lucia paiano - javadevil.altervista.orgjavadevil.altervista.org/sbob/3a1sc/patologia/12 - teti -...
TRANSCRIPT

Lezione Patologia 06-11-2013 Prof.ssa Diana Teti
[Digitare il sottotitolo del documento]
Meccanismi Apoptotici Patologia cellula
neoplastica
Lucia Paiano

1
(la prof.ssa Teti inizia la lezione riassumendo in breve qualcosa riguardo la lezione precedente)
Riprendiamo oggi l’argomento dell’apoptosi trattato nella lezione precedente!!
Sono chiari i vari domini del complesso di APAF-1? E le modalità con ci esso modifica la sua
conformazione? Ricordiamo che il primo cambiamento conformazionale avviene grazie al legame
con il citocromo C mentre il secondo cambiamento conformazionale è dato dal legame con l’ATP.
Infatti l’apoptosi è un azione cellulare che RICHIEDE ENERGIA proprio perché l’ATP serve per
modificare la conformazione di APAF in modo che si dispieghi la molecola, in modo che possano
associarsi 7 molecole di APAF-1 con 7 MOLECOLE DI PRO-CASPASI 9 formando il complesso
dell’APOPTOSOMA! …………………………………
La prof.ssa proietta una slide nella
quale sono rappresentate le due
vie INTRINSECA ED ESTRINSECA
DELL’APOPTOSI.
Come notate dall’immagine,
abbiamo una rappresentazione
delle due vie dell’apoptosi,
intrinseca ed estrinseca.
VIA ESTRINSECA è
caratterizzata
dall’impegno di
RECETTORI DI
SUPERFICIE. Sono
essenzialmente di tre tipi:
- RECETTORI FAS/ FAS-L - RECETTORI TNFR1-TNFR2 (famiglia di recettori che lega il TNF alfa) - RECETTORI PER I TRIAL
VIA INTRINSECA mediata da alterazioni portate alla cellula soprattutto a livello del DNA.

2
(LA PROF.ssa inizia a fare esempi per ciascuna via ma questi saranno poi successivamente
ripetuti durante la lezione e approfonditi)
ESEMPIO VIA ESTRINSECA :
Qui è rappresentato principalmente la via estrinseca mediata dal recettore FAS chiamato
anche CD95. È il recettore per il LIGANDO FAS (FAS-LIGAND O FAS-L) che è un componente
della famiglia del TNF. Il legame del ligando con il suo recettore (lo vedremo meglio più tardi
durante la lezione) comporta il fatto che a livello del recettore che si lega con il FAS-L si debba
inserire una MOLECOLA ADATTATRICE. La cellula infatti prima di inoltrare il programma
apoptotico deve rendersi conto se esso risulta essere necessario o meno. Per cui risulterà rallentato
il pathway che porta alla morte cellulare mediante appunto l’inserimento di queste MOLECOLE
ADATTATRICI. Quella che interviene legandosi al recettore dopo che esso si è a sua volta legato con
il FAS-L si chiama FADD (FAS associated DEATH DOMAIN). Questi recettori e le molecole
adattatrici presentano dei domini particolari chiamati DOMINI DELLA
MORTE (DD) perché implicati nella realizzazione del programma di morte cellulare.
Per cui FADD è una PROTEINA CHE CONTIENE DOMINI DELLA MORTE (DD) , associata al recettore FAS. Essendo una molecola adattatrice ha due regioni diverse:
Regione che si lega a FAS
Regione che lega la PRO-CASPASI 8 che è coinvolta appunto nella via di attivazione
estrinseca. In questo caso i domini non saranno più DD ma DE (death effector ) che
interagiscono appunto con la pro-caspasi otto che attivata a CASPASI 8 ATTIVERA’ LA
PROCASPASI-3 . La pro-caspasi 8 fa parte della caspasi INIZIATRICI mentre la 3 delle
EFFETTRICI.
3
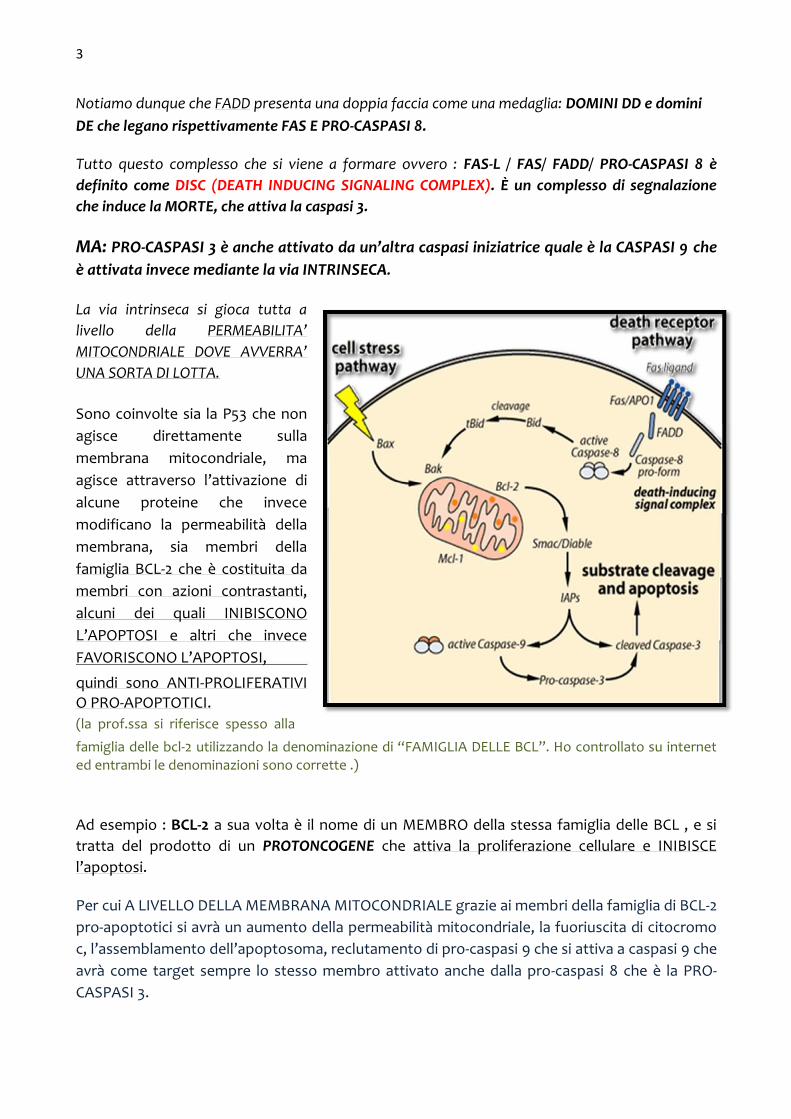
Notiamo dunque che FADD presenta una doppia faccia come una medaglia: DOMINI DD e domini DE che legano rispettivamente FAS E PRO-CASPASI 8.
Tutto questo complesso che si viene a formare ovvero : FAS-L / FAS/ FADD/ PRO-CASPASI 8 è
definito come DISC (DEATH INDUCING SIGNALING COMPLEX). È un complesso di segnalazione
che induce la MORTE, che attiva la caspasi 3.
MA: PRO-CASPASI 3 è anche attivato da un’altra caspasi iniziatrice quale è la CASPASI 9 che
è attivata invece mediante la via INTRINSECA.
La via intrinseca si gioca tutta a
livello della PERMEABILITA’
MITOCONDRIALE DOVE AVVERRA’
UNA SORTA DI LOTTA.
Sono coinvolte sia la P53 che non
agisce direttamente sulla
membrana mitocondriale, ma
agisce attraverso l’attivazione di
alcune proteine che invece
modificano la permeabilità della
membrana, sia membri della
famiglia BCL-2 che è costituita da
membri con azioni contrastanti,
alcuni dei quali INIBISCONO
L’APOPTOSI e altri che invece
FAVORISCONO L’APOPTOSI, quindi sono ANTI-PROLIFERATIVI O PRO-APOPTOTICI. (la prof.ssa si riferisce spesso alla famiglia delle bcl-2 utilizzando la denominazione di “FAMIGLIA DELLE BCL”. Ho controllato su internet ed entrambi le denominazioni sono corrette .) Ad esempio : BCL-2 a sua volta è il nome di un MEMBRO della stessa famiglia delle BCL , e si
tratta del prodotto di un PROTONCOGENE che attiva la proliferazione cellulare e INIBISCE
l’apoptosi.
Per cui A LIVELLO DELLA MEMBRANA MITOCONDRIALE grazie ai membri della famiglia di BCL-2
pro-apoptotici si avrà un aumento della permeabilità mitocondriale, la fuoriuscita di citocromo
c, l’assemblamento dell’apoptosoma, reclutamento di pro-caspasi 9 che si attiva a caspasi 9 che
avrà come target sempre lo stesso membro attivato anche dalla pro-caspasi 8 che è la PRO-
CASPASI 3.

4
CASPASI E PROCASPASI 3
>> L’azione su PRO-CASPASI 3 che diventa CASPASI 3 è mediata anche da un altro composto
che prende il nome di PAC-1 (si legge in inglese pac one) che sta per first procaspase activating
compound il quale agirà su residui di ACIDO ASPARTICO presenti sulla PRO-CASPASI 3 .
PRO-CASPASI 3 nella sua forma lineare presenta: (immagine in basso )
PRO-DOMINIO che viene scisso nella sua attivazione da PRO-CASPASI 3 a CASPASI3
Domini P17-P12 che vengono scissi in mezzo da PAC-1 a livello dell’acido aspartico in posizione 175 . Una volta liberati, i monomeri p17 e p12 si assembleranno in
ETEROTETRAMERI e diventa così la caspasi 3 attiva
SITO SAFETY CATH composto da 3 residui di acido aspartico (TRA 179-181) , a livello dei quali agisce sempre PAC-1 È importante ricordare anche la CISTEINA 163 che rappresenta il sito catalitico della PRO-caspasi 3. Ricordiamo che le caspasi sono cistein proteasi perchè appunto hanno il sito attivo costituito dalla cisteina; in questo caso si tratta della cisteina 163 che d{ l’azione.

5
QUALI SONO I SUBSTRATI DELLA CASPASI 3?
Sono presi qui in considerazione soprattutto quelli che agiscono a livello del DNA:
di inattivato)
o COMPLESSO CARD- ICAD (proteina chaperon di CAD).
L’acronimo CAD sta
CASPASE ACTIVATED
DESOSSIRIBONUCLEASE.
Essa frammenta il DNA
MANIERA REGOLARE ed è
ATTIVATA DALLE CASPASI
Infatti la caspasi libera CAD
dalla sua molecola inibitrice,
che la blocca ovvero I-CAD
(INHIBITOR OF CAD).
Per cui soltanto idrolizzando
I-CAD, CAD viene liberato
può agire a livello inter nucleosomale. Questo è uno degli enzimi coinvolti nella frammentazione precisa e regolare del DNA in quanto ce ne sono altri.
o DFF (FATTORE DI FRAMMENTAZIONE DNA) costituit0 in maniera simile a CAD ed I-CAD
da DUE SUBUNITA’ : 45 E 40. Sotto questa forma eterodimerica è inattivo. La caspasi 3
idrolizza il fattore di frammentazione 45, liberando il fattore di frammentazione del DNA
40 che si OLIGOMERIZZA unendosi ad altre molecole di DF40 e da ciò esso sarà in grado
di IDROLIZZARE IL DNA.
Anche qui così (come in CAD) abbiamo un discorso di regolazione dovuta al fatto che in
condizioni NON DI MORTE CELLULARE, le desossiribonucleasi sono INATTIVE: una CAD,
bloccata da I-CAD, idrolizzata da caspasi 3 ed il DFF perché sotto forma di eterodimero inattivo.

6
MODELLI FAMIGLIE RECETTORI : VIA ESTRINSECA APOPTOSI
1. RECETTORE FAS/FAS-L :
Quando FAS interagisce con il suo ligando, si assiste ad un processo di
TRIMERIZZAZIONE di FAS PRIMA E DI FADD DOPO. Tutti i recettori, anche quelli che
hanno come ligandi i fattori di crescita, quando legano il ligando tendono a dimerizzare,
questo del FAS è un processo simile solo che invece di una dimerizzazione avremo una
TRIMERIZZAZIONE del recettore.
A questo punto (come già detto in precedenza) vi sarà il reclutamento di FADD che si
legherà al FAS mediante i domini DD (della morte). Vi è un certo numero di molecole
FADD che interagiscono con il DEATH DOMAIN POSSEDUTO ANCHE DA FAS.
A livello di FADD sono messi in evidenza anche i DOMINI EFFETTORI DELLA MORTE (DE) che reclutano la pro-caspasi 8, formando il complesso DISC.
Questo è il modello in cui FADD si lega sulla faccia ESTERNA della coda citoplasmatica di FAS ma potrebbe anche legarsi sulla superficie interna.
PUNTI DI CONTATTO TRA VIA INTRINSECA ED ESTRINSECA
Abbiamo parlato di apoptosi e di via intrinseca ed estrinseca come se fossero due
processi separati; in realtà ci sono sempre delle interazioni tra le due. Così come avviene
nella coagulazione dove avrò vari punti di contatto tra via intrinseca e via estrinseca,
così anche nella cascata dell’apoptosi troveremo diversi punti di contatto. (fare
riferimento all’immagine nella pagina seguente)

7
Quando Caspasi 8 viene attivata dal legame con FADD; abbiamo visto
poco fa che attiva PRO-CASPASI 3 IN CASPASI 3 ma, non solo, la CASPASI
8 è anche in grado di agire IDROLIZZANDO UN MEMBRO DELLA
FAMIGLIA BCL chiamato BID (bcl-2 interacting domain) che è un DOMINIO DI INTERAZIONE CON BCL-2.
Succede che BID idrolizzato da CASPASI 8 diventa una piccola molecola
detta T-BID (truncated bid), trattasi della porzione C-TERMINALE, che è in
grado di interagire con la plasmamembrana mitocondriale , alterandone la
permeabilità e quindi ATTIVANO LA VIA INTRINSECA. Quindi vedete che
c’è uno stretto legame tra l’una e l’altra via poichè attraverso caspasi 8 e
BID possiamo avere un’ interazione con la via intrinseca.
A LIVELLO DELLA VIA INTRINSECA agiscono i vari membri della famiglia BCL. Essa è
costituita da membri che ATTIVANO L’APOPTOSI (bax-bad-bim) che dal citosol si
localizzano sulla MEMBRNA MITOCONDRIALE permeabilizzandola (RILOCALIZZAZIONE), e la
cui azione è antagonizzata da BCL-2 (un altro membro) che è appunto un ANTI-
APOPTOTICO, poiché impedisce la fuoriuscita del citocromo C e quindi la formazione
dell’apoptosoma.
L’altro punto di congiunzione tra via intrinseca e estrinseca è la IDROLISI-
ATTIVAZIONE DI PRO-CASPASI 3 che può avvenire sia ad opera della CASPASI 9 che della pro-caspasi 8.

8 Caspasi 3 poi agirà a livello a livello del DFF (fattore di frammentazione del DNA) idrolizzando la
subunità 45 e liberando la subunità 40 che entra nel nucleo e cambierà la sua conformazione
diventando un OLIGOMERO. Questo cambiamento di conformazione provoca un’acquisizione
dell’attivit{ DNA-asica e quindi è in grado di determinare la frammentazione del DNA.
Oltre al recettore FAS, di cui abbiamo appena parlato, esistono altri recettori della via ESTRINSECA OVVERO:
2. RECETTORE TNFR-1 e TNFR-2 (recettori per i membri della famiglia
del TNF (tumor necrosis factor)) :
A livello di questi recettori si possono verificare due risposte cellulari diverse e addirittura OPPOSTE!!!
Si può verificare infatti una risposta cellulare che porta alla MORTE (APOPTOSI) ed una che porta alla SOPRAVVIVENZA (PROLIFERAZIONE). Sono quindi due eventi completamente OPPOSTI TRA LORO, OPPOSTI FINO ALL’ESCLUSIONE RECIPROCA!!
Questo a dimostrazione di come una cellula prima di andare incontro a MORTE valuti
attentamente le condizioni, l’ambiente, la situazione in cui si trova per poter prendere la
DECISIONE! COME FA LA CELLULA A PRENDERE UNA DECISIONE?
MEDIANTE LE MOLECOLE ADATTATRICI che sono quelle che regolano il DESTINO DELLA
CELLULA DOPO IL LEGAME DEI RECETTORI CON IL LIGANDO. A livello di questi recettori si
pensava che interagisse direttamente la molecola FADD che appunto era quella molecola
adattatrice di FAS (vista sopra). Se fosse così però la cellula non avrebbe altro destino che
l’apoptosi, invece:
- A livello di questi recettori (TNFR-1 e TNFR-2) si inserisce una diversa molecola adattatrice chiamata TRADD (TNFR-1 associated death domain protein)
che a sua volta può:
Se la cellula deve andare incontro ad Se la cellula NON deve andare incontro
APOPTOSI, legarsi a FADD ad apoptosi, legarsi ad un’altra molecola
ovvero RIP – 1 (receptor interacting
Protein), una serin-treonin chinasi
contentente DD (death domain)

9
Tutto dipende dalle CONDIZIONI AMBIENTALI, DIPENDE SE CI SONO STIMOLI DI
SOPRAVVIVENZA CHE SUPERANO QUELLI DI MORTE E VICEVERSA. Ad esempio nei tumori succede che i segnali di morte cellulare sono meno intensi e forti
rispetto a quelli della sopravvivenza (citochine, fattori di crescita) che naturalmente
sposteranno l’equilibrio verso la SOPRAVVIVENZA!!!
Quindi tutto ciò (si riferisce alla doppia possibilità di risposta) si può realizzare nella cellula poiché al recettore si vanno a legare appunto queste molecole adattatrici.
In particolare :
Dal legame di TRADD CON FADD avremo che: FADD
legherà la PRO-CASPASI 8 E QUINDI SI ATTIVA
CLASSICA CASCATA CHE PORTA ALL’APOPTOSI.
Dal legame di TRADD CON RIP -1 (RECEPTOR
INTERACTIVE PROTEIN, trattasi di una proteina che
interagisce con il recettore) avremo che: a RIP-1
legher{ un’altra PROTEINA ADATTATRICE CHIMATA
TRAF (TNF ASSOCIATED FACTOR: precisamente saranno implicate TRAF-1 e TRAF-2 ) che ATTIVA DI TRASDUZIONE DEL SEGNALE rappresentate
JANUS KINASE ed NF-KB CHE ATTIVANO PROLIFERAZIONE CELLULARE.
Ricapitolando per raccogliere le idee:
(riferirsi all’immagine nella pag seguente)
Al recettore si lega una MOLECOLA ADATTATRICE POLIVALENTE chiamata TRADD.
A TRADD si può legare FADD e quindi CASPASI 8 e attivazione dell’apoptosi
A TRADD non si può legare solo FADD ma , ad esso si può legare una proteina che
interagisce con il recettore ovvero RIP-1 che a sua volta legherà TRAF (TNF associated
factor).
A questo punto TRAF sarà in grado DI ATTIVARE DELLE VIE DI SOPRAVVIVENZA
mediate da alcune chinasi come la JANUS CHINASE (JNK= N-terminal Janus chinase) . Le
chinasi hanno come ruolo fondamentale quello di FOSFORILARE ed in questo caso così
come nella maggior parte dei casi le fosforilazioni sono eventi che attivano fattori di

10
crescita e trasduzione del segnale di MITOGENESI. Tutto avviene attraverso le MAP-
CHINASI e quindi attiveranno la proliferazione cellulare.
Le vie di attivazione della proliferazione si attivano anche mediante l’attivazione di un
FATTORE DI CRESCITA NUCLEARE: NF-KB . Trattasi di un fattore di crescita che è stato
trovato per la prima volta nei LINFOCITI B dove era necessario per la sintesi delle catene
leggere K. Poi si è visto però che si tratta di un FATTORE DI TRASCRIZIONE
UBIQUITARIO CHE ATTIVA MOLTI GENI PRO-INFIAMMATORI. (Durante l’infiammazione
ad esempio i macrofagi, per produrre le citochine pro-infiammatorie, sono stimolati
mediante i Toll like recetor, ad attivare l’NF-KB che si và a legare a dei particolari SITI DI
CONSENSO per esso presenti in quasi tutti i geni PRO-INFIAMMATORI ). Tutto ciò non
basta poiché NF-KB è in grado anche di attivare l’espressione dei geni che regolano la
PROLIFERAZIONE CELLULARE. Per quanto concerne sempre NF-KB, trattasi di un
sistema che presuppone la presenza di un INIBITORE DI NF-KB che viene FOSFORILATO
ED INVIATO ALLA DISTRUZIONE NELL’AMBITO DEL PROTEASOMA. Rimarranno le
subunit{ dell’NK-FB che migrano verso p50 e p65 che migrano nel nucleo dove agiranno
come fattori di trascrizione per geni coinvolti nella proliferazione cellulare.
Quindi le vie DI TRASDUZIONE CELLULARE che attivano NF-KB E JNK SONO VIE CHE ATTIVANO LA PROLIFERAZIONE CELLULARE.

11
Per cui paradossalmente siamo partiti da un LIGANDO DI MORTE con il
TNFR-1 e si arriva all’attivazione della proliferazione cellulare. Questo ACCADE PREVALENTEMENTE NEI TUMORI!!!
3. TRAIL-R : RECETTORE PER ligando TRIAL
TRAIL è un ligando: TNF RELATED APOPTOSIS INDUCING LIGAND, che riconosce essenzialmente 4 tipi di RECETTORI però ce ne sono anche di più.
Questi ultimi prendono il nome di TRAIL-R1, TRAIL-R 2, TRAIL-R 3, TRAIL-R-4. Questa nomenclatura però è stata sostituita da una più snella. Infatti conosciamo:
DR4= TRAIL-R 1 e DR5 =
TRAIL-R-2. Questi due recettori hanno una coda citoplasmatica
costituita dai DEATH DOMAIN (DD)
che quindi interagendo con il
TRAIL possono trasmettere il
segnale di morte cellulare.
DcR1 = TRAIL-R 3 e DcR2=
TRAIL-R 4, altri due recettori che o non hanno il DEATH
DOMAIN oppure lo hanno molto
molto CORTO e quindi sono
INCAPACI DI TRASMETTERE IL
SEGNALE DELLA MORTE
ALL’INTERNO DELLA CELLULA.
DcR2 ha una porzione citoplasmatica piccolissima (DD);
DcR1 non ha questa porzione citoplasmatica.

12
Perché sono stati denominati DcR? Questo acronimo “Dc” riguarda il
soprannome che è stato dato a questi recettori ovvero DECOY RECEPTORS, IN
QUANTO SI TRATTA DI RECETTORI CHE INGANNANO I LIGANDI DI MORTE
CELLULARE QUALI IL TRAIL. Il TRAIL infatti interagisce con DCR1 e DCR2 convinto
di indurre l’APOPTOSI. In realt{ l’apoptosi NON VIENE INDOTTA perché la
porzione citoplasmatica di questi recettori è appunto piccola o non presente
totalmente. Allora la cellula sta ricevendo un segnale di morte cellulare che non
può essere trasmesso e allora questi recettori sono stati così paragonati a delle
TRAPPOLE CHE INGANNANO IL SEGNALE DI MORTE CELLULARE IN QUANTO VI E’
L’INTERAZIONE CON IL SEGNALE DI MORTE CELLULARE MA NON VI POTRA’
ESSERE LA RISPOSTA APOPTOTICA E QUINDI DI MORTE.
NON SI TRATTA DI UN FEED-BACK, poiché il feed back avviene quando qualcosa
viene attivato e poi il prodotto stesso andrà ad agire sulla struttura che ha
generato la risposta. Qui più che feed-back, si ritorna al concetto di EQUILIBRIO
OMEOSTATICO e regolazione dell’apoptosi.
È un processo regolato attraverso lo stesso meccanismo!!! È difficile scappare a
questa regolazione poiché il meccanismo è lo stesso ci saranno recettori che
trasmetteranno il segnale di apoptosi, mentre altri no.
NON SI TRATTA NEANCHE DI AFFINITA’!!! Non è che DCR1-2 siano più o meno
affini al TRAIL. TUTTI E QUATTRO I RECETTORI HANNO LA STESSA IDENTICA
AFFINITA’ solo che DcR1 e DcR2 non hanno o hanno una corta coda
citoplasmatica e quindi non possono trasmettere il segnale quindi si ritorna
ancora al fatto che
“ALCUNI RECETTORI PER LA LORO CONFORMAZIONE TRASMETTONO IL
SEGNALE ED ALTRI NO” e ciò comporta UN EQUILIBRIO in quanto anche in
presenza di segnali apoptotici viene impedita una TOTALE MORTE CELLULARE
poiché appunto sono presenti alcuni recettori che non solo non trasmettono il
segnale della morte MA DCR1 E DCR2 INTERAGENDO CON IL TRAIL LO
SOTTRAGGONO DAL POSSIBBILE LEGAME CON DR4 E DR5 che indurrebbero
apoptosi.!! È PROPRIO QUI CHE SI GIOCA L’EQUILIBRIO ed e’ per questo motivo
che SONO CHIAMATI RECETTORI DECOY OVVERO TRAPPOLA!!!
Questo equilibrio è importante sia nelle cellule normali che nelle CELLULE
NEOPLASTICHE in quanto in queste troveremo una DIVERSA ESPRESSIONE di
questi recettori. A parte il fatto che è molto complicato il discorso poiché anche a
livello di questi recettori possono essere attivati vie di sopravvivenza cellulare
quali NF-KB E JNK come a livello di TNFR.

13
Quindi DCR1-DCR2 rispetto a DR4-DR5, svolgono un RUOLO OPPOSTO!!! L’ESPRESSIONE di
DCR1-DCR2 dovrebbe essere esaltata nei tumori poiché si oppongono all’apoptosi invece,
stranamente, nella maggior parte dei casi sono IPO-ESPRESSI IN QUANTO IPERMETILATI. (noi
lo stiamo studiando in laboratorio in alcune linee tumorali e anche in alcuni campioni di
tumori). Questo non si concilia con un processo di TRASFORMAZIONE NEOPLASTICA ma :
in corso di neoplasie la presenza di fattori di crescita e di attivazione può determinare lo
slittamento verso la via di proliferazione piuttosto che verso un’altra. Quindi si può
CAPOVOLGERE il ruolo di DcR1- DcR-2 con quello di DR-4, DR-5. Determinante è quindi il
diverso ambiente cellulare in cui le cellule si trovano a persistere.
A livello di TRAIL-R in particolare DR4-DR5 non abbiamo parlato di PROTEINA ADATTATRICE;
ne abbiamo parlato invece per quanto concerneva TNFR1-2, ovvero TRADD, FADD, RIP-1.
Diciamo meglio che la molecola che funge da PROTEINA ADATTATRICE per DR4-5 è
PRESENTE MA E’ SCONOSCIUTA e probabilmente si ritiene che a livello di questa
proteina che ancora non è stata individuata ma che comunque si va a legare al recettore
DR4-DR5, venga ATTIVATA la PRO-CASPASI 10 in caspasi 10 che comunque agisce
sempre attivando la PRO-CASPASI 8 in caspasi 8. Da queste cascate iniziatrici poi si
attiverà la cascata delle varie caspasi che porterà all’apoptosi e che è regolata in senso
negativo dalle proteine della famiglia delle BCL.
IAP protein
Nel meccanismo di regolazione dell’apoptosi sono
importanti alcune proteine INIBITRICI DELL’APOPTOSI
ovvero le INHIBITORS OF APOPTOSIS o IAPs che
antagonizzano l’azione degli attivatori dell’apoptosi
liberati secondariamente dai mitocondri ovvero le
SMAC/DIABLO (ricordiamo che i mitocondri nella via
intrinseca sono coinvolti non solo perché liberano il
CITOCROMO C ma anche perché liberano queste
SMAC/DIABLO). Ma, le IAPs agiscono anche
DIRETTAMENTE, bloccando le CASPASI.
Le IAPs sono delle proteine un po’ particolari in quanto
hanno un alto grado di omologia con alcune sequenze di
proteine prodotte da alcuni virus chiamati
BACULOVIRUS. Queste proteine sintetizzate dai baculovirus, vengono prodotte proprio per evitare che le cellule da essi infettate vadano
incontro a MORTE CELLULARE, perché questi virus hanno tutto l’interesse a MANTENERE IN
VITA la cellula per poter essi stessi sopravvivere. La natura è veramente mirabile!!! Queste

14
proteine si chiamano proprio IAPs. Queste ultime sono state scoperte naturalmente anche
nell’uomo, hanno un alto grado di omologia per quanto riguarda soprattutto una sequenza di
70 amminoacidi che si chiama BIR (Baculovirus IAP repeat).
La prima proteina IAP scoperta nell’uomo si è vista esser codificata a livello di una regione del CROMOSOMA X per cui le IAPs umane prendono il nome di X-IAPs.
In questa IMMAGINE possiamo denotare tutte le vie di interazione delle IAP a livello del meccanismo dell’apoptosi.
IAP agisce sia a livello delle caspasi iniziatrici quale la CASPASI 9 (caspasi iniziatrice per eccellenza) BLOCCANDOLA
IAP agisce anche a livello delle caspasi effettrici quali CASPASI 3 e CASPASI 7 BLOCCANDOLE.
Riguardando questo schema possiamo mettere in evidenza a parte il ruolo di IAP anche lo
schema di azione di TRADD, una molecola adattatrice che si lega a TNFR1-2 e alla quale, come
abbiamo gi{ detto in precedenza, può legarsi FADD che porta all’attivazione di PRO-CASPASI 8
IN CASPASI 8 e probabilmente a questo livello interviene appunto

15
IAP che si lega e blocca l’azione della CASPASI 8 (questo fenomeno è ancora in dubbio,
più sicura è l’azione sulla CASPASI 9 accennata poco fa)
OPPURE agir{ su TRADD. Infatti, mediante l’intervento di RIP-1, può legarsi la molecola
TRAF dal quale vengono attivate le vie di fosforilazione mediante delle chinasi, tra le
quali JNK (JANUS CHINASI) che portano alla fosforilazione del complesso AP1.
Quest’ultimo è appunto un complesso formato da vari fattori di trascrizione
quali c-fos, c-jun, Fra 1, Fra 2, Jun B, Jun D che possono formare omodimeri ed
eterodimeri. In questo caso non dobbiamo citare c-myc (non dobbiamo fare la cantilena c-
fos, c-jun, c-myc in quanto questi tre sono dei fattori di trascrizione attivati nella prima parte
della fase G1, quindi sono i geni cosiddetti precoci) poiché si tratta di un prodotto di un
proto-oncogene che noi troviamo attivato nel LINFOMA DI BURKITT. Nel complesso AP1
fosforilato dalle JANUS CHINASI NON ABBIAMO LA PARTECIPAZIONE DI c-myc.
Ritornando allo schema, TRAF una volta legatosi a TRADD può portare alla fosforilazione
dell’INIBITORE DI NF-KB (INF-kB) che sarà ubiquitinato, idrolizzato a livello del proteasoma
mentre si libera NF-KB che andrà ad attivare vari geni coinvolti nella proliferazione tra i quali
alcuni geni della famiglia BCL-2, ovvero BCL-XL che attiveranno la proliferazione.
Anche a questo livello probabilmente (ancora non se ne ha anche qui la certezza) intervengono le IAPs: non solo esse bloccheranno le caspasi 3, 7, 9 ma
PROBABILMENTE intervengono anche nell’attivazione della via di sopravvivenza
ATTIVANDO LA VIA DI TRAF e le JANUS CHINASI implicate dirottando la risposta
cellulare verso la sopravvivenza piuttosto che verso l’apoptosi.
MEMBRI DELLA FAMIGLIA BCL-2
Trattasi di una numerosa famiglia, i cui membri non vanno molto d’accordo, poiché alcuni agiscono in un modo ed altri in un’altra.
Vediamo cosa succede a livello di questi membri della famiglia BCL quando una cellula avvia il suo programma di morte cellulare.
Cominciamo però dalla:
Cellula in CONDIZIONI DI RIPOSO.
Tutto si gioca a livello della MEMBRANA MITOCONDRIALE dove sono appunto
localizzate le proteine BCL-2 che sono prodotti di un protoncogene che attiva la
proliferazione impedendo l’apoptosi, impedendo l’azione di un altro membro che si
chiama BAK che a sua volta è posizionato a livello della membrana mitocondriale.

16
Per cui a livello della membrana mitocondriale vi sono 2 ANTAGONISTI:
- BAK che facilita e dunque favorisce l’aumento della permeabilit{ mitocondriale
- BCL-2 che impedisce l’aumento della
permeabilità mitocondriale impedendo
BAK che quindi sarà TENUTO IN STALLO e
BLOCCATO da BCL-2
A livello del citosol ci sono altri membri della famiglia BCL-2:
- BAX che sono tenute a livello del citosol
da BCL-2 che quindi avrà doppia funzione,
da un lato bloccherà BAK a livello della
membrana mitocondriale, dall’altra
bloccherà nel citosol le molecole di BAX
che fa sempre parte di questa famiglia. Il
meccanismo di questo sequestro a livello del citosol ancora è sconosciuto, probabilmente BCL-2 complesserà con BAX così come fa con BAC.
- BAD = mantenuto a livello del citosol da un meccanismo di FOSFORILAZIONE. Se
infatti BAD è FOSFORILATO è SEQUESTRATO NEL CITOSOL e non può migrare a
livello mitocondriale per alterare la membrana mitocondriale . BAD è fosforilato nel
caso in cui la cellula sia esposta a stimoli mitogenici, poiché questi attivano le MAP-
KINASI che tra le altre cose fosforilano appunto BAD.
Per cui RIASSUMENDO IN CONDIZIONI DI RIPOSO ABBIAMO:
BAD fosforilato anche da qualche piccolo segnale di sopravvivenza che la cellula riceve
BAX bloccato nel citosol da BCL-2
BAK che sta sempre sulla membrana mitocondriale ma è reso inoffensivo da BCL-2.
Succede che ad un certo punto che la
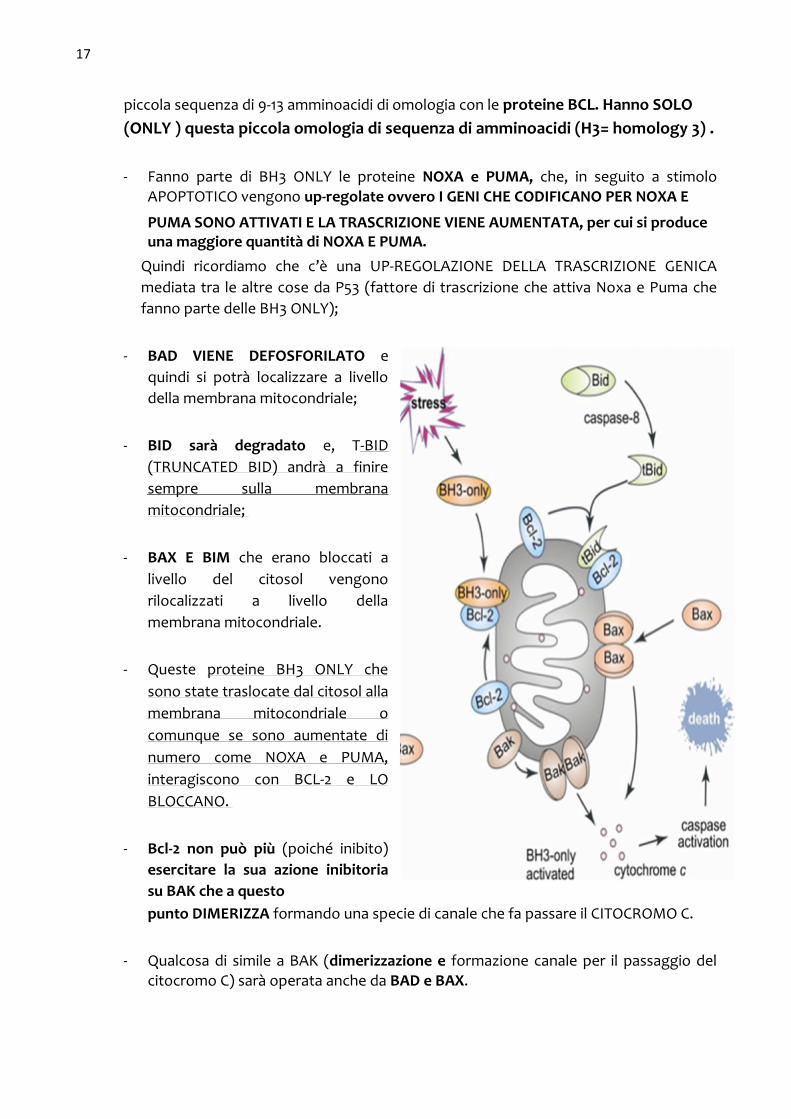
cellula riceve un SEGNALE DI APOPTOSI Vengono attivati tutti i membri della famiglia BCL-2 ad attivita’ pro-apoptotica che fanno parte di un gruppo che prende il nome di BH3-ONLY. Si tratta di proteine che hanno una
17
piccola sequenza di 9-13 amminoacidi di omologia con le proteine BCL. Hanno SOLO (ONLY ) questa piccola omologia di sequenza di amminoacidi (H3= homology 3) .
- Fann0 parte di BH3 ONLY le proteine NOXA e PUMA, che, in seguito a stimolo APOPTOTICO vengono up-regolate ovvero I GENI CHE CODIFICANO PER NOXA E
PUMA SONO ATTIVATI E LA TRASCRIZIONE VIENE AUMENTATA, per cui si produce una maggiore quantità di NOXA E PUMA.
Quindi ricordiamo che c’è una UP-REGOLAZIONE DELLA TRASCRIZIONE GENICA
mediata tra le altre cose da P53 (fattore di trascrizione che attiva Noxa e Puma che
fanno parte delle BH3 ONLY);
- BAD VIENE DEFOSFORILATO e
quindi si potrà localizzare a livello
della membrana mitocondriale;
- BID sarà degradato e, T-BID
(TRUNCATED BID) andrà a finire
sempre sulla membrana
mitocondriale;
- BAX E BIM che erano bloccati a
livello del citosol vengono
rilocalizzati a livello della
membrana mitocondriale.
- Queste proteine BH3 ONLY che
sono state traslocate dal citosol alla
membrana mitocondriale o
comunque se sono aumentate di
numero come NOXA e PUMA,
interagiscono con BCL-2 e LO
BLOCCANO.
- Bcl-2 non può più (poiché inibito)
esercitare la sua azione inibitoria
su BAK che a questo punto DIMERIZZA formando una specie di canale che fa passare il CITOCROMO C.
- Qualcosa di simile a BAK (dimerizzazione e formazione canale per il passaggio del citocromo C) sarà operata anche da BAD e BAX.

18
Ci sono le varie vie di FAS e altri stimoli della morte tramite recettori che non sono il FAS. Però
la cellula è sottoposta anche a fattori di sopravvivenza.
Partiamo dalla situazione di SOPRAVVIVENZA perché se la cellula non muore, deve vivere e deve avere dei SEGNALI DI SOPRAVVIVENZA.
(ho inserito in basso l’immagine effettivamente proiettata dalla prof. Quindi basta seguirla nei minimi dettagli per comprendere la seguente spiegazione che ad essa è riferita)
Vedete come tutti i segnali di sopravvivenza : PKC,ERK1/2 PI3K, sono tutti fattori che
ATTIVANO LE MAP KINASI. Ne esiste anche un altro di segnale che è PKB che assieme a
PKC dà vie di sopravvivenza e di fosforilazione di tirosin chinasi molto importanti.
BAD in seguito all’azione di queste chinasi VIENE FOSFORILATO su degli specifici residui
amminoacidici, indicati con dei numeretti, e quindi RIMANE SEQUESTRATO NEL
CITOSOL.
Nel caso in cui NON C’E’ LO STIMOLO DI SOPRAVVIVENZA (riferirsi sempre all’immagine precedente) (Si parte da uno stress genotossico, da un danno al DNA!!! )
BAD NON sarà FOSFORILATO (senza i numeretti), dunque si porterà alla membrana mitocondriale dove bloccherà BCL-XL (una isoforma di un membro della famiglia delle

19
BCL ovvero BCL-XL ; l’altra isoforma è BCL-XS). Abbiamo infatti due fattori ANTI-
APOPTOTICI : BCL-2 e BCL-XL.
Inoltre, se c’è l’attivazione della caspasi 8, da BID si ricava T-BID e quest’ultimo si muover{ a livello anch’esso della membra mitocondriale BLOCCANDO BCL-XL.
T-BID facilità la dimerizzazione di BAC e quindi la formazione di un canale a livello della membrana mitocondriale con fuoriuscita di CITOCROMO C.
Grazie all’azione di P53 avremo l’UP-REGOLAZIONE di NOXA E PUMA che vanno anche esse alla membrana mitocondriale dove BLOCCANO BCL-2
BIM blocca BCL-2
BAX è attivato anche da p53 e paradossalmente può essere attivato anche dalle JANUS
KINASI che in questo caso hanno azione PRO-APOPTOTICA. Però SE PREVALE BCL-2
allora BAX risulterà bloccato.
Tutto il complesso della risposta in generale NON E’ LINEARE. Parlavo del discorso di
recettori DECOY che bloccano l’apoptosi, e invece no, ci sono condizioni che possono
comportare un meccanismo completamente opposto.
(La prof.ssa fa una sorta di riepilogo appoggiandosi ad un’immagine)
Come vedete nella cellula apoptotica , BAD non è fosforilato da parte delle chinasi come
AKT. Esso andrà a livello della membrana mitocondriale bloccando le due molecole pro-
apoptotiche BCL-2 e BCL-XL e anche qui avremo la formazione di un canale formato da Bax
che consente la fuoriuscita del citocromo C.
Mentre in una cellula normale, con segnale di sopravvivenza, BAX è fosforilato e quindi BCL-2 e BCL-XL evitano che BAX possa formare il canale in questione.

20
Da tenere presente è ancora il CROSS-TALK tra la via intrinseca e la via estrinseca mediato sia a
livello della caspasi 9 su caspasi 3 che a livello della caspasi otto su BID che agirà già a livello
mitocondriale.
SINTESI MAGGIORE O MINORE DEI MEMBRI DELLA FAMIGLIA BCL-2 coinvolti
nella proliferazione e nell’apoptosi.
Vedete che quando normalmente BAX e BCL-2 sono in equilibrio tra di loro.
Quando aumenta la sintesi di BAX per cui si formano degli omodimeri di BAX
allora preverr{ l’apoptosi, viceversa se abbiamo un aumento dell’espressione del
gene che codifica per BCL-2 allora si formeranno degli omodimeri di BCL-2 che
avranno un’azione ANTI-APOPTOTICA. Tutto si gioca su un EQUILIBRIO DI
ESPRESSIONE DI GENI.
Ci sarà normalmente anche un equilibrio tra l’espressione delle isoforme del
gene BCL-X. Dello stesso gene esistono infatti le due isoforme BCL-XS, BCL-XL.
L’isoforma BCL-XL (isoforma large) si comporta come BCL-2 nel BLOCCARE L’APOPTOSI. Se prevale essa infatti avremo un blocco dell’APOPTOSI
L’ISOFORMA BCL-XS (isoforma small) invece se prevale porterà ad un’ATTIVAZIONE DELL’APOPTOSI.

21
Anche questa è la
dimostrazione di COME UNA
CELLULA POSSA RISPONDERE
ad uno STRESS FISIOLOGICO O
AMBIENTALE mediante l’espressione dei GENI
DELLO STRESS , quali P38 (CHE è UNA PROTEIN CHINASI), JANUS CHINASI, GADD 45
(Growth Arrest and DNA
Damage) con le tre isoforme
che viene attivato proprio dal
danno al DNA.
Le tre isoforme di Gadd 45 ad esempio interagiranno con PCNA , questo ANTIGENE di risposta
nucleare favorendo la riparazione del DNA, oppure legando P21 e quindi interviene a livello
dell’arresto del ciclo cellulare in G1, oppure a livello del legame con la ciclina B1- con la chinasi
ciclino dipendente.
PROF.SSA TETI : Io, di solito, quando facevo queste lezioni parlavo anche delle degenerazioni però ho
pensato di completare invece con le alterazioni dei meccanismi che regolano la proliferazione cellulare con
qualche accenno riguardo il comportamento della cellula neoplastica. Comunque noi quest’anno purtroppo
abbiamo poche lezioni (meno dell’anno scorso) , quindi ho paura che non riusciremo a fare tutto. Le
degenerazioni non so se riusciremo a farle, ma sono cose abbastanza codificate in quanto fanno parte della
vecchia patologia anche se ci sono questioni relative agli enzimi che sono leggermente più complicate.
Intanto ho pensato che fosse utile focalizzarci un attimino su ciò che concerne la trasformazione
neoplastica.

22
I tumori, sia che siano maligni, sia che siano benigni, sono conseguenze di alterazioni geniche. Queste alterazioni nella maggior parte dei casi sono dovute a mutazioni geniche, e in un’ altra percentuale dei casi,che stanno diventando sempre più numerosi, ad alterazioni epigenetiche cioè chimiche delle basi del DNA.
Si tratta comunque di un’alterazione genica che,
nel caso delle mutazioni geniche è irreversibile
mentre, nel caso delle alterazioni epigenetiche potrebbe diventare reversibile ma non con l’allontanamento dello stimolo che lo ha indotto ma con l’intervento di sostanze e terapie tese a modificare l’alterazione epigenetica mentre invece tutte le altre forme di alterazione della proliferazione sono tutte forme che sono REVERSIBILI IN SEGUITO ALL’ALLONTANAMENTO DELLO STIMOLO CHE LO HA PRODOTTO. (Questo CONCETTO è fondamentale per capire lo sviluppo dei tumori!!!)
Se l’alterazione NEOPLASTICA riguarda i geni e quindi il nucleo ( e da qui si parte, non c’è
dubbio) la struttura della cellula che subisce modificazioni significative nel senso della
patogenesi della trasformazione neoplastica è la PLASMAMEMBRANA.
In realtà TUTTA la cellula subisce delle modificazioni, diventa una cellula ATIPICA (solo nella
cellula neoplastica possiamo utilizzare questo aggettivo). Saranno alterate le strutture dal
punto di vista funzionale, morfologico della cellula , ma tra tutte le strutture cellulari quella che
ha un significato patogenetico da cui derivano altre patologie di alterazione della cellula
neoplastica è senza dubbio la MEMBRANA PLASMATICA perché tramite essa la cellula
comunica con le altre cellule. Quindi l’importanza delle alterazioni della membrana plasmatica

23
sta nel fatto che la cellula neoplastica si comporta in maniera diversa rispetto alle altre
cellule proprio per le alterazioni che ha a livello della plasma-membrana.
È vero che i geni codificano per proteine coinvolte nella proliferazione ma, la membrana
plasmatica REGOLA la proliferazione, si parla di controllo del CONTATTO CELLULA-CELLULA, in
quanto le cellule possono comunicare tra di loro e inviare dei segnali di inibizione della
proliferazione e del movimento. Quindi le modifiche a livello della membrana plasmatica sono
strettamente correlate, come evidenziano alcuni esperimenti, con l’invasivit{ e la crescita
patologica.
Sono coinvolte nella trasformazione neoplastica tutte le giunzioni cellulari dalle aderenti, alle comunicanti, alle occludenti. Esse presenteranno grado variabile a secondo delle caratteristiche del tumore ovvero invasività e malignità.
Come sapete le giunzioni intercellulari aderenti formano legami forti tra cellula e cellula, quelle
comunicanti consentono passaggio di ioni, di messaggi e molecole che regolano la proliferazione
mentre le occludenti formano una barriera selettiva alla DIFFUSIONE di varie sostanze. Il fatto
che siano alterate le giunzioni occludenti vuol dire che la cellula è più esposta all’aggressione di
sostanze nocive e lesive che altrimenti non arriverebbero. Quindi tutti e tre i tipi di giunzione
intervengono in modo diverso a seconda della loro funzione nella patogenesi della
trasformazione.

24
RUOLO DELLE SINGOLE GIUNZIONI NEI TUMORI:
GIUNZIONI ADERENTI (maculae adherentes - desmosomi)
In questa immagine sono mostrate le principali componenti che partecipano alla
formazioni delle giunzioni aderenti. L’alterazione della CADERINA è frequentissima nelle
alterazioni tumorali. L’espressione della E-CADERINA è in quasi tutti i tumori
RIDOTTISSIMA. La E- CADERINA è infatti fondamentale nell’adesione CELLULA-CELLULA
per cui una delle cause della diminuita adesione cellulare nelle trasformazioni
neoplastiche sarà una ridotta espressione della E-CADERINA che fisiologicamente,
forma una sorta di CERNIERA MOLECOLARE poiché i dimeri di caderina di una
cellula con quelli dell’altra cellula vanno a formare,
unendosi, una struttura che a vista d’occhio somiglia
molto ad una cerniera. La cosa interessante non è solo
il fatto che la E-CADERINA formi la molecola base per
l’adesione cellula-cellula ma è rilevante il fatto che la E-
CADERINA svolge un ruolo importante anche nella
regolazione della proliferazione cellulare.
Attraverso la E-CADERINA avremo il legame con il
CITOSCHELETRO quindi con tutte le proprietà motrici
e funzionali della cellula. La E-caderina è legata alla
BETA-CATENINA, legata a sua volta alla ALFA-
CATENINA che interagisce e si lega ai FILAMENTI DI
ACTINA. O ancora la caderina può essere ancorata ad
altri componenti come la vinculina o la radexina e
così via. In alcuni tipi cellulari (non in tutti) la E-CADERINA tramite la BETA-CATENINA è legata ad una
proteina detta APC, coinvolta nell’adesione cellulare e nella trasmissione del messaggio
all’interno della cellula, che è codificata da un GENE ONCOSOPPRESSORE.

25
In alcuni tumori soprattutto in quelli del colon poliposici si è visto che c’è un deficit di
espressione di questo gene per cui una minor produzione di APC. L’acronimo APC sta infatti per
ADENOMATUS POLYPOSIS COLI, gene che è alterato, soppresso, deleto e quindi il suo
prodotto non viene prodotto in questo tipo di tumori dove appunto ne rappresenta una causa
principale.
Si è visto ancora che nei tumori invasivi più che maligni, le giunzioni aderenti ed in particolare le maculae adherentes e i desmosomi sono ridotti di numero e di grandezza!
Vi è una correlazione diretta tra la diminuzione di grandezza e del numero e quindi della
quantità delle giunzioni aderenti e l’INVASIVITA’ DEL TUMORE. Meno sono presenti è più
invasivo è il tumore. Pare che la correlazione sia più con l’invasivit{ piuttosto che con la
malignit{ del tumore, anche se la malignit{ è abbinata all’invasivit{.
Nelle cellule maligne l’adesione tra cellula e cellula risulta quindi essere ridotta. Questa
diminuzione dell’adesione è alla base della PRIMA FASE DELLE METASTASI. Noi tra le
caratteristiche dei tumori maligni parliamo di TENDENZA ALLA METASTASI intendendola come
una conseguenza del fatto vero del tumore maligno che è l’alterazione dei geni che codificano
per PRODOTTI DI DIFFERENZIAZIONE e che codificano per prodotti della proliferazione. Questa
è una CONSEGUENZA poiché le alterazioni della plasma-membrana sono CONSEGUENZA DEL
FATTO CHE NEI TUMORI MALIGNI SONO ALTERATI I GENI CHE SONO RESPONSABILI NON SOLO DELLA PROLIFERAZIONE MA ANCHE DELLA DIFFERENZIAZIONE.
Risulta evidente come le cellule normali siano di loro unite mediante le E-CADERINE mentre
nelle cellule maligne vi è una riduzione delle
CADERINE per cui esse non sono più unite tra
loro. In poche parole nelle cellule NEOPLASTICHE
viene a mancare L’ADESIONE OMOTIPICA (ADESIONE TRA CELLULE DELLO STESSO TIPO).
Ma, nonostante nelle cellule neoplastiche a causa della diminuzione della E-CADERINA viene a
mancare l’adesione OMOTIPICA, dall’altro si ha un AUMENTO DELL’ADESIONE ETEROTIPICA
poiché le cellule tumorali subiscono una transizione dal tipo EPITELIALE AL TIPO
MESENCHIMALE che ha affinità con cellule non omotipiche; si tratta di una transizione definita TEM (transizione epitelio-mesenchimale).

26
Le cellule neoplastiche NON SONO PIU’
UNITE TRA DI LORO, sono separate, sono
cellule definite ASOCIALI (questo termine è
perfetto in quanto riassume quello che è
l’aspetto della INCOMUNICABILITA’ delle
cellule tra di loro perché non solo mancano le E-caderine ma mancano anche le giunzioni comunicanti).
La cellula neoplastica PERO’ DIVENTA CAPACE DI ADERIRE ALLA MATRICE EXTRACELLULARE.
GIUNZIONI COMUNICANTI
In questa immagine viene mostrata sia una emi-
connessione (metà di un canale di un canale di
connessione tra due cellule ), sia una connessione
completa con la formazione del canale. Notiamo
come le due emi-connessioni portino alla
formazione di un UNICO CANALE DI CONNESSIONE
attraverso il quale è consentito il passaggio di IONI
E ANCHE PICCOLE MOLECOLE.
L’alterazione delle giunzioni comunicanti nelle cellule tumorali è gravissima in quanto
viene ad essere alterato uno dei componenti di un intero sistema di comunicazione tra
cellula e cellula ovvero il CANALE. Magari la cellula produce il segnale che però non può
essere inviato perché manca appunto il mezzo di trasmissione.
Nelle cellule tumorali si è visto che c’è una ridottissima correlazione tra riduzione delle
giunzioni comunicanti e condizione di metaplasia o displasia. Man mano che la cellula
infatti si avvia a diventare neoplastica si assiste ad una riduzione graduale delle giunzioni
GAP che sono ridottissime nel carcinoma (tumore conclamato) e sono invece solo
diminuite nella displasia e nella metaplasia.
Ovviamente le giunzioni GAP se mancano o sono alterate, impediscono l’accoppiamento ionico tra cellule dello stesso tipo in quanto esse non conservano la formazione di
COPPIE CHE GARANTISCONO UNA LIBERA OSMOSI TRA LE DUE CELLULE CON PASSAGGIO ANCHE DI IONI (si parla di ACCOPPIAMENTO IONICO).

27
Anche in questo caso le giunzioni
comunicanti sono realizzate SOLO CON LE
CELLULE DISPLASTICHE ALTERATE DELLO
STESSO CITOTIPO e non con quelle
normali circostanti, un po’ come avveniva
per le giunzioni comunicanti. Si assiste
infatti alla formazione di COMUNITA’
CELLULARI AVULSE, AUTONOME E
INDIPENDENTI DAL CONTESTO, per
questo si parla di isolamento completo.
Le cellule displastiche formano delle
COMUNITA’ TRA DI LORO, SI DISTACCANO DAL CONTESTO DELLE CELLULE NORMALI, e
questo le rende PIU’ SUSCETTIBILI ALL’AZIONE DI AGENTI CANCEROGENI (è più facile,
ma non è detto, che evolvano verso un fenotipo neoplastico).
THE END
LUCIA PAIANO