libro petrología y mineralogía
TRANSCRIPT


UNIVERSIDAD DE SANTIAGO DE CHILE FACULTAD DE INGENIERIA
DEPARTAMENTO INGENIERIA DE MINAS
PETROLOGÍA Y MINERALOGIA
MINERALOGIA

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 1
MINERALOGÍA
Mineral es un sólido inorgánico, de composición definida, poseedor de una estructura cristalina ordenada, y que resulta de un proceso de formación o transformación geológico. La definición anterior; si bien es cierto, es clara y precisa, no da cuenta de la importancia del entendimiento de la mineralogía.
Aún cuando el reconocimiento de las distintas especies minerales resulta llamativo, principalmente en términos del conocimiento. No es hasta cuando se investiga el medio en que se encuentran inmersos estos minerales, que se toma conciencia de su verdadera importancia. Este medio complejo es roca, la cual constituye en gran parte el ambiente en donde se desarrollan actividades relativas a las ciencias de la Tierra.
Áreas multidisciplinarias, tales como Geología, Geografía, Obras Civiles, Metalurgia,
Topografía y Minería, entre otras, desarrollan sus actividades sobre este material. Debido a lo anterior, el estudio detallado debe comenzar desde la estructura más pequeña, para luego, en base al conocimiento, extrapolar estas condiciones hacia el macizo rocoso en evaluación.
Una vez entendida y configurada la estructura básica que posee cada mineral, es
posible reconstruir el ambiente de su formación, y cómo estos minerales reaccionarán frente a la exposición de las distintas condiciones ambientales. A partir de esto, se tiene una mejor idea de una serie de parámetros y propiedades físicas de cada mineral.
En el estado actual de la minería, se explotan reservas a partir de un modelo
geológico construido. Para el desarrollo de tal construcción, se deben considerar todos los eventos geológicos ocurridos a través del tiempo. Esta información es aportada en gran parte por la composición mineralógica que tiene el yacimiento. Por lo tanto la Evaluación de Recursos, es un área influenciada altamente por la mineralogía. Por otro lado, el diseño minero puede ser agresivo, pero asegurando la estabilidad de la construcción. Para llevarlo acabo entonces, se debe tener en cuenta una serie de consideraciones al respecto del comportamiento mecánico que presenta la roca. Claro es que este comportamiento se encuentra gobernado por los minerales constituyentes de este material. Es por esto, que cada diseño debe incorporar una caracterización del macizo rocoso a nivel Geológico, Geotécnico y Geomecánico. Para finalizar el proceso productivo, se debe pasar de roca a metal, o a no metal. Para realizar esto, deben ser estudiadas y optimizadas las recuperaciones que se obtienen tras cada proceso, y estas recuperaciones son función de los elementos químicos que componen a cada mineral, i.e. la estructura interna de los minerales. En este sentido, la Metalurgia también se ve condicionada por el entendimiento de la Mineralogía. Por ello la planificación del consumo de reservas se hace considerando dominios geometalúrgicos, para así lograr una alimentación homogénea a la planta de tratamiento, desde un yacimiento heterogéneo.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 2
En el presente escrito, se estudia la mineralogía comenzando desde el análisis más básico, pasando por su influencia en el comportamiento mecánico y la descripción de estructuras de los minerales formadores de roca, para finalmente tratar algunos de los procesos mediante los cuales se conforman las distintas asociaciones mineralógicas.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs
CONTENIDOS
1. PERIODICIDAD Y SIMETRÍA 1
1.1. La Red, Celda Unitaria y el Motivo 2 1.2. Simetría de redes bi-dimensionales 3 1.3. Grupos bi-dimensionales puntuales y espaciales 4 1.4. Redes y sistemas cristalinos tridimensionales 7 1.5. Grupos puntuales tri-dimensionales y su representación 9 1.6. Grupos espaciales tri-dimensionales 9 1.7. Planos y direcciones en un cristal 12
2. ANISOTROPÍA Y PROPIEDADES FÍSICAS 14
2.1. Anisotropía - una analogía mecánica 14 2.2. Propiedades de un tensor de segundo orden y sus
variaciones con la dirección 16 2.3. Control de la simetría sobre las propiedades físicas 17 2.4. Propiedades ópticas de los minerales 20
2.4.1. Variación del índice de refracción en un cristal - indicatriz óptica 20
2.4.2. Control de la simetría en la variación del índice de refracción en un cristal 20
2.4.3. Birrefringencia 22 2.4.4. Colores de interferencia 25
2.5. Una nota en las propiedades elásticas de los minerales 25 2.6. Relación entre anisotropía y estructura cristalina 26 2.7. Anisotropía y la forma externa de los minerales 26
3. LA ESTRUCTURA CRISTALINA DE LOS MINERALES I 28
3.1. Enlaces en estructuras cristalinas 28 3.2. Descripción de estructuras cristalinas 28
3.2.1. Estructuras de Empaquetamiento Cerrado (EC). ECCúbico y ECHexagonal 28
3.2.2. Sitios intersticiales en Estructuras de Empaquetamiento Cerrado 30
3.2.3. Estructuras tipo basadas en empaquetamiento cerrado 33 3.2.4. Minerales con estructura basada en Empaquetamiento
Cerrado 35 3.2.5. Estructuras construidas de poliedros 38
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs
4. LA ESTRUCTURA CRISTALINA DE LOS MINERALES II – SILICATOS 41
4.1. El tetraedro [SiO4] 41 4.2. Algunas generalizaciones respecto de la composición y
estructura de los silicatos 43 4.3. Nesosilicatos - Silicatos con Tetraedro [SiO4] aislado 44
4.3.1. Los minerales de Olivino 44 4.3.2. Los minerales de Granate 46 4.3.3. Silicatos de Aluminio: Kianita, Andalusita y Silimanita 46
4.4. Sorosilicatos 47 4.5. Inosilicatos - Silicatos de Cadena Simple 47
4.5.1. Piroxenos 48 4.5.2. Piroxenoides 50
4.6. Inosilicatos - Silicatos de cadena doble 51 4.7. Filosilicatos - Silicatos en capas 52
4.7.1. Politipismo en las micas 54 4.8. Tectosilicatos - Silicatos en Armazón 56
4.8.1. Minerales de Sílice 56 4.8.2. Kalsilita-Nefelina: Tridimita distorsionada 58 4.8.3. Los feldespatos 59
5. DEFECTOS EN MINERALES 64
5.1. Defectos puntuales 64 5.1.1. Defectos de Schottky y Frenkel 65 5.1.2. Impurezas y substitución atómica 66 5.1.3. Centros de color en los cristales 66 5.1.4. Difusión 66 5.1.5. Estequiometría alterada 67
5.2. Defectos lineares 68 5.2.1. El vector de Burges 68 5.2.2. Dislocación de filo y dislocación de tornillo 69 5.2.3. Bucle dislocacional - Interacción entre defectos
puntuales y lineares 69 5.2.4. Dislocaciones parciales y fallas de empaquetamiento 69
5.3. Defectos planares 70 5.3.1. Fallas de empaquetamiento 70 5.3.2. Defectos de Wadsley 70 5.3.3. Defectos planares y reacciones químicas en estado
sólido 70 5.3.4. Fronteras de dominio antifase 71 5.3.5. Fronteras de maclas 71
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs
6. ENERGÉTICA EN LA ESTABILIDAD DE MINERALES I - CONCEPTOS BÁSICOS 72
6.1. Algunos conceptos básicos de la termodinámica 72 6.2. Cambios de entalpía en transformaciones y reacciones en
minerales 73 6.3. Entropía y desorden 73
6.3.1. Entropía configuracional 74 6.3.2. Entropía electrónica 75 6.3.3. Entropía vibracional 75
6.4. Energía libre de Gibbs G, y el equilibrio 76 6.4.1. Procesos reversibles e irreversibles. Metaestabilidad 77 6.4.2. Transiciones de fase de primer y segundo orden 79
6.5. Determinación de cantidades termodinámicas 80 6.5.1. Medidas experimentales directas 80 6.5.2. Estudios de equilibrio de fase 80 6.5.3. Determinación de parámetros termodinámicos a partir
de simulaciones computacionales 81
7. ENERGÉTICA EN LA ESTABILIDAD DE MINERALES II - SOLUCIÓN SÓLIDA, ORDENAMIENTO Y EXSOLUCIÓN 82
7.1. Solución sólida 82
7.1.1. Energía libre de una solución sólida 82 7.1.2. Diagrama de fase de equilibrio para una solución
sólida - el solvus 84 ANEXO 86 Diagrama de fase en sistemas binarios simples
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 1
1. PERIODICIDAD Y SIMETRÍA
La fotografía (Figura 1.1) representa una proyección bidimensional de la estructura atómica de la cordierita. Esta muestra claramente la característica fundamental de todas las estructuras cristalinas. La distribución de átomos es periódica.
Además de la simetría de translación, los patrones de distribución de átomos guardan relaciones de simetría con los átomos vecinos y con los enlaces que los unen. En la Figura 1.1, se aprecian elementos de simetría tales como ejes de simetría senaria, ternaria, binaria, y líneas especulares.
En un cristal bien formado, la simetría interna de la distribución atómica se refleja en la
relación simétrica entre la orientación de las caras externas (Figura 1.2). Lo importante de la simetría y la estructura interna de un mineral, es que influencian fuertemente sus propiedades físicas (e.g. mecánicas, eléctricas, ópticas, magnéticas y térmicas) además de reflejar el comportamiento que el mineral puede tener bajo cambios en las condiciones de presión y temperatura del medio.
Figura 1.2. Versuvianita (Ca(Mg,Fe)2Al4(SiO4)5(Si2O7)2(OH,F)4). (a) Vista isométrica, ejes de simetría y planos especulares, (b) Vista superior, eje cuaternario.
Figura 1.1. Estructura atómica de la cordierita (Mg2Al4Si5O18), Proyección de una capa delgada (≈ 200 Å) de mineral. Arreglo hexagonal, en donde los sitios negros representan canales a través de la estructura, mientras las zonas blancas constituyen una alta densidad de materia.
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Mauricio Domcke G. M. Pilar Espinoza
1.1. La Red, Celda Unitaria y el Motivo
Las operaciones de simetría son mucho más fáciles de entender en el plano, pero las
mismas ideas se extienden para las tres dimensiones. Un conjunto de puntos ordenados se puede generar repitiendo un motivo en las
direcciones correspondientes a las dimensiones de este. Elegir el motivo se basa en la
simpleza (no necesariamente en los nodos que contengan materia). La Figura 1.3 muestra tres formas de representar el orden de la cordierita (Figura 1.1). Se opta por a = b y γ = 120°, la que corresponde a la más pequeña celda unitaria que describe a la estructura. Esta celda se denomina hexagonal primitiva (primitiva dado que sólo contiene puntos en los vértices de la figura).
Página 2
En otras ocasiones, es preferible describir las estructuras en base a celdas no
primitivas. La Figura 1.4 es similar a la Figura 1.3, pero debido a que a ≠ b γ ≠ 120°, se prefiere la celda rectangular centrada (el punto en su centro hace que no sea primitiva), la cual tiene los atributos de simetría del tipo ortorrómbico.
Considerando lo anterior, se define celda
unitaria como la manera más simple que se tiene para representar el motivo de cualquier estructura, ya sea en el espacio o en un plano, y que mediante simetría de translación y de rotación en las direcciones cristalográficas, permiten construir la totalidad de la distribución de puntos.
Para describir una estructura cristalina, se comienza con la red y luego se especifica el arreglo atómico, su espaciamiento, y los ángulos de enlace, i.e. las coordenadas de los átomos relativas a cada punto de la red. El patrón de átomos asociado a cada nodo de la red, es lo que se denomina motivo. La Figura 1.5 muestra lo anterior usando una red bi- dimensional similar al patrón mostrado en la
Figura 1.3. Tres posibles celdas para la cordierita. Se opta por a = b y γ = 120°
Figura 1.4. Celda unitaria no primitiva.
Figura 1.5. Celda hexagonal con origen en “O” Átomos blancos con coordenadas (2/3, 1/3) y (1/3,2/3) representan el motivo de la estructura.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 3
Figura 1.1. La estructura, puede ser definida como la suma de la red (la cual define la periodicidad) y el motivo existente en su interior.
1.2. Simetría de redes bi-dimensionales
Cualquier grupo ordenado de puntos en el espacio puede ser representado por alguno de los 4 sistemas cristalinos bi-dimensionales, los cuales se muestran en la Figura 1.6.
Figura 1.6. Las cinco redes bi-dimensionales, y las simetrías de la distribución de puntos de la red. Para cada una, se muestra la celda unitaria a la izquierda, y los elementos de simetría operando en la celda, a su derecha.
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
Todo arreglo de puntos en las dos dimensiones puede representarse mediante uno de estos 4 sistemas cristalinos bi-dimensionales.
F. Michael Dobbs D. Página 4 Mauricio Domcke G. M. Pilar Espinoza
Además de la translación,
del deslizamiento invertido y de las líneas especulares, existen otros operadores de simetría. Estos se muestran en su totalidad en la Figura 1.7, y corresponden, además de lo anterior, a ejes de rotación 1, 2, 3, 4 y 6. i.e. la figura se repite 1, 2, 3, 4 y 6 veces respectivamente, en un giro de 360°.
1.3. Grupos bi-dimensionales puntuales y espaciales
Figura 1.7. Elementos de simetría operando en las dos dimensiones.
La simetría completa de una estructura no se define solamente por la red, dado que el
arreglo de átomos alrededor de cada punto de la red puede también ser simétrico, tal como en la Figura 1.1. Considerando la posible combinación de los elementos de simetría de tales arreglos de átomos, se necesita obtener la cantidad de combinaciones entre los ejes de rotación 1, 2, 3, 4 y 6 y las líneas especulares. Cada combinación diferente entre los elementos de simetría de los átomos que rodean a cada nodo de la red -sin considerar la repetición de translación ni el deslizamiento invertido-, y los operadores de simetría de la celda, define un grupo puntual, por lo que este último puede ser descrito y entendido, como la manera en que los átomos se relacionan al nodo de la red. La Figura 1.8 muestra los diez grupos puntuales existentes para las dos dimensiones.
Figura 1.8. Los 10 grupos puntuales bi-dimensionales. Estos representan el número total de combinaciones de las líneas especulares con los ejes de rotación, alrededor de un punto.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 5
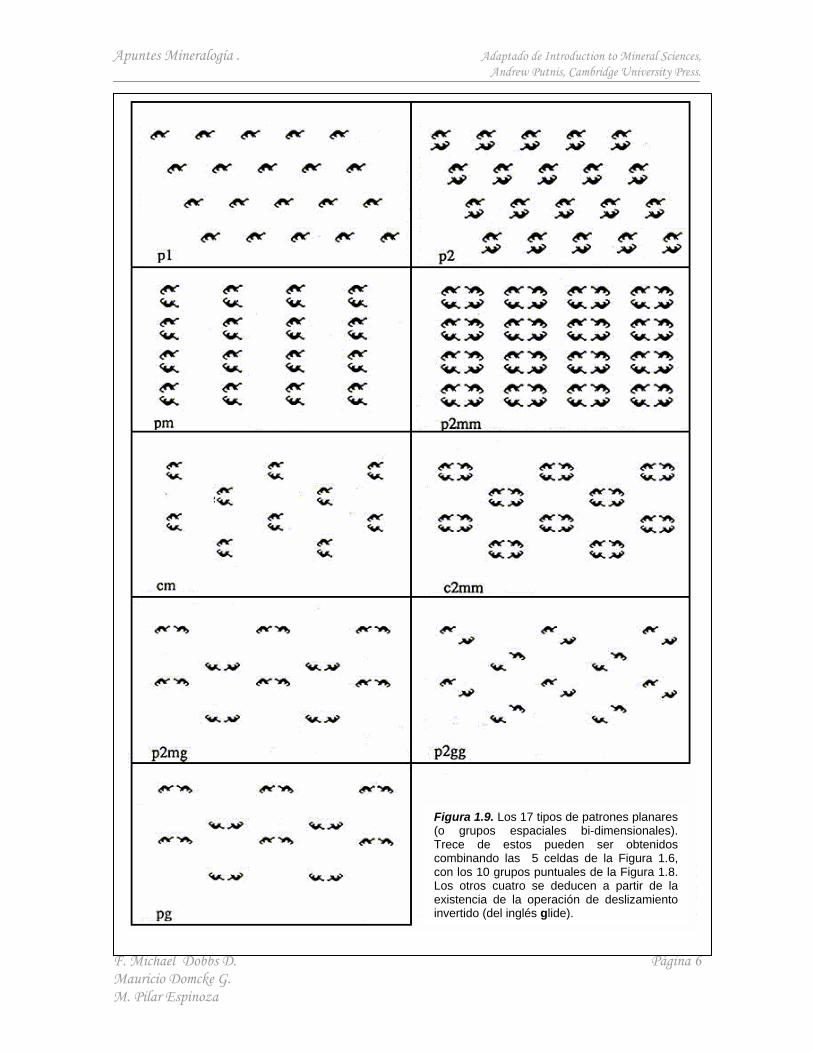
Para completar el análisis de la simetría bi-dimensional, se combinan los 10 grupos puntuales con las 5 redes o sistemas cristalinos descritos anteriormente. Aquí se introduce el deslizamiento invertido (una operación compuesta, que como se muestra en la Figura 1.7, involucra una reflexión a través de un plano, más un desplazamiento en medio vector a partir del origen), logrando un total de 17 grupos espaciales bi-dimensionales. Estos se muestran en la Figura 1.9. Todo patrón o arreglo repetitivo de puntos ordenados debe basarse en uno de esos grupos espaciales.
Mauricio Domcke G. M. Pilar Espinoza
Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 6
concah¿asdasd
Figura 1.9. Los 17 tipos de patrones planares (o grupos espaciales bi-dimensionales). Trece de estos pueden ser obtenidos combinando las 5 celdas de la Figura 1.6, con los 10 grupos puntuales de la Figura 1.8. Los otros cuatro se deducen a partir de la existencia de la operación de deslizamiento invertido (del inglés glide).
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 7
Figura 1.9. (Continuación)
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 8
Figura 1.9. (Continuación)
1.4. Redes y sistemas cristalinos tridimensionales
El desarrollo del concepto de redes, grupos puntuales y grupos espaciales para las tres dimensiones, se realiza de la misma manera en que se efectuó anteriormente para las dos dimensiones, pero asumiendo la complejidad extra que significa aumentar la cantidad de combinaciones entre los operadores de simetría. Esto hace a la simetría tri-dimensional más difícil de visualizar, aún cuando los principios son los mismos.
Los tipos de mallas tri-dimensionales fueron derivados por Bravais en 1848, quien
encontró 14 redes distintas inmersas dentro de 7 sistemas cristalinos. Las redes se ilustran en Figura 1.10, mientras que la Tabla 1.1 resume su agrupamiento dentro de los sistemas cristalinos.
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 9
TABLA 1.1
LOS 7 SISTEMAS CRISTALINOS Y LAS 14 REDES DE BRAVAIS
Sistema cristalino Dimensión celdas unitarias Simetría esencial Redes de Bravais
Triclínico a ≠ b ≠ c α ≠ β ≠ γ Ninguna o eje 1 P
Monoclínico a ≠ b ≠ c α = γ = 90 ° ≠ β Un eje 2 P, C
Ortorrómbico a ≠ b ≠ c α = γ = β = 90 °
Tres ejes 2 mutuamente perpendiculares P, C, I, F
Tetragonal a = b ≠ c α = γ = β = 90 ° Un eje 4 P, I
Cúbico a = b = c α = γ = β = 90 ° 4 ejes 3 P, I, F
Trigonal a = b = c 120° > α=γ =β > 90 ° 1 eje 3 P
a = b ≠ c 1 eje 6 P Hexagonal α = β = 90 °; γ = 120°
Figura 1.10. Las 14 redes de Bravais. Todos los sólidos cristalinos pueden ser descritos por celdas unitarias que pertenecen a uno de estos tipos. Al respecto de la notación para cada celda, se nota que en primer lugar, se menciona el sistema cristalino, y luego, el tipo de red, donde; “P” es Primitiva, “I“ centrada en el Interior, “C” es centrada en las caras bases y “F” es centrada en las todas caras.
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 10
1.5. Grupos puntuales tri-dimensionales y su representación
A los 10 grupos puntuales planares se le agrega una tercera dimensión y se derivan los 32 grupos puntuales tri-dimensionales, los que definen la relación de los átomos que se disponen alrededor de un nodo de la red. Se debe considerar que la línea especular se transforma en un plano y que la rotación alrededor de un punto se convierte en la rotación alrededor de un eje. Además se introduce un nuevo elemento de simetría llamado eje de roto-inversión. Se opera con el eje y después se invierte por el centro. Visualizar las relaciones angulares de los grupos puntuales resulta complejo, por esto es que su representación se realiza mediante una proyección estereográfica. La Figura 1.11 muestra esta representación para todos los sistemas cristalinos de las 32 clases de simetría.
1.6. Grupos espaciales tri-dimensionales
La combinación de los 32 grupos puntuales con las 14 redes de Bravais generan el conjunto completo de grupos espaciales tridimensionales, mediante un procedimiento análogo a los grupos bidimensionales. Todo el ordenamiento estructural de los minerales se puede describir asociándolo a alguno de los 230 grupos espaciales resultantes. Su derivación realizada al final del siglo XIX, fue hecha por Schoenflies, Barlow y Federov.
Un grupo espacial es entonces, la última sub-división dentro de la clasificación de los
elementos de simetría. Cada grupo espacial se encuentra asociado a un grupo puntual, el cual a su vez pertenece a una de las 14 redes de Bravais, que como se vio se encuentran inmersas en alguno de los 7 sistemas cristalinos. Cualquier material cristalino tiene una estructura que pertenece a uno de estos 230 grupos espaciales.
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 11
Figura 1.11. Los 32 grupos puntuales. Cada uno es ilustrado con 2 estereogramas, uno muestra la simetría, y el otro muestra como esta simetría opera en una dirección general o polo.
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 12
Figura 1.11. (Continuación).
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 13
1.7. Planos y direcciones en un cristal
La posición de un plano respecto de los ejes cristalográficos se expresa mediante los índices de Miller. Los planos de un cristal se determinan de la manera siguiente. Consideremos la cara de un cristal, como el de la Figura 1.12, para el cual las constantes de la red son a, b y c. Si un conjunto de planos intercepta estos ejes y divide a a en “h” partes, a b en “k” partes, y a c en “l” partes, cualquiera de los planos es denotado entre paréntesis redondo, y con las dimensiones (hkl). Otra manera de decirlo es que el plano (hkl) tiene interceptos de a/h, b/k y c/l en los tres ejes cristalográficos, i.e. los índices de Miller son los recíprocos de los interceptos fraccionales a cada eje. En la Figura 1.12, el conjunto de caras paralelas corta a a en dos distancias unitarias, a b también en dos, mientras que a c en una; por lo tanto los índices son (1/2 1/2 1). Debido a que los índices son enteros, los correspondientes al ejemplo son (1 1 2).
Figura 1.12. Esta gráfica muestra un set de planos equiespaciados (de los cuales el primero se encuentra sombreado) en los cuales se repiten las constantes a, b y c a través de los ejes cristalográficos x, y y z respectivamente. Los círculos negros representan los
De manera de familiarizar el concepto de los índices de Miller, en la Figura 1.13 se muestran ejemplos de planos y sus direcciones, notados mediante los índices señalados.
puntos de la red a través de los 3 ejes cristalográficos. Los índices de Miller para este set de planos son (112).
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 14
Figura 1.13. Ejemplos de planos y sus índices de Miller.
Las direcciones en un cristal se describen de manera similar que los planos, pero son denotadas por números enteros entre paréntesis cuadrados. Una dirección general [UVW] corresponde a un vector con proyecciones U
a, Vb y Wc en los respectivos ejes cristalográficos. (Figura 1.14). Para ilustrar esto con mayor claridad, la Figura 1.15 muestra una estructura bi-dimensional en el plano xy con varias direcciones [UVW] denotadas. Se puede identificar también en esta figura, que una dirección tiene similares índices que los planares, sólo cuando se trabaja en el sistema cúbico. En la Figura, se observa claramente que la dirección [110], no es perpendicular al plano (110), el cual está dibujado con línea segmentada.
Figura 1.14. Dirección [UVW] en relación a sus componentes a lo largo de sus ejes cristalográficos
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs D. Página 15
Figura 1.15. Ejemplos que muestran direcciones en una malla bi-dimensional. Se puede notar que en general, una dirección no es perpendicular al plano indicado con los mismos índices. Es el caso particular del plano (110) con la dirección [110].
Mauricio Domcke G. M. Pilar Espinoza

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 14
2. ANISOTROPÍA Y PROPIEDADES FÍSICAS La mayoría de los materiales cristalinos son anisotrópicos, eso significa que la magnitud de varias propiedades físicas dependerá de la dirección en que se midan con respecto de la del cristal.
Algunas propiedades de los minerales son claramente no direccionales. Por ejemplo, la densidad o la capacidad calórica pueden medirse sin tomar una dirección referencial. Estas propiedades son denominadas propiedades escalares o tensores de rango cero. Otras propiedades, tales como la energía superficial y por lo tanto la reactividad química, dependen de la orientación cristalográfica de la superficie; pero no son en sí mismas cantidades que dependan de la dirección en que se midan. Estas propiedades se denominan propiedades vectoriales o tensores de primer rango. Por otro lado, propiedades como la conductividad térmica, la cual relaciona dos cantidades vectoriales, son denominadas tensores de segundo rango y deben ser especificadas de una manera distinta. Con un sistema de ejes se puede especificar la propiedad por sus tres valores principales. Esta clasificación de las propiedades físicas, también involucran a tensores de tercer y cuarto rango, pero estas escapan al objetivo de este escrito. Propiedades elásticas, pertenecen a los tensores de cuarto rango, por lo que su descripción es claramente más compleja.
El propósito de este análisis es ilustrar el fenómeno de la anisotropía y por otro lado,
proveer una base de entendimiento de las propiedades ópticas de los minerales. Las propiedades elásticas sólo se mencionarán brevemente.
2.1. Anisotropía - una analogía mecánica
Previo a analizar las propiedades físicas de los minerales, una simple analogía mecánica bi-dimensional ayudará a describir generalmente los conceptos de anisotropía, y como especificar una propiedad cuando varía con la dirección.
Se considera un sistema mecánico donde un
anillo central es sostenido por medio de dos pares de resortes ortogonales, a un marco (Figura 2.1). Los resortes a los lados opuestos del anillo son idénticos, pero tienen diferentes constantes de elasticidad al par ortogonal. Se aplica una fuerza general F al anillo; vector causa, y se obtiene un desplazamiento; vector efecto. La relación entre la causa y el efecto, define la rigidez de los resortes.
Cuando se aplica una fuerza general F al
sistema, la intuición lleva a pensar que el desplazamiento del anillo dependerá de la rigidez relativa de ambos pares de resortes, relación causa-efecto. i.e. el desplazamiento dependerá de la orientación de la fuerza. De todas maneras, el vector desplazamiento no se torna paralelo al vector fuerza debido a la diferencia en las constantes de elasticidad de los resortes, i.e. debido a la anisotropía.
Figura 2.1. En este sistema, una anillo central, es sostenido por dos pares de resortes, los cuales están fijos a un marco.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 15
La componente de la fuerza F aplicada en las direcciones x e y son Fcosθ y Fsinθ respectivamente (Figura 2.2.a). Si la rigidez (definida como el desplazamiento por fuerza unitaria aplicada) de los resortes a largo del eje x es k1, y a lo largo de y K2, entonces los desplazamientos serán x = k1 Fcosθ e y = K2 Fsinθ. El desplazamiento resultante es mostrado en la Figura 2.2.b, y el ángulo (φ) resultante entre este desplazamiento y el eje x está dado por
tan φ = k2/k1 (tan θ)
Figura 2.2. Análisis del sistema anisotrópico de la Figura 2.1. (a): Componentes de la fuerza F aplicada en los ejes principales. (b): Componentes del desplazamiento en los ejes principales.
Entonces en general, la dirección del desplazamiento no será la misma que la de la aplicación de la fuerza. Sólo cuando θ es igual a 0° ó 90°, el desplazamiento (vector efecto) será paralelo a la fuerza aplicada (vector causa). Basándose en la reciente analogía, se deben realizar una serie de observaciones acerca de la anisotropía:
i. En un sistema anisotrópico, el vector efecto en general no es paralelo al vector causa aplicado.
ii. En el ejemplo bi-dimensional mencionado, existen dos direcciones ortogonales en
las cuales el efecto se vuelve paralelo a la causa. Para las tres dimensiones, se puede demostrar que estas direcciones son tres y ortogonales entre sí.
iii. Un sistema anisotrópico puede ser analizado en términos de las componentes a lo
largo de estas direcciones ortogonales, llamadas ejes principales. Sobre estos ejes principales, los valores de la propiedad física son denominados valores principales. En el caso descrito esta era la rigidez de ambos pares de resortes (k1 y k2). Para las tres direcciones, esto se extiende a k1, k2 y k3
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 16
Para las tres dimensiones, una dirección general puede ser representada mediante sus cosenos direccionales, los cuales se ilustran en la Figura 2.3.a. Considere un sistema en el cual se aplica una fuerza F en una dirección general, resultando en un desplazamiento D desfasado de esta fuerza en ϕ (Figura 2.3.b). La componente de este desplazamiento en la dirección de F se relaciona a la fuerza aplicada F por una constante K (la cual en la analogía mecánica depende de la rigidez de los tres resortes).
Figura 2.3.a. Los cosenos direccionales l, m, n de la dirección de la fuerza F, están dados por los cosenos de los ángulos entre F y los ejes x, y, z respectivamente.
Dado que la componente de D en la dirección de F es DF = D cosϕ K = D cosϕ / F = DF / F Es posible calcular el valor de K en términos de k1, k2, y k3. Las componentes de F en los ejes principales son: Fx = Fl Fy = Fm Fz = Fn Las componentes del desplazamiento en los ejes principales son entonces: Dx = k1Fl Dy = k2Fm Dz = k3Fn La magnitud D resultante del desplazamiento en la dirección de F (DF) puede ser escrita en términos de sus componentes como: DF = Dxl + Dym + Dzn Resultando; DF = k1Fl2 + k2Fm2 + k3Fn2
Luego; K = DF / F Entonces K = k1l2 + k2m2 + k3n2 (Ec. 2.1) Esta última ecuación, define el valor de la propiedad K en cualquier dirección, en términos de los tres valores principales k1, k2, k3 y sus cosenos direccionales l, m, n.
Figura 2.3.b. La componente de D en la dirección de F está definida como DF = D cosϕ
2.2. Propiedades de un tensor de segundo orden y sus variaciones con la dirección
En un sistema de tres ejes ortogonales, es posible visualizar la variación de una propiedad K con la dirección, al representar la magnitud de K como la distancia medida desde un origen. El resultado será una superficie tridimensional cuya forma expresa
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 17
fácilmente la anisotropía de la propiedad. Entonces, en un material isótropo la propiedad en análisis será igual en todas las direcciones, por lo tanto la superficie que la representa es una esfera.
Para obtener la forma de esa superficie de representación, se considera un punto con
coordenadas x,y,z a una distancia r del origen en la dirección de F (Figura 2.4). Los cosenos direccionales l,m,n pueden ser escritos en términos de x,y,z como
Figura 2.4. Esquema utilizado para la derivación de las superficies representativas que describen la variación de una propiedad perteneciente a un tensor de segundo rango.
Figura 2.5. Elipsoide con semiejes a, b y c.
l = x/r, m = y/r, n = z/r
Substituyendo estos valores en Ec. 2.1, se obtiene que
r2K = 1 Es decir, r = 1/√K, entonces
k1x2 + k2y2 + k3z2 = 1 (Ec. 2.2) Si los tres valores principales, k1, k2, k3 son positivos, entonces la Ec. 2.2 describe una elipsoide. La ecuación característica para una elipsoide es
x2/a2 + y2/b2 + z2/c2 = 1 Donde a, b y c son los semiejes (Figura 2.5). los semiejes para la superficie que representa la propiedad anisótropa son
1/√ k1, 1/√ k2, 1/√ k3 En cualquier dirección general, el radio es igual al valor de 1/√K en esa dirección. 2.3. Control de la simetría sobre las propiedades físicas
Se denominará X, Y, Z a los ejes principales de la elipsoide, siempre ortogonales. Y para describir el control de la simetría sobre las propiedades físicas, se debe analizar como están relacionados estos ejes a los ejes cristalográficos del mineral.
Principio de Neumann: Los elementos de simetría de cualquier propiedad física de un cristal deben incluir los elementos de simetría del grupo puntual del cristal. Entonces, a
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 18
partir de la analogía mecánica, y el conocimiento de la descripción de propiedades a partir de una elipsoide, se tiene para los distintos sistemas cristalográficos.
i. Para un cristal triclínico, dado que no hay restricciones de simetría, la elipsoide puede tener cualquier orientación respecto de los ejes cristalográficos.
ii. En el sistema monoclínico, ya que existe un eje binario a lo largo de uno de los
ejes cristalográficos, uno de los ejes de la elipsoide debe ser paralelo a este eje.
iii. En el sistema ortorrómbico, los tres ejes de la elipsoide deben ser paralelos a los ejes cristalográficos.
iv. Cuando se consideran los sistemas tetragonal, hexagonal y trigonal se hace
evidente que la elipsoide es de menor simetría, sin embargo si la elipsoide general tiene k1 = k2, tendrá entonces una sección circular en el plano XY. (elipsoide de revolución). Este plano es entonces, perpendicular al eje de mayor simetría del cristal, ejes de 4, 6 y 3 repeticiones.
v. En el sistema cúbico la propiedad de tensor de segundo orden es isótropo.
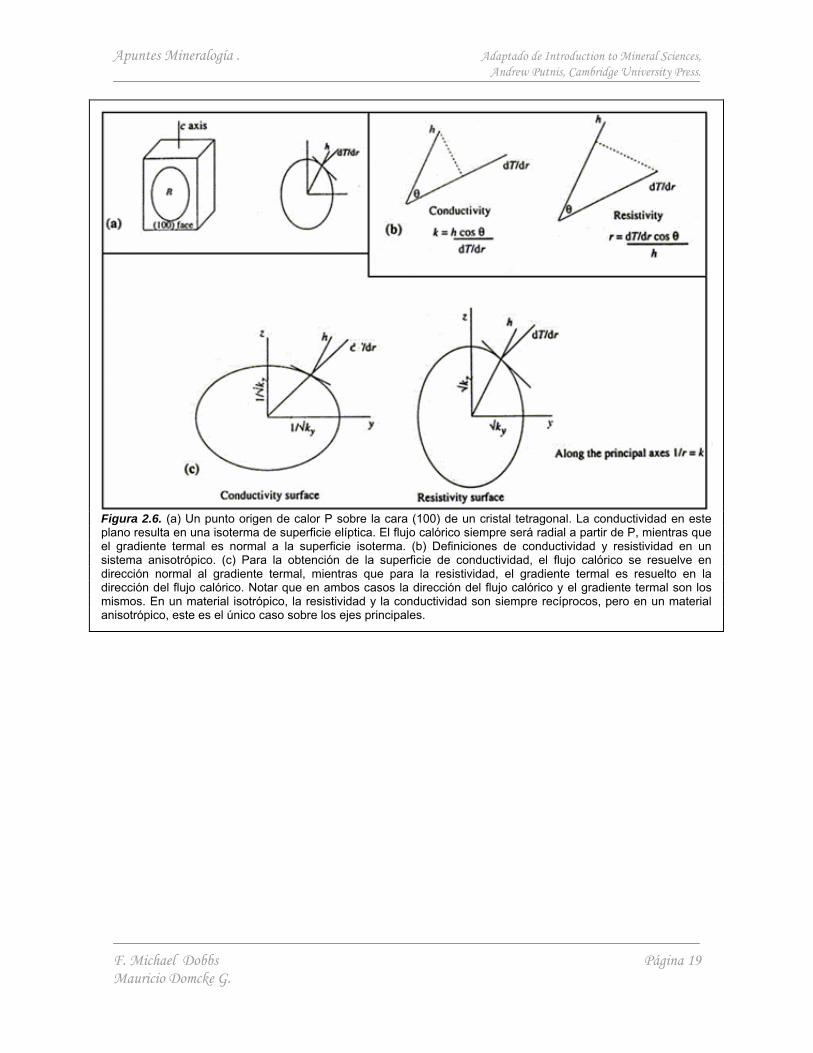
Un par de ejemplos del control que ejerce la simetría sobre las propiedades físicas de los minerales se muestran a continuación. La Figura 2.6 indica como se propaga un flujo calórico sobre un cristal, y la Figura 2.7 muestra la difusión anisotrópica del Ni en el Olivino.
Mauricio Domcke G.
Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 19
Figura 2.6. (a) Un punto origen de calor P sobre la cara (100) de un cristal tetragonal. La conductividad en este plano resulta en una isoterma de superficie elíptica. El flujo calórico siempre será radial a partir de P, mientras que el gradiente termal es normal a la superficie isoterma. (b) Definiciones de conductividad y resistividad en un sistema anisotrópico. (c) Para la obtención de la superficie de conductividad, el flujo calórico se resuelve en dirección normal al gradiente termal, mientras que para la resistividad, el gradiente termal es resuelto en la dirección del flujo calórico. Notar que en ambos casos la dirección del flujo calórico y el gradiente termal son los mismos. En un material isotrópico, la resistividad y la conductividad son siempre recíprocos, pero en un material anisotrópico, este es el único caso sobre los ejes principales.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 20
2.4. Propiedades ópticas de los minerales
Figura 2.7. Las dos secciones principales de la superficie de representación que describe la variación del coeficiente de difusión en el mineral ortorrómbico olivino. En esta ocasión a : b ; c toman los valores de 0.48 : 0.55 : 0.09 respectivamente.
2.4.1. Variación del índice de refracción en un cristal - indicatriz óptica Las propiedades ópticas, deben ser expresadas en términos que permitan relacionar el vector causa con el vector efecto. El índice de refracción (n) de un medio isotrópico, se define como la razón entre la velocidad de la luz en el vacío (c), y la velocidad de la luz del medio (v). i.e., n = c/v. En un mineral, este índice de refracción se representa como cualquier tensor de segundo rango, con todas las propiedades, restricciones y alcances mencionados anteriormente. La forma más general corresponde a una elipsoide denominada indicatriz óptica. 2.4.2. Control de la simetría en la variación del índice de refracción en un cristal La variación del índice de refracción en un cristal se analiza de forma análoga a la realizada en la sección 2.2 de este escrito.
i. En el sistema cúbico, no existe variación con la dirección del índice de refracción y entonces la elipsoide se transforma en una esfera que tiene como eje único el valor de “n”.
ii. Para cristales que pertenecen al sistema tetragonal, hexagonal y trigonal, la
indicatriz óptica se transforma en una elipsoide de revolución, con sección circular normal a los ejes 3 ó 4 ó 6. Se deben indicar dos valores principales para los ejes, e y o (de extraordinario y ordinario respectivamente). Figura 2.8.a.
iii. Para cristales de menor simetría se deben indicar tres valores principales para los
índices de refracción. Estos son α < β < γ. Figura 2.8.b.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 21
Frecuentemente resulta útil imaginar la indicatriz óptica, como una superficie inmersa dentro del cristal. Gracias a lo anterior, se ha conseguido identificar con claridad la relación existente entre los ejes cristalográficos y la indicatriz óptica. La Figura 2.9 muestra dos claros ejemplos de visualización. En el cuarzo (quartz), la indicatriz es más extendida en la dirección paralela al eje c, debido a que e > o. Para tales casos, se refiere a cristales ópticos positivos. Para la calcita (calcite), la cual representa el caso contrario, se denomina cristal óptico negativo.
Figura 2.8. La variación con la dirección del índice de refracción en un cristal, es descrita por una elipsoide denominada indicatriz óptica.
Figura 2.9. Relación entre la orientación óptica de una indicatriz uniaxial y la orientación cristalográfica.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 22
El eje normal a una sección isotrópica es denominado eje óptico. En un cristal uniaxial, el eje óptico es perpendicular al plano xy¸ ordinario. Para cristales biaxiales, existen dos secciones isotrópicas, una de ellas se muestra en Figura 2.9.a. El ángulo entre los dos ejes ópticos es denominado 2vγ (Figura 2.9.b). Si 2vγ < 90°, el cristal es óptico positivo y si 2vγ > 90°, el cristal es óptico negativo. 2.4.3. Birrefringencia El comportamiento óptico de un cristal es consecuencia de las propiedades
anisotrópicas que posee. Dicho con mayor simpleza, si un rayo de luz incidente atraviesa un cristal anisotrópico, da origen a dos rayos refractados, en vez de uno. Estos dos rayos refractados siempre se polarizan en planos perpendiculares entre si, por lo que se tienen dos índices de refracción diferentes. Si los índices de refracción son bastante distintos, y el cristal es suficientemente grueso, los dos rayos pueden ser fácilmente separados, debido a la diferencia en el ángulo de refracción. La doble refracción de la calcita (Figura 2.11) ilustra claramente esta situación. La
diferencia numérica entre los dos índices de refracción es denominada birrefringencia. La calcita, es uno de los minerales que posee mayor birrefringencia.
Figura 2.10. Ejes ópticos para un cristal biaxial
Figura 2.11. Doble refracción en la calcita
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 23
Resulta importante interpretar la birrefringencia en términos de la relación que existe entre la orientación del rayo de luz incidente y la orientación de la indicatriz, y entonces visualizar cómo la birrefringencia depende de la orientación. Notar que el vector luz eléctrica E, el cual interactúa con el cristal, es siempre perpendicular a la dirección del rayo, debido a esto, los dos rayos refractados vibran en direcciones ortogonales entre sí. El plano en que estos dos rayos refractados vibran, es el plano normal a la dirección del rayo incidente. La Figura 2.12 ilustra la anterior situación con un rayo incidente en una indicatriz uniaxial. El plano normal al rayo incidente (e’o) define una sección elíptica dentro de la indicatriz (línea segmentada), Las vibraciones E de los rayos refractados se deben encontrar dentro de la sección e’o. Los dos ejes principales de esta sección definen las dos direcciones de vibración, y las dimensiones de los semiejes mayor y menor definen los dos índices de refracción.
Figura 2.12. Rayo incidente en una indicatriz uniaxial. El vector E se encuentra inmerso en el plano e’o.
En la Figura 2.12., se puede ver, que si el rayo incide a través del eje óptico, los dos rayos refractados tendrán direcciones o y o, e índices de refracción de igual magnitud, por lo que la birrefringencia es nula. Por otro lado, si el rayo incidente entra a través de la dirección de o (cualquiera de los dos semiejes), los rayos refractados se propagarán a través de e y o (este último ortogonal a la dirección del rayo incidente), y la diferencia de los índices de refracción (birrefringencia) es la máxima que se puede tener dentro de la indicatriz. Finalmente, se debe considerar un último aspecto al respecto de la doble refracción. Hasta ahora, no se ha impuesto ninguna restricción del estado de polarización del rayo incidente. Sin embargo, Si deseamos describir las relaciones de fase entre los dos rayos refractados, el rayo incidente debe ser polarizado plano. Esto se consigue haciendo pasar luz ordinaria a través de un filtro polaroide (polarizador) antes de que entre al cristal. (Figura 2.13).
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 24
Figura 2.13. Una rayo de luz no polarizado, el vector eléctrico E oscila en todas las direcciones normales a la dirección del rayo incidente. Cuando este pasa a través de un polarizador, sólo la componente de E a lo largo del eje de transmisión del polaroide es transmitido, resultando un rayo de luz polarizado.
Cuando este rayo incidente polarizado entra a una sección del mineral, es dividido en dos rayos refractados, cada uno inicialmente de igual fase. A medida que los dos rayos pasan a través del mineral se va produciendo una diferencia entre ellos que deja a uno más retrasado, ya que experimentan índices de refracción diferentes y por lo tanto velocidades de propagación distintas. Estos salen del mineral vibrando en direcciones ortogonales, las que generarán componentes en un segundo filtro polaroide (analizador) cuya dirección de polarización es perpendicular al polarizador (primer filtro). Esta situación se denomina luz cruzada, y sirve para analizar las direcciones de polarización de la luz que pasan por un cristal. (Figura 2.14) Si un cristal es isótropo, el vector E incidente pasará a través de él sin cambiar su orientación por lo tanto será bloqueado por el analizador. Se dice que el cristal esta en extinción; un cristal isótropo está siempre en extinción. En un cristal anisótropo el vector incidente E será dividido en dos componentes ortogonales. Sin embargo si este es paralelo a una de esas direcciones solo uno de los componentes será transmitido, el estado de polarización permanecerá sin cambios y por tanto el analizador bloqueará al rayo. Esto es una posición de extinción. Existe otra posición de extinción al rotar 90º el cristal de tal forma que el vector incidente E es paralelo al otro componente.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 25
Figura 2.14. Esquema de polarización de luz similar al Microscopio.
2.4.4. Colores de interferencia
La mayoría de los minerales muestran una birrefringencia menor que la calcita, por tanto esta propiedad no es notable sin utilizar un microscopio petrográfico. Un rayo de luz polarizada plana que atraviesa un mineral anisótropo se parte en dos rayos de distinto índice de refracción, distinta velocidad de transmisión. Entonces si recorren la misma distancia (espesor del corte de roca, 30 μm), emergerán en una posición diferente en la onda, desfasados. La extensión del desfase depende de la diferencia de índices de refracción. Los dos rayos que vibran ortogonales, vectores E ortogonales, con diferencia de fase entre ellos, se recombinan en el analizador formando una sola onda que es el resultado de la interferencia entre las dos que emergen del cristal. En el caso de una iluminación blanca la interferencia resulta de la destrucción de ciertas longitudes de onda, dando como resultado un color de interferencia característico. Si los minerales son examinados en una sección transparente estándar, los colores de interferencia se relacionan directamente con la birrefringencia. 2.5. Una nota en las propiedades elásticas de los minerales
Aún cuando al inicio de este capítulo se mencionó que las propiedades elásticas se encuentran en una categoría distinta a las propiedades de segundo rango, se hará una breve referencia a ellas.
En el análisis del comportamiento de los minerales ante las solicitaciones de esfuerzo
diferencial se usa la relación existente entre el tensor de esfuerzos σ y el tensor de deformaciones λ (elipsoides). En el ámbito de la deformación elástica estos tensores
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 26
están linealmente relacionados por las constantes elásticas del cristal. El caso más general, el triclínico, necesita de 21 de ellas, las que se reducen a 3 en los minerales cúbicos. Para agregados sólidos isótropos, vidrios y rocas policristalinas, hay dos constantes elásticas, el Módulo de Young (σ/ε) y el de Poisson (-ε'/ε, donde ε corresponde a la deformación en la dirección del esfuerzo y ε' la deformación en las direcciones ortogonales).
2.6. Relación entre anisotropía y estructura cristalina La relación entre estas es evidente. En algunos minerales que presentan una extrema
anisotropía como el grafito, este tiene una estructura conformada por capas de carbono fuertemente enlazados a una distancia de 1.42Ǻ, (Figura 2.15.a) mientras las capas se enlazan débilmente a una distancia de 3.35Ǻ. Estas capas son fácilmente separadas, lo que le confiere la suavidad característica. Esta fuerte anisotropía explica que la resistividad eléctrica perpendicular a las capas sea de 5 * 10-3 Ωm y la paralela a las capas sea de 5 * 10-6 Ωm, es decir, el grafito es semiconductor perpendicular a las capas y es conductor metálico paralelo a estas. Similar a la conductividad térmica que es 28.3*10-6K-1 perpendicular a las capas y virtualmente cero paralelo a las capas.
Para la calcita es evidente con los índices de refracción, dado que si el vector E vibra
paralelo a los planos CO3 el índice de refracción es de 1,658 y si E vibra perpendicular a estos, entonces el índice es de 1.486, debido a la perturbación del campo eléctrico provocado por los oxígenos vecinos en las capas. (Figura 2.15.b).
Figura 2.15. (a) Estructura del grafito. Capas de átomos de carbono dispuestos en las esquinas de una malla hexagonal. (b) Estructura de la Calcita.
2.7. Anisotropía y la forma externa de los minerales La interpretación científica de la morfología de los minerales comienza con Bravais
alrededor de 1850, quien postuló que las caras de un cristal deberían ser aquellas que mostraban una mayor densidad de puntos de red. A principios del siglo XX, se introdujeron sofisticaciones a estos postulados; pero sin tomar en cuenta los arreglos
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 27
atómicos ni los tipos de enlaces que participan, por tanto no son aplicables al tratar de predecir la estructura de superficie.
Hartman y Perdok en 1955 relacionan la morfología a direcciones estructurales importantes, esto es, direcciones que contienen una cadena de elementos fuertemente enlazados, llamadas cadenas de enlace periódico. Asbestos fibrosos, micas en capas son ejemplos de esto. En estructuras más complejas la relación no es tan evidente lo que es expresado por la introducción de impurezas en el ambiente de crecimiento.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 28
3. LA ESTRUCTURA CRISTALINA DE LOS MINERALES I 3.1. Enlaces en estructuras cristalinas Un enlace es la fuerza cohesiva que mantiene unidos a los átomos dentro de una estructura, existen en los sólidos cristalinos enlaces en las tres dimensiones y son esencialmente cuatro.
i. Metálico: En un átomo de elementos metálicos usualmente hay una, dos o raramente tres valencias de electrones exteriores débilmente ligadas al núcleo, por lo que pueden ser removidas para formar un ion positivo. En una estructura metálica, cada átomo aporta estos iones los cuales se mueven libremente como en un “mar” de electrones, el cual provee el “pegamento” necesario para enlazar a otros iones y formar el enlace.
ii. Covalente: Comparten uno o más electrones del orbital externo, provocando un
traslape de orbitales, que define la dirección del enlace. La coordinación entre átomos vecinos con enlaces covalentes está restringida y bien definida
iii. Iónico: El llenado del faltante del orbital externo es realizado por transferencia de
electrones desde un átomo al otro, tal que resulta un anión y un catión con simetría esférica. Fuerza electrostática y empaquetamiento cerrado.
iv. Van der Waals: Es la débil atracción universal existente entre átomos y moléculas
neutras que se encuentran muy cerca. La explicación de este enlace yace en que aún en una carga neutra existe una distribución de cargas la que fluctúa rápidamente produciendo esta pequeña vibración lo que posibilita el enlace.
3.2. Descripción de estructuras cristalinas Existen distintas formas de describir una estructura cristalina. La más precisa es especificar la forma y tamaño de la celda unitaria (es decir, el sistema cristalino y los parámetros de la red) y alistar las coordenadas de cada tipo de átomo que se encuentra dentro de la celda. Esta forma no es clara en la visualización de la estructura y la comparación con otras, por lo que existen maneras alternativas de describir el arreglo de átomos en la estructura y la más simple es la de empaquetamiento cerrado. 3.2.1. Estructuras de Empaquetamiento Cerrado (EC). ECCúbico y ECHexagonal
Se trata de describir la estructura como si fuesen esferas idénticas ordenadas en una caja de tres dimensiones. Este concepto es directamente aplicable a las estructuras de los metales, pero también es útil para muchas otras estructuras donde la ocupación del espacio por átomos es de importante consideración. La Figura 3.1muestra un empaquetamiento de esferas (radio = r) bidimensional, en el cual cada esfera se encuentra en contacto con seis otras. Dentro de este
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 29
empaquetamiento, existen 3 direcciones de empaquetamiento a 60°. La celda unitaria que repite este patrón bi-dimensional es hexagonal, con parámetros de red a = 2r. Para construir una estructura tri-dimensional, se superponen capas idénticas una sobre otra, y la mejor forma de hacer esto, es que las esferas de la capa superior, llenen los espacios que deja la capa inferior. Refiriéndose a la primera capa como la capa A, se puede apreciar que existen dos sitios que la capa posterior puede ocupar, denominados B y C en la Figura 3.1. La segunda capa puede ocupar todos los sitios B (con lo que se transforma en la capa B), o todos los sitios C (con lo que se transforma en la capa C). En superponer la capa B o C, la diferencia yace meramente en rotar la capa con una esfera como pivote en un ángulo de 180°, por lo que no hay diferencia geométrica. Se escoge por conveniencia alfabética la capa B, obteniendo un arreglo de capas AB. Sin embargo, la elección de la tercera capa tiene altas consecuencias. Nuevamente existen dos posibilidades; la capa se puede posicionar directamente por sobre la capa A, o lo puede hacer por sobre las posiciones C. La primera opción provoca una secuencia de capas ABA, la que si se repite de la misma forma proporciona un arreglo ABABAB... el cual es llamado Empaquetamiento Cerrado Hexagonal (ECH). La segunda opción, conduce a una secuencia de capas ABCABC... la cual es llamada Empaquetamiento Cerrado Cúbico (ECC). Se verá cada una a continuación en más detalle. La Figura 3.2 ilustra un ECH. La celda unitaria es descrita por la repetición hexagonal en la capa A (ah = 2r) con la dimensión de c (ch) igual al doble del ancho de la capa. Inspecciones de la superposición de capas revelan que los átomos en la capa B no poseen el mismo entorno que los de la capa A, (la orientación de los enlaces es diferente), por lo que la celda es primitiva con puntos de la red sólo en las esquinas. Los planos de empaquetamiento cerrado son paralelos a (001). Muchos metales poseen esta estructura de empaquetamiento cerrado, incluyendo al magnesio (a = 3.21Ǻ y c = 5.21Ǻ), zinc (a = 2.66Ǻ y c = 4.95Ǻ) y el titanio (a = 2.51Ǻ y c = 4.68Ǻ).
Figura 3.1. Una capa simple de esferas de radio r . Existen dos tipos de sitios en esta capa denominados B y C respectivamente.
Figura 3.2. ECH genera esta estructura, con átomos en las posiciones coordenadas 0,0,0 y 1/3,2/3,1/2 en cada celda unitaria. El conjunto de átomos a la altura z = ½ genera la capa B en el empaquetamiento cerrado ABAB... y es mostrado con color negro en la Figura.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 30
La Figura 3.3 ilustra el ECC. Se podría describir la celda unitaria de esta estructura de la misma manera que se hizo para el ECH, es decir, basándose en la celda unitaria hexagonal de la capa A, pero ahora existe una repetición de tres capas. Por lo anterior, esta estructura se describe en base a la celda unitaria hexagonal pero con repetición de tres capas en la dirección de c; A, B y C, las que tienen todas idéntico ambiente definiendo una celda unitaria cúbica centrada en las caras. El Cu (a = 3.61Ǻ), Ag (a = 4.08Ǻ), Au (a = 4.07Ǻ) y Al (a = 4.05Ǻ) son algunos de los muchos metales que presentan estructuras ECC.
Otra estructura comúnmente adoptada por los metales es la cúbica de cuerpo centrado, la que contiene un átomo en el centro del cubo cuyo ambiente es idéntico al de los vértices y por ello cada átomo está en un nodo de la red. Esta estructura (Figura 3.4) la encontramos en el Na (a = 4.22Ǻ), Cr (a = 2.88Ǻ) y Fe (a = 2.87Ǻ).
Figura 3.4. Estructura cúbica de cuerpo centrado con átomos en las posiciones 0,0,0 y 1/2,1/2,1/2 en cada celda unitaria.
Figura 3.3. (a) ECC ABCABC... mostrado en la misma orientación que en la Figura 3.2, con los átomos A en la posición 0,0,0; Los átomos B en la posición 1/3,2/3,1/3 y los átomos C a 2/3,1/3,2/3 en una celda unitaria. (b) Muestra la celda cúbica convencional, es el mismo arreglo, pero organizado en las tres dimensiones. El sombreado de los átomos es congruente con el de la Figura (a) para mostrar la relación entre las dos orientaciones. Se observa que se disponen en planos del tipo (111).
3.2.2. Sitios intersticiales en Estructuras de Empaquetamiento Cerrado Varias estructuras simples de minerales pueden ser descritas en términos de aniones ordenados en un empaquetamiento cerrado aproximado, en donde pequeños cationes ocupan los espacios intersticiales.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 31
Lo primero entonces, es describir las formas de los sitios producidos entre tales iones. Estos sitios se encuentran entre dos capas empaquetadas y son los mismos tanto para ECC como para ECH. (Figura 3.5)
(a)
(b) (c) (d)
Figura 3.5. En (a), dos capas con empaquetamiento cerrado muestran los dos tipos de sitios intersticiales entre átomos con estructura tipo empaquetamiento cerrado. La capa de átomos sombreados se encuentra bajo la sin sombra.
En Figura 3.5, los intersticios etiquetados con “x” están envueltos por cuatro átomos empaquetados, los cuales forman un tetraedro apuntando hacia “afuera”, tal como se muestra en (3.5c). Existen otros sitios similares a los anteriores, pero con el tetraedro formado por los átomos apuntando hacia “adentro,” este tipo se muestra en (3.5b). Los sitios etiquetados con “•”, son octaédricos y su número de coordinación es seis (un octaédro lo forman 6 vértices), tal como se muestra en (3.5d). Considerando lo anterior, se debe hacer notar que en una estructura de empaquetamiento cerrado, ya sea cúbico o hexagonal, existe el doble de sitios tetraédricos al respecto de los octaédricos.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 32
Las Figuras 3.6 y 3.7 muestran la orientación y posición de octaedros y tetraedros para ECC y ECH. Por simplicidad, se muestra un solo sitio para cada caso.
Figura 3.6. Empaquetamiento cerrado cúbico. En (a) se muestran las posiciones de los sitios tetraédricos, mientras que en (b), las de los octaédricos. Ambos para una celda unitaria.
(a) (b)
Figura 3.7. Empaquetamiento cerrado hexagonal. En (a) se muestran las posiciones de los sitios tetraédricos, mientras que en (b), las de los octaédricos. Ambos para una celda unitaria.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 33
Aunque los sitios intersticiales están presentes en la misma proporción en la ECC y ECH, sus orientaciones en la celda unitaria en relación al empaquetamiento cerrado es distinta en cada caso.
En las figuras anteriores (3.6 y 3.7) se muestran los sitios en relación a las celdas
unitarias en los que se ha destacado solo uno de ellos. En la estructura ECC hay cuatro iones de empaquetamiento cerrado por celda unitaria, entonces, existen cuatro sitios octaédricos y ocho sitios tetraédricos por cada una. En la estructura ECH hay dos iones de empaquetamiento cerrado por tanto existen dos sitios octaédricos y cuatro tetraédricos por celda unitaria.
Una consideración importante para este tipo de análisis es la regla de la razón de
radios: r / R, para un sitio tetraédrico r / R ≥ 0,225 y para uno octaédrico r / R, = 0,414 3.2.3. Estructuras tipo basadas en empaquetamiento cerrado
Si se consideran estructuras simples basadas en iones ordenados en empaquetamiento cerrado, los cuales serán usualmente aniones (X) y cationes (M) en los sitios intersticiales, se pueden definir algunos tipos estructurales importantes.
i. Empaquetamiento cerrado cúbico (ECC) con todos los sitios octaédricos ocupados. Esta es la estructura de la bien estudiada halita.
Halita (NaCl): Su celda unitaria es cúbica con a = 5.62Å y su red de Bravais es F, por lo que se tiene un octaedro en el centro (juntando los seis puntos del “centrado en las caras”). Los aniones Cl- se encuentran en las esquinas y en el centro “a 1”, y en los puntos medios “a 1/2”. Por el contrario, los cationes Na+ se encuentran “a 1/2” en las esquinas y en el centro, y “a 1” en los puntos medios. Los enlaces Na-Cl se realizan entre las aristas del cubo y dada la coordenada z de los iones, la distancia del enlace es a/2 = 2.81 Å. (La escritura “a 1” ó “a 1/2” es una manera de mostrar la coordenada z de los iones). (Figura 3.8)
Figura 3.8. Estructura de la Halita
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 34
ii. Empaquetamiento cerrado cúbico (ECC) con la mitad de los sitios tetraédricos ocupados. La estequiometría de la estructura es MX y se llama estructura de la Esfalerita ya que es una de las estructuras del ZnS.
Esfalerita o Blenda de Zinc (ZnS): Su celda unitaria es cúbica con a = 5.41Å y su red de Bravais es F, por lo que existen 8 tetraedros (uno por cada vértice del cubo). En los puntos de la red se encuentran los azufres (S-) y los cationes (Zn+) ocupan sólo la mitad de los tetraédros (2 por arriba en la diagonal y al inverso los de abajo). Por esto se puede representar como ECC semi-tetraédrico donde los aniones ocupan las esquinas y el centro “a 1”, y los puntos medios “a 1/2”, mientras que para los cationes las coordenadas son (1/4,1/4,1/4); (1/4,3/4,3/4); (3/4,1/4,3/4); (3/4,3/4,1/4). La distancia del enlace Zn-S es de 2.34 Å. (Figura 3.9)
Figura 3.9. Estructura de la Esfalerita
iii. Empaquetamiento cerrado cúbico (ECC) con todos los sitios tetraédricos ocupados. La estequiometría es ahora M2X o MX2 dependiendo del ion que esté en empaquetamiento cerrado. La estructura es llamada estructura de la Fluorita, CaF2, en donde el Ca2+ está en arreglo de empaquetamiento cerrado y el F- en los sitios intersticiales. El caso contrario se conoce como estructura Anti- Fluorita.
Fluorita (CaF2): Su celda unitaria es cúbica con a = 5.46Å y su red de Bravais es F, por lo que existen 8 tetraedros (uno por cada vértice del cubo). En los puntos de la red se encuentran los cationes Ca2+ (a excepción de los demás ECC) y ocupando todos los sitios tetraédricos los aniones F-. Debido a lo anterior, es que se puede describir la estructura como ECC tetraédrico con los cationes ocupando los vértices y el centro “a 1”, y los puntos medios “a 1/2”, y los aniones en filas y columnas “a 1/4“ y “a 3/4“, y alternando en c también “a 1/4“ y “a 3/4“ (en total ocho aniones). La distancia del enlace Ca-F es de 2.36 Å. (Figura 3.10)
Figura 3.10. Estructura de la Fluorita
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 35
iv. Empaquetamiento cerrado hexagonal
(ECH) con todos los sitios octaédricos ocupados. El ejemplo tipo es la estructura del NiAs (Arseniuro de Ni).
Arseniuro de Níquel (NiAs): Puede ser descrito como un ECH octaédrico con parámetros a = 3.6Å y c = 5.0Å, con los aniones (As) en las esquinas “a 1” y “a (1/3, 2/3,1/2)”, y los cationes Ni “a (2/3,1/3,1/4)” y “a (2/3,1/3,3/4)”. La distancia del enlace Ni-As es 2.43Å. (Figura 3.11)
Figura 3.11. Estructura del Arseniuro de Níquel
v. Empaquetamiento cerrado hexagonal (ECH) con la mitad de los sitios tetraédricos ocupados. Llamada estructura de la Wurtzita, otra de las formas del ZnS.
Wurtzita (ZnS): Este polimorfo de la Esfalerita es descriptible mediante un ECH semi-tetraédrico, con parámetros a = 3.81Å y c =6.23 Å. Los aniones de azufre se encuentran en las mismas posiciones que en el NiAs, pero los cationes de Zn se disponen en todas las esquinas “a 5/8” y en la misma posición que el otro azufre “a 1/8”. La distancia del enlace Zn-S es de 2.34 Å. (Figura 3.12).
Figura 3.12. Estructura de la Wurtzita
3.2.4. Minerales con estructura basada en Empaquetamiento Cerrado
La Periclasa (MgO) y la Galena (PbS) tienen la estructura del NaCl, y así existen muchos otros óxidos y sulfuros cuyas estructuras se basan en los 5 tipos antes descritos, o en algunas de sus variantes.
i. La estructura de la Calcopirita
La composición es CuFeS2 y tiene una estructura basada en la blenda de zinc, con los átomos de S en ECC y con la mitad de los sitios tetraédricos ocupados, todos con sus ápices apuntando en la misma dirección. Si los átomos de Cu y Fe tuviesen una probabilidad de 0,5 en los sitios tetraédricos, entonces la estructura tendría desorden y su estequiometría sería (Cu,Fe)S2. Sin embargo, el ordenamiento de los cationes en ella, resulta del doblamiento de la celda cúbica en una dirección, formando una celda tetragonal. (Figura 3.13).
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 36
La probabilidad de variaciones de la razón Cu/Fe es mayor a alta temperatura, ya que existe un mayor desorden, el que disminuye en baja temperatura al adoptar la estructura doblada de la Esfalerita. A temperaturas altas la Calcopirita forma una solución sólida restringida, con una variación composicional entre Cu y Fe la que disminuye al bajar la temperatura hasta la fórmula inicial. Este proceso es común en varios minerales. (Figura 3.14).
ii. La solución sólida Troilita-Pirrotita (FeS-Fe1-xS)
La estructura de la Troilita, FeS, está basada en del NiAs, con un ECH de los S y con el Fe ocupando los sitios tetraédricos. Bajo los 140ºC, la estructura sufre una pequeña distorsión, por tanto no puede seguir siendo descrita con el uso de una celda hexagonal, sino con una celda unitaria de tipo monoclínica y de mayor tamaño. (Figura 3.15)
En altas temperaturas, la
estructura NiAs del FeS puede variar sobre un rango de composiciones Fe1-xS produciendo vacantes en los sitios octaédricos. Los sulfuros de hierro con esta fórmula general se denominan Pirrotita, los que varían entre FeS y Fe7S8, hasta una vacante cada ocho átomos. El orden de estos sitios vacantes tiene una fuerte dependencia de la temperatura y la fugacidad de S.
Figura 3.13. Estructura de la Calcopirita
Figura 3.15. Troilita, FeS posee la estructura tipo NiAs cuando se encuentra sobre los 140°C.
Figura 3.14. Posible variación composicional en la solución sólida de la parte central del sistema Cu-Fe-S. Las zonas sombreadas denotan tres soluciones sólidas existentes. La primera es cercana a la Bornita Cu FeS5 4 (bornite), luego la cercana a la Calcopirita CuFeS (iss) y finalmente la solución sólida de la Pirrotita (po) 2
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 37
iii. La estructura del Corindón, Hematita e Ilmenita
Los minerales Corindón (α-Al2O3), Hematita (α-Fe2O3) e Ilmenita (FeTiO3) tienen estructura derivada del NiAs, en en la cual los oxígenos presentan un empaquetamiento aproximadamente hexagonal y los cationes ocupando sitios octaédricos. Se distingue en la fórmula que la razón catión/anión no es 1, por lo que sólo dos tercios de los sitios intersticiales están ocupados, y dependiendo el ordenamiento de esta ocupación resulta la diferencia en la celda unitaria. (Figura 3.16).
En la Ilmenita y Hematita la situación se complica ya que puede existir una sustitución de Fe por Ti, de hecho en la Ilmenita la mitad de los Fe están reemplazados por Ti, dependiendo de la temperatura podrá existir una solución sólida entre estos extremos la cual es limitada a baja temperatura.
Figura 3.16. La estructura del Corindón. (a) Una capa de átomos de oxígeno empaquetados (círculos sin relleno) que muestran la ocupación ocatédrica de los sitios ocupados (átomos con relleno). (b) Los átomos de oxígeno en ECH, representados por las líneas horizontales denominadas ABAB...
iv. La estructura de la Espinela
Las Espinelas son un grupo de minerales del tipo AB2O4, cuyo nombre viene del
MgAl2O4 y es descrita como una estructura en que los oxígenos son ECC y los cationes A y B ocupan un octavo de los sitios tetraédricos, y la mitad de los sitios octaédricos. La celda unitaria usual de ellos es A8BB16O32, la que es difícil de ilustrar y presenta una gran flexibilidad de ocupación por distintos cationes. Lo anterior la hace una estructura común dentro de un centenar de compuestos, incluidos varios minerales e importantes óxidos magnéticos comerciales. (Figura 3.17).
La oxidación de la magnetita, Fe3O4, es provocada por el paso del ión Fe+2 a Fe+3, cuando todos los iones son trivalentes se provoca una estructura de Espinela defectuosa originando el mineral Maghemita, Fe21,67O32, esto tiene importancia en la propiedad magnética que pierde esta Espinela defectuosa, la que finalmente se convierte en Hematita.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 38
La estructura de la Espinela es adoptada por algunos sulfuros, llamados Thioespinelas, las que tienen una composición variable en el rango de Fe-Co-Ni-S.
Figura 3.17. Estructura de la Espinela AB2O4. La celda unitaria cúbica contiene 32 átomos de oxígeno en ECC. Para mostrar la distribución de los sitios catiónicos, la estructura puede ser descrita en términos de pequeños cubos denominados “A” y “B” en (a). “A” representa un tetraedro AO4 y “B” el cubo B4O4 los cuales albergan a los cationes B en sitios octaédricos. Para completar la estructura, la anterior distribución de cubos AO4 y B4O4 se ubican dentro de los octantes de un cubo mayor en los cuales existen átomos de A en las esquinas y centrados en las caras, como se muestra en (b).
3.2.5. Estructuras construidas de poliedros A menudo, resulta bastante conveniente describir una estructura en términos de la coordinación de un catión dentro de un poliedro, y la manera en la cual estos poliedros se conectan entre ellos, ya sea compartiendo ápices, aristas o caras. En las estructuras de empaquetamiento cerrado mencionadas anteriormente, se dio cuenta de la coordinación de varios cationes. Por ejemplo, al mencionar que en la Esfalerita los cationes Zn ocupaban sitio tetraédricos, se aclaraba que su coordinación era 4. Por otro lado, en la Halita, al mencionar que el sodio se encontraba en un sitio octaédrico, se hacía mención tácita de que su número de coordinación era 6. (Un hexaedro regular (cubo) lo conforman 8 vértices, por lo tanto su coordinación es 8, por otro lado, el octaedro lo conforman 6 vértices por lo que su número de coordinación es 6).
La estructura del NaCl que es de tipo ECC puede ser descrita también como octaedros que comparten aristas. Cada arista es compartida por dos octaedros; al centro del octaedro hay un átomo de Na y en cada vértice hay uno de Cl . La Figura 3.19 muestra que dentro de la celda unitaria del NaCl los sitios tetraédricos no están ocupados.
Figura 3.19. Estructura de la Halita, descrita
en términos del compartimiento de aristas de octaedros
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 39
La estructura de la blenda de Zn
puede ser descrita como tetraédros que comparten vértices. Cada vértice es compartido por cuatro tetraédros en cuyos centros se ubican los átomos de Zn. (Figura 3.20).
Por otro lado, la estructura de la
Espinela puede ser vista como fila de octaedros que comparten aristas, y que corren a lo largo de una de las diagonales del cubo, los que a su vez están unidos con las siguientes líneas de octaedros a través de tetraedros unidos por un vértice a los octaedros. Entonces, cada vértice esta compartido por tres octaedros y un tetraedro. (Figura 3.21).
Figura 3.20. Estructura de la Esfalerita, descrita en términos de tetraédros, los cuales comparten vértices.
i. Estructura de la Perovskita
La Perovskita CaTiO3 tiene una estructura que es adoptada por muchos compuestos con fórmula general ABO3. La estructura ideal es cúbica con octaedros en cada vértice en un arreglo infinito de vértices compartidos. El catión A está al centro del cubo y coordinado a doce átomos de O, ubicados estos en la mitad de cada una de las aristas del cubo. En ésta disposición ideal, la razón de largos de enlace A-O a B-O debe ser igual a √2, (Figura 3.22). Cuando ésta condición no puede ser satisfecha la estructura se distorsiona cambiando la forma del octaedro o torciendo los enlaces entre los octaedros. Esto provoca una pérdida de simetría, la que en la mayoría de los minerales se recupera a muy altas presiones. Es por esto que se infiere que el manto inferior de la Tierra está conformado por perovskitas, MgSiO3, de alta simetría, (Figura 3.23).
Figura 3.21. Parte de la estructura de la Espinela, en donde se muestra la relación existente entre los tetraedros AO4 y los octaedros BO6 en una cara (100) de la celda unitaria cúbica
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 40
ii. La estructura del Rutilo
En el rutilo, la estructura forma cadenas de octaedros TiO6 a lo largo del eje c los cuales comparten aristas con los octaedros vecinos y están unidos mediante aristas opuetas con el octaedro del frente. La celda unitaria es tetragonal con parámetros a = b = 4.59 Å, y c=2.96 Å. A altas presiones, el SiO2 posee la estructura del rutilo y el polimorfo de la Sílice se llama estishovita. Es una de las pocas estructuras donde el Si no aparece en coordinación tetraédrica con el O. (Figura 3.24).
Figura 3.23. Dos modificaciones de la estructura de la Perovskita. (a) Rotación alternada de los octaédros BO64. (b) Desplazamiento de los Figura 3.22. La estructura cúbica cationes B
de la Perovskita ABO3.
Figura 3.24. La estructura del rutilo TiO6. (a) arreglo de cadenas de octaedros TiO que comparten aristas 6vistas a su largo. (b) Vista de la estructura a lo largo del eje a, mostrando la cadena de octaedros. (c) Una vista de la estructura a través de [110] que muestra como las cadenas de octaedros se encuentran unidas lateralmente a través de sus esquinas.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
4. LA ESTRUCTURA CRISTALINA DE LOS MINERALES II - SILICATOS
El silicio es el segundo elemento en abundancia en la corteza y el manto después del oxígeno, y entre ellos forman el enlace más fuerte que pueda encontrarse en un óxido. No es de extrañar entonces, que el 95% de las rocas estén formadas por solo unos pocos grupos de minerales, tales como feldespatos, cuarzo, anfíbolas, piroxenos y micas.
Para poder entender los grupos de silicatos, y considerando el capítulo anterior, nos
basaremos en una razón meramente geométrica: poliedros. Estos, en compañía de otros elementos como los átomos Al3+, Fe3+, Mg2+, Fe2+, Na+, Ca2+ y K+ entre otros, y sus radios iónicos, nos ayudarán a clasificar a los silicatos en diferentes asociaciones.
Los distintos grupos mostrarán diversos comportamientos, los cuales se encuentran
condicionados por sus estructuras en una gran medida. La descripción de la estructura de los silicatos depende del modelo que se use para el
enlace Si-O. En un modelo iónico puro, formado de iones Si4+ y O2-, estos se mantienen unidos por fuerzas electrostáticas no direccionales. Los iones O2- presentan empaquetamiento cerrado y con el balance de carga provistos por el Si4+ y otros cationes en los sitios intersticiales de tamaño apropiado. Una estructura como la anterior presenta una densidad que no es propia de la mayoría de los silicatos. En el modelo covalente la descripción del enlace como orbitales traslapados es congruente con el hecho de que en la mayoría de los silicatos, el Si se encuentra en coordinación tetraédrica y el enlace que se ha medido difiere muy poco del ángulo ideal del tetraedro, 109.5º. Sin embargo, ninguno de los modelos explica todos los distintos aspectos, por tanto se establece un consenso de que el enlace es hasta un 50% iónico.
La descripción geométrica de los silicatos se refiere principalmente a como están
asociados los tetraedros [SiO4], o están conectados a otros poliedros catiónicos o comparten vértices con otros tetraedros [SiO4]. El arreglo de tetraedros que comparten vértices forma el esqueleto de la estructura de los silicatos, con los otros cationes ocupando sitios apropiados.
Otra característica que es importante destacar, es la substitución de Si4+ por Al3+ y
considerando que el oxígeno es O2-. Entonces, este reemplazo debe ser equilibrado por la introducción de otros cationes para mantener la neutralidad eléctrica. 4.1. El tetraedro [SiO4]
Para entender la configuración de los silicatos, se comenzará con su esqueleto básico,
i.e., el tetraedro [SiO4]. La geometría del tetraedro está definida por el largo del enlace Si-O y el ángulo de enlace O-Si-O y cómo estos son afectados por la estructura en que están insertos. (Figura 4.1)
Las dimensiones del tetraedro [SiO4] han sido estudiadas mediante numerosas
técnicas, incluida la difracción de rayos X, obteniéndose las siguientes conclusiones principales:
F. Michael Dobbs Página 41 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
i. La longitud media del enlace Si-O es 1.62Å. La resistencia del enlace limita la
variación de esta distancia entre 1.60Å y 1.64Å.
ii. Cuando el tetraedro [SiO4] se
encuentra dentro de una estructura, la distancia de enlace entre los átomos de silicio Si, y los átomos de puente de oxígeno Op son en promedio mayor por cerca de 0.025Å comparados con los enlaces Si-O en los cuales no existe puente de oxígeno. El ángulo del enlace Op-Si-Op es ligeramente menor comparado con aquel en que los oxígenos no se comparten. Esto sugiere que los Si son desplazados de los centros de los tetraédros.
iii. Cuando los tetraédros comparten
esquinas, el ángulo de enlace Si-O-Si define la orientación de la estructura (Figura 4.1c). Este ángulo puede variar entre 120° y 180° (Figura 4.2) dependiendo las condiciones del medio, principalmente de presión y temperatura, en las que se encuentre la estructura. Libre de deformación, este ángulo es de 140°.
Por otro lado,
cuando un Al substituye a un Si, el ángulo de enlace aumenta a 1.75Å. Esta diferencia de tamaño se acomoda con un cambio en el enlace T-O-T.
Figura 4.1. Los silicatos son construidos en base al tetraedro SiO4. (a). Un modelo isométrico en donde se muestran los cuatro oxígenos ubicados en las esquinas del tetraedro. (b) Un modelo menos real, pero aclaratorio, en el que se muestran los enlaces Si-O. (c) Dos tetraedros SiO4 que comparten una esquina para formar un par Si2O7. El ángulo θ, es el ángulo el enlace SI-O-Si.
Figura 4.2. Diagrama de contorno, el cual muestra como la Energía Potencial de un par Si2O7 varía con el largo del enlace Si-O del puente de oxígeno, y el ángulo de enlace Si-O-Si.
F. Michael Dobbs Página 42 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
4.2. Algunas generalizaciones respecto de la composición y estructura de los silicatos
Debido a la complejidad de la composición de los silicatos, resulta beneficioso señalar que existe una relación general entre la fórmula química, y la manera en como se unen los tetraedros de [SiO4].
Los silicatos son clasificados de acuerdo al grado de polimerización que presenten,
y teniendo en cuenta que: • Los tetraedros se unen compartiendo vértices.
• No más de dos tetraedros pueden compartir un vértice, es decir, un oxígeno como
puente.
• La asignación de cargas formales al Si4+ y O2-, es una conveniente manera de asegurar el balance de carga de la fórmula, dando al tetraedro una carga neta negativa, es decir, [SiO4]4-.
i. La estructura más simple corresponde
a tetraedros aislados, i.e., no existe puente de oxígeno. La razón Si:O es 1:4 lo que le da al tetraedro una carga neta negativa [SiO4]4-, un ejemplo es el olivino Mg2SiO4. (Figura 4.1b)
ii. Cuando dos tetraedros forman pares
unidos por el puente de oxígeno, cada uno de ellos tiene tres enlace no conectados por lo que el par tiene una razón de 2/7, entonces una carga [Si2O7]6-, un ejemplo es la rankinita Ca3Si2O7. (Figura 4.1c)
iii. En los silicatos de cadenas simples,
(Figura 4.3a) cada tetraedro tiene dos puentes de oxígeno y dos enlaces libres, lo que le da una razón Si:O=1:3. Las infinitas cadenas tienen una carga neta de [SiO3]n2n-, un ejemplo es la enstatita MgSiO3
iv. En los silicatos de cadena doble,
(Figura 4.3b) dos cadenas simples se encuentran unidas de tal forma que la mitad de los tetraedros tienen dos oxígenos compartidos y dos libres (Si:O=1:3), mientras que la otra mitad tiene tres compartidos y uno libre (Si:O=1:2.5), lo que da una razón neta Si:O=2:5.5. Las cadenas tienen una carga neta de [Si4O11]n6n-, un ejemplo es la
Figura 4.3. (a) Parte de una cadena infinita simple de tetraédros SiO4, en la cual cada tetraedro comparte dos vértices. (b) Parte de una cadena infinita doble de tetraédros SiO4, en la cual la mitad de los tetraedros comparten dos vértices y la otra, tres vértices. (c) Parte de una armazón infinito de tetraédros SiO4, en el cual cada tetraedro comparte tres vértices.
F. Michael Dobbs Página 43 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
antofilita Mg7Si8O22(OH)2. Los grupos OH en las anfíbolas son independientes de los tetraedros.
v. Cuando los tetraedros forman hojas de anillos, (Figura 4.3c) cada tetraedro tiene
tres enlaces compartidos y uno libre, razón Si:O=1:2.5. Las hojas tienen una carga neta de [Si2O5]n2n-, un ejemplo es el talco Mg6Si8O20 (OH)4. Nuevamente, los grupos OH son independientes de los tetraedros.
vi. Finalmente, cuando todos los tetraedros comparten sus vértices con otros
tetraedros, se forma una malla tridimensional, con los cuatro oxígenos compartidos por tetraedro y una razón Si:O=1:2, como el cuarzo SiO2.
Aún cuando esta forma de relacionar la estructura con la geometría posee ventajas,
también existe sus complicaciones:
i. Cuando el Al substituye al Si en el tetraedro, se mantiene la razón Si:O, contrastándola con la razón (Si+Al)/O.
ii. En otros casos el Al puede estar formando parte de los cationes que no
reemplazan al Si, en este caso esto se regulariza en la fórmula química, como por ejemplo en la muscovita K2Al4[Si6Al2O20](OH)4.
iii. En los polimorfos Al2SiO5 (Kianita, Andalucita o Silimanita), el Al se encuentra en
sitios octaédricos, esto provoca que algunos oxígenos no se encuentren conectados al tetraedro aislado, esto se traduce en una razón Si:O menor que 1:4.
4.3. Nesosilicatos - Silicatos con Tetraedro [SiO4] aislado Formados por tetraedros unidos por cationes que yacen entre ellos. Los más comunes
de este grupo son los Olivinos, Granates y los Silicatos de Aluminio. Además de esto el circón, esfeno, topacio, estaurolita y cloritoide.
4.3.1. Los minerales de Olivino
Estos minerales, formadores de roca, tienen una fórmula general M2SiO4, donde M es
Mg2+, Fe2+ o Ca2+.
La estructura puede ser descrita desde varios puntos de vista. La Figura 4.4 muestra el arreglo de tetraedros [SiO4] en una proyección a través del eje a de una celda unitaria ortorrómbica. Los tetraedros aislados apuntan alternadamente hacia arriba y hacia abajo a lo largo de filas paralelas al eje c; en esta proyección hay filas en dos niveles de la celda unitaria, el inferior con valor a=0 y el superior a=1/2. En cada nivel las filas están separadas por el parámetro b de la celda. También en cada nivel, los tetraedros se encuentran unidos por sitios octaédricos que contienen los cationes M. Hay dos tipos de estos sitios, denominados M1 y M2. Los M1 forman cintas de octaedros paralelas al eje c, que comparten aristas. Las cintas en una capa están conectadas a los de la capa de arriba por los octaedros M2 que también comparten aristas. Los octaedros M1 y M2 tomados juntos forman un armazón tridimensional. En esta descripción idealizada los
F. Michael Dobbs Página 44 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
poliedros son regulares; en la estructura real ambos sitios están algo distorsionados siendo M2 un poco más grande y distorsionado que M1.
La estructura del olivino puede ser,
también, descrita como un arreglo aproximado ECH de átomos de oxígeno con los cationes M y Si ocupando la mitad de los sitios octaédricos y 1/8 de los sitios tetraédricos respectivamente. Las capas de EC se ubican en el plano (100); si los átomos de oxígeno estuviesen perfectamente empaquetados, entonces, los octaedros M1 y M2 serían regulares.
La mayoría de los olivinos naturales tienen composiciones en el rango Mg2SiO4 (forsterita) a Fe2SiO4 (fayalita), y estos forman una solución sólida completa a temperatura ambiente.
En los olivinos de Ca, el Ca2+ ocupa los sitios M2 en tanto que la solución sólida entre monticellita y kirschtonita sería CaMgSiO4 y CaFeSiO4. Esto involucra una distribución azarosa de Mg2+ y Fe2+ sobre los sitios M1. No existe solución sólida entre este grupo y el anterior mencionado, dada la diferencia de tamaño de los cationes y la distorsión estructural por estos provocada.
A muy alta presión en el manto de La
Tierra, el olivino de Fe o Mg se transforma a una estructura de espinela con un aumento de la densidad del 10%, lo cual provoca una discontinuidad de las ondas sísmicas a una profundidad aproximada de 400 Km.
Figura 4.4. La estructura del olivino, M2SiO4, (a) Una proyección sobre el eje a de la celda ortorrómbica (línea segmentada), en la cual se observan los tetraedros aislados apuntando alternadamente hacia arriba y hacia abajo y formando filas a lo largo del eje c. Los cationes M, unen a los tetraédros y se ubican formando también filas sobre el eje c. Estas filas se encuentran en dos niveles dentro de la celda unitaria: a la altura a=0, dibujada con línea gruesa, y a la altura a=1/2, dibujada con línea fina. Los cationes M1 se muestran como círculos rellenos, caso contrario de los cationes M2. (b) Nivel inferior de la celda unitaria, en donde se observa la relación entre los octaedros M1 y M2 y las filas de tetraédros SiO4.
F. Michael Dobbs Página 45 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
4.3.2. Los minerales de Granate
Los granates son una solución sólida multicomposicional de fórmula general (A3
2+BB23+(SiO4)3), donde A puede
ser Ca , Mg , Fe o Mn y B; Al , Fe o Cr . El grupo de los granates se puede separar en dos: los que en los sitios A contienen Ca y los que en los sitios B contienen Al. El primer grupo es llamado Piralspita y está compuesto por el Piropo (Mg), la Almandina (Fe ) y la Spessartina (Mn), el otro grupo es denominado Ugrandita y está compuesto por Uvarovita (Cr), Grossular (Al), Andradita (Fe ).
2+ 2+ 2+ 2+
3+ 3+ 3+
2+
3+
La celda unitaria es cúbica
para todos los tipos de granates y está conformada por el tetraedro SiO4, que comparte esquina con octaedros BO6, el cual a su vez comparte arista con cubos distorsionados AO8, y estos, compartiendo vértices con dos tetraedros SiO4. (Figura 4.5) 4.3.3. Silicatos de Aluminio: Kianita, Andalusita y Silimanita
Estos tres minerales tienen la misma composición química (Al2SiO5) pero poseen estructuras diferentes determinadas por las distintas condiciones físicas (Presión y Temperatura) del medio en que se forman (polimorfismo). Estos polimorfos son importantes ya que sirven para establecer las condiciones de ambientales del metamorfismo en la corteza. (Figura 4.6)
Estructuralmente los tres minerales poseen cadenas
de octaedros AlO6 que comparten aristas a lo largo del eje c (Figura 4.7). Estos octaedros contienen la mitad de los Al en su fórmula estructural. La otra mitad de los Aluminios están en coordinación que es distinta para los tres tipos; coordinación 6 para la Kianita, coordinación 5 para las Andalucita, y 4 para la Silimanita. Estos poliedros de Aluminio alternan con los tetraedros SiO4.
Figura 4.5. Parte de la estructura cúbica del granate, donde se aprecia la relación entre los tetraédros aislados SiO4, y los octaedros BO6.
Figura 4.6. Diagrama de estabilidad P-T. En él se muestran las condiciones ambientales para la existencia del polimorfo aluminosilicatado.
F. Michael Dobbs Página 46 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
4.4. Sorosilicatos
Estos silicatos están formados por pares de tetraedros, los cuales comparten una esquina. (Figura 4.1c). Los pares de tetraedros se encuentran entrelazados por cationes de valencia 2, tal como el Ca que forma la Rankinita (Ca3Si2O7). 4.5. Inosilicatos - Silicatos de Cadena Simple
A continuación, se describirán dos grupos de minerales; piroxenos y piroxenoides. En ambos, el tetraedro SiO4 forma cadenas lineares simples, compartiendo dos oxígenos por tetraedro, como se muestra en Figura 4.8. En los piroxenos la periodicidad de la cadena es dos, i.e., la cadena se repite cada dos tetraédros. En los piroxenoides, se suceden largas cadenas, tal como en la wollastonita CaSiO3, donde la periodicidad es 3. En ambos, la razón Si:O=1:3, con una carga neta de (SiO3)n
2n- Las cadenas están unidas por cationes ubicados generalmente en sitios octaédricos. La flexibilidad de las cadenas permite que existan configuraciones geométricas estables para una amplia variedad de cationes y dentro de un gran rango de condiciones de presión y temperatura.
Figura 4.7. La estructura de los tres polimorfos Al2SiO5. En cada figura se puede apreciar la posición de los octaedros AlO6 (sombreados), los cuales forman cadenas a lo largo del eje c. La posición de los otros poliedros de Al y los tetraédros SiO4 se muestran en círculos. La coordinación del Al se observa en el superíndice adyacente a la notación del ion.
Figura 4.8. Comparación de las cadenas simples de tetraédros. (a) Piroxenos con una periodicidad de dos tetraédros. (b) Piroxenoides con una periodicidad de tres tetraédros.
F. Michael Dobbs Página 47 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
4.5.1. Piroxenos La mejor manera de describir las variadas modificaciones de la estructura de los
piroxenos, es con un modelo idealizado que muestre la topología básica, para luego considerar el efecto del tamaño del catión y de la temperatura dentro de esta topología. La Figura 4.9 muestra una cadena simple, extendida -como en todos los piroxenos- a lo largo del eje c, definiendo este parámetro dentro de la celda unitaria (c ≈ 5.2Å). Las cadenas son apiladas “espalda con espalda” formando capas paralelas a los planos (100). El parámetro b está dado por el arreglo del apilamiento (b ≈ 8.9Å). Las posiciones de los cationes en los piroxenos son de dos tipos; sitios M1 y M2. Los primeros ubicados entre los ápices de tetraédros opuestos, y los segundos ubicados entre sus bases. Los sitios M1 son menores, con la forma muy similar a un octaédro regular. Los sitios M2 son mayores y más distorsionados, octaédricos cuando contienen a un pequeño catión, pero pueden cambiar su coordinación de 6 a 8, para albergar a cationes con un mayor radio iónico. Los sitios M forman cadenas que comparten aristas, las cuales se enfilan paralelas a las cadenas de silicatos.
Figura 4.9. (a) Una cadena simple de piroxenos extendida a través del eje c. (b) Arreglo de los tetraédros SiO4 en la estructura de los piroxenos vista a lo largo del eje c.
i. Clinopiroxenos
La Figura 4.10 muestra el arreglo de las cadenas de silicatos a lo largo de su eje mayor en los clinopiroxenos y define la orientación y periodo del eje a (≈ 9.7Å). La celda unitaria es monoclínica, con el ángulo β cercano a los 106°, de ahí proviene la denominación clinopiroxeno. Un mecanismo para acomodar a distintos tamaños de cationes, es la rotación de los tetraedros en sentidos opuestos de las cadenas que albergan a cationes más grandes, tal como el Ca, el cual se ubica en los sitios M2 cambiando su coordinación de 6, a 8 distorsionado. Según el ángulo que formen respecto del extendido, las rotaciones son de tipo “S” o de tipo “O”. (Figura 4.11). Por esta rotación los piroxenos albergan una amplia variedad de cationes sometidos a diferentes condiciones de presión y temperatura.
F. Michael Dobbs Página 48 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
Las rotaciones son causadas por la ocurrencia de cationes de gran radio iónico, con el fin de lograr su acomodo. Los más importantes de los minerales piroxenos, representados por la fórmula general ABSi2O6, tienen composiciones en la cual A puede ser Ca2+ , Mg2+, Fe2+ o Na+, en el sitio M2; y B puede ser Mg2+, Fe2+, Fe3+ o Al3+ en el sitio M1. Las composiciones de los piroxenos de Ca, Fe, Mg se representan generalmente como un cuadrilátero. (Figura 4.12).
Figura 4.10. Una vista de la estructura de los clinopiroxenos a lo largo del eje b.
Figura 4.11. Relación entre las cadenas simples de tetraédros SiO4. (a) Una cadena extendida. (b) y (c). Disminución en el tamaño de los tetraédros debido a la disminución de la temperatura o la ocupación de un catión.
Figura 4.12. Cuadrilátero composicional de los piroxenos.
Existen piroxenos ricos en Ca, diópsido y hedenbergita. Estos forman una solución
sólida cuyo elemento intermedio es la augita. También existen otros piroxenos, los cuales son pobres en Ca, llamados pigeonita. Estos presentan variaciones estructurales dependientes de la temperatura. Ambas soluciones sólidas existen como una sola a alta temperatura.
ii. Ortopiroxenos
En los piroxenos que prácticamente no contienen Ca2+, se produce una reorganización estructural fundamental, dado que los sitios M1 y M2 son ocupados por cationes pequeños. Esto genera una celda unitaria de tipo ortorrómbica. La relación entre ésta y la celda de los clinopiroxenos es aproximadamente 2a senβ, en tanto que los parámetros b y c permanecen iguales (Figura 4.13). El sitio M2 en los ortopiroxenos es muy pequeño
F. Michael Dobbs Página 49 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
para contener Ca2+, por lo que virtualmente no existe una solución sólida entre ortopiroxenos y clinopiroxenos. 4.5.2. Piroxenoides
En la wollastonita, típico piroxenoide, las cadenas de tetraedros se unen a las cadenas de octaedros,
los cuales contienen cationes de gran diámetro, ya que tres aristas de tetraedros [SiO4] son equivalentes a dos aristas de octaedros [CaO6]. La periodicidad de la cadena de tetraedros es de 3 y se enfilan paralelos a una columna de cationes. (Figura 4.14)
Por la diferencia de tamaño entre el Ca2+ y el Mg2+, no existe una solución sólida entre wollastonita y diópsido. El Ca2+ pude ser reemplazado por Mn2+ o Fe2+, cationes más pequeños. Esto lo realiza combinando estructuras de tipo piroxeno y tipo piroxenoide. Es decir, repite cada dos tetraedros, y luego tres, tipo wollastonita. De esta forma, se pueden llegar a construir minerales como la
Ferrosilita III, el cual posee un periodo de 9 tetraedros. (Figura 4.15).
Esta situación de reemplazo por iones
de características geométricas distintas, está controlada por la presión y la temperatura del ambiente. Incrementando la presión y/o disminuyendo la temperatura, el efecto es el equivalente a la disminución del tamaño del catión, y por esto es que MnSiO3 forma la piroximangita y luego toma la estructura de piroxeno a medida que se incrementa la presión. (Figura 4.16).
Figura 4.13. Relación entre la celda unitaria de los clinopiroxenos y de los ortopiroxenos.
Figura 4.14. Relación entre una cadena de tetraédros y una cadena de octaedros en la wollastonita.
Figura 4.15. Triángulo composicional de los más comunes de los piroxenoides dentro del sistema Ca-Mn-Fe.
F. Michael Dobbs Página 50 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
4.6. Inosilicatos - Silicatos de cadena doble
i. Anfíbolas
En cuanto al comportamiento y estructura de los minerales de anfíbolas, se debe señalar su alta similitud con los piroxenos, pero con un mayor grado de complejidad, particularmente debido al amplio rango de composiciones químicas que en ellas se observan.
La característica estructural esencial de
las anfíbolas, corresponde a una cadena doble de tetraédros [SiO4], la cual puede ser vista como dos cadenas simples unidas por vértices compartidos, con un plano especular a lo largo de la juntura. La mitad de los tetraédros tiene dos oxígenos compartidos y dos libres, mientras que la otra mitad tiene tres oxígenos compartidos y uno libre, entonces la carga neta es (Si4O11)n
6n-. Las cadenas se extienden a lo largo del eje c, con un valor unitario común a todos los tipos de estructuras anfíbolas (c ≈ 5.2Å). El apilamiento de las cadenas es análogo al de los piroxenos, formando capas en los planos (100), con ápices alternadamente apuntando hacia arriba y hacia abajo, definiendo el valor del eje unitario b. Los sitios catiónicos de la estructura, se encuentran definidos por su posición relativa a los ápices y bases de los tetraédros [SiO4]. Los sitios entre las bases son llamados M4, y los sitios más pequeños, entre ápices, M1, M2 y M3. La coordinación de los sitios M4 es ocho (i.e. un hexaedro), con un Ca2+; y seis (i.e. un octaedro), con un Mg2+ u otro catión pequeño. Los sitios M1, M2 y M3, son todos octaédricos. La cadena doble tiene un tercer tipo de sitio catiónico, denominado A, y ubicado entremedio de los sitios M4, pero ligeramente desplazado. Este sitio A, puede ser una vacante, puede estar parcialmente lleno, o completamente ocupado por Na+ y/o Ca2+. Finalmente, el (OH)- o F-, ocupa el centro del anillo hexagonal, al nivel de los ápices de los tetraédros. (Figura 4.17).
Figura 4.16. Diagrama Presión-Temperatura, mostrando las distintas zonas de estabilidad para la rodonita, piroximangita y clinopiroxeno.
Figura 4.17. Representación esquemática de la estructura de las anfíbolas.
F. Michael Dobbs Página 51 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
La composición general de las anfíbolas puede ser descrita por la fórmula:
A0-1BB2C5T8O22(OH,F)2 Donde: A : Na+, Ca2+
B : Ca2+,Na+;Fe2+,Mg2+
C : Fe2+, Mg2+, Fe3+, Al3+
T : Si4+, Al3+, hasta un máximo de Al2Si6.
Debido al amplio rango composicional de las anfíbolas, estas cristalizan dentro de una gran variedad de rocas. La anfíbola más común en rocas ígneas y metamórficas es la serie de la hornblenda, con extremos NaCa2(Mg4,Al)(Al2Si6)O22(OH)2 y Ca2Mg5Si8O22(OH)2. Las relaciones básicas entre el tipo de estructura, su composición, la temperatura y presión de formación de los minerales de anfíbolas, son análogas a las de los minerales de piroxenos. Al igual que en estos últimos, existen dos tipos de anfíbolas; unas ricas en Ca2+ y otras pobres en él, definiendo así clinoanfíbolas y ortoanfíbolas respectivamente. La relación entre sus parámetros es, al igual que en los piroxenos, aorto = 2aclinosenβ . Para la primera situación, existe una solución sólida completa entre tremolita Ca2Mg5Si8O22(OH)2 y ferroactinolita Ca2Fe2+
5Si8O22(OH)2, pasando por la actinolita que es de composición intermedia entre Mg2+ y Fe2+. En las sin Ca2+, existe una solución sólida no completa, entre el extremo magnésico y el férrico, comenzando por la antofilita Mg7Si8O22(OH)2, para llegar a la grunerita Fe2+
7Si8O22(OH)2. (Figura 4.18).
Por otro lado, aún cuando las estructuras son muy similares entre anfíbolas y piroxenos, el mecanismo esencial para albergar cationes en las anfíbolas contempla sólo una rotación de tipo “O”, comparada con las de tipo “O” y ”S” existentes en los piroxenos. En las anfíbolas, el largo de la doble cadena disminuye debido a la rotación de sus tetraédros. De todas maneras, el operador de simetría especular se sigue manteniendo, ya que la rotación se realiza de la misma manera para tetraédros enfrentados. (Figura 4.19). 4.7. Filosilicatos - Silicatos en capas Estos silicatos, están conformados por dos bloques básicos constituyentes. Una capa de tetraedros [SiO4] (T), y otra capa de octaedros (O) ligada por sus aristas. En la capa de tetraedros [SiO4], cada uno de ellos comparte tres oxígenos, formando una capa continua con carga neta (Si2O5)n
2n-. Los oxígenos sin enlazar, apuntan
Figura 4.18. Cuadrilátero composicional de los minerales de anfíbolas.
Figura 4.19. Rotación en “O” de los tetraédros en las anfíbolas.
F. Michael Dobbs Página 52 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 53 Mauricio Domcke G.
todos en la misma dirección (Figura 4.20) y comparten ese vértice con la capa de octaedros. En estos últimos, los vértices son átomos de O, o grupos de OH.
En cada capa de tetraédros T, se forman anillos, y por cada uno de ellos, existen tres octaedros de la capa inferior O (Figura 4.21). Si los iones que ocupan estos octaedros son divalentes, tal como el Mg2+ o Fe2+, para obtener el balance de carga, se ocupan los tres sitios octaédricos; en esta ocasión, se denomina a la capa como trioctaédrica. Por el contrario, si los iones que ocupan los octaedros son trivalentes, tal como Al3+ o Fe3+, el balance de carga se logra ocupando dos de los tres sitios, por lo que se denomina a la capa como dioctaédrica. (Figura 4.22). La manera en que se apilan estas capas T con las capas O, determinan los tres tipos de Filosilicatos.
Figura 4.20. Parte de una capa infinita de tetraédros[SiO4], todos apuntando en la misma dirección.
Figura 4.22. (a) Capa trioctaédrica. (b) Capa dioctaédrica distorsionada por los octaedros sin ocupar.
Figura 4.21. Conexión entre las capas básicas de tetraédros y octaedros en los filosilicatos. En la figura se aprecia que por cada anillo de tetraédros, existen tres octaedros por debajo.
Los filosilicatos son construidos, tal como se muestra en Figura 4.21, por la combinación de sus dos capas principales. Por otro lado, a partir de la misma figura, se puede notar que existen limitaciones al respecto de los tamaños de los cationes que ocupan estos sitios. Un ajuste ideal entre la capa de tetraédros y la de octaedros, requiere de cationes con un radio de aproximadamente 0.70Å, para los sitios octaédricos; i.e., más pequeños que el Mg2+. En muchos silicatos ocurre la substitución del Al3+ por Si4+, tanto

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
como cationes M en el octaédro, esto provoca una variación en el tamaño de la malla. Una consecuencia de este ajuste de tamaño, es el limitado crecimiento de algunos cristales, como por ejemplo las arcillas. Existen tres grupos principales de filosilicatos, estos se esquematizan y ejemplifican en Figura 4.23 de página siguiente.
i. Capa de silicatos 1:1 Grupo formado por una capa de tetraedros [SiO4],
combinada con otra de octaedros, agrupación tipo T-O. Estas capas se mantienen balanceadas, y sólo enlaces débiles las mantienen unidas.
ii. Capa de silicatos 2:1 En este grupo de filosilicatos, una capa de octaedros, se
sitúa entre los ápices de dos capas de tetraedros [SiO4], agrupación tipo T-O-T. Estas capas se unen mediante enlaces débiles, tipo Van der Waals si son neutras, o se unen por un catión, si ha ocurrido substitución en alguna capa, dejando como resultado una carga residual en esta.
iii. Capa de silicatos 2:1:1 En este tipo de silicatos, una capa de tetraedros se
dispone entre dos capas T-O-T, a esta agrupación se le denomina T-O-T-O, debido a su período.
4.7.1. Politipismo en las micas
En las micas, filosilicatos T-O-T, existe substitución tanto en los tetraedros (Al3+ por Si4+) como en los octaedros (M+ por M2+). Debido a esto, resulta una carga negativa en las capas, la cual es compensada por iones ubicados entre éstas y cargados positivamente. Los más comunes de estos cationes son K+, Na+ o Ca2+. La muscovita, KAl2(Si3Al)O10(OH)2 es un típico ejemplo. (Figura 4.24).
Figura 4.24. Estructura de la muscovita, se observa que las capas de tetraédros tiene un desplazamiento en su empaquetamiento, indicado por la flecha. Este desplazamiento es denominado vector de stagger.
F. Michael Dobbs Página 54 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
Dos características de esta estructura llevan al politipismo observado en las micas. Primero, en el “sándwich T-O-T”, las dos capas de tetraédros no se encuentran opuestas directamente entre sí. Existe un desplazamiento -vector de stagger” de la Figura 4.24- necesario para producir la coordinación octaédrica en la capa de octaedros. Segundo, la simetría hexagonal de la capa de tetraédros significa que el desplazamiento producido se desarrolla en cualquiera de las seis direcciones. El punto importante es cómo se relaciona este desplazamiento entre capas sucesivas. Existen 6 posibles ángulos de empaquetamiento entre capas: 0°, 60°, 120°, 180°, 240° y 300°. Figura 4.25 muestra las diferentes secuencias de estos ángulos de empaquetamiento, configurando los seis politipos estándares en las micas.
Figura 4.23. Representación esquemática de los tres grupos de filosilicatos, formados a partir de las distintas uniones entre sus dos capas básicas constituyentes.
Figura 4.25. Politipismo en las micas. Los seis politipos son producidos por diferentes combinaciones de la orientación del vector de stagger.
F. Michael Dobbs Página 55 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
4.8. Tectosilicatos - Silicatos en Armazón En los silicatos en armazón, todos los vértices de los tetraédros [SiO4] se encuentran compartidos, formando de esta manera una trama tridimensional relativamente abierta. Cuando no existe substitución del ion Si4+, la composición de esta trama es SiO2, con todas las valencias de enlace satisfechas. Por otro lado, cuando el Al3+ substituye al Si4+ dentro del tetraedro, se requiere de cationes intersticiales para mantener el balance de carga. La amplitud de esta estructura tridimensional da como resultado sitios intersticiales catiónicos grandes, comparados con los de los inosilicatos. Entonces, cationes tales como Na+, Ca2+, son considerados “pequeños” comparados con K+; mientras que el Mg2+ es muy pequeño como para cumplir un rol dentro de estas estructuras. Los aluminosilicatos son, lejos, los minerales más abundantes en la corteza terrestre, los feldespatos constituyen cerca del 65% en volumen de ella. Mineralógicamente son interesantes y desafiantes, desde el punto de vista de la respuesta de la estructura a los cambios de temperatura, presión y composición. 4.8.1. Minerales de Sílice La sílice SiO2, ocurre en un número de formas diferentes en la Tierra. El cuarzo, el más común de los polimorfos cristalinos, es estable hasta una temperatura de 857°C, la tridimita es estable entre los 857°C y los 1470°C y la cristobalita desde los 1470°C, hasta la temperatura de fusión a 1713°C. Las formas de alta presión son coesita, estable en la corteza profunda, y estishovita, la que se piensa estable en el manto de la Tierra (Figura 4.26). La estishovita tiene estructura del rutilo, y es uno de los escasos materiales en que el Si ocurre en coordinación octaédrica. En la superficie de la tierra, y en el suelo oceánico, la sílice también ocurre en formas amorfas y parcialmente cristalinas.
Figura 4.26. Diagrama de estabilidad Presión-Temperatura en donde se muestran los polimorfos de la sílice.
Las estructuras del cuarzo, tridimita y cristobalita, requieren de una transformación reconstructiva -romper enlaces y crear unos nuevos- para pasar de una estructura a otra. Los minerales antes mencionados tienen formas estructurales “de alta y de baja” . Las formas de alta poseen una mayor simetría, mientras que las de baja podrían ser consideradas como la versión “arrugada” de la primera, con enlaces doblados y simetría reducida. Estas transformaciones no requieren de una transformación reconstructiva, sino que de una denominada transformación por desplazamiento (Figura 4.27). Estas últimas tienen una mucho más alta probabilidad de ocurrencia.
F. Michael Dobbs Página 56 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
La forma de baja de la tridimita y la cristobalita son posibles sólo cuando la transformación reconstructiva de tridimita a cuarzo no sucede.
i. Cuarzo
El cuarzo de alta, mayor simetría; posee una estructura basada en cadenas espirales de tres tetraédros (posiciones 0, 1/3 y 2/3 en Figura 4.28), apretadas y paralelas al eje c. Estos espirales están unidos por sus vértices, para formar un doble espiral con “paso izquierdo” o con “paso derecho”. Este hecho, da lugar al crecimiento de cristales con características iguales a su forma estructural, por lo que cada cristal es la imagen especular de otro. Este suceso es denominado enantiomorfismo.
Figura 4.27. Relación entre cuarzo de alta y cuarzo de baja. (a) Cuarzo de alta, se observan sólo los átomos de Si dentro de la proyección en el plano ab. (b) La distorsión en la estructura, para la obtención de cuarzo de baja. Esto significa una rotación de los tetraédros y una disminución en la simetría desde hexagonal a trigonal.
Figura 4.28. Estructura del cuarzo de alta, proyectada sobre el plano ab.
El cuarzo de baja, conserva las relaciones principales de simetría, pero con una rotación de los tetraédros reduciendo esta de simetría hexagonal a simetría trigonal.
F. Michael Dobbs Página 57 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
ii. Tridimita y cristobalita
Estos minerales poseen similitudes en el patrón de ordenamiento de tetraédros y es por eso que conviene analizarlos al mismo tiempo. Ambos pueden ser descritos en términos de capas de tetraédros, similar a los filosilicatos, pero con sus ápices apuntando alternadamente hacia arriba y hacia abajo. Estas capas se apilan una sobre otra, unidas por los ápices de los tetraédros. La simetría hexagonal de cada capa da lugar a la posibilidad de relacionarlas en diferentes secuencias de empaquetamiento. En la tridimita, existe una repetición de dos capas: ABABABABAB (o mejor escrito A∀ A∀ A∀ A∀ A∀, debido a la capa inferior invertida) y una estructura hexagonal con capas paralelas a los planos (100). La cristobalita tiene una repetición de tres capas ABCABC y una estrutura cúbica centrada en las caras con capas paralelas a los planos (111).
La transformación de una estructura en otra es reconstructiva, y por el hecho de que
ambas están conformadas por capas, es normal encontrar entre éstas entrecrecimiento a escala fina. A temperaturas menores de 150°C y 230°C, respectivamente para tridimita y cristobalita, existen las formas de baja, derivadas de sus homólogas de alta mediante transformación por desplazamiento.
iii. Sílice en ambientes marinos
Los depósitos de sílice en suelo oceánico, se forman a partir de la acumulación de restos de diatomeas, radolarios y otros organismos marinos los cuales segregan conchas de sílice amorfo. Esta forma de sílice se denomina Ópalo-A, la cual al recristalizar por el enterramiento y la diagénesis forma chert, una roca compuesta por cuarzo cristalino muy fino. La secuencia diagenética hacia el cuarzo tiene un elemento intermedio, el Ópalo-CT, una mezcla fina de tridimita y cristobalita.
4.8.2. Kalsilita-Nefelina: Tridimita distorsionada
Cuando parte de los Si4+ son reemplazados por Al3+ dentro de una estructura de tectosilicatos, es necesario que existan cationes intersticiales para proveer el correspondiente balance de carga electrónica. Estos cationes, ocupan los sitios mayores del armazón y también la distorsión dentro de la estructura. Las estructuras de la kalsilita KalSiO4 (Figura 4.29a) y la nefelina (K,Na)AlSiO4 (Figura 4.29b), son derivadas de la tridimita de alta, en donde la mitad de los Si4+ son reemlazados por Al3+, y por otro lado, los cationes compensadores K+ o Na+, ocupan los canales entre los anillos de tetraédros. Estos canales tienen diferentes formas, hexagonal, ditrigonal y oval, dependiendo del tipo de catión presente.
Figura 4.29a. Estrucutura de la kalsilita KAlSiO4, con los cationes K ubicados en el centro de los anillos distorsionados.
F. Michael Dobbs Página 58 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
4.8.3. Los feldespatos La estructura en armazón comprimida de los feldespatos, es una exitosa solución para acomodar a cationes grandes tales como K+, Na+ y Ca2+ en los minerales aluminosilicatados . Los feldespatos tienen una fórmula general MT4O8, con 25% y 50% de Si4+ reemplazado por Al3+ en los sitios T, y los sitios M ocupados por Na+, K+, Rb+, Ca2+, Sr2+ o Ba2+. Las composiciones más naturales de los feldespatos se ubican en la zona achurada del triángulo de la Figura 4.30, KAlSi3O8 - NaAlSi3O8 - CaAl2Si2O8 y representan la solución sólida de alta temperatura.
Todos los feldespatos tienen un patrón de unión de los tetraédros similar, el cual puede aceptar distorsiones y por lo tanto reducción de simetría. El origen de estas modificaciones estructurales y la interrelación entre varios factores que juegan un rol en la disminución de simetría, han sido una preocupación en la mineralogía por décadas. No es sólo que los feldespatos sean lejos los minerales más comunes, sino que el amplio rango de sus posibles estados estructurales que pueden se encontrados en diferentes rocas con diferentes historias de enfriamiento, proveen una oportunidad única para estudiar cómo una estructura responde a un ambiente geológico que cambia lentamente.
Figura 4.29b. Estrucutura de la nefelina (K0.25,Na0.75)AlSiO4, en donde óvalos y anillos distorsionados contienen a los cationes Na y K respectivamente.
Figura 4.30. Triángulo composicional, el cual muestra la extensión de las soluciones sólidas para los feldespatos alcalinos y los feldespatos plagioclasa.
Se comienza con la estructura ideal de alta temperatura de la sanidina KAlSi3O8. El
silicio y el aluminio están distribuidos azarosamente de tal forma que el promedio de ocupación es 25% Al3+ y 50% Si4+. La estructura es bastante complicada, por lo que esta se describirá mediante unidades idealizadas para la obtención de sus características esenciales. La construcción básica del entramado está conformada por anillos de cuatro tetraédros con pares alternados en sus vértices, apuntando estos en direcciones opuestas (Figura 4.31a). Los anillos son agrupados en capas, produciendo espacios ovales rodeados de cuatro anillos. (Figura 4.31b).
F. Michael Dobbs Página 59 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 60 Mauricio Domcke G.
En la tercera dimensión, los anillos están unidos unos a otros formando una cadena tipo cremallera paralela al eje a. (Figura 4.32). En la Figura 4.32b, la cremallera es representada por líneas que unen los centros de los tetraedros. La manera en que se relacionan estas cremalleras con otras se observa en Figura 4.33. Aquí, los cationes K+ se encuentran entre planos especulares ubicados entre las cremalleras, ocupando el mayor espacio dentro de la estructura.
Las figuras recientemente mostradas, esquematizan una descripción idealizada de la estructura, en tanto la real muestra alguna rotación de los tetraédros, manteniéndose la simetría monoclínica C2/m, la más alta de los feldespatos. Esta es la estructura parental, desde la cual todas las otras estructuras de los feldespatos pueden ser derivadas; en ellas se distinguen dos conjuntos de tetraédros T1 y T2. Cualquier distorsión que cambie la forma de la celda unitaria reducirá la simetría, así como el orden y distribución de los cationes Al3+ y Si4+.
Figura 4.31. (a) Estructura básica de los feldespatos. (b) Anillos con simetría cuaternaria que forman una capa en la cual los anillos están relacionados mediante simetría especular paralela a los planos (010). Existen dos conjuntos de tetraédros individuales simétricos entre ellos y denominados como T1 y T2. Los cationes ocupan los sitios ovales formados entre anillos.
Figura 4.32. (a) La relación entre los anillos de simetría cuaternaria, mostrada en plano perpendicular al observado en Figura 4.31. (b) Esquema obtenido al mostrar sólo los centros que contienen Si dentro de la estructura tipo cremallera.
Figura 4.33. Una vista en perspectiva para la configuración de los feldespatos, en la cual se puede apreciar la estructura tipo cremallera reflejada mediante planos especulares (010).

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
i. Distorsiones estructurales y ordenamiento Si-Al en la estructura de los feldespatos
Antes de describir las variadas modificaciones en la estructura de los feldespatos, es necesario realizar algunas observaciones sobre los factores que provocan el cambio desde la estructura de alta de la sanidina C2/m, hasta la estructura de menos simetría. Aunque estos factores se describen separadamente, es necesario señalar que su acción no es excluyente.
• La estructura expandida de alta temperatura, tiende a colapsar alrededor del sitio intersticial catiónico, a medida que la temperatura desciende. Un catión suficientemente grande, como el Ba o K, es capaz de impedir el colapso, y por lo tanto la estructura permanece extendida. Por otro lado, un catión pequeño tal como Na o Ca, provocará una distorsión en la estructura, reduciendo su simetría a triclínica C1. La Figura 4.34 muestra como la distorsión alrededor de los sitios catiónicos cambia el largo del enlace catión-oxígeno, y el ángulo interaxial α, desde 90° en la estructura monoclínica, hasta los 93.4° en la triclínica.
• Otra tendencia en las estructuras de aluminosilicatos, es el ordenamiento de los
tetraédros Al-Si a bajas temperaturas. En comparación con la distorsión estructural vista en el punto anterior, este proceso de distorsión es lento y envuelve el quiebre de fuertes enlaces Si-O y Al-O. Las razones Si :Al en los feldespatos son variables entre 1:3 y 2:2, lo que provoca como consecuencia la distorsión de la estructura parental, debido a que los tetraédros [AlO4] son de mayor tamaño que los [SiO4]. Esto se observa en Figura 4.35, la cual es una proyección un poco diferente de un par de cremalleras, lo que permite observar de mejor manera la distribución de Si, Al.
Figura 4.34. (a) En la sanidina, KAlSI3O8, la coordinación del oxígeno alrededor del K es simétrica y la estructura es monoclínica. (b) En la albita de alta, NaAlSi3O8, la coordinación del oxigeno alrededor del Na está distorsionada y la estructura es triclínica.
Figura 4.35. Perspectiva de estructura tipo cremalleras para los feldespatos.
F. Michael Dobbs Página 61 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
• El tercer método de reducción de simetría, corresponde a la relación conjunta entre los dos puntos anteriores, i.e., distorsión y ordenamiento. En este tipo de reducción, si existe algún grado de orden entre Al-Si, la reducción no se produce hasta que todo el orden se haya perdido.
ii. Los feldespatos alcalinos
La albita NaAlSi3O8 es monoclínica C2/m sobre los 980° C (monoalbita), pero su
estructura se rompe bajo esta temperatura, debido al pequeño átomo de Na, convirtiéndose en una estructura triclínica C1. En esta etapa, mientras existe un pequeño orden Al-Si, se le conoce como albita de alta, pero bajo los 700° C, comienza el ordenamiento y la distorsión que convierten a la estructura en una albita de baja. (Figura 4.36). La sanidina es la forma de KAlSi3O8 de alta temperatura, monoclínica C2/m, la que se mantiene estable debido a que el gran átomo de K previene las transformaciones estructurales. Lo mismo ocurre para la ortoclasa, que es una forma de feldespato potásico que se enfría a una rapidez moderada, y debido a que la albita tiene la misma forma estructural, es posible encontrar una solución sólida completa entre Na Y K a alta temperatura. La microclina tienen una estructura triclínica C1, y posee un menor grado de ordenamiento que los anteriores. Bajo los 300° C, no existe posibilidad de substitución entre Na y K, lo cual originaría a las pertitas, desmezcla de la solución sólida de alta temperatura.
iii. Los feldespatos plagioclasa
Entre la albita NaAlSi3O8 y la anortita
CaAl2Si2O8, la estructura debe combinar las complejidades de los cambios estructurales de los miembros finales, con una solución sólida en la cual la razón Si :Al es variable. En la anortita pura, el pequeño átomo de Ca es incapaz de soportar la estructura monoclínica expandida C2/m. A cualquier temperatura bajo el punto de fusión, y dada la igual proporción de Si y Al, esta tiene una estructura triclínica I1ordenada. (Figura 4.37). En general, los intermedio poseen una estructura tipo triclínica C1, con una mezcla submicroscópica de plagioclasas de distinta composición y orden.
Figura 4.36. Diagrama de fase estructural, en el cual se muestra la variación de la estructura a medida que se disminuye la temperatura.
Figura 4.37. Esquema en que se muestran las regiones de estabilidad en los feldespatos plagioclasa.
F. Michael Dobbs Página 62 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
iv. Cordierita
La cordierita, (Fe,Mg)2Si4Al5O18 es un importante mineral, debido a que nos muestra la
relevancia de las variables termodinámicas en la estructuración del orden interno cristalino. Se encuentra en rocas metamórficas y sobre los 1450° C posee una estructura hexagonal, bajo esta temperatura existe una transformación por desplazamiento, haciendo que la estructura disminuya su simetría a una celda ortorrómbica. Las Figuras 4.38 y 4.39, así como la Figura 1.1, muestran el ordenamiento interno de la estructura de la cordierita.
Figura 4.38. Distribución de los iones de silicio y aluminio en la cordierita. La celda hexagonal de la estructura desordenada se esquematiza con línea segmentada, mientras que la celda ortorrómbica de la estructura con mayor simetría está dibujada con línea continua.
Figura 4.39. Tres orientaciones equivalentes de la celda ortorrómbica (línea continua), derivadas de la celda hexagonal (línea segmentada) en la cordierita.
F. Michael Dobbs Página 63 Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 64
5. DEFECTOS EN MINERALES
En una estructura cristalina ideal, cada celda unitaria es similar a las otras en forma, tamaño y contenido. Esta celda presenta una composición química y un arreglo atómico que es estable en un ambiente particular en la Tierra. Sin embargo, en los minerales existen siempre violaciones locales o defectos en este arreglo perfecto. Sitios vacantes, reemplazos por átomos impuros, desorden en el reemplazo de los sitios catiónicos o errores de empaquetamiento, son algunos de los defectos que influyen en las propiedades del mineral.
La importancia de los defectos estriba en la derivación de ciertas propiedades
mineralógicas, tales como la reactividad química y la facilidad de difusión al interior de la estructura; esto considerando aún la escasa presencia de ellos. Por ejemplo, el defecto del sitio vacante, defecto puntual, puede afectar a una de cada 10.000 celdas unitarias y provocar un aumento considerable de la rapidez de reacción de un mineral. La presencia de impurezas en una estructura, puede producir el cambio de color del mineral (rubíes rojos, esmeraldas verdes y zafiros azules deben su color a la presencia de impurezas). Por otro lado, la deformación de minerales es controlada por la presencia y el desplazamiento de defectos lineares o dislocaciones. A escala geológica, estas dislocaciones forman montañas y mantienen a los continentes moviéndose. Los defectos planares son superficies dentro de una estructura cristalina, en donde la estructura a un lado de la superficie está relacionada a la del otro lado por un desplazamiento o una rotación de simetría. Existen varios tipos de defectos planares, algunos de ellos curvos o con superficies complejas. La forma en como se mueven los defectos en una estructura, y la distribución y abundancia de ellos; controla las transformaciones de una estructura a otra, dentro del estado sólido. 5.1. Defectos puntuales Cualquier cristal a temperatura sobre el cero absoluto, contiene algún defecto puntual, el cual generalmente es un sitio vacante o una impureza. Hasta cierta concentración, la presencia de defectos reduce la energía libre de Gibbs G del cristal, lo cual favorece su estabilidad. La energía libre de Gibbs G, es una cantidad termodinámica, la cual es mínima cuando un cristal se encuentra en equilibrio con su entorno. Está definida por la siguiente expresión.
(Ec 5.1)G = H – TS Donde la entalpía H, es la energía interna a presión constante, la cual es la suma de todas las energías electroestáticas de los enlaces interatómicos y la energía cinética, debida a la vibración atómica. La entropía S, es una medida del estado de desorden en el cristal y T, es la temperatura en grados Kelvin. La creación de una vacancia u otro defecto puntual, requiere de energía. Entonces existe un incremento de entalpía en el cristal bajo su ocurrencia. Debido a que los defectos aumentan el grado de desorden dentro del cristal, también existe un aumento en
Mauricio Domcke G.
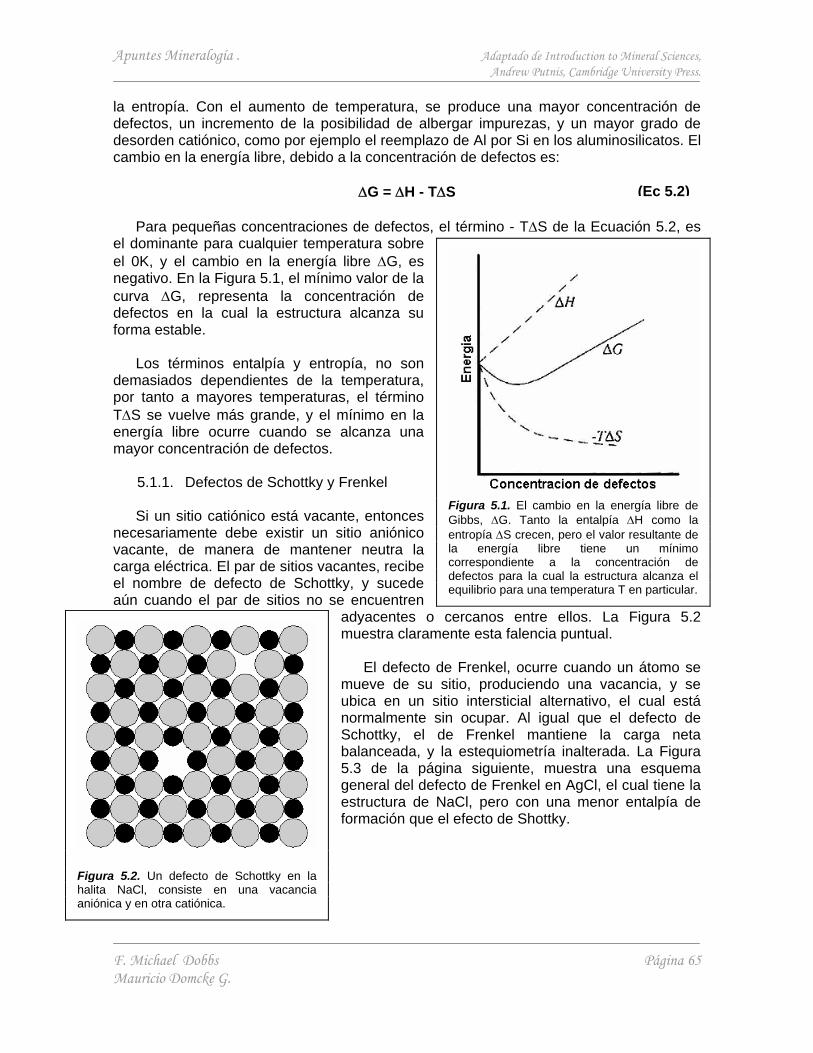
Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 65
la entropía. Con el aumento de temperatura, se produce una mayor concentración de defectos, un incremento de la posibilidad de albergar impurezas, y un mayor grado de desorden catiónico, como por ejemplo el reemplazo de Al por Si en los aluminosilicatos. El cambio en la energía libre, debido a la concentración de defectos es:
ΔG = ΔH - TΔS Para pequeñas concentraciones de defectos, el término - TΔS de la Ecuación 5.2, es el dominante para cualquier temperatura sobre el 0K, y el cambio en la energía libre ΔG, es negativo. En la Figura 5.1, el mínimo valor de la curva ΔG, representa la concentración de defectos en la cual la estructura alcanza su forma estable. Los términos entalpía y entropía, no son demasiados dependientes de la temperatura, por tanto a mayores temperaturas, el término TΔS se vuelve más grande, y el mínimo en la energía libre ocurre cuando se alcanza una mayor concentración de defectos.
5.1.1. Defectos de Schottky y Frenkel
Si un sitio catiónico está vacante, entonces necesariamente debe existir un sitio aniónico vacante, de manera de mantener neutra la carga eléctrica. El par de sitios vacantes, recibe el nombre de defecto de Schottky, y sucede aún cuando el par de sitios no se encuentren
adyacentes o cercanos entre ellos. La Figura 5.2 muestra claramente esta falencia puntual.
El defecto de Frenkel, ocurre cuando un átomo se
mueve de su sitio, produciendo una vacancia, y se ubica en un sitio intersticial alternativo, el cual está normalmente sin ocupar. Al igual que el defecto de Schottky, el de Frenkel mantiene la carga neta balanceada, y la estequiometría inalterada. La Figura 5.3 de la página siguiente, muestra una esquema general del defecto de Frenkel en AgCl, el cual tiene la estructura de NaCl, pero con una menor entalpía de formación que el efecto de Shottky.
Figura 5.1. El cambio en la energía libre de Gibbs, ΔG. Tanto la entalpía ΔH como la entropía ΔS crecen, pero el valor resultante de la energía libre tiene un mínimo correspondiente a la concentración de defectos para la cual la estructura alcanza el equilibrio para una temperatura T en particular.
Figura 5.2. Un defecto de Schottky en la halita NaCl, consiste en una vacancia aniónica y en otra catiónica.
(Ec 5.2)
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 66
5.1.2. Impurezas y substitución atómica
Los compuestos minerales son impuros, ya que existe una amplia gama de elementos substitutos. Estas impurezas se pueden tratar como defecto puntual, debido a que existe una concentración de impurezas para la cual la estructura alcanza el equilibrio. La concentración y dispersión de estas impurezas dependen de la entalpía y la entropía asociadas a la substitución. A esto generalmente se le llama entalpía y entropía de mezclado. Algunos minerales sólo permiten pequeñas variaciones dentro de su composición pura, por ejemplo el cuarzo SiO2; mientras que para otros, existe una solución sólida completa entre dos extremos composicionales, por ejemplo, en los minerales de olivino, desde Mg2SiO4 (Forsterita) hasta Fe2SiO4 (Fayalita).
Figura 5.3. Un defecto de Frenkel en AgCl, consiste en un sitio catiónico vacante, y átomo de Ag, ocupando un intersticio que no está normalmente considerado en la estructura ideal.
5.1.3. Centros de color en los cristales
En algunas estructuras los sitios vacantes pueden provocar una coloración dentro del
cristal. Por ejemplo en la halita, las vacancias de Na producen un coloración amarillenta, ya que los aniones circundantes cuentan con varios niveles de energía por los cuales enlazarse.
En minerales como el cuarzo, el cual acepta muy poca substitución, los sitios vacantes
de O, o las substituciones por cationes trivalentes, pueden causar cambios de color.
5.1.4. Difusión
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 67
La existencia de defectos puntuales fue propuesta como una manera de explicar la difusión volumétrica de iones y átomos a través de estructuras cristalinas simples. En tales estructuras, es claro que para que se produzca el mecanismo de difusión, debe existir; o un sitio vacante, o un intersticio sobre el cual se produzca el desplazamiento. En la Figura 5.4 se muestran los distintos mecanismos de difusión, siendo el del sitio vacante el más probable.
Figura 5.4. Varios mecanismos de difusión atómicos. (a) y (b) no involucran vacancias. (c) es un mecanismo de migración con sitio vacante. (d) y (e) son migraciones intersticiales.
Figura 5.5. Tres etapas en la migración desde un sitio a otro, del átomo sombreado Las posiciones (1) y (3) representan posiciones de equilibrio, con la mínima energía libre. La posición intermedia requiere de distorsiones de la red, por lo que involucra un incremento en la energía ,dando lugar a la barrera energética.
Independiente del mecanismo de difusión, para que se produzca el desplazamiento de un átomo desde un sitio a otro, este debe superar una barrera energética E. Esta energía es denominada como energía de activación para la difusión. La Figura 5.5 esquematiza y explica esta situación.
5.1.5. Estequiometría alterada
Los defectos puntuales son estequiométricos, pero no alteran la razón catión:anión. Aun así, algunos compuestos pueden acomodar una gran número de sitios vacantes, especialmente a altas temperaturas, y esto tiene el efecto de cambiar significativamente la razón nominal catión:anión. La Pirrotita Fe1-xS, puede existir a altas temperaturas con 0 < x < 0,125 y una distribución azarosa de sitios vacantes. Con altas concentraciones de sitios vacantes, la ganancia de entropía debido al desorden es más importante para la estabilidad de la estructura, por tanto se puede conseguir un estado desordenado de sitios vacantes. En bajas temperaturas, la distribución de los sitios vacantes es ordenada por el equilibrio electroestático de ellos.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 68
Una situación similar ocurre con la wustita, Fe1-xO, la cual posee estructura tipo NaCl y sitios vacantes en las posiciones del Fe. En este caso, las posiciones forman grupos, y estos grupos son semejantes a fragmentos de estructura de espinela Fe3O4. Entonces la wustita puede ser considerada como una estructura parcialmente ordenada con microdominios de espinela. La oxidación de la magnetita es otro ejemplo donde los defectos acomodan un cambio en la fugacidad de oxígeno, provocando la transformación de sitios vacantes en la estructura de la espinela, hasta la composición Fe21.67O32 denominada Maghemita. Esta composición es similar a la de la hematita Fe2O3. La transformación de estructura de espinela a la de hematita sucede fácilmente sobre los 500° C. A menores temperaturas, este cambio estructural mayor es difícil, pudiendo existir indefinidamente la estructura de espinela defectuosa, a pesar de ser termodinámicamente menos estable que la hematita.
Muchos otros grupos importantes de
minerales que contienen Fe, varían su población de defectos, dependiendo en alguna medida, de la presión parcial de oxígeno (fugacidad de oxígeno, ƒ(O2)). Cualquier propiedad física que dependa de la densidad de defectos, es por tanto, altamente dependiente de la ƒ(O2). La Figura 5.6 da cuenta de la variación del coeficiente de difusión D de Mg,Fe en los olivinos a partir de ƒ(O2).
Figura 5.6. Variación del coeficiente de difusión con la fugacidad de oxígeno.
5.2. Defectos lineares
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 69
Los procesos dinámicos que continuamente toman lugar en las profundidades de la Tierra, a altas temperaturas, producen esfuerzos diferenciales sobre los minerales componentes de las rocas, provocando en ellos una deformación permanente mediante un proceso de deslizamiento, en el cual parte del cristal se mueve, en relación a la fracción adyacente. La Figura 5.7., muestra de forma exagerada como ocurre este desplazamiento, esquematizando deformaciones por deslizamiento, en planos paralelos, producidas bajo la acción de esfuerzos de tracción en un simple cristal.
Por otro lado, el movimiento de minerales
individuales, tanto como el de granos minerales sobre otros, son responsables -a gran escala- de la traslación de material en la corteza y el manto terrestre.
5.2.1. El vector de Burges
El desplazamiento y la dirección de una deformación asociada con una dislocación, está definida por el vector de Burges b, como se describe en Figura 5.8.
En una dislocación perfecta (o unitaria), el vector de Burges es igual al vector de traslación de la celda, y la dislocación que ocupa menos energía, representa al menor vector de Burges Materiales como los metales son dúctiles, incluso a bajas temperaturas, debido a que tienen enlaces no direccionales y movimientos de fácil dislocación. En el otro extremo, materiales como el diamante, de enlaces covalentes, son muy duros, ya que el desplazamiento alrededor de líneas de dislocación es muy localizado, y requiere de fuerzas muy altas para realizarlo.
Figura 5.7. Esquematización de deformaciones en planos paralelos producidas por la aplicación de esfuerzos de tracción dinámicos en un simple cristal.
Figura 5.8. Determinación del vector de Burges b de una dislocación mediante la creación de un circuito el cual empieza en S, recorre 7 pasos atómicos en ambas direcciones, para terminar en F. El vector de Burges es representado por la distancia FS, la cual indica la falla para cerrar el circuito, y por lo tanto la dislocación.
5.2.2. Dislocación de filo y dislocación de tornillo
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 70
Una dislocación se describe mediante la especificación del vector de Burges y la dirección de la línea de dislocación. Cuando el vector de Burges es perpendicular a la dirección de la dislocación, se denomina a esta última dislocación de filo. Por otro lado, si el vector de Burges es paralelo a la dirección de dislocación, se denomina con el nombre de dislocación de tornillo. 5.2.3. Bucle dislocacional - Interacción entre defectos puntuales y lineares
Cuando existe una suficientemente alta temperatura como para permitir la difusión atómica, defectos puntuales, tales como sitios vacantes, pueden propagarse dentro de la estructura y entonces interactuar con líneas de dislocación. Esto permite a una dislocación de filo moverse desde un plano de deslizamiento a otro. Cuando las dislocaciones se mueven a lo largo de un solo plano de deslizamiento se les denomina dislocaciones de deslizamiento. Por otro lado, cuando el movimiento de una dislocación de filo se realiza fuera de su plano de deslizamiento, se denomina bucle dislocacional. 5.2.4. Dislocaciones parciales y fallas de empaquetamiento Cuando un dislocación perfecta, en la cual el vector de Burges es igual al vector traslación de la celda unitaria, ocurre en un cristal, deja la estructura meramente intacta trasladando parte de la estructura en una distancia igual a una celda unitaria. En la red cúbica, el vector de Burges, para una dislocación perfecta, es siempre un eje o vector cristalográfico, pero en estructuras más complejas y reales, este no es el caso necesariamente, y el vector de Burges es sólo una fracción del vector cristalográfico. Esta dislocación parcial no reemplaza perfectamente la estructura, como para conformar la estructura ideal, aunque desplazada. La Figura 5.9, muestra una dislocación parcial, en la cual el vector de Burges es ½[010].
5.3. Defectos planares
Figura 5.9. Dislocación parcial ocurrida en una celda no cúbica, si no que más compleja y real. Esto genera fallas en el empaquetamiento, provocando un quiebre en la periodicidad y simetría de la estructura En este caso, el empaquetamiento provoca una error en el orden de los átomos sombreados con color más claro.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 71
Los defectos planares representan la falta de mayor orden dentro de la simetría de un
cristal. No todos los defectos en dos dimensiones son planos, sino que algunas veces están constituidos por complejas superficies curvas. Los defectos planares pueden ser fallas en el empaquetamiento, defectos de Wadsley (también denominados planos cristalográficos de corte), maclas y fronteras de dominio antifase.
5.3.1. Fallas de empaquetamiento
Este tipo de falla involucra un deslizamiento de la estructura a través de un vector
desplazamiento R, el cual se encuentra ubicado en el plano de falla. El deslizamiento de una dislocación parcial, como se observa en Figura 5.9, es el que provoca esta falla de apilamiento. El vector de Burges b, se convierte en el vector desplazamiento R. Este tipo de defectos se producen en estructuras que pueden ser descritas en términos de capas.
5.3.2. Defectos de Wadsley
Cuando el vector desplazamiento R asociado a un defecto planar forma un ángulo con el plano de falla, la composición química alrededor de la falla es distinta a la de toda la estructura. (Figura 5.10). La presencia de tales defectos cambian la estequiometría del cristal. En la sección 5.1.5. se da cuenta de cómo en un cristal con su estequiometría alterada es capaz de ordenarla mediante una alta concentración de defectos. Similarmente, los defectos puntuales son eliminados de manera de resolver la problemática química-estructural, mediante la creación de los defectos de Wadsley.
5.3.3. Defectos planares y reacciones químicas en
estado sólido
En cierta cantidad de sistemas cristalinos, el mecanismo de reacción en el estado sólido involucra un cambio en la composición bajo ciertas condiciones ambientales, mediante la propagación de defectos planares desde la superficie del cristal hacia su interior. El mejor ejemplo para este tipo de reacciones es el intercambio hídrico en los inosilicatos, pasando de cadenas simples a cadenas dobles.
El reemplazo de piroxenos anhídridos por
anfíbolas es un fenómeno común cuando tales minerales se encuentran en contacto con fluidos hidrotermales. La figura 5.11 muestra este acontecimiento.
Figura 5.10. Dirección no paralela del vector R relativa al plano de falla, provocando un cambio en la composición química alrededor del defecto.
Figura 5.11. En la Figura se observa el reemplazo de cadenas dobles por las cadenas simples, generando una cadena de defectos en la estructura ideal de los piroxenos.
5.3.4. Fronteras de dominio antifase
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 72
Una frontera antifase es una interfase que no contiene cristales y en la cual existe un
error en la simetría traslacional. La interfase, la cual es generalmente curva, aparece cuando una estructura se transforma desde su forma de alta a la de baja, reteniendo sus características configuracionales principales, pero con la perdida de algunos elementos de simetría traslacional.
i. Transformación de alta-baja de la pigeonita.
La pigeonita de alta temperatura tiene grupo espacial C2/c con todas las cadenas de piroxenos equivalentes. A menores temperaturas, las cadenas sufren una distorsión debido a la rotación individual de los tetraédros, reduciendo su simetría al grupo espacial P21/c (Figura 5.12). Si la rotación de las cadenas en una parte del cristal no está relacionada con la otra, cabe la posibilidad de la formación de una interfase en la cual la secuencia de cadenas es incorrecta. El vector desplazamiento que relaciona las dos regiones del cristal a lo largo de la frontera antifase es ½[110], el cual guarda relación con la transformación C � P. Otra manera de describir el origen de la frontera antifase, es considerando que la celda primitiva de la estructura de baja temperatura puede tener dos posibles orígenes; (0,0,0) o (1/2,1/2,1/2), llevando a la formación de dos tipos de regiones en el cristal, relacionadas por el vector ½[110]. Estos dominios son a veces referidos como variantes traslacionales producidas por la pérdida de la simetría. A estos dominios se les conoce como dominios antifase.
5.3.5. Fronteras de maclas
Las fronteras de maclas pueden ser explicadas en un sentido similar a las fronteras de dominio antifase, en donde las fronteras relacionan dos dominios por un vector desplazamiento provocando así un error en la simetría traslacional. Las fronteras de maclas separan dominios los cuales están relacionados por un operador de simetría elemental, i.e., un plano especular un eje de rotación, o un eje de roto inversión. (Figura 5.13).
Figura 5.12. En la transformación de su estructura de alta a baja, la pigeonita forma dos conjuntos de cadenas (sombreados y sin sombrear)
Figura 5.13. Deformación especular.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 72
6. ENERGÉTICA EN LA ESTABILIDAD DE MINERALES I - CONCEPTOS BÁSICOS
Uno de los temas centrales en la mineralogía, es estudiar como reaccionan los minerales bajo los distintos cambios físico-químicos en el ambiente. Cualquier estructura sometida a estos cambios responde buscando su mínima energía libre, sea cual sea su mecanismo de transformación. i.e., está en búsqueda de un estado de equilibrio.
Debido a lo anterior, es que en este capítulo se definirán las funciones necesarias para
describir la estabilidad de un mineral, sin afán de buscar una profundización en conceptos de termodinámica.
6.1. Algunos conceptos básicos de la termodinámica
La energía interna U de una estructura mineral, corresponde a la suma de la energía potencial almacenada en los enlaces interatómicos y la energía cinética debida a la vibración de sus elementos constituyentes. Al aumentar el calor, se incrementa la energía cinética (y por ende la temperatura), lo que también hace crecer su energía interna. Este proceso de aumento de la temperatura, implica una expansión de la estructura cristalina por lo cual ésta efectúa un trabajo hacia su entorno. Por esto, y tomando en consideración la conservación de la energía (1ª ley de la termodinámica) se tiene:
dU = dQ – PdV
Donde dQ es el cambio en el contenido calórico y PdV es la cantidad de trabajo realizado en la expansión. Con P presión y dV cambio en el volumen.
Otras funciones termodinámicas que se deben definir son entalpía H y capacidad
calórica C.
H = U + PV
C = dQ/dT En general, los procesos geológicos ocurren a presión constante, por lo que:
Cp = (dQ/dT)p = (dH/dT)p
A bajas temperaturas, cuando T se aproxima a 0 K, tanto Cp como Cv tienden a 0, la forma en que Cp varía con la temperatura se muestra en Figura (6.1). De todas formas, en los sólidos, la diferencia entre Cp y Cv es despreciable, llegando a ser significativa sobre los 1500 K. El cambio en la entalpía entre temperaturas T1 y T2, puede ser resuelto integrando la Ecuación 6.4, obteniendo:
Figura 6.1. Variación de la capacidad calórica, con la temperatura.
Ec 6.4
Ec 6.3
Ec 6.2
Ec 6.1
ΔH = ∫ CpdT
T2
T1
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 73
Entonces, la entalpía total para cualquier temperatura T1 es:
H = H0 + ∫ CpdT T1
0
Ec 6.5 Donde H0 incluye solo la entalpía debida a la energía potencial del cristal a 0 K, dado que la energía vibracional a esta temperatura es nula, cero absoluto.
6.2. Cambios de entalpía en transformaciones y reacciones en minerales
Cuando un mineral cambia su estructura en una transformación polimórfica, o una asociación de minerales cambia a otra, existirá un cambio en la entalpía ΔH. Si existe una reducción de la entalpía en la reacción, el proceso se llama exotérmico. Por el contrario, si ΔH es positivo, el proceso es denominado como endotérmico.
Un criterio para predecir el cambio, sería entonces la minimización de la entalpía, la
fase estable debería ser la que posee la mínima entalpía. Un ejemplo lo constituyen los polimorfos aragonito y calcita (CaCO3). A 25° C, temperatura ambiente, la entalpía del aragonito es H = -1207,74 KJ/mol, mientras que para la calcita H = -1207,34 KJ/mol. Se observa que para la última fase, la entalpía es levemente mayor, y aún bajo estas condiciones, la fase que más prevalece en condiciones de intemperismo es la calcita. Es decir, la entalpía por sí sola no sirve para predecir la estabilidad de una estructura. Por esto se define una nueva cantidad denominada entropía S.
6.3. Entropía y desorden
Cuando un mineral cambia de una estructura a otra, de forma implícita intercambia
calor con su entorno. La entropía se define como la cantidad que mide el cambio en el estado de ordenamiento asociado al proceso. El cambio total de entropía es igual a la suma del cambio de entropía en el mineral (i.e. el sistema en consideración) y el cambio de entropía en el entorno, esto es:
dS = dSsistema + dSentorno Para una reacción reversible dS = 0, pero los procesos naturales son irreversibles, y según la 2ª ley de la termodinámica, tienden a aumentar el desorden, por lo que dS>0. Esto significa que si un mineral se vuelve más ordenado en un proceso de transformación, y por lo tanto reduce su entropía, el calor liberado hacia el entorno, debe incrementar la entropía del entorno de manera de satisfacer la segunda ley de la termodinámica. El cambio en la entropía está definido por: Ec 6.6dS > dQ/T En un sistema cerrado, y sin intercambio de calor, la entropía y la entalpía se relacionan mediante la ecuación:
dS > dH/T
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 74
Se puede notar que a presión constante dQ = dH. Entonces el criterio para que suceda la transformación mineral o el proceso de reacción es:
dH - TdS < 0 Si dH - TdS = 0, el sistema se encuentra en equilibrio. Por otro lado, si dH - TdS > 0, la reacción no se producirá. Desde este punto de vista, el término dH - TdS puede ser usado como un criterio para predecir la dirección de la reacción, o una definición del estado de equilibrio. Esta cantidad es conocida como cambio en la energía libre de Gibbs dG del sistema.
dG = dH - TdS o,
G = H - TS Cuando se considera el sistema con volumen constante, dU - TdS se transforma en el criterio determinante, éste es conocido como la variación en la energía libre de Helmholtz dF, o:
F = U - TS
6.3.1. Entropía configuracional
En una expresión estadística, de acuerdo a Boltzmann, la entropía de un sistema para un estado dado es relativa a la probabilidad de existencia de tal estado. Entendiendo como estado, a la probabilidad de que cierto arreglo estructural de átomos ocurra (Figura 6.2).
Figura 6.2. Estados de orden y desorden para un igual número de átomos de A y B (sombreados y sin sombrear) en una malla cúbica simple.
Ec 6.8
Ec 6.7
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 75
Matemáticamente esto se expresa como:
S = κ * Log ω Donde ω, es la probabilidad de que cierto estado dado exista y κ es la constante de Boltzmann (1,38 x 10-23 JK-1). En una estructura mineral compleja, puede existir más de un sitio sobre el cual el desorden puede ocurrir, la expresión mas general para esta situación es
S = -nR(xALogxA + xBLogxB BB)
La ecuación 6.10 es conocida como entropía de mezclado, donde n es el número de sitios en los cuales ocurre la mezcla. Dado que tanto xA como xB son fracciones atómicas, S es siempre positivo, como se muestra en Figura 6.3. i.e., sin importar el aporte atómico, siempre existirá algún grado de desorden configuracional.
B
6.3.2. Entropía electrónica
Esta configuración aparece cuando electrones de
orbitales no llenos se distribuyen sobre orbitales degenerados. En los metales de transición, como el Ti3+, el cual tiene sus orbitales 3d sin llenar, esta forma de entropía es una importante contribución a la entropía total.
Cuando un cambio en la estructura de un mineral
resulta en un cambio en la coordinación de un ion metálico de transición, la configuración electrónica también cambia. Las consecuencias termodinámicas pueden envolver tanto variaciones en la entropía electrónica como en la entalpía, debido a la variación en el campo de energía de estabilización del cristal.
Figura 6.3. Forma general de la entropía configuracional, asociada a al desorden de los átomos A y B. Se puede observar que el máximo desorden se alcanza para fracciones de átomos A = B = 0,5.
Ec 6.10
Ec 6.9
6.3.3. Entropía vibracional
Un fonón, es un quantum de energía vibracional. El espectro de fonones se define
como el número de fonones en cada rango de frecuencia. Esta función es la densidad fonónica de los estados, la que depende de Cp, y del desorden en la distribución de los elementos, los que son difíciles de mejorar a bajas temperaturas por lo complicado de la difusión. Esto implica “congelar” el desorden y por tanto aumentar el nivel de fonones en la estructura.
La entropía vibracional a cualquier temperatura T1 está dada por la expresión:
S = S0 + ∫ CpdT/T T1
0
Ec 6.11
Mauricio Domcke G.
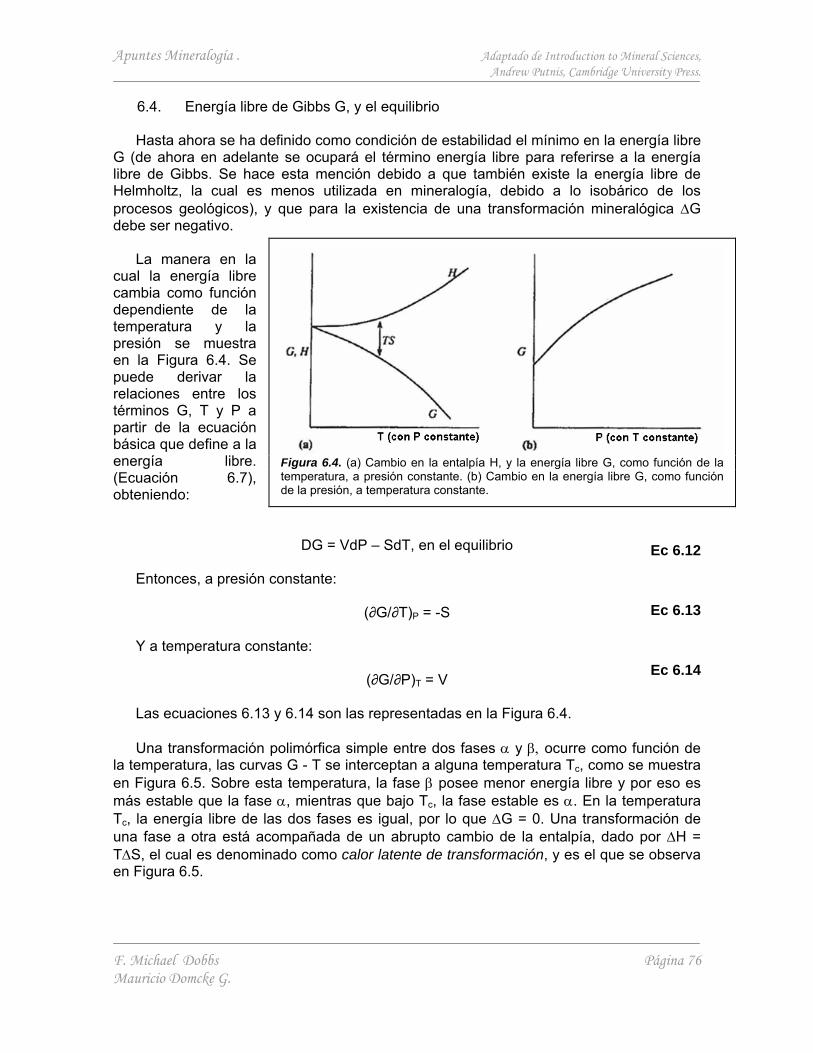
Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 76
6.4. Energía libre de Gibbs G, y el equilibrio
Hasta ahora se ha definido como condición de estabilidad el mínimo en la energía libre G (de ahora en adelante se ocupará el término energía libre para referirse a la energía libre de Gibbs. Se hace esta mención debido a que también existe la energía libre de Helmholtz, la cual es menos utilizada en mineralogía, debido a lo isobárico de los procesos geológicos), y que para la existencia de una transformación mineralógica ΔG debe ser negativo.
La manera en la
cual la energía libre cambia como función dependiente de la temperatura y la presión se muestra en la Figura 6.4. Se puede derivar la relaciones entre los términos G, T y P a partir de la ecuación básica que define a la energía libre. (Ecuación 6.7), obteniendo:
DG = VdP – SdT, en el equilibrio
Figura 6.4. (a) Cambio en la entalpía H, y la energía libre G, como función de la temperatura, a presión constante. (b) Cambio en la energía libre G, como función de la presión, a temperatura constante.
Ec 6.12
Entonces, a presión constante:
Ec 6.13(∂G/∂T)P = -S Y a temperatura constante: Ec 6.14(∂G/∂P)T = V Las ecuaciones 6.13 y 6.14 son las representadas en la Figura 6.4. Una transformación polimórfica simple entre dos fases α y β, ocurre como función de la temperatura, las curvas G - T se interceptan a alguna temperatura Tc, como se muestra en Figura 6.5. Sobre esta temperatura, la fase β posee menor energía libre y por eso es más estable que la fase α, mientras que bajo Tc, la fase estable es α. En la temperatura Tc, la energía libre de las dos fases es igual, por lo que ΔG = 0. Una transformación de una fase a otra está acompañada de un abrupto cambio de la entalpía, dado por ΔH = TΔS, el cual es denominado como calor latente de transformación, y es el que se observa en Figura 6.5.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 77
Si ahora se considera tanto a la temperatura como la presión y se observan los campos de estabilidad para los dos polimorfos α y β, la curva de energía libre se transforma en una superficie en el espacio G-T-P (Figura 6.6a) y su intersección define el equilibrio entre α y β. La pendiente de esta línea intersección proyectada sobre el plano P-T, es conocida como la relación de Clapeyron, la cual se obtiene de hacer ΔG = 0 en la ecuación 6.12.
ΔVdP = ΔSdT o,
dP/dT = ΔS/ΔV
La Figura 6.6b muestra una relación de Clapeyron típica para una transformación polimórfica. Una pendiente linear implica que tanto ΔS como ΔV son independientes de la presión y la temperatura, lo cual en muchas reacciones constituye una suposición razonable, debido a que muchas reacciones ocurren entre sólidos de composición constante.
Figura 6.5. Variación de la energía libre G, y la entalpía H para dos fases α y β como función de la temperatura.
Figura 6.6. (a) Superficies de energía libre para dos fases α y β, en el espacio G-P-T, su intersección define una curva equilibrio entre ellas. La proyección de esta línea en el plano P-T se denota como AB. (b) El equilibrio entre α y β definido por la línea AB en el plano P-T.
6.4.1. Procesos reversibles e irreversibles. Metaestabilidad
Las curvas Gα y Gβ de la Figura 6.7a indican que para mantener el equilibrio, la fase β, se debe transformar a la α a partir de Tc, si la temperatura desciende. Si esta última aumenta, la transición de fases es inversa, i.e., desde α hasta β. En la Figura 6.7a, las fases estables se muestran con curvas marcadas con flechas. A través de estas curvas, el sistema se encuentra en un continuo estado de equilibrio, por lo que el cambio en la entropía ΔS = 0. Por otro lado, sobre estas curvas el proceso es reversible, debido a que la misma curva existe tanto para el aumento como para el descenso de la temperatura.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 78
Sin embargo, en un ambiente más real, las
transformaciones no ocurren exactamente a la temperatura de equilibrio, debido a que en este punto, la energía libre de ambas fases es la misma. Sólo cuando haya una reducción la energía libre podrá ocurrir la transformación desde β a α. Para que esto ocurra, se requiere de algún grado de enfriamiento ΔT, tal como se muestra en Figura 6.7b. La cantidad de enfriamiento requerida para que la transformación ocurra, depende de la variación estructural entre ambas fases, i.e., la barrera energética que debe atravesar la estructura de alta para llegar a la de baja. Una pequeña barrera implica una pequeña variación en la temperatura. De todas maneras, ΔT es mayor para pasar desde β a α, que desde α a β. Esto, debido a que al aumentar la temperatura, la movilidad atómica crece. (Comparación ΔT Figura 6.7b y 6.7c).
Un efecto de la disminución en la movilidad
atómica, es que los cambios estructurales se vuelven más complicados. Un ejemplo bien conocido lo constituyen los polimorfos diamante y grafito, cuyos campos de estabilidad se muestran en la Figura 6.8. Los diamantes crecen en las profundidades del manto terrestre y son transportados rápidamente a la superficie -fuera de su estado de equilibrio- en magmas kimberlíticos de emplazamiento sub-volcánico. Su tasa de transformación en condiciones ambientales a grafito, es prácticamente despreciable, a pesar del hecho de que su energía libre ha disminuido en cerca de 3 KJ/mol. En estas condiciones, la fase diamante es considerada como metaestable.
Figura 6.7. Curvas de energía libre para dos fases α y β interceptadas en la temperatura de equilibrio Tc.
Figura 6.8. Diagrama de estabilidad. Diamante - Grafito.
Figura 6.9. A temperatura ambiente, el diamante no se transforma a grafito debido a la falta de energía de activación ΔG.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 79
(Figura 6.9). Otro hecho importante de resaltar, es que si una estructura mineral compleja no se
transforma a la fase termodinámicamente estable, puede hacerlo en otra fase que sea de más fácil transformación, tal como lo representa la Figura 6.10. En esta esquema, la línea segmentada representa la energía libre para la fase γ, la cual es termodinámicamente inestable bajo cualquier temperatura, pero puede ser cinéticamente más accesible que la fase α. Si la transformación β ⇒ γ es más fácil (requiere de menos energía de activación) que la transformación β ⇒ α, la transformación β ⇒ γ recibe el nombre de equilibrio metaestable, a la temperatura T2.
Figura 6.10. Curvas de energía libre para las fases α, β y γ.
6.4.2. Transiciones de fase de primer y segundo orden
Las transiciones estructurales de fase entre polimorfos, pueden ser clasificadas a
partir del comportamiento de sus funciones termodinámicas. Estas transiciones de fase ocurren como respuesta de la estructura a los cambios en el ambiente externo al que están expuestas. Las transiciones de fase son clasificadas de acuerdo a la discontinuidad de las derivadas parciales de su energía libre. Así, la energía libre para las transiciones de primera fase son discontinuas en su primera derivada. Por otro lado, las de segundo orden lo son en su segunda derivada. El comportamiento de las funciones termodinámicas para ambos tipos de transición se muestra en la Figura 6.11.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 80
6.5. Determinación de cantidades termodinámicas
Figura 6.11. Variación en las funciones termodinámicas G, H y S, y Cp, en función de la temperatura para: (a) Transición de fase de primer orden. (b) Transición de fase de segundo orden.
Hasta ahora, se han estudiado una serie de funciones termodinámicas, principalmente
G, H, S y Cp, analizando la variación de estas como función de la presión y la temperatura. En esta sección se describe brevemente la forma en que estas funciones son obtenidas.
Existen varias maneras de obtener información termodinámica. Las propiedades
volumétricas son relativamente fáciles de medir, utilizando para esto técnicas de difracción de rayos-x. Las propiedades térmicas son un poco más complicadas y a estas se les hará mención en lo que continúa de este escrito. Los principales mecanismos de medición son: (i) medidas experimentales directas; (ii) estudios de equilibrio de fase y (iii) métodos teóricos como las simulaciones computacionales.
6.5.1. Medidas experimentales directas
Mediciones directas de la capacidad calórica sobre un amplio rango de temperatura, puede ser utilizadas para calcular tanto la entropía como la entalpía. Estas medidas pueden ser realizadas para diferentes rangos de temperatura.
i. Para bajas temperaturas (< a 400 K), la capacidad calórica puede ser medida con alta precisión (error < 0,2%), utilizando técnicas de calorimetría adiabática. La muestra es térmicamente aislada y se registra el alza de la temperatura debida a la aplicación de calor eléctrico durante un tiempo fijo para una determinada cantidad de voltaje.
Mauricio Domcke G.
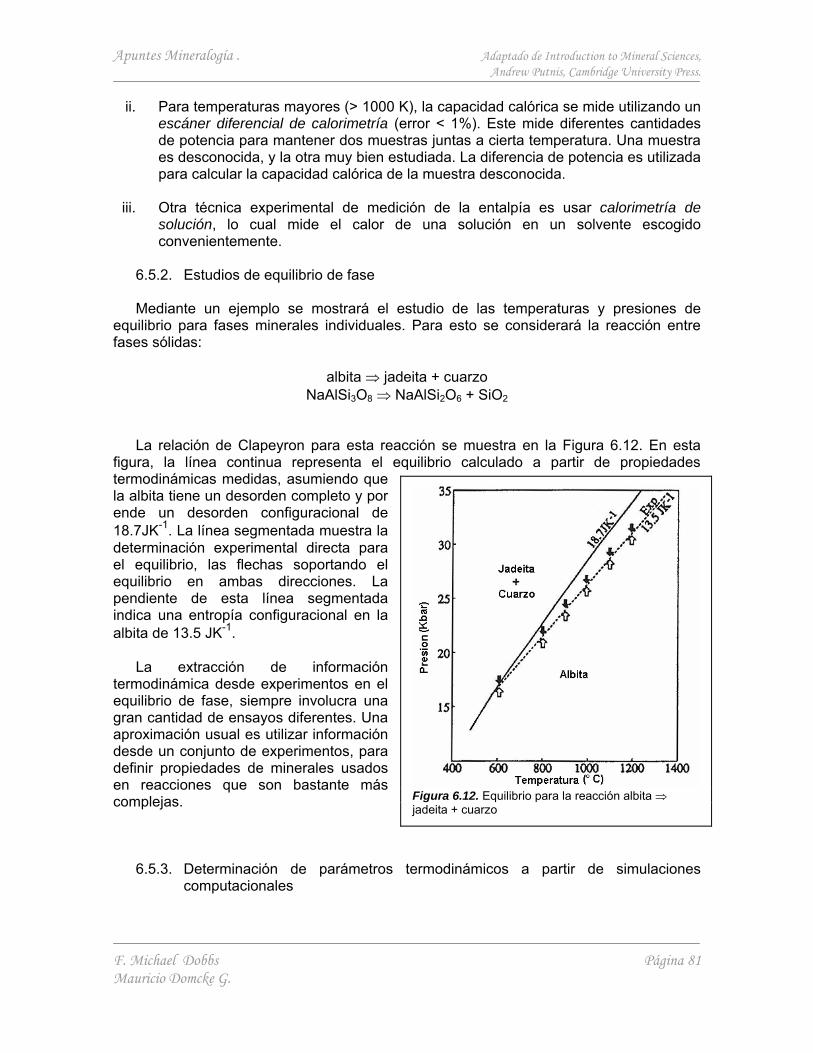
Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 81
ii. Para temperaturas mayores (> 1000 K), la capacidad calórica se mide utilizando un escáner diferencial de calorimetría (error < 1%). Este mide diferentes cantidades de potencia para mantener dos muestras juntas a cierta temperatura. Una muestra es desconocida, y la otra muy bien estudiada. La diferencia de potencia es utilizada para calcular la capacidad calórica de la muestra desconocida.
iii. Otra técnica experimental de medición de la entalpía es usar calorimetría de
solución, lo cual mide el calor de una solución en un solvente escogido convenientemente.
6.5.2. Estudios de equilibrio de fase
Mediante un ejemplo se mostrará el estudio de las temperaturas y presiones de
equilibrio para fases minerales individuales. Para esto se considerará la reacción entre fases sólidas:
albita ⇒ jadeita + cuarzo NaAlSi3O8 ⇒ NaAlSi2O6 + SiO2
La relación de Clapeyron para esta reacción se muestra en la Figura 6.12. En esta figura, la línea continua representa el equilibrio calculado a partir de propiedades termodinámicas medidas, asumiendo que la albita tiene un desorden completo y por ende un desorden configuracional de 18.7JK-1. La línea segmentada muestra la determinación experimental directa para el equilibrio, las flechas soportando el equilibrio en ambas direcciones. La pendiente de esta línea segmentada indica una entropía configuracional en la albita de 13.5 JK-1. La extracción de información termodinámica desde experimentos en el equilibrio de fase, siempre involucra una gran cantidad de ensayos diferentes. Una aproximación usual es utilizar información desde un conjunto de experimentos, para definir propiedades de minerales usados en reacciones que son bastante más complejas. Figura 6.12. Equilibrio para la reacción albita ⇒
jadeita + cuarzo
6.5.3. Determinación de parámetros termodinámicos a partir de simulaciones computacionales
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 82
La simulación computacional de propiedades constituye el último avance dentro de la “experimentación” en las propiedades termodinámicas que gobiernan a los minerales. Un computador es configurado con información al respecto de los átomos constituyentes, y las fuerzas que los relacionan.
i. Cálculo estático de la energía de la red
Aquí el problema es determinar la configuración de mínima energía de un conjunto de átomos, minimizando la energía potencial debida a los enlaces interatómicos. Se comienza con un conjunto particular de coordenadas atómicas y un modelo de potencial atómico, el cual expresa la energía potencial de esta estructura como función de las coordenadas atómicas. Mediante ajustes a las coordenadas atómicas, el computador encuentra las condiciones de mínima energía para una determinada topología y simetría.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 82
7. ENERGÉTICA EN LA ESTABILIDAD DE MINERALES II - SOLUCIÓN SÓLIDA, ORDENAMIENTO Y EXSOLUCIÓN
La existencia de soluciones sólidas juega un rol importante en la determinación de la composición química de los minerales. Como se ha visto hasta ahora, el rango composicional de estos, depende en gran medida la temperatura, y de cómo el aumento ésta puede hacer más flexible la estructura, en orden de albergar una mayor cantidad de cationes y facilitar la posible substitución que exista entre ellos.
7.1. Solución sólida
La mayoría de los grupos minerales existen sobre un rango de composición química, el cual generalmente es expresado en términos de la composición de sus miembros finales. Por ejemplo el olivino (Mg,Fe)2SiO4, puede tener cualquier composición entre forsterita Mg2SiO4 y fayalita Fe2SiO4, mediante la substitución entre Fe2+ y Mg2+. Entonces el olivino constituye una solución sólida substitucional. Esto es posible debido a que la diferencia entre los radios iónicos de Fe2+ y Mg2+ no es muy marcada (aproximadamente menor al 15%). En otros casos, los límites composicionales son dependientes en gran medida de las condiciones ambientales, principalmente temperatura, la cual a mayor grado, da cabida a un mayor rango composicional. Por ejemplo, en el reemplazo de Na+ por K+ en los feldespatos alcalinos, la considerable diferencia de los radios introduce deformaciones y limita a la solución sólida; sin embargo, a altas temperaturas, la solución sólida es virtualmente completa.
Por otro lado, las substituciones no deben ser necesariamente entre dos elementos, y
esto depende de la habilidad que tenga la estructura para acomodar a los nuevos cationes, y las restricciones que tenga para mantener su carga eléctrica balanceada. Por ejemplo en los feldespatos plagioclasa existe una doble substitución para la solución sólida de los extremos albita NaAlSi3O8 y anortita CaAl2Si2O8. la cual puede ser escrita como:
Al3+ + Ca2+ ⇔ Si4+ + Na+
Otra forma menos común por la cual los minerales pueden cambiar su composición, es mediante una solución sólida por omisión. En las pirrotitas Fe1-xS, las vacancias catiónicas generan un rango composicional variable entre FeS y Fe7S8, donde el balance total de carga dentro de la estructura es mantenido convirtiendo algunos de los Fe2+ en Fe3+. Finalmente, en la solución sólida intersticial, sitios estructurales normalmente no ocupados son utilizados. Por ejemplo en la tridimita SiO2, la composición puede variar a nefelina debido a la ocupación de Na+ en sitios intersticiales, provocando la substitución de Al3+ por Si4+, de manera de mantener la carga total neutra.
7.1.1. Energía libre de una solución sólida La estabilidad de una solución sólida dependerá del mínimo valor de la energía libre de mezclado Gmix. El cambio en la energía libre de mezclado ΔGmix está dado por:
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 83
ΔGmix = ΔHmix - TΔSmix Ec. 7.1 Resulta claro que la entropía de mezclado, la cual siempre es positiva, tiende a reducir la energía libre y por lo tanto, favorece la formación de una solución sólida. La limitación en el grado de solución sólida proviene entonces de la entalpía de mezclado. Cuando la estructura de los dos miembros finales de una solución sólida es la misma, la energía libre puede ser representada por una curva a lo ancho de todo el rango composicional, es decir, la solución sólida sólo existe si se mantiene la configuración atómica elemental. En la discusión próxima se considerará que la solución sólida es completa entre sus dos miembros finales, i.e., poseen la misma estructura. De todas maneras, los mismos principios son aplicables a soluciones sólidas con miembros finales de diferentes estructuras. Como se mencionó con anterioridad, el término limitante para que la solución ocurra es el cambio en la entalpía de mezclado. Conforme a eso, se discutirán dos casos:
i. Solución sólida ideal, ΔHmix = 0 La solución sólida es estabilizada en todo el rango composicional por el cambio en la entropía de mezclado, esto es:
Figura 7.1. (a) Energía libre de mezclado ΔGmix de una solución sólida ideal entre sus miembros finales A y B, como función de su composición. (b) La energía libre total de una solución está dada por G1 + Gmix, donde G1 es la energía libre de una mezcla entre A y B.
ΔGmix = - TΔSmix = RT(xALogxA + xBLogxB BB)
como se muestra en la Figura 7.1a. El efecto de la temperatura es meramente cambiar el mínimo valor de la energía libre. La energía total de una solución sólida ideal dependerá también de las energías libres de A y B. Una mezcla mecánica de A y B, con fracciones molares xA y xB respectivamente, tiene una energía libre de:
B
Ec. 7.2 G1 = GAxA + GBxB BB
Entonces, la energía total de una solución sólida está dada por:
GT = G1 GAmix Por lo que, como se muestra en Figura 7.1b:
GT = GAxA + GBxB BB + RT(xALogxA + xBLogxB BB) Ec. 7.3
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 84
ii. Solución sólida regular, ΔHmix > 0 El caso más regular, es que exista un cambio en la entalpía, debido a la interacción atómica entre los componentes A y B. Dependiendo de los valores relativos de ΔH y -TΔS, la energía libre de mezclado puede ser positiva o negativa a lo largo de todo el rango composicional (Ecuación 7.1), tal como lo muestra la Figura 7.2. La Figura 7.3 muestra la dependencia descendiente para la curva ΔGmix, a medida que la temperatura disminuye. A altas temperaturas, la entropía se hace dominante, y la curva ΔGmix es siempre cóncava hacia arriba, es decir, una solución sólida homogénea es estable sobre todo el rango composicional. A medida que la temperatura crece, el término TΔSmix es mayor que ΔHmix sólo para soluciones diluidas, entonces existe una región de composición intermedia, entre los dos mínimos valores de ΔGmix, en la cual la solución sólida puede reducirle valor de su energía libre mediante una desmezcla en dos composiciones separadas, a esta coexistencia de dos fases separadas se le conoce como exsolución. A temperaturas mucho menores, sólo soluciones sólidas anchas y muy diluidas son estables.
Figura 7.2. Energía libre de mezclado ΔGmix de una solución sólida regular.
Dependiendo la magnitud del término ΔHmix, el modelo de solución sólida regular muestra que sobre cierta temperatura, una solución sólida completa es estable, mientras que a menor temperatura, el rango de variación composicional de la solución se vuelve más restringido. Esto se ilustra en un diagrama de fase de equilibrio, el cual muestra el rango de estabilidad como función de la temperatura y la composición.
7.2. Diagrama de fase de equilibrio para una solución sólida - el solvus El diagrama de fases de equilibrio para una solución sólida regular, se obtiene trazando una curva con los sitios de mínima energía libre de mezclado, tal como se observa en Figura 7.3. El resultado es una curva de solubilidad denominada solvus, la cual define los límites de solubilidad de A en B y de B en A, y el campo, sombreado en la figura, en el cual la coexistencia de dos fases es más estable que la solución sólida. Sobre la temperatura crítica Ts, la solución sólida es estable sobre todo el rango composicional. Bajo esta temperatura, existe una falencia en la miscibilidad dentro de la cual la composición estable se obtiene por la desmezcla y la coexistencia de dos fases. En la Figura 7.3, a la temperatura T3, cualquier solución sólida entre x3 y x4, se exsolverá en dos fases de composición x3 y x4 respectivamente. El comportamiento termodinámico para una solución sólida de alta temperatura mientras se enfría, se ilustra en la Figura 7.4. En ésta, se tiene inicialmente una sola fase de composición Co, al llegar a la temperatura T3, esta composición encuentra el solvus, por lo que está en equilibrio con C1. Si la solución sólida de alta temperatura se enfría
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 85
hasta T4, la energía libre que lleva a la desmezcla aumenta, y si existe la suficiente cantidad de tiempo, comenzará a aparecer una pequeña cantidad de material con composición C3, cambiando el resto de la composición de la solución sólida a C2, de forma de mantener el balance másico. La fase de composición C3 forma precipitados dentro de la composición sólida de C2.
La determinación de las proporciones relativas para la exsolución de fases se explicará a continuación, como forma anexa a este capítulo.
Figura 7.3. Esta figura ilustra la relación entre la dependencia de la temperatura de las curvas de energía libre y el diagrama de fase de equilibrio. (a) Variación en la energía libre de mezclado como función de la temperatura para una solución sólida regular en la cual la entalpía de mezclado es positiva. A medida que la temperatura se disminuye desde T1 hasta T5, la influencia de la decreciente entropía genera dos mínimos en la curva de la energía libre, x3 y x4. (b) La posición de estos mínimos define la curva llamada solvus. Sobre Ts existe una solución sólida completa y estable, pero dentro del área sombreada, el equilibrio involucra la coexistencia de dos fases separadas.
Figura 7.4. Secuencia de enfriamiento para una fase de composición inicial Co.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 86
ANEXO Diagrama de fase en sistemas binarios simples
• Fase: Cantidad de materia existente en cualquiera de sus estados, poseedora de una composición química definida, y físicamente distinta de cualquier otra parte inmersa dentro de un sistema.
El motivo de la definición anterior es que en este apéndice, se aplicarán sistemas binarios precisamente a diagramas de fases. El hecho que sean binarios quiere decir que se describirá la estabilidad para dos miembros finales A y B. El punto inicial para lograr entender la forma de los diagramas de fase, lo constituye la curva energía libre v/s composición (en adelante, curva G - x. Para una solución sólida ideal, la curva G - x es continua con un mínimo en x = 0,5 -fase α en la Figura 7.5. Otras soluciones sólidas mas restringidas son representadas también en esta figura, y denotadas como β y γ.
Figura 7.5. Representación esquemática de curvas G – x, para tres fases distintas denominadas α, β y γ.
A continuación, se analizarán distintos procesos de cristalización mediante varios ejemplos.
• Ejemplo 1: Cristalización de una solución sólida ideal Se considerará un fundido de composición binaria en alta temperatura, el cual se enfría para formar una solución sólida continua a baja temperatura. En ambas fases -solución sólida y fundido- las curvas G-x son continuas (Figura 7.6a). A altas temperaturas, como T1, el fundido es estable por sobretodo el rango composicional y su energía libre es menor que la del sólido. Sin embargo, en el enfriamiento, las posiciones relativas de las curvas G- x, tanto para el sólido como el fundido, cambian, y es así que a la temperatura más baja T5, la fase estable para cualquier composición es la sólida (Figura 7.6e). A temperaturas intermedias, ambas curvas deben cruzarse. La Figura 7.6b muestra que el primer toque entre ambas curvas, para una composición pura de A, se produce a la temperatura T2. En la Figura 7.6c, cuando ambas curvas se entrecruzan, las composiciones de coexistencia (c1 y c2) de la solución sólida y el fundido están definidas por la tangente en común. A medida que la temperatura sigue descendiendo, estos puntos de tangencia se mueven dentro del diagrama hacia la derecha, hasta llegar al último punto en que se interceptan, el cual es la temperatura de fusión T4 para la composición 100% B. El diagrama de equilibrio resultante se obtiene mediante la unión de estos puntos de tangencia, tal como lo muestra la Figura 7.6f. Este diagrama puede ser utilizado tanto para entender el comportamiento de un sólido al fundirse, o de un fundido al cristalizar.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 87
Figura 7.6. Secuencia de curvas G – x para un fundido y una solución sólida completa, a medida que la temperatura desciende desde T1 hasta T5.
Para entender el proceso de cristalización se seguirá el siguiente ejemplo. Se tiene un fundido inicial de composición intermedia “x” (Figura 7.6f). A medida que desciende la temperatura, la línea segmentada que sale de x en la Figura 7.6f se topará con otra denominada liquidus. La composición del primer sólido en cristalizar se deduce trazando un línea horizontal a partir de este tope, la cual intercepta otra curva, denominada solidus. Esta composición es considerablemente más rica en A que todo el
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 88
volumen involucrado en el proceso. A medida que la temperatura desciende, la cristalización a lo largo del solidus continúa. La composición de este cristal se deduce de la misma forma que anteriormente, es decir, se continúa por la curva del liquidus y se trazan líneas horizontales hasta interceptar la del solidus. La cristalización ocurrirá hasta que la cantidad de fundido se acabe. Esto ocurre para una cristalización de sólido de composición idéntica al fundido inicial. (Figura 7.7). Puede ocurrir también, que la cinética del proceso no permita que el equilibrio se mantenga a lo largo del enfriamiento. Esto produce, por ejemplo en los feldespatos plagioclasa, zonas de cristales con núcleo de una composición y matriz de otra. El diagrama de fase para esta solución sólida de alta temperatura se muestra en la Figura 7.8.
La anortita tiene mayor punto de fusión que la albita, por lo que los feldespatos plagioclasa de composición intermedia tienden a tener núcleos ricos en Ca, y matrices ricas en Na.
Figura 7.7. Un fundido inicial de composición x topa la línea de liquidus a la temperatura T3, a esta temperatura comienza a cristalizar sólido de composición c1. Debido a que empieza a cristalizar material de composición rica en A, el fundido pierde elementos de esta composición y desciende por la línea del líquidos en la dirección de B. Así, para la composición de fundido igual a c4, la cristalización es de composición c3 (ambos menos ricos en A). Finalmente, el proceso de cristalización culmina en el punto en que se acaba la cantidad de fundido. En este punto cristaliza un sólido de composición idéntica a la inicial del fundido. (c2 = x)
Figura 7.8. Diagrama de fase de alta temperatura, para describir la fusión y cristalización dela solución sólida de las plagioclasas.
• Ejemplo 2: Cristalización en la cual los dos miembros finales tienen soluciones
sólidas limitadas Si existen dos curvas separadas de energía libre, para dos soluciones sólidas limitadas α y β, la secuencia de curvas de energía libre es la que se muestra en Figura 7.9, juntas al diagrama de fase. Bajo los puntos de fusión de los dos miembros finales, las curvas de liquidus y solidus definen regiones de dos fases (fundido + α, fundido + β) en cualquier lado del diagrama. Existe una cierta temperatura en donde las curvas G – x de
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 89
ambas fases comparten sólo una tangente, y entonces todo está en equilibrio. Esta temperatura es conocida como temperatura eutéctica Te. El punto E en el diagrama de fase es el punto eutéctico, en el cual el fundido está en equilibrio tanto con las fases α y β, como con sus composiciones dadas por los finales de sus líneas horizontales de empate.
Figura 7.9. Secuencia de curvas G – x para un fundido y dos fases sólidas α y β, como función de la disminución de la temperatura desde T1 a T5, y el resultante diagrama de fase (f). Las curvas de energía libre para los dos sólidos puede ser igualmente bien reemplazada por una solución sólida con una falta en la miscibilidad.
Mauricio Domcke G.
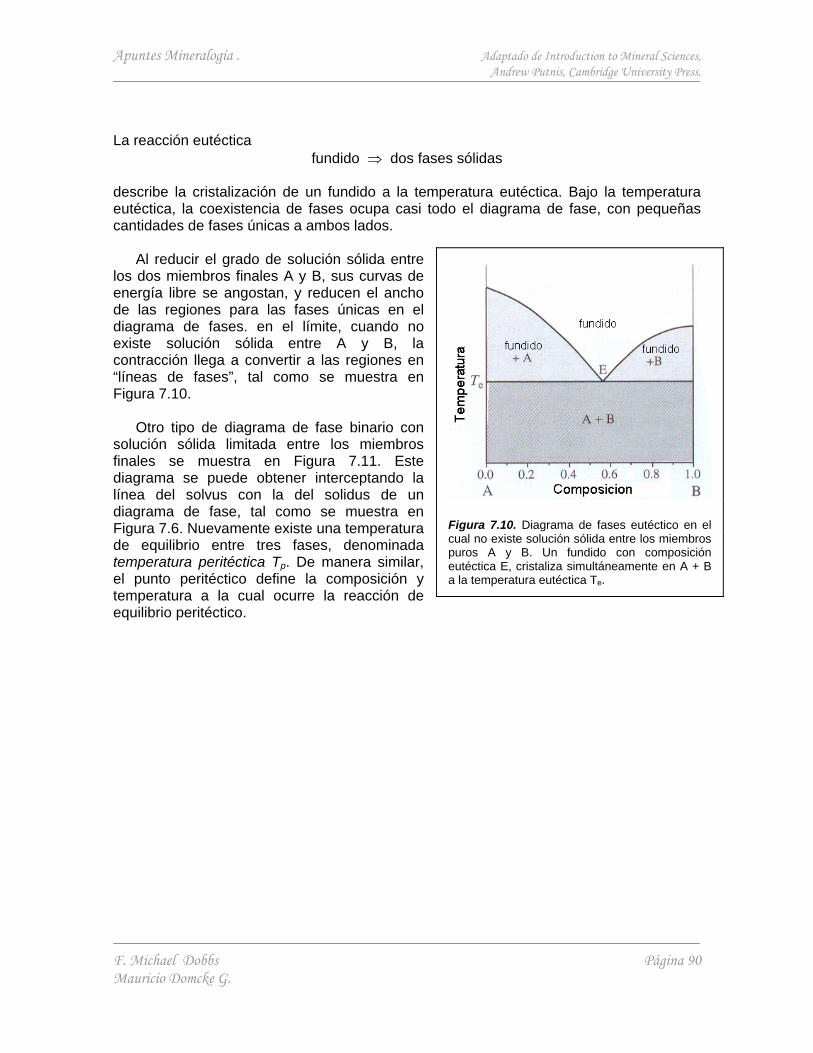
Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 90
La reacción eutéctica
fundido ⇒ dos fases sólidas describe la cristalización de un fundido a la temperatura eutéctica. Bajo la temperatura eutéctica, la coexistencia de fases ocupa casi todo el diagrama de fase, con pequeñas cantidades de fases únicas a ambos lados. Al reducir el grado de solución sólida entre los dos miembros finales A y B, sus curvas de energía libre se angostan, y reducen el ancho de las regiones para las fases únicas en el diagrama de fases. en el límite, cuando no existe solución sólida entre A y B, la contracción llega a convertir a las regiones en “líneas de fases”, tal como se muestra en Figura 7.10. Otro tipo de diagrama de fase binario con solución sólida limitada entre los miembros finales se muestra en Figura 7.11. Este diagrama se puede obtener interceptando la línea del solvus con la del solidus de un diagrama de fase, tal como se muestra en Figura 7.6. Nuevamente existe una temperatura de equilibrio entre tres fases, denominada temperatura peritéctica Tp. De manera similar, el punto peritéctico define la composición y temperatura a la cual ocurre la reacción de equilibrio peritéctico.
Figura 7.10. Diagrama de fases eutéctico en el cual no existe solución sólida entre los miembros puros A y B. Un fundido con composición eutéctica E, cristaliza simultáneamente en A + B a la temperatura eutéctica Te.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 91
fundido + sólido ⇒ sólido
• Ejemplo 3: Diagramas de fase complejos
Figura 7.11. (a) Diagrama de fases con un intervalo simple de fundido a alta temperatura y el solvus a baja temperatura. (b) La intersección de la curva del solidus con el solvus genera un punto P, denominado punto peritéctico, en el cual la composición sólida peritéctica P, está en equilibrio con el fundido M y el sólido S.
Hasta ahora, se han ejemplificado tipos de cristalización para solución sólida, cristalización eutéctica y peritéctica. En la práctica, diagramas de fase más complejos se obtienen de la combinación de éstos, en los cuales a composición intermedia de los miembros finales contiene otras fases.
Mauricio Domcke G.

Apuntes Mineralogía . Adaptado de Introduction to Mineral Sciences, Andrew Putnis, Cambridge University Press.
F. Michael Dobbs Página 92
Para ejemplificar estos diagramas, se considerará el sistema de alta temperatura MgO - SiO2, en el cual existen las siguientes fases sólidas : periclasa, MgO; forsterita, Mg2SiO4; enstatita, MgSiO3; y sílice SiO2 tanto como cristobalita como tridimita. (Figura 7.12). Ninguna de esta fases existe como solución sólida dentro del diagrama, por lo que aparecen como líneas de fases dentro de éste. Además, el fundido el cual a altas temperaturas existe como una única fase, bajo los 1950° C se vuelve inmiscible (2 líquidos) a la composición rica en sílice. Esta falta en la miscibilidad es análoga a la del solvus en una solución sólida, y determina la separación de fundidos ricos en SiO2 y Mg. Existen dos eutécticos dentro de este sistema, uno cercano a los 1850°C, en el cual un fundido de composición cercana al 40% SiO2 cristaliza en periclasa + forsterita; y otro cercano a los 1540° C donde un fundido de composición cercana al 65% SiO2 cristaliza en cristobalita + enstatita. La cristobalita pura se funde a una temperatura cercana a los 1900° C, a un fundido de la misma composición, i.e., ocurre una fusión congruente. Por otro lado, en la enstatita ocurre una fusión incongruente a los 1557° C, debido a que se transforma en forsterita más un fundido rico en sílice.
Figura 7.12. Diagrama de fase para el sistema MgO – SiO2.
La transformación desde cristobalita hasta tridimita se indica meramente por las líneas horizontales entre la enstatita y la sílice marcando el cambio de fase.
Mauricio Domcke G.