i tumori ematologici: inquadramento clinico · •these leukemias do not fit easily into the who...
TRANSCRIPT

I tumori ematologici: inquadramento clinico
Nicola CascavillaEmatologia – San Giovanni Rotondo

1. Le emopatie neoplastiche, a complessità multidisciplinare ed a rischio dicondizioni di variabilità, talvolta sono gestite con approcci personali, nonscevri da errori.
2. C’è necessità di un inquadramento clinico complessivo e di percorsicontrollati concordi con i sistemi classificativo/prognostici e le linee guidaterapeutiche.
L’inquadramento clinico e l’analisi epidemiologica permettono di:a) Costruire processi orientati a continuità, integrazione, completezzab) Definire percorsi diagnostico-terapeutici più semplificatic) Personalizzare approcci diagnostici e trattamenti finalizzati ai risultati programmatid) Condividere criteri diagnostici clinici, biologici e strumentalie) Registrare incidenza e relazioni con fattori ambientali, sociali, personalif) Programmare spesa sanitaria ed investimenti per nuovi farmaci
Inquadramento clinico delle emopatie neoplastiche

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche

Pazienti < 65 anni: 35%
Pazienti tra 65 e 74 anni: 28%
Pazienti > 75 anni: 37%
Età Media: 77 – 83 anni
1% di tutte le neoplasie maligne
15.000 nuovi casi per anno in Europa
Sopravvivenza Mediana: circa 33 mesi primadell’avvento delle nuove terapie.
Recenti progressi terapeutici grazie ai nuoviagenti (Talidomide, Bortezomib, Lenalidomide)da soli o in associazione, con raddoppiamentodella sopravvivienza globale e libera da malattia.
Ulteriori miglioramenti sono attesi con i farmacipiù recenti (nuovi immunomodulanti, nuoviinibitori del proteasoma, anticorpi monoclonali).
Incidenza annuale/100.000
Totale Maschi Femmine
Tutte le età 5,7 7,0 4,7
< 65 anni 2,1 2,5 1,8
≥ 65 anni 30,0 38,2 24,8
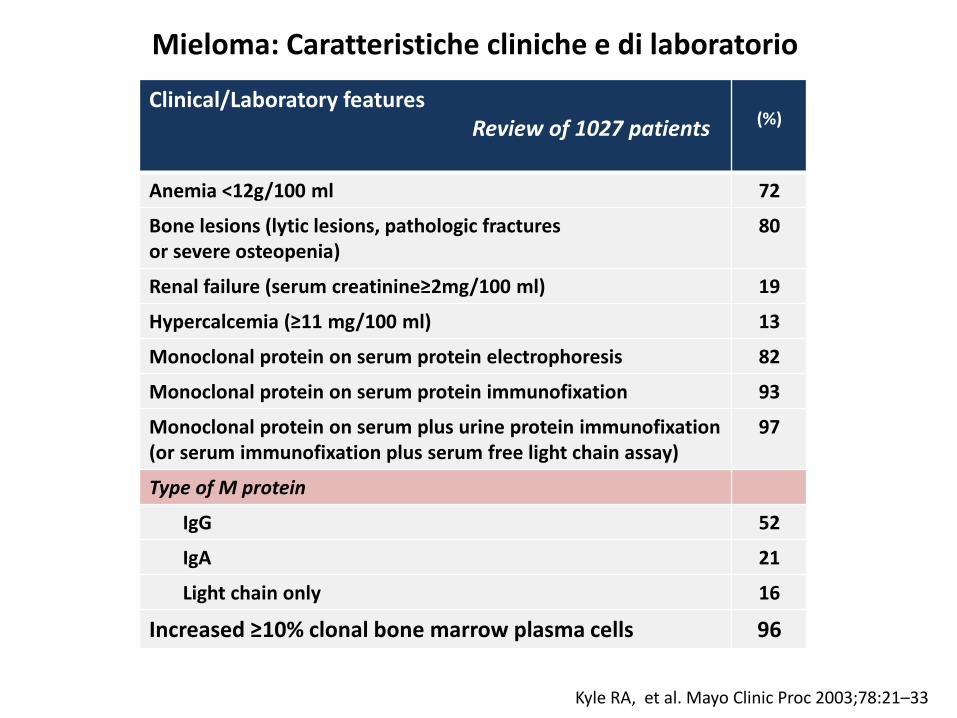
Mieloma: Caratteristiche cliniche e di laboratorio
Kyle RA, et al. Mayo Clinic Proc 2003;78:21–33
Clinical/Laboratory featuresReview of 1027 patients (%)
Anemia <12g/100 ml 72
Bone lesions (lytic lesions, pathologic fracturesor severe osteopenia)
80
Renal failure (serum creatinine≥2mg/100 ml) 19
Hypercalcemia (≥11 mg/100 ml) 13
Monoclonal protein on serum protein electrophoresis 82
Monoclonal protein on serum protein immunofixation 93
Monoclonal protein on serum plus urine protein immunofixation(or serum immunofixation plus serum free light chain assay)
97
Type of M protein
IgG 52
IgA 21
Light chain only 16
Increased ≥10% clonal bone marrow plasma cells 96

Rajkumar SV et al, Lancet Oncology, vol. 15, 2014
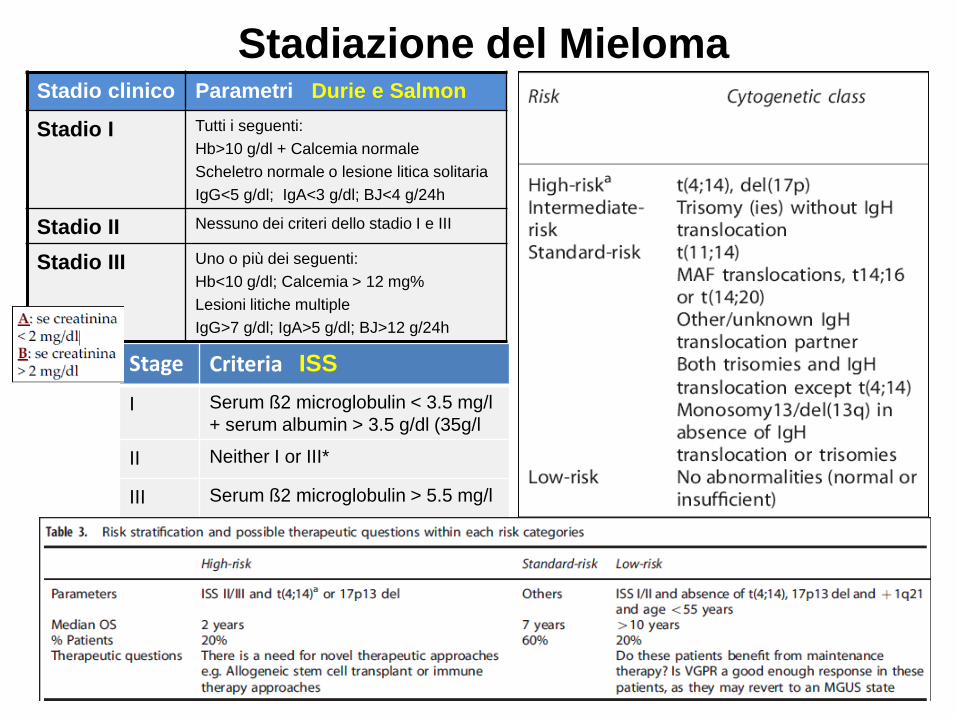
Stage Criteria ISS
I Serum ß2 microglobulin < 3.5 mg/l
+ serum albumin > 3.5 g/dl (35g/l
II Neither I or III*
III Serum ß2 microglobulin > 5.5 mg/l
Stadiazione del MielomaStadio clinico Parametri Durie e Salmon
Stadio I Tutti i seguenti:
Hb>10 g/dl + Calcemia normale
Scheletro normale o lesione litica solitaria
IgG<5 g/dl; IgA<3 g/dl; BJ<4 g/24h
Stadio II Nessuno dei criteri dello stadio I e III
Stadio III Uno o più dei seguenti:
Hb<10 g/dl; Calcemia > 12 mg%
Lesioni litiche multiple
IgG>7 g/dl; IgA>5 g/dl; BJ>12 g/24h
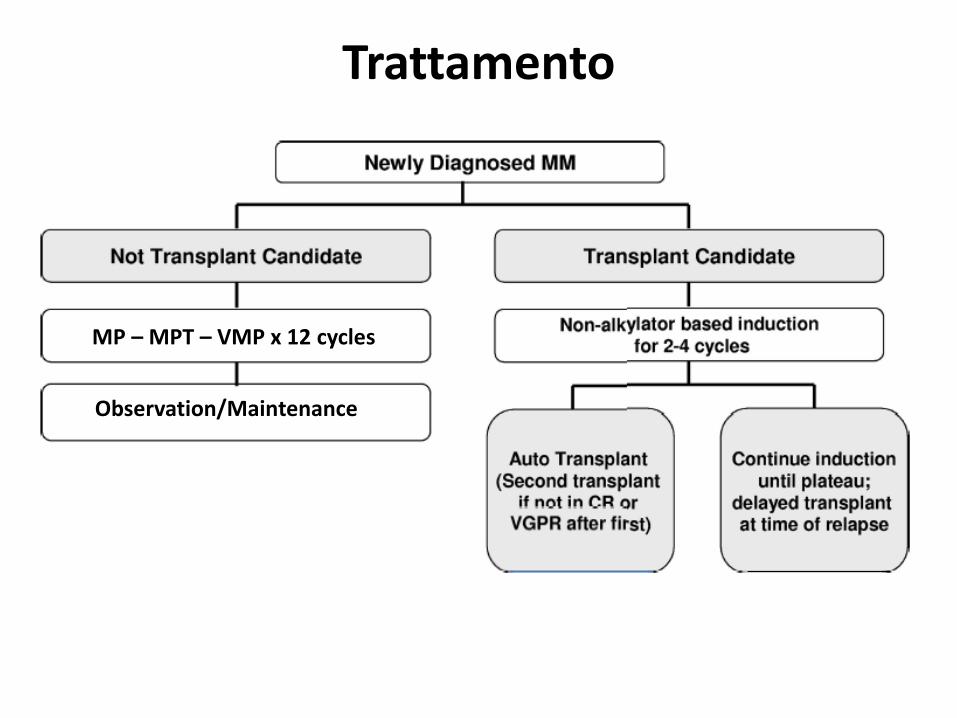
MP – MPT – VMP x 12 cycles
Observation/Maintenance
Trattamento

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche
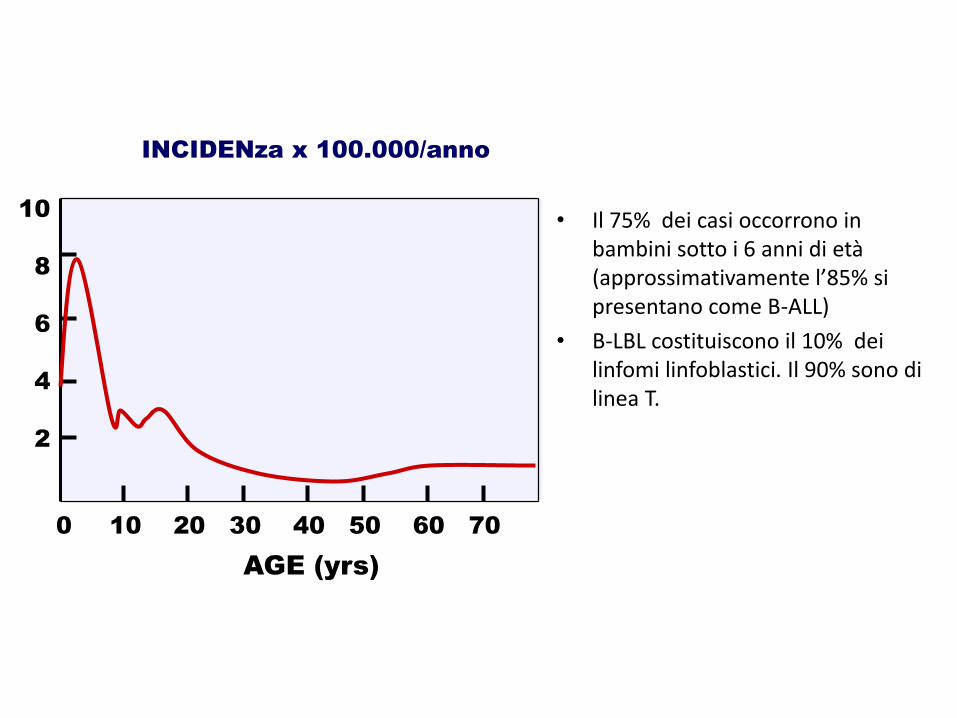
AGE (yrs)
INCIDENza x 100.000/anno
0 10 20 30 40 50 60 70
10
8
6
4
2
• Il 75% dei casi occorrono in bambini sotto i 6 anni di età (approssimativamente l’85% si presentano come B-ALL)
• B-LBL costituiscono il 10% dei linfomi linfoblastici. Il 90% sono di linea T.

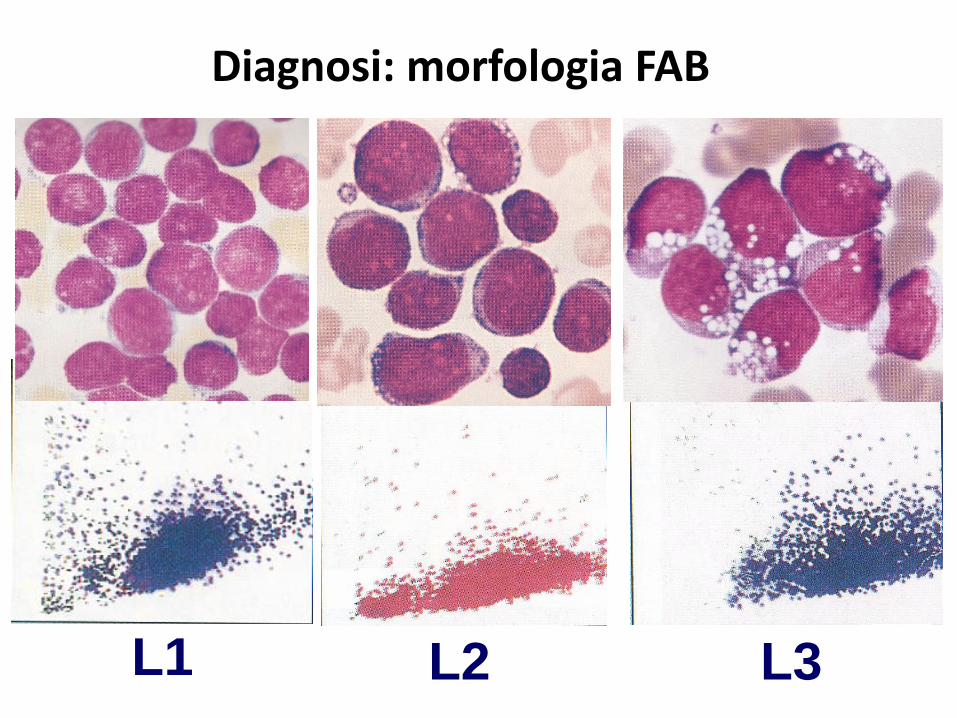
Diagnosi: morfologia FAB
L1 L2 L3
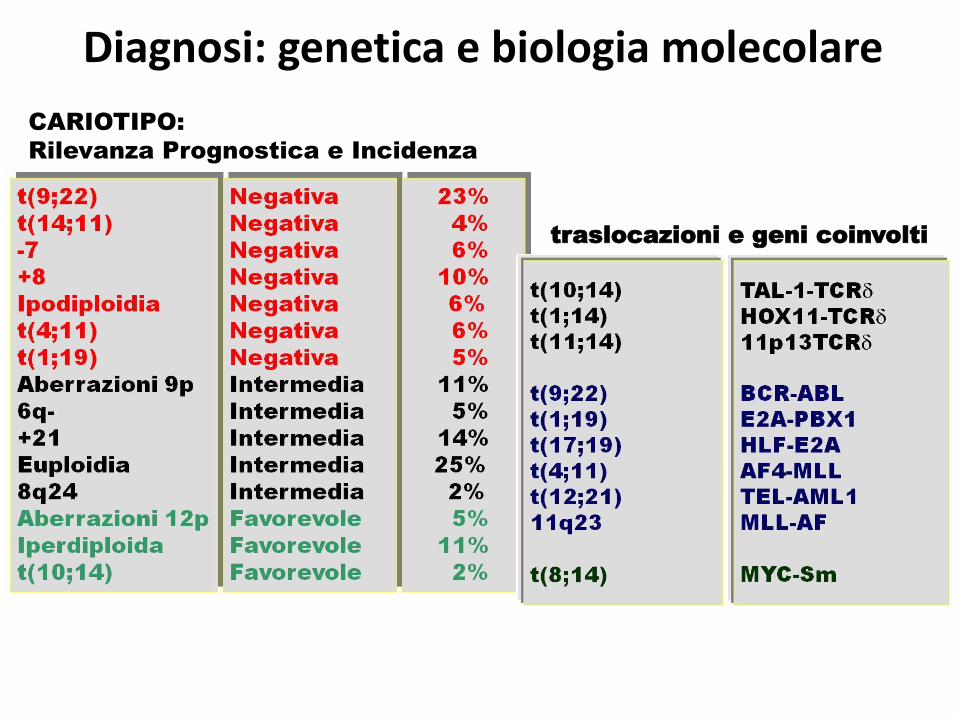
CARIOTIPO:
Rilevanza Prognostica e Incidenza
traslocazioni e geni coinvolti
Diagnosi: genetica e biologia molecolare
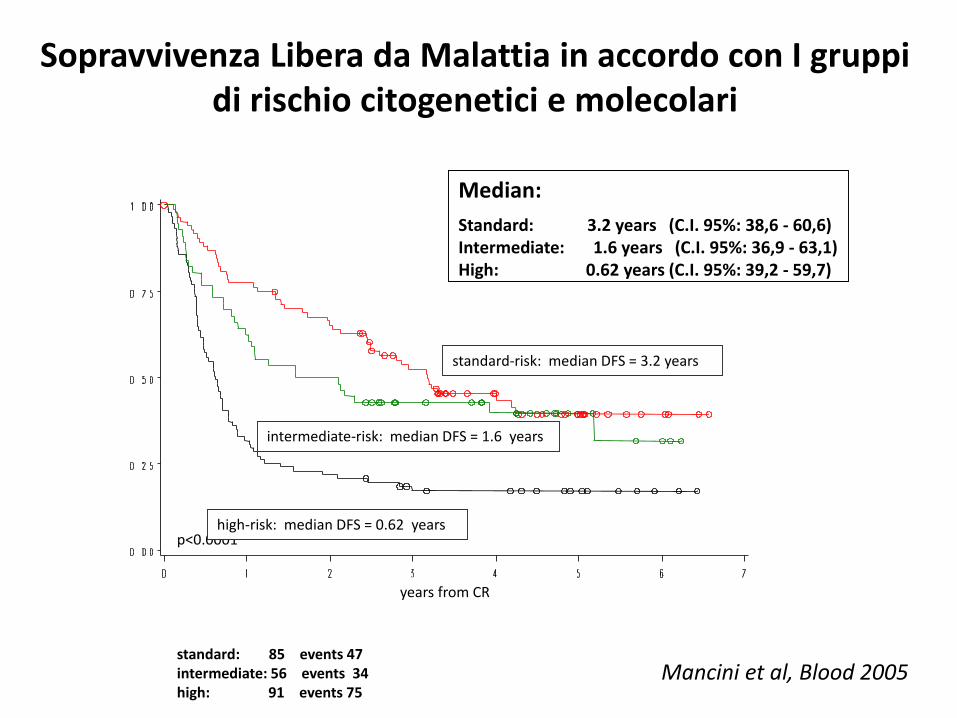
standard-risk: median DFS = 3.2 years
years from CR
p<0.0001
standard: 85 events 47intermediate: 56 events 34high: 91 events 75
intermediate-risk: median DFS = 1.6 years
high-risk: median DFS = 0.62 years
Sopravvivenza Libera da Malattia in accordo con I gruppi di rischio citogenetici e molecolari
Median:
Standard: 3.2 years (C.I. 95%: 38,6 - 60,6) Intermediate: 1.6 years (C.I. 95%: 36,9 - 63,1) High: 0.62 years (C.I. 95%: 39,2 - 59,7)
Mancini et al, Blood 2005
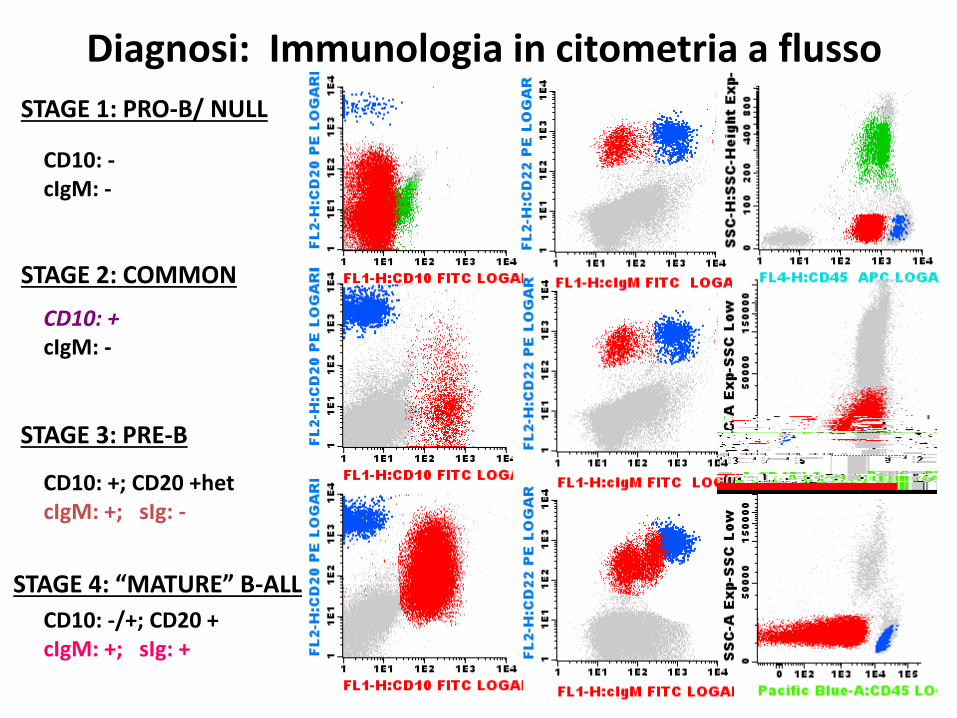
STAGE 1: PRO-B/ NULL
STAGE 2: COMMON
STAGE 3: PRE-B
STAGE 4: “MATURE” B-ALL
CD10: -cIgM: -
CD10: +cIgM: -
CD10: +; CD20 +hetcIgM: +; sIg: -
CD10: -/+; CD20 +cIgM: +; sIg: +
Diagnosi: Immunologia in citometria a flusso
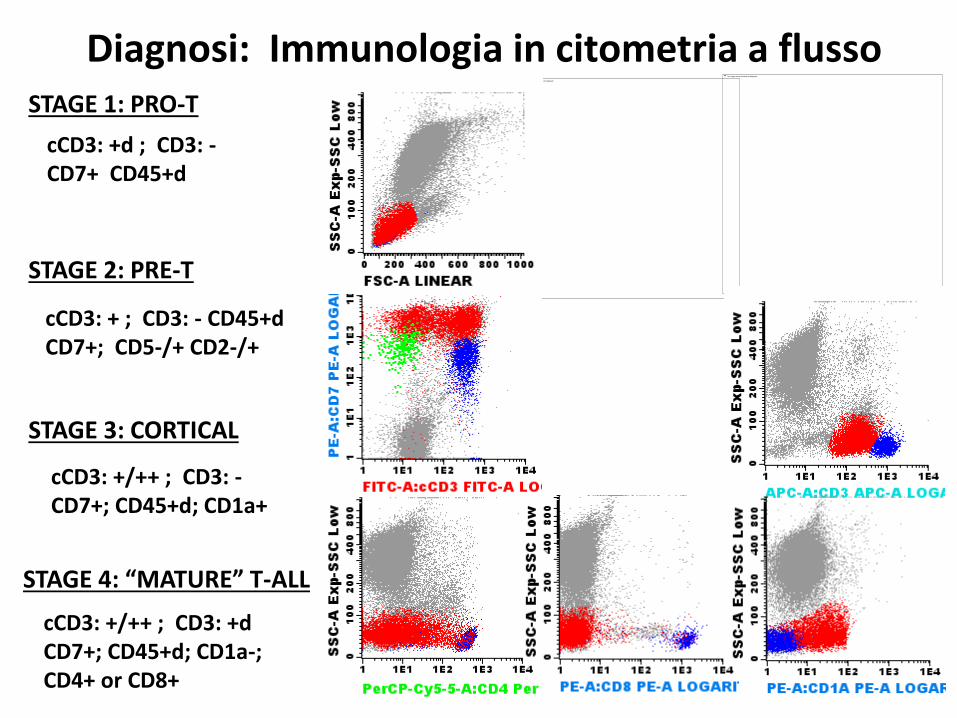
STAGE 1: PRO-T
STAGE 2: PRE-T
STAGE 3: CORTICAL
STAGE 4: “MATURE” T-ALL
cCD3: +d ; CD3: -CD7+ CD45+d
cCD3: + ; CD3: - CD45+dCD7+; CD5-/+ CD2-/+
cCD3: +/++ ; CD3: -CD7+; CD45+d; CD1a+
cCD3: +/++ ; CD3: +dCD7+; CD45+d; CD1a-;CD4+ or CD8+
Diagnosi: Immunologia in citometria a flusso

B-ALL Ph+25% of adult ALL and 2-4% of childhood ALLPatients may have organ involvementThe worst prognosis among ALL patients
B- ALL with t(v;11q23)The most common leukemia in infants < 1 yr of ageFrequently with high WBCs and CNS involvementMLL-AF4 translocation have a poor prognosis
B-ALL with t(12;21)25% of cases of B-ALL, Frequency decreases in older childrenNo characteristic clinical featuresVery favourable prognosis in > 90% children
B-ALL with hyperdiploidy(>50 and < 66 chromosomes)25% of B ALLNo characteristic clinical featuresVery favourable prognosis in > 90% children
B-ALL with hypodiploidy• < 46 chromosomes• 5% of ALL • Poor prognosis
B-ALL with t(5;14)• < 1% ALL• Prognosis similar to other ALL
B-ALL with t(1;19)• < 1% of ALL• Poor prognosis
B-ALL con «recurrent genetic abnormalities»
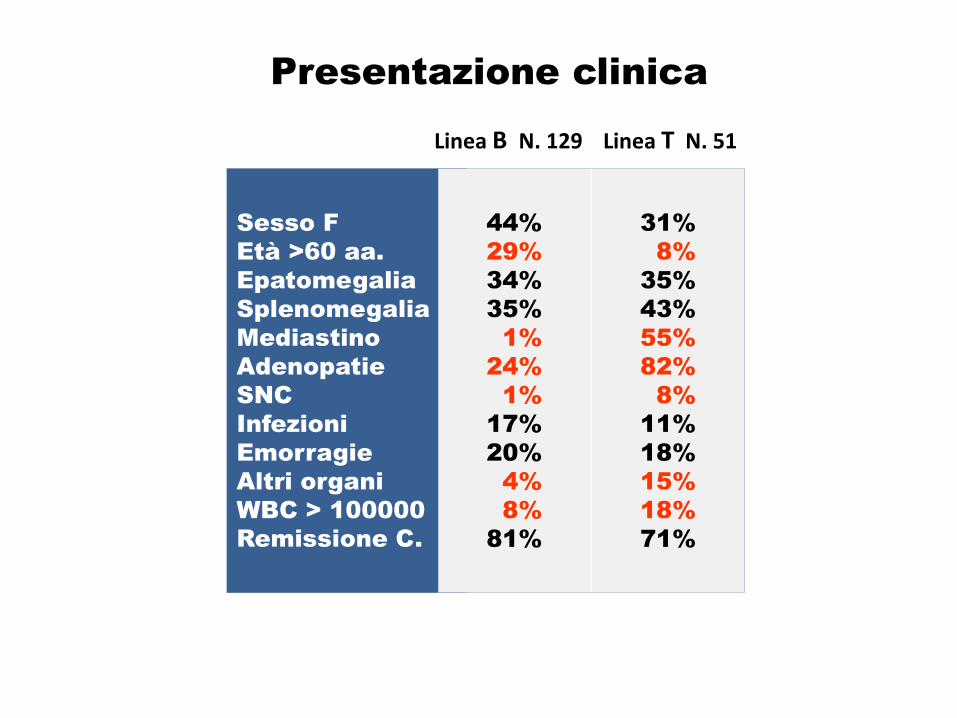
31%
8%
35%
43%
55%
82%
8%
11%
18%
15%
18%
71%
Presentazione clinica
Sesso F
Età >60 aa.
Epatomegalia
Splenomegalia
Mediastino
Adenopatie
SNC
Infezioni
Emorragie
Altri organi
WBC > 100000
Remissione C.
Linea B N. 129 Linea T N. 51
44%
29%
34%
35%
1%
24%
1%
17%
20%
4%
8%
81%
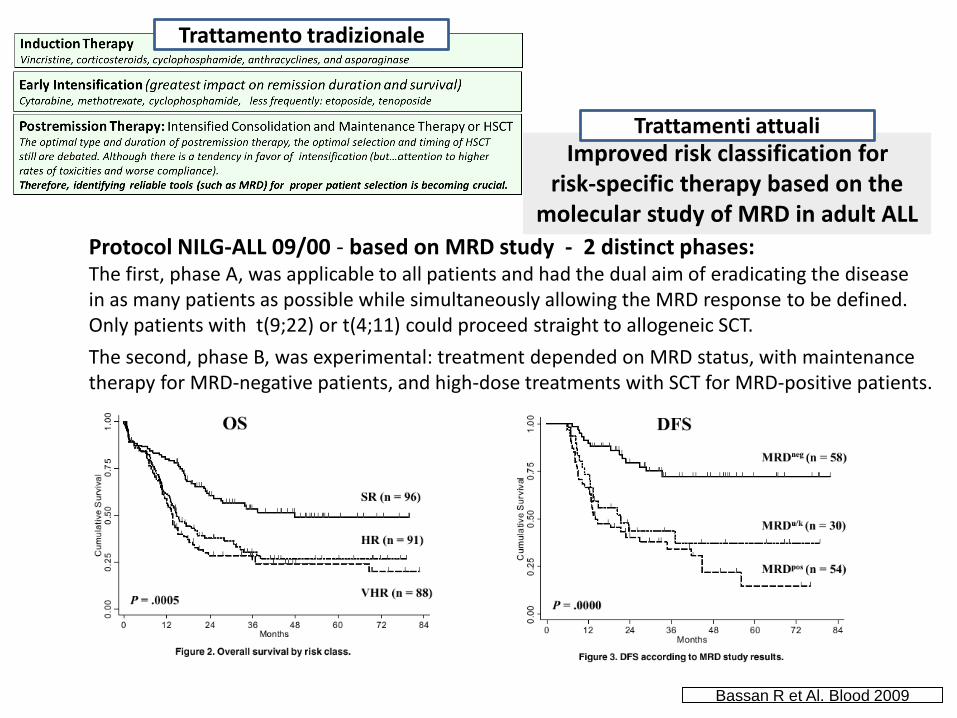
Protocol NILG-ALL 09/00 - based on MRD study - 2 distinct phases:The first, phase A, was applicable to all patients and had the dual aim of eradicating the diseasein as many patients as possible while simultaneously allowing the MRD response to be defined. Only patients with t(9;22) or t(4;11) could proceed straight to allogeneic SCT.
The second, phase B, was experimental: treatment depended on MRD status, with maintenance therapy for MRD-negative patients, and high-dose treatments with SCT for MRD-positive patients.
Improved risk classification for risk-specific therapy based on the
molecular study of MRD in adult ALL
Bassan R et Al. Blood 2009
Trattamento tradizionale
Trattamenti attuali

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche

CRITERI DIAGNOSTICI INDISPENSABILI
Un gruppo di disordini maligni della cellula staminale emopoietica caratterizzati da:– Insufficienza del midollo osseo con citopenie e relative complicanze– Alterazioni displastiche della emopoiesi– Tendenza ad evoluzione leucemica
Incidenza globale 3.7-4.8/100.000 x annoEtà mediana: 70 anni; incidenza: 34-47/100.000 > 75 anni
Sindromi Mielodisplastiche
1
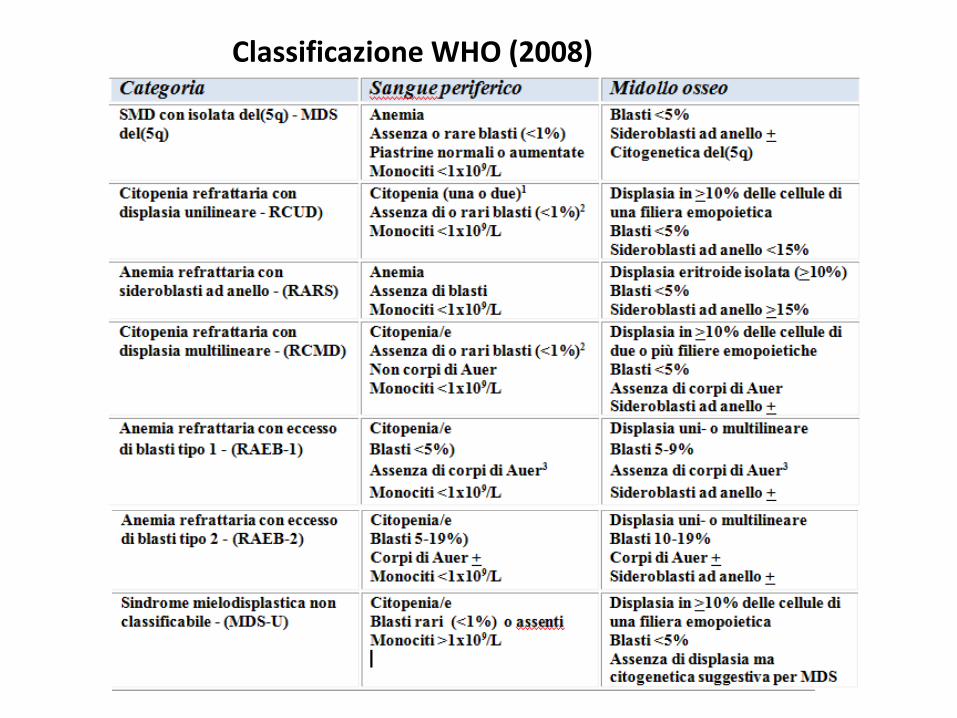
Classificazione WHO (2008)
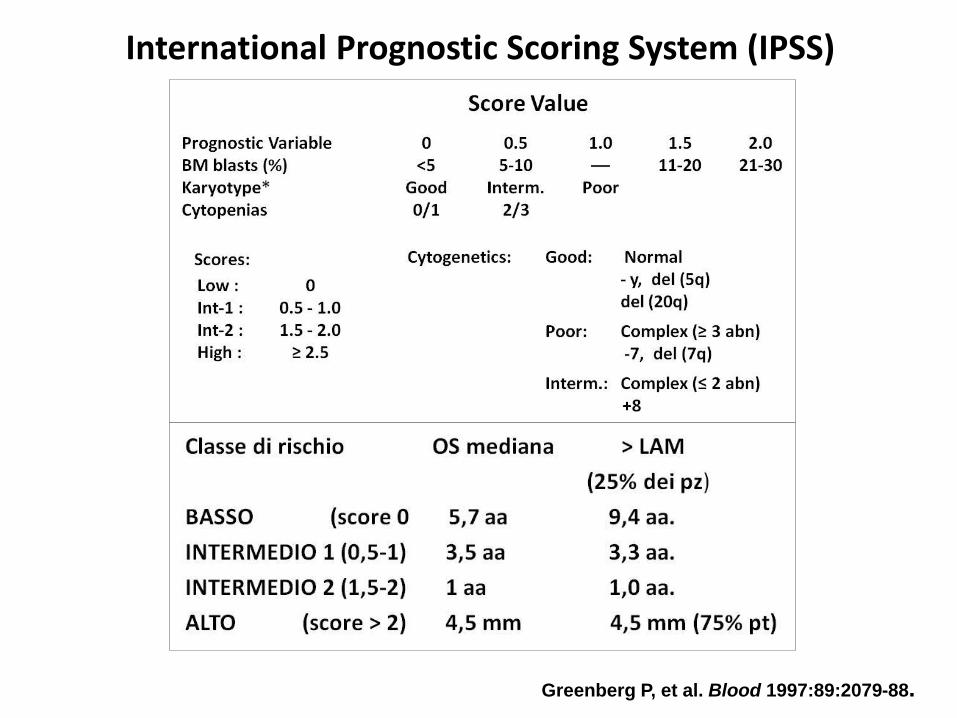
Greenberg P, et al. Blood 1997:89:2079-88.
International Prognostic Scoring System (IPSS)
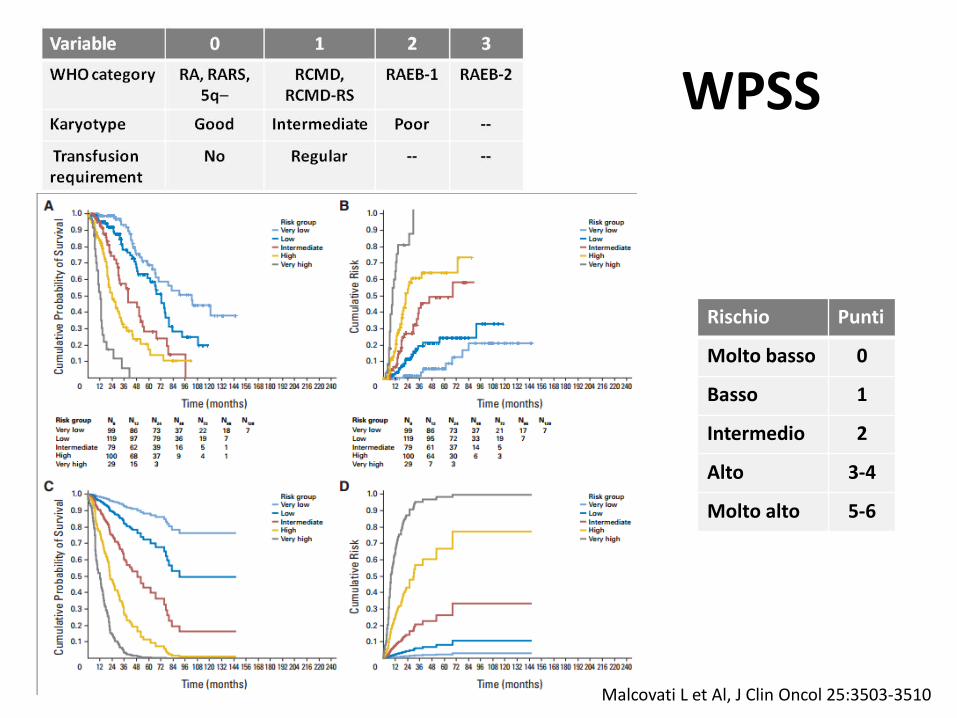
WPSS
Rischio Punti
Molto basso 0
Basso 1
Intermedio 2
Alto 3-4
Molto alto 5-6
Malcovati L et Al, J Clin Oncol 25:3503-3510
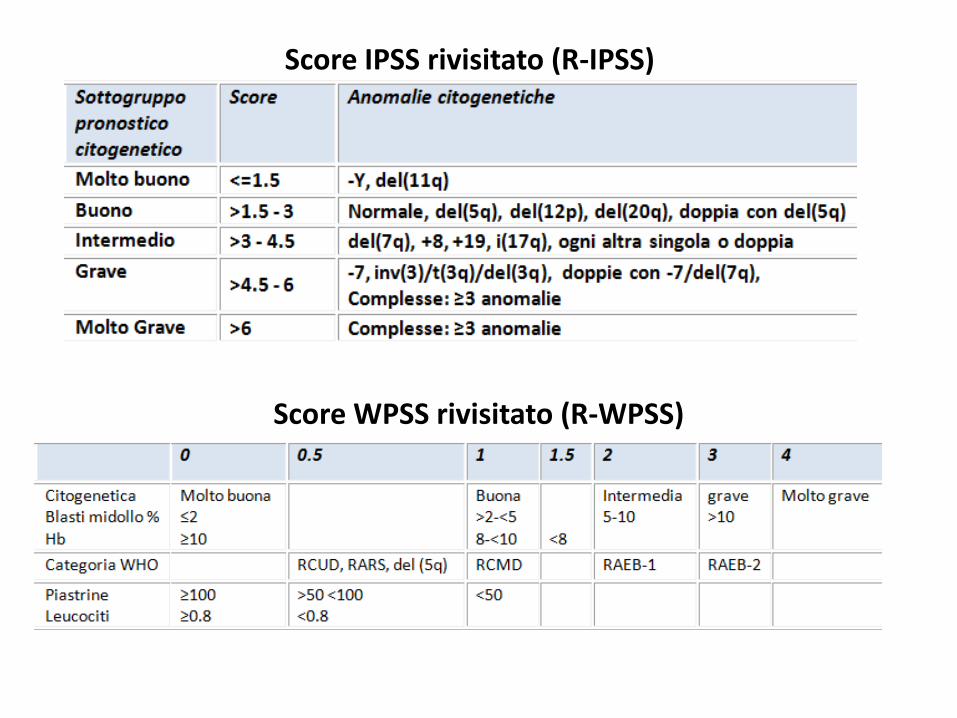
Score WPSS rivisitato (R-WPSS)
Score IPSS rivisitato (R-IPSS)
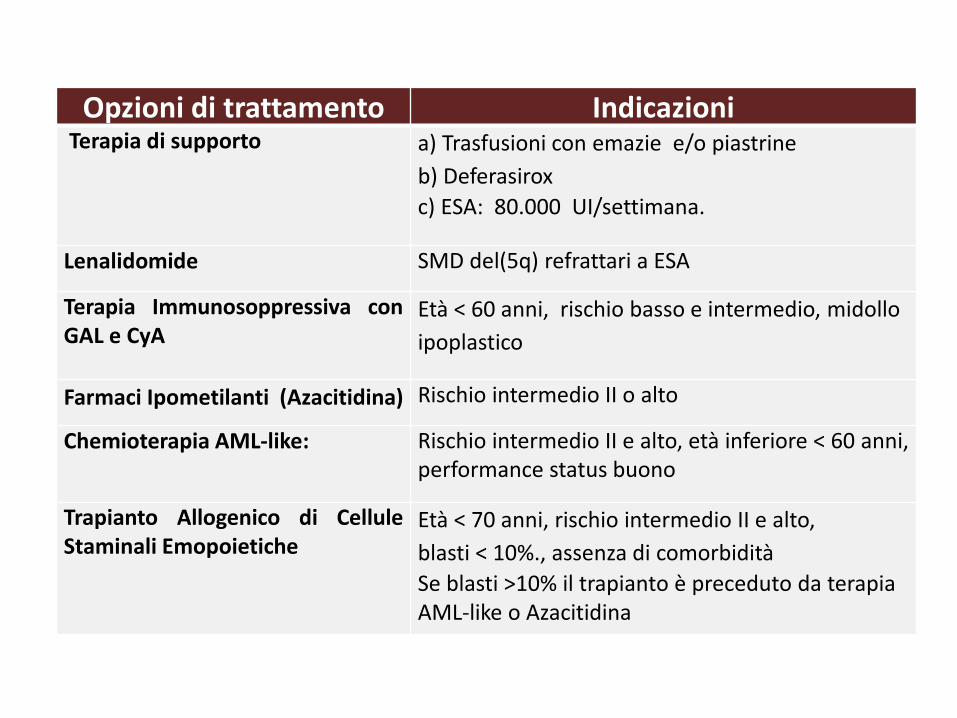
Opzioni di trattamento IndicazioniTerapia di supporto a) Trasfusioni con emazie e/o piastrine
b) Deferasirox
c) ESA: 80.000 UI/settimana.
Lenalidomide SMD del(5q) refrattari a ESA
Terapia Immunosoppressiva conGAL e CyA
Età < 60 anni, rischio basso e intermedio, midollo
ipoplastico
Farmaci Ipometilanti (Azacitidina) Rischio intermedio II o alto
Chemioterapia AML-like: Rischio intermedio II e alto, età inferiore < 60 anni, performance status buono
Trapianto Allogenico di CelluleStaminali Emopoietiche
Età < 70 anni, rischio intermedio II e alto,
blasti < 10%., assenza di comorbidità
Se blasti >10% il trapianto è preceduto da terapia AML-like o Azacitidina

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche
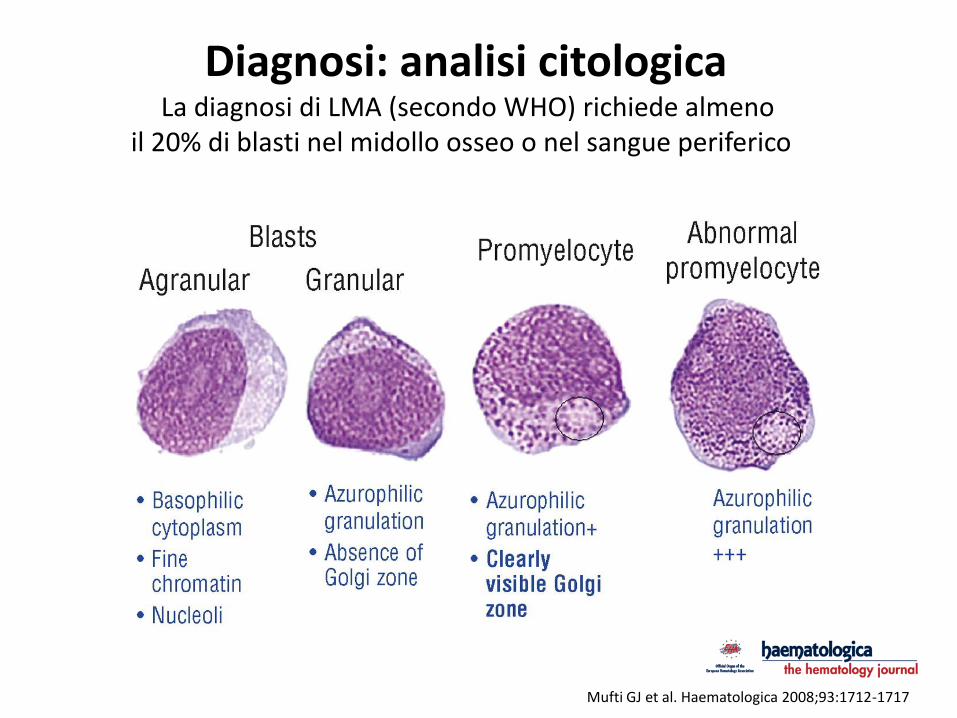
Mufti GJ et al. Haematologica 2008;93:1712-1717
Diagnosi: analisi citologica La diagnosi di LMA (secondo WHO) richiede almeno
il 20% di blasti nel midollo osseo o nel sangue periferico

Dohner D et Al, Blood 2010; 115: 453-474
Diagnosi: analisi immunologica in citometria a flusso
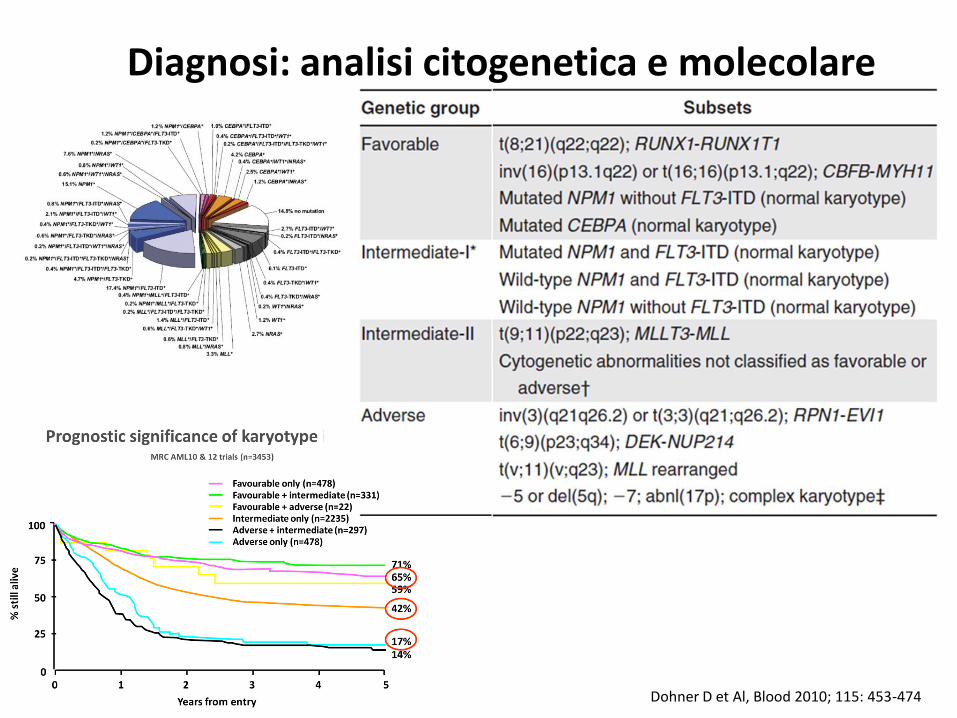
Dohner D et Al, Blood 2010; 115: 453-474
Diagnosi: analisi citogenetica e molecolare

1. Acute myeloid leukemia with recurrent genetic abnormalities
2. Acute myeloid leukemia with myelodysplasia-related changes
3. Therapy-related myeloid neoplasms
4. Acute myeloid leukemia, not otherwise specified
5. Myeloid sarcoma
6. Myeloid proliferations related to Down syndrome
7. Blastic plasmacytoid dendritic cell neoplasm
8. Acute leukemias of ambiguous lineage
AML classificationWorld Health Organization Classification (2008)
Vadiman JW et Al. Blood 2008; 114: 937-51
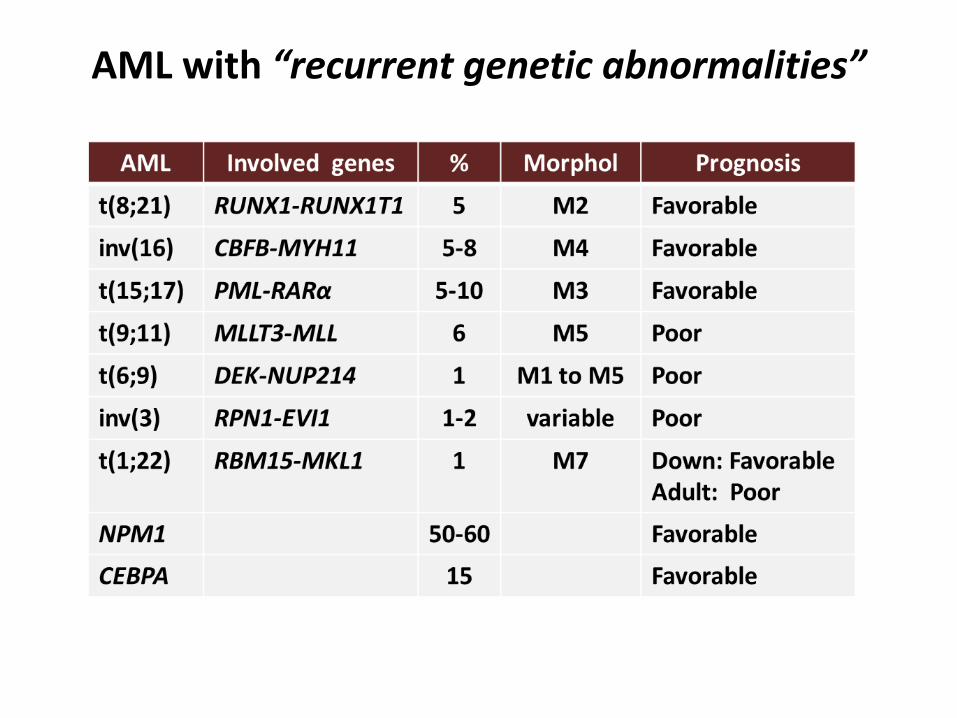
AML with “recurrent genetic abnormalities”

World Health Organization (WHO) classification
Vadiman JW et Al. IARC; 2008
• These leukemias do not fit easily into the WHO subtypes
• This category accounts for about 25% of AML, but as more genetic subgroupsare reconized, the number will diminish
Acute myeloid leukemia, not otherwise specified

Acute leukemias of ambiguous lineage
Acute undifferentiated leukemia Mixed phenotype acute leukemia with t(9;22)(q34;q11.2); BCR-ABL1 Mixed phenotype acute leukemia with t(v;11q23); MLL rearranged Mixed phenotype acute leukemia, B-myeloid, NOS Mixed phenotype acute leukemia, T-myeloid, NOS
Show no clear evidence of differentiation along a single lineage:1. No lineages specific antigens are present2. The blasts express antigens of more than one lineage to such a degree that it
is not possible to assign the leukemia to a specific lineage-related category.
My lineage: MPO (flow cytometry, immunohistochemistry, or cytochemistry), orMonocytic differentiation (at least 2 of: NSE, CD11c, CD14, CD64, lysozyme)
T lineage: CyCD3 (flow cytometry or immunohistochemistry). orSurface CD3 (rare in mixed phenotype acute leukemia)
B lineage: Strong CD19 with at least 1 of: CD79a, cyCD22, CD10, orWeak CD19 with at least 2 of the: CD79a, cyCD22, CD10
Requirements for assigning more than one lineage to a single blast population in mixed phenotype acute leukemia (MPAL):
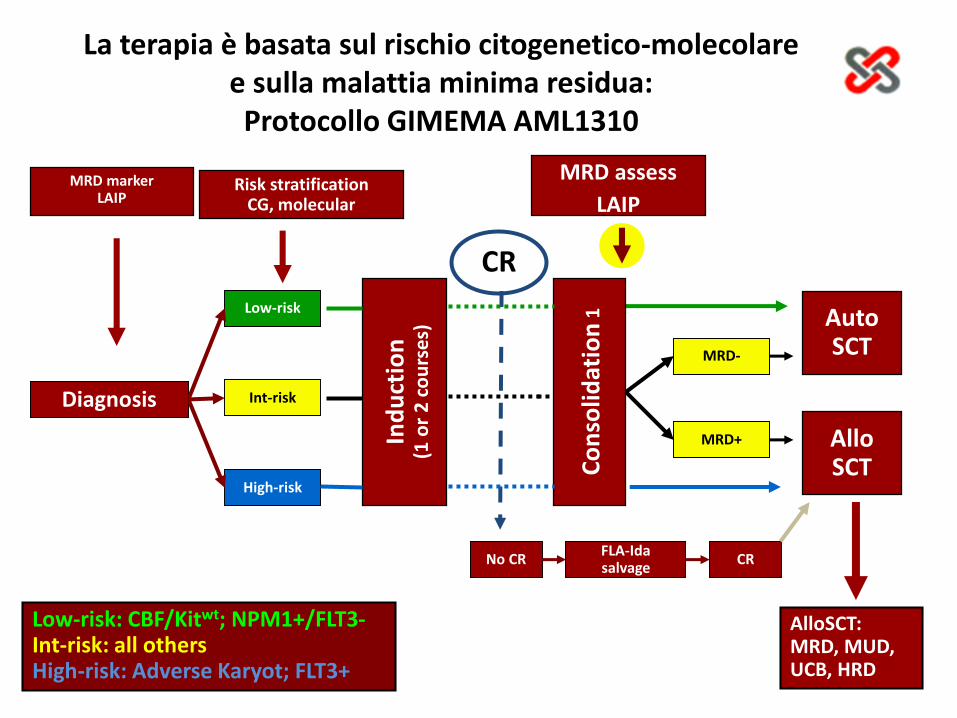
Low-risk: CBF/Kitwt; NPM1+/FLT3-Int-risk: all othersHigh-risk: Adverse Karyot; FLT3+
Diagnosis
Low-risk
Int-risk
High-risk
MRD-
MRD+
MRD markerLAIP
Risk stratificationCG, molecular
MRD assess
LAIP
FLA-Ida salvage
No CR CR
CR
Ind
uct
ion
(1 o
r 2
co
urs
es)
Co
nso
lidat
ion
1 AutoSCT
AlloSCT
AlloSCT: MRD, MUD, UCB, HRD
La terapia è basata sul rischio citogenetico-molecolare e sulla malattia minima residua: Protocollo GIMEMA AML1310

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche

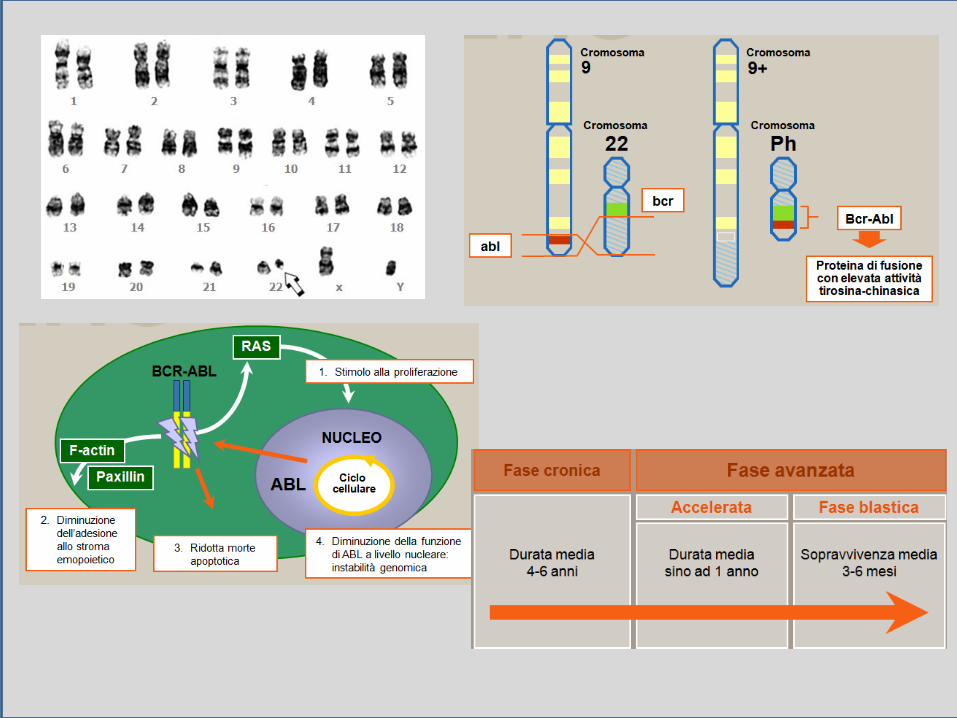
Malattia clonale della cellula staminale emopoietica. Incidenza x anno: 1-5 casi su 100.00. Età media: 45-60 anni .
Per la conferma diagnostica ha valore il riconoscimento di: a) Cromosoma Philadelphia (Ph’) o t(9;22)(q34;q1) con 9q+ e 22q-b) Gene di fusione BCR/ABL
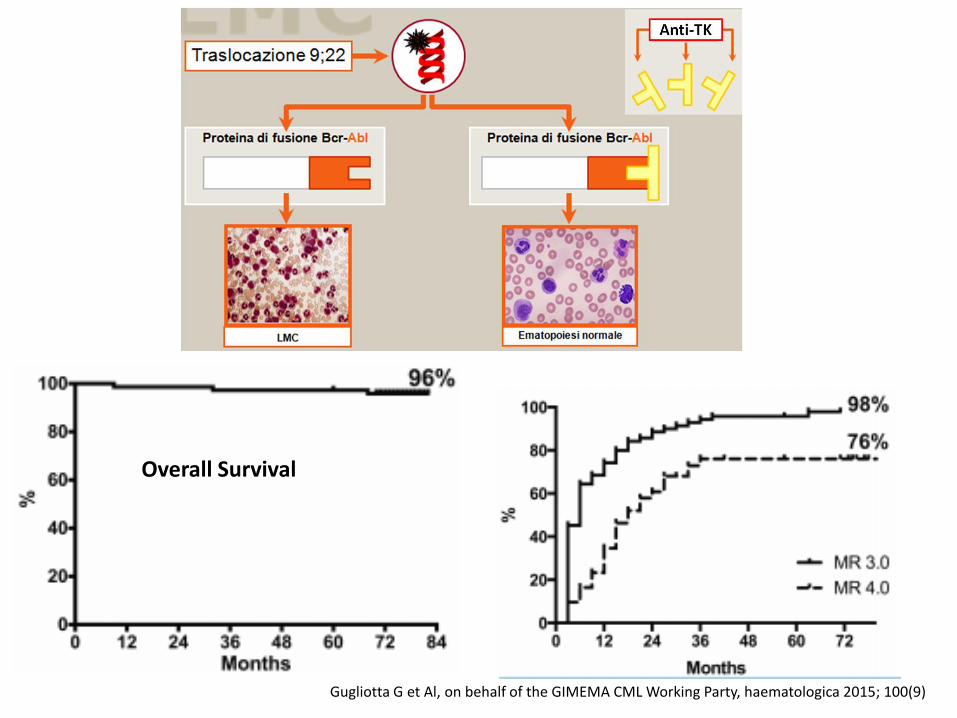
Overall Survival
Gugliotta G et Al, on behalf of the GIMEMA CML Working Party, haematologica 2015; 100(9)

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche

• Disordini neoplastici (clonali) della cellula staminale emopoietica• Iperplasia di tutte le linee cellulari, ma solitamente di una in particolare• La fibrosi midollare costituisce un evento secondario• Posibile evoluzione in Leucemia Mieloide Acuta
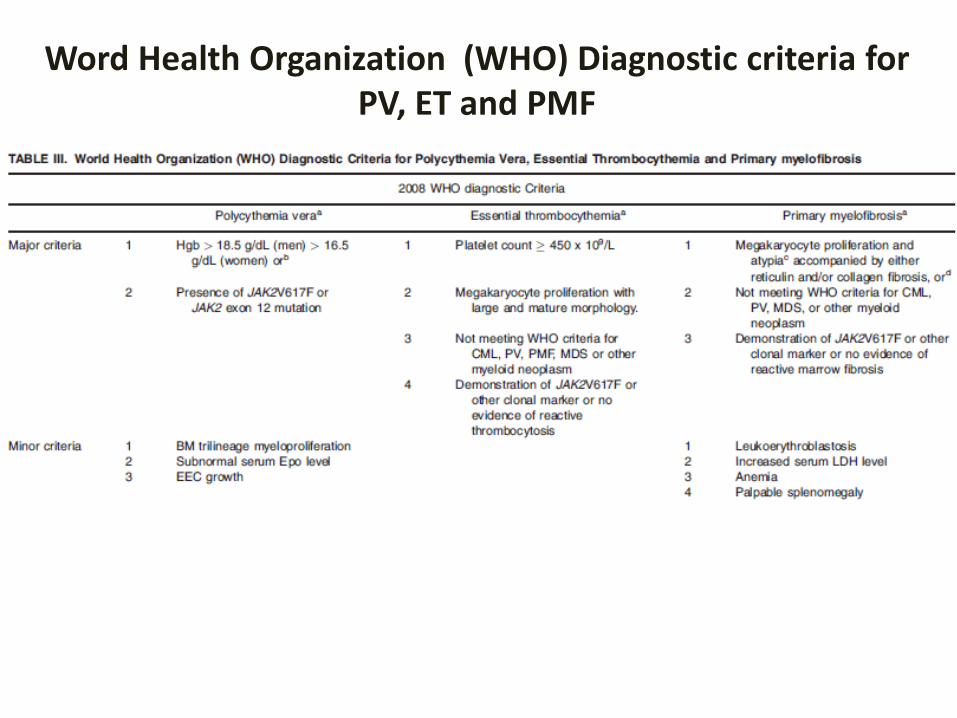
Word Health Organization (WHO) Diagnostic criteria forPV, ET and PMF
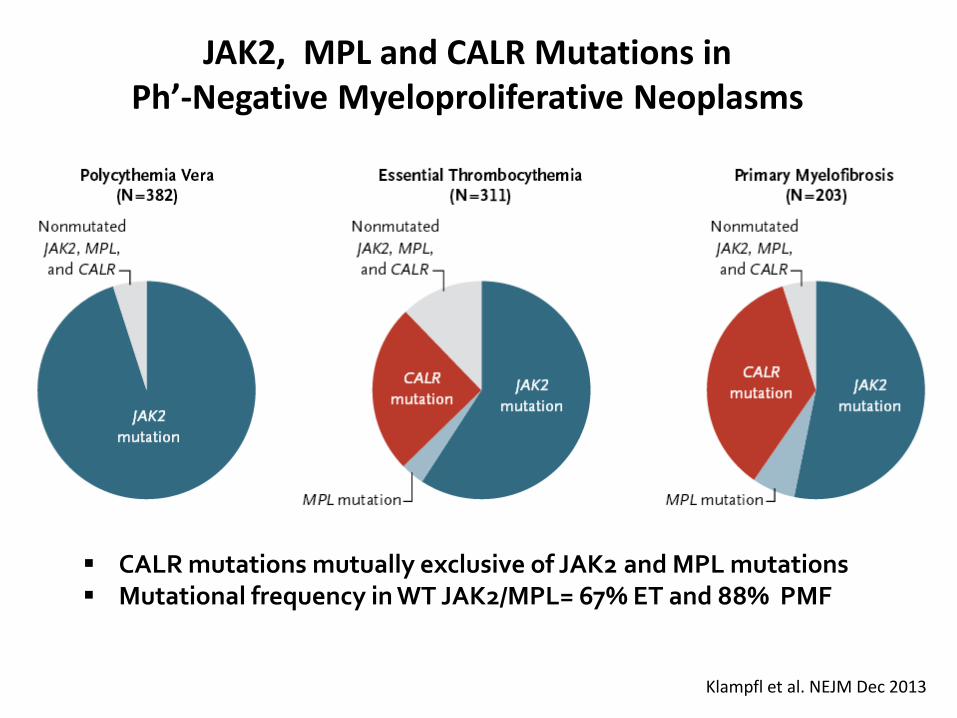
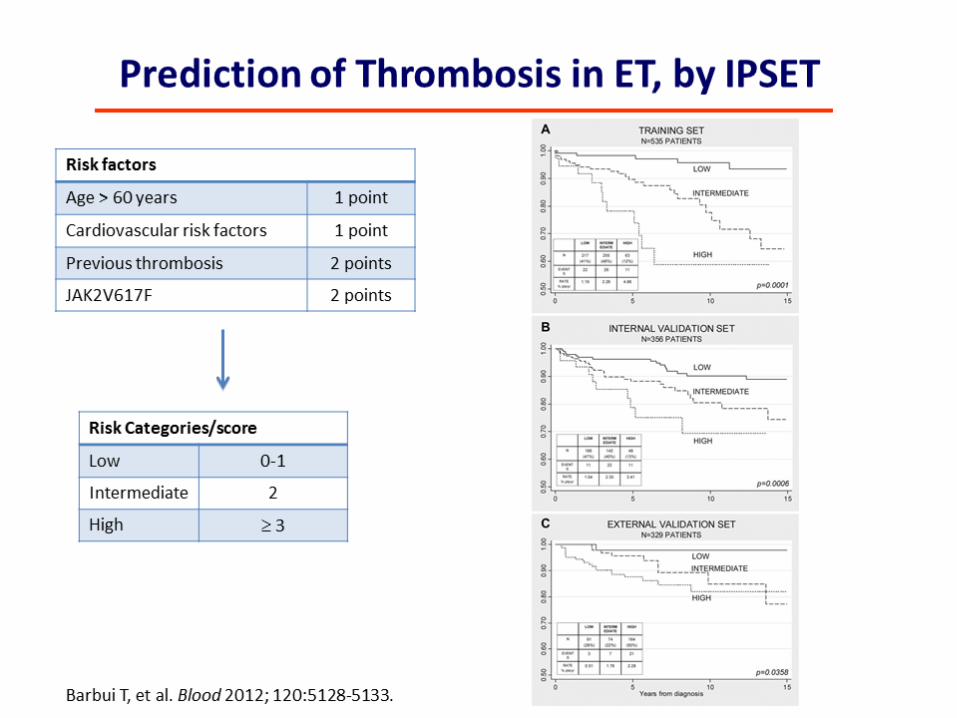
JAK2, MPL and CALR Mutations in Ph’-Negative Myeloproliferative Neoplasms
Klampfl et al. NEJM Dec 2013
CALR mutations mutually exclusive of JAK2 and MPL mutations Mutational frequency in WT JAK2/MPL= 67% ET and 88% PMF
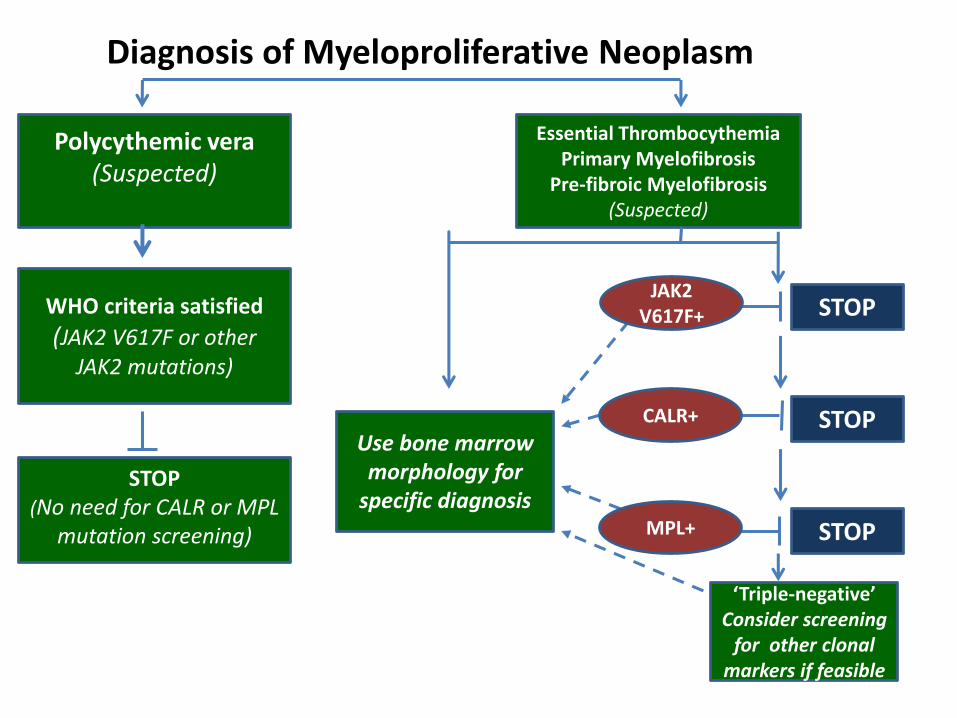
Diagnosis of Myeloproliferative Neoplasm
STOP
STOP
STOP
‘Triple-negative’Consider screening
for other clonalmarkers if feasible
Use bone marrowmorphology for
specific diagnosisSTOP
(No need for CALR or MPL mutation screening)
WHO criteria satisfied
(JAK2 V617F or otherJAK2 mutations)
Polycythemic vera(Suspected)
Essential ThrombocythemiaPrimary Myelofibrosis
Pre-fibroic Myelofibrosis(Suspected)
CALR+
JAK2 V617F+
MPL+
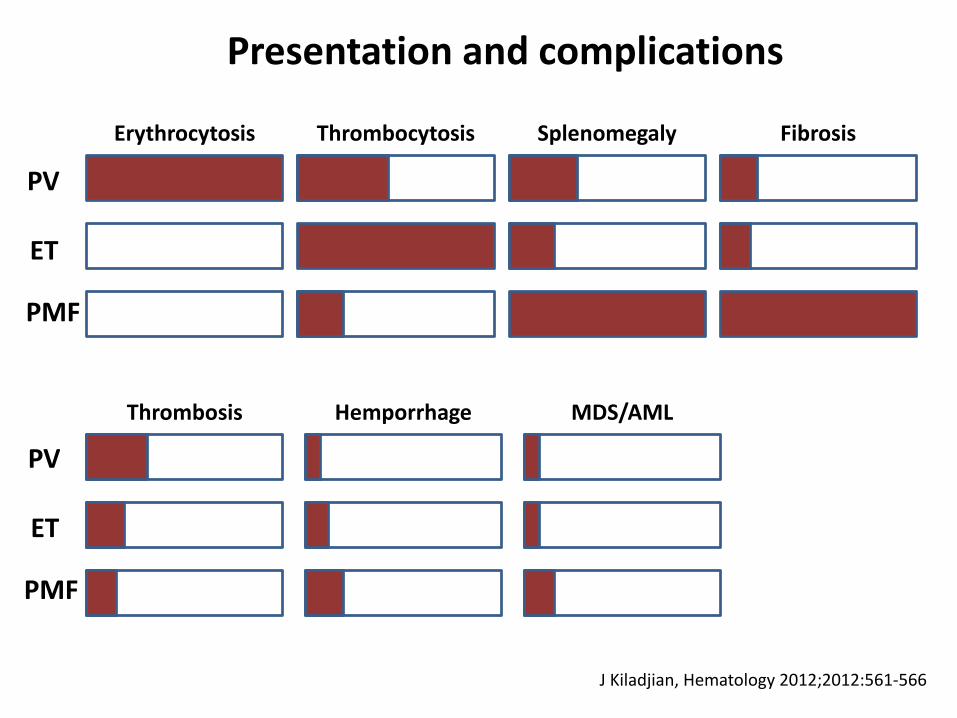
PV
ET
PMF
PV
ET
PMF
Erythrocytosis FibrosisSplenomegalyThrombocytosis
Thrombosis MDS/AMLHemporrhage
Presentation and complications
J Kiladjian, Hematology 2012;2012:561-566
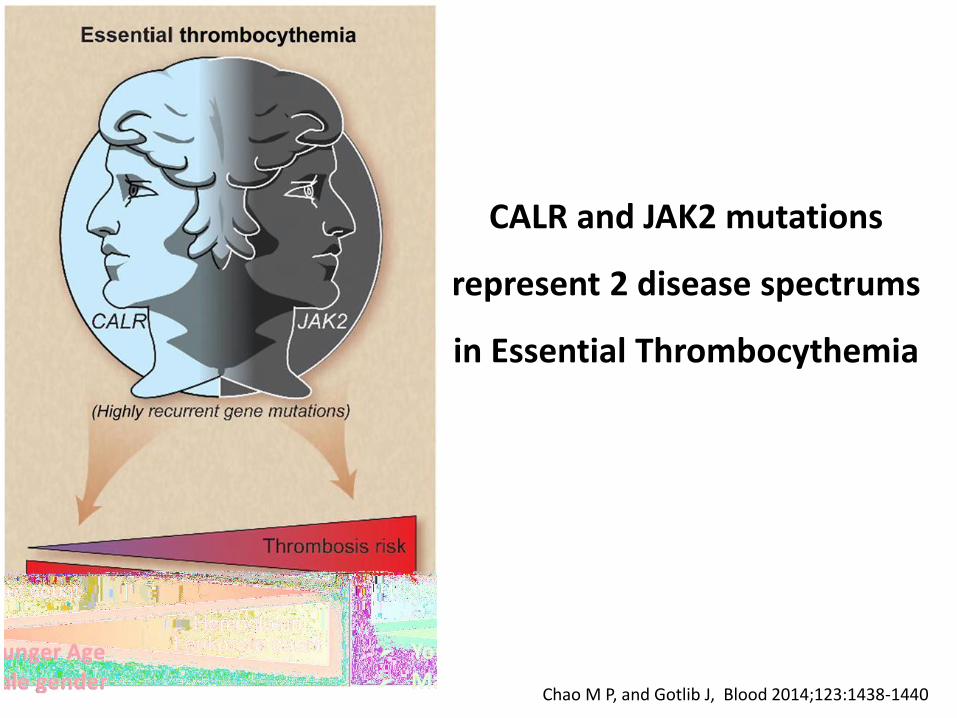
CALR and JAK2 mutations
represent 2 disease spectrums
in Essential Thrombocythemia
Chao M P, and Gotlib J, Blood 2014;123:1438-1440
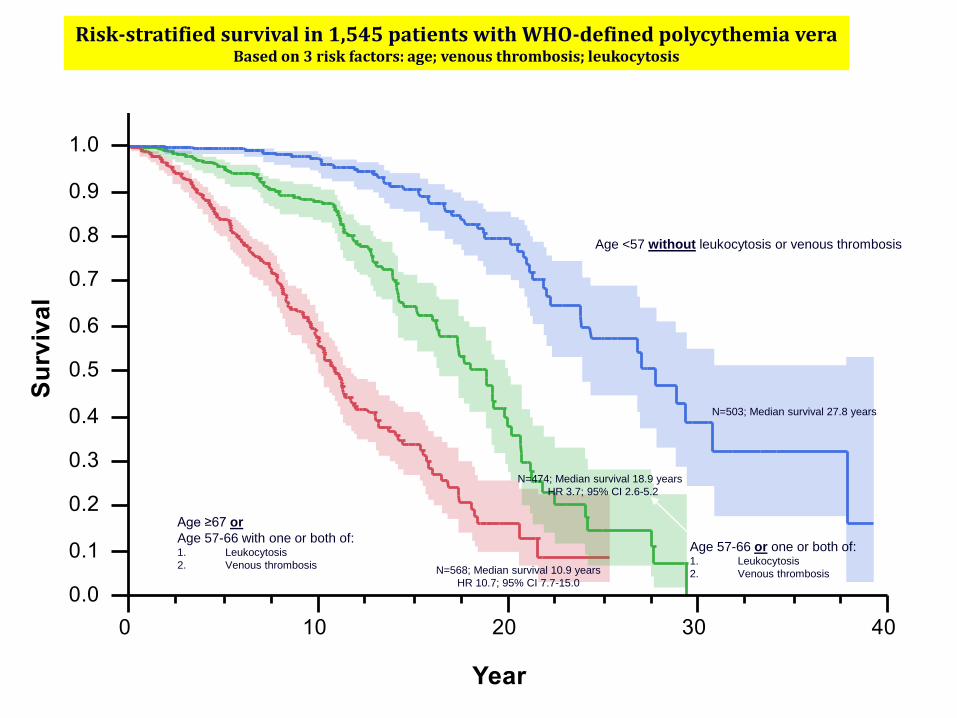
Risk-stratified survival in 1,545 patients with WHO-defined polycythemia veraBased on 3 risk factors: age; venous thrombosis; leukocytosis
N=568; Median survival 10.9 years
HR 10.7; 95% CI 7.7-15.0
N=474; Median survival 18.9 years
HR 3.7; 95% CI 2.6-5.2
N=503; Median survival 27.8 years
Age <57 without leukocytosis or venous thrombosis
Age 57-66 or one or both of: 1. Leukocytosis
2. Venous thrombosis
Age ≥67 or
Age 57-66 with one or both of: 1. Leukocytosis
2. Venous thrombosis
Tefferi et al. Leukemia. 2013;27:1874
Tefferi et al. Leukemia. 2013;27:1874
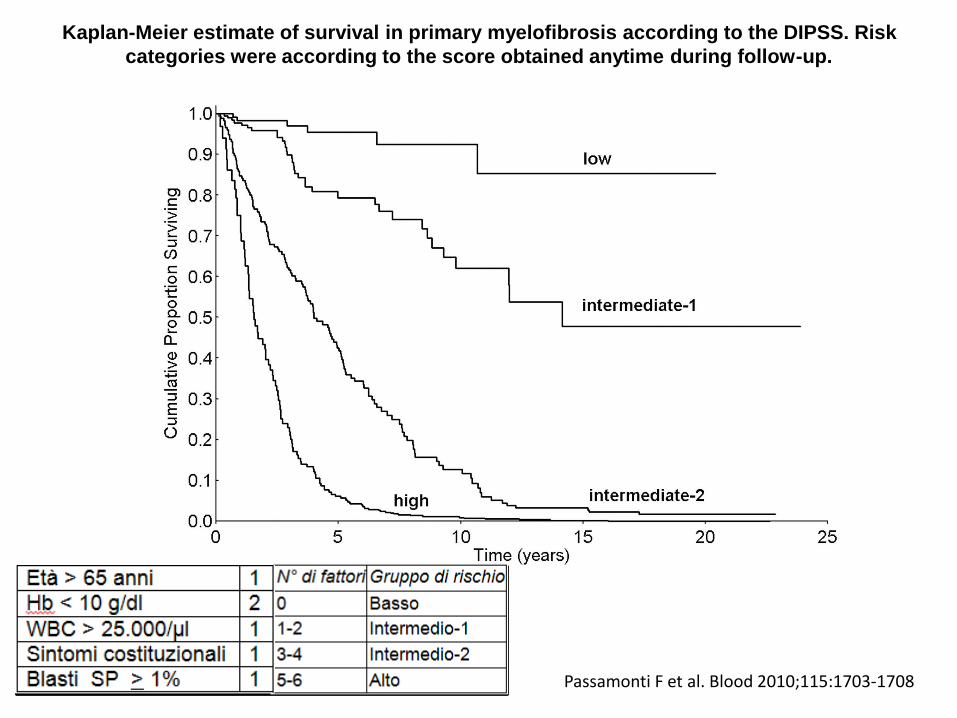
Kaplan-Meier estimate of survival in primary myelofibrosis according to the DIPSS. Risk
categories were according to the score obtained anytime during follow-up.
Passamonti F et al. Blood 2010;115:1703-1708
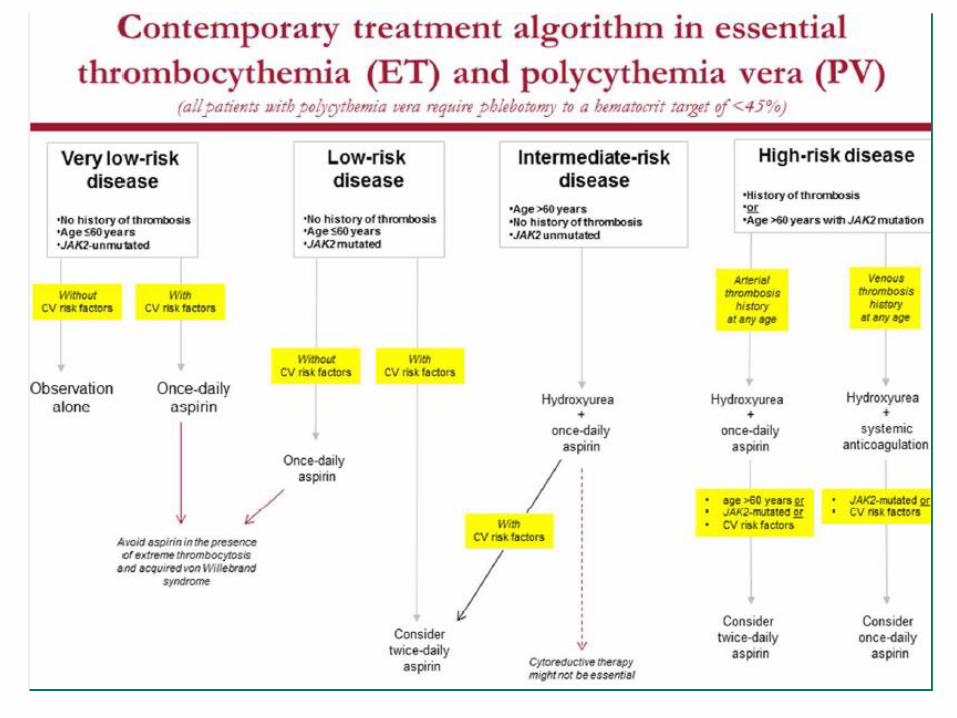

• Ruxolitinib provides relevant clinical benefits for MF patientssuch as rapid reductions in splenomegaly and improvementsin survival, symptoms and QoL, that were sustained for ≥ 3years of treatment in a large number of patients
• Different studies (INCB18424 - 251, COMFORT I & II, JUMP) andlonger follow-ups continue to suggest a survival advantagewith ruxolitinib treatment compared with historical controls,placebo and BAT, independently from the JAK2 mutationalstatus
Current therapy in Myelofibrosis

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche

INCIDENZALa LLC è la leucemia più comune nel mondo occidendate con una incidenza di 4.2/100 000/anno. L’incidenza aumenta a >30/100 000/anno nei pazienti con età >80 anni. L’età mediana alla diagnosi è di 72 anni.Circa il 10% dei pazienti con LLC sono più giovani di 55 anni.
DIAGNOSILa diagnosi di LLC è stabilita dai seguenti criteri:1. La presenza nel sangue periferico di > 5000 Linfociti B monoclonali/μL per la durata di almeno 3 mesi.2. I linfociti sono piccoli, maturi, con nucleo a cromatina addensata e senza un evidente nucleolo.3. Le cellule della LLC co-esprimono l’antigene CD5 ed antigeni B di superficie come CD19, CD20 e CD23. I livelli delle sIg, CD20 e CD79b sono poco espressi rispetto ai linfociti B normali. Ciascun clone di LLC presenta restrizione delle catene leggere λ o k.
Eichhorst B et Al, Annals of Oncology 22 (Suppl 6): vi50–vi54, 2011
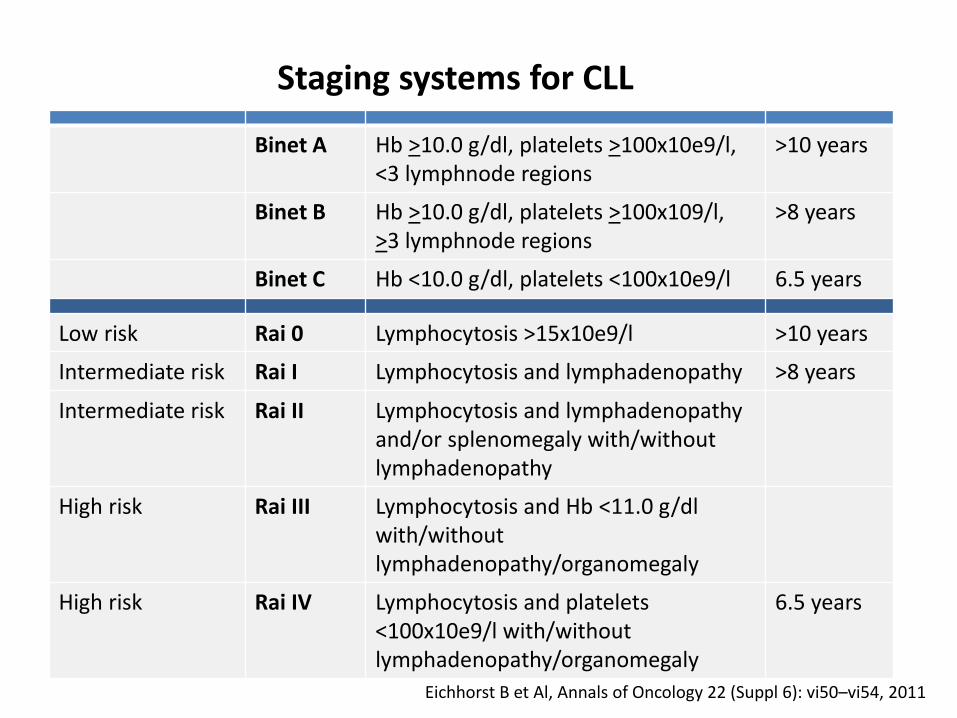
Binet A Hb >10.0 g/dl, platelets >100x10e9/l, <3 lymphnode regions
>10 years
Binet B Hb >10.0 g/dl, platelets >100x109/l, >3 lymphnode regions
>8 years
Binet C Hb <10.0 g/dl, platelets <100x10e9/l 6.5 years
Low risk Rai 0 Lymphocytosis >15x10e9/l >10 years
Intermediate risk Rai I Lymphocytosis and lymphadenopathy >8 years
Intermediate risk Rai II Lymphocytosis and lymphadenopathyand/or splenomegaly with/withoutlymphadenopathy
High risk Rai III Lymphocytosis and Hb <11.0 g/dl with/withoutlymphadenopathy/organomegaly
High risk Rai IV Lymphocytosis and platelets <100x10e9/l with/withoutlymphadenopathy/organomegaly
6.5 years
Staging systems for CLL
Eichhorst B et Al, Annals of Oncology 22 (Suppl 6): vi50–vi54, 2011
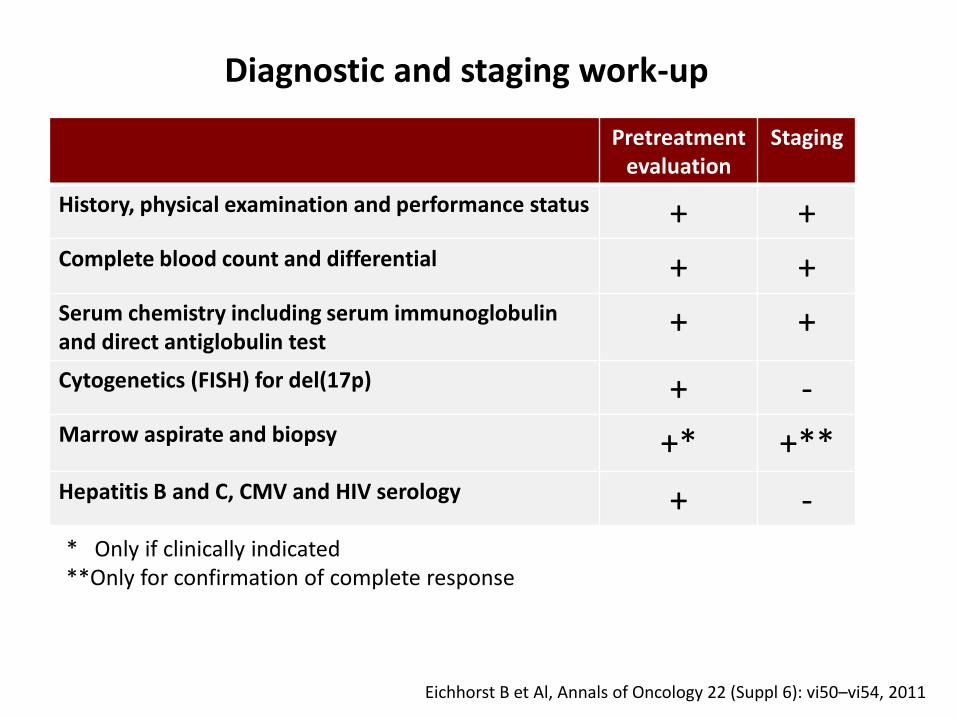
+ ++ ++ –
Pretreatmentevaluation
Staging
History, physical examination and performance status + +Complete blood count and differential + +Serum chemistry including serum immunoglobulinand direct antiglobulin test
+ +
Cytogenetics (FISH) for del(17p) + -Marrow aspirate and biopsy +* +**Hepatitis B and C, CMV and HIV serology + -* Only if clinically indicated**Only for confirmation of complete response
Diagnostic and staging work-up
Eichhorst B et Al, Annals of Oncology 22 (Suppl 6): vi50–vi54, 2011
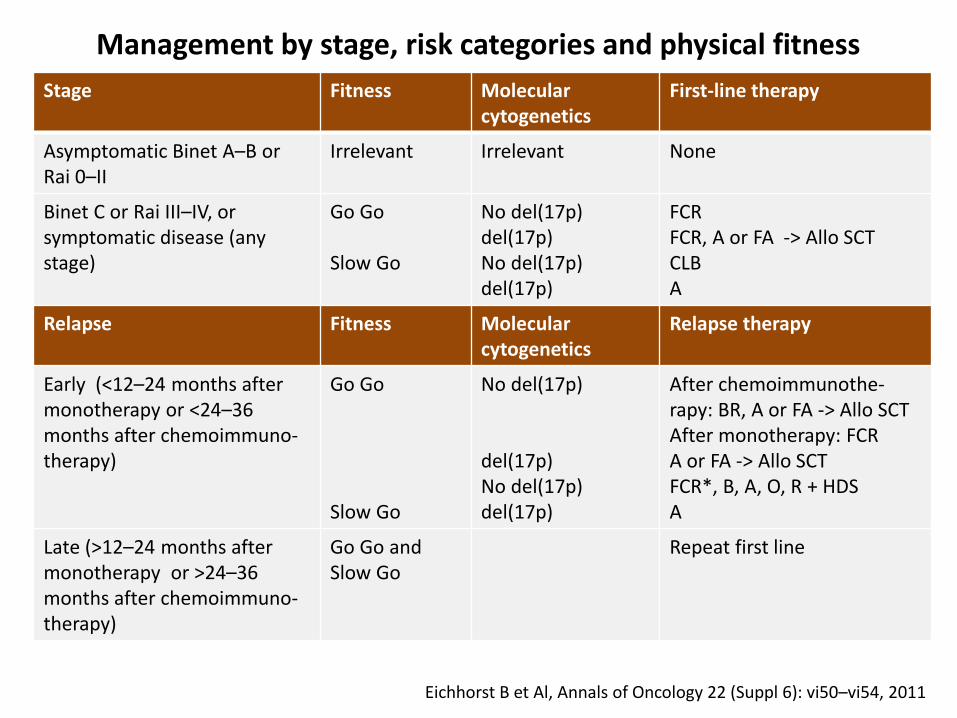
Stage Fitness Molecularcytogenetics
First-line therapy
Asymptomatic Binet A–B orRai 0–II
Irrelevant Irrelevant None
Binet C or Rai III–IV, orsymptomatic disease (anystage)
Go Go
Slow Go
No del(17p)del(17p)No del(17p)del(17p)
FCRFCR, A or FA -> Allo SCTCLBA
Relapse Fitness Molecularcytogenetics
Relapse therapy
Early (<12–24 months aftermonotherapy or <24–36months after chemoimmuno-therapy)
Go Go
Slow Go
No del(17p)
del(17p)No del(17p) del(17p)
After chemoimmunothe-rapy: BR, A or FA -> Allo SCT After monotherapy: FCRA or FA -> Allo SCTFCR*, B, A, O, R + HDSA
Late (>12–24 months aftermonotherapy or >24–36months after chemoimmuno-therapy)
Go Go and Slow Go
Repeat first line
Management by stage, risk categories and physical fitness
Eichhorst B et Al, Annals of Oncology 22 (Suppl 6): vi50–vi54, 2011
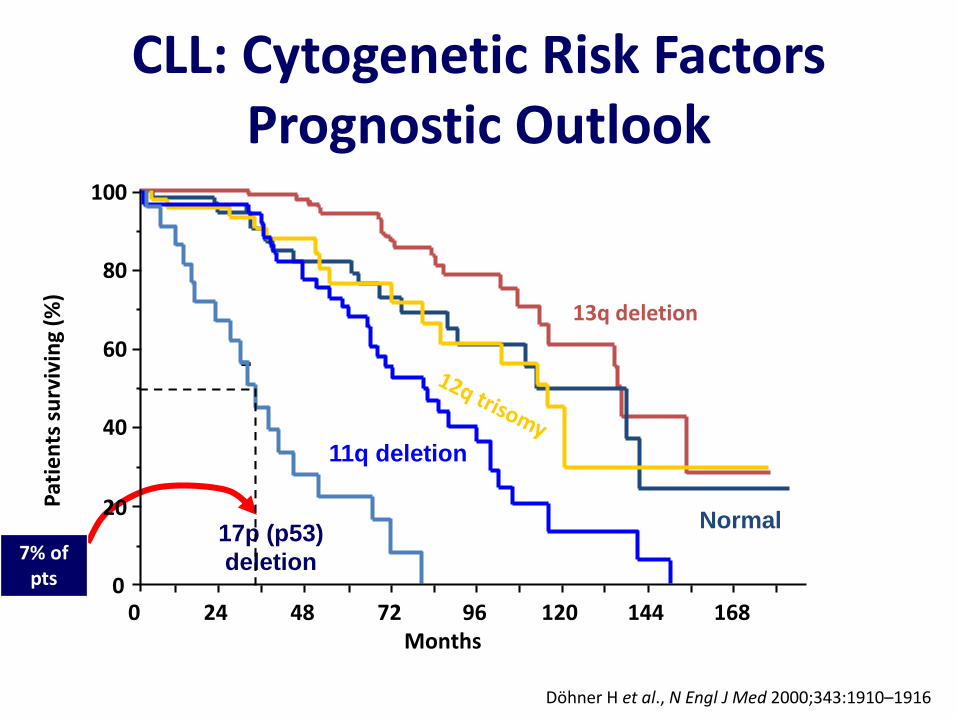
CLL: Cytogenetic Risk Factors Prognostic Outlook
Döhner H et al., N Engl J Med 2000;343:1910–1916
Pat
ien
ts s
urv
ivin
g (%
)
100
80
60
40
20
00 24 48 72 96 120 144 168
Months
13q deletion
17p (p53)
deletion
11q deletion
Normal
7% of pts

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche


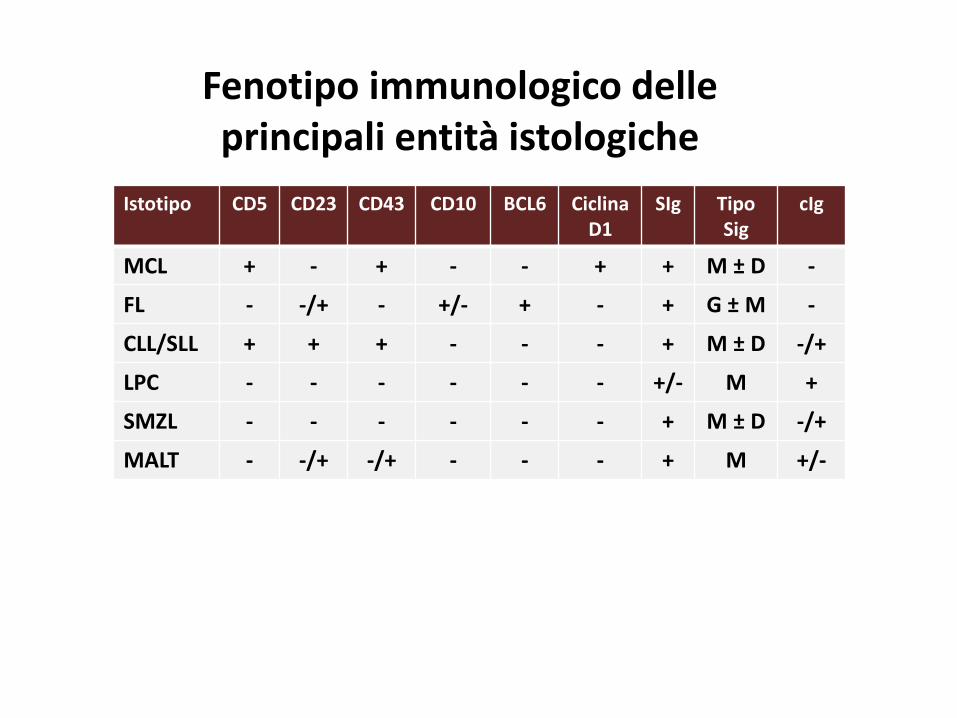
Istotipo CD5 CD23 CD43 CD10 BCL6 CiclinaD1
SIg Tipo Sig
cIg
MCL + - + - - + + M ± D -
FL - -/+ - +/- + - + G ± M -
CLL/SLL + + + - - - + M ± D -/+
LPC - - - - - - +/- M +
SMZL - - - - - - + M ± D -/+
MALT - -/+ -/+ - - - + M +/-
Fenotipo immunologico delle principali entità istologiche
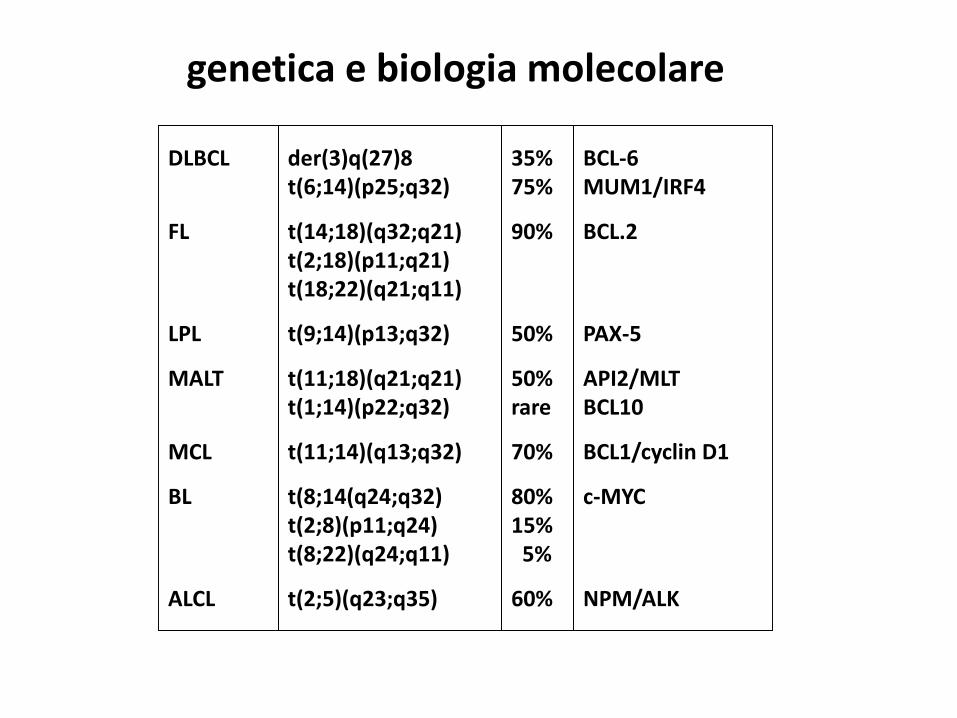
der(3)q(27)8t(6;14)(p25;q32)
t(14;18)(q32;q21)t(2;18)(p11;q21)t(18;22)(q21;q11)
t(9;14)(p13;q32)
t(11;18)(q21;q21)t(1;14)(p22;q32)
t(11;14)(q13;q32)
t(8;14(q24;q32)t(2;8)(p11;q24)t(8;22)(q24;q11)
t(2;5)(q23;q35)
DLBCL
FL
LPL
MALT
MCL
BL
ALCL
35%75%
90%
50%
50%rare
70%
80%15%
5%
60%
BCL-6MUM1/IRF4
BCL.2
PAX-5
API2/MLTBCL10
BCL1/cyclin D1
c-MYC
NPM/ALK
genetica e biologia molecolare
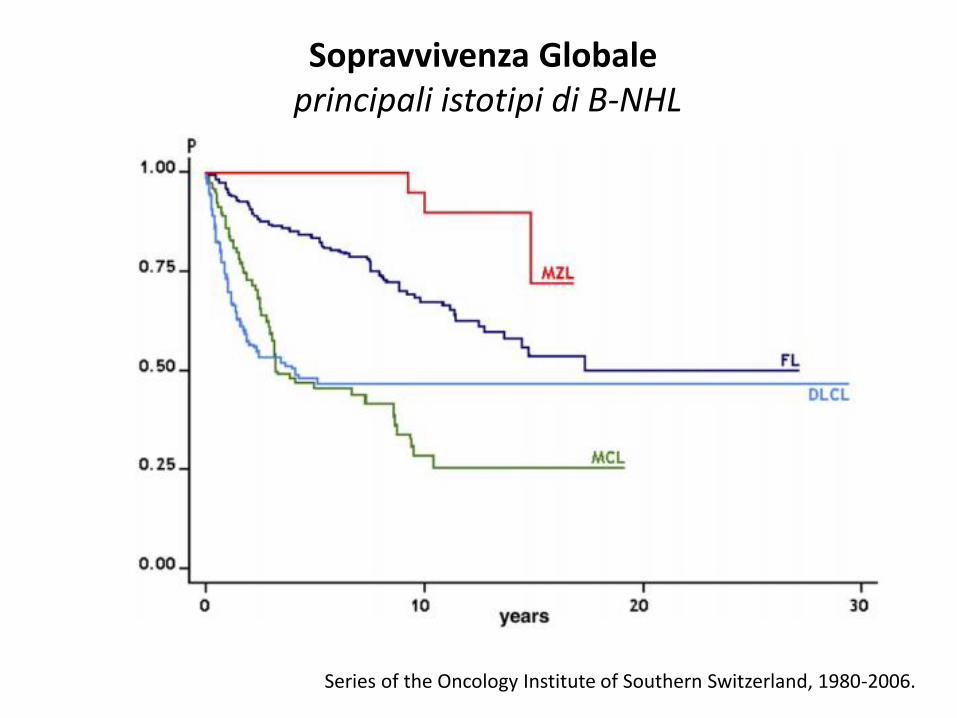
Series of the Oncology Institute of Southern Switzerland, 1980-2006.
Sopravvivenza Globale principali istotipi di B-NHL
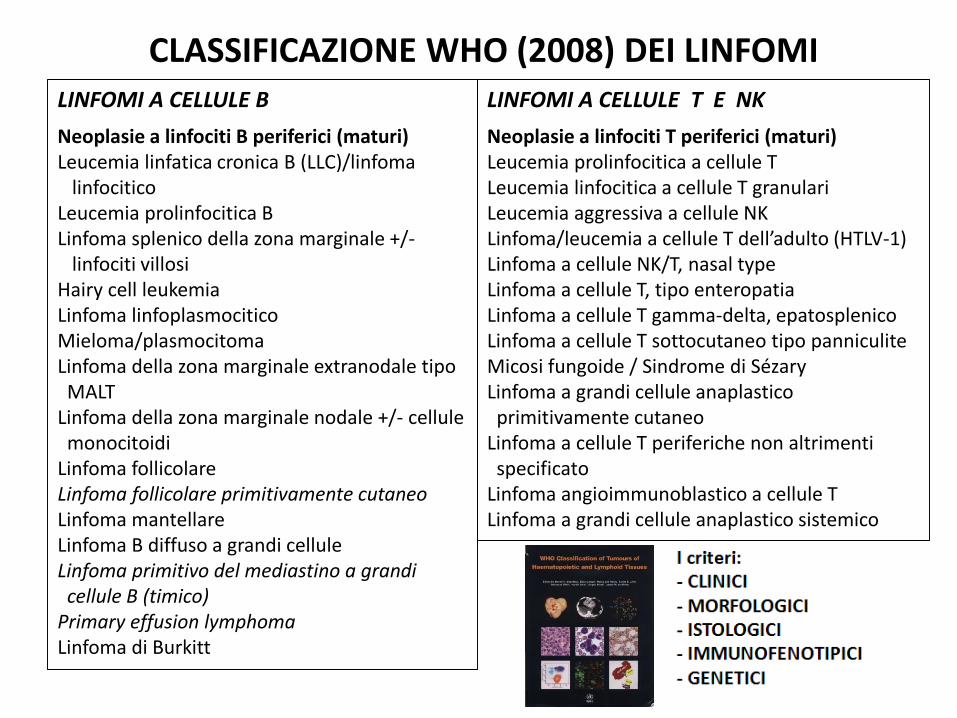
LINFOMI A CELLULE B
Neoplasie a linfociti B periferici (maturi)Leucemia linfatica cronica B (LLC)/linfoma
linfociticoLeucemia prolinfocitica BLinfoma splenico della zona marginale +/-
linfociti villosiHairy cell leukemiaLinfoma linfoplasmociticoMieloma/plasmocitomaLinfoma della zona marginale extranodale tipo MALT
Linfoma della zona marginale nodale +/- cellule monocitoidi
Linfoma follicolareLinfoma follicolare primitivamente cutaneoLinfoma mantellareLinfoma B diffuso a grandi celluleLinfoma primitivo del mediastino a grandi cellule B (timico)
Primary effusion lymphomaLinfoma di Burkitt
LINFOMI A CELLULE T E NK
Neoplasie a linfociti T periferici (maturi)Leucemia prolinfocitica a cellule TLeucemia linfocitica a cellule T granulariLeucemia aggressiva a cellule NKLinfoma/leucemia a cellule T dell’adulto (HTLV-1)Linfoma a cellule NK/T, nasal typeLinfoma a cellule T, tipo enteropatiaLinfoma a cellule T gamma-delta, epatosplenicoLinfoma a cellule T sottocutaneo tipo panniculiteMicosi fungoide / Sindrome di SézaryLinfoma a grandi cellule anaplastico primitivamente cutaneo
Linfoma a cellule T periferiche non altrimenti specificato
Linfoma angioimmunoblastico a cellule TLinfoma a grandi cellule anaplastico sistemico
CLASSIFICAZIONE WHO (2008) DEI LINFOMI

General symptoms


Diagnostic work-up

Diffuse Large B-cell LynphomaInternational Prognostic Index (IPI)
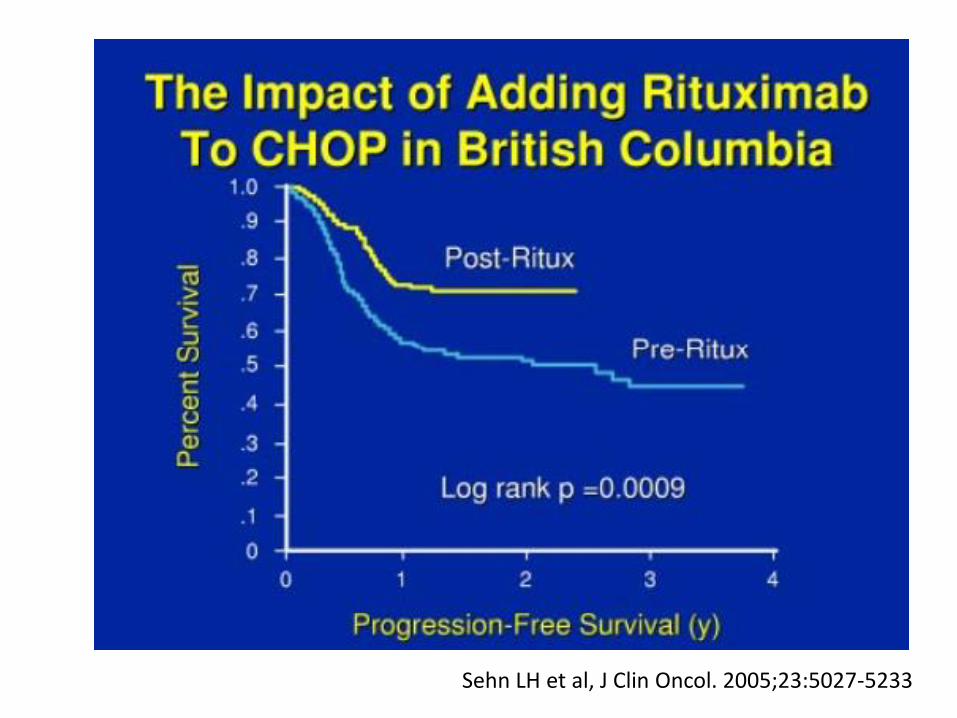
Sehn LH et al, J Clin Oncol. 2005;23:5027-5233
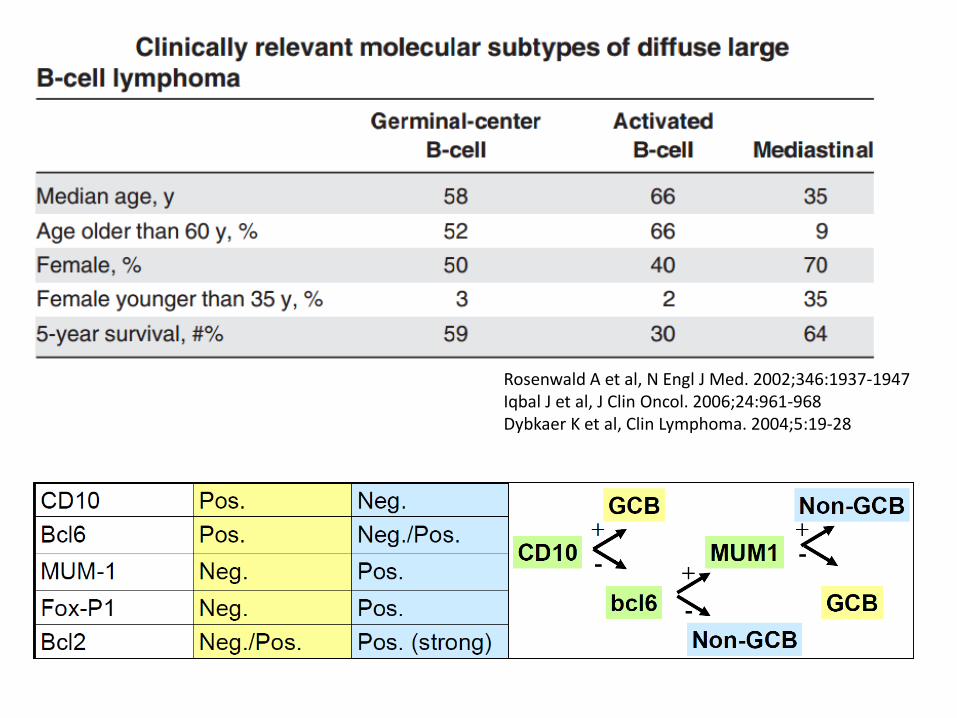
Rosenwald A et al, N Engl J Med. 2002;346:1937-1947 Iqbal J et al, J Clin Oncol. 2006;24:961-968Dybkaer K et al, Clin Lymphoma. 2004;5:19-28

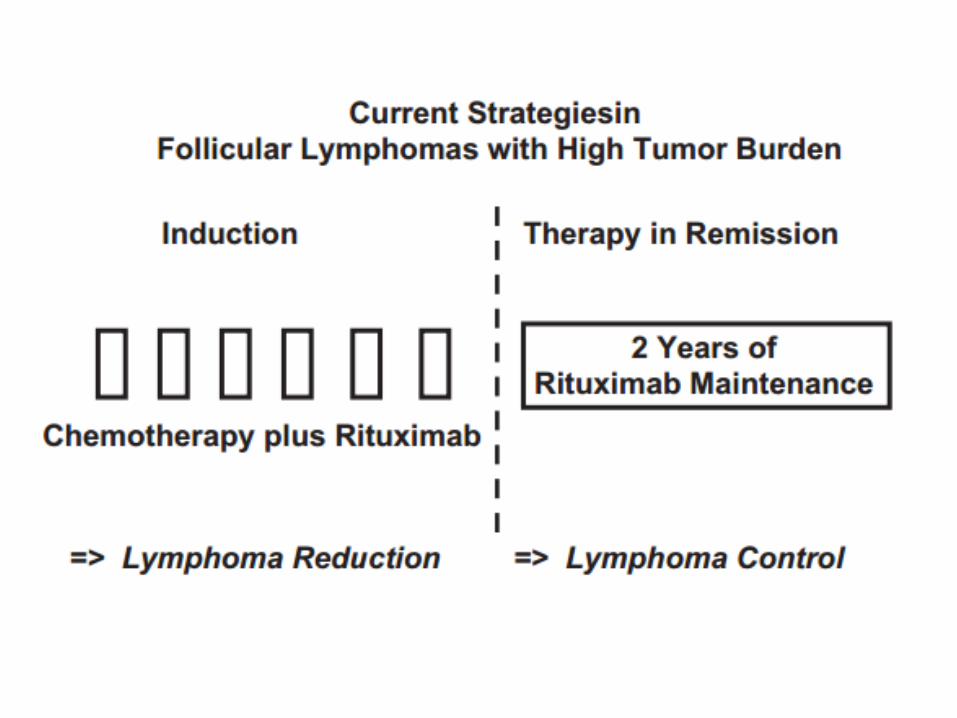
Follicular lymphoma
With 0-1 risk factors, low risk; 2 intermediate risk; 3-5 high risk.
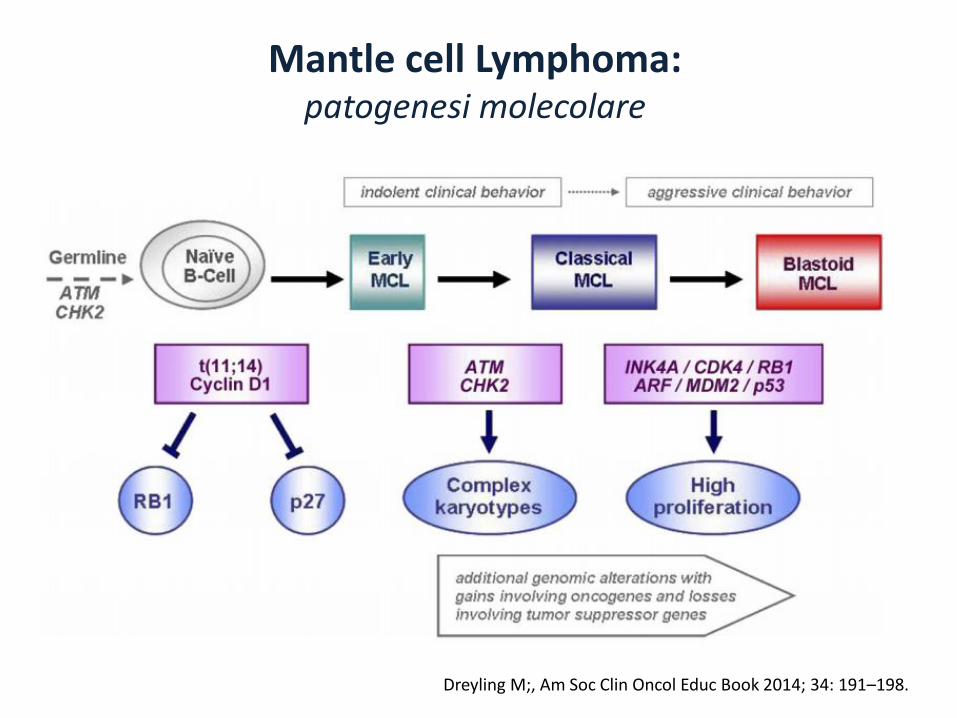
Mantle cell Lymphoma:patogenesi molecolare
Dreyling M;, Am Soc Clin Oncol Educ Book 2014; 34: 191–198.
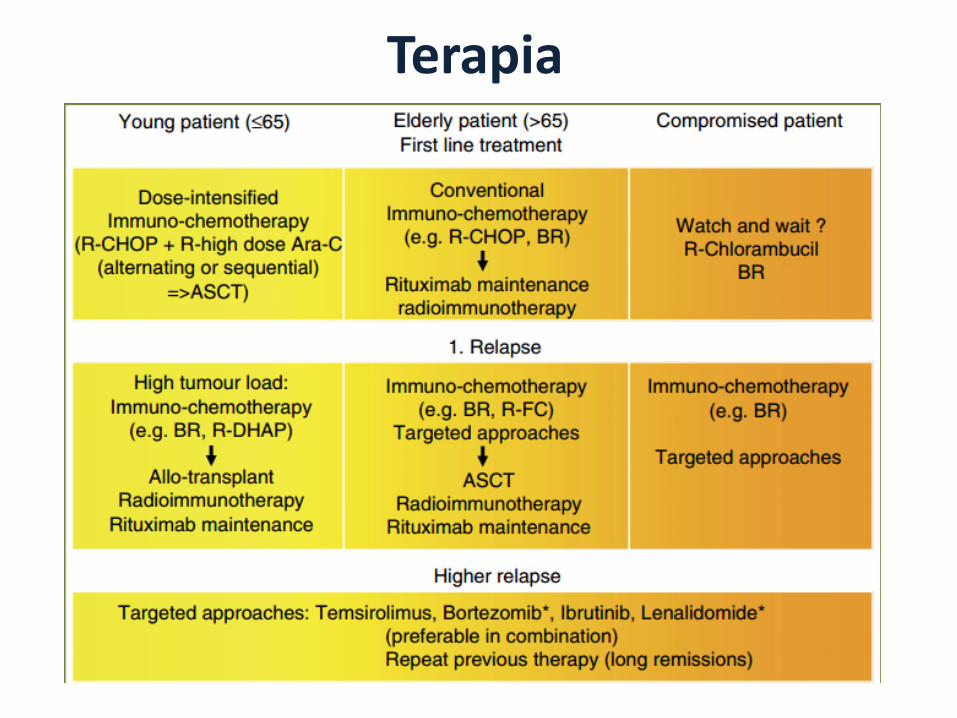
Terapia

Epidemiologia PTCLs originano linfociti T periferici post-timici o da cellule NK mature. Rappresentano il 10-15% di tutti i Linfomi non Hodgkin.Nei EATL c’è una più alta incidenza dell’aplotipo “human leukocyte antigen (HLA)” associato alla malattia celiaca. Altri sottotipi PTCL sono associati con disordini cronici autoimmuni come la malattia Crohn. Rapporto M/F = 2:1. Età mediana alla diagnosi: 60-70 anni.
DiagnosiIn accordo con la classificazione WHO (2008), la distinzione tra le diverse entità di PTCL richiede l’integrazione fra quadro clinico, morfologia, immunoistochimica, citofluorimetria, citogenetica e biologia molecolare.Nei PTCL la conferma della natura neoplastica di una popolazione T cellulare è basata sulla morfologia, fenotipo linfocitario T aberrante e riarrangiamento clonale dei geni T-cell receptor (TCR) (genotipi αβ versus γδ).
LINFOMI A CELLULE T E NK
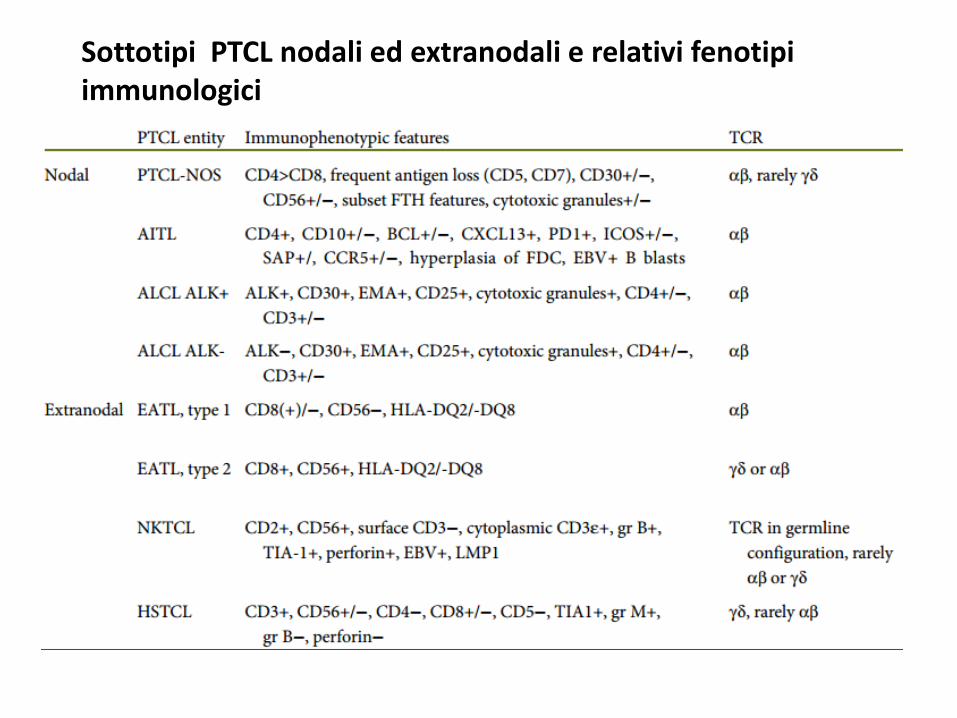
Sottotipi PTCL nodali ed extranodali e relativi fenotipi immunologici
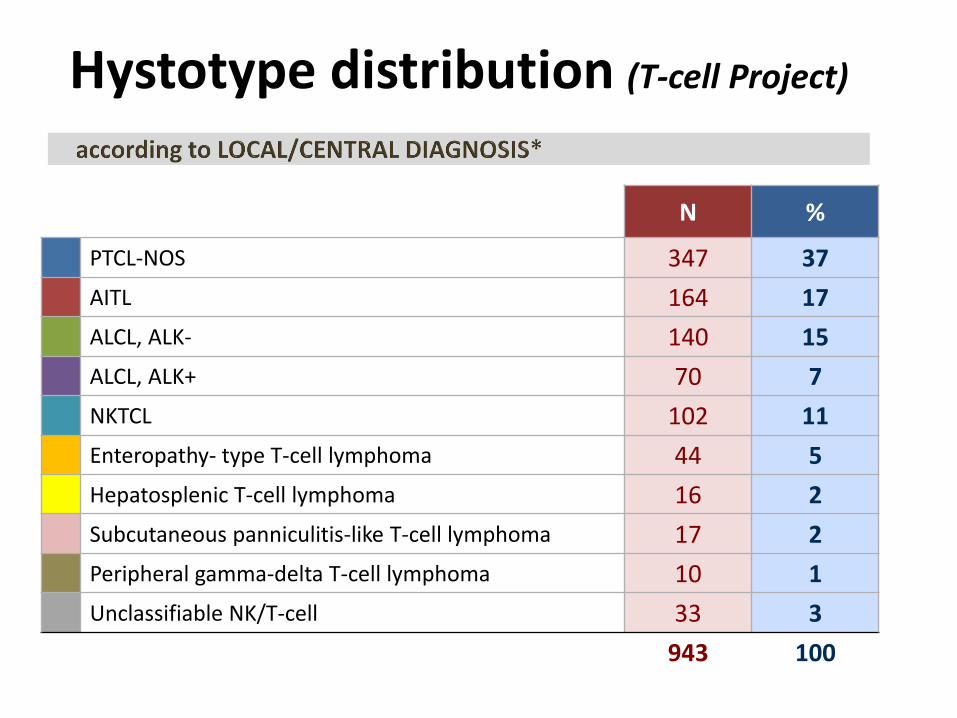
Hystotype distribution (T-cell Project)
N %
PTCL-NOS 347 37
AITL 164 17
ALCL, ALK- 140 15
ALCL, ALK+ 70 7
NKTCL 102 11
Enteropathy- type T-cell lymphoma 44 5
Hepatosplenic T-cell lymphoma 16 2
Subcutaneous panniculitis-like T-cell lymphoma 17 2
Peripheral gamma-delta T-cell lymphoma 10 1
Unclassifiable NK/T-cell 33 3
943 100
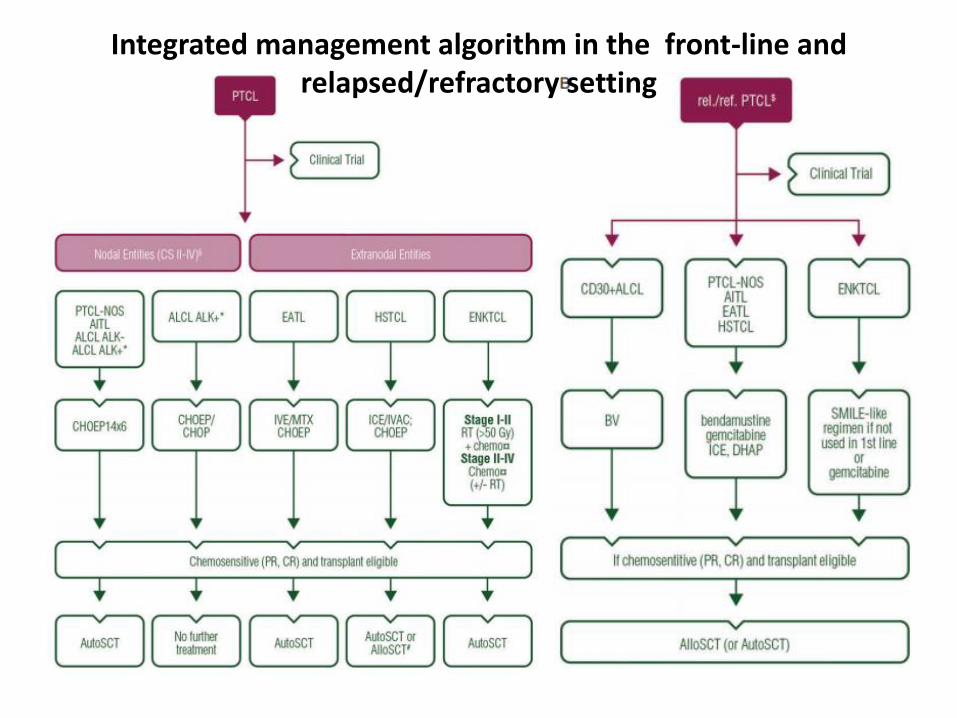
Integrated management algorithm in the front-line and relapsed/refractory setting

1. Mieloma Multiplo2. Leucemia Linfoblastica Acuta3. Sindromi Mielodisplastiche4. Leucemie Mieloidi Acute5. Leucemia Mieloide Cronica 6. Neoplasie Mieloproliferative Ph-7. Leucemia Linfatica Cronica8. Linfomi non-Hodgkin9. Linfomi di Hodgkin
Le emopatie neoplastiche

Linfoma di Hodgkin a predominanza linfocitaria nodulare(NLPHL)
Linfoma di Hodgkin classico (cHL):a sclerosi nodularericco di linfocitia cellularità mistaa deplezione linfocitaria
WHO 2008 Classificazione dei Linfomi di Hodgkin
Diagnosi immunofenotipicaL’immunofenotipo delle cellule neoplastiche nel cHL e NLPHL differiscesignificativamente. In contrasto con le cellule di Red Sternberg che sonoconsistentemente positive per CD30 e CD15,occasionalmente positive perCD20 e negative per CD45, le cellule LP sono caratterizzate dallaespressione del CD20 e CD45 e mancano del CD15 e CD30.
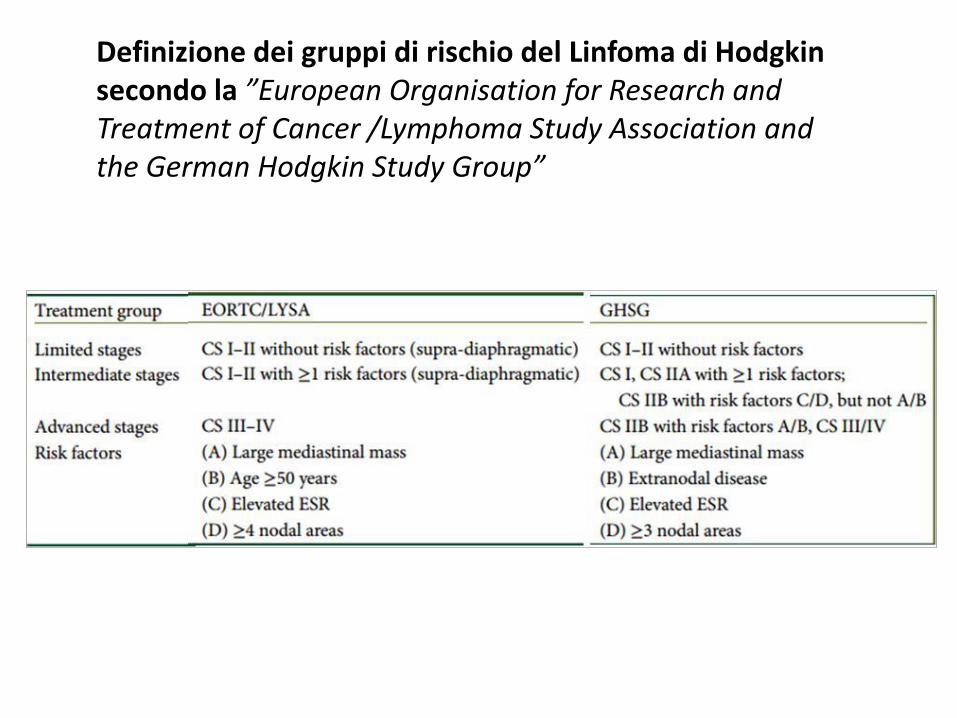
Definizione dei gruppi di rischio del Linfoma di Hodgkin secondo la ”European Organisation for Research and Treatment of Cancer /Lymphoma Study Association and the German Hodgkin Study Group”
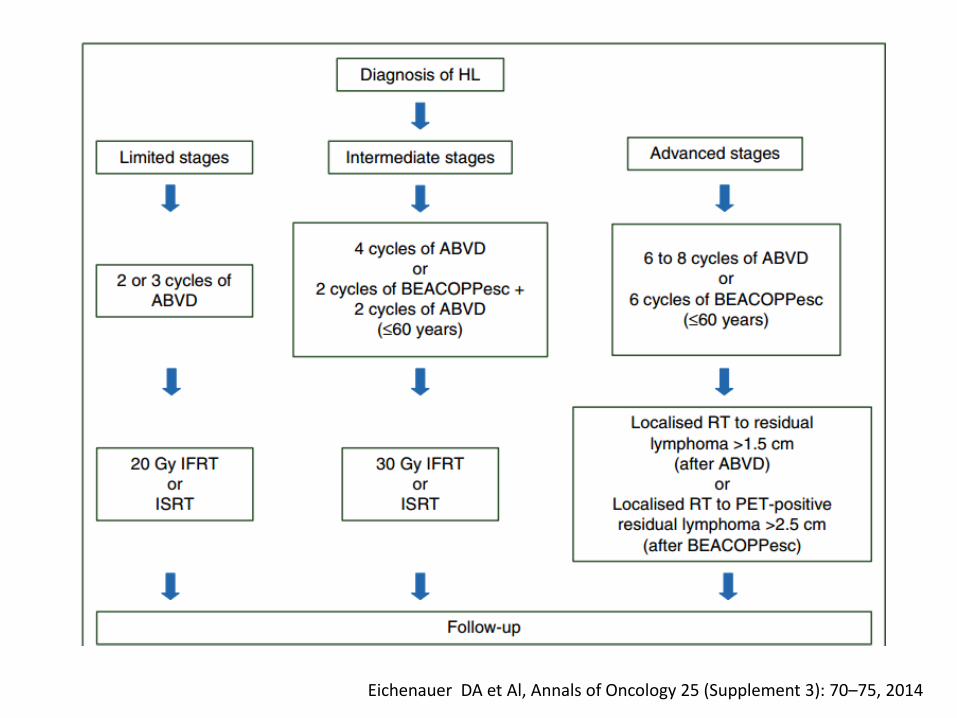
Eichenauer DA et Al, Annals of Oncology 25 (Supplement 3): 70–75, 2014