identification of the principal aflatoxin b1-dna adduct formed in vivo in rat liver
TRANSCRIPT

Identification of the Principal Aflatoxin B1-DNA Adduct Formed in vivo in Rat LiverAuthor(s): R. G. Croy, J. M. Essigmann, V. N. Reinhold and G. N. WoganSource: Proceedings of the National Academy of Sciences of the United States of America,Vol. 75, No. 4 (Apr., 1978), pp. 1745-1749Published by: National Academy of SciencesStable URL: http://www.jstor.org/stable/68088 .
Accessed: 06/05/2014 08:51
Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at .http://www.jstor.org/page/info/about/policies/terms.jsp
.JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range ofcontent in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new formsof scholarship. For more information about JSTOR, please contact [email protected].
.
National Academy of Sciences is collaborating with JSTOR to digitize, preserve and extend access toProceedings of the National Academy of Sciences of the United States of America.
http://www.jstor.org
This content downloaded from 130.132.123.28 on Tue, 6 May 2014 08:51:38 AMAll use subject to JSTOR Terms and Conditions

Proc. Natl. Acad. Sci. USA Vol. 75, No. 4, pp. 1745-1749, April 1978 Biochemistry
Identification of the principal aflat in vivo in rat liver
(high-pressure liquid chromatography/carcinogen-nucleic acid ii
R. G. CROY*, J. M. ESSIGMANN*, V. N. REINHOLDt, Als * Department of Nutrition and Food Science, Massachusetts Institute of Technolol Harvard Medical School, Boston, Massachusetts 02115
Contributed by G. N. Wogan, February 6, 1978
ABSTRACT The products of in vivo covalent binding of activated aflatoxin B1 (AFB1) to DNA have been investigated in rats. The principal covalent product formed in liver DNA of rats treated with AFB1 has been identified as 2,3-dihydro-2- (N7-guanyl)-3-hydroxy-aflatoxin Bl. This compound was isolated from the liver DNA of rats dosed with AFBi (2.0 mg/kg) in suf- ficient quantity for characterization by physicochemical tech- niques, including field-desorption mass spectrometry. This in- formation together with results of chemical methylation of the compound proved that the major adduct formed between DNA and AFB1 in vivo is identical to that produced in vitro when AFBi is incubated with DNA in the presence of a rat liver mi- crosomal activating system. Quantitative studies of formation of this compound revealed a dose-dependent relationship be- tween the level of its occurence in liver DNA and AFBi doses over the range 0.125-1.0 mg/kg.
Aflatoxin B1 (AFB1), a secondary fungal metabolite of several species of the genus Aspergillus, is acutely toxic to most animal species, although sensitivity varies widely. The rat, rainbow trout and duck are among the most susceptible to its toxic ef- fects. AFB1 is also a potent hepatocarcinogen in these species (1) and is a potent mutagen in mammalian and bacterial cells (2-4). Epidemiological investigations in several human popu- lations have revealed an association of increased incidences of hepatocellular carcinoma with increasing dietary contamina- tion by AFB1 (5).
Biological effects of AFB1 have been extensively studied in the rat. Acute doses of the toxin in this species produce both morphological and biochemical alterations primarily associated with the liver (6). Reversible inhibition of RNA synthesis has been demonstrated in vivo (7, 8) and has been shown to be temporally associated with nucleolar segregation (9). DNA synthesis is also reversibly inhibited in normal (10) and partially hepatectomized (8, 11) animals following acute doses of the toxin.
Investigations of various in vitro models have demonstrated the requirement for metabolic conversion of AFB1 to an active derivative which interacts covalently with DNA (12) and results in alterations of normal biochemical processes asso- ciated with it such as transcription and replication (8). Covalent binding of AFB1 to DNA in vitro is dependent on microsomal conversion of AFB1 to the reactive 2,3-exo-epoxide. Although this labile molecular species has not been isolated, much indirect evidence has accumulated to substantiate its transient existence (13, 14). Covalent binding in vivo to nu- cleophilic atoms in proteins and nucleic acids is also thought to be mediated through metabolic activation to this reactive
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U. S. C. ?1734 solely to indicate this fact.
17
oxin B1-DNA adduct formed
iteraction/field desorption mass spectrometry)
[D G. N. WOGAN*
;y, Cambridge, Massachusetts 02139; and t Department of Biochemistry,
compound (15). Identification of the reactive sites in these macromolecules and the covalent compounds formed with AFB1 may provide a more complete understanding of its bio- logical effects and provide further information on which to base hypotheses concerning the mechanisms underlying its carci- nogenic properties.
The nucleophilic sites of interaction with proteins have not been identified. However, recently, the N7 atom of guanine has been identified independently by two groups (16, 17) as the major site of reaction of the 2,3-epoxide with DNA in vitro. The product of this reaction, 2,3-dihydro-2-(N7-guanyl)-3- hydroxy-aflatoxin B1 (AFB1-N7-Gua) was isolated from DNA that had been incubated with AFB1 in the presence of a mi- crosomal activation system.
By using the preparative and analytical chromatographic procedures previously developed for isolation of the in vitro adduct, we have isolated the major adduct formed between AFB1 and rat liver DNA in vivo. Preliminary chromatographic and spectral evidence indicated that this product was similar to the compound formed in vitro. Application of the techniques of field desorption mass spectrometry and chemical methyl- ation has established its identity to the in vitro product. Evi- dence of the identity of the principal AFB1-DNA adducts formed in vitro and in vivo has also been provided by Lin et al. (17) who compared the chromatographic behavior of several derivatives of these compounds.
Quantitative determinations revealed a dose-dependent relationship between the amount of N7-adduct formed in DNA by AFBi at 0.125-1.0 mg/kg.
MATERIALS AND METHODS
Male Fischer rats were obtained from Charles River Breeding Laboratories (North Wilmington, MA) and fed a semisynthetic agar gel diet (18) ad lib. Rats (130-150 g) were injected intra- peritoneally with AFB1 at 2 mg/kg, purified by the method of Asao et al. (19) in 50 jil of dimethyl sulfoxide (Burdick and Jackson Laboratories, Muskegon, MI). Rats were stunned and decapitated 2 hr after dosing. Their livers were quickly per- fused with 0.05 M Tris-HCl, pH 7.5, and excised.
Isolation of Nucleic Acids. Nuclei were isolated by a method modified from that of Hymer and Kuff (20). All procedures were performed at 2? unless otherwise noted. After removal of connective tissue with a tissue press, livers were homogenized in 24 ml of 0.25 M sucrose/2 mM CaC12/O.01 M Tris-HCl, pH 7.5. The homogenate was filtered through 250-jm nylon mesh and then 100-jim nylon mesh. Additional buffer and 25% Triton
Abbreviations: AFB1, aflatoxin B1; AFB1-N7-Gua, 2,3-dihydro-2- (N7-guanyl)-3-hydroxy-aflatoxin B1; HPLC, high-pressure liquid chromatography.
45
This content downloaded from 130.132.123.28 on Tue, 6 May 2014 08:51:38 AMAll use subject to JSTOR Terms and Conditions

1746 Biochemistry: Croy et al.
X-100 in homogenizing buffer were slowly added to obtain a final concentration of 5% Triton X-100 in a volume that was 9 times the liver weight. The homogenate was centrifuged at 1000 X g for 10 min. The supernatant was removed by aspi- ration and the crude nuclear pellet was resuspended in ho- mogenizing buffer and frozen for storage at -70?.
Nucleic acids were extracted from the crude nuclear prep- aration by a procedure modified from that of Marmur (21). Nuclei were thawed and suspended in 0.05 M Tris-HCl, pH 6.5. Appropriate volumes of 5% sodium dodecyl sulfate and 4 M NaCl were added to the suspended nuclei which resulted in concentrations of 1% sodium dodecyl sulfate and 1 M NaCl; the final concentration of DNA was 0.2-0.3 mg/ml. An equal volume of CHCl3/isoamyl alcohol, 24:1 (vol/vol), was added and the two phases were shaken vigorously for 20 min at room temperature. The aqueous and organic phases were separated by centrifugation at 7000 X g for 10 min, and the aqueous phase was isolated and extracted a second time by using the same procedure. Nucleic acids were precipitated from the aqueous phase with 3 volumes of cold ethanol, spun onto glass rods, washed two times in ethanol, and dried in vacuo.
Hydrolysis of Nucleic Acids. The nucleic acids were dis- solved at 0.8-0.9 mg/ml in 0.05 M KAc, pH 5.2/0.1 mM ZnC12. DNA was denatured by heating at 95? for 15 min and rapidly cooling on ice. Nuclease P1 (Yamasa Shoyu Co., Ltd. Choshi, Japan), 1 mg/200 mg of nucleic acid, was added to the solution and incubated overnight at 40?. The solution was deproteinized by shaking with an equal volume of CHC13 and the two phases were separated by centrifugation at 700 X g for 10 min.
Chromatography. AFB1 adducts were isolated by high- pressure liquid chromatography (HPLC) using a Micromeritics model 7000 chromatograph equipped with a Waters Associates model 440 detector (254 and 365 nm). The deproteinized hy- drolysate was adjusted to 10% methanol and loaded directly onto a 0.8 X 60 cm column packed with C18-Corasil B (Waters Associates) as described (16). The column was eluted at ambient temperature with 10% methanol at 3 ml/min until all material absorbing at 254 nm was removed. A 20-min gradient of 10- 80% methanol was then used to elute the less polar aflatoxin- containing material.
Analytical chromatography was performed on a jBondapak C18 column (Waters Associates) eluted at 50? and 1.0 ml/min with 35% methanol/H20.
Methylation of Adduct. Methylation of microgram amounts of aflatoxin-containing material was accomplished by modi- fication of a method previously described (16). Approximately 1 jg of material was mixed with 5 j1 of dimethyl sulfate in 100 jil of N,N-dimethylacetamide at room temperature for 6 hr. After evaporation to dryness overnight in vacuo, the sample was hydrolyzed with 50 1l of 70% perchloric acid at 95? for 1 hr. The solution was neutralized with 3 M KOH and adjusted to pH 4.0 with 88% formic acid. After centrifugation, a portion of the sample was analyzed by cation-exchange HPLC on a 25 X 0.22 cm Durrum DC-4A column eluted with 0.1 M ammo- nium formate, pH 4.1 at 0.4 ml/min and 63?.
Spectrometry. Field-desorption mass spectra were obtained on a Varian model 731 spectrometer using a combined field- ionization/field-desorption/electron-impact ion source. Sam- ples dissolved in water/methanol mixtures were applied to the emitter with a microsyringe and air-dried. Oscillograph re- cording was initiated when ions were detected in the molecular ion region and then the magnetic field was scanned down while the emitter current was increased.
Ultraviolet-visible spectra were taken in 0.1 M HC1 and 0.1 M NaOH using a Beckman model 25 UV spectrometer.
Proc. Natl. Acad. Sci. USA 75 (1978)
Quantitative Determinations. Quantitative determinations of the level of N7-Gua substitution in DNA were made using [3H]AFB1, specific activity 5 Ci/mmol (Moravek Biochemi- cals), diluted with unlabeled material to specific activity of 100 mCi/mmol. Doses (50 jl) of 0.125, 0.25, 0.5, and 1.0 mg of AFBi in dimethyl sulfoxide per kg were administered intra- peritoneally to 140 g male Fischer rats. The DNA isolated from single rat livers was hydrolyzed and analyzed chromato- graphically as described above with the exceptions that the gradient elution phase was 40 min long and isocratic separation of peaks I and II was not performed; 3-ml fractions were col- lected throughout the load, wash, and gradient phases.
The level of formation of AFBi-N7-Gua in DNA was cal- culated by dividing the amount (mmol) of DNA bases in the hydrolysate applied to the column, as determined by the di- phenylamine reaction, by the amount (mmol) of AFB1-N7-Gua determined through the measurement of 3H activity. 3H ex- change was determined using [14C]AFBL (Makor Chemicals, Ltd., Jerusalem, Israel) at 180 mCi/mmol. Animals were in- jected with AFB1 at 0.5 mg/kg containing 35 jCi of [3H]AFB1 and 5 jiCi of [14C]AFBi. The ratio 3H/14C was determined in the isolated adduct peak and the percentage exchange of the 3H label was calculated from the decrease in this ratio relative to that determined for the administered dose.
DNA was determined by the method of Burton (22) as modified by Giles and Myers (23). RNA was determined by the orcinol reaction (24).
RESULTS
Isolation of Adduct. The crude nuclear fraction isolated from 60 rat livers was used for preparation of nucleic acid ad- ducts. The yield of nuclei, measured by DNA content, was 95-100%. RNA, associated with the nuclear fraction, was not separated from DNA during the isolation procedure. Under conditions used for RNA digestion (i.e., pH 7.5 and 37?) a 10% or greater loss of bound material from DNA was noted in pre- vious experiments. Approximately 450 mg of DNA and 80 mg of RNA were obtained by using the isolation procedure de- scribed in Materials and Methods.
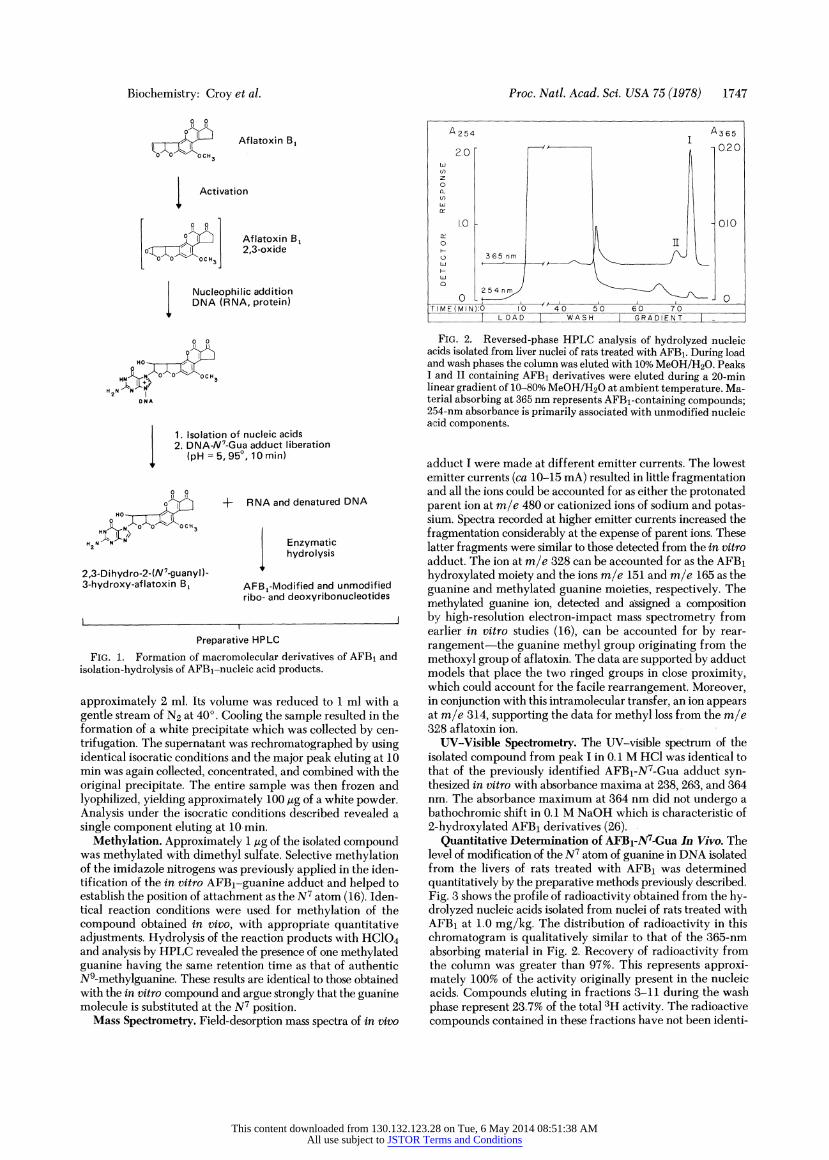
Fig. 1 summarizes the postulated activation pathway and outlines the isolation method. Denaturation of the DNA in acidic buffer liberated the 7-substituted purine deoxyribo- nucleotide derivatives as modified bases (25) and rendered the DNA susceptible to digestion by nuclease P1. Digestion of RNA and denatured DNA by nuclease Pi resulted in efficient and rapid hydrolysis of RNA and denatured DNA to nucleotide derivatives. Ribo- and deoxyribonucleotides were separated from AFBi-modified bases by preparative scale reversed-phase HPLC. A typical separation is shown in Fig. 2. Up to 100 mg of nucleic acid hydrolysate was processed in one run. Nucleotide components were eluted during the load and wash phases. AFBl-ribonucleotide derivatives which are not hydrolyzed during the heat denaturation step are eluted in this portion of the chromatogram. These polar compounds are easily separated from the relatively nonpolar N7-guanine derivative which is eluted at 74 min during the gradient phase. Complete separa- tion of this peak from a minor peak eluting at 71 min was not possible using this preparatory column with large amounts of material. These two peaks were isolated together and evapo- rated to 2 ml at 45? under reduced presssure. Components of these peaks were separated by using isocratic conditions with a Waters jBondapak C18 column eluted with methanol/water, 35:65 (vol/vol), at 50? and 1.0 ml/min. Under these conditions, a major peak was seen at 10 min and several minor ones at 8, 6, and 3 min. The major peak (I) was collected in a volume of
This content downloaded from 130.132.123.28 on Tue, 6 May 2014 08:51:38 AMAll use subject to JSTOR Terms and Conditions
Biochemistry: Croy et al.
0
10 ? Aflatoxin B,
OCH3
Activation
ill Aflatoxin B1 to"^~,[ 1 2,3-oxide
Nucleophilic addition DNA (RNA, protein)
0 Ho
DNA
1. Isolation of nucleic acids 2. DNA-N7-Gua adduct liberation
sI (pH=5,95?,10min)
poyS- + RNA and denatured DNA HO
HN'o" OCH3
H2 N N Enzymatic hydrolysis
2,3-Dihydro-2-(N7_guanyl )- 3-hydroxy-aflatoxin B1 AFB,-Modified and unmodified
ribo- and deoxyribonucleotides
Preparative HPLC
FIG. 1. Formation of macromolecular derivatives of AFB1 and isolation-hydrolysis of AFBl-nucleic acid products.
approximately 2 ml. Its volume was reduced to 1 ml with a gentle stream of N2 at 40?. Cooling the sample resulted in the formation of a white precipitate which was collected by cen- trifugation. The supernatant was rechromatographed by using identical isocratic conditions and the major peak eluting at 10 min was again collected, concentrated, and combined with the original precipitate. The entire sample was then frozen and lyophilized, yielding approximately 100 ug of a white powder. Analysis under the isocratic conditions described revealed a single component eluting at 10 min.
Methylation. Approximately 1 jig of the isolated compound was methylated with dimethyl sulfate. Selective methylation of the imidazole nitrogens was previously applied in the iden- tification of the in vitro AFBi-guanine adduct and helped to establish the position of attachment as the N7 atom (16). Iden- tical reaction conditions were used for methylation of the compound obtained in vivo, with appropriate quantitative adjustments. Hydrolysis of the reaction products with HC104 and analysis by HPLC revealed the presence of one methylated guanine having the same retention time as that of authentic N9-methylguanine. These results are identical to those obtained with the in vitro compound and argue strongly that the guanine molecule is substituted at the N7 position.
Mass Spectrometry. Field-desorption mass spectra of in vivo
Proc. Natl. Acad. Sci. USA 75 (1978) 1747
A 254 A 365
2.0 0.20
o
1.0IME MIN): 40 50 60 7 0.10
I LOAD I WASH I GRADIENT I
FIG. 2. Reversed-phase HPLC analysis of hydrolyzed nucleic acids isolated from liver nuclei of rats treated with AFB1. During load and wash phases the column was eluted with 10% MeOH/H20. Peaks I and II containing AFB1 derivatives were eluted during a 20-min linear gradient of 10-80% MeOH/H20 at ambient temperature. Ma- terial absorbing at 365 nm represents AFBi-containing compounds; 254-nm absorbance is primarily associated with unmodified nucleic acid components.
adduct I were made at different emitter currents. The lowest emitter currents (ca 10-15 mA) resulted in little fragmentation and all the ions could be accounted for as either the protonated parent ion at m/e 480 or cationized ions of sodium and potas- sium. Spectra recorded at higher emitter currents increased the fragmentation considerably at the expense of parent ions. These latter fragments were similar to those detected from the in vitro adduct. The ion at m/e 328 can be accounted for as the AFB1 hydroxylated moiety and the ions m/e 151 and m/e 165 as the guanine and methylated guanine moieties, respectively. The methylated guanine ion, detected and assigned a composition by high-resolution electron-impact mass spectrometry from earlier in vitro studies (16), can be accounted for by rear- rangement-the guanine methyl group originating from the methoxyl group of aflatoxin. The data are supported by adduct models that place the two ringed groups in close proximity, which could account for the facile rearrangement. Moreover, in conjunction with this intramolecular transfer, an ion appears at m/e 314, supporting the data for methyl loss from the m/e 328 aflatoxin ion.
UV-Visible Spectrometry. The UV-visible spectrum of the isolated compound from peak I in 0.1 M HCI was identical to that of the previously identified AFBi-N7-Gua adduct syn- thesized in vitro with absorbance maxima at 238, 263, and 364 nmrn. The absorbance maximum at 364 nm did not undergo a bathochromic shift in 0.1 M NaOH which is characteristic of 2-:hydroxylated AFB1 derivatives (26).
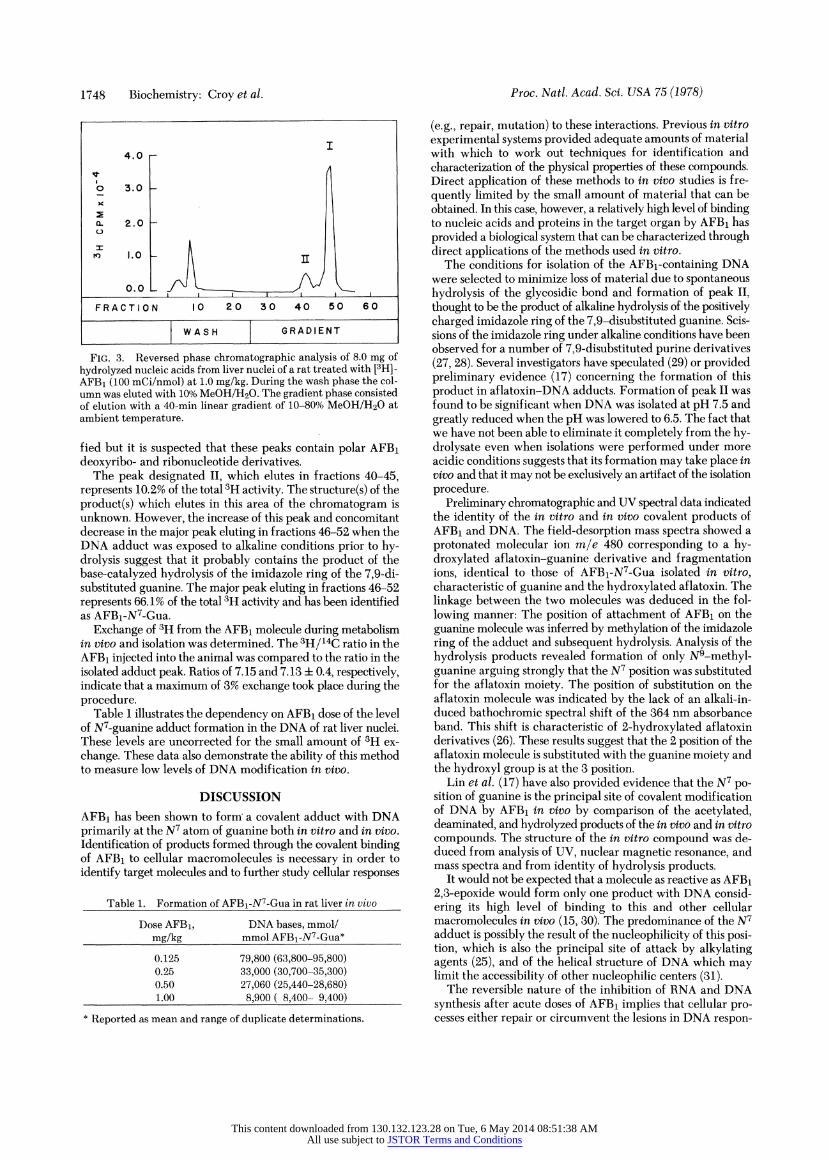
Quantitative Determination of AFBi-N7-Gua In Vivo. The level of modification of the N7 atom of guanine in DNA isolated from the livers of rats treated with AFB1 was determined quantitatively by the preparative methods previously described. Fig. 3 shows the profile of radioactivity obtained from the hy- drolyzed nucleic acids isolated from nuclei of rats treated with AFB1 at 1.0 mg/kg. The distribution of radioactivity in this chromatogram is qualitatively similar to that of the 365-nm absorbing material in Fig. 2. Recovery of radioactivity from the column was greater than 97%. This represents approxi- mately 100% of the activity originally present in the nucleic acids. Compounds eluting in fractions 3-11 during the wash phase represent 23.7% of the total 3H activity. The radioactive compounds contained in these fractions have not been identi-
This content downloaded from 130.132.123.28 on Tue, 6 May 2014 08:51:38 AMAll use subject to JSTOR Terms and Conditions
1748 Biochemistry: Croy et al.
4.0 -
0 3.0-
- 2.0 - 0
r- ro 1.0 I
0.0
FRACTION 10 20 30 40 50 60
WASH GRADIENT
FIG. 3. Reversed phase chromatographic analysis of 8.0 mg of
hydrolyzed nucleic acids from liver nuclei of a rat treated with [3H]- AFB1 (100 mCi/nmol) at 1.0 mg/kg. During the wash phase the col- umn was eluted with 10% MeOH/H20. The gradient phase consisted of elution with a 40-min linear gradient of 10-80% MeOH/H20 at ambient temperature.
fied but it is suspected that these peaks contain polar AFBi deoxyribo- and ribonucleotide derivatives.
The peak designated II, which elutes in fractions 40-45, represents 10.2% of the total 3H activity. The structure(s) of the product(s) which elutes in this area of the chromatogram is unknown. However, the increase of this peak and concomitant decrease in the major peak eluting in fractions 46-52 when the DNA adduct was exposed to alkaline conditions prior to hy- drolysis suggest that it probably contains the product of the base-catalyzed hydrolysis of the imidazole ring of the 7,9-di- substituted guanine. The major peak eluting in fractions 46-52 represents 66.1% of the total 3H activity and has been identified as AFBi-N7-Gua.
Exchange of 3H from the AFBi molecule during metabolism in vivo and isolation was determined. The 3H/14C ratio in the
AFB1 injected into the animal was compared to the ratio in the
isolated adduct peak. Ratios of 7.15 and 7.13 i 0.4, respectively, indicate that a maximum of 3% exchange took place during the
procedure. Table 1 illustrates the dependency on AFB1 dose of the level
of NV-guanine adduct formation in the DNA of rat liver nuclei.
These levels are uncorrected for the small amount of 3H ex-
change. These data also demonstrate the ability of this method
to measure low levels of DNA modification in vivo.
DISCUSSION
AFB1 has been shown to form' a covalent adduct with DNA
primarily at the N7 atom of guanine both in vitro and in vivo. Identification of products formed through the covalent binding of AFB1 to cellular macromolecules is necessary in order to
identify target molecules and to further study cellular responses
Table 1. Formation of AFB1-N7-Gua in rat liver in vivo
Dose AFB1, DNA bases, mmol/
mg/kg mmol AFBi-N7-Gua*
0.125 79,800 (63,800-95,800) 0.25 33,000 (30,700-35,300) 0.50 27,060 (25,440-28,680) 1.00 8,900 ( 8,400- 9,400)
* Reported as mean and range of duplicate determinations.
Proc. Natl. Acad. Sci. USA 75 (1978)
(e.g., repair, mutation) to these interactions. Previous in vitro experimental systems provided adequate amounts of material with which to work out techniques for identification and characterization of the physical properties of these compounds. Direct application of these methods to in vivo studies is fre- quently limited by the small amount of material that can be obtained. In this case, however, a relatively high level of binding to nucleic acids and proteins in the target organ by AFB1 has provided a biological system that can be characterized through direct applications of the methods used in vitro.
The conditions for isolation of the AFBi-containing DNA were selected to minimize loss of material due to spontaneous hydrolysis of the glycosidic bond and formation of peak II, thought to be the product of alkaline hydrolysis of the positively charged imidazole ring of the 7,9-disubstituted guanine. Scis- sions of the imidazole ring under alkaline conditions have been observed for a number of 7,9-disubstituted purine derivatives (27, 28). Several investigators have speculated (29) or provided preliminary evidence (17) concerning the formation of this product in aflatoxin-DNA adducts. Formation of peak II was found to be significant when DNA was isolated at pH 7.5 and greatly reduced when the pH was lowered to 6.5. The fact that we have not been able to eliminate it completely from the hy- drolysate even when isolations were performed under more acidic conditions suggests that its formation may take place in vivo and that it may not be exclusively an artifact of the isolation procedure.
Preliminary chromatographic and UV spectral data indicated the identity of the in vitro and in vivo covalent products of AFBi and DNA. The field-desorption mass spectra showed a protonated molecular ion m/e 480 corresponding to a hy- droxylated aflatoxin-guanine derivative and fragmentation ions, identical to those of AFB1-N7-Gua isolated in vitro, characteristic of guanine and the hydroxylated aflatoxin. The linkage between the two molecules was deduced in the fol- lowing manner: The position of attachment of AFB1 on the guanine molecule was inferred by methylation of the imidazole ring of the adduct and subsequent hydrolysis. Analysis of the hydrolysis products revealed formation of only N9-methyl- guanine arguing strongly that the N7 position was substituted for the aflatoxin moiety. The position of substitution on the aflatoxin molecule was indicated by the lack of an alkali-in- duced bathochromic spectral shift of the 364 nm absorbance band. This shift is characteristic of 2-hydroxylated aflatoxin derivatives (26). These results suggest that the 2 position of the aflatoxin molecule is substituted with the guanine moiety and the hydroxyl group is at the 3 position.
Lin et al. (17) have also provided evidence that the N7 po- sition of guanine is the principal site of covalent modification of DNA by AFB1 in vivo by comparison of the acetylated, deaminated, and hydrolyzed products of the in vivo and in vitro compounds. The structure of the in vitro compound was de- duced from analysis of UV, nuclear magnetic resonance, and mass spectra and from identity of hydrolysis products.
It would not be expected that a molecule as reactive as AFBi 2,3-epoxide would form only one product with DNA consid- ering its high level of binding to this and other cellular macromolecules in vivo (15, 30). The predominance of the N7 adduct is possibly the result of the nucleophilicity of this posi- tion, which is also the principal site of attack by alkylating agents (25), and of the helical structure of DNA which may limit the accessibility of other nucleophilic centers (31).
The reversible nature of the inhibition of RNA and DNA synthesis after acute doses of AFB1 implies that cellular pro- cesses either repair or circumvent the lesions in DNA respon-
This content downloaded from 130.132.123.28 on Tue, 6 May 2014 08:51:38 AMAll use subject to JSTOR Terms and Conditions

Biochemistry: Croy et al.
sible for template inhibition. Identification and quantitation of these compounds will enable further studies on mechanisms of their repair by cellular processes and of their role in carci- nogenesis and mutagenesis.
We thank Paul Donahue and Michael Couch for their contributions to this work. Contributions made by Professor Klaus Biemann and the staff of the Massachusetts Institute of Technology Mass Spectrometry Facility, which is operated under Grant RR00317 from the Biotech- nology Resources Branch, Division of Research Resources, National Institutes of Health, are also gratefully acknowledged. Financial support for this research was provided by Grants 5 PO1 ES 00597-06 and 5 T32 ES07020-02 and by Contract NO1-CP 43265 from the National Institutes of Health.
1. Wogan, G. N. (1973) in Methods in Cancer Research, ed. Busch, H. (Academic Press, New York), Vol. 7, pp. 309-344.
2. Krahn, D. F. & Heidelberger, C. (1977) Mutat. Res. 46, 27- 44.
3. Garner, R. C. & Wright, C. M. (1973) Br. J. Cancer 28, 544- 551.
4. Wong, J. J. & Hsieh, D. P. (1976) Proc. Natl. Acad. Sci. USA 73, 2241-2244.
5. Wogan, G. N. (1976) in Liver Cell Cancer, eds. Cameron, H. M., Linsell, D. A. & Warwick, G. P. (Elsevier/North-Holland Bio- medical Press, New York), pp. 121-151.
6. Butler, W. H. (1964) Br. J. Cancer 18, 756-762. 7. Friedman, M. A. & Wogan, G. N. (1970) Life Sci. 9, 741-747. 8. Lafarge, C. & Frayssinet, C. (1970) Int. J. Cancer 6, 74-83. 9. Pong, R. S. & Wogan, G. N. (1970) Cancer Res. 30, 294-304.
10. Rogers, A. E. & Newberne, P. M. (1967) Cancer Res. 27, 855- 864.
11. DeRecondo, A. M., Frayssinet, C. H., Lafarge, C. & LeBreton, E. (1966) Biochim. Biophys. Acta 119,322-330.
12. Garner, R. C. (1973) Chem. Biol. Interact. 6, 125-129.
Proc. Natl. Acad. Sci. USA 75 (1978) 1749
13. Swenson, D. H., Miller, J. A. & Miller, E. C. (1973) Biochem. Biophys. Res. Commun. 53, 1260-1267.
14. Swenson, D. H., Miller, E. C. & Miller, J. A. (1974) Biochem. Biophys. Res. Commun. 60, 1036-1043.
15. Swenson, D. H., Lin, J., Miller, E. C. & Miller, J. A. (1977) Cancer Res. 37, 172-180.
16. Essigmann, J. M., Croy, R. G., Nadzan, A. M., Busby, W. F., Reinhold, V. N., Biichi, G. & Wogan, G. N. (1977) Proc. Natl. Acad. Sci. USA 74, 1870-1874.
17. Lin, J., Miller, J. A. & Miller, E. C. (1977) Cancer Res. 37, 4430-4438.
18. Wogan, G. N. & Newberne, P. M. (1967) Cancer Res. 27, 2370-2376.
19. Asao, T., Biuchi, G., Abdel-Kader, M. M., Chang, S. B., Wick, E. L. & Wogan, G. N. (1965) J. Am. Chem. Soc. 87, 882-886.
20. Hymer, W. C. & Kuff, E. L. (1964) J. Histochem. Cytochem. 12, 359-363.
21. Marmur, J. (1961) J. Mol. Biol. 3,208-218. 22. Burton, K. (1956) Biochem. J. 62, 315-323. 23. Giles, K. W. & Meyers, A. (1965) Nature 206,93. 24. Albaum, H. G. & Umbreit, W. W. (1947) J. Biol. Chem. 167,
369-376. 25. Lawley, P. D. (1976) in Screening Tests in Chemical Carcino-
genesis, eds. Montesano, R., Barsch, H. & Tomatis, L. (Interna- tional Agency for Research on Cancer, Lyon, France), pp. 181-208.
26. Biiehi, G., Foulkes, D. M., Kurono, M., Mitchell, G. F. & Schneider, R. S. (1967) J. Am. Chem. Soc. 89,6745-6753.
27. Lawley, P. D. & Brookes, P. (1963) Biochem. J. 89, 127-138. 28. Hecht, S. M., Adams, B. L. & Kozarich, J. W. (1976) J. Org.
Chem. 41, 2303-2311. 29. Martin, C. N. & Garner, R. C. (1977) Nature 267,863-865. 30. Garner, R. C. & Wright, C. M. (1975) Chem. Biol. Interact. 11,
123-131. 31. Singer, B. (1975) in Progress in Nucleic Acid Research and
Molecular Biology, ed. Cohn, W. E. (Academic Press, New York), Vol. 15, pp. 219-284.
This content downloaded from 130.132.123.28 on Tue, 6 May 2014 08:51:38 AMAll use subject to JSTOR Terms and Conditions