investigating the function of anaplastic lymphoma kinase
TRANSCRIPT

1
Umeå University Medical Dissertations
New Series nr. 1235 ISSN: 0346-6612 ISBN: 978-91-7264-708-4
Investigating the function of
Anaplastic Lymphoma Kinase
Emma Vernersson Lindahl
Department of Medical Bioscience, Pathology
Umeå University, Sweden
Umeå 2008

2
Umeå University Medical Dissertations
New Series nr. 1235 ISSN: 0346-6612 ISBN: 978-91-7264-708-4
Cover: wild type mouse embryo, developmental day 13.5. ALK is visualized (red) in part of
the diencephalon. Immunohistochemistry conducted 041215, picture acquired 041216
by Emma Vernersson Lindahl
Copyright© 2008 Emma Vernersson Lindahl
Printed by Arkitektkopia, Umeå, Sweden
Umeå 2008

3
Abstract
naplastic Lymphoma Kinase (ALK) was discovered in 1994, as a chromosomal
translocation, t(2;5)(p23;q35), often seen in Anaplastic Large Cell Lymphomas (ALCL).
Since then ALK has been extensively studied in this disease as well as in different model
organisms. Due to its expression pattern within the central and peripheral nervous system
ALK has been implicated in neuronal development. This hypothesis has been further
strengthened by studies from Drosophila which have shown ALK to have an important role in
optic lobe development. A recently described ALK mouse knockout model do not indicate an
essential role for ALK in development, although a potential role within the central nervous
system was strengthened. This since ALK-/-
animals has an increased number of progenitor
cells in the hippocampus and display altered behavior.
The overall aim of the studies included in this thesis was to elucidate the function of ALK in
the mouse. As a first step toward this goal we conducted an analysis of ALK mRNA and
protein expression patterns during development. The strong expression of ALK in neuronal
structures supports a role for ALK in neuronal development during embryogenesis.
To further investigate the function of ALK in a physiological context we have developed two
different ALK knockout strains, the ALK Kinase knockout (KO) and the ALK exon1 KO. The
only visible phenotype in these strains is a reduction of total body weight which is apparent in
the ALK-/-
population when compared to wild type littermates. This size difference seems to
take place after birth and is not due to an alteration in food consumption. We have also
extensively studied the ALK Kinase KO with respect to gross development, the
gastrointestinal canal and the olfactory system. ALK displays a very distinct expression
pattern within the gastrointestinal canal being confined to enteric neuron precursors during
embryogenesis and enteric nerves in the adult tissue. From these studies we conclude that
ALK is not needed for development and viability in mice although it does play a role in
regulation of body weight via a presently unknown mechanism.
In addition, we have investigated the relationship between the Drosophila and mouse ALK
receptor by examining the ability of the Drosophila Alk ligand Jelly-Belly, Jeb, to activate
mouse ALK. Using different in vivo and in vitro techniques, we have shown that activation of
mouse ALK cannot be accomplished by Drosophila Jeb. From this study we draw the
conclusion that during development ligands for the Drosophila and mouse ALK has diverged
to a level at which they can no longer substitute for each other.
Keywords: ALK, nervous system, knockout, development, Jelly-Belly
Author: Emma Vernersson, Department of Medical Bioscience, Pathology, Building 6M, 2nd
floor, Umeå University, S-901 85 UMEÅ, Sweden. E-mail:
[email protected]; phone: +46 (0)90 785 12 96
A

4
Table of Content
Papers Included in this Thesis 6 1. Published Articles 6
2. Manuscripts 6
Abbreviations 7
Introduction 10 1. Receptor Tyrosine Kinases 10
2. Receptor Tyrosine Kinases in Cancer 10 3. Discovery of the RTK Anaplastic Lymphoma Kinase 11
4. Structural Features of Anaplastic Lymphoma Kinase 11
5. Function of Anaplastic Lymphoma Kinase 13 5.1 Drosophila Anaplastic lymphoma kinase 13
5.2 C. elegans Anaplastic Lymphoma Kinase 14
5.3 Mammalian Anaplastic Lymphoma Kinase 14
6. Anaplastic Lymphoma Kinase in Disease 19
6.1 Anaplastic Lymphoma Kinase Fusion Proteins in Cancer 19
6.1.1 Anaplastic Large Cell Lymphoma 19
6.1.2 Mouse Models for ALK Driven ALCL 20
6.1.3 ALK Fusion Proteins in ALCL 21
6.1.4 Oncogenic Signaling via ALK Fusion Proteins 22
6.1.5 Inflammatory Myofibroblastic Tumour 27
6.1.6 Non-Small Cell Lung Cancer 27
6.1.7 ALK Positive Diffuse B-cell lymphoma 28 6.2 Anaplastic Lymphoma Kinase Overexpression in Cancer 28
6.3 Anaplastic Lymphoma Kinase Gain-of-Function in Cancer 29
6.3.1 Neuroblastoma 29 7. Potential Treatments Strategies for ALK Positive Cancers 31

5
Aim 33 1. Overall Aim 33
1.1 Specific Aim: Article I 33
1.2 Specific Aim: Article II 33 1.3 Specific Aim: Manuscript I 33
1.4 Specific Aim: Manuscript II 33
Results and Discussion 34
1. Article I: ALK Expression during Development 34
2. Article II: Can Drosophila Jeb activate mouse ALK? 35
2.1 Why is Drosophila Jeb unable to activate mouse ALK? 36
2.2 What about the LDLa domain? 36
3. Manuscript I and II: What is the physiological
consequence of loss of ALK function? 37 3.1 The ALK Kinase KO and the ALK exon1 KO 37
3.2 What function might ALK have during mouse 37
embryogenesis?
3.3 Is there a role for ALK in the adult olfactory bulb? 38
3.4 The ALK Kinase KO and the ALK exon1 KO are small…
…but why? 39
3.5 Why are animals smaller? Lessons from the literature. 40
3.6 Why is ALK not vital for life in the mouse? 40
Conclusions 42 1. Article I 42
2. Article II 42 3. Manuscript I and II 42
Acknowledgement 43
References 45
Original Papers and Manuscripts 60

6
Papers Included in this Thesis
1. Published Articles
I. Vernersson E, Khoo NK, Henriksson ML, Roos G, Palmer RH, Hallberg B. (2006)
Characterization of the expression of the ALK receptor tyrosine kinase in mice.
Gene Expr. Patterns, 6(5):448-61*
II. Yang HL, Eriksson T, Vernersson E, Vigny M, Hallberg B, Palmer RH. (2007)
The ligand Jelly Belly (Jeb) activates the Drosophila Alk RTK to drive PC12 cell
differentiation, but is unable to activate the mouse ALK RTK. J. Exp. Zoolog. B
Mol. Dev. Evol, 308(3):269-82**
2. Manuscripts
I. Vernersson E, Khoo NK, Roos G, Palmer RH, Hallberg B. (2008) The ALK KO
display an idiopathic reduction of total body weight. Manuscript
II. Vernersson E, Berghard A, Bohm S, Roos G, Palmer RH, Hallberg B. (2008) ALK
expression within the olfactory bulb. Manuscript
*Reprinted from Gene Expression Patterns, 6(5), Vernersson E, Khoo NK, Henriksson ML, Roos G, Palmer RH, Hallberg B, Characterization of the expression of the ALK receptor tyrosine kinase in mice, page 448-461,
copyright (2006), with permission from Elsevier
**Reprinted from Journal of experimental zoology. part B, molecular and developmental evolution, 308(3), Yang
HL, Eriksson T, Vernersson E, Vigny M, Hallberg B, Palmer RH, The ligand Jelly Belly (Jeb) activates the Drosophila Alk RTK to drive PC12 cell differentiation, but is unable to activate the mouse ALK RTK, page 269-
282, copyright (2007), with permission of Wiley-Liss, Inc. a subsidiary of John Wiley & Sons, Inc.

7
Abbreviations
AKT AKR Thymoma
ALCL Anaplastic Large Cell Lymphoma
ALK Anaplastic Lymphoma Kinase
ALO17 ALK lymphoma oligomerization partner on chromosome 17
ATIC 5-aminoimidazole-4-carboxamide ribonucleotide
formyltransferase/IMP cyclohydrolase
ATP Adenosinetriphosphate
Bin Biniou
BMP4 Bone Morphogenetic Protein 4
CARS Cysteinyl-tRNA synthetase
CD4 Cluster of Differentiation 4
CD30 Cluster of Differentiation 30
CDC42 Cell Division Cycle 42
CLTC1 Clathrin heavy chain-like 1
CML Chronic Myeloid Leukemia
c-Myc cellular-myelocytomatosis viral oncogene
CSN Central nervous system
dALK Drosophila Anaplastic Lymphoma kinase
daf3 and 5 abnormal dauer formation 3 and 5
DBA Dolichos biflorus agglutinin
DDR1 Discoidin Domain Receptor 1
DIG Digoxigenin
Dpp Decapentaplegic
duf Dumbfounded
EGF-like Epidermal Growth Factor-like
EML4 Echinoderm microtubule-associated protein like 4
ERK Extracellular signal Regulated Kinase
FGFR1-4 Fibroblast Growth Factor Receptor 1-4
FoxF1 Forkhead box F1
FOXO3a Forkhead box O3A
fsn-1 F-box protein at the synapse
GIST Gastrointestinal Stromal Tumour
Grb2 Growth Factor Receptor-Bound protein 2
HARP Heparin Affin Regulatory Peptide
Hen-1 Hesitation-1
HB-GAM Heparin Binding Growth-Associated Molecule
HBNF Heparin-binding Neurotrophic Factor
IgG Immunoglobulin G
IL-1ra InterLeukin 1 Receptor Antagonist
IMT Inflammatory Myofibroblastic Tumor
IR insulin receptor
IRS-1 Insulin receptor substrate 1
JAK3 Janus Kinase-3
Jeb Jelly-Belly

8
kirrel1 and 2 Kin of IRREgular chiasm-Like 1 and 2
LDL Low-Density Lipoprotein
LDLa Low-Density Lipoprotein receptor class A,
LDLR Low-Density Lipoprotein Receptor
LTK Leukocyte Tyrosine Kinase
LRP low-density Lipoprotein Related protein
MAM Meprin A-5 protein and receptor protein tyrosine phosphatase Mu
MAPK Mitogen-Activated Protein Kinase
MEK MAPK/ERK Kinase
mig-13 MIGration
Miple 1 and 2 MIdkine and PLEiotrophin
MK Midkine
mKirre3 mammalian Kin of IRREgular chiasm-Like 3
MSN Moesin
mTOR mammalian Target Of Rapamycin
MYH9 Non muscle myosin heavy chain
NEPH1 Nephrosis 1 homolog
NFκB Nuclear Factor Kappa B
NPM Nucleophosmin
OB Olfactory Bulb
OSF-1 Osteoblast-Specific Factor-1
OSN Olfactory sensory Neurons
p130Cas p130 Crk-associated substrate
PC12 Pheocytochroma cells 12
p.c. post coitum
PDGFR Platelet-Derived Growth Factor Receptor
PI3K Phosphoinositide-3 Kinase
PKB Protein Kinase B
PLCγ Phospholipase C γ
PP2 Protein Phosphatase 2
PTK Protein Tyrosine Kinase
PTN Pleiotrophin
RANBP2 Ran-binding protein 2
RAS rat sarcoma
RET Rearranged during Transfection
RIHB Retinoic Acid-inducible Heparin Binding protein
RPTPβ/δ Receptor Protein Tyrosine Phosphatase β/δ
RTK Receptor Tyrosine Kinase
SCD-2 Suppressor of Constitutive Dauer 2
SH2 domain Src homology 2 domain
Shc Src homology 2 containing
SDS-PAGE Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis
SEC31L1 SEC31 homologue A
SF steel factor
Shp-1 and 2 SH2 domain-containing phosphatase 1 and 2
SMαA Smooth Muscle alpha Actin

9
sma-5 small body size
soc-1 Suppressor Of Clr
Sox8 SRY-box containing gene 8
STAT3 and 5 Signal Transducer and Activator of Transcription 3 and 5
TACE TNFα Converting Enzyme
TBX1/2/3/10 T BoX family 1/2/3/10
TFG TRK-fused gene
TGFβ Transforming Growth Factor-beta
TMP3 and 4 Tropomyosin 3 and 4
vSrc viral Sarcoma

10
Introduction
1. Receptor Tyrosine Kinases
he enzymatic phosphorylation reaction was first described in 1954 by Burnett and
Kennedy with their discovery that a protein present in rat liver mitochondria could
catalyze the transfer of a phosphate group from ATP to serine hydroxyl groups of the protein
Casein [1]. However the phenomenon of phosphate in protein was demonstrated already in
1906, almost 50 years earlier, so the concept was not entirely new [2]. From the discovery of
Burnett and Kennedy it took almost 25 years before modification of tyrosine via phosphate
would be identified [3], and in 1980 Hunter and Sefton demonstrated the first protein with
tyrosine phosphorylation activity, vSrc [4].
There are approximately 520 different protein kinases in the human genome that can be
subdivided into 9 different groups (not including the atypical protein kinases) [5]. One of these
is the tyrosine kinase group containing 90 different members further subdivided into two
major classes, the receptor tyrosine kinases (RTK) and the non-receptor tyrosine kinases [5,
6].
All RTKs share a common domain structure with 1) an extracellular ligand binding domain
that is usually glycosylated; 2) a transmembrane domain and; 3) a cytoplasmic domain
containing a highly conserved kinase domain with the catalytic function of the protein [7].
The general activation of an RTK is envisioned to occur via ligand mediated receptor
dimerisation leading to autophosphorylation of specific tyrosine‟s in the intracellular region
followed by conformational changes of the kinase domain allowing ATP and substrate to bind
and transmit downstream signals [7]. There are different variations of this scenario depending
on which ligand/receptor pair is being considered. For example all known RTKs are
monomers in the absence of ligand, except the insulin receptor (IR) family of RTKs with the
IR being a disulfide linked dimer of two polypeptide chains forming a heterodimer in the cell
membrane [8]. The ligand for the IR - insulin - functions as a monomer in activating the
receptor [9]. Conversely, other ligands, such as Steel factor (SF), need to be present as dimers
to activate their target receptor [10].
2. Receptor Tyrosine Kinases in Disease
n their article from 1979, where Eckhart, Hutchinson and Hunter establish tyrosine
phosphorylation as an event occurring within mammalian cells they stated in their
discussion that “The phosphorylation of tyrosine is intriguing…and suggests that purification
of the activity would be worthwhile” [3]. Almost 30 years later tyrosine phosphorylation is
acknowledged as a key event regulating diverse cellular functions such as differentiation,
proliferation, migration and metabolism. Furthermore since the discovery of the first tyrosine
kinase [4], they have notoriously been found deregulated in numerous oncogenic malignancies
[11, 12].
T
I

11
More than half of the known RTKs have been implicated in mediating cellular transformation
via either of three main principles firstly) RTK overexpression; secondly) gain of function
mutations or; thirdly) genomic re-arrangement [11].
The Fibroblast Growth Factor Receptor (FGFR) family contains 4 members, FGFR1, FGFR2,
FGFR3 and FGFR4, which are all being implicated in different oncogenic processes via either
of the above principles [13]. Stem-cell myeloproliferative disorder is caused via genomic re-
arrangements resulting in fusion of the FGFR1 kinase domain to various partner protein
creating constitutively kinase active chimeric proteins with oncogenic potential, such as
CEP110-FGFR1 [14] and FIM-FGFR1 [15]. Over expression of FGFR1 and FGFR3 have
been implicated in breast cancer [16] and thyroid carcinoma [17] respectively while activating
point mutations in FGFR3 have been indicated in bladder carcinoma development [18]. These
activating mutations for FGFR3 occur either in the kinase domain, promoting a more open
(active) conformation, or in the extracellular domain leading to ligand-independent receptor
dimerisation [19, 20]. Regardless of alteration the outcome is the same with unregulated
downstream signaling via the receptor.
As well as being implicated in oncogenic development, deregulated RTKs can also be found
in developmental syndromes. Indeed, we can continue to utilize the FGFR family to exemplify
this. Mutations within the extracellular domain of FGFR3, conferring ligand independent
activation [21], are causative for one form of dwarfism [22]. Other mutations generate active
versions of FGFR1 and FGFR2 which cause Pfeiffer syndrome [23-25]. Pfeiffer syndrome is a
genetic disorder characterized by premature fusion of certain skull bones and depending on
type (I, II or III) the affected individual present with more or less severe symptoms [23].
3. Discovery of the RTK Anaplastic Lymphoma Kinase
naplastic Lymphoma Kinase (ALK) was discovered by two groups in 1994 [26, 27].
Shiota et. al. characterized a previously unknown tyrosine kinase protein from an
Anaplastic Large Cell Lymphoma (ALCL) cell line, terming it p80 due to its molecular size of
80 kDa. They speculated that this protein could be a chimeric protein created via a fusion
event [26]. This would indeed be verified later the same year when Morris and colleagues
demonstrated that the translocation, t(2;5)(p23:q35), often seen in ALCL, created a fusion
protein (NPM-ALK), of approximately 80 kDa, containing the amino-terminal portion of the
nucleophosmin (NPM) protein and the kinase domain of a tyrosine kinase protein that they
termed ALK after the disease [27].
4. Structural Features of Anaplastic Lymphoma Kinase
eing an RTK, ALK display the classical structural features of ligand binding extracellular
domain, a transmembrane spanning region and an intracellular tyrosine kinase domain.
Based on homology ALK is grouped with Leukocyte Tyrosine Kinase (LTK) and together
they form their own subgroup within the IR super family [28, 29].
The human ALK gene encodes a protein of 1620 amino acids giving rise to a protein of
approximately 180 kDa. However, as a result of posttranslational modifications (N-linked
A
B

12
glycosylations) ALK migrates at approximately 220 kDa in a conventional SDS-PAGE [28,
29]. Mouse ALK consists of 1621 amino acids [28, 29], and human ALK of 1620, while
Drosophila Alk [30] and C.elegans ALK [31, 32] are 1701 and 1421 amino acids in length
respectively.
Characterizing both mouse and human ALK, both Morris and Iwahara defined a signal peptide
in the amino-terminus of ALK, thought to be important for localization of the protein to the
cell membrane, one EGF-like cysteine rich motif, one large glycine rich domain in the
extracellular region and the kinase domain within the C-terminal region [28, 29] (figure 1).
The EGF-like structure, also noted in LTK [33], plays a role in ligand binding [34], although,
so far, no such function have been reported for this motif in ALK. Beside the EGF-like
domain Morris and colleagues also identified a second cysteine rich motif [28] that would be
further characterized in Drosophila Alk (dAlk) by Loren et. al. in 2001 and identified as an
LDLa domain [30]. Within ALK the LDLa (Low-density lipoprotein receptor class A) domain
has an unknown function. However this module mediates the binding between the LDL-
receptor and LDL [35, 36] implying a role for this domain of ALK in ligand binding.
In the work by Loren and colleagues they also identified one MAM (Meprin A-5 protein and
receptor protein tyrosine phosphatase Mu) domain within the amino-terminal region of dALK
[30]. Further work lead to the identification of a second MAM domain in the extracellular
region of dALK [37]. Using Pfam [38] or SMART [39], one MAM domain is denoted in
human ALK localized at amino acid 480-636. Using PROSIT [40] a second MAM domain is
localized ranging from amino acid 264-427 within the extracellular region in human, however
it displays weaker homology with the core MAM domain relative to the one denoted at
position 480-636. In mouse, both MAM domains, localized at amino acid position 270-428
and 484-635, are readily displayed using either of the above domain prediction programs.
MAM domains are thought to participate in cell/cell interactions [41] but their significance for
ALK is not determined. The importance of the MAM domain is nevertheless emphasized via
work from Drosophila which show that a point mutation at amino acid 681 (in Alk2) altering a
highly conserved aspartic acid in the MAM domain to arginine renders dAlk inactive [37].
During this study they also demonstrated the functional significance of the glycine rich
domain since in four independent dAlk mutants (Alk4, Alk
5, Alk
6 and Alk
7) a single glycine
within the glycine rich domain was found to be mutated to an acidic amino acid rendering
dALK inactive [37].
The ALK receptor is structurally highly conserved throughout evolution with the overall
protein homology between dAlk and mouse ALK being approximately 30%. However
conserved domains exhibit a higher degree of similarity e.g. the kinase domains showing 58%
similarity. This can be compared with the mouse and human ALK, which show 87% overall
homology on protein level and within the kinase domains differs in only 4 amino acids. It
should be emphasized that mouse and human ALK are highly similar; however, human ALK
contains one extra tyrosine, Y1604. Site-directed mutagenesis of this tyrosine residue in NPM-
ALK, a fusion protein of NPM and ALK found in the disease ALCL (discussed in more
details in coming sections), results in loss of oncogenic potential [42].

13
5. Function of Anaplastic Lymphoma Kinase
5.1 Drosophila Anaplastic lymphoma kinase
he in vivo function of Alk has been most thoroughly studied in Drosophila melanogaster
were Alk was first characterized in 2001 and shown to drive ERK activation in vivo [30].
Additional studies would illustrate a vital role for dAlk in formation of the visceral
musculature of the gut [37]. This phenotype would later be attributed to a lack of founder cells
in Drosophila embryos deficient in dAlk signaling [43, 44]. In a wild type context founder
cells are specified in the visceral mesoderm, thereafter fusing with fusion competent cells to
give rise to the multinucleated visceral musculature of the gut [45, 46]. Since dAlk signaling
specifies the founder cells, it follows that in the absence of founder cell no fusion event occurs
and as a consequence no assembly of a functional gut musculature occurs in dALK mutant
flies.
The Jelly-belly (Jeb) protein has been reported as the activating ligand for dAlk, and as such is
also required for founder cell specification [43, 44]. Jeb is a secreted protein of approximately
61 kDa containing a secretory signal and an LDLa domain [47]. The interaction of Jeb and
dAlk appears to be mediated via the LDLa domain in Jeb, since Jeb mutant protein lacking the
Figure 1: Mouse and human ALK display
87% overall homology on protein level
with identical domain layout. The amino
acid (a.a) position for human ALK is
entered below. The positioning of mouse
ALK domains is nearly identical. Human
ALK include a signal peptide (s.s) at a.a 1-
26, two Meprin A-5 protein and receptor
protein tyrosine phosphatase Mu (MAM)
domains at a.a 264-427 and 480-626, one
Low-density lipoprotein receptor class A
(LDLa) domain at a.a 453-471, one
Glycine rich (G-rich) domain at aa 816-940
and one epidermal growth factor (EGF) -
like domain at a.a 994-1021. A
transmembrane spanning segment (TM)
connects the extracellular region with the
protein tyrosine kinase (PTK) domain at
a.a 1116-1383. The closest family member,
LTK, is depictured with the coresponding
regions denoted. The s.s at a.a 1-16, G-
rich domain at a.a 63-334, the EGF-like
domain at a.a 400-428 and the kinase
domain at a.a 510-777.
extr
acel
lula
rS.S
MAM
MAM
LDLa
G-rich
TM
PTK
intr
acel
lula
r
hALK hLTK
EGF-like
PTK
G-rich
S.S
EGF-like
T

14
LDLa domain is unable to bind dAlk [44]. Activation of the Jeb-Alk signaling pathway leads
to ERK activation and further downstream transcription of target molecules such as, Duf/Kirre
[43, 44], Org [44] and Hand [48] (figure 2).
Recent data have implicated dAlk in two additional events. First Shirinian et. al. established
the importance of dAlk in development of the endoderm, showing that dAlk activity in the
visceral mesoderm is a requirement for TGFβ signaling in the adjacent endoderm [49].
Secondly Bazigou and colleagues demonstrated a role for the Jeb-Alk signaling pathway
during optic lobe development, where lack of either protein leads to miss targeting of the R-8
axons during later maturation of the optic lobe neuropil [50].
5.2 C. elegans Anaplastic Lymphoma Kinase
n C. elegans ALK has been implicated in neuronal control of entry of dauer and synapse
stabilization [31, 32]. In 2004 Liao et. al. characterized the C. elegans protein Fsn-1 as
being part of a ubiquitin ligase complex stabilizing synapse formation in GABAergic
neuromuscular junctions. This function was partly mediated via down regulation of T10H9.2
the C. elegans homologue of mammalian ALK [31]. More recently mutants of T10H9.2, or
SCD-2 as it´s now formally known were characterized. These mutants display a temperature
sensitive adaptation for entry of dauer stage. Epistasis analysis places SCD-2 in the TGFβ
pathway upstream of Daf3/Daf5. Furthermore, Hen-1 was identified as the SCD-2 ligand [32].
Hen-1 is the homologue of the Drosophila ALK ligand Jeb, and is also a secreted ligand
containing an LDLa domain [51].
5.3 Mammalian Anaplastic Lymphoma Kinase
ammalian ALK has been postulated to play a role in the normal development and
function of the nervous system. This dogma within the field had its birth in the
extensive expression of ALK mRNA throughout the nervous system during mouse
embryogenesis [28, 29]. Further, this pattern of ALK mRNA expression is recapitulated in the
developing central nervous system of the chicken were ALK localizes to a subset of spinal
motor neurons, the sympathetic ganglia and dorsal root ganglia [52]. In mice the intensity of
the ALK transcript diminishes after birth, reaching minimum levels at 3 weeks of age, and is
thereafter maintained at low levels in the adult animal [29]. Immunohistochemical studies of
adult human tissue by Pulford et. al. have shown a pattern consistent with that earlier reported
for mouse ALK with a week ALK signal observed in scattered neuronal cells of the
hypothalamus, basal ganglia and thalamic nuclei [53]. While Pulford and colleagues did not
detect ALK protein in any other tissue studied, including the testis and duodenum [53], Morris
et. al observed ALK mRNA transcripts of different sizes in testis, small intestine and brain of
adult human material [27]. This suggesting, that differential splicing of ALK occurs.
Interestingly, the most extensive ALK expression was noted in the testis and small intestine
[27] implicating a function for ALK in these structures.
In vitro studies have also suggested a role for ALK in neuronal development. Souttou et. al. in
2001, substituted the extracellular region of ALK with the mouse IgG 2b Fc domain, resulting
in the creation of a constitutively active membrane bound ALK:IgG 2b Fc hybrid protein,
I
M
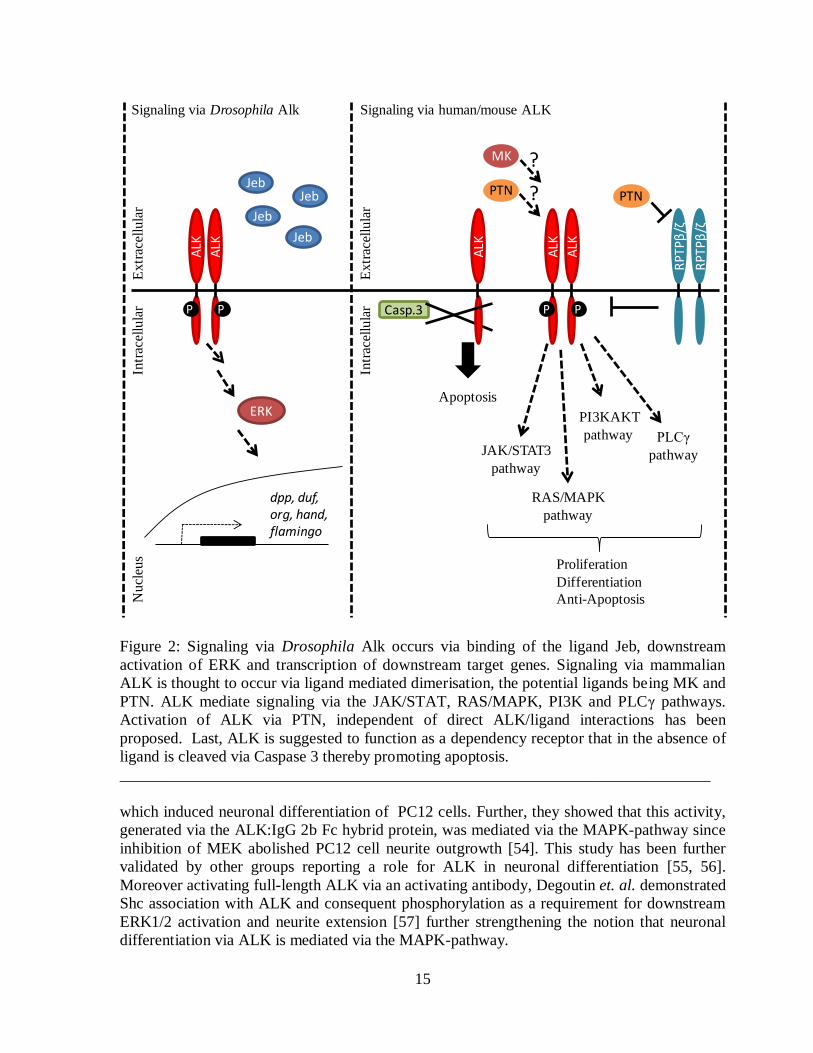
15
Figure 2: Signaling via Drosophila Alk occurs via binding of the ligand Jeb, downstream
activation of ERK and transcription of downstream target genes. Signaling via mammalian
ALK is thought to occur via ligand mediated dimerisation, the potential ligands being MK and
PTN. ALK mediate signaling via the JAK/STAT, RAS/MAPK, PI3K and PLCγ pathways.
Activation of ALK via PTN, independent of direct ALK/ligand interactions has been
proposed. Last, ALK is suggested to function as a dependency receptor that in the absence of
ligand is cleaved via Caspase 3 thereby promoting apoptosis.
__________________________________________________________________________
which induced neuronal differentiation of PC12 cells. Further, they showed that this activity,
generated via the ALK:IgG 2b Fc hybrid protein, was mediated via the MAPK-pathway since
inhibition of MEK abolished PC12 cell neurite outgrowth [54]. This study has been further
validated by other groups reporting a role for ALK in neuronal differentiation [55, 56].
Moreover activating full-length ALK via an activating antibody, Degoutin et. al. demonstrated
Shc association with ALK and consequent phosphorylation as a requirement for downstream
ERK1/2 activation and neurite extension [57] further strengthening the notion that neuronal
differentiation via ALK is mediated via the MAPK-pathway.
Signaling via Drosophila Alk
Jeb
Jeb
JebJeb
ERK
dpp, duf, org, hand,flamingo
Signaling via human/mouse ALK
PTN
RPTPβ/ζ
RPTPβ/ζ
RAS/MAPK
pathway
Proliferation
Differentiation
Anti-Apoptosis
PLCγ
pathway
PI3KAKT
pathwayJAK/STAT3
pathway
Casp.3
Apoptosis
PTN
MK ?
?
Intr
acel
lula
rE
xtr
acel
lula
rN
ucl
eus
Intr
acel
lula
rE
xtr
acel
lula
r
ALK
ALK
P P
ALK
ALK
ALK
P P

16
Using a similar approach, replacing the extracellular domain of ALK with the epidermal
growth factor receptor extracellular region, Piccinini and coworkers demonstrated that ALK
had the capacity to phosphorylated and activate PLCγ and PI3K. Activation of the chimeric
EGFR/ALK protein effectively transformed NIH3T3 cells [58] illustrating the potential
oncogenic properties of ALK when deregulated.
In mammals Pleiotrophin (PTN) also known as heparin binding growth-associated molecule
(HB-GAM) [59], osteoblast-specific factor-1 (OSF-1) [60], heparin affin regulatory peptide
(HARP) [61] and heparin-binding neurotrophic factor (HBNF) [62] and Midkine (MK) also
known as retinoic acid-inducible heparin binding protein (RIHB) [63], have been postulated
to be the activating ligands for ALK [64, 65]. In 2001, using PTN as bait to screen a phage
display cDNA library prepared from human foetal brain, the Wellstein group would identify a
putative PTN binding sequence that they matched to the extracellular region of ALK [64].
Subsequently they reported the identification of a second ligand, the PTN related protein MK.
Using cross-competing assays they showed that MK and PTN competed for the binding to
ALK. In addition, using antibodies directed toward the extracellular region of ALK they were
able to block the in vitro ligand/receptor interaction [65] suggesting a role of MK and PTN in
binding ALK. MK and PTN are small, heparin binding, growth factors implicated in diverse
processes such as neural development, cell migration and angiogenesis [66, 67]. They are
conserved throughout evolution and are found in species ranging from Drosophila to man
[67]. In addition to ALK there are a number of other proposed receptors for MK and PTN. To
date, MK and PTN have been shown to bind and signal via the RPTPβ/δ [68, 69] and N-
syndecan [70, 71] while MK can also bind LRP [72] as well as the α4β1- and α6β1-integrins
[73].
Enhanced proliferation is a commonly observed phenomenon when investigating ALK
activation via PTN [64, 74, 75]. This function seems to be dependent on protein kinase B
(PKB)/AKT activation implicating the PI3K pathway as a target of ALK mediated signaling
[74, 75]. Indeed, Bowden et. al. demonstrated a role for ALK, after PTN activation, as an anti-
apoptotic agent functioning via the MAPK-pathway in NIH3T3 cells [74]. This role has been
further emphasized by studies utilizing glioblastoma cell lines expressing full length ALK
were ribozyme mediated reduction of ALK lead to increased apoptosis [75].
The MK/ALK signaling pathway has been reported, as with PTN/ALK, to be important for
proliferation [65, 76]. Kuo et. al. demonstrated that ALK after activation with MK, interacts
with IRS-1 leading to further downstream signaling and ultimately activation of NF-κB
thereby inducing cell growth. On knockdown of the p65 subunit of NF-κB the proliferation
noted after ALK activation via MK was abolished [76].
Even though several groups have shown that PTN and MK are able to activate ALK [64, 65,
74-77] there are several reports of the opposite [55, 56, 78-80] leading to a degree of
controversy in the field. Moog-Lutz et. al. have clearly shown that agonist monoclonal
antibodies directed toward ALK mediate efficient ERK1/2 phosphorylation and differentiation
of PC12 cells. However, this effect could not be reproduced with PTN which failed to induce
phosphorylation of ALK [56]. The group of Tadashi Yamamoto also showed a dependence on
ALK signaling for neuronal differentiation of SK-H-SH cells using activating antibodies

17
toward ALK. The neuritogenic signal could be blocked with the MEK inhibitor PD98059, thus
implicating the MAPK pathway. Using MK and PTN there were unable to reproduce the
activation of ALK [55].
In 2005 Lu and colleagues demonstrated the presence of two different species of PTN, PTN15
and PTN18. Upon stimulation with PTN15 ALK promoted glioblastoma growth. Conversely
PTN18 promoted glioblastoma migration in an RPTPβ/δ dependent manner [77]. As
mentioned above, RPTPβ/δ has also been suggested as a receptor for PTN [68]. The existence
of two different PTN isoforms was hypothesized to explain the discrepancy in result presented
previously concerning the ability of PTN to activate ALK. However, Mathivet et. al. was
unable to reproduce the effects of either PTN15 or PTN18 on ALK, although they did agree
with Lu et. al. that PTN18 indeed promoted migration of glioblastoma cells via the RPTPβ/δ
receptor [78]. Thus the physiological significance of ALK activation via PTN and MK is still
a matter of debate and investigation within the field
One new theory concerning ALK activation via PTN has arisen recently from the finding of
Perez-Pinera et. al. They showed that PTN indirectly lead to phosphorylation of ALK via
binding to and inactivation of the phosphatase RPTPβ/δ [81]. This phosphorylation of ALK
was not dependent on the ALK extracellular region since a membrane bound construct
containing the ALK intracellular region was phosphorylated equally as well as the full length
ALK receptor [81].
It is important to note when discussing MK and PTN as ligands for ALK that they show no
homology toward the Drosophila Alk ligand Jeb. Jeb harbors a signal peptide and an LDLa
domain [47], while MK and PTN are composed of two domains, one N-terminal N-domain
and one C-terminal C-domain [82, 83], which contain heparin binding modules important for
their activity [82, 84]. In Drosophila the homologues for MK and PTN are the orphan ligands
Miple1 and Miple2 (Miple for Midkine and Pleiotrophin) [85]. The combined expression
pattern of Miple1 and Miple2 complement the expression pattern of dAlk indicating that a
role as activating ligands for dAlk is possible [85]. However, this has so far not been formally
tested at the genetic level and remains a hypothesis.
The group of Allouche recently suggested that ALK might have a function independent of
activation, being a dependency receptor. They showed that ALK was cleaved, via caspase 3
activity, thereby exposing a proapoptotic domain, within the intracellular region, leading to
enhanced apoptosis in cells of lymphoid (Jurkat) or neuronal (13.S.1.24) origin exposed to
apoptosis inducing agents. This proapoptotic function was counteracted via activation of the
ALK receptor using activating antibodies [79] (figure 2).
Throughout the ALK literature a number of comments within different reports can be found
regarding an investigation of a mouse ALK knockout (KO) strain generated by the Morris
group, stating that it is viable without any gross alterations [86-90]. A recent publication has
described an independently generated mouse ALK KO [91]. This strain lack the
transmembrane and intracellular region of ALK, and display increased hippocampal
progenitor proliferation, increased performance in hippocampal associated tasks and an
increased level of dopamine within the basal cortex [91]. Further, the authors also high-lighted

18
an increase in the number of calretinin positive cells within hippocampus [91], a phenotype
also noted in the MK knockout. In this report, Nakamura et. al. characterize the MK KO and
describes a delay in hippocampus development illustrated by the increased/prolonged stain
with calretinin [92]. Within the hippocampus PTN has been shown to act as a regulator of long
term potential in a PTN KO mouse model [93, 94]. While not conclusive, the results of both
the receptor ALK and the putative ligands MK and PTN in the context of hippocampal
development and function suggest a possible link between ALK and PTN/MK.
MK and PTN have similar expression patterns in the mouse [95-98] and studies of MK-/-
embryos, which display a strong up regulation of PTN expression during development,
suggest that PTN and MK are functionally redundant [99]. Indeed, the MK/PTN double KO
shows a more severe phenotype then either of the single KOs. These phenotypes include
female infertility [100] and hearing impairments [101]. If MK and PTN are true ligands for
ALK there is a strong probability they could counteract the loss of ALK via signaling through
any of the other known MK/PTN receptors. The only known ALK family member, LTK, is
expressed in pre-B-cells and adult neurons of hippocampus and cerebral cortex [33]. Since the
ALK KO phenotype is related to hippocampus [91], the idea that LTK might compensate for
ALK loss is intriguing and relevant. Furthermore Yamada and coworkers have demonstrated a
role for LTK in promoting neuronal differentiation of PC12 cells [102], underscoring the fact
that ALK and LTK may potentially compensate for each other. A role for LTK during
development was recently demonstrated by Lopes and coworkers. They could show that
during zebrafich embryogenesis LTK specifies iridophores, from the neural crest linage,
needed for correct pigmentation patterns [103]. Thus far, however mouse LTK appears to only
be expressed in the adult and not during development [33], suggesting a clear distinction from
the ALK receptor, which is extensively expressed during embryogenesis [28, 29]. In addition,
even though LTK and ALK share a highly similar intracellular tyrosine kinase domain there
are notable differences in the extracellular region. LTK lacks several domains found in ALK,
such as the MAM and LDLa domains (figure 1). There is no published LTK knockout (in
higher mammals) to date that might give clues to its role in an in vivo context and the question
regarding whether or not LTK might substitute for loss of ALK will have to await future
scientific investigations.
Upon investigation of the human ALK protein product several groups have reported the
presence of a 140 kDa protein in addition to the 220 kDa full-length ALK species [29, 55-57,
78]. The 140 kDa species is thought to be the result of a cleavage within the extracellular
region of full-length ALK generating an 80 kDa form present in culture medium [56].
Moreover the 140 kDa ALK protein is phosphorylated in response to activation of ALK [55,
57, 78]. The presence of an 85 kDa ALK protein in NIH3T3 cells expressing full-length ALK
was reported by Motegi and coworkers. This protein was phosphorylated in response to
activating antibodies directed toward the extracellular region of ALK [55]. However, to date
no one has been able to show any physiological function for these shorter ALK variants. It is
possible that the shorter forms of ALK are generated after activation of the receptor as part of
the downregulation/degradation processes within the cell. It would be interesting to investigate
whether the shorter variants of ALK show signs of increased ubiquination pattern or
increased/decreased signaling properties compared with the full length ALK receptor. Future

19
work should lead to the definition of the cleavage sites, and the functional relevance of these
forms of ALK in vivo.
6. Anaplastic Lymphoma Kinase in Disease
LK was first discovered as a fusion protein, NPM-ALK, in patients suffering from the
disease ALCL [27]. ALK fusion proteins have also been denoted in inflammatory
myofibroblastic tumour [104], non-small cell lung cancer [105] and diffuse B-cell lymphoma
[106]. In comparison with other tyrosine kinases ALK has also been implicated in oncogenic
processes via overexpression [107] and gain-of function mutations [108-112].
As well as being described as an oncogene, ALK has also been implicated in one case of
disease, schizophrenia [113]. Here Kunugi et. al. reported that a polymorphism at
Glu1529Asp present within the intracellular region of ALK was positively correlated with
susceptibility towards schizophrenia. However they were unable to rule out the possibility that
other polymorphisms, in linkage disequilibrium with the ALK polymorphism, were
responsible for the susceptibility noted in the patient population [113]. One additional study
correlates ALK to schizophrenia, the ALK KO mouse described by Bilsland and colleagues
display an increased level of the dopamine metabolite HVA, together with specific behavioral
changes. In fact, similar phenomenon are observed in schizophrenia patients receiving
antipsychotic treatment, which may suggest a potential involvement of ALK in modulating
schizophrenia [91]. However this is far from being understood at the molecular level.
MK has also been implicated in schizophrenia. Shimizu et. al. detected both increased and
decreased levels of MK in blood serum of schizophrenia patients and suggested that there are
two different entities of MK alterations within schizophrenia [114].
6.1 Anaplastic Lymphoma Kinase Fusion Proteins in Cancer
6.1.1 Anaplastic Large Cell Lymphoma
LK has mostly been studied in the context of ALCL. This disease was denoted already in
1985 [115] and with today‟s nomenclature for lymphomas it can be subdivided into three
different subtypes based on morphology. ALCL is a Non-Hodgkin‟s lymphoma arising from
T-cells characterized by the expression of CD30. ALK positive ALCL is most common in
young adults and children and account for 3% of total Non-Hodgkin‟s lymphoma and 10-30%
of all Non-Hodgkin‟s lymphoma in children [116]. A combinatorial chemotherapy treatment,
CHOP (Cyclophosphamide, Hydroxydaunorubicin (Doxorubicin), Oncovin (Vincristine) and
Prednisone) is applied in Sweden for treatment of ALCL patients [117]. Cyclophosphamide is
an alkylating agent [118] while Doxorubicin intercalates DNA thereby inhibiting cell growth
[119]. Oncovin is a mitotic inhibitor that mediates arrest of mitosis in the metaphase [120]
while Prednisone is a synthetic corticosteroid drug [121]. There is no treatment for ALK
positive ALCL aimed at directly targeting ALK.
ALK expression in ALCL is a prognostic factor used to predict clinical outcome for patients
presenting with ALCL. These patients have a higher 5-year survival rate as compared to ALK
A
A

20
negative cases [122, 123]. Within the ALK positive cohort levels of NPM-ALK transcripts can
also predict outcome of disease with a more unfavorable outcome being apparent in the cohort
having high expression of NPM-ALK [124]. ALK positive ALCL occurs to a higher extent in
children and young adults [122, 123]. However, ALK expression in ALCL is a positive
prognostic factor independent of patient age [125].
Currently there is insufficient information to explain the discrepancy observed between ALK
positive and ALK negative ALCL. However, a few plausible explanations have been
proposed. ten Berge et. al. demonstrated that active caspase 3 expression in ALCL was a
strong prognostic indicator for favorable outcome. The activity of caspase 3 was strongly
correlated with expression of ALK [126]. Since caspase 3 is a key-player in executing
apoptosis the authors suggested that this might partially explain the more favorable outcome
of ALK positive ALCL in correlation to ALK negative ALCL [126]. Khoury and coworkers
detected STAT3 in both ALK positive and ALK negative ALCL cases, leading them to
suggest activated STAT3 as a negative prognostic factor independent of ALK expression in
ALCL [127]. Survivin [128] and MUC-1 [129] are two other marker that indicate a poorer
outcome in ALCL regardless of ALK status. However within the ALK positive cohort of
ALCL MUC-1 expression is indicative of a poorer prognosis with a decrease in overall
survival [129]. The exact action of MUC-1 in modulating ALK positive ALCL is not
determined. However, MUC-1 is commonly over-expressed in oncogenic processes and via its
adhesive properties thought to modulate both metastatic capacity and provide hindrance for
immune cells trying to access the tumour cells [130].
There have been a number of reports regarding expression of ALK fusion protein in healthy
individuals. Trumper et. al. detected minor amounts of NPM-ALK transcript in blood from
healthy donors [131] while Maes and colleagues detected NPM-ALK fusion transcripts in
healthy lymphoid tissue via RT-PCR [132]. Both groups raised the question of whether the
fusion transcript on its own is enough for inducing oncogenic transformation or if secondary
events are needed.
6.1.2 Mouse Models for ALK Driven ALCL
PM-ALK has been established as the causative protein in ALCL by a number of groups
both in vitro and in vivo. For example, Kuefer et. al. in 1997, could induce lymphoid
malignancies in lethally irradiated mice receiving bone marrow expressing human NPM-ALK
[133]. Thereby, lending in vivo support for NPM-ALK as an oncogenic protein as suggested
by in vitro experiments [134, 135]. Further, Chiarle et. al. created transgenic animals
expressing NPM-ALK under the control of the CD4 promoter. This strain developed CD30
positive NPM-ALK T-cell lymphomas as well as plasma cell tumors [136]. Other strategies of
investigating NPM-ALK within the development of ALCL using animal models have been
reviewed recently by Turner et. al. [137].
N

21
6.1.3 ALK Fusion Proteins in ALCL
ue to a translocation event, already established during the late 80th [138-140], the kinase
domain of ALK is fused to the 5´end of NPM creating the constitutively expressed fusion
protein NPM-ALK [27] within the disease ALCL. NPM is a multifunctional protein, which
serves as a molecular chaperone involved in ribosome biogenesis with shuttling of pre-
ribosome subunits from the nucleus to the cytoplasm, in addition to playing a role in DNA
repair, transcription and genomic stability [141]. NPM-ALK is constitutively phosphorylated
as a result of the oligomerization domain within NPM bringing two ALK tyrosine kinase
domains together thereby stimulating autophosphorylation and activation [134]. Bischof and
coworkers further explored this dependency upon NPM for oncogenic potential via NPM-
ALK. By using different deletion mutants of NPM-ALK they demonstrated that the complete
NPM portion was required for dimerisation and phosphorylation of the fusion protein and
subsequent transformation of NPM-ALK transfected Fr3T3 cells [142]. NPM-ALK appears to
be present in both the cytoplasm and in the nucleus. However, the nuclear localization seems
to be of no importance for transformation via NPM-ALK. Instead the nuclear localization is a
consequence of the internal properties of NPM as demonstrated by Bischof et. al. [142] and
Mason et. al. [143] and it is the presence of NPM-ALK in the cytoplasm that induces
malignant transformation.
Beside the NPM-portion the ALK kinase domain of NPM-ALK is an absolute requirement for
transforming activity. This is illustrated via a mutation in the ATP binding region of the ALK
catalytic domain, K210R, which renders NPM-ALK kinase dead and eliminate the malignant
phenotype of this protein [142].
In addition to NPM, numerous other fusion partners exist for ALK in ALCL namely; ALO17
(ALK lymphoma oligomerization partner on chromosome 17) [144], TFG (TRK-fused gene)
[145, 146], MSN (moesin) [147], TPM3 and 4 (Tropomyosin 3 and 4) [148-150], ATIC (5-
aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase) [151,
152], MYH9 (Non-muscle myosin heavy chain) [153] and CLTC1 (Clathrin heavy chain-like
1) [154](table 1). Furthermore, all partner proteins of ALK share common characteristics.
Firstly) the transcription of the fusion protein is driven via the promoter of the ALK partner
protein; secondly) the localization of the fusion protein is determined via the ALK partner
protein and; finally) the ALK partner protein has some mode of oligomerization thereby
inducing phosphorylation and activation of the ALK kinase domain [88].
As mentioned above, NPM is responsible for the dimerisation essential for
autophosphorylation of and downstream signaling via the NPM-ALK tyrosine kinase domain
[134, 142]. The TRP3 and TFG fusion partners contain dimeric α-coiled-coil structures that
are hypothesized to be responsible for the dimerisation of TPM3-ALK [148] and TFG-ALK
[145] respectively. Since ATIC exists as a homodimer [155] this property is assumed to be
responsible for the activation of ATIC-ALK [151]. For Moesin, MYH9 and CLTC the reason
behind dimerisation is slightly more complex. CLTC is a component of Clathrin coated
vesicles [156] and Touriol et. al. proposed that clathrin coat formation activate the kinase
domain of ALK via close proximity of the CLTC-ALK fusion proteins. This hypothesis is
supported by data concerning the localization of CLTC-ALK showing a punctuated pattern,
D

22
within the cell, consistent with clathrin coated vesicles [154]. The theory of “close proximity”
as a mean of activating a tyrosine kinase domain is also implemented by Tort and coworkers
[147]. They speculate that MSN-ALK is activated via the ability of moesin to crosslink the
plasma membrane with the actin cytoskeleton [147]. Myosin heavy chain forms as a dimer
however the MYH9 sequence that is fused to ALK is missing the dimerisation domain of the
full-length protein. Still the MYH9-ALK fusion protein is phosphorylated in vivo, which
indicates either an uncharacterized dimerisation domain or, as suggested by the authors, that
MYH9 potentially interacts with other proteins leading to multimerization and consequent
activation of ALK [153].
6.1.4 Oncogenic Signaling via ALK Fusion Proteins
PM-ALK signals via the PLCγ, the PI3K , the RAS/MAPK and the JAK/STAT pathways
[157] as described below in more detail.
PLCγ catalyses the generation of diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3)
from phosphatidylinositol-4,5-biphosphate (PIP2). DAG and IP3 activate protein kinase C and
trigger the release of Ca2+
[158], respectively. Via its SH2 domain PLCγ has been shown to
directly interact with NPM-ALK. This interaction is of importance for the transforming
potential of NPM-ALK since mutated NPM-ALK which lacks the NPM-ALK/PLCγ
interaction site fails to induce transformation in transfected cell culture systems [42].
Generating NPM-ALK constructs with tyrosine to phenylalanine mutants of all possible
autophosphorylation sites (figure 3), Y664 (corresponding to Y1604 in human full-length
ALK) was noted as responsible for the PLCγ interaction. Of note is that all other mutants
(except the kinase dead Y338, Y342 and Y343 mutants) retained their ability to confer IL3
independent growth of Ba/F3 cells [42] indicating a key role for the PLCγ pathway in ALK
dependent transformation.
PI3K consists of two different subunits, the p110 catalytic subunit and the p85 regulatory
subunit [159]. NPM-ALK interacts with the PI3K p85 subunit [160, 161] thereby activating
the catalytic subunit of PI3K [160, 161]. Inactivation of the PI3K pathway induces apoptosis
in NPM-ALK positive cells [160, 162], indicating a role for the PI3K/AKT pathway in anti-
apoptotic signaling. Further, PKB/AKT activation appears to be a critical step for the
malignant transformation potential by NPM-ALK. This is supported by the work of Slupianek
et. al. who inoculated mice with NPM-ALK positive cells expressing dominant negative
PKB/AKT, leading to a strong impairment of tumour growth, and a delay in tumour formation
as compared to control animals [162].
FOXO3a is a transcription factor which regulates a number of genes involved in cell cycle
progression, promoting cell-cycle arrest when active [163]. NPM-ALK can, via the
PI3K/PKB/AKT pathway, deregulate FOXO3a. Active PKB/AKT phosphorylates FOXO3a,
leading to retention of FOXO3a in the cytoplasm. This results in alteration in the level of
transcription of several FOXO3a target genes in NPM-ALK expressing lymphoma [164], such
as cyclin D2, Bin-1 and p27kip1
, which thereby promote cell survival and cell cycle
progression.
N

23
Table 1: Fusion Partners of Anaplastic Lymphoma Kinase in Disease
Disease Fusion Protein Chromosomal Abnormality References
ALCL NPM-ALK t(2;5)(p23;q35) [27]
ALCL ALO17-ALK t(2;17)(p23;q25) [144]
ALCL TFG-ALK t(2;3)(p23;q21) [145, 146]
ALCL MSN-ALK t(2;X)(p23;q11-12) [147]
ALCL TMP3-ALK t(1;2)(q25;p23) [148, 149]
ALCL TMP4-ALK t(2;19)(p23;p13) [150]
ALCL ATIC-ALK inv(2)(p23;q35) [151, 152]
ALCL MYH9-ALK t(2;22)(p23;q11.2) [153]
ALCL CLTC1-ALK t(2;17)(p23;q23) [154]
IMT TMP4-ALK t(2;19)(p23;p13) [165]
IMT TMP3-ALK t(1;2)(q25;p23) [165]
IMT CLTC1-ALK t(2;17)(p23;q23) [166, 167]
IMT SEC31L1-ALK t(2;4)(p23;q21) [168]
IMT RANBP2-ALK t(2;2)(p23;q13)
inv(2)(p23;q11-13)
[169]
IMT CARS-ALK t(2;11;2)(p23;p15;q31) [144, 170]
NSCLC EML4-ALK inv(2)(p21;p23) [105]
DLBCL NPM-ALK t(2;5)(p23;q35) [171]
DLBCL CLTC-ALK t(2;17)(p23;q23) [172]
Table 1; Fusion Partners of Anaplastic Lymphoma Kinase in Disease; NPM; nucleophosmin,
ALO17; ALK lymphoma oligomerization partner on chromosome 17, TFG; TRK-fused gene,
MSN; moesin, TPM3 and 4; Tropomyosin 3 and 4, ATIC; 5-aminoimidazole-4-carboxamide
ribonucleotide formyltransferase/IMP cyclohydrolase, MYH9; Non-muscle myosin heavy
chain, CLTC1; Clathrin heavy chain-like 1, SEC31L1; SEC31 homologue A, RANBP2; Ran-
binding protein 2, CARS; Cysteinyl-tRNA synthetase, EML4; Echinoderm microtubule--
associated protein like 4.
________________________________________________________________________

24
Exclusion of FOXO3a from the nucleus due to phosphorylation via PKB/AKT leads to the
repression of p27kip1
[164], a negative regulator of cell cycle progression [173]. Rassidakis et.
al. would further demonstrate that up-regulating p27kip1
via inactivation of PKB/AKT induces
cell-cycle arrest in NPM-ALK positive cells [174].
A second target for PI3K/PKB/AKT signaling in ALK positive ALCL is mTOR. Vega et. al.,
presented data showing that direct inhibition of PKB/AKT mediates a reduced
phosphorylation of mTOR. However, phosphorylation of mTOR was not completely
abrogated and the authors concluded that additional pathways must function in regulating
mTOR [175]. Marzec et. al. validated the results implicating the PI3K/PKB/AKT pathway in
regulating mTOR. They also showed a stronger dependency on the RAS/MAPK pathway as
an upstream regulator of mTOR in NPM-ALK expressing cells [176].
The RAS/MAPK pathway as a downstream target of NPM-ALK was first reported by the
Yamamoto group. They demonstrated an interaction between NPM-ALK and three different
adaptor proteins IRS-1, Shc and Grb2 [134]. Since Shc and Grb2 are required for recruiting
RAS to the plasma membrane [177], this implicated the RAS/MAPK pathway as a
downstream target of NPM-ALK activity. Interactions with IRS-1 and Shc were however not
essential for transformation since NPM-ALK mutants unable to interact with Shc and IRS-1
still had the ability to transform NIH3T3 cells [134]. A more recent study has shown that
NPM-ALK seems to activate MEK directly. Further, depletion of ERK1 and ERK2, which are
downstream targets of MEK, simultaneously impaired cell proliferation while ERK1
depletion alone induced apoptosis in an ALCL derived ALK positive cell line [178],
indicating that ERK1/2 are involved in survival and proapoptotic functions.
The last major pathway, which is engaged in survival mechanisms of NPM-ALK expressing
ALCL is the JAK/STAT pathway. Several reports have demonstrated a direct interaction
between NPM-ALK and the phosphorylation status of STAT3 and consequent increased
activation of STAT3 [179-181]. The fusion protein NPM-ALK also interacts with and
activates JAK3 [179, 182], the receptor associated tyrosine kinase responsible for STAT3
activation [183]. However whether JAK3 is necessary for the activation of STAT3 in ALCL is
unclear. Zamo et. al., reported strong phosphorylation of STAT3, using truncated NPM-ALK,
which lacks the interaction site for JAK3. This indicates that the JAK3 interaction with NPM-
ALK is not required for STAT3 phosphorylation [179]. Conversely Amin et. al. obtained a
direct interaction between NPM-ALK and JAK3 and through inhibition of JAK3 they
demonstrated a clear reduction in STAT3 activation [182]. This dependency of STAT3
activation by JAK3 phosphorylation in ALK positive ALCL was further supported by Qiu and
coworkers [184], who observed similar results. In a recent study Marc et. al. showed that
depleting JAK3, using inhibitors and RNA knockdown techniques, had no effect on STAT3
phosphorylation in cells expressing NPM-ALK [185]. These data then arguing against the data
presented by Amin [182] and Qiu [184]. Further, the methods used and conclusions drawn by
Marc and colleagues [185] was strongly questioned by Amin and coworkers in editorial
correspondence [186]. However, JAK3 activity is strongly associated with ALK expression
and STAT3 phosphorylation in vivo [187] arguing for a role of JAK3 in ALK positive ALCL
pathogenesis. Disregarding the conflicting data concerning STAT3 phosphorylation it is clear

25
__________________________________________________________________________
that down-regulation of active STAT3 is incompatible with a transforming phenotype in ALK
positive ALCL with cells displaying increased apoptosis and cell-cycle arrest [182, 184, 185,
188].
Several groups have reported additional information of the importance of the JAK/STAT
pathway in ALCL [180, 188]. One finding is the loss of Shp1 in ALK positive ALCL due to
DNA methylation [189]. Shp1 is a negative regulator for the JAK/STAT pathway [183] and
by restoring Shp1 levels in NPM-ALK expressing cells the pathway is inactivated and cell-
cycle progression halted [188]. In addition, PP2, a protein interacting with STAT3 and
necessary for sustained phosphorylation, was found to be over-expressed in ALK positive
ALCL by Zhang et. al. Inhibition of PP2 lead to the abrogation of STAT3 phosphorylation
independently of NPM-ALK activity [180].
Figure 3: The intracellular region of
human and mouse ALK contains the
protein tyrosine kinase (PTK) domain
and span amino acid (aa) 1058-1620 and
1062-1621 respectively. Herein all
potential autophosphorylation sites
within human and mouse ALK are
depictured. Note that there is no
equivalent tyrosine in mouse ALK for
human ALK Y1604. Tyrosines within the
activation loop are depictured in bold
(ref). Localization of tyrosines within the
intracellular region is not depictured at
accurate scale. TM, transmembrane
domain.
hALK mALK
Hum
an A
LK
PT
K d
om
ain
aa 1
116-1
38
3
Mo
use
AL
K P
TK
do
mai
n a
a 11
20
-13
87
TM
Y1139
Y1278
Y1282
Y1283
Y1359
Y1385
Y1135
Y1282
Y1286
Y1287
Y1363
Y1389
Y1063
Y1082
Y1096
Y1059
Y1078
Y1092
Y1405
Y1590
Y1592
Y1401
Y1584
Y1586
Y1604

26
STAT3 has been shown to play a role in NPM-ALK mediated proliferation and anti-apoptotic
actions. However another member of the STAT family, STAT5, displays a more unclear role
in the pathogenesis of ALK positive ALCL. Several reports indicate that STAT5 is
undetectable in ALCL using both in vitro cell culture and in vivo patient samples [179, 180].
However Nieborowska-Skorska et. al. demonstrate an interaction between STAT5 and NPM-
ALK. Further, expressing a dominant negative STAT5B construct they report induction of
apoptosis and cell-cycle arrest in ALCL derived cell lines expressing NPM-ALK [190]. A
more intriguing result was observed after the initial down-regulation of STAT5B, and the
concomitant apoptosis and cell-cycle arrest. Here, a small population of cells regained their
ability to proliferate and induce tumour formation although with a slower growth rate
compared to the parental cell line [190]. This implicates STAT5B as a target for NPM-ALK
mediated proliferation, although perhaps not as an obligatory player. The phosphorylation of
STAT5B in ALCL has also been demonstrated by Zhang and coworkers [191]. In their study
they demonstrated a tumour suppressor function for STAT5A in ALK positive ALCL derived
cell lines. In these cells STAT5A was silenced via methylations in a STAT3 dependent
manner and up-regulation of STAT5A, mediated by inhibition of methylation, resulted in
decreased transcription of NPM-ALK due to the ability of STAT5A to interact with the NPM-
ALK promoter region [191].
Beside the above mentioned players there a number of other novel proteins implicated in
NPM-ALK mediated malignancy. Cdc42, a protein clearly established in regulating cell shape
and migration [192], is regulated via NPM-ALK in ALCL. Upon depleted of Cdc42 cell cycle
arrest and apoptosis are induced [193]. Another protein, which has been implicated is
p130Cas; NPM-ALK can no longer cause transformation on depletion of p130Cas protein.
This dependency on p130Cas has been suggested to be modulated via Grab2 in NPM-ALK
positive ALCL cells [194]. Further, knockdown of Shp2 reduces the migratory capacity of
cells expressing NPM-ALK [195] and Src kinases, especially pp60Src, have been suggested to
be important for the proliferative ability of NPM-ALK positive ALCL cells [196].
Few of the other known ALK fusion proteins have been studied as extensively as NPM-ALK
with respect to downstream signaling mediated by the ALK fusion protein. Nevertheless they
are assumed to function in similar manners as the NPM-ALK fusion protein. This assumption
is supported by studies executed by Hernández et. al. [146] and Trinei et. al. [152]. Trinei et.
al. show an association between ATIC-ALK and Grb2 and Shc [152]. Similar observations
were demonstrated by Hernández et. al. for the TFG-ALK fusion protein. Moreover, in this
study they also demonstrated an association between TFG-ALK and PLCγ [146].
Armstrong and colleagues made an extensive study were they evaluated NPM-ALK, TPM3-
ALK, TFG-ALK, CLTC-ALK and ATIC-ALK with respect to their oncogenic potential both
in vivo and in vitro. They demonstrated, in an in vitro assay, that TPM3-ALK displayed higher
migratory and invasive capacity when compared with the four other ALK fusion proteins
examined. Conversely, when grafting transfected cells subcutaneously in nude mice, NPM-
ALK and TFG-ALK mediated tumours were readily detected 6 days after inoculation, while
TPM3-ALK mediated tumours were detected a few days later [197]. The invasive properties
of the ALK fusion proteins were further evaluated, by an in vivo lung metastasis assay. Here
the TPM3-ALK fusion product shows an increased capacity to form metastases in comparison

27
to the other ALK fusion proteins [198]. Whether or not this increased ability for TPM-ALK
transfected cells to induce metastases will have any clinical relevance is currently unclear.
Employing patient samples, Khoury and coworkers, observed that activated STAT3 was only
expressed in approximately 85% of the ALK positive ALCL cases. The authors speculate that
this could be due to differences in signaling via the different ALK fusion proteins [127]. This
theory is supported by Armstrong et. al. who demonstrated effective phosphorylation of
STAT3 via NPM-ALK, TPM3-ALK, TFG-ALK and ATIC-ALK while showing that CLTC-
ALK was incapable of phosphorylating STAT3 [197].
6.1.5 Inflammatory Myofibroblastic Tumour
nflammatory Myofibroblastic Tumour (IMT) is a benign malignancy of mesenchymal
origin with a prominent inflammatory component consisting of plasma cells and
lymphocytes [199]. First described in the lung these “inflammatory pseudotumours” was
considered a post inflammatory condition rather than a malignant process [200]. They can
manifest in almost any anatomical site [199] although the most common places are the lung,
abdomen and retroperitoneum [201]. In 1999, the 2p23 chromosomal rearrangement and
expression of ALK was reported by Griffin et. al. as a sign that IMTs, or a subpopulation of
IMTs, were neoplastic in origin rather than reactive [104]. Further studies have confirmed the
presence of several different ALK fusion proteins containing the kinase domain of ALK and
partner proteins responsible for dimerisation (table 1).
35-60% of all IMTs appear to exhibit ALK rearrangements [202, 203] with ALK positive
IMTs preferentially affecting young individuals [165, 202, 203]. This pattern of occurrence is
similar to that noted for ALK positive ALCL. Moreover the prognosis appears to be better for
ALK positive IMTs [204] as with ALK positive ALCL.
6.1.6 Non-Small Cell Lung Cancer
ung cancer is the most common cancer in the world and the leading cause of cancer
deaths [205]. Lung carcinoma can be subdivided into two major groups, small-cell lung
carcinoma and non-small cell lung carcinoma (NSCLC) with NSCLC constituting the larger
proportion of cases. As a result of a transformation screen Soda et. al. identified the ALK
fusion protein EML4-ALK in NSCLC [105]. In further investigations employing ALK
inhibitors, they observed a reduction of cell growth in BA/F3 cells expressing EML4-ALK,
supporting an oncogenic role for the fusion protein [105]. Other fusion partners of ALK have
so far not been reported for NSCLC [206] and the EML4-ALK event seem to be a rare
occurrence present in only 3% of all NSCLC cases [207]. However, activating EGFR
mutations have also been found in a small proportion of NSCLC responsive to treatment with
gefitinib (Iressa) [208, 209], an EGFR kinase inhibitor [210, 211]. ALK, being an
uncontrolled activated tyrosine kinase in NSCLC just like EGFR, may prove to be an effective
target in the treatment of NSCLC cases presenting with EML4-ALK. In fact a recent study has
demonstrated an effect of the ALK inhibitor TAE684 in EML4-ALK NSCLC cell lines [212].
However this effect was only seen in one out of three tested cell lines demonstrating a need to
further characterize the EML4-ALK subpopulation of NSCLC.
I
L

28
6.1.7 ALK Positive Diffuse B-cell lymphoma
iffuse large B-cell lymphoma (DLBCL) is of interest from the perspective that there
seem to be an ALK positive subpopulation expressing either full-length ALK [213] or
ALK fusion protein, NPM-ALK [106, 171] or CLTC-ALK [172] respectively. Arber et. al. in
1996 reported the occurrence of NPM-ALK transcript in large cell lymphomas of B-cell origin
[106] which was subsequently verified by Adam et. al. [171]. CLTC-ALK was reported by
several groups as a rare finding in DLBCL [172, 214, 215] and full-length ALK protein have
so far only been detected in 7 cases of DLBCL [213].
Expression of ALK, either as full-length or as a fusion protein, in DLBCL appears to be a rare
event with only 33 reported cases to date. However, when summarized they seem to occur
preferentially in males spanning all age groups and be correlated with a more aggressive
clinical outcome and a poor response to chemotherapy [216].
6.2 Anaplastic Lymphoma Kinase Overexpression in Cancer
verexpression of ALK in different tumour materials has been evaluated by several
groups. Dirks et. al., evaluated ALK expression in a large panel of human tumour cell
lines and reported expression of the ALK kinase domain in thyroid carcinoma, NSCLC, breast
cancer, melanoma, neuroblastoma, glioblastoma, astrocytoma, retinoblastoma, ewing sarcoma
and rhabdomyosarcoma [80]. Besides these tumour types, ALK expression has also been
noted in leiomyosarcoma, malignant peripheral nerve sheath tumours and malignant fibrous
histocytoma [107]. Some of these - NSCLC, glioblastoma and neuroblastoma - have been
evaluated with respect to ALK activation. However, several of these cancer types are
uncharacterized at the molecular level.
In the case of rhabdomyosarcoma, leiomyosarcoma and malignant fibrous histocytomas an
increased copy number of the chromosomal region 2p23 might explain the overexpression of
ALK [107]. Furthermore, in rhabdomyosarcoma ALK expression appears to be preferentially
found in the alveolar subtype [107, 217].
In glioblastoma ALK phosphorylation and subsequent downstream PKB/AKT activation
appears to occur via PTN. Moreover, in vitro experiments demonstrate a dose dependent role
for ALK with glioblastoma cell lines depleted of ALK growing at a reduced rate compared to
the parental cell line [75].
In breast cancer, a clear role for ALK has not been firmly established, however several line of
evidence point toward a role for ALK in this disease. Firstly; ALK is strongly expressed in
several different subtypes of human breast cancers in a pattern not consistent with wt tissue
[218]. Secondly; PTN, the proposed mammalian ALK ligand, is extensively expressed in
breast cancer [219, 220] and expression of truncated PTN in a human breast cancer cell line
abrogated tumours formation in nude mice, implicating PTN as a player in the malignant
phenotype of breast cancer [221]. Thirdly; the PTN receptor RPTPβ/δ is strongly expressed in
different subtypes of human breast cancer [218]. These three observations together with the
data from Perez-Pinera et. al. which suggest that ALK is indirectly activated via
D
O

29
PTN/RPTPβ/δ signaling [81] have lead to the proposal that ALK may be activated via the
PTN/RPTPβ/δ pathway and potentially harbor an oncogenic potential in breast cancer
development.
6.3 Anaplastic Lymphoma Kinase Gain-of-Function in Cancer
6.3.1 Neuroblastoma
euroblastoma is derived from neural crest cells of the sympaticoadrenal lineage and can
therefore arise throughout the sympathetic nervous system. It is the most common solid
tumour in childhood and accounts for 15% of all pediatric oncology deaths [222].
Expression of full-length ALK in neuroblastoma was first demonstrated by Lamant and
coworkers in 2000 [223]. Further studies from the group of Sakai show that the ALK locus is
amplified in neuroblastoma cell lines and also in primary patient samples [224, 225].
Furthermore, they found a physical association between ALK and ShcC [224]. Through
disruption of ALK expression in the neuroblastoma cell lines, NB-39-nu and Nagai,
employing siRNA, ShcC phosphorylation (as well as PKB/AKT activation) was down
regulated with concomitant apoptosis of the disrupted cells [225].
During the present year (2008) 5 groundbreaking studies have been published regarding the
presence of activating ALK mutations in both familiar and sporadic cases of neuroblastoma
[108-112]. All mutations (except one) are localized within the kinase domain of ALK and are
assumed to be activating in nature (table 2). Further, neuroblastoma patients who are positive
for ALK mutations appear to have a worse prognosis [108-112].
The initial report by Mossé et. al. [108] resulted from the investigation of familiar cases of
neuroblastoma. A number of different mutations in the ALK protein kinase domain were
described (table 2). In addition, several neuroblastoma cell lines were shown to contain
activating ALK mutations. Knockdown of ALK in these cell lines resulted in an inhibition of
proliferation [108]. The data of Chen et. al. verified the oncogenic potential of the activating
ALK mutants, F1174L and K1062M. They further expressed these mutants in NIH3T3 cells
and employing grafts of ALK mutant expressing cells onto nude mice observed rapid
formation of subcutaneous tumours [111].
Different mutants of ALK found in neuroblastoma patients seem to respond differently to
inhibition of ALK via the chemical inhibitor, TAE684. Using this ALK inhibitor, George et.
al. observed a reduction of cell proliferation and increased apoptosis in a neuroblastoma line
harboring the ALK F1174L mutation, while a neuroblastoma cell line containing the ALK
R1274Q mutant did not respond. Nevertheless when expressed in NIH3T3 cells, both the ALK
R1274Q and the ALKF1174 mutants induced a transformed phenotype and apoptosis was
initiated when cells were exposed to TAE684. Moreover the ALK T1151M mutant protein
was not phosphorylated when expressed in NIH3T3 cell and neither did the cells survive when
grown in IL3 depleted medium. The authors concluded that potential ALK inhibitors should
be investigated with regard to the complete panel of ALK mutants as well as the cellular
milieu in vivo [109].
N

30
Table 2: Activating Mutations of Human Anaplastic Lymphoma Kinase in Neuroblastoma
Nucleotide Changes aa Mutation Targeted Region References
3260 C T T1087I Juxtamembrane domain [111]
3271 G A D1091N Juxtamembrane domain [108]
3383 G C G1128A P loop [108]
3452 C T T1151M Kinase domain [109]
3497 T G M1166R C helix [108]
3512 T A I1171N C helix [108]
3520 T A F1174I End of C helix [108, 109, 111,
112]
3521 T G F1174C End of C helix [110, 111]
3520 T G F1174V End of C helix [111]
3522 C A F1174L End of C helix [108, 111, 112]
3575 G C R1192P β4 strand [108]
3700 G A A1234T Catalytic loop? [109]
3733 T G F1245V Catalytic loop [108]
3733 T A F1245I Catalytic loop [112]
3735 C A F1245L Catalytic loop [111, 112]
3734 T G F1245C Catalytic loop [108, 109]
3749 T C I1250T Catalytic loop [108]
3824 G T R1275L Activation loop [110]
3824 G A R1275Q Activation loop [108-112]
3833 A C Y1278S Activation loop? [110] Table 2; Activating mutations within the ALK kinase domain in neuroblastoma patients

31
7. Potential Treatments Strategies for Anaplastic Lymphoma Kinase
Positive Cancers
he development of tyrosine kinase inhibitors for use in cancer therapy have proven
effective in several cases. Most well-known is Gleevec which target the ABL PTK in
chronic myeloid leukemia (CML) [226]. In CML, ABL is present as a fusion protein (BCR-
ABL) due to a translocation event t(9;22) [227]. In addition to inactivating ABL Gleevec also
targets the c-Kit RTK and PDGFR [228, 229]. c-Kit is highly expressed in gastrointestinal
stromal tumours (GIST) [230] and clinical trials in 2001 and 2002 established that Gleevec
could be used in treatment of the malignancy [231, 232]. Other examples of RTK inhibitors
are Gefitinib and Erlotinib, which are selective inhibitors for EGFR (ErbB1) and used in
treatment of NSCLC [233].
The Settleman group is one of many that have investigated the potential clinical application
for ALK inhibitors in ALK positive malignancies. Using the ALK inhibitor TAE684 in more
the 600 different cell lines derived from human cancers they show inhibition of proliferation
in ALK positive ALCL, neuroblastoma and NSCLC cell lines [234]. TAE684 is a 5-chloro-
2,4-diaminophenylpyrimidine which most likely targets the ALK ATP binding pocket thereby
blocking ATP binding [235]. Subsequent administration of TAE684 after injections of NPM-
ALK positive ALCL cells (Karpas 299) in mice prevented development of disease as well as
regression of preinduced ALCL [235]. PF-2341066, an ATP competitive compound, targeting
c-MET and ALK [236] has also been shown to reduce ALK positive ALCL development in an
animal model [237].
UNC-01, a staurosporine derivative, inhibiting PKC [238] and chk1 [239] has been reported to
cause disease regression in one patient with ALK positive ALCL however the effect on ALK
was never validated [240]. Further, Gunby and colleagues when investigating potential
inhibitor of ALK kinase activity, could not confirm a direct relationship between UNC-01 and
ALK [241]. Currently there is a phase 1 clinical trial using UNC-1 in patients with relapsed or
refractory systemic ALCL or mature T-cell lymphoma with the secondary intent to investigate
its effect in ALK positive ALCL (www.ClinicalTrial.gov).
Besides using inhibitors, ALK has also been the target of strategies aimed at reducing the
mRNA thereby depleting the protein. Employing RNA interference to silence ALK Piva et. al.
could induce cell death in ALK positive ALCL cell lines. More importantly in an in vivo
animal model ALCL tumour growth was severely impaired by injection of adenovirus
expressing shRNA targeting ALK [242].
Direct targeting of ALK may be the preferential way to influence ALK positive malignancies
since downstream signaling might be redundant. Yet, by targeting different ALK related
proteins several authors have demonstrated reduced viability and tumour forming capacity of
NPM-ALK positive cell lines. Bonvini et. al. showed an interaction between the chaperone
Hsp90 and NPM-ALK. Using 17-allyl-amino-demethoxygeldanamycin (17-AAG), a
compound specifically inactivating Hsp90 they induced degradation of NPM-ALK and
subsequent apoptosis in ALCL cell lines [243]. Another example of possible downstream
targets in ALK positive ALCL is PKB/AKT. Injecting mice with cells expressing NPM-ALK
T

32
and a dominant negative PKB/AKT construct, Slupianek et. al. demonstrated a severely
impaired tumour forming capacity as compared to animals receiving NPM-ALK expressing
cells with an active PKB/AKT pathway [162].
ALK therapy in the form of vaccination has been brought forward as a possible treatment
since a few reports indicate immunological responses towards ALK in patients [244, 245].
Indeed, Chiarle et. al. tested this hypothesis, vaccinating animals toward human ALK prior to
challenge them with ALK expressing lymphoma cells. They observed protection against
disease in vaccinated animals and in already diseased animals the vaccination in combination
with standard treatment increased the cure rate [246].
Currently, there are no approved treatments for oncogenic malignancies caused via aberrant
ALK activation that directly targets ALK or the downstream ALK activated signaling
pathways. However ALK directed treatments would most likely benefit a number of different
cancer patient populations and is an area of intense research.

33
Aim
1. Overall Aim
he overall aim of this thesis has been to evaluate the function of the Receptor Tyrosine
Kinase Anaplastic Lymphoma Kinase. This is however a rather large area to cover and
there are several different directions available for a PhD student to enter, so for each project a
specific aim have been employed.
1.1 Specific Aim: Article I
o evaluate the mRNA and protein expression pattern of ALK during mouse development
to gain new insight regarding possible structures dependent on ALK signaling
1.2 Specific Aim: Article II
o explore the potential conservation of the ALK signaling pathway by evaluating the
ability of Drosophila Jeb in activating mouse ALK.
1.3 Specific Aim: Manuscript I
o investigate the potential phenotype in two different mouse models lacking functional
Anaplastic Lymphoma Kinase thereby exploring the in vivo function of the receptor.
1.4 Specific Aim: Manuscript II
o elucidate the functional role of Anaplastic Lymphoma Kinase in the olfactory system of
the adult mice by clarifying the localization of Anaplastic Lymphoma Kinase mRNA and
protein.
T
T
T
T
T

34
Results and Discussion
1. Article I: ALK Expression during Development
e have conducted an extensive investigation of ALK mRNA and protein expression
during mouse embryogenesis focusing on developmental days 10.5 to 16.5. Prior to this
study two other groups have independently described ALK expression at the mRNA level [28,
29] however ALK protein expression had not been reported before. Throughout the
investigated developmental stages ALK was strongly expressed, both at the mRNA and
protein level, in structures of the central nervous system (CNS) e.g. the diencephalon, medulla
oblongata, pons and spinal cord.
In the spinal cord ALK mRNA can be visualized from day 11.5 to 14.5 throughout the ventral
compartment. However, between days 12.5 to 14.5 ALK mRNA in the spinal cord displays a
peculiar pattern. In the most caudal part of the spinal cord there is a shift in expression from
the ventral to the dorsal side. This shift in expression is also observed during developmental
day 13.5 in rat for PTN [96], a potential ALK ligand [64]. Within the spinal cord a
subpopulation of the ALK positive cells most likely represents the future motor neurons as
depicted using the markers Isl1 and nkx6.2 [247]. Hurley et. al., investigating ALK expression
during chicken development, also noted ALK expression in the Isl1 positive subpopulation of
spinal motor neurons [52].
In the gastrointestinal canal we detected ALK mRNA and protein in the anterior part of the
structure and with increasing developmental stages the expression progressed posterior.
However ALK was never observed in the hindgut, regardless of the stage investigated. This
may be due to regional differences in the structures examined, and there is also the possibility
that ALK might be expressed in the hindgut during later developmental stages. In addition,
ALK appears to be expressed in a layer just proximal to the musculature presumably
representing the developing enteric neurons.
Beside the above mentioned structures we could also detect ALK in a number of different
ganglions as well as in potentially non neuronal areas including the testis and ovary.
The ALK mRNA and protein expression patterns are in general overall agreement. However,
in some structures such as the diencephalon and spinal cord ALK mRNA and protein, even
though localized within the same structure, do not overlap. We assume this is due to the
subcellular localization of ALK in developing nerve cells with mRNA and protein being
localized in different compartments of the nerve (e.g. cell body and axon).
In conclusion we detect ALK mRNA in numerous neuronal compartments verifying and
extending the result presented in 1997 by Morris et. al. and Iwahara et. al. [28, 29]. Further we
present comprehensive novel data addressing the localization of ALK protein during mouse
development.
W

35
2. Article II: Can Drosophila Jeb activate mouse ALK?
hen discussing ALK function one important and somewhat controversial question is;
how is mammalian ALK activated? Being an RTK the presumed first step toward
activation of ALK would be postulated to occur via ligand binding to the extracellular region
[7]. For dALK this is indeed the case, with the secreted protein Jeb being an established
activating ligand [43, 44]. For mammalian ALK the picture is more complex. Thus far two
different ligands have been proposed for mammalian ALK, MK and PTN [64, 65], however
their assumed role as ligands for ALK are strongly debated (discussed in detail in section 5.3).
Whether or not there is a vertebrate “Jeb-like” ligand capable of activating ALK is a question
of great interest in the light of establishing the in vivo function of ALK as well as clarifying
the signaling pathways driven by ALK. Thus far no “Jeb-like” molecule has been described in
vertebrates that might function as an ALK ligand. To circumvent the issue of not finding
suitable Jeb candidates in vertebrates we reasoned that if there is a Jeb-like molecule in
vertebrates serving the same function as Jeb in Drosophila then in theory Jeb should be able to
activate mouse ALK (if the Jeb/dALK interaction is conserved throughout evolution). To
investigate this theory we employed several different strategies as discussed below.
In Drosophila lack of functional Alk is lethal [43, 44]. This phenotype can be rescued via
expression of the full length dAlk. However expression of full-length mouse ALK fails to
rescue the dAlk mutant phenotype indicating that mouse ALK is unable in substituting for loss
of dAlk. This further suggests that Jeb, even if it associates with mouse ALK, is ineffective in
inducing the appropriate signals needed to rescue the dAlk mutant. Expression of a dominant
negative dAlk protein in a wild type Drosophila background phenocopies the dAlk mutant
phenotype. Conversely, expression of the dominant negative mouse ALK construct does not
interfere with normal gut formation confirming that Jeb lacks the ability to activate mouse
ALK in an in vivo Drosophila context.
To further investigate the potential Jeb/mouse ALK relationship we used a cell based system
were we are able to induce either dAlk or mouse ALK using tetracycline. Stimulating mouse
ALK using an activating antibody directed toward mammalian ALK we could demonstrate
ALK phosphorylation, however dAlk was not phosphorylated. Moreover using recombinant
purified Jeb we could strongly stimulate tyrosine phosphorylation of dAlk, yet no effect on
mouse ALK was apparent. Thus, this further supports our conclusion that Jeb is not able to
activate mouse ALK. Further, in PC12 cells recombinant Jeb could initiate differentiation of
dAlk expressing, but not, mouse ALK expressing cells. This is in perfect agreement with the
above results and we conclude that Drosophila Jeb protein is incapable at inducing signaling
via the mouse ALK RTK. Furthermore, we were able to show that Jeb/dALK signaling could
efficiently induce neurite outgrowth in PC12 cells. This suggest a certain degree of
conservation at the level of functional readout between the two receptors.
W

36
2.1 Why is Drosophila Jeb unable to activate mouse ALK?
iven that Drosophila Jeb is unable to activate mouse ALK, we naturally raised the
question of why? What evolutionary processes have rendered mouse ALK insensitive to
Drosophila Jeb given the conservation of the RTK domain structure? It is theoretically
possible that during evolution either mouse ALK on its own or the vertebrate “Jeb”/ALK pair
have evolved to such an extent that substituting either with the Drosophila counterpart is no
longer feasible. A similar situation has been reported for the RTK RET where the mammalian
ligand is unable in activating the Drosophila Ret receptor although both human and
Drosophila Ret can induce PC12 cell differentiation [248].
The overall amino acid identity between Drosophila and mouse ALK is approximately 30%.
So the idea of mouse ALK evolving to the extent of no longer recognizing the Drosophila Jeb
ligand is not without context. However this raises another interesting topic, is the function
described for Drosophila Alk conserved for mouse ALK? Within Drosophila, Alk has an
essential role in visceral muscle development [43, 44] and lacking Alk signaling is not
compatible with life. However the recently published mouse ALK knockout strain, and our
own ALK knockout mouse, is viable without any notable intestinal defects [91]. Yet the
distribution of ALK protein throughout the gastrointestinal tract during mouse embryogenesis
as illustrated by us [249] and others [28, 29] indicate ALK as a potential player during
developmental processes in the gastrointestinal tract. The idea of redundancy between ALK
and its family member LTK [28, 29] is an interesting question that might explain the non
lethal phenotype of the ALK-/-
mice.
Beside the critical requirement for dAlk function in muscle development it is also of
importance for correct targeting of axon originating in the adult Drosophila optic lobe [50].
The published mouse ALK knockout display an overproduction of neuronal progenitor cells
within the hippocampus [91] supporting a neuronal function for ALK. This study, by Bilsland
et. al., did not address the question of axonal targeting and to date there is no available
information regarding this area in mammalian systems. It would be intriguing to address this
topic in an ALK knockout mouse model especially as we have demonstrated previous that
ALK is heavily expressed in the optic nerve during embryogenesis [249].
2.2 What about the LDLa domain?
he Jeb LDLa domain is important for its interaction with dAlk [44]. This implies that
possible Jeb-like molecules in other organisms responsible for activating ALK
presumably contain the LDLa domain. We proposed that Hen-1 [51] and Mig-13 [250] from
C. elegans might be possible ligands for ALK on the basis of their LDLa domain. Indeed,
Hen-1 has recently been confirmed as an ALK (SCD-2) ligand experimentally by Reiner et.
al. [32] suggesting that the concept of the Jeb/ALK interaction is conserved, at least in lower
organisms.
G
T

37
3. Manuscript I and II: What is the physiological consequence of loss
of ALK function?
3.1 The ALK Kinase KO and the ALK exon1 KO
hen the project described in the sections below was initiated there was no available
mouse models for investigating the in vivo function of ALK. During the course of this
project Bilsland et. al. published a study regarding a mouse ALK knockout strain [91]. This
strain will be discussed below were appropriate. In addition, over recent years the group of
Stephan Morris has mentioned, in the discussion of several articles, that a mouse ALK
knockout exhibit no apparent phenotypes [86-90].
We were primarily interested in exploring the physiological function of ALK in the context of
higher vertebrates. The mouse ALK gene consists of 29 transcribed exons, spanning 730 kbp
of genomic DNA. By adopting a strategy to delete exons 20 to 23, we created a mutant ALK
locus with a disrupted ATP binding site within the kinase domain [90]. ALK transcript and
protein can still be detected in this strain, however the mutant ALK protein expressed should
be kinase dead.
In the process of generating the ALK Kinase KO we discovered that a company (Lexicon
Pharmaceutical) had designed an ALK knockout lacking the first exon of ALK. This region
contains the starting ATG and the signal peptide and the hypothesis was that the ALK protein
should not be expressed in this mutant, or if expressed the mutant ALK protein should not
localize to the cell membrane. Examination of this ALK knockout strain (referred to as the
ALK exon1 KO) revealed expression of ALK protein. We have not addressed the issue of
whether or not the ALK protein produced localizes to the right compartment in this animal.
The ALK protein produced, although truncated, could possibly contain a functional tyrosine
kinase domain and technically be able to transmit signals downstream.
The ALK kinase KO and the ALK exon1 KO mice are viable. They can be kept as
homozygous stocks and display no gross morphological defects neither during development
nor as adult animals.
3.2 What function might ALK have during mouse embryogenesis?
o investigate the ALK homozygous population in greater detail we examined ALK
homozygous mouse embryos, comparing them to wild type littermates. In Drosophila
org-1 and duf/kirre are transcriptionally regulated via Alk signaling and in the absence of Alk
they are not expressed [43, 44]. The mammalian counterparts are TBX1 and mkirre3
respectively. mKirre3 is thought to participate in axonal pathfinding and synapse formation
during embryogenesis [251], moreover it is heavily expressed in structures that correlate with
ALK expression during development [251-253]. TBX1 is expressed during embryogenesis
[254] and is important for cardio and craniofacial development [255]. However, the ALK
Kinase KO homozygous embryos displayed no alterations in expression pattern for either
TBX1 or mkirre3.
W
T

38
Kirrel1 (NEPH1) and Kirrel2 are closely related to mKirre3. Kirrel2 modulates neuronal
targeting within the olfactory bulb [256] while Kirrel1 has been implicated in synapse
formation during embryogenesis [257]. The possibility that either of these could be regulated
via ALK was investigated in ALK Kinase KO mice. As for mkirre3, both kirrel1 and kirrel2
exhibited wild type expression in ALK-/-
mice.
TBX1 belongs to a family of 16 different members in mammals [255]. TBX2 and TBX3 are
functionally important in heart, limb and mammary gland development [258, 259] and since
their expression patterns overlap with ALK during development [254] we employed them in
our development study. For both TBX2 and TBX3 we observed a strong overlap with ALK
during development. However, in the ALK Kinase KO homozygote embryos there were no
alterations in expression detected.
Beside the above discussed markers we also investigated the potential ALK ligands PTN and
MK [64, 65]. We could show a close relationship between PTN and ALK protein expression
in the developing central nervous system consistent with the hypothesis that PTN may be a
ligand for ALK. In contrast, MK was not expressed in any areas that could be correlated with
ALK either directly or by close proximity. Furthermore there were no alterations in PTN or
MK protein localization in ALK-/-
embryos. Finally, we examined three different markers,
pSMAD, SMαA and Sox8. Sox8 was used since the expression pattern of this protein is
similar to that of ALK [260]. SMαA was utilized to investigate the visceral musculature of the
gastrointestinal canal while pSMAD was employed due to its dependency on functional ALK
signaling in Drosophila [49]. In conclusion none of these three markers displayed any
irregular expression patterns when examined in ALK Kinase KO homozygous embryos.
Furthermore, results obtained with the ALK exon1 KO results were similar to those with the
ALK Kinase KO. Thus, we were unable to define any apparent defect during embryogenesis
indicating that ALK is not important for vital embryological developmental stages.
3.3 Is there a role for ALK in the adult olfactory bulb?
n the published ALK knockout mouse strain, β-Gal was inserted upon removal of part of the
ALK kinase domain, thereafter visualizing ALK positive cells via expression of β-gal.
Utilizing this approach the areas of strongest β-gal expression was within the olfactory bulb
(OB) [91]. This prompted us to investigate this structure more closely in our ALK Kinase KO
model. In adult animals we detected ALK mRNA within the mitral cell layer and in scattered
cells of the external plexiform layer, with very strong ALK expression throughout the
glomerular layer of the OB. The expression of ALK in the glomerular layer is indicative of the
ALK protein being present in axonal projections. ALK could also be detected both at the RNA
and protein level in the basal part of the nasal epithelium.
From the olfactory epithelium, olfactory sensory neurons (OSN) project to structures termed
glomeruli within the OB leading to establishment of the olfactory map [261]. In order to
investigate the role of ALK in setting up the olfactory map we utilized the markers Kirrel2 and
DBA. Kirrel2 was analyzed since it has been shown to be important for targeting of OSN to
glomeruli [256]. DBA was analyzed since it recognizes specific subpopulations of glomeruli
I

39
[262]. However, within ALK Kinase KO homozygote animals these markers display wild type
expression patterns suggesting that ALK is not involved in establishing the olfactory map.
mKirre3 is expressed in the adult olfactory bulb [253]. This, together with the fact that
Drosophila ALK regulates transcription of duf/kirre (the mammalian counterpart being
mKirre3) [43, 44], encouraged us to investigated mKirre3 in an ALK-/-
context. However,
mKirre3 levels were unaltered between ALK+/+
and ALK-/-
animals.
Using tyrosine hydroxylase and cFos, two markers displaying increased intensity within the
OB when olfactory sensory neurons are activated, no alterations could be noted when
examining ALK kinase KO homozygous adult animals as compared with wild type controls.
This indicates that the olfactory neurons of the nasal epithelium can efficiently converge on
the olfactory bulb even in the absence of ALK activity.
In conclusion, we suggest that ALK does not play an essential role in establishing the
olfactory map or in converging odor information from the OSN to the OB. Nevertheless, the
apparent expression of ALK in areas of synaptic contact suggests that it may have a role in
neuronal transmission. Furthermore, ALK can be detected in the olfactory sensory area of the
nasal epithelium and since the MK/PTN double knockout presents down regulation of odor
receptors [101] it would be intriguing to investigate the possible relationship between ALK
and the odor receptors.
3.4 The ALK Kinase KO and the ALK exon1 KO are small…but why?
ne obvious phenotype is apparent in the ALK Kinase KO and the ALK exon1 KO
homozygous populations; they both exhibit a reduction of total body weight.
Comparing the two strains, ALK-/-
females seem to be affected to a higher extent than males.
Since disrupting ALK gives a more pronounced phenotype in the female population we
suggest that ALK is gender specific. A phenotype dependent on gender has been proposed for
the MK/PTN double knockout were MK-/-
PTN-/-
females are sterile while the corresponding
males can breed. Also there is a distortion in the gender ratio with fewer MK-/-
PTN-/-
females
being born compared to MK-/-
PTN-/-
males [100].
This decrease in total body weight is not apparent before birth and investigating nutritional
intake in the ALK kinase KO strain did not reveal any significant alterations between the
ALK-/-
and ALK+/+
animals indicating that the ALK-/-
reduced weight is not due to a lack of
food intake.
In mouse, ALK is expressed throughout the gastrointestinal canal during embryogenesis [28,
29, 249]. Furthermore Morris et. al. detected strong expression of ALK in the small intestine
of adult human tissue [27]. This motivated us to investigate the intestine in the ALK Kinase
KO strain comparing ALK-/-
and ALK+/+
littermates as alterations in this structure might
explain the reduction in weight noted for the ALK-/-
population. We could identify ALK
positive cells in the gastrointestinal canal of both developing embryos and adult animals, as
developing enteric neuron precursors and mature enteric neurons respectively. No obvious
O

40
morphological defects were detected in the ALK-/- population, and markers employed to
visualize specialized structures of the intestine displayed similar patterns in ALK-/-
and wild
type littermates. Even though we cannot detect any obvious alterations there might be subtle
defects present that might explain the reduced weight noted. In addition, we note that the
enteric nerve system is responsible for control and coordination of intestinal motility, secretion
and blood flow [263] needed for proper digestion. Thus, further analysis will be required to
shed light on the mechanism behind the reduced weigh observed in ALK-/- mutant mice. At
the present time, we state that the ALK knockout display an idiopathic reduction in weight.
3.5 Why are animals smaller? Lessons from the literature
hat are the reasons in the literature for genetically modified animals presenting a
smaller size then there wild type counterparts? Altering fat deposits and regulation of
fat cells is one way to affect body size. This is apparent in the Interleukin 1 receptor antagonist
(IL-1ra) knockout were defective regulation of adiposive tissue give a lean mice phenotype
[264]. Another example is the acetylating enzyme, spermidine/spermine N1-acetyltransferase,
that when overexpressed in mice results in a lean phenotype due to alterations of fat
homeostasis [265]. The skeleton is another area often targeted when size is disrupted as
apparent for both the transcription factor Sox8 and the RTK discoidin domain receptor 1
(DDR1). Loss of Sox8 is concurrent with a small body size due to premature osteoblast
differentiation and reduced bone mass [266] while mice deficient in DDR1 display dwarfism
due to reduced chondrocyte proliferation and bone growth [267]. Besides the above mentioned
examples several other mouse models displaying a small body size are found in the literature.
TNFα converting enzyme (TACE) is responsible for cleavage of the TNFα precursor to
generate the mature cytokine TNFα. Mice deficient in TACE fail in controlling energy
homeostasis since they cannot respond to energy depletion with the appropriate behavior (e.g.
increased food consumption and decrease of energy expenditure) [268]. c-Myc is embryonic
lethal when depleted. However in a c-Myc+/-
context animals are viable yet smaller when
compared to wild type littermates, this due to multiorgan hypoplasi due to decreased amount
of cells [269]. Last it is of interest that the MK/PTN double knockout has a reduced body
weight, although the molecular mechanisms behind this phenotype have not been established
[100]. Presently we can only speculate on the mechanism involved in ALK-/-
animals,
however, it is clear that ALK has an important role in the regulation of total body weight.
3.6 Why is ALK not vital for life in the mouse?
he ALK knockouts presented here and the one published by Bilsland et. al. [91] do not
display any severe phenotypes and can be maintained via homozygous breeding. Since
Drosophila mutants lacking functional ALK signaling have such an apparent and deleterious
phenotype [44] the question arise of why this not the case in mouse?
There are several different theories which may address the above question of which a few are
discussed below. Firstly, the ALK knockout strain established by Bilsland and coworkers, just
like our ALK Kinase KO strain, lacks the kinase domain and presents residual ALK mutant
transcript in the adult mouse brain [91]. Expression of the mutant ALK proteins can also be
detected in our ALK Kinase KO and ALK exon1 KO strains. This suggests that the function
W
T

41
of ALK may not be completely abrogated although the protein is altered. Furthermore, for the
ALK exon1 KO, it is possible that the mutant protein made contains a functional kinase
domain. This may explain the milder phenotype seen in the ALK exon1 KO-/-
populations
when compared with the ALK Kinase KO strain, indicating that it still retains some ALK
signaling properties. Thus, while impossible to address with the current mouse mutants, it
could be that complete deletion of the ALK locus – or at least complete abrogation of the
production of ALK protein - may result in a lethal phenotype. Secondly, ALK has one close
family member, LTK, and it is very possible that LTK function is able to rescues the loss of
ALK. While this is still an unaddressed question, it should be noted that ALK is highly
expressed during embryogenesis [28, 29, 249], while LTK is absent [33]. Conclusion of this
open question will require generation of an LTK knockout, together with a combined
LTK/ALK double knockout. Thirdly, if PTN and MK are the ligand for ALK as proposed by
Stoica et. al. [64, 65] it is possible that they could compensate for loss of ALK. Such a
scenario can be envisioned, since there are multiple receptors proposed for MK and PTN.
Fourthly, ALK may not be essential for life in mouse. However, even if ALK is not vital for
“staying alive” in a mouse context one must remember that laboratory animals are kept in a
safe and secluded environment. Maybe if subjected to the natural environment loss of ALK
will prove incompatible with life from the perspective of „survival of the fittest‟.

42
Conclusions
1. Article I
ALK mRNA and protein is strongly expressed during development mostly confined
to the CNS
2. Article II
Mouse ALK is unable to substitute for the loss of dALK in an in vivo Drosophila
assay
Drosophila Jeb cannot associate with and signal via mouse ALK neither in an in vivo
Drosophila context or in different in vitro cell based assays
Drosophila Alk, when activated via recombinant purified Jeb protein, induces neurite
outgrowth in PC12 cells.
3. Manuscript I and II
ALK-/-
animals are viable, fertile and can be maintained as a homozygous stock
ALK is not vital for developmental processes during embryogenesis
ALK is expressed within the adult olfactory bulb however it does not have an essential
role in establishing the odor map or converging odor information from the olfactory
sensory neurons to the olfactory bulb
ALK-/-
knockout animals display a reduction in total body weight that is not dependent
on food intake, organ weight or fat content
ALK is expressed in the enteric nerve system of the gastrointestinal canal in the adult
and developing gastrointestinal canal.

43
Acknowledgement
First and most importantly, my supervisor Bengt Hallberg. I´m grateful for the opportunity
you presented me with several years ago, to do my PhD in your lab. Over the years you have
been a guiding light pointing me in the right direction (never saying directly what my mistake
was – but rather making sure I know where to find the answer and have me report it back). I
can only say thank you, for all the knowledge and encouragement you have given me.
Secondly, my co-supervisor Ruth Palmer, your guidance and support in all issues has been
deeply appreciated and acknowledged. Thirdly , my second co-supervisor Göran Roos. We
may not have had such a close supervisor/student relationship but I have appreciated you input
in my work
To all people contributing to the results presented in this thesis (beside the once presented
above), Professor Marc Vigny, Dr. Maria Henriksson, Dr. Nelson Khoo, Dr. Hai-Ling
Yang, Therese Eriksson, Professor Anna Berghard and Professor Staffan Bohm.
Special thanks to Christina Schönherr for giving me her table with ALK point mutations to
use in my thesis
Special thanks to the staff in the animal house especially Ulla-Britt, Kerstin, Carola and Maria
for excellent caretaking of my animals and always helping out when needed. Without you life
in the animal house would not be fun.
To all members of the laboratory that have passed over the years. Maria, Anna, Charlotte
(without you me English have deteriorated to “Svengelska”), Lubna (I will not forget you in
USA), Hai-Ling (you are always present in my mind), Christina (my one and only labmate)
and all student non mentioned and none forgotten
Everyone in the laboratory of Ruth Palmer, walking over and having a chat/FIKA/gossip have
made the working day slightly more endurable (when my motivation have been low).
To Åsa, Terry and Karin for all help over the years with administrative issues as well as all
other questions I would not have known have to solve if not asking you
To everyone at the department of Medical Bioscience (floor 2 and 3). A workplace is nothing
without your workmates - You have all made the working day more than just work.
To my friends online and irl, Zarkan, Zarkana and Lich. I appreciate all moments spent with
you, both in front and away from the computer screen. Snittaren, lunch is an always
appreciated moment to gossip about work, family and foremost online playing . Maria and
Anne-May, I may be extremely bad at keeping contact but I have valued our lunch dates.
Till min familj. Mary (mamma) och Bert (pappa) utan erat stöd hade jag inte varit den jag är
idag. Ni är ovärdeligt, älskar er. Frida, Lisa och Kajsa, jag kunde inte ha fått bättre
småsystrar, älskar er

44
Johnny, tack för att du finns där, för inhandlad risen när blodsockernivån var låg, för att du
inte klagat när jag spenderat aldeles för många timmar på jobbet, och sköt all markservice
medans jag skrivit min avhandling, för att du finns där när jag behöver dig som mest och
accepterar mig för den jag är, älskar dig
And remember it can be hard to differentiate between people…
“Magicians and scientists are, on the face of it, poles apart. Certainly, a group of people who
often dress strangely, live in a world of their own, speak a specialized language and frequently
make statements that appear to be in flagrant breach of common sense have nothing in
common with a group of people who often dress strangely, speak a specialized language, live
in ... er ...”
Terry Pratchet, Ian Stewart and Jack Cohen
The science of Discworld

45
References
1. Burnett, G. and E.P. Kennedy, The enzymatic phosphorylation of proteins. J Biol
Chem, 1954. 211(2): p. 969-80.
2. Levene, P.A. and C.L. Alsberg, The cleavage products of Vitellin. J Biol Chem, 1906.
2: p. 127-133.
3. Eckhart, W., M.A. Hutchinson, and T. Hunter, An activity phosphorylating tyrosine in
polyoma T antigen immunoprecipitates. Cell, 1979. 18(4): p. 925-33.
4. Hunter, T. and B.M. Sefton, Transforming gene product of Rous sarcoma virus
phosphorylates tyrosine. Proc Natl Acad Sci U S A, 1980. 77(3): p. 1311-5.
5. Manning, G., et al., The protein kinase complement of the human genome. Science,
2002. 298(5600): p. 1912-34.
6. Robinson, D.R., Y.M. Wu, and S.F. Lin, The protein tyrosine kinase family of the
human genome. Oncogene, 2000. 19(49): p. 5548-57.
7. Schlessinger, J., Cell signaling by receptor tyrosine kinases. Cell, 2000. 103(2): p.
211-25.
8. Van Obberghen, E., Signalling through the insulin receptor and the insulin-like growth
factor-I receptor. Diabetologia, 1994. 37 Suppl 2: p. S125-34.
9. Luo, R.Z., et al., Quaternary structure of the insulin-insulin receptor complex. Science,
1999. 285(5430): p. 1077-80.
10. Lemmon, M.A., et al., Kit receptor dimerization is driven by bivalent binding of stem
cell factor. J Biol Chem, 1997. 272(10): p. 6311-7.
11. Blume-Jensen, P. and T. Hunter, Oncogenic kinase signalling. Nature, 2001.
411(6835): p. 355-65.
12. Lengyel, E., K. Sawada, and R. Salgia, Tyrosine kinase mutations in human cancer.
Curr Mol Med, 2007. 7(1): p. 77-84.
13. Eswarakumar, V.P., I. Lax, and J. Schlessinger, Cellular signaling by fibroblast
growth factor receptors. Cytokine Growth Factor Rev, 2005. 16(2): p. 139-49.
14. Guasch, G., et al., FGFR1 is fused to the centrosome-associated protein CEP110 in the
8p12 stem cell myeloproliferative disorder with t(8;9)(p12;q33). Blood, 2000. 95(5): p.
1788-96.
15. Popovici, C., et al., Fibroblast growth factor receptor 1 is fused to FIM in stem-cell
myeloproliferative disorder with t(8;13). Proc Natl Acad Sci U S A, 1998. 95(10): p.
5712-7.
16. Yoshimura, N., et al., The expression and localization of fibroblast growth factor-1
(FGF-1) and FGF receptor-1 (FGFR-1) in human breast cancer. Clin Immunol
Immunopathol, 1998. 89(1): p. 28-34.
17. Onose, H., et al., Overexpression of fibroblast growth factor receptor 3 in a human
thyroid carcinoma cell line results in overgrowth of the confluent cultures. Eur J
Endocrinol, 1999. 140(2): p. 169-73.
18. Cappellen, D., et al., Frequent activating mutations of FGFR3 in human bladder and
cervix carcinomas. Nat Genet, 1999. 23(1): p. 18-20.
19. Naski, M.C., et al., Graded activation of fibroblast growth factor receptor 3 by
mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet, 1996.
13(2): p. 233-7.

46
20. Knowles, M.A., Role of FGFR3 in urothelial cell carcinoma: biomarker and potential
therapeutic target. World J Urol, 2007. 25(6): p. 581-93.
21. Webster, M.K. and D.J. Donoghue, Constitutive activation of fibroblast growth factor
receptor 3 by the transmembrane domain point mutation found in achondroplasia.
EMBO J, 1996. 15(3): p. 520-7.
22. Rousseau, F., et al., Mutations in the gene encoding fibroblast growth factor receptor-
3 in achondroplasia. Nature, 1994. 371(6494): p. 252-4.
23. Vogels, A. and J.P. Fryns, Pfeiffer syndrome. Orphanet J Rare Dis, 2006. 1: p. 19.
24. Schell, U., et al., Mutations in FGFR1 and FGFR2 cause familial and sporadic
Pfeiffer syndrome. Hum Mol Genet, 1995. 4(3): p. 323-8.
25. Cornejo-Roldan, L.R., E. Roessler, and M. Muenke, Analysis of the mutational
spectrum of the FGFR2 gene in Pfeiffer syndrome. Hum Genet, 1999. 104(5): p. 425-
31.
26. Shiota, M., et al., Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase
similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene, 1994. 9(6): p.
1567-74.
27. Morris, S.W., et al., Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM,
in non-Hodgkin's lymphoma. Science, 1994. 263(5151): p. 1281-4.
28. Morris, S.W., et al., ALK, the chromosome 2 gene locus altered by the t(2;5) in non-
Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly
related to leukocyte tyrosine kinase (LTK). Oncogene, 1997. 14(18): p. 2175-88.
29. Iwahara, T., et al., Molecular characterization of ALK, a receptor tyrosine kinase
expressed specifically in the nervous system. Oncogene, 1997. 14(4): p. 439-49.
30. Loren, C.E., et al., Identification and characterization of DAlk: a novel Drosophila
melanogaster RTK which drives ERK activation in vivo. Genes Cells, 2001. 6(6): p.
531-44.
31. Liao, E.H., et al., An SCF-like ubiquitin ligase complex that controls presynaptic
differentiation. Nature, 2004. 430(6997): p. 345-50.
32. Reiner, D.J., et al., C. elegans anaplastic lymphoma kinase ortholog SCD-2 controls
dauer formation by modulating TGF-beta signaling. Curr Biol, 2008. 18(15): p. 1101-
9.
33. Bernards, A. and S.M. de la Monte, The ltk receptor tyrosine kinase is expressed in
pre-B lymphocytes and cerebral neurons and uses a non-AUG translational initiator.
EMBO J, 1990. 9(7): p. 2279-87.
34. Appella, E., I.T. Weber, and F. Blasi, Structure and function of epidermal growth
factor-like regions in proteins. FEBS Lett, 1988. 231(1): p. 1-4.
35. Russell, D.W., M.S. Brown, and J.L. Goldstein, Different combinations of cysteine-
rich repeats mediate binding of low density lipoprotein receptor to two different
proteins. J Biol Chem, 1989. 264(36): p. 21682-8.
36. Hussain, M.M., Structural, biochemical and signaling properties of the low-density
lipoprotein receptor gene family. Front Biosci, 2001. 6: p. D417-28.
37. Loren, C.E., et al., A crucial role for the Anaplastic lymphoma kinase receptor tyrosine
kinase in gut development in Drosophila melanogaster. EMBO Rep, 2003. 4(8): p.
781-6.
38. Finn, R.D., et al., Pfam: clans, web tools and services. Nucleic Acids Res, 2006.
34(Database issue): p. D247-51.

47
39. Schultz, J., et al., SMART, a simple modular architecture research tool: identification
of signaling domains. Proc Natl Acad Sci U S A, 1998. 95(11): p. 5857-64.
40. Hulo, N., et al., The 20 years of PROSITE. Nucleic Acids Res, 2008. 36(Database
issue): p. D245-9.
41. Beckmann, G. and P. Bork, An adhesive domain detected in functionally diverse
receptors. Trends Biochem Sci, 1993. 18(2): p. 40-1.
42. Bai, R.Y., et al., Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic
lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-
gamma to mediate its mitogenicity. Mol Cell Biol, 1998. 18(12): p. 6951-61.
43. Englund, C., et al., Jeb signals through the Alk receptor tyrosine kinase to drive
visceral muscle fusion. Nature, 2003. 425(6957): p. 512-6.
44. Lee, H.H., et al., Jelly belly protein activates the receptor tyrosine kinase Alk to specify
visceral muscle pioneers. Nature, 2003. 425(6957): p. 507-12.
45. Klapper, R., et al., The formation of syncytia within the visceral musculature of the
Drosophila midgut is dependent on duf, sns and mbc. Mech Dev, 2002. 110(1-2): p.
85-96.
46. Martin, B.S., et al., A distinct set of founders and fusion-competent myoblasts make
visceral muscles in the Drosophila embryo. Development, 2001. 128(17): p. 3331-8.
47. Weiss, J.B., et al., Jelly belly: a Drosophila LDL receptor repeat-containing signal
required for mesoderm migration and differentiation. Cell, 2001. 107(3): p. 387-98.
48. Varshney, G.K. and R.H. Palmer, The bHLH transcription factor Hand is regulated by
Alk in the Drosophila embryonic gut. Biochem Biophys Res Commun, 2006. 351(4):
p. 839-46.
49. Shirinian, M., et al., Drosophila Anaplastic Lymphoma Kinase regulates Dpp
signalling in the developing embryonic gut. Differentiation, 2007. 75(5): p. 418-26.
50. Bazigou, E., et al., Anterograde Jelly belly and Alk receptor tyrosine kinase signaling
mediates retinal axon targeting in Drosophila. Cell, 2007. 128(5): p. 961-75.
51. Ishihara, T., et al., HEN-1, a secretory protein with an LDL receptor motif, regulates
sensory integration and learning in Caenorhabditis elegans. Cell, 2002. 109(5): p.
639-49.
52. Hurley, S.P., et al., Anaplastic lymphoma kinase is dynamically expressed on subsets
of motor neurons and in the peripheral nervous system. J Comp Neurol, 2006. 495(2):
p. 202-12.
53. Pulford, K., et al., Detection of anaplastic lymphoma kinase (ALK) and nucleolar
protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the
monoclonal antibody ALK1. Blood, 1997. 89(4): p. 1394-404.
54. Souttou, B., et al., Activation of anaplastic lymphoma kinase receptor tyrosine kinase
induces neuronal differentiation through the mitogen-activated protein kinase
pathway. J Biol Chem, 2001. 276(12): p. 9526-31.
55. Motegi, A., et al., ALK receptor tyrosine kinase promotes cell growth and neurite
outgrowth. J Cell Sci, 2004. 117(Pt 15): p. 3319-29.
56. Moog-Lutz, C., et al., Activation and inhibition of anaplastic lymphoma kinase
receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of
pleiotrophin. J Biol Chem, 2005. 280(28): p. 26039-48.

48
57. Degoutin, J., M. Vigny, and J.Y. Gouzi, ALK activation induces Shc and FRS2
recruitment: Signaling and phenotypic outcomes in PC12 cells differentiation. FEBS
Lett, 2007. 581(4): p. 727-34.
58. Piccinini, G., et al., A ligand-inducible epidermal growth factor receptor/anaplastic
lymphoma kinase chimera promotes mitogenesis and transforming properties in 3T3
cells. J Biol Chem, 2002. 277(25): p. 22231-9.
59. Merenmies, J. and H. Rauvala, Molecular cloning of the 18-kDa growth-associated
protein of developing brain. J Biol Chem, 1990. 265(28): p. 16721-4.
60. Tezuka, K., et al., Isolation of mouse and human cDNA clones encoding a protein
expressed specifically in osteoblasts and brain tissues. Biochem Biophys Res
Commun, 1990. 173(1): p. 246-51.
61. Courty, J., et al., Mitogenic properties of a new endothelial cell growth factor related
to pleiotrophin. Biochem Biophys Res Commun, 1991. 180(1): p. 145-51.
62. Kovesdi, I., et al., Heparin-binding neurotrophic factor (HBNF) and MK, members of
a new family of homologous, developmentally regulated proteins. Biochem Biophys
Res Commun, 1990. 172(2): p. 850-4.
63. Vigny, M., et al., Identification of a new heparin-binding protein localized within chick
basement membranes. Eur J Biochem, 1989. 186(3): p. 733-40.
64. Stoica, G.E., et al., Identification of anaplastic lymphoma kinase as a receptor for the
growth factor pleiotrophin. J Biol Chem, 2001. 276(20): p. 16772-9.
65. Stoica, G.E., et al., Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a
growth factor for different cell types. J Biol Chem, 2002. 277(39): p. 35990-8.
66. Muramatsu, T., Midkine and pleiotrophin: two related proteins involved in
development, survival, inflammation and tumorigenesis. J Biochem, 2002. 132(3): p.
359-71.
67. Kadomatsu, K. and T. Muramatsu, Midkine and pleiotrophin in neural development
and cancer. Cancer Lett, 2004. 204(2): p. 127-43.
68. Meng, K., et al., Pleiotrophin signals increased tyrosine phosphorylation of beta beta-
catenin through inactivation of the intrinsic catalytic activity of the receptor-type
protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A, 2000. 97(6): p.
2603-8.
69. Maeda, N., et al., 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-
like protein-tyrosine phosphatase zeta/RPTPbeta, binds pleiotrophin/heparin-binding
growth-associated molecule (HB-GAM). J Biol Chem, 1996. 271(35): p. 21446-52.
70. Nakanishi, T., et al., Expression of syndecan-1 and -3 during embryogenesis of the
central nervous system in relation to binding with midkine. J Biochem, 1997. 121(2):
p. 197-205.
71. Raulo, E., et al., Isolation of a neuronal cell surface receptor of heparin binding
growth-associated molecule (HB-GAM). Identification as N-syndecan (syndecan-3). J
Biol Chem, 1994. 269(17): p. 12999-3004.
72. Muramatsu, H., et al., LDL receptor-related protein as a component of the midkine
receptor. Biochem Biophys Res Commun, 2000. 270(3): p. 936-41.
73. Muramatsu, H., et al., alpha4beta1- and alpha6beta1-integrins are functional
receptors for midkine, a heparin-binding growth factor. J Cell Sci, 2004. 117(Pt 22): p.
5405-15.

49
74. Bowden, E.T., G.E. Stoica, and A. Wellstein, Anti-apoptotic signaling of pleiotrophin
through its receptor, anaplastic lymphoma kinase. J Biol Chem, 2002. 277(39): p.
35862-8.
75. Powers, C., et al., Pleiotrophin signaling through anaplastic lymphoma kinase is rate-
limiting for glioblastoma growth. J Biol Chem, 2002. 277(16): p. 14153-8.
76. Kuo, A.H., et al., Recruitment of insulin receptor substrate-1 and activation of NF-
kappaB essential for midkine growth signaling through anaplastic lymphoma kinase.
Oncogene, 2007. 26(6): p. 859-69.
77. Lu, K.V., et al., Differential induction of glioblastoma migration and growth by two
forms of pleiotrophin. J Biol Chem, 2005. 280(29): p. 26953-64.
78. Mathivet, T., P. Mazot, and M. Vigny, In contrast to agonist monoclonal antibodies,
both C-terminal truncated form and full length form of Pleiotrophin failed to activate
vertebrate ALK (anaplastic lymphoma kinase)? Cell Signal, 2007. 19(12): p. 2434-43.
79. Mourali, J., et al., Anaplastic lymphoma kinase is a dependence receptor whose
proapoptotic functions are activated by caspase cleavage. Mol Cell Biol, 2006.
26(16): p. 6209-22.
80. Dirks, W.G., et al., Expression and functional analysis of the anaplastic lymphoma
kinase (ALK) gene in tumor cell lines. Int J Cancer, 2002. 100(1): p. 49-56.
81. Perez-Pinera, P., et al., Anaplastic lymphoma kinase is activated through the
pleiotrophin/receptor protein-tyrosine phosphatase beta/zeta signaling pathway: an
alternative mechanism of receptor tyrosine kinase activation. J Biol Chem, 2007.
282(39): p. 28683-90.
82. Muramatsu, H., et al., Localization of heparin-binding, neurite outgrowth and
antigenic regions in midkine molecule. Biochem Biophys Res Commun, 1994. 203(2):
p. 1131-9.
83. Fabri, L., et al., Characterization of bovine heparin-binding neurotrophic factor
(HBNF): assignment of disulfide bonds. Biochem Int, 1992. 28(1): p. 1-9.
84. Kilpelainen, I., et al., Heparin-binding growth-associated molecule contains two
heparin-binding beta -sheet domains that are homologous to the thrombospondin type
I repeat. J Biol Chem, 2000. 275(18): p. 13564-70.
85. Englund, C., et al., Miple1 and miple2 encode a family of MK/PTN homologues in
Drosophila melanogaster. Dev Genes Evol, 2006. 216(1): p. 10-8.
86. Morris, S.W., et al., Alk+ CD30+ lymphomas: a distinct molecular genetic subtype of
non-Hodgkin's lymphoma. Br J Haematol, 2001. 113(2): p. 275-95.
87. Pulford, K., et al., The emerging normal and disease-related roles of anaplastic
lymphoma kinase. Cell Mol Life Sci, 2004. 61(23): p. 2939-53.
88. Pulford, K., S.W. Morris, and F. Turturro, Anaplastic lymphoma kinase proteins in
growth control and cancer. J Cell Physiol, 2004. 199(3): p. 330-58.
89. Allouche, M., ALK is a novel dependence receptor: potential implications in
development and cancer. Cell Cycle, 2007. 6(13): p. 1533-8.
90. Duyster, J., R.Y. Bai, and S.W. Morris, Translocations involving anaplastic lymphoma
kinase (ALK). Oncogene, 2001. 20(40): p. 5623-37.
91. Bilsland, J.G., et al., Behavioral and neurochemical alterations in mice deficient in
anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications.
Neuropsychopharmacology, 2008. 33(3): p. 685-700.

50
92. Nakamura, E., et al., Disruption of the midkine gene (Mdk) resulted in altered
expression of a calcium binding protein in the hippocampus of infant mice and their
abnormal behaviour. Genes Cells, 1998. 3(12): p. 811-22.
93. Amet, L.E., et al., Enhanced hippocampal long-term potentiation in mice lacking
heparin-binding growth-associated molecule. Mol Cell Neurosci, 2001. 17(6): p. 1014-
24.
94. Pavlov, I., et al., Role of heparin-binding growth-associated molecule (HB-GAM) in
hippocampal LTP and spatial learning revealed by studies on overexpressing and
knockout mice. Mol Cell Neurosci, 2002. 20(2): p. 330-42.
95. Matsumoto, K., et al., Localization of pleiotrophin and midkine in the postnatal
developing cerebellum. Neurosci Lett, 1994. 178(2): p. 216-20.
96. Vanderwinden, J.M., et al., Cellular distribution of the new growth factor pleiotrophin
(HB-GAM) mRNA in developing and adult rat tissues. Anat Embryol (Berl), 1992.
186(4): p. 387-406.
97. Fan, Q.W., T. Muramatsu, and K. Kadomatsu, Distinct expression of midkine and
pleiotrophin in the spinal cord and placental tissues during early mouse development.
Dev Growth Differ, 2000. 42(2): p. 113-9.
98. Mitsiadis, T.A., et al., Expression of the heparin-binding cytokines, midkine (MK) and
HB-GAM (pleiotrophin) is associated with epithelial-mesenchymal interactions during
fetal development and organogenesis. Development, 1995. 121(1): p. 37-51.
99. Herradon, G., et al., Midkine regulates pleiotrophin organ-specific gene expression:
evidence for transcriptional regulation and functional redundancy within the
pleiotrophin/midkine developmental gene family. Biochem Biophys Res Commun,
2005. 333(3): p. 714-21.
100. Muramatsu, H., et al., Female infertility in mice deficient in midkine and pleiotrophin,
which form a distinct family of growth factors. Genes Cells, 2006. 11(12): p. 1405-17.
101. Zou, P., et al., Mice doubly deficient in the midkine and pleiotrophin genes exhibit
deficits in the expression of beta-tectorin gene and in auditory response. Lab Invest,
2006. 86(7): p. 645-53.
102. Yamada, S., et al., Expression of a chimeric CSF1R-LTK mediates ligand-dependent
neurite outgrowth. Neuroreport, 2008. 19(17): p. 1733-8.
103. Lopes, S.S., et al., Leukocyte tyrosine kinase functions in pigment cell development.
PLoS Genet, 2008. 4(3): p. e1000026.
104. Griffin, C.A., et al., Recurrent involvement of 2p23 in inflammatory myofibroblastic
tumors. Cancer Res, 1999. 59(12): p. 2776-80.
105. Soda, M., et al., Identification of the transforming EML4-ALK fusion gene in non-
small-cell lung cancer. Nature, 2007. 448(7153): p. 561-6.
106. Arber, D.A., L.H. Sun, and L.M. Weiss, Detection of the t(2;5)(p23;q35) chromosomal
translocation in large B-cell lymphomas other than anaplastic large cell lymphoma.
Hum Pathol, 1996. 27(6): p. 590-4.
107. Cessna, M.H., et al., Expression of ALK1 and p80 in inflammatory myofibroblastic
tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol, 2002. 15(9): p.
931-8.
108. Mosse, Y.P., et al., Identification of ALK as a major familial neuroblastoma
predisposition gene. Nature, 2008. 455(7215): p. 930-5.

51
109. George, R.E., et al., Activating mutations in ALK provide a therapeutic target in
neuroblastoma. Nature, 2008. 455(7215): p. 975-8.
110. Janoueix-Lerosey, I., et al., Somatic and germline activating mutations of the ALK
kinase receptor in neuroblastoma. Nature, 2008. 455(7215): p. 967-70.
111. Chen, Y., et al., Oncogenic mutations of ALK kinase in neuroblastoma. Nature, 2008.
455(7215): p. 971-4.
112. Caren, H., et al., High incidence of DNA mutations and gene amplifications of the ALK
gene in advanced sporadic neuroblastoma tumours. Biochem J, 2008. 416(2): p. 153-
9.
113. Kunugi, H., et al., Possible association between nonsynonymous polymorphisms of the
anaplastic lymphoma kinase (ALK) gene and schizophrenia in a Japanese population.
J Neural Transm, 2006. 113(10): p. 1569-73.
114. Shimizu, E., et al., Two clusters of serum midkine levels in drug-naive patients with
schizophrenia. Neurosci Lett, 2003. 344(2): p. 95-8.
115. Stein, H., et al., The expression of the Hodgkin's disease associated antigen Ki-1 in
reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and
histiocytic malignancies are derived from activated lymphoid cells. Blood, 1985.
66(4): p. 848-58.
116. Jaffa, E.S., Harris, N. L., Stein, H., Vardiman, J. W., ed. World Health Organization
Classification of Tumours: Tumours of Haematopoietic and lymphoid tissues. 2001,
IARC Press: Lyon.
117. Hagberg, H., et al., Vårdprogram for Lymfom 2007. 2007, Reklam & Katalogtryck,
Uppsala, Sweden. p. 34.
118. Brock, N., The history of the oxazaphosphorine cytostatics. Cancer, 1996. 78(3): p.
542-7.
119. Momparler, R.L., et al., Effect of adriamycin on DNA, RNA, and protein synthesis in
cell-free systems and intact cells. Cancer Res, 1976. 36(8): p. 2891-5.
120. Sieber, S.M., J.A. Mead, and R.H. Adamson, Pharmacology of antitumor agents from
higher plants. Cancer Treat Rep, 1976. 60(8): p. 1127-39.
121. Tissing, W.J., et al., Molecular determinants of glucocorticoid sensitivity and
resistance in acute lymphoblastic leukemia. Leukemia, 2003. 17(1): p. 17-25.
122. Shiota, M., et al., Anaplastic large cell lymphomas expressing the novel chimeric
protein p80NPM/ALK: a distinct clinicopathologic entity. Blood, 1995. 86(5): p. 1954-
60.
123. Nakamura, S., et al., Anaplastic large cell lymphoma: a distinct molecular pathologic
entity: a reappraisal with special reference to p80(NPM/ALK) expression. Am J Surg
Pathol, 1997. 21(12): p. 1420-32.
124. Li, C., et al., Prognostic significance of NPM-ALK fusion transcript overexpression in
ALK-positive anaplastic large-cell lymphoma. Mod Pathol, 2007. 20(6): p. 648-55.
125. Gascoyne, R.D., et al., Prognostic significance of anaplastic lymphoma kinase (ALK)
protein expression in adults with anaplastic large cell lymphoma. Blood, 1999. 93(11):
p. 3913-21.
126. ten Berge, R.L., et al., Expression levels of apoptosis-related proteins predict clinical
outcome in anaplastic large cell lymphoma. Blood, 2002. 99(12): p. 4540-6.

52
127. Khoury, J.D., et al., Differential expression and clinical significance of tyrosine-
phosphorylated STAT3 in ALK+ and ALK- anaplastic large cell lymphoma. Clin
Cancer Res, 2003. 9(10 Pt 1): p. 3692-9.
128. Schlette, E.J., et al., Survivin expression predicts poorer prognosis in anaplastic large-
cell lymphoma. J Clin Oncol, 2004. 22(9): p. 1682-8.
129. Rassidakis, G.Z., et al., Prognostic significance of MUC-1 expression in systemic
anaplastic large cell lymphoma. Clin Cancer Res, 2003. 9(6): p. 2213-20.
130. Hattrup, C.L. and S.J. Gendler, Structure and function of the cell surface (tethered)
mucins. Annu Rev Physiol, 2008. 70: p. 431-57.
131. Trumper, L., et al., Detection of the t(2;5)-associated NPM/ALK fusion cDNA in
peripheral blood cells of healthy individuals. Br J Haematol, 1998. 103(4): p. 1138-44.
132. Maes, B., et al., The NPM-ALK and the ATIC-ALK fusion genes can be detected in
non-neoplastic cells. Am J Pathol, 2001. 158(6): p. 2185-93.
133. Kuefer, M.U., et al., Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid
malignancy in mice. Blood, 1997. 90(8): p. 2901-10.
134. Fujimoto, J., et al., Characterization of the transforming activity of p80, a
hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal
translocation t(2;5). Proc Natl Acad Sci U S A, 1996. 93(9): p. 4181-6.
135. Wellmann, A., et al., The activated anaplastic lymphoma kinase increases cellular
proliferation and oncogene up-regulation in rat 1a fibroblasts. FASEB J, 1997.
11(12): p. 965-72.
136. Chiarle, R., et al., NPM-ALK transgenic mice spontaneously develop T-cell lymphomas
and plasma cell tumors. Blood, 2003. 101(5): p. 1919-27.
137. Turner, S.D. and D.R. Alexander, What have we learnt from mouse models of NPM-
ALK-induced lymphomagenesis? Leukemia, 2005. 19(7): p. 1128-34.
138. Fischer, P., et al., A Ki-1 (CD30)-positive human cell line (Karpas 299) established
from a high-grade non-Hodgkin's lymphoma, showing a 2;5 translocation and
rearrangement of the T-cell receptor beta-chain gene. Blood, 1988. 72(1): p. 234-40.
139. Le Beau, M.M., et al., The t(2;5)(p23;q35): a recurring chromosomal abnormality in
Ki-1-positive anaplastic large cell lymphoma. Leukemia, 1989. 3(12): p. 866-70.
140. Rimokh, R., et al., A translocation involving a specific breakpoint (q35) on
chromosome 5 is characteristic of anaplastic large cell lymphoma ('Ki-1 lymphoma').
Br J Haematol, 1989. 71(1): p. 31-6.
141. Okuwaki, M., The structure and functions of NPM1/Nucleophsmin/B23, a
multifunctional nucleolar acidic protein. J Biochem, 2008. 143(4): p. 441-8.
142. Bischof, D., et al., Role of the nucleophosmin (NPM) portion of the non-Hodgkin's
lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in
oncogenesis. Mol Cell Biol, 1997. 17(4): p. 2312-25.
143. Mason, D.Y., et al., Nucleolar localization of the nucleophosmin-anaplastic lymphoma
kinase is not required for malignant transformation. Cancer Res, 1998. 58(5): p. 1057-
62.
144. Cools, J., et al., Identification of novel fusion partners of ALK, the anaplastic
lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory
myofibroblastic tumor. Genes Chromosomes Cancer, 2002. 34(4): p. 354-62.

53
145. Hernandez, L., et al., TRK-fused gene (TFG) is a new partner of ALK in anaplastic
large cell lymphoma producing two structurally different TFG-ALK translocations.
Blood, 1999. 94(9): p. 3265-8.
146. Hernandez, L., et al., Diversity of genomic breakpoints in TFG-ALK translocations in
anaplastic large cell lymphomas: identification of a new TFG-ALK(XL) chimeric gene
with transforming activity. Am J Pathol, 2002. 160(4): p. 1487-94.
147. Tort, F., et al., Molecular characterization of a new ALK translocation involving
moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab Invest, 2001. 81(3): p.
419-26.
148. Lamant, L., et al., A new fusion gene TPM3-ALK in anaplastic large cell lymphoma
created by a (1;2)(q25;p23) translocation. Blood, 1999. 93(9): p. 3088-95.
149. Siebert, R., et al., Complex variant translocation t(1;2) with TPM3-ALK fusion due to
cryptic ALK gene rearrangement in anaplastic large-cell lymphoma. Blood, 1999.
94(10): p. 3614-7.
150. Meech, S.J., et al., Unusual childhood extramedullary hematologic malignancy with
natural killer cell properties that contains tropomyosin 4--anaplastic lymphoma kinase
gene fusion. Blood, 2001. 98(4): p. 1209-16.
151. Ma, Z., et al., Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive
anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an
enzyme involved in purine nucleotide biosynthesis. Blood, 2000. 95(6): p. 2144-9.
152. Trinei, M., et al., A new variant anaplastic lymphoma kinase (ALK)-fusion protein
(ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res,
2000. 60(4): p. 793-8.
153. Lamant, L., et al., Non-muscle myosin heavy chain (MYH9): a new partner fused to
ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer, 2003. 37(4): p.
427-32.
154. Touriol, C., et al., Further demonstration of the diversity of chromosomal changes
involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to
CLTCL (clathrin chain polypeptide-like). Blood, 2000. 95(10): p. 3204-7.
155. Beardsley, G.P., et al., Structure and functional relationships in human pur H. Adv
Exp Med Biol, 1998. 431: p. 221-6.
156. Young, A., Structural insights into the clathrin coat. Semin Cell Dev Biol, 2007.
18(4): p. 448-58.
157. Chiarle, R., et al., The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat
Rev Cancer, 2008. 8(1): p. 11-23.
158. Carpenter, G. and Q. Ji, Phospholipase C-gamma as a signal-transducing element.
Exp Cell Res, 1999. 253(1): p. 15-24.
159. Wymann, M.P. and L. Pirola, Structure and function of phosphoinositide 3-kinases.
Biochim Biophys Acta, 1998. 1436(1-2): p. 127-50.
160. Bai, R.Y., et al., Nucleophosmin-anaplastic lymphoma kinase associated with
anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt
antiapoptotic signaling pathway. Blood, 2000. 96(13): p. 4319-27.
161. Polgar, D., et al., Truncated ALK derived from chromosomal translocation
t(2;5)(p23;q35) binds to the SH3 domain of p85-PI3K. Mutat Res, 2005. 570(1): p. 9-
15.

54
162. Slupianek, A., et al., Role of phosphatidylinositol 3-kinase-Akt pathway in
nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res,
2001. 61(5): p. 2194-9.
163. Huang, H. and D.J. Tindall, Dynamic FoxO transcription factors. J Cell Sci, 2007.
120(Pt 15): p. 2479-87.
164. Gu, T.L., et al., NPM-ALK fusion kinase of anaplastic large-cell lymphoma regulates
survival and proliferative signaling through modulation of FOXO3a. Blood, 2004.
103(12): p. 4622-9.
165. Lawrence, B., et al., TPM3-ALK and TPM4-ALK oncogenes in inflammatory
myofibroblastic tumors. Am J Pathol, 2000. 157(2): p. 377-84.
166. Patel, A.S., et al., RANBP2 and CLTC are involved in ALK rearrangements in
inflammatory myofibroblastic tumors. Cancer Genet Cytogenet, 2007. 176(2): p. 107-
14.
167. Bridge, J.A., et al., Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in
inflammatory myofibroblastic tumor. Am J Pathol, 2001. 159(2): p. 411-5.
168. Panagopoulos, I., et al., Fusion of the SEC31L1 and ALK genes in an inflammatory
myofibroblastic tumor. Int J Cancer, 2006. 118(5): p. 1181-6.
169. Ma, Z., et al., Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in
inflammatory myofibroblastic tumor. Genes Chromosomes Cancer, 2003. 37(1): p. 98-
105.
170. Debelenko, L.V., et al., Identification of CARS-ALK fusion in primary and metastatic
lesions of an inflammatory myofibroblastic tumor. Lab Invest, 2003. 83(9): p. 1255-65.
171. Adam, P., et al., A case of a diffuse large B-cell lymphoma of plasmablastic type
associated with the t(2;5)(p23;q35) chromosome translocation. Am J Surg Pathol,
2003. 27(11): p. 1473-6.
172. De Paepe, P., et al., ALK activation by the CLTC-ALK fusion is a recurrent event in
large B-cell lymphoma. Blood, 2003. 102(7): p. 2638-41.
173. Abukhdeir, A.M. and B.H. Park, P21 and p27: roles in carcinogenesis and drug
resistance. Expert Rev Mol Med, 2008. 10: p. e19.
174. Rassidakis, G.Z., et al., Inhibition of Akt increases p27Kip1 levels and induces cell
cycle arrest in anaplastic large cell lymphoma. Blood, 2005. 105(2): p. 827-9.
175. Vega, F., et al., Activation of mammalian target of rapamycin signaling pathway
contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic
large cell lymphoma. Cancer Res, 2006. 66(13): p. 6589-97.
176. Marzec, M., et al., Oncogenic tyrosine kinase NPM/ALK induces activation of the
rapamycin-sensitive mTOR signaling pathway. Oncogene, 2007. 26(38): p. 5606-14.
177. McKay, M.M. and D.K. Morrison, Integrating signals from RTKs to ERK/MAPK.
Oncogene, 2007. 26(22): p. 3113-21.
178. Marzec, M., et al., Oncogenic tyrosine kinase NPM/ALK induces activation of the
MEK/ERK signaling pathway independently of c-Raf. Oncogene, 2007. 26(6): p. 813-
21.
179. Zamo, A., et al., Anaplastic lymphoma kinase (ALK) activates Stat3 and protects
hematopoietic cells from cell death. Oncogene, 2002. 21(7): p. 1038-47.
180. Zhang, Q., et al., Multilevel dysregulation of STAT3 activation in anaplastic lymphoma
kinase-positive T/null-cell lymphoma. J Immunol, 2002. 168(1): p. 466-74.

55
181. Amin, H.M., et al., Selective inhibition of STAT3 induces apoptosis and G(1) cell cycle
arrest in ALK-positive anaplastic large cell lymphoma. Oncogene, 2004. 23(32): p.
5426-34.
182. Amin, H.M., et al., Inhibition of JAK3 induces apoptosis and decreases anaplastic
lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene, 2003. 22(35):
p. 5399-407.
183. Xu, D. and C.K. Qu, Protein tyrosine phosphatases in the JAK/STAT pathway. Front
Biosci, 2008. 13: p. 4925-32.
184. Qiu, L., et al., Autocrine release of interleukin-9 promotes Jak3-dependent survival of
ALK+ anaplastic large-cell lymphoma cells. Blood, 2006. 108(7): p. 2407-15.
185. Marzec, M., et al., Inhibition of ALK enzymatic activity in T-cell lymphoma cells
induces apoptosis and suppresses proliferation and STAT3 phosphorylation
independently of Jak3. Lab Invest, 2005. 85(12): p. 1544-54.
186. Amin, H.M., Q. Lin, and R. Lai, Jak3 contributes to the activation of ALK and Stat3 in
ALK(+) anaplastic large cell lymphoma. Lab Invest, 2006. 86(4): p. 417-9; author
reply 420-1.
187. Lai, R., et al., Jak3 activation is significantly associated with ALK expression in
anaplastic large cell lymphoma. Hum Pathol, 2005. 36(9): p. 939-44.
188. Han, Y., et al., Restoration of shp1 expression by 5-AZA-2'-deoxycytidine is associated
with downregulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell
lymphoma. Leukemia, 2006. 20(9): p. 1602-9.
189. Khoury, J.D., et al., Methylation of SHP1 gene and loss of SHP1 protein expression
are frequent in systemic anaplastic large cell lymphoma. Blood, 2004. 104(5): p. 1580-
1.
190. Nieborowska-Skorska, M., et al., Role of signal transducer and activator of
transcription 5 in nucleophosmin/ anaplastic lymphoma kinase-mediated malignant
transformation of lymphoid cells. Cancer Res, 2001. 61(17): p. 6517-23.
191. Zhang, Q., et al., STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK
and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat
Med, 2007. 13(11): p. 1341-8.
192. Ridley, A.J., Rho family proteins: coordinating cell responses. Trends Cell Biol, 2001.
11(12): p. 471-7.
193. Ambrogio, C., et al., The anaplastic lymphoma kinase controls cell shape and growth
of anaplastic large cell lymphoma through Cdc42 activation. Cancer Res, 2008.
68(21): p. 8899-907.
194. Ambrogio, C., et al., p130Cas mediates the transforming properties of the anaplastic
lymphoma kinase. Blood, 2005. 106(12): p. 3907-16.
195. Voena, C., et al., The tyrosine phosphatase Shp2 interacts with NPM-ALK and
regulates anaplastic lymphoma cell growth and migration. Cancer Res, 2007. 67(9): p.
4278-86.
196. Cussac, D., et al., Nucleophosmin-anaplastic lymphoma kinase of anaplastic large-cell
lymphoma recruits, activates, and uses pp60c-src to mediate its mitogenicity. Blood,
2004. 103(4): p. 1464-71.
197. Armstrong, F., et al., Differential effects of X-ALK fusion proteins on proliferation,
transformation, and invasion properties of NIH3T3 cells. Oncogene, 2004. 23(36): p.
6071-82.

56
198. Armstrong, F., et al., TPM3-ALK expression induces changes in cytoskeleton
organisation and confers higher metastatic capacities than other ALK fusion proteins.
Eur J Cancer, 2007. 43(4): p. 640-6.
199. Gleason, B.C. and J.L. Hornick, Inflammatory myofibroblastic tumours: where are we
now? J Clin Pathol, 2008. 61(4): p. 428-37.
200. Umiker, W.O. and L. Iverson, Postinflammatory tumors of the lung; report of four
cases simulating xanthoma, fibroma, or plasma cell tumor. J Thorac Surg, 1954. 28(1):
p. 55-63.
201. Coffin, C.M., et al., Extrapulmonary inflammatory myofibroblastic tumor
(inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of
84 cases. Am J Surg Pathol, 1995. 19(8): p. 859-72.
202. Coffin, C.M., et al., ALK1 and p80 expression and chromosomal rearrangements
involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol, 2001. 14(6): p.
569-76.
203. Cook, J.R., et al., Anaplastic lymphoma kinase (ALK) expression in the inflammatory
myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol,
2001. 25(11): p. 1364-71.
204. Chun, Y.S., et al., Pediatric inflammatory myofibroblastic tumor: anaplastic
lymphoma kinase (ALK) expression and prognosis. Pediatr Blood Cancer, 2005. 45(6):
p. 796-801.
205. Kamangar, F., G.M. Dores, and W.F. Anderson, Patterns of cancer incidence,
mortality, and prevalence across five continents: defining priorities to reduce cancer
disparities in different geographic regions of the world. J Clin Oncol, 2006. 24(14): p.
2137-50.
206. Shinmura, K., et al., EML4-ALK fusion transcripts, but no NPM-, TPM3-, CLTC-,
ATIC-, or TFG-ALK fusion transcripts, in non-small cell lung carcinomas. Lung
Cancer, 2008. 61(2): p. 163-9.
207. Perner, S., et al., EML4-ALK fusion lung cancer: a rare acquired event. Neoplasia,
2008. 10(3): p. 298-302.
208. Lynch, T.J., et al., Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med,
2004. 350(21): p. 2129-39.
209. Paez, J.G., et al., EGFR mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science, 2004. 304(5676): p. 1497-500.
210. Ward, W.H., et al., Epidermal growth factor receptor tyrosine kinase. Investigation of
catalytic mechanism, structure-based searching and discovery of a potent inhibitor.
Biochem Pharmacol, 1994. 48(4): p. 659-66.
211. Barker, A.J., et al., Studies leading to the identification of ZD1839 (IRESSA): an orally
active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to
the treatment of cancer. Bioorg Med Chem Lett, 2001. 11(14): p. 1911-4.
212. Koivunen, J.P., et al., EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor
in lung cancer. Clin Cancer Res, 2008. 14(13): p. 4275-83.
213. Delsol, G., et al., A new subtype of large B-cell lymphoma expressing the ALK kinase
and lacking the 2; 5 translocation. Blood, 1997. 89(5): p. 1483-90.
214. Gascoyne, R.D., et al., ALK-positive diffuse large B-cell lymphoma is associated with
Clathrin-ALK rearrangements: report of 6 cases. Blood, 2003. 102(7): p. 2568-73.

57
215. Chikatsu, N., et al., ALK+, CD30-, CD20- large B-cell lymphoma containing
anaplastic lymphoma kinase (ALK) fused to clathrin heavy chain gene (CLTC). Mod
Pathol, 2003. 16(8): p. 828-32.
216. Reichard, K.K., R.W. McKenna, and S.H. Kroft, ALK-positive diffuse large B-cell
lymphoma: report of four cases and review of the literature. Mod Pathol, 2007. 20(3):
p. 310-9.
217. Pillay, K., D. Govender, and R. Chetty, ALK protein expression in
rhabdomyosarcomas. Histopathology, 2002. 41(5): p. 461-7.
218. Perez-Pinera, P., et al., Anaplastic lymphoma kinase is expressed in different subtypes
of human breast cancer. Biochem Biophys Res Commun, 2007. 358(2): p. 399-403.
219. Garver, R.I., Jr., et al., Midkine and pleiotrophin expression in normal and malignant
breast tissue. Cancer, 1994. 74(5): p. 1584-90.
220. Riegel, A.T. and A. Wellstein, The potential role of the heparin-binding growth factor
pleiotrophin in breast cancer. Breast Cancer Res Treat, 1994. 31(2-3): p. 309-14.
221. Zhang, N., et al., Human breast cancer growth inhibited in vivo by a dominant
negative pleiotrophin mutant. J Biol Chem, 1997. 272(27): p. 16733-6.
222. Maris, J.M., et al., Neuroblastoma. Lancet, 2007. 369(9579): p. 2106-20.
223. Lamant, L., et al., Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J
Pathol, 2000. 156(5): p. 1711-21.
224. Miyake, I., et al., Activation of anaplastic lymphoma kinase is responsible for
hyperphosphorylation of ShcC in neuroblastoma cell lines. Oncogene, 2002. 21(38): p.
5823-34.
225. Osajima-Hakomori, Y., et al., Biological role of anaplastic lymphoma kinase in
neuroblastoma. Am J Pathol, 2005. 167(1): p. 213-22.
226. Druker, B.J., et al., Effects of a selective inhibitor of the Abl tyrosine kinase on the
growth of Bcr-Abl positive cells. Nat Med, 1996. 2(5): p. 561-6.
227. Shtivelman, E., et al., Fused transcript of abl and bcr genes in chronic myelogenous
leukaemia. Nature, 1985. 315(6020): p. 550-4.
228. Buchdunger, E., et al., Selective inhibition of the platelet-derived growth factor signal
transduction pathway by a protein-tyrosine kinase inhibitor of the 2-
phenylaminopyrimidine class. Proc Natl Acad Sci U S A, 1995. 92(7): p. 2558-62.
229. Buchdunger, E., et al., Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro
signal transduction mediated by c-kit and platelet-derived growth factor receptors. J
Pharmacol Exp Ther, 2000. 295(1): p. 139-45.
230. Hirota, S., et al., Gain-of-function mutations of c-kit in human gastrointestinal stromal
tumors. Science, 1998. 279(5350): p. 577-80.
231. van Oosterom, A.T., et al., Safety and efficacy of imatinib (STI571) in metastatic
gastrointestinal stromal tumours: a phase I study. Lancet, 2001. 358(9291): p. 1421-3.
232. Demetri, G.D., et al., Efficacy and safety of imatinib mesylate in advanced
gastrointestinal stromal tumors. N Engl J Med, 2002. 347(7): p. 472-80.
233. Arora, A. and E.M. Scholar, Role of tyrosine kinase inhibitors in cancer therapy. J
Pharmacol Exp Ther, 2005. 315(3): p. 971-9.
234. McDermott, U., et al., Genomic alterations of anaplastic lymphoma kinase may
sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res, 2008. 68(9): p.
3389-95.

58
235. Galkin, A.V., et al., Identification of NVP-TAE684, a potent, selective, and efficacious
inhibitor of NPM-ALK. Proc Natl Acad Sci U S A, 2007. 104(1): p. 270-5.
236. Zou, H.Y., et al., An orally available small-molecule inhibitor of c-Met, PF-2341066,
exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic
mechanisms. Cancer Res, 2007. 67(9): p. 4408-17.
237. Christensen, J.G., et al., Cytoreductive antitumor activity of PF-2341066, a novel
inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of
anaplastic large-cell lymphoma. Mol Cancer Ther, 2007. 6(12 Pt 1): p. 3314-22.
238. Takahashi, I., et al., UCN-01 and UCN-02, new selective inhibitors of protein kinase
C. II. Purification, physico-chemical properties, structural determination and
biological activities. J Antibiot (Tokyo), 1989. 42(4): p. 571-6.
239. Graves, P.R., et al., The Chk1 protein kinase and the Cdc25C regulatory pathways are
targets of the anticancer agent UCN-01. J Biol Chem, 2000. 275(8): p. 5600-5.
240. Sausville, E.A., et al., Phase I trial of 72-hour continuous infusion UCN-01 in patients
with refractory neoplasms. J Clin Oncol, 2001. 19(8): p. 2319-33.
241. Gunby, R.H., et al., An enzyme-linked immunosorbent assay to screen for inhibitors of
the oncogenic anaplastic lymphoma kinase. Haematologica, 2005. 90(7): p. 988-90.
242. Piva, R., et al., Ablation of oncogenic ALK is a viable therapeutic approach for
anaplastic large-cell lymphomas. Blood, 2006. 107(2): p. 689-97.
243. Bonvini, P., et al., Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel
Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine
phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-
allylamino,17-demethoxygeldanamycin. Cancer Res, 2002. 62(5): p. 1559-66.
244. Pulford, K., et al., Immune response to the ALK oncogenic tyrosine kinase in patients
with anaplastic large-cell lymphoma. Blood, 2000. 96(4): p. 1605-7.
245. Passoni, L., et al., In vivo T-cell immune response against anaplastic lymphoma kinase
in patients with anaplastic large cell lymphomas. Haematologica, 2006. 91(1): p. 48-
55.
246. Chiarle, R., et al., The anaplastic lymphoma kinase is an effective oncoantigen for
lymphoma vaccination. Nat Med, 2008. 14(6): p. 676-80.
247. Melton, K.R., A. Iulianella, and P.A. Trainor, Gene expression and regulation of
hindbrain and spinal cord development. Front Biosci, 2004. 9: p. 117-38.
248. Abrescia, C., et al., Drosophila RET contains an active tyrosine kinase and elicits
neurotrophic activities in mammalian cells. FEBS Lett, 2005. 579(17): p. 3789-96.
249. Vernersson, E., et al., Characterization of the expression of the ALK receptor tyrosine
kinase in mice. Gene Expr Patterns, 2006. 6(5): p. 448-61.
250. Sym, M., N. Robinson, and C. Kenyon, MIG-13 positions migrating cells along the
anteroposterior body axis of C. elegans. Cell, 1999. 98(1): p. 25-36.
251. Komori, T., et al., Expression of kin of irregular chiasm-like 3/mKirre in
proprioceptive neurons of the dorsal root ganglia and its interaction with nephrin in
muscle spindles. J Comp Neurol, 2008. 511(1): p. 92-108.
252. Morikawa, Y., et al., Expression of mKirre in the developing sensory pathways: its
close apposition to nephrin-expressing cells. Neuroscience, 2007. 150(4): p. 880-6.
253. Tamura, S., et al., Expression of mKirre, a mammalian homolog of Drosophila kirre,
in the developing and adult mouse brain. Neuroscience, 2005. 133(3): p. 615-24.

59
254. Chapman, D.L., et al., Expression of the T-box family genes, Tbx1-Tbx5, during early
mouse development. Dev Dyn, 1996. 206(4): p. 379-90.
255. Naiche, L.A., et al., T-box genes in vertebrate development. Annu Rev Genet, 2005.
39: p. 219-39.
256. Serizawa, S., et al., A neuronal identity code for the odorant receptor-specific and
activity-dependent axon sorting. Cell, 2006. 127(5): p. 1057-69.
257. Gerke, P., et al., Neuronal expression and interaction with the synaptic protein CASK
suggest a role for Neph1 and Neph2 in synaptogenesis. J Comp Neurol, 2006. 498(4):
p. 466-75.
258. Harrelson, Z., et al., Tbx2 is essential for patterning the atrioventricular canal and for
morphogenesis of the outflow tract during heart development. Development, 2004.
131(20): p. 5041-52.
259. Davenport, T.G., L.A. Jerome-Majewska, and V.E. Papaioannou, Mammary gland,
limb and yolk sac defects in mice lacking Tbx3, the gene mutated in human ulnar
mammary syndrome. Development, 2003. 130(10): p. 2263-73.
260. Sock, E., et al., Idiopathic weight reduction in mice deficient in the high-mobility-
group transcription factor Sox8. Mol Cell Biol, 2001. 21(20): p. 6951-9.
261. Mori, K., Grouping of odorant receptors: odour maps in the mammalian olfactory
bulb. Biochem Soc Trans, 2003. 31(Pt 1): p. 134-6.
262. Lipscomb, B.W., H.B. Treloar, and C.A. Greer, Cell surface carbohydrates reveal
heterogeneity in olfactory receptor cell axons in the mouse. Cell Tissue Res, 2002.
308(1): p. 7-17.
263. Grundy, D. and M. Schemann, Enteric nervous system. Curr Opin Gastroenterol, 2005.
21(2): p. 176-82.
264. Somm, E., et al., Decreased fat mass in interleukin-1 receptor antagonist-deficient
mice: impact on adipogenesis, food intake, and energy expenditure. Diabetes, 2005.
54(12): p. 3503-9.
265. Jell, J., et al., Genetically altered expression of spermidine/spermine N1-
acetyltransferase affects fat metabolism in mice via acetyl-CoA. J Biol Chem, 2007.
282(11): p. 8404-13.
266. Schmidt, K., et al., The high mobility group transcription factor Sox8 is a negative
regulator of osteoblast differentiation. J Cell Biol, 2005. 168(6): p. 899-910.
267. Labrador, J.P., et al., The collagen receptor DDR2 regulates proliferation and its
elimination leads to dwarfism. EMBO Rep, 2001. 2(5): p. 446-52.
268. Gelling, R.W., et al., Deficiency of TNFalpha converting enzyme (TACE/ADAM17)
causes a lean, hypermetabolic phenotype in mice. Endocrinology, 2008. 149(12): p.
6053-64.
269. Trumpp, A., et al., c-Myc regulates mammalian body size by controlling cell number
but not cell size. Nature, 2001. 414(6865): p. 768-73.