lecture 4 and 5 leukemia and chronic myeloproliferative disorders abdulkarim aldosari
TRANSCRIPT

Lecture 4 and 5
Leukemia and Chronic myeloproliferative disorders
Abdulkarim Aldosari

Objectives
• Define leukemia
• Compare and contrast acute versus chronic leukemia, and leukemia versus lymphoma
• Compare and contrast acute myeloid and acute lymphoblastic leukemia
• Describe FAB and WHO classifications for ALL and AML
• Describe characteristic morphology and cytochemical staining patterns for each of the subtypes of AML
• Correctly identify blast on a peripheral smear and distinguish between the features of a lymphoblast and a myeloblast
• Name and describe the characteristics of each of the chronic myeloproliferative disorders

Definitions and Overview
• Neoplasms = “new growth” due to dysregulated proliferation of a single transformed cell
• External growth factors that regulate proliferation are reduced or eliminated due to genetic
mutations in the transformed cell
• Benign neoplasm = differentiated cells that do not spread or invade surrounding tissue
Can progress with further mutations to a malignant neoplasms
• Malignant neoplasms = “deadly” “having the potential to produce death”
Proliferating cells with potential to metastasize Only malignant tumors are correctly termed as cancer Cancer is actually a malignancy of epithelial tissue, it is also commonly used to include all malignant
neoplasms.

Definitions and Overview• Leukemia = lymphoid and myeloid malignant bone marrow neoplasms
when abnormal cells are seen in both the bone marrow and the peripheral blood
• Lymphoma = abnormal proliferation of lymphoid cells within the lymphatic
tissue or lymph nodes
• Spectrum
Benign Malignant
Reactive leukocytosis MPD MDS Acute leukemia
Leukemoid reaction

Leukemia – definition and overview
• A malignant disease that affects the hematopoietic tissue
• Normal bone marrow is replaced by abnormal blood cells (neoplastic cells)
• Cells sometimes found in the PB, in the reticuloendothelial organs, and other
organs
• Prognosis – poor > death
• Incidence of leukemia in the USA = 8-10 new cases/100,000 individuals per year
• Approximately every 10 minutes, someone in the US dies from a blood cancer =
nearly 150 people each day or more than six people every hour.

Leukemia statistics• There are an estimated 310,046 people living with, or in remission from,
leukemia in the US.
• In 2013, 48,610 people are expected to be diagnosed with leukemia.
• In 2013, 23,720 people are expected to die from leukemia.
• Approximately 33 percent more males are living with leukemia than females.
• More males than females are diagnosed with leukemia and die of leukemia.
• Leukemia causes almost one-third of all cancer deaths in children and adolescents younger than 15 years.
• www.lls.org/diseaseinformation/getinformationsupport/factsstatistics

Leukemia – definition and overview• Acute leukemia = rapid onset with abnormal expansion of immature
cells/blasts
• Chronic leukemia = slow progression with abnormal expansion of mature
cells
• Divided by two cell types: myeloid and lymphoid
Acute myeloid leukemia (AML), or chronic myeloid leukemia (CML)
Acute lymphoid leukemia (ALL), or chronic lymphoid leukemia (CLL)
• Chronic leukemia (CML or CLL) generally associated with adults
• Most common form in children = ALL

Leukemia – definition and overview• Most cases affect adults = 10:1 compared to children
Most common = AML (34% of cases) and CLL (29%
• CLL is extremely rare in children – unusually before the age of 40
• 50% of all cases occur after the age of 64
• Adults 70-90 yrs. old > CML
• Higher rates in males than females
• Higher in European descent than African, lowest in American Indians and
Alaskan

Comparison of acute and chronic leukemia
Acute – Affects all ages, sudden onset
Becomes fatal in 6 months if untreated
Loss of BM function
Mild to severe anemia and/or thrombocytopenia,
WBC count- variable
Immature neoplastic cells
Chronic - Affects mostly adults, can last 2-6 years,
Early diagnosis = ↑ survival
Elevated WBC count > 50,000/µL
Mature neoplastic cells
Mild anemia, normal platelet count,
Prominent and massive organomegaly

Advances• Advances in diagnosis and treatment > improvement in survival
• Laboratory analysis – cytochemical cytogenetic, immunologic, molecular techniques > id specific categories of leukemia > distinct treatment protocols
• Bone marrow and stem cell transplantation
• Cytotoxic drugs, radiation
• Targeted approaches – tyrosine kinase inhibitors, protease inhibitors
• Genetic mutations - altered expressions of oncogenes and tumor suppressor genes > unregulated cellular proliferation

Factors that predispose or increase the incidence of leukemia
Host Factors
Hereditary – congenital chromosomal disorders
Abnormal chromosomal number
Immunodeficiency
Chronic marrow dysfunction
Environmental Factors
Radiation
Chemicals
Drugs
Viruses

Classification of leukemias
Classified according to cell type - cell maturity and/ or cell lineage
French-American- British (FAB)
World Health Organization (WHO)
Divide leukemias into ALL and AML

Acute leukemia
ALL more common in children = 75% of childhood leukemias
AML more common in adults = 80%
Incidents of AML increases with age
Median age 63 yrs.

Clinical onset of acute leukemia• Within a few weeks pts. present with weakness, bleeding abnormalities, flu-like
symptoms
• Due to proliferation and accumulation of abnormal cells >
• BM failure > anemia, Granulocytopenia, thrombocytopenia and their sequelae
• Organ infiltration > marrow expansion, spleen, liver, lymph nodes, CNS,
gums, mouth
Symptoms
• Bleeding, DIC, infections, gingival hypertrophy = swelling of the gums, oral lesions
• Bone and joint pain, neurologic conditions - CNS > intracranial pressure> nausea,
vomiting, headache

Laboratory evaluation of acute leukemia
Clinical history, physical examination,
CBC and PB smear
• Mild anemia – normocytic normochromic
• ↓ platelet count
• Variable WBC
• Blasts or other immature cells (may be rare or absent)
• nRBC
• Myelodysplastic features - Pseudo-Pelger-Huet
• Hypogranular neutrophils
Diagnosis can be established with PB smear but BM is the preferred
specimen

Laboratory evaluation of acute leukemiaBM aspirate and biopsy – required to establish diagnosis
• >20% blast in BM or PB = minimum WHO classification requirement for acute leukemia
• >30% for FAB classification
Morphological examination – cell lineage, guide to further studies
Immunologic cell markers studies – flow cytometry for blast immunophenotype
Cytochemical staining – differentiate granulocytic from monocytic leukemias
Karyotyping
Molecular studies
DNA flow cytometry
Electron microscopy

Specimen and evaluation of morphology
Properly collected samples - In EDTA tubes
Adequate amount of BM material collected and prepared
Morphology
• Where cells are not overcrowded, or shape distorted
• Distinguishing features of myeloid and lymphoid cells
Size, Nuclear chromatin, cytoplasm, nucleoli
Auer Rods – (+) in 60% of AML patients
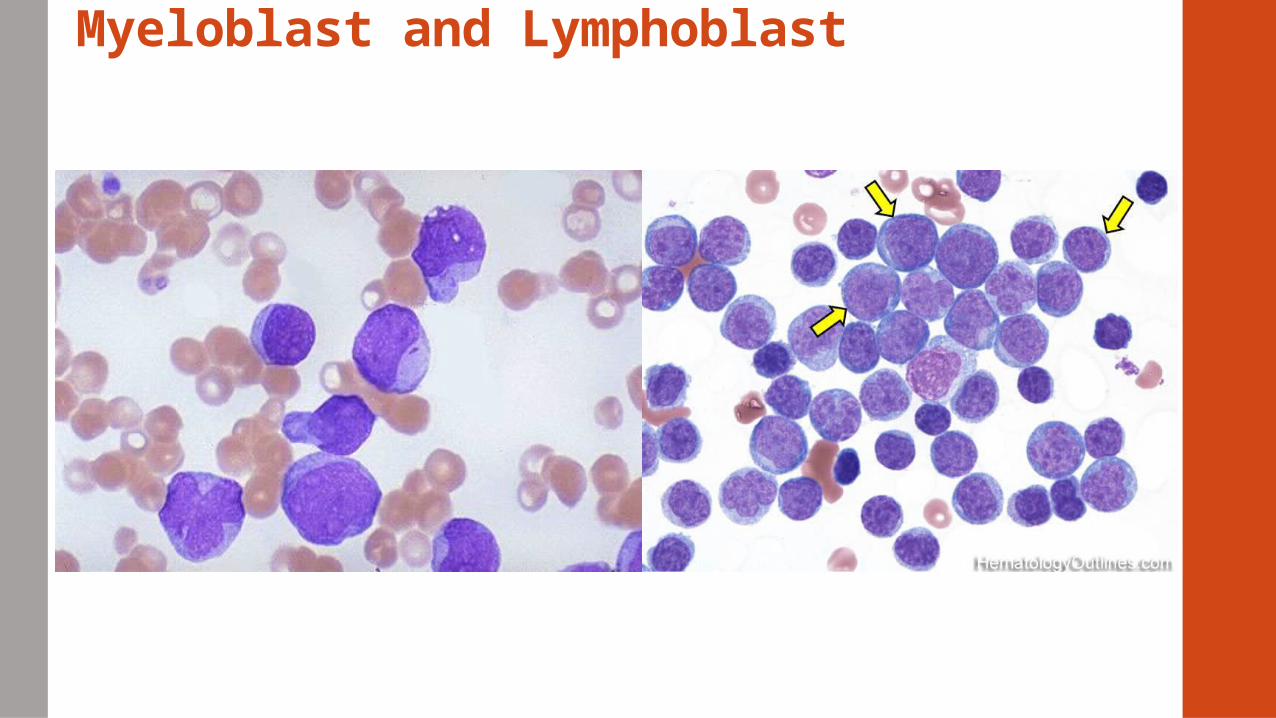
Myeloblast and Lymphoblast
Special Stains
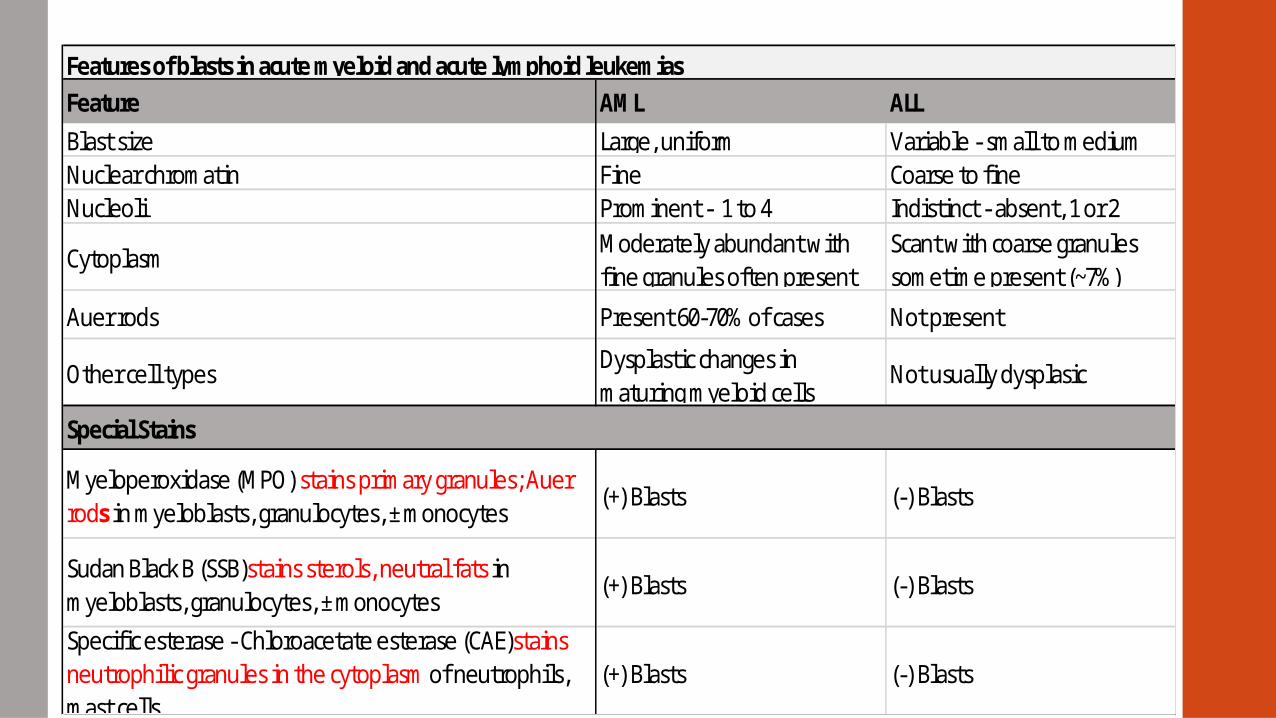
Features of blasts in acute myeloid and acute lymphoid leukemias
Variable - small to mediumCoarse to fineIndistinct - absent, 1 or 2Scant with coarse granules sometime present (~7%)
Not present
Not usually dysplasic
(-) Blasts
(-) Blasts
(-) Blasts
ALL
Large, uniformFineProminent - 1 to 4Moderately abundant with fine granules often present
Present 60-70% of cases
Other cell types
Myeloperoxidase (MPO) stains primary granules; Auer rods in myeloblasts, granulocytes, ± monocytes
Sudan Black B (SSB)stains sterols, neutral fats in myeloblasts, granulocytes, ± monocytes
Specific esterase - Chloroacetate esterase (CAE)stains neutrophilic granules in the cytoplasm of neutrophils, mast cells
AML
Dysplastic changes in maturing myeloid cells
(+) Blasts
(+) Blasts
(+) Blasts
Feature
Blast sizeNuclear chromatinNucleoli
Cytoplasm
Auer rods

Specimen and evaluation of morphology
Cytochemical staining
• Identify chemical components of the cells – enzymes or lipids
• Distinguish between AML and ALL
Sub-classify AML
• Fresh preparations preferred, control smears
• Look for (+) reaction in leukemic blasts rather than in mature cells
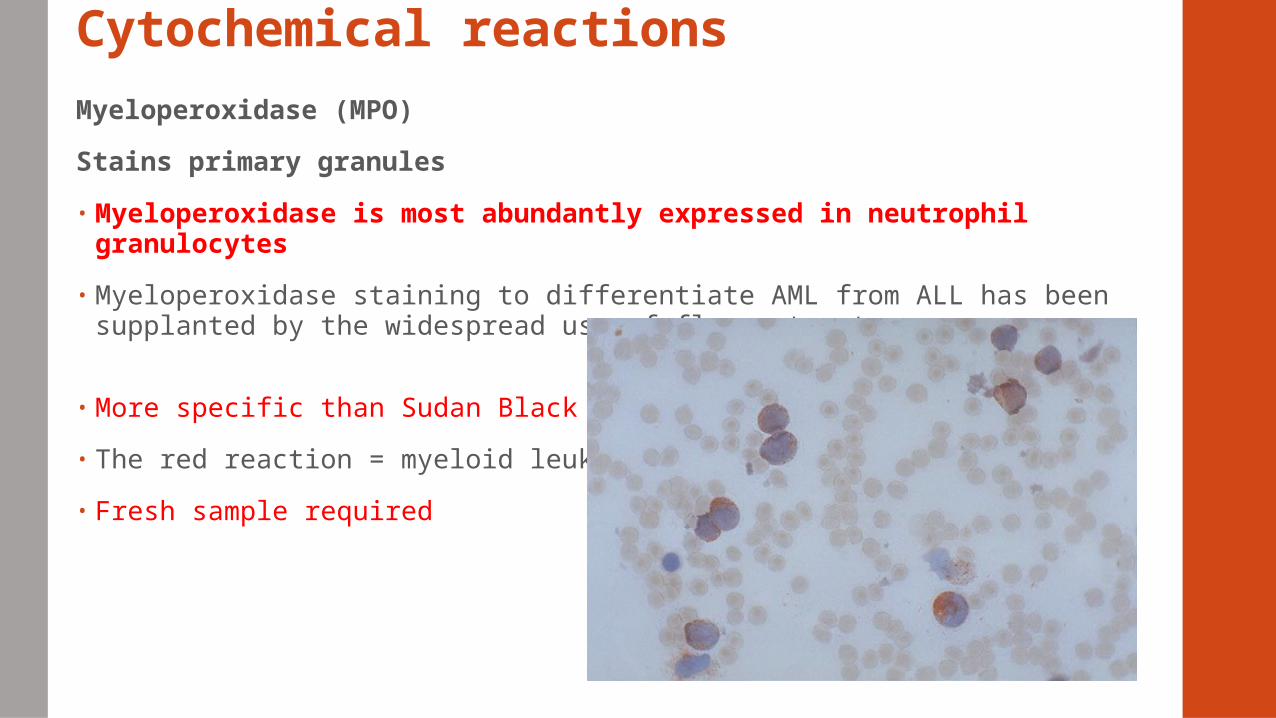
Cytochemical reactionsMyeloperoxidase (MPO)
Stains primary granules
• Myeloperoxidase is most abundantly expressed in neutrophil granulocytes
• Myeloperoxidase staining to differentiate AML from ALL has been supplanted by the widespread use of flow cytometry
• More specific than Sudan Black B
• The red reaction = myeloid leukemia
• Fresh sample required

Cytochemical reactionsSudan Black B (SBB)
Stains primary and secondary granules
• Stains phospholipids, neutral fats, sterols
• Most sensitive for granulocytic precursors
• Less specific than MPO
• Does not diminish with time
• Useful for samples that are not freshly drawn

Cytochemical reactions
Nonspecific esterase (alpha-naphthyl butyrate) (NSE)
Most specific for monocytes
• Monocytes and their precursors stain a diffusely (+) pretty red color, T-lymphocytes may have some dot-like (+)staining, may also be (+) in megakaryocytes
• Neutrophils and their precursors and most of the other types of cells in the bone marrow are negative
Specific esterase (chloroacetate esterase CAE)
• (+) for myeloblasts

Cytochemical reactionsPeriodic acid-Schiff (PAS)
Stains glycogen, glycoproteins, glycolipids
Does not distinguish between ALL and AML
• (+) Lymphocytes – granular pattern
• (+) Granulocytes – diffuse staining pattern
• (-) Normal erythroid precursors
• (+) Erythroblasts
Differentiate erythroleukemia from pernicious anemia
Erythroleukemia - rare form of acute myeloid leukemia where the myeloproliferation is of erythroblastic precursors.
PA- Megaloblastic anemias = blood smear shows large, fragile, immature erythrocytes, known as megaloblasts

Cytochemical reactionsAcid Phosphatase
• Constituent of lysosomes present in most cells
• Characteristic for T cell ALL > focal polarized acid phosphatase activity
• (+) Hairy cell leukemia uninhibited by tartrate resistant acid phosphatase Other leukocytes inhibited by tartrate
Terminal deoxynucleotidyl transferase (TdT)
• Nuclear enzyme that differentiates ALL from AML
• DNA polymerase present in both T and B lymphocyte progenitors,
• Absent in normal myeloid cells
• Performed by flow cytometry, immunofluorescent microscopy, immunohistochemical methods

Immunologic markers
Antibodies used to identify specific antigens characteristic to a particular cell
line
Antigens present on blast cell surface or within the cytoplasm
Two techniques:
• Flow cytometry – done on PB and BM aspirate
• Immunohistochemistry – paraffin sections of core biopsy
Paraffin section - a histologic section cut from tissue that has been embedded in
paraffin wax

Cell surface markers• Different cell lines and different stages express different surface proteins
• Highly specific monoclonal antibodies - discriminate various stages of human lymphocyte and granulocyte differentiation (see table 16-9)
• CD = cluster designation – a protocol used for the identification and investigation of cell surface molecules provide targets for immunotyping cells
• Used when leukemic cells are poorly differentiated by cytochemical stains
• Replacing conventional cytochemical methods
• Use a panel of markers that includes abs to several myeloid-associated
antigensNo single marker defines all forms of AML
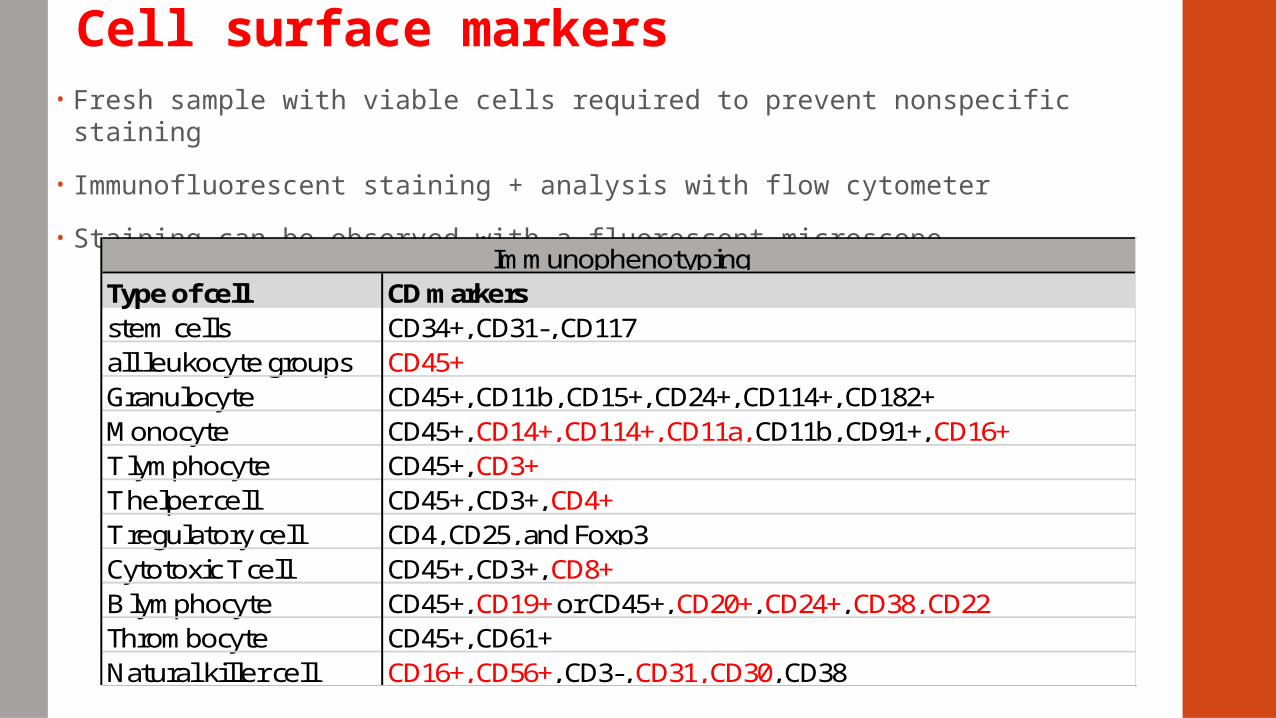
Cell surface markers• Fresh sample with viable cells required to prevent nonspecific staining
• Immunofluorescent staining + analysis with flow cytometer
• Staining can be observed with a fluorescent microscope
Type of cellstem cellsall leukocyte groupsGranulocyteMonocyteT lymphocyteT helper cellT regulatory cellCytotoxic T cellB lymphocyteThrombocyteNatural killer cell
Immunophenotyping
CD45+, CD3+, CD4+CD4, CD25, and Foxp3CD45+, CD3+, CD8+CD45+, CD19+ or CD45+, CD20+, CD24+, CD38, CD22CD45+, CD61+CD16+, CD56+, CD3-, CD31, CD30, CD38
CD markersCD34+, CD31-, CD117CD45+CD45+, CD11b, CD15+, CD24+, CD114+, CD182+CD45+, CD14+, CD114+, CD11a, CD11b, CD91+, CD16+CD45+, CD3+

Cytoplasmic markersMarkers directed at cytoplasmic antigens
For flow cytometry – additional step to fix cells to allow abs to enter the cytoplasm
Quantity of surface ags and cytoplasmic ags varies
Especially useful in assessing cell lineage in ALL
CD3 – T-cell ALL
CD22, CD79a – aid in defining B-cell lineage In ALL
Cytoplasmic IgM heavy chain
Pre-B-cell ALL (+)
Early-pre-B-cell ALL (-)
Pre-B-cell ALL has worse prognosis than early-pre-B-cell ALL
Cytoplasmic heavy chain staining no longer routinely preformed > FISH now used

CytogeneticsCytogenetics refers to the microscopic analysis of chromosomes in individual cells
• Play a role in diagnosis
• Sub-classification of leukemia
• Prognosis
• Selection and monitoring of therapy
Cytogenetics studies can be performed on
• fresh blood
• bone marrow
• prenatal specimens
• solid tissue specimens
• fixed specimens

Cytogenetic analysisFISH –(fluorescent in situ hybridization)
• Fluorescently labeled DNA probes that are capable of hybridizing to
complementary chromosomal regions
• To view the chromosomal location of a particular gene or DNA sequence
through a microscope
• Used to identify a translocation of chromosome 1 to 19 - t(1;19) and other
cytogenic abnormalities
Translocation t(1;19) in pre-B-cell ALL linked to poor prognosis of pre-B-cell ALL 2nd most common structural abnormality Translocation = reciprocal interchange of portions of two chromosomes,

Cytogenetic analysis
Karyotype is a test to identify and evaluate chromosomes
• Metaphase chromosomes + stains = distinctive banding patterns →
chromosome pairs are then arranged into a standardized format known as a
karyotype.
• Hybridization probe corresponded to a segment of chromosome
• Permits the simultaneous tracking of all human chromosomes
• Detect chromosomal rearrangements; translocations, deletions, duplications,
isochromosomes
• Fails to detect chromosomal abnormalities in 40-50% of AML
• Automated spectral karyotyping has ↑ the sensitivity and accuracy of
karyotypic analysis

Cytogenetics
Chromosomal abnormalities associated with different forms of
leukemia
• Philadelphia chromosome t(9;22) > CML
• Translocation t(15;17) > acute promyelocytic leukemia (M3)
• t(8;21) > M2 – favorable prognosis, good response to treatment
• Table 16-10

Molecular genetics
Cloning of genes: Ig genes, T-cell receptor genes, other genes
DNA FISH probes
PCR assays – more sensitive than FISH
Microarray assays

FAB classification of AMLM0 through M7
• based on the type of cell from which the leukemia developed
• how mature the cells are
• based on how the leukemia cells looked under the microscope after routine staining
M0 through M5 all start in precursors of white blood cells
M6 starts in very early forms of red blood cells
M7 starts in early forms of cells that make platelets
Subtypes are linked with certain symptoms
• Bleeding or blood clotting problems are often a problem for patients with the M3= acute promyelocytic leukemia (APL).

FAB classification

Alternate names for AML
acute nonlymphoblastic leukemia (ANLL
acute myelogenous leukemia
acute myeloblastic leukemia
acute granulocytic leukemia

Main features of AML
M0
• 5% of adult AML
• (-) for all cytochemical stains
• poor prognosis compared to other types
M1
• 15%
• Aggressive leukemia
M2
• 25% - most common type

Main features of AMLM3
• 10%
• Auer rods present
• Bleeding or blood clotting problems (DIC)
• t(15:17)
M4
• 20%
• (+) NSE
• Myeloid and monocytic antigens
M5
• Excess of monocytic cells
• Skin and gum involvement

Main features of AMLM6
• 5%
• Erythroid precursors
• Megaloblastic, ineffective erythropoiesis, ringed sideroblast
• Difficult to differentiate from Myelodysplastic syndromes M6 = ≥ 50% erythroblasts and myeloblasts > 30%
M7
• 5%
• Proliferation of megakaryoblasts

WHO classificationNewer system that includes factors that help to better classify cases of AML based on a patient’s outlook
AML with recurrent genetic abnormalities
• AML with a translocation between chromosomes 8 and 21
• AML with a translocation or inversion in chromosome 16
• APL (M3), which usually has translocation between chromosomes 15 and 17
• AML with changes in chromosome 11
AML with multilineage dysplasia
• more than one abnormal myeloid cell type is involved
AML related to previous chemotherapy or radiation

WHO classificationAML not otherwise specified
• Includes cases of AML that don’t fall into one of the above groups
• Similar to the FAB classification Undifferentiated AML (M0) AML with minimal maturation (M1) AML with maturation (M2) Acute myelomonocytic leukemia (M4) Acute monocytic leukemia (M5) Acute erythroid leukemia (M6) Acute megakaryoblastic leukemia (M7) Acute basophilic leukemia Acute panmyelosis with myelofibrosis Myeloid sarcoma
also known as granulocytic sarcoma or chloroma

Treatment of AML and response to treatmentCytogenetic and molecular abnormalities provide best information for predicting clinical outcome and therapy
Subgroups to determine therapy
• Acute promyelocytic (APL) -DIC > risk of bleeding
• AML with inv(16) or t(8;21)
• AML in the elderly
Targeted drug treatments
• Based on genetic profile

Acute lymphoblastic leukemia
• 75% of ALL occur in children
• Precursor B-cell ALL
• Usually fatal > 80% may be cured
• Classification based on immunophenotypic, molecular, cytogenetic and morphologic characteristics
WHO - immunophenotypic, molecular, cytogenetic
FAB – blast morphologic characteristics
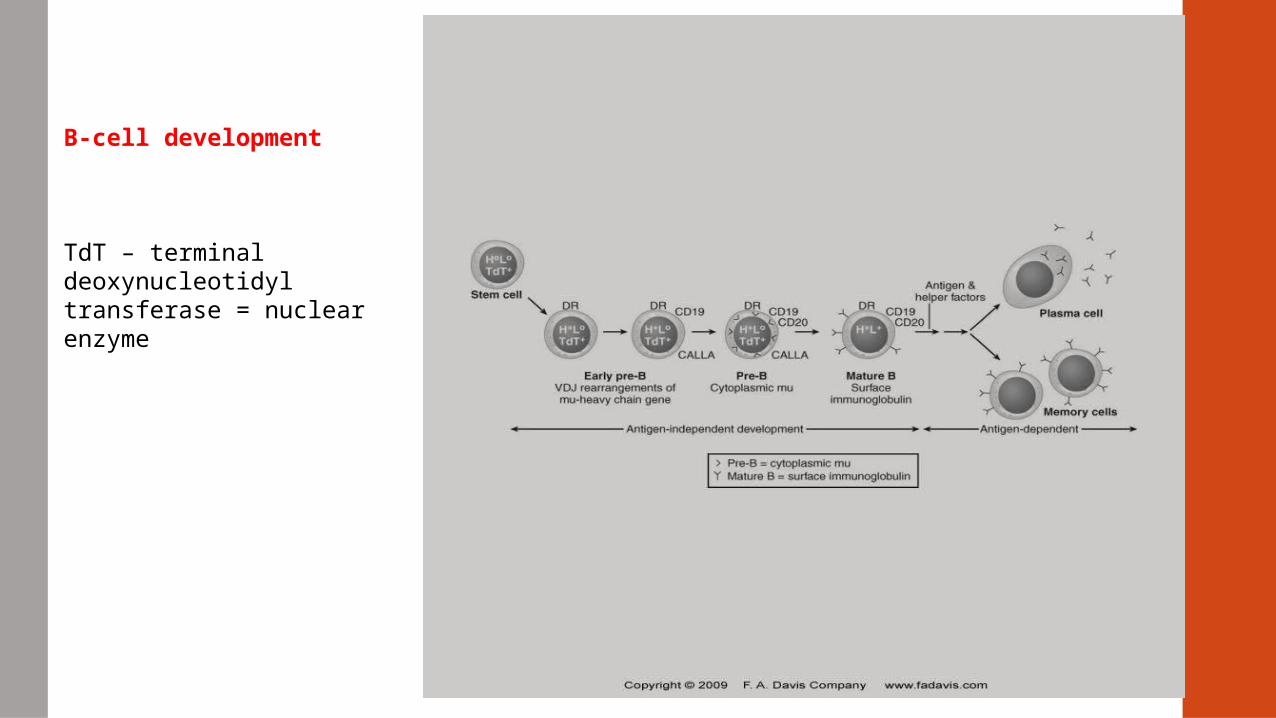
B-cell development
TdT – terminal deoxynucleotidyl transferase = nuclear enzyme
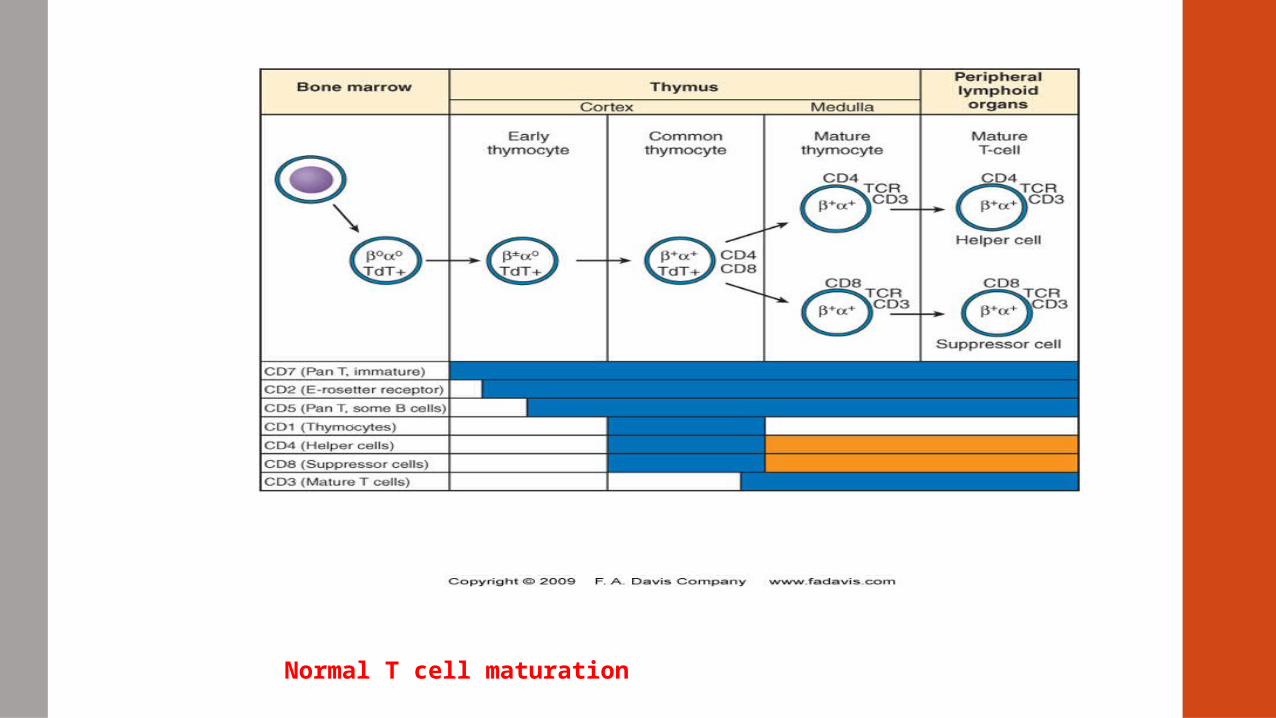
Normal T cell maturation

FAB classification of ALLSeparated into three morphological groups
L1
• Small uniform lymphoblasts
• Scanty basophilic cytoplasm
• Variable vacuoles
• Inconspicuous nucleoli
• Regular nuclear shape, occasional clefting
• Homogenous chromatin
• N:C - high

FAB classification of ALLL2
• Large heterogeneous/pleomorphic blasts
• Moderate cytoplasm, often intensely basophilic,
• Variable vacuoles
• Large nucleus, irregular shape with clefting and indentation,
• Large nucleoli
• Variable nuclear chromatin
• N:C – lower than L1

FAB classification of ALLL3
• Medium to large homogenous cells
• Moderate cytoplasm that is intensely basophilic
• Prominent cytoplasmic vacuoles
• At least one prominent nucleoli (may be 2-4),
• Round to oval nucleus
• Finely stippled homogenous chromatin
• Cytologically identical to Burkitt’s and Burkitt’s like lymphoma
• Has mature phenotype (i.e. expresses surface immunoglobulin)
• Fat vacuoles are Sudan black+, Oil red O+ and PAS
• L3 was considered worst prognosis based on morphology

Burkitt’s Leukemia/Lymphoma (Mature B-Cell
ALL)
• Similar to L3 but lacks the immunophenotypic characteristics of early B cell
The TdT is negative; CD19, CD20, HLA-DR (+); most cases (-) CD10
• WHO classification does not include the FAB L3 subtype
• Burkitt leukemia is rare
• Characterized by rearrangement of the c-myc gene on chromosome 8 by one of three chromosomal translocations – t(8;14), t(2;8), t(8;22)
• Poor prognosis, worse than pre-B ALL

WHO classification of ALL
• Based on the clonal population of either B- or T-lymphocyte lineage
• Based on whether population is early lymphocyte or lymphoblast
• Relies heavily on immunophenotyping of blast population
• Lymphoblasts are phenotyped using monoclonal antibodies
• Nuclear enzyme (TdT) used to diagnose lymphoblastic leukemia

WHO classification of ALLPrecursor B lymphoblastic leukemia / lymphoblastic lymphoma:
ALL with t(12;21) (TEL-AML1)
Most common translocation – 16-28%
2 -5 years old
CD10 (+), CD20 (-)
ALL with t(1;19) (PBX1-E2A)
CD10 (+), CD34 (-)
Poor prognosis
ALL with t(9;22); BCR-ABL (Philadelphia chromosome)
Fatal in all age groups
25% of adult ALL
CD10 (+), CD34 (+)
Treatment Imatinib

WHO classification of ALL
ALL with t(4;11) (AF4-MLL)
• ALL1 and HRX on chromosome 11q23
• 11q23 abnormalities associated with poor prognosis
• Most common form of infant ALL
Burkitt’s leukemia(Mature B-Cell ALL) – not included in WHO
classification of precursor B cell ALL

WHO classification of ALL
Precursor T lymphoblastic leukemia / lymphoma
15-25% of all patients
TdT (+)
CD1,CD2,CD3,CD4/CD8
T-cells used to be identified by – E-rosette formation
• Lymphocytes incubated with sheep RBC and observe a rosette formation

Treatment of ALL and response to treatment
• Greatest success – pediatric ALL > 80%
• Adults – 20-40%
• Due to different treatment- In clinical trials most children are treated
• CNS prophylactic treatment – standard in ALL; chemotherapy
• Allogenic stem cell transplantation (SCT) – high risk ALL
• Targeted therapies – Imatinib, tyrosine kinase inhibitor, chemotherapy, genetic interventions

Chronic myeloproliferative disorders Heterogeneous group of disorders
Clonal disorders of the BM > excessive proliferation of one or more non-lymyphoid cell lines > ↑ in cell in PB
CML – Chronic Myelogenous Leukemia
PV –Polycythemia Vera
ET – Essential Thrombocytopenia
IMF – Chronic Idiopathic Myelofibrosis

Chronic myeloproliferative disorders Recent discoveries
BCR-ABL = fusion gene in CML
JAK2 = Janus Kinase 2 gene in myeloproliferative disorders
Addition of new disorders by WHO
Chronic Neutrophilic leukemia
Chronic eosinophilic leukemia/ hypereosinophilic syndrome
Unclassifiable myeloproliferative disease

Chronic myelogenous leukemiaFirst human disease to be traced to a specific chromosomal abnormality – in
1960
Philadelphia (Ph) chromosome – ID at Upenn
Translocation between the long arms of chromosome 9 and 22 = t(9;22)
Chronic granulocytic leukemia
20% of adult leukemia , mostly male
Median age = 45-55
Unusual in children
↑risk – exposure to radiation
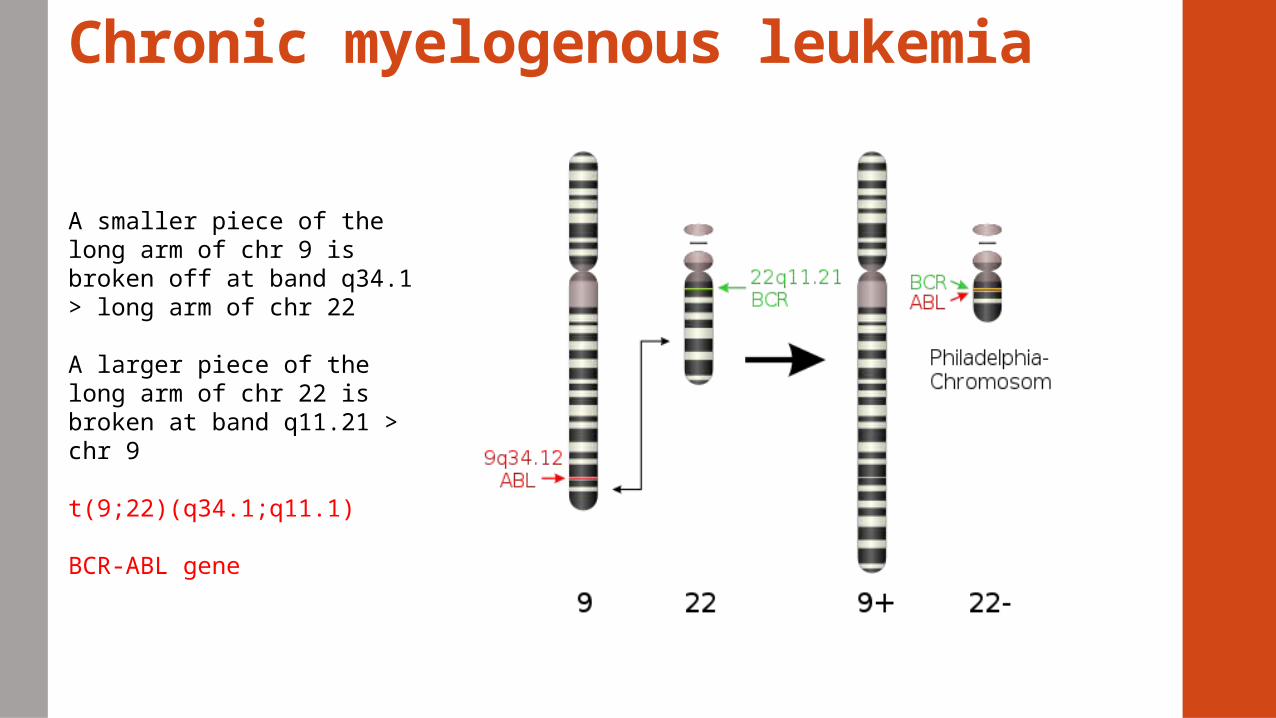
Chronic myelogenous leukemia
A smaller piece of the long arm of chr 9 is broken off at band q34.1 > long arm of chr 22
A larger piece of the long arm of chr 22 is broken at band q11.21 > chr 9
t(9;22)(q34.1;q11.1)
BCR-ABL gene

Pathogenesis of CMLBCR-ABL gene gives abnormal CML clone a growth advantage over normal cells >
• Allows them to replace the normal BM cells
• Causes uncontrolled cell growth
• Slight delay in maturation
• Unresponsiveness to normal growth regulators
• Prevent apoptosis in CML clone
• Early release of immature forms into blood stream

Clinical featuresAsymptomatic or symptomatic
Discovered during routine physical
Common complaints – general malaise, loss of appetite, fullness in abdomen, weight loss, night sweats, bone tenderness
Splenomegaly, hepatomegaly, lymphadenopathy
Bleeding complications

Clinical featuresCharacterized by three phases
Chronic
• When most patients are diagnosed
• Stable, unresponsive to chemotherapy
• Progression occurs 3-5 yrs. after onset
• Have only mild symptoms of fatigue,
• Left side pain, joint and/or hip pain,
• Abdominal fullness
• Absence of treatment > progresses to an accelerated phase

Clinical featuresAccelerated
• Unexplained fevers; Significant weight loss;
• Progressive leukocytosis; Worsening splenomegaly
• Require higher doses of myelo-suppressive drugs
• Bone and joint pain; Bleeding; infections
• Persistent thrombocytosis or thrombocytopenia
• 10–19% myeloblasts in the blood or bone marrow
• >20% basophils in the blood or bone marrow
• Platelet count <100,000, unrelated to therapy
• Platelet count >1,000,000, unresponsive to therapy
• Cytogenetic evidence of clonal evolution in addition to the Philadelphia chromosome

Clinical features
Blast
• Conversion from CML to aggressive form of acute leukemia
• Difficult to treat
• Worst prognosis
• >20% blasts in the blood or bone marrow
• Large clusters of blasts in the bone marrow on biopsy
• Development of a granulocytic sarcoma (extramedullary blast proliferation)

Laboratory findingsIn Peripheral Blood In Bone marrow
↑ WBC >100,000/µL Myeloid hyperplasia
<10% blasts in PB <10% blasts
Myelocytes ↑megakaryocytes
Pseudo-Pelger- Huet – in late stage myelofibrosis
↑basophils Psuedo-Gaucher cells
Thrombocytosis
Anemia- normocytic Genetic studies
↓Leukocyte alkaline phosphatase (LAP) Philadelphia Chromosome
Eosinophilia/monocytosis (+) 90-95%
BCR-ABL (+) >95%
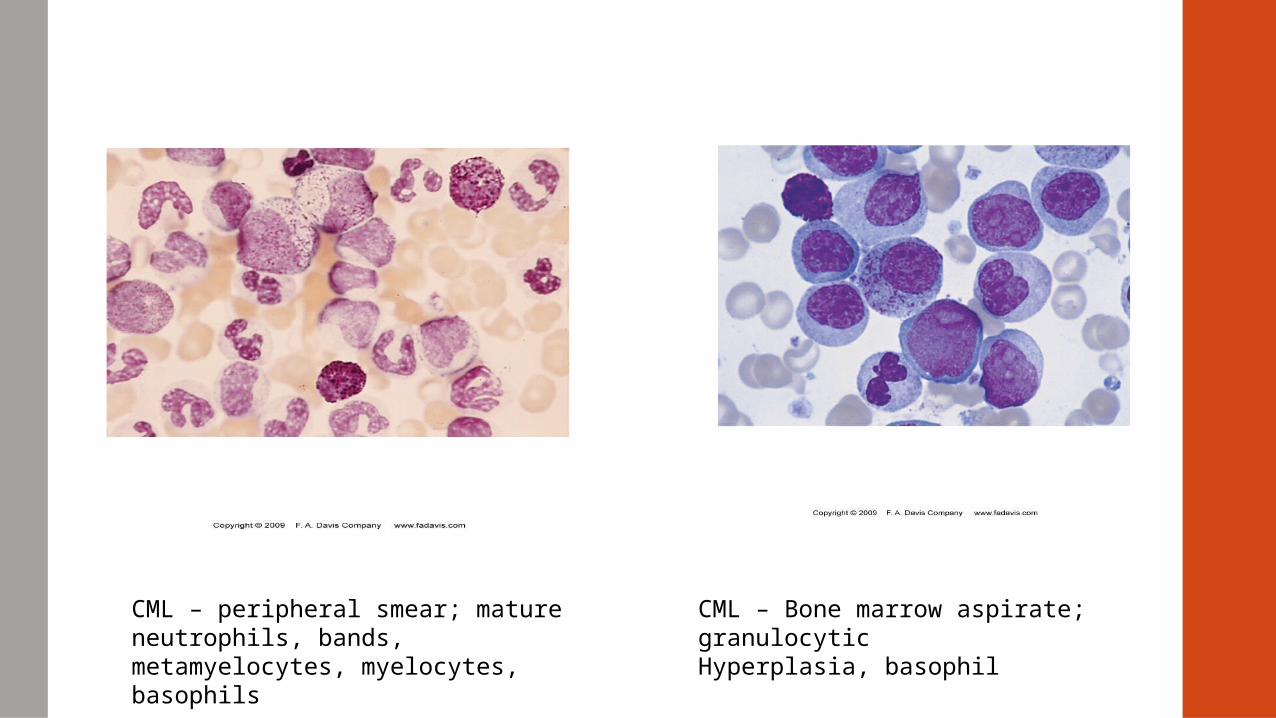
CML – peripheral smear; mature neutrophils, bands, metamyelocytes, myelocytes, basophils
CML – Bone marrow aspirate; granulocyticHyperplasia, basophil

Differential diagnosis CML Leukemoid reaction CNL CEL aCML
Ph Chr + (-) (-) (-) (-)
LAP score low High High
Basophilia 1-3+ 0 0 0
0
Myelocyte bulge + (-) (-) (-)
(+)
aCML – atypical Chronic myeloid leukemia

Treatment for CMLImatinib mesylate (Gleevec)
• a potent inhibitor of the oncogenic tyrosine kinase BCR-ABL
• has shown remarkable clinical activity in patients (CML)
• Produce long term remissions
Allogeneic bone marrow transplants = only cure for CML
• HLA-matched donor
• Improved if performed earlier in course of disease
• Survival rates – 78%

Other chronic myeloproliferative neoplasms (MPNs)
• Clonal hematopoietic stem cell malignancy
• Characterized by excessive production of blood cells
• Associated with thrombosis, hemorrhage, splenomegaly and transformation to
AML
• 3 most common BCR-ABL negative MPNs
PV –Polycythemia Vera
ET – Essential Thrombocytopenia
IMF – Chronic Idiopathic Myelofibrosis

Diagnosis of BCR-ABL negative MPNs, MPDs
• Adopted by WHO
• ID clonal marker – JAK V617F, single point mutation G1849T in exon 14
• Gene codes for a cytoplasmic Tyrosine Kinase
• Gain of function mutation > clonal proliferation
Substitution of Valine to Phenylalanine at position 617 > constitutive activation of
tyrosine kinase signalling

Diagnosis of BCR-ABL negative MPNs, MPDs• JAK2 burden↓ with successful therapy, disappears in some pts. and can
reappear during relapse
• Quantitative analysis – useful for diagnosis and management
• If JAK2 V617F negative > JAK2 exon 12 mutation
• JAK2 exon 12 mutation (+) 2-4% of PV patients
• Myeloproliferative leukemia gene (MPL) also identified
• Found at chr 1p34 – encodes the thrombopoietin receptor > platelet production
• Acquired MPL mutations associated with severe anemia (W515L and W515K) (+) in ET and MF, (-) in PV
• Tested after JAK2 V617F is ruled out
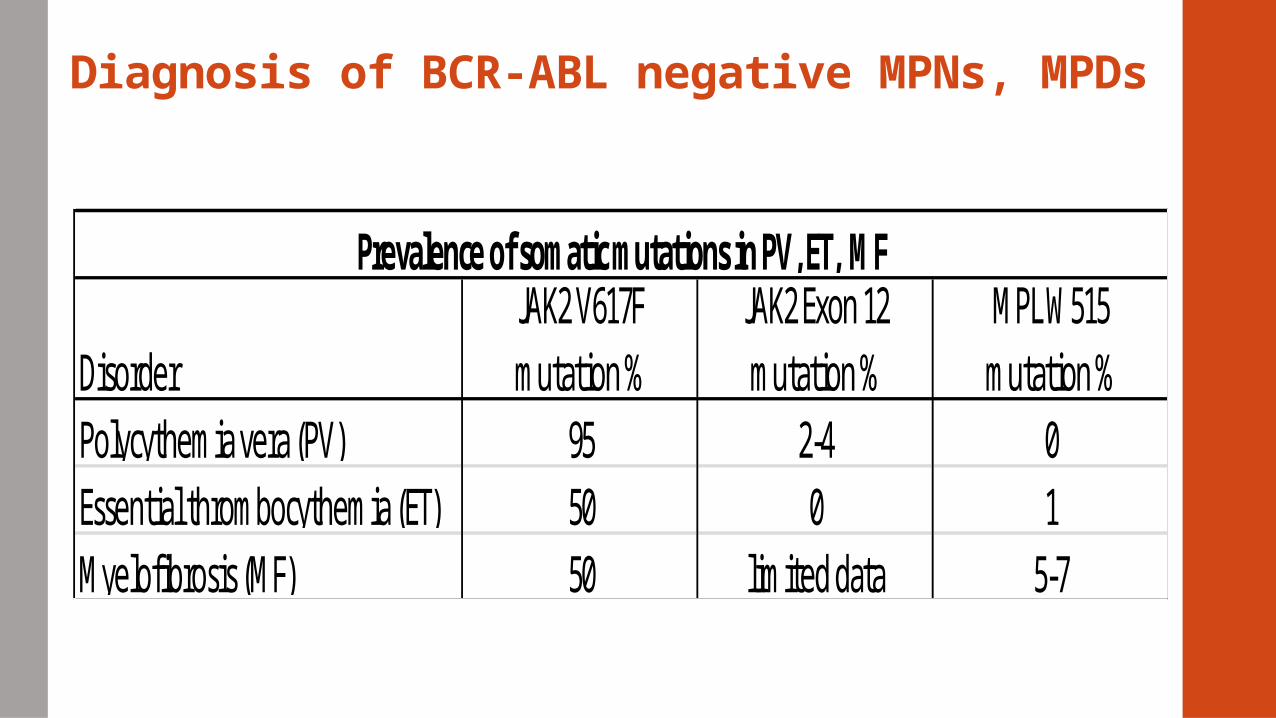
Diagnosis of BCR-ABL negative MPNs, MPDs
Prevalence of somatic mutations in PV, ET, MF
95 01
5-7
DisorderPolycythemia vera (PV)Essential thrombocythemia (ET)Myelofibrosis (MF)
JAK2 V617F mutation %
JAK2 Exon 12 mutation %
MPL W515 mutation %
5050
2-40
limited data

Polycythemia Vera (PV)• ↑ Production of red blood cells
• ↑ Production of white blood cells and platelets.
• Median age of onset – 60yrs, mostly men
• Chronic disease
• Asymptomatic
• Thrombocytosis; bleeding – secondary to erythroid expansion, hyper-viscosity, ↑platelets
• Headaches; hemorrhagic stroke, angina, MI, cramping or pain in legs
• Thrombosis = most common complication of PV

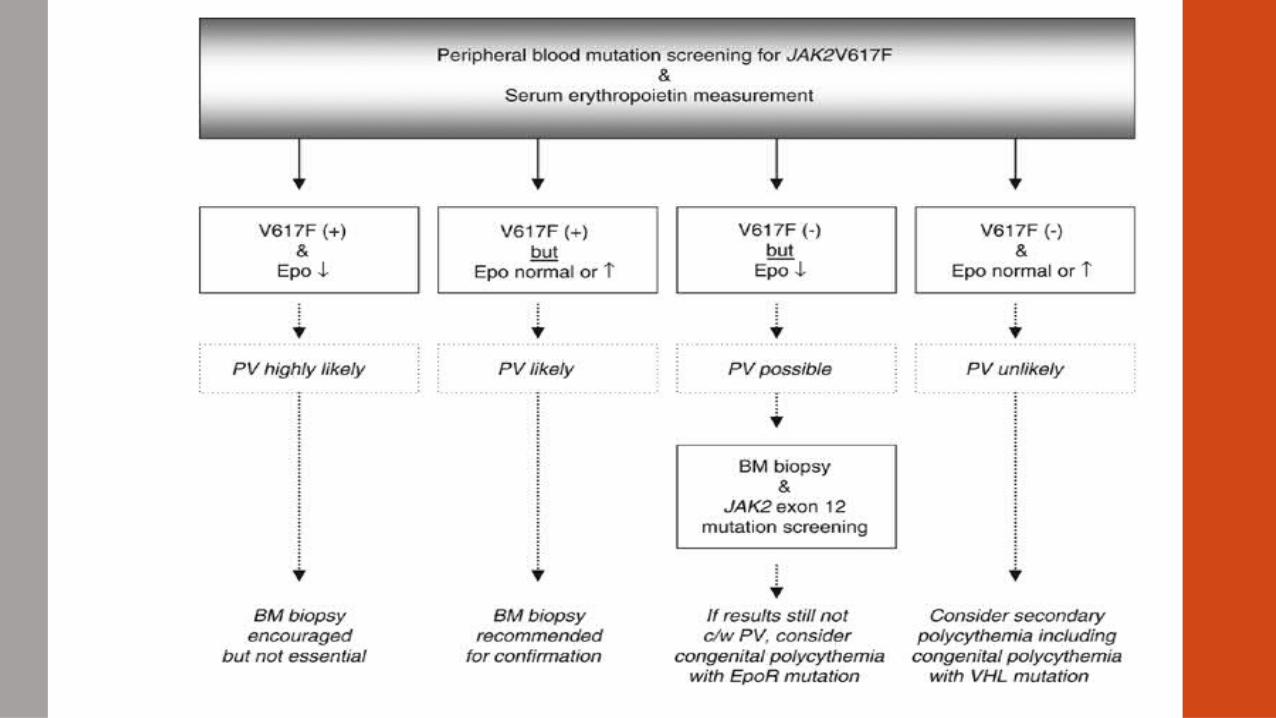
Polycythemia Vera• ↑ Hct >58% males, >52% females
• ↑ RBC mass; splenomegaly
• ↑ LAP
• ↓ Erythropoietin
• Normal arterial O2 saturation
• 90% of patients have a mutation in JAK2 Kinase gene (V617F) > activation of downstream pathways (STATS)

Polycythemia VeraTreatment
Phlebotomy
Hydroyurea
Interferon α2b

Secondary polycythemia• An absolute increase in red blood cell mass that is caused by enhanced
stimulation of red blood cell production
• May be acquired or congenital
• driven by circulating factors that are independent of the function of hematopoietic stem cells
• The frequency of secondary polycythemia depends on the underlying disease
• Relative polycythemia or erythrocytosis > decreased plasma volume with a normal red blood cell mass


Essential thrombocythemia (ET)
• Body produces too many platelet cells → blood to clot → block blood vessels
→ heart attack or stroke
• Platelet count markedly ↑ -( > 600 x 109/L to > 1000 x 109/L)
• Dysfunctional platelets → hemorrhage, thrombosis
• Splenomegaly
• Neurologic manifestations

Essential thrombocythemia (ET)
• Must be differentiated from reactive thrombocytosis
ET Reactive Thrombocytosis
Platelet ct>1,000,000 Frequent Infrequent
Leukocytosis Frequent may be present
Anemia normocytic,normochromic microcytic, hypochromic
Splenomegaly <50% not present
Clustering in BM common not present
BM fibrosis 20% not present

Essential thrombocythemia (ET)• Some people have no signs or symptoms
• Clots in the brain → strokes or temporary stroke-like episodes known as transient ischemic attacks
• Thrombosis in the legs → leg pain, swelling, or both
• Clots in the lungs (pulmonary embolism) → blocking blood flow in the lungs and causing chest pain and difficulty breathing (dyspnea)
• Uncontrolled ET can cause pregnancy complications, including: Spontaneous abortion (miscarriage) Fetal growth retardation Premature delivery Placental abruption (premature separation of the placenta and uterus

Essential Thrombocythemia (ET)• Affects an estimated 1 to 24 per 1 million people worldwide
• Mutations in the JAK2, MPL, THPO, and TET2.
• Familial essential thrombocythemia - inherited in an autosomal dominant
pattern
• Acquired from gene mutations that occur after conception = somatic
mutation
• Other names
essential thrombocytosis
primary thrombocythemia
primary thrombocytosis

Essential Thrombocythemia (ET)• The proteins produced from the JAK2, MPL, and THPO genes are part of a
signaling pathway - JAK/STAT pathway - transmits chemical signals from outside the cell to the cell's nucleus
• Through JAK/STAT pathway - proteins promote the proliferation of platelets and their precursor cells, megakaryocytes
• Mutations in the JAK2, MPL, and THPO genes > over-activation of the JAK/STAT pathway > overproduction of megakaryocytes > an increased number of platelets > thrombosis > many signs and symptoms of ET

Treatment for ET• The drugs most commonly used to treat ET
Hydroxyurea (Hydrea®)—This myelosuppressive drug (an agent that suppresses
the marrow’s production of blood cells) – High risk patients
Anagrelide (Agrylin®)- decreases platelet formation in most patients and is given by
mouth
Low dose aspirin - Patients with < 1,000,000 /µL
Interferon alfa (immediate-release preparations Intron® A and Roferon-A® and
sustained-release preparations PEG-Intron® and Pegasys®)
Plateletpheresis— to skim platelets from a patient’s blood and then return the
plasma and red cells to the patient; used only in emergency situations

Idiopathic myelofibrosis • IM
• PMF –primary myelofibrosis
• Agnogenic myeloid metaplasia (AMM)
• chronic idiopathic myelofibrosis (CIMF)
• myelofibrosis with myeloid metaplasia

Idiopathic myelofibrosis • Characterized by abnormalities in hematopoiesis and buildup of scar tissue
(fibrosis) in the bone marrow
• Impairs the ability to generate new blood cells resulting in a progressive
pancytopenia.
• May occur as a secondary characteristic PV or ET
• Affects approximately 1 in 500,000 people worldwide
• Not inherited but from gene mutations that occur after conception = somatic
mutation

Idiopathic myelofibrosis • Mutations in the JAK2, MPL, and TET2 genes are associated with most cases
of primary myelofibrosis
• Develops slowly and is mainly observed in people over the age of 50
• May also develop as a side-effect of treatment with some drugs that target
hematological disorders

Idiopathic myelofibrosis symptoms
• Symptoms vary - related to the abnormalities affecting blood cell production
• Asymptomatic, symptom-free for many years.
• May develop fatigue, fever, frequent infections, pale skin, night sweats and
unexplained weight loss
• Splenomegaly, hepatomegaly- due to extramedullary hematopoiesis
• 50% of cases have a mutation of the JAK2 gene

Treatment for IM• In November 2011, the FDA approved ruxolitinib (Jakafi) (inhibitor of JAK 1
and 2) as a treatment for myelofibrosis
• The one known treatment is allogeneic stem cell transplantation
• Folic acid, allopurinol or blood transfusions Dexamethasone, alpha-interferon
and Hydroxyurea