leucemias y linfomas en pediatria
TRANSCRIPT

LEUCEMIAS Y LINFOMAS EN PEDIATRIAGABRIELA VELÁSQUEZ ARTEAGA
TUTOR: DRA. GUILLERMINA YONG
UNIVERSIDAD DE GUAYAQUIL FACULTAD DE CIENCIAS MEDICAS
INTERNADO ROTATIVOHOSP. PEDIATRICO ROBERTO GILBERT
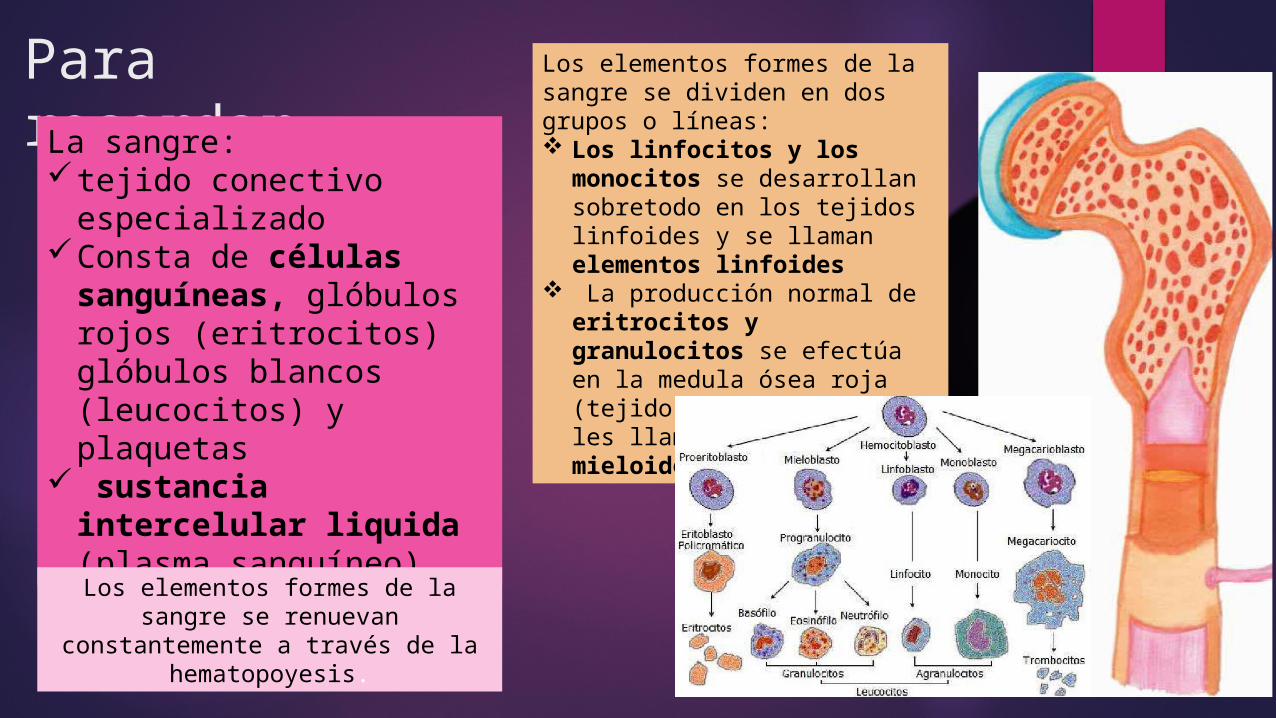
Para recordar….La sangre: tejido conectivo
especializadoConsta de células
sanguíneas, glóbulos rojos (eritrocitos) glóbulos blancos (leucocitos) y plaquetas
sustancia intercelular liquida (plasma sanguíneo)
Los elementos formes de la sangre se renuevan constantemente a
través de la hematopoyesis.
Los elementos formes de la sangre se dividen en dos grupos o líneas: Los linfocitos y los
monocitos se desarrollan sobretodo en los tejidos linfoides y se llaman elementos linfoides
La producción normal de eritrocitos y granulocitos se efectúa en la medula ósea roja (tejido mieloide) y se les llama elementos mieloides

LEUCEMIAS EN PEDIATRIA La leucemia es un cáncer que se origina en las células primitivas
productoras de sangre de la médula ósea.
La proliferación incontrolada y progresiva de dichas células da lugar a una sustitución de las células normales de la médula ósea, invadiendo la sangre y los diferentes órganos y tejidos.
Se han identificado leucemias derivadas de cada una de las series celulares de la médula ósea: leucemia linfoblástica (LL), leucemia mieloblástica, leucemia monocítica, leucemia mielomonocítica, etc
Además, según su curso clínico se clasifican en: agudas, sin tratamiento llevan a la muerte del paciente en semanas o meses, y crónicas, sin tratamiento pueden causar la muerte en varios meses o años.

La causa es desconocida. Lo que se conoce son factores de riesgo que parecen estar implicados en su aparición:
_Factores genéticos: hay mayor riesgo en sd de down, en la anemia de Fanconi, en individuos con ataxia-telangiectasia, etc.
– Exposición amplia a productos químicos: benceno, productos derivados del petróleo y determinados pesticidas.
– Radiaciones ionizantes: por ejemplo, en los supervivientes de las explosiones atómicas de y Nagasaki.
– Quimioterapia por un tumor anterior: el uso de determinados fármacos en el tratamiento del cáncer se asocia a un mayor riesgo de leucemias.
– Alteraciones inmunológicas: hay un aumento de incidencia de linfomas en individuos con enfermedades autoinmunes (lupus eritematoso diseminado, síndrome de Sjögren, artritis reumatoide juvenil, etc.), tratados con inmunosupresores durante bastante tiempo.

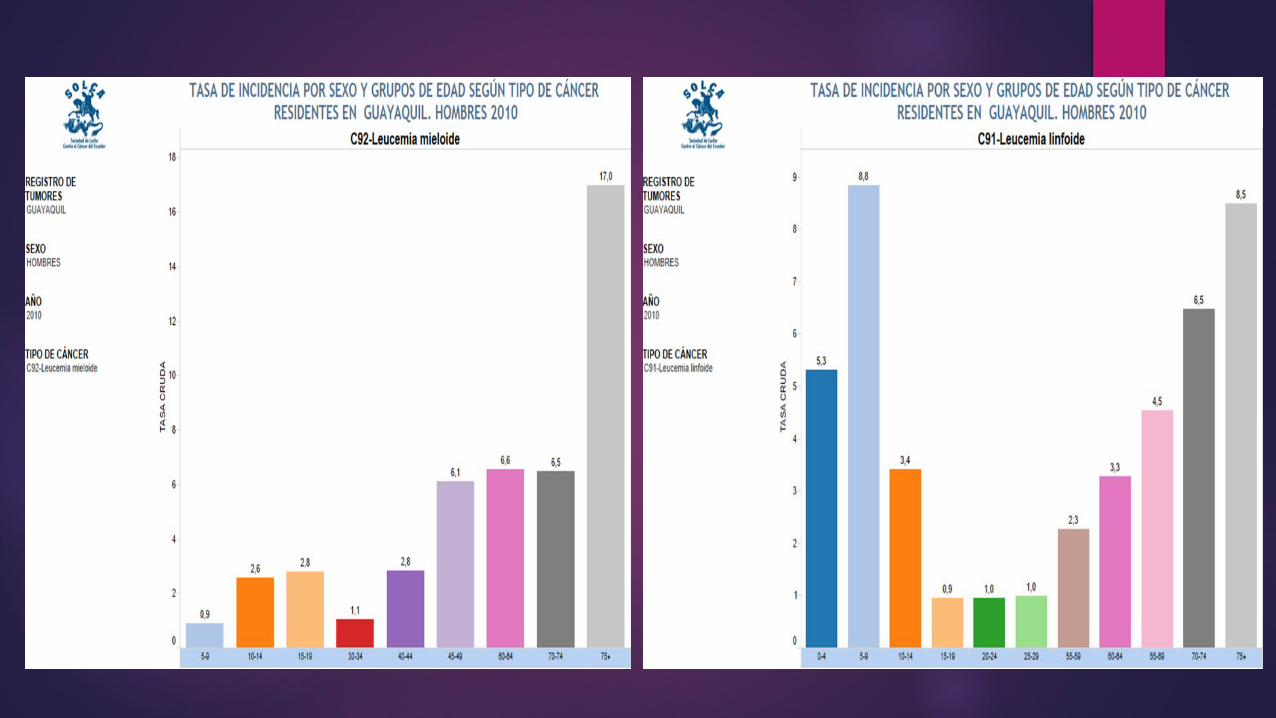
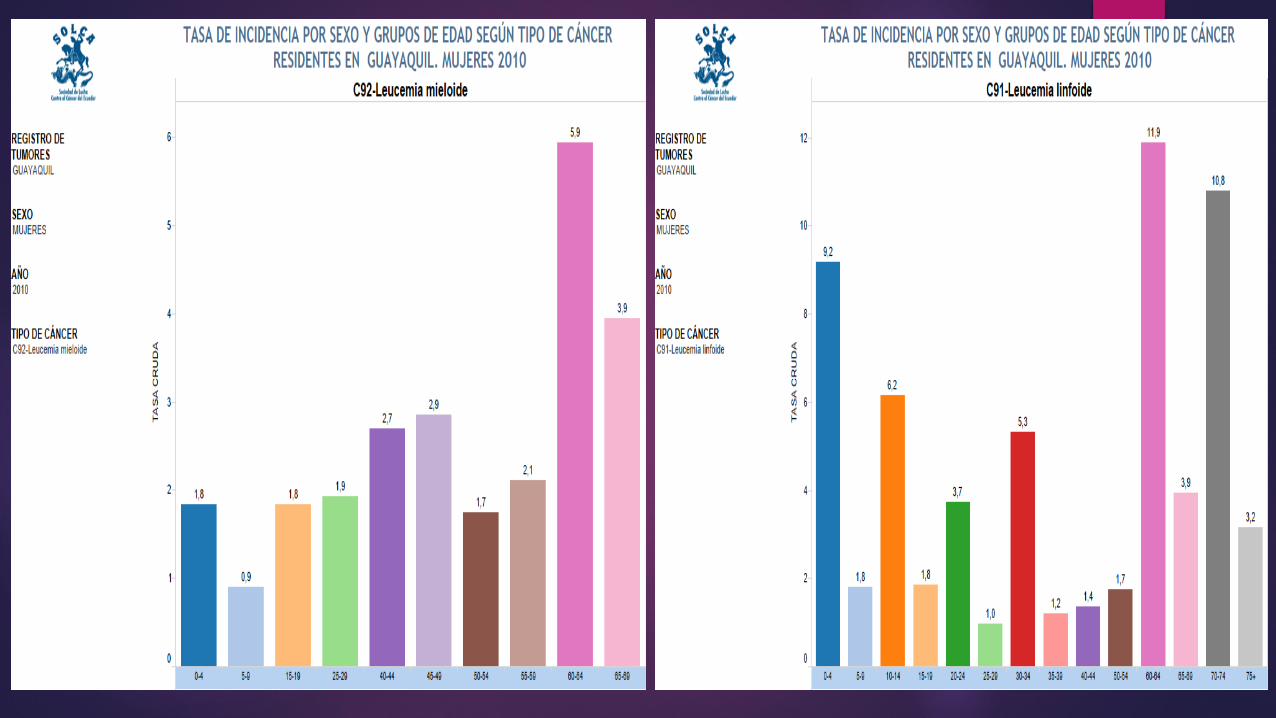
EPIDEMIOLOGIALa leucemia representa un 25-30% de
las neoplasias en menores de 14 años, siendo el cáncer más frecuente
en la infancia
Más de un 95% de las leucemias infantiles son
Agudas LLA (80%) LMA (20%)
La ALL es ligeramente más común entre los niños blancos e hispanos que entre los niños afroamericanos y americanos asiáticos, y es más común entre los niños que entre las niñas.
La AML ocurre casi igualmente entre niños y niñas de todas las razas.
Las leucemias crónicas son poco comunes en los niños. La mayoría de los casos son leucemia mieloide crónica (CML), la cual tiende a ocurrir más en adolescentes que en niños de menor edad.

LEUCEMIAS AGUDAS
Los dos tipos principales de leucemia aguda son: · Leucemia linfocítica aguda (linfoblástica) (acute lymphocytic leukemia,
ALL) 80%:Alrededor de tres de cuatro leucemias en niños son ALL. Esta leucemia se origina de formas tempranas de linfocitos en la médula ósea. · Leucemia mieloide aguda (acute myelogenous leukemia, AML) 20%:
este tipo de leucemia, también llamada leucemia mieloide aguda, leucemia mielocítica aguda o leucemia no linfocítica aguda representa la mayoría de los casos remanentes. La AML se inicia a partir de las células mieloides que forman los glóbulos blancos (que no son linfocitos), los glóbulos rojos o las plaquetas
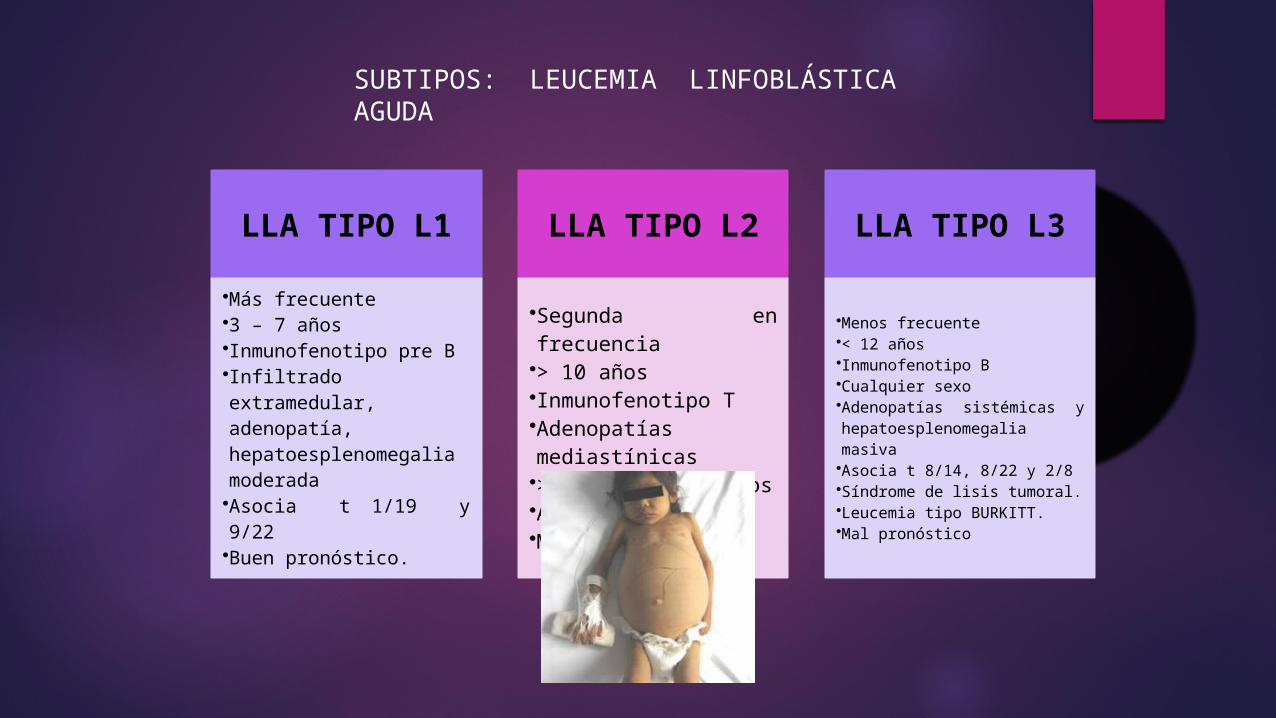
LLA TIPO L1
•Más frecuente•3 – 7 años•Inmunofenotipo pre B•Infiltrado extramedular, adenopatía, hepatoesplenomegalia moderada•Asocia t 1/19 y 9/22 •Buen pronóstico.
LLA TIPO L2
•Segunda en frecuencia•> 10 años•Inmunofenotipo T•Adenopatías mediastínicas•> 50 000 leucocitos•Asocia t 11/14•Mal pronóstico
LLA TIPO L3
•Menos frecuente•< 12 años•Inmunofenotipo B•Cualquier sexo•Adenopatías sistémicas y hepatoesplenomegalia masiva•Asocia t 8/14, 8/22 y 2/8•Síndrome de lisis tumoral.•Leucemia tipo BURKITT.•Mal pronóstico
SUBTIPOS: LEUCEMIA LINFOBLÁSTICA AGUDA


El cáncer pediátrico no es prevenible, pero se puede detectar oportunamente. La demora en la remisión de un paciente con
cáncer y la iniciación tardía o suspensión del tratamiento pueden significar la diferencia entre la vida y la muerte.
Ante cualquier sintomatología indicativa de cáncer es esencial iniciar el abordaje con una buena anamnesis.Debemos interrogar sobre antecedentes familiares de tumores, muertes y causas de éstas, así como sobre la posible exposición materna a rayos X.En los antecedentes patológicos, es relevante registrar la presencia de enfermedades genéticas que predispongan a presentar tumores (síndrome de Down, anemia de Fanconi, neurofibromatosis, etc.), o de algunas situaciones en las que la incidencia de cáncer es superior a la habitual como inmunodeficiencias congénitas, enfermedades autoinmunes o malformaciones congénitas
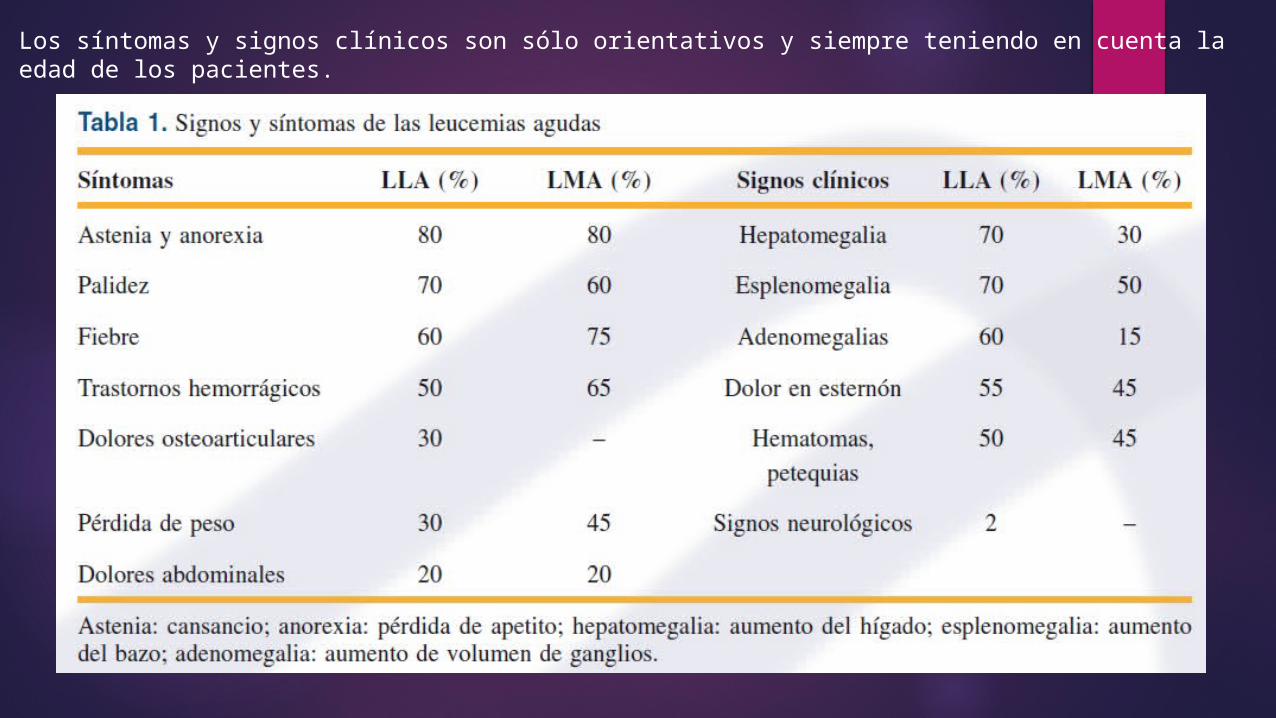
Los síntomas y signos clínicos son sólo orientativos y siempre teniendo en cuenta la edad de los pacientes.
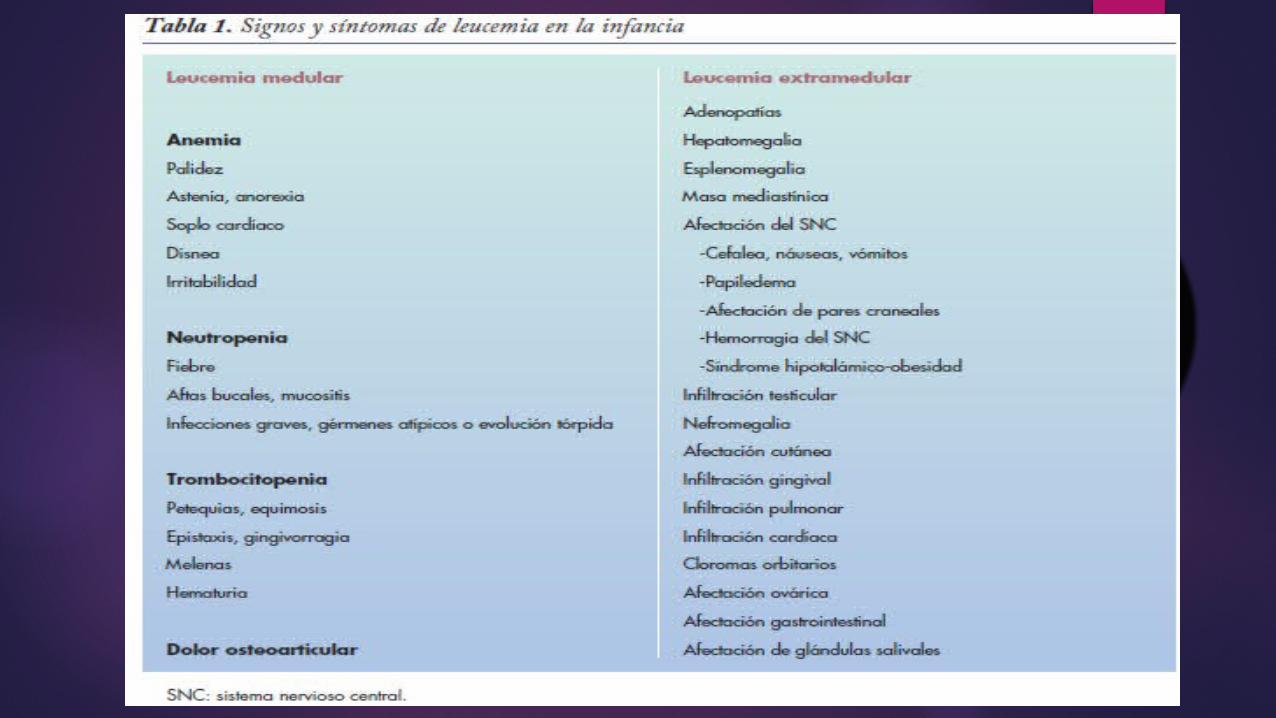

En la práctica clínica pediátrica, las adenopatías cervicales, inguinales o axilares son frecuentes y están presentes en numerosas infecciones virales.
Una adenopatía debe hacernos sospechar malignidad si:— Tiene un diámetro superior a 2,5 cm.— No responde al tratamiento antiinflamatorio y antibiótico durante > 2 semanas.— Está situada en una localización menos frecuente: supraclavicular, retroauricular o epitroclear.— Se asocia a síndrome tóxico y/o a pérdida de más de un 10% del peso inicial.— La radiografía de tórax es patológica con ensanchamiento mediastínico.

Se sugiere que ante la clasificación de posible cáncer o sospecha diagnóstica de leucemia aguda se realice remisión inmediata a un centro asistencial de tercer o cuarto nivel, de alta complejidad que cuente con servicio y especialistas en oncología, hematología, oncohematologia pediátrica

DIAGNOSTICO:CLÍNICO:• Los datos clínicos nos sirven de
sospecha.
LABORATORIO:•Nos sirve para confirmar el diagnóstico.•En sangre periférica:•Glóbulos rojos : anemia normocítica normocrómica•Plaquetas: trombocitopenia – 50.000 por mm3 (Sínd Hemorrágico)•Leucocitos: 20.000- 50.000 por mm3 (promedio). Cuando presenta + 50.000 la leucemia es grave y maligna
Aumento del ácido úrico (Hiperuricemia): Cuando se inicia la quimioterapia hay un aumento del catabolismo celular lo cual produce una lisis celular con aumento de ácido úrico; esto puede llevar al paciente a una insuficiencia renal por bloqueo tubular y producir la muerte del paciente. Por este motivo antes de comenzar el tratamiento con quimioterapia se debe administrar alopurinol para disminuir los niveles de ácido úrico.

EXAMEN PARA CONFIRMAR EL DIAGNOSTICO:
PUNCIÓN Y ASPIRADO DE LA
MÉDULA ÓSEA: Da el diagnóstico definitivo
• Blastos -30%, se hace seguimiento y nuevo aspirado después de un tiempo
• 30- 50%, caso probable de leucemia
• +50%, positivo para leucemia
PUNCIÓN LUMBAR: Se estudia el LCR en
caso de infiltración del SNC. • Pleocitosis
moderada con presencia de blastos
• Aumento de proteínas
• Disminución de glucosa
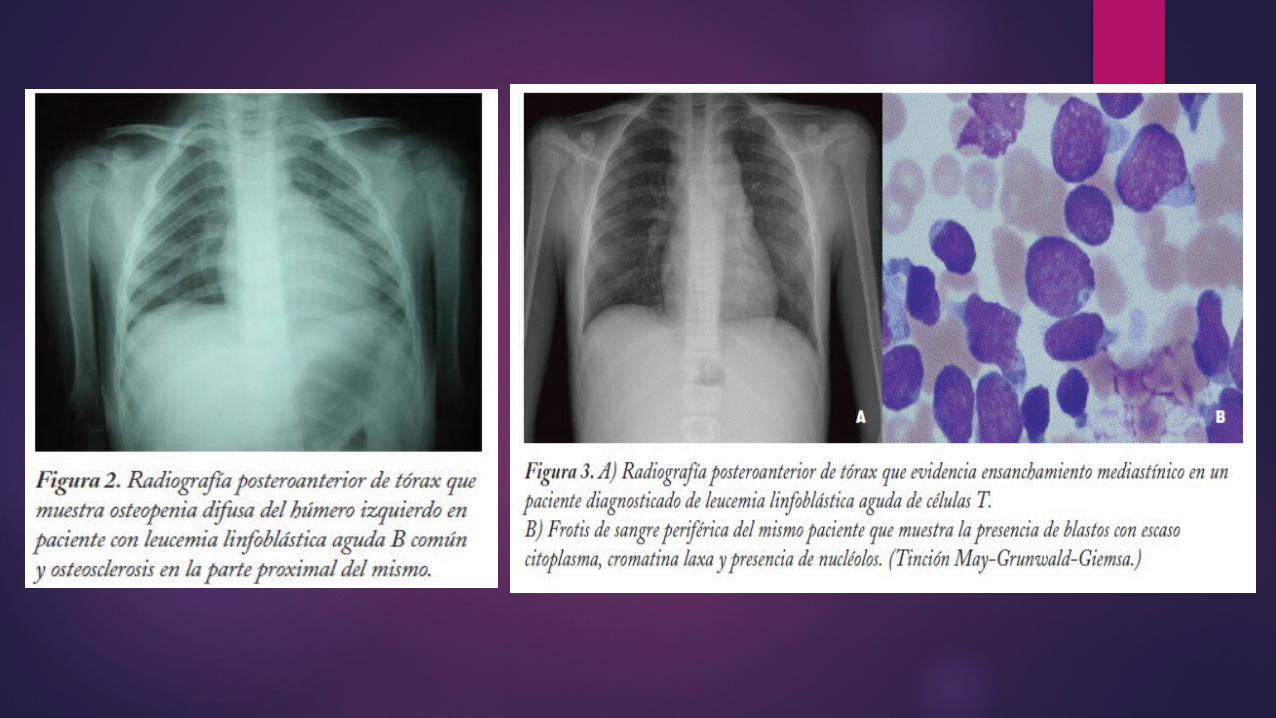
DATOS RADIOLOGICOS
:
• Bandas radiolúcidas transversas localizadas en metáfisis
• Lesiones osteolíticas
• Lesiones de osteoporosis
• Lesiones de osteoesclerótica
• Elevación subperióstica con neoformación ósea
TRATAMIENTOMEDIDAS GENERALES• Alimentación• Higiene adecuada• Apoyo psicológico y
emocional• Glóbulos rojos
concentrados 5ml/Kg, si la Hb es <7g.
• Concentrado de plaquetas 5 U/Kg, si hay <20.000 mm3
• En el caso de septicemia se da: Ampicilina + Aminoglucósidos + Cefalosporinas
TRATAMIENTO ESPECÍFICO• Fase de inducción de
la remisión• Fase de profilaxis• Fase se
mantenimiento

FASE DE INDUCCIÓN DE LA REMISIÓN
Esta fase del tratamiento dura de 6- 8 semanas y obtenemos el 90% de la remisión completa clínica y de laboratorio, por lo tanto hay que iniciar la fase de profilaxis del SNC para evitar la leucemia meníngea
Hidratar al paciente
Administrar Alopurinol 100 mg/m2 de superficie corporal en 3 dosis
Prednisona 40 mg/m2 de superficie corporal/día VO + Vincristina 1.5 mg/m2 de superficie corporal/ semanal IV + Asparaginasa 100.000 U/m2 de superficie corporal cada 2 semanas IM
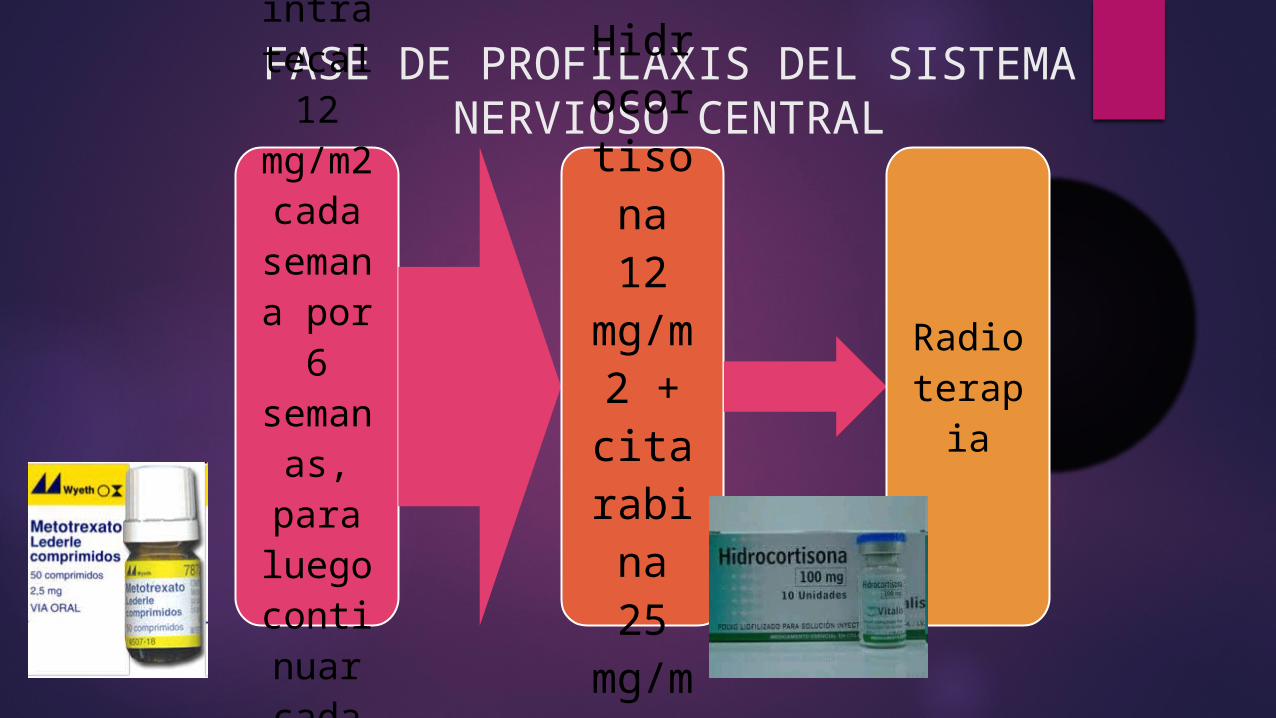
FASE DE PROFILAXIS DEL SISTEMA NERVIOSO CENTRAL
Metotrexate por vía
intratecal 12 mg/m2 cada semana por
6 seman
as, para luego contin
uar cada 8 semanas por
2 años.
Hidrocortisona 12
mg/m2 +
citarabina 25
mg/m2
Radioterapia

FASE DE MANTENIMIEMTO
Debe durar 2 años y
medio a 3 años
6 Mercaptopu
rina 50 mg/m2/día
VO + Metotrexate 20 mg/m2
cada semana VO
o IV
Luego un refuerzo
mensual de Vincristina 1.5mg/m2
cada mes IV +
Prednisona 40 mg/m2 VO los 7 primeros días de
cada mes.
“Si a pesar del
tratamiento el
paciente no
responde como último recurso queda el
trasplante medular”.

Leucemia linfocítica aguda (ALL)La tasa de supervivencia a 5 años para los niños con ALL ha aumentadosignificativamente con el tiempo y en general ahora es mayor de 85%.
Leucemia mieloide aguda (AML)También la tasa de supervivencia a cinco años general para niños con AML ha aumentado con el tiempo y ahora está entre 60% y 70%. Sin embargo, las tasas de supervivencia varían dependiendo del subtipo de AML y de otros factores. Por ejemplo, la mayoría de los estudios sugieren que la tasa de curación de la leucemia promielocítica aguda (APL), un subtipo de AML, es ahora superior al 80%, aunque las tasas son menores para algunos otros subtipos de AML.
LINFOMAS EN PEDIATRIA
LINFOMA NO HODGKIN
LINFOMA DE HODGKIN

Los linfomas son un tipo de cáncer que se desarrolla en el sistema linfático.
El sistema linfático incluye la red de vasos linfáticos y los ganglios linfáticos distribuidos por todo el organismo y en conexión con el sistema sanguíneo.
Por los vasos linfáticos circulan los linfocitos que combaten las infecciones. Otras estructuras del sistema linfático son: bazo, timo, amígdalas y médula ósea.
También se encuentra en la mucosa del estómago e intestino y en la propia piel, lo que explica que los linfomas puedan aparecer en cualquiera de estas áreas, en especial los linfomas no Hodgkin

EPIDEMIOLOGIA
Los linfomas (Hodgkin y no Hodgkin) ocupan el tercer lugar en frecuencia de los cánceres en pediatría.
El linfoma no Hodgkin (LNH) comprende aproximadamente entre 7 al 10% de las causas de cáncer en niños y jóvenes menores de 20 años.
En general ocurren más frecuentemente en la segunda década de la vida y no es común en niños menores de 3 años. La incidencia en las dos últimas décadas ha aumentado en los grupos de edad entre 15 y 19 años y se ha mantenido estable en menores de 15 años (Pery, 1999).

• El linfoma de Hodgkin (LH), también llamado enfermedad de Hodgkin (EH), constituye el 6% de las neoplasias de los niños (Reiter, 2006).
• Es poco frecuente en menores de 5 años
• Los adolescentes son el grupo pediátrico donde se presenta con mayor frecuencia; predomina en varones con una razón 4:1 entre 3 y 7 años, de 3:1 entre 7 y 9 años y de 1,3:1 en mayores de 10 años (Tarbell, 1993).
• Se ha reportado mayor frecuencia de enfermedad en los parientes en primer grado de consanguinidad
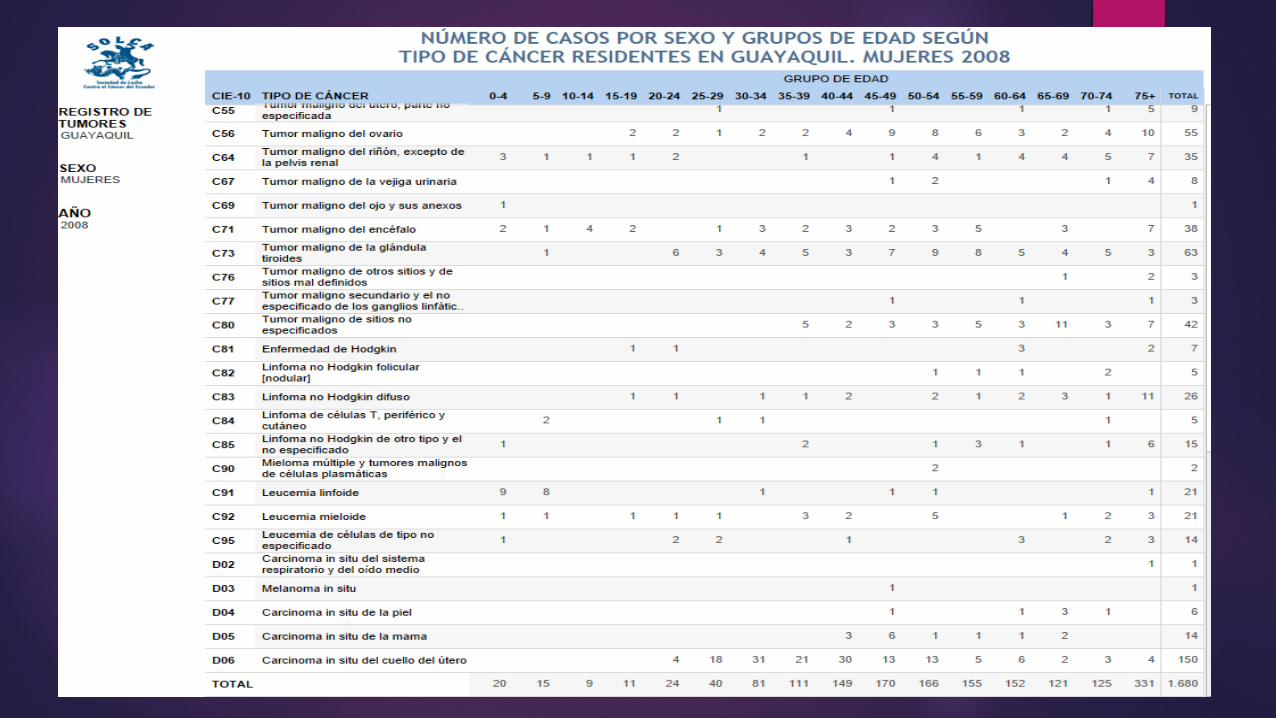
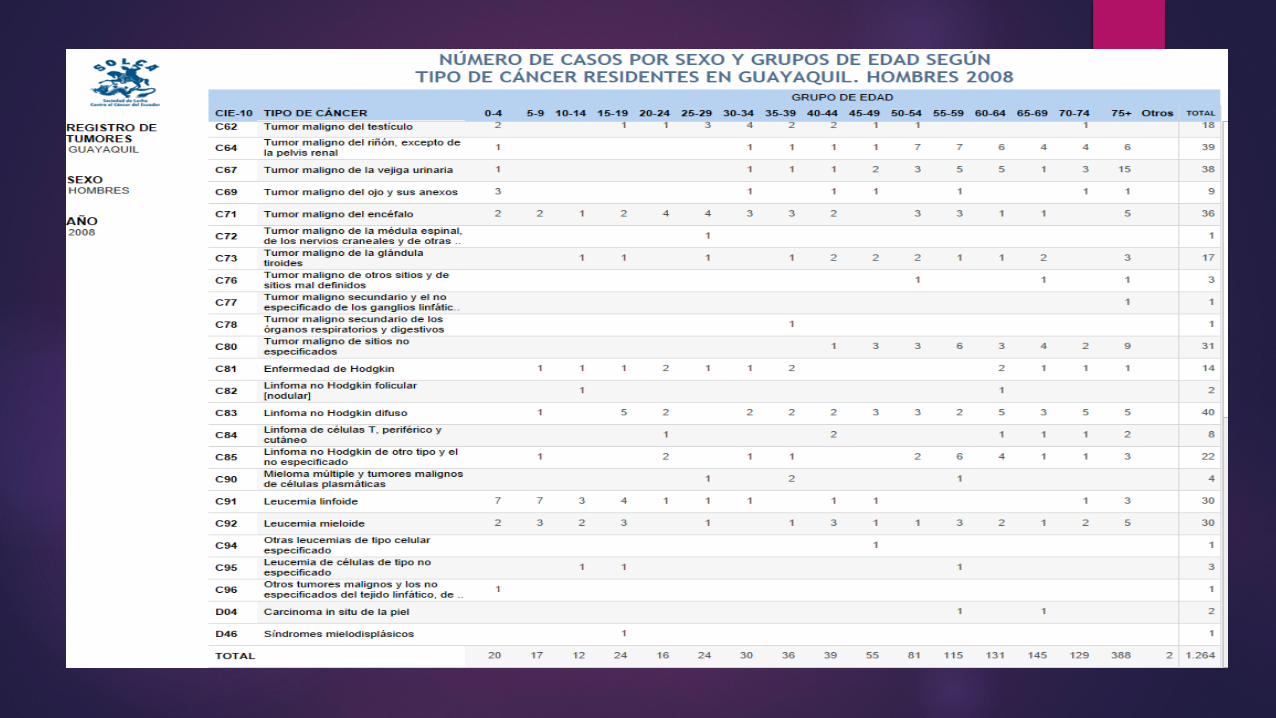
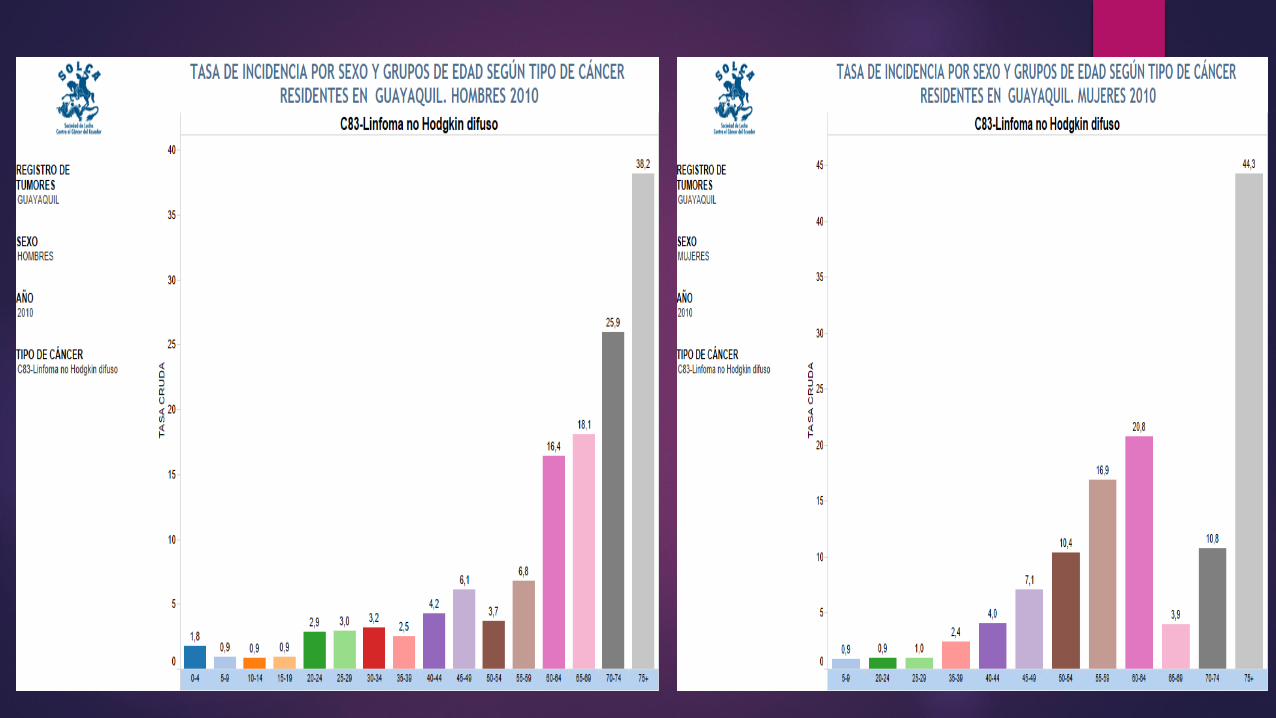

LINFOMA O ENFERMEDAD DE HODGKIN La causa etiológica es desconocida; recientemente se ha
relacionado con el virus de Epstein-Barr. Otro hecho interesante es que, en individuos positivos para
el VIH con linfoma de Hodgkin, la inmensa mayoría son positivos para el virus de Epstein-Barr, responsable de la mononucleosis infecciosa.
Los pacientes que han presentado mononucleosis infecciosa tienen un mayor riesgo de padecer este tipo de linfoma.
Los signos y síntomas más comunes del linfoma de Hodgkin en edad pediátrica son: fiebre adenopatía cervical indolora, pérdida de peso >10%, adenopatía supraclavicular, sudoración, adenopatía axilar, epitroclear anorexia masa mediastinal,.
fatiga, dificultad para respirar, circulación colateral, tos persistente, ingurgitación yugular, dolor torácico, síndrome de vena cava superior, prurito, esplenomegalia, hepatomegalia

La biopsia por escisión de la adenopatía sospechosa. El diagnóstico debe completarse con una historia clínica y una
exploración física cuidadosa de todas las regiones linfáticas accesibles, valorando el tamaño del hígado y bazo.
Se realizarán pruebas de laboratorio, estudios radiológicos (TC o PET) y biopsia/aspirado de la médula ósea.
Todo esto conduce a conocer los estadios de extensión que tienen un valor pronóstico y son importantes a la hora de la planificación terapéutica.
Los estadios van desde formas localizadas a formas extensas
DIAGNOSTICO

CLASIFICACIÓN ANATÓMICA ANN ARBORUbica a los pacientes en estados clínicos de la enfermedad (Howard, 2007)
Estado I Compromiso de una zona ganglionar o un sitio extraganglionar.
Estado II Compromiso de 2 o más zonas ganglionares en el mismo lado del dia- fragama o compromiso extranodal localizado en el mismo lado del diafragma.
Estado III Compromiso ganglionar de 2 o más zonas ganglionares o compromiso de órganos extraganglionares en ambos lados del diafragma.
Estado IV Enfermedad no localizada, difusa o diseminada que compromete uno o más órganos extralinfáticos con o sin compromiso ganglionar. Incluye cualquier compromiso de hígado, médula ósea, pulmones o de LCR.
Zonas del sistema linfático: Ganglios, bazo, timo, anillo de Waldeyer, apéndice y placas de Peyer
Sufijo A Sin síntomas B, Sufijo B Con síntomas B
Sufijo E para compromiso extranodal
Sufijo S para compromiso de bazo
Sufijo X para masa voluminosa

TRATAMIENTO Los elementos básicos del tratamiento son la radioterapia, la quimioterapia, la combinación
de quimio/radioterapia y el tratamiento de apoyo.
La elección del tratamiento viene dada en gran parte por extensión de la enfermedad.
La radioterapia es un tratamiento local, por lo que puede plantearse como único tratamiento en estadios localizados.
La quimioterapia es el tratamiento de elección en la mayoría de los casos, especialmente en los estadios avanzados (III-IV).
En todos los casos se puede contemplar el uso de radioterapia sobre áreas afectadas inicialmente, tras seis ciclos de quimioterapia, especialmente si existen dudas de la existencia de enfermedad residual.
Dado el alto porcentaje de curaciones, es esencial valorar sus efectos secundarios a la hora de decidir el tratamiento. Las toxicidades inmediatas comprenden las hematológicas, náuseas/vómitos, neuropatías, mucositis, alopecia, etc., que se pueden prevenir o minimizar.
En ocasiones pueden aparecer efectos secundarios tardíos como leucemias agudas, cáncer de pulmón, mama, melanomas. Otros efectos secundarios son la infertilidad, afectación cardíaca, hipotiroidismo, etc. Todo esto hace que el seguimiento sea importante en la asistencia integral de los pacientes

LINFOMA NO HODGKINLos signos y síntomas más comunes de LNH en edad pediátrica son: • masa abdominal, • crecimiento de los ganglios, • adenomegalias de cualquier localización, • Inflamación del cuello o la mandíbula, • ingurgitación venosa del cuello, • dificultad para deglutir, • asimetria de las amígdalas, • dificultad respiratoria, • tos, • sibilancias, • ortopnea, • hipoventilacion, • dolor óseo, fiebre, • hepatomegalia y/o esplenomegalia, • pérdida de peso,• palidez
• asimetría facial, • parálisis facial central o
periférica, • sudoración nocturna, • masa maxilar y/o
orbitaria, • cefalea, • debilidad para la marcha, • paraplejia/paraparesia • petequias, • sangrado, • cambios de pigmentación, • aumento del tamaño
testicular

DIAGNOSTICO
En la población pediátrica esto permite considerar dos grandes grupos de patologías:
Las neoplasias de precursores B o T dentro de las que están los linfomas linfoblásticos (LL), y
El grupo de las neoplasias de células B maduras periféricas dentro de las que están los Linfomas de Burkitt (LB) y tipo Burkitt, los linfomas de célula B grande y difusos, los linfomas anaplásicos de célula grande y otras variedades menos frecuentes como los linfomas T periféricos
Biopsia por escisión de la adenopatía sospechosa
Se sugiere tomar hemograma completo, extendido de sangre periférica, análisis bioquímico completo: creatinina, nitrogeno uréico, transaminasas, bilirrubinas, ácido úrico, sodio, potasio, calcio, fosforo, magnesio, LDH, radiografía de tórax y ecografía abdominal a todo paciente con sospecha de LNH

Es importante realizar pruebas radiológicas y de laboratorio que nos ayudarán a estimar el riesgo.
Permite agrupar en: bajo riesgo, en riesgo intermedio-bajo, intermedio-alto y alto, que serían los de peor pronóstico y tendrían implicaciones en el tratamiento.
La planificación terapéutica depende del tipo de linfoma, del estadio de extensión de la enfermedad, así como del índice pronóstico. En la estrategia terapéutica se contempla la cirugía, la radioterapia, la quimioterapia, interferón, anticuerpos monoclonales y la radioinmunoterapia.
La cirugía desempeña un papel principal en la toma de biopsias, pero en este tipo de linfomas puede tener indicación terapéutica en localizaciones extraganglionares como en estómago, tiroides, etc.
La radioterapia puede valorarse en estadios localizados (I-II), pero en general se utiliza quimioterapia.
Los anticuerpos monoclonales como rituximab constituyen uno de los avances terapéuticos más importantes en los LNH de tipo B que expresan en antígeno CD20, utilizándose en combinación con quimioterapia. Otras posibilidades son interferón y radioinmunoterapia.
El interferón puede presentar en el transcurso del tratamiento prolongado alteraciones psíquicas y neurológicas, toxicidad hematológica y hepática, astenia y náuseas y vómitos como lo más destacable, de los que se recupera el paciente al suspender el tratamiento.