in situ activation of sting pathway with polymeric sn38
TRANSCRIPT

Biomaterials 268 (2021) 120542
Available online 21 November 20200142-9612/© 2020 Elsevier Ltd. All rights reserved.
In situ activation of STING pathway with polymeric SN38 for cancer chemoimmunotherapy
Jiayu Zhao a,b, Sheng Ma a,b, Yudi Xu a,c, Xinghui Si a,b, Haochen Yao a,d, Zichao Huang a,b, Yu Zhang a,e, Haiyang Yu a,e, Zhaohui Tang a,b,e,**, Wantong Song a,e,*, Xuesi Chen a,b,e
a Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China b University of Science and Technology of China, Hefei, 230026, China c University of Chinese Academy of Sciences, Beijing, 100039, China d Department of Pathogenobiology, The Key Laboratory of Zoonosis, Chinese Ministry of Education, College of Basic Medical Science, Jilin University, Changchun, 130012, China e Jilin Biomedical Polymers Engineering Laboratory, Changchun, 130022, China
A R T I C L E I N F O
Keywords: DNA-Targeting agents SN38 Exosome STING pathway Chemoimmunotherapy
A B S T R A C T
STING (stimulator of interferon genes) signaling pathway has attracted considerable attention in cancer immunotherapy due to its capacity to boost vigorous antitumor immunity. However, the shortage of effective STING agonists limits the promotion of STING pathway in cancer treatment. Herein, we present an approach for in situ activation of STING pathway with nanoparticles delivered DNA-targeting chemo agents, based on the understanding that cytosol DNA is a pre-requisite for STING pathway activation. Through in vitro screening among several DNA-targeting chemo agents, we identified 7-ethyl-10-hydroxycamptothecin (SN38) as the most potent drug for stimulating interferon (IFN)-β secretion and proved that this process is mediated by the passage of DNA-containing exosomes from treated tumor cells to bone marrow-derived dendritic cells (BMDCs) and sub-sequent activation of the STING pathway. Furthermore, we designed a polymeric-SN38 conjugate that could self- assemble into nanoparticles (SN38-NPs) for in vivo application. The SN38-NPs formulation reduced toxicity of free SN38, effectively stimulated the activation of STING pathway in E0771 tumors, and resulted in a tumor suppression rate (TSR%) of 82.6%. Our results revealed a new mechanism of SN38 in cancer treatment and should inspire using more DNA-targeting agents, especially in nanoformulation, for activating STING pathway and cancer chemoimmunotherapy.
1. Introduction
Cancer immunotherapy aiming to stimulate the host’s immune sys-tem for cancer control is eliciting a revolution in cancer treatment [1–4]. Representative immunotherapeutic means like immune checkpoint in-hibitors (ICI) for relieving the immune negative regulation between tumor cells and the immune effector cells have gained great success in the clinic and been approved for the treatment of nearly 20 cancer types [5–7], however, only a small fraction of patients response to ICI treat-ment [8,9]. As a result, various means were developed for stimulating the activation of tumor-specific immune responses, including cancer
vaccines [10,11], immunogenic cell death [12–15], photodynamic therapy [16,17] or stimulators for pattern-recognition receptors (PRRs) [18,19]. STING is a cytosolic PRR that acts as an innate immune sensor for cyclic dinucleotides (CDNs) and an adaptor molecule for cytosolic DNA through the cyclic GMP-AMP synthase (cGAS)-STING signaling axis [20–23]. Recent studies identified the critical role of STING in cancer immune surveillance as STING-deficient mice have a higher susceptibility to tumor formation while activation of STING triggers robust antitumor immunity [24–26]. As a result, STING pathway is becoming an important research interest in recent cancer immuno-therapy studies.
* Corresponding author. Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. ** Corresponding author. Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022,
China. E-mail addresses: [email protected] (Z. Tang), [email protected] (W. Song).
Contents lists available at ScienceDirect
Biomaterials
journal homepage: www.elsevier.com/locate/biomaterials
https://doi.org/10.1016/j.biomaterials.2020.120542 Received 13 July 2020; Received in revised form 3 November 2020; Accepted 15 November 2020

Biomaterials 268 (2021) 120542
2
Currently, major available STING agonists are naturally occurring CDNs, including cGAMP, c-di-GMP, and c-di-AMP [27], which could bind with the STING dimer and stimulate STING-related type I IFN secretion [28–30]. Limited by the poor stability and poor bioavailability, direct usage of CDNs is weak in immune stimulation, and various nanoformulations were developed for enhancing the antitumor immu-nity of CDNs [31–33]. Vadimezan (DMXAA) is a small molecular STING agonist firstly discovered by Prantner et al. [34]. Although great tumor inhibition effect was observed in mouse models, DMXAA failed in clin-ical trials due to the differences in STING structure between mice and humans [35]. More recently, Ramanjulu et al. reported another small molecule as STING receptor agonist which is systemically efficacious for treating tumors in mice [36]. This diABZIs was designed with high-throughput screening of small molecules that compete with the binding of radio-labeled cGAMP to human STING and a linking strategy to synergize the effect of two symmetry-related ABZI. Overall, agonists for STING receptors are still lacking and the potential of stimulating STING pathway in the cancer immunotherapy field is still to be explored.
Since STING is an immune sensor for cytosol DNA, cytosol delivery or stimulating the production of cytosol DNA may be an alternative way for activation of the STING pathway. It has been known that some chemotherapeutic and radiotherapeutic approaches would induce DNA damage and DNA cytosol leakage, suggesting these treatments or these agents may have potential in activating the cGAS-STING pathway and inducing powerful immune responses. For instance, doxorubicin and daunorubicin have been demonstrated to induce IFN production and trigger the cGAS-STING pathway for circumventing Ebola virus immune evasion [37–39]. DNA damage from topoisomerase I inhibition with low-dose camptothecin was also reported to trigger strong antiviral immune response through cGAS detection of cytoplasmic DNA [40,41].
More recently, Kitai et al. reported that cancer cells treated with top-otecan would release exosomes containing DNA and activate DCs via STING signaling [42]. The above findings suggest that some specific DNA-targeting agents may be applied for in situ activation of the STING pathway and reinforce antitumor immunity inside the tumor microen-vironment if they are properly delivered to the tumor site. Unlike directly injecting STING agonists into the tumor, the method of deliv-ering DNA-targeting chemo drugs to tumor sites may combine chemo-therapy and immunotherapy for cancer treatment and maybe quite attractive for cancer chemoimmunotherapy.
In this study, we screened a quantity of DNA-targeting agents in vitro for stimulating the STING pathway activation. SN38, a topoisomerase I inhibitor, was identified as the top candidate among these drugs to stimulate the cytosol DNA transfer from tumor cells to antigen- presenting cells and elicit robust type I IFN production and STING pathway activation. For prolonging the blood circulation and reducing the adverse effects on the immune system, we designed a polymeric SN38 nanoformulation for in vivo delivery, and the SN38-NPs markedly inhibited murine breast cancer E0771 tumor growth (Scheme 1). We believe this study will inspire using nanotechnology to deliver DNA- targeting agents to in situ motivating STING pathway for cancer chemoimmunotherapy.
2. Materials and methods
2.1. Materials
γ-Benzyl-L-glutamate-N-carboxylic anhydride (BLG-NCA) was pur-chased from Shanghai Yeexin Biochem&Tech Co., Ltd. Methoxy poly (ethylene glycol) with 5000 Da (mPEG5K) was purchased from Sigma-
Scheme 1. Schematic illustration of SN38-NPs for in situ activating STING pathway and cancer chemoimmunotherapy. Tumor-bearing mice were injected with self- assembled SN38-NPs, and the following processes occurred in the tumor microenvironment: (1) SN38-NPs were internalized by tumor cells, and the released DNA- targeted agents SN38 caused DNA damage and leakage to cytosol of the tumor cells; (2) Cytosol DNA was transferred from tumor cells to dendritic cells (DCs) through exosomes; (3) STING pathway was stimulated in situ in DCs and DCs were activated; (4) Innate immune responses including type I IFN (IFN-I) and activated NK cells suppressed tumor growth. The SN38 induced tumor cell apoptosis and STING-related innate immunity activation achieve synergistic chemoimmunotherapy for solid tumors.
J. Zhao et al.

Biomaterials 268 (2021) 120542
3
Aldrich. 7-Ethyl-10-hydroxycamptothecin (SN38) was obtained from Tokyo Chemical Industry (TCI) Co., Ltd. Doxorubicin hydrochloride (DOX⋅HCl) was purchased from Beijing Huafeng United Technology Corporation. Topotecan hydrochloride (TPT⋅HCl) was obtained from Dalian Meilun Biotechnology Co., Ltd. IR, CDDP and Cy5-NHS ester were obtained from Dalian Meilun biotechnology Co., Ltd. 3-(4,5- dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) and Cell Counting Kit-8 (CCK8) were obtained from Solarbio Co., Ltd. 2,4,6- trichlorobenzoylchloride was bought from Beijing J&K Co., Ltd. Dime-thylaminopyridine (DMAP) was purchased from Aladdin Reagent Co., Ltd. Mouse IFN-beta DuoSet ELISA (DY8234) and Mouse IL-6 DuoSet ELISA (DY406) were obtained from R&D Systems Co., Ltd. Antibodies used for Western Blot, including interferon regulatory factor 3 (IRF-3) Rabbit mAb, Phospho-IRF-3 (Ser396) Rabbit mAb, cGAS Rabbit mAb, cGAS Rabbit mAb, Phospho-STING (Ser365) Rabbit mAb and STING Rabbit mAb were purchased from Cell Signaling Technology Co., Ltd. Roswell Park Memorial Institute (RPMI-1640), TNF alpha Mouse ELISA Kit, IFN gamma Mouse ELISA Kit and BCA protein assay kit were pur-chased from Thermal Fisher Scientific Co., Ltd. Fetal Bovine Serum (FBS) was obtained from Biological Industries. Murine GM-CSF and Murine IL-4 were obtained from Pepro Tech Co., Ltd. Antibodies used for flow cytometry were all purchased from BioLegend Co., Ltd.
2.2. Cell lines and cell culture
The murine medullary breast cancer E0771 cells and murine breast cancer 4T1 cells were purchased from Shanghai Guan Dao Co., Ltd. and cultured in RPMI-1640 (containing 10% fetal bovine serum (FBS), 50 U/ mL penicillin and 50 U/mL streptomycin) at 37 ◦C in an atmosphere of 5% CO2.
Bone marrow derived dendritic cells (BMDCs) were prepared ac-cording to Son’s method [43]. Briefly, femurs and tibias were removed out from 4 to 6 weeks old C57BL/6 mice and isolated the bone marrow cells. The bone marrow cells were incubated in RPMI-1640 containing 20 ng/mL GM-CSF and 10 ng/mL IL-4 for 6 days after lysis of erythro-cytes, during which half of the media were replaced with fresh RPMI-1640 twice and cytokines were supplemented, then the BMDCs were harvested at 7 days for further study.
2.3. Animals and tumor models
All animal studies were carried out according to the guidelines approved by the Animal Care and Use Committee of Jilin University. Female C57BL/6 mice (4–6 weeks or 6–8 weeks) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Female SD rats were purchased from Liaoning Changsheng biotechnology Co., Ltd.
The subcutaneous E0771 tumor model was established by injecting 1.0 × 106 E0771 cells into the right flank of 6–8 weeks old female C57BL/6 mice in 100 μL of PBS. When tumors sizes reached ~60 mm3, mice were treated with PBS, IR, SN38 or SN38-NPs intravenously.
2.4. Synthesis of PLG-g-mPEG/SN38 conjugates
Poly-(L-glutamic acid) graft poly-(ethylene glycol) (PLG-g-mPEG) was synthesized according to our previous work [44], with an average of 160 L-glutamic acid repeating units and 8.3 mPEG on side chains. SN38 was directly conjugated to the PLG-g-mPEG through the Yamaguchi reaction. Briefly, PLG-g-mPEG (0.8 g, 0.0.27 mmol) was added in a schlenk flask and dispersed in 16.0 mL anhydrous DMF under N2 con-dition, then the 2,4,6-trichlorobenzoylchloride (0.244 g, 1 mmol) and TEA (0.101 g, 1 mmol) were added in and the mixture was heated up to 60 ◦C for activating carboxyl groups. After 1 h, SN38 (0.2 g, 0.509 mmol) and DMAP (0.025 g, 0.2 mmol) were dissolved in 4.0 mL anhy-drous DMF and then injected into the PLG-g-mPEG reaction system for another 12 h reaction at 35 ◦C. The mixture solution was precipitated in excess diethyl ether and washed twice with ether, primrose yellow solid
was obtained with further dialysis and lyophilization. Polymer compo-sitions were characterized via 1HNMR in D2O or DMSO‑d6 on a Bruker AV-500 or Bruker AV-300 NMR spectrometer at 22 ◦C.
The samples of drug loading capacity (DLC) detection were prepared as follows: PLG-g-mPEG/SN38 was completely dissolved in 1 M NaOH solution for hydrolysis in a 37 ◦C shaker, 1 h later, the same volume of 1.2 M TFA was added. Then, DMSO was added in for dissolving the hydrolyzed SN38. The absorbance of SN38 was detected by fluo-rospectrophotometer on a microplate reader (Tecan Spark, Switzerland) with excitation wavelength of 270 nm and emission wavelength of 410 nm. The DLC was calculated referred to the equation below:
DLC =Amount of SN38 in NPs
Amount of NPs× 100%
2.5. Synthesis of Cy5-labeled PLG-g-mPEG/SN38
Cy5-NHS ester (5 mg, 0.0632 mmol) was dispersed in 1.5 mL DMSO, then the diehtylamine (7.59 mg, 0.13 mmol) was injected in system and stirred in darkness at 35 ◦C for 12 h. Take another reaction bottle, PLG-g- mPEG/SN38 (50 mg), EDC⋅HCl (2.88 mg, 0.015 mmol) and NHS (1.73 mg, 0.015 mmol) were dissolved with 1.5 mL DMSO. The system was stirred at room temperature for 3 h, following transferred to Cy5 reac-tion system for further reaction of 48 h. Later, the whole mixture system was removed in a dialysis bag (MWCO 1000 Da) for redundant Cy5 molecules elimination, obtaining aquamarine blue solid after lyophilization.
2.6. In vitro drug release of PLG-g-mPEG/SN38
The release buffer was made up of 0.1 M PB buffer containing 0.5% Tween 80. The release assays were carried out at different conditions: (1) pH 7.4, (2) pH 6.8, (3) pH 6.8 with 20 U/mL esterase. PLG-g-mPEG/ SN38 (containing 0.5 mg SN38) was dissolved in 5.0 mL release buffer and transferred into a dialysis bag (MWCO 30 KDa), then immersed into the 45.0 mL release buffer. At each scheduled time point, 1 mL of dialysis buffer was collected and supplemented with 1 mL fresh buffer. The amount of total released SN38 was evaluated with fluorospec-trophotometer consistent as described above.
2.7. In vitro cytotoxicity assays
The in vitro cytotoxicities of free SN38, TPT, DOX, CDDP and SN38- NPs to tumor cells were evaluated by MTT assay and cytotoxicities to BMDCs were calculated using CCK8 kits. Typically, 4000 tumor cells or 15000 BMDCs were seeded in 96-well culture plates per well with 200 μL RPMI-1640 for overnight incubation, then the culture media were removed out and supplemented with fresh media containing drugs (0.01–100 μM). After incubation for 48 h or 12 h, 20 μL 5% MTT solution or 20 μL CCK8 solution were injected in each well. For MTT assays, after 4 h incubation, abandoned culture medium and filled in 150 μL DMSO for detection. For CCK8 tests, the plates were directly tested after 1–2 h of incubation. The absorbance of each well was measured at 490 nm for MTT assays and 450 nm for CCK8 assays on a TECAN microplate reader. The percent of cell viability was calculated by comparing the optical density (OD) values of sample wells with that of control wells.
2.8. Exosomes isolation and DNA extraction
1.5 × 106 E0771 cells were seeded on three 100-mm dishes and incubated overnight, then the media was removed and supplemented with fresh media containing 5 μM SN38 or SN38-NPs. Collected the E0771 supernatant after 48 h incubation and removed out the cell debris via centrifugation at a speed of 3000 g for 10 min. Exosomes were extracted and purified by an exosome isolation kit (Umibio Co., LtD., UR52121). All operations were carried out following the product
J. Zhao et al.

Biomaterials 268 (2021) 120542
4
instructions. Then the content of exosomes was quantified by BCA as-says. About 3 mg of exosomes were obtained after the process. DNA was extracted and purified from exosomes by a TIANamp Genomic DNA Kit. The content of dsDNA was detected by a microplate reader. Purified DNA was stained with ethidium bromide (EB) following agarose gel electrophoresis and visualized by a UV transilluminator (Beijing Junyi Co., LtD., JY04S-3C).
2.9. Cellular uptake
2 × 105 E0771 cells were seeded per well in a 6-well plate and incubated overnight. The culture media were replaced with fresh media containing Cy5-labeled SN38-NPs for another 3 or 6 h incubation. Then the E0771 cells were washed with PBS for 3 times and incubated with 0.1% CSFE for 10 min. Next the E0771 cells were washed with PBS for 3 times fixed with PBS containing 4% (w/v) formaldehyde. After washed with PBS for another 3 times, the cells were stained with DAPI. Images were taken by a confocal laser scanning microscope (CLSM, Carl Zeiss LSM 700, Germany).
2.10. Pharmacokinetics study
SD rats (average weight 200 g) were treated with SN38 (8 mg/kg, dissolved in the 8:1:1 mixture of PBS and ethanol, Cremophor EL®) or SN38-NPs (8 mg/kg equivalent (eq.) to SN38, dissolved in PBS) intra-venously. Blood samples were collected from the orbital cavity through a capillary tube at each desired time point (5 min, 0.5, 1, 2, 4, 6, 10, 24 and 49 h), mixed with heparin sodium, and the plasmas were obtained after centrifugation for 10 min × 3000 rpm/min. Then 100 μL plasma was treated with 80 μL 1 M NaOH at 37 ◦C for 1 h, followed with 80 μL 1.2 M phosphoric acid was added in to adjust pH value. SN38 was extracted from plasmas by using 300 μL acetonitrile and then detected by high performance liquid chromatography (HPLC) with EClassical 3100, which is equipped with Supersil ODS2 5 μm 4.6 × 200 mm chromatographic column, P3100 high pressure constant current pump and UV3100 detector. Tests were conducted at a flow rate of 1.0 mL/min with mobile phases of acetonitrile (solvent A) and water (solvent con-taining 0.1% TFA). UV detection for SN38 was performed at 378 nm. The half-life of the drug (t1/2) and area under the drug concen-tration–time curve from 0 to 49 h in plasma (AUC0-t) were calculated using PKSolver [45].
2.11. Biodistribution of SN38-NPs
1 × 106 E0771 cells were injected subcutaneously at the female C57BL/6 mice (6–8 weeks old) right flank. Once the tumors reached 80 mm3, Cy5-labeled SN38-NPs (30 mg/kg eq. to SN38) was intravenously injected to the mice. At 4 h and 24 h post-injection, major organs (heart, liver, spleen, lung, kidney) and tumor were removed out for fluores-cence imaging. The tissue fluorescence intensity was visualized by the Davinch-Invivo HR imaging system (Davinch, Korea), and the excitation wavelength is at 650 nm and the emission wavelength is at 700 nm.
2.12. In vivo antitumor efficiency
The female C57BL/6 mice (6–8 weeks old) were subcutaneously embedded with E0771 cells (1 × 106) at the right flank. Once the tumor volume reached approximately 60 mm3, the mice were assigned into 4 groups at random (n = 9) and received the treatment of PBS, IR (30 mg/ kg), SN38 (10 mg/kg), SN38-NPs (30 mg/kg eq. to SN38). SN38 stock solution was prepared by firstly dissolve in DMSO, and then added 85% PEG-400 as an adjuvant, and finally mixed with 10% PBS to reduce the viscosity. The mice were received 4 treatments in a total dosage of 120 mg/kg. Body weights and tumor volumes changes were recorded. The day when the treatment started was designated as day 0. The tumor volumes were measured with vernier caliper and calculated as follows:
tumor volume (V) = a × b2/2, where a represents the major axis, and b represents the minor axis of the tumor. The tumor suppression rate (TSR) was calculated as the formula: TSR (%) = [(Vc - Vx)/Vc] × 100%, where the Vc and Vx represent the mean tumor volume of the PBS group and treatment group, respectively.
2.13. Flow cytometry analysis
To analyze the contents of the immune cell in the tumor microen-vironment, the mice were sacrificed three days after the final treatment. The obtained tumors were cut in pieces and digested in tumor dissoci-ation buffer (RPMI-1640 containing collagenase, hyaluronidase, and DNase). Single-cell suspensions were obtained after filtration with nylon mesh and incubated with various antibodies. FACS tests were carried out by a flow cytometer (BD FACSCanto II) and the results were analyzed with the FlowJo.
2.14. ELISA test
E0771 or 4T1 tumor cells were treated with various drugs at a con-centration of 5 μM for 48 h, then the drug pretreated tumor cells su-pernatant used as conditioned media (CM) were obtained after centrifugation at a speed of 12000 rpm/min for 5 min. BMDCs were cultured with 50% CM plus 50% fresh media for 12 h. After that, the BMDCs supernatant was removed out and centrifuged for cytokines analysis using ELISA kits, which were carried out as manufacturer’s instructions.
To test the cytokines levels in tumor tissues, 100 mg tumor tissues were grinded in 1 mL PBS by a tissue homogenizer on ice. The super-natants were extracted for ELISA tests through centrifuging at a speed of 12000 rpm/min for 5 min. The test procedures strictly followed the manufacturer’s instructions.
2.15. Western blot analysis
The tumor cells supernatant was obtained by the same treatment as described above and used to treat BMDCs 1 h or 4 h. Then the BMDCs were collected and washed with 0.9% NaCl, RIPA lysis buffer with protease and phosphatase inhibitors was added in for 30 min incubation on ice. The proteins of BMDCs were extracted through centrifuge at a speed of 12000 rpm for 5 min. Protein content quantification was car-ried out by the BCA protein assay kit. The proteins were boiled at 100 ◦C for 10 min with the loading buffer added in. Then the electrophoreses process was conducted through SDS-PAGE by a gel-electrophoretic apparatus (Bio-Rad mini, USA), and the proteins were transferred to the PVDF films and incubated with the antibodies against various pro-teins overnight on a shaker at 4 ◦C. Subsequently, the PVDF films were washed 5 times and incubated with HRP conjugated antibodies for 1 h. The Western blot images were obtained by Amersham Imager 600 (AI600, General Electric Co., Ltd., USA) with 300 μL of ECL chemilu-minescent reagent (Beyotime biotechnology Co., Ltd., P0018AS) added on the top of the membrane. And the quantification was performed with Image J.
2.16. Statistical analysis
All experiments were performed at least three times and expressed as means ± standard deviation (s.d.). For comparison between two groups, student’s t-test was used. For comparison between multiple groups, one- way ANOVA was used.
3. Results and discussion
3.1. Screening chemo agents for stimulating type I IFN secretion
Cells may release DNA fragments-containing exosomes at certain
J. Zhao et al.

Biomaterials 268 (2021) 120542
5
conditions to maintain cellular homeostasis, and these events may provoke innate immune responses in antigen-presenting cells captured the exosomes [40]. With this in mind, we designed an experiment to evaluate the capacity of different DNA-targeting chemo agents in inducing inflammatory cytokines secretion from BMDCs, by culturing BMDCs with the supernatants of E0771 and 4T1 tumor cells treated with various drugs, including doxorubicin (DOX), cisplatin (CDDP), top-otecan (TPT) and SN38.
Firstly, we tested the cytotoxicity of different drugs to tumor cells and BMDCs to identify an appropriate drug exposure concentration. MTT and CCK8 assay was applied to tumor cells and BMDCs testing respectively because BMDCs are suspension cells not suitable for MTT assays. As shown in Fig. 1A, the tumor cells and BMDCs had distin-guished responses to various drugs. 4T1 cells were more resistant to SN38 and TPT treatment, while E0771 cells were more resistant to CDDP treatment. DOX showed similar strong toxicity to all tumor cells. Specially, DOX exhibited much stronger cytotoxicity to BMDCs than other drugs. As a result, 5 μM of various drugs were used to treat 4T1 and E0771 cells for 48 h, and then the cell culture supernatants (Conditioned Media; CM) were transferred to BMDCs with an equal volume of fresh culture medium for another 12 h incubation (final drug concentration for BMDCs was 2.5 μM) and the supernatants were subjected to ELISA tests (Fig. 1B). As shown in Fig. 1C, single treatment of tumor cells and BMDCs with various drugs mentioned above resulted in weak IL-6
secretion changes compared with the control group, while treatment of BMDCs with 4T1 or E0771 CM resulted in obvious changes in IL-6 secretion. Especially, E0771 cells treated with SN38 and TPT lead to a 67-fold and 72-fold increase in the IL-6 secretion from BMDCs compared to the control group. The IL-6 secretion from BMDCs incubated with 4T1 CM was relatively lower than that incubated with E0771 CM, which may be attributed to the relative resistance of 4T1 cells to SN38 and TPT. The DOX treated 4T1 CM resulted in decreased IL-6 secretion from the BMDCs. This could be attributed to the high cytotoxicity of DOX to BMDCs and decreased cell viability. Further, we analyzed the IFN-β secretion levels at various conditions, because type I IFNs are key cy-tokines in stimulating the innate and adaptive immune responses and correlates closely with the STING-related TANK-binding kinase 1 (TBK1)-interferon regulatory factor 3 (IRF3) inflammatory pathway [24, 25,46]. As shown in Fig. 1D, SN38 is the top drug in significantly enhancing the IFN-β secretion from BMDCs in both 4T1 and E0771 groups, suggesting SN38 may be a good candidate in stimulating the STING pathway activation.
3.2. SN38 treated tumor cells activate STING pathway in BMDCs through exosome secretion
To verify the hypothesis that SN38-treated tumor cells can success-fully stimulate the STING pathway activation in immune cells, we
Fig. 1. Screening chemo drugs for stimulating type I IFN secretion. (A) In vitro cytotoxicity of various drugs on 4T1, and E0771 at 48 h detected by MTT assays, and on BMDCs at 12 h detected by CCK8 tests; n = 4. (B) Schematic representing drugs screening process for inducing cytokine secretion from BMDCs. (C) IL-6 con-centration (Conc.) in supernatants of cells incubated with free drugs, and IL-6 concentration in supernatants of BMDCs incubated with E0771 CM or 4T1 CM; n = 4. (D) IFN-β concentration in supernatants of cells incubated with free drugs, and IFN-β concentration in supernatants of BMDCs incubated with E0771 CM or 4T1 CM; n = 4. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, ns = not significant. All statistical data are presented as means ± s.d.
J. Zhao et al.

Biomaterials 268 (2021) 120542
6
measured the STING pathway-related proteins expression in the BMDCs after various treatments via western blot assay. As shown in Fig. 2A, the phosphorylated STING (PSTING) and the downstream phosphorylated IRF-3 (PIRF-3) protein expression in BMDCs were much increased at 1 h and 4 h after SN38 CM treatment, confirming that the supernatant of E0771 cells treated with SN38 could elicit STING pathway activation in BMDCs. To further verify the activation of BMDCs, flow cytometry was applied to detect the BMDC surface markers. As shown in Fig. 2B, the expression of costimulatory molecule CD80 (B7-1) on BMDCs surface was significantly upregulated after coculture with the supernatant from SN38 treated E0771 cells, suggesting increased BMDCs activation upon incubation with SN38-treated E0771 cells CM. Similar STING pathway activation and surface marker changes were also observed in BMDCs cocultured with SN38-treated 4T1 cells CM (Fig. S1).
To investigate the underlying mechanism of SN38 in stimulating the IFN-β secretion and STING pathway activation, we conducted a series of following experiments. There are reports showing that some DNA- targeting agents may lead to DNA leakage to the cytosol and secretion of DNA-containing exosomes to the extracellular space [42]. These DNA fragments-containing exosomes could be captured by antigen-presenting cells and elicit STING pathway activation and cyto-kine secretion. SN38 is the active metabolite of irinotecan and a topo-isomerase I inhibitor, which blocks the ligation step of the cell cycle and generate DNA single- or double-strand breaks in the cell [47]. Therefore, SN38 may induce the production of DNA-containing exosomes from the treated cells. To prove this point, we collected the supernatants of E0771 cells and further purified the exosomes from the supernatants with an exosome isolation kit (Fig. 2C). As shown in Fig. 2D, no sig-nificant increase in exosome production was observed after SN38 treatment. However, the DNA content in these exosomes was much
increased (Fig. 2E). This difference was confirmed in the DNA agarose gel electrophoresis image, with a clear DNA band observed in the SN38 treated group, while no DNA band observed in the control PBS group (Fig. 2F). The above results indicated that SN38 could induce the pro-duction of DNA-containing exosomes from E0771 tumor cells, possibly by generating DNA breaks in the cells. To further verify the effect of these exosomes on STING pathway activtion in BMDCs, we treated BMDCs directly with the isolated exosomes, and conducted Western blot assays with the extracted proteins. As shown in Fig. 2G, obviously increased PIRF-3 and PSTING expression was observed after the exosome treatment. The above results explained that SN38 treatment could induce STING pathway activation in BMDCs through the passage of DNA-containing exosomes from tumor cells to BMDCs.
3.3. Synthesis and characterization of PLG-g-mPEG/SN38
Previous reports showed that nanoformulation is meaningful in enhancing the immunogenic responses of chemo agents by reducing the toxicity and inhibition effect of chemo agents to the immune system [13]. Considering the possible toxicity of free SN38 to the immune system [48], we designed a polymeric formulation of SN38 by conju-gating SN38 to poly(L-glutamic acid)-g-methoxy poly(ethylene glycol) (PLG-g-mPEG) for tumor-targeted delivery and drug release. PLG-g-mPEG was synthesized according to our previous work [44], and SN38 was conjugated to the side chains via the Yamaguchi esterification (Fig. 3A) [49]. The chemical structure of PLG-g-mPEG/SN38 was veri-fied by 1H NMR, with typical peaks of SN38 at δ 7.65 (1H, d), δ 7.41 (1H, s), δ 7.07 (1H, d), δ 6.85 (1H, s), δ 1.14 (3H, t) and δ 0.91 (3H, t) ppm, indicating the successful conjugation of SN38 to the PLG-g-mPEG (Fig. S2). The four characteristic peaks of SN38 in PLG-g-mPEG/SN38 at
Fig. 2. SN38-treated tumor cells CM stimulates STING pathway activation in BMDCs via exosome secretion. (A) Western blot images and quantitative analysis of proteins extracted from BMDCs after treated with PBS or E0771 SN38 CM for 1 h and 4 h. (B) The expression of CD80 on BMDCs surface after the incubation of PBS or SN38-treated E0771 CM for 12 h, analyzed by flow cytometry; n = 3. (C) Schematic representing isolation of exosomes from SN38-treated E0771 supernatant, followed by DNA extraction from exosomes. (D) BCA quantification of exosomes extracted from E0771 supernatants after treatment with PBS or SN38; n = 5. (E) Quantification of DNA extracted and purified from exosomes; n = 5. (F) Image of DNA agarose gel electrophoresis by UV imaging. (G) Western blot images of proteins extracted from BMDCs after incubation with exosomes isolated from CM of E0771 cells treated with PBS or SN38. *p < 0.05. All statistical data are presented as means ± s.d.
J. Zhao et al.
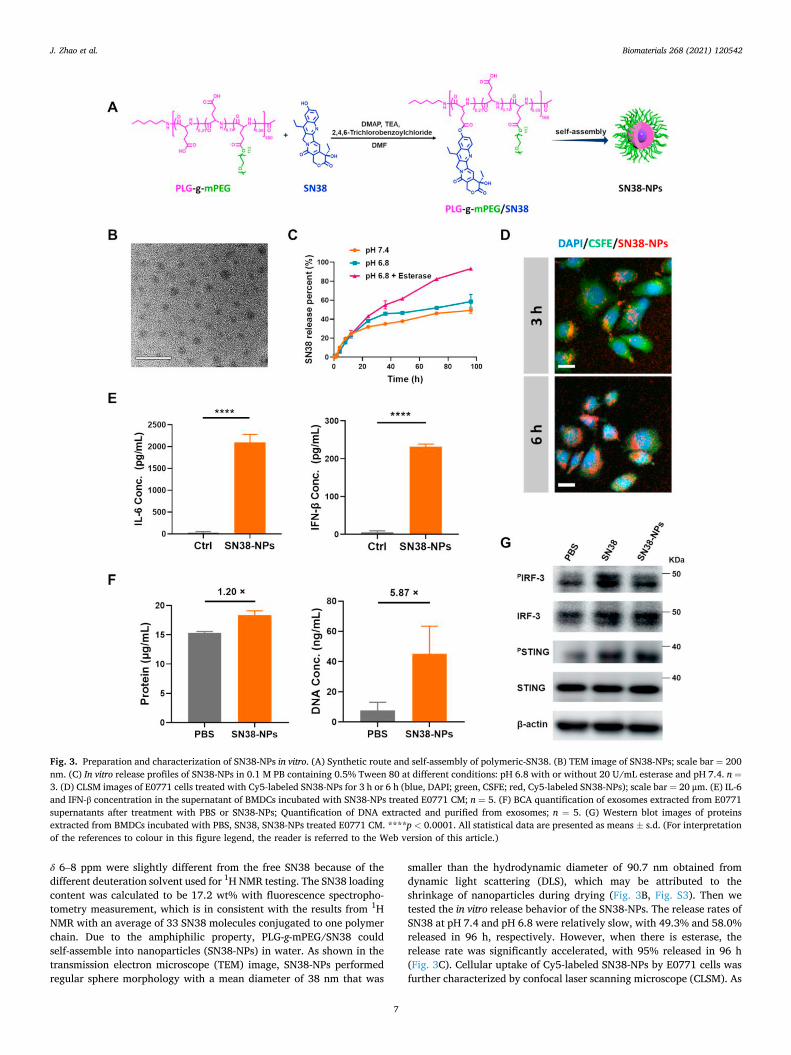
Biomaterials 268 (2021) 120542
7
δ 6–8 ppm were slightly different from the free SN38 because of the different deuteration solvent used for 1H NMR testing. The SN38 loading content was calculated to be 17.2 wt% with fluorescence spectropho-tometry measurement, which is in consistent with the results from 1H NMR with an average of 33 SN38 molecules conjugated to one polymer chain. Due to the amphiphilic property, PLG-g-mPEG/SN38 could self-assemble into nanoparticles (SN38-NPs) in water. As shown in the transmission electron microscope (TEM) image, SN38-NPs performed regular sphere morphology with a mean diameter of 38 nm that was
smaller than the hydrodynamic diameter of 90.7 nm obtained from dynamic light scattering (DLS), which may be attributed to the shrinkage of nanoparticles during drying (Fig. 3B, Fig. S3). Then we tested the in vitro release behavior of the SN38-NPs. The release rates of SN38 at pH 7.4 and pH 6.8 were relatively slow, with 49.3% and 58.0% released in 96 h, respectively. However, when there is esterase, the release rate was significantly accelerated, with 95% released in 96 h (Fig. 3C). Cellular uptake of Cy5-labeled SN38-NPs by E0771 cells was further characterized by confocal laser scanning microscope (CLSM). As
Fig. 3. Preparation and characterization of SN38-NPs in vitro. (A) Synthetic route and self-assembly of polymeric-SN38. (B) TEM image of SN38-NPs; scale bar = 200 nm. (C) In vitro release profiles of SN38-NPs in 0.1 M PB containing 0.5% Tween 80 at different conditions: pH 6.8 with or without 20 U/mL esterase and pH 7.4. n =3. (D) CLSM images of E0771 cells treated with Cy5-labeled SN38-NPs for 3 h or 6 h (blue, DAPI; green, CSFE; red, Cy5-labeled SN38-NPs); scale bar = 20 μm. (E) IL-6 and IFN-β concentration in the supernatant of BMDCs incubated with SN38-NPs treated E0771 CM; n = 5. (F) BCA quantification of exosomes extracted from E0771 supernatants after treatment with PBS or SN38-NPs; Quantification of DNA extracted and purified from exosomes; n = 5. (G) Western blot images of proteins extracted from BMDCs incubated with PBS, SN38, SN38-NPs treated E0771 CM. ****p < 0.0001. All statistical data are presented as means ± s.d. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
J. Zhao et al.

Biomaterials 268 (2021) 120542
8
shown in Fig. 3D, much SN38-NPs could be observed intracellularly, and the uptake was increased as time prolonged. We further compared the cytotoxicity of SN38-NPs and free SN38. As shown in Fig. S4, SN38-NPs showed slightly lower toxicity than free SN38 in E0771 cells. These re-sults indicated that the SN38-NPs could protect the SN38 from quick release but result in similar cytotoxicity to tumor cells due to the esterase-assisted quick drug release. Importantly, the cytotoxicity of SN38-NPs is much weaker than free SN38 to BMDCs, with IC50 values of 41.7 μM and 4.2 μM (Fig. S4), suggesting the SN38-NPs could reduce the toxicity of SN38 to BMDCs and perform preference cytotoxicity to tumor cells than BMDCs.
To confirm the capacity of SN38-NPs in inducing STING activation in BMDCs, we proceeded a similar experiment as free SN38 mentioned above. As shown in Fig. 3E, much increased IL-6 and IFN-β secretion was detected in BMDCs culture medium incubated with the supernatants of E0771 cells treated with SN38-NPs. In addition, we detected slightly increased exosome secretion but much increased DNA content in the exosomes extracted from E0771 cells supernatant after the treatment with SN38-NPs as compared to the PBS group (Fig. 3F). Meanwhile, increased expression of PSTING and PIRF3 in BMDCs was also observed in the SN38-NPs treatment group (Fig. 3G). These results suggested that SN38-NPs possess the capacity of SN38 in stimulating the STING pathway activation and inducing related cytokine secretion.
3.4. Pharmacokinetics and biodistribution of SN38-NPs
The pharmacokinetics and biodistribution of SN38-NPs were tested in Sprague Dawley (SD) rats. Free SN38 and SN38-NPs were intrave-nously injected into SD rats, the orbital venous blood was collected at defined time points, and the SN38 concentrations in plasma were analyzed by HPLC. As shown in Fig. 4A, free SN38 quickly disappeared from the blood, while SN38-NPs maintained a much higher SN38 con-centration and longer circulation time in the blood. Notably, SN38-NPs showed a half-life (t1/2) of 10.6 h which was remarkedly longer than that of free SN38 which was less than 5 min. The area under the curve (AUC0-
t) of SN38-NPs was 308 μg⋅mL-1⋅h, which was much higher than that of free SN38 with an AUC of only 0.098 μg⋅mL-1⋅h. We then analyzed the biodistribution of SN38-NPs in E0771 tumor-bearing C57BL/6 mice. Cy5-labeled SN38-NPs were injected into mice, and major organs and the tumors were excised for optical imaging at 4 h and 24 h post in-jection. As shown in Fig. 4B, liver and tumor were the major organs for SN38-NPs accumulation, and there was still strong signal in the tumor at 24 h after injection.
3.5. In vivo tumor inhibition studies of SN38-NPs
Although there have been several reports on using polymeric-SN38 conjugates for in vivo tumor therapy [50–53], we think there may be an unrevealed mechanism based on our observation that SN38 and SN38-NPs could induce STING pathway activation by passage the DNA-containing exosomes from tumor cells to DCs. As a result, SN38-NPs therapy may perform in a chemoimmunotherapy way by not only killing tumor cells but also in situ activating the STING pathway-related antitumor immunity. To clarify this aspect, we per-formed the in vivo anti-tumor study of the SN38-NPs in an E0771 tumor model, and detailedly analyzed the tumor immune status after the treatment.
The subcutaneous E0771 tumor model was established by injecting 1.0 × 106 E0771 cells into the right flank of female C57BL/6 mice. When the tumor volume reached around 60 mm3, the mice were separated into four groups randomly. Then the mice received four injections of PBS, IR (30 mg/kg), SN38 (10 mg/kg) and SN38-NPs (30 mg/kg eq. based on SN38). Free SN38 was given at a lower dose because mice immediately died after injection with 30 mg/kg or 20 mg/kg SN38. Irinotecan (IR) was supplemented as another control group since SN38 is the active metabolite of IR and IR is used in clinical practice with better tolerance [54–56]. The body weights and tumor volumes were recorded every two days for 14 days (Fig. 5A). As shown in Fig. 5B, SN38-NPs effectively inhibited the E0771 tumor growth, and resulted in a tumor suppression rate (TSR%) of 82.6% on day 14, while the free SN38 treatment resulted in a TSR% of 54.1%, and IR treatment only resulted in a TSR% of 11.3%. No obvious body weight changes were observed in all the treatment groups, however, 3 out of 9 mice died in the SN38 group during the treatmenet, suggesting the acute toxicity of free SN38 (Fig. 5C). Several reasons may account for the better in vivo performance of SN38-NPs over free SN38 and IR treatment: SN38-NPs have longer blood circulation time and better accumulation in the tumor compared to free drugs, thus resulting in higher drug concentration in the tumor and better chemo-therapeutic efficacy - this is in consistent with previously observed nanocarrier-based SN38 therapy for murine tumors; in addition, SN38-NPs may be more efficient in eliciting STING-related immune re-sponses inside the tumor.
To investigate the alterations in tumor immune microenvironment after treatment, we analyzed the tumor tissues on day 15 at 4 days after the final drug administration. As shown in Fig. 5D, various pro- inflammatory cytokines including IL-6, TNF-α, IFN-γ, and IFN-β were much increased after SN38-NPs treatment, while no significant changes
Fig. 4. In vivo characterization of pharmacokinetics and biodistribution of SN38-NPs. (A) The concentration of SN38 in plasma of SD rats after the intravenous injection of free SN38 or SN38-NPs (8 mg/kg eq. to SN38). The total amount of SN38 in the plasma was extracted and detected by HPLC. Half-life (t1/2) and the area under the curve (AUC0-t) were listed in the graph. n = 3. (B) Fluorescence images of major organs and tumors at 4 h and 24 h post-injection of Cy5-labeled SN38-NPs. ****p < 0.0001, ns = no significant. All statistical data are presented as means ± s.d.
J. Zhao et al.
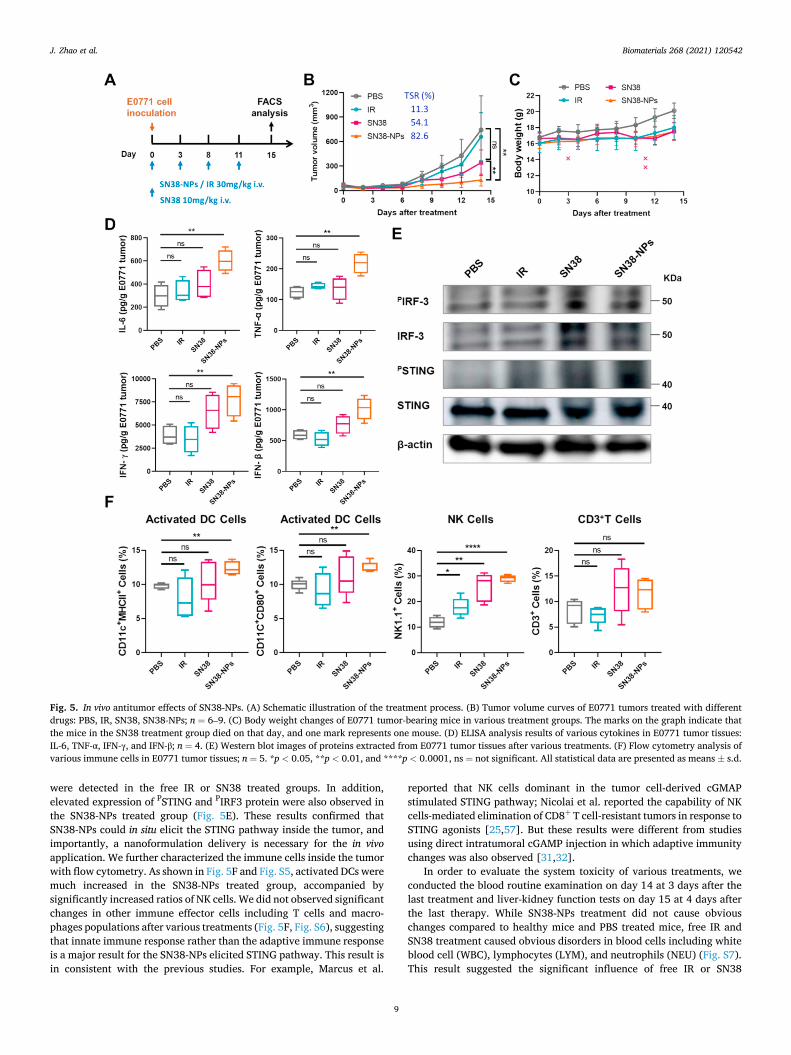
Biomaterials 268 (2021) 120542
9
were detected in the free IR or SN38 treated groups. In addition, elevated expression of PSTING and PIRF3 protein were also observed in the SN38-NPs treated group (Fig. 5E). These results confirmed that SN38-NPs could in situ elicit the STING pathway inside the tumor, and importantly, a nanoformulation delivery is necessary for the in vivo application. We further characterized the immune cells inside the tumor with flow cytometry. As shown in Fig. 5F and Fig. S5, activated DCs were much increased in the SN38-NPs treated group, accompanied by significantly increased ratios of NK cells. We did not observed significant changes in other immune effector cells including T cells and macro-phages populations after various treatments (Fig. 5F, Fig. S6), suggesting that innate immune response rather than the adaptive immune response is a major result for the SN38-NPs elicited STING pathway. This result is in consistent with the previous studies. For example, Marcus et al.
reported that NK cells dominant in the tumor cell-derived cGMAP stimulated STING pathway; Nicolai et al. reported the capability of NK cells-mediated elimination of CD8+ T cell-resistant tumors in response to STING agonists [25,57]. But these results were different from studies using direct intratumoral cGAMP injection in which adaptive immunity changes was also observed [31,32].
In order to evaluate the system toxicity of various treatments, we conducted the blood routine examination on day 14 at 3 days after the last treatment and liver-kidney function tests on day 15 at 4 days after the last therapy. While SN38-NPs treatment did not cause obvious changes compared to healthy mice and PBS treated mice, free IR and SN38 treatment caused obvious disorders in blood cells including white blood cell (WBC), lymphocytes (LYM), and neutrophils (NEU) (Fig. S7). This result suggested the significant influence of free IR or SN38
Fig. 5. In vivo antitumor effects of SN38-NPs. (A) Schematic illustration of the treatment process. (B) Tumor volume curves of E0771 tumors treated with different drugs: PBS, IR, SN38, SN38-NPs; n = 6–9. (C) Body weight changes of E0771 tumor-bearing mice in various treatment groups. The marks on the graph indicate that the mice in the SN38 treatment group died on that day, and one mark represents one mouse. (D) ELISA analysis results of various cytokines in E0771 tumor tissues: IL-6, TNF-α, IFN-γ, and IFN-β; n = 4. (E) Western blot images of proteins extracted from E0771 tumor tissues after various treatments. (F) Flow cytometry analysis of various immune cells in E0771 tumor tissues; n = 5. *p < 0.05, **p < 0.01, and ****p < 0.0001, ns = not significant. All statistical data are presented as means ± s.d.
J. Zhao et al.

Biomaterials 268 (2021) 120542
10
treatment on the mice white blood cells and the necessity of using the SN38-NPs formulation. In addition, no significant increase in alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (CRE) were detected, suggesting there is no damage to liver and kidney after various treatments (Fig. S8).
4. Conclusion
In conclusion, based on the understanding of cytosol DNA is a pre- requisite for STING pathway activation, we proposed a strategy using DNA-targeting chemo drugs for stimulating STING signals and cancer chemoimmunotherapy. Through in vitro screening of several DNA- targeting chemo drugs, we identified SN38 as the top candidate for stimulating the STING signals in BMDCs, and proved that the stimula-tion was realized by the passage of DNA-containing exosomes from the treated tumor cells to BMDCs. Furthermore, we proved that a nano-formulation of SN38 is necessary for in situ STING activation, and the polymeric-SN38 conjugate mainly induced innate immune responses including DCs and NK cells activation. Although there have been several SN38 nanoformulations developed in pre-clinical or clinical process, our study revealed a new mechanism for the in vivo anti-tumor mechanisms of SN38. Additionally, the preprared SN38-NPs provides a new option for activation of STING pathway for cancer therapy and should inspire using more DNA-targeting agents, especially in nanoformulations, for in situ activation of the STING pathway and cancer chemoimmunotherapy.
Credit author statement
Jiayu Zhao: Methodology, Validation, Investigation, Writing – orig-inal draft. Sheng Ma: Investigation, Methodology. Yudi Xu: Data cura-tion, Methodology. Xinghui Si: Data curation, Methodology. Haochen Yao: Investigation. Zichao Huang: Investigation. Haiyang Yu: Method-ology. Wantong Song: Conceptualization, Project administration, Writing – review & editing, Funding acquisition. Zhaohui Thang: Re-sources, Supervision, Funding acquisition. Xuesi Chen: Resources, Su-pervision, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (51973215, 51673185, 51673189, 51829302, 52003268, 51833010 and 51520105004), the Program of Scientific Development of Jilin Province Science (20190103112JH), as well as the support from the Youth Innovation Promotion Association of Chinese Academy of Sciences (2020232).
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi. org/10.1016/j.biomaterials.2020.120542.
References
[1] Daniel S. Chen, I. Mellman, Oncology meets immunology: the cancer-immunity cycle, Immunity 39 (1) (2013) 1–10.
[2] I. Mellman, G. Coukos, G. Dranoff, Cancer immunotherapy comes of age, Nature 480 (7378) (2011) 480–489.
[3] M.F. Sanmamed, L. Chen, A paradigm shift in cancer immunotherapy: from enhancement to normalization, Cell 175 (2) (2018) 313–326.
[4] W. Song, M. Das, X. Chen, Nanotherapeutics for immuno-oncology: a crossroad for new paradigms, Trends Canc. (2020) 288–298.
[5] D.M. Pardoll, The blockade of immune checkpoints in cancer immunotherapy, Nat. Rev. Canc. 12 (4) (2012) 252–264.
[6] A. Ribas, J.D. Wolchok, Cancer immunotherapy using checkpoint blockade, Science 359 (6382) (2018) 1350–1355.
[7] P. Sharma, J.P. Allison, Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential, Cell 161 (2) (2015) 205–214.
[8] A. Haslam, V. Prasad, Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs, JAMA Netw Open 2 (5) (2019) e192535.
[9] M. Yarchoan, A. Hopkins, E.M. Jaffee, Tumor mutational burden and response rate to PD-1 inhibition, N. Engl. J. Med. 377 (25) (2017) 2500–2501.
[10] Z. Hu, P.A. Ott, C.J. Wu, Towards personalized, tumour-specific, therapeutic vaccines for cancer, Nat. Rev. Immunol. 18 (3) (2018) 168–182.
[11] W. Song, S.N. Musetti, L. Huang, Nanomaterials for cancer immunotherapy, Biomaterials 148 (2017) 16–30.
[12] S. Ma, W. Song, Y. Xu, X. Si, S. Lv, Y. Zhang, Z. Tang, X. Chen, Rationally designed polymer conjugate for tumor-specific amplification of oxidative stress and boosting antitumor immunity, Nano Lett. 20 (4) (2020) 2514–2521.
[13] J.J. Moon, Elimination of established tumors with nanodisc-based combination chemoimmunotherapy, Sci. Adv. 4 (4) (2018) eaao1736.
[14] W. Song, L. Shen, Y. Wang, Q. Liu, T.J. Goodwin, J. Li, O. Dorosheva, T. Liu, R. Liu, L. Huang, Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap, Nat. Commun. 9 (1) (2018) 1–11.
[15] Q. Wang, Y. Wang, J. Ding, C. Wang, X. Zhou, W. Gao, H. Huang, F. Shao, Z. Liu, A bioorthogonal system reveals antitumour immune function of pyroptosis, Nature 579 (7799) (2020) 421–426.
[16] T. Luo, K. Ni, A. Culbert, G. Lan, Z. Li, X. Jiang, M. Kaufmann, W. Lin, Nanoscale metal-organic frameworks stabilize bacteriochlorins for type I and type II photodynamic therapy, J. Am. Chem. Soc. 142 (16) (2020) 7334–7339.
[17] Z. Meng, X. Zhou, J. Xu, X. Han, Z. Dong, H. Wang, Y. Zhang, J. She, L. Xu, C. Wang, Z. Liu, Light-triggered in situ gelation to enable robust photodynamic- immunotherapy by repeated stimulations, Adv. Mater. 31 (24) (2019) e1900927.
[18] W. Song, M. Das, Y. Xu, X. Si, Y. Zhang, Z. Tang, X. Chen, Leveraging biomaterials for cancer immunotherapy: targeting pattern recognition receptors, Mater. Today Nano 5 (2019) 100029.
[19] J.P. Cerliani, S.R. Stowell, I.D. Mascanfroni, C.M. Arthur, R.D. Cummings, G. A. Rabinovich, Expanding the universe of cytokines and pattern recognition receptors: galectins and glycans in innate immunity, J. Clin. Immunol. 31 (1) (2011) 10–21.
[20] X. Cai, Y.H. Chiu, Z.J. Chen, The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling, Mol. Cell. 54 (2) (2014) 289–296.
[21] J. Tao, X. Zhou, Z. Jiang, cGAS-cGAMP-STING, The three musketeers of cytosolic DNA sensing and signaling, IUBMB Life 68 (11) (2016) 858–870.
[22] Q. Chen, L. Sun, Z.J. Chen, Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing, Nat. Immunol. 17 (10) (2016) 1142–1149.
[23] G.N. Barber, STING-dependent cytosolic DNA sensing pathways, Trends Immunol. 35 (2) (2014) 88–93.
[24] L. Deng, H. Liang, M. Xu, X. Yang, B. Burnette, A. Arina, X.D. Li, H. Mauceri, M. Beckett, T. Darga, X. Huang, T.F. Gajewski, Z.J. Chen, Y.X. Fu, R. R. Weichselbaum, STING-dependent cytosolic DNA sensing promotes radiation- induced type I interferon-dependent antitumor immunity in immunogenic tumors, Immunity 41 (5) (2014) 843–852.
[25] A. Marcus, A.J. Mao, M. Lensink-Vasan, L. Wang, R.E. Vance, D.H. Raulet, Tumor- derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response, Immunity 49 (4) (2018) 754–763, e4.
[26] S.R. Woo, M.B. Fuertes, L. Corrales, S. Spranger, M.J. Furdyna, M.Y. Leung, R. Duggan, Y. Wang, G.N. Barber, K.A. Fitzgerald, M.L. Alegre, T.F. Gajewski, STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors, Immunity 41 (5) (2014) 830–842.
[27] E.J. Diner, D.L. Burdette, S.C. Wilson, K.M. Monroe, C.A. Kellenberger, M. Hyodo, Y. Hayakawa, M.C. Hammond, R.E. Vance, The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING, Cell Rep. 3 (5) (2013) 1355–1361.
[28] X. Zhang, H. Shi, J. Wu, X. Zhang, L. Sun, C. Chen, Z.J. Chen, Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING, Mol. Cell. 51 (2) (2013) 226–235.
[29] G.J. Shang, C.G. Zhang, Z.J.J. Chen, X.C. Bai, X.W. Zhang, Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP, Nature 567 (7748) (2019) 389–393.
[30] C.G. Zhang, G.J. Shang, X. Gui, X.W. Zhang, X.C. Bai, Z.J.J. Chen, Structural basis of STING binding with and phosphorylation by TBK1, Nature 567 (7748) (2019) 394–398.
[31] S.T. Koshy, A.S. Cheung, L. Gu, A.R. Graveline, D.J. Mooney, Liposomal delivery enhances immune activation by STING agonists for cancer immunotherapy, Adv Biosyst. 1 (1–2) (2017) 1600013.
[32] D. Shae, K.W. Becker, P. Christov, D.S. Yun, A.K.R. Lytton-Jean, S. Sevimli, M. Ascano, M. Kelley, D.B. Johnson, J.M. Balko, J.T. Wilson, Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy, Nat. Nanotechnol. 14 (3) (2019) 269–278.
[33] N. Cheng, R. Watkins-Schulz, R.D. Junkins, C.N. David, B.M. Johnson, S. A. Montgomery, K.J. Peine, D.B. Darr, H. Yuan, K.P. McKinnon, Q. Liu, L. Miao, L. Huang, E.M. Bachelder, K.M. Ainslie, J.P. Ting, A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer, JCI Insight 3 (22) (2018), e120638.
J. Zhao et al.

Biomaterials 268 (2021) 120542
11
[34] D. Prantner, D.J. Perkins, W. Lai, M.S. Williams, S. Sharma, K.A. Fitzgerald, S. N. Vogel, 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential, J. Biol. Chem. 287 (47) (2012) 39776–39788.
[35] J. Conlon, D.L. Burdette, S. Sharma, N. Bhat, M. Thompson, Z. Jiang, V.A. K. Rathinam, B. Monks, T. Jin, T.S. Xiao, S.N. Vogel, R.E. Vance, K.A. Fitzgerald, Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid, J. Immunol. 190 (10) (2013) 5216–5225.
[36] J.M. Ramanjulu, G.S. Pesiridis, J. Yang, N. Concha, R. Singhaus, S.Y. Zhang, J. L. Tran, P. Moore, S. Lehmann, H.C. Eberl, M. Muelbaier, J.L. Schneck, J. Clemens, M. Adam, J. Mehlmann, J. Romano, A. Morales, J. Kang, L. Leister, T.L. Graybill, A. K. Charnley, G. Ye, N. Nevins, K. Behnia, A.I. Wolf, V. Kasparcova, K. Nurse, L. Wang, Y. Li, M. Klein, C.B. Hopson, J. Guss, M. Bantscheff, G. Bergamini, M. A. Reilly, Y. Lian, K.J. Duffy, J. Adams, K.P. Foley, P.J. Gough, R.W. Marquis, J. Smothers, A. Hoos, J. Bertin, Design of amidobenzimidazole STING receptor agonists with systemic activity, Nature 564 (7736) (2018) 439–443.
[37] G. Pepin, C. Nejad, J. Ferrand, B.J. Thomas, H.J. Stunden, E. Sanij, C.H. Foo, C. R. Stewart, J.E. Cain, P.G. Bardin, B.R.G. Williams, M.P. Gantier, Topoisomerase 1 inhibition promotes cyclic GMP-AMP synthase-dependent antiviral responses, mBio 8 (5) (2017) e01611-e01617.
[38] P. Luthra, S. Aguirre, B.C. Yen, C.A. Pietzsch, M.T. Sanchez-Aparicio, B. Tigabu, L. K. Morlock, A. Garcia-Sastre, D.W. Leung, N.S. Williams, A. Fernandez-Sesma, A. Bukreyev, C.F. Basler, Topoisomerase II inhibitors induce DNA damage- dependent interferon responses circumventing Ebola virus immune evasion, mBio 8 (2) (2017), 00368-17.
[39] H. Imai, H. Dansako, Y. Ueda, S. Satoh, N. Kato, Daunorubicin, a topoisomerase II poison, suppresses viral production of hepatitis B virus by inducing cGAS- dependent innate immune response, Biochem. Biophys. Res. Commun. 504 (4) (2018) 672–678.
[40] A. Takahashi, R. Okada, K. Nagao, Y. Kawamata, A. Hanyu, S. Yoshimoto, M. Takasugi, S. Watanabe, M.T. Kanemaki, C. Obuse, E. Hara, Exosomes maintain cellular homeostasis by excreting harmful DNA from cells, Nat. Commun. 8 (2017) 15287.
[41] J. Ahn, T. Xia, A. Rabasa Capote, D. Betancourt, G.N. Barber, Extrinsic phagocyte- dependent STING signaling dictates the immunogenicity of dying cells, Canc. Cell 33 (5) (2018) 862–873 e5.
[42] Y. Kitai, T. Kawasaki, T. Sueyoshi, K. Kobiyama, K.J. Ishii, J. Zou, S. Akira, T. Matsuda, T. Kawai, DNA-containing exosomes derived from cancer cells treated with topotecan activate a STING-dependent pathway and reinforce antitumor immunity, J. Immunol. 198 (4) (2017) 1649–1659.
[43] Y.I. Son, S. Egawa, T. Tatsumi, R.E. Redlinger Jr., P. Kalinski, T. Kanto, A novel bulk-culture method for generating mature dendritic cells from mouse bone marrow cells, J. Immunol. Methods 262 (1–2) (2002) 145–157.
[44] H. Yu, Z. Tang, D. Zhang, W. Song, Y. Zhang, Y. Yang, Z. Ahmad, X. Chen, Pharmacokinetics, biodistribution and in vivo efficacy of cisplatin loaded poly(l- glutamic acid)-g-methoxy poly(ethylene glycol) complex nanoparticles for tumor therapy, J. Contr. Release 205 (2015) 89–97.
[45] Y. Zhang, M. Huo, J. Zhou, S. Xie, PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel, Comput. Methods Progr. Biomed. 99 (3) (2010) 306–314.
[46] I. Banerjee, B. Behl, M. Mendonca, G. Shrivastava, A.J. Russo, A. Menoret, A. Ghosh, A.T. Vella, S.K. Vanaja, S.N. Sarkar, K.A. Fitzgerald, V.A.K. Rathinam, Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis, Immunity 49 (3) (2018) 413–426 e5.
[47] W. Voigt, S. Matsui, M.B. Yin, W.C. Burhans, H. Minderman, Y.M. Rustum, Topoisomerase-I inhibitor SN-38 can induce DNA damage and chromosomal aberrations independent from DNA synthesis, Anticancer Res. 18 (5a) (1998) 3499–3505.
[48] Z. Nagheh, S. Irani, R. Mirfakhraie, R. Dinarvand, SN38-PEG-PLGA-verapamil nanoparticles inhibit proliferation and downregulate drug transporter ABCG2 gene expression in colorectal cancer cells, Prog. Biomater. 6 (4) (2017) 137–145.
[49] H. Dhimitruka, J. SantaLucia, Investigation of the Yamaguchi esterification mechanism. Synthesis of a Lux-S enzyme inhibitor using an improved esterification method, Org. Lett. 8 (1) (2006) 47–50.
[50] Q. Sun, X. Sun, X. Ma, Z. Zhou, E. Jin, B. Zhang, Y. Shen, E.A. Van Kirk, W. J. Murdoch, J.R. Lott, T.P. Lodge, M. Radosz, Y. Zhao, Integration of nanoassembly functions for an effective delivery cascade for cancer drugs, Adv. Mater. 26 (45) (2014) 7615–7621.
[51] J. Wang, L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu, Y. Shen, The role of micelle size in tumor accumulation, penetration, and treatment, ACS Nano 9 (7) (2015) 7195–7206.
[52] H. Zhang, J. Wang, W. Mao, J. Huang, X. Wu, Y. Shen, M. Sui, Novel SN38 conjugate-forming nanoparticles as anticancer prodrug: in vitro and in vivo studies, J. Contr. Release 166 (2) (2013) 147–158.
[53] T. Hamaguchi, T. Doi, T. Eguchi-Nakajima, K. Kato, Y. Yamada, Y. Shimada, N. Fuse, A. Ohtsu, S. Matsumoto, M. Takanashi, Y. Matsumura, Phase I study of NK012, a novel SN-38-incorporating micellar nanoparticle, in adult patients with solid tumors, Clin. Canc. Res. 16 (20) (2010) 5058–5066.
[54] W.H. Li, J.M. Xu, L. Shen, T.S. Liu, W.J. Guo, W. Zhang, Z.Y. Chen, X.D. Zhu, J. Li, Phase II study of weekly irinotecan and capecitabine treatment in metastatic colorectal cancer patients, BMC Canc. 14 (1) (2014) 986.
[55] Y. Guan, Y. Shen, Y. Xu, C. Li, J. Wang, W. Gu, P. Lian, D. Huang, S. Cai, Z. Zhang, J. Zhu, An expansion study of genotype-driven weekly irinotecan and capecitabine in combination with neoadjuvant radiotherapy for locally advanced rectal cancer with UGT1A1 *1*1 genotype, Therap. Adv. Gastroenterol 12 (2019), 1756284819852293.
[56] H. Wang, L. Zhou, K. Xie, J. Wu, P. Song, H. Xie, L. Zhou, J. Liu, X. Xu, Y. Shen, S. Zheng, Polylactide-tethered prodrugs in polymeric nanoparticles as reliable nanomedicines for the efficient eradication of patient-derived hepatocellular carcinoma, Theranostics 8 (14) (2018) 3949–3963.
[57] C.J. Nicolai, N. Wolf, I.C. Chang, G. Kirn, A. Marcus, C.O. Ndubaku, S. M. McWhirter, D.H. Raulet, NK cells mediate clearance of CD8+ T cell-resistant tumors in response to STING agonists, Sci. Immunol. 5 (45) (2020) eaaz2738.
J. Zhao et al.