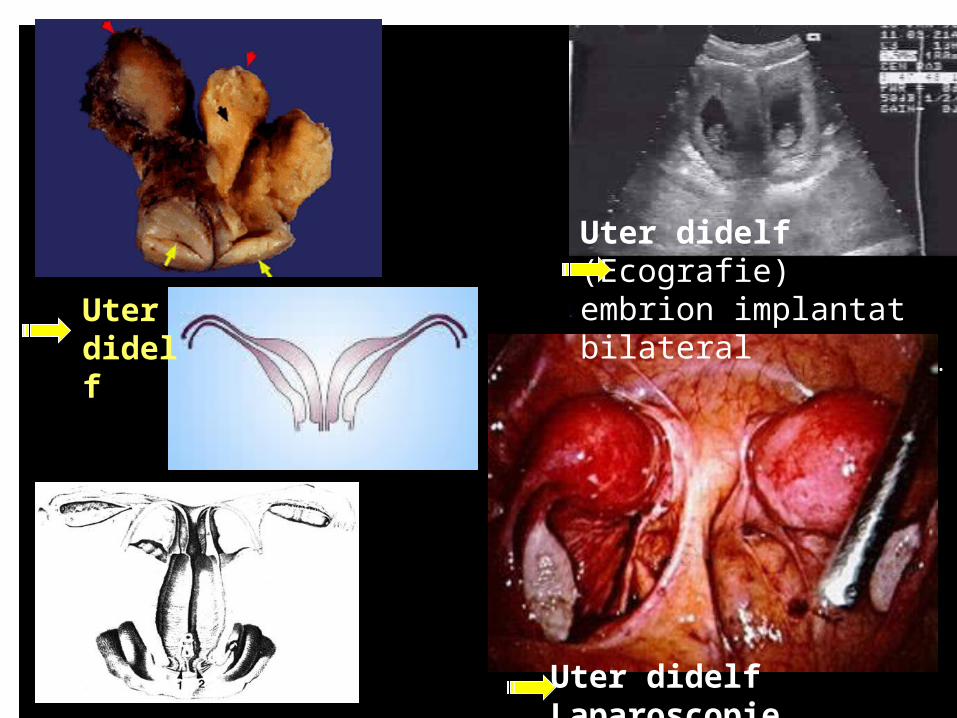
.Uter didelf
Uter didelf (Ecografie)embrion implantat bilateral
Uter didelf Laparoscopie
.
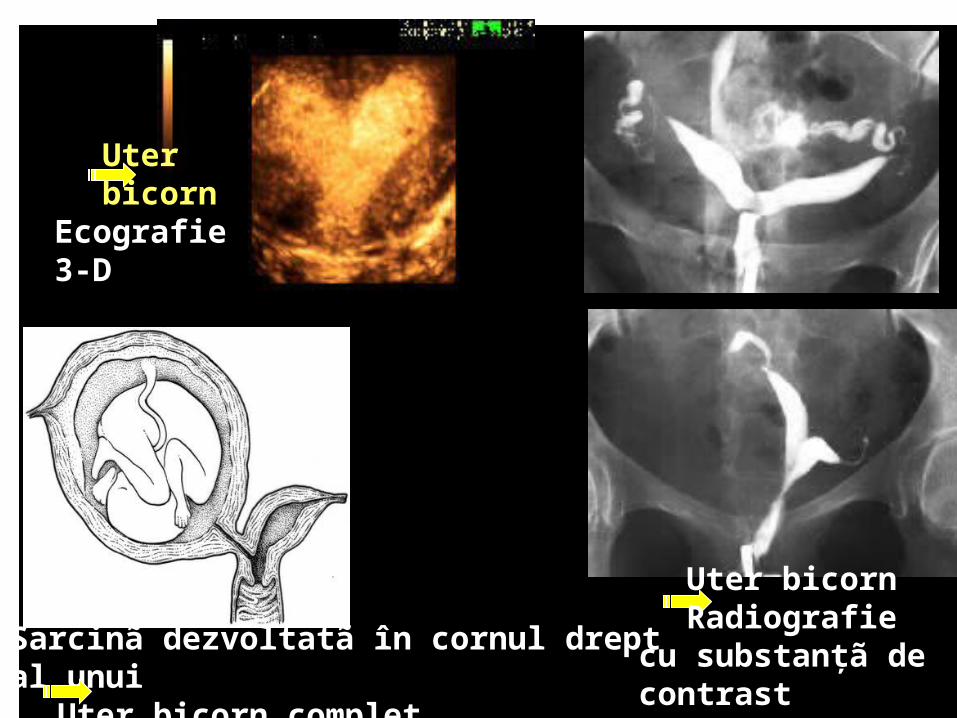
Sarcinã dezvoltatã în cornul drept al unui Uter bicorn complet
Uter bicornRadiografie cu
substanţã de contrast
Uter bicorn
Ecografie 3-D
.
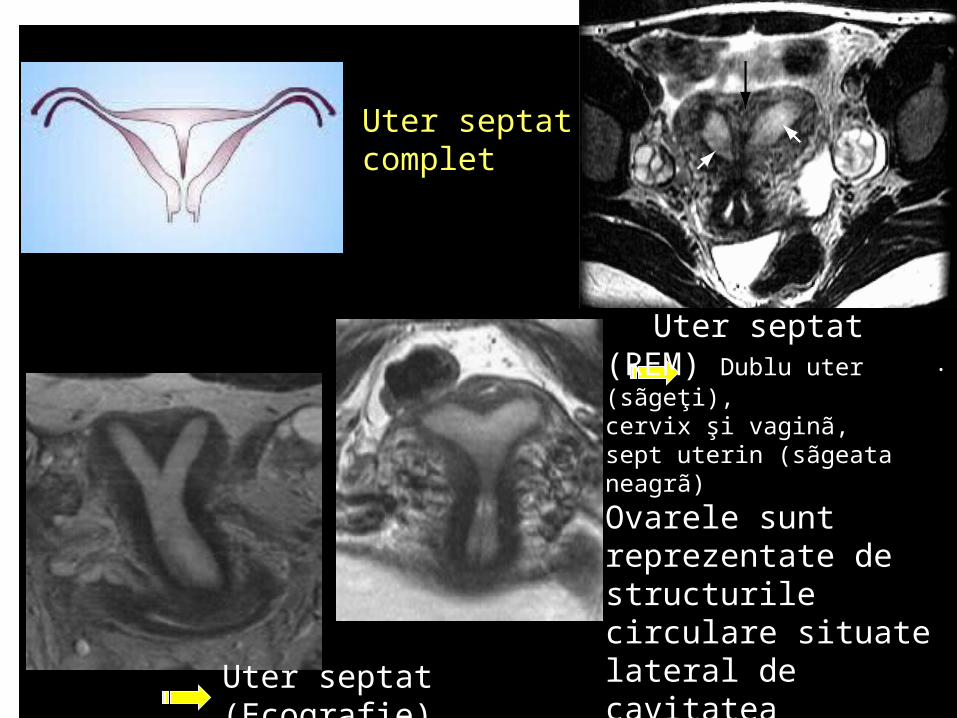
Uter septat complet
Uter septat (Ecografie)
Uter septat (REM) Dublu uter (sãgeţi), cervix şi vaginã, sept uterin (sãgeata neagrã)
Ovarele sunt reprezentate de structurile circulare situate lateral de cavitatea uterinã.

. Uter septat. Laparoscopie Dublu cervix şi vaginã. continuitatea fundului uterin
Uter septat Piesã anatomicã formol 10%

.
Glanda mamară
Evolueazãdependent de influenţe hormonale în trei etape i.u: S4-6: îngroşare a ectodermului/zona toracică:creasta mamarã. S6 – Luna 5 îngroşarea schiţei mamare urmată de invaginare în mezenchimul peretelui toracic, creştere tridimensională, invazia mezenchimului; în S12-16 celulele mezenchimale se diferenţiază în celule musculare mamelonare şi areolare. Dupã luna a 5-a: 15-20 muguri plini, ţesut adipos. Pânã în luna a8-a:mugurii se ramificã, capãtã lumen, devin Canale Lactifere. Schiţa mamară secundară: glande sebacee, sudoripare, foliculi piloşi. Dupã naştere: 3 etape evolutive:
La pubertate/definitiveazã acinii glandei. La finele sarcinii/rãspuns la estrogen-progesteron.Dupã menopauzã: atrofia ţesutului glandular.

.
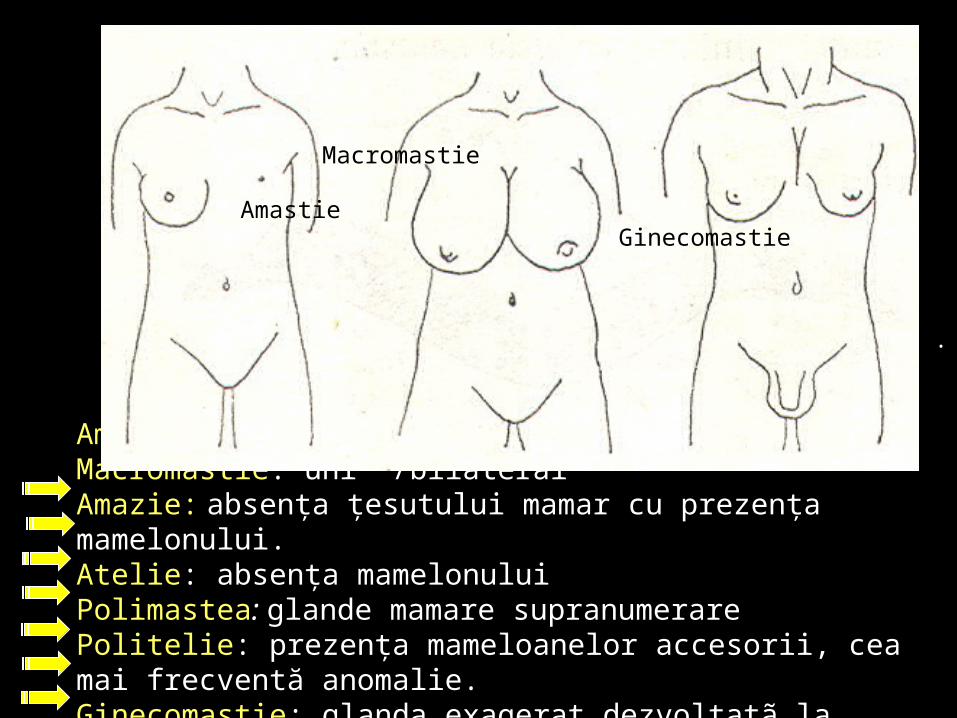
A. Anomalii de sediu: Glandã mamarã axilarã. F. la nivelul toracelui între coasta a 7-a şi a 9-a. B. Anomalii de volum (glandã rudimentarã). C. Anomalii numerice: glande mamare accesorii. D.glandã dublã. H. Glandã mamarã supranumerarã E. Mamelon dedublat. G. Glandã mamarã supranumerarã situatã la nivelul coapsei stângi.
Anomalii în dezvoltarea glandei mamare
.
Amastie – lipsa glandei mamareMacromastie: uni /bilateral Amazie: absenţa ţesutului mamar cu prezenţa mamelonului. Atelie: absenţa mamelonului Polimastea: glande mamare supranumerare Politelie: prezenţa mameloanelor accesorii, cea mai frecventă anomalie.Ginecomastie: glanda exagerat dezvoltatã la indivizi de sex masculin.
GinecomastieAmastie
Macromastie

.
Polimastia

.
Asimetrie congenitalã benignã a glandei mamare
PolitelieSânul drept prezintã 2 mameloane
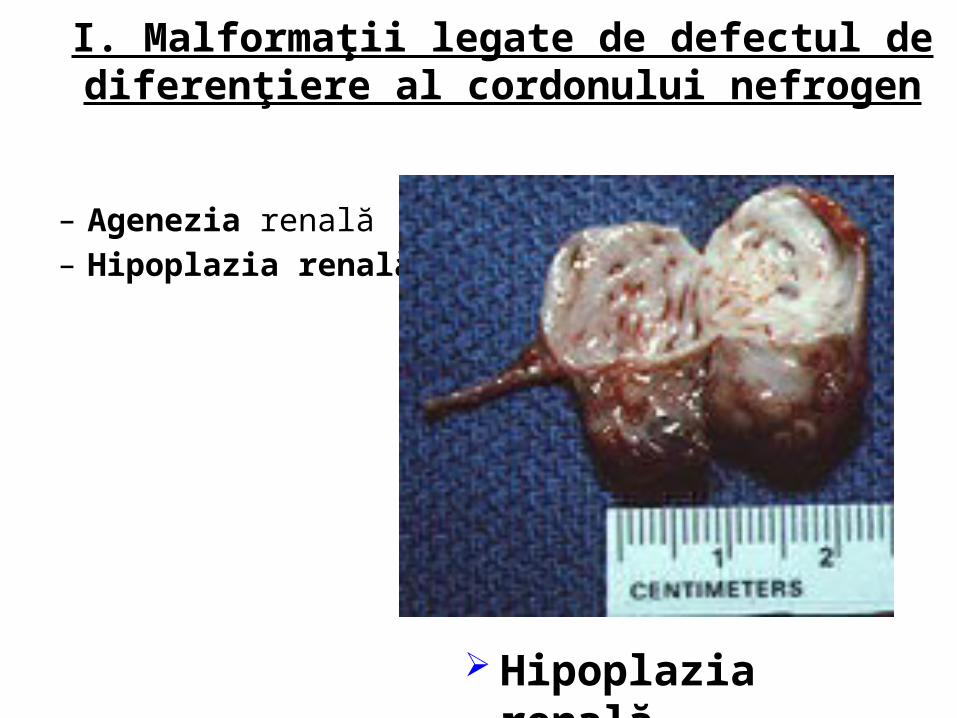
I. Malformaţii legate de defectul de diferenţiere al cordonului nefrogen
– Agenezia renală– Hipoplazia renală
Hipoplazia renală
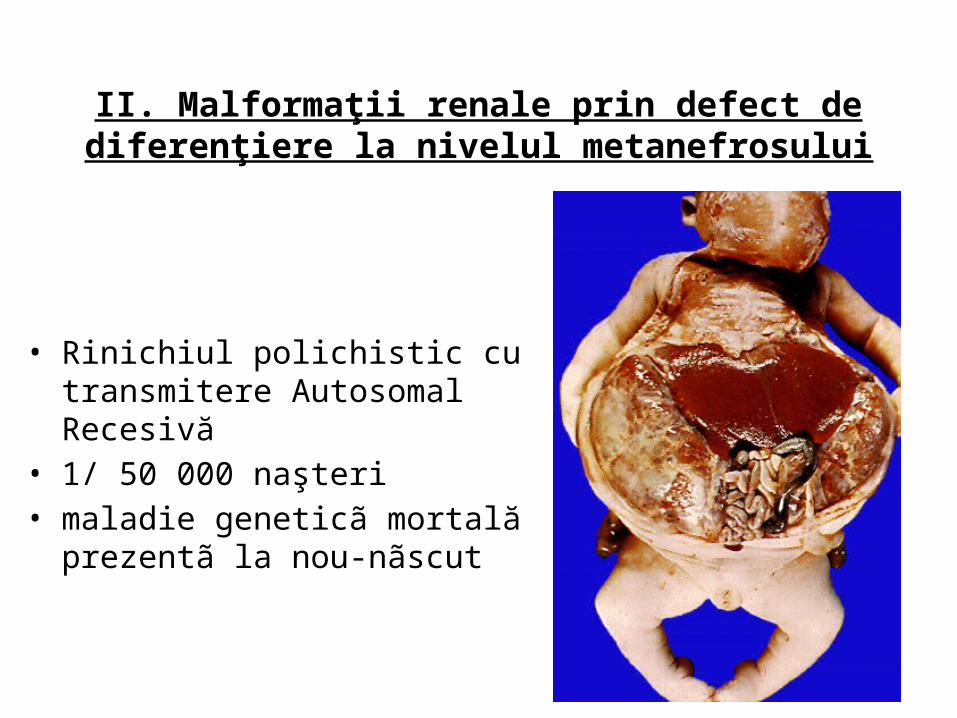
II. Malformaţii renale prin defect de diferenţiere la nivelul metanefrosului
• Rinichiul polichistic cu transmitere Autosomal Recesivă
• 1/ 50 000 naşteri• maladie geneticã mortală prezentã la
nou-nãscut

Rinichiul polichistic cu transmitere Autosomal Recesivă
Chisturile apar prin defecte ale joncţiunii tubilor colectori cu tubii contorţi distali

Chistul solitar Chisturile parapielice situate în sinusul renal, fără comunicare cu bazinetul sau
calicele Rinichiul spongios (maladia chistică a piramidelor renale) – în zona medulară,
în vecinătatea papilelor apar formaţiuni chistice. Este bilaterală şi mai puţin frecventă la femei. Clinic se manifestă prin infecţii ale tractului urinar superior.
Nefroza congenitală infantilă – apare în special la prematuri; incidenţă familială, transmisă autosomal recesiv
• Rinichiul polichistic cu transmitere Autosomal Dominantã
• 1/400-1/1000 naşteri.
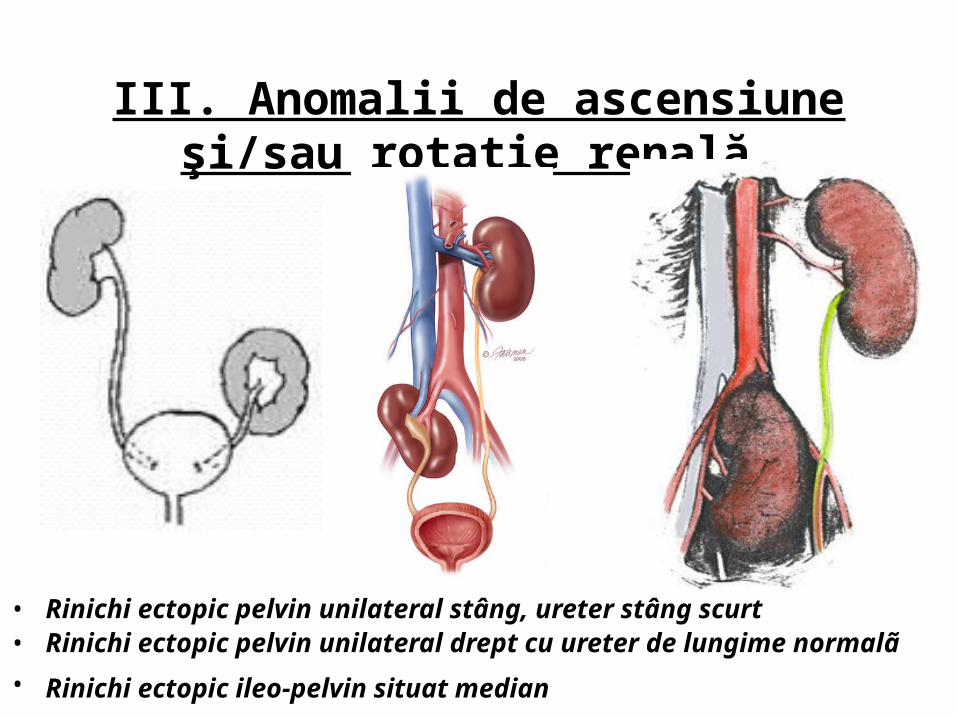
III. Anomalii de ascensiune şi/sau rotaţie renală
• Rinichi ectopic pelvin unilateral stâng, ureter stâng scurt • Rinichi ectopic pelvin unilateral drept cu ureter de lungime normalã • Rinichi ectopic ileo-pelvin situat median

• Rinichi ectopic pelvin supranumerar
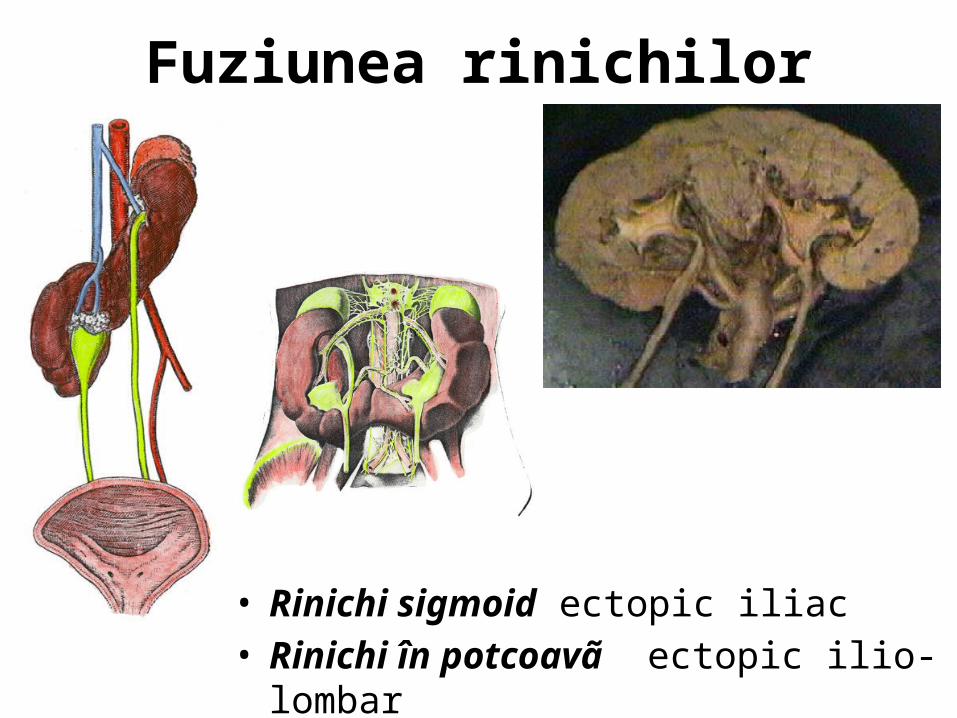
Fuziunea rinichilor
• Rinichi sigmoid ectopic iliac• Rinichi în potcoavã ectopic ilio-lombar

– Rinichi ectopic bilateral (anomalie de sediu şi rotaţie), uretere torsionate

Anomalii renale vasculare
• Artere renale supranumerare • Pe mãsurã ce rinichii ascensioneazã sunt revascularizaţi progresiv de cãtre vase
care iau naştere din aorta dorsalã: artera iliacã, porţiunea distalã a aortei şi în final din artera renalã ramurã din aorta abdominalã.
• Ocazional, unele ramuri vasculare caudale persistã dupã ascensiunea rinichilor. • Artrele accesorii iau naştere din porţiunile aortei adiacente arterei renale
principale sau uneori chiar din artera iliacã.

• Rinichiul stâng prezintã un ureter bifid.
• De reţinut existenţa venelor renale multiple, dintre care una se divide pentru a permite trecerea aortei abdominale.

IV. Malformaţii ureterale
– atrezie ureterală (forma bilaterală este incompatibilă cu viaţa) – duplicaţie ureterală.

– Ureter bifid bilateral, – 4 orificii de deschidere la
nivelul vezicii urinare. Ureterele care dreneazã urina aceluiaşi rinichi sunt rãsucite între ele (torsiune).

– Ureter dublu. – Ureterul normal situat se deschide în vezica urinarã – ureterul ectopic se poate deschide în uretrã, în vestibulul vaginei
sau în porţiune superioarã a vaginei

– Stenoza joncţiunii pielo-ureterale cu distensia bazinetului stâng şi a calicelor– hidronefroză.
– Aspect normal al rinichiului drept. Urografie cu substanţã de contrast

– Megaureter bilateral, hidronefrozã (dreapta), valvã de uretrã posterioarã– Megaureter bilateral, valvã de uretrã posterioarã, ureterocel. – Vârsta gestaţionalã: 7 sãptãmâni

– Megaureter stâng cu distensia bazinetului, hidronefrozã pre- şi retropielicã– Megaureter drept

– Megaureter stâng, obstrucţie în zona de implantare a ureterului în vezica urinarã prin anomalii în dezvoltarea musculaturii ureterului
Megaureter drept cu hidronefrozã. Megadolicoureter stâng cu reflux vezico-ureteral
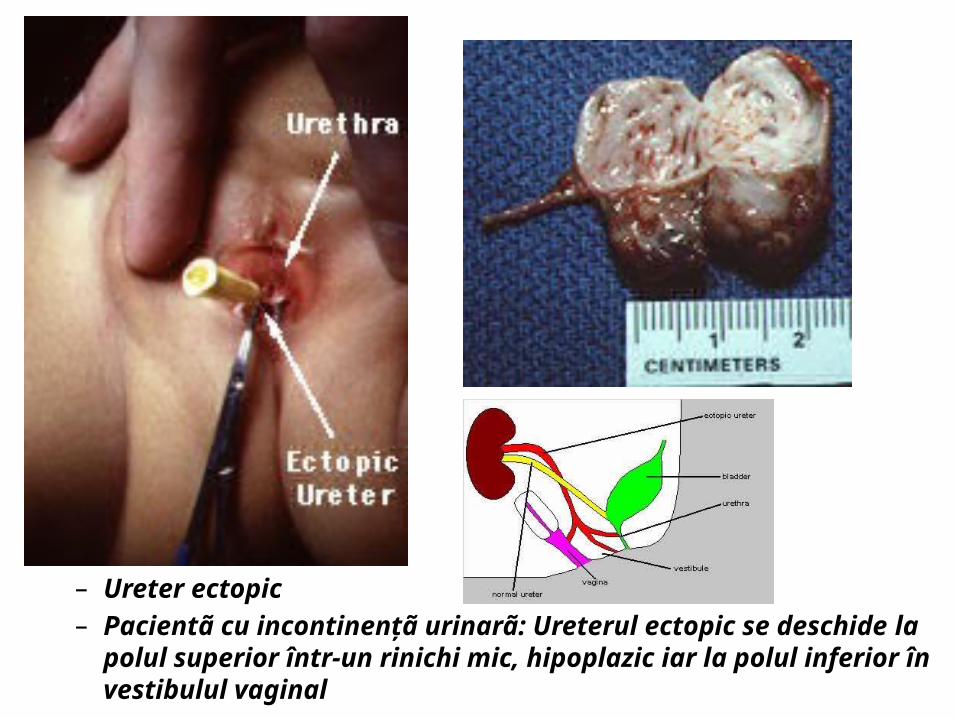
– Ureter ectopic– Pacientã cu incontinenţã urinarã: Ureterul ectopic se deschide la polul
superior într-un rinichi mic, hipoplazic iar la polul inferior în vestibulul vaginal
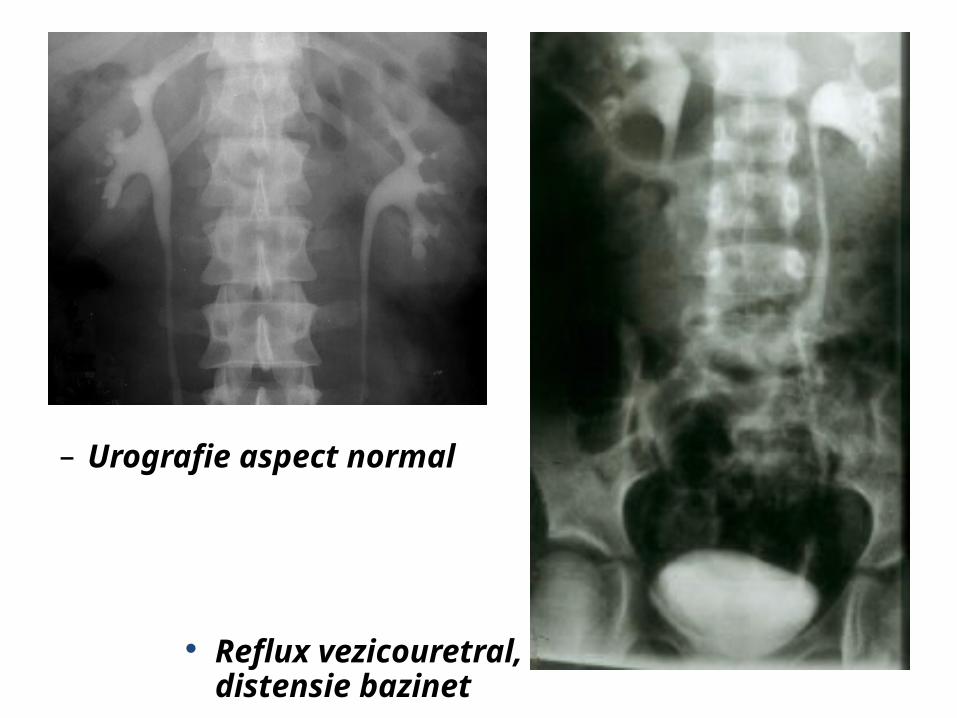
– Urografie aspect normal
Reflux vezicouretral, distensie bazinet

V. Malformaţii congenitale de uretrã
• A. duplicaţie completã a uretrei – cea mai rarã anomalie de uretrã.
• B. duplicaţia porţiunii distale a uretrei, cu pãstrarea uretrei proximale de aspect normal.
• C. uretra accesorie (dorsal sau ventral faţã de uretra normalã); se terminã orb la extremitatea proximalã.
• D Bifurcaţia uretrei în porţiunea proximalã; uretra accesorie se deschide la joncţiunea scrotului cu penisul.
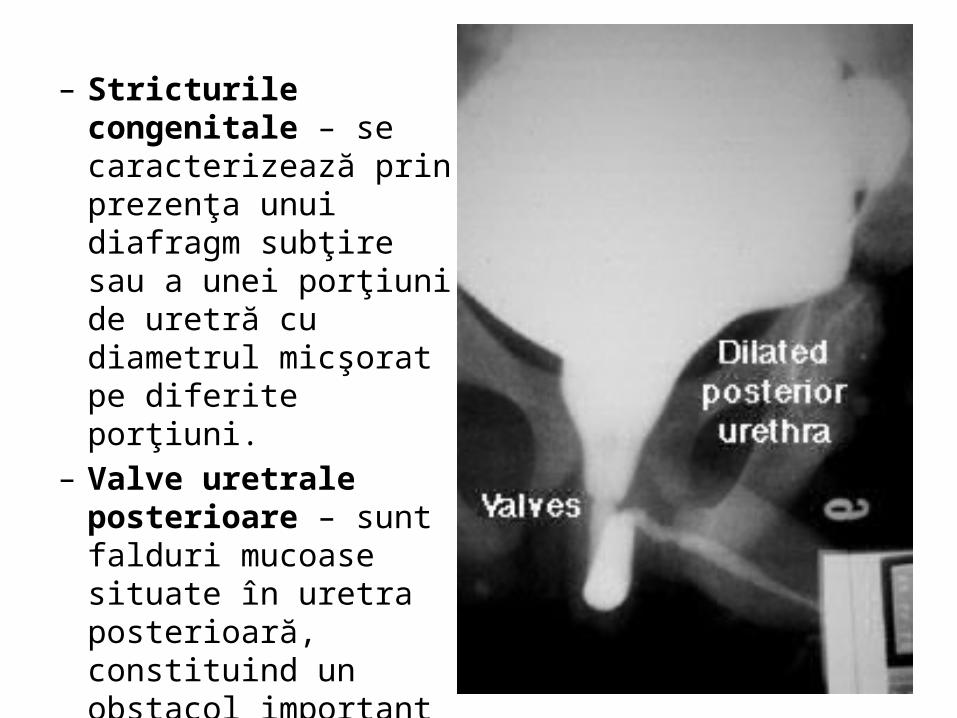
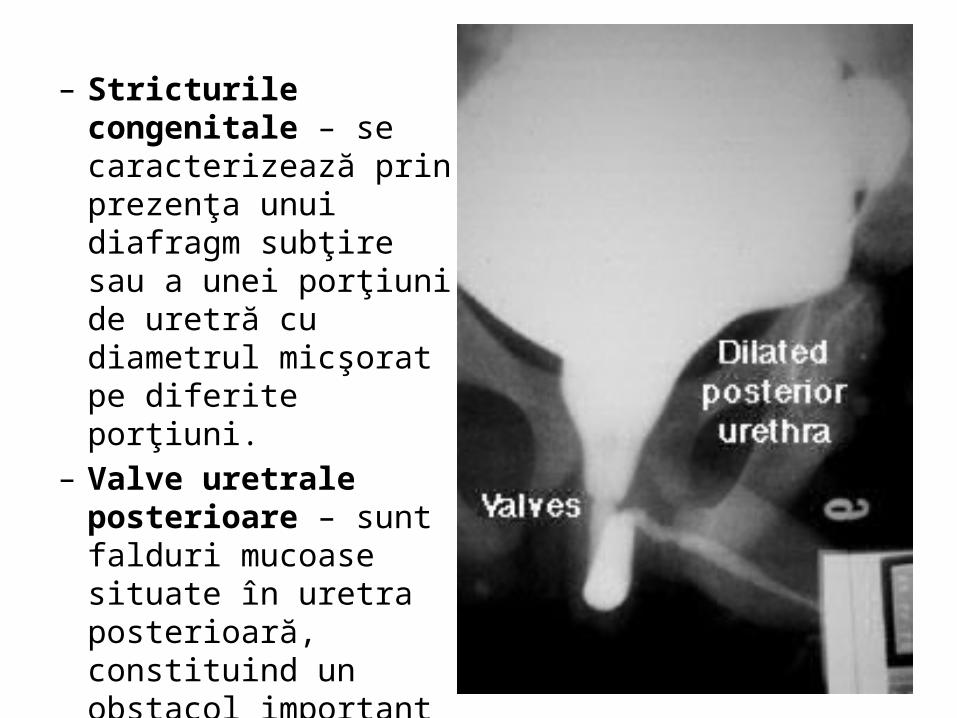
– Stricturile congenitale – se caracterizează prin prezenţa unui diafragm subţire sau a unei porţiuni de uretră cu diametrul micşorat pe diferite porţiuni.
– Valve uretrale posterioare – sunt falduri mucoase situate în uretra posterioară, constituind un obstacol important în eliminarea urinii în timpul micţiunii.

VI. Malformaţii congenitale ale vezicii urinare
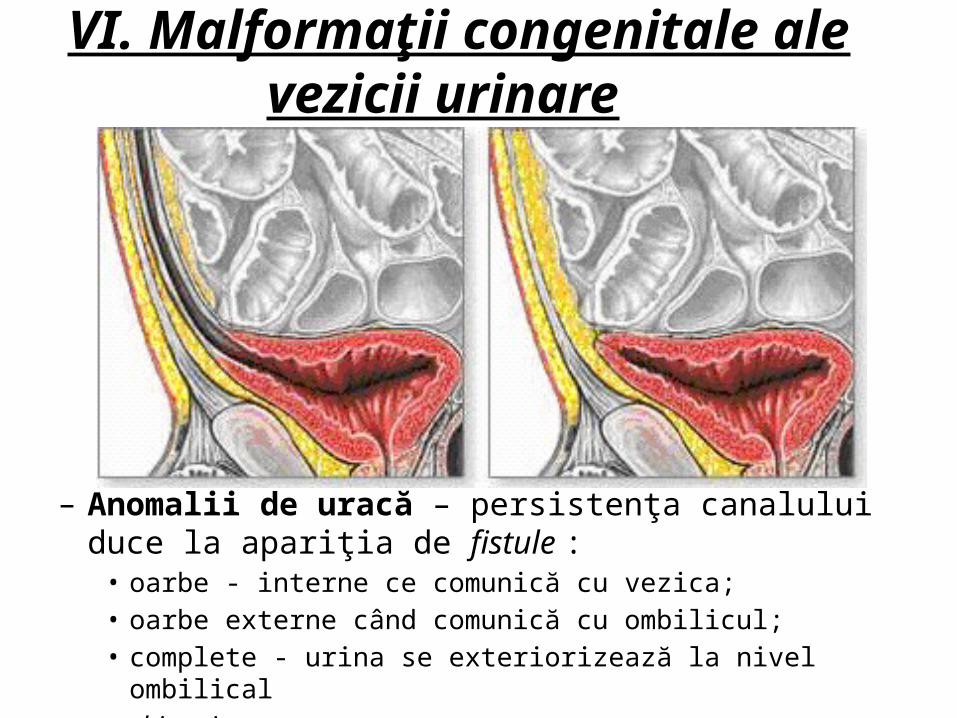
– Anomalii de uracă – persistenţa canalului duce la apariţia de fistule : -oarbe - interne ce comunică cu vezica;
-oarbe externe când comunică cu ombilicul; -complete - urina se exteriorizează la nivel ombilical
-chisturi
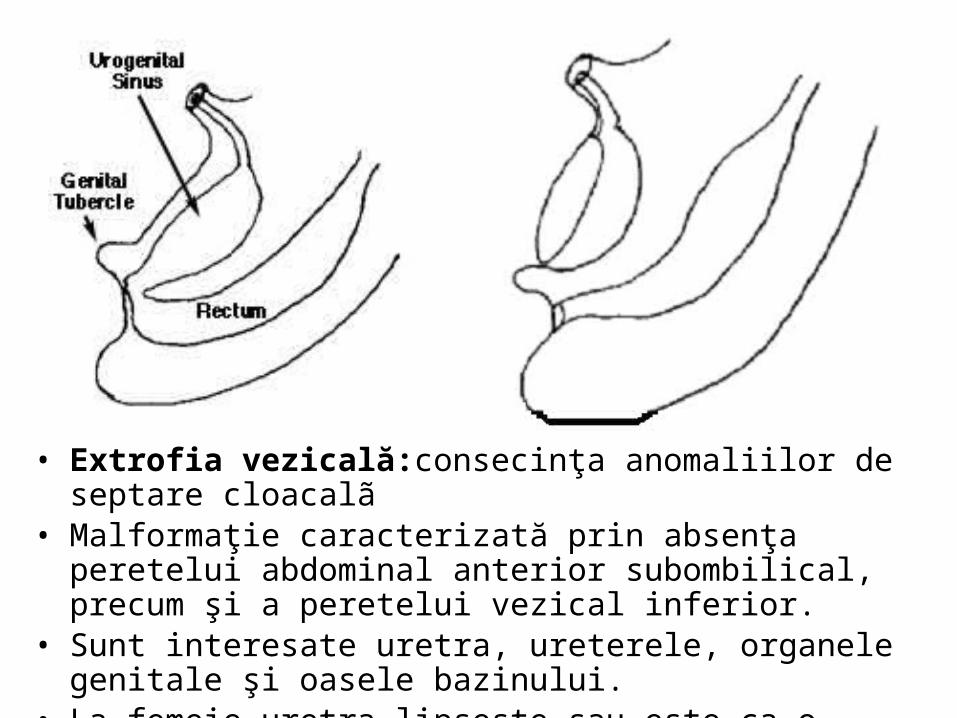
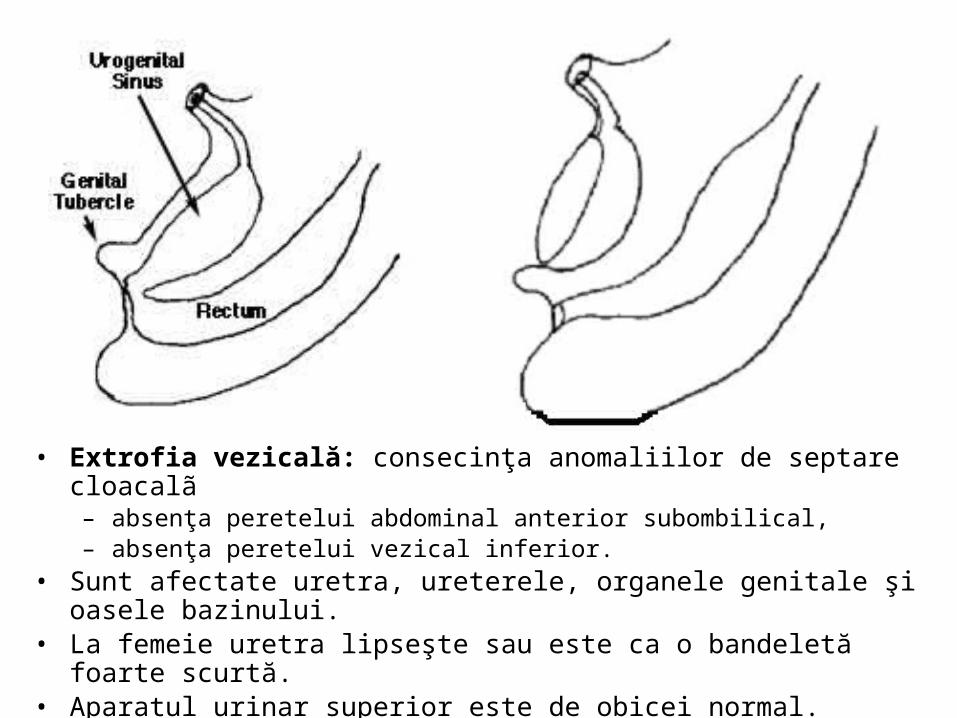
• Extrofia vezicală:consecinţa anomaliilor de septare cloacalã • Malformaţie caracterizată prin absenţa peretelui abdominal
anterior subombilical, precum şi a peretelui vezical inferior. • Sunt interesate uretra, ureterele, organele genitale şi oasele
bazinului. • La femeie uretra lipseşte sau este ca o bandeletă foarte scurtă.
Aparatul urinar superior este de obicei normal.

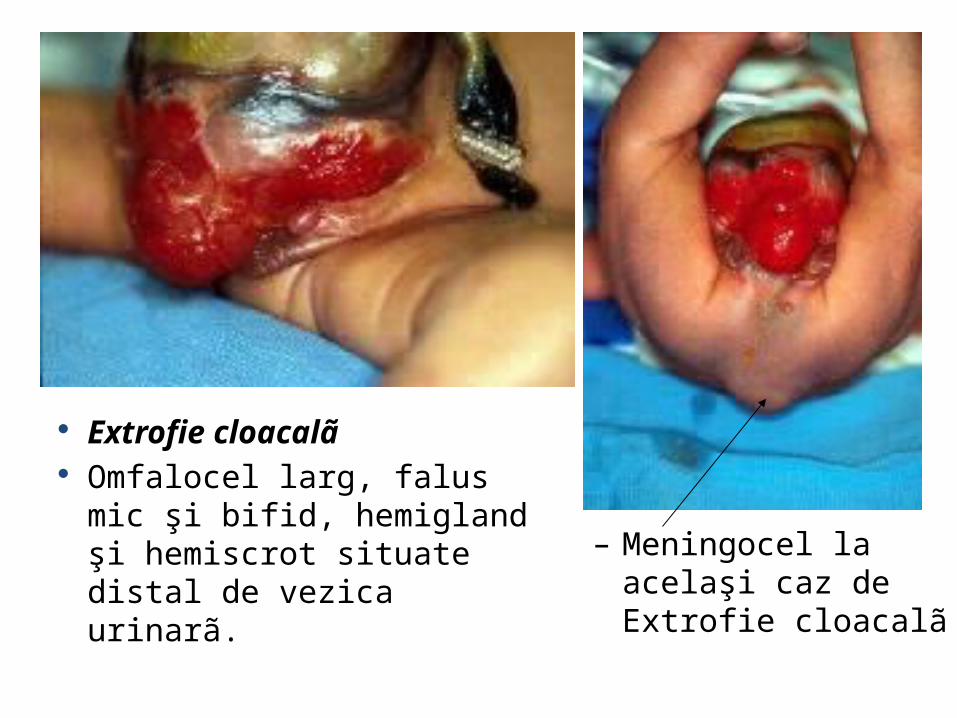
– Meningocel la acelaşi caz de Extrofie cloacalã
Extrofie cloacalã Omfalocel larg, falus mic şi bifid,
hemigland şi hemiscrot situate distal de vezica urinarã.
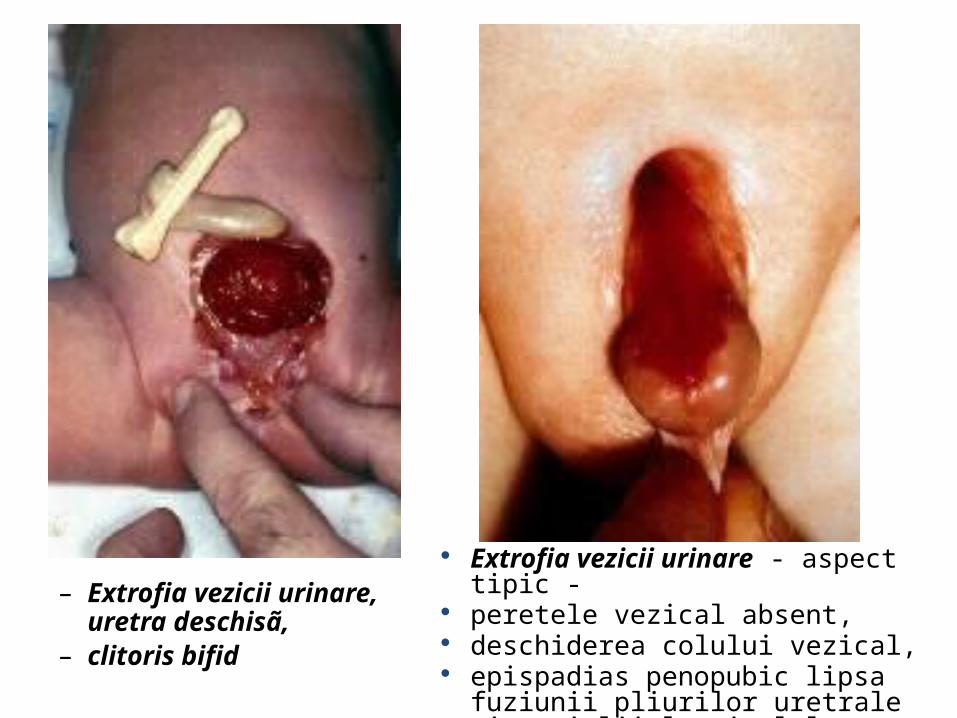
– Extrofia vezicii urinare, uretra deschisã,
– clitoris bifid
Extrofia vezicii urinare - aspect tipic - peretele vezical absent, deschiderea colului vezical, epispadias penopubic lipsa fuziunii pliurilor
uretrale şi a pielii la nivelul glandului penian
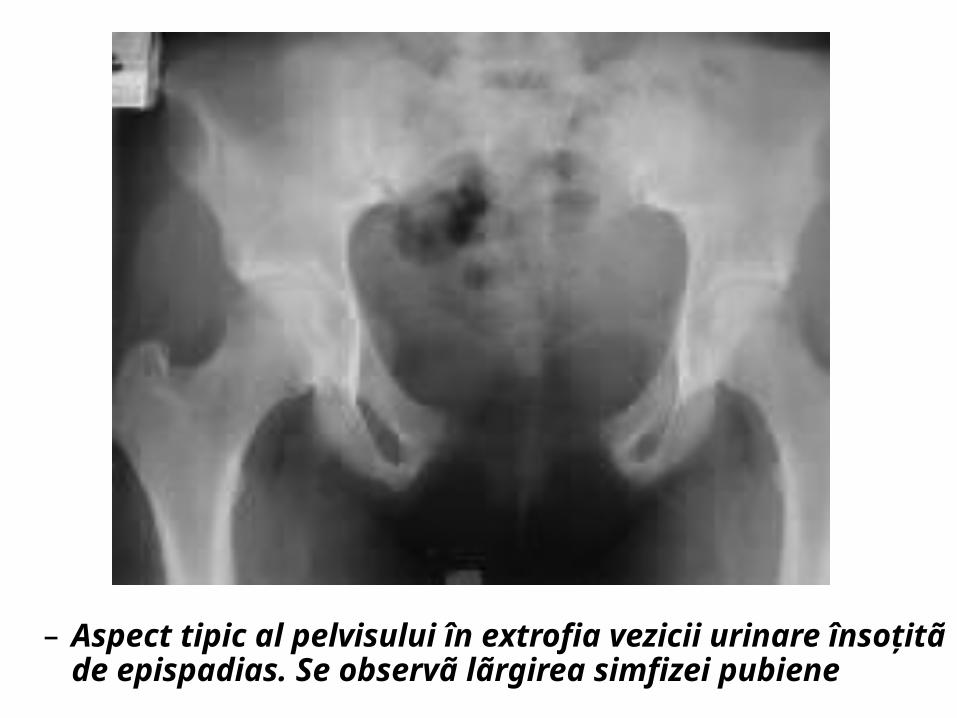
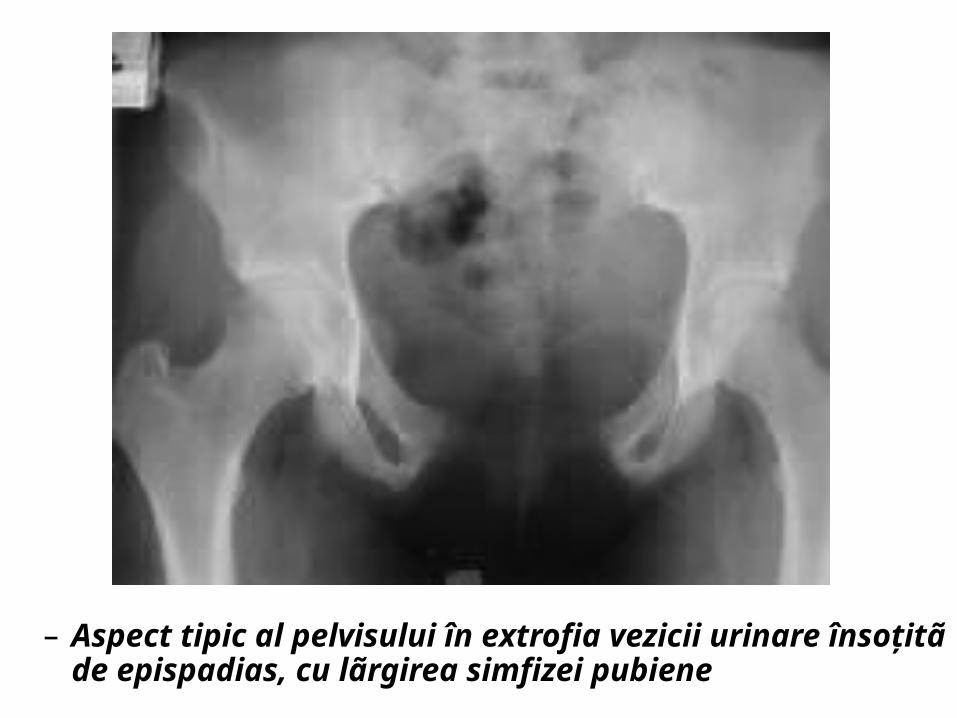
– Aspect tipic al pelvisului în extrofia vezicii urinare însoţitã de epispadias. Se observã lãrgirea simfizei pubiene

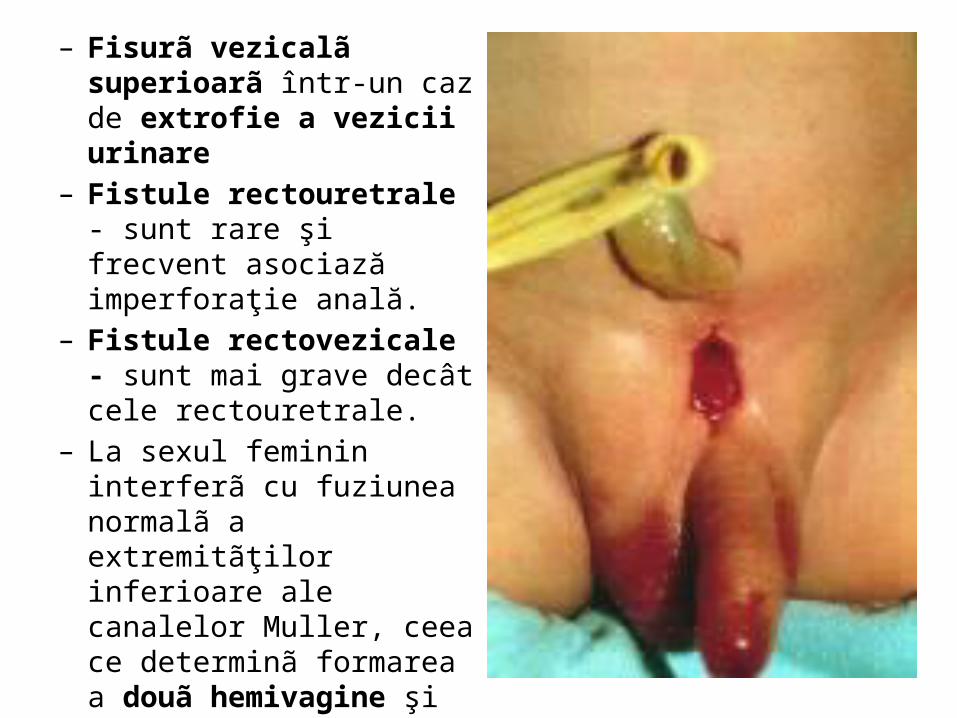
– Fisurã vezicalã superioarã într-un caz de extrofie a vezicii urinare
– Fistule rectouretrale - sunt rare şi frecvent asociază imperforaţie anală.
– Fistule rectovezicale - sunt mai grave decât cele rectouretrale.
– La sexul feminin interferã cu fuziunea normalã a extremitãţilor inferioare ale canalelor Muller, ceea ce determinã formarea a douã hemivagine şi douã hemiutere separate, ale cãror cavitãţi comunicã direct cu lumenul vezicii urinare.
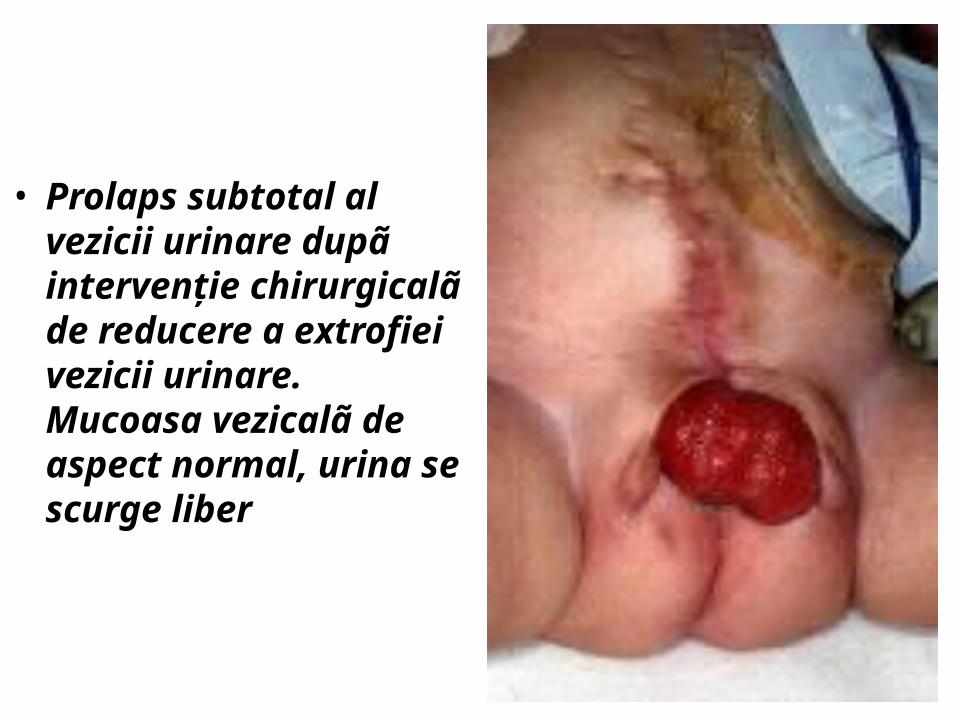
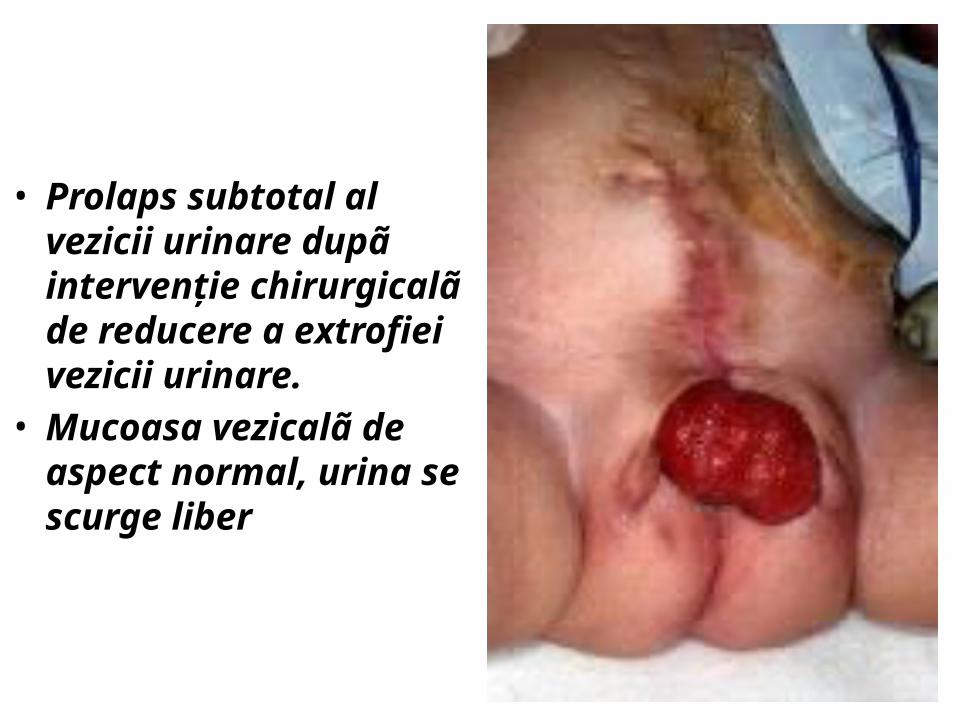
• Prolaps subtotal al vezicii urinare dupã intervenţie chirurgicalã de reducere a extrofiei vezicii urinare. Mucoasa vezicalã de aspect normal, urina se scurge liber


Bibliografie • Ronald W Dudek BRS Embryology Edit.
Lippincott Williams Wilkins 2014
• Keith Moore & T. V. N. Persaud & Mark Torchia Before We Are Born, ESSENTIALS OF EMBRYOLOGY AND BIRTH DEFECTS WITH STUDENT CONSULT ONLINE ACCESS Edit Elsevier 2015
• Bruce Carson Human Embryology and Developmental Biology, with STUDENT CONSULT ONLINE ACCESS Edit Elsevier 2014
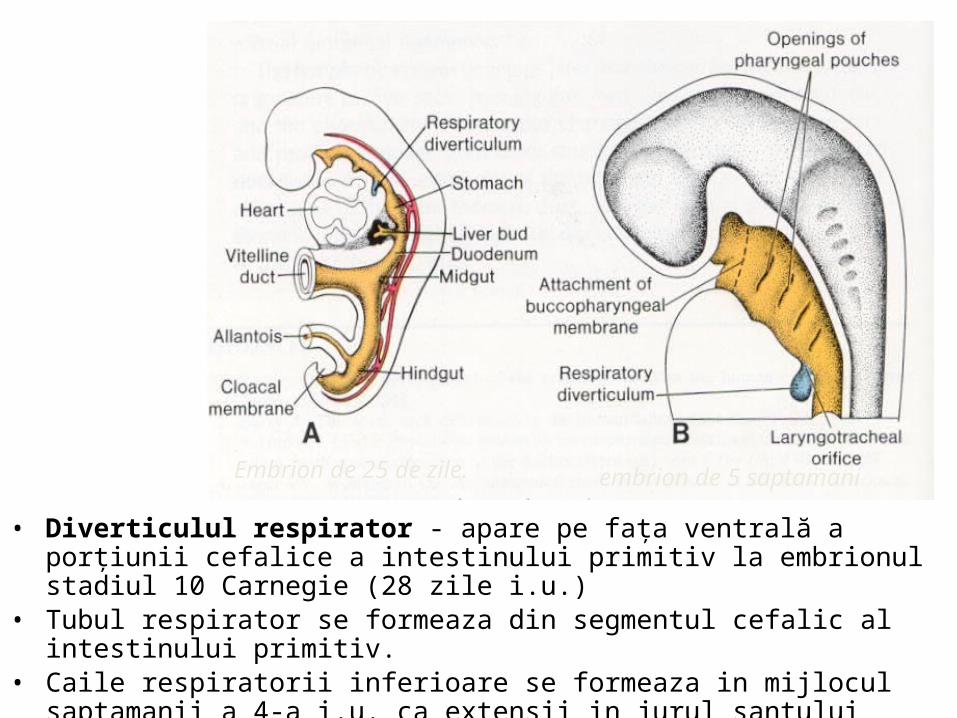
• Diverticulul respirator - apare pe faţa ventrală a porţiunii cefalice a intestinului primitiv la embrionul stadiul 10 Carnegie (28 zile i.u.)
• Tubul respirator se formeaza din segmentul cefalic al intestinului primitiv. • Caile respiratorii inferioare se formeaza in mijlocul saptamanii a 4-a i.u. ca extensii
in jurul santului laringotraheal.
Embrion de 25 de zile. embrion de 5 saptamani

• diverticulul respirator se formează din intestinul primitiv cefalic (podeaua faringelui) • Alungire caudală • Separare de intestin prin apariţia a două pliuri longitudinale, eso-traheale, care vor fuziona
ulterior formand septul septul traheo-esofagian • Resorbţia septului traheo-esofagian în săptămâna a 5-a, cu separarea traheei de esofag• Esofagul devine situat dorsal iar traheea şi mugurii pulmonari ventral.• Primordiul respirator menţine comunicarea cu faringele prin orificiul laringeal

• Laringele se diferenţiază În săptămânile 6-7 i.u.
Portiunea ventrală a faringelui
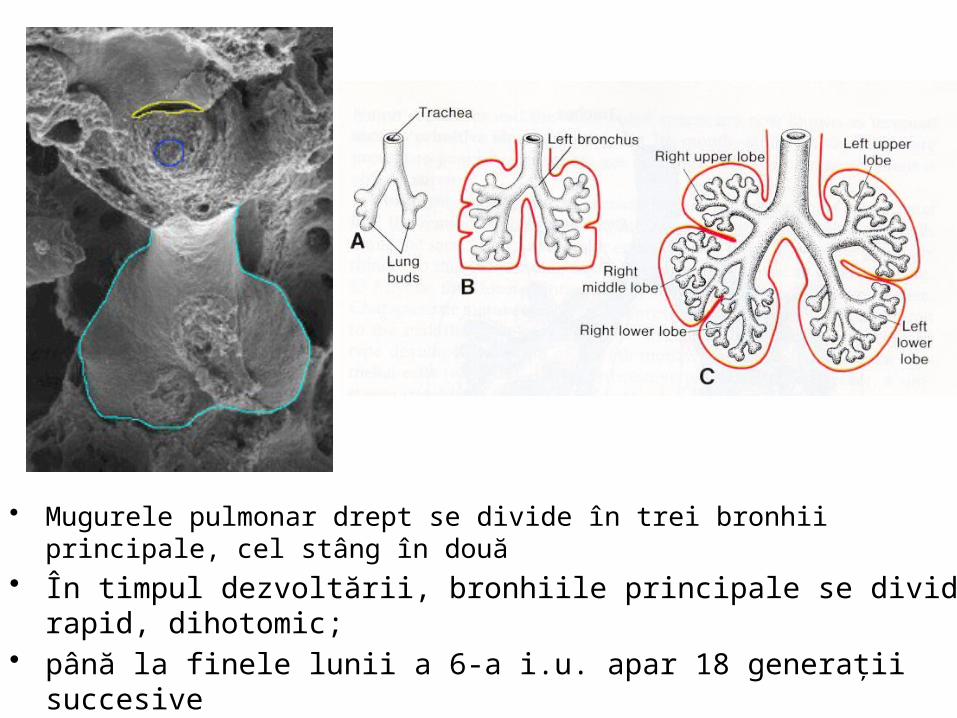
• Mugurele pulmonar drept se divide în trei bronhii principale, cel stâng în două • În timpul dezvoltării, bronhiile principale se divid rapid, dihotomic; • până la finele lunii a 6-a i.u. apar 18 generaţii succesive • postnatal apar 6 generaţii; • arborele bronşic atinge forma finală la 7-8 ani după naştere.

Principalele faze ale ontogenezei pulmonareFAZA DE CREŞTERE INTERVALUL
DE TIMP CARACTERISTICI
EMBRIONARĂ Zilele 26-52 Dezvoltarea: traheei, bronhiilor principale, lobare, segmentare
PSEUDOGLANDULARĂ Ziua 52-săptămâna 16 Dezvoltarea: arborelui bronşic în continuare până la brohiole respiratorii
CANALICULARĂ Săptămânile 17-28 Dezvoltarea patului vascular, schiţarea acinului alveolar, aplatizarea şi diferenţierea epiteliului respirator
SACCULARĂ Săptămânile 29-30 Dezvoltarea prealveolelor
ALVEOLARĂ Săptămâna 36 -termen Dezvoltarea alveolelor
PLĂMÂNII

• Primordiul pulmonar apare în săptămâna a 4-a i.u. ca diverticul endodermal laringo-traheal prezent la nivelul feţei ventrale a esofagului primitiv, situat într-o atmosferă mezenchimală
• se bifurcă la inferior in doi muguri bronho-pulmonari• Prin ramificare mugurii bronho-pulmonari cresc (în grosimea mezocardului dorsal)
lateral şi caudal pătrunzând în lumenul canalelor pleuro-peritoneale, unde formează mugurii pulmonari.

• mugurii pulmonari cresc caudal şi lateral, în cavitatea coelomică • Spaţiul îngust situat pe ambele părţi ale intestinului primitiv cefalic este denumit canal
pericardioperitoneal; • Pliurile pleuropericardice separă cavităţile pleurale primitive • Pliurile pleuroperitoneale separă cavitatea pericardică şi peritoneală. • Mezodermul splanhnic intra-embrionar (în contact direct cu plămânii) dezvoltă pleura
viscerală. • Mezodermul somatic intra-embrionar formează pleura parietală.

• Tesut pulmonar la nou-nascut.
STADIUL SACCULAR / SÃPTÃMÂNILE 29-36 I.U.• arborele bronşic schiţează acinul prin:
– formarea ductelor alveolare, – continuarea procesului de aplatizare şi diferenţiere a
epiteliului luminal.
• În timpul ultimele 2 luni prenatală şi în primii ani postnatal numărul de saci terminali creşte continuu. Celulele care căptuşesc sacii denumite celule epiteliale alveolare de tip I devin aplatizate aşa încât capilarele care le înconjură proemină în sacii alveolari. Se realizează astfel un contact intim cunoscut ca barieră
aer-sânge. • Diferenţierea celulelor alveolare de tip I este urmată
de apariţia celulelor alveolare de tip II care-şi păstrează aspectul cubic (granuloase) şi sunt responsabile de secreţia de surfactant (substanţă capabilă să scadă tensiunea superficială a aerului alveolar).
• După luna a 6-a i.u şi până la naştere se dezvoltă reţeaua limfatică pulmonară.

• Alveolele pulmonare mature nu sunt prezente înainte de naştere. • la naştere sunt prezente 17-20 milioane alveole = 1/6 din alveolele adultului• După naştere, în primii 10 ani, se formează 300 milioane alveole =5/6 din alveolele
adultului
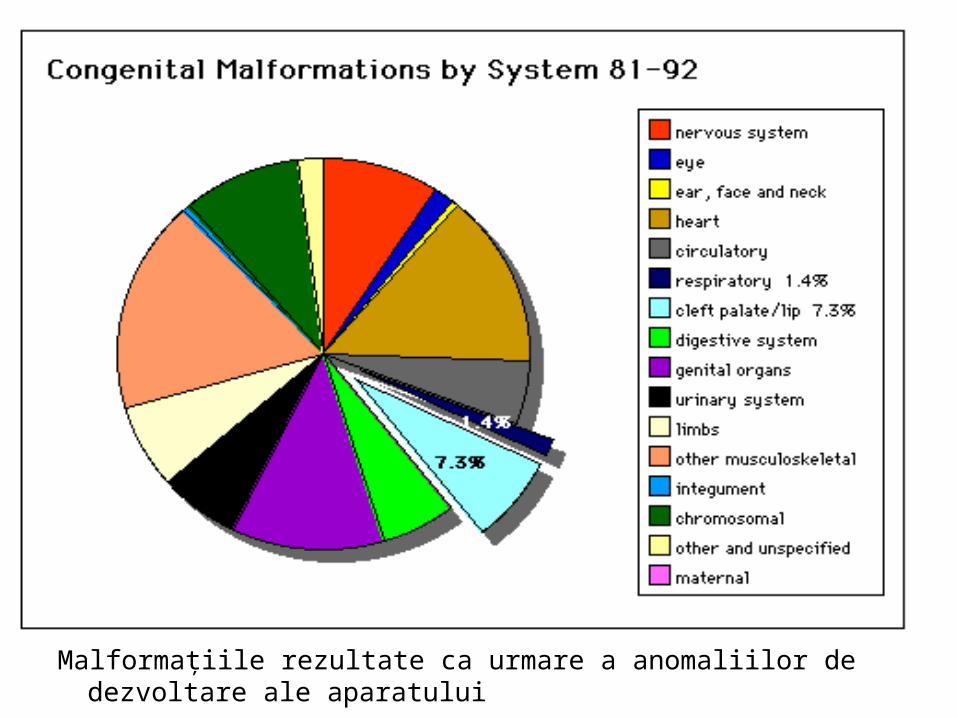
Malformaţiile rezultate ca urmare a anomaliilor de dezvoltare ale aparatuluirespirator reprezintă 1,4% din totalul malformaţiilor pe aparate şi sisteme.

• Hernia diafragmatica congenitala • Malformatii congenitale ale Laringelui
– atrezia laringelui– atrezia partiala a laringelui:
– supralotic– glotic– subglotic
– stenoza subglotica– laringomalacia – chist glotic – laringocel – fistula laringo-traheo-esofagiana – despicatura laringo-traheo-esofagiana (laringe despicat, peristenta comunicarii eso-
traheale) – obstructii laringiene extrinseci congenitale – laringocel extern
• Anomalii Arteriale– arc aortic dublu – compresie arteriala
• Malformatii congenitale ale Traheei – traheomalacia
• Malformatii Congenitale ale Căilor pulmonare • Malformatii Congenitale ale Plamanilor

• Incidenţa 1:1200-1500 de nou-născuţi; 1:3000 nasteri • Apare
– Sporadic– Recurenta familiala
• Efect al hipoplaziei pulmonare• Asociaza hipertensiune pulmonara.• Urmare a lipsei de dezvoltare a portiunii posterolaterale a diafragmei (rezulta
persistenta foramen Bochdalek ) • 90% dintre cazuri este afectata partea stanga a difragmei. • Rata de supravietuire aproximativ 50%. • Diagnostic Antenatal ultrasunete. • Factori de risc crescut:
– polihidramnion – Trisomia 18 – Trisomia 21.
• Approximativ 40% din cazuri asociaza alte anomalii. • Rar diagnosticata incidental, la copilul mare.
Hernia diafragmatică congenitala

Hernia Diafragmatică Congenitală

• Hernie diafragmatica
congenitala stângă
• Deplasarea mediastinului la
dreapta;
Aspect normal dupa interventie chirurgicala
• Examen Clinic: • cianoza severa, • abdomen scafoid (excavat), • reducerea sau absenta aerului de prtea
afectata,• ocazional zgomote hidro-aerice in torace.
• Hernie diafragmatica congenitala stângă• Stomacul situat in hemithoracele stâng
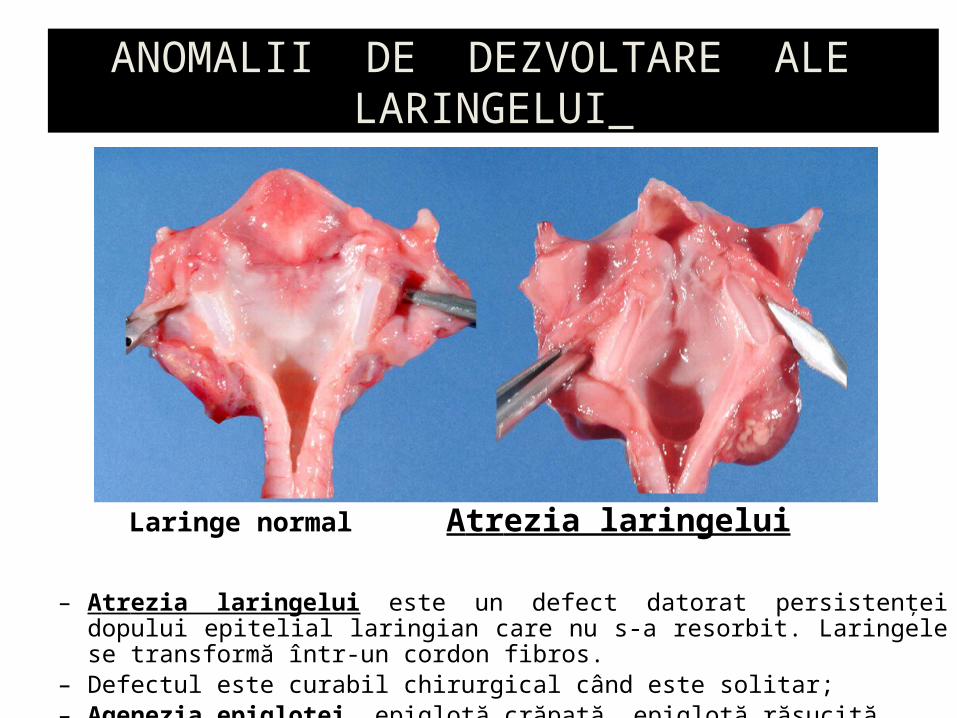
ANOMALII DE DEZVOLTARE ALE LARINGELUI
– Atrezia laringelui este un defect datorat persistenţei dopului epitelial laringian care nu s-a resorbit. Laringele se transformă într-un cordon fibros.
– Defectul este curabil chirurgical când este solitar;– Agenezia epiglotei, epiglotă crăpată, epiglotă răsucită
Laringe normal Atrezia laringelui

Laringomalacia (Condromalacia laringelui)
• consecinţa lipsei de condrificare a modelelor fibroase ale cartilajelor laringelui.
• Procesul se poate întinde şi la nivelul traheei şi bronhiilor.
• Nefiind rigide, aceste organe se colabează în timpul inspiraţiei, provocând accidente asfixice.
• Clinic: stridor evident din primele luni
• Tratament: Se impune intervenţie chirurgicală intre 18 luni – 2 ani
• Asocieri: disfagie, hipotrofie
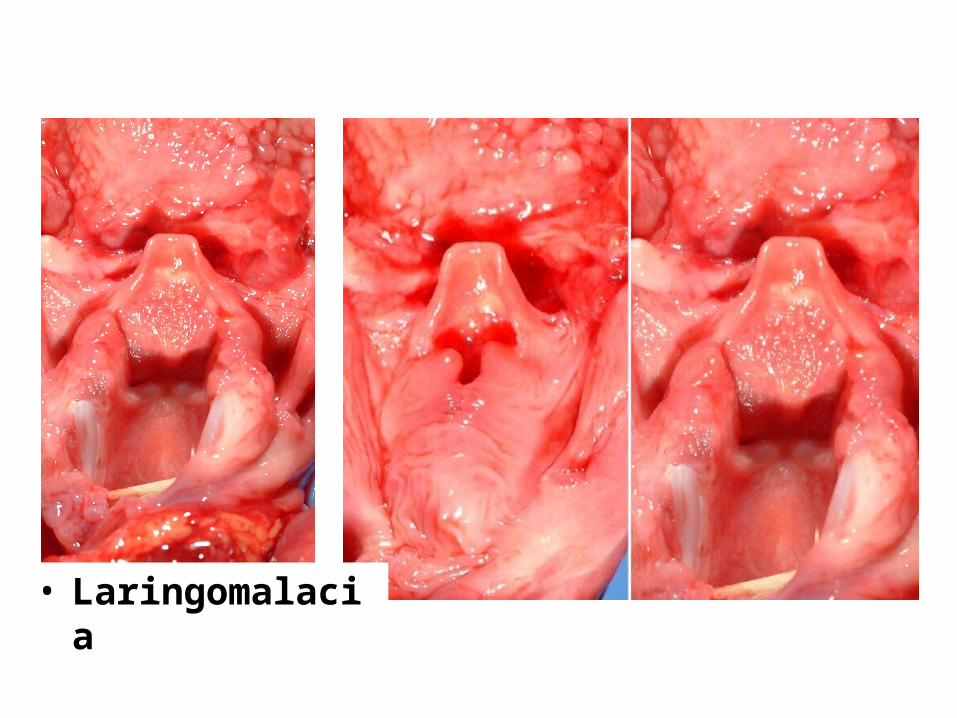
• Laringomalacia
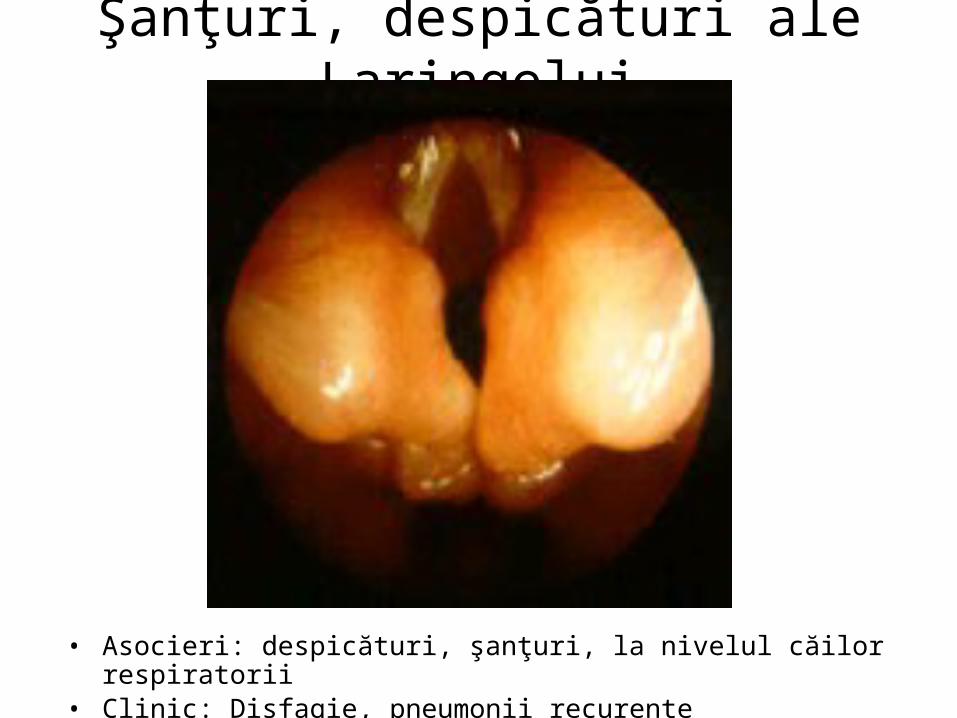
Şanţuri, despicături ale Laringelui
• Asocieri: despicături, şanţuri, la nivelul căilor respiratorii• Clinic: Disfagie, pneumonii recurente

ANOMALII DE DEZVOLTARE ALE TRAHEEI
- Condromalacia traheei- Atrezie traheală

– Fistula eso-traheală, – Prin separare incompletă a traheei de intestin– Asocieri:– fisura mediană posterioară a cartilajului cricoid, – esofag atrezic şi divizat transversal,– traheea se deschide în segmentul esofagian
inferior, – segmentul esofagian superior se termină în fund
de sac.

– Fistula eso-traheală, – Prin separare incompletă a
traheei de intestin– Asocieri:– fisura mediană posterioară a
cartilajului cricoid, – esofag atrezic şi divizat
transversal,– traheea se deschide în
segmentul esofagian inferior, – segmentul esofagian superior
se termină în fund de sac.
AtreziaEsofaguluiasociata cuFistulaEsotraheală87%
AtrezieEsofagianaizolata8%
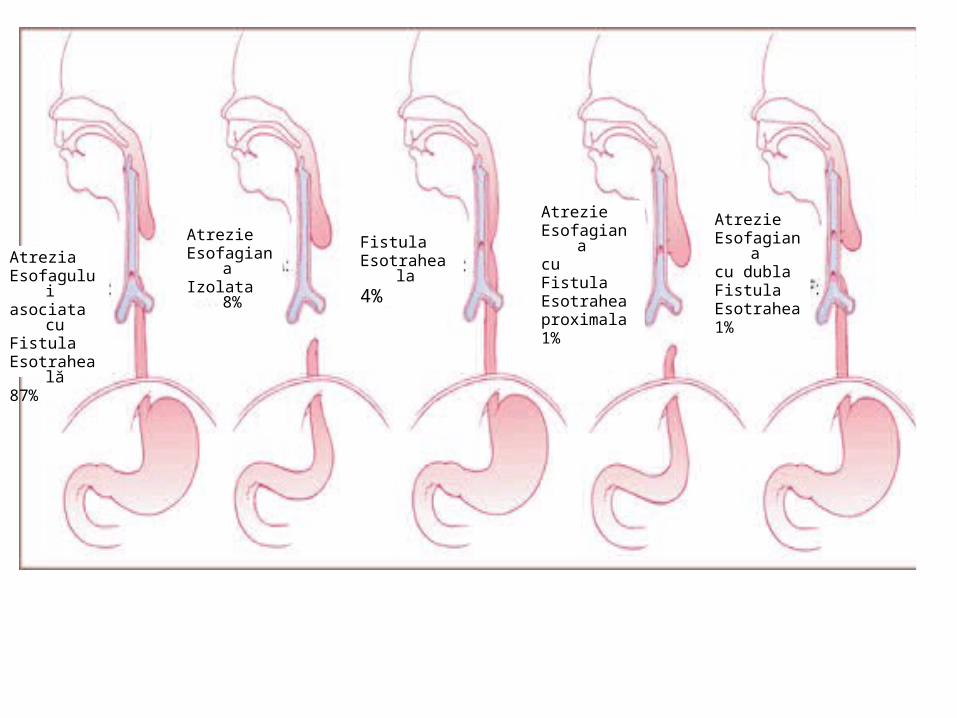
AtreziaEsofaguluiasociata cuFistulaEsotraheală87%
AtrezieEsofagianaIzolata 8%
Fistula Esotraheala4%
AtrezieEsofagianacu dubla Fistula Esotrahea1%
AtrezieEsofagianacuFistula Esotraheaproximala1%

Diverticulul laringo -traheal creste in zonaventro-caudala afaringelui primitiv
Stadii succesive in dezvoltarea septului traheo-esofagian
PlicileLongitudinaletraheo-esofagienefuzioneaza medio - sagital alcatuindseptul traheo - esofagian.
Septul traheoesofagiancomplet format
Devierea posterioara aseptului traheo - esofagian determinaatrezia esofagului sidezvoltarea fistulei eso –t raheale.

Despicatura laringo-traheo-esofagiana congenitala
• Sugar intubat pentru insuficienta respiratorie.
• Sonda nazogastrica trece dar nu poate patrunde in stomac.
• Despicatura laringo-traheo-esofagiana congenitala este Vizibila pe Rx toracelui.
• Radiografia toracelui: Sonda nazogastrica situata in bronhia principala stanga, patrunde in lobul inferior stang
• Laringoscopie: confirma prezenta unei despicaturi laringo-traheo-esofagiene extinse, laringe - carina.
ANOMALII ALE ARBORELUI BRONŞIC
• Îngustări: – agenezie, – stenoze, – atrezie
• Bronhomalacia reprezintă obstrucţia difuză a bronhiei prin anomalie a structurii inelelor cartilaginoase.
• Bronşiectazia congenitală este dilataţia anormală, permanentă a uneia sau mai multor bronhii însoţită de defecte ale peretelui bronşic.În etiologia ei se recunosc cauze infecţioase şi afecţiuni cu determinare genetică
• Traheobronhomegalia Este o afecţiune congenitală cu transmitere autosomal recesivă ce afectează ţesutul muscular şi elastic traheo- bronhic în care traheea poate egala calibrul coloanei vertebrale, iar bronhiile principale sunt mult dilatate.

ANOMALII IN DEZVOLTAREA PLĂMÂNILOR
– Anomaliile pulmonare pot interesa:-Plămânul în ansamblu: agenezie pulmonară, hipoplazie
pulmonară, modificarea numărului de lobi (fisuri anormale);-Doar arborele bronşic: stenoză congenitală intrinsecă (asociată
cu atelectazie), stenoză intrinsecă, bronhomalacie, bronşiectazie, chisturi bronhice;
-Relaţia morfo-funcţională arbore bronşic-artere pulmonare: sechestraţie bronhopulmonară;
-Vasele pulmonare: anomalii izolate ale arterelor pulmonare, anomalii arterio-venoase, anomalii ale venelor pulmonare, anomalii ale limfaticelor pulmonare.
-În lipsa stratului de surfactant are loc colaps alveolar în timpul expiraţiei(Maladia Membranelor Hialine)

• Anomalii care interesează plămânul în ansamblu
• Agenezia şi aplazia pulmonară • Sunt rezultatul lipsei de formare a mugurilor pulmonari (agenezia) sau înlocuirii cu un
bont (aplazia). • Aplazia pulmonară unilaterală permite supravieţuirea.
• Hipoplazia pulmonară • Diminuarea numărului diviziunilor bronhice şi volumului pulmonar,• Reducerea numărului de saci terminali. • Incidenţa este de aproximativ 10% din nou-născuţii examinaţi post mortem; 2%o nou-
născuţi vii. • În multe cazuri numărul generaţiilor bronşice este redus, fapt care sugerează o injurie
apărută în intervalul săptămâna 10 - 14 când plămânul dezvoltă o puternică ramificaţie a arborelui bronşic.
• Forma cea mai frecventă este hipoplazia pulmonară secundară unor factori care acţionează prin efect restrictiv asupra plămânilor

• Hipoplazia pulmonară secundară insuficienţei cutiei toracice (omfalocel). • Hipoplazia pulmonară secundară prin supresia mişcărilor respiratorii. indusă prin
consumul matern de: – Etanol – Barbiturice, – Tutun

• Chisturi pulmonare congenitale unice / multiple, uni sau bilaterale;• Emfizemul lobar congenital
– prima afecţiune pulmonară congenitală în ordinea frecvenţei. – Constă în formarea uneia sau mai multor bule realizate:
• printr-o anomalie a peretelui bronşic (componentă cartilaginoasă) sau • printr-o obstrucţie determinată de falduri ale mucoasei bronşice (mecanism de
“supapă”).

Malformaţia chistică
• Epidemiologie: • 25% dintre malformaţiile
congenitale pulmonare.• incidenţă : 1/25000-35000
sarcini. • 4-26% in asocieri
plurimalformative
• incidenţă : 1/25 000-1/35 000 sarcini

Chist congenital adenomatoid malformativ pulmonar
• Anomalie pulmonara structurala,• Vizibila antenatal ca leziune chistica • Poate asocia hidrops fetal daca este destul de voluminos pentru a obstrua
intoarcerea venoasa catre inima. • Exista mai multe Tipuri, identificate pe criteriul componentelor leziunii:
– solid, (tipul lezional solid (prognostic mai grav decat tipul chistic)– microchistic, – macrochistic.
• Evolutie:“– regresii" ale leziunii in utero, cu Rx torace aspect normal la nastere – Insuficienta respiratorie severa la nastere, necesitatea interventiei chirurgicale de
urgenta– Poate asocia malignitati ulterioare, pe parcursul vietii
• Se recomanda – CT scaner la 3-6 luni dupa interventia chirurgicala– rezectia chirurgicala in perioada copilariei, chiar daca starea sugarului / copilului este
buna.
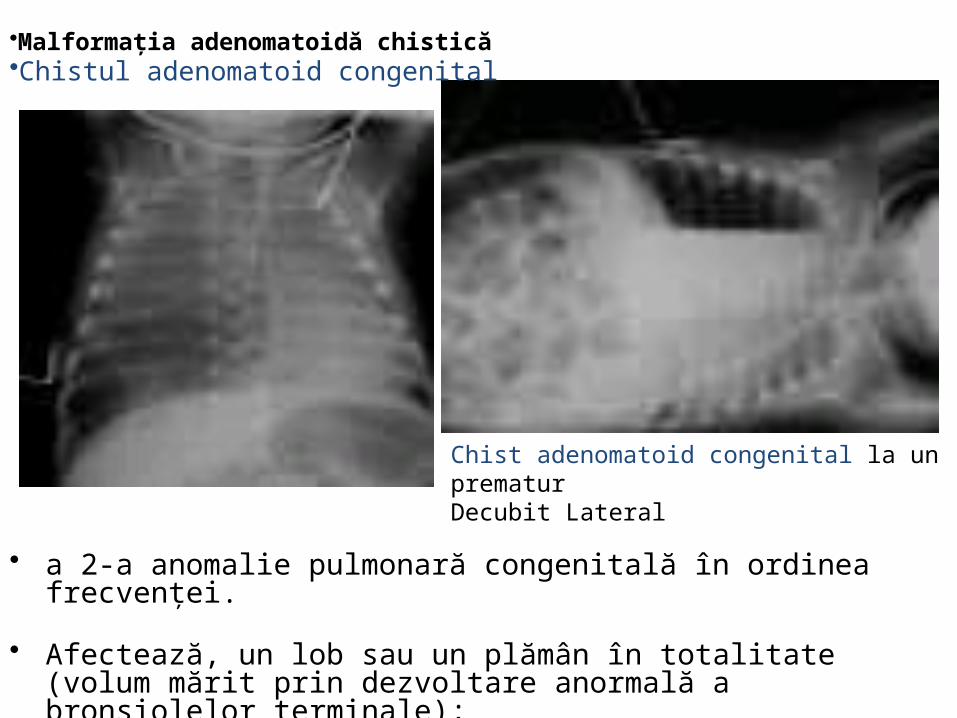
• a 2-a anomalie pulmonară congenitală în ordinea frecvenţei.
• Afectează, un lob sau un plămân în totalitate (volum mărit prin dezvoltare anormală a bronşiolelor terminale);
• Risc crescut pentru neoplasm pulmonar primitiv.
•Malformaţia adenomatoidă chistică •Chistul adenomatoid congenital
Chist adenomatoid congenital la un prematurDecubit Lateral

Abces Pulmonar Congenital
• Poate asocia malformaţii structurale: – sechestrarea lobului pulmonar, – chist pulmonar congenital
chist pulmonar congenital / pl. stg

• Pulmonii supranumerari se formeaza– din expansiuni separate,
supranumerare ale intestinului cefalic
– din segmente ale pulmonilor aflati in dezvoltare, segmente care pierd legatura cu restul plamanului.
• Lobul accesor format inainte de formarea pleurei este invelit de aceeasi pleura ca si pulmonul normal, fiind considerat intralobar.
• Lobul accesor format dupa constituirea pleurei are propria pleura , fiind considerat extralobar.
Pulmon Sechestrat
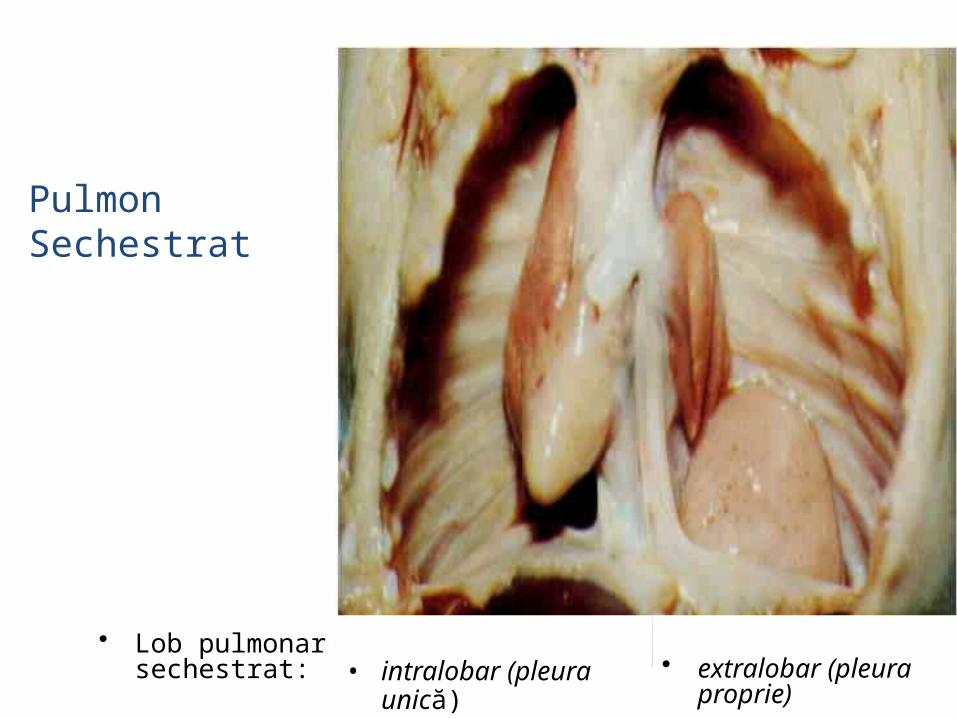
Pulmon Sechestrat
• intralobar (pleura unică)
• extralobar (pleura proprie)
• Lob pulmonar sechestrat:

• Anomalii Asociate : 50-60% din cazuri– 1.Hernia Diafragmatica congenitala,– 2.Pectus excavatum congenital, – 3.Fistula traheo esofagiana congenitala,– Duplicatie esofagiana, chist esofagian,– Chist bronsic congenital, – Megacolon congenital – Malformatii cardiace congenitale.
• Diagnostic Diferential : – malformatii Chistice adenomatoidepulmonare,– teratom mediastinal,– hernia diafragmatica congenitala.
• Prognostic: • Rata ameliorata de supravietuire dupa
interventie chirurgicala• Mortalitate 100% in asocierea cu hidrops• Risc de recurenta: necunoscut, sporadic.• Management: terapie In utero• Esential: Diagnosticul antenatal si interventia
chirurgicala, pentru a preveni:– Hidropsul fetal– Hipoplazia pulmonara.
Pulmon Sechestrat
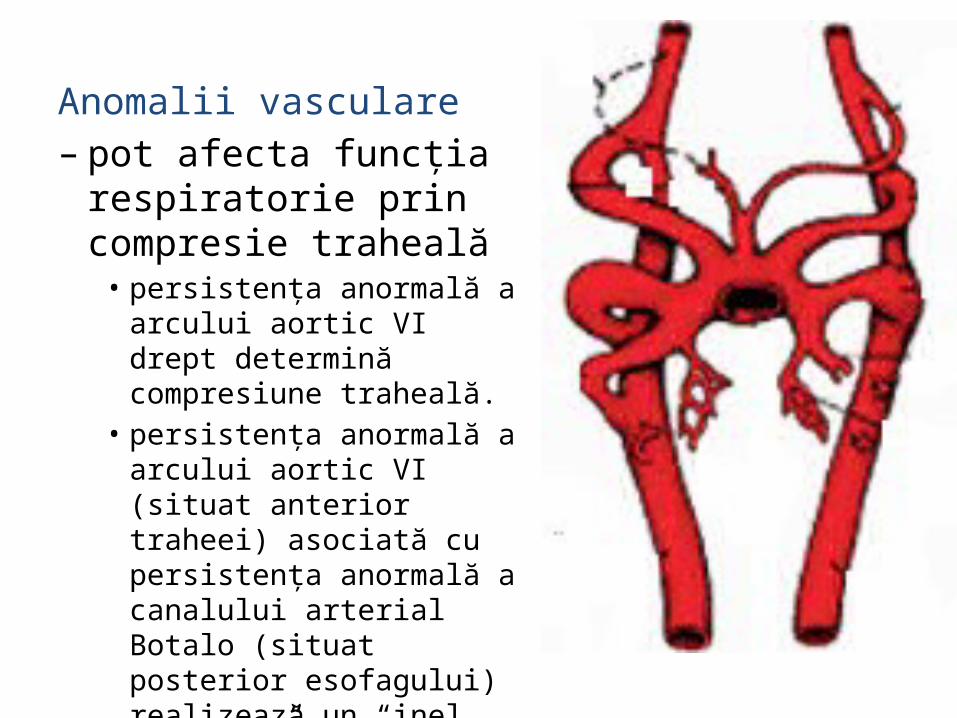
Anomalii vasculare – pot afecta funcţia
respiratorie prin compresie traheală
• persistenţa anormală a arcului aortic VI drept determină compresiune traheală.
• persistenţa anormală a arcului aortic VI (situat anterior traheei) asociată cu persistenţa anormală a canalului arterial Botalo (situat posterior esofagului) realizează un “inel vascular” constrictiv în jurul traheei şi esofagului

3 Malformaţii arteriovenoase / Plămânul drept. Tratament: embolizare.
Malformaţii arteriovenoase

Fibroza chisticã de pancreas (Mucoviscidozã)
• Transmitere ereditarã • Recesivã • 1/3300 populaţia infantilã (Caucazieni)• 1/15300 (Afroamericani)• sex ratio 1/1• gena controleazã producerea unei proteine care
regleazã transportul transmembranar al ionilor de Na şi Cl – întreruperea transportului transmembranar de
Na şi Cl, – deshidratare, – vâscozitate crescutã a secreţiilor.
• 3% dintre Caucazieni poartã 1 genã anormalã (purtãtori sãnãtoşi),
• 3/10 000 dintre Caucazieni moştenesc ambele gene anormale (bolnavi de fibrozã chisticã de pancreas),
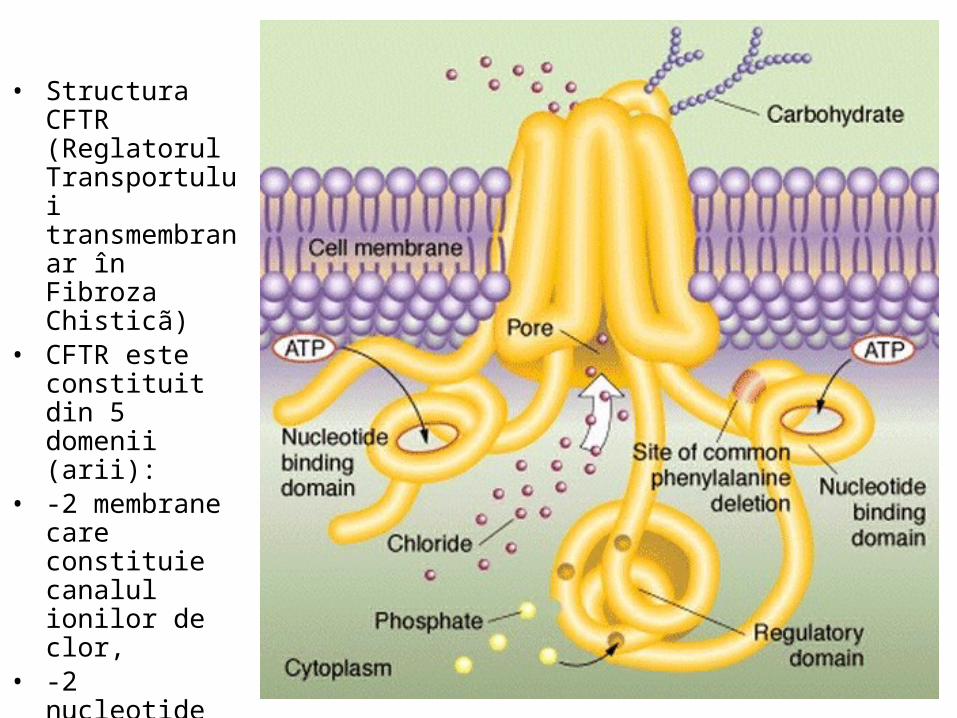
• Structura CFTR (Reglatorul Transportului transmembranar în Fibroza Chisticã)
• CFTR este constituit din 5 domenii (arii):
• -2 membrane care constituie canalul ionilor de clor,
• -2 nucleotide care atrag ATP şi-l hidrolizeazã,
• -1 reglator.

• Infecţii respiratorii,• Absorbţie redusã: grãsimi, proteine, vitamine.• Deficit nutriţional şi staturoponderal• Episoade de ocluzie intestinalã:15-20% ileus meconial, toate cazurile de ileus
meconial dezvoltã ulterior Mucoviscidozã
Fibroza chisticã de pancreas

Tutunul
Substanţa Acţiunea Efectul
Nicotinã contractã vaselearteriale şi venoaseombilicale; trece în laptelematern
Hipoxie,Hipotrofie
Monoxid de
Carbon
altereazã peretelea. ombilicale
Hipoxie
Altesubstanţe
împiedicã funcţiilemetabolice şi detransport aleplacentei
Hipotrofie
“o femeie care fumeazã 2 pachete ţigãri / zi scade fluxul sanguin placentar cu 41% în timpul cât fumeazã” (Christen 1998)
Fumatul în timpul sarcinii creşte riscul de:
• Avort spontan, • Naştere prematurã, • Anomalii placentare:
– Abruptio placenta, – Placenta previa

• Fumatul în timpul sarcinii creşte semnificativ riscul de: • Dismaturitate (12 % dintre nou-nãscuţii mamelor fumãtoare sunt dismaturi - 7.2%
/ mame nefumãtoare). • reducerea taliei şi greutãţii corporale, volumului plãmânilor, dimensiunilor capului,
– greutatea micã la naştere poate asocia: paralizie cerebralã, retard mental. – la 5 ani dupã naştere talia acestor copii este mai redusã comparativ cu copii mamelor
nefumãtoare• Despicãturi labio-palatine congenitale (risc de 1,5 – 2 ori mai mare pentru mamele
fumãtoare), • ”moarte subitã a nou-nãscutului” - risc de 2-6 ori mai mare pentru mamele
fumãtoare, comparativ cu cele nefumãtoare. • 60% dintre mamele copiilor decedaţi prin ”moarte subitã a nou-nãscutului” sunt
fumãtoare.
Conţinut ţigarete Nr
Substanţe chimice > 4000
Substanţe carcinogene 50
Substanţe care induc anomalii de dezvoltare 6
Fumatul Pasiv
ţ
Fumatul Activ

• Inhalarea fumului rezultat din combustia biomasei in primele 18 luni de viata– scade rata de dezvoltare a plamanilor, – afecteaza cresterea si dezvoltarea plamanilor, – creste semnificativ riscul de reactii
sensibilizarii la aeroalergeni, – scade cresterea somatica.
• combustia biomasei genereaza particule fine de compusi organici care – induc stress oxidativ, cresc productia de
cytokine (efect inflamator) – produc inflamare cronica a cailor rspiratorii la
copii care inhaleaza fumului rezultat din combustia biomasei .
Poluarea
Aparatul urinar are legături strânse anatomo-funcţionale cu aparatul genital atât în timpul dezvoltării embriologice cât şi în morfologia lor definitivă.
Componentele ambelor aparate, urinar şi genital, derivã din mezodermul intermediar
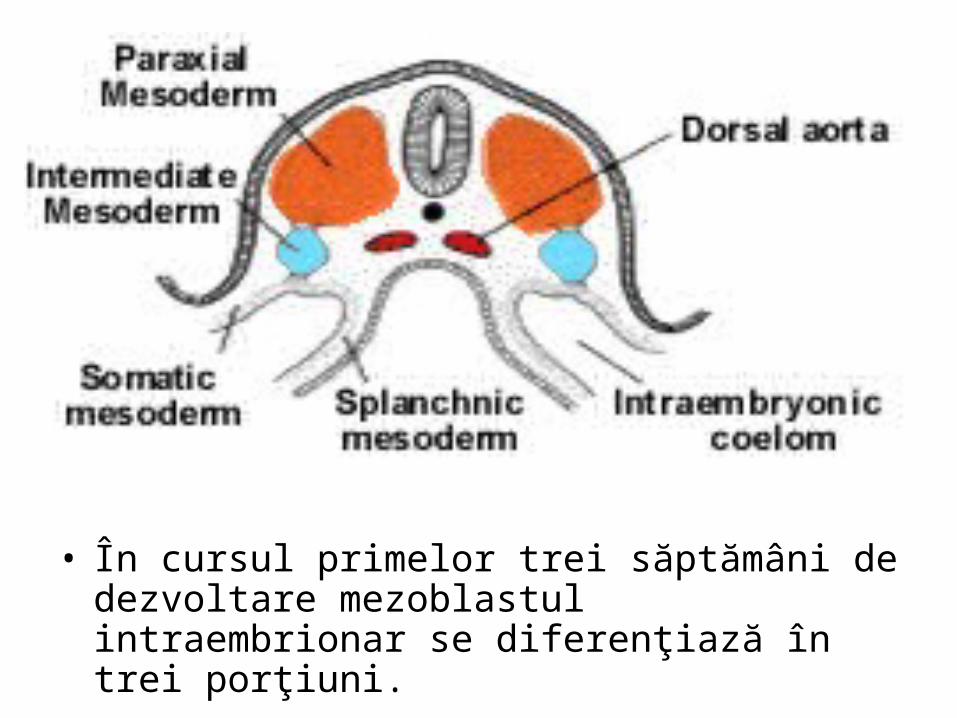
• În cursul primelor trei săptămâni de dezvoltare mezoblastul intraembrionar se diferenţiază în trei porţiuni.
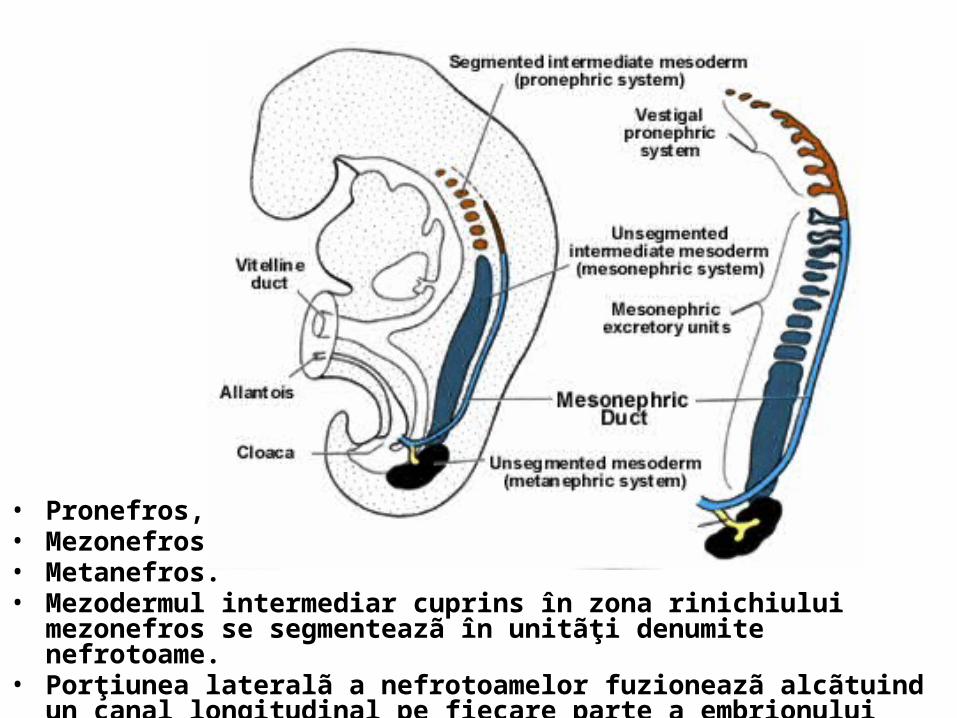
• Pronefros, • Mezonefros, • Metanefros.• Mezodermul intermediar cuprins în zona rinichiului mezonefros se segmenteazã
în unitãţi denumite nefrotoame. • Porţiunea lateralã a nefrotoamelor fuzioneazã alcãtuind un canal longitudinal pe
fiecare parte a embrionului canalul mezonefrotic
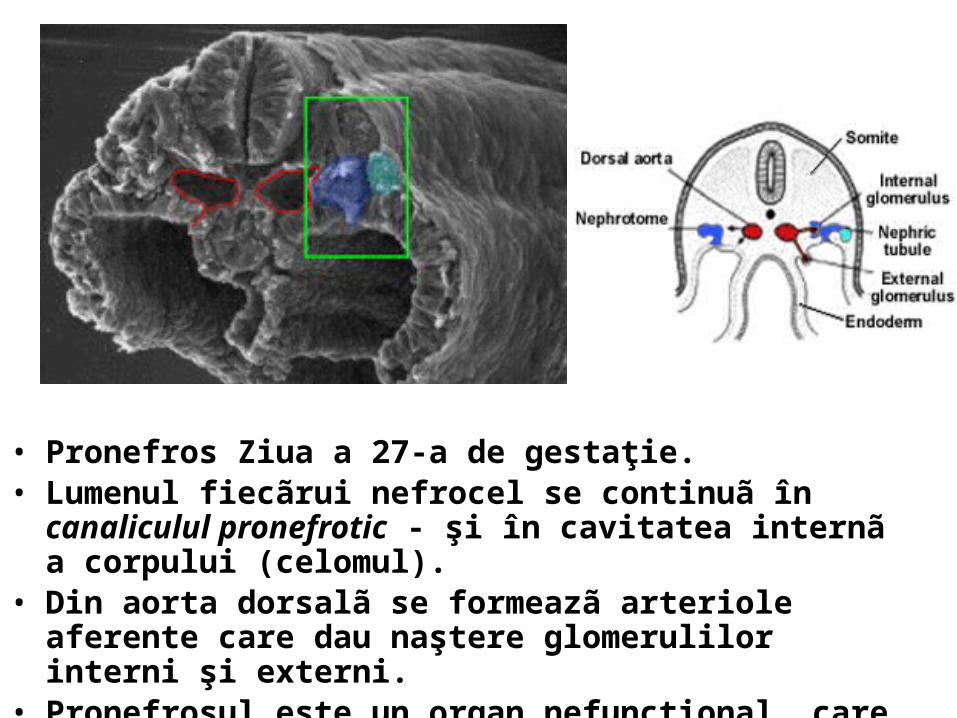
• Pronefros Ziua a 27-a de gestaţie. • Lumenul fiecãrui nefrocel se continuã în canaliculul pronefrotic -
şi în cavitatea internã a corpului (celomul). • Din aorta dorsalã se formeazã arteriole aferente care dau
naştere glomerulilor interni şi externi. • Pronefrosul este un organ nefuncţional, care involueazã dupã ce
a indus formarea mezonefrosului.
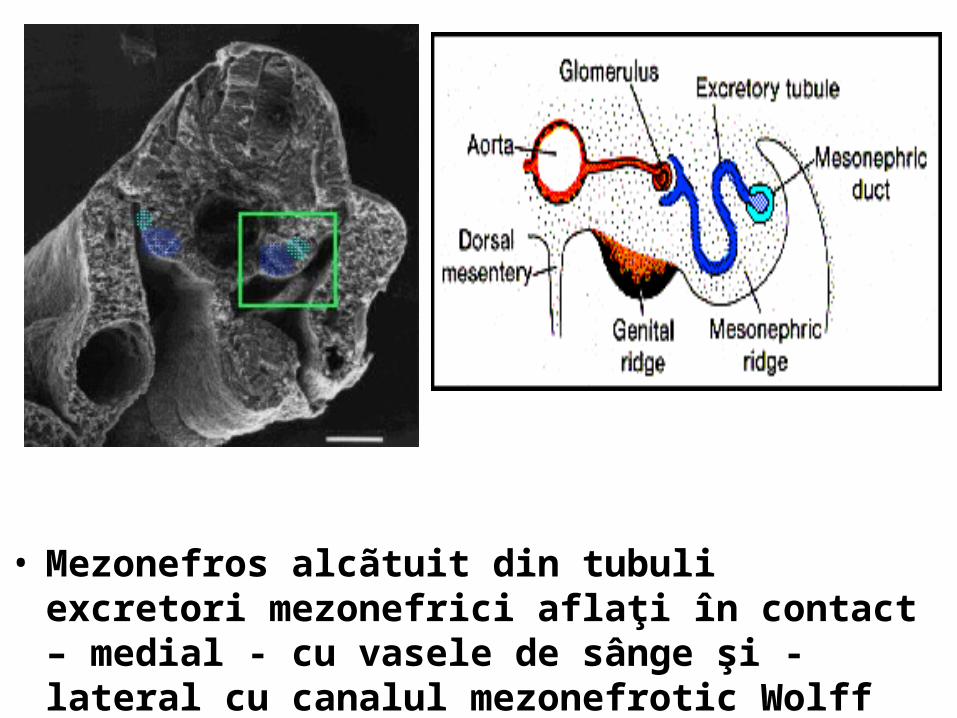
• Mezonefros alcãtuit din tubuli excretori mezonefrici aflaţi în contact – medial - cu vasele de sânge şi - lateral cu canalul mezonefrotic Wolff

• Relaţia spaţialã dintre glomerulul vascular, tubulul excretor mezonefric şi canalul mezonefrotic Wolff Imagine 3-D microscop electronic

• Ziua 28 de gestaţie. Pe faţa medialã a mezonefrosului se dezvoltã creasta genitalã. Canalul mezonefrotic Wolff este situat în partea lateralã a rinichiului mezonefros
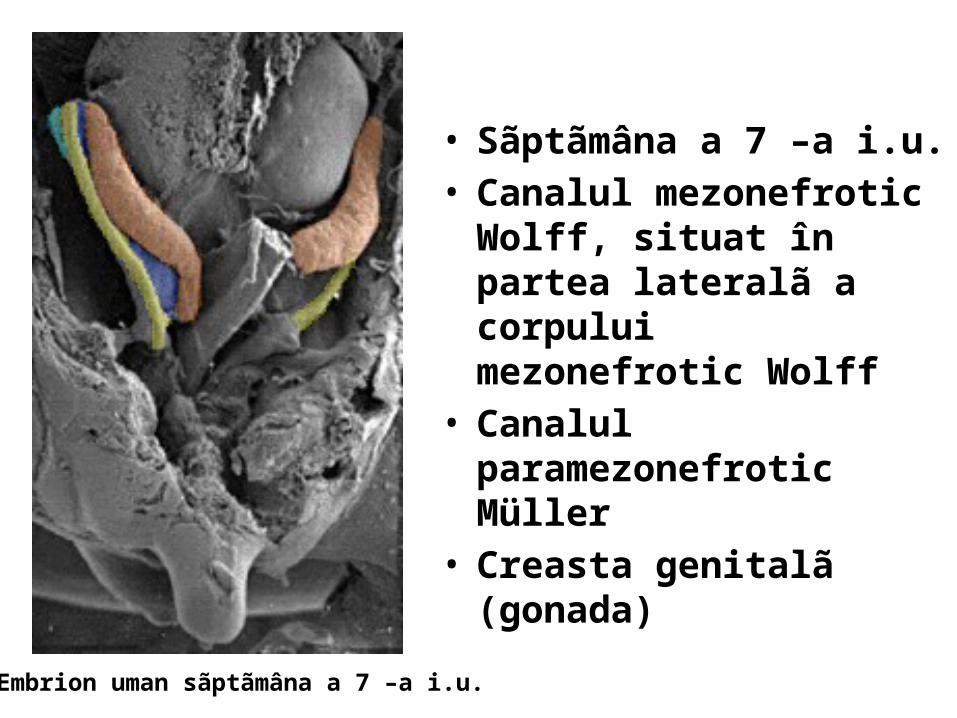
• Sãptãmâna a 7 –a i.u. • Canalul mezonefrotic Wolff,
situat în partea lateralã a corpului mezonefrotic Wolff
• Canalul paramezonefrotic Müller
• Creasta genitalã (gonada)
Embrion uman sãptãmâna a 7 –a i.u.

• Rinichiul Mezonefros • Funcţionează ca organ
excretor tranzitor, • Induce dezvoltarea rinichiului
metanefros (rinichiul definitiv)• Induce dezvoltarea canalelor
paramezonefrotice Müller • La bărbat se diferenţiază în
sistemul canalelor sexuale;

• La bãrbat canalul mezonefrotic Wolff devine canal deferent, vizibil în imaginea 3-D între testicul şi prostatã.

• La femeie • Canalul mezonefrotic Wolff degenereazã • Canalele paramezonefrotice Müller formeazã tubele
uterine şi uterul.

• Pe imaginea 3-D se observã gonada situatã medial de corpul Wolff

• Metanefros (rinichi definitiv) se dezvoltã sub influenţa inductivã a rinichiului mezonefros.
• La joncţiunea canalului Wolff cu cloaca se formeazã mugurele ureteral
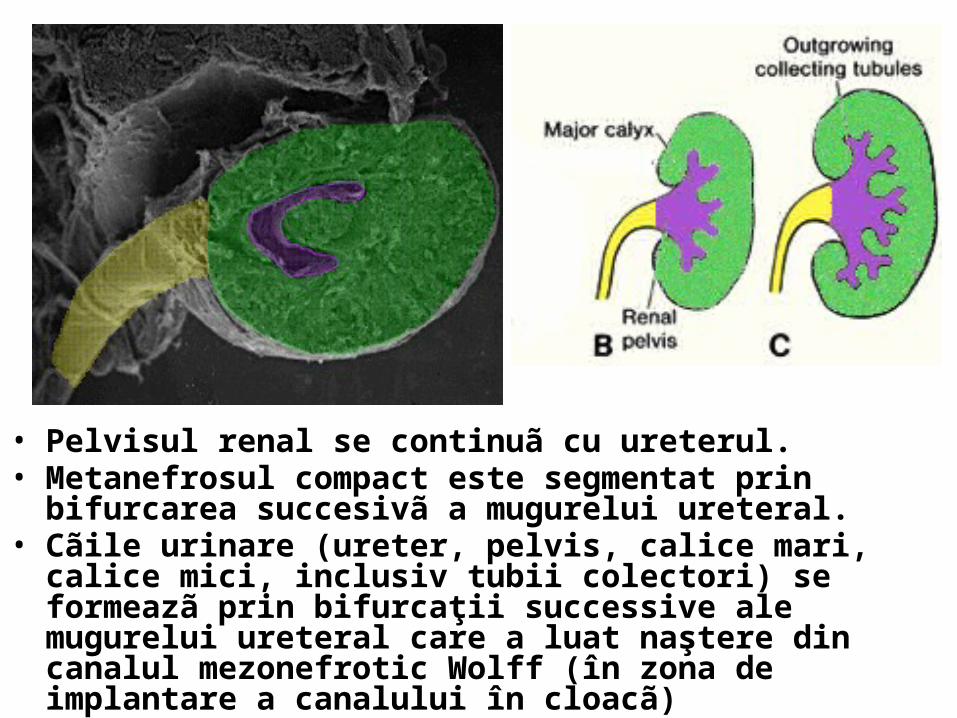
• Pelvisul renal se continuã cu ureterul. • Metanefrosul compact este segmentat prin bifurcarea succesivã a
mugurelui ureteral.• Cãile urinare (ureter, pelvis, calice mari, calice mici, inclusiv tubii
colectori) se formeazã prin bifurcaţii successive ale mugurelui ureteral care a luat naştere din canalul mezonefrotic Wolff (în zona de implantare a canalului în cloacã)

• Ascensiunea rinichilor are loc în 2 etape:• Ascensiune rapidă – Săptămâna 8i.u.
datorită creşterii în lungime a peretelui abdominal şi coloanei vertebrale; – în aceastã etapã rinichii întâlnesc artera
mezentericã inferioarã; dacă nu o depăşesc (cazul rinichiului în potcoavã) devin ectopici
• Ascensiune lentă până la 11 ani

• Cloaca se divide (septul Tourneaux) în: – sinus urogenital, – canal anorectal.
• Sinusul urogenital – Superior - se continuã cu alantoida – inferior - închis temporar de membrana urogenitalã.
• În sinusul urogenital se deschide canalul mezonefrotic Wolff; la joncţiunea lor canalul Wolff dã naştere mugurelui ureteral
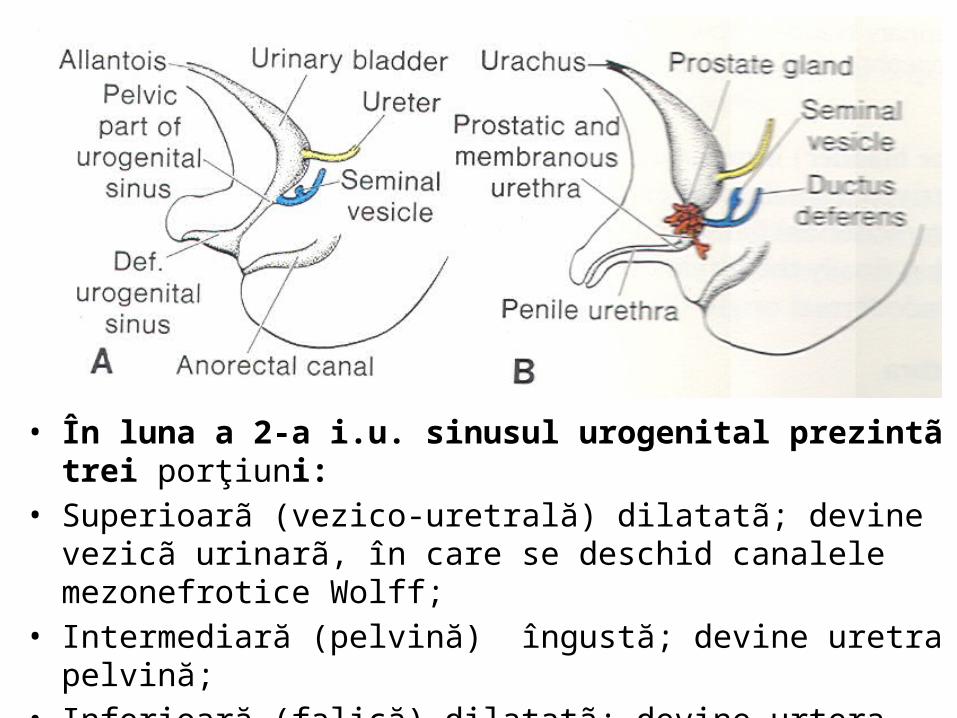
• În luna a 2-a i.u. sinusul urogenital prezintã trei porţiuni: • Superioarã (vezico-uretrală) dilatatã; devine vezicã urinarã, în care
se deschid canalele mezonefrotice Wolff;• Intermediară (pelvină) îngustă; devine uretra pelvină;• Inferioară (falică) dilatatã; devine urtera falică.

• La bărbat:• Poziţia ureterului faţã de canalul mezonefrotic Wolff:
– În sãptãmâna a 5-a i.u. ureterul apare ca un mugure al canalului Wolff – În sãptãmâna a 7-a i.u. (în urma înglobãrii progresive a porţiunii caudale a
canalului Wolff în peretele vezicii urinare) ureterul se deschide în vezica urinarã printr-un orificiu propriu
• Trigonul vezical se formeazã din mezodermul canalului mezonefrotic Wolff iar restul vezicii urinare ia naştere din endodermul sinusului urogenital

La femeie:• Porţiunea sinusului urogenital situată caudal de vezică,
până în dreptul tuberculului Müller formeazã uretra• Porţiunile pelvină şi falică formeazã vestibulul vaginal în
care se deschid separat vagina şi uretra.
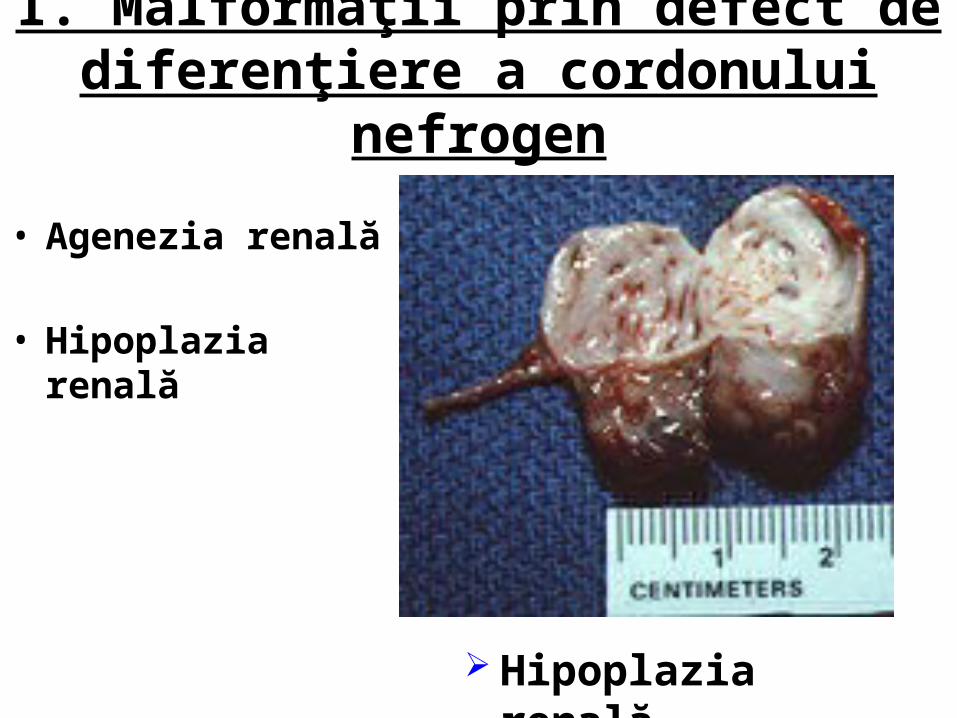
I. Malformaţii prin defect de diferenţiere a cordonului nefrogen
• Agenezia renală
• Hipoplazia renală
Hipoplazia renală

II. Malformaţii renale prin defect de diferenţiere a rinichiului metanefros
• Rinichiul polichistic cu transmitere Autosomal Recesivã
• maladie geneticã mortală prezentã la nou-nãscut
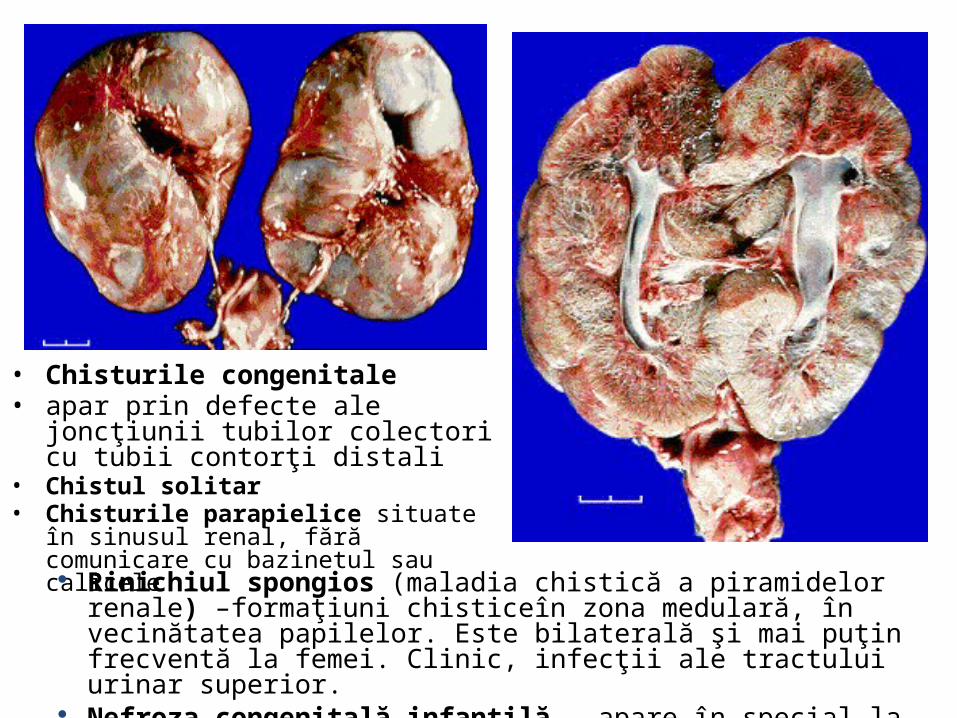
• Chisturile congenitale • apar prin defecte ale joncţiunii tubilor
colectori cu tubii contorţi distali• Chistul solitar • Chisturile parapielice situate în sinusul renal,
fără comunicare cu bazinetul sau calicele
Rinichiul spongios (maladia chistică a piramidelor renale) –formaţiuni chisticeîn zona medulară, în vecinătatea papilelor. Este bilaterală şi mai puţin frecventă la femei. Clinic, infecţii ale tractului urinar superior.
Nefroza congenitală infantilă – apare în special la prematuri; incidenţă familială, transmisă autosomal recesiv

III. Anomalii de ascensiune şi/sau rotaţie a rinichiului
• Rinichi ectopic pelvin unilateral stâng, ureter stâng scurt • Rinichi ectopic pelvin unilateral drept cu ureter de lungime normalã • Rinichi ectopic ileo-pelvin situat median

• Rinichi ectopic pelvin supranumerar

Fuziunea rinichilor
• Rinichi sigmoid ectopic iliac• Rinichi în potcoavã ectopic ilio-lombar

– Rinichi ectopic bilateral (anomalie de sediu şi rotaţie), uretere torsionate

• Variante de poziţie ale rinichiului în potcoavã şi anomalii numerice de ureter

Anomalii renale vasculare
• Artere renale supranumerare • Pe mãsurã ce rinichii ascensioneazã sunt revascularizaţi progresiv de cãtre vase
care iau naştere din aorta dorsalã: artera iliacã, porţiunea distalã a aortei şi în final din artera renalã ramurã din aorta abdominalã.
• Ocazional, unele ramuri vasculare caudale persistã dupã ascensiunea rinichilor. • Artrele accesorii iau naştere din porţiunile aortei adiacente arterei renale
principale sau uneori chiar din artera iliacã.

• Ureter bifid stâng. • Vene renale multiple,
dintre care una se divide pentru a permite trecerea aortei abdominale.
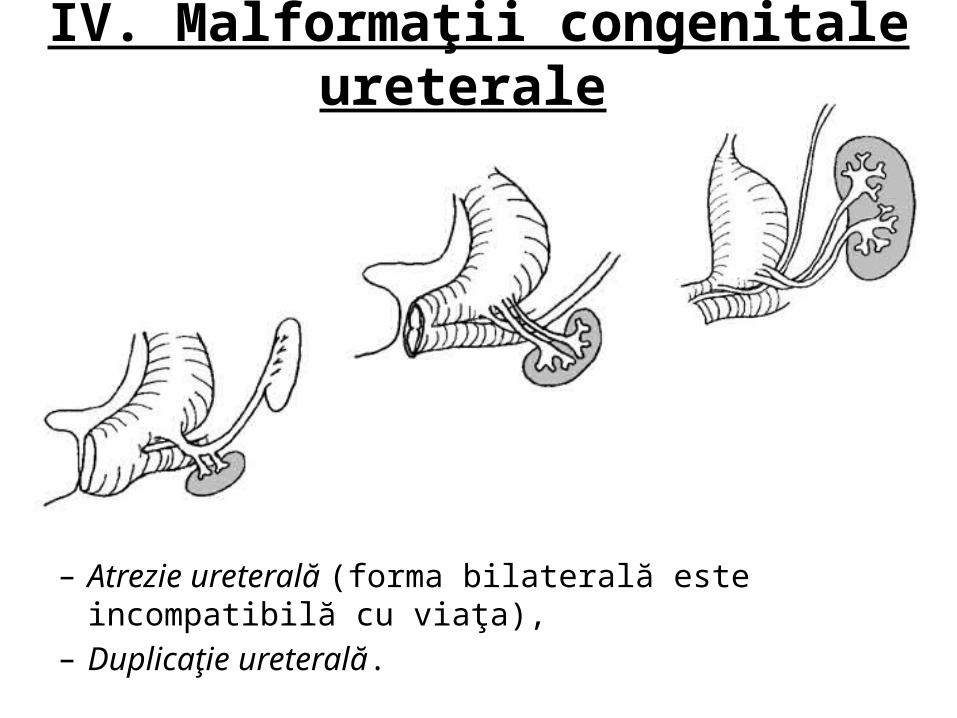
IV. Malformaţii congenitale ureterale
– Atrezie ureterală (forma bilaterală este incompatibilă cu viaţa),– Duplicaţie ureterală.
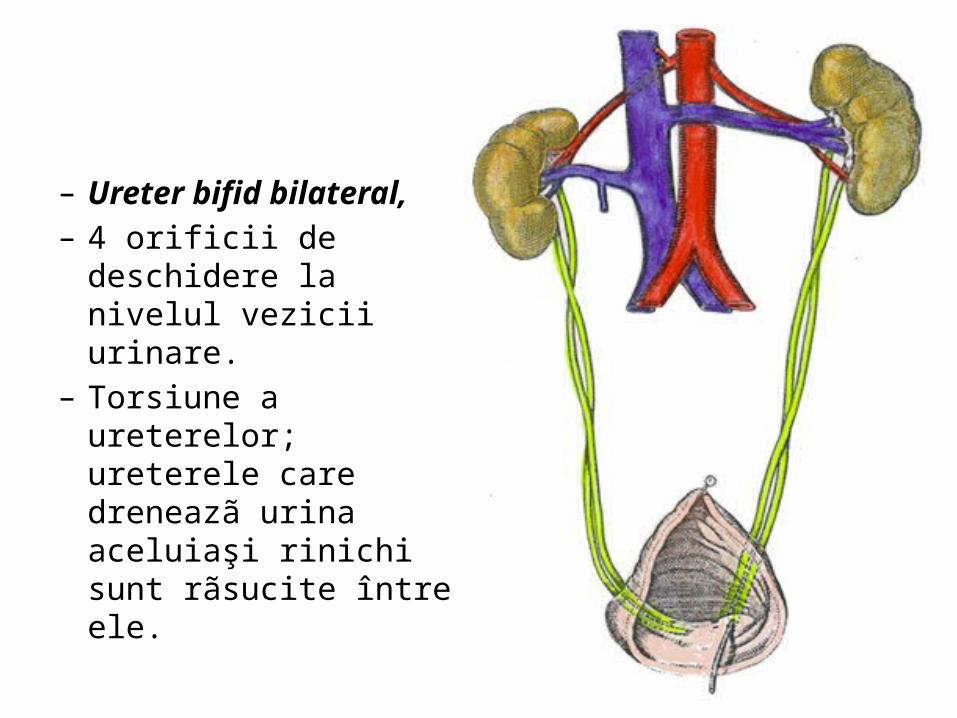
– Ureter bifid bilateral, – 4 orificii de deschidere la
nivelul vezicii urinare.– Torsiune a ureterelor;
ureterele care dreneazã urina aceluiaşi rinichi sunt rãsucite între ele.
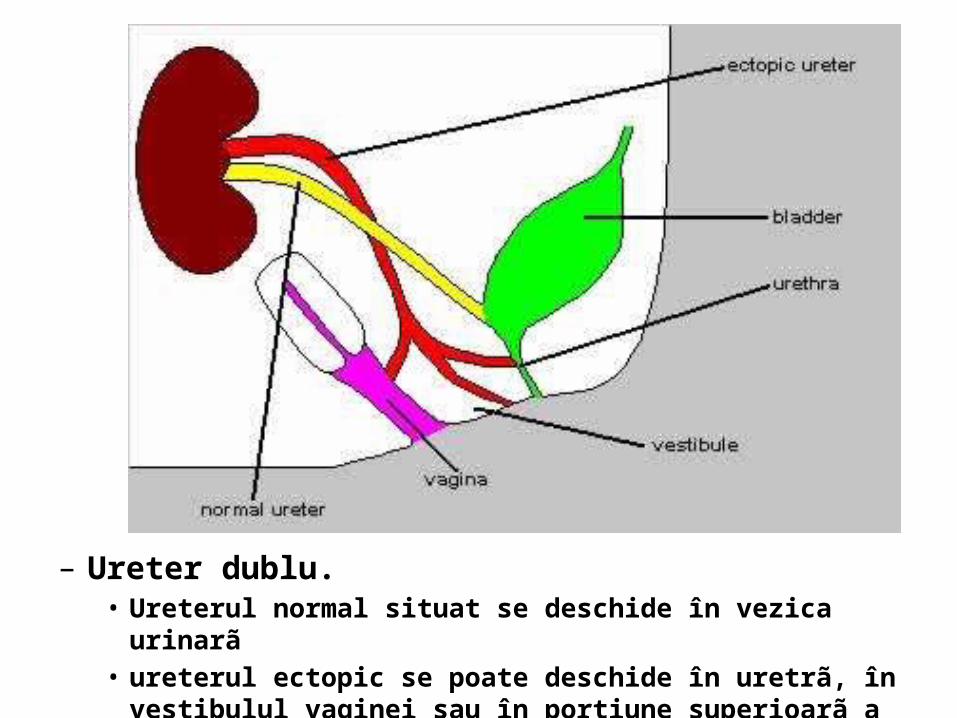
– Ureter dublu. • Ureterul normal situat se deschide în vezica urinarã • ureterul ectopic se poate deschide în uretrã, în vestibulul vaginei sau în
porţiune superioarã a vaginei

– Stenoza joncţiunii pielo-ureterale cu distensia bazinetului stâng şi a calicelor– hidronefroză.
– Aspect normal al rinichiului drept. Urografie cu substanţã de contrast

– Megaureter bilateral, hidronefrozã (dreapta), valvã de uretrã posterioarã– Megaureter bilateral, valvã de uretrã posterioarã, ureterocel. – Vârsta gestaţionalã: 7 sãptãmâni

– Megaureter stâng cu distensia bazinetului, hidronefrozã pre- şi retropielicã– Megaureter drept

– Megaureter stâng, obstrucţie în zona de implantare a ureterului în vezica urinarã prin anomalii în dezvoltarea musculaturii ureterului
Megaureter drept cu hidronefrozã. Megadolicoureter stâng cu reflux vezico-ureteral
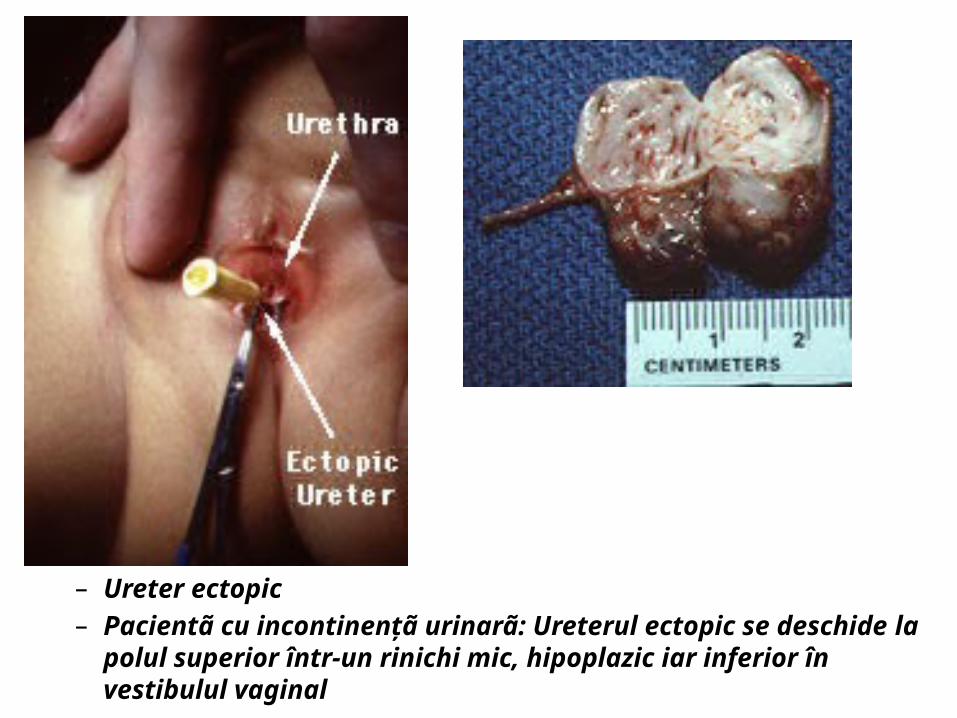
– Ureter ectopic– Pacientã cu incontinenţã urinarã: Ureterul ectopic se deschide la polul
superior într-un rinichi mic, hipoplazic iar inferior în vestibulul vaginal

– Urografie aspect normal
Reflux vezicouretral, distensie bazinet

V. Malformaţii congenitale de uretrã
• Duplicaţie completã a uretrei – cea mai rarã anomalie de uretrã.
• Duplicaţia porţiunii distale a uretrei, cu pãstrarea uretrei proximale de aspect normal.
• Uretra accesorie (dorsal sau ventral faţã de uretra normalã); se terminã orb la extremitatea proximalã.
• Bifurcaţia uretrei în porţiunea proximală; uretra accesorie se deschide la joncţiunea scrotului cu penisul.
duplicaţie completã
duplicaţie distală
uretra accesorie
bifurcaţia proximală
– Stricturile congenitale – se caracterizează prin prezenţa unui diafragm subţire sau a unei porţiuni de uretră cu diametrul micşorat pe diferite porţiuni.
– Valve uretrale posterioare – sunt falduri mucoase situate în uretra posterioară, constituind un obstacol important în eliminarea urinii în timpul micţiunii.
VI. Malformaţii congenitale ale vezicii urinare
– Anomalii de uracă – persistenţa canalului duce la apariţia de fistule :
• oarbe - interne ce comunică cu vezica; • oarbe externe când comunică cu ombilicul; • complete - urina se exteriorizează la nivel ombilical • chisturi

– Agenezia vezicală – cu deschideri ectopice ureterale
– Vezica urinarã dublă – se manifestă clinic micţional
– Diverticuli vezicali –defecte de dezvoltare ale musculaturii vezicale, caracteristic peretele cu structurã completă ca şi vezica urinarã normală.
– Maladia de col vezical (boala Marion) – contractura congenitală a colului vezical Duckett, afecţiune obstructivă caracterizată prin hipertrofie şi scleroză a muşchiului neted al colului, care duce la importante leziuni ale aparatului urinar supraiacent.
Acumulare de urină în diverticulul vezical
• Extrofia vezicală: consecinţa anomaliilor de septare cloacalã – absenţa peretelui abdominal anterior subombilical, – absenţa peretelui vezical inferior.
• Sunt afectate uretra, ureterele, organele genitale şi oasele bazinului. • La femeie uretra lipseşte sau este ca o bandeletă foarte scurtă. • Aparatul urinar superior este de obicei normal.
– Meningocel la acelaşi caz de Extrofie cloacalã
Extrofie cloacalã Omfalocel larg, falus mic şi bifid,
hemigland şi hemiscrot situate distal de vezica urinarã.

– Extrofia vezicii urinare, uretra deschisã,
– clitoris bifid
Extrofia vezicii urinare - aspect tipic - peretele vezical absent, deschiderea colului vezical, epispadias penopubic prin lipsa fuziunii
pliurilor uretrale şi a pielii la nivelul glandului penian
– Aspect tipic al pelvisului în extrofia vezicii urinare însoţitã de epispadias, cu lãrgirea simfizei pubiene
– Fisurã vezicalã superioarã într-un caz de extrofie a vezicii urinare
– Fistule rectouretrale - sunt rare şi frecvent asociază imperforaţie anală.
– Fistule rectovezicale - sunt mai grave decât cele rectouretrale.
– La sexul feminin interferã cu fuziunea normalã a extremitãţilor inferioare ale canalelor Muller, ceea ce determinã formarea a douã hemivagine şi douã hemiutere separate, ale cãror cavitãţi comunicã direct cu lumenul vezicii urinare.
• Prolaps subtotal al vezicii urinare dupã intervenţie chirurgicalã de reducere a extrofiei vezicii urinare.
• Mucoasa vezicalã de aspect normal, urina se scurge liber

• Care malformaţie congenitală este cel mai frecvent asociată cu oligohidramnion:
a). rinichi ectopicb). agenezie renalac). rinichi in potcoavad). rinichi supranumerare). rinichi polichistic
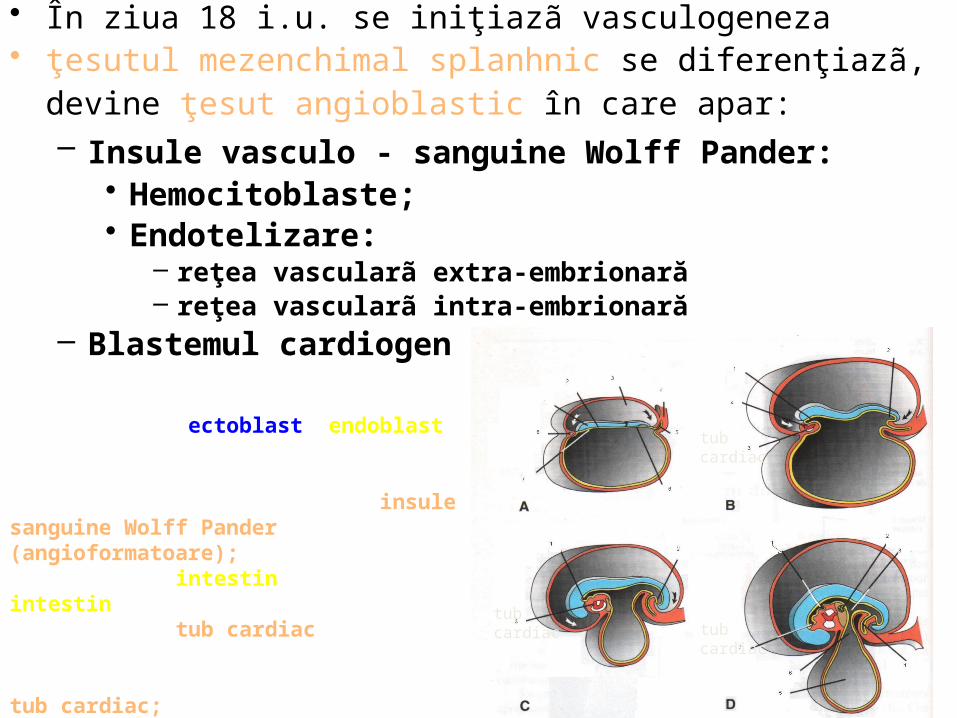
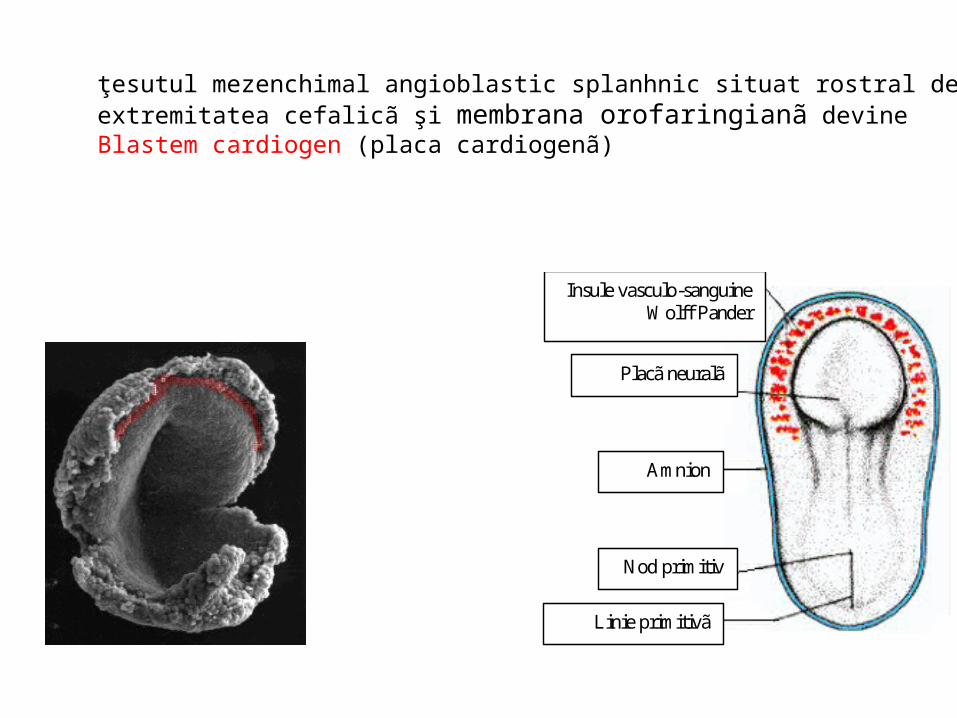
A (18 zile): -ectoblast; endoblast; -cavitate amniotică; - pedicul embrionar; -alantoidă; membrana cloacală;-placa precordală; -insule sanguine Wolff Pander (angioformatoare);B (20 zile): intestin anterior; intestin caudal; cavitate pericardică; tub cardiac;C (21 zile): membrană bucofaringiană; membrană cloacală; tub cardiac; D (22 zile): mugure pulmonar; mugure hepatic; intestin mijlociu; alantoidă; vezicula ombilicală; canal vitelin; rest de membrană bucofaringiană
• În ziua 18 i.u. se iniţiazã vasculogeneza • ţesutul mezenchimal splanhnic se diferenţiazã, devine ţesut
angioblastic în care apar: – Insule vasculo - sanguine Wolff Pander:
• Hemocitoblaste; • Endotelizare:
– reţea vascularã extra-embrionară– reţea vascularã intra-embrionară
– Blastemul cardiogen
tub cardiac tub
cardiac
tub cardiac
Linie primitivã
Nod primitiv
Amnion
Placã neuralã
Insule vasculo-sanguine Wolff Pander
ţesutul mezenchimal angioblastic splanhnic situat rostral de extremitatea cefalicã şi membrana orofaringianã devine Blastem cardiogen (placa cardiogenã)

Din Blastemul cardiogen situat iniţial la extremitatea anterioarã a discului embrionar se dezvoltã 2 tuburi endocardice.
CelomIntraembrionar
Mezoderm splanhnic
Endoderm

Între învelişul mezenchimatos, miocardic al tubului cardiac şi tubul endocardic/endoteliu se aflã un spaţiu plin cu matrice extracelularã (reticul subendotelial, ţesut subendocardic, gelatina cardiacã)
Miocitele diferenţiate din blastemul cardiogen prezintã spontan activitate electricã,
(potenţiale de acţiune). Impulsurile simpatice şi
parasimpatice modificã ritmul dar nu iniţiazã bãtãile inimii.
Epicardul seros (pericardul visceral) este format din celule mezoteliale derivate independent din mezodermul splanhnic

În ziua 24, înainte de definitivarea ansei cardiace, se observã (vedere frontalã): bulbul cordului (bulb arterial), strâmtoarea ventriculobulbarã; ventricul primitiv; strâmtoarea atrioventricularã; atriul primitiv, strâmtoarea sinoatrialã; sinusul venos Perechea de aorte dorsale formate din mezenchimul dorsal al discului embrionar se conecteazã cu tubii cardiaci înaintea curbãrii;
La vârsta de 20 zile i.u. inima, situatã între septul transvers şi membrana orofaringianã, este animatã de contracţii
..
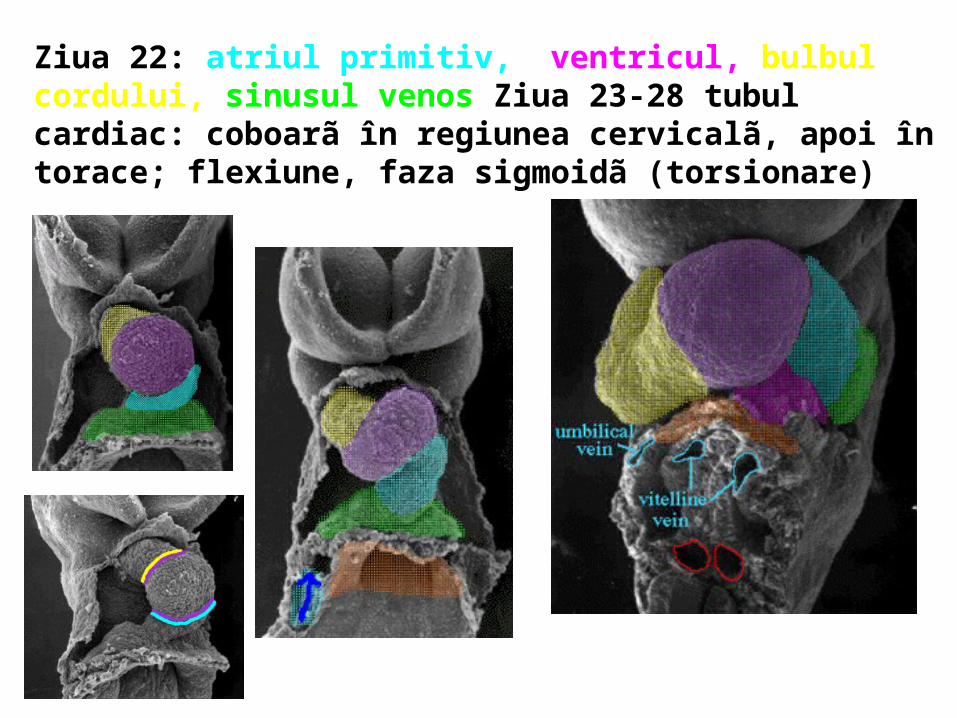
Ziua 22: atriul primitiv, ventricul, bulbul cordului, sinusul venos Ziua 23-28 tubul cardiac: coboarã în regiunea cervicalã, apoi în torace; flexiune, faza sigmoidã (torsionare)
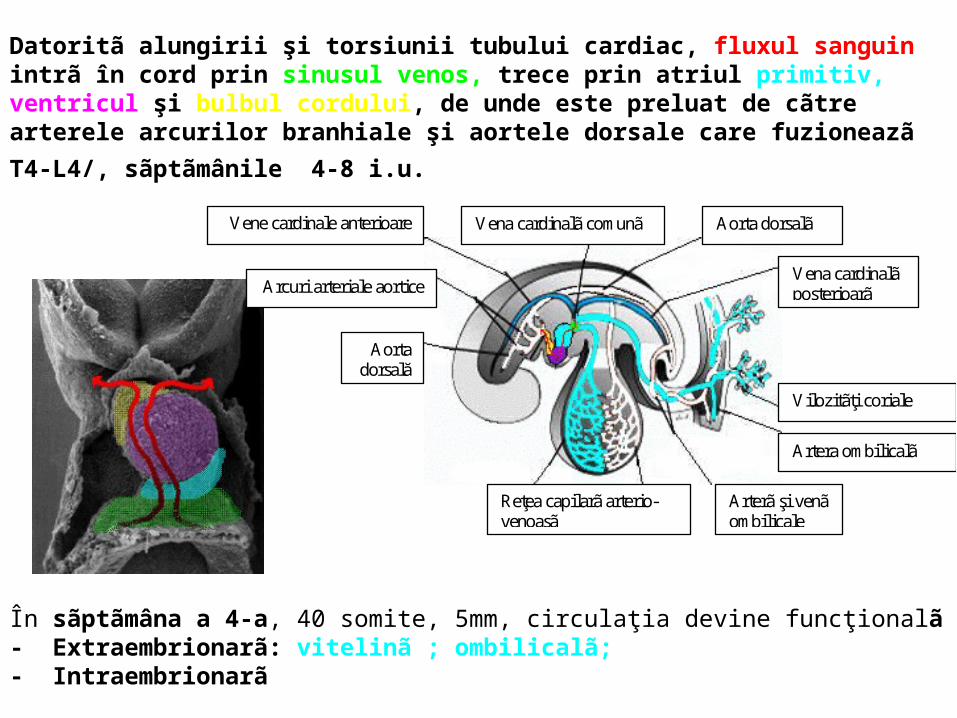
În sãptãmâna a 4-a, 40 somite, 5mm, circulaţia devine funcţionalã- Extraembrionarã: vitelinã ; ombilicalã; - Intraembrionarã
Datoritã alungirii şi torsiunii tubului cardiac, fluxul sanguin intrã în cord prin sinusul venos, trece prin atriul primitiv, ventricul şi bulbul cordului, de unde este preluat de cãtre arterele
arcurilor branhiale şi aortele dorsale care fuzioneazã T4-L4/, sãptãmânile 4-8 i.u.
Vene cardinale anterioare
Arcuri arteriale aortice
Aorta dorsală
Vena cardinalã comunã Aorta dorsalã
Vena cardinalã posterioarã
Vilozitãţi coriale
Artera ombilicalã
Arterã şi venã ombilicale
Reţea capilarã arterio- venoasã

În sinusul venos se deschid trei perechi de vase: venele cardinale care dreneazã capul, gâtul, pereţii corpului; venele viteline care iniţial dreneazã sacul vitelin; venele ombilicale, care aduc sânge oxigenat de la placentã
Sinus venos
Mugure hepatic
Duoden
V. cardinalã
V vitelinã stângã
V ombilicalã
Ziua 25 i.u.
Cavitate pericardicã
Atriu
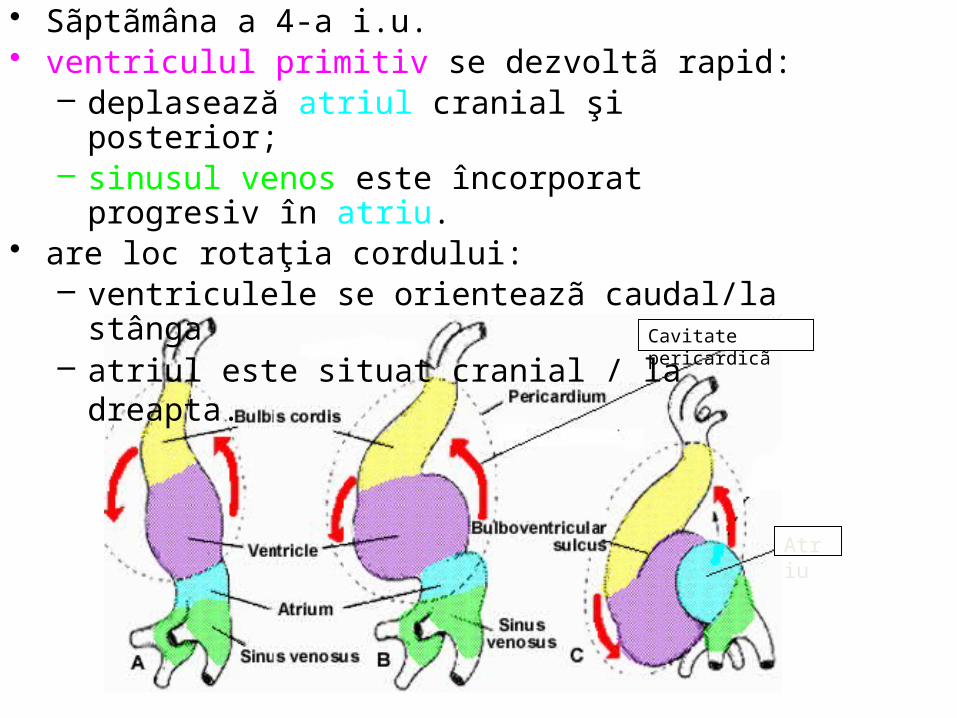
• Sãptãmâna a 4-a i.u. • ventriculul primitiv se dezvoltã rapid:
– deplasează atriul cranial şi posterior; – sinusul venos este încorporat progresiv în atriu.
• are loc rotaţia cordului: – ventriculele se orienteazã caudal/la stânga – atriul este situat cranial / la dreapta.

Bulbul, plasat cranial, prezintã conul arterial (conus) şi trunchiul bulbului arterial (truncus), fixat de regiunea branhialã, de la care pornesc 2 aorte ventrale care dau naştere arcurilor aortice care se vor reuni înapoia faringelui în aortele dorsale
Cavitate pericardicã
Atriu

• 2 artera carotidă internă stângă • 3 Arcul aortic I • 4 schiţa globului ocular• 5Cono truncus
6 arcul aortic VI (arcul pulmonar) • 7Aorta dorsală 8 Sacul Aortic 9 Ventricul drept
aortele ventrale dau naştere arcurilor aortice care străbat arcurile branhiale apoi se reunesc în aortele dorsale
• celule din crestele neurale migrează prin arcurile branhiale III, IV, VI ajung la cord, participă la formarea cseptului ventricular membranos. .
Gena pax3 este responsabilă de migrarea normală; absenţa genei pax3 (experimental) determină apariţia MC severe.
Creste Neurale
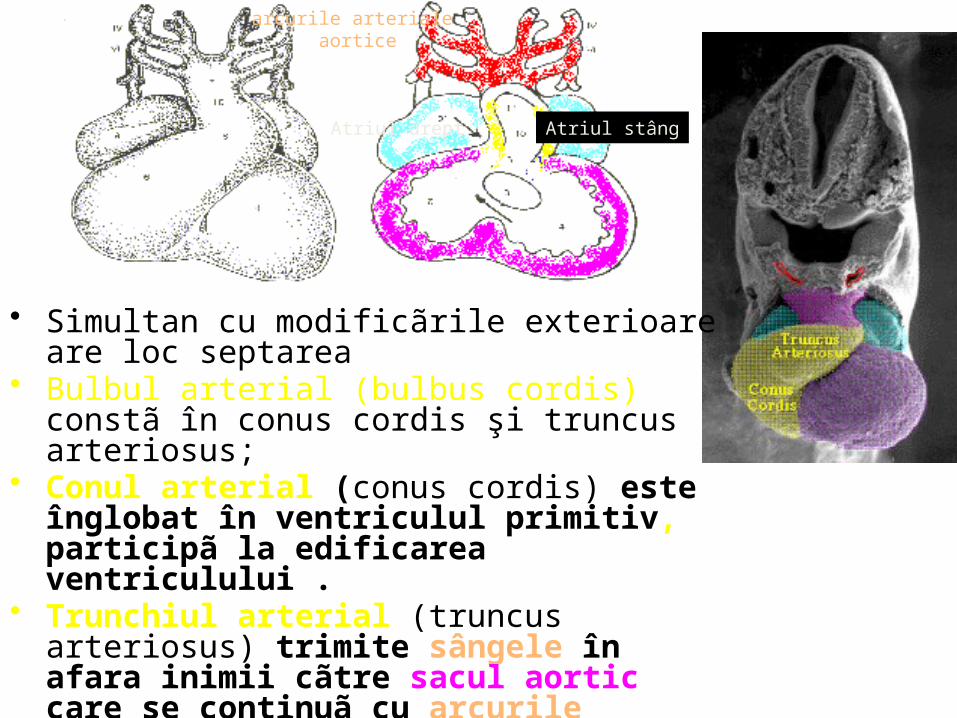
• Simultan cu modificãrile exterioare are loc septarea • Bulbul arterial (bulbus cordis) constã în conus cordis
şi truncus arteriosus; • Conul arterial (conus cordis) este înglobat în
ventriculul primitiv, participã la edificarea ventriculului .
• Trunchiul arterial (truncus arteriosus) trimite sângele în afara inimii cãtre sacul aortic care se continuã cu arcurile arteriale aortice.
• ventriculul şi atriul sunt evidente. • Atriul comunicã cu ventriculul prin canalul
atrioventricular.
Atriul stângAtriul drept
arcurile arteriale aortice

Endocardul (derivat din endoteliul vascular) situat la periferia canalului atrio-ventricular formeazã doi muguri endocardici (ventral şi dorsal) care se apropie, fuzioneazã, alcãtuiesc perniţa endocardicã a canalului atrio-ventricular
.
.
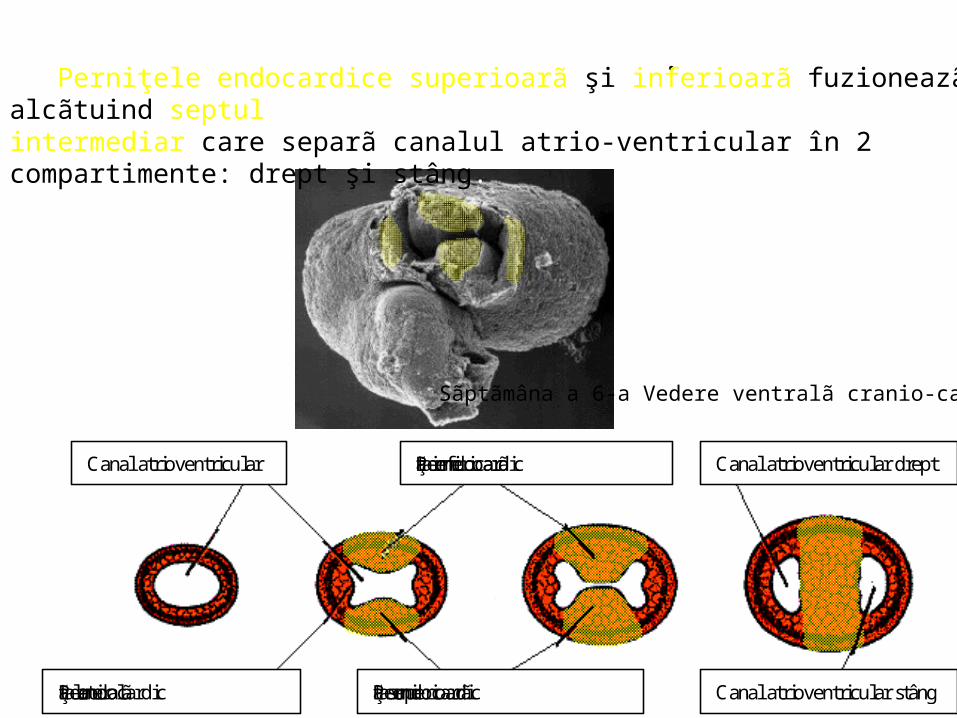
Perniţele endocardice superioarã şi inferioarã fuzioneazã alcãtuind septul intermediar care separã canalul atrio-ventricular în 2 compartimente: drept şi stâng.
Canal atrioventricular Perniţa endocardicã inferioarã Canal atrioventricular drept
Perniţa endocardicã superioarãPerniţa endocardicã lateralã Canal atrioventricular stâng
Sãptãmâna a 6-a Vedere ventralã cranio-caudalã.

.
Orificiul atrioventricular stâng este strãjuit de Valva mitralã Orificiul atrioventricular drept are de Valva tricuspidã
.
Cuspid anterior al valvei mitrale
Cuspid septal alValvei tricuspide

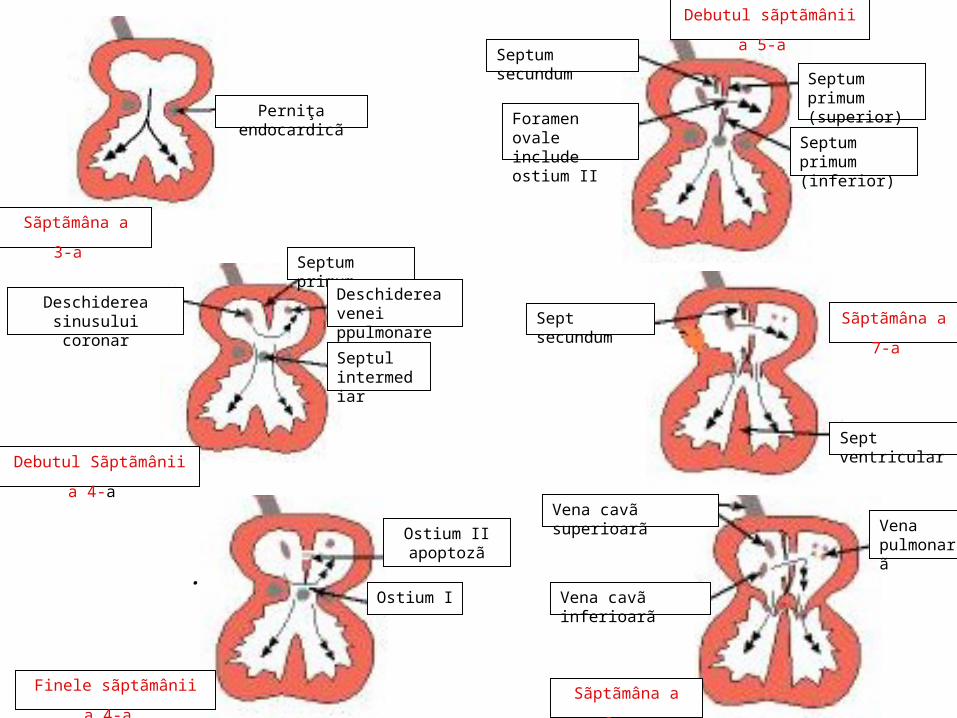
.La finele sãptãmânii a 4-a, din partea posterosuperioarã a tavanului atriului primitiv se dezvoltã o creastã semilunarã, septum primum Born, cu direcţie de creştere antero-inferioarã, spre septul intermediar, împreunã cu care delimiteazã ostium I;
.
Septum I
Ostium I
Foramen interventricular

.Ostiul I dispare prin fuziunea septului I cu septul intermediar; Formarea ostiumului II are loc concomitent, prin proces de apoptozã (moarte celularã dirijatã) în partea superioarã a septului I
.
Septum I
Ostium I
Foramen interventricular
Foramen interventricular
Ostium II
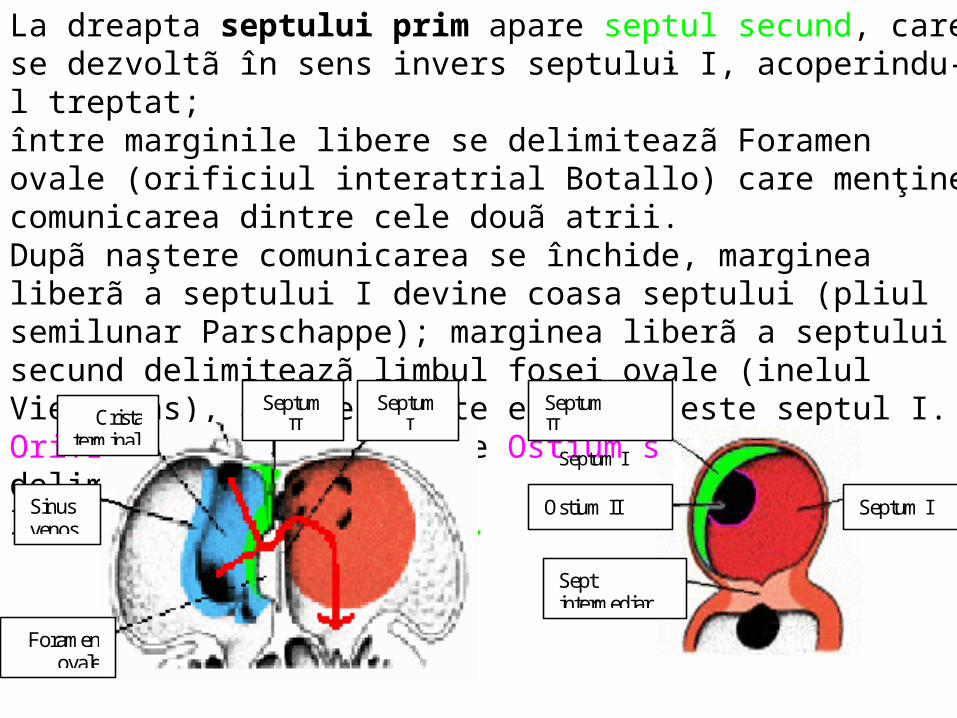
.La dreapta septului prim apare septul secund, care se dezvoltã în sens invers septului I, acoperindu-l treptat; între marginile libere se delimiteazã Foramen ovale (orificiul interatrial Botallo) care menţine comunicarea dintre cele douã atrii. Dupã naştere comunicarea se închide, marginea liberã a septului I devine coasa septului (pliul semilunar Parschappe); marginea liberã a septului secund delimiteazã limbul fosei ovale (inelul Vieussens), a cãrei parte excavatã este septul I. Orificiul Botallo include Ostium sccundum, delimitat de marginile libere ale septului prim.
.
S ep tumII
S ep tum I
S ep tum IO stiu m II
S ep tin te rm ed iar
S ep tumI
I
S ep tumIIC ris ta
te rm in alis
F o ram eno vale
S in u sven o s
.
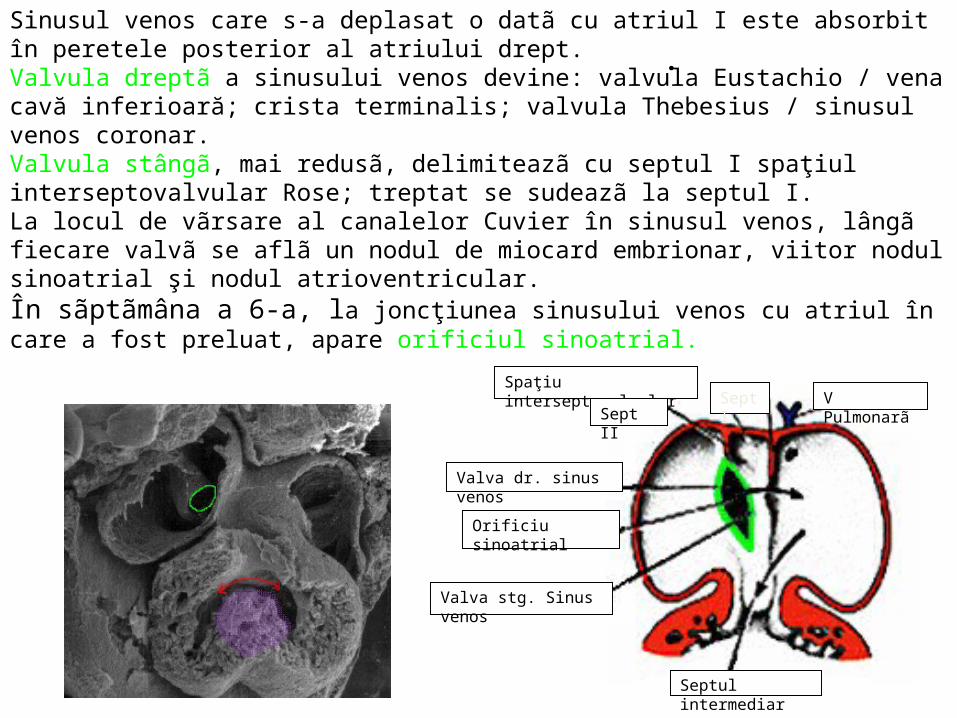
Sinusul venos care s-a deplasat o datã cu atriul I este absorbit în peretele posterior al atriului drept. Valvula dreptã a sinusului venos devine: valvula Eustachio / vena cavă inferioară; crista terminalis; valvula Thebesius / sinusul venos coronar. Valvula stângã, mai redusã, delimiteazã cu septul I spaţiul interseptovalvular Rose; treptat se sudeazã la septul I. La locul de vãrsare al canalelor Cuvier în sinusul venos, lângã fiecare valvã se aflã un nodul de miocard embrionar, viitor nodul sinoatrial şi nodul atrioventricular. În sãptãmâna a 6-a, la joncţiunea sinusului venos cu atriul în care a fost preluat, apare orificiul sinoatrial.
Sept I V Pulmonarã
Septul intermediar
Spaţiu interseptovalvular
Sept II
Valva dr. sinus venos
Orificiu sinoatrial
Valva stg. Sinus venos

.Atriile se definitiveazã prin preluarea sinusului venos în atriul drept şi integrarea pãrţii stângi a sinusului venos, unde se varsã trunchiul venos pulmonar, în peretele posterior al
atriului stâng
.
SeptumII
Septum I
Septum I
Sinus venos
Vena cavãSuperioarã
Cristaterminalis
Foramenovale
.
.
Perniţa endocardicã
Deschiderea sinusului coronar
Septum primum
Deschiderea venei ppulmonare
Septul intermediar
Debutul Sãptãmânii a 4-a
Finele sãptãmânii a 4-a
Ostium I
Ostium IIapoptozã
Debutul sãptãmânii a 5-a
Septum primum(superior)
Septum primum(inferior)
Septum secundum
Foramen ovaleinclude ostium II
Sãptãmâna a 3-a
Sãptãmâna a 7-a
Sept ventricular
Sept secundum
Sãptãmâna a 8-a
Vena cavã superioarã
Vena cavã inferioarã
Vena pulmonarã
.
Sept aorticopulmonar
AortaArtera pulmonarã
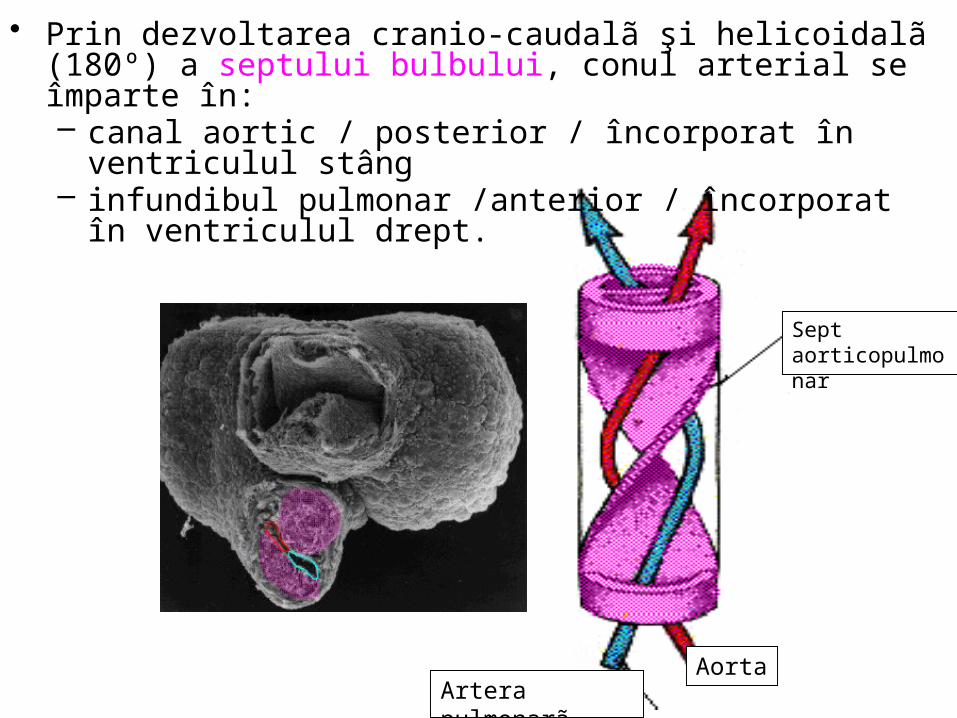
• Prin dezvoltarea cranio-caudalã şi helicoidalã (180º) a septului bulbului, conul arterial se împarte în: – canal aortic / posterior / încorporat în ventriculul stâng – infundibul pulmonar /anterior / încorporat în ventriculul drept.

.
Sept aorticopulmonar
AortaArtera pulmonarã
• Septul bulbului trece prin mijlocul valvelor laterale ale bulbului arterial, separã cãile de ejecţie ventriculare: – dreaptã / artera pulmonarã,– stângã / artera aortã
• valvulele arterei pulmonare: – anterioarã, postero-lateralã dreaptã, postero-lateralã stângã
• valvulele semilunare ale aortei: – posterioarã, antero-lateralã dreaptã, antero-lateralã stângã
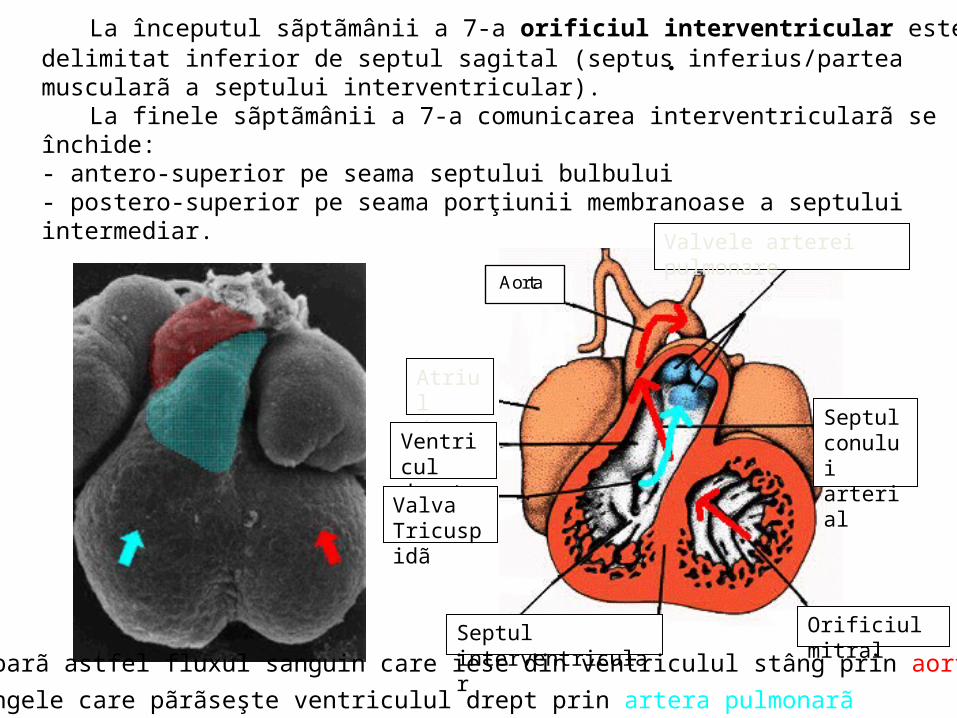
.La începutul sãptãmânii a 7-a orificiul interventricular este delimitat inferior de
septul sagital (septus inferius/partea muscularã a septului interventricular). La finele sãptãmânii a 7-a comunicarea interventricularã se închide:
- antero-superior pe seama septului bulbului - postero-superior pe seama porţiunii membranoase a septului intermediar.
.
Aorta
Valvele arterei pulmonare
Atriul drept
Ventricul drept
Valva Tricuspidã
Septul conului arterial
Orificiul mitralSeptul interventricularSe separã astfel fluxul sanguin care iese din ventriculul stâng prin aortã,
de sângele care pãrãseşte ventriculul drept prin artera.pulmonarã

CIRCULAŢIA SÂNGELUI ÎNAINTE DE NAŞTERE
.

CIRCULAŢIA SÂNGELUI DUPÃ NAŞTERE
.

Incidenţa generalã a cardiopatiilor congenitale:8,8/1000 n-nãscuţi vii. A 2-a cauzã de mortalitate. Mortalitatea prin cardiopatii congenitale 1,2%o nou – nãscuţi; 80% din totalul deceselor de nou – nãscuţi. Defect Septal Ventricular 16,1% /CC. Tetralogie Fallot 9,2% /CC Transpoziţia vaselor mari 9,1% /CC. Coarctaţia aortei 7,8% /CC
Persistenţa canalului arterial 5,7%/CC Stenozã pulmonarã 3,8%/CC
Anomalii în dezvoltarea cordului
0
2
4
6
8
10
12
14
16
18
20
1 2 3 4 5 6 7 8 9 10 11 12
Mortalitatea prin malformaţii congenitale la populaţia 0-1 an în Judeţul Constanţa în ultimii 12 ani

• I Anomalii de poziţie
– I. 1. Ectopie cardiacă:• cervicală;• cervicotoracică;• sternală;• abdominală
– I. 2. Dextrocardie
Anomalii în dezvoltarea cordului

• II Anomalii interne - ale structurii • II. 1. Anomaliile valvulelor semilunare şi aortice• II. 2. Anomaliile canalului atrioventricular
– Canal atrio-ventricular persistent; – Atrezia tricuspidei;– Maladia Ebstein
• II. 3. Anomaliile septului interatrial– Defect septal interatrial– Persistenţa ostiului secund, – Atriul comun
• II. 4. Anomaliile septului interventricular– Cord trilocular biatrial – Defect septal ventricular
• II. 5. Anomaliile bulbului arterial– Persistenţa trunchiului arterial– Transpoziţia completă a vaselor mari– Trilogia Fallot; – Tetralogia Fallot

Malformaţie relativ rarã 2 – 6% dintre malformaţiile congenitale Manifestãri clinice Evidente din primele luni de viaţã
Persistenţa Canalului Atrio-Ventricular
: hipotrofie staturo-ponderalã, paloare, polipnee, infecţii pulmonare recidivante. Examenul radiologic Cord mãrit de volum (cardiomegalie), artere pulmonare dilatate, vascularizaţie pulmonarã crescutã. Tratament Prognostic spontan sumbru. Tratament chirurgical între 2-6 ani dar şi/sugar; mortalitate operatorie 2%.

-a 3-a ca frecvenţã
dintre cardiopatiile cianogene
congenitale. - prognostic
spontan grav. Atriul drept mãrit de volum, cu pereţi îngroşaţi. -Ventricul drept hipoplazic 1,5-5% în medie 2,3%/CC;
Manifestãrile clinice CC cianogenã cu toleranţã clinicã mediocrã: cianozã precoce, dispnee de efort. Radiologic dilatarea Atriului drept, hipoplazie ventricul drept, vascularizaţie pulmonarã scãzutã. Evoluţie spontanã 40% deces/primul an de viaţã. Tratament chirurgical paleativ.
Atrezia tricuspidei
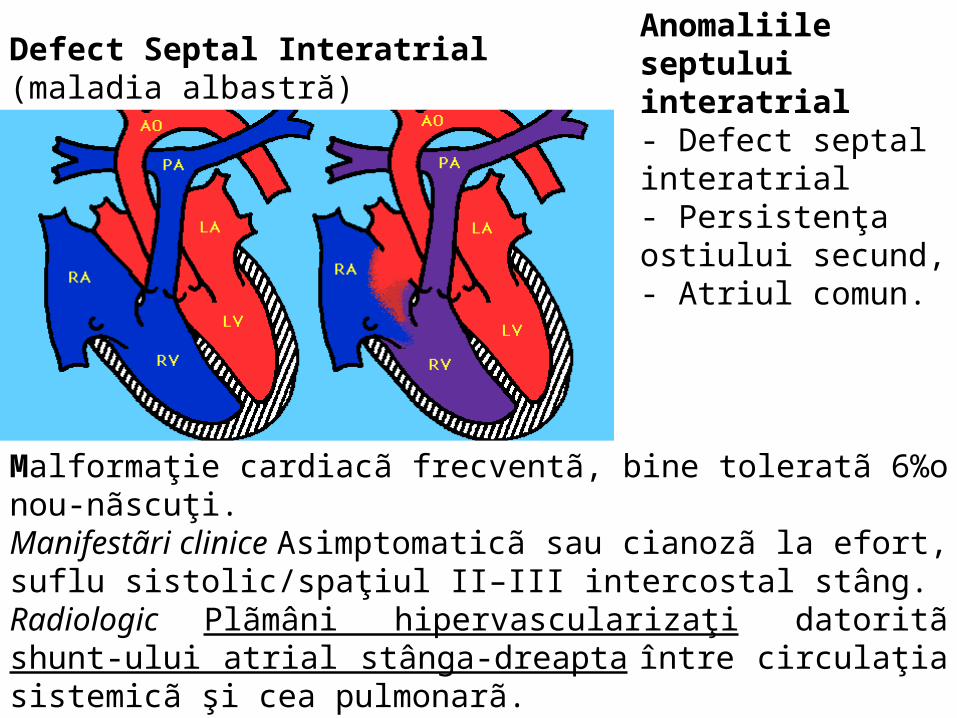
Defect Septal Interatrial (maladia albastră)Anomaliile septului interatrial- Defect septal interatrial- Persistenţa ostiului secund, - Atriul comun.
Malformaţie cardiacã frecventã, bine toleratã 6%o nou-nãscuţi. Manifestãri clinice Asimptomaticã sau cianozã la efort, suflu sistolic/spaţiul II–III intercostal stâng. Radiologic Plãmâni hipervascularizaţi datoritã shunt-ului atrial stânga-dreapta între circulaţia sistemicã şi cea pulmonarã.Tratament chirurgical intervenţie de preferinţã între 5-12 ani, rezultate excelente în timp. Mortalitate postoperatorie micã nedepãşind 1-2%.

• Defect Septal Atrial • Ostium Primum
Normal

• Defect Septal Atrial • Ostium Secundum /foramen ovale patent
Normal
Defect septal ventricular
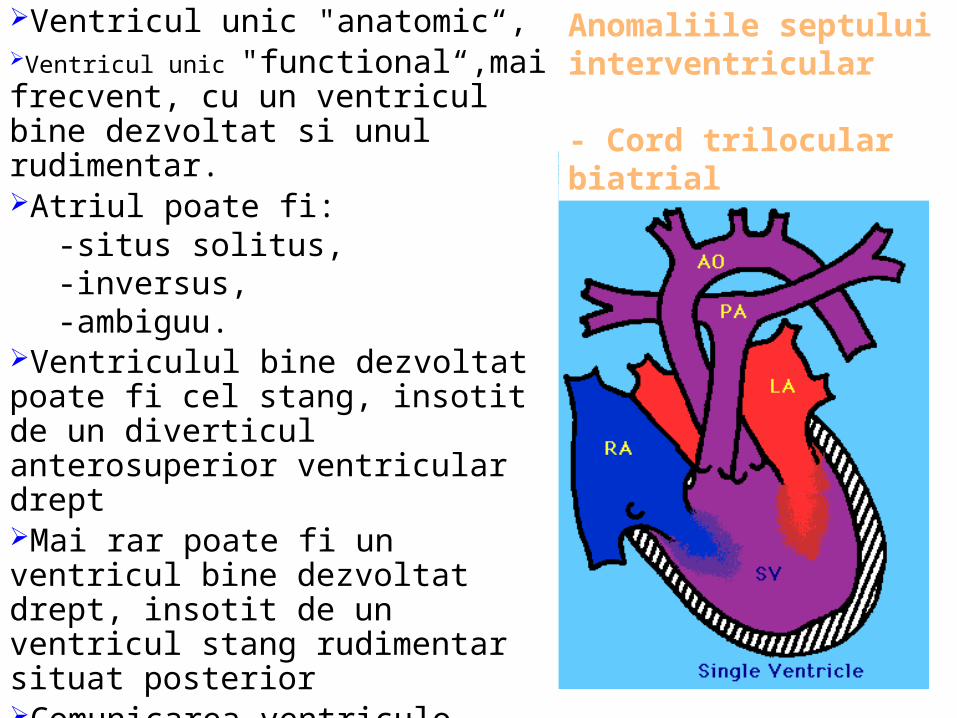
Ventricul unic "anatomic“, Ventricul unic "functional“,mai frecvent, cu un ventricul bine dezvoltat si unul rudimentar.Atriul poate fi:
-situs solitus, -inversus, -ambiguu.
Ventriculul bine dezvoltat poate fi cel stang, insotit de un diverticul anterosuperior ventricular dreptMai rar poate fi un ventricul bine dezvoltat drept, insotit de un ventricul stang rudimentar situat posteriorComunicarea ventriculo-arteriala poate fi concordanta sau discordanta,Arterele mari au originea in acelasi ventricul, pot fi normale sau stenozate
Anomaliile septului interventricular
- Cord trilocular biatrial
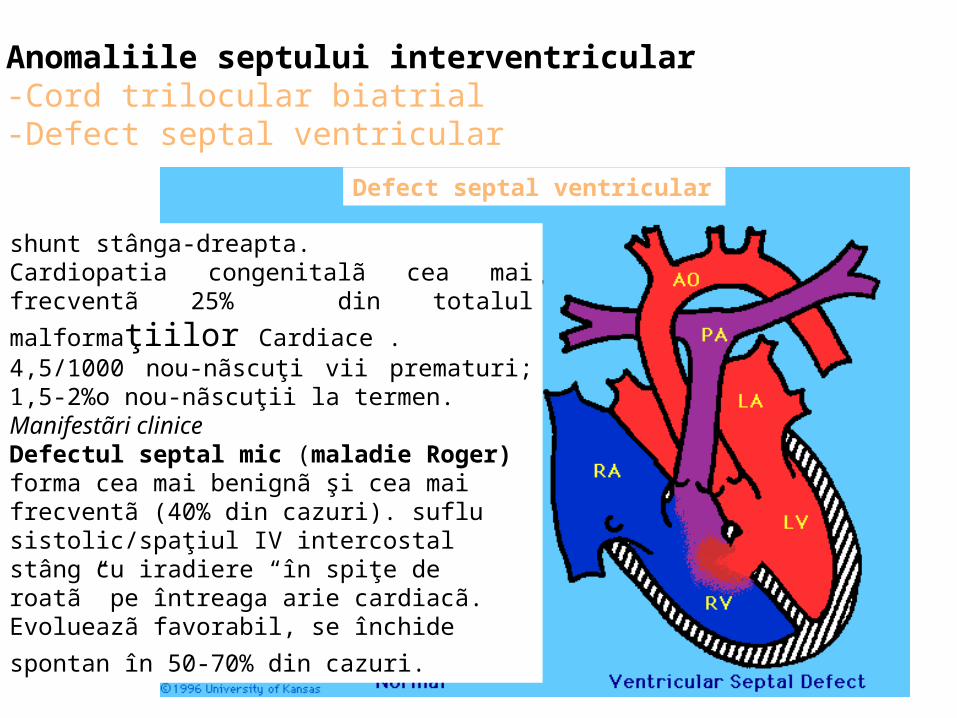
Anomaliile septului interventricular-Cord trilocular biatrial -Defect septal ventricular
shunt stânga-dreapta. Cardiopatia congenitalã cea mai frecventã 25% din
totalul malformaţiilor Cardiace . 4,5/1000 nou-nãscuţi vii prematuri; 1,5-2%o nou-nãscuţii la termen. Manifestãri cliniceDefectul septal mic (maladie Roger) forma cea mai benignã şi cea mai frecventã (40% din cazuri). suflu sistolic/spaţiul IV intercostal stâng cu iradiere “în spiţe de roatã” pe întreaga arie cardiacã. Evolueazã favorabil, se închide spontan în 50-70% din
cazuri.
Defect septal ventricular
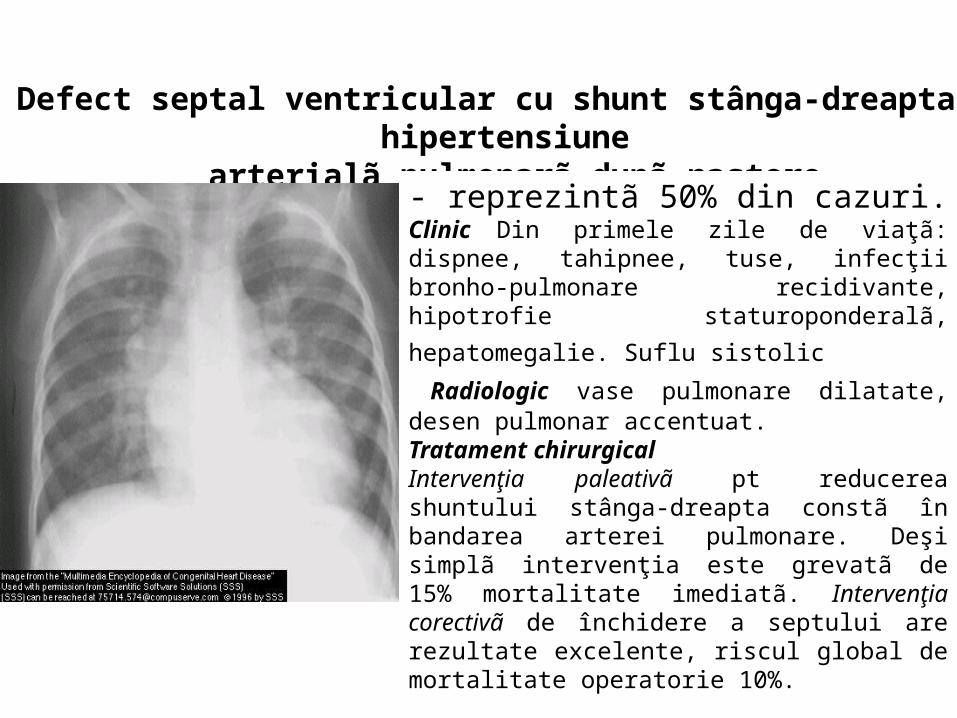
Defect septal ventricular cu shunt stânga-dreapta şi hipertensiune arterialã pulmonarã dupã naştere
- reprezintã 50% din cazuri. Clinic Din primele zile de viaţã: dispnee, tahipnee, tuse, infecţii bronho-pulmonare recidivante, hipotrofie
staturoponderalã, hepatomegalie. Suflu sistolic Radiologic vase pulmonare dilatate, desen pulmonar accentuat. Tratament chirurgical Intervenţia paleativã pt reducerea shuntului stânga-dreapta constã în bandarea arterei pulmonare. Deşi simplã intervenţia este grevatã de 15% mortalitate imediatã. Intervenţia corectivã de închidere a septului are rezultate excelente, riscul global de mortalitate operatorie 10%.
Malformaţie cardiacã gravã, include DSV Frecvenţã 1-4% / din Cardiopatiile Congenitale Manifestãri clinice cianozã şi semne de shunt stânga-dreapta: dispnee, tahipnee, transpiraţii profuze, hipotrofie ponderalã. suflu sistolic/parasternal stâng cu maximum de intensitate/spaţiul 3-4 intercostal stâng.Tratament Cu preţul unei mortalitãţi crescute, peste 50% din cazuri, vârstele mici şi îndeosebi sugarul beneficiazã de intervenţia paleativã. Dupã vârsta de 4-5 ani se face intervenţia corectivã a DSV şi implantarea unei valve proteze între ventricolul drept şi aretrele pulmonare.
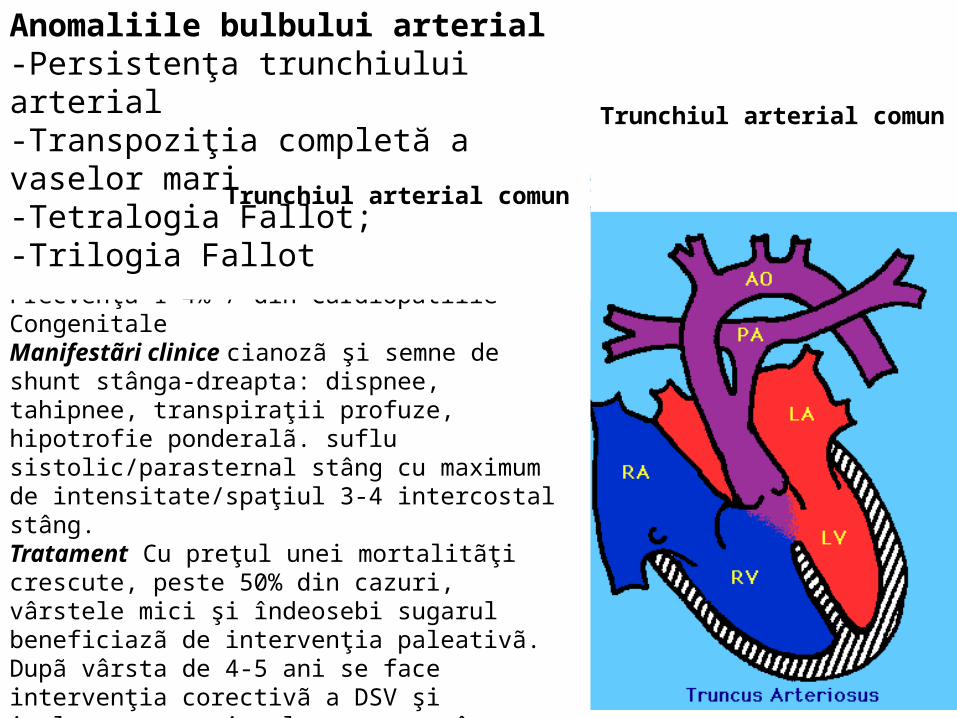
Trunchiul arterial comun
Anomaliile bulbului arterial-Persistenţa trunchiului arterial-Transpoziţia completă a vaselor mari-Tetralogia Fallot; -Trilogia Fallot Trunchiul arterial comun

Sângele din V.stang trece în circulaţia pulmonarã iar din V.drept în circulaţia sistemicã. Clinic Cianozã precoce în primele 48 ore de viaţã, hipoxie, tahipnee, dispnee. 50% fãrã modificãri la auscultaţie. Radiologic volum cardiac crescut, vascularizaţie pulmonarã crescutã.
Transpoziţia completă a vaselor maricardiopatie congenitalã frecventã, 5 din 10 000 % nou-nãscuţi, deosebit de gravã, /copii sex masculin; la 1 an 9% / CC; >2 ani 5% CC

Evoluţie spontan mortalã > 90%. Pentru asigurarea supravieţuirii este necesarã crearea artificialã a unei comunicãri la nivel atrial. Anomalii sociate - 98% din cazuri : DSV (75% din cazuri), stenoza pulmonara sau subpulmonara (75% din cazuri), anomalii ale valvei tricuspide (Ebstein-like in 30% din cazuri). Bloc Congenital complet in 5% din cazuri. Tratament între câteva luni şi 2-3 ani se realizeazã detranspozarea vaselor mari. 70% şanse de viaţã normalã unui nou-nãscut cu transpoziţie de mari vase fãrã alte malformaţii asociate, operat la momentul oportun.
Normal Transpoziţia completă a marilor vase
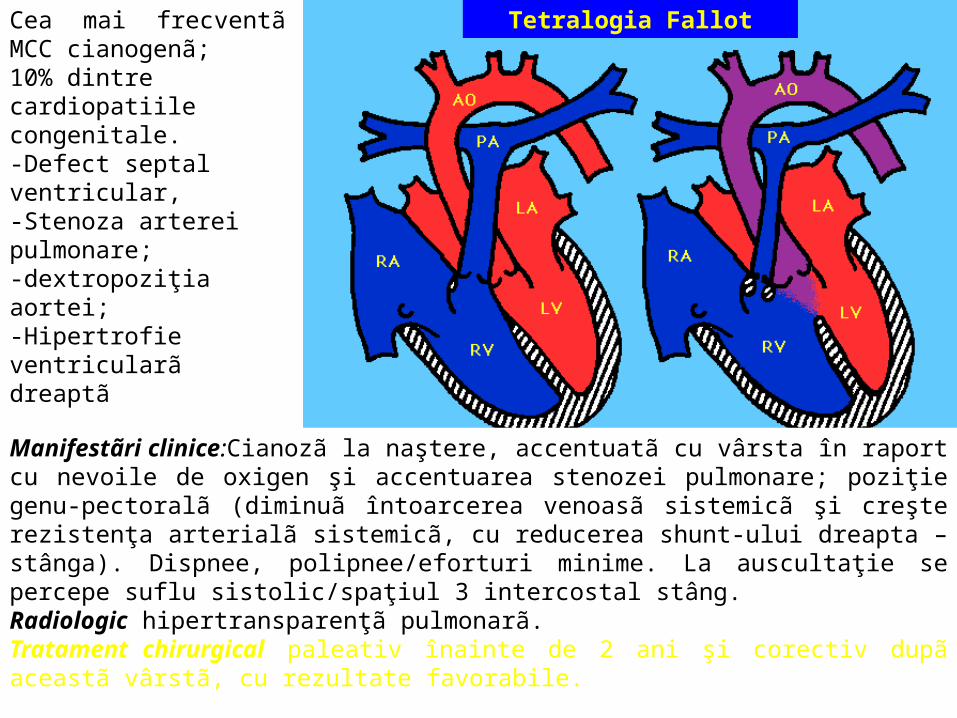
Tetralogia FallotCea mai frecventã MCC cianogenã;10% dintre cardiopatiile congenitale. -Defect septal ventricular, -Stenoza arterei pulmonare; -dextropoziţia aortei; -Hipertrofie ventricularã dreaptã
Manifestãri clinice:Cianozã la naştere, accentuatã cu vârsta în raport cu nevoile de oxigen şi accentuarea stenozei pulmonare; poziţie genu-pectoralã (diminuã întoarcerea venoasã sistemicã şi creşte rezistenţa arterialã sistemicã, cu reducerea shunt-ului dreapta – stânga). Dispnee, polipnee/eforturi minime. La auscultaţie se percepe suflu sistolic/spaţiul 3 intercostal stâng. Radiologic hipertransparenţã pulmonarã. Tratament chirurgical paleativ înainte de 2 ani şi corectiv dupã aceastã vârstã, cu rezultate favorabile.

• Canalul arterial Botallo (Ductus ateriosus) existent in viata fetala intre artera pulmonara si aorta descendenta, asigura comunicarea dintre sangele venos al circulatiei pulmonare si sangele arterial al circulatiei sistemice.
Persistenta canalului arterial Botallo• Determina un shunt stanga – dreapta datorat presiunii sangelui in
sistemul arterial sitemic, mai mare ca presiunea sanguina din artera pulmonara.
• Imagistica: Persistenta canalului arterial determina • Shunt vascular stanga – dreapta,• Dilatarea
– ventriculului stang, – atriului stang, – aortei.
• Existenta arcului aortic proeminent ajuta diagnosticul diferential cu DSA si DSV unde arcul aortic are aspect normal sau putin proeminent.
• In cazul obstructiei ventriculului stang sau al scaderii fluxului sanguin pulmonar Persistenta canalului arterial poate mentine viata nou-nascutului.


Coarctatia aortei• Ingustarea valvei aortice sau a portiunii situate imediat sub - sau - deasupra valvei aortice, cu cresterea rezistentei la la eliminarea sangelui din ventriculul stang.
• proces progresiv, cu efecte progresive.
• Initial poate fi asimptomatica. • Simptomele apar cand cordul nu mai
poate compensa obstructia. • Stenoza poate afecta doar valva
aortica sau pot coexista si alte tipuri de stenoze.
• Exista 3 tipuri de Stenoza aortica congenitala
• 1) Valvular (obstructia valvei) • 2) Subaortic (stenoza subaortica) • 3) Supra-aortic (stenoza supra-
aortica)
•Ingustarea sau constrictia aortei. Gradul de obstructie variaza de la redus la sever.
Stenoza aortica congenitala
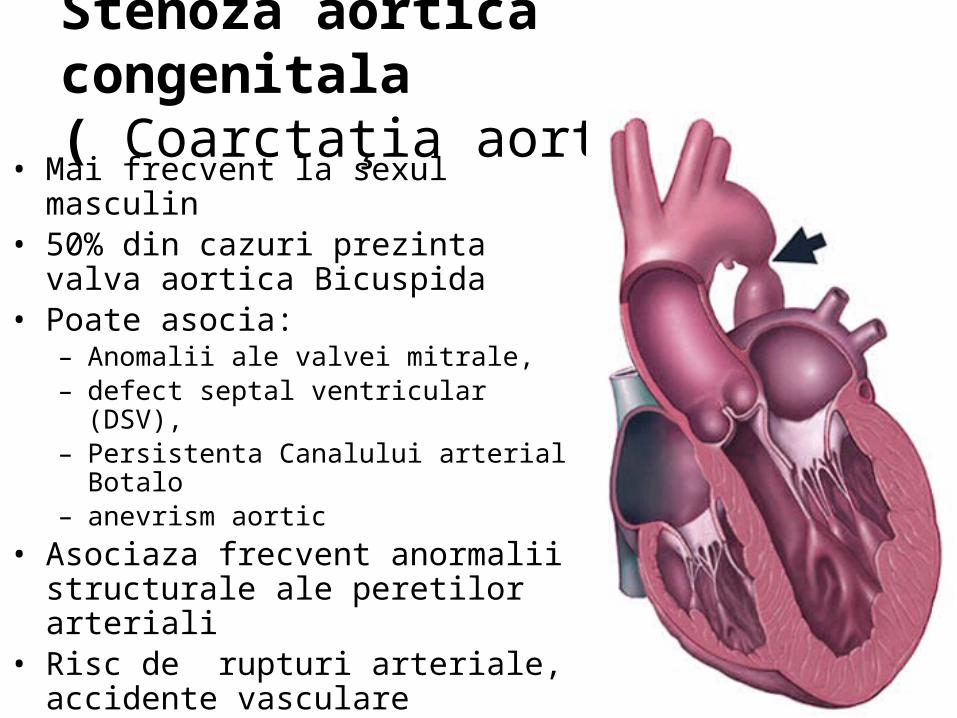
( Coarctaţia aortei )• Mai frecvent la sexul masculin• 50% din cazuri prezinta valva aortica
Bicuspida• Poate asocia:
– Anomalii ale valvei mitrale, – defect septal ventricular (DSV), – Persistenta Canalului arterial Botalo– anevrism aortic
• Asociaza frecvent anormalii structurale ale peretilor arteriali
• Risc de rupturi arteriale, accidente vasculare cerebrale, chiar si dupa tratamentul chirurgical; pentru preventia acestor complicatii este necesar controlul presiunii arteriale
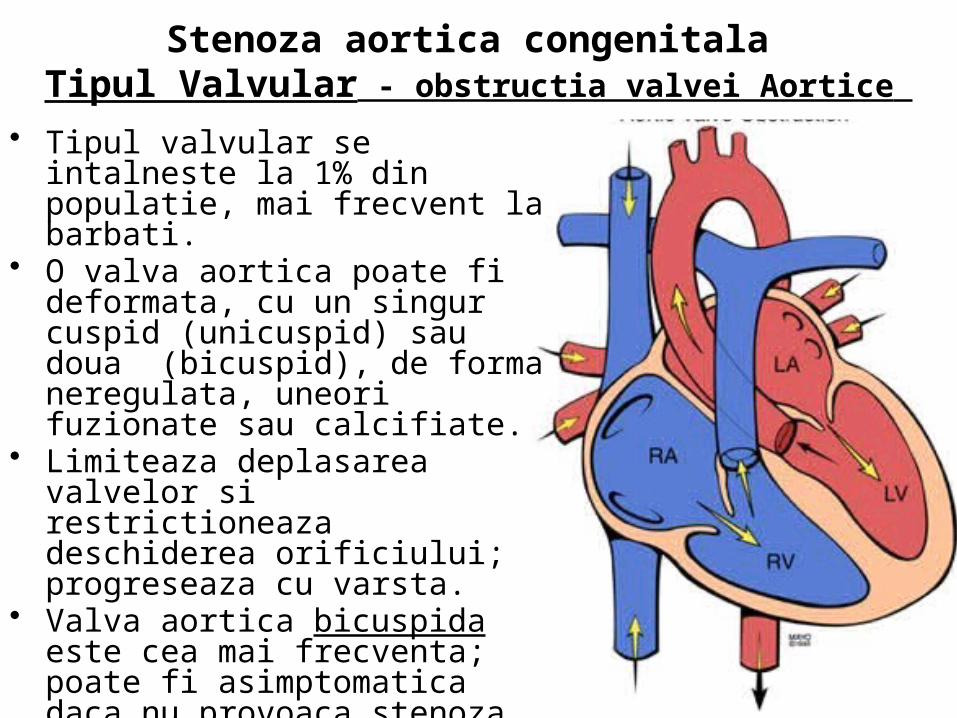
• Tipul valvular se intalneste la 1% din populatie, mai frecvent la barbati.
• O valva aortica poate fi deformata, cu un singur cuspid (unicuspid) sau doua (bicuspid), de forma neregulata, uneori fuzionate sau calcifiate.
• Limiteaza deplasarea valvelor si restrictioneaza deschiderea orificiului; progreseaza cu varsta.
• Valva aortica bicuspida este cea mai frecventa; poate fi asimptomatica daca nu provoaca stenoza (ingustarea) sau regurgitarea. Frecvent se calcifiaza devenind dura, ceea ce determina progresiv aparitia stenozei sau a regurgitarii.
• Valva unicuspida provoaca tulburari precoce in primele zile dupa nastere.
Stenoza aortica congenitala Tipul Valvular - obstructia valvei Aortice
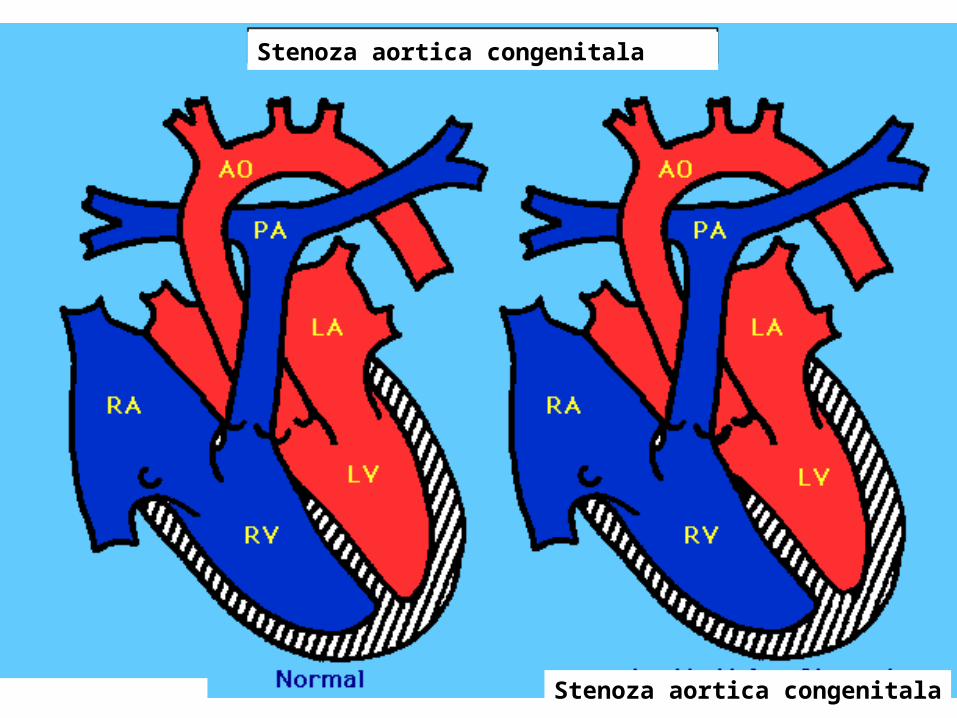
Stenoza aortica congenitala
Stenoza aortica congenitala
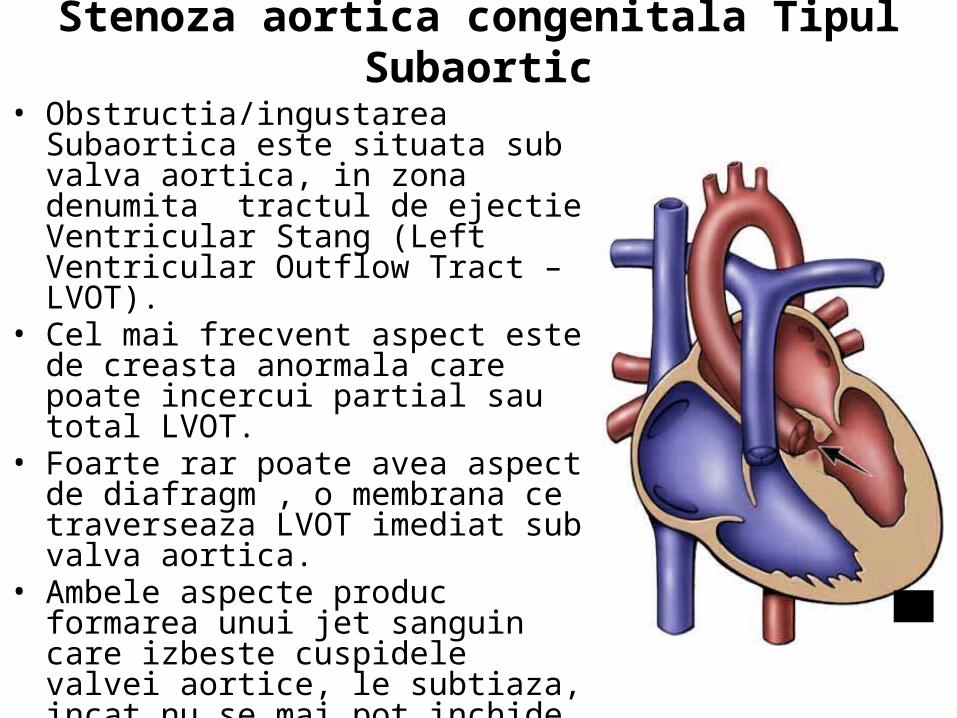
• Obstructia/ingustarea Subaortica este situata sub valva aortica, in zona denumita tractul de ejectie Ventricular Stang (Left Ventricular Outflow Tract – LVOT).
• Cel mai frecvent aspect este de creasta anormala care poate incercui partial sau total LVOT.
• Foarte rar poate avea aspect de diafragm , o membrana ce traverseaza LVOT imediat sub valva aortica.
• Ambele aspecte produc formarea unui jet sanguin care izbeste cuspidele valvei aortice, le subtiaza, incat nu se mai pot inchide si sangele regurgiteaza. Se solicita astfel mai mult ventriculul stang (hipertrofie ventricul stang).
• Stenoza Subvalvulara exista la 10% dintre pacientii cu stenoza aortica si este de 2 ori mai frecventa la barbati.
Stenoza aortica congenitala Tipul Subaortic
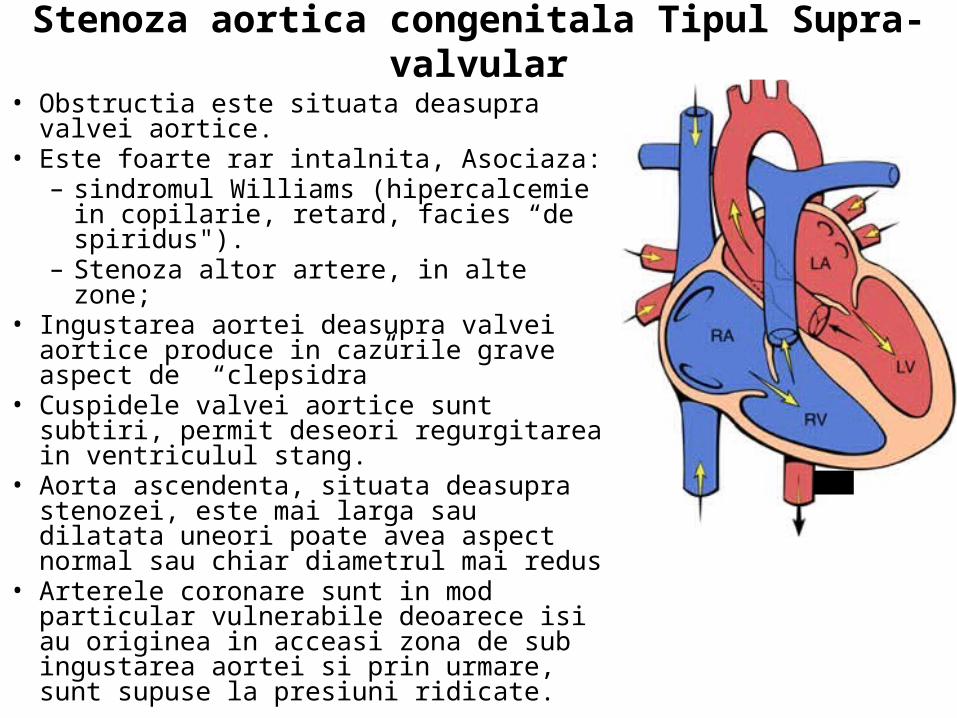
• Obstructia este situata deasupra valvei aortice.
• Este foarte rar intalnita, Asociaza: – sindromul Williams (hipercalcemie in
copilarie, retard, facies “de spiridus"). – Stenoza altor artere, in alte zone;
• Ingustarea aortei deasupra valvei aortice produce in cazurile grave aspect de “clepsidra”
• Cuspidele valvei aortice sunt subtiri, permit deseori regurgitarea in ventriculul stang.
• Aorta ascendenta, situata deasupra stenozei, este mai larga sau dilatata uneori poate avea aspect normal sau chiar diametrul mai redus
• Arterele coronare sunt in mod particular vulnerabile deoarece isi au originea in acceasi zona de sub ingustarea aortei si prin urmare, sunt supuse la presiuni ridicate.
Stenoza aortica congenitala Tipul Supra-valvular

Stenoza aortica congenitala Tipul Supra-valvular
Stenoza aortica Tipul Supra-valvular
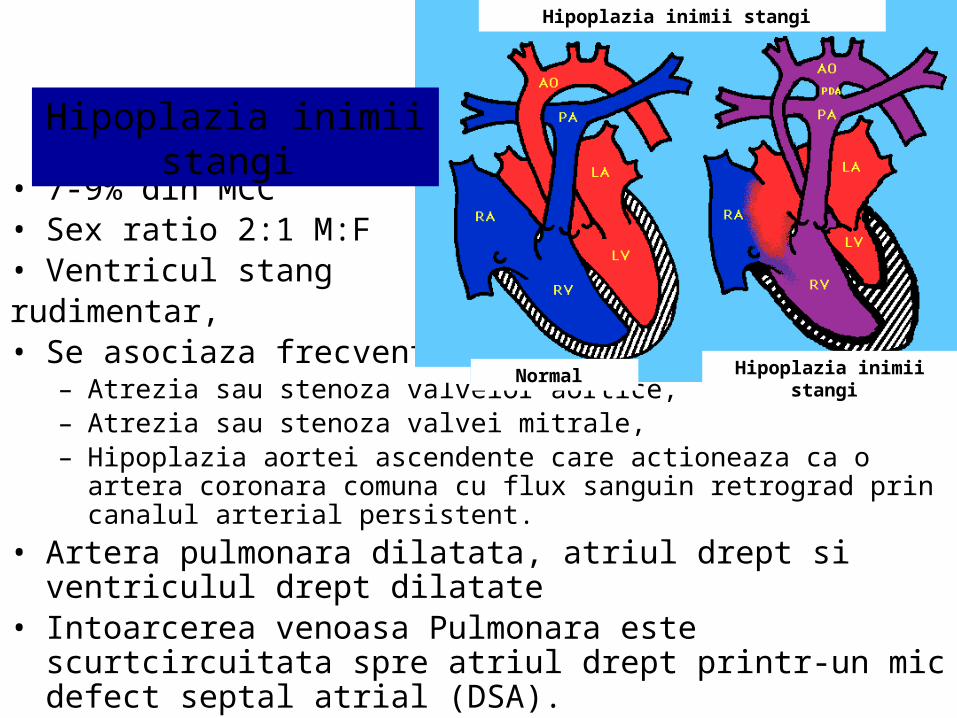
• 7-9% din MCC• Sex ratio 2:1 M:F• Ventricul stang rudimentar, • Se asociaza frecvent:
– Atrezia sau stenoza valvelor aortice, – Atrezia sau stenoza valvei mitrale,– Hipoplazia aortei ascendente care actioneaza ca o artera coronara comuna cu
flux sanguin retrograd prin canalul arterial persistent.• Artera pulmonara dilatata, atriul drept si ventriculul drept dilatate• Intoarcerea venoasa Pulmonara este scurtcircuitata spre atriul drept
printr-un mic defect septal atrial (DSA).• Cianoza se datoreaza perfuziei periferice reduse si congestiei
pulmonare.
Hipoplazia inimii stangi
Hipoplazia inimii stangi
Hipoplazia inimii stangi Normal
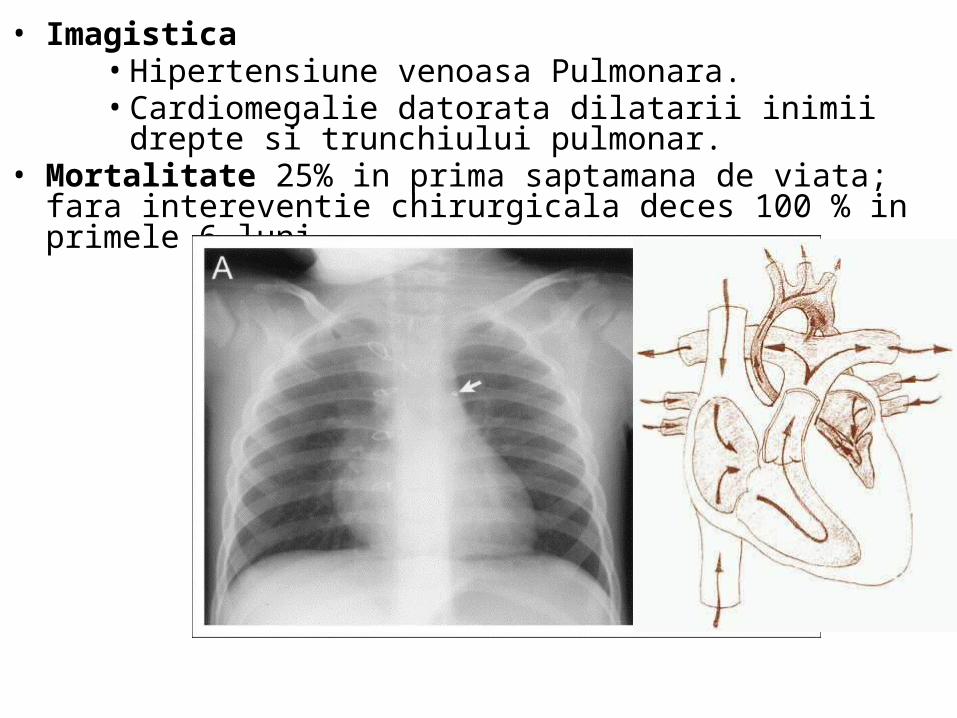
• Imagistica• Hipertensiune venoasa Pulmonara.• Cardiomegalie datorata dilatarii inimii drepte si trunchiului
pulmonar.• Mortalitate 25% in prima saptamana de viata; fara intereventie
chirurgicala deces 100 % in primele 6 luni

DEZVOLTAREA CRANIULUI
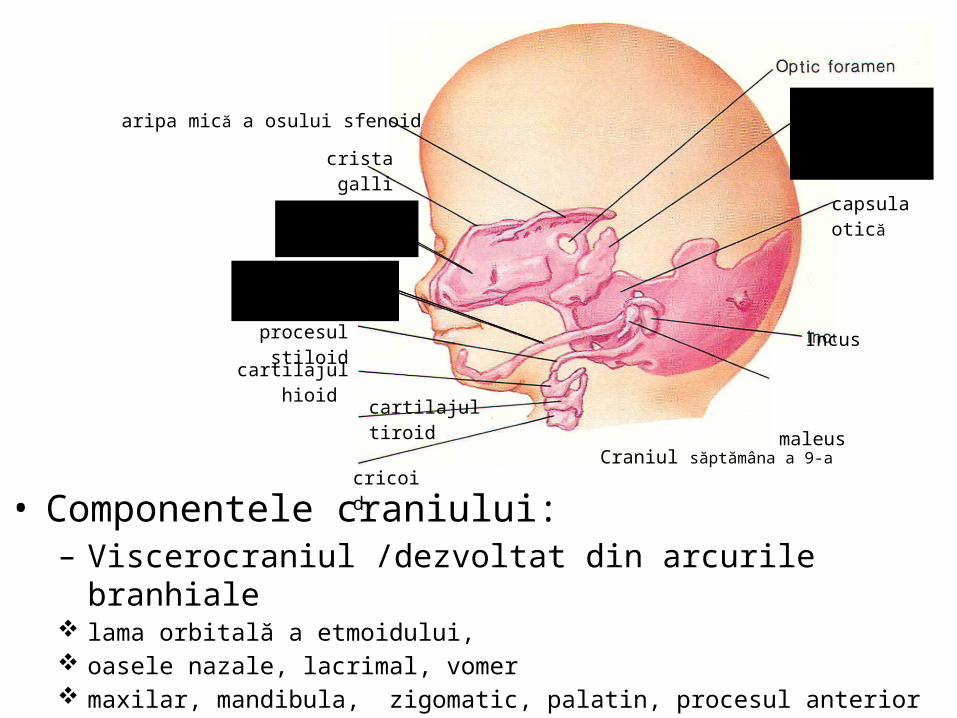
• Componentele craniului: – Viscerocraniul /dezvoltat din arcurile branhiale lama orbitală a etmoidului, oasele nazale, lacrimal, vomer maxilar, mandibula, zigomatic, palatin, procesul anterior al ciocanului.
maleus
Capsula nazală
cricoid
capsula otică
Incus
aripa mică a osului sfenoid
crista galli
cartilajul Meckel
procesul stiloid
cartilajul hioid
cartilajul tiroid
Craniul săptămâna a 9-a
aripa mare a osului sfenoid

Neurocraniul: Calota / osificare desmală
-Temporal: partea scuamoasă şi timpanică, -Parietal , -Frontal, -Occipital: partea superioară a scuamei;
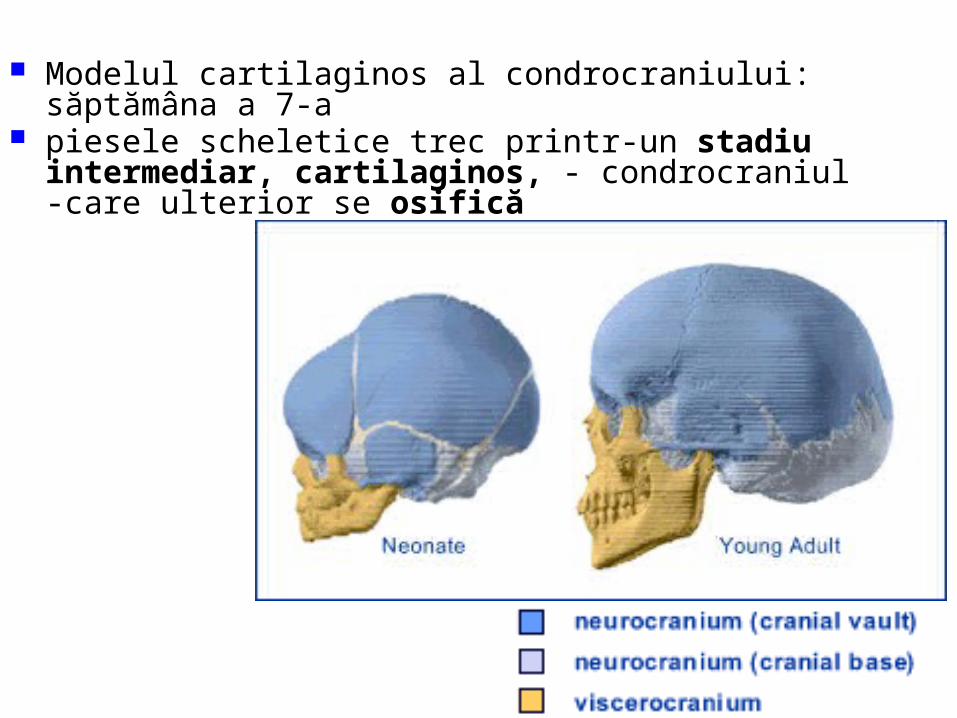
Modelul cartilaginos al condrocraniului: săptămâna a 7-a piesele scheletice trec printr-un stadiu intermediar,
cartilaginos, - condrocraniul -care ulterior se osifică
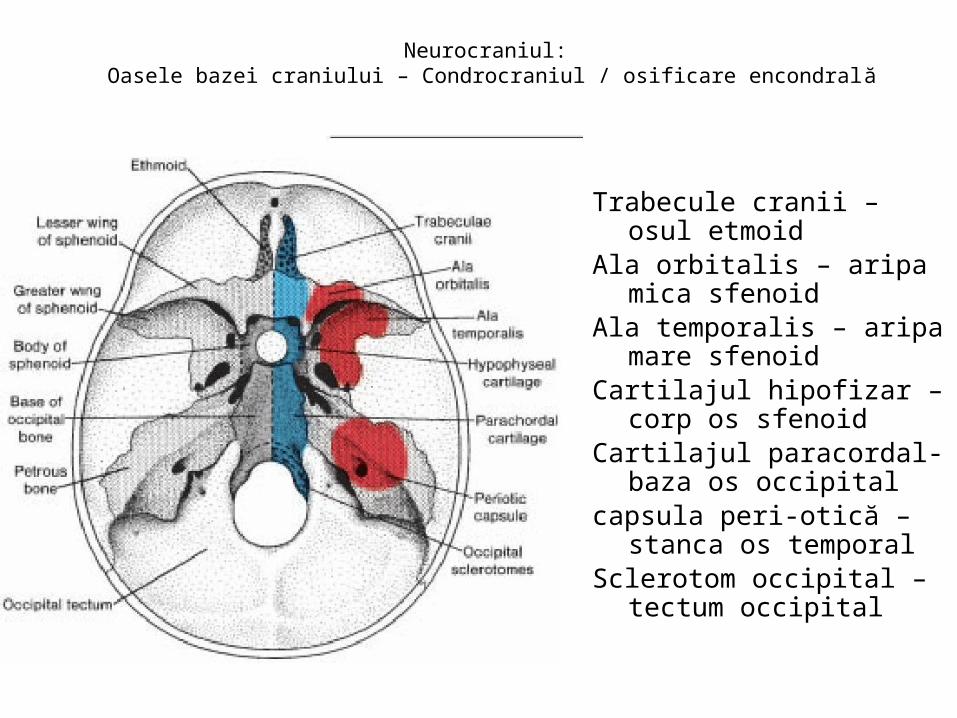
Neurocraniul: Oasele bazei craniului – Condrocraniul / osificare encondrală
Trabecule cranii – osul etmoidAla orbitalis – aripa mica sfenoidAla temporalis – aripa mare
sfenoidCartilajul hipofizar – corp os
sfenoidCartilajul paracordal- baza os
occipitalcapsula peri-otică – stanca os
temporal Sclerotom occipital – tectum
occipital
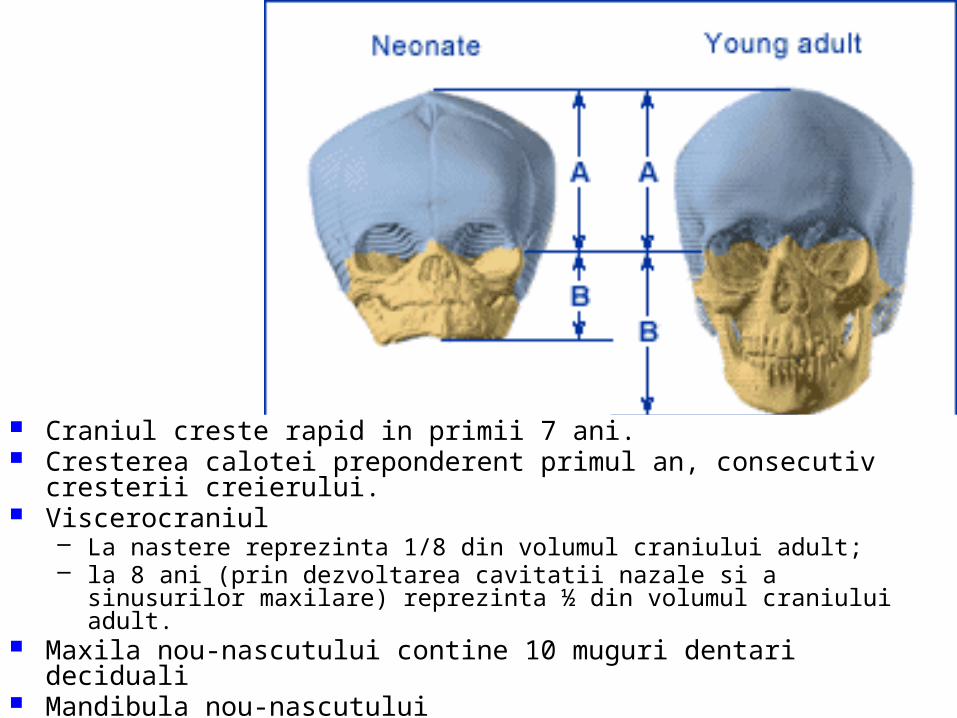
Craniul creste rapid in primii 7 ani. Cresterea calotei preponderent primul an, consecutiv cresterii creierului. Viscerocraniul
– La nastere reprezinta 1/8 din volumul craniului adult; – la 8 ani (prin dezvoltarea cavitatii nazale si a sinusurilor maxilare) reprezinta ½ din
volumul craniului adult. Maxila nou-nascutului contine 10 muguri dentari deciduali Mandibula nou-nascutului
– contine 10 muguri dentari deciduali – este alcatuita din 2 jumatati unite printr-o sutura mediana.

• 5 suturi craniene majore:• Metopica sau frontala. • Sagitala • Coronara• Lambdoida• Scuamoasa.
– Marginea Superioara a portiunii scuamoase a osului temporal.
– Anterior se articuleaza cu aripa mare a osului sfenoid;
– Superior se articuleaza cu osul parietal;
– Posterior si inferior se articuleaza cu osul occipital.

• 6 fontanele :• 1 anterioara, se inchide la varsta de 1,2 - 2 ani • 2 sfenoidale (anterolaterale), • 2 mastoidiene (posterolaterale), • 1 posterioara, se inchide la 3 luni dupa nastere
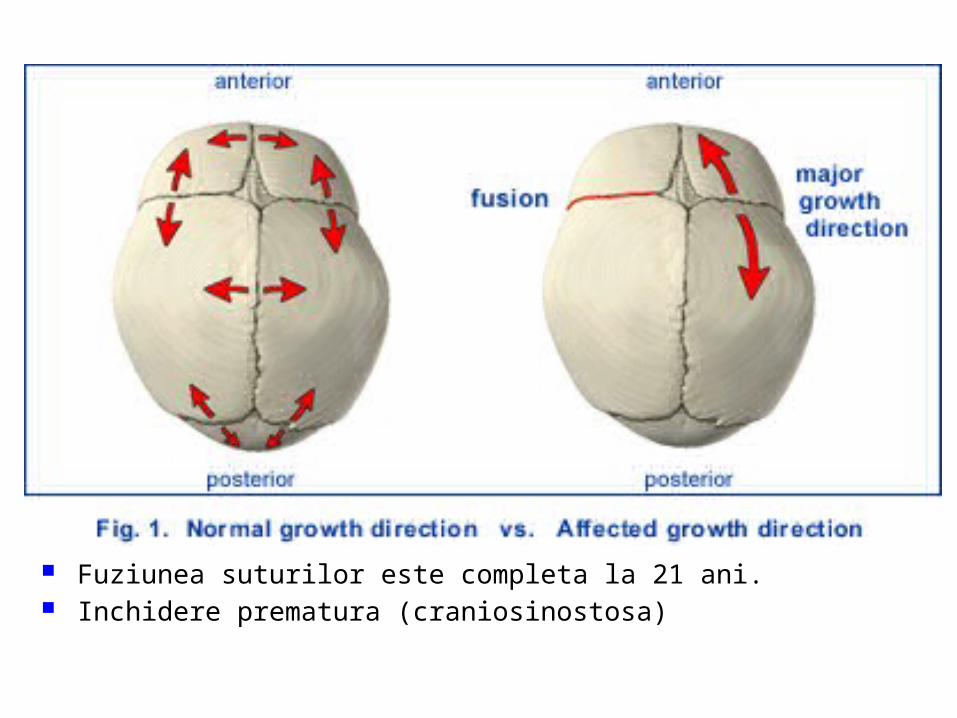
Fuziunea suturilor este completa la 21 ani. Inchidere prematura (craniosinostosa)
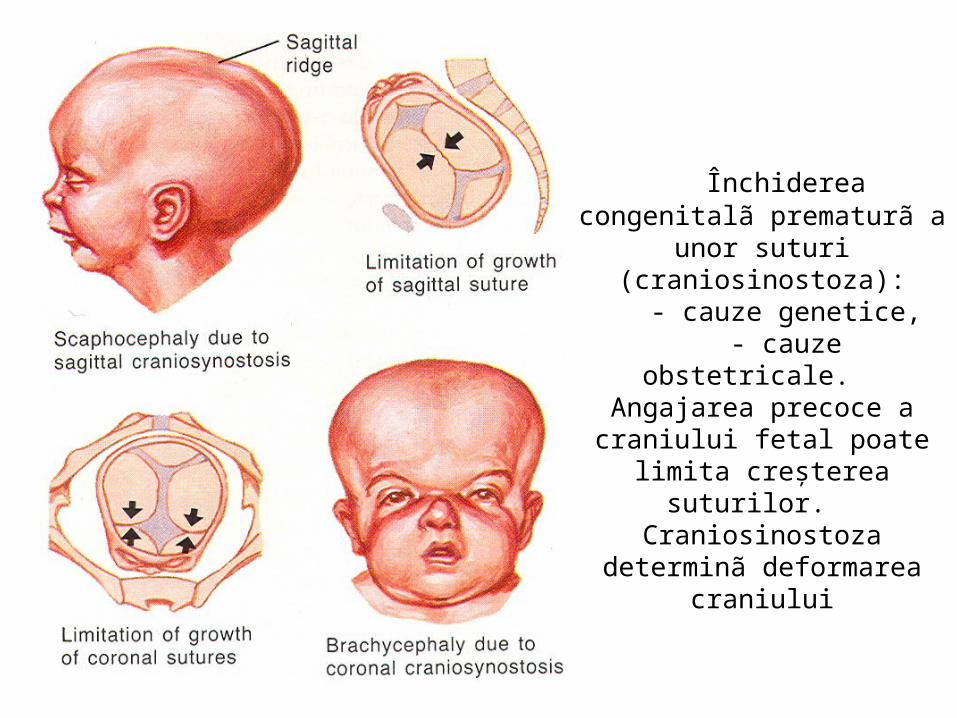
Închiderea congenitalã prematurã a unor suturi
(craniosinostoza):- cauze genetice,
- cauze obstetricale. Angajarea precoce a
craniului fetal poate limita creşterea suturilor. Craniosinostoza determinã
deformarea craniului

Sinostoza Sagitala
Inainte dupa interventia chirurgicala, la 1 an 6 luni

● Malformaţii congenitale
Bolta palatina ogivalã : întârziere în dezvoltarea mugurilor maxilari şi a proceselor palatine; -izolată, -sindroame: alcoolic fetal, - Cornelia de Lange, - sindrom Treacher Collins - disostoza mandibulo-facială Francescheti, hipoplazia facială laterală a strucuturilor; - sindromul Apert -
Sindromul Apert-hipoplazia mediofacială-sinostoza coronară

• În perioada de creştere ţesutul fibros care se interpune între piesele osoase are rol generator, osteogen, favorizând creşterea oaselor perpendicular pe linia de sutură.
• Închiderea precoce a suturilor determină microcefalia.
sindrom Down - trisomie 21:-microcefalie, -retard psiho- motor, facies tipic: fante palpebrale mongoloide,hipoplazia nasului, rădăcina nasului aplatizată şi jos situată, urechi mici, jos situate

-Anencefalia - datoratã neînchiderii neuroporului anterior -caracterizatã prin lipsa porţiunii cefalice a tubului neural.
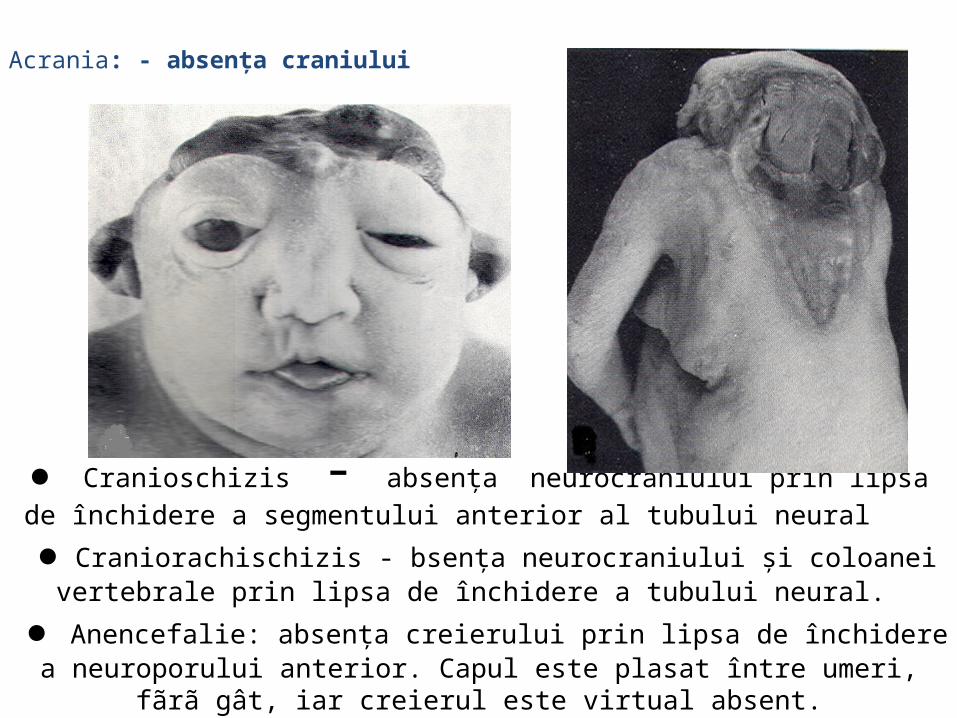
● Cranioschizis - absenţa neurocraniului prin lipsa de închidere a segmentului anterior al tubului neural
● Craniorachischizis - bsenţa neurocraniului şi coloanei vertebrale prin lipsa de închidere a tubului neural.
● Anencefalie: absenţa creierului prin lipsa de închidere a neuroporului anterior. Capul este plasat între umeri, fãrã gât, iar creierul este virtual absent.
● Acrania: - absenţa craniului

DEZVOLTAREA FEŢEI

DEZVOLTAREA FEŢEI
• între săptămâna a 4-a (embrion de 5mm) şi a 8-a (embrion de 14,5mm),
• procese de inducţie, creştere şi diferenţiere inegalã, în urma cãrora apar mugurii faciali.
• La sfârşitul săptămînii a 4-a extremitatea cefalică prezintă pe faţa ventrală o mare cavitate denumită stomodeum (gura primitivă), dispusă în sens transversal, delimitatã superior de mugurele frontonazal iar inferior de mugurele mandibular.

• sǎptǎmâna a 4-a • celule ale crestelor neurale migreazǎ în ectomezenchimul
regiunii faciale
Human Age: 28 days

• Ectomezenchimul facial este colonizat de celule provenite din crestele neurale
• În sãptãmâna a 4-a i.u.extremitatea cefalică este centrată de o cavitate numită gura primitivă (stomodeum);
• în partea infero-lateralã a mugurelui frontonazal, Epiteliul ectodermal ce înveleşte mugurele frontonazal se diferenţiază de o parte şi alta a liniei mediane, alcătuind cele douã placode olfactive (viitorul epiteliu olfactiv al foselor nazale).
• În sãptãmâna a 5-a, gura primitivă (stomodeum) este delimitată lateral de doi muguri maxilari formaţi pe seama mugurelui mandibular.
• la începutul sãptãmânii a 6-a, placodele olfactive se adâncesc devenind canale olfactive;’ în jurul placodelopr olfactive apar mugurii nazali mediali şi laterali
la începutul sãptãmânii a 6-a, în jurul placodelor olfactive care se adâncesc devenind canale olfactive, se edificã mugurii nazali mediali şi mugurii nazali laterali

• În săptămînile 6-7 mugurii nazali mediali fuzionează alcătuind un segment intermediar din care se vor forma:
– Filtrum al buzei superioare, – osul intermaxilar, – gingia superioară a regiunii respective,– palatul primar.
• Între mugurele nazal lateral şi cel maxilar se delimitează şanţul nazo-lacrimal în a cărui extremitate lateralã se găseşte cupa optica.
• În decursul săptămînii a 6-a mugurele nazal lateral fuzionează cu cel maxilar iar şantul lacrimo-nazal se închide.
• Se stabileşte continuitatea feţei laterale a nasului cu partea laterală a buzei superioare. Concomitent mugurele nazal medial trimite o prelungire denumitã proces globos, care fuzionează cu mugurele maxilar transformând şantul olfactiv în canal olfactiv.
În săptămînile 6-7 mugurele nazal medial trimite o prelungire denumitã proces globos, care fuzionează cu mugurele maxilar

• Fiecare canal olfactiv are un orificiu anterior numit narina primitivă, posterior fiind închis până în săptămâna a7-a de membrana oro-nazalã (faza de sac olfactiv).
• Prin resorbţia membranei oro-nazale se formeazã coana primitivă.
• Coanele primitive sunt foarte apropiate de narine şi buza superioarã, iar peretele despărţitor dintre cei doi saci olfactivi formează un sept nazal median.
• Acesta devine partea medianã a plafonului gurii primitive - palatul primitiv incomplet - deci un început de separare a gurii primitive de cavitatea nazală -“faza amfibiană” a dezvoltării feţei, în timpul căreia canalul respirator se deschide în plafonul gurii primitive.
Dezvoltarea feţei în timpul fazei “piscinã”: gura primitivã (stomodeum) are aspectul unei cavitãti comune respiratorii şi bucale. Pereţi sãi laterali sunt constituiţi din mugurii maxilari. Procesele palatine ale mugurilor maxilari privesc caudal.

• Mugurii maxilari cresc şi alunecă spre linia mediană, medial şi caudal de mugurii nazali laterali, pe care-i împinge în profunzime şi cu care fuzionează.
• La finele săptămânii a 6-a stomodeum-ul are un contur neregulat, este limitat lateral de mugurii maxilari şi cei mandibulari
• Pe măsură ce mugurii maxilari cresc şi se apropie de linia medianã, dimensiunile gurii se reduc.
• Locul de fuziune al mugurelui maxilar cu cel nazal medial este indicat de crestătura unghiulară a buzei superioare finele săptămânii a 6-a.
stomodeum are un contur neregulat, este limitat lateral de mugurii maxilari; buza superioarã se formeazã pe seama materialului mugurilor maxilari şi nazali mediali.
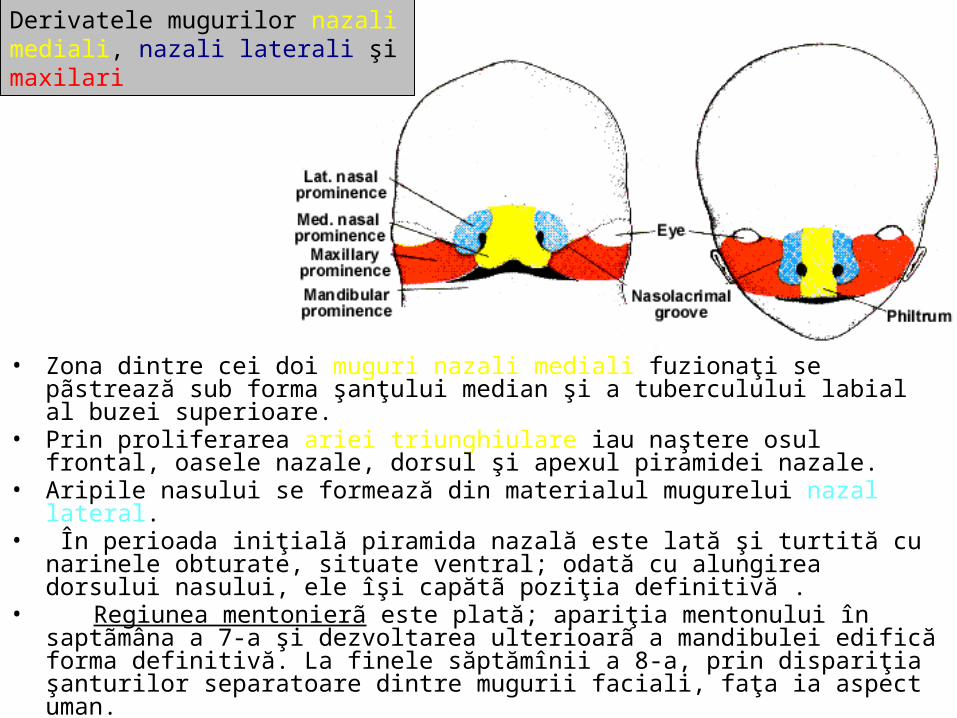
• Zona dintre cei doi muguri nazali mediali fuzionaţi se pãstrează sub forma şanţului median şi a tuberculului labial al buzei superioare.
• Prin proliferarea ariei triunghiulare iau naştere osul frontal, oasele nazale, dorsul şi apexul piramidei nazale.
• Aripile nasului se formează din materialul mugurelui nazal lateral.• În perioada iniţială piramida nazală este lată şi turtită cu narinele obturate, situate ventral;
odată cu alungirea dorsului nasului, ele îşi capătã poziţia definitivă .• Regiunea mentonierã este plată; apariţia mentonului în saptãmâna a 7-a şi
dezvoltarea ulterioarã a mandibulei edifică forma definitivă. La finele săptămînii a 8-a, prin dispariţia şanturilor separatoare dintre mugurii faciali, faţa ia aspect uman.
• Etapa de modelare a feţei constă în: • înalţarea şi alungirea dorsului nasului, dispariţia urmelor şanţurilor separatoare, aducerea
ochilor în poziţie ventralã (sãptãmâna 5-7), micşorarea proeminenţelor cerebrale, apariţia mentonului (sãptãmâna a 7-a).
Derivatele mugurilor nazali mediali, nazali laterali şi maxilari

Derivatele mugurilor faciali • Mugurii nazali mediali: partea membranoasă
a septului nazal, osul incisiv, premaxilar, cei patru incisivi, gingia, papila incisivă, frâul buzei superioare, braţul medial al cartilajului alar cartilajul paraseptal, Vomerul, lama perpendiculară a etmoidului.
• Mugurii nazali laterali: aripile nasului, peretele lateral al piramidei nazale, bolta fosei nazale, capsula nazală, lama ciuruită şi masele laterale ale etmoidului cu concha nazală superioară şi mijlocie, concha inferioară, braţul lateral al cartilajului alar şi osul lacrimal.
• Mugurele frontal formează osul frontal şi porţiunea rostrală a septului nazal osos.
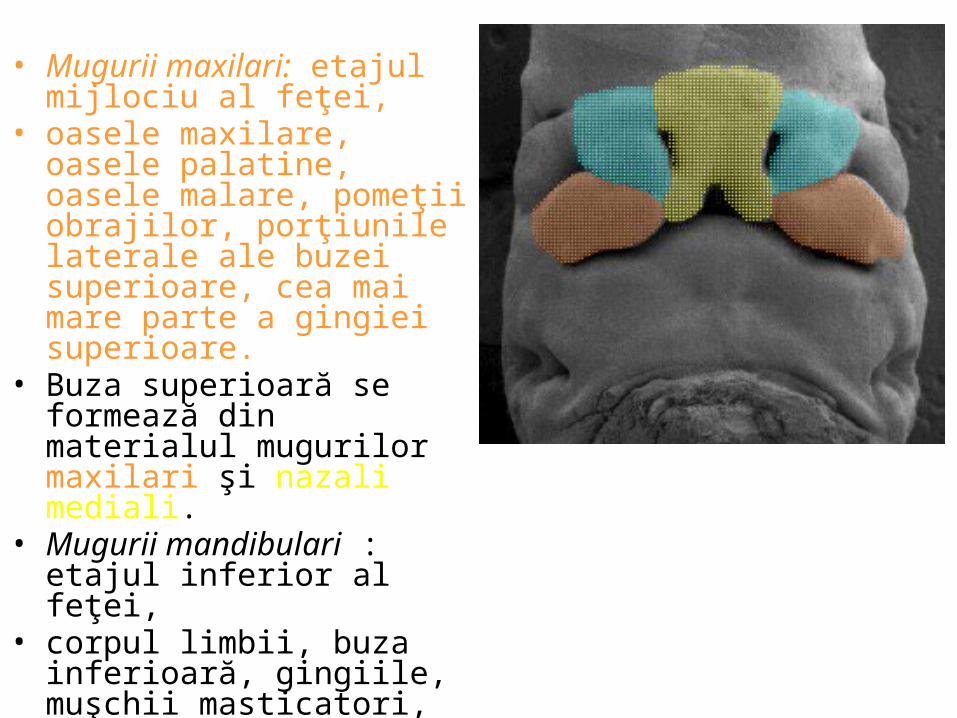
• Mugurii maxilari: etajul mijlociu al feţei,
• oasele maxilare, oasele palatine, oasele malare, pomeţii obrajilor, porţiunile laterale ale buzei superioare, cea mai mare parte a gingiei superioare.
• Buza superioară se formează din materialul mugurilor maxilari şi nazali mediali.
• Mugurii mandibulari : etajul inferior al feţei,
• corpul limbii, buza inferioară, gingiile, muşchii masticatori, m. milohioidian, pântecele anterior al digastricului, m. tensor al valului palatin, m. tensor al timpanului, mandibula

Malformaţii induse în perioada embrionară precoce, săptămânile a 3-a - a 4-a de gestaţie.
1 la 16 000 născuţi vii. Urechi fuzionate pe linia mediană, absenţa mandibulei . Anomalie de dezvoltare a prozencefalului asociată cu hipotelorism şi hipoplazia piramidei nazale.
Otocefalie

Ciclopie (fantă palpebrală unică)Proboscis (tub nazal unic rezultat prin fuziunea în exces a mugurilor nazali interni).

Diprosopia - dedublarea feţei.
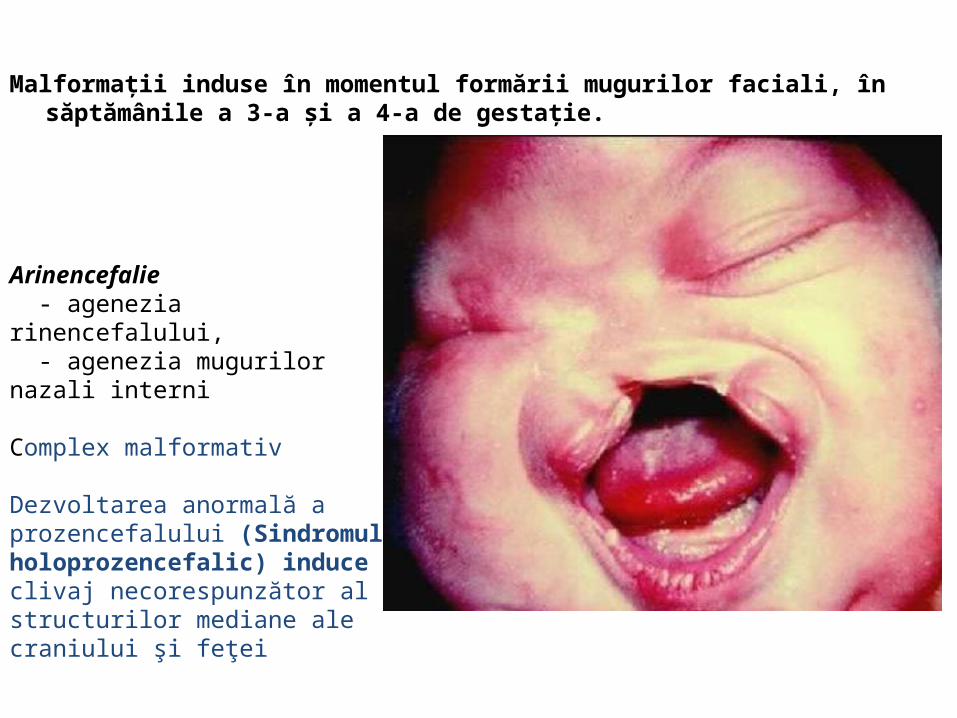
Malformaţii induse în momentul formării mugurilor faciali, în săptămânile a 3-a şi a 4-a de gestaţie.
Arinencefalie - agenezia rinencefalului, - agenezia mugurilor nazali interni
Complex malformativ
Dezvoltarea anormală a prozencefalului (Sindromul holoprozencefalic) induce clivaj necorespunzător al structurilor mediane ale craniului şi feţei
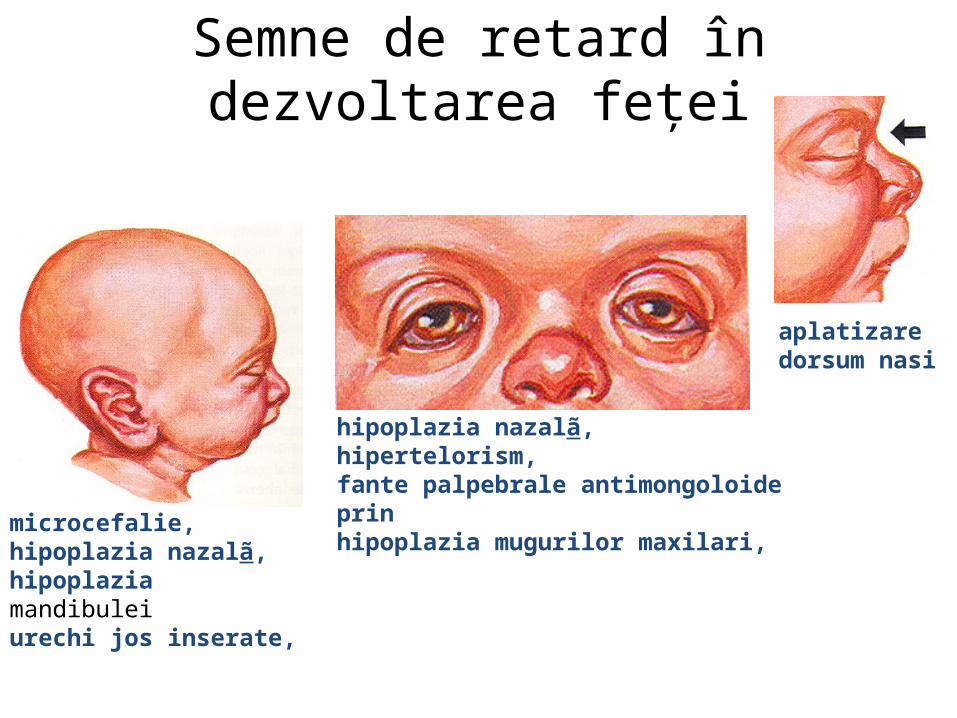
Semne de retard în dezvoltarea feţei
microcefalie, hipoplazia nazalã,hipoplazia mandibulei urechi jos inserate,
aplatizare dorsum nasi
hipoplazia nazalã,hipertelorism, fante palpebrale antimongoloide prin hipoplazia mugurilor maxilari,

Macrostoma: hipoplazia mugurilor maxilari
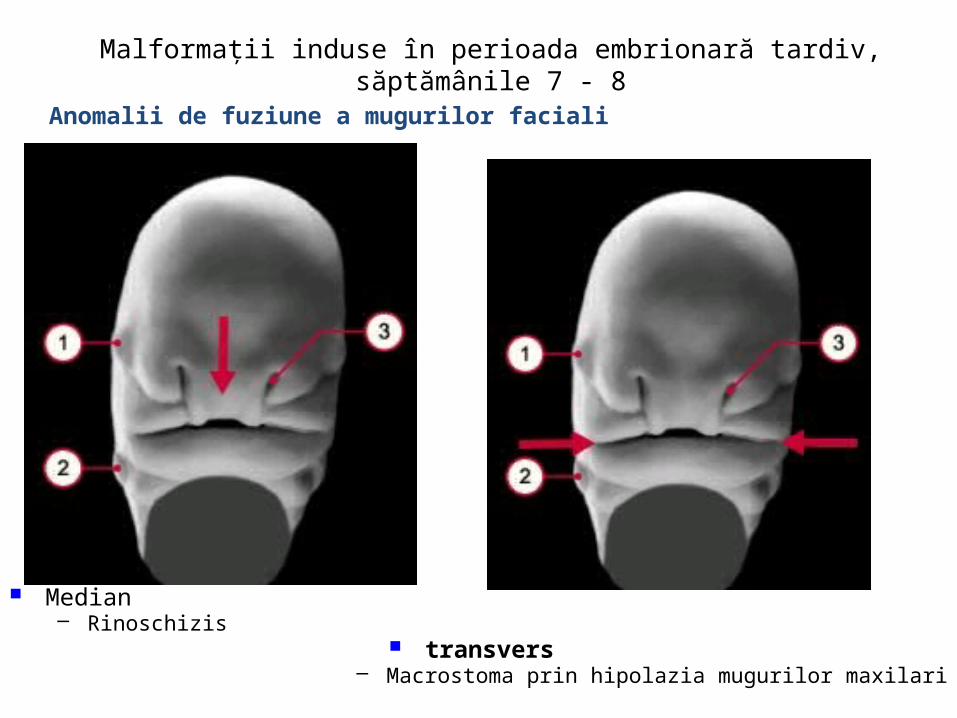
Median– Rinoschizis
transvers – Macrostoma prin hipolazia mugurilor maxilari
Malformaţii induse în perioada embrionară tardiv, săptămânile 7 - 8
Anomalii de fuziune a mugurilor faciali
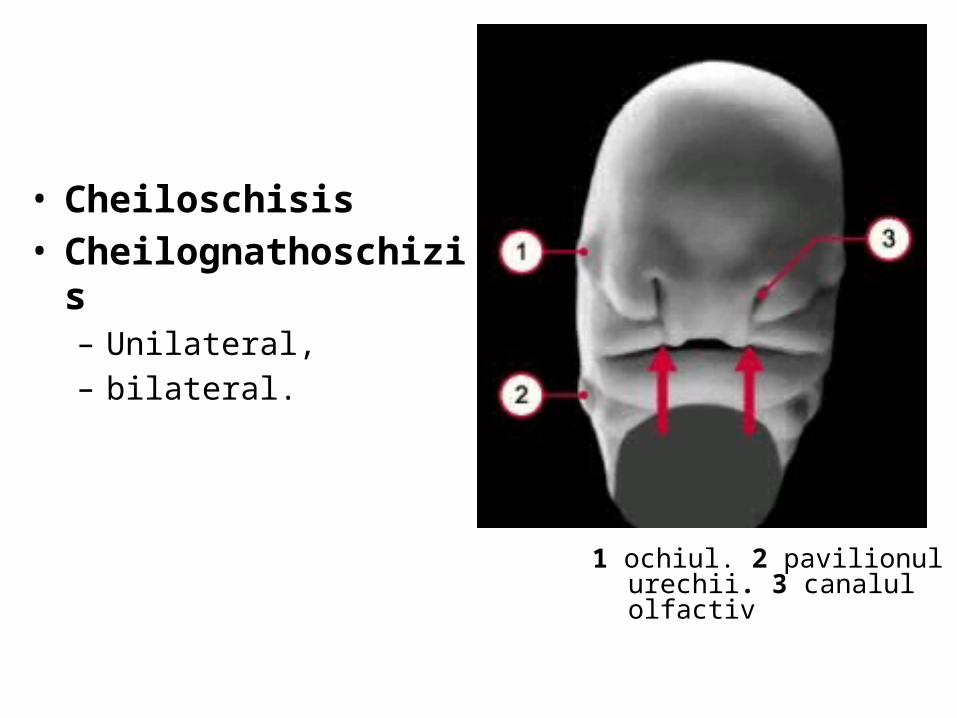
• Cheiloschisis• Cheilognathoschizis
– Unilateral,– bilateral.
1 ochiul. 2 pavilionul urechii. 3 canalul olfactiv

Cheilognatoschizis bilateral
Cariotip normal: 46, XY

• Cheilognatoschizis bilateral• Anual/SUA 227 500 sau 7% nn- defecte ale extremitatii cefalice :• 1/700 nn. • Sex ratio 2:1.

• Coloboma facialis • Aplazia căilor lacrimale
1 ochiul. 2 pavilionul urechii. 3 canalul olfactiv
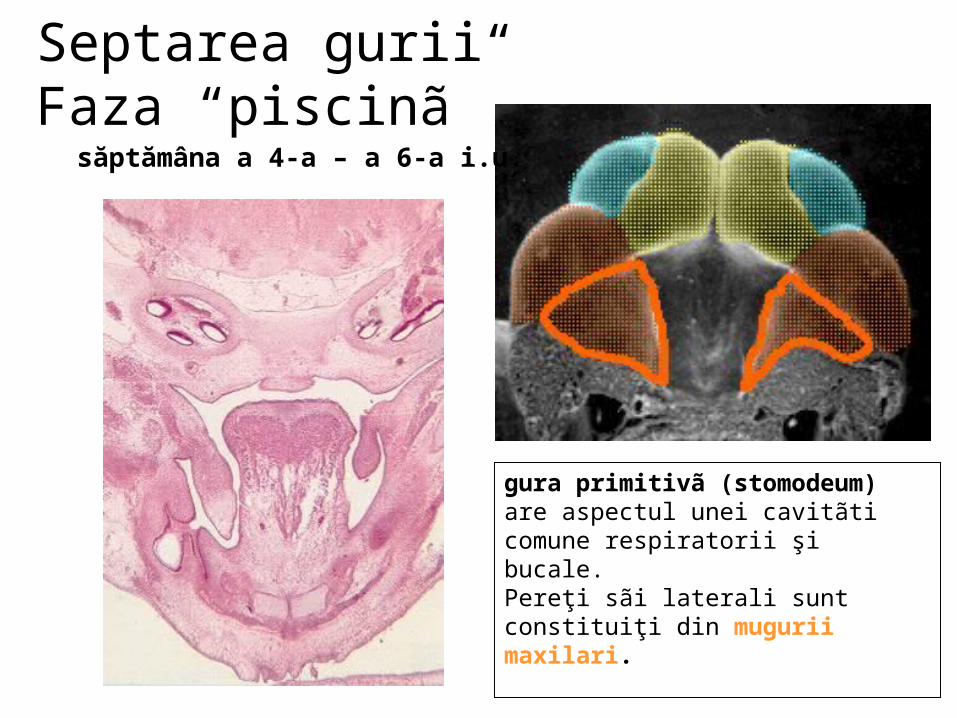
Septarea gurii Faza “piscinã”
gura primitivã (stomodeum) are aspectul unei cavitãti comune respiratorii şi bucale. Pereţi sãi laterali sunt constituiţi din mugurii maxilari.
săptămâna a 4-a – a 6-a i.u.

• Pe faţa internă a mugurilor maxilari iau naştere două procese palatine asemãnãtoare cu două lamele ce se dezvoltă oblic şi spre posterior de o parte şi de alta a limbii.
• Procesele palatine ale mugurilor maxilari privesc caudal.
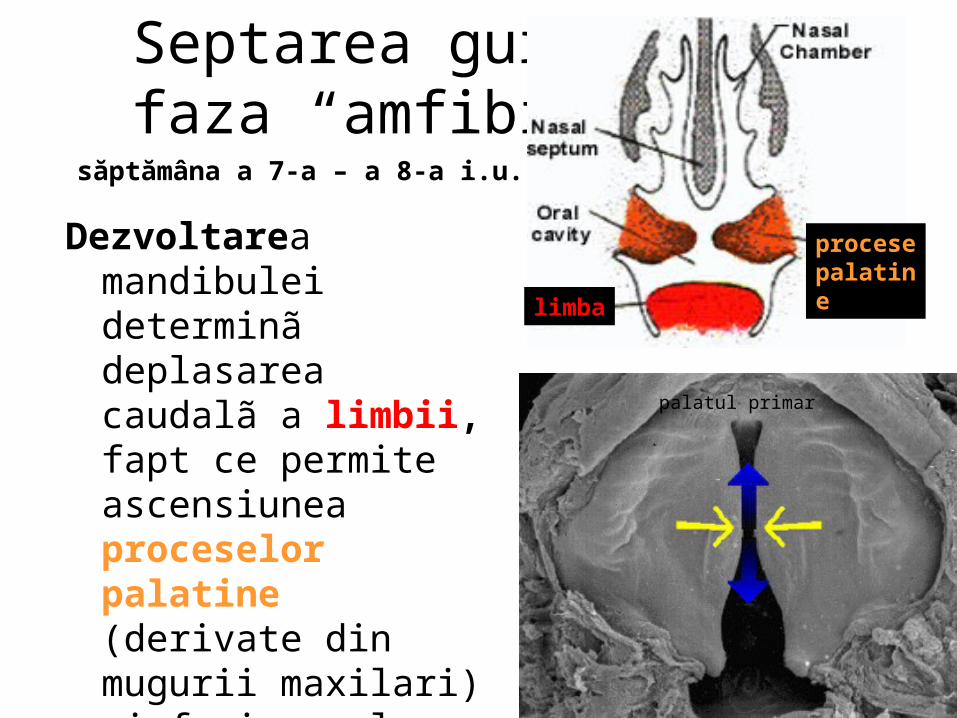
Septarea gurii faza “amfibian”
Dezvoltarea mandibulei determinã deplasarea caudalã a limbii, fapt ce permite ascensiunea proceselor palatine (derivate din mugurii maxilari) şi fuziunea lor, cu formarea palatului primar;
săptămâna a 7-a – a 8-a i.u.
palatul primar
procese palatine
limba
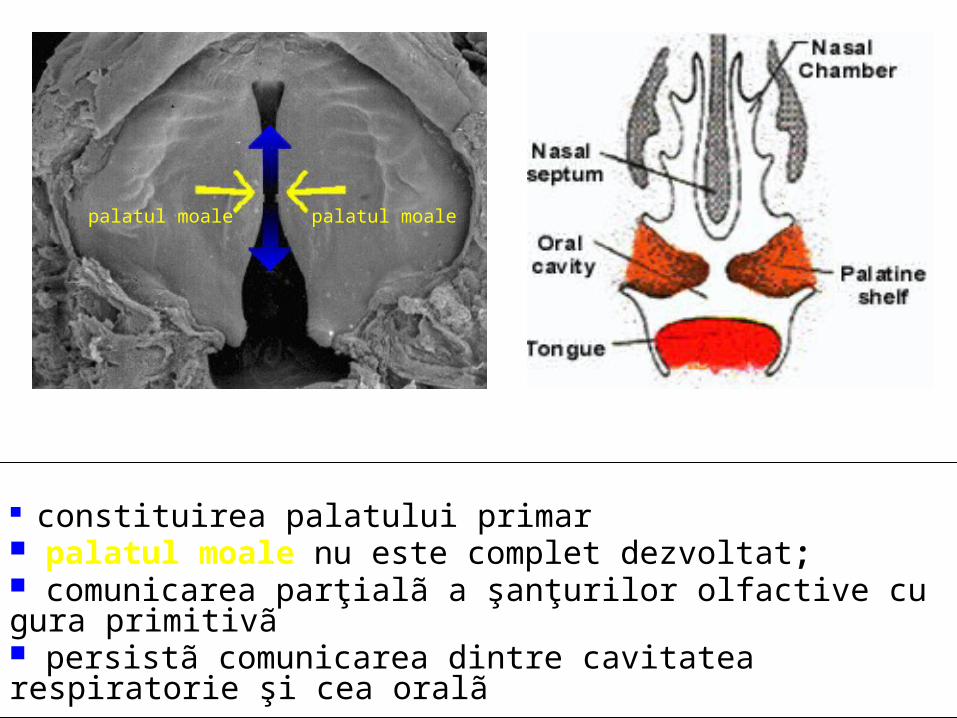
constituirea palatului primar palatul moale nu este complet dezvoltat; comunicarea parţialã a şanţurilor olfactive cu gura primitivã persistã comunicarea dintre cavitatea respiratorie şi cea oralã
palatul moale palatul moale

Septarea gurii faza “mamifer ”
• Anterior, procesele palatine fuzionează cu palatul primar, la acest nivel foramen incisive rămâne ca o demarcaţie între palatul primar şi cel secundar.
• În perioada fuziunii proceselor palatine, septul nazal coboară pentru a se uni cu palatul nou format.
• Formarea palatului secundar şi fuziunea acestuia cu septul nazal separã cavitatea oralã de cea respiratorie.
săptămâna a 8-a - a 10-a i.u.
septul nazal
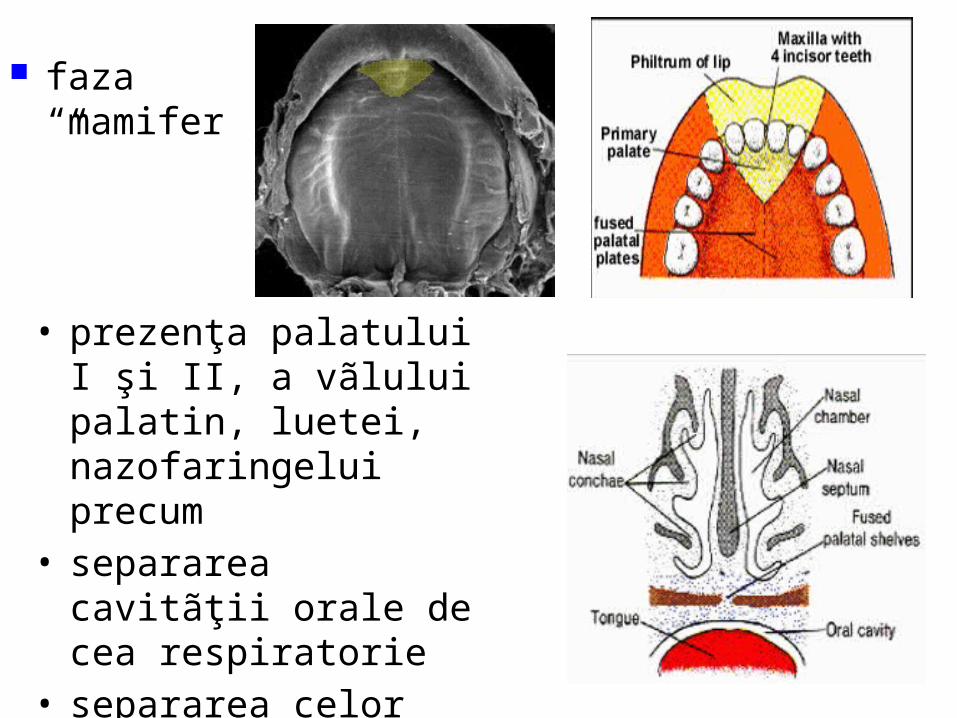
faza “mamifer ”
• prezenţa palatului I şi II, a vãlului palatin, luetei, nazofaringelui precum
• separarea cavitãţii orale de cea respiratorie
• separarea celor douã canale olfactive între ele.

• Factori teratogeni exogeni
• Fizici– emisii de radiaţii radioactive, mai ales în stadii incipiente de
dezvoltare.• Chimici
– droguri, factori alimentari (carenţe alimentare/vitamina A, avitaminoze E, hipervitaminoza A, acid folic, acid pantotenic, nicotinamida)
– hipoxia.• Biologici
– infecţii materne, infecţii fetale.• ● Factori teratogeni endogeni
– anomalii cromozomiale numerice (trisomia 21, trisomia 13, trisomia 18)

Malformaţii care apar în săptămânile 5-8 de gestaţie
• în momentul creşterii şi fuziunii mugurilor faciali:– persistenţa unora dintre şanţurile existente între
mugurii faciali,– dezvoltare redusă sau în exces a mugurilor
faciali
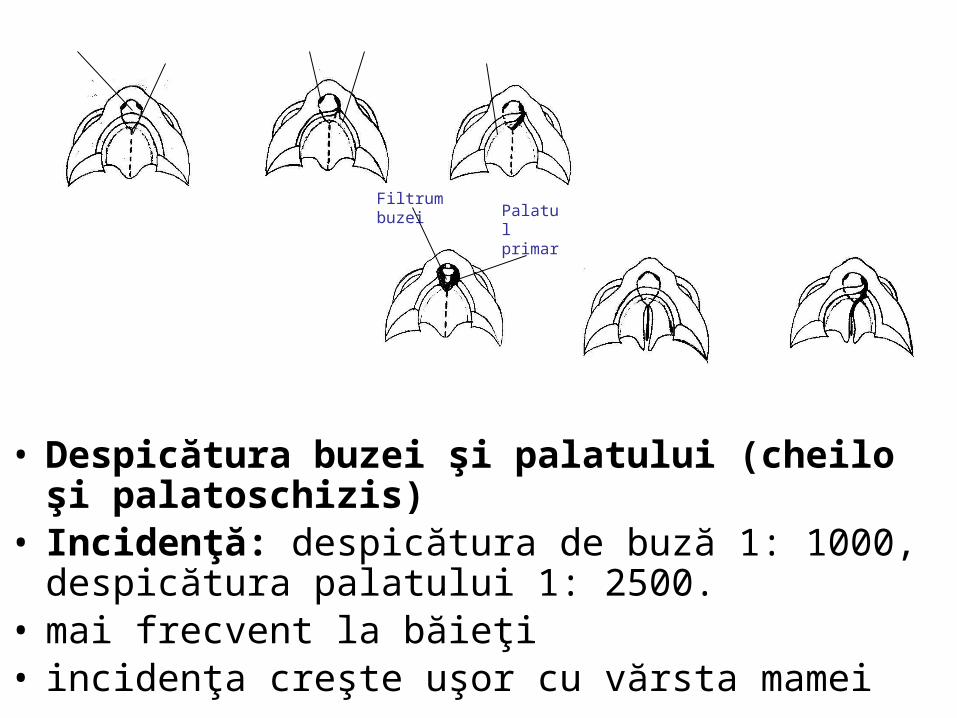
• Despicătura buzei şi palatului (cheilo şi palatoschizis) • Incidenţă: despicătura de buză 1: 1000, despicătura
palatului 1: 2500.• mai frecvent la băieţi • incidenţa creşte uşor cu vărsta mamei
Palatul primar Foramenul incisivilor Nara Buza
Gingia
Filtrum buzeiPalatul primar

• Părinţii sănătoşi, I-ul copil cu despicătură debuză, al II-ea are risc 4% pentru acelaşi defect
• ambii copii afectaţi, riscul pentru al III-lea este 9%. • un părinte cu despicătură de buză şi un copil afectat, probabilitatea ca următorul
copil să fie afectat 17%. • părinţii sănătoşi, un copil cu despicătură palatină, următorul copil risc 2%. • un părinte şi unul din copii afectaţi simultan: probabilitatea ca următorul copil să
aibă despicătură palatină 17%.
Palatul primar Foramenul incisivilor Nara Buza
Gingia
Filtrum buzeiPalatul primar

Buza de iepure, frecvenţã: 1%0 naşteri-lipsa de dezvoltare şi coalescenţã a mugurilor nazali interni cu mugurii maxilari. -bilaterală , simetrică sau asimetrică. -simplă, cheiloschizis sau cheilognatoschizis -asociată cu fisura palatină, Cheilognatopalatoschizis bilateral - gura de lup
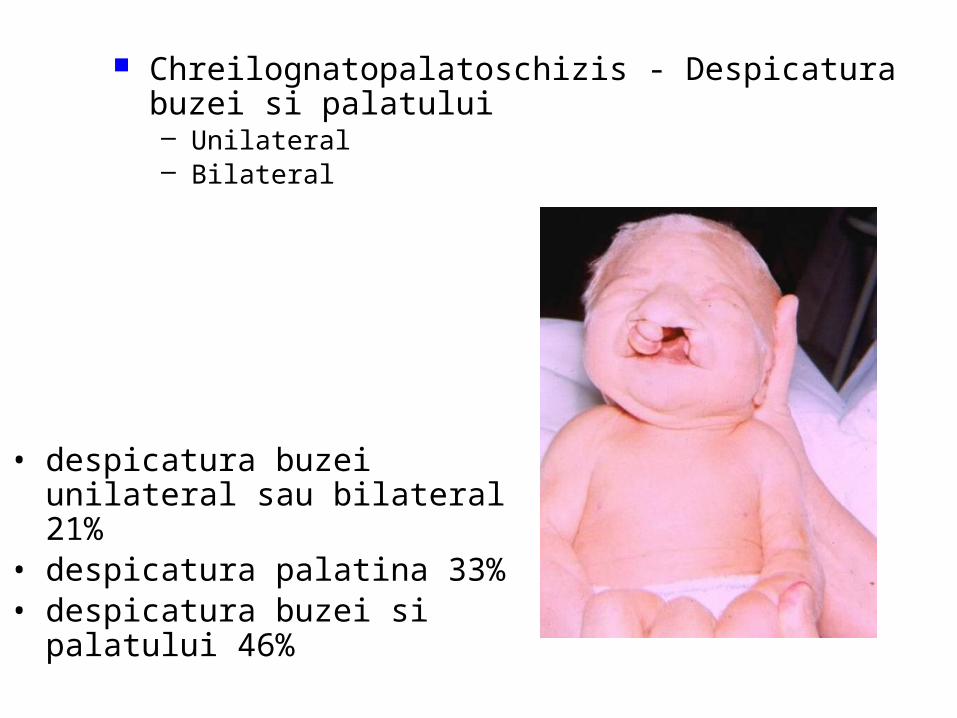
• despicatura buzei unilateral sau bilateral 21%
• despicatura palatina 33%• despicatura buzei si palatului 46%
Chreilognatopalatoschizis - Despicatura buzei si palatului – Unilateral – Bilateral

- microcefalie, - hipoplazie nazalã, - urechi jos inserate, - fante palpebrale antimongoloide, hipoplazia mugurilor maxilari - hipertelorism, - aplatizarea dorsum nasi.
Semne patognomonice de retard în dezvoltarea feţei

Sindroame care asociază malformaţii congenitale ale feţei

Sindromul Apert ( Acro-cefalo-sindactilia)
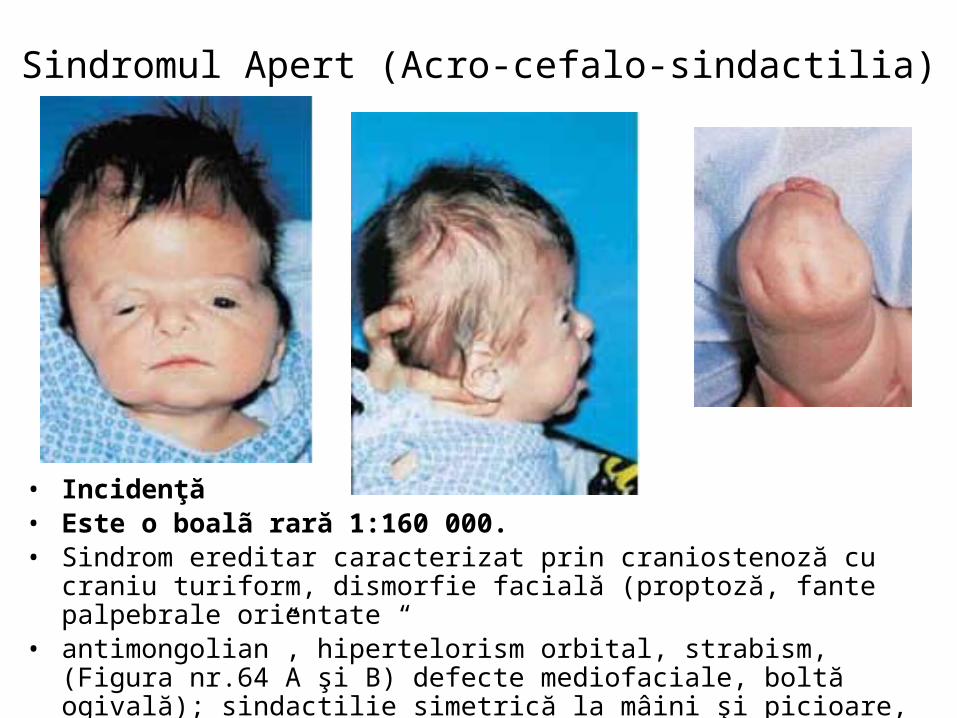
Sindromul Apert (Acro-cefalo-sindactilia)
• Incidenţă• Este o boalã rară 1:160 000.• Sindrom ereditar caracterizat prin craniostenoză cu craniu turiform, dismorfie
facială (proptoză, fante palpebrale orientate “• antimongolian”, hipertelorism orbital, strabism, (Figura nr.64 A şi B) defecte
mediofaciale, boltă ogivală); sindactilie simetrică la mâini şi picioare, afectând degetele II,III şi IV

Sindromul de Langeafecteazã 1/30 000 - 1/60 000 din numărul total de nou născuţi, cu repartiţie egală între cele două sexe.Etiopatogenie: necunoscută. Se crede că la baza acestui sindrom stau anomalii genetice, însă aceste modificări sunt greu de pus în evidenţă.Tablou clinic Constant se observã hipertelorism, fante antimongoloide, narine subţiri, comisuri “căzute”, micrognaţie, păr jos implantat, microbrahicefalie

Factori endogeni: boli cromozomale
• Sindromul Down (trisomia 21)• cel mai frecvent sindrom malformativ determinat de o anomalie cromozomală. • Incidenţă 1:650 ;variazã direct proporţional cu vârsta maternã.• sindromul Down însumează > 5% din copii cu retard mental.• Retardul mental variazã de la debilitate mintală la imbecilitate. • QI între 25 şi 50 dar, ocazional, pot fi educaţi şî învaţă să citească şi să scrie
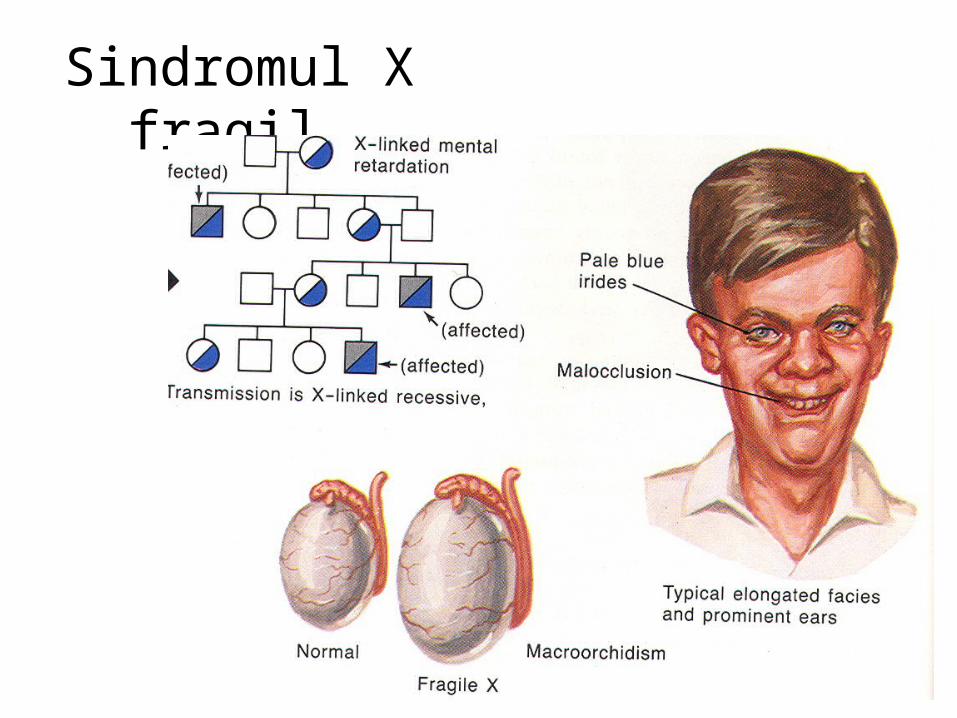
Sindromul X fragil
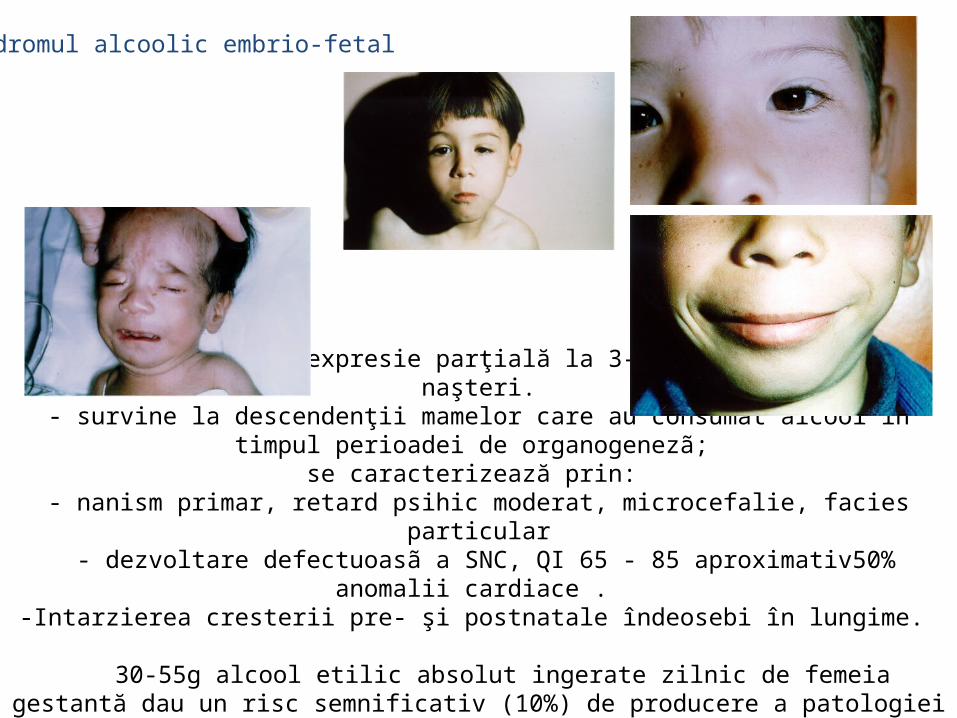
- frecvenţă de 2‰ expresie parţială la 3-5 născuţi vii/1000 naşteri.- survine la descendenţii mamelor care au consumat alcool în timpul perioadei de organogenezã;
se caracterizează prin: - nanism primar, retard psihic moderat, microcefalie, facies particular
- dezvoltare defectuoasã a SNC, QI 65 - 85 aproximativ50% anomalii cardiace . -Intarzierea cresterii pre- şi postnatale îndeosebi în lungime.
30-55g alcool etilic absolut ingerate zilnic de femeia gestantă dau un risc semnificativ (10%) de producere a patologiei fetale. Pe loturi de femei gravide s-a demonstrat că, la cantităţi
mai mari, riscul creşte la 20%.
Sindromul alcoolic embrio-fetal

HIV congenital
Asoect tipic de retard în dezvoltarea feţei: -microcefalie, -hipoplazie nazalã,-urechi jos inserate, -hipertelorism, -aplatizarea bazei piramidei nazale,-nas în şa.

Retard în dezvoltarea staturo-ponderalã :
-Hipotrofie -Sclere de culoare bleu /prin lipsa de oxigen, -Bose frontale / prin deficit vitamina D, calciu.

Morfogeneza dinţilor
• Dezvoltarea dintilor este iniţiată de interacţiunea dintre celule epiteliului cavităţii bucale primitive şi mezechimul subiacent colonizat de celule migrate din crestele neurale. Originea embrionară a dinţilor este dublă:
– emailul provine din epiteliul gurii primitive, de origine ectodermală;– dentina, pulpa şi cementul au origine mezenchimală, iau naştere din mezodermul subiacent.
• Prima schiţă în formarea dinţilor apare ca lamă dentară, în săptămâna a 7-a de gestaţie, prin condensarea stratului bazal al epiteliului ce tapetează cavitatea orală, între schiţa buzei şi a gingiei.
• Lama dentară formează naştere, de fiecare parte 10 proeminenţe ovoidale denumite muguri dentari, ce constituie primordiile componentei ectodermale a dinţilor.
• Mugurii dentari se cavitează,i iau aspect de cupă dentară constituită din:– strat extern, epiteliul dentar extern, ce acoperă cupa – strat intern, epiteliul dentar intern. – reticulul stelat. Situat deasupra epiteliului dentar extern
• Mezenchimul din interiorul cupei dentare va forma papila dentară, din care iau naştere dentina şi pulpa dentară.
• Cupa dentară se adânceşte devenind clopot dentar. • În primele 2 – 11 luni de viaţă după naştere coroana dinţilor de lapte nu este complet
formată, iar rădăcina abia începe să se dezvolte.

• Evoluţia dentiţiei deciduale
Dinte decidual Iniţierea calcificării coroanei
Erupţia Calcificarea rãdãcinii
Atriţie
Incisiv Central Inferior
Luna a 4-a de gestaţie 6-8 luni 6 luni 7 ani
Incisiv Central Superior
Luna a 4-a de gestaţie 7-9 luni 7 luni 7 ani
Incisiv Lateral Inferior
Luna a 4-a – a 5-a de gestaţie
7-10luni la 9 luni 8 ani
Incisiv Lateral Superior
Luna a 4-a – a 5-a de gestaţie
8-10luni la 9 luni 8 ani
Canini Luna a 5-a de gestaţie 1,5 ani 3 ani, 9 luni 12 ani
Molar I Luna a 5-a de gestaţie 2 ani 2 ani, 6 luni 10 ani
Molar II Luna a 6-a de gestaţie 2 ani 3 ani 11 ani

• Evoluţia dentiţiei definitive
Dinte definitiv Începerea calcificării coroanei
Terminarea calcificãrii coroanei
Erupţia Terminarea calcificãrii rãdãcinii
Incisiv Central 3-4 luni 4 – 5 ani 6 – 8 ani 9 - 10 ani
Incisiv Lateral 3-12 luni 4 – 5 ani 7 – 9 ani 10 – 11 ani
Canin 4-5 luni 6 – 7 ani 11 - 13 ani 12 – 15 ani
Premolar I 1 ½ – 2 ani 5 – 6 ani 12 – 13ani 12 – 13 ani
Premolar II 2 – 2 ½ ani 6 – 7 ani 11 – 13ani 12 – 14 ani
Molar I La naştere 2 ½ - 3 ani 6 – 7 ani 9 – 10 ani
Molar II 2 ½ - 3 ani 7 – 8 ani 12 – 14ani 14 – 16 ani
Molar III 7 – 10 ani 12 – 16 ani 18 ani 18 – 25 ani

Anomalii şi malformaţii congenitale ale dinţilor
• Dentinogeneza imperfectã (odontogeneza imperfectă) • Administrarea tetraciclinei în perioada de formare şi
creştere a dinţilor sau în timpul primelor luni de gestaţie poate determina anomalii dentare ale dentiţiei definitive şi/sau deciduale, cu depunerea tetraciclinei în dentină, la nivelul liniei de creştere (Owen).
• -Amelogeneza imperfectã hipocalcefiantã ereditarã • -Displazia cleido-cranianã ereditară (disostoza cleido-
cranio-digitalã Jansen) se caracterizează prin absenţa incisivilor laterali superiori permanenţi, discranie, agenezie clavicularã şi uneori anomalii scheletice; se transmite autozomal dominant.

•Dislpazia condroectodermală (sindromul Ellis van Creveld) asociază:
–absenţa dinţilor în porţiunea inferioară a mandibulei şi multiple malformaţii şi anomalii dentare de formă şi mărime.–nanismul datorat condrodistrofiei,–anomalii ale membrelor
Displazia ectodermală hipohidrotică Anodonţie

• Anomaliile numerice ale dinţilor– în plus (hiperdonţie), – în minus (hipodonţie), până la lipsa tuturor
dinţilor (anodonţie) în varianta parţială sau totală.
• Modificãrile de direcţie : – anteversie, – posteroversie, – lateroversie.
• Modificãrile de sediu – Transpoziţia constã în inversiunea sediului
unor dinţi, mai frecvent caninul în locul premolarului
– Heterotopia constã în erupţia dinţilor în altã zonã decât cea a crestei alveolare, cea mai frecventã fiind la nivelul palatului osos.
• Anomaliile de formã şi volum – dinţi mici (microdentism), – dinţi mari (macrodentism), coroanã dublã,
rãdãcini multiple, rãdãcini fuzionate, dinţi torsionaţi.
– Dinţii Hutchinson apar în sifilisul congenital tardiv;, microdonţia incisivilor
• -Anomaliile de erupţie
Malformaţii congenitale ale dentiţiei definitive: anodonţie
şi hipodonţie.

Microdonţia incisivului lateral şi a caninului, lipsa molarilor.

Sistemul branhial

Sistemul branhial

Arculbranhial
Pãrţile moi derivate Pãrţile dure derivate Nervul arculuibranhial
I mandibular
Muşchii masticatori, m.milohioidian, pânteculanterior alm. digastric, m.tensor al palatului, m.tensor timpani
Cartilajul Meckel, mandibula,ligamentul sfeno-mandibular,ciocanul (maleus), nicovala(incus), lig. anterior al ciocanului
V trigemen

Arculbranhial
Pãrţile moi derivate Pãrţile dure derivate Nervul arcului branhial
II hioidian
Muşchii mimicii(buccinator, auricular,frontal, platysma,orbicularul ochiului şigurii), pântecul posterior alm. digastric, m.stilohioidian, m. scăriţei
ScăriţaProcesele stiloideLigamentul stilohioidCornul mic şi porţiuneasuperioară a corpului osuluihioid
VII facial
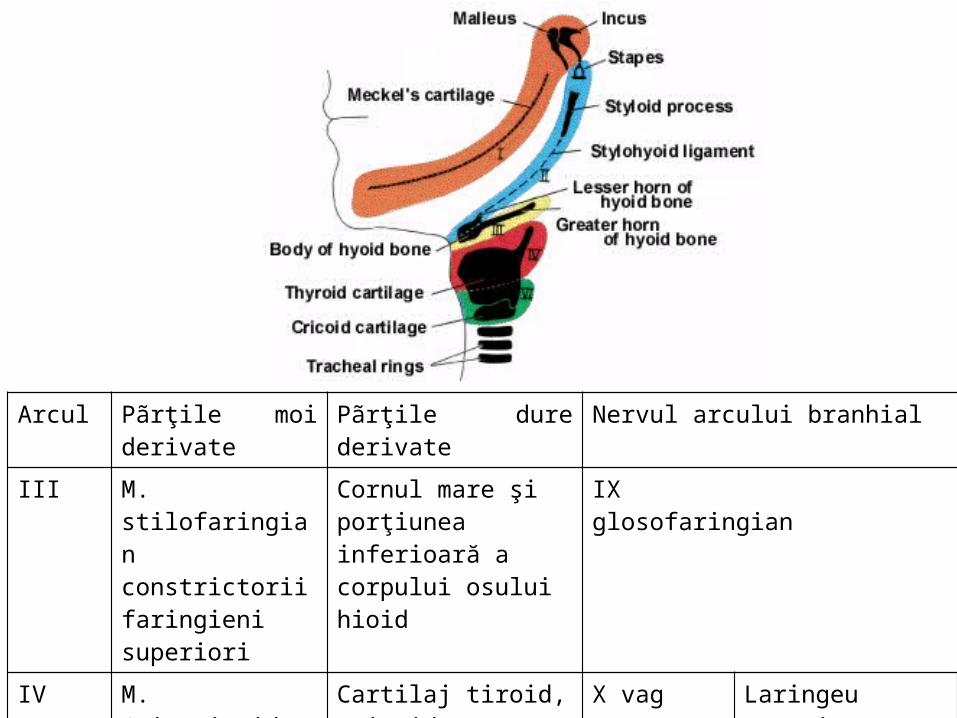
Arcul Pãrţile moi derivate Pãrţile dure derivate Nervul arcului branhial
III M. stilofaringianconstrictorii faringieni superiori
Cornul mare şi porţiunea inferioară a corpului osului hioid
IX glosofaringian
IV
VI
M. CricotiroidM. Ridicător palatinM. constrictori ai faringelui
Cartilaj tiroid, cricoid, aritenoid, corniculat, cuneiform
X vag
X vag
Laringeu superior
M. intrinseci ai laringelui
Laringeu recurent
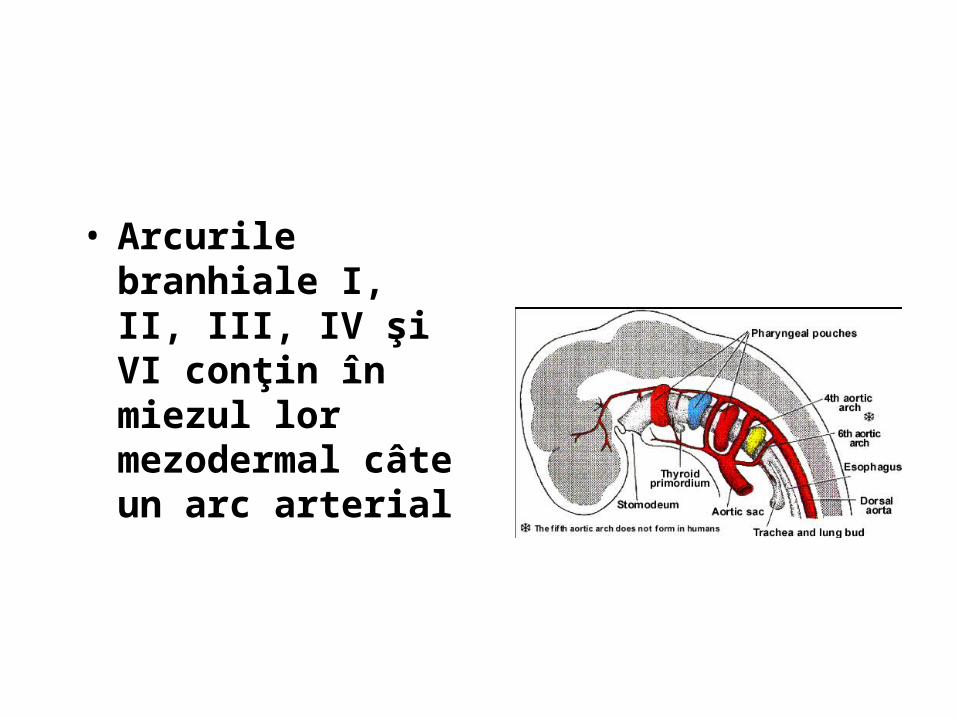
• Arcurile branhiale I, II, III, IV şi VI conţin în miezul lor mezodermal câte un arc arterial

Derivatele arcurilor aortice
Arcul I: artera maxialarã; arc II: a. hioidianã;
arc III: a. carotidã internã; arcIV: drept: a. suvclavie dreaptã;
arc IV stâng: crosa aortei; arc VI drept: a. pulmonarã dreaptã;
arc VI stâng: a. pulmonarã stângã şi canalul arterial Botalo.
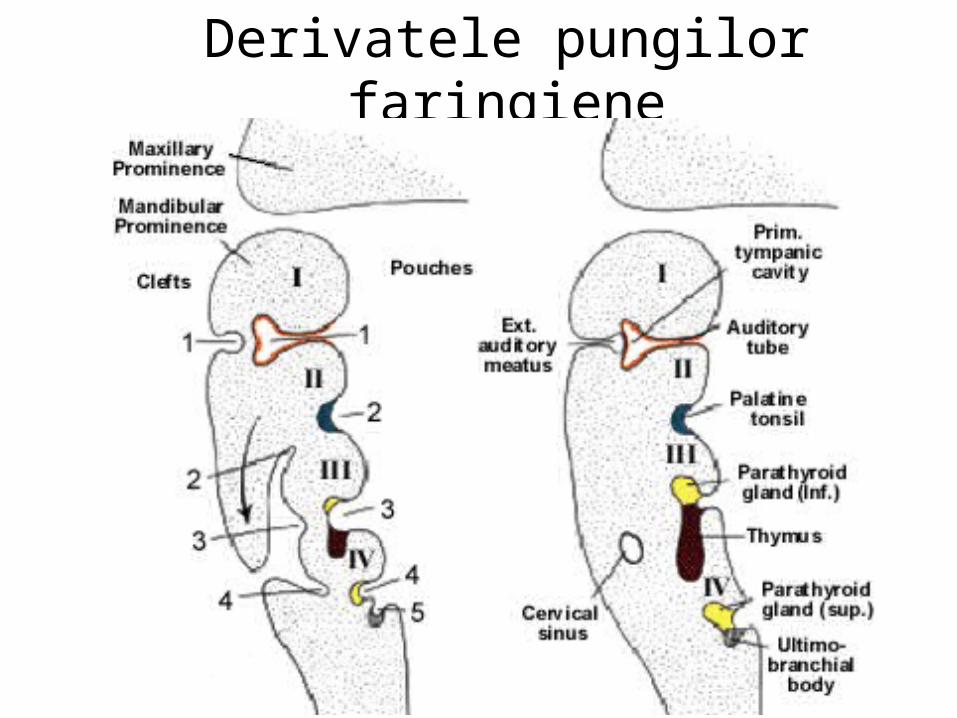
Derivatele pungilor faringiene

Ontogeneza urechii medii• Primul şanţ branhial se
adânceşte din luna a 2-a pînă în luna a 4-a ca un canal (canalul tubo timpanic Koelliker);
• extremitatea sa internă va comunica cu prima pungă faringiană;
• Orificiul de comunicare va deveni ostiul faringian al tubei auditive Eustachio.

Compartimentul timpanomastoidian
•3 săpt. – recesul tubotimpanic (1-a pungă faringiană - endoderm) •6 săpt. – mezenchimul osicular separă şanţul extern de punga faringiană.•20 săpt. – cavitatea timpanică se dezvoltă şi înglobează oscioarele•22 săpt. – se formează antrumul mastoidian•33 săpt. – pneumatizarea primară a antrumul mastoidian

• Extremitatea laterală a canalului începe să se lărgească în luna a 3-a, deplasează mezenchimul înconjurător şi înglobează o serie de condensări ale acestuia.
• Condensările iau naştere din:– cartilajul Meckel al primului arc branhial
(ciocanul), – cartilajul Reichter al celui de al 2 –lea arc
(nicovala)– dintr-un cartilaj propriu (scăriţa)
• Ele vor forma oscioarele urechii medii.
• Nucleul scăriţei se organizează în jurul arterei stapediene (din al doilea arc branhial) care ulterior dispare.

• A. Secţiune transversă printr-un embrion de 7 sapt. • La nivelul regiunii rombencefalice, se observă:
– recesul tubotimpanic dezvoltat din prima pungă faringiană,– condensarea mezenchimală, din care se vor dezvolta oscioarele urechii medii.
• B. la nivelul urechii medii se observă precursorii cartiloginoşi ai oscioarelor auditive. Linia galbenă de la nivelul mezenchimului indică viitoare expansiune a cavităţii timpanice primitive tapetata de endodermul dezvoltat din prima pungă faringiană

• Urechea medie si tuba auditiva derivă din punga faringiana 1; sunt tapetate de epiteliu de origine endodermală din punga 1 faringiana.
• Osicioarele care conduc şi amplifică sunetul de la nivelul membranei timpanice către fereastra ovală deriva din – arcul branhial 1 (ciocanul şi nicovala) – Arcul branhial 2 (scăriţa).

• Meatul auditiv extern se dezvoltă din şanţul branhial extern 1.• Membrana timpanică este alcatuită din epiteliul ectodermal al
santului branhial 1 extern, fîşii de ţesut mezenchimatos din arcul branhial 1, ţesut endodermal din epiteliul endodermal al santului branhial 1 intern.
• Pavilionul urechii se dezvoltă din ţesutul mezenchimal al arcurilor branhiale 1 si 2.
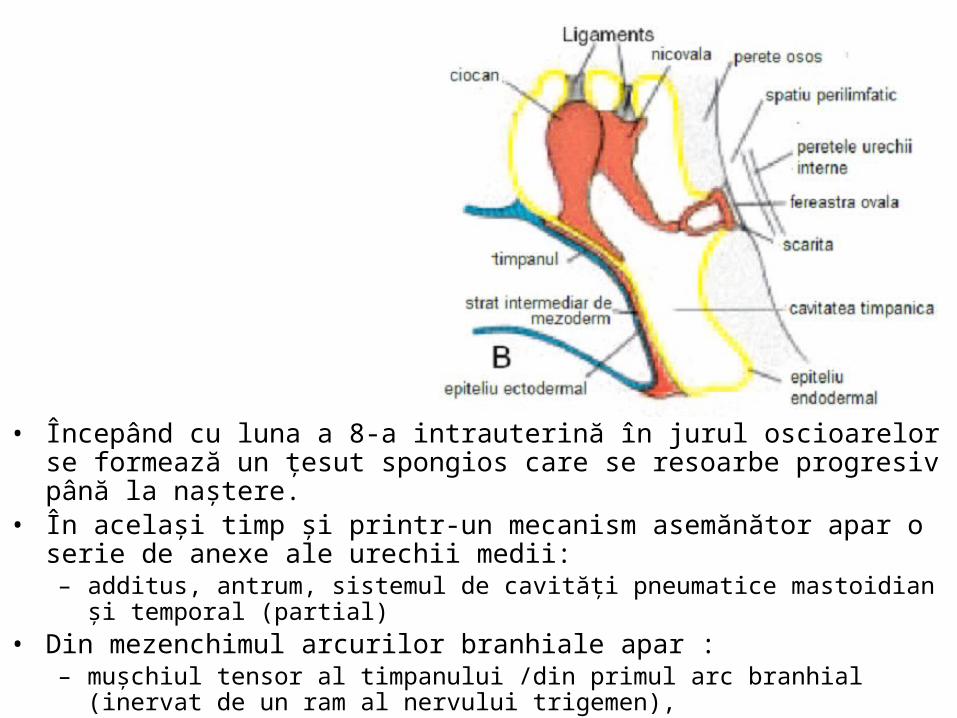
• Începând cu luna a 8-a intrauterină în jurul oscioarelor se formează un ţesut spongios care se resoarbe progresiv până la naştere.
• În acelaşi timp şi printr-un mecanism asemănător apar o serie de anexe ale urechii medii:– additus, antrum, sistemul de cavităţi pneumatice mastoidian şi temporal (partial)
• Din mezenchimul arcurilor branhiale apar : – muşchiul tensor al timpanului /din primul arc branhial (inervat de un ram al nervului
trigemen), – muşchiul scăriţei /din al doilea arc branhial (inervat de nervul facial).

fistule cervicale congenitale,
chisturi cervicalecongenitale
m. Sternocleidomastoidian
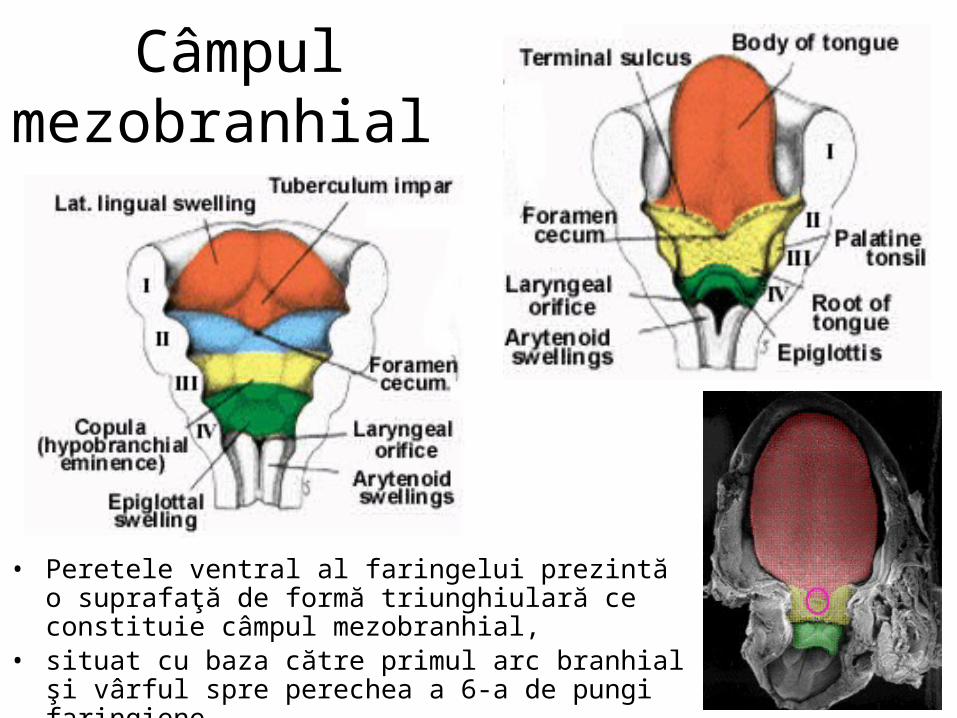
Câmpul mezobranhial
• Peretele ventral al faringelui prezintă o suprafaţă de formă triunghiulară ce constituie câmpul mezobranhial,
• situat cu baza către primul arc branhial şi vârful spre perechea a 6-a de pungi faringiene.
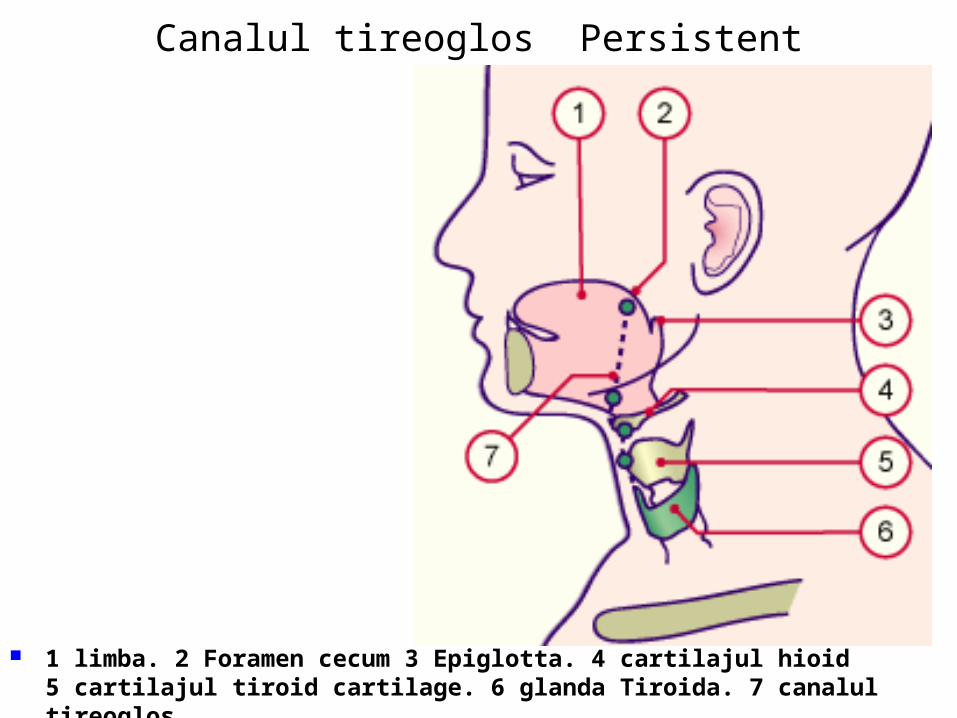
Canalul tireoglos Persistent
1 limba. 2 Foramen cecum 3 Epiglotta. 4 cartilajul hioid 5 cartilajul tiroid cartilage. 6 glanda Tiroida. 7 canalul tireoglos
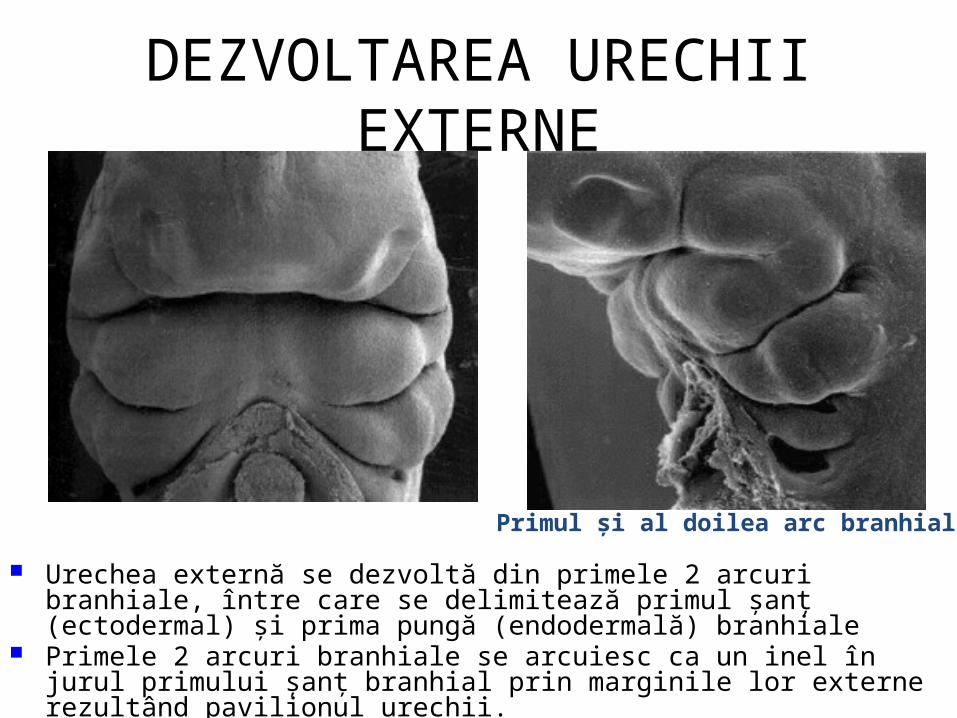
DEZVOLTAREA URECHII EXTERNE
Primul şi al doilea arc branhial
Urechea externă se dezvoltă din primele 2 arcuri branhiale, între care se delimitează primul şanţ (ectodermal) şi prima pungă (endodermală) branhiale
Primele 2 arcuri branhiale se arcuiesc ca un inel în jurul primului şanţ branhial prin marginile lor externe rezultând pavilionul urechii.

La sfârşitul săptămânii a 6-a embrionare apar în schiţa pavilionului nodului mezenchimali condensaţi – corniculii His – care în luna a 3-a se condrifică.
Pe primul arc apar mugurii tragusului, rădăcinii helixului şi helixul, iar pe arcul al doilea antihelixul, antitragusul şi lobulul.

• vedere laterală a capului unui embrion, • cei 6 muguri auriculari înconjură capătul dorsal a primului şanţ branhial• Se observă poziţia urechii în raport cu cavitatea bucală şi orbitele• Dezvoltarea mandibulei şi a regiunii cervicale plasează pavilionul într-o poziţie defivinitvă
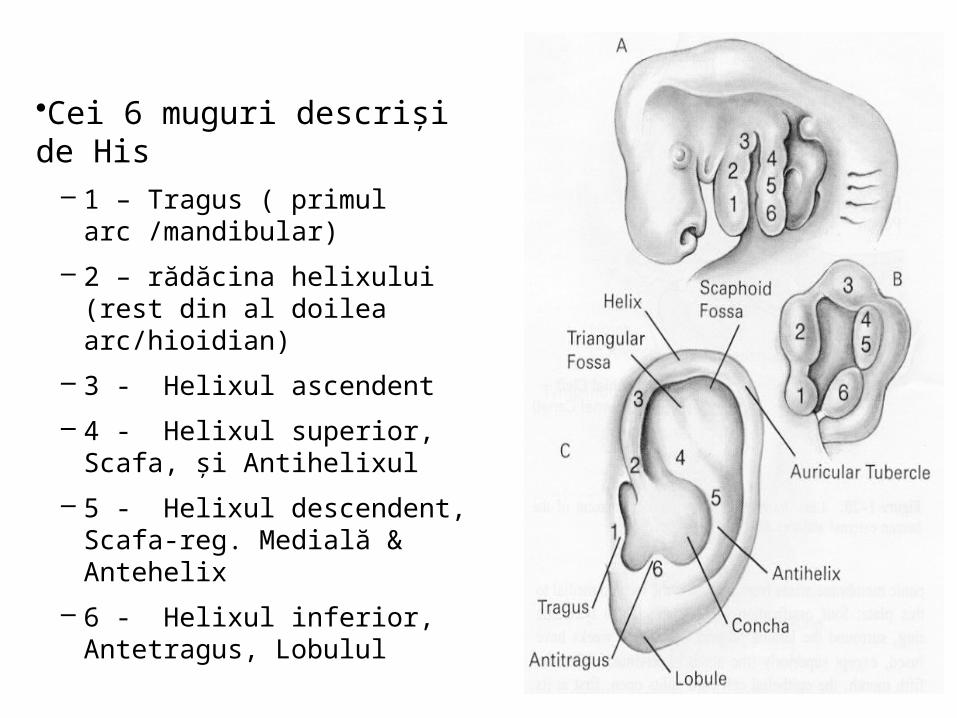
•Cei 6 muguri descrişi de His– 1 – Tragus ( primul arc
/mandibular)
– 2 – rădăcina helixului (rest din al doilea arc/hioidian)
– 3 - Helixul ascendent
– 4 - Helixul superior, Scafa, şi Antihelixul
– 5 - Helixul descendent, Scafa-reg. Medială & Antehelix
– 6 - Helixul inferior, Antetragus, Lobulul
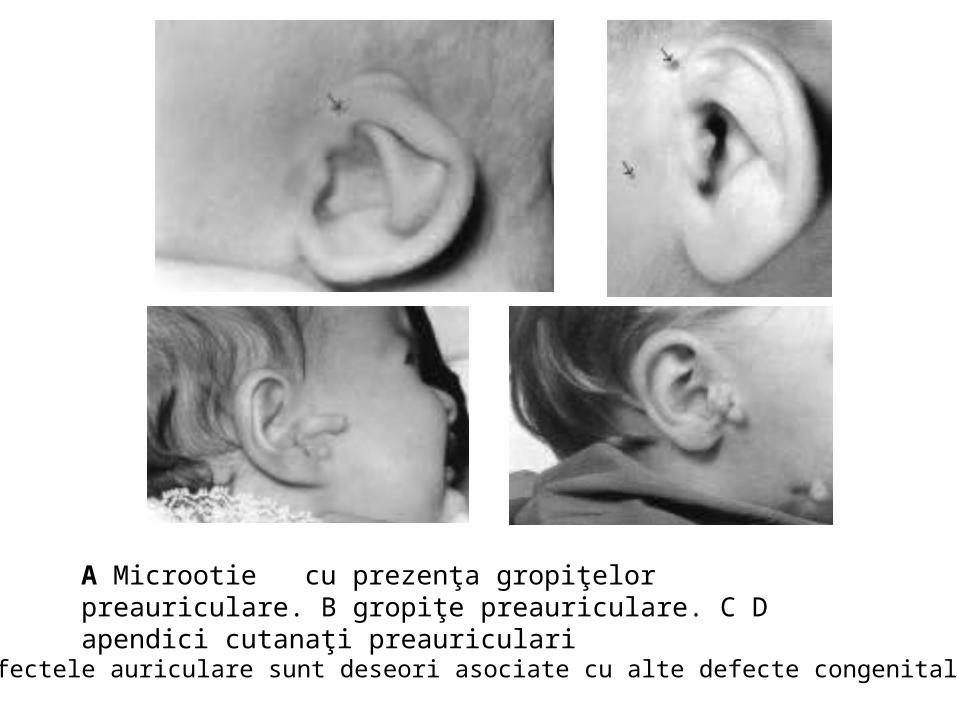
A Microotie cu prezenţa gropiţelor preauriculare. B gropiţe preauriculare. C D apendici cutanaţi preauriculari
Defectele auriculare sunt deseori asociate cu alte defecte congenitale.
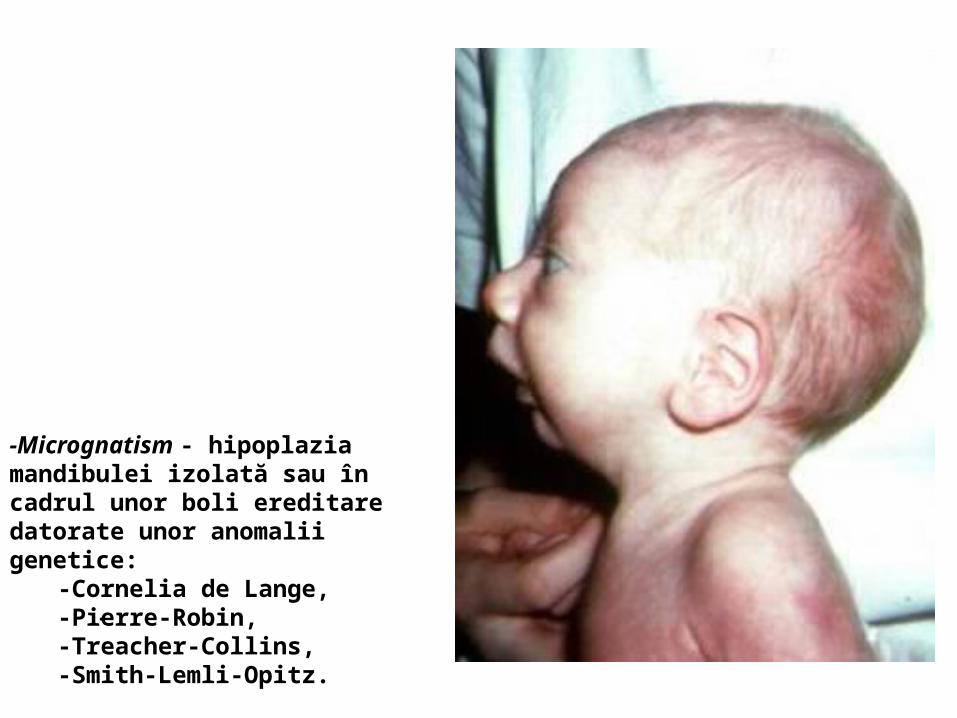
.
.
-Micrognatism - hipoplazia mandibulei izolată sau în cadrul unor boli ereditare datorate unor anomalii genetice:
-Cornelia de Lange, -Pierre-Robin, -Treacher-Collins, -Smith-Lemli-Opitz.
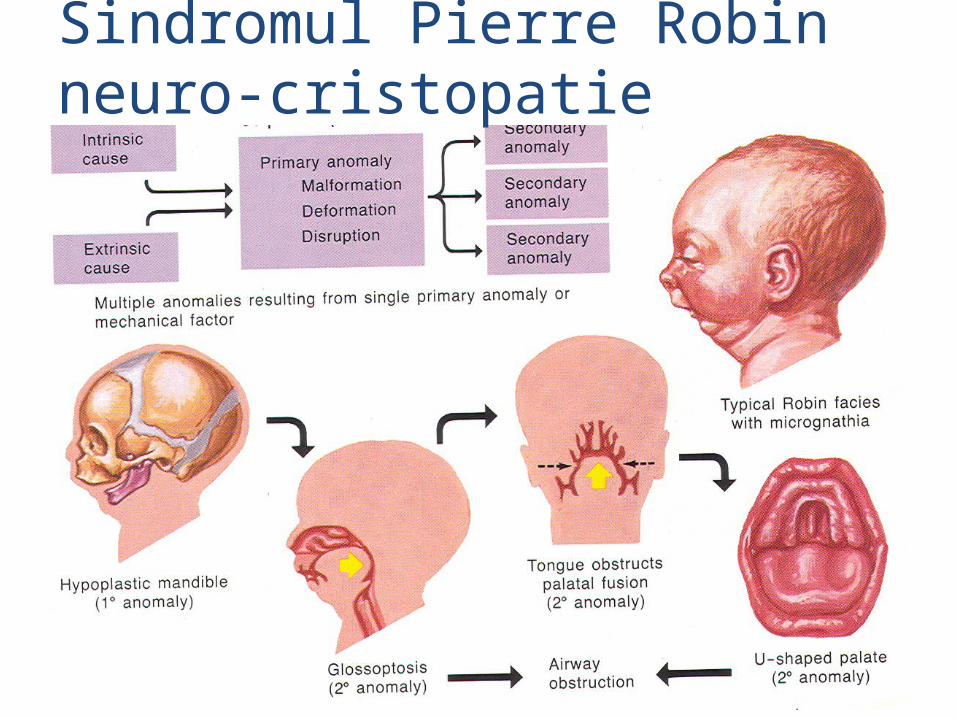
Sindromul Pierre Robin neuro-cristopatie

• Sindrom Pierre Robin • -hipoplazie mandibulară
(micrognaţie severă), • -glosoptoză• -palatoschizis.• Incidenţă: 2% dintre nou-
născuţii vii.
Sindrom Pierre Robin Neuro-cristopatie

Complex malformativ dominat de asociarea: coloboma pleoapelor, anomalii ale dezvoltării urechii externe, micrognaţie şi hipoplazia regiunilor zigomaticomalare ale
craniului
Disostoza mandibulofaciala (Sindromul Franceschetti-Klein)
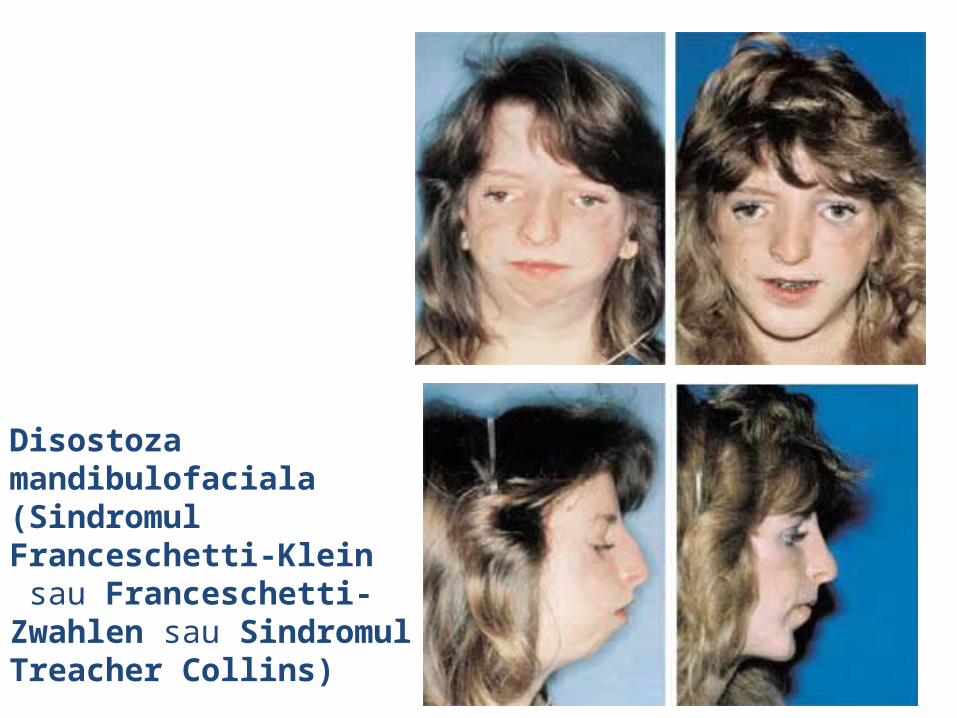
Disostoza mandibulofaciala (Sindromul Franceschetti-Klein sau Franceschetti-Zwahlen sau Sindromul Treacher Collins)

• Dr Angelo Di George, endocrinolog, Spitalul St-Christopher, Philadelphia:
• 1981 Dr De La Chapelle: transmis genetic• 1991, Dr Driscol - deletia 22q • Sindromul Di George:- Incidenta: 1 / 2000-4000 nou-născuţi vii- Intarzie migrarea neuronala din cresteleneurale
• 80% cardiopatii congenitale: • 47% anomalii ale arcului aortic, • 12% tetralogie Fallot• 16% anomalii palat – cavitate bucala • 28% Absenta timus, • 21% hipocalcemi prin Aplazia sau
deficienta glandelor paratiroide • 12% anomalii ale sistemului nervos
central. • Fenotipul poate fi foarte putin afectat la
unul dintre parinti si foarte sever la copilul de acelas sex cu parintele afectat.
Sindromul Di George sindromul velo-cardio-facial

• Diverticulul respirator - apare pe faţa ventrală a porţiunii cefalice a intestinului primitiv la embrionul stadiul 10 Carnegie (28 zile i.u.)
• Tubul respirator se formeaza din segmentul cefalic al intestinului primitiv. • Caile respiratorii inferioare se formeaza in mijlocul saptamanii a 4-a i.u. ca extensii
in jurul santului laringotraheal.
Embrion de 25 de zile. embrion de 5 saptamani

• diverticulul respirator se formează din intestinul primitiv cefalic (podeaua faringelui) • Alungire caudală • Separare de intestin prin apariţia a două pliuri longitudinale, eso-traheale, care vor fuziona
ulterior formand septul septul traheo-esofagian • Resorbţia septului traheo-esofagian în săptămâna a 5-a, cu separarea traheei de esofag• Esofagul devine situat dorsal iar traheea şi mugurii pulmonari ventral.• Primordiul respirator menţine comunicarea cu faringele prin orificiul laringeal

• Laringele se diferenţiază În săptămânile 6-7 i.u.
Portiunea ventrală a faringelui

• Mugurele pulmonar drept se divide în trei bronhii principale, cel stâng în două • În timpul dezvoltării, bronhiile principale se divid rapid, dihotomic; • până la finele lunii a 6-a i.u. apar 18 generaţii succesive • postnatal apar 6 generaţii; • arborele bronşic atinge forma finală la 7-8 ani după naştere.
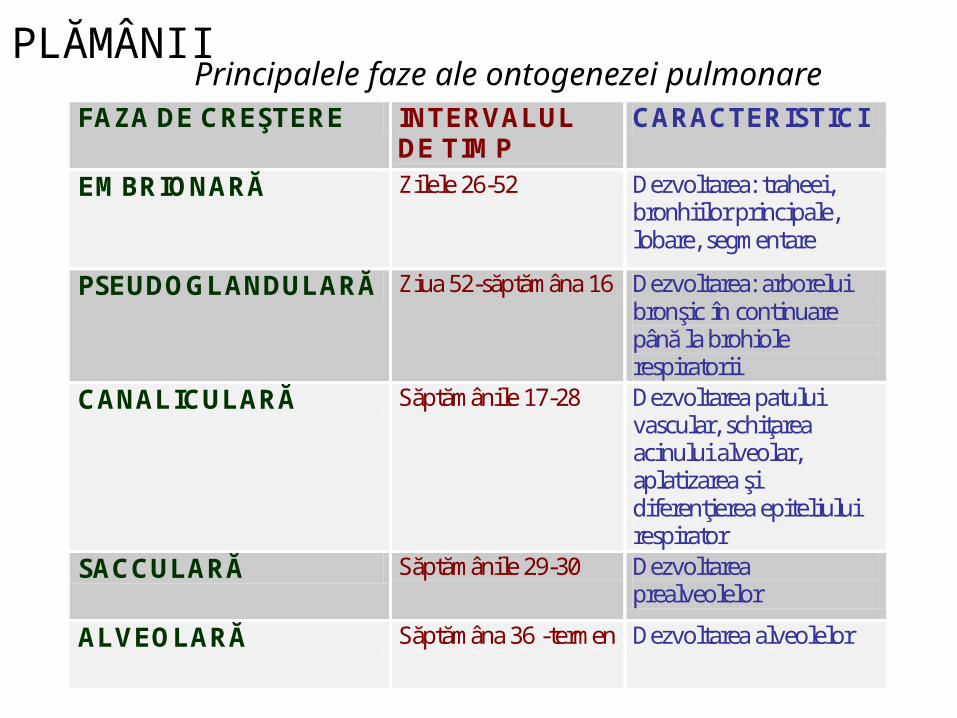
Principalele faze ale ontogenezei pulmonareFAZA DE CREŞTERE INTERVALUL
DE TIMP CARACTERISTICI
EMBRIONARĂ Zilele 26-52 Dezvoltarea: traheei, bronhiilor principale, lobare, segmentare
PSEUDOGLANDULARĂ Ziua 52-săptămâna 16 Dezvoltarea: arborelui bronşic în continuare până la brohiole respiratorii
CANALICULARĂ Săptămânile 17-28 Dezvoltarea patului vascular, schiţarea acinului alveolar, aplatizarea şi diferenţierea epiteliului respirator
SACCULARĂ Săptămânile 29-30 Dezvoltarea prealveolelor
ALVEOLARĂ Săptămâna 36 -termen Dezvoltarea alveolelor
PLĂMÂNII

• Primordiul pulmonar apare în săptămâna a 4-a i.u. ca diverticul endodermal laringo-traheal prezent la nivelul feţei ventrale a esofagului primitiv, situat într-o atmosferă mezenchimală
• se bifurcă la inferior in doi muguri bronho-pulmonari• Prin ramificare mugurii bronho-pulmonari cresc (în grosimea mezocardului dorsal)
lateral şi caudal pătrunzând în lumenul canalelor pleuro-peritoneale, unde formează mugurii pulmonari.
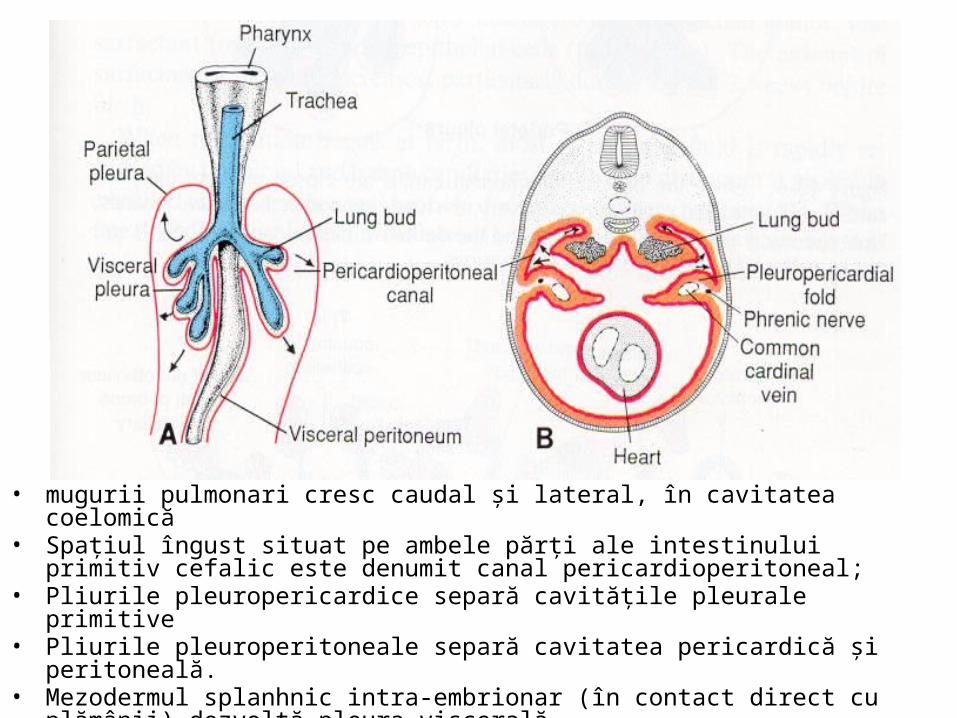
• mugurii pulmonari cresc caudal şi lateral, în cavitatea coelomică • Spaţiul îngust situat pe ambele părţi ale intestinului primitiv cefalic este denumit canal
pericardioperitoneal; • Pliurile pleuropericardice separă cavităţile pleurale primitive • Pliurile pleuroperitoneale separă cavitatea pericardică şi peritoneală. • Mezodermul splanhnic intra-embrionar (în contact direct cu plămânii) dezvoltă pleura
viscerală. • Mezodermul somatic intra-embrionar formează pleura parietală.
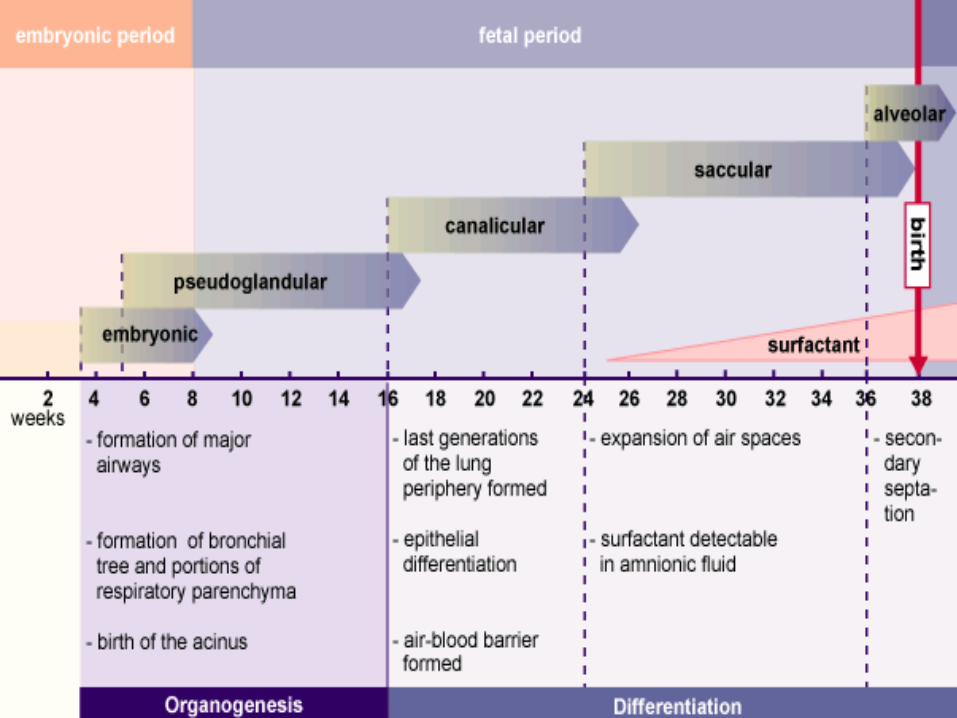
• Tesut pulmonar la nou-nascut.
STADIUL SACCULAR / SÃPTÃMÂNILE 29-36 I.U.• arborele bronşic schiţează acinul prin:
– formarea ductelor alveolare, – continuarea procesului de aplatizare şi diferenţiere a
epiteliului luminal.
• În timpul ultimele 2 luni prenatală şi în primii ani postnatal numărul de saci terminali creşte continuu. Celulele care căptuşesc sacii denumite celule epiteliale alveolare de tip I devin aplatizate aşa încât capilarele care le înconjură proemină în sacii alveolari. Se realizează astfel un contact intim cunoscut ca barieră
aer-sânge. • Diferenţierea celulelor alveolare de tip I este urmată
de apariţia celulelor alveolare de tip II care-şi păstrează aspectul cubic (granuloase) şi sunt responsabile de secreţia de surfactant (substanţă capabilă să scadă tensiunea superficială a aerului alveolar).
• După luna a 6-a i.u şi până la naştere se dezvoltă reţeaua limfatică pulmonară.

• Alveolele pulmonare mature nu sunt prezente înainte de naştere. • la naştere sunt prezente 17-20 milioane alveole = 1/6 din alveolele adultului• După naştere, în primii 10 ani, se formează 300 milioane alveole =5/6 din alveolele
adultului

Malformaţiile rezultate ca urmare a anomaliilor de dezvoltare ale aparatuluirespirator reprezintă 1,4% din totalul malformaţiilor pe aparate şi sisteme.

• Hernia diafragmatica congenitala • Malformatii congenitale ale Laringelui
– atrezia laringelui– atrezia partiala a laringelui:
– supralotic– glotic– subglotic
– stenoza subglotica– laringomalacia – chist glotic – laringocel – fistula laringo-traheo-esofagiana – despicatura laringo-traheo-esofagiana (laringe despicat, peristenta comunicarii eso-
traheale) – obstructii laringiene extrinseci congenitale – laringocel extern
• Anomalii Arteriale– arc aortic dublu – compresie arteriala
• Malformatii congenitale ale Traheei – traheomalacia
• Malformatii Congenitale ale Căilor pulmonare • Malformatii Congenitale ale Plamanilor

• Incidenţa 1:1200-1500 de nou-născuţi; 1:3000 nasteri • Apare
– Sporadic– Recurenta familiala
• Efect al hipoplaziei pulmonare• Asociaza hipertensiune pulmonara.• Urmare a lipsei de dezvoltare a portiunii posterolaterale a diafragmei (rezulta
persistenta foramen Bochdalek ) • 90% dintre cazuri este afectata partea stanga a difragmei. • Rata de supravietuire aproximativ 50%. • Diagnostic Antenatal ultrasunete. • Factori de risc crescut:
– polihidramnion – Trisomia 18 – Trisomia 21.
• Approximativ 40% din cazuri asociaza alte anomalii. • Rar diagnosticata incidental, la copilul mare.
Hernia diafragmatică congenitala

Hernia Diafragmatică Congenitală
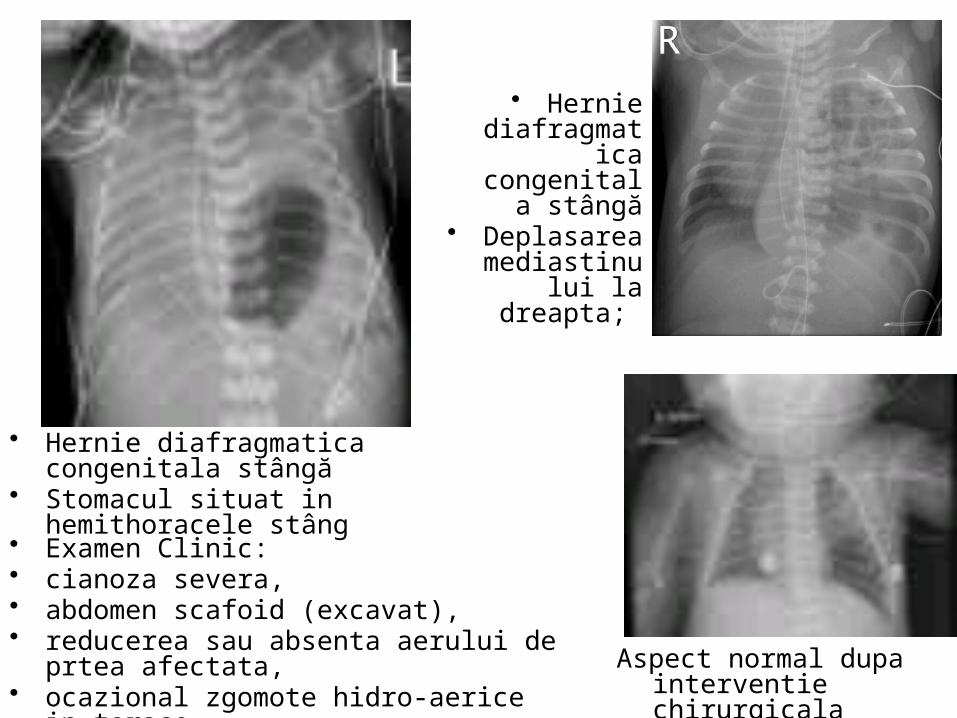
• Hernie diafragmatica
congenitala stângă
• Deplasarea mediastinului la
dreapta;
Aspect normal dupa interventie chirurgicala
• Examen Clinic: • cianoza severa, • abdomen scafoid (excavat), • reducerea sau absenta aerului de prtea
afectata,• ocazional zgomote hidro-aerice in torace.
• Hernie diafragmatica congenitala stângă• Stomacul situat in hemithoracele stâng
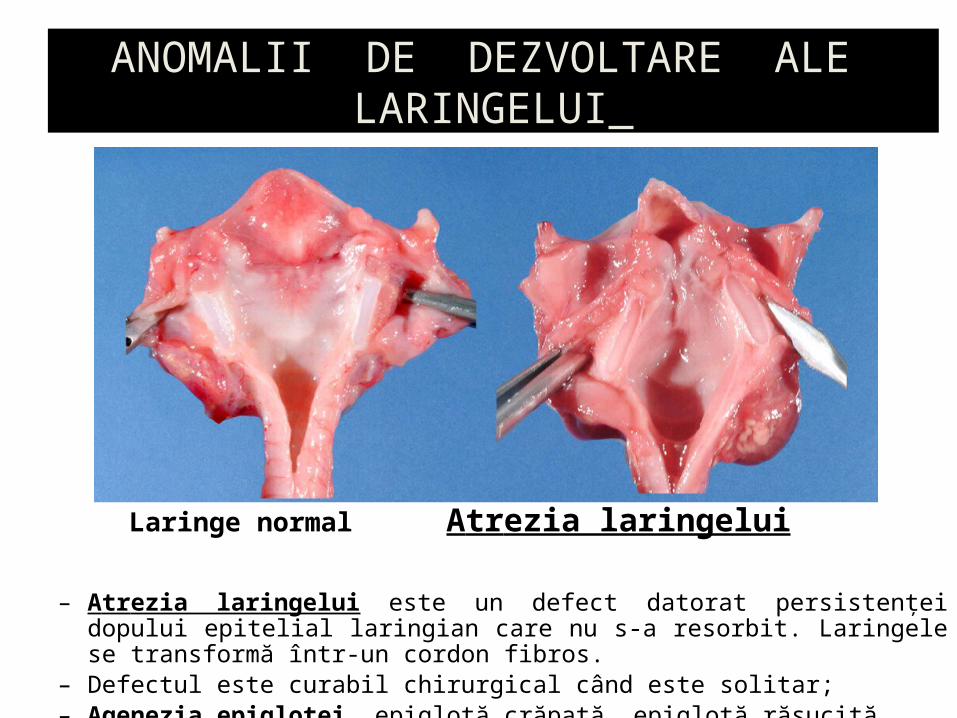
ANOMALII DE DEZVOLTARE ALE LARINGELUI
– Atrezia laringelui este un defect datorat persistenţei dopului epitelial laringian care nu s-a resorbit. Laringele se transformă într-un cordon fibros.
– Defectul este curabil chirurgical când este solitar;– Agenezia epiglotei, epiglotă crăpată, epiglotă răsucită
Laringe normal Atrezia laringelui
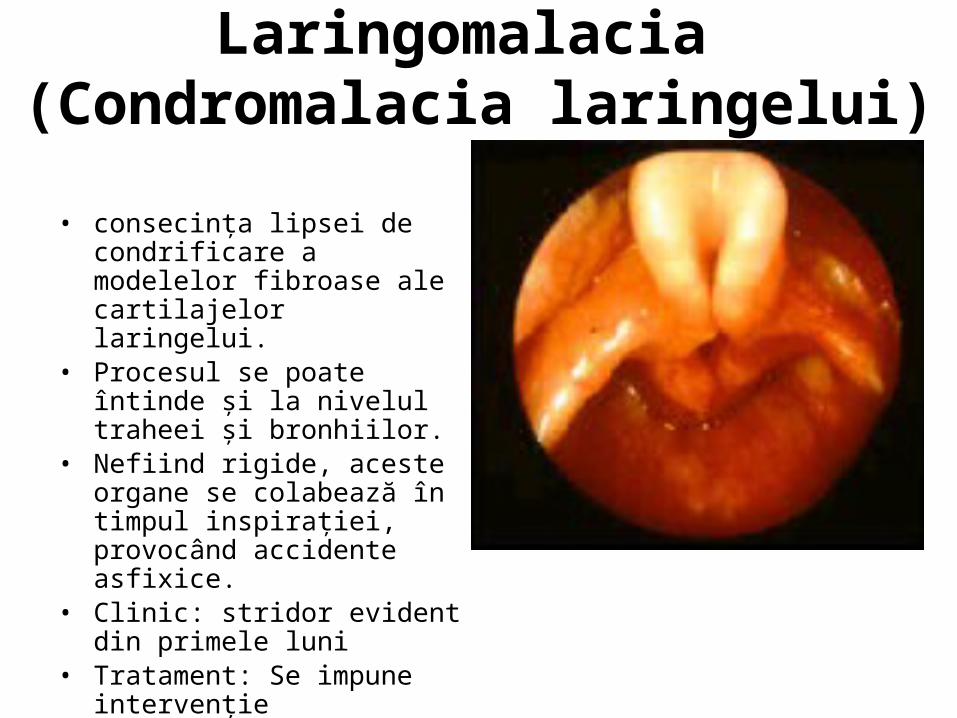
Laringomalacia (Condromalacia laringelui)
• consecinţa lipsei de condrificare a modelelor fibroase ale cartilajelor laringelui.
• Procesul se poate întinde şi la nivelul traheei şi bronhiilor.
• Nefiind rigide, aceste organe se colabează în timpul inspiraţiei, provocând accidente asfixice.
• Clinic: stridor evident din primele luni
• Tratament: Se impune intervenţie chirurgicală intre 18 luni – 2 ani
• Asocieri: disfagie, hipotrofie

• Laringomalacia
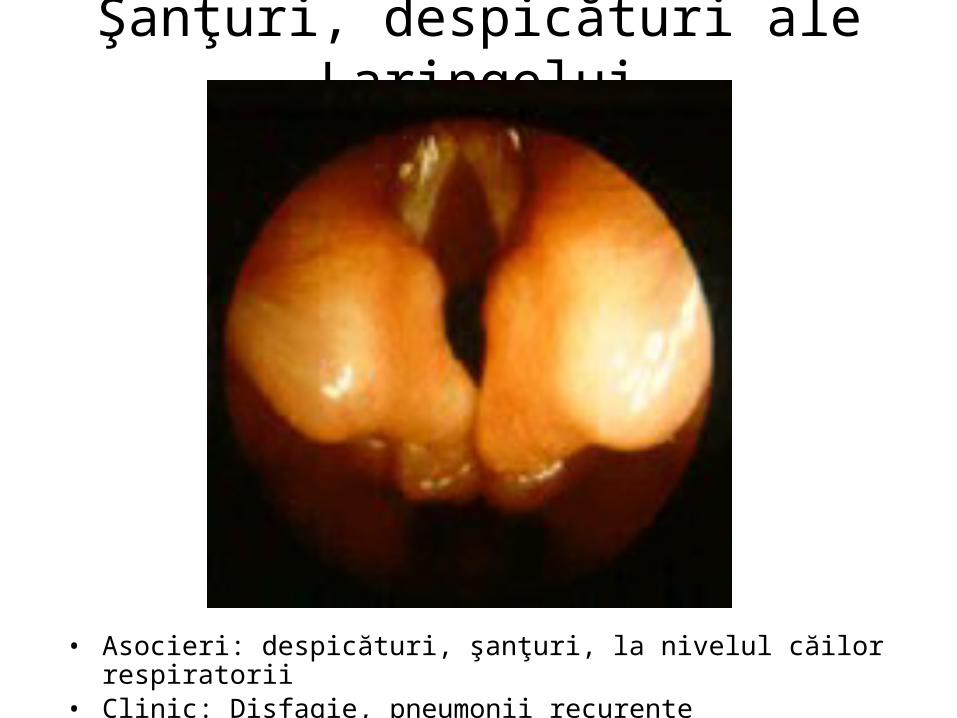
Şanţuri, despicături ale Laringelui
• Asocieri: despicături, şanţuri, la nivelul căilor respiratorii• Clinic: Disfagie, pneumonii recurente
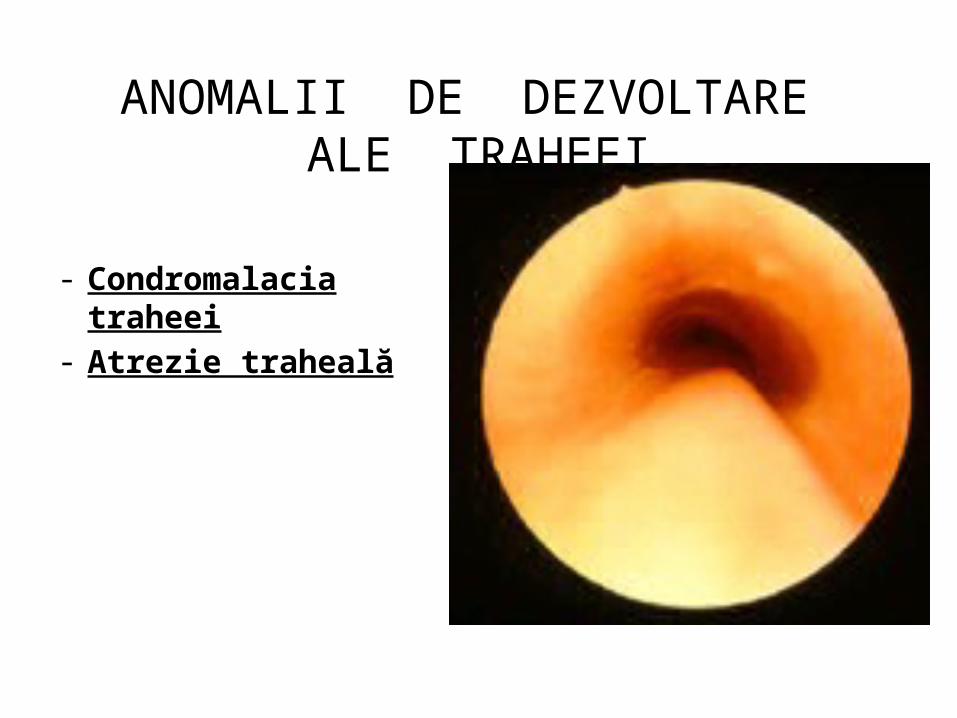
ANOMALII DE DEZVOLTARE ALE TRAHEEI
- Condromalacia traheei- Atrezie traheală

– Fistula eso-traheală, – Prin separare incompletă a traheei de intestin– Asocieri:– fisura mediană posterioară a cartilajului cricoid, – esofag atrezic şi divizat transversal,– traheea se deschide în segmentul esofagian
inferior, – segmentul esofagian superior se termină în fund
de sac.

– Fistula eso-traheală, – Prin separare incompletă a
traheei de intestin– Asocieri:– fisura mediană posterioară a
cartilajului cricoid, – esofag atrezic şi divizat
transversal,– traheea se deschide în
segmentul esofagian inferior, – segmentul esofagian superior
se termină în fund de sac.
AtreziaEsofaguluiasociata cuFistulaEsotraheală87%
AtrezieEsofagianaizolata8%
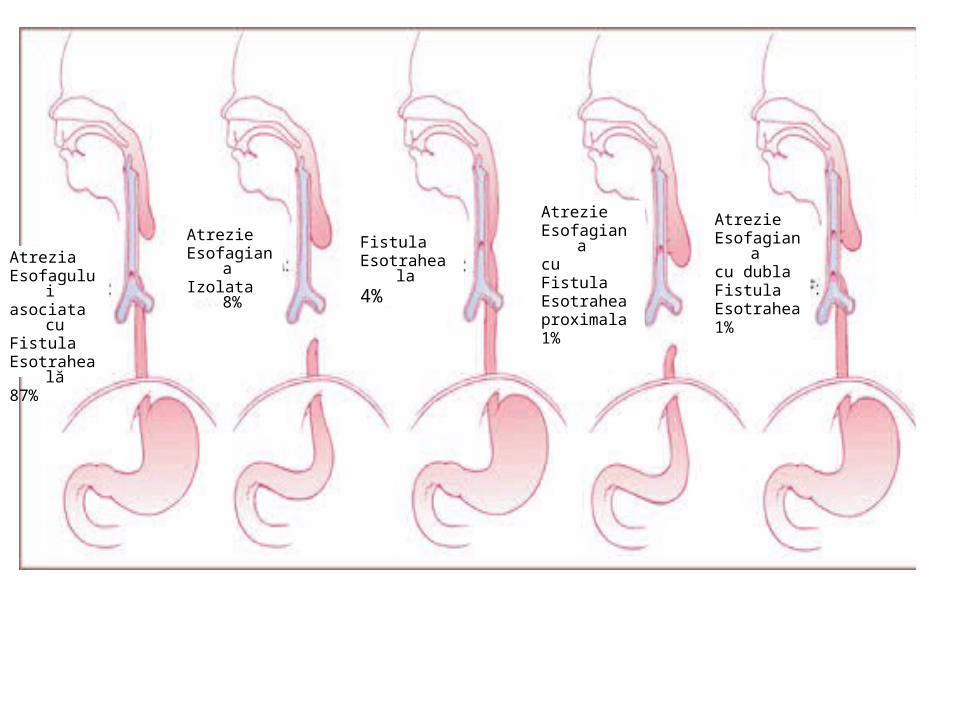
AtreziaEsofaguluiasociata cuFistulaEsotraheală87%
AtrezieEsofagianaIzolata 8%
Fistula Esotraheala4%
AtrezieEsofagianacu dubla Fistula Esotrahea1%
AtrezieEsofagianacuFistula Esotraheaproximala1%
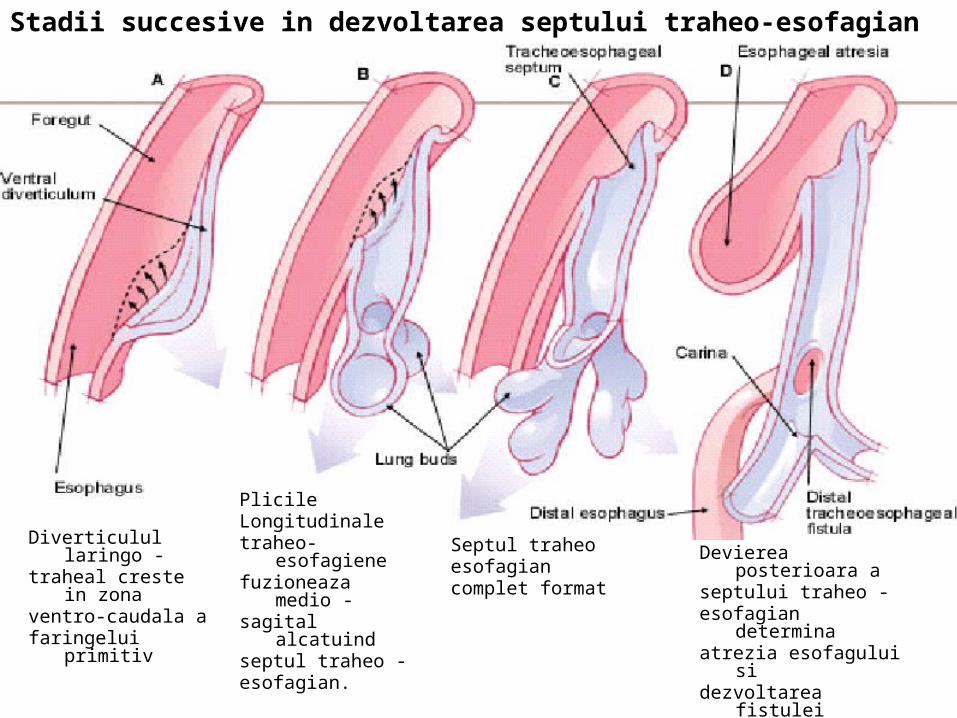
Diverticulul laringo -traheal creste in zonaventro-caudala afaringelui primitiv
Stadii succesive in dezvoltarea septului traheo-esofagian
PlicileLongitudinaletraheo-esofagienefuzioneaza medio - sagital alcatuindseptul traheo - esofagian.
Septul traheoesofagiancomplet format
Devierea posterioara aseptului traheo - esofagian determinaatrezia esofagului sidezvoltarea fistulei eso –t raheale.
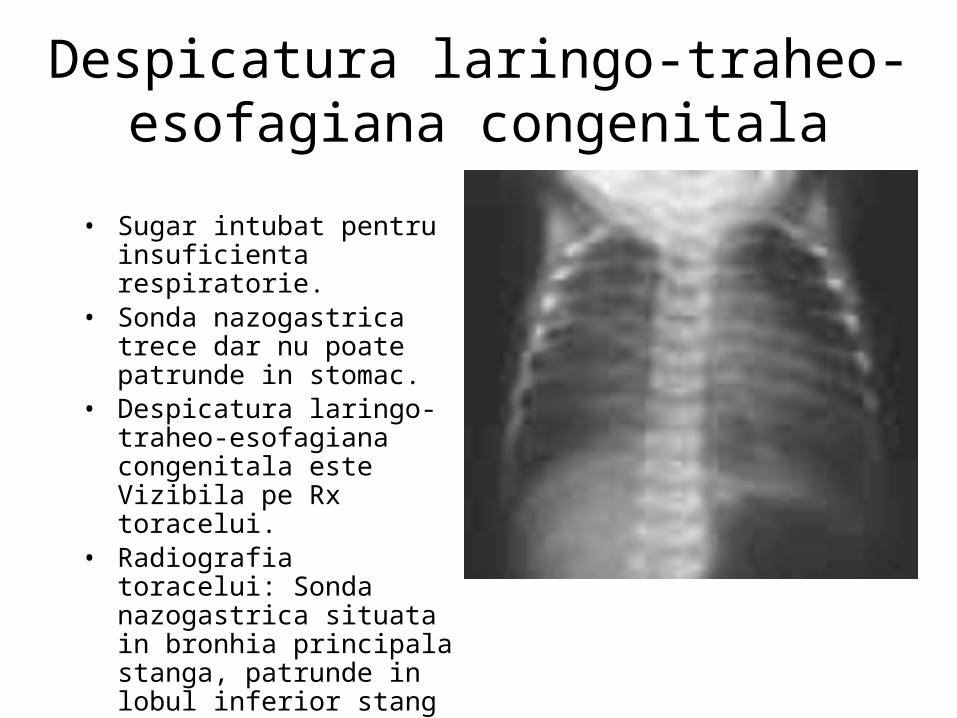
Despicatura laringo-traheo-esofagiana congenitala
• Sugar intubat pentru insuficienta respiratorie.
• Sonda nazogastrica trece dar nu poate patrunde in stomac.
• Despicatura laringo-traheo-esofagiana congenitala este Vizibila pe Rx toracelui.
• Radiografia toracelui: Sonda nazogastrica situata in bronhia principala stanga, patrunde in lobul inferior stang
• Laringoscopie: confirma prezenta unei despicaturi laringo-traheo-esofagiene extinse, laringe - carina.
ANOMALII ALE ARBORELUI BRONŞIC
• Îngustări: – agenezie, – stenoze, – atrezie
• Bronhomalacia reprezintă obstrucţia difuză a bronhiei prin anomalie a structurii inelelor cartilaginoase.
• Bronşiectazia congenitală este dilataţia anormală, permanentă a uneia sau mai multor bronhii însoţită de defecte ale peretelui bronşic.În etiologia ei se recunosc cauze infecţioase şi afecţiuni cu determinare genetică
• Traheobronhomegalia Este o afecţiune congenitală cu transmitere autosomal recesivă ce afectează ţesutul muscular şi elastic traheo- bronhic în care traheea poate egala calibrul coloanei vertebrale, iar bronhiile principale sunt mult dilatate.

ANOMALII IN DEZVOLTAREA PLĂMÂNILOR
– Anomaliile pulmonare pot interesa:-Plămânul în ansamblu: agenezie pulmonară, hipoplazie
pulmonară, modificarea numărului de lobi (fisuri anormale);-Doar arborele bronşic: stenoză congenitală intrinsecă (asociată
cu atelectazie), stenoză intrinsecă, bronhomalacie, bronşiectazie, chisturi bronhice;
-Relaţia morfo-funcţională arbore bronşic-artere pulmonare: sechestraţie bronhopulmonară;
-Vasele pulmonare: anomalii izolate ale arterelor pulmonare, anomalii arterio-venoase, anomalii ale venelor pulmonare, anomalii ale limfaticelor pulmonare.
-În lipsa stratului de surfactant are loc colaps alveolar în timpul expiraţiei(Maladia Membranelor Hialine)
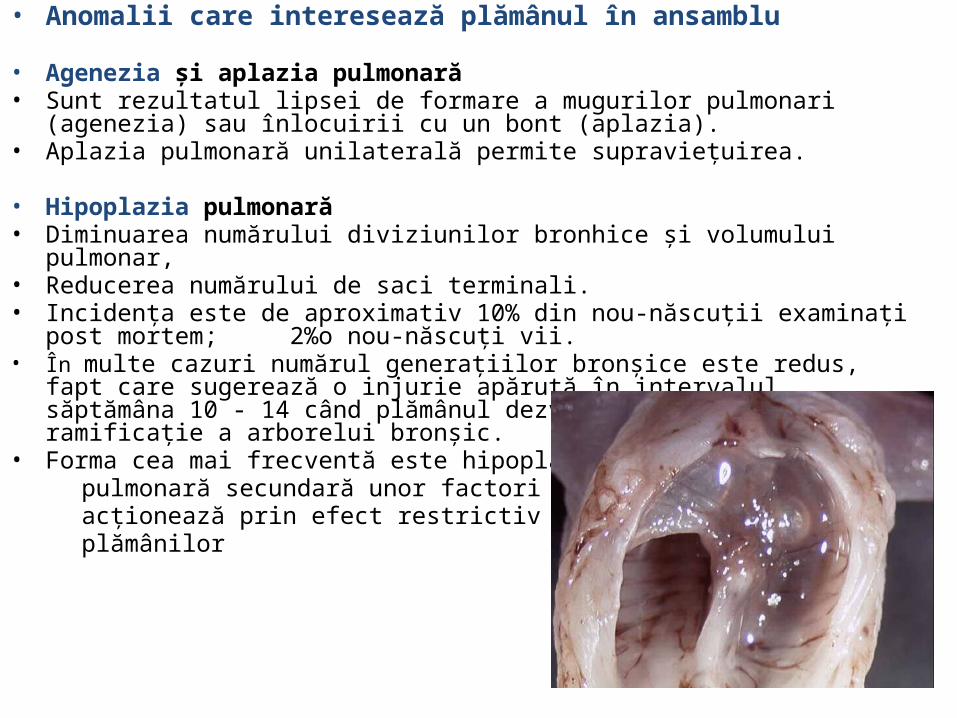
• Anomalii care interesează plămânul în ansamblu
• Agenezia şi aplazia pulmonară • Sunt rezultatul lipsei de formare a mugurilor pulmonari (agenezia) sau înlocuirii cu un
bont (aplazia). • Aplazia pulmonară unilaterală permite supravieţuirea.
• Hipoplazia pulmonară • Diminuarea numărului diviziunilor bronhice şi volumului pulmonar,• Reducerea numărului de saci terminali. • Incidenţa este de aproximativ 10% din nou-născuţii examinaţi post mortem; 2%o nou-
născuţi vii. • În multe cazuri numărul generaţiilor bronşice este redus, fapt care sugerează o injurie
apărută în intervalul săptămâna 10 - 14 când plămânul dezvoltă o puternică ramificaţie a arborelui bronşic.
• Forma cea mai frecventă este hipoplazia pulmonară secundară unor factori care acţionează prin efect restrictiv asupra plămânilor

• Hipoplazia pulmonară secundară insuficienţei cutiei toracice (omfalocel). • Hipoplazia pulmonară secundară prin supresia mişcărilor respiratorii. indusă prin
consumul matern de: – Etanol – Barbiturice, – Tutun

• Chisturi pulmonare congenitale unice / multiple, uni sau bilaterale;• Emfizemul lobar congenital
– prima afecţiune pulmonară congenitală în ordinea frecvenţei. – Constă în formarea uneia sau mai multor bule realizate:
• printr-o anomalie a peretelui bronşic (componentă cartilaginoasă) sau • printr-o obstrucţie determinată de falduri ale mucoasei bronşice (mecanism de
“supapă”).

Malformaţia chistică
• Epidemiologie: • 25% dintre malformaţiile
congenitale pulmonare.• incidenţă : 1/25000-35000
sarcini. • 4-26% in asocieri
plurimalformative
• incidenţă : 1/25 000-1/35 000 sarcini

Chist congenital adenomatoid malformativ pulmonar
• Anomalie pulmonara structurala,• Vizibila antenatal ca leziune chistica • Poate asocia hidrops fetal daca este destul de voluminos pentru a obstrua
intoarcerea venoasa catre inima. • Exista mai multe Tipuri, identificate pe criteriul componentelor leziunii:
– solid, (tipul lezional solid (prognostic mai grav decat tipul chistic)– microchistic, – macrochistic.
• Evolutie:“– regresii" ale leziunii in utero, cu Rx torace aspect normal la nastere – Insuficienta respiratorie severa la nastere, necesitatea interventiei chirurgicale de
urgenta– Poate asocia malignitati ulterioare, pe parcursul vietii
• Se recomanda – CT scaner la 3-6 luni dupa interventia chirurgicala– rezectia chirurgicala in perioada copilariei, chiar daca starea sugarului / copilului este
buna.

• a 2-a anomalie pulmonară congenitală în ordinea frecvenţei.
• Afectează, un lob sau un plămân în totalitate (volum mărit prin dezvoltare anormală a bronşiolelor terminale);
• Risc crescut pentru neoplasm pulmonar primitiv.
•Malformaţia adenomatoidă chistică •Chistul adenomatoid congenital
Chist adenomatoid congenital la un prematurDecubit Lateral

Abces Pulmonar Congenital
• Poate asocia malformaţii structurale: – sechestrarea lobului pulmonar, – chist pulmonar congenital
chist pulmonar congenital / pl. stg
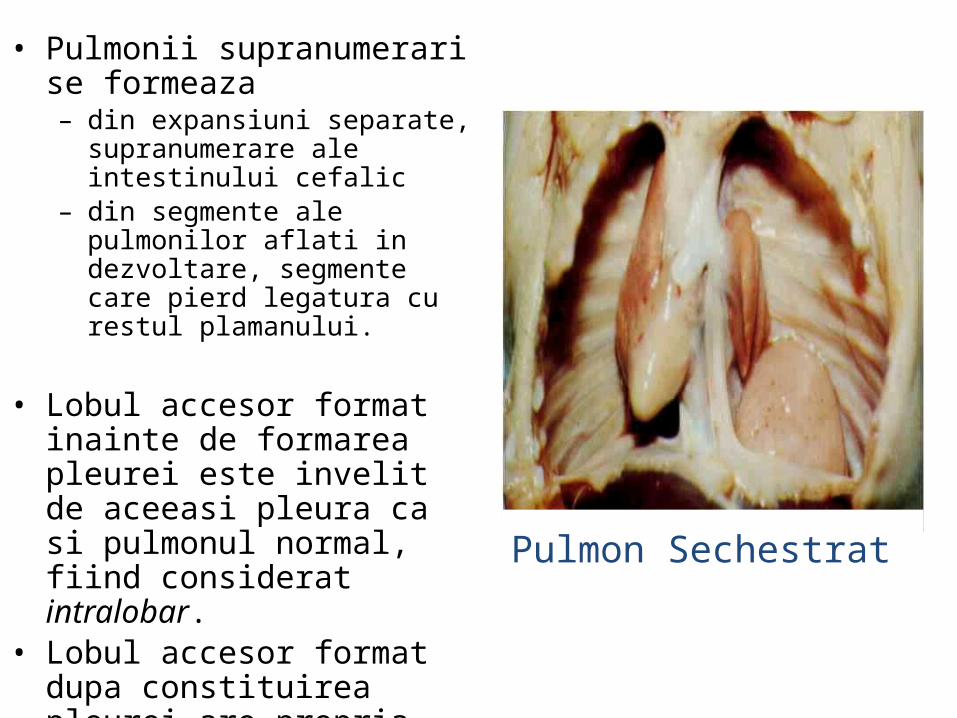
• Pulmonii supranumerari se formeaza– din expansiuni separate,
supranumerare ale intestinului cefalic
– din segmente ale pulmonilor aflati in dezvoltare, segmente care pierd legatura cu restul plamanului.
• Lobul accesor format inainte de formarea pleurei este invelit de aceeasi pleura ca si pulmonul normal, fiind considerat intralobar.
• Lobul accesor format dupa constituirea pleurei are propria pleura , fiind considerat extralobar.
Pulmon Sechestrat

Pulmon Sechestrat
• intralobar (pleura unică)
• extralobar (pleura proprie)
• Lob pulmonar sechestrat:

• Anomalii Asociate : 50-60% din cazuri– 1.Hernia Diafragmatica congenitala,– 2.Pectus excavatum congenital, – 3.Fistula traheo esofagiana congenitala,– Duplicatie esofagiana, chist esofagian,– Chist bronsic congenital, – Megacolon congenital – Malformatii cardiace congenitale.
• Diagnostic Diferential : – malformatii Chistice adenomatoidepulmonare,– teratom mediastinal,– hernia diafragmatica congenitala.
• Prognostic: • Rata ameliorata de supravietuire dupa
interventie chirurgicala• Mortalitate 100% in asocierea cu hidrops• Risc de recurenta: necunoscut, sporadic.• Management: terapie In utero• Esential: Diagnosticul antenatal si interventia
chirurgicala, pentru a preveni:– Hidropsul fetal– Hipoplazia pulmonara.
Pulmon Sechestrat

Anomalii vasculare – pot afecta funcţia
respiratorie prin compresie traheală
• persistenţa anormală a arcului aortic VI drept determină compresiune traheală.
• persistenţa anormală a arcului aortic VI (situat anterior traheei) asociată cu persistenţa anormală a canalului arterial Botalo (situat posterior esofagului) realizează un “inel vascular” constrictiv în jurul traheei şi esofagului

3 Malformaţii arteriovenoase / Plămânul drept. Tratament: embolizare.
Malformaţii arteriovenoase

Fibroza chisticã de pancreas (Mucoviscidozã)
• Transmitere ereditarã • Recesivã • 1/3300 populaţia infantilã (Caucazieni)• 1/15300 (Afroamericani)• sex ratio 1/1• gena controleazã producerea unei proteine care
regleazã transportul transmembranar al ionilor de Na şi Cl – întreruperea transportului transmembranar de
Na şi Cl, – deshidratare, – vâscozitate crescutã a secreţiilor.
• 3% dintre Caucazieni poartã 1 genã anormalã (purtãtori sãnãtoşi),
• 3/10 000 dintre Caucazieni moştenesc ambele gene anormale (bolnavi de fibrozã chisticã de pancreas),
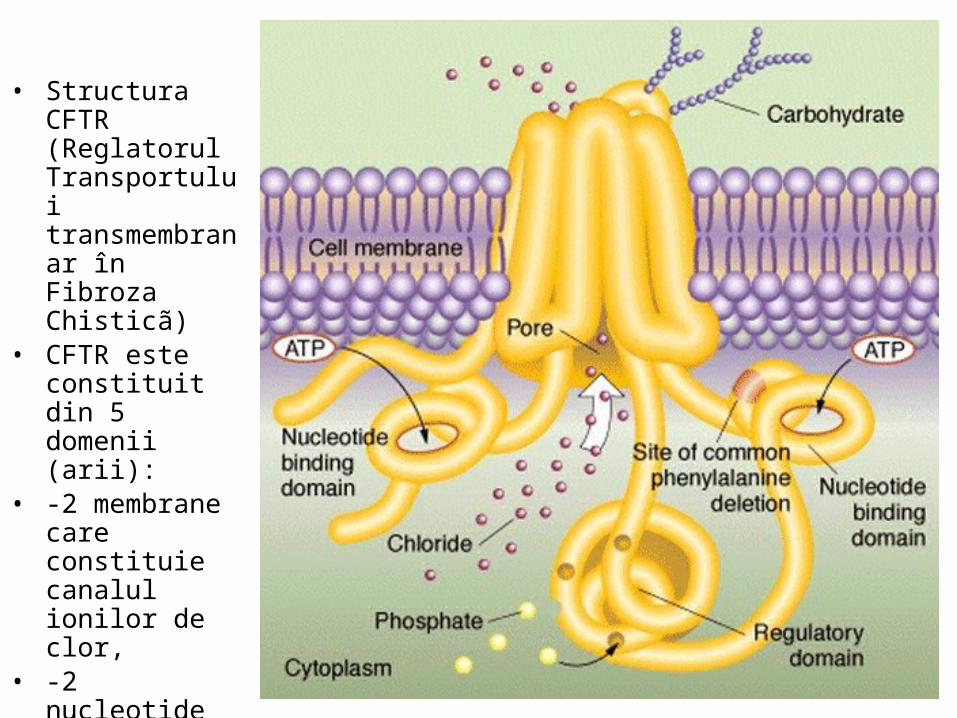
• Structura CFTR (Reglatorul Transportului transmembranar în Fibroza Chisticã)
• CFTR este constituit din 5 domenii (arii):
• -2 membrane care constituie canalul ionilor de clor,
• -2 nucleotide care atrag ATP şi-l hidrolizeazã,
• -1 reglator.
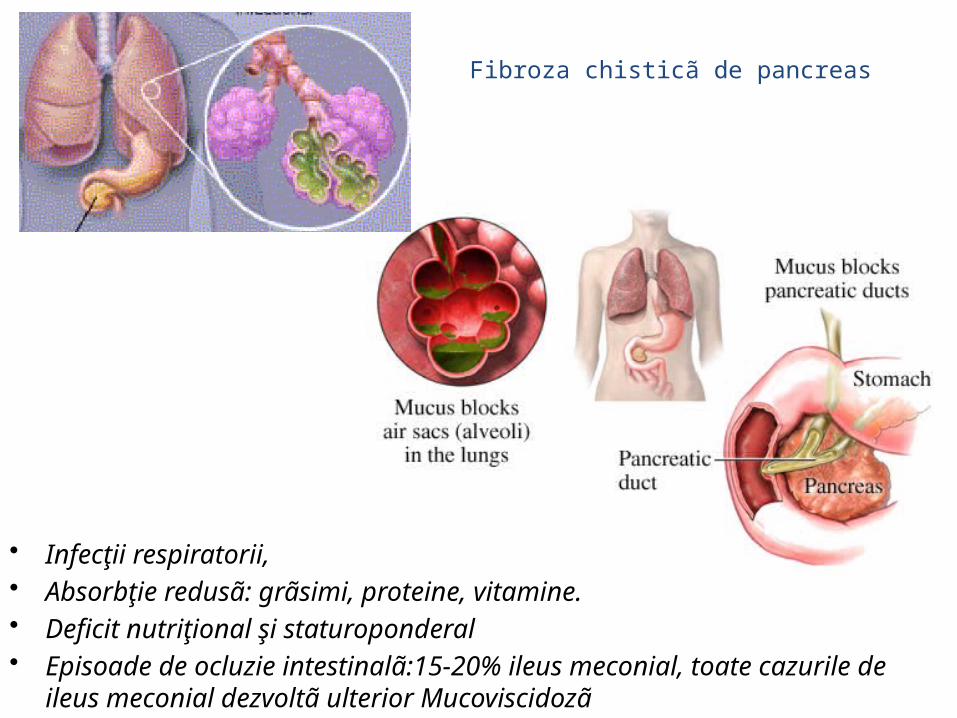
• Infecţii respiratorii,• Absorbţie redusã: grãsimi, proteine, vitamine.• Deficit nutriţional şi staturoponderal• Episoade de ocluzie intestinalã:15-20% ileus meconial, toate cazurile de ileus
meconial dezvoltã ulterior Mucoviscidozã
Fibroza chisticã de pancreas

Tutunul
Substanţa Acţiunea Efectul
Nicotinã contractã vaselearteriale şi venoaseombilicale; trece în laptelematern
Hipoxie,Hipotrofie
Monoxid de
Carbon
altereazã peretelea. ombilicale
Hipoxie
Altesubstanţe
împiedicã funcţiilemetabolice şi detransport aleplacentei
Hipotrofie
“o femeie care fumeazã 2 pachete ţigãri / zi scade fluxul sanguin placentar cu 41% în timpul cât fumeazã” (Christen 1998)
Fumatul în timpul sarcinii creşte riscul de:
• Avort spontan, • Naştere prematurã, • Anomalii placentare:
– Abruptio placenta, – Placenta previa

• Fumatul în timpul sarcinii creşte semnificativ riscul de: • Dismaturitate (12 % dintre nou-nãscuţii mamelor fumãtoare sunt dismaturi - 7.2%
/ mame nefumãtoare). • reducerea taliei şi greutãţii corporale, volumului plãmânilor, dimensiunilor capului,
– greutatea micã la naştere poate asocia: paralizie cerebralã, retard mental. – la 5 ani dupã naştere talia acestor copii este mai redusã comparativ cu copii mamelor
nefumãtoare• Despicãturi labio-palatine congenitale (risc de 1,5 – 2 ori mai mare pentru mamele
fumãtoare), • ”moarte subitã a nou-nãscutului” - risc de 2-6 ori mai mare pentru mamele
fumãtoare, comparativ cu cele nefumãtoare. • 60% dintre mamele copiilor decedaţi prin ”moarte subitã a nou-nãscutului” sunt
fumãtoare.
Conţinut ţigarete Nr
Substanţe chimice > 4000
Substanţe carcinogene 50
Substanţe care induc anomalii de dezvoltare 6
Fumatul Pasiv
ţ
Fumatul Activ

• Inhalarea fumului rezultat din combustia biomasei in primele 18 luni de viata– scade rata de dezvoltare a plamanilor, – afecteaza cresterea si dezvoltarea plamanilor, – creste semnificativ riscul de reactii
sensibilizarii la aeroalergeni, – scade cresterea somatica.
• combustia biomasei genereaza particule fine de compusi organici care – induc stress oxidativ, cresc productia de
cytokine (efect inflamator) – produc inflamare cronica a cailor rspiratorii la
copii care inhaleaza fumului rezultat din combustia biomasei .
Poluarea