integrative modeling of transcriptional regulation in response to antirheumatic therapy
TRANSCRIPT

BioMed CentralBMC Bioinformatics
ss
Open AcceMethodology articleIntegrative modeling of transcriptional regulation in response to antirheumatic therapyMichael Hecker*1, Robert Hermann Goertsches2, Robby Engelmann2, Hans-Juergen Thiesen2 and Reinhard Guthke1Address: 1Leibniz Institute for Natural Product Research and Infection Biology – Hans-Knoell-Institute, Beutenbergstr. 11a, D-07745 Jena, Germany and 2University of Rostock, Institute of Immunology, Schillingallee 70, D-18055 Rostock, Germany
Email: Michael Hecker* - [email protected]; Robert Hermann Goertsches - [email protected]; Robby Engelmann - [email protected]; Hans-Juergen Thiesen - [email protected]; Reinhard Guthke - [email protected]
* Corresponding author
AbstractBackground: The investigation of gene regulatory networks is an important issue in molecularsystems biology and significant progress has been made by combining different types of biologicaldata. The purpose of this study was to characterize the transcriptional program induced byetanercept therapy in patients with rheumatoid arthritis (RA). Etanercept is known to reducedisease symptoms and progression in RA, but the underlying molecular mechanisms have not beenfully elucidated.
Results: Using a DNA microarray dataset providing genome-wide expression profiles of 19 RApatients within the first week of therapy we identified significant transcriptional changes in 83 genes.Most of these genes are known to control the human body's immune response. A novel algorithmcalled TILAR was then applied to construct a linear network model of the genes' regulatoryinteractions. The inference method derives a model from the data based on the Least AngleRegression while incorporating DNA-binding site information. As a result we obtained a scale-freenetwork that exhibits a self-regulating and highly parallel architecture, and reflects the pleiotropicimmunological role of the therapeutic target TNF-alpha. Moreover, we could show that ourintegrative modeling strategy performs much better than algorithms using gene expression dataalone.
Conclusion: We present TILAR, a method to deduce gene regulatory interactions from geneexpression data by integrating information on transcription factor binding sites. The inferrednetwork uncovers gene regulatory effects in response to etanercept and thus provides usefulhypotheses about the drug's mechanisms of action.
BackgroundThe molecular interactions within a biological system giverise to the function and behavior of that system. In sys-tems biology, one aims to formulate the complex interac-
tions of biological processes by mathematical models. Amajor focus of the field is the uncovering of the dynamicand intertwined nature of gene regulation.
Published: 24 August 2009
BMC Bioinformatics 2009, 10:262 doi:10.1186/1471-2105-10-262
Received: 10 February 2009Accepted: 24 August 2009
This article is available from: http://www.biomedcentral.com/1471-2105/10/262
© 2009 Hecker et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Page 1 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
Gene expression is mainly regulated at the level of mRNAtranscription by proteins called transcription factors(TFs), that specifically bind the DNA at the regulatoryregion of their target genes. A number of collections ofexperimentally defined TF binding sites (TFBS) have beenassembled. The most commonly used is the Transfac data-base, which catalogs eukaryotic TFs and their knownbinding sites [1]. The expression level of a gene usuallydepends on the occupancy states of multiple TFBS. How-ever, gene regulation is much more complex and includesdifferent layers of post-transcriptional control. Theentirety of gene regulatory processes constitutes a networkof genes, regulators, and the regulatory connectionsbetween them – namely a gene regulatory network(GRN). In the past, various modeling approaches havebeen proposed to (partially) reconstruct GRNs fromexperimental data on the basis of different mathematicalconcepts and learning strategies, and distinct degrees ofabstraction [2-4]. A graph is always the basic modelingscheme for a GRN, with nodes symbolizing regulatory ele-ments (e.g. genes and proteins) and edges representing(activatory and inhibitory) relationships between them.Common mathematical formalisms of such a graph areBoolean networks, Bayesian networks, association net-works and systems of equations. Boolean networksassume that genes are simply on or off, and apply Booleanlogic to model dynamic regulatory effects. In contrast,Bayesian networks model gene expression by random var-iables and quantify interactions by conditional probabili-ties. Interactions in association networks are typicallyundirected and derived by analyzing pairs of genes for co-expression e.g. using mutual information as a similaritymeasure. Systems of equations describe each gene'sexpression level as a function of the levels of its putativepredictors. For specific types of functions they could drawon well developed statistical techniques to efficiently fittheir model parameters. However, GRN inference isalways a challenging task because of incomplete knowl-edge of the molecules involved, the combinatorial natureof the problem and the fact, that often available data arelimited and inaccurate. Microarray gene expression dataare typically used to derive rather phenomenological GRNmodels of how the expression level of a gene is influencedby the expression level of other genes, i.e. the model alsoincludes indirect regulatory mechanisms. Obviously, theincorporation of other types of data in addition to geneexpression data (e.g. gene functional annotations,genome sequence data, protein-protein and protein-DNAinteraction data) as well as the integration of prior biolog-ical knowledge (e.g. from scientific literature) supportsthe inference process. Moreover, it is necessary to utilizebiological plausible assumptions considering the networktopology (e.g. structural sparseness). The integration ofdiverse types of biological information and modelingconstraints allows for more accurate GRN models and is a
current challenge in network reconstruction. Bayesian net-works and systems of linear equations have been moststudied for such combined analyses [3-5].
Organizing biological data in network models may helpunderstanding complex diseases such as human autoim-mune diseases [6]. Many studies implicate hundreds ofgenes in the pathogenesis of autoimmune diseases, but westill lack a comprehensive conception of how autoimmu-nity arises. Understanding structure and dynamics ofmolecular networks is critical to unravel such complexdiseases. Network analyses may not only support theinvestigation of autoimmune diseases but also the optimi-zation of their treatment. Here, we focus on rheumatoidarthritis (RA), which is a multifactorial polygenic diseaseand might be termed a systems biology disease. RA is achronic inflammatory disorder primarily afflicting thesynovial joints, and autoimmunity plays a pivotal role inits chronicity and progression. The disease is characterizedby autoreactive behavior of immune cells and the induc-tion of enzymes which lead to the destruction of cartilageand bone [7]. The inflammatory processes are triggered bycytokines and other immune system-related genes thatform a complex network of intra- and intercellular molec-ular interactions. A number of cytokine proteins play acritical role as mediators of immune regulation. In RA, thetwo cytokines TNF-alpha and IL-1 are considered masterregulators that act in a complementary and synergisticmanner [8,9]. By blocking TNF-alpha, etanercept inter-venes this molecular network and thus is thought to re-balance the immune system's dysregulation [10-12].Etanercept therapy in RA patients has been proven to slowdisease progression, but the precise molecular mecha-nisms remained unclear. To investigate the therapeuticeffects on transcriptional regulation, GRN inference tech-niques can be applied. This could lead to a better under-standing of the modes of action of etanercept as well asthe pathogenesis underlying RA. We may also understandwhy the drug fails to control the disease in about 30% ofthe patients (non-responders).
We studied a group of 19 patients suffering from RA forwhich DNA microarrays were used to obtain genome-wide transcriptional profiles within the first week ofetanercept administration [13]. A set of etanercept respon-sive genes was attained. The majority of these genes areknown to control the body's immune response. SeveralTFBS were identified as overrepresented in the genes' reg-ulatory regions and we used the corresponding informa-tion on TF-gene interactions as a template for modelingthe underlying GRN. A system of linear equations waschosen to mathematically describe the regulatory effectsbetween the genes and TFs (i.e. the network nodes). Weused a hybrid of the Least Angle Regression (LARS) andthe Ordinary Least Squares regression (OLS) to find the
Page 2 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
model structure and estimate the coefficients. In doing so,the modeling is constrained to include only a subset of theputative TF-gene interactions. That way, our approachconsiders that genes usually regulate other genes indi-rectly through the activity of one or more TFs, whichmakes the model straightforward to interpret in terms oftrue molecular interactions. The resulting GRN was fur-ther analyzed using e.g. gene ontology (GO) and clinicalinformation (figure 1). We found that our integrativemodeling strategy, namely the TFBS-integrating LARS(TILAR), is able to reconstruct GRNs more reliably thanother established methods. This is one of the first studiesthat utilizes network analysis to investigate transcriptionalregulation in response to a therapeutic drug in humans[14].
Results and discussionEffects of etanercept therapy on gene expressionWe used the Affymetrix microarray dataset from Koczan etal. [13] which provided expression levels of peripheralblood mononuclear cells (PBMC) measured in 19patients suffering from RA. For each patient, blood sam-ples were taken before treatment (baseline) as well as 72(day 3) and 144 hours (day 6) after start of immuno-therapy by etanercept. Clinical response was assessed over3 months and revealed 7 patients with persistent diseaseactivity (non-responders).
We analyzed the DNA microarray data in respect to com-mon gene expression changes observed in the wholegroup of patients after therapy onset. First of all, we pre-processed the data to correct for systematic effects. Moreimportantly, signal intensities were calculated by applyinga custom chip definition file by Ferrari et al. that is com-posed of custom-probesets including only probes match-ing a single gene [15]. As such, a one-to-onecorrespondence between genes and custom-probesets is
preserved, which deeply improves gene-centered analysisof human Affymetrix data [16]. Finally, the data pre-processing yields expression levels of 11,174 differentgenes for each of the 55 microarrays in the dataset (fordetails see the methods section).
Afterwards, we identified a set of genes significantly up- ordown-regulated in response to etanercept. It is importantto note that the filtering of genes is a crucial step in GRNinference as there is a tight relationship between modelcomplexity (i.e. network size and level of detail of themodel), the amount of data required for inference and thequality of the results. On the one hand, a small anddetailed network model might better fit the given data,but only a sufficiently large model can capture the funda-mental properties that constitute a GRN including scale-freeness, redundancy and self-regulation. In this study, weutilized a t-statistic in conjunction with an MA-plot-basedsignal intensity-dependent fold-change criterion (referredto as MAID filtering) to select genes with expressionchanges in the first week of therapy (see methods).Through this filtering we identified 37 genes as differen-tially expressed at day 3 versus baseline, and 57 genes atday 6. Altogether, 48 genes were found down-regulatedand 35 genes up-regulated, comprising a set of 83 genes intotal (additional files 1 and 2).
We searched for overrepresented terms of the GO biolog-ical process ontology in the list of 83 selected genes andfound that most of the genes are known to control thebody's immune response (additional file 3, see methods).Remarkably, genes of the I-kappaB kinase/NF-kappaB cas-cade (GO:0007249) are enriched in the gene set and rep-resented by 5 genes (NFKBIA, TNFRSF1A, TLR8, NOD2,HMOX1). NF-kappaB is a key factor in the transcription ofmany inflammatory genes and has been implicated in thepathological processes of RA. The NF-kappaB cascade is
Workflow used to study gene regulatory effects in response to etanercept therapyFigure 1Workflow used to study gene regulatory effects in response to etanercept therapy. A network model of transcrip-tional regulation is inferred by integrating transcription factor binding site information.
Page 3 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
mainly activated by the proinflammatory cytokines IL-1and TNF-alpha. As was shown, TNF-blocking agents suchas etanercept prevent TNF-alpha from binding to its recep-tors, induction of signal transduction cascades and activa-tion of TFs including NF-kappaB [10]. Here, we foundNFKBIA, that inactivates NF-kappaB by trapping it in thecytoplasm, up-regulated early after therapy initiation. Onthe other hand, genes known to activate the NF-kappaBprotein, e.g. NOD2 and TNFRSF1A (a TNF receptor), weredown-regulated after therapy onset. Thus, the result of ourfiltering indicates the expected suppression of NF-kappaBactivity by etanercept. In addition, we found evidence fora modulation of B cell mediated immunity. The corre-sponding GO category (GO:0019724) comprises thegenes C1QB, CLU and TLR8 whose mean expression wassignificantly lower at day 3 and 6 compared to baseline,respectively. Interestingly, TLR8 signaling is linked to thecontrol of CD4+ regulatory T (Treg) cells. Treg cellsactively suppress host immune responses and, as a conse-quence, play an important role in preventing autoimmu-nity [17]. TLR8 is thought to initiate immune processes byreversing the suppressive function of Treg cells [18]. Itsdown-regulation by etanercept might be an important fac-tor to control the disease.
Genes responsive to etanercept administration are proba-bly under control of certain TFs, whose activities are(maybe indirectly) affected by this drug. Therefore, weanalyzed the regulatory regions around the respectivetranscription start sites (TSS) of these genes for occurrenceof overrepresented TFBS (see methods). Identifying TFBS,particularly in higher eukaryotic genomes, is an enormouschallenge and cross-species sequence conservation isoften used as an effective filter to improve the predictions.We found evolutionarily conserved binding sites enrichedfor 12 TFs (represented by 19 Transfac binding profiles).
These 12 TFs connect 52 out of the 83 genes through 96TF-gene interactions, whereas each TF is linked to at least4 genes (table 1). The list of TFs includes C/EBP-beta,which is an important transcriptional activator in the reg-ulation of genes involved in immune and inflammatoryresponses, including the cytokines IL-6, IL-8 and TNF-alpha [19]. Binding sites for the TATA binding protein(TBP) were detected in 14 genes. TBP binds DNA at theTATA-element, and as a subunit of the TFIID complexcoordinates the initiation of transcription by RNApolymerase. Although TBP is always involved, its TATA-binding activity is dispensable for the positioning of theRNA polymerase. In fact, approximately 76% of humancore promoters lack TATA-like elements [20]. However, inthe set of 83 genes, those genes having the TATA box wereoverrepresented. The two TFs ZIC1 and ZIC3 were consid-ered as one TF entity, as they have highly similar DNAbinding properties. None of the 12 TFs showed significanttranscriptional changes in the data. Nevertheless, theinformation on predicted TF-gene interactions can beused as a GRN template during inference. Before describ-ing how this is done by TILAR we will outline the generalprinciples of the modeling approach.
Linear network modelingWe chose a system of equations to model the regulatoryinteractions among the genes affected by etanercept ther-apy. The concept of modeling gene regulation by a systemof equations is to approximate gene expression levels as afunction of the expression of other genes and environ-mental factors. Modeling GRNs by systems of equationshas several benefits as they can describe regulatory effectsin a flexible, quantitative, directed manner, and take intoaccount that gene regulators act in combination. With sys-tems of equations one can easily model positive and neg-ative feedback loops, and describe even non-linear and
Table 1: Evolutionarily conserved binding sites were found to be enriched for 12 TFs.
TF Name Transfac ID Official Full Name P-value Expected Count Count
TBP, TFIID V$TBP_01, V$TATA_C, V$TATA_01 TATA box binding protein 0.0042 6.41 14C/EBPbeta V$CEBPB_01, V$CEBPB_02 CCAAT/enhancer binding protein beta 0.0112 5.03 11Zic1, Zic3 V$ZIC1_01, V$ZIC3_01 Zic family member 1/3 0.0183 6.13 12AP-2rep V$AP2REP_01 Kruppel-like factor 12 0.0264 1.68 5HNF-1, HNF-1A V$HNF1_01, V$HNF1_C HNF1 homeobox A 0.0274 2.30 6Lmo2 V$LMO2COM_01, V$LMO2COM_02 LIM domain only 2 0.0352 5.98 11SRY V$SRY_02 sex determining region Y 0.0374 1.85 5ATF-2 V$CREBP1_01 activating transcription factor 2 0.0408 1.30 4Cart-1 V$CART1_01 ALX homeobox 1 0.0415 1.30 4COMP1 V$COMP1_01 cooperates with myogenic proteins 1 0.0422 3.23 7Hlf V$HLF_01 hepatic leukemia factor 0.0470 1.97 5NF-1, NF-1/L V$MYOGNF1_01, V$NF1_Q6 nuclear factor I 0.0492 7.10 12
= 96
The column "Count" denotes the number of genes that possess a TFBS for the respective TF. All in all, 96 TF-gene interactions were predicted (GRN template).
Page 4 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
dynamic phenomena of biological systems. However, asmore complex models require higher amounts of accuratedata to learn their parameters reliably, researchers oftenutilize systems of linear equations (linear models). Linearmodels have been successfully employed in many appli-cations, e.g. to reconstruct GRNs relevant for developmentof the central nervous system in rats [21], osteoblast dif-ferentiation in mice [22,23], galactose regulation in yeast[24] and immune response of human blood cells to bac-terial infection [25]. Linear models assume that gene reg-ulatory effects are limited to be linear and additive and asimple one can be written as:
where vector xi contains the M expression levels measuredfor gene i, N is the number of genes in the network, andthe weights wij define relationships between the genes.When inferring a linear model we need to estimate theweights wij (i.e. the model parameters) from the data. Theweights specify the existence of regulatory relationshipsbetween genes, their nature (activation or inhibition) andrelative strength. If wij > 0 gene xj activates gene xi, if wij < 0xj inhibits xi, and wij = 0 implies that xi is not under controlof xj. This simplicity makes linear models easy to interpret,even if the encoded relationships have a wide range ofmeanings: edges in the network might represent directphysical interactions (e.g. when a gene encodes a TF regu-lating another gene) or rather conceptual interactions(e.g. when the expression levels of two genes merely cor-relate).
Linear models can also be used to describe the dynamicsof the network. In this case, the model is a system of lineardifference equations that approximates the change of geneexpression in time. However, this approach is inappropri-ate for our application as the time-series in the microarraydataset consist of only 3 time-points and the timebetween two subsequent measurements is rather long (3days). Nevertheless, the modeling strategy illustrated herecan be easily adapted for the inference of dynamic mod-els.
To fit the (static) linear model to the data, equation (1)can be written in matrix form as follows:
These N systems can be coupled as:
Now, a GRN model can be inferred by estimating (com-prising all the model parameters in w) from input matrixX (having M' = MN rows and N' = (N-1)N columns) andoutput vector y using OLS regression.
However, despite the fact that linear models are a strongsimplification of the true GRN, equation (3) is already anunderdetermined system of linear equations in our partic-ular study as the number of genes in the network (N = 83)is greater than the number of measurements (M = 55).That means, infinitely many solutions exist. Therefore,biologically motivated constraints have to be included totackle this problem. The most commonly used modelingconstraint is the sparseness of GRNs. Sparseness reflectsthe fact that genes are regulated only by a limited numberof regulators. The sparseness constraint minimizes thenumber of edges, i.e. reduces the effective number ofmodel parameters. Sparse linear models can be recon-structed via the Lasso (Least absolute shrinkage and selec-tion operator) method [26], which effectively performssimultaneous parameter estimation and variable selec-tion. The Lasso is a version of OLS that constrains the sumof the absolute regression coefficients :
The Lasso penalizes model complexity by shrinking thecoefficients j (and hence wij) toward 0, more so for smallvalues of s. A modification of the LARS algorithm imple-ments the Lasso [27]. LARS builds up estimates for insuccessive steps, each step adding one covariate to themodel, so that gradually model parameters are set non-zero. In simple terms, LARS is a less greedy version of tra-ditional forward selection methods. LARS and its variantsare computationally efficient. The algorithm requires onlythe same order of magnitude of computational effort asOLS to calculate the full set of Lasso estimates (i.e. for alls 0).
The Lasso approach was first introduced to infer regula-tory interactions by van Someren et al. [28] and has sincebeen applied in several GRN studies [22,29]. However,even if the network connectivity is constrained, there is alimitation in inferring GRNs using gene expression dataonly. Hence, there is a need to incorporate different typesof information during network reconstruction. Variousdata and information from biomedical literature and
ˆ ,,
x w xi ij j
j j i
N
== ≠∑1
(1)
ˆ ,
, ( , ,
( )
( )
y X
y X
iM Ni i
ii M N
ii
i N
x x x
= ⋅ =
= =
× −
× − −
1
1 1
1 for where
11 1 1 1 1, , , ), ( , , , , , ) .x x w w w wi Ni
i i i i i i NT
+ − +=
(2)
ˆ : ˆ .( )y X y X= ⋅ = ⋅× −MN N N1 or in shorter form
(3)
ˆ arg min
= − ⋅⎛
⎝
⎜⎜
⎞
⎠
⎟⎟
⎧
⎨⎪
⎩⎪
⎫
⎬⎪
⎭⎪=
′
=
′
∑∑ y xi ij j
j
M
i
N
1
2
1
subject to j
j
M
s≤=
′
∑1
.
(4)
Page 5 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
databases can be utilized in combination with geneexpression levels to increase model accuracy.
An integrative learning strategy usually consists of twosteps. First, a template of the network is built, e.g. basedon known TF-DNA interactions or molecular interactionsautomatically extracted from the literature by text mining.This template represents a supposition of the true networkstructure, that might be uncertain and incomplete. Sec-ond, an inference algorithm is applied that fits the modelto the measured data while taking the template intoaccount, trading off data-fit and template-fit. When infer-ring linear models, such template information can beincluded by adapting the Lasso method. This is possibleby introducing additional weights on the coefficients of the constraint in equation (4):
A relatively low weight j provokes that the edge corre-sponding to j is preferred to be in the final model. Hence,the modeler is able to incorporate partial prior knowledgeby setting the weights appropriately. Recently, this con-cept was applied to integrate human microarray data withregulatory relationships obtained by literature mining bydefining each j as a constant [23] and as weight functionof j [13], respectively.
TILAR – a TFBS-integrating linear modeling approachHere, we propose TILAR – a TFBS-integrating inferencetechnique that differs from the adaptive Lasso approach,and employs TF binding information as prior knowledge.As we will show, it is even possible to combine the adap-tive Lasso and TILAR. According to our modeling scheme,we distinguish two types of network nodes: genes (thatwere selected for inferring regulatory relationshipsbetween them) and TFs (for which respective binding sitesare overrepresented in the gene set). Expression levels ofthe gene set (that possibly includes genes encoding TFs)are required for the modeling. The algorithm then aims toassign (directed) TF-gene and gene-TF interactions (net-work edges). A TF-gene interaction represents a physicalinteraction, i.e. a TF binds the region that encompassesthe TSS of a certain gene and thus regulates its transcrip-tion. In contrast, gene-TF interactions can have differentmeanings: the gene itself might encode a transcriptionalregulator of the TF, or the gene product controls the activ-ity of the TF at the proteomic level, or the gene triggers sig-naling cascades that affect the TF, etc. Using both types ofinteractions, the model reflects that genes regulate othergenes indirectly through a combination of TFs (figure 2).As a reminder, the TFBS overrepresentation analysis
revealed 96 putative TF-gene interactions. Now, the idea isto use this information as a GRN template by constrainingthe modeling to include only a subset of these TF-geneinteractions. As the inference method not necessarily usesall the given TF-gene interactions, we consider the fact thatthey are computationally predicted and therefore not allof them might refer to biologically functional bindingsites. In practice, the algorithm starts with the entire set ofTF-gene interactions and then iteratively removes avoida-ble interactions through a backward stepwise selectionprocedure (see methods). The information on (the cur-rent set of included) TF-gene interactions is written inmatrix B, which is defined as:
ˆ arg min
= − ⋅⎛
⎝
⎜⎜
⎞
⎠
⎟⎟
⎧
⎨⎪
⎩⎪
⎫
⎬⎪
⎭⎪=
′
=
′
∑∑ y xi ij j
j
M
i
N
1
2
1
subject to j j
j
M
s≤=
′
∑1
.
(5)
Illustration of our proposed modeling conceptionFigure 2Illustration of our proposed modeling conception. Here, we aim to reconstruct a gene regulatory network con-sisting of 4 genes (dark blue) and 2 transcription factors (light gray). For simplicity, we assume that only one gene expres-sion measurement was performed. The expression level of each gene is given in the gene nodes. (A) The GRN template: In this example, two genes possess at least one TF binding site in their regulatory region as indicated by 3 TF-gene inter-actions (purple). (B) In that case, there are 5 possible gene-TF interactions (i.e. model parameters ) in the network (dashed, orange). If available, we might consider prior knowl-edge on gene-TF interactions during inference (adaptive TILAR). (C) A possible inference result including 3 gene-TF interactions (solid, orange). Here, the model perfectly fits the data (e.g. "8" = 2.0·"4") with two nominal model parameters set to zero. (D) We can use the inferred model to derive gene-gene relationships from the edges between genes and TFs (gray). The benchmarking was conducted on such gene-gene interactions.
Page 6 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
In our particular study, 96 entries in B were set to 1 in thefirst iteration. TILAR then assigns the parameters in themodel to gene-TF interactions, as follows:
where F is the number of TFs. For modeling the transcrip-tional regulation in response to etanercept we have N = 83genes, that showed significantly changed expression levelsafter therapy onset, and F = 12 TFs, whose binding sitesare overrepresented in the regulatory regions of theselected genes. In the model, each gene can exhibit a reg-ulatory effect on each TF, except those TFs that hold a TF-gene interaction to this gene (this restriction is dispensa-ble when inferring a dynamic model). If wkj = 0, there is nogene-TF interaction between gene j and TF k. Otherwise,gene j controls the activity of TF k and thus regulates allthe genes that possess a TFBS for TF k. In this case, theexpression levels of gene j explain the expression of thegenes regulated by TF k. Again, to infer the GRN model, weneed to estimate the model parameters wkj from the geneexpression data while constraining the model to be sparse.Similar to equation (3), we can couple the subsystems, asfollows:
where Nr is the number of genes possessing at least oneoverrepresented TFBS, and #B is the number of TF-geneinteractions considered at the current iteration. The coef-ficients of this equation now correspond to gene-TFinteractions in the model. Finally, a sparse solution toequation (8) can be found using the Lasso according toequation (4).
The TILAR modeling approach proposed here is advanta-geous for several reasons. First, TF expression levels arenot required, since the activity of TFs is modeled implic-itly (like a hidden node). This is beneficial, as mRNA lev-els of TFs are often low and do not necessarily correlatewith TF activity. In fact, TF proteins often need to be acti-vated by phosphorylation. Second, the nominal numberof model parameters w in equation (7) is generally lowerthan in equation (1). Therefore, our method tackles theproblem of having too many parameters in comparison tolimited amounts of experimental data. In our particularapplication, equation (8) is an overdetermined system oflinear equations (as M·Nr = 55·52 = 2860 >F·N-#B =
12·83-#B = 996-#B, #B 96), i.e. we are able to infer acomplex network of 83 genes (and 12 TFs) without beingin conflict with the data requirements. Third, by using TFbinding predictions as prior knowledge we can recon-struct GRNs more reliably. Besides, the inferred modelsare relatively easy to interpret. Finally, the integration ofTFBS information is accomplished by simply specifyingthe regression equation (i.e. input matrix X and outputvector y) adequately. Therefore, we can combine theTILAR approach with the adaptive LARS, i.e. solve equa-tion (8) according to equation (5) if prior knowledge ongene-TF interactions is available (adaptive TILAR).
Modeling the gene regulatory response to etanerceptTo examine the early transcriptional effects of etanerceptwe applied the TILAR algorithm to construct a GRN modelon the basis of gene expression data and knowledge onTF-gene interactions obtained by TFBS analysis. For thispurpose, the GRN inference problem was formulatedaccording to equation (8). The essential part of our mod-eling approach is the LARS algorithm that is used toobtain all possible Lasso solutions for this linear regres-sion equation.
TILAR iteratively applies LARS in a backward stepwiseselection procedure in order to refuse TF-gene interactionsthat do not fit the data well (see methods). Hence, thelearning strategy takes into account that the prediction ofTFBS might be error-prone. In this study, 12 out of 96 pre-dicted TF-gene interactions were discarded. These 12interactions may result from false positive TFBS predic-tions, or the magnitude of the TF-gene interactions wasnot enough for being confirmed based on the gene expres-sion levels.
After we identified the subset of 84 TF-gene interactions,we used LARS to define which gene-TF interactions haveto be included at different degrees of network connectiv-ity. That means we used LARS only for variable selection,but the actual coefficients were estimated by OLS (seemethods). This LARS/OLS hybrid technique usuallyachieves sparser estimates and more accurate predictions,and thus outperforms the ordinary Lasso [27,30]. Finally,we selected the most parsimonious estimate with low 10-fold cross-validation error (additional file 4). In this way,the method avoids overfitting to the data and conse-quently yields a sparse GRN model. The final model con-sists of 22 inferred gene-TF interactions and 84 TF-geneinteractions, and was visualized using Cytoscape 2.6.0(figure 3, additional file 5).
Model interpretationSystems biological models need to be interpretable inorder to be useful. In general, the modeling goals of accu-rate prediction and interpretation are contradictory since
bj k
kj =⎧⎨
1
0
,
, .
if gene possesses a binding site for TF
else⎩⎩(6)
ˆ ( ) ,x b w x bi kj kj j ki
j
N
k
F
= −==
∑∑ 111
(7)
ˆ ,( # )y X= ⋅× −MN FN Br (8)
Page 7 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
interpretable models should be simple, but more accuratemodels might be quite complex. The model reconstructedhere seems to satisfy both requirements. On the one hand,the network model is fairly complex as it consists of 95nodes and 106 edges while using 22 model parameters wij(specifying the strength of the gene-TF interactions). Yetthe model is readily interpretable due to the intuitive lin-ear modeling scheme.
Apparently, the inferred model is sparse, i.e. each networknode is under control of only few regulators. The maxi-mum in-degree in the GRN is 5 (on average 1.12). Never-theless, some nodes (named hubs) are highly connectedin the network, e.g. TBP which has an out-degree of 12. Afurther characteristic and biologically meaningful prop-erty of the network is its scale-free structure. Scale-freenessdenotes the phenomenon that the degree distribution inbiological networks often follows a power law, i.e. thefraction P(k) of nodes in the network having k connec-tions goes as P(k)~k-, where is a constant. This means
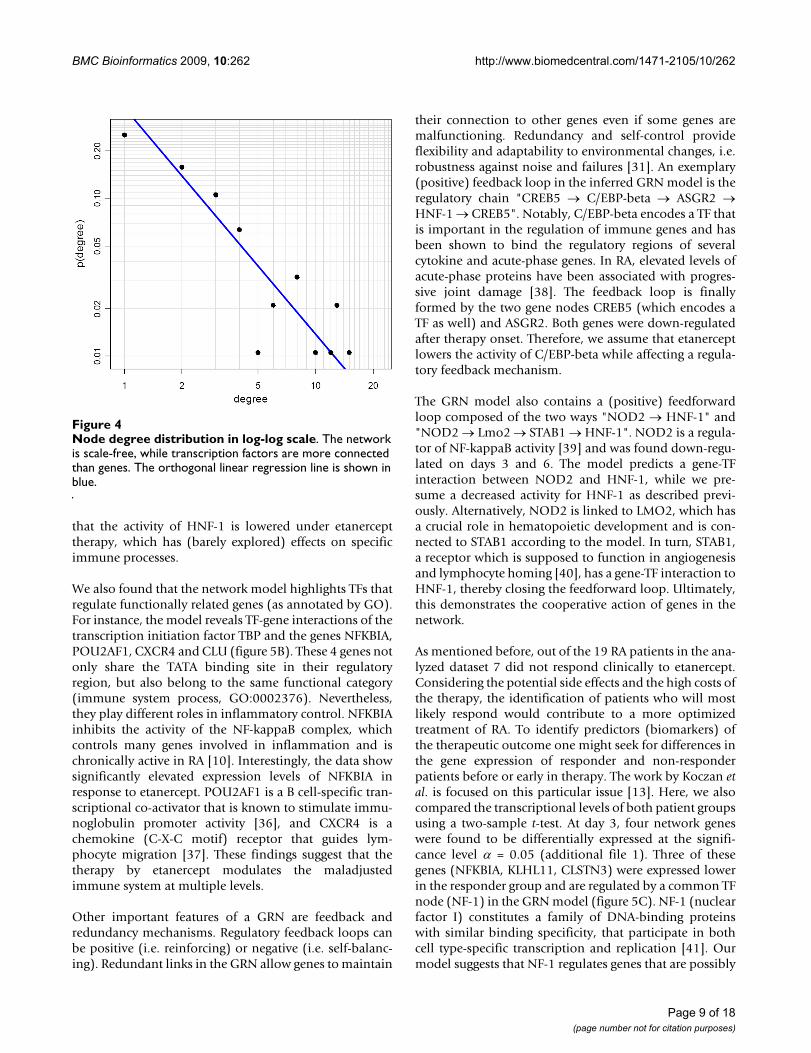
that in scale-free networks most of the nodes are lowlyconnected, while a few are relatively highly connected.Scale-freeness indicates a network's decentralization andstructural stability, and in consequence its robustnessagainst random fluctuations [31]. The scale-free design ofGRNs is well studied in literature [32,33], and the GRNreconstructed here is scale-free with = 2.22 (as calculatedaccording to Clauset et al. [34], see figure 4).
A closer look at the interactions in the network revealedgene sets co-regulated by a common TF. For example, 6TF-gene interactions were assigned to the transcriptionalactivator HNF-1 in the GRN template (table 1). Two ofthem were not considered in the final model as they wereeliminated during backward stepwise selection. However,the 4 remaining genes that are predicted to be under con-trol of HNF-1 (AQP9, TCN2, CREB5, C4orf18) are alldown-regulated in the patients during first week of ther-apy (figure 5A). AQP9 is assumed to have some role inimmunological response [35]. Hence, we can hypothesize
Reconstructed gene regulatory network of genes up- or down-regulated during first week of therapyFigure 3Reconstructed gene regulatory network of genes up- or down-regulated during first week of therapy. The TILAR algorithm used gene expression data and transcription factor binding predictions to infer a network of 84 TF-gene and 22 gene-TF interactions. The size of the nodes corresponds to their degree of connectivity. Three parts of the network model are shown in detail in figure 5. The full model is available as a Cytoscape session file of (additional file 5).
Page 8 of 18(page number not for citation purposes)
BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
that the activity of HNF-1 is lowered under etanercepttherapy, which has (barely explored) effects on specificimmune processes.
We also found that the network model highlights TFs thatregulate functionally related genes (as annotated by GO).For instance, the model reveals TF-gene interactions of thetranscription initiation factor TBP and the genes NFKBIA,POU2AF1, CXCR4 and CLU (figure 5B). These 4 genes notonly share the TATA binding site in their regulatoryregion, but also belong to the same functional category(immune system process, GO:0002376). Nevertheless,they play different roles in inflammatory control. NFKBIAinhibits the activity of the NF-kappaB complex, whichcontrols many genes involved in inflammation and ischronically active in RA [10]. Interestingly, the data showsignificantly elevated expression levels of NFKBIA inresponse to etanercept. POU2AF1 is a B cell-specific tran-scriptional co-activator that is known to stimulate immu-noglobulin promoter activity [36], and CXCR4 is achemokine (C-X-C motif) receptor that guides lym-phocyte migration [37]. These findings suggest that thetherapy by etanercept modulates the maladjustedimmune system at multiple levels.
Other important features of a GRN are feedback andredundancy mechanisms. Regulatory feedback loops canbe positive (i.e. reinforcing) or negative (i.e. self-balanc-ing). Redundant links in the GRN allow genes to maintain
their connection to other genes even if some genes aremalfunctioning. Redundancy and self-control provideflexibility and adaptability to environmental changes, i.e.robustness against noise and failures [31]. An exemplary(positive) feedback loop in the inferred GRN model is theregulatory chain "CREB5 C/EBP-beta ASGR2 HNF-1 CREB5". Notably, C/EBP-beta encodes a TF thatis important in the regulation of immune genes and hasbeen shown to bind the regulatory regions of severalcytokine and acute-phase genes. In RA, elevated levels ofacute-phase proteins have been associated with progres-sive joint damage [38]. The feedback loop is finallyformed by the two gene nodes CREB5 (which encodes aTF as well) and ASGR2. Both genes were down-regulatedafter therapy onset. Therefore, we assume that etanerceptlowers the activity of C/EBP-beta while affecting a regula-tory feedback mechanism.
The GRN model also contains a (positive) feedforwardloop composed of the two ways "NOD2 HNF-1" and"NOD2 Lmo2 STAB1 HNF-1". NOD2 is a regula-tor of NF-kappaB activity [39] and was found down-regu-lated on days 3 and 6. The model predicts a gene-TFinteraction between NOD2 and HNF-1, while we pre-sume a decreased activity for HNF-1 as described previ-ously. Alternatively, NOD2 is linked to LMO2, which hasa crucial role in hematopoietic development and is con-nected to STAB1 according to the model. In turn, STAB1,a receptor which is supposed to function in angiogenesisand lymphocyte homing [40], has a gene-TF interaction toHNF-1, thereby closing the feedforward loop. Ultimately,this demonstrates the cooperative action of genes in thenetwork.
As mentioned before, out of the 19 RA patients in the ana-lyzed dataset 7 did not respond clinically to etanercept.Considering the potential side effects and the high costs ofthe therapy, the identification of patients who will mostlikely respond would contribute to a more optimizedtreatment of RA. To identify predictors (biomarkers) ofthe therapeutic outcome one might seek for differences inthe gene expression of responder and non-responderpatients before or early in therapy. The work by Koczan etal. is focused on this particular issue [13]. Here, we alsocompared the transcriptional levels of both patient groupsusing a two-sample t-test. At day 3, four network geneswere found to be differentially expressed at the signifi-cance level = 0.05 (additional file 1). Three of thesegenes (NFKBIA, KLHL11, CLSTN3) were expressed lowerin the responder group and are regulated by a common TFnode (NF-1) in the GRN model (figure 5C). NF-1 (nuclearfactor I) constitutes a family of DNA-binding proteinswith similar binding specificity, that participate in bothcell type-specific transcription and replication [41]. Ourmodel suggests that NF-1 regulates genes that are possibly
Node degree distribution in log-log scaleFigure 4Node degree distribution in log-log scale. The network is scale-free, while transcription factors are more connected than genes. The orthogonal linear regression line is shown in blue.
Page 9 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
relevant for the individual success of etanercept therapy,while the prognostic value of NFKBIA mRNA levels isalready under discussion [13]. However, even if thishypothesis still has to be verified, the analysis clearly dem-onstrates the use of correlating clinical features withmolecular network structures [6,42].
The GRN model provides many testable hypotheses andthus may be a starting point for new experiments. Thosecould aim to study the expression changes of specificgenes in more detail, or to analyze their regulatory effectsthereby validating parts of the inferred network. Take forinstance the previously mentioned subnetwork of NF-1and its target genes (figure 5C). The model suggests thatmembers of the NF-1 family bind the promoters of 10etanercept-responsive genes. This might be tested by elec-trophoretic mobility shift assays or chromatin immuno-precipitation techniques. One could also investigate thegenes' transcriptional changes during therapy in a largercohort of RA patients by generating expression profilesusing real-time polymerase chain reaction. This would beparticularly useful for the three genes that were found sig-nificantly lower expressed in the responder group. As anext step, levels of the respective proteins and protein iso-forms could be quantified using western plots andenzyme-linked immunosorbent assays. For example, it
would be interesting to measure the amount of HNF-1proteins as we postulate a lowered activity of this TF as amolecular therapeutic effect. In the model all target genesof HNF-1 are down-regulated in response to etanerceptadministration (figure 5A). Similarly, other parts of thenetwork are worth to study, e.g. the inferred regulatoryfeedback loop including C/EBP-beta and CREB5. Tran-script and protein levels of in vitro cultures of PBMC cellsmay also be analyzed in a time-dependent manner. Thisallows for controlled perturbation experiments such assiRNA mediated knock-down of NF-1 expression with orwithout the presence of etanercept. Last but not least, onecould examine the cell type-specific expression of genes inthe network. Recent studies point out a functional impair-ment of Treg cells in RA [17]. It would be attractive to fur-ther elucidate the altered immunosuppressive capacity ofTreg cells, their role in the treatment of RA and the modu-lation of Treg cells by Toll-like receptors such as TLR8 thatwas down-regulated in the dataset.
Performance evaluationTo demonstrate the benefit of the TILAR modelingapproach, we tried to evaluate how reliable the structureof the underlying GRN can be inferred. The assessment ofthe GRN inference performance is a challenging task, asevidently, true regulatory interactions are barely known
Detail views of the network model shown in figure 3Figure 5Detail views of the network model shown in figure 3. (A) The modeling strategy takes into account that target genes of a transcription factor are often co-expressed. For example, all the genes that are regulated by HNF-1 are down-regulated after therapy onset. Outer parts are shown with lower opacity. (B) A set of genes associated with the GO category "immune sys-tem process" is predicted to contain TATA-like elements in their regulatory regions. (C) Three genes were expressed lower in responders at day 3 and all of them are regulated by NF-1 according to the model.
Page 10 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
and curated datasets for benchmarking are missing,though there are attempts to remedy this shortcoming[43,44]. A further difficulty is that the knowledge used tovalidate a GRN model must be different from the knowl-edge integrated during modeling.
Here, we utilized gene-gene interaction informationobtained by text mining for performance evaluation (seemethods). By assessing the inference quality on literature-derived (undirected) gene-gene links, we were also able tocompare our method with other inference techniqueswhich do not incorporate prior knowledge. It is importantto note that regulatory gene-gene interactions are implic-itly defined in our network model by gene-TF and TF-geneinteractions, as genes are constrained to regulate othergenes via one or more TFs (figure 2D). For the inferrednetwork 158 gene-gene interactions can be deduced fromthe 22 gene-TF and 84 TF-gene edges. However, literaturemining reports only 5 gene-gene relationships betweenthe 83 genes in the network, which is not nearly enoughfor validation purposes. Since the biological role of manyselected genes remains to be investigated, we assume thatthe lack of text mining information is mainly due to theliterature bias, by which genes that have been intensivelystudied for many years (e.g. TNF-alpha) are cited moreoften than less prominent genes.
To overcome this issue, we sought for genes well describedin the literature. For them we could expect many knowngene regulatory interactions, so that a systematic evalua-tion of the performance of network reconstructionsbecomes feasible. We finally chose genes that are most fre-quently co-mentioned in the context of RA in PubMed. Arespective list of genes was obtained from the Autoim-mune Disease Database (version 1.2 as of August 19,2008), which is a literature-based database that providesgene-disease associations of all known or suspectedautoimmune diseases [45]. Out of the top 50 genes catal-oged for the disease term "rheumatoid arthritis", 42 geneswere measured in the Affymetrix dataset (additional file6). We will denote the network of these 42 genes as thebenchmarking GRN in the following.
Genes in the benchmarking network include severalmatrix-metallo-proteinases and a vast number ofcytokines, in particular interleukins and the therapeutictarget TNF-alpha. Overall, 389 gene-gene interactionsbetween these genes could be retrieved through text min-ing. These interactions constitute a text mining network inwhich all but two genes are connected. The genes with themost connections are IL-6 (37), TNF-alpha (37) and IL-1(33). We analyzed the regulatory regions of all the 42genes and found overrepresented DNA-binding sites of 10TFs (additional file 7). Amongst others, TFBS of NF-kap-paB and AP-1 are significantly enriched, which is not sur-
prising as both TF complexes play central roles in immuneregulation and are proven to be involved in the pathogen-esis of RA [10,46]. In the resulting GRN template these 10TFs are linked to 31 genes by means of 67 TF-gene interac-tions. When constructing a linear model of the bench-marking GRN using our novel inference algorithm TILAR,13 TF-gene interactions were discarded during the back-ward stepwise selection procedure, i.e. 54 TF-gene interac-tions remained in the model. LARS then provided modelpredictions for different degrees of network connectivity(in successive LARS steps representing the dependency onparameter s).
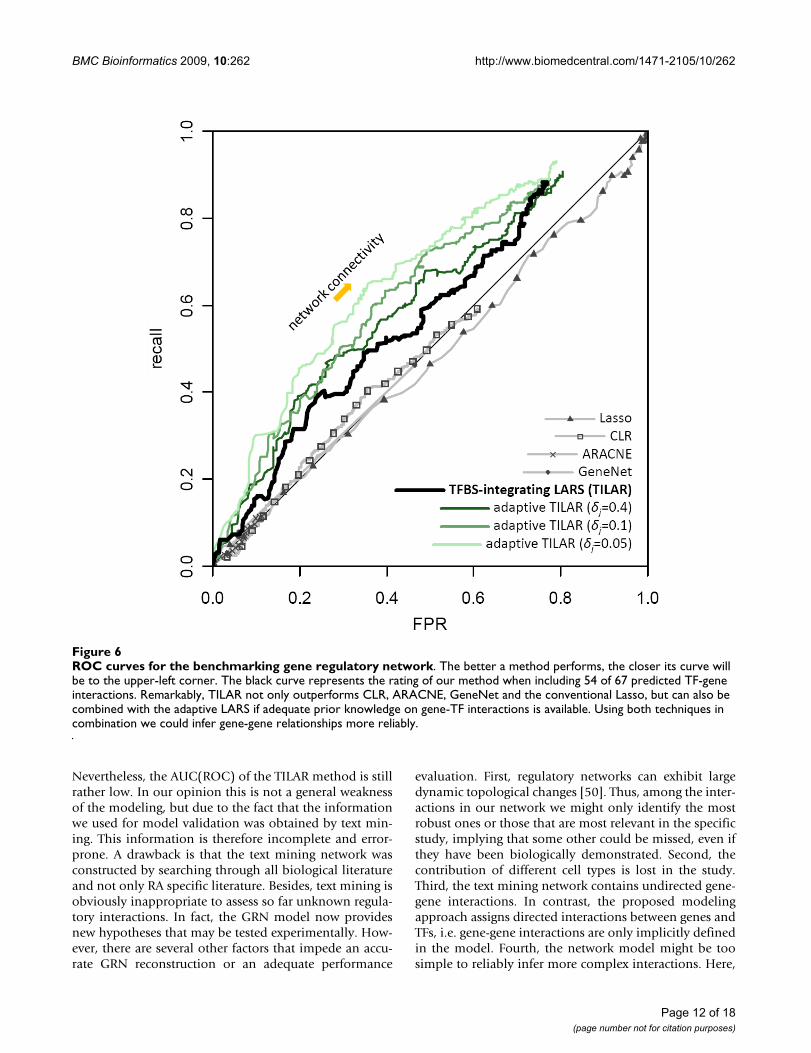
Next, we tested whether the inferred edges between genesexist or not in the text mining network containing 389gene-gene interactions. For this purpose, we calculated themeasures recall, precision and false positive rate (FPR) fordifferent network connectivities. A plot of the precisionversus the recall performance of a method (in case of LARSas a function of s) and the ROC (receiver operating char-acteristic) curve, where recall is plotted against FPR, aretwo widely used visualizations for performance evalua-tion [43,44]. The ROC analysis allows comparison of theinference quality against a random prediction by calculat-ing the area under the curve (AUC), while an AUC(ROC)close to 0.5 corresponds to a random forecast.
We utilized both recall-precision and ROC curves to assessand compare the performance of our algorithm and fourdifferent popular GRN inference methods: the conven-tional Lasso approach, CLR [47], ARACNE [48], andGeneNet [49] (see methods). While CLR and ARACNE usemutual information, GeneNet computes a partial correla-tion network. The resulting performance curves show thatthe proposed TILAR algorithm outperforms the othermodeling algorithms (figure 6, additional file 8). Whenusing AUC(ROC) as a single metric for benchmarking, theapplied methods score as follows: Lasso – 0.478, ARACNE– 0.500, GeneNet – 0.503 and CLR – 0.504, whereasTILAR achieves an AUC(ROC) of 0.581. Next, we checkedwhether the algorithms performed significantly betterthan a random GRN prediction (RAND, see methods). Wefound, that the predictions of our approach were signifi-cantly better than RAND at the level = 0.05 (P-value =1.674e-05), while this was not the case for CLR, ARACNE,GeneNet and Lasso. Interestingly, gene-TF-RAND,another random algorithm that predicts gene regulatoryinteractions by including all 67 putative TF-gene interac-tions (i.e. the prior knowledge) into the model withoutconsidering the gene expression data (see methods), alsoyields a relatively high AUC(ROC) of 0.549 (P-value =0.006). This suggests that TILAR performs well because ofboth the quality of TFBS predictions and data-fitting usingLARS (figure 7).
Page 11 of 18(page number not for citation purposes)
BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
Nevertheless, the AUC(ROC) of the TILAR method is stillrather low. In our opinion this is not a general weaknessof the modeling, but due to the fact that the informationwe used for model validation was obtained by text min-ing. This information is therefore incomplete and error-prone. A drawback is that the text mining network wasconstructed by searching through all biological literatureand not only RA specific literature. Besides, text mining isobviously inappropriate to assess so far unknown regula-tory interactions. In fact, the GRN model now providesnew hypotheses that may be tested experimentally. How-ever, there are several other factors that impede an accu-rate GRN reconstruction or an adequate performance
evaluation. First, regulatory networks can exhibit largedynamic topological changes [50]. Thus, among the inter-actions in our network we might only identify the mostrobust ones or those that are most relevant in the specificstudy, implying that some other could be missed, even ifthey have been biologically demonstrated. Second, thecontribution of different cell types is lost in the study.Third, the text mining network contains undirected gene-gene interactions. In contrast, the proposed modelingapproach assigns directed interactions between genes andTFs, i.e. gene-gene interactions are only implicitly definedin the model. Fourth, the network model might be toosimple to reliably infer more complex interactions. Here,
ROC curves for the benchmarking gene regulatory networkFigure 6ROC curves for the benchmarking gene regulatory network. The better a method performs, the closer its curve will be to the upper-left corner. The black curve represents the rating of our method when including 54 of 67 predicted TF-gene interactions. Remarkably, TILAR not only outperforms CLR, ARACNE, GeneNet and the conventional Lasso, but can also be combined with the adaptive LARS if adequate prior knowledge on gene-TF interactions is available. Using both techniques in combination we could infer gene-gene relationships more reliably.
Page 12 of 18(page number not for citation purposes)
BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
we assumed that gene regulatory effects are linear andadditive, and precluded auto-regulation, i.e. a gene is notallowed to control a TF for which it possesses a TFBS. Thelatter because the data were not well suited for inferring adynamic model. Moreover, the inference algorithm isbased on co-expression at the transcriptional level, even ifthe amount of mRNA may not correspond to the level and(regulatory) activity of the proteins. However, more datawould be required to infer more accurate models.
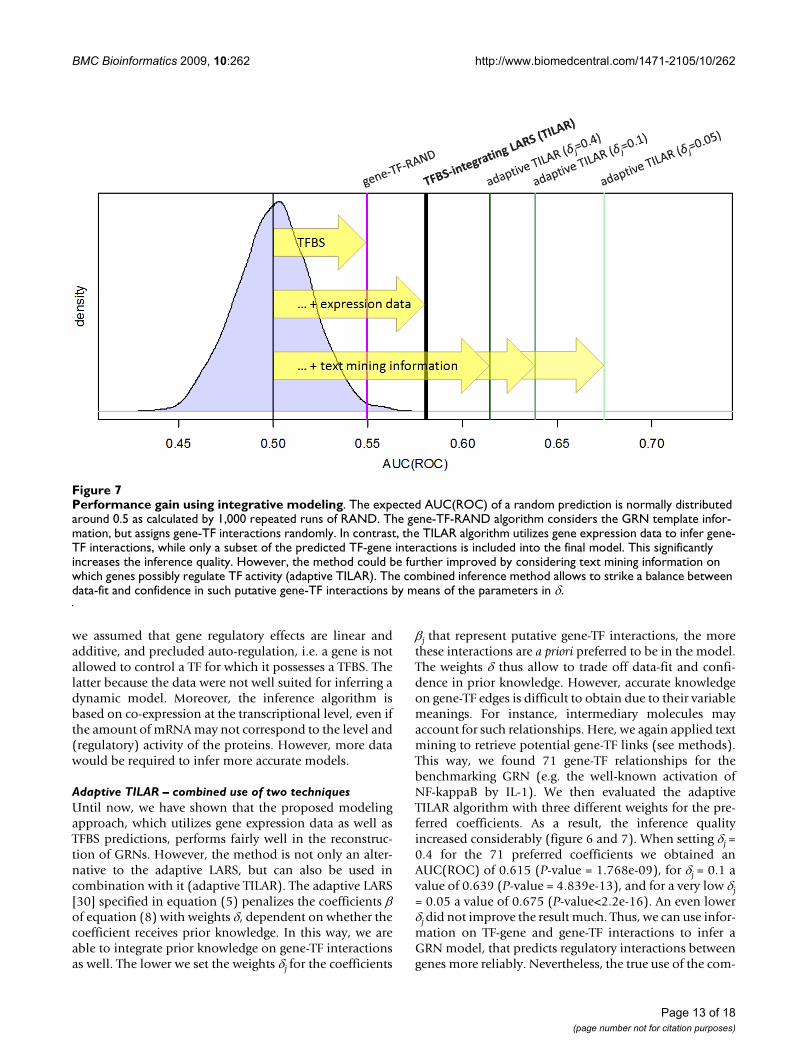
Adaptive TILAR – combined use of two techniquesUntil now, we have shown that the proposed modelingapproach, which utilizes gene expression data as well asTFBS predictions, performs fairly well in the reconstruc-tion of GRNs. However, the method is not only an alter-native to the adaptive LARS, but can also be used incombination with it (adaptive TILAR). The adaptive LARS[30] specified in equation (5) penalizes the coefficients of equation (8) with weights , dependent on whether thecoefficient receives prior knowledge. In this way, we areable to integrate prior knowledge on gene-TF interactionsas well. The lower we set the weights j for the coefficients
j that represent putative gene-TF interactions, the morethese interactions are a priori preferred to be in the model.The weights thus allow to trade off data-fit and confi-dence in prior knowledge. However, accurate knowledgeon gene-TF edges is difficult to obtain due to their variablemeanings. For instance, intermediary molecules mayaccount for such relationships. Here, we again applied textmining to retrieve potential gene-TF links (see methods).This way, we found 71 gene-TF relationships for thebenchmarking GRN (e.g. the well-known activation ofNF-kappaB by IL-1). We then evaluated the adaptiveTILAR algorithm with three different weights for the pre-ferred coefficients. As a result, the inference qualityincreased considerably (figure 6 and 7). When setting j =0.4 for the 71 preferred coefficients we obtained anAUC(ROC) of 0.615 (P-value = 1.768e-09), for j = 0.1 avalue of 0.639 (P-value = 4.839e-13), and for a very low j= 0.05 a value of 0.675 (P-value<2.2e-16). An even lowerj did not improve the result much. Thus, we can use infor-mation on TF-gene and gene-TF interactions to infer aGRN model, that predicts regulatory interactions betweengenes more reliably. Nevertheless, the true use of the com-
Performance gain using integrative modelingFigure 7Performance gain using integrative modeling. The expected AUC(ROC) of a random prediction is normally distributed around 0.5 as calculated by 1,000 repeated runs of RAND. The gene-TF-RAND algorithm considers the GRN template infor-mation, but assigns gene-TF interactions randomly. In contrast, the TILAR algorithm utilizes gene expression data to infer gene-TF interactions, while only a subset of the predicted TF-gene interactions is included into the final model. This significantly increases the inference quality. However, the method could be further improved by considering text mining information on which genes possibly regulate TF activity (adaptive TILAR). The combined inference method allows to strike a balance between data-fit and confidence in such putative gene-TF interactions by means of the parameters in .
Page 13 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
bination of adaptive and TFBS-integrating LARS requiresmore investigation. For instance, the determination of theweights is not straightforward, as we might set differentweights for each gene-TF interaction and even relativelyhigh weights (i.e. j>1.0) if certain gene-TF relationshipscan be excluded a priori. However, this is beyond the scopeof this paper.
ConclusionWe developed TILAR – a method for deriving transcrip-tional regulatory networks from gene expression data byintegrating TF binding predictions. The algorithm is alsoable to incorporate prior knowledge on the putative regu-lators of TF activity (adaptive TILAR). Our linear, additivemodeling approach distinguishes genes and TFs in thenetwork, and identifies the connections between thembased on the fast LARS regression algorithm and specificconstraints on the network structure. The major advantageof this modeling strategy is that only few model parame-ters are sufficient for a complex network, which is still easyto interpret. When applied on short-term gene expressionprofiles of RA patients treated with etanercept, themethod uncovers molecular immunotherapeutic effectsand thus provides testable hypotheses about the drugs'mechanisms of action. A closer look on the modelrevealed genes co-regulated by a common TF and TFs thatregulate functionally related genes. Moreover, the recon-structed GRN exhibits a scale-free, self-regulating andmassively parallel architecture.
We evaluated the inference quality using a text miningnetwork and found that our modeling method outper-forms all other algorithms tested. Notably, TILAR allowsfor a higher prediction accuracy than using just geneexpression data or TF binding information alone. Moreefforts are needed to study different configurations ofTILAR, e.g. we could analyze a larger DNA region for over-represented TFBS, and to assess the benefit of combiningthis method with the adaptive LARS. Besides, furtherexperiments need to be performed to verify specific inter-actions that were predicted by the model. However, evenif significant theoretical and experimental challengesremain, we could demonstrate that organizing heteroge-neous data and prior biological knowledge in systemsbiological models can strongly support the investigationof autoimmune diseases and their therapies. Supplemen-tary materials including R codes are available at http://www.hki-jena.de/index.php/0/2/490.
MethodsDNA microarray data pre-processingWe used the human DNA microarray dataset from Koczanet al. [13] including expression profiles of 19 etanercept-treated RA patients. Blood samples were taken for eachpatient before treatment as well as 72 and 144 hours after
first application of etanercept. Transcriptional levels ofPBMC were then measured using Affymetrix HumanGenome U133A arrays. As for 2 patients the third time-point is missing, the dataset consists of 55 microarrayexperiments. In the applied Affymetrix microarrays mostprobesets include probes matching transcripts from morethan one gene and probes which do not match any tran-scribed sequence. Therefore, we utilized a custom chipdefinition file (CDF), that is based on the informationcontained in the GeneAnnot database [15,51]. GeneAn-not-based CDFs are composed of probesets includingonly probes matching a single gene and thus allow for amore reliable determination of expression levels. We usedversion 1.4.0 of the custom CDF and the MAS5.0 algo-rithm to pre-process the raw probe intensities. Data nor-malization was performed by a loess fit to the whole datawith span = 0.05 (using R package affy). Finally, the dataprocessing yields mRNA abundances of 11,174 differentgenes.
Filtering differentially expressed genesThe filtering aims to identify a subset of genes significantlyup- or down-regulated within the first week of therapy. Awidely used filter criterion is the (logarithmized) fold-change from baseline. However, a fixed fold-changethreshold ignores the inherent structure of DNA microar-ray data. Therefore, we applied an MA-plot-based signalintensity-dependent fold-change criterion (MAID filter-ing) to select genes. The MAID filtering takes into accountthat the variability in the log fold-changes increases as themeasured signal intensity decreases [52]. First, the filter-ing procedure calculates for each gene the values A and M,which are commonly used for visualizing microarray datain an MA-plot. A is the log signal intensity of a gene aver-aged over all patients, while M is the mean intensity log-ratio between the baseline levels and the expression levelsat day 3 and 6, respectively. Then, the intensity-dependentvariability in the data is estimated by computing the inter-quartile range (IQR) of the M values in a sliding window.Afterwards, an exponential function f(x) = a·e-bx+c is fittedto the IQR's by a non-linear robust regression, which inturn is used to calculate so-called MAID-scores by dividingeach M value by f(A). As a consequence, the absolutevalue of a gene's MAID-score is higher, the more itsexpression level is altered after start of therapy. Further-more, we assessed which genes are differentially expressedaccording to a paired t-test comparing the expression lev-els at day 3 and 6 versus baseline, respectively. Finally, weselected the genes having |MAID-score|>2.5 and t-test P-value < 0.05.
GO analysisOverrepresented GO terms were found using GOstats, aBioconductor package written in R. Each GO term is testedwhether it is significantly associated to the list of filtered
Page 14 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
genes out of the 11,174 measured genes. The analysis wasperformed for gene functional annotations of the biolog-ical process GO category.
Identification of TF-gene interactions (GRN template)TFBS were derived from the UCSC database build hg18[53]. The database provides a TFBS conserved (tfbsCons-Sites) track, that contains the location and score of TFBSconserved in the human/mouse/rat alignment. The dataare purely computational and were generated using posi-tion weight matrices (PWMs) for TFBS contained in thepublic Transfac Matrix and Factor databases created byBiobase. For the whole human genome 3,837,187 TFBSpredictions associated to 258 different PWMs (184unique TF identifiers) can be found in the tfbsConsSitestrack. We defined the regulatory region of each gene as the1,000 bp up- and downstream of the TSS (as stated inGeneCards database 2.38). This specification is in agree-ment with current findings by the ENCODE pilot projectwhich revealed that regulatory sequences are symmetri-cally distributed around the TSS with no bias towardsupstream regions [54]. Then, we scanned the regulatoryregions of the selected genes for overrepresented TFBS. Indoing so, each TF is tested whether its binding site occursin this region for more genes than would be expected bychance. To take into account the inherent redundancy ofthe Transfac database, a TF is supposed to regulate a gene(TF-gene interaction) if any PWM for this TF matches theDNA sequence at the gene's regulatory region. Using ahypergeometric test analyzing the TF binding predictionsfor all the 11,174 genes measured, we can identify a subsetof TFs associated to the genes in the network at the signif-icance level = 0.05. This leads to a list of predicted TF-gene interactions that can serve as a template for GRNmodeling.
TFBS-integrating GRN inference (TILAR algorithm)First, the expression levels of each gene were standardizedso that they have variance 1 and mean 0. Given these data,we then defined a regression equation according to equa-tion (8), while considering the full set of putative TF-geneinteractions. Afterwards, we calculated all LARS estimates(steps) for this equation using the R package lars withdefault settings. Each LARS estimate specifies a subset ofcovariates, i.e. states which gene-TF interactions arepresent in the model and which are not (in the latter casethe corresponding model parameter is set to zero). Toselect a single estimate, we chose the model that mini-mizes Mallows' Cp statistic [55], thereby preventing over-fitting and ensuring sparseness. The whole procedure wasthen repeated in a backward stepwise selection scheme inwhich TF-gene interactions were iteratively eliminated (orreinserted) if this allowed for a model that exhibits asmaller residual sum of squares (RSS). In this way, a sub-set of TF-gene interactions was found. For the regression
equation including this subset all possible Lasso estimates(see equation (4)) are provided by LARS. We then calcu-lated for each LARS step the OLS fit using only the respec-tive covariates (LARS/OLS hybrid [27]). Hence, we usedLARS for variable selection, but not to estimate the modelcoefficients. Moreover, we evaluated the 10-fold cross-val-idation error (CVerror) for each LARS/OLS solution andfinally selected the most parsimonious model within 1standard deviation from the CVerror minimum (additionalfile 4). It should be noted that we used the Cp statistic asa crude selection criterion during the backward stepwiseselection procedure, because the Cp is much faster to com-pute than the CVerror.
To integrate prior knowledge on gene-TF interactions (aswe did for the benchmarking GRN) we strictly followedthe above learning strategy, except that we employed theadaptive variant of the LARS algorithm according to Zou[30]. The adaptive LARS assigns weights to each coeffi-cient as written in equation (5). We penalized coefficientsj for which we have no prior knowledge with a neutralweight j = 1.0. If literature mining suggested a gene-TFinteraction we penalized the corresponding coefficientwith a smaller j (0.4, 0.1 and 0.05, respectively) toimprove variable selection.
The learning strategy of the (adaptive) TILAR is summa-rized as follows:
1. Define D as the given (standardized) gene expres-sion data
2. Define P as the given set of putative TF-gene interac-tions (GRN template)
3. Use D to specify regression equation L subject to Paccording to equation (8)
4. Solve L using (adaptive) LARS and calculateRSS(Cpmin), i.e. the RSS of the LARS estimate that min-imizes Cp
5. Optional: Perform a backward stepwise selection onP, i.e. iteratively and exhaustively remove or reinsertelements in P and repeat 3. and 4., and stop when alocal minimum for RSS(Cpmin) is found
6. Recompute the regression coefficients to L in termsof a LARS/OLS hybrid and return the most parsimoni-ous estimate within 1 standard deviation of the 10-fold CVerror minimum
Performance evaluationWe used gene-gene interaction information for bench-marking. The software PathwayArchitect 2.0.1 was
Page 15 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
employed to automatically extract such gene-gene linksfrom the literature. We retrieved only gene-gene interac-tions of context type "expression" and "regulation" aslabeled by PathwayArchitect. To obtain putative gene-TFlinks (which were used for the adaptive TILAR) we alsoconsidered interactions of type "protein modification".The gene-gene information was applied to assess the infer-ence quality of our and a total of four other easy-to-applyGRN inference methods, namely CLR, ARACNE,GeneNet, and the conventional Lasso, while we did nottake into account the directions of the relationships. CLR,ARACNE and GeneNet are thought to build (undirected)gene association networks and have been implemented byuse of the R packages minet and GeneNet. To compute theentire set of Lasso solutions to equation (3) we used theLARS modification (R package lars). All methods were runon standardized gene expression levels with default set-tings. Moreover, a random inference algorithm calledRAND was implemented, which randomly assigns con-nections between genes until a fully connected network isformed. The RAND method was further adapted to infernetworks of TF-gene and gene-TF interactions similar tothe proposed modeling scheme (gene-TF-RAND). Morespecifically, gene-TF-RAND utilizes all the TF-gene inter-actions predicted by the TFBS overrepresentation analysisand randomly adds gene-TF edges to the network. As forTILAR, gene-TF interactions were not allowed when geneand TF were already connected by a TF-gene interaction,and gene-gene links result implicitly. The AUC(ROC)value of gene-TF-RAND was obtained by the mean of1,000 repeated runs. Apart from that, we tested whetherany inference technique performed significantly betterthan a random prediction. For this purpose, we calculatedP-values which specify the probability that an AUC(ROC)value computed by RAND will be higher than theAUC(ROC) value of the particular inference algorithm.The P-values are calculated by 1 minus the cumulativeprobabilities, which are evaluated at the AUC(ROC) valueof the respective method, of the normal distribution hav-ing the mean and standard deviation of 1,000 RAND-cal-culated AUC(ROC) values.
AbbreviationsTF: transcription factor; GRN: gene regulatory network;RA: rheumatoid arthritis; TFBS: TF binding site; LARS:least angle regression; OLS: ordinary least squares; GO:gene ontology; TILAR: TFBS-integrating LARS; PBMC:peripheral blood mononuclear cells; MAID: MA-plot-based signal intensity-dependent fold-change criterion;TSS: transcription start site; Lasso: least absolute shrinkageand selection operator; FPR: false positive rate; ROC:receiver operating characteristic; AUC: area under thecurve; CDF: chip definition file; IQR: interquartile range;PWM: position weight matrix; RSS: residual sum ofsquares.
Conflict of interestsThe authors declare that they have no competing interests.
Authors' contributionsRGu and H-JT directed the study. MH carried out the anal-yses on the data and wrote the paper. RGu, RGo and REassisted in interpretation of the results and contributed towriting the paper. All authors read and approved the finalmanuscript.
Additional material
Additional file 1List of 83 genes with significant expression changes during first week of therapy. The table provides diverse types of information for each gene, e.g. Entrez ID, official full name and the calculated MAID-scores.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2105-10-262-S1.xls]
Additional file 2Filtering of genes regulated in response to etanercept therapy. (A) Super-imposed MA-plot visualizing the applied gene filtering method. Here, gene expression levels measured 3 days after therapy onset are compared with baseline levels. The MAID filtering takes into account that the variability in the mean log-fold changes (M) depends on the mean log signal inten-sity (A). 37 genes showed an up- or down-regulation at day 3 (green). (B) In a similar manner, 57 genes were found higher or lower expressed at day 6 in comparison to baseline. In this way, 83 different genes were selected in total. (C) Mean time-courses of these 83 genes. 25 genes were found up- or down-regulated at day 3 (left), 45 at day 6 (middle) and 13 at day 3 and 6 (right).Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2105-10-262-S2.png]
Additional file 3Overrepresented terms of the GO biological process ontology. P-values were computed for each GO term based on the hypergeometric distribu-tion. Only functional categories with P-value < 0.01 and where at least 3 out of 83 genes are associated ("Count") are shown.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2105-10-262-S3.xls]
Additional file 4Model selection using cross-validation. Training error (scaled by 10) and 10-fold CVerror (RSS mean of 10 subsets) are shown for the LARS/OLS solutions of the first 300 LARS steps. The blue area represents the stand-ard deviation of CVerror. The red line shows the LARS step selected for the final model, i.e. the most parsimonious model within 1 standard deviation from the CVerror curve minimum, for which 22 model parameters are non-zero.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2105-10-262-S4.png]
Page 16 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
AcknowledgementsWe thank Peter Lorenz and Dirk Koczan for helpful discussions. Thanks to Steven John Smith for critical proofreading of the manuscript.
Funding: This study was supported by grants from the German Federal Min-istry of Education and Research (BMBF, BioChancePlus, 0313692D).
References1. Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, Hor-
nischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S,Lewicki-Potapov B, Michael H, Münch R, Reuter I, Rotert S, Saxel H,Scheer M, Thiele S, Wingender E: TRANSFAC: transcriptionalregulation, from patterns to profiles. Nucleic Acids Res 2003,31(1):374-378.
2. Filkov V: Identifying Gene Regulatory Networks from GeneExpression Data. In Handbook of Computational Molecular BiologyEdited by: Aluru S. Chapman & Hall/CRC Press; 2005:27.1-27.29.
3. Hecker M, Lambeck S, Toepfer S, van Someren EP, Guthke R: Generegulatory network inference: Data integration in dynamicmodels – A review. Biosystems 2009, 96(1):86-103.
4. Cho KH, Choo SM, Jung SH, Kim JR, Choi HS, Kim J: Reverse engi-neering of gene regulatory networks. IET Syst Biol 2007,1(3):149-163.
5. Werhli AV, Husmeier D: Reconstructing gene regulatory net-works with bayesian networks by combining expression datawith multiple sources of prior knowledge. Stat Appl Genet MolBiol 2007, 6:Article 15.
6. Benson M, Breitling R: Network theory to understand microar-ray studies of complex diseases. Curr Mol Med 2006,6(6):695-701.
7. Kavanaugh AF, Lipsky P: Rheumatoid arthritis. In Inflammation:Basic Principles and Clinical Correlates 3rd edition. Edited by: Gallin JI,Snyderman R. Philadelphia, PA, USA: Lippincott Williams & Wilkins;1999:1017-1037.
8. McInnes IB, Schett G: Cytokines in the pathogenesis of rheuma-toid arthritis. Nat Rev Immunol 2007, 7(6):429-442.
9. Brennan FM, Maini RN, Feldmann M: Role of pro-inflammatorycytokines in rheumatoid arthritis. Springer Semin Immunopathol1998, 20(1–2):133-147.
10. Smolen JS, Steiner G: Therapeutic strategies for rheumatoidarthritis. Nat Rev Drug Discov 2003, 2(6):473-488.
11. Feldmann M, Maini RN: Lasker Clinical Medical ResearchAward. TNF defined as a therapeutic target for rheumatoidarthritis and other autoimmune diseases. Nat Med 2003,9(10):1245-1250.
12. Glocker MO, Guthke R, Kekow J, Thiesen HJ: Rheumatoid arthri-tis, a complex multifactorial disease: on the way toward indi-vidualized medicine. Med Res Rev 2006, 26(1):63-87.
13. Koczan D, Drynda S, Hecker M, Drynda A, Guthke R, Kekow J,Thiesen HJ: Molecular discrimination of responders and non-responders to anti-TNFalpha therapy in rheumatoid arthri-tis by etanercept. Arthritis Res Ther 2008, 10(3):R50.
14. Fernald GH, Knott S, Pachner A, Caillier SJ, Narayan K, Oksenberg JR,Mousavi P, Baranzini SE: Genome-wide network analysis revealsthe global properties of IFN-beta immediate transcriptionaleffects in humans. J Immunol 2007, 178(8):5076-5085.
15. Ferrari F, Bortoluzzi S, Coppe A, Sirota A, Safran M, Shmoish M, Fer-rari S, Lancet D, Danieli GA, Bicciato S: Novel definition files forhuman GeneChips based on GeneAnnot. BMC Bioinformatics2007, 8:446.
16. Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE,Myers RM, Speed TP, Akil H, Watson SJ, Meng F: Evolving gene/transcript definitions significantly alter the interpretation ofGeneChip data. Nucleic Acids Res 2005, 33(20):e175.
17. Boissier MC, Assier E, Biton J, Denys A, Falgarone G, Bessis N: Reg-ulatory T cells (Treg) in rheumatoid arthritis. Joint Bone Spine2009, 76(1):10-14.
18. Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y,Wang HY, Wang RF: Toll-like receptor 8-mediated reversal ofCD4+ regulatory T cell function. Science 2005,309(5739):1380-1384.
19. Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, NakajimaT, Hirano T, Kishimoto T: A nuclear factor for IL-6 expression(NF-IL6) is a member of a C/EBP family. EMBO J 1990,9(6):1897-1906.
20. Yang C, Bolotin E, Jiang T, Sladek FM, Martinez E: Prevalence of theinitiator over the TATA box in human and yeast genes andidentification of DNA motifs enriched in human TATA-lesscore promoters. Gene 2007, 389(1):52-65.
21. D'haeseleer P, Wen X, Fuhrman S, Somogyi R: Linear modeling ofmRNA expression levels during CNS development andinjury. Pac Symp Biocomput 1999, 4:41-52.
22. van Someren EP, Vaes BL, Steegenga WT, Sijbers AM, Dechering KJ,Reinders MJ: Least absolute regression network analysis of themurine osteoblast differentiation network. Bioinformatics 2006,22(4):477-484.
23. Yong-A-Poi J: Adaptive least absolute regression networkanalysis improves genetic network reconstruction byemploying prior knowledge. In Master's thesis Delft University ofTechnology, Department of Mediamatics; 2008.
24. Thorsson V, Hörnquist M, Siegel AF, Hood L: Reverse engineeringgalactose regulation in yeast through model selection. StatAppl Genet Mol Biol 2005, 4:Article 28.
25. Guthke R, Möller U, Hoffmann M, Thies F, Töpfer S: Dynamic net-work reconstruction from gene expression data applied toimmune response during bacterial infection. Bioinformatics2005, 21(8):1626-1634.
26. Tibshirani R: Regression shrinkage and selection via the lasso.J Royal Statist Soc B 1996, 58:267-288.
Additional file 5Zip-archived Cytoscape session file of the reconstructed GRN. The net-work model contains predicted regulatory interactions of genes responsive to etanercept therapy in RA. A simplified visualization of the network is shown in figure 3, while detail views are shown in figure 5.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2105-10-262-S5.zip]
Additional file 6List of the 50 most frequently mentioned genes in the context of RA. 42 of these genes were measured in the dataset and used to evaluate the per-formance of our modeling approach.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2105-10-262-S6.xls]
Additional file 7Overrepresented binding sites for the benchmarking gene regulatory net-work. 10 transcription factors represented by 20 Transfac binding profiles were found to be enriched, providing 67 predicted TF-gene interactions in total.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2105-10-262-S7.xls]
Additional file 8Recall-precision curves for the benchmarking GRN. We evaluated the per-formance of different modeling strategies based on gene-gene relationships found by text mining. The black curve represents the rating of our method when including 54 of 67 predicted TF-gene interactions. The TILAR approach outperforms CLR, ARACNE, GeneNet and the conventional Lasso. When used in combination with the adaptive LARS we could fur-ther increase the inference quality.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2105-10-262-S8.png]
Page 17 of 18(page number not for citation purposes)

BMC Bioinformatics 2009, 10:262 http://www.biomedcentral.com/1471-2105/10/262
Publish with BioMed Central and every scientist can read your work free of charge
"BioMed Central will be the most significant development for disseminating the results of biomedical research in our lifetime."
Sir Paul Nurse, Cancer Research UK
Your research papers will be:
available free of charge to the entire biomedical community
peer reviewed and published immediately upon acceptance
cited in PubMed and archived on PubMed Central
yours — you keep the copyright
Submit your manuscript here:http://www.biomedcentral.com/info/publishing_adv.asp
BioMedcentral
27. Efron B, Hastie T, Johnstone I, Tibshirani R: Least angle regres-sion. Ann Statist 2004, 32(2):407-499.
28. van Someren EP, Wessels LFA, Reinders MJT, Backer E: Regulariza-tion and noise injection for improving genetic network mod-els. In Computational And Statistical Approaches To Genomics Edited by:Zhang W, Shmulevich I. Boston, MA, USA: Kluwer Acadmic Publish-ers; 2002:211-226.
29. Gustafsson M, Hörnquist M, Lombardi A: Constructing and ana-lyzing a large-scale gene-to-gene regulatory network – lasso-constrained inference and biological validation. IEEE/ACMTrans Comput Biol Bioinform 2005, 2(3):254-261.
30. Zou H: The adaptive lasso and its oracle properties. J Am StatAssoc 2006, 101:1418-1429.
31. Kitano H: Foundations of Systems Biology Cambridge, MA, USA: MITPress; 2001.
32. Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, CalifanoA: Reverse engineering of regulatory networks in human Bcells. Nat Genet 2005, 37(4):382-390.
33. Jordan IK, Mariño-Ramírez L, Wolf YI, Koonin EV: Conservationand coevolution in the scale-free human gene coexpressionnetwork. Mol Biol Evol 2004, 21(11):2058-2070.
34. Clauset A, Shalizi CR, Newman MEJ: Power-law distributions inempirical data. 2007 [http://arxiv.org/abs/0706.1062].
35. Ishibashi K, Kuwahara M, Gu Y, Tanaka Y, Marumo F, Sasaki S: Clon-ing and functional expression of a new aquaporin (AQP9)abundantly expressed in the peripheral leukocytes permea-ble to water and urea, but not to glycerol. Biochem Biophys ResCommun 1998, 244(1):268-274.
36. Strubin M, Newell JW, Matthias P: OBF-1, a novel B cell-specificcoactivator that stimulates immunoglobulin promoter activ-ity through association with octamer-binding proteins. Cell1995, 80(3):497-506.
37. Rey M, Vicente-Manzanares M, Viedma F, Yáñez-Mó M, Urzainqui A,Barreiro O, Vázquez J, Sánchez-Madrid F: Cutting edge: associa-tion of the motor protein nonmuscle myosin heavy chain-IIAwith the C terminus of the chemokine receptor CXCR4 in Tlymphocytes. J Immunol 2002, 169(10):5410-5414.
38. O'Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B:Acute-phase serum amyloid A production by rheumatoidarthritis synovial tissue. Arthritis Res 2000, 2(2):142-144.
39. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G: Nod2,a Nod1/Apaf-1 family member that is restricted to mono-cytes and activates NF-kappaB. J Biol Chem 2000,276(7):4812-4818.
40. Salmi M, Koskinen K, Henttinen T, Elima K, Jalkanen S: CLEVER-1mediates lymphocyte transmigration through vascular andlymphatic endothelium. Blood 2004, 104(13):3849-3857.
41. Qian F, Kruse U, Lichter P, Sippel AE: Chromosomal localizationof the four genes (NFIA, B, C, and X) for the human tran-scription factor nuclear factor I by FISH. Genomics 1995,28(1):66-73.
42. Goertsches RH, Hecker M, Zettl UK: Monitoring of multiple scle-rosis immunotherapy: From single candidates to biomarkernetworks. J Neurol 2008, 255(S6):48-57.
43. Stolovitzky G, Monroe D, Califano A: Dialogue on reverse-engi-neering assessment and methods: the DREAM of high-throughput pathway inference. Ann N Y Acad Sci 2007,1115:1-22.
44. Stolovitzky G, Prill RJ, Califano A: Lessons from the DREAM2Challenges. Ann N Y Acad Sci 2009, 1158:159-195.
45. Karopka T, Fluck J, Mevissen HT, Glass A: The Autoimmune Dis-ease Database: a dynamically compiled literature-deriveddatabase. BMC Bioinformatics 2006, 7:325.
46. Kinne RW, Bräuer R, Stuhlmüller B, Palombo-Kinne E, Burmester GR:Macrophages in rheumatoid arthritis. Arthritis Res 2000,2(3):189-202.
47. Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G,Kasif S, Collins JJ, Gardner TS: Large-scale mapping and valida-tion of Escherichia coli transcriptional regulation from acompendium of expression profiles. PLoS Biol 2007, 5(1):e8.
48. Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, DallaFavera R, Califano A: ARACNE: an algorithm for the recon-struction of gene regulatory networks in a mammalian cellu-lar context. BMC Bioinformatics 2006, 7(Suppl 1):S7.
49. Opgen-Rhein R, Strimmer K: From correlation to causation net-works: a simple approximate learning algorithm and its
application to high-dimensional plant gene expression data.BMC Syst Biol 2007, 1:37.
50. Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, GersteinM: Genomic analysis of regulatory network dynamics revealslarge topological changes. Nature 2004, 431(7006):308-312.
51. Chalifa-Caspi V, Shmueli O, Benjamin-Rodrig H, Rosen N, Shmoish M,Yanai I, Ophir R, Kats P, Safran M, Lancet D: GeneAnnot: interfac-ing GeneCards with high-throughput gene expression com-pendia. Brief Bioinform 2003, 4(4):349-360.
52. Yang IV, Chen E, Hasseman JP, Liang W, Frank BC, Wang S, Sharov V,Saeed AI, White J, Li J, Lee NH, Yeatman TJ, Quackenbush J: Withinthe fold: assessing differential expression measures andreproducibility in microarray assays. Genome Biol 2002,3(11):research0062.
53. Kuhn RM, Karolchik D, Zweig AS, Wang T, Smith KE, RosenbloomKR, Rhead B, Raney BJ, Pohl A, Pheasant M, Meyer L, Hsu F, HinrichsAS, Harte RA, Giardine B, Fujita P, Diekhans M, Dreszer T, ClawsonH, Barber GP, Haussler D, Kent WJ: The UCSC GenomeBrowser Database: update 2009. Nucleic Acids Res2009:D755-761.
54. ENCODE Project Consortium: Identification and analysis offunctional elements in 1% of the human genome by theENCODE pilot project. Nature 2007, 447(7146):799-816.
55. Mallows C: Some comments on cp. Technometrics 1973,15:661-675.
Page 18 of 18(page number not for citation purposes)