hydrologie des régions arides: progrès récents; recherches...
TRANSCRIPT

HYDROLOGIE DES RÉGION
PROGRÈS RECENTS
Par H. SCHOELLER
professeur d’hydrogéologie et de géologie à la Faculté des sciences de Bordeaux
U N E S C O

RECHERCHES SUR LA ZONE ARIDE - XII HYDROLOGIE DES RBGIONS ARIDES
PROGRBS RgCENTS

Dans cette collection :
I. II. III. IV. V.
VI. VII.
Compte rendu des recherches relatives à l’hydrologie de la zone aride. Actes du colloque d’Ankara sur l’hydrologie de la zone ande. Directory of Institutions Engaged in Arid Zone Research (en anglais seulement). Utilisation des eaux salines : compte rendu de recherches. Plant Ecology: Proceedings of the Montpellier Symposium / Écologie végétale : Actes du colloque de Montpellier. Plant Ecology, Reviews of Research / lhologie végétale : compte rendu de recherches. Wind and Solar Energy: Proceedings of the N e w Delhi Symposium / Énergie solaire et éolienne : Actes du colloque de N e w Delhi / Energia solar y eólica: Actas del coloquio celebrado en Nueva Delhi. Human and Animal Ecology: Reviews of Research/ lhologie humaine et animale : compte rendu de recherches. Guide des travaux de recherche sur la mise en valeur des régions arides. Climatologie : compte rendu de recherches. Climatology and Microclimatology: Proceedings of the Canberra Symposium / Climatologie et microclimatologie : Actes du colloque de Canberra. Hydrologie des régions arides. Progrès récents.
VIII.
IX. X. XI.
XII.
A partir de 1955, les comptes rendus de recherches sont publiés sous couverture jaune, les Actes des colloques sous couverture grise.

Publié en 1959 par l’organisation des Nations Unies pour l’éducation, la science et la culture, place de Fontenoy, Paris-70
Imprimeries Oberthur
0 Unesco 1959 NS. 58/III. 16/F

A V A N T - P R O P O S
E programme de la zone aride de l’Unesco, établi en 1951, a été transformé en projet majeur lors de la neuvihme session de la Conférence générale en 1956. Cette modi- $cation a eu pour conséquence un accroissement substantiel des moyens qui sont
accordés à l’organisation pour encourager les recherches sur la zone aride et, en particulier, pour apporter une aide directe à certaines institutions de la région qui s’étend de l’Afrique du Nord au Moyen-Orient et à l’Asie méridionale. Mais dans le cadre du projet majeur, le rassemblement et la diffusion des informations scientifiques résultant des études sur les problèmes des zones arides demeurent un des objectifs essentiels.
Onze volumes ont paru jusqu’ici dans la collection de l’Unesco intitulée Recherches sur la zone aride qui comprend des comptes rendus de recherches sur des sujets tels que l’hydro- logie, l’écologie végétale, l’utilisation des eaux salines, l’écologie humaine et animale, la climatologie, ainsi que les actes des colloques consacrés à ces mêmes sujets dans le cadre du programme. Dans cette série, le présent ouvrage inaugure un genre un peu diflérent de publications
qui seront soit des mises à jour de comptes rendus déjà parus,’soit des monographies concernant les recherches effectuées dans certains domaines, qui présentent une importance particulière mais n’ont pas donné lieu à des travaux assez étendus pour justifier de plus amples développements. Depuis la publication de comptes rendus de recherches sur l’hydrologie de la zone aride
et des actes du colloque d’Ankara, l’hydrologie et tout spécialement l’hydrogéologie ont enregistré des progrès très importants dans des branches telles que l’exploitation des eaux souterraines, leur géochimie, l’utilisation des traceurs radio-actifs, etc. Le professeur Schoeller a bien voulu, dans ce volume, analyser ces progrès récents et réunir une documen- tation bibliographique étendue portant sur les travaux publiés depuis 1952. En offrant cet ouvrage aux spécialistes de l’hydrologie et aux autres chercheurs qui
s’occupent des problèmes de la zone aride, le Secrétariat de l’Unesco tient à exprimer sa reconnaissance à l’auteur et à tous ceux qui lui ont fourni les renseignements les plus récents dont il avait besoin.

T A B L E D E S M A T I R R E S
INTRODUCTION . . . . . . . . . . . . . . .
CHAPITRE PREMIER . Considérations générales sur les conditions he .formation des eaux souterraines. leur alimentation et les ressources dans les régions arides . . Le bilan de l’eau dans les nappes Les conditions de formation des eaux souterraines dans les régions Les ressources en eaux souterraines . . . . . . .
Ressources naturelles . . . . . . . . . Ressources régulatrices . . . . . . . . . Réserves séculaires . . . . . . . . . . Ressources exploitables . . . . . . . . .
.
. . . . . . .
L’évaluation des ressources naturelles dans un vaste territoire
. . arides . . . . . . . . . . . .
. .
. .
. .
. .
. .
. .
. .
. .
CHAPITRE II . Recherche et exploitation des nappes dans les régions arides . Les conditions dans la zone d’alimentation . . . . . . . . . Les conséquences dues au puisage de l’eau h l’aide de puits et forages Les débits à demander aux nappes. débits de sécurité
. . . . . . . . .
Prise dans les réserves . . . . . . . . . . . . . Prise dans le débit de la nappe. dans les ressources naturelles . . . . Disposition des puits . . . . . . . . . . . . . Relation entre le débit des puits et forages et le débit des nappes . . .
. Conditions critiques . . . . . . . . . . . . . .
. . . . . . . . . . . . . .
. . . . .
Exemple d’étude d’alimentation en eau et de drainage dans une région aride
Ordre de priorité . . . . . . . . . . . . . . Alimentation en eau et drainage Exploitation des nappes d’eau souterraines et des sources Utilisation des eaux des montagnes et de l’avant-pays
L’emmagasinement des eaux . . . . . . . . . . . .
CHAPITRE III . Calcul de la perméabilité et de la transmissivité à l’aide de pompage en régime transitoire . . . . . . . . . . . . . . Méthode de Theis . . . . . . . . . . . . . . . . Méthode approximative . . . . . . . . . . . . . . . Méthode de Boulton pour les puits en nappe libre . . . . . . . . .
. . . . . . . . . Détermination numérique de V . . . . . . . . . . . . Calcul du niveau de pompage dans le puits
9
11
11 13 20 20 20 21 21 21
23
24 24 26 26 26 27 27 30 31 32 32 33 33 34
35
35 38 40 42 44

Méthodes tenant compte du débit retardé. provenant du n specific yield )) dans le cas des nappes libres. ou de la drainance dans le cas des nappes captives . . . .
Méthode de Boulton . . . . . . . . . . . . . . Méthode de Hantush . . . . . . . . . . . . . .
Cas de régime permanent . . . . . . . . . . . . Cas de régime non permanent . . . . . . . . . . . .
CHAPITRE IV . Géochimie des eaux souterraines . . . . . . . . . Mise en solution . . . . . . . . . . . . . . . . Eaux des différents terrains . . . . . . . . . . . . . .
Terrains calcaires . . . . . . . . . . . . . . . Gypse et terrains salifères . . . . . . . . . . . . . Eaux en contact avec des marnes et des argiles . . . . . . . . Eaux des sables et grès normaux Eaux des sables et des grès purement siliceux . . . . . . . . Eaux en contact avec des matières organiques . . . . . . . . Granites et gneiss . . . . . . . . . . . . . . . Basaltes . . . . . . . . . . . . . . . . .
Les phénomènes modificateurs . . . . . . . . . . . . . Les phénomènes réducteurs . . . . . . . . . . . . . Les échanges de base . . . . . . . . . . . . . .
Valeurs absolues et produits . . . . . . . . . . . . . Valeurs relatives . . . . . . . . . . . . . . . Graphiques . . . . . . . . . . . . . . . . . Diagrammes de Collins en colonnes Le diagramme semi4ogarithmique . . . . . . . . . . . .
. . . . . . . . . . .
L a concentration . . . . . . . . . . . . . . . Les représentations des analyses : les rapports . . . . . . . . .
. . . . . . . . . . .
L a composition chimique des eaux dans les terrains
Les zonalités de la composition chimique des eaux
. . . . . . . . L’évolution de la composition chimique dans une même nappe . . . . . .
. . . . . . . . Zonalité géologique . . . . . . . . . . . . . . . Zonalité verticale . . . . . . . . . . . . . . . Zonalité climatérique . . . . . . . . . . . . . . Résidu sec . . . . . . . . . . . . . . . . . Teneur en bicarbonates . . . . . . . . . . . . . . SO4 et C1 . . . . . . . . . . . . . . . . . Ca. Mg. N a . . . . . . . . . . . . . . . .
L a composition chimique des eaux dans les déserts. d’après Kounine . . . . La formation et la composition chimique des eaux souterraines dans les régions arides. d’après Sie-Bektchourine . . . . . . . . . . . . . .
Première étape . . . . . . . . . . . . . . . . Deuxième étape . . . . . . . . . . . . . . . . Troisième étape . . . . . . . . . . . . . . . .
Quelques caractères généraux des déserts et semi-déserts du point de vue géochimique .
CHAPITRE V . Les différents traceurs. la circulation microscopique de l’eau dans les
Les caractères d‘un traceur idéal . . . . . . . . . . . . . Les différentes sortes de traceurs non radio-actifs
Traceurs solides . . . . . . . . . . . . . . . . Traceurs chimiques solubles . . . . . . . . . . . . . Traceurs colorants . . . . . . . . . . . . . . .
L a circulation de l’eau dans les terrains . . . . . . . . . . . Différents trajets . . . . . . . . . . . . . . . Trajet général . . . . . . . . . . . . . . . . Trajets détournés . . . . . . . . . . . . . .
terrains. les traceurs radio-actqs . . . . . . . . . . .
. . . . . . . .
44 44 48 49 50
53
53 55 56 56 56 57 57 57 58 58 59 59 60 62 67 67 68 68 68 69 69 72 73 73 73 75 76 76 77 77 79
80 82 82 83 83
84
84 85 85 86 87 88 88 89 89

Trajets directs . . . . . . . . . . . . . . . . Trajets en mouvement turbulent . . . . . . . . . . . Trajet en régime laminaire dans un capillaire . . . . . . . . Trajet dans un sable de dimensions latérales illimitées . . . . . .
L’adsorption et la rétention des traceurs . . . . . . . . . . .
Méthode d‘injection continue. méthode par dilution . . . . . . . Méthode d‘injection continue. méthode de vitesse . . . . . . . . Méthode d‘injection par bouffée . . . . . . . . . . . . Absence de phénomènes d’adsorption . . . . . . . . . . . Succession des pics . . . . . . . . . . . . . . . Présence de phénomènes d’adsorption . . . . . . . . . . .
Les entraîneurs . . . . . . . . . . . . . . . . Utilisation des traceurs radio-actifs . . . . . . . . . . . .
Dsérents traceurs radio-actifs possibles . . . . . . . . . . Ilmetteurs p purs . . . . . . . . . . . . . . . hetteurs p et y . . . . . . . . . . . . . . . Isotopes susceptibles d’être adsorbés et de réagir avec l’eau ou avec le terrain . . Isotopes non facilement adsorbables . . . . . . . . . . . Méthodes d’emploi des traceurs radio-actifs . . . . . . . . .
Les méthodes d’injection des traceurs dans l’aquifère . . . . . . . .
Différentes expériences où ont été utilisés les traceurs radio-actifs . . . . Essai du Laboratoire de recherches de Californie . . . . . . . Essai de Serre-Ponçon (France) . . . . . . . . . . . Essai à Cauterets et à Luz (France) . . . . . . . . . . Essais dans des terrains pétrolifères de l’Oklahoma (Nowata County) et du Kansas (Anderson County). hats-Unis d‘Amérique . . . . . . . . . Essai du Wadi Rayan. dans le désert de Libye . . . . . . . . Étude des bilans d’eau . . . . . . . . . . . . .
BIBLIOGRAPHIE . . . . . . . . . . . . . . .
90 90 90 91 93 95 95 96 96 97 97 98 98 99 101 101 101 102 102 102 106 106 108 108
108 111 112
116

I N T R O D U C T I O N
EAU - et tout particulièrement l’eau souterraine - conditionne la vie dans les régions semi-arides et arides. I1 n’est pas douteux que l’hydrologie - et notamment l’hydrogéologie - a un rôle primordial à y jouer. Sans elle toute
autre étude serait vaine. Or, depuis le colloque d’Ankara en 1952, cette discipline a fait des progrès rapides
dans toutes les directions imaginables, et dans tous les pays. Exposer l’ensemble de ces progrès en quelques pages est impossible; ils sont trop
nombreux et intéressent trop de domaines; on risquerait de n’en donner que des aperçus peu féconds. Aussi a-t-on été conduit à ne traiter que des questions nouvelles, les moins connues,
ou de celles qui avaient été jusqu’alors particulièrement négligées. On a laissé volon- tairement de côté certains sujets, souvent importants, c o m m e celui de l’évapotrans- piration, gui n’est pas encore suffisamment au point. Certes l’évapotranspiration a son importance dans les bilans hydrologiques de tout un bassin fluvial. Mais comment évaluer sa part dans le bilan des nappes, toutes différentes les unes des autres? Dans le chapitre premier est exposé l’un des problèmes essentiels de l’hydrologie
des pays arides : les conditions de formation des eaux souterraines, leur alimentation, les ressources en eau.
Le chapitre II est consacré à la recherche des eaux et à leur exploitation. Rechercher les nappes est en effet un véritable travail scientifique. Les exploiter n’est pas possible sans une étude générale de la région, sans une étude détaillée de points particuliers. On ne peut s’y risquer, au hasard, sans programme précis. Le chapitre III traite de la détermination de la transmissivité des terrains aquifères,
à l’aide de certaines méthodes récentes. En effet la détermination du débit des nappes constitue une des tâches essentielles de l’hydrogéologie des régions arides. Mais pour calculer ce débit il faut connaître la perméabilité ou mieux la transmissivité des terrains aquifères. Certainement l’une des meilleures méthodes de détermination de la trans- missivité est celle du débit des puits et forages en régime transitoire. Mais ces méthodes sont peu connues en dehors des fitats-Unis d’Amérique; elles peuvent cependant rendre d‘immenses services. Le chapitre IV donne un aperçu général de la géochimie des eaux souterraines,
particulièrement importante dans les régions arides puisque la salure est liée à l’aridité. I1 montre comment les eaux acquièrent leur composition chimique, comment cette composition chimique évolue dans les nappes, dans les terrains et aussi suivant les climats.
9

Enh le chapitre v traite de l’utilisation des traceurs, en particulier des traceurs radio-actifs, pour déterminer la direction et la vitesse de circulation de l’eau souterraine. Trois traceurs radio-actifs, le brome-82, l’iode-131 et le tritium, présentent des avan- tages certains. Cependant le besoin se fait sentir de traceurs ayant une durée de vie, non de quelques jours (comme l’iode-131) ou de quelques années (comme le tritium), mais de quelques semaines ou de quelques mois. L’usage des traceurs demande une connaissance plus approfondie de la circulation microscopique de l’eau dans les terrains. Aussi avons-nous en partie exposé cette question.
10

C H A P I T R E P R E M I E R
Considkrations générales sur les conditions de formation des eaux souterraines,
leur alimentation et les ressources dans les régions arides
Nous donnons ici quelques aperçus de travaux relatifs aux conditions de formation des eaux souterraines, à leur alimentation et aux ressources dans les régions arides. Nous les avons choisis parmi les nombreuses études qui ont paru dans le monde entier, en raison de leur caractère général, d’une part, de leur intérêt pour les régions arides, d’autre part. Nous avons volontairement laissé de côté un certain nombre d’études classiques.
LE BILAN D E L’EAU D A N S LES RAPPES
L e bilan de l’eau dans les nappes représente encore, le plus souvent, une terrible inconnue de l’hydrogéologie. Et pourtant ce bilan est de première importance, puisqu’en dernier ressort l’hydrogéologie a pour but pratique de déterminer les ressources disponibles en eau souterraine. C’est pourquoi il est bon de citer ici les travaux de M. A. Velikanov 1 et de B. I. Koudéline [19, 201 z.
Le bilan des eaux souterraines est naturellement lié à celui des eaux d’un bassin fluvial qui peut être établi par exemple pour une année et doit comporter toutes les variations susceptibles d’entrer en jeu.
Le bilan fluvial comprend la pluviométrie, x, le run-off, y, des rivières. Mais ce dernier peut se décomposer en run-off superficiel, yl. (c’est-à-dire le ruissellement), et en run-off souterrain, ya, en provenance des nappes libres.
Outre les éléments précédents, il y a lieu de tenir compte de l’évaporation, z (dont on aura déduit la condensation), des valeurs u, s o m m e de toutes les variations posi- tives ou négatives des réserves d‘eau, augmentation ou diminution de la couverture de neige, élévation ou abaissement du niveau d’eau des nappes libres, du niveau de l’eau des fleuves, des lacs, etc. Mais le bassin fluvial ne se superpose pas nécessairement à une structure hydro-
géologique. Des nappes phréatiques d’un bassin adjacent peuvent se déverser dans le bassin considéré et vice versa. Nous en connaissons. Mais, il faut le dire, ces cas ne sont pas fréquents.
Ils le sont déjà plus pour les nappes non phréatiques, peu profondes. On aura donc à tenir compte de la valeur fw, de cette circulation de l’eau souterraine entre le bassin considéré et les bassins adjacents. Le signe + correspond à une circulation vers les bassins adjacents, le signe - vers le bassin étudié. 1. M. A. VELIKANOV, HydrogPologie de la terre, 1948. 2. Les cbXreB entre crochets renvoient A la bibliographie en fin de volume.
11

Hydrologie des régions arides
S’il n’y a pas de nappes artésiennes profondes, le bilan se formule ainsi :
Mais si l’on établit le bilan pour un grand nombre d’années, les variations tantôt positives, tantôt négatives, arrivent à s’annuler, ce qui simplifie beaucoup le calcul du bilan général, en évitant des incertitudes majeures. On établit donc le bilan général en prenant la moyenne de n années.
Dans ce cas,
et l’on a :
avec
Yo = Yo7 + Yos yo étant le run-off total de la rivière.
I1 arrivera assez souvent que wo soit négligeable. Mais ce serait une lourde faute que de ne pas en tenir compte systématiquement. Une vérification s’impose toujours, car l’alimentation d’un bassin par des eaux souterraines originaires d’un autre bassin est parfois très importante. Cela a lieu tres fréquemment, par exemple, dans les pays calcaires. Ainsi, la Garonne prend bien l‘eau de sa source, non sur le versant français mais sur le versant espagnol.
U n e autre complication intervient. Parfois, dans un bassin fluvial, des eaux arté- siennes profondes s’alimentent et déversent leur eau dans un m ê m e bassin, Mais ces eaux artésiennes appartiennent à un cycle hydrologique de durée beaucoup plus longue que celui des nappes peu profondes. Cette durée peut être de l’ordre de dizaines de milliers ou m ê m e de centaines de milliers d’années. Ainsi, dans le bassin Aquitain - lequel n’a pourtant qu’une faible étendue - l’auteur a calculé que la durée de circulation était de l’ordre de 25 O00 à 30 O00 ans, en se fondant sur la perméabilité et l’inclinaison de la surface piézométrique. L’eau qui en sort est entrée dans la nappe à la période du Wurmien. Mais du Wurmien à l’époque actuelle, la pluviométrie a dû subir de profonds changements et, en toute logique, nous devrions tenir compte de toutes ses variations et de toutes celles du débit des nappes artésiennes. En ce qui concerne la nappe dite de 1’Albien du Sahara, un calcul effectué de la m ê m e manière donne, pour un trajet de 300 km, une durée de parcours de 500 O00 à plus d’un million d’années. L e bilan ne doit donc plus être établi en fonction du nombre n d’années précédentes, mais d’après un nombre N, bien plus élevé et d’un ordre géologique. Puisque les nappes libres et les nappes peu profondes participent à une alimentation
qui s’exprime en un cycle de courte durée, tandis que les eaux artésiennes profondes appartiennent à un autre régime de circulation, remontant à une période beaucoup plus éloignée, non de l’ordre des variations climatériques de courte durée, mais d’un ordre géologique, on voit qu’il est nécessaire de dissocier les bilans de ces deux caté- gories d’eau souterraine. I1 doit y avoir une indépendance de principe entre les deux régimes. Nous avons donc à tenir compte de ya, le run-off d’origine artésienne, et de la
variation (z, - x)Sa de la pluviométrie qui lui est attachée, sur les affleurements du bassin artésien.
12

Formation et alimentation des eaux souterraines
D’autre part, un bassin peut, soit recevoir de l’eau artésienne alimentée par un bassin voisin, soit en déverser dans un bassin voisin. I1 faut donc tenir compte de la quantité f wu On obtient ainsi l’équation suivante :
x + ( X u - 4 sa =y, + Ys + Y U + 2 f 2, f wu f wg Si l’on prend un nombre d’années, N :
c:vm ---f 0
1 c (xu--.) --+ o N
Et en prenant les moyennes sur N années, on a : x1 = Y17 + Y18 + YlU + 21 f WlU zt 2%
S’il n’y a pas de communication d’un bassin B un autre x1 = Y1r + YlS + YlU + 21
ou
z1= Y1 + 21 Telles sont les équations qu’on devrait toujours considérer, en prenant un nombre d’années N, d’ordre géologique. Mais en réalité, le régime des nappes artésiennes profondes doit être relativement constant parce que c’est la différence d’altitude entre la zone d’alimentation et les zones de décharge qui règle le débit de ces nappes qui ont toujours des longueurs très grandes. On peut admettre en effet qu’en raison de leur faible vitesse de circulation, c’est-à-dire de leur faible débit par unité de front, dû à la faible pente de leur surface piézométrique, ces nappes débordent aux affleu- rements. Elles ont donc toujours la m ê m e charge et le m ê m e débit. Dès lors on n’a plus besoin de prendre la moyenne générale relative à un temps N, il suffit de celle qui est relative au temps n beaucoup plus court. C’est-à-dire qu’on peut employer l’équation :
xo = Yo7 + Yos + You + 20 f wo, f WO8
xo = Y o 7 + Y o s + Y o a f ~ o
Et s’il n’y a pas communication entre les bassins
ou
xo =yo + 20
LES CONDITIONS D E FORMATION DES E A U X SOUTERRAINES DANS LES RÉGIONS ARIDES
Nous exposerons ici, en les complétant, les vues générales de Kounine [22] sur les condi- tions de formation des eaux souterraines dans les déserts. Kounine fait remarquer que, dans les régions désertiques, la nature du cheminement et de l’alimentation des eaux et les autres caractères du premier aquifère dépendent essentiellement des conditions géographiques. Si l’on considère des couches de plus en plus profondes, cette liaison décrott graduel-
lement. C’est pourquoi les eaux profondes captives, alimentées dans des régions très éloignées des déserts, où elles arrivent par cheminement souterrain, ne présentent
13

Hydrologie des régions arides
qu’une influence désertique minime, voire nulle, que l’on considère leur régime, leur composition chimique, ou tout autre facteur. L a nappe du Crétacé inférieur continental du Sahara, dite nappe albienne, qui a
son alimentation hors du désert, dans l’Atlas, constitue un bon exemple à cet égard. Pour comprendre les caractères hydrogéologiques spécifiques des déserts, il faut
donc étudier en premier lieu les eaux influencées par les conditions géologiques; c o m m e l’indique Kounine, ce fait est très important du point de vue pratique, car, bien souvent, les autres eaux sont inaccessibles. Mais il y a des cas d’espèce. Par exemple, dans la majeure partie du Sahara, ce sont au contraire les eaux profondes qu’il est plus facile de capter.
Kounine fait remarquer qu’en réalité, en regardant les choses d’un point de vue tout à fait général, aucun caractère ne permet de distinguer les eaux profondes des eaux non profondes, c’est-à-dire les eaux sans caractères désertiques des eaux à carac- tèry désertiques.
Evidemment, cela tient à ce que les phénomènes sont beaucoup plus complexes qu’il n’apparaît au premier abord - remarque qu’on a mille fois l’occasion de formuler en hydrogéologie. C o m m e nous le verrons dans la partie géochimique de cet ouvrage, les eaux ont tendance à présenter une concentration de plus en plus élevée à mesure qu’elles sont plus profondes. Or, les eaux profondes des régions désertiques ont elles aussi des eaux très concentrées. Et rien ne permet de distinguer les causes diverses de concentration des eaux : on aboutit toujours aux mêmes caractéristiques chimiques. Dans les plaines alluviales, formées d’accumulations puissantes de sédiments sablo-
argileux, seules les premières nappes libres, généralement superposées les unes aux autres, pourront être considérées c o m m e eaux (( supérieures N. Dans les régions de basses altitudes, les eaux (( supérieures )) comprennent un complexe d‘horizons aquifères, débutant aux zones élevées érodées et finissant aux bassins artésiens locaux.
Ainsi la division en eau (( peu profonde )) et en eau (( profonde )) est déterminée par les particularités structurales, lithologiques et géomorphologiques.
Les conditions d’alimentation des eaux souterraines des déserts sont très différentes de celles de n’importe quelle autre région climatique. Cette différence dépend des proportions des diverses parties du bilan entrant en ligne de compte pour l’alimenta- tion. Par exemple, l’infiltration directe des précipitations a une importance plus grande dans les déserts pierreux que dans les autres. Dans les déserts argileux, elle se réduit presque à zéro. Dans les régions situées à la limite des déserts sableux et argileux, l’importance de l’infiltration de l’eau des lacs temporaires s’accroît fortement. Dans les déserts sableux, l’accumulation d’eau condensée devient particulièrement importante. Enfin, lorsque l’on considère encore ce bilan du régime des nappes, le côté sortie
de l’eau est très différent dans les régions désertiques. Nous pouvons étendre cette notion aux régions arides. A la différence de ce qui se passe dans les régions non déser- tiques, la perte d’eau par évaporation à partir de la surface de la nappe, par l’inter- médiaire de la frange capillaire - et cela d’après Kounine à toute profondeur - par suite du transport de la vapeur par transpiration du sol, revêt une importance considé- rable. A titre d’exemple, Kounine examine les déserts d’Asie centrale où il distingue
deux types de formations : a) des plaines de piedmont alluviales, ordinairement situées dans les zones de subsidence, au pied des montagnes ou dans les plates-formes margi- nales; b) des plaines structurales, généralement rapportées à des plates-formes.
Les premières plaines sont caractérisées par la présence d’accumulations très épaisses de sédiments meubles. Les eaux souterraines à surface libre y occupent de très vastes étendues. Leurs zones d’alimentation sont généralement éloignées et leur salure est élevée. Les alimentations locales interviennent peu dans la balance des eaux. Mais, du point de vue pratique, elles sont particulièrement importantes, car elles donnent
14

Formation et alimentation des eaux souterraines
des eaux douces de types lenticulaires dans une région où les eaux sont en majorité salées. Les secondes plaines, les plaines structurales, sont caractérisées par le grand dévelop-
pement de roches marines consolidées ou de roches métamorphiques, renfermant de nombreux horizons aquifères, de structure semblable et de faible extension. Les alimentations locales prédominent ici, tandis que les alimentations lointaines sont médiocres. L a composition chimique des eaux est très variée. Avec N. K. Guirski [17], nous diviserons les eaux souterraines des déserts et des
semi-déserts en deux grands groupes : lo les eaux souterraines qui naissent dans le domaine des déserts et des semi-déserts; 20 les eaux souterraines qui se forment prin- cipalement dans les régions externes, mais voisines, et qui arrivent aux déserts et semi-déserts par l’intermédiaire de couches perméables. Voyons en particulier les conditions d’alimentation des eaux souterraines dans les
déserts eux-mêmes [l?, 291. L’alimentation peut être assurée par condensation de l’humidité de l’air, par les
chutes de pluies et les inatrations d’eaux superficielles : rivières permanentes, lacs, rivières temporaires.
L’alimentation par condensation ne joue qu’un rôle insignifiant. I1 y a certes une condensation dans les roches sableuses, les roches fissurées, les roches poreuses com- pactes, les formations de blocs ou de galets; m ê m e dans les conditions géologiques les plus favorables, elle n’atteindrait, d’après Siline-Bektchourine et Plotnikov [29], qu’une hauteur d’eau de 4 à 8 mm. M ê m e d’après Guirski 1171, l’alimentation par condensation serait pratiquement nulle. I1 faut en effet, c o m m e je l’ai moi-même indiqué [27], tenir compte des différences
de pression de vapeur de l’atmosphère de l’air et de l’atmosphère du sol. C’est en Bté que les conditions de condensation sont les plus favorables. U n e vague de froid descend dans le sol jusqu’à la zone neutre de température. C’est donc à ce moment que la tension de vapeur de l’atmosphère est la plus faible. Or, sauf au voisinage de la mer, cette tension n’est inférieure à celle de l’atmosphère de l’air que très peu de jours dans les régions arides, en Tunisie par exemple. Le raisonnement de Guirski le confime. M ê m e si, dans les conditions les plus favorables, une condensation se produit dans
la partie tout à fait supérieure du sol, en raison de sa sécheresse, l’eau ainsi condensée retourne, au bout de très peu de temps, dans l’atmosphère. Ce phénomène s’observe très bien dans les déserts, au Sahara par exemple dans les dunes où, en hiver, par suite du rayonnement nocturne, le sol se refroidit au point qu’une condensation se forme et humecte une couche de quelques millimètres à un centimètre d’6paisseur. Cette humidité s’évapore au cours des premières heures de la journée 1.
Quant à l’alimentation par les pluies, elle ne prend une certaine importance que dans les régions arides des latitudes N. où les précipitations ont lieu en hiver, au moment où l’humidité générale est élevée. Dans les régions arides où la pluie tombe pendant l’été et en petite quantité, l’alimentation est pratiquement nulle en raison de la reprise par évaporation. En tout cas, l’alimentation dépend essentiellement de la nature des roches, ainsi
que je l’ai montré. Lorsque la pluie tombe sur des roches fissurées et dépourvues de végétation - et il y en a dans les déserts et les semi-déserts - elle ne subit pas l’évapo- transpiration et gagne les nappes qu’elle alimente en eau douce.
Mais lorsque les pluies tombent sur des roches à porosité d’interstices, l’infiltration est moins facile. Si la perméabilité est assez grande, l’eau descend rapidement et se trouve ainsi plus ou moins à l’abri des phénomènes d’évaporation tandis que, lorsque la perméabilité est très faible, l’eau reste longtemps dans le sol au voisinage de la surface et l’évaporation la reprend entièrement.
1. H. SCAOELLER, m L’hydrogéologie d‘une partie de la vaIlSe de la Saoura et du Grand Erg occidental n, Bull. Soc. ghol. Fr. (5), t. 15, 1945, p. 563-585.
15

Hydrologie des régions arides
Outre les phénomènes précédents, il faut tenir compte (Guirski) des mouvements dans le sol et de l’humidité à l’état de vapeur. En été quand la température de la surface du sol atteint sa valeur maximum, l’eau se déplace de la partie inférieure de la zone humide du sol amenée par la pluie vers la zone de température constante, ce qui peut lui permettre de gagner la nappe. En hiver, le mouvement est inverse. Si, au cours de l’année, la quantité d’eau qui descend excède la quantité d‘eau qui
se déplace vers le haut, l’humidité due à la pluie alimentera en eau douce les nappes phréatiques. Si le contraire se produit, l’eau s’évaporera à travers le sol et le sol se chargera en sels.
Nous n’avons que peu de données sur la valeur de l’alimentation par les pluies. D’ailleurs elle dépend essentiellement de la nature des terrains, si l’on ne tient pa8 compte de l’influence de la végétation. Guirski [l?] a calculé une alimentation de 1 à 2 mm dans la région qu’il a étudiée. L’alimentation par les rivières temporaires peut, dans certains cas, voire dans la
majorité des cas lorsqu’il s’agit de régions vraiment désertiques, dépasser l’alimentation par les pluies.
Elle se produit, soit B la suite de simples crues remplissant le lit, soit à la suite d’inondations. N’insistons pas sur l’infiltration des crues; elle dépend essentiellement de la nature
lithologique du lit et des berges, de la hauteur et de la durée de la crue. Toujours est-il que, dans des conditions favorables (perméabilité, charge et durée importantes), une grande quantité d’eau peut gagner les nappes, surtout s’il n’y a qu’une reprise négligeable par évapotranspiration, la végétation arbustive étant peu abondante le long du cours d’eau.
Les inondations des cours d‘eau temporaires - des oueds - ainsi que l’accumulation des eaux de ruissellement dans les dépressions topographiques, doivent, pour une grande part, rendre compte de l’alimentation des nappes dans les régions désertiques. C’est ainsi que Guirski [17] estime qu’elle atteint jusqu’à 10 mm par an, là où il admet une alimentation de 1 à 2 mm par la pluie. C’est d’ailleurs la conclusion à laquelle était aussi arrivé Dubief Cl41 pour le Sahara : (( Seule la fraction des eaux de pluie soumise au ruissellement, grâce à sa concentration en des zones restreintes sous une épaisseur notable et pendant une durée assez longue, pourra donner lieu à une infill- tration profonde susceptible d’alimenter les nappes souterraines du désert. ))
Cela dépend évidemment aussi de la nature du terrain. Si la perméabilité est suffi- sante, cette eau gagne en grande partie les nappes, tandis que, si elle est faible, l’eau infiltrée dans le sol s’évapore, amenant une concentration en sels.
Cependant il ne faudrait pas conclure que seules les eaux de ruissellement alimentent les nappes des déserts. En effet, si les pluies sont suffisantes et le terrain assez perméable, l’alimentation des nappes par la pluie a bien lieu. On ne pourrait expliquer autrement, par exemple, l’alimentation de la nappe du Grand Erg occidental, près de Béni-Abbès, au Sahara, et la très faible teneur en sels (300 mg/l) de ses eaux1.
L’alimentation par les rivières permanentes et par l’irrigation doit évidemment être prise en considération quand elle existe, c o m m e en figypte et en U.R.S.S., dans l’Asie moyenne et au Kazakstan [29]. L’irrigation amène là une surélévation d’eau douce, en forme de dôme à la surface des nappes. Mais, dès que l’irrigation cesse, le dôme s’applatit et les eaux salées sous-jacentes réapparaissent bientôt.
L’évapotranspiration, conjointement avec la faiblesse de la pluviométrie, joue le rôle essentiel dans l’alimentation des nappes d‘eau souterraines, c o m m e nous le reverrons dans le chapitre concernant la géochimie.
Elle crée un déficit de capacité de rétention dans la zone d’évaporation.
1. H. SCHOELLBB, op. rit.
16

Formation et alimentation des eaux souterraines
Comme le fait remarquer J. Tixeront [30], on ne possède pas de renseignements suffisants sur ce déficit de capacité de rétention d’eau des sols arides. En Tunisie, il existe une grande différence à cet égard entre les régions à plu-
viométrie supérieure à 200 mm par an et lee régions à pluviométrie inférieure. Dans la région de l’oued el Kebir (pluie annuelle 500 mm) le sol est saturé quand
la pluie atteint 100 mm environ. Dans la région de Sfax, la pluie atteint 200 mm. L’épaisseur du sol est parfois
considérable et l’évaporation peut se faire sentir à grande profondeur, ce qui peut amener des déficits de saturation importants. I1 faut ajouter que le déficit de capacité de rétention dépend certainement, non
seulement du facteur évapotranspiration, mais aussi de la nature du terrain. Dans la région de l’oued el Kébir (Tunisie) les terrains sont essentiellement argilo-schisteux, tandis que, dans celle de Sfax, les formations sableuses jouent un grand rôle. Dans les régions à pluviométrie inférieure à 200 mm et surtout dans les déserts,
la capacité de rétention peut être très faible, en raison du ralentissement des processus de formation du sol par manque d‘eau. En fait, la roche est à nu sur de vastes étendues. Cela s’applique évidemment fort bien aux calcaires et à toute roche consolidée dans les fissures desquels l’eau de pluie peut s’engouffrer et se soustraire à l’évaporation. I1 n’en est pas tout à fait de même dans les roches meubles, sableuses ou dans les roches argileuses, possédant une capacité élevée de rétention et dans lesquelles la simple évaporation peut créer des déficits très importants d’eau de rétention, qui devront être comblés avant que toute infiltration allant aux nappes puisse se produire. Et ces phénomènes se reflètent dans la composition chimique des eaux. Ainsi, dans le Sahara occidental, dans la région de Béni-Abbès, les eaux sortant
des quartzites ordoviciens ont des teneurs en sels de 900 mg/l dont 190 à 320 mg/l de chlore, tandis que les eaux du quaternaire meuble en ont de 5 à 7 811 et plus avec des teneurs de 1800 à 2 500 mg/l et plus de chlore1. Dans les régions à pluviométrie annuelle de moins de 200 m m , la quantité de pluie
est si réduite qu’elle tombe en général sur des terrains non saturés; cependant, rares sont les cours d’eau qui restent plusieurs années sans couler, même en plein désert. I1 faut donc admettre qu’une crue s’est produite, soit que le sol ait tout de même été saturé dans des régions limitées du bassin, soit que le facteur principal de l’écou- lement ait été l’intensité de la précipitation. Dans bien des cas, c’est effectivement ce facteur qui conditionne l’écoulement. D’ailleurs, d’après Dubief, une crue se pro- duirait dans le Sahara central, lorsque la cadence arrive à dépasser 5 mm avec une intensité supérieure à 0,5 mm. Cela nous amène à distinguer deux sortes de ruissellement, c’est-à-dire deux sortes
d‘excès momentanés de rétention, des (( ruissellements de saturation )) provoquée par une saturation du sol à la suite de pluies prolongées et des (( ruissellements d’in- tensité )) dus à ce que l’intensité de la pluviométrie est plus grande que la vitesse d’infiltration, ces deux dernières expressions étant employées par J. Tixeront. Les eaux de pluie ne peuvent ainsi s’infiltrer que dans certains cas, lorsque le déficit
de rétention n’est pas trop fort et que l’infiltration de la pluie suffit à le combler. On conçoit dès lors que dans les régions semi-arides, et n fortiori dans les régions andes, il puisse y avoir des discontinuités périodiques de l’alimentation des nappes d’eau souterraines. Ainsi, pendant un certain nombre d’années, il arrive m ê m e que les pluies ne
descendent qu’en très petite quantité, et qu’aucune pluie ne parvienne jusqu’aux nappes; soit que les chutes de pluie aient 8th trop faibles, soit que le deficit de capacité de rétention du sol ait été trop grand, l’alimentation des nappes est alors plus faible que l’écoulement et les nappes se vident c,omme se videraii, un réservoir
1. Schoellcr, ob-rivdtian, nun publites.
17

Hydrologie des r6gions arides
insuffisamment alimenté. Généralement la loi de vidange peut s’exprimer par l’équation suivante :
(1)
q étant le débit de la nappe au temps t, po le débit au temps to et e l’alimentation de la nappe. L a hauteur de la surface de la nappe peut être rattachée au débit par une relation
simple. Aussi les variations du niveau de la nappe suivent une équation semblable ?i l’équation (1) ci-dessus.
Précisément, ce qui caractérise les régions andes, ce sont les périodes généralement espacées et assez brèves où sont réunies les conditions de possibilités de réalimentation des nappes.
Pendant cette période, l’eau traverse la zone d’évapotranspiration et de là gagne facilement Ia nappe à travers le restant de la zone d’aération, le niveau de la nappe remonte brusquement et par conséquent aussi son débit.
(q - c) = (no - c)e--a(c-t.)
A cette période d’alimentation succède à nouveau une période de vidange.
1945 1946 1947 1948 1949 1950 1951 1952 1953 1954 Anndes
e Profondeur obswee. - Mouvement observ6. - - - - Mouvement supposé. FIG. 1. Mouvement du plan d’eau au puits Larue (Tixeront [29]).
On ne peut mieux l’illustrer que par le graphique ci-joint de la variation de la hauteur d’eau d’un puits de la plaine de Grombalia en Tunisie (voir fig. 1). Dans la pratique, les choses ne se présentent pas toujours de manière aussi tranchée. I1 est certain que, dans les formations meubles, les périodes de non-réalimentation
peuvent être très longues, en raison des grands déficits de capacité de rétention des sols. Mais dans les roches consolidées fissurées, ces périodes sont beaucoup plus brèves et presque directement reliées aux périodes de pluie, l’évapotranspiration ne jouant plus qu’un r81e accessoire.
Ainsi, dans les régions arides, c’est seulement lors de certaines années que sont alimentées les nappes des roches non fissurées. I1 doit, à plus forte raison, en être de m ê m e dans les régions désertiques. Toutefois, pour les roches bien fissurées, le tableau est moins sévère. En somme, en allant des régions pluvieuses aux régions arides, on passe d’un régime
d’alimentation presque régulier, à des régimes périodiques, puis à des régimes discon- tinus.
Mais cela est fonction de la nature du terrain. L a tendance A la discontinuité est d’autant moins marquée que la zone d’aération est plus épaisse et la perméabilité de cette zone plus faible. Lorsqu’il n’y a pas discontinuité dans les alimentations annuelles des nappes, c o m m e
18

Formation et alimentation des eaux souterraines
cela peut se produire dans les terrains très fissurés des régions andes, ou c o m m e cela se produit dans tous les terrains des régions pluvieuses, il y a malgré tout des périodi- cités du taux d‘alimentation des nappes. Ces p6riodicités sont surtout liées aux 0uc- tuations climatiques et pluviométriques, et aux variations de l’évapotranspiration; toutes ces fluctuations présentent d’ailleurs elles-mêmes une certaine périodicité. L. J. Tison [58, 591 et G. Tison [56, 571 ont relié la périodicité de l’alimentation
à la périodicité du débit des nappes. Soit une nappe aquifère alimentée selon une fluctuation périodique et C le débit
d’alimentation, on peut poser la formule suivante :
(1) . 2x2 C = C, + c, sin -
t0
C, étant le débit moyen entrant et c z la demi-amplitude d’une quelconque des
Le débit q sortant est lié à la hauteur d’eau par la loi oscillations de période to.
me étant la porosité effective, c’est-à-due le coefficient d’emmagasinement, et ß une constante.
Cela suppose évidemment une nappe de grande longueur dans laquelle dh/dx reste constant. On a par conséquent :
Les équations (1) et (2) donnent :
e, intégrée, donne :
7 est le retard to 2 xs 7 = - arctg -
2x to P On montre qu’en général, pour les nappes très étendues, il vaut un quart de la période. Dans le cas particulier des oscillations, le retard des oscillations de la nappe sur celles de l’alimentation est de trois mois.
qo est le débit à l’instant initial; q, la demi-amplitude du débit sortant; S l’étendue de la nappe; p = - ‘ est le coefficient angulaire de la variation du débit en fonction de la
m dh hauteur et de la porosité effective.
1. Le premier terme, C,, correspond au débit moyen. 2. Le deuxième terme
L’expression (4), co m m e on le voit, comprend 3 termes :
. (2: 2:) qm sin - - -
19

Hydrologie des régions arides
peut être obtenu en décalant
3.
2 m to
C, sin-
d’un temps T (le retard), et en réduisant les ordonnées dans le rapport a (l’amor- tissement). L e troisième terme
Pt i to
intervient surtout dans le cas d’une nappe qui a subi une dénivellation importante par rapport à son niveau d’équilibre. Si l’on prend un temps assez long après une telle dénivellation, ce terme disparaît pratiquement. Dénotant une baisse expo- nentielle du débit, et une descente exponentielle du niveau de l’eau, il correspond à la vidange de la nappe dont il était question plus haut. On a ainsi
to étant ici le temps de l’origine.
LES RESSOURCES E N E A U X SOUTERRAINES
C o m m e nous l’avons déjà dit, le but pratique de l’hydrogéologie est de connaître les ressources disponibles en eau dans une région et de savoir comment on peut les exploiter.
Mais en ce qui concerne les ressources en eau souterraine, il y a lieu de faire quelques distinctions, car elles ne sont pas toutes de m 2 m e type, ni de m ê m e origine. Et elles ne peuvent s’exploiter les unes c o m m e les autres. N. A. Plotnikovl et Bogomolov et Plotnikov [6] divisent les ressources en quatre
groupes : ressources naturelles, ressource8 régulatrices, réserves séculaires, ressources d’exploitation.
Ressources naturelles. Celles-ci concernent les débits des nappes d‘eau souterraine dans leur état naturel. C’est le débit de l’écoulement naturel.
Les moyens pratiques de déterminer les débits de ces nappes se divisent en deux catégories : 1. Détermination par application de la loi de Darcy au mouvement de l’eau de la
nappe elle m ê m e : soit en déterminant la pente et le coefficient de Darcy, soit en déterminant la vitesse de terrain de l’eau et la porosité.
2. Détermination à l’aide de forages et de puits : détermination du coefficient de per- méabilité par la méthode de Thiem (ou mieux celle de Theis) et pente de la nappe; détermination du débit unitaire de la nappe par calcul du rayon d’influence.
Ressources régulatrices. Plotnikov entend par ressources régulatrices toutes les quantités d’eau qui s’accumulent dans l’horizon aquifère pendant les périodes d’ali- mentation, au printemps, à la suite de la fonte des neiges ou lors des chutes intenses de pluie. Les ressources régulatrices sont localisées dans la zone d’oscillation naturelle de la surface des nappes libres d’eau souterraine.
1. N. A. PLOTNIKOV; G. N. BOGOMOLOV; G. M. KAMENSKI, u Classification des ressources des eaux souterraines pour l’ali- mentation totale en eau et mdthodes de calcul >>, in SILINE-BERTCHOURINE, Géologie spéciale, Moscou, Gosgeolizda t, 1946.
20

Formation et alimentation de eaux souterraines
Si donc W, est le volume de l’épaisseur de terrain entre le niveau le plus élevé et le niveau le plus bas de la surface libre de la nappe, et p la porosité effective, les res- 8ources régulatrices sont :
Réserves séculaires. Celles-ci sont constituées par les quantités d’eau situées dans l’horizon aquifère sous la zone d’oscillation de la surface de la nappe libre, lorsqu’il s’agit d’une nappe libre, ou dans le cas des nappes captives, par toute la quantité d’eau située dans l’aquifère. C’est l’eau qu’on pourrait obtenir par assèchement total de la couche. Si donc p est la porosité libre, et V le volume de la couche aquifère dans le cas
d’une nappe captive, ou le volume de la partie saturée d’eau dans le cas d’une nappe libre, la réserve séculaire est
Q = PV Ressources exploitables. Ce sont les quantités d’eau qui peuvent être obtenues des horizons aquifères pour l’alimentation au moyen d’ouvrages de captage.
Pour calculer les ressources exploitables QE des nappes libres, on détermine le débit des ressources Q, naturelles, situées au-dessus de l’enceinte du captage, et le débit Q, des ressources naturelles passant sous l‘enceinte de captage
Q E = Q,-Q, Quand il s’agit de nappes captives, on emploie en U.R.S.S. la méthode des (( entonnoirs régionaux de dépression 1). Ce terme d’ (( entonnoirs régionaux de dépression )) a été introduit par Plotnikov. I1 estime que, dans la plupart des cas, la baisse de pression des eaux artésiennes est le résultat de l’extraction de l’eau dans une série de forages régionaux. C’est pourquoi on peut parler d‘extraction régionale d’eau et d’entonnoirs de dépression ou de rabattement régionaux d’eau.
L a profondeur et la forme de l’entonnoir régional de dépression dépendent non seulement de l’extraction de l’eau, mais aussi des surfaces des zones où sont effectués les forages.
D’après Plotnikov, le plus grand rabattement a lieu dans la zone centrale de l’ex- ploitation régionale des eaux souterraines, et peut être déterminé à l’aide de la formule
dans laquelle S est la baisse régionale au lieu central d’exploitation régionale, en mètres; Q le débit régional de l’eau souterraine, en m3/jour; et u la dépression régionale spécifique, c’est-&-dire la baisse du niveau par 1 O00 ms/jour d’extraction régionale d’eau.
Par conséquent, on tire de la formule précédente : 1 O00 s
Q=- a
ÉVALUATION DES RESSOURCES NATURELLES DANS U N VASTE TERRITOIRE
D e toutes les ressources, les plus importantes sont les ressources naturelles, car ce sont les seules, dans un régime permanent de mouvement et d’alimentation des eaux, qu’on doit considérer pour déterminer les ressources exploitables. Koudeline [19,20,21] a donné quelques principes pour l’estimation régionale de ces ressources.
Les méthodes par prospection hydrogéologique et recherches expérimentales localisées dont on se sert pour calculer les ressources naturelles en eau souterraine de régions
21

Hydrologie des r6gwns arides
limitées, ne peuvent être facilement appliquées pour déterminer les ressources naturelles de vastes territoires.
L a meilleure méthode est d’utiliser les hydrographes des rivières, hydrographes qu’on décompose pour déterminer la partie de l’écoulement de la rivière correspondant au run-off souterrain. C’est ce run-off souterrain qui représente les ressources en eau souterraine du bassin.
Cette manière d’opérer a donné des résultats satisfaisants en U.R.S.S. Pour cela on se sert des équations des bilans dont il a été question plus haut. On peut ainsi, pour de grandes régions, dresser des cartes du bilan d’eau d’un grand
nombre d’années. I1 y a naturellement lieu d’utiliser les particularités structurales et hydrogéologiques. Ces cartes comprendront non pas trois (comme sur les cartes modernes) mais cinq isolignes :
Isolignes du taux des prdcipitations Xo. Isolignes de run-off Yo. Isolignes d’hvaporation Zo. Isolignes d’infiltration vers les aquifères profonds + Wo. Isolignes de run-off artésien (d’enrichissement) dans un bassin de riviere - Wo. Ces méthodes, qui donnent une estimation régionale des ressources en eau, non seulement des nappes libres, mais aussi des nappes artésiennes, n’excluent évidemment pas les travaux hydrogéologiques détaillés de prospection et d’expérimentation. Elles ne s’y substituent nullement quand il s’agit de résoudre les Problemes d’alimentation locale en eau.
22

C H A P I T R E I I
Recherche et exploitation des nappes dans les régions arides
L a recherche des nappes et des sources ainsi que leur exploitation n’offrent aucune difficulté hydrodynamique. Leur captage n’apporte qu’une amélioration des pointe de sortie de l’eau. I1 peut être souvent techniquement diflìcile, mais il introduit raremeut des perturbations dans l’hydraulique générale des nappes. I1 n’en est pas de m ê m e de l’exploitation des nappes par puits et forages. I1 faut
alors tenir compte de tous les facteurs essentiels qui interviennent dans la pertur- bation du régime de la nappe par suite de l’extraction de l’eau en des points s o m m e toute anormaux. Une connaissance de tous ces facteurs est de première importance pour interpréter les observations faites sur les niveaux des nappes à la suite de l’éta- blissement de puits et de forages, et formuler des prévisions.
Nous verrons que les facteurs essentiels qui contrôlent l’action des puits sont : lo les caractéristiques de l’alimentation des nappes; 20 la distance des puits à la zone d’alimentation; 30 la distance des puits à la zone d’évacuation naturelle; 40 le caractère du cône de dépression du puits.
Toutes les eaux souterraines d’importance économique sont en mouvement. Une nappe immobile est dénuée d’intérêt, absence de mouvement signifiant absence d’alimentation. Exploiter une telle nappe reviendrait simplement à tirer l’eau d’une réserve épuisable.
L a pente de la surface piézométrique a donc une importance aussi considérable que la perméabilité ou aussi la transmissivité de l’aquifère. Ces deux éléments - pente et transmissivité ou perméabilité - sont A égalité dans la formule de Darcy.
Les vitesses le plus souvent enregistrées sont de quelques dizaines à quelques cen- taines de mètres par an dans les nappes phréatiques : dans certaines nappes artésiennes, elles sont le plus souvent encore beaucoup plus faibles, par exemple 2 à 3 mètres par an dans la nappe des sables paléocènes de l’Aquitaine, un demi-mètre par an dans la nappe dite de 1’Albien du Sahara. I1 faut en outre tenir compte d’un autre point de vue. Les nappes d’eau souterraine
subissent des perturbations : u) des oscillations dues aux variations saisonnières d’ali- mentation; b) des oscillations de longue durée, dues aux alternances de périodes multiannuelles sèches et humides; c) des oscillations de durée géologique, c o m m e celles qui ont affecté le Quaternaire.
Nous laissons de côté les autres perturbations provoquées par les variations de la pression atmosphérique, les marées terrestres et toutes autres perturbations de très courte durée, qui n’interviennent pas dans les problèmes d’alimentation des nappes.
Cela dit, on peut admettre que les nappes sont approximativement en équilibre dynamique, c’est-à-dire qu’elles débitent autant d’eau qu’elles en reçoivent.
23

Hydrologie des régions arides
Toute modification dans Ia décharge de la nappe par captage de puits, dans l’ali- mentation par recharge ou par évapotranspiration, amènera une perturbation dans le régime naturel de l’écoulement.
L a prise d’eau de la nappe par captage ou par puits, ou bien diminuera le débit des autres points d’eau, ou bien nécessitera une nouvelle augmentation de l’alimentation. Toute diminution ou toute augmentation de l’alimentation amènera une diminution ou un accroissement du débit des points d’eau.
LES CONDITIONS D A N S LA ZONE D’ALIMENTATION
L’alimentation permanente peut être due : lo à la pluie; 20 à l’infiltration d’eau de rivière; 30 à une alimentation indirecte par une autre nappe.
D’une manière générale, on ne prend pas suffisamment en considération ce dernier facteur. I1 ne peut être négligé, surtout lorsqu’on abaisse la surface piézométrique de la nappe exploitée. Une alimentation indirecte de cet ordre survient quand existent des nappes adjacentes ou bien des nappes supérieures ou inférieures ayant une zone de contact avec la nappe. La baisse de potentiel se transmet alors à ces nappes qui livrent leur eau. M & m e lorsqu’il y a des nappes superposées ou sous-jacentes, séparées de la nappe exploitée par un imperméable, la baisse de pression fait suinter de l’eau à travers le toit ou le mur. Si peu perméables que soient ce toit et ce mur, la quantité d’eau qui arrive en supplément à la nappe peut constituer une fraction non négligeable de l’alimentation de la nappe, car le débit unitaire est à multiplier par la grande sur- face du toit et du mur. En ce qui concerne l’alimentation, il faut considérer deux cas :
1. L’intensité de l’alimentation potentielle annuelle ou saisonnière est supérieure à la quantité d‘eau que peut recevoir la nappe en un temps donné. L’aquifère se remplit complètement puis déborde. I1 y a des zones marécageuses aux affleurements, et des Bources de trop pleh I1 est donc possible d’augmenter le débit de la nappe en puisant de l’eau à l’aval,
c’est-à-dire d’augmenter les ressources qu’on demande à la nappe. 2. L’intensité de l’alimentation annuelle ou saisonnière est inférieure à la capacité
d’absorption de la nappe. I1 s’établit un équilibre hydrodynamique. L a pente de la surface piézométrique s’abaisse de manière que la décharge égale la recharge.
L’alimentation est alors fonction de la quantité d’eau reçue par la pluie sur le sol, des infiltrations d’eau de rivière, de la vitesse de descente de l’eau infiltrée, du déficit de capacité d’eau de rétention du sol, provoqué par l’évapotranspiration. Dans ce dernier cas, il est impossible de faire débiter à la nappe plus que ce
qu’elle débitait, à moins de créer une suralimentation aux affleurements. L a seule augmentation de débit qui pourrait se produire serait un drainage d’une autre nappe ou une alimentation par le toit ou le mur, à la suite de la baisse de pression de la nappe.
LES CONSÉQUENCES D U E S A U PUISAGE D E L’EAU A L’AIDE DE PUITS ET F O R A G E S
Lorsqu’on pompe dans un puits ou que l’eau fait débiter un forage, on crée un cône de dépression qui se propage, il ne faut pas l’oublier, jusqu’aux limites externes de l’aqui- fère. Mais il n’atteint ces limites qu’au-delà d’un certain temps.
L a formule de Theis :
24

Recherche et exploitation des nappes
dans laquelle A est le rabattement en mètres à tout point, Q le débit en m3/s du puits et T le coefficient de transmissivité en m3/s/m7 montre que, pendant la période transitoire de propagation du cône, sa dimension horizontale est indépendante du débit. Si l’on double celui-ci, le rabattement en chaque point est doublé. Mais le cône n’a pas pour cela un diamètre plus grand. L’extension du cône ne dépend que des propriétés du ter- rain aquifère et du temps.
I1 est extrêmement important à noter que, tant que le cône continue à s’étendre, c’est-à-dire tant qu’il n’a pas atteint les limites de l‘aquifère, il ne s’établit pas de nouvel équilibre dans le régime de la nappe à l’extérieur du cône, c’est-à-dire entre l’alimentation de la nappe et la décharge. On se borne en effet à puiser de l’eau dans des réserves. Le nouvel équilibre ne se
produit qu’une fois que le cône a rejoint la zone d’alimentation et la zone d’émergence. L a vitesse de propagation du cône est inversement proportionnelle au coefficient
d’emmagasinement, S. Dans les nappes libres, S = n x 10-l. D’une manière générale, il y Correspond
sensiblement au specijic yield. L’extension est alors très lente, le cône de dépression n’atteignant les limites du système aquifere qu’au bout d’un temps très long. Et le nouvel équilibre de la nappe ne peut s’établir qu’au bout de ce temps.
Dans les nappes captives, le coefficient d’emmagasinement ne correspond plus au specijc yield, mais à la compressibilité de l’aquifère et à la dilatation de l’eau. C’est dire que S a toujours des-valeurs très petites, de l’ordre de Le cône s’étend donc très rapidement : 100, 1000, 10 O00 fois plus vite que dans le cas des nappes libres; il atteint très rapidement les limites du système aquifère, de sorte que l’équilibre nouveau peut se réaliser en un temps relativement court et un nouveau régime de circulation s’établit.
loA4,
I1 est bon de donner ici une idée des vitesses de propagation du cône : Ainsi, dans une nappe libre ayant une transmissivité T = 1’25 x m3/s, et un
coefficient d’emmagasinement de 0’2, l’extension du cône en fonction du temps s’effectue très lentement.
m3/s et un coefficient d’emmagasinement S = 1 x l’extension du cône en fonction du temps est beaucoup plus rapide.
Au contraire, dans une nappe captive ayant une transmissivité T = 1’25 x
Extension du cône (en mètre*).
1 mn I h 1 jonr 10 jours 100 jours 1000 jours
Nappe libre 0,91 7,11 34,8 110 348 1100
Nappe captive 41 318 1558 4 930 15580 49 300
Lorsque le cône a atteint la zone d’alimentation, il est modifié par les effets de l’apport d’eau dans cette zone. De même, lorsqu’il a atteint la zone d’émergence, il est modifié par l’effet de l’arrêt ou de la diminution du débit des émergences. Si le taux de pompage ne dépasse pas le débit d’alimentation et celui d’écoulement
hors de la nappe, le cône arrivera à un état d’équilibre, en même temps que la nappe atteindra son nouvel équilibre. Si le taux de pompage est plus élevé, la nappe s’affaiblit, sa surface piézométrique
s’abaisse continuellement et les puits ou forages atteignent la réserve de la nappe, réserve qui n’a qu’une durée limitée.
25

Hydrologie des régions arides
LES DÉBITS A DEMANDER A U X NAPPES, DÉBITS DE SÉCURITÉ
Dès lors, la question suivante se pose : quels débits peut-on demander aux nappes et quel est le débit maximum, que nous appellerons (( débit de sécurité D, au-del8 duquel on provoquerait son épuisement ?
C o m m e nous l’avons dit, lorsqu’on soustrait de l’eau d’une nappe à l’aide de puits ou de forages, on commence par puiser dans ses réserves mais on capte ensuite une partie du débit m ê m e de la nappe. Nous avons donc à examiner tout d’abord la sous- traction initiale de l’eau des réserves puis l’utilisation normale, en régime permanent, de la nappe.
Prise dans les réserves.
Les conditions ne sont pas les mêmes suivant qu’il s’agit d’une nappe libre ou d’une nappe captive.
Dans une nappe captive, la quantité d’eau qu’on tire de la réserve est fournie par la diminution de pression de la nappe; or cette diminution de la pression augmente d’autant l’action de la pression de la couverture de la nappe sur le toit de celle-ci, d’où compression de l’aquifère. Mais, la compressibilité des terrains étant relativement faible, la quantité d’eau ainsi prise dans la réserve est minime. D’autre part, c o m m e le cône s’étend rapidement, la réserve est très vite épuisée.
Néanmoins la quantité d’eau ainsi libérée n’est pas aussi négligeable qu’on pourrait le croire a priori. Si nous admettons donc un coefficient d’emmagasinement de une baisse courante de pression de 10 m d’eau livrera un volume de m3, soit 1 litre par mètre carré de surface de nappe, et 1 O00 O00 de mètres cubes pour une nappe normale d’une superficie de 1 O00 km2. On obtiendrait un débit de 2 700 m3/jour, ou 310 l/s, si l’on retirait cette eau en un an. Dans les nappes libres, la quantité d’eau prise dans la réserve est considérablement
plus grande, puisqu’elle est égale à la porosité effective, multipliée par le volume d’aquifère existant entre la nouvelle et l’ancienne surface piézométrique. Le coefficient d’emmagasinement en porosité effective peut atteindre facilement 0,ZO. M ê m e si l’on a une nappe libre dix fois moins grande, soit de 100 km2, en abaissant la surface piézo- métrique de 3 m seulement, on retirera de la réserve 60 O00 O00 mètres cubes. Mais c o m m e le cône ne s’étend que très lentement, le puisage dans la réserve se
fait pendant un temps très long, sans affecter sensiblement le régime d’ensemble de la nappe.
Prise dans le de’hit de la nappe, dans les (( ressou.rces naturelles )I.
Ainsi, on prend dès lors très rapidement l’eau à l’écoulement propre de la nappe, dans le cas des nappes artésiennes, Cela n’a lieu que très tardivement au contraire, souvent de nombreuses années après, dans le cas des nappes libres.
Ce supplément d’eau disponible ne doit pas faire illusion sur les possibilités ultérieures de la nappe. On n’avait fait que puiser dans une importante réserve qu’on n’aura plus à sa disposition. On ne saurait trop insister sur cet aspect du problème de l’utilisation des nappes.
Une fois ce régime établi, on ne peut prendre à la nappe plus que ce qu’elle débite. I1 faut donc établir un débit de sécurité. Ce débit, c’est celui de la nappe. I1 est dif-
ficile de le calculer 8 l’aide de la surface d’alimentation, car ce dernier ne peut jamais être déterminé a priori d’une manière suffisamment précise.
Le meilleur moyen est encore d’estimer le débit de la nappe à partir de la pente de la surface piéeométrique, de la transmissivité et de la longueur du front de la nappe qui peuvent être plus aisément calculées.
26

Recherche et exploitation des nappes
Mais il y a deux sortes de débits d’alimentation à considérer : lo le débit primitif, avant tout établissement de captage; 20 le débit nouveau, débit de la nappe plus grand que le débit primitif car il s’y ajoute : a) l’eau qui s’infiltre en plus grande quantité par la zone d’alimentation, par suite de la suppression des trop-pleins, lorsque ceux-ci existent; b) l’eau non évapotranspirée en raison de la baisse de la surface piézométrique qui empêche les plantes de retirer de grandes quantités d’eau par évapotranspiration, là où la surface piézométrique de la nappe était au voisinage du sol; c) l’eau drainée des autres nappes par suite de la baisse de pression de la nappe qui introduit une plus grande différence de pression entre celle-ci et les nappes adjacentes. 11 y a aussi suralimenta- tion par des nappes latérales ou à travers le toit ou le m u r de la nappe. On voit donc qu’on a parfois intérêt à demander à la nappe un débit plus grand que
le débit primitif, afin de récupérer l’eau qui aurait pu être perdue, dans les trop-pleins, par évaporation et afin de soutirer l’eau à des nappes voisines.
Mais naturellement il faudra estimer le nouveau débit de sécurité afin de ne pas le dépasser.
Disposition des puits.
Reste à savoir quelles sont les meilleures dispositions à adopter pour exploiter la nappe. kvidemment, le premier facteur à prendre en considération est celui de l’utilisation
des puits. Mais ce facteur mis à part, il y a tout avantage à disperser les points de captage, de manière à ne pas provoquer de baisse trop localisée de la surface piézo- métrique. Cette baisse doit être étalée aussi largement que possible. Ensuite, ces points de captage seront disposés le long d’un front de la nappe, plutôt que parallèle- ment à la direction des íilets liquides. L a nappe doit être exploitée dans toute sa largeur. I1 est préférable de placer les zones de captage dans les zones ou au voisinage des zones où la nappe affleure vraiment ou presque, par exemple : a) dans les zones alimentées par u n trop-plein; on permet ainsi à la nappe de recuperer le débit des sources de trop-plein et l’eau évapotranspirée ; b) dans les zones d’émergences; on récupère les filets perdus des émergences et l’eau évapotranspirée; e) dans les zones où Ia nappe affleure presque, on récupère encore l’eau évapotranspirée. On voit ainsi comment on peut agir sur l’économie de la nappe, afin d’en obtenir
le meilleur rendement. On pourrait penser que si l’on retire de la nappe, à l’aide de puits ou de forages, la quantité d’eau que celle-ci reçoit de la zone d’alimentation, on asséchera sa partie aval. Mais on a reconnu que le débit des puits n’arrêtait pas le débit des émergences naturelles.
Relation entre le débit des puits et forages et le débit des nappes.
Une des questions les plus importantes de l’hydrogéologie est de connaître la relation qui existe entre le débit des puits et forages et le débit des nappes. I1 ne s’agit pas, en effet, de creuser n’importe oh des puits et forages et d’en extraire des débits quelconques. Il y a intérêt à savoir quel débit on peut retirer d’une nappe et de connaître le m a x i m u m de ce débit, m a x i m u m au-delà duquel on risque d’épuiser la nappe. S’il s’agit d’une nappe sans écoulement, le problème est simple; dans ce cas, on
n’a affaire qu’à un réservoir souterrain sans alimentation. La quantité d’eau qu’on retirera au m a x i m u m sera celle du volume d’eau de gravité, renfermée dans la couche aquifère dont on mesurera les dimensions. I1 est à remarquer que ces volumes sont souvent énormes, beaucoup plus grands qu’une estimation à première vue ne l’indique. I1 est toujours bon de se livrer à quelques calculs très simples. Ainsi supposons une couche aquifère de 10 m d’épaisseur, ayant une surface de 10 km2, chiffre qui n’est pas très élevé. Si le specijc yield de cette couche est de 0’2, on pourra retirer un volume de 20 millions de mètres cubes d’eau, soit 10 l/s pendant soixante-trois années.
27
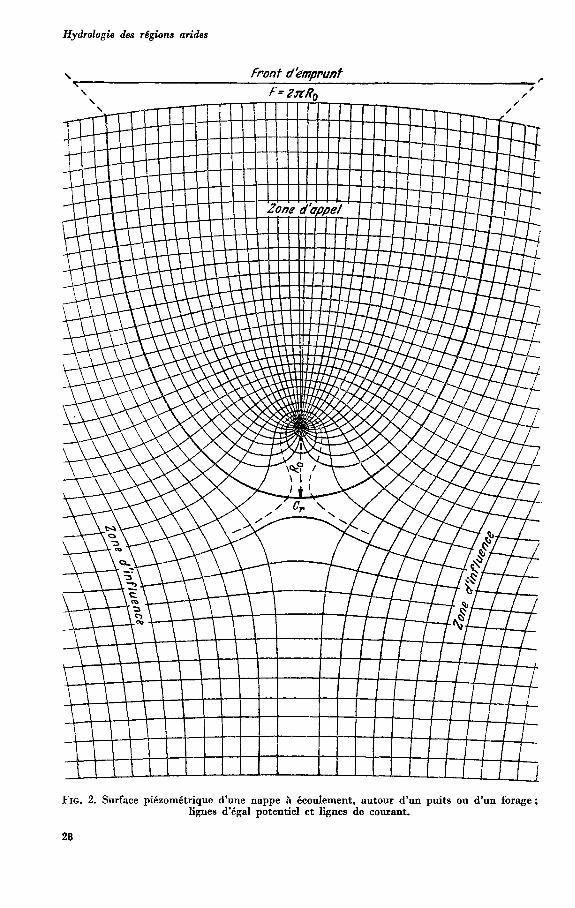
Hydrologie des régions arides
FIG. 2. Surface piézométrique d'une nappe à écoulement, autour d'un puits ou d'un forage; lignes d'égal potentiel et lignes de courant.
28

Recherche et exploitation des nappes
Mais la sagesse conseille de ne pas puiser l’eau dans un capital qui doit servir de volant; il faut puiser, non dans les réserves, mais dans les ressources naturelles. Ces ressources sont données par l’écoulement de la nappe.
Alors que toute nappe sans écoulement a nécessairement une surface piézométrique horizontale, toute nappe qui s’alimente et qui débite, c’est-à-dire toute nappe en mou- vement, à une surface inclinée.
Lorsque l’on pompe dans un puits ou dans un forage, on produit un cône ou plus exactement une zone d‘abaissement de pression autour du puits ou du forage. Dans le cas d’une nappe libre, cette zone de dépression se traduit par un abaissement de la surface de la nappe, tandis que dans une nappe captive, il n’y a qu’un abaissement de la surface piézométrique. Mais nous pouvons y distinguer (voir fig. 2) un cône d’appel ou une zone d’appel, constitués par toute la partie de la nappe dont les filets liquides se dirigent vers le forage ou vers le puits.
Ce cône ou zone d’appel comprend donc un rayon d’appel et un périmètre d’appel ou périmètre d’emprunt. Au-delà de cette zone d‘appel existe une zone de la nappe, influencée par l’appel du forage ou du puits, mais dont les Nets échappent à cet appel Nous appellerons cette zone : cône ou zone d’influence ou d’action. Enfin nous appellerons rayon fictif un rayon qui, du point de vue hydraulique, se
comporte c o m m e le rayon d’action. U n puits [67] ou un forage prélève dans la nappe une partie de son écoulement,
passant à travers une longueur F de front de nappe. Si i est la pente de la surface piézométrique de la nappe, K la perméabilité, T la transmissivité, q le débit du puits, le front F d’emprunt du puits est
Or, le débit d’un puits ordinaire est :
comme
H2 - h2 In Rflr
q - K
F z= 2xRf (3)
H2 - h2 Rf In Rf = ~
2 Hi (4)
Cette dernière équation permet, sans connaître la perméabilité, de déterminer le rayon fictif du puits et par là, à l’aide de l’équation (3)’ F; et à l’aide de l’équation (l), K ou T. Si l’on connaît le front total L de la nappe, on a donc pour débit total de la nappe :
Q = TiL
S’il s’agit d’une nappe captive, le débit d’un forage est : A
In Rf/r q = 2xKH ~
on a donc :
On peut de la nieme manière déterminer le rayon fictif Rf, le front d’emprunt F, la Perméabilité K ou la transmissivité T, puis le débit total de la nappe.
29

Hydrologie des r6gwns arides
L’auteur [68] a appliqué cette méthode à l’étude des rapports entre les débits d’ex- traction des forages puisant l’eau dans la nappe des sables paléocènes du bassin Aqui- tain, et l’alimentation de cette nappe.
Les fronts d’emprunt de chaque puits au forage sont situés en amont de chacun d’eux. On voit donc, c o m m e nous l’avons déjà indiqué, qu’on a intérêt à aligner les forages de manière que les fronts d’emprunt, eux aussi, se succèdent le long de la zone d’alimentation, sans se chevaucher.
Par la méthode précédente, il est ainsi possible d’adapter au débit naturel des nappes les débits des puits et des forages.
Le débit ainsi calculé est le débit naturel, c’est-à-dire la ressource naturelle de la nappe, celui que fournit l’alimentation annuelle. En réalité, on peut, dans un grand nombre de cas, tirer davantage sans épuiser la nappe. En effet, lorsqu’on pompe dans une nappe, on abaisse la pression de celle-ci dans un domaine de plus en plus grand autour du forage. Lorsqu’on pompe dans la nappe un débit du m ê m e ordre de grandeur que celui de la nappe, la baisse de pression s’étend sur un domaine du m ê m e ordre de grandeur que celui de la nappe. D e ce fait, on crée ou on augmente la diffé- rence de potentiel entre la nappe et des nappes adjacentes. Ces nappes adjacentes livreront donc à la nappe un débit supplémentaire. D e m ê m e on créera entre la nappe et les nappes inférieures ou supérieures une différence de potentiel qui assurera une filtration supplémentaire à travers l’imperméable, et par conséquent de nouvelles ressources. Ensuite la baisse du niveau piéaométrique dans la zone d’alimentation annulera les sources de trop-plein et, en m ê m e temps, permettra une infiltration plus facile en évitant partiellement l’évapotranspiration des ressources supplémentaires en eau qui se seraient inutilement perdues. Enfin, dans bien des cas, on récupérera des eaux qui, normalement, échappent aux sources dans les zones de sortie des nappes. On aura donc, en plus, ce qu’on pourrait appeler un débit artificiel supplémentaire
de drainage. Mais la recherche de l’équilibre entre l’alimentation naturelle plus le débit artificiel
supplémentaire et le débit d’exploitation est difficile à établir. Ainsi, lorsqu’on veut établir les bilans devant être pris en considération pour l’ex-
ploitation des nappes, il faut tenir compte de toutes les remarques précédentes. C o m m e on le voit, le bilan n’est pas aussi simple B établir qu’on aurait pu le penser d’abord.
EXEMPLE D’ÉTUDE D’ALIMENTATION EN E A U ET DE DRAINAGE D A N S UNE RÉGION ARIDE
lhablir les bilans, rechercher les quantités d’eau disponible et les quantités d’eau à drainer dans une région ne peut se faire suivant un plan général applicable à toutes les régions. Autant de régions, autant de cas d’espèce. L a façon d‘opérer dépend non seulement des conditions géographiques, géologiques et hydrologiques, mais aussi du but visé. Nous citerons néanmoins le cas envisagé par Loehnberg et dont on retrouve très
souvent l’application. Loehnberg [65], dans un article très suggestif, a étudié les conditions d’exploitation
des eaux dans une région semi-aride, conditions qui se rencontrent souvent. I1 s’agit d’une région de plaines, bordée de montagnes, au pied desquelles s’alignent
des collines. Les montagnes sont formées de roches modérément perméables, à pendage relativement élevé en direction de la plaine. L a plaine argileuse est composée de matériaux de remplissage, alluvions reposant sur des argiles, des marnes, des shales. Les collines adossées au pied des montagnes sont constituées par des éboulis et des cônes de déjection. Le contact entre les montagnes et les plaines peut être une simple faille, un système compliqué de failles, ou encore une simple discordance stratigraphique. On admet une pluviométrie d’environ 300 mm en plaine, de 600 mm en montagne.
30

Recherche et exploitation des nappes
L a concentration des pluies, dans une période de trois à cinq mois, provoquera des ruissellements périodiques importants, des inondations et des conditions maréca- geuses, en particulier aux niveaux inférieurs. L a saturation en eau de ces terrains décroît pendant les mois secs d’été, mais peut se maintenir dans les sols lourds.
1. Crues soudaines et brèves, provenant directement de la concentration de la pluie. 2. Run-off résultant de précipitations prolongées, de pluies directes sur le sol, aussi
bien que du drainage d‘un emmagasinement de courte durée dans les terrains (drainage, accroissement du débit de petites et de grandes sources).
3. Run-off provoqué par des précipitations prolongées, mais causé par un écoulement retardé à travers les sources principales, après le remplissage de l’aquifère majeur.
Les sources peuvent se grouper en deux types : 1. Sources de montagne, dues à des conditions locales favorables, tectoniques et
lithologiques. 2. Sources situées au pied des collines, au contact des collines et des plaines, provenant
par exemple d’un cône de déjection ou d’une terrasse. Les eaux souterraines qu’on peut espérer rencontrer sont : 1. L’eau souterraine dans les formations modérément perméables de la région mon-
2. L’eau souterraine dans le remplissage des vallées et dans les cônes de déjection. 3. L’eau souterraine se déplaçant tres lentement dans les sédiments argileux de la
Les marais des régions montagneuses sont cantonnés aux eiivirons des émergences et des sources. Et il en est de m ê m e dans la partie supérieure de la plaine en raison de la texture lâche des terrains. En bordure des terrains argileux, les marais sont disposés en larges taches, en liaison
avec des émergences. Dans la partie la plus basse de la plaine, faite d’argile et de marnes, les marais prennent un grand développement et se maintiennent plus ou moins fortement d’une manière permanente. I1 est bien évident, c o m m e nous l’avons dit plus haut, que ce schéma de Loehnberg
ne s’applique pas uniformément à toute contrée semi-aride. Par exemple, la région montagneuse peut fort bien ne pas être plissée. Mais cela ne change pas beaucoup les conditions hydrogéologiques. L a région de bordure de la chaîne plissée peut être formée de collines à stratifications horizontales, s’étendant plus ou moins vers la plaine (c’est m ê m e là un type très fréquent). Et, surtout, la plaine peut être formée, non de couches relativement argileuses,
mais de formations très perméables, sableuses et autres, sur de très grandes épaisseurs, ce qui modifie alors du tout au tout le schéma de Loehnberg. D e tels types sont extrêmement fréquents dans les régions arides.
Enfin, le substratum de ces terrains alluviaux peut renfermer des couches plus ou moins perméables, aquifères, alimentées dans la région haute, et qui donnent naissance à des nappes captives, souvent très puissantes. Dans cette zone des plaines, il y aura donc lieu de considérer à la fois les nappes libres, dites phréatiques, et les nappes captives. Si le schéma de Loehnberg ne peut s’appliquer directement à toute contrée semi-
aride, il donne néanmoins des directives très utiles et très précieuses sur la façon de rechercher et d’exploiter les nappes dans les régions semi-arides.
Le run-off se classe en trois catégories suivant son volume et sa durée :
tagneuse.
partie basse de la plaine.
Conditions critiques.
Pour développer le pays le mieux possible, il faut tirer parti de toutes les ressources en eau et récupérer le maximum de terrains cultivables. En conséquence : 10 on captera toutes les ressources possibles en eau, pour les utiliser au m a x i m u m pendant la période
31

Hydrologie des régions arides
sèche; 20 on éliminera toutes les conditions marécageuses dues aux écoulements des sources, à l’émergence de la surface piézométrique des nappes, au ruissellement, aux chutes de pluie et à la remontée de la surface des nappes en hiver. Mais les conditions de recherche et d’exploitation conduisent à des contra-
dictions. 1. Les captages des sources et les installations faites à partir de celles-ci à l’aval des
vallées des montagnes peuvent devenir superflus, lorsque l’on capte ultérieurement ou au m ê m e moment les sources situées plus en amont, surtout si les sources d’aval ne sont qu’une résurgence des sources d’amont.
2. Les installations faites aux sources et à partir des sources situées en plaine, à la bordure amont du remplissage argileux épais, deviennent en partie inutiles lorsque l’on capte la nappe en amont ou en aval à l’aide de puits, annulant ou diminuant ainsi le débit de ces sources.
3. Pour contrôler les inondations, on est amené à approfondir les oueds et à redresser leurs cours. Mais, plus tard, l’exploitation de ces crues demande une diminution de la pente d’écoulement et un allongement du parcours de l’eau pour favoriser l’infiltration et réalimenter ainsi les nappes d’eau souterraine.
4. D e m ê m e que l’exploitation des sources, celle de puits situés à différents niveaux de la plaine est de nature à provoquer un abaissement de la surface piézométrique, ce qui rend nécessaires des rajustements coûteux des installations de pompage, des conduites et des réservoirs.
5. Les relations quantitatives entre l’eau fossile et l’eau cyclique souterraine ne peuvent être toujours déterminées avant les campagnes systématiques de forages et de pompages. I1 est donc très difficile, lors des phases de mise en exploitation, de calculer les débits de sécurité et les rabattements finals de la nappe. Les puits origi- nellement bons ne le seront souvent plus et les rapports entre les débits moyens optimums à la fin de la saison pluvieuse et les débits optimums à la fin de la saison sèche ne peuvent être établis que très lentement, ce qui rend la tâche plus ardue.
6. Les canaux de drainage, d’abord creusés à des profondeurs faibles ou moyennes, deviendront insuffisants lorsque, à la suite de l’exploitation des nappes, la surface de celles-ci s’abaissera.
&idemment, certaines de ces complications sont prévisibles. I1 n’en est pas moins vrai que plusieurs problèmes restent insolubles : 10 le désir d’exploiter le surplus de l’eau de ruissellement d’hiver et de l’eau souterraine d’hiver conduirait à emmagasiner. ces eaux à grand frais, dans des réservoirs où l’évaporation est très élevée en été; 20 le souci d’éliminer les conditions marécageuses dans les sols lourds, saturés non seulement en hiver, mais également en été, conduirait à retirer de l’eau de cette région pendant toute l’année et en particulier en hiver, ce qui reviendrait à un gaspillage.
L a seule solution satisfaisante serait de créer des emmagasinements d’eau dans des réservoirs souterrains.
Ordre de priorité.
Quel est maintenant l’ordre de priorité à adopter pour les travaux de recherche et d’exploitation ?
Alimentation en eau et drainage. L a priorité de l’alimentation en eau sur le drainage doit d’abord être établie. 1. Dans les contrées semi-arides, l’irrigation est le facteur primordial d’expansion de
l’agriculture. Si elle ne bénéficie que de la pluie, la plus grande partie du pays reste en friche pendant presque toute la saison sèche, tandis que sa valeur peut être multipliée grâce à l’irrigation.
32

Recherche et exploitation des nappes
2. Dans les contrées semi-arides, les demandes d’eau pour l’irrigation dépassent généralement les ressources aisément accessibles en été.
3. Toute extraction d’eau pour l’usage domestique ou pour l’agriculture n’est en s o m m e qu’un processus de drainage, dans la mesure où elle diminue l’écoulement vers les régions de sols lourds.
Le contrôle des inondations, en raison de ses nombreuses incertitudes et du fait qu’il dépend partiellement des sources normales et de l’écoulement souterrain, doit être effectué plus tard, c o m m e le drainage. L’utilisation et l’emmagasinement de l’eau des sources et de l’eau souterraine pour l’irrigation, en particulier si cette utilisation a lieu également en saison humide, abaissent le niveau des nappes, permettent une infiltration supplémentaire et diminuent de la m ê m e quantité le volume d’eau des crues.
C’est pourquoi une augmentation de l‘exploitation des sources et des eaux souter- raines représente déjà un début de contrôle des crues et de la formation des marais.
Exploitation des nappes d’eau souterraines et des sources. L a priorité de l’exploitation des eaux des nappes souterraines sur celles des sources doit être établie ensuite. En effet, les sources représentent, soit une évacuation normale de la nappe, soit un trop- plein de la couche aquifère. Le puisage optimum de l’eau de la nappe abaissera tempo- rairement, ou d‘une manière permanente, la surface de la nappe ou la surface piézo- métrique de celle-ci, ce qui aura pour effet de diminuer et même, dans certains cas extrêmes, d’annuler le débit des sources.
Mais la priorité du captage des eaux souterraines sur celui des sources ne doit pas être un principe appliqué aveuglément, car il y a lieu de tenir compte des conditions hydrogéologiques, qui sont différentes en montagne, en plaine et dans les régions intermédiaires.
L a région de montagnes ou de collines possède diverses nappes aquifères. En raison des grandes différences d’altitude entre les zones d’alimentation et les points d’éva- cuation, il y a souvent un gradient de pression élevé. D e plus, la surface de la nappe et la surface piézométrique sont à une grande profondeur. Il est donc difficile d’exploiter les nappes autrement que par captage de sources, dont les unes peuvent être des sources de trop-plein ou de débordement, et les autres des sources de déversement, c’est-à-dire de fin de nappe, une partie de l’eau pouvant provenir aussi des régions plus élevées.
Mais il faut ajouter aux observations de Loehnberg, le cas d’une plaine formée de terrains très perméables. I1 n’y a plus alors de sources de trop-plein dans la partie haute de la plaine. L a surface piézométrique de la nappe y est au contraire très pro- fonde, et cette surface piézométrique se rapproche de plus en plus de la surface du sol vers l’aval. Dès lors, le développement du captage des eaux de la nappe par puits ou forage
n’a pas à tenir compte du captage de sources qui n’existent pas ou qui, du moins, seraient alors indépendantes de la nappe.
D e m ê m e il faut envisager le cas d’une plaine pourvue de nappes profondes dont le captage peut n’avoir aucune influence sur le régime amont des eaux. Mais seule une étude hydrogéologique serrée peut permettre de l’affirmer.
Utilisation des eaux des montagnes et de l’avant-pays. L a priorité des captages et de l’utilisation des eaux appartient à l’avant-pays pour les raisons suivantes : 1. Cet avant-pays est le principal réceptacle des ressources régionales en eaux sou-
terraines. Plus bas, l’eau souterraine ne peut être captée en raison de la nature argileuse des terrains à perméabilité trop faible. Plus haut, dans les collines, il y a de l’eau souterraine, mais celle-ci rejoint les nappes de l’avant-pays, tant qu’elle ne sort pas en sources ou ne se dissipe pas dans des marais localisés.
33

Hydrologie des régions arides
2. Dans cet avant-pays, le coût des forages et des pompages est moins élevé et le
3. L’extraction de l’eau dans cet avant-pays draine directement et plus effectivement
Mais nous devons aussi considérer le cas d’une plaine perméable. Dès lors, ce n’est pas dans l’avant-pays qu’on aura intérêt à capter l’eau, mais dans lee parties plus basses où cette eau est moins profonde et o& les sols conviennent mieux à l’irrigation.
Puisque ces régions reçoivent, en partie du moins, de l’eau des nappes des régions situées en amont des collines, on peut se demander s’il y a lieu de creuser d’autres puits et forages dans les collines. On y a souvent intérêt. En effet, en déprimant les nappes, on facilite l’augmentation de l’emmagasinement souterrain des eaux souter- raines pendant les périodes pluvieuses, en évitant les écoulements de trop-plein ou de ddbordement des nappes. Cela diminue d’autant le run-off des périodes pluvieuses et par conséquent les pertes d’eau qu’on peut ainsi récupérer pendant les périodes sèches. Et on a avantage à créer les stockages souterrains dans les régions amont, plutôt qu’en aval, surtout si en aval la surface des nappes, trop proches du sol, a tendance à former des marécages.
L’ordre établi pour le captage de l’eau dans les montagnes dépend tout a fait des interdépendances des systèmes hydrogéologiques des collines et de ceux des montagnes. Le plus souvent, il y a séparation complète. L’ordre de captage des sources et des nappes dans les montagnes dépend des relations mutuelles de celles-ci.
transport de l’eau est facilité vers les régions d’utilisation.
que dans les collines l’eau des 801s saturés de la plaine.
L ’EMMAGASINEMENT DES EAUX
En saison pluvieuse, il y a un excédent d’eau que, de toute évidence, on a avantage à conserver jusqu’à la période de sécheresse pendant laquelle les besoins sont les plus importants.
I1 est certain que les réservoirs souterrains, surtout dans les régions arides ou semi- arides, sont bien supérieurs aux réservoirs aériens.
Ils empêchent les pertes par évaporation, souvent considérables dans les régions sèches (de 0,8 à 3 m sur les surfaces libres).
Ils sont le plus souvent moins coûteux, et risquent moins d’être détruits par les tremblements de terre, les guerres, etc.
Les sites de construction possibles sont plus nombreux et souvent plus étendus que pour les barrages d’eau superficielle.
Cependant, ils présentent certains désavantages Bvidents : il est plus di%icile de projeter avec précision et de construire un barrage souterrain qu’un barrage aérien. Mais, à cet égard, les progrès sont de plus en plus nombreux.
Pour reprendre l’eau du barrage souterrain, il faut dépenser de I’dnergie, alors que dans un barrage aérien, l’eau s’écoule par gravit&
34

C H A P I T R E I I I
Calcul de la perméabilité et de la transmissivité à l’aide de pompage en régime transitoire
L a détermination du débit des nappes est une des tâches essentielles de l’hydrogéologie des régions arides. Mais il est impossible de calculer ce débit si l’on ne connaît pas la perméabilité ou mieux la transmissivité des couches aquifères.
L a perméabilité, on le sait, peut être déterminée en laboratoire, à l’aide de la granu- lométrie ou, ce qui est mieux, à l’aide de perméamètres. Mais pour mesurer la perméabi- lité non d’échantillons mais de l’ensemble d’un terrain, rien ne vaut les pompages de puits ou de forages. On connaît la méthode de Dupuit-Thiem. Nous en avons également proposé une [67] que nous analysons plus loin et qui permet de calculer la perméabilité par la simple connaissance du rabattement de l’eau dans le forage, le débit correspondant et la pente de la nappe. Mais tout cela suppose que l’équilibre du nouveau régime introduit à la suite du
pompage est atteint. Or ce régime est extrêmement lent à s’établir, en particulier lorsqu’il s’agit d’une nappe libre. L a méthode par mesure en régime transitoire, inau- gurée par Theis, est bien meilleure mais malheureusement ignorée encore en bien des contrées. Nous rappellerons donc ici brièvement cette méthode qui s’applique aux nappes
captives et aussi aux nappes libres pourvu que dans ces nappes libres on ne considère pas les points situés au voisinage immédiat du puits. Nous exposerons ensuite les méthodes de Boulton et celles de Hantush, qui tiennent compte de la quantité d’eau passant à travers le toit.
MÉTHODE DE THE IS^
L a formule de non-équilibre donne le débit q constant (en m3/s) d’un puits ou d’un forage, en fonction de la transmissivité T (en m3/s) et du rabattement A (en mètres) du niveau piézométrique de la nappe au temps t (en secondes) du pompage. Ce temps est mesuré depuis l’origine du pompage à débit constant. Le rabattement A est observé à la distance R (en mètres) du puits ou du forage. Le débit q de pompage est constant. Rappelons que la transmissivité T est le produit du coefficient de perméabilité K en m3/s, par l’épaisseur E de la nappe. T = K x E. Dès lors si i est la pente de la
1. C. V. THEIS, u The relation between the lowering of the piezometric surface and the rate and duration of diacharge of a well using ground water storage n. Trans. Amcr. geophya. LI”., 161h Annual Meeting, 1935, p. 519-524. -, u The signification and nature of the cone of depression in groundwater bodies n, Econ. GsoI., vol. 33, no 8, 1938. p. 889-900. L K. WENZEL, Marhods of determining permeability of water-bearing materials. 1942. (U. S. geological mrwy, water aupply paper 887.)
35

Hydrologie des régions arides
surface piézométrique de la nappe, le débit de la nappe par mètre de front de celle-ci est Q = Ti. L’équation fondamentale est :
R2S u=- 4Tt
dans laquelle S est le coefficient d’emmagasinement. Dans le cas des nappes captives, c’est le volume d’eau libéré d’une colonne verticale de terrain ayant la hauteur A de l’aquifère et une section horizontale de 1 m2, et cela sous l’action d’une baisse de pression de 1 m d’eau. C’est le volume de l’eau libérée par la compression du terrain et l’expansion de l’eau, sous l’effet de la diminution de la pression de la nappe : il est de 1’0d1e de à lo4. Dans le cas des nappes libres, il est égal au spscijic yield, c’est-à-dire à la capacité de libre écoulement (eau de gravité). S est exprimé en fraction décimale. On a
u4 u2 u2 2.2! 3.3! 4.4!
w(u) = - 0,577 216 - lnu + u - ~ -k -- - -~ 4nt
d‘où 0,0797 T
A = _- q W(U)
UTt RZ S = 0,004 ~
Les valeurs de W(u) en fonction de U sont données par une table1 ou par un graphique B échelle log-log. On peut déterminer la transmissivité de deux manières : lo soit en observant les variations du rabattement A dans un m ê m e tube piézométrique, en fonction du temps; on construit alors un graphique de log A de ce puits, en fonction de log l/t; 20 soit en observant à un m ê m e instant t les rabattements A qui ont lieu dans une m ê m e rangée de tubes piézométriques, situés à des distances R du forage de pompage; on construit alors un graphique de log A en fonction de log Rz/t de chaque
Ces graphiques sont établis sur papier transparent à la même échelle que le graphique log W(u) - log U. On obtient ainsi, dans chacun de ces deux cas, une courbe qui n’est autre qu’une portion de la courbe log W(u) en fonction de log U. On superpose le graphique log A - log RZ/t (ou le graphique log A /log l/t) au graphique
log W(u) - log U. La superposition est possible en un seul emplacement. On choisit alors dans cette superposition une valeur de A sur la courbe de papier transparent. L a coordonnée de A se superpose à la coordonnée de W(u) et U de l’autre graphique et l’on porte A et W(u,) dans l’équation (4) d’où l’on tire T. Et en portant U et T dans l’équation (5), on en tire le coefficient d’emmagasinement
S. D’une manière générale, il vaut mieux utiliser la relation log A - log RZ/t relative aux observations faites sur plusieurs piézomètres, à un même moment, que la relation
puits.
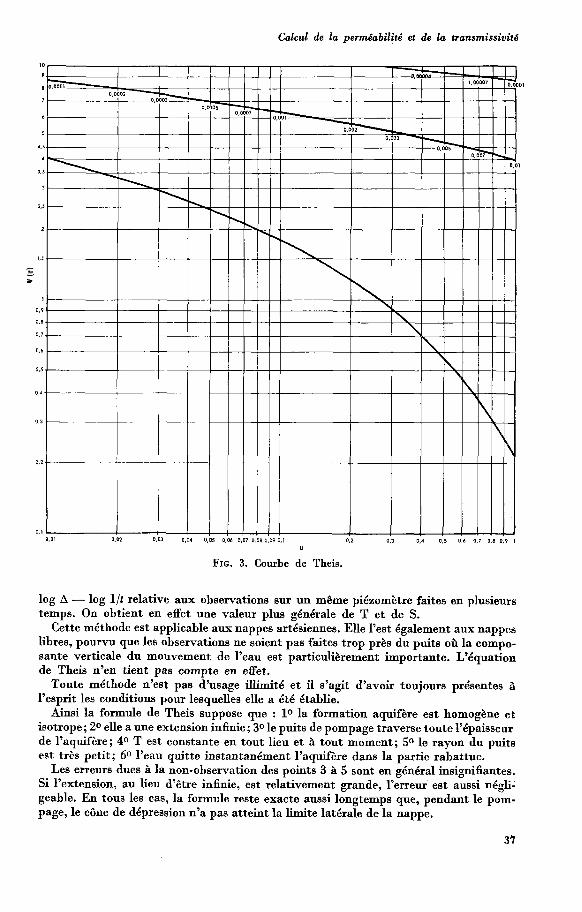
Calcul de la perméabilitB et de la transmissivité
o1
1
Lu 0.8 0.9 I
FIG. 3. Courbe de Theis.
log A - log l/t relative aux observations sur un m ê m e piézomètre faites en plusieurs temps. On obtient en effet une valeur plus générale de T et de S.
Cette méthode est applicable aux nappes artésiennes. Elle l’est également aux nappes libres, pourvu que les observations ne soient pas faites trop près du puits où la compo- sante verticale du mouvement de l’eau est particulièrement importante. L’équation de Theis n’en tient pas compte en effet. Toute méthode n’est pas d’usage illimité et il s’agit d’avoir toujours présentes à
l’esprit les conditions pour lesquelles elle a été établie. Ainsi la formule de Theis suppose que : lo la formation aquifère est homogène et
isotrope; 20 elle a une extension infinie; 30 le puits de pompage traverse toute l’épaisseur de l’aquifère; 40 T est constante en tout lieu et à tout moment; 50 le rayon du puits est très petit; 60 l’eau quitte instantanément l’aquifère dans la partie rabattue.
Les erreurs dues à la non-observation des points 3 à 5 sont en général insignifiantes. Si l’extension, au lieu d’être infinie, est relativement grande, l’erreur est aussi négli- geable. En tous les cas, la formule reste exacte aussi longtemps que, pendant le pom- page, le cône de dépression n’a pas atteint la limite latérale de la nappe.
37

Hydrologie. des régwns arides
Les erreurs proviennent surtout, dans le cas des nappes libres, de ce que le départ de l’eau du terrain prend un certain temps. L’erreur est d’autant plus forte que le terrain a plus de difficulté à s’égoutter, c’est-à-dire que les pores sont plus petits.
Une règle à calcul a été imaginée par Theis et Brown [lo21 et des nomographes établis par Remson et V a n Hylckama [98] pour faciliter les calculs rendus nécessaires par la méthode de Theis, sans avoir à se servir du graphique.
MÉTHODE APPROXIMATIVE
Lorsque l’on a pompé suffisamment longtemps, la série alternée des équations 1 et 2 devient négligeable devant le logarithme et le terme constant. D e ce fait, l’équation se simplifie et l’on peut, au bout d’un certain temps, au moins quarante-huit heures de pompage à débit constant, appliquer une méthode plus pratique. Les équations 1 et 2 s’écriront :
2,303 q 2,25Tt (6) A = --log ~
4xT R2S A 2,303 2.25Tt q 4xT R2S log ~
-~ - _
On peut alors procéder de plusieurs manières : 1. On observe les rabattements A de plusieurs tubes piézométriques à un m ê m e moment;
t est alors constant. Aussi l’équation (6) peut-elle s’écrire : 2,303 q (8) ~ - _ _ - dA
d(1og R) 2xT
0,376 (8’) d(A/q) d(1og R)
T =
On établit une courbe A/q en fonction de log R (fig. 4)’ d’oh l’on tire T. 2. On observe les rabattements A dans un seul tube en fonction du temps; R est alors
constant. Aussi l’équation (6) donne-t-elle : d A 2,303q -
d(1ogt) 4xT d’où
0,183 (9) d(Aln) d(10g t)
T=-
On établit alors une courbe de A/q en fonction de log t (fig. 5), ce qui permet de cal- culer T a l’aide de la portion rectiligne asymptotique du graphique. Ce graphique peut être appliqué au puits de pompage de rayon r.
situés B des distances R différentes. Or on a : 3. On observe les rabattements A/q de plusieurs puits A divers temps t. Ces puits sont
0,183 T = d(A1q)
d(1og R2/t)
1. H. Y. COOPER; C. E. JACOB, u A generalized graphical method for evaluating formation constants and aummariaing well- field history n, Trons. Amer. gaophys. Un., vol. 27, no 4. 1946, p. 526-534. C. E. JACOB, u Flow of ground water n, in Ronsx. Hunter snginaering hydraulics. N e w York, John Wiley and Sons, 1950. p. 321-386. E. DE GEUS [90].
38

Calcul de la perméabilité et de la transmissivite'
FIG, 4. Courbe A/q - log R
FIG. 5. Courbe A/q - log z
R2 FIG. 6. Courbe A/q - log - 1
39

Hydrologie des rigiom arides
On porte donc les points de A/q en fonction de log R2/t, pour chaque puits (fig. 6). Les points obtenus pour chaque puits à différents temps t doivent s’aligner avec les mêmes points obtenus sur les autres puits. On a ainsi un graphique synthétique donnant la moyenne des perméabilités des différents points à des moments variés. On donne évidemment aux points correspondant à un m ê m e puits un signe distinctif.
On obtient ainsi une courbe dont la portion rectiligne permet, de la m ê m e manière, de déterminer T. L a droite asymptotique coupe l’axe des temps en un point défini par :
log z - to S R2 2,25T
On en tire donc facilement : TI R2
S = 2,25 après avoir calculé T.
Cette méthode de Theis est directement applicable aux puits en nappe captive, lorsqu’on n’a pas à tenir compte du suintement par le toit. Mais, pour les puits en nappe libre, Jacob fait remarquer qu’il y a certaines restrictions. Si le rabattement au puits de pompage est de moins de 2 % de l’épaisseur de l’aquifère, les calculs précédents de S ne sont pas trop entachés d’erreur du fait du temps demandé pour que le drainage du cône puisse s’effectuer. On peut corriger les erreurs de la manière suivante. On détermine d’abord T d’après l’une des équations (8) ou (9). Puis on calcule S
pour différents temps t et différentes valeurs de A à l’aide de l’équation (6) et pour différentes valeurs de R. On obtient alors des valeurs en fonction de t. Pour avoir S on extrapole jusqu’à des t très grands. Si l’on soustrait des A observés A2/2H (H étant l’épaisseur de l’aquifère), on obtient
des valeurs plus exactes de T, et les valeurs de S sont plus proches de la réalité. On obtient des résultats tout à fait conformes si A > 0’25 H, pourvu que S soit partout le même.
MÉTHODE DE BOULTON P O U R LES PUITS EN NAPPE LIBRE
On a vu que l’intégrale exponentielle de Theis n’est pas applicable aux nappes libres, du fait qu’au voisinage des puits il y a une composante verticale dont il n’est pas tenu compte dans la formule de Theis. Cependant on peut appliquer cette formule si :
Kt Tt SH Sa2 est > 5
Boulton [87] donne une méthode qui permet de se servir de l’intégrale m ê m e pour les points situés au voisinage du puits, et d’utiliser des rabattements du puits très grands et s’approchant de l’imperméable.
1. L’aquifère est homogène et isotrope. I1 descend jusqu’à l’imperméable. I1 est
2. Le puits est parfait, c’est-à-dire qu’il descend jusqu’à l’imperméable. 3. Le coefficient d’emmagasinement est constant. 4. L’écoulement obéit à la loi de Darcy. L a perméabilité est constante. 5. L a surface piézométrique de la nappe est horizontale, avant pompage. I1 n’y a pas
6. On pompe à débit constant, depuis l’instant t = O. Boulton donna la formule suivante du rabattement :
Les conditions sont les suivantes :
d’extension latérale infinie. L’imperméable est horizontal.
d’alimentation de la nappe par la pluie au voisinage du puits.
40

Calcul de la perméabilité et de la transmissivité
CD est la pression plus le potentiel en tout point de l’aquifère saturé en eau.
@ = + z étant le poids spécifique de l’eau.
JO la fonction de Bessel de la première espèce d’ordre zéro. R la distance horizontale à partir de l’axe du puits. K le coefficient de perméabilité. z la hauteur d‘eau de la nappe, à la distance R. S le coefficient d’emmagasinement.
Y
Pour l’exécution des calculs, il est commode d’introduire les quantités sana dimen- sions :
R Kt ‘P=K et T=- SH
Et en adoptant A = SH pour variable, on a
A étant le rabattement au point R.
transmissivité T = KH, l’équation précédente devient : En désignant par V (p, t) l’intégrale définie et en introduisant le coefficient de
qui est l’équation proposée par Boulton pour le rabattement de la surface de la nappe. Quand le facteur T est assez grand, c’est-à-dire aussi lorsque t est suffisamment
grand, tanh peut être remplacé par h dans l’équation (3) sans qu’il y ait de changement appréciable de la valeur de l’intégrale. En se servant de la première intégrale de Weber on montre que :
V(~,T)=-- Ei - - -Xi : ( 4p:) ou
(5)
Ei est l’intégrale exponentielle utilisée par Theis : W(u) = - Ei(--). X, est un terme de correction petit quand 7 est grand. Ainsi l’intégrale exponen-
tielle s’applique à des nappes libres, lorsqu’on n’utilise que des temps de pompage suffisamment. grands.
L a fonction V de l’équation (4) n’a pas encore été tabulée. On trouvera ci-dessous un rudiment de tableau (voir tableau 3).
Pour de faibles valeurs de T, T < 0,05, la fonction V est donnée d’une manière approchée par l’équation.
X, étant un facteur de correction, petit quand T est lui-même petit. On a alors
P 1 T A = - ’ sinh-1 - + sinh-1 - -
2xK1 ( p P 1 = étant la hauteur d’eau dans le puits quand q = O, dans le cas d’un aquifère de hauteur illimitée.
41

Hydrologie des régions arides
Détermination numérique de V.
Pour déterminer les valeurs de V, on trouvera par quadrature les valeurs de X, pour les plus petites valeurs de 7 et les valeurs de X, pour les plus grandes valeurs de T. Les valeurs de V peuvent être déterminées à partir des équations (7) et (5)’ en se
servant des tables publiées de sinh-l z et de Ei(--). On pourra déterminer les valeurs intermédiaires de X, et X, des tableaux par inter-
polatioe linéaire. Pour V l’interpolation est plus difficile. On portera les valeurs de V dans l’équation (4) qui donnera T en fonction de A et q
d’un piézomètre.
TABLEAU 1. Valeur de X, (p, 7).
0,05 0,0150 0,0143 0,0125 0,0101 0,0077 0,0056 0,0021 0.20 0,0564 0,0541 0,0480 0,0398 0,0312 0,0234 0,0099
TABLEAU 2. Valeurs de X, (p, 7).
7 p=o.o p=o,z p=0.4 p=0,6 p=0,8 p=1.0 psl.5
l,oo 0,1810 0,1747 0,1575 0,1331 0,1057 0,0787 0,0250 5,OO 0,0344 0,0343 0,0338 0,0330 0,0320 0,0306 0,0264
TABLEAU 3. Valeurs de V(p, 7).
7 pz0.2 p=0.4 p=0,6 p=o,a p= 1.0 pel.5
0,05 0,214 0,092 0,051 0,032 0,021 0,008 0,20 0,756 0,358 0,207 0,132 0,088 0,835 l,oo 1,844 0,183 0,826 0,599 0,443 0,220 5,OO 2,785 2,096 1,696 1,416 1,203 0,832
On voit, d’après le tableau 1, que, pour des valeurs de T = 0,05, l’erreur de calcul de A ne dépasse pas 6 cm lorsque qj2nT = 10, si 1’011 néglige X,. Le tableau 2 montre que, pour une valeur de T = 5, l’erreur de calcul de A commise
en utilisant l’intégrale exponentielle à la place de V ne dépasse pas 3 %. En conclusion, pour calculer les rabattements A, on se servira de l’équation (4) :
1.
2.
3.
42
Lorsqu’on a des valeurs de T < 0’05, on calcule V (p, T) à l’aide de l’équation :
Mais on ajoutera au rabattement A de (4) les corrections données par le graphique de Boulton (voir fig. 7). Lorsquel’on a des valeurs de 0’05 Q T < 5, on calcule V(p, T) à l’aide du tableau 3, et l’on porte ces valeurs dans l’équation (4). Lorsque l’on a des valeurs de T > 5, on calcule V (p, 7) à l’aide de l’équation :
V(p, 7) = - - Ei - ( 4:) que l’on porte dans l’équation (4). Mais il faut corriger A en y ajoutant les valeure données par le graphique de Boulton (voir fig. 8).
Calcul de la permdabilité et de la transmissivit6
O. 3 O, 5 p (dchelle logarithmique)
FIG. 7. Courbes de Boulton pour la correction du rabattement de la surface libre quand T < 0.05.
Type de TW h, point h. h:
x 0.0066 0.424 O 0.0252 0.403
0,50 1.00 3.00 p (&helle logarithmique)
FIG. 8. Courbe de Boulton pour la correction du rabattement de la surface libre quand T > 5.
43

Hydrologie des r6gwns arides
I1 convient de signaler que, par cette méthode, la détermination de la transmissivité n’est pas aisée.
Calcul du niveau de pompage dans le puits.
Le calcul du niveau dans le puits de pompage lui-même doit être fait à l’aide de la formule :
r p. =
r étant le rayon du puits, H l’épaisseur de l’aquifère, m une valeur à choisir dans le tableau 4 en fonction des différentes valeurs de 7
TABLEAU 4.
7 m
0,05 l,oo l,oo 5,OO
- 0,043 + 0,087 + 0,512 + 1,228
MÉTHODES T E N A N T COMPTE DU DÉBIT RETARDÉ, PROVENANT DU (( SPECIFIC YIELD )) D A N S LE CAS DES NAPPES LIBRES, O U D E LA DRAINANCE D A N S LE CAS DES NAPPES CAPTIVES
Dans les formules précédentes, il n’est pas tenu compte du débit retardé qui provient de l’égouttement originaire de la partie du terrain situé au-dessus du cône de dépression lorsque 1,011 pompe dans les nappes libres. I1 n’est pas non plus tenu compte, dans le cas des nappes captives, de l’eau qui traverse le toit ou le m u r de l’aquifère par suite de la chute de pression autour du forage.
Certes, dans le cas des nappes libres, ce débit retardé n’est pas un facteur important lorsque l’aquifère est formé de sables et graviers dans lesquels l’égouttement à lieu très rapidement. D’apri% Boulton il peut être négligé pour des sables dont les grains ont de 0,15 à 0,85 mm et à plus forte raison pour des sables aux grains plus gros.
Méthode de Boulton.
Nous devons à Boulton [88] une première étude. Boulton a considéré deux cas.
1. Un aquifère formé de sable grossier repose sur une couche horizontale impermdable. Mais, au-dessus du sable grossier, s’étend une couche de sable très fin et de limon. U n puits traverse complètement l’aquifère. Lorsqu’on pompe, le cône de dépression
reste dans la couche de sable fin et de limon, sans descendre dans les sables grossiers. On voit qu’un tel cas est exactement celui de nombreuses nappes d’eau dans les
alluvions récentes des fleuves et rivières. C o m m e la quantité d’eau qui passe de la conche supérieure de sable fin et de
limon dans l’aquifère à travers l’aire du cône de dépression est très faible, surtout
44

Calcul de la perméabilité et de la transmissivité
si le puits est tube ou cimenté à la traversée de la couche supérieure, on peut utiliser la formule du débit des puits en nappe captive. I1 faudra tenir compte dans le débit du puits de l’écoulement retardé provenant du toit semi-perméable à la suite de la baisse de pression existant dans le domaine du cône.
2. I1 s’agit d’un aquifère, formé par exemple de sable grossier, compris entre un toit et un mur, imperméables mais compressibles et d’épaisseur constante, de sables fins ou limons, donnant alors un débit retardé. Le puits traverse completement l’aquifère, dont ]la nappe reste captive m ê m e
pendant le pompage. Boulton montre que l’équation différentielle établissant la relation entre le rabat-
tement, la distance et le temps est :
JT oh A est le rabattement à Ia distance R; t le temps compté depuis le début du pompage; T le temps compté depuis le commencement du débit retardé;
c=cr -;
T S
S’ T
a = - ;
T représente la transmissivité; S le coefficient d’emmagasinement ; S’ le débit retardé par unité de surface et par unité de rabattement.
L’intégration de l’équation (1) donne :
où ac s + S’ ? = I + - = - U S
et JO se rapporte à la fonction de Bessel de la première espèce d’ordre zéro.
de l’intégrale exponentielle bien connue Lorsque R est suffisamment petit, on peut obtenir une bonne approximation à l’aide
Q 4xT A1 -A = - [In? + Ei(- rt) - Ei(- qat)]
soit
(3)
utilisable lorsque l’on a les tables de Ei(-%) - Inx. L a quantité Al - A eat la 45

Hydrologie des régions arides
f~
~~
E~
~$
~~
~$~~~~~~~~ j
0000000000 0000000000
< ..............................

Calcul de la perméabilité et de la
transmissivit6
..
..
...................................................

Hydrologie des régwns arides
correction à déduire du rabattement Al donné par la formule de Theis, utilisant T et S. On obtient alors le rabattement réel A, tenant compte du débit retardé.
s+ S’ ?=s Pour résoudre l’équation, on commence par se servir de la formule de Theis qui donne la transmissivité T et S. Mais avec la formule de Theis, les valeurs de S peuvent varier d’un point à un autre,
ou m ê m e en fonction du temps, du fait que cette la formule ne tient pas compte de la drainance S’. On retient de la formule de Theis le coefficient de transmissivité qui en général
varie peu d’un point à un autre. On se sert du coe5cient obtenu par pompage prolongé, qu’on porte dans les équations (3) et (4) ci-dessus. Puis on donne à CL et -q des valeurs d’essai qu’on porte dans les équations (3) et (4),
jusqu’à ce que les valeurs de A ainsi calculées correspondent aux valeurs de A observées pour le différents temps.
S’ S
S’ c = u -
T
- Avec -q on obtient ainsi :
Avec a on calcule :
Mais, avant d’accepter les valeurs de GL, 3 et a, on doit les introduire dans l’équation (2) et on calcule l’intégrale en se servant des données des plus grandes valeurs de R où un rabattement a été constaté.
L a méthode de Boulton donne ainsi le drainance S’.
M6thode de Hantush.
Les études concernant l’apport d’eau par drainance au cours de pompage ont été poursuivies par Hantush [94].
Supposons une nappe pouvant être considérée c o m m e ayant une extension inhie, une épaisseur constante, et située dans un aquifère parfaitement élastique. Provenant du toit ou du m u r semi-imperméable, il se produit une drainance proportionnelle au rabattement. U n puits prend l’eau à débit constant dans cet aquifère; Hantush et Jacob [92,93]
ont donné les équations des rabattements en régime non permanent :
A=-W U,- 4 L ( .”)
dans lesquelles : R2S 4Tt
R2 Tt
u = -
q = - - - - 4B2u SB2

Calcul de la perméabilité et de la transmissivité
A étant le rabattement; q le débit du puits; T la transmissivité; S le coefficient d’emmagasinement ; B = a / ~ b ’ / ~ ’ , le facteur de drainance; M’ et b’ les conductivités hydrauliques et l’épaissenr de la couche semi-imperméable, à travers laquelle la drainance a lieu;
R les distances à partir de l‘axe du foragc; t ]le temps compté à partir du début du pompage, ou, s’il s’agit d’une remontée, à partir du début de I’arret du pompage;
KO la fonction modifiée de Bessel de seconde espèce et d’ordre zéro; W (u, R/B) peut etre appelé la fonction du puits pour le système drainant. La constante relative à la drainance, c’est-à-dire au volume d’eau qui traverse une unité de surface du toit ou du mur de l’aquifère pour une différence de pression d’une unité entre l’aquifère et le semi-imperméable, peut-&tre représentée par :
Considérons le débit du puits et les rabattements en régime permanent, c’est-à-dire d’équilibre. I1 suat de donner 2 t des valeurs tendant à l’infini. L’équation (lb) s’établit alors ainsi :
A, exprimant le rabattement maximum.
Cas de régime permanent. Le chne de &pression a cess6 de s’approfondir et de s’étendre. Le régime du puits et de la nappe est alors en équilibre dynamique. On porte les données du pompage sur un papier semi-logarithmique, les différentes distances, R, sur l’échelle logarithmique en abscisse et les rabattements A correspondant à chacune des distances R en ordonnée arithmétique (voir fig. 9). La plupart des points doivent alors se situer le long d’une droite. On calcule ensuite la transmissivité T et le facteur de drainance B de la manière suivante. Pour trouver T, on porte dans l’équation
l’inclinaison dA/d loglOR de la droite la plus satisfaisante passant par les points du graphique. Pour trouver B, on choisit un point quelconque sur cette droite. On en porte les coordonnées A, soit A, et R, dans l’équation
loglo (,,,, $) A, z ~ 2,303 q 2xT (5)
d’où l’on tire B. ou bien l’on prolonge la droite jusqu’à R, de coordonnée A = O et on tire B de la relation B = 0,89 Ra. La drainance
T I<‘ y=-=- B2 b’
peut être alors calculée.
49

Hydrologie des régions arides
Cas du regime non permanent (dit transitoire). Régime pendant lequel le rabattement du cône de dépression continue à crottre. Les calculs peuvent être conduits d’après les observations faites soit à l’aide d’un seul piézomètre à des moments dsérents, soit 1 l’aide de plusieurs au m ê m e moment.
1. Observations faites ci l’aide d’un seul pi&zomJtre. La méthode ne convient que lorsqu’il est possible d’extrapoler les valeurs de A, de manière 1 obtenir la valeur A,,, maximum. On reporte sur un graphique les différentes valeurs du rabattement A observées
aux divers temps t, A sur une échelle arithmétique et t sur une échelle logarithmique. Et l’on construit la courbe (voir fig. 10). L a courbe présente un point d’inflexion dont la coordonnée Ai est égale à :
L a pente mi de la courbe en ce point d’inflexion est égale à :
Et B ce m ê m e point : R2S R
’ 4Tti 2B u . = - - = -
fi étant le temps correspondant. Enfin la relation entre le rabattement Ai et la pente mi au point d’inflexion est :
a) On calcule R/B l’aide de l’équation (9). Pour cela : lo on calcule d’abord Ai; on l’obtient en calculant la valeur du rabatte- ment maximum Am par extrapolation de la courbe; on a en effet :
ou bien on détermine sur le graphique la valeur de Ai au point d’infiexion; 20 on en profite pour noter sur le graphique le temps ti correspondant ; 30 on calcule la pente mi de la courbe au point d’inflexion; 40 on porte Ai et mi dans l’équation (9) ci-dessus. On calcule alors R/B à l’aide du tableau de la fonction :
6) On calcule B à l’aide de R/B et R. c) On calcule T. Pour cela on reporte les valeurs de q, Ai, mi et R/B dans l’une
des deux équations : 2,303 q (-
m( = -e 4 xT
A(= 2 4 xT KO(:)
d) On calcule S en portant T, tir R et R/B dans l’équation (8) qui s’écrit également :
50
Calcul de la permbbilité et de la transmissivire
A
1
FIG. 9. Graphique A - log R. Régime permanent.
FIG. 10. Graphique A - log t. Régime non permanent. Un seul piézomhtre.
FIG. 11. Graphique A - log t. Régime non permanent. Plusieurs piézomhtres.
logt
FIG. 12. Graphique R - log mi .
51

Hydrologie des régions arides
e) On calcule la drainance S’ : K T sr = = - b’ B2
En reportant les diverses valeurs ainsi calculées dans l‘équation (l), ou devrait obtenir les rabattements A observés sur le terrain. Mais cela n’est pas toujours le cas. On porte les coefficients calculés plus haut dans l‘équation (1). Quelquefois la valeur de A, est mal extrapolée. 11 faut alors l’ajuster ainsi que mi.
2. Observations faites à l’aide de plusieurs piézomètres. On peut d’abord faire sur chacun des piézomètres les observations et calculs du paragraphe précédent. On obtient ainsi pour chaque piézomètre des valeurs de T, B, S, K’lb’. Et l’on prend les valeurs les plus convenables. Cette méthode exige naturellement qu’on puisse obtenir par extrapolation les
rabattements maximum A . Avec la méthode suivante ce n’est pas nécessaire. I1 suffit que la courbe semi-loga-
rithmique A - log t présente une portion suffisamment développée de la partie rectiligne (voir fig. 11) correspondant au point d’inflexion. I1 faut au moins deux piézo- niètres d‘observation. On procède alors de la manière suivante : a) Pour chaque piézomètre on construit la courbe A - log t sur un même papier semi-
logarithmique. b) On calcule a3 =
-~ Tb’/M’ à l’aide de la relation
B = 0,434 [dR/d(loglo mi)] (10
et cela pour chacun des piézomètres. A cet effet : lo on calcule la pente mi de la portion rectiligne des courbes A -log t
de chacun des piézomètres; 20 sur papier semi-logarithmique on construit une courbe en portant en abscisse logarithmique les points mi et en ordonnée arithmétique les distances R des piézomètres correspondants (voir fig. 12). On doit obtenir une droite, car on a :
(11) 2,303 q R = 2,303 B loglo - - loglo mi [ 4xT
on calcule la pente dR/d(log,, mi) qu’on porte dans l’hquation (40) ci-dessus. c) On calcule la transmissivité T à l’aide de la relation
2,303 q (13) T=- 4xmi,
mio est la valeur de mi lorsque R = O; on détermine cette valeur à l’aide du graphique log mi - R en prolongeant la droite jusqu’à R = O.
d) On calcule la drainance S’ : K’ T y = - = - b‘ B2
e) Calcul du coefficient d’emmagasinement S; on utilise la relation (8) qui donne :
I1 faut donc encore calculer ti.
l’équation (6) : Pour chaque piézomètre, on calcule A, à l’aide de q, T, R, B, que l’on porte dans
Ai = 4xT KO(:)
On reporte ensuite Ai sur le graphique du piézomètre correspondant, ce qui permet d’obtenir ti. On a ainsi des valeurs de S pour chaque piézomètre et l’on prend la moyenne.
52

C H A P I T R E I V
Géochimie es eaux souterraines
L a géochimie des eaux souterraines a une importance considérable en hydrogéologie, particulièrenient dans les régions arides. Le tout n’est pas de trouver Ileau. Encore faut-d que cette eau soit utilisable. Or,
dans les régions arides, les eaux souterraines sont très fréquemment salées. Rien souvent le facteur de contr8le n’est plus alors la quantité, mais la qualité.
Pl importe donc de connaître les lois de la géochimie des eaux souterraines, per- mettant d’éclairer la composition chimique et les causes de la salure des eaux. D e plus, cette géochimie donne de précieux renseignements sur les conditions de
gisement et le mouvement des eaux souterraines. Certes la composition chimique des eaux a fait l’objet d’abondantes études. Mais
peu sont consacrées aux phénomènes géochimiques en eux-mêmes. Nous ne po~voiis donner ici qu’un aperçu général en nous contentant d’aborder
Iles problèmes. La principale question est de savoir comment les eaux souterraines acquièrent leur
composition chimique, puis comment celle-ci peut évoluer. Pl est certain que la premihre transformation a lieu dans le sol cultivable qui n’est
lui-même qu’un résidu de l’attaque des roches par les eaux d’infiltration. Au cours de l’infiltration dans la zone d’aération, peu de modifications ont l’occasion
d’intervenir. C’est un lieu de passage rapide. Dans les nappes, au contraire, le cheminement de l’eau est le plus souvent très lent,
de telle sorte que les réactions entre l’eau et la roche ont le temps de s’accomplir plus complètement. Des phénomènes tels que les échanges de bases et les concentrations peuvent inter-
venir secondairement et donner aux eaux des caractéristiques tout à fait différentes.
M I S E EN SOLUTION^ [164]
La composition chimique des eaux souterraines résulte en premier lien de l’attaque de la partie supérieure de la roche mère des sols cultivables, dont la zone d’altération, plus 03 moins épaisse suivant les terrains géologiques et les climats, se rhduit, dans certains cas, à une simple surface de corrosion. Mais les phénomènes de mise en solution sont complexes, car, dans la zone d’évapotranspiration, la descente de l’eau vers la nappe souterraine est la résultante dcs phénomènes d’infiltration et d’évapotranspi-
1. W. SCIIOELLER, Cours d’itydro&logie, Paris, Inrtitut iianpis du p6trole. 1949, 1 vol., 364 p3ger.
55

Hydrologie des régions arides
ration et, par conséquent, la composition chimique des eaux d’infiltration qui gagnent les nappes est aussi la résultante de phénomènes d’attaque chimique, de dissolution d’une part, et de précipitation d’autre part.
Sous les climats tempérés, le sol présente surtout des phénomènes d’attaque dans la zone éluviale A et des phénomènes d’attaque, mais aussi de précipitation, dans la zone illuviale B. Il faut bien que l’attaque principale ait lieu dans la zone d’altération et que les précipitations dans la zone B ne représentent qu’un phénomène transitoire, dans l’équilibre dynamique, si nous considérons la permanence des phénomènes.
Dans les régions arides et les régions désertiques, il y a toujours attaque principale à la partie supérieure de la roche mère. Mais les phénomènes transitoires de précipita- tion dans le sol sont beaucoup plus importants par suite de l’évapotranspiration plus intense et de la pluviométrie plus faible.
Cependant, dans les régions arides et plus particulièrement dans les déserts, l’aridité peut être telle que sur certains terrains, c o m m e les calcaires massifs, la végétation ne puisse pousser et engendrer du sol.
L’attaque de la roche mère se trouve alors limitée par la quasi-absence de l’a- gent principal d’attaque, le gaz carbonique produit dans et par le sol agricole.
Dès qu’elles pénètrent dans le sol, les eaux dissolvent les gaz N,, O,, H,, He, CO,, NH,. Lorsqu’on étudie la solubilité de ces gaz, on constate : 10 que la plupart des gaz les plus communs en contact avec l’eau, N,, O,, H,, He, ont une solubilité. 21 peu près égale, ne variant que du simple au double ; 20 que d’autres gaz très fréquents, CO,, H,S, NH,, ont une solubilité 40 à 200 fois plus grande, voire 60 O00 fois pour NH,.
Dans le sol et dans la zone d’altération de la roche mère, les eaux peuvent déjà dissoudre certains éléments de roches, le NaCl, le gypse Caso4 2H,O, l’anhydrite CaSO,, le calcaire CaCO,, les dolomies et calcaires dolomitiques (CaMg)CO,, etc. Mais toutes ces substances ne sont pas également solubles. Leurs coefficients de solu- bilité sont les suivants :
g/kg de solugion a IO0 C g/kg de solurion a 100 C
0,014 071
753 107
1,926
236
394 349 263
82,5
En réalité, la mise en solution dans le sol et dans la zone d’altération est essen- tiellement due à des attaques chimiques, dans lesquelles entrent en ligne de compte les phénomènes suivants : 1. Hydratation, par exemple de la biotite, de l’anhydrite, de l’oligiste; 2. Hydrolyse, en particulier dans les silicates; 3. Oxydation des sulfures, des oxydes incomplètement pourvus d’oxygène, ferreux,
manganeux, etc.; il est à remarquer que l’oxydation des sulfures produit de l’acide sulfurique, puissant agent d’attaque.
L’oxydation est m a x i m u m au voisinage de la source de l’oxygène, l’air atmo- sphérique, c’est-&-dire dans la partie supérieure du sol. Le plus souvent l’oxygène ne peut pénétrer tres profondément, car il est consommé au fur et à mesure de son trajet descrndant, de telle sorte que les roches peuvent conserver leur pouvoir réducteur.
4. L’attaque chimique. Le gaz carbonique est certainement le facteur le plus im- portant de l’attaque des minéraux. Une très petite partie du CO, provient de celui de l’air au-dessus du sol dissous par l’eau de pluie. L a tension de 0,0003 du CO, de l’atmosphère ne permet qu’une dissolution de 50 mg de C0,Ca. En réalité, la quasi-totalité du CO, a son origine dans l’air du sol où la tension de ce gaz est de 0,001 à 0,Ol. I1 provient de la combustion biologique et chimique des matières orga-
54

Géochimie des eaux souterraines
niques du sol, de la respiration des racines des végétaux et des divers organismes qui vivent dans le sol cultivable. A ce gaz carbonique s’ajoute celui qui résulte de l’attaque par les acides organiques du sol, par l’acide sulfurique provenant de l’oxyda- tion des sulfures et parl’acide nitrique forméau cours des phénomènesde nitrification.
Dans les eaux souterraines normales, les teneurs en HCO, varient entre 180 et 550 mg/l, dépassant rarement 600 mg/l. Les valeurs les plus courantes sont comprises entre 180 et 360 mg, correspondant donc à des tensions en CO, semblables à celle des sols agri- coles. C’est ce gaz carbonique qui est l’agent actif de l’altération des roches, sans qu’on
puisse négliger l’acide sulfurique, l’acide nitrique et les acides organiques précédemment mentionnés.
L’attaque du calcaire peut se réduire à l’équation suivante :
k, klk,
[H,CO,] = - [HCOJ2[Ca++]
dans lesquelles 6 represente la tension du CO,; a le coe5cient de dissolution du CO, libre dans l’eau; [HsCOJ la concentration en mols de CO, libre dissous dans l’eau, 6quilibrant la dissolution de [HCO;] et de [Ca++]; [HCO;], [Ca] la concentration en mols; k,, k,, k, les constantes de première et de deuxième dissociation de l’acide carbonique, et le produit de solubilité du CaCO,
L’attaque des silicates est elle aussi, en définitive, une action plue ou moins directe du gaz carbonique. I1 y a certes, au début, une mise en solution vraie des silicates (Correm). Mais l’attaque
dépend surtout de l’acidité ou de la forte alcalinité de l’eau. Or l’acidité de l’eau est essentiellement due au gaz carbonique. Pour l’orthose on aurait :
8 H,O + K [AISi30,J + K+ + 3Si4+ + AIJ+ + 16 OH- Ala++ 3OH- + Al(OH), Si4+ + 3 OH- 3 SiO, + 3 H+ SiOs=+ H,O + SiO, + 2 OH-
L’aluminium et la silice réagissent ensuite pour donner en milieu acide de la kaolinite, en milieu basique de la montmorillonite ou des illites.
Les feldspaths mettent ainsi en solution des ions K, Na, Ca et de la silice molé- culaire, mais peu de silice colloïdale.
Les minéraux ferromagnésiens apportent aussi les éléments précédents, mais avec en plus Mg et Fe. C o m m e les silicates mettent en solution infiniment moins de C1 que d’alcalins, on voit qu’il existe un déséquilibre chlore-alcalin d’origine, caractéristique des eaux issues des roches cristallines et cristallophylliennes, qu’il ne faut pas confondre avec le déséquilibre dû à des échanges de bases.
E A U X D E S D I F F É R E N T S T E R R A I N S
L a composition chimique dépend ainsi de la nature des terrains. I. H. SCEIOE~X, a L’id3uence du climat sur la composition chimique des eaux souterraines vadoses B, Bull. Soc. g6d. Fr. (5).
t. II, 1941. p. 267-289; et Cours d’hydrogéologie, Paris, Institut franqaiu du p6trole. 1949, 1 vol.. 364 pages.
55

Hydrologie des régions arides
Terrains calcaires.
Le CO, dissous est rapidement saturé en C0,Ca. Mais, comme nous l’avons vu, le gaz carbonique du sol a une tension ne variant que fort peu. En conséquence, la teneur en COSH et Ca correspondant de l’eau n’oscille qu’entre les limites étroites que nous avons indiquées plus haut. L a dissolution du carbonate de calcium des calcaires se trouvera donc limitée. Mais
celle des autres sels contenus dans les calcaires dépend de la nature de ces roches. D’une manière générale, la circulation par fissures, même si elle n’exclut pas d’autres
modes de circulation, est prédominante dans les calcaires. La surface d’attaque par rapport au volume d’eau qui circule se trouve ainsi considérablement réduite. D e plus, les sels les plus solubles, les chlorures, les sulfates, sont emprisonnés dans la
matière calcaire. Le manque de pores dans les calcaires compacts et dans les calcaires cristallins ne permet pas à l’eau d’aller les chercher en profondeur, tout au plus à quelques décimètres lorsque le calcaire subit une altération. I1 n’en va évidemment pas de même dans les calcaires très poreux. Mais, dans l’un
comme dans l’autre cas, ces sels ne peuvent être mis en liberté que par dissolution préalable des carbonates qui les emprisonnent. Aussi la composition chimique des eaux doit-elle avoir tendance à refléter la composition chimique des calcaires. Comme ceux-ci sont, d’une manière générale, pauvres en chlorures et en sulfates, les eaux des calcaires seront surtout riches en carbonates et pauvres en chlorures et en sulfates ; un faible résidu sec en sera une autre conséquence. Dans les dolomies les phénomènes sont du même genre que dans les calcaires. Mais,
d’une manière génbrale, il semble que le rapport rMg/rCa* soit plus petit dans l’eau que dans les roches, en particulier dans les calcaires dolomitiques. La calcite est, en effet, plus soluble que la dolomite.
Gypse et terrains salifhres.
Les eaux ~e chargent rapidement en sels non pas à cause de l’étendue ou de la durée du contact eau-roche, mais en raison de la grande solubilit6 du gypse et de la grande teneur en autres sels très solubles, amenant une mise en solution rapide. Les eaux traversant le gypse atteignent très rapidement de hautes teneurs en
Caso,, allant très souvent jusqu’à saturation. D e même, les eaux des terrains salifères contiennent une très grande quantité de chlorures. I1 est à remarquer que l’augmentation du SO, entraîne non seulement une augmen-
tation du calcium, mais aussi du magnésium, le gypse renfermant toujours une propor- tion plus ou moins grande de Mg. D’ailleurs, une fois la saturation de CaS0, atteinte, Ca ne peut plus augmenter. Seul MgSO, peut encore se dissoudre. Les eaux arrivent ainsi à présenter des teneurs extrêmement élevées en SO, et C1, en
Ca, Mg et Na, conduisant à des résidus secs qui peuvent atteindre plus de 200 gjl. I1 est à remarquer que la concentration du CO, combiné reste voisine de la normale
et se tient même plutôt en dessous qu’au-dessus lorsque SO, est élevé par suite du jeu du produit de solubilité S0,Ca. Enfin, la solubilité du sulfate de chaux augmentant avec la teneur en chlorures, les
eaux chlorurées sodiques peuvent avoir une teneur en SO, et Ca plus élevée que les eaux seulement sulfatées calciques. Le rapport sMg/rCa a alors tendance à croître.
Eaux en contact avec des marnes et des argiles.
Les eaux souterraines peuvent être en contact avec des marnes, des argiles ou bien des shales (mamolites, argilitea). Ces roches ont souvent une très grande porosité,
Le symbole I indique que 1’616ment devant lequel il est placé est exprime en milliéquivalents
56

Géochimie des eaux souterraines
dépassant parfois 50 yo. Leurs pores sont très ténus, donc aussi très abondants. La surface de contact eau-roche est extrêmement grande. D e plus, la vitesse de circulation est excessivement faible à l’intérieur de ces roches considérées comme imperméables. Enfin les marnes et les argiles ont pu retenir par adsorption, comme en témoignent les analyses chimiques, une quantité notable de sels (chlorures, sulfates), grâce à la nature colloïdale d’une partie de leurs constituants et à la finesse du grain de la roche. Cette rétention s’est faite, soit par adsorption lors de la précipitation des sédiments, soit par emprisonnement d’eau de mer, une fois ces argiles et marnes précipitées. Ces sédiments pouvaient emprisonner plus de 50 yo d’eau de mer rapportée à leur volume. Aussi, les eaux en contact avec les roches argileuses sont-elles chargées en sels, avec très souvent des résidus secs de plusieurs grammes. Le CO, combiné garde sa valeur ordinaire. Mais les teneurs en SO, et C1 sont supé-
rieures à celles des eaux des autres terrains, exception faite des terrains gypseux et salifères. Généralement, elles dépassent la concentration en HCO;. Tantôt le SO, l’emporte sur le CI, tantôt c’est l’inverse. Naturellement, les hautes valeurs de SO, amènent de hautes valeurs de Ca et Mg et les hautes valeurs de C1, de hautes valeurs de Na. Les échanges de bases sont très fréquents au contact des roches argileuses, les argiles
sodiques échangeant des ions N a contre des ions Ca et Mg de l’eau, les argiles calciques échangeant leurs ions Ca contre des ions N a et Mg. La teneur en SiQ, est plus grande que dans les autres eaux.
Eaux des sables et grès normaux.
Les sables et beaucoup de grès ont de nombreux piores dkveloppant une très grande surface d’attaque du terrain, ce qui permek, par conséquent, la mise en solution rapide d’une grande quantité de cations et d’anions. Et cette quantité est d’autant plus élevée que l’eau, par suite de sa circulation beaucoup plus lente, se trouve plus longtemps en contact avec les terrains. I1 en résulte que les eaux des sables et des grès sont nor- malement plus chargées en sels (SO,, Cl, Na, Mg, Ca) que celles des calcaires. La teneur en CO, est celle de toutes les eaux de la même région climatique.
Eaux des sables et des grès purement siliceux.
Ici, puisqu’il n’y a guère que du quartz, l’eau ne pourra se charger que de très peu d’éléments, même dans les régions arides. Le CO, dissous, le m ê m e qu’ailleurs, donne certes un certain nombre d’ions HCO, et
CO, par dissolution. Mais ceux-ci ne sont pas augmentés de ceux que fournirait la dissolution des carbonates, absents de ces roches. La teneur en ions HCO, est donc faible, quelques milligrammes à quelques dizaines de milligrammes seulement. Comme il n’y a que très peu d’ions Ca et Mg, tout l’acide carbonique d’dquilibre n’est pas saturé. I1 reste, par conséquent, du CO, agressif. Enfin la pauvreté en ions HCO, donne des pH faibles, de l’ordre de 6 et 5. La teneur en C1 et en SO,, quoique souvent plus faible que dans les eaux des terrains calcaires - il n’y en a que quelques dizaines de milli- grammes au plus - peut devenir égale k la teneur en HCO,, ou même, et c’est le cas le plus fréquent, la surpasser en raison de la faiblesse de HCO,. Ca et Mg sont faibles; N a peut dépasser Ca.
Eaux en contact avec de5 matières organiques.
Les eaux peuvent être en contact avec des matières organiques, telles quc des tourbes, des lignites, des charbons, des hydrocarbures, qui constituent un milieu réducteur et provoquent, par l’intermédiaire de bactéries, une réduction de sulfates. I1 en résulte :
57

Hydrologie des régions arides
10 un abaissement de la teneur en SO, et par conséquent aussi de rSO$rCl, par rapport aux eaux de m ê m e origine, mais n’ayant pas été en contact avec les matières organiques; 20 une production d’H,S; 30 une élévation de la teneur en CO, combiné.
Cette dernière provient de la production importante de CO, libre par suite des com- bustions chimiques et biologiques de la matière organique.
Les eaux circulant dans les roches cristallines et cristallophylliennes sont toutes différentes de celles des roches sédimentaires. L a mise en solution de ces roches est en effet extrêmement difficile, incomparablement plus faible que celle des principales roches sédimentaires.
Granites et gneiss.
L a décomposition des minéraux apportera : quartz, SiO, - (traces); orthose, SiO, + K; plagioclases, SiO, + N a + + Ca++; biotite, K + Fe; muscavite, K.
Les eaux renfermeront donc une grande quantité de silice, d’alcalins, de calcium et d’ions HCO, provenant du CO, de l’atmosphère du sol. L a proportion alcalin-calcium dépendra de la teneur en calcium des plagioclases et de leur quantité relative par rapport à l’orthose. Dans les granites ordinaires, le rapport
K,O + Na,O Ca0
est > 3 par conséquent élevé, ce qui rend les eaux surtout alcalines. L’attaque du granite par les eaux est toujours difficile. Autrement dit, les eaux issues du granite seront très peu chargées en divers ions et présenteront toujours un excès de CO,, puisque ce dernier ne sera pas contrebalancé par des bases. Ces eaux seront donc très acides à l’origine, mais pourront, par départ du gaz carbonique, devenir alcalines, les alcalis prédominant. C o m m e pour toutes les roches éruptives, le résidu sec est faible, par suite de la lenteur des phénomènes d’attaque. Lors de la carbonatation des granites, le départ de beaucoup le plus important de
la roche est celui de la silice. Mais celle-ci ne peut se maintenir dans les eaux qu’en quantité relativement faible (10 à 40 mg). D e m ê m e le fer, dont le départ de la roche est souvent le plus important après celui de la silice, ne peut rester en solution qu’en une quantité restreinte, qui est fonction de l’acidité, de la teneur en CO, et du potentiel d’oxydo-réduction de l‘eau. Il n’y en a donc que quelques dizièmesde milligrammes à quelques milligrammes par litre.
Les alcalins par contre, qui sont éliminés de Ia roche dans une proportion voisine ou parfois m ê m e supérieure à celle du fer, pourront être mis en solution en plus grande quantité. L a teneur des eaux en alcalins est fonction de la vitesse de carbonatation. Elle est par conséquent très faible, inférieure à 2 milliéquivalents. Le N a prédomine sur le K retenu par adsorption dans les produits de décomposition, malgré la prédo- minance du K dans les roches.
Les alcalins terreux sont éliminés des roches granitiques en moins grande quantité que les alcalins, sauf dans les granites à amphibole. Ca et Mg ont donc tendance à être en moins grande quantité que Na. Mais la différence est faible, rMg/rCa < 1.
Les ions C1 et SO, sont toujours en petite quantité : moins de 2 milliéquivalents. L a teneur en HCO, reste faible. Le pH est bas comme pour toutes les roches éruptives. Les eaux sont en général agressives. I1 y a, d’une manière très générale, plus de rNa que de rC1, déséquilibre d’origine
et non dû à un échange de bases.
Basaltes.
La décomposition des basaltes amène un départ moins important de silice que dans 58

Géochimie des eaux souterraines
les granites. Cependant les eaux des basaltes sont, d’une manière générale, plus riches en silice que les eaux des granites, en moyenne 20 à 30 mg/l. Si les basaltes éliminent plus de fer que les granites, leurs eaux n’en contiennent pas plus que les autres eaux dans les mêmes conditions de p H , de [HCO,], de potentiel d’oxydo-réduction.
Ici, par suite du grand départ de Ca et de M g des roches, Ca et Mg prédo- minent dans les eaux. Mais il n’y a guère que 3 milliéquivalents de Ca, 2 de Mg, tandis que N a est toujours faible, ne dépassant normalement pas 2 milliéquivalents. SO, et C1 sont aussi très faibles et HCO, également, mais ce dernier un peu plus
fort que dans les eaux des granites, d’où un p H moins acide. Le résidu sec est naturellement peu élevé (il ne dépasse guère 400 mg/l). En somme, les eaux des roches éruptives présentent les caractères suivants :
Résidu sec faible par suite de la mise en saturation très difficile des éléments. Alcalins prédominants en général. Ca très faible, ne prend une valeur notable que dans les diorites et les amphibolites. Mg toujours plus ou moins faible. Fe apporté par les minéraux ferromagnésiens, tels que la magnétite, et mis en solution
C1 très faible, par suite de la rareté des chlorures : rC1 < rNa. SO, peut être apporté par la pyrite, mais toujours en petite quantité. SiO, abondante. p H originellement faible. Mais les eaux qui laissent échapper le CO, libre, par exemple
celles qui ont été longtemps en contact avec l’air atmosphérique, ont un p H élevé, par suite de la prédominance des alcalins qui peuvent se maintenir en solution.
grâce à l’acidité des eaux.
LES P H É N O M ~ N E S MODIFICATEURS [168]
Au cours de leur trajet souterrain et m ê m e dès leur origine dans le sol, les eaux souter- raines subissent fréquemment certaines modifications dans leur composition chimique; les plus importantes sont les réductions, les échanges de bases et la concentration.
Les phénomènes réducteurs.
Nous n’insisterons pas sur ces phénomènes qui ont fait l’objet de maints travaux. L a réduction la plus importante à considérer est celle des sulfates. Néanmoins, il ne faut pas oublier qu’elle peut également porter sur les nitrates et les nitrites.
Certaines eaux souterraines présentent une teneur anormalement faible ou nulle en SO,, contrairement aux autres eaux issues des mêmes terrains ou des mêmes nappes. Par contre, elles renferment fréquemment de l’hydrogène sulfuré, des sulfures, de l’hyposulfite, etc. Et cette faible teneur est toujours accompagnée de la présence de matière organique et parfois également de cel1.e d’un des éléments réduits précédents. L a matière organique en question peut être des débris végétaux ou animaux en décomposition, de la tourbe, des lignites, des charbons, du pétrole. On pensait autrefois que la réduction des sulfates était provoquée par la matière
organique elle-même. Mais on sait maintenant qu’elle est due à des micro-organismes anaérobies bien spécifiques (Sporovibrio desulfuricans, avec ses variétés aestuarii, et Sporovibrio rubentschicki).
Dans l‘activité biochimique de ces micro-organismes, le donateur d’hydrogène est un composé organique ou m ê m e l’hydrogène moléculaire. Le composé organique est oxydé anaérobiquement par l’action d’une déshydrogénase. L’hydrogène moléculaire est activé directement grâce à une hydrogénase. L’accepteur d’hydrogène est d’abord l’oxygène dissous dans l’eau. Et une fois tout cet oxygène utilisé, les accepteurs sont
59

Hydrologie des régions arides
successivement : SO,, SO,, SO,, SO, soit quatre étapes conformément au schéma de Kluyver :
§O,H, + SO,R, + §O,H, --f SOH, + SH,
Et l’on aura comme résultante totale :
SO, + 8H+ -+ 4H,O + S’ + 8e et les équilibres suivants :
[SO,] [ H+] * Eh = EO + 0,0075 log
v5-1
avec EO = 0,P4 à 25” s- + 2H+ Tf H,S Ca++ + S= Tf C ~ S Cas + B,S CaSB,S H,O + CO, 2 M,CO,
H,CO, H+ + HCO, €KO; Tf €I+ + CO,
-+ Ca++ + CO: + Caco, II,O H+ + OH-
Cette réduction est accompagnée d‘une oxydation des composCs orgeniques et, par conséquent, d‘une production de CO, qui donne naissance à des ioiis CO,, ECO, et H+ en grand nombre. I1 en résulte que la reduction des sulfates, d’une part diminue la teneur en SO,, et d’autre part apporte des ions S,O,, S:, HCS,, H-1- et par consé- quent H,S.
Les échutiges de bases.
Les eaux sont également susceptibles d’entrer en contact avec différentes substances qui ont la propriété d’échanger leurs ions contre ceux de ces eaux. Ces substances ont des pouvoirs d’adsorption d’ailleurs assez variables : on peut
distinguer deux types extrêmes d’adsorption avec tous les intermédiaires : une adsorp- tion physique ou adsorption de Van der Waals à faibles liaisons entre l’adsorbant et l’adsorbé, et une adsorption chimique à liaisons énergiques de valence. D’ailleurs, ces deux adsorptions peuvent se produire simultanément. I1 peut y avoir ainsi non seulement fixation à la surface ou même à l’intérieur de
ces substances, mais aussi échange de ces cations contre des cations de l’eau. I1 s’effectue donc un échange de bases. I1 est à remarquer que les mêmes phénomènes peuvent se produire pour les anions, si les conditions physiques de l’adsorbant le permettent. Les substances contenues dans les terrains et susceptibles d’adsorber et d’échanger
sont : lo les mineraux argileux, la glauconie; 20 les minéraux zéolithiques; 30 les substances organiques, l’humus par exemple. Les argiles et les humus donnent des colloïdes électropositifs, donc capables de
fixer et d’échanger des cations, c’est-&-dire des bases. L’alumine donne des colloides positifs. Enfin leg colloides d’hydroxyde ferrique ont un caractère amphotère, c’est- à-dire qu’ils peuvent être, soit positifs, soit négatifs, suivant le pH de l’eau, fixant donc soit des cations soit des anions. Dans les minéraux argileux, tels que la kaolinite, l’halloysite, les illites, les chlorites,
etc., oti les fixations des cations ont surtout lieu sur les bords, la capacité d’échanger est relativement faible. I1 n’en va pas de même avec les minéraux tels que la montmo- rillonite, la vermiculite, à l’intérieur des mailles desquels il peut également se produire
60

Géochimie des eaux soutelraines
des substitutions, tandis qu’à leur surface les fixations sont très importantes. L a capacité d’échange y est très grande. Les échanges peuvent être également produits par des zéolithes, la glauconie, les
matières organiques. Comme il est souvent difficile de savoir quelles substances ont échangé leurs bases contre celles de l’eau, nous les appellerons des permutolites. Le degré de fixation ne dépend pas seulement de la roche, mais également de la
nature des cations. Ceux-ci sont d’autant plus énergiquement fixés que l’ion est moins hydrat6. Les ions bivalents se fixent plus énergiquement que les ions monovalents à degré d’hydratation égale. Le pouvoir de fixation f est ainsi d’une manière générale :
fH ) fRb ) fBa)fSr )fCa)fMg ) fK) [Na ) f’Li Le potassium joue un rôle particulier. 11 est très difficilement déplaçable, énergi-
quement fixé dans les illites. D e plus, il a les dimensions exactes (diamètre ionique de 2’66 A) qui lui permettent de se placer dans les cavités de la couche oxygène. NH, a d’ailleurs un rale similaire à K. L’adsorption est également fonction des concentrations respectives du cation dans
l’adsorbant et dans le liquide. La concentration dans l’adsorbant varie beaucoup moins vite que la concentration dans la solution, et l’adsorption est naturellement beaucoup plus intense pour les solutions diluées que pour les solutions concentrées. D’après Wiegener et Seeny, le rapport qui existe à l’équilibre entre la concentration
initiale a, en milli6quivalents du cation dans le liquide, et la concentration x, en milliéquivalents après équilibre, est le suivant :
où a - x représente la quantité de cations échangés, passant du liquide 5 l’argile et vice uersa. Si nous exprimons par la formule
l’indice d’échange de bases, tel que nous le définirons plus loin, on a :
Enfin nous noterons que l’indice d’échange de bases est d’autant plus complet, c’est- à-dire d’autant plus proche de l’équilibre, que la solution a étt plus longtemps en contact avec l’échangeur. En conclusion : lo la quantité absolue de sels contenus dans une eau est d‘autant plus
grande que l’eau était plus chargée en éléments échangeables ; 20 la quantité relative est d’autant plus grande, autrement dit l’échange est d’autant plus complet en un temps donné, que la concentration est plus faible et que ce temps de contact est plus grand. Considérons maintenant des échanges de plusieurs cations entre les permutolites et
l’eau, par exemple des échanges de Ca et de Mg; on a :
Des échangeurs de bases ayant été en équilibre avec une eau ayant des rapports : rMg,/rCa,, rNa,/rCa,, rNa,/rMg,
ou rNae
rCa, + rMg,
61

Hydrologie des régwns arides
de valeurs d o ~ é e s posséderont donc des rapports rMgv/rCap, rNaplrCay, rNav/rMgP
ou rNa,
rCap + rMgp _____
à valeurs bien définies. Si une autre eau entre en contact avec des échangeurs, un nouvel équilibre tendra
à s’établir entre ces échangeurs et la nouvelle eau. Et il est bien évident que la modi- fication des rapports ci-dessus mentionnés de la nouvelle eau se fera de telle sorte que ces rapports tendront vers les valeurs de ceux de la première eau.
Ces notions fondamentales permettent d’étudier les relatiom entre ces échanges de bases et l’origine des eaux. Les eaux renferment N a 7, K+, Ca++, Mg++, H com m e principaux cations. Nous
pourrons donc avoir les échanges suivants : -+ perm. 2Na + Ca++ +perm. Ca + 2Na+
perm. 2Na + Mg++ Tf perm. Mg + 2Na+ perm. Ca++ + M g + + z p e r m . M g + Ca++
perm. 2Na + 2K + perm. 2K + 2Na+ -+
Les échanges de bases sont donc de nature B modifier complètement les rapports des cations dans les eaux et en particulier les rapports K/Na, Na/Ca, Na/Mg, Mg/Ca. Pour apprécier le degré d’échange de bases, on peut adopter les indices suivants
lorsqu’il y a échange de N a et de K de l’eau contre Mg et Ca de la permutolite (i.e.b. est alors positif) :
C1- (Na + K) c1
C1- (Na + K) SO, + HCO, + NO,
i.e.b.=r et i.d. := r -
lorsque l’échange s’effectue en sens inverse, i.e.6. est négatif et l’on peut parler alors d’indice de déséquilibre.
Chlore alcalin. Nous abandonnerons le terme d’indice d’échange de bases, car il peut déjà y avoir un déséquilibre d’origine, c o m m e dans l’eau de mer oii rC1 > rNa - rK, com m e dans les eaux issues des roches cristallines où rC1 < rNa + rK, sans qu’il y ait eu échange des bases.
La concentration l.
L a concentration peut se faire par évaporation ou par dissolution. Dans la concen- tration par évaporation, il y a une intluence essentiellement climatique. L’évaporation a en effet, surtout lieu dans les zones d’alimentation des nappes. L’eau de pluie, ayant imbibé le sol, est à fiouveau reprise par évaporation dans la zone d’évaporation du 801. L’eau du sol se concentre ainsi progressivement. L a pluie suivante, permettant une infiltration profonde, entraine, plus ou moins diluées, ces solutions concentrées du sol jusqu’aux nappes d’eau. On conçoit aisément que plus l’intervalle entre deux pluies pénétrantes alimentantes est grand, moins les pluies sont abondantes, et plus la température et le déficit de saturation de l’air sont élevés, plus les eaux d’infiltration sont alors concentrées. C’est ce qui explique que la concentration des eaux souter- raines augmente des régions tempérées jusqu’aux régions tropicales, pour diminuer ensuite vers les régions équatoriales 2.
1. H. SCHOELLER, a Les modifications de la composition cbimique de l’eau dans une même nappe n, Association internationale
2. -, a L’inBuenee du climat nur Ia composition chimique des eaux souterraines vadoses m. Bull. Soc. ghl. (9, t. XI, 1941. d’hydrologie acientijique, Assemblke d’Oslo, 1948, p., 124-129.
p. 261-209.
62

Gdochimie des eaux souterraines
I1 est toutefois à noter qu’il peut y avoir concentration par évaporation dans les nappes profondes, lorsque des gaz s’échappent de ces nappes, en entraînant de la vapeur d’eau. C’est le cas en particulier des eaux de certains gisements pétrolifères d’oh partent des hydrocarbures gazeux et du gaz carbonique l. En ce qui concerne la concentration par dissolution, les facteurs principaux sont
la température, la pression, l’étendue de la surface de contact, le volume de l’eau en présence, le temps. Et l’on sait, d’après la loi de Nernst, que (( la vitesse de disso- lution d’un corps solide est proportionnelle au déficit de saturation D. Par conséquent, plus une nappe sera profonde, c’est-à-dire plus son eau aura une température élevée, plus le terrain aquifère sera à grain fin, dans le cas de porosité d’interstices, ou diaclasé, dans le cas de terrain à porosité de fissures, plus les pores ou les fissures seront étroits, plus la vitesse de circulation de l’eau sera faible et plus la nappe sera longue, plus alors les eaux seront concentrées. L a concentration par dissolution ne peut évidemment dépasser un certain degré,
car les eaux tendent à être en équilibre physicochimique avec les roches dans lesquelles elles circulent. Cet équilibre lui-même ne tend à se réaliser qu’après une durée plus ou moins longue (et sa réalisation complète se situerait à l’infini) - durée qui est fonction de la nature du terrain et de la concentration des sels dans l’eau, confor- mément à la loi de Nernst.
Examinons maintenant quelles sont les principales modifications de la composition chimique qui résultent de la concentration par dissolution. Les principaux radicaux contenus dans les eaux sont les suivants : Ca, Mg, Na, C1, SO,, CO,, HCO,. Nous avons donc à examiner les solubilités et les produits de solubilité des sels résultant de la combinaison de ces divers ions. Ces sels peuvent être rangés suivant leur solu- bilit6, S, dans l’ordre suivant :
Nombre de grammes par kg d’eau Produit de solubili16
Cacos CaSO, MgCO,, 3H20 NaHCOs NaBS04 Na,CO, NaCl W O 4 QC12 CaC12
0,013 à 18°C 2,016 à 18°C
96 à 20°C 193 B 20°C 213 ZI 20°C 358 B 2OoC 355 B 2O0C 546 à 20°C 745 à 20°C
0,48 x à 25” C 6,l x à 18” C 1,4 x lo-* à 16’ C
D’un autre côté, les principaux sels qu’on rencontre dans les roches et qui peuvent par conséquent y 6tre dissous en grande quantité sont CaCO,, CaSO,, MgCO, et NaCl. Les autres ne se trouvent qu’à l’état de traces ou tout à fait exceptionnellement dans certains gisements. Déjà nous pouvons nous rendre compte que certains sels ne seront jamais précipités hors des eaux souterraines vadoses, par exemple NaHCO, et Na2C0,. I1 faudrait en effet avoir dans ces eaux souterraines d’origine météorique des valeurs de CO, combiné de 2 500 à 7 500 mg par litre au moins, valeurs qui n’y sont jamais atteintes.
Les eaux souterraines, sauf celles de terrains exclusivement siliceux ou silicatés, sont d6jà sensiblement saturées en carbonate et en bicarbonate de chaux. D’un autre côté, la tension du gaz carbonique des eaux souterraines reste toujours à peu près la m ê m e 2
1. R. VAN A. MILLS: Roger C. WELLS, a The evaporation and concentration of waters associated with petroleum end natura 1
2. EI. SCEOELLER. a L’influence du climat SUP la composition chimique dea eaux souterraines vadoses n, op. cit., p. 284. gas n. U. S. Geological Survey Bull. 623, 1919, 104 pl., + 5 fig.
63

Hydrologie des régions arides
que celle du gaz carbonique de l’air du sol cultivable, c’est-à-dire qu’elle est comprise entre 0,005 et 0,06. I1 ne peut donc y avoir, par suite de la concentration par dissolu- tion, une augmentation de la teneur en carbonate et en bicarbonate, sauf évidemment si les eaux se chargent en ions autres que Ca, CO, et HCQ,, ce qui a pour effet d’aug- menter le produit de solubilité, mais jamais dans de grandes proportions. D’un autre côté, la dissolution de sels renfermant des ions Ca, par exemple CaSQ,, aura pour effet de diminuer la teneur en ions CO, et HCO,. En somme, la teneur en CO, combiné se maintiendra en général entre certaines limites, une limite supérieure fonction de la tension du CO, pouvant légèrement s’élever suivant la teneur en ions autres que Ca, CO, et HCO,, et une limite inférieure pouvant s’abaisser suivant la teneur en ions Ca. D’une manière générale, le CO, combiné oscille entre 75 et 240 mg. En faisant abstraction des eaux acides issues des terrains cristallins ou gréseux purs,
on peut donc conclure, puisque les eaux sont à peu près saturées en carbonate de chaux, que si une eau a une concentration égale ou inférieure à la solubilité normale du CaCO,, soit 300 à 400 mg, elle doit être presque exclusivement bicarbonatée calcique, le carbonate de chaux étant le premier sel à être dissous dans le sol cultivable. Le gypse est le sel suivant, le moins soluble et parmi les plus fréquents. En dissolvant
le gypse, les eaux apportent des ions Ca qui, par conséquent, diminuent la teneur en CO, combiné de l’eau. Et il est remarquable de constater que les eaux très chargées en CaSO, ont toujours des teneurs en CO, combiné en dessous de la normale. La limite de solubilité en CaSO, peut être facilement atteinte en raison de l’abondance de ce sel, et même naturellement surélevée si les eaux dissolvent des sels renfermant des ions autres que Ca et SO,. Les eaux chargées en NaCl peuvent ainsi avoir des teneurs en CaSO, beaucoup plus grandes. L a solubilité du CaS042H,0 (comptée en CaSQ,), de 2,l g à 200 C pour une concentration nulle de NaC1, croît jusqu’à 7,3 g pour une concentration de NaCl = 146,2 g, puis décroît pour des concentrations supérieures. L a solubilité pourrait être diminuée par une nouvelle dissolution de nouveaux sels de calcium. Mais parmi ceux-ci, le CaC1, n’existe pour ainsi dire pas dans les roches et le CaCO, a déjà atteint sa concentration maximum. Mais un apport de sulfates, tels que Na,SO, ou DlgSO,, peut amener le dépassement du produit de solubilité et, par conséquent, provoquer une précipitation de ce dernier sel. D e tels précipités de sulfate de calcium ont été observés dans les roches. La dissolution du sulfate de magnésie est certaine dans les terrains gypsifères, car on constate que les eaux souterraines, à grandes valeurs de SO,, ont des teneurs toujours très élevées en Mg, ce qui entraîne une augmentation des rapports Mg/Ca et Na/Ca. Le sel le plus commun des eaux souterraines, et qui possède une solubilité supérieure
aux précédentes, est le NaCl. Un long parcours, un temps de contact tres grand, une vaste surface de contact augmentent la teneur en NaCl de l’eau, mais sans amener la saturation qui n’a lieu que tout à fait exceptionnellement, la concentration en NaCl se mettant seulement en équilibre avec la concentration en NaCl du terrain. Pour qu’il y ait des valeurs voisines de la saturation, il faut que le terrain aquifère soit salifère. Ce n’est donc qu’exceptionnellement qu’on observe de telles valeurs. En conclusion, on voit que, plus une eau se charge de sels, au-dessus d’une certaine
concentration correspondant à la saturation du sulfate de chaux, plus le rapport SO,/Cl doit diminuer et plus les rapports NafCa, NafMg et m ê m e MgfCa, doivent augmenter. La concentration par évaporation obéit à peu près aux mêmes règles que précé-
demment; et les variations des rapports des divers radicaux se font suivant le même ordre. Mais ici, il peut y avoir des précipitations plus nombreuses. La première préci- pitation dans le sol est celle du CaCO,, sous forme de concrétions calcaires, observables dans les régions tempérées, et de tuf dans les régions steppiques ou prédésertiques; la seconde est celle du gypse dans les régions désertiques. Puis viennent les précipi- tations de sels de soude, carbonate de soude ou sulfate de soude, suivant la teneur en radicaux CO, et SO, de l’eau.
64

Géochimie des eaux souterraines
D e tout ce qui précède, il résulte, en admettant que le CO, combiné ne dépasse pas 300 mg, qu’on doit généralement avoir dans les eaux1 d’abord :
rCO, > rC1 ou rSO, et au-dessus d’une certaine concentration totale des sels dissous qui, en général, est approximativement de 60 milliéquivalents :
rC1 ou rSO, > rC0, et au-dessus d’une concentration encore plus élevée, généralement voisine de 180 milliéquivalents :
rC1 > rSO, > rCO, si les eaux n’ont pas dissous de sels de magnésie.
Évidemment s’il y a des sels de magnésie, SO, peut prendre des valeurs plus élevées. Nous avons constaté que c’est pratiquement au-dessus d’une concentration supérieure à 290 milliéquivalents qu’on a toujours :
rC1> rS0, > KO, On voit donc que, si pour une raison quelconque une eau souterraine se concentre, que ce soit par évaporation d’une nappe arrivant tout près de la surface du sol, que ce soit dans le sol m ê m e de la zone d’alimentation, que ce soit par enrichissement progressif, par circulation très lente, ou par suite de très longs cheminements où les questions de temps et de surface entrent en jeu, elle se transforme complètement, tendant toujours vers une eau de formule :
rC1> rSO, > rC0, et rNa > rMg > rCa formule qui se rapproche de celle de l’eau de mer.
Le tableau de la page 66 donne quelques exemples d’eaux très voisines de leur point de saturation, soit en NaCl, soit en S0,Ca.
Il existe des nappes d’eau à l’intérieur desquelles ne s’effectue aucune circulation, soit qu’il y ait égalité de pression hydrostatique sur tout le pourtour de la nappe, soit que la nappe soit complètement fermée. D e m ê m e des portions de nappe peuvent avoir de l’eau stagnante, la circulation de l’eau ne se faisant que dans une partie de la nappe. Dans de telles eaux de stagnation, la concentration des sels, n’ayant pas été limitée par le temps, a pu être totale. L’eau doit donc être en équilibre ou sensiblement en équilibre avec la roche. I1 suffit d’ailleurs d’une très petite quantité de sels solubles dans la roche et d’une très petite quantité d’éléments chimiquement attaqués par l’eau pour donner une concentration élevée à cette eau.
Soit par exemple une roche de densité d contenant n yo en poids de NaCl, m étant la porosité. I1 y a donc dans un mètre cube de roche 1 O00 dn(1 - m) kg de NaCl, et dans l’eau d’imbibition 1 O00 dn(1 - m) g de sels par litre, en admettant qu’il y ait équilibre de telle façon que la concentration dans l’eau soit la m ê m e que dans la roche, naturellement à volume égal. Si donc une roche ayant une porosité de 0,20 et une densité réelle de 2,65, renferme seulement 2 “loo de NaCl, l’eau en équilibre avec la roche renfermerait 4,240 g de NaCl par litre. Les argiles peuvent renfermer 1 à 2 % de NaCl. Avec d = 2,2, m = 0,40, le calcul donne pour l’eau une concentration de 13’2 à 26,4 g de NaCl par litre. Ces proportions correspondent à celles des roches d’affleurement déjà lessivées. Par
conséquent, les eaux profondes des terrains non lessivés peuvent présenter des concen- trations de sels beaucoup plus élevées. Or, la plupart du temps, la grande teneur en NaCl des eaux souterraines est choisie c o m m e critère de la reconnaissance des eaux connées, c’est-à-dire des eaux emprisonnées par les sédiments lors de leur précipitation et devenant par la suite fossiles. En réalité, c o m m e on le voit, ce ne peut en être un, puisqu’il est c o m m u n à d’autres eaux. Une grande teneur en NaCl ne désigne pas
1. H. SCHOELLER, a Sur la conaentration des sels dissous dans les eaux souterraines n, C. R. du congrch d’Erfoud du Comi16 d’étude des enux souterraines, Rabat. 1934. p. 46-54.
65
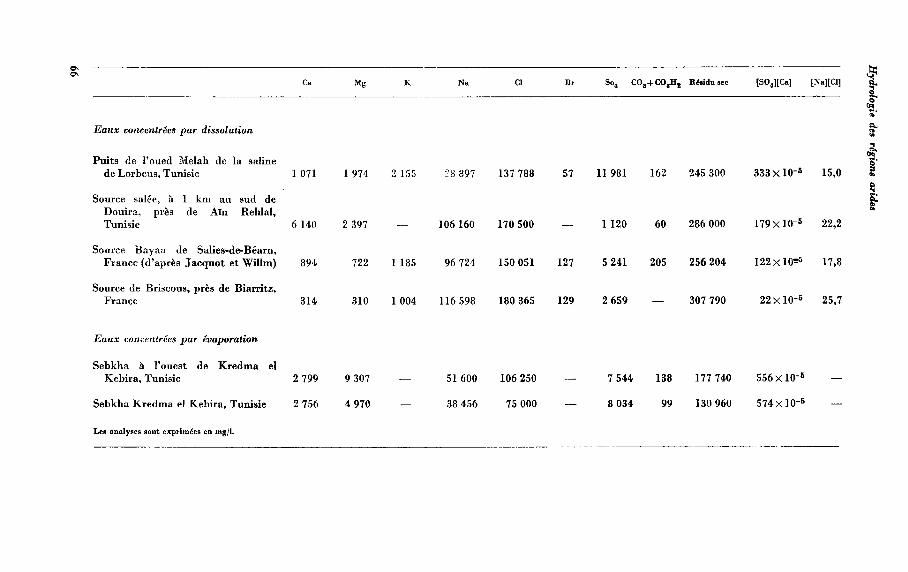
Hydrologie des régions arides
0. In 3
19
z X n
m m
O O
m
In d
01
0.1 W 3
3
02
3 3
m
t- In
Co OD t- t- m 3
r- ot OD
56 c1
ro ro r3
Cl
*
r- ot 3
3
t- O
r3
s .I m m ß
o
m
a
so
; ff
Se
w2
cl
SW
.a -a e, '3 mo
I 19
2 X
W
In In
O
d
t- t- t- 3
OD m 3
d d
In t- I O
In N
W O 3
O O
\o
ln 3
I r- O
m
01
ot o\
t- E3
3
8 i! B
-a U .2 p .2 P o
-I
k
1m ..
c: .- et A$ 3g
cn
I Lo
O - X e t- In O
\o Q, O
m
.-.I
Q, 0
9
m O
m I O O O
In P-
W
In d
CO m I O
t- QI
d
W
In t- 0.1
B
." i? 5 2 2 2 4 .p, w m z m
66

Gdochimie des eaux souterraines
forcément une eau de mer fossile, mais peut tout simplement signifier une concentration par évaporation ou seulement par dissolution, dans ce dernier cas souvent par des eaux souterraines stagnantes. L’étude de la concentration et de ses effets sur la composition chimique deseaux
ne peut être faite qu’en considérant les produits de solubilité : Ainsi pour le carbonate de calcium, il s’agira d’examiner le produit [CO,] [Ca++]= k,
Mais comme la teneur en CO, de l’eau dépend de la tension du CO, dissous, on consi- dérera l’équation
22,4 6 éq. = ~ [H2C03]éq.
2
Pour le sulfate de calcium, on examinera le produit [SO,] [Ca] = k,
Dana certains cas où la saturation en NaCl peut être atteinte, on examinera le produit
[Na1 [Cl1 = kNaa Ces produits varient, il ne faut pas l’oublier, avec la température, la force ionique de la solution.
LES REPRÉSENTATIONS DES A N A L Y S E S : LES RAPPORTS
D’après ce qui précède, on voit qu’il y a lieu d’attacher de l’importance à certaines valeurs absolues, à certains rapports et à certains produits.
Valeurs absolues et produits.
HCO:, La valeur absolue la plus importante à considérer est celle du HCO,. D’une manière générale, elle est assez constante, car elle dépend de la tension du CO, de l’atmospvere des terrains, qui, elle, ne varie que peu. HCO, a des valeurs inférieures à la normale lorsque l’eau entre en contact avec une atmosphère à tension en CO, inférieure à celle qui existe dans le sol ou les terrains. C’est le cas des eaux des mers, des lacs et des fleuves, surmontés de l’air atmosphérique dont la tension du CO, n’est que de 0,0003. HCO, a des valeurs au-dessus de la normale lorsque du CO, arrive en proportion
anormale au contact de l’eau. Ce CO, a une origine volcanique, métamorphique ou bien provient de matières organiques (lignite, charbon, hydrocarbures, etc.) qui ont tou- jours tendance à s’oxyder.
Mais, lorsqu’on examine la valeur absolue de HCO,, il ne faut pas oublier de tenir compte du produit de solubilité [CO,] [Ca], car un apport de Ca, par exemple par échange de bases ou par dissolution de gypse, peut avoir pour effet d’abaisser la teneur en HCO,, si le produit est atteint.
so,, Là aussi la valeur absolue est à prendre en considération. L a dissolution des sulfates, du gypse en particulier, donne des concentrations très élevées en SO,. Mais, là encore, pour bien juger du phénomène, il faut examiner le produit [SO,] [Ca].
Ainsi une teneur extrêmement faible en SO,, paraissant anormalement basse, peut tres bien être due à un apport très important de calcium dans l’eau. Elle n’est pas nécessairement due à une réduction des sulfates par échange de bases.
67

Hydrologie des régions arides
c1 L a valeur absolue du chlore est un des éléments les plus importants témoignant du degré de stagnation des eaux, du temps de contact avec les terrains, de la longueur du trajet, du degré d’évapotranspiration.
Quant aux valeurs absolues de Ca, M g et Na, elles dépendent des valeurs absolues des anions et des échanges de bases.
Valeurs relatives.
Les valeurs relatives ne sont pas moins importantes.
rSO,/rCl Est souvent caractéristique d’une m ê m e nappe, tant que la concentration du SO, n’a pas atteint le maximum permis par le produit de solubilité du S0,Ca.
Lorsque ce dernier est dépassé, le rapport ne peut que diminuer. Lorsque les eaux sont très peu concentrées, moins de 0,5 rSO, ou rC1, rSO,/Cl tend
21 varier dans un sens ou dans un autre, car un très faible apport de SO, ou de C1 entraîne une très forte variation du rapport.
rMg/rCa Est aussi souvent caractéristique d’une m ê m e nappe. Cependant, lorsque SO, et C1 augmentent, le rapport augmente généralement, puisque [COJ [Ca] reste fixe et que SO, amène souvent M g avec Ca.
Na Na Na
Mg Ca Ca+ Mg r-9 r- et r ~ sont aussi souvent précieux à considérer [164].
C1- N a r- c1 Indice de déséquilibre chlore-alcalin, indique soit un déséquilibre d’origine (eaux des roches cristallines), soit un échange de bases. On voit donc que toute représentation des analyses dont les éléments ont été réduits
en pourcentages ne peut convenir, puisqu’on se prive des indications fournies par les valeurs absolues. D e plus, une telle représentation pourrait conduire à des interpré- tations erronées. Ainsi les eaux de gisements de pétrole sont caractérisées par des valeurs absolues
élevées en HCOP Or une analyse ou un graphique ramenant les milliéquivalents en pourcentages indiquerait des valeurs extrêmement faibles, et cela tout simplement parce que les eaux de gisements de pétrole sont, le plus souvent, très chargées en sels, en NaCl en particulier. Analyses réduites en pourcentages et graphiques correspon- dants sont donc absolument B proscrire. I1 faut donc s’en tenir aux analyses où les anions et cations sont exprimés en grammes
ou milligrammes par litre ou par kilogramme, analyses qu’on transforme en millié- quivalents. Comment opérer cette transformation?
Graphiques.
Les graphiques à conseiller sont les suivants.
D i a g r a m m e de Collins e n colonnes. Le diagramme comporte deux colonnes juxta- posées. Sur la colonne de gauche on porte successivement de bas en haut rCa, rMg, rNa + 2K et sur la colonne de droite, de bas en haut, rHC0; + rCO& rSO,, rC1. On aperçoit tout de suite le déséquilibre chlore-alcalin.
68

Géochimie des eaux souterraines
Le diagramme semi-logarithmique 1. On utilise un papier semi-logarithmique. Sur l’axe arithmétique des abscisses, on dispose successivement à intervalles réguliers, et de gauche à droite, le dixième de la concentration, les radicaux rCa, rMg, rNa, rC1, rSO,, rHCO,+, KO, et l’on porte sur l’axe des ordonnées, gradué suivant une échelle logarithmique, le nombre de milliéquivalents de chacun de ces éléments (voir fig. 13). Les points obtenus sont reliés par des droites. I1 en résulte que si les eaux ont des concen- trations différentes, les graphiques se placent les uns au-dessus des autres. D e plus, dans un tel graphique, deux droites parallèles réunissant, l’une deux éléments A et B d’une m ê m e eau, l’autre les mêmes éléments A’ et B‘ d’une autre eau, indiquent un m ê m e rapport de ces éléments : A/B = A’IB’.
Ces graphiques permettent également, d’après l’inclinaison des traits, de déterminer les rapports des éléments entre eux et en particulier les éléments que nous considérons c o m m e caractéristiques ou de première importance,
CI - Na CI
[rMg/rCa rSO,/rCl r ~ rNa/Ca rNa/Mg
D e plus, il est possible de voir si les produits de solubilité sont atteints. Par exemple, pour le produit [SO,] [Ca], il suffit de joindre les points rSO, et rCa par
une droite : si cette droite coupe la verticale CaSO, (placée à mi-chemin entre rSO, et rCa) au-dessous du point de saturation S, la saturation n’est pas atteinte. Si, au contraire, le point d’intersection est au-dessus de S, il y a sursaturation. Ce point S est situé sur la verticale de telle sorte qu’on ait :
- 1 1 2 2
log S = - log (rSO,) (da) = - log (4[SO,] [Ca]) x 10-8
[SO,] [Ca] = K, du produit de solubilité. Pour la solubilité du C03Ca, on utilise la relation
k2 k, k,
rHC0,)2 (rCa) = k, = 2 (H,CO,) éq/ __ X 10,
On joint alors rHC0, et rCa par une droite qui coupe une verticale placée à un tiers de chemin de rHCO, et à deux tiers de chemin de rCa. Si le point d’intersection est situé au-dessous de k,, il n’y a pas saturation. S’il est situé au-dessus, il y a sursaturation. On tiendra naturellement compte de la température et de la force ionique. Pour plus de détails nous renvoyons à [168].
LA COMPOSITION CHIMIQUE DES E A U X DANS LES TERRAINS
On peut tirer quelques règles des observations faites sur la composition chimique des eaux souterraines. 1. Les eaux issues d’une roche de m ê m e nature pétrographique, quel que soit l’âge du
terrain ou quelle que soit la nappe issue de ce terrain, peuvent présenter des carac- tères communs. Ainsi, les eaux des calcaires auront généralement rC03>rS04 OU rC1 et rCa>rMg ou rNa; les eaux des grès et sables purs, de m ê m e que les eaux des roches cristallines, auront un résidu sec faible, un CO, combiné au-dessous de la normale; les eaux en contact avec des éléments argileux ou marneux, un résidu sec Blevé et une teneur plus grande en rC1 et rNa de sorte qu’on aura rC1 OU rS04>rCOs et rNa>rMg ou rCa; et les eaux issues des terrains gypseux, un résidu sec élevé et rSO,>rCO,.
1. H. SCHOELLER, u Utilité de la notion des échanges de bases pour la comparaison dee eaux souterraines vadoses n, Bull. soc. gehl. Fr. (5). t. 5. 1935, p. 651-657.
69

Hydrologie des r6gions arides

Ghchimis dea eaux soumainss
A 2 3
0,025
FIG. 14. &helle logarithmique de solubilités. 1. fichelle de la solubilité du CaSO,, 2 HeO pour diverses forces ioniques et diverses températures. 2. fichelle des k,, calcul des tensions. 3. &helle des k; en fonction de la force ionique et de la température. 4. &helle des p H d’équilibre.
71

Hydrologie des régwns arides
Mais toutes les eaux issues d’une roche de m ê m e nature pétrographique n’ont pas nécessairement exactement la m ê m e composition chimique, avec des rapports caractéristiques identiques, car la diversité de l’alimentation de la nappe, la lon- gueur du trajet des filets liquides et le climat entraînent des modifications de compo- sition chimique.
2. Les eaux issues de terrains de m ê m e nature pétrographique, de m ê m e âge, dans une m ê m e région, ont généralement des caractères communs. Et entre elles il y a une parenté beaucoup plus grande qu’entre les eaux issues de terrains de m ê m e nature pétrographique, mais d’âge différent et appartenant à des nappes différentes.
3. Mais les eaux de deux nappes différentes, situées dans ce m ê m e terrain, peuvent avoir des compositions chimiques différentes, m ê m e si ces deux nappes sont situées côte à cate. Plus les nappes sont éloignées les unes des autres, plus il risque d’y avoir de différences dans la composition chimique. En effet, l’alimentation des nappes peut ne pas être partout chimiquement la même. Les faciès eux-mêmes peuvent changer d’un point à un autre. Les différentes nappes peuvent avoir des trajets plus ou moins longs, ce qui provoque des variations dans les rapports caractéristiques. Dans certaines nappes, la circulation peut se faire de préférence, soit au sein m ê m e du terrain aquifère, soit au contact du mur, soit au contact du toit, causes encore de modifications de la composition chimique.
4. Par contre, les eaux d’une m ê m e nappe située dans un m ê m e terrain ont des carac- tères chimiques relativement constants, beaucoup plus constants que ceux des eaux de deux nappes distinctes, m ê m e si celles-ci sont issues d’un terrain de m ê m e nature pétrographique, de m ê m e âge et de m ê m e assise géologique locale.
L’ÉVOLUTION D E LA COMPOSITION CHIMIQUE D A N S U N E M Ê M E N A P P E 1
I1 ne faut pas croire que la composition chimique de l’eau d’une nappe reste constante de l’amont à l’aval. Tout au contraire, elle subit le plus souvent une très nette évolution. Tout d’abord on observe une augmentation de la concentration totale des sels
dissous. Cette augmentation est due aux nouvelles dissolutions. Naturellement, plus le trajet est long, la circulation de l’eau lente, le temps de contact important, les pores petits, autrement dit, plus les surfaces de contact entre eau et roches sont grandes, plus alors I’accroissement de la concentration générale est sensible. Cette augmentation générale détermine en grande partie le sort des autres éléments. Ainsi, d’une maniere générale, le rapport rS04/rC1 diminue de l’amont à l’aval.
En effet, la vitesse de dissolution est plus grande pour les chlorures que pour les sulfates alcalino-terreux, puisque cette vitesse est proportionnelle au déficit de saturation. Cependant, il est bien évident que, si le terrain a w e r e est riche en sulfates et rela-
tivement pauvre en chlorures, l’inverse se produit et augmente le rapport rS04/rC1. Mais la concentration s’accroît toujours plus à l’aval. L a saturation en S0,Ca est bientôt atteinte. A partir de ce moment, le rapport rSO,/rCl doit obligatoirement diminuer. Le rapport rMg/rCa a, en général, tendance B diminuer de l’amont à l’aval. En
effet, l’apport de Ca par dissolution du C0,Ca cesse immédiatement, car les eaux en sont saturées dès l’origine. D e plus, la dissolution du CaS0, est moins rapide que celle du MgSO, et du MgCl,.
L’indice de déséquilibre, lui aussi, se modifie de l’amont à l’aval. L’indice peut d’abord être positif dans la zone d’alimentation, diminuer progressivement, puis devenir négatif et ce de plus en plus vers l’aval. Mais, dans certains cas, lorsque la concen- tration en chlore s’élève fortement, en général au-delà de 500 milliéquivalents, l’indice d’échange reprend de nouveau une valeur positive. Ces phénomènes sont d’autant plus intenses que la durée de contact eau-terrain est plus grande. Autrement dit, 1. H. SCAOELLER, K Les variations de la composition chimique de l’eau dans les nappes souterraines n, Association intermario-
M l e d’hydrologie scientijique, Assemblde d’Oslo, 1948, p. 130-144.
72

Géochimie des euw souterraines
l’intensité de l’échange dépend non seulement de la longueur du trajet, mais aussi de la lenteur du cheminement et de la grandeur des surfaces de contact eau-terrain.
L a réduction des sulfates qui peut s’opérer s’il y a des matières organiques est, elle aussi, d’autant plus complète que le temps de contact est plus grand. Mais un terrain aquifère n’est pas toujours homogène dans son étendue. Aussi
conçoit-on que des filets liquides assez voisins, circulant dans des matériaux différents, puissent dissoudre différemment les substances et, par là, varier leur composition chimique. Enfin, dans un terrain aquifère, l’eau circule le plus souvent le long de zones privi-
légiées, rarement d’une manière active, dans toute la masse du terrain aquifère. Ceci permet une individualisation des filets liquides, s’oppose à leur mélange et empêche la masse liquide d’avoir une composition parfaitement homogène.
I1 faut enfin ajouter le cas d’une nappe libre souterraine se rapprochant très for- tement de la surface du sol (moins de 2 mètres ou m ê m e moins d’un mètre). I1 se pro- duira, en particulier dans les régions arides, une concentration par évaporation et par le jeu des eaux, alternativement ascendant, par remontée capillaire, et descendant, par infiltration. L e résidu sec augmente; en m ê m e temps, rS04/rC1 diminue et rMg/rCa augmente à la suite de la précipitation de S0,Ca et de C03Ca. Pour bien saisir l’évolution de la composition chimique, on a intérêt à tracer sur dea
cartes des isocones ou courbes d’égale concentration, des isochlores ou courbes d’égale
C1- N a
c1 teneur en C1, des courbes d’égale valeur de rSO,/Cl, rMg/rCa, T , etc.
LES ZONALITÉS DE LA COMPOSITION CHIMIQUE DES EAUX
Zonalité géologique.
Ainsi, les éléments des eaux peuvent s’enrichir par attaque chimique ou par disso- lution. Cet enrichissement dépend de la nature du terrain, puisque la mise en solution du NaCl est plus rapide que celle du S04Ca, et l’attaque du calcaire plus rapide que celle des roches cristallines et cristallophylliennes. L a concentration dépend ainsi non seulement de la longueur du trajet, du temps de contact de l’eau avec le terrain, mais aussi de la nature de la roche. I1 y a donc là des facteurs essentiellement géologiques.
L a nature elle-même de la composition chimique de l’eau dépend des substances chimiques attaquables et solubles de la roche, elle dépend aussi des possibilités des réactions secondaires qui peuvent se produire dans les terrains : par exemple, les échanges de bases dus à la présence de permutolites, telles que certaines argiles, la glauconie, etc.; la réduction des sulfates provoquée par la présence de matières orga- niques dans les terrains. On a donc affaire là encore à des causes essentiellement géo- logiques. C o m m e la répartition des assises géologiques se fait suivant certaines plages, disons certaines zones, on conçoit que l’on puisse avoir des zones oh les eaux souter- raines présentent les mêmes caractères. Ainsi il y a des zones d’eaux à faible teneur en calcium, à faible résidu sec, eaux généralement agressives. Ces zones se superposent à des massifs cristallins, ou bien à des sables siliceux très purs (exemple : les sables des Landes en France, les sables des dunes sahariennes). A côté de ces zones on en trouve d’autres, aux eaux très sulfatées et chlorurées par
exemple, qui correspondent à des affleurements de Trias. On aura donc une certaine zonalité géologique.
Zonalit6 verticale.
Les eaux souterraines circulent d’autant plus lentement et sont d’autant plus dif-
73
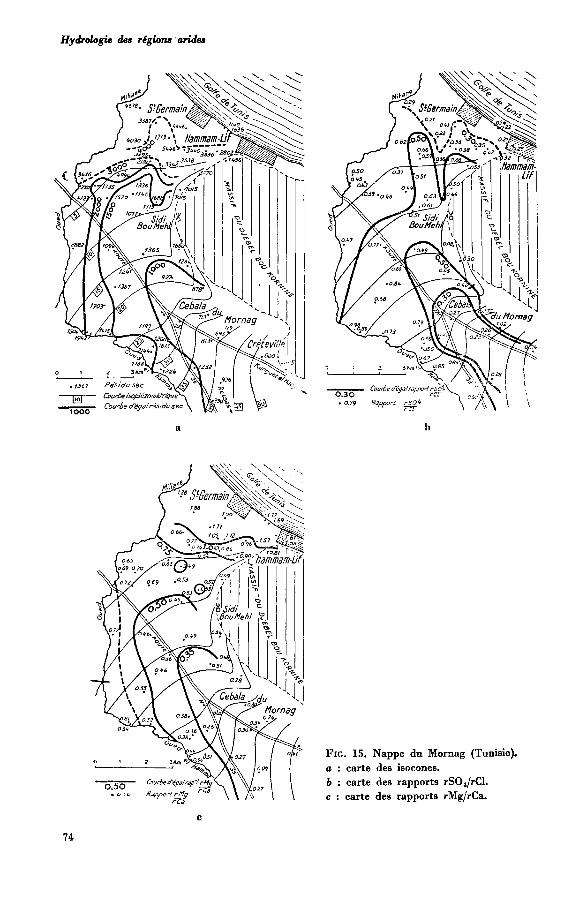
Hydrologie des régions aridss
a b
FIG. 15. Nappe du Momag (Tunisie). u : carte des isocones. b : carte des rapports rS04/rCI. e : carte des rapports rMg/rCa.
C
74

Wchìmie des eaux souterraines
ficilement déplaçables par d’autres eaux (eaux d’infiltration superficielles en parti- culier) qu’elles sont plus profondes.
Plus les terrains sont profonds, moins ils sont lessivés et plus l’eau peut y séjourner longtemps. On conçoit dès lors que la concentration de l’eau en sels dissous augmente avec la profondeur. C’est ce qui a été constaté depuis fort longtemps, notamment dans les bassins pétrolifères (Rogers 1917, Minor 1934, Torrey 1934, Case 1934 in [168]). On a donc une certaine zonalité verticale. I1 est tout naturel, dès lors, qu’avec la profondeur les eaux se transforment et que, hy-
drocarbonatées en surface, elles deviennent chlorurées en profondeur, selon le schema suivant d’Ignatovitch et de Souline dans l’ouvrage de Siline-Bektchourine [171].
HCO; -+ HCO;, SO, + SO, ---f SoiCl- + Cl-SO: -+ C1-
Ce schéma exprime tout simplement la variation de la composition chimique, imposée par la concentration I.
Les trois premiers groupes se trouvent dans la zone supérieure de circulation intense des eaux souterraines.
Les eaux sulfatées-chlorurées et chlorurées-sulfatées existent habituellement dans la zone moyenne de circulation ralentie, les eaux chlorurées dans la zone de circulation très ralentie. Naturellement, cette zonalité est influencée par la zonalité climatérique, de m ê m e
qu’elle est perturbée par des conditions géologiques.
Zonalité climatirique.
Nous avons vu qu’il pouvait y avoir une concentration par évaporation et que, suivant le degré de concentration, la composition chimique de l’eau présentait des caractères tout différents.
La concentration est d‘autant plus importante que le pouvoir évaporant est plus grand, c’est-à-dire que la température de l’air et sa sécheresse sont plus élevées. La concentration est également d’autant plus grande que la pluviométrie est plus faible. Elle est donc d’autant plus importante que le climdt est plus aride. Nous reviendrons plus loin sur le mécanisme de cette concentration. I1 y a là une influence essentiellement climatérique qui entraine une certaine zonalité. Bien qu’il n’y ait encore que peu d’études sur cette zonalité climatérique, nul n’ignore
que la composition chimique des eaux des sources et des puits de l’Afrique et du Sahara et des autres régions arides diffère de la composition chimique des eaux souter- raines des régions tempérées et des régions froides. Et chacun sait que les eaux des montagnes sont plus pures que celles de la plaine. L a composition chimique des eaux est donc bien influencée par le climat 2.
Quels sont les facteurs climatériques qui régissent la composition chimique des eaux et comment agissent-ils ?
L a pluviométrie joue un rôle prédominant. Elle provoque un lessivage des terrains, entrainant les sels les plus solubles dans les circuits souterrains et superficiels, c’est-à- dire essentiellement le NaCl et le CaSO, des eaux. Le CaCO, suit un autre régime; car, très faiblement soluble, il nécessite l’intervention d’un autre facteur, tout à fait indépendant de la pIuviométrie pour être mis en solution, la teneur en CO, libre de l’eau.
I1 restera donc d’autant moins de sels solubles dans les terrains et la dilution des sels restants, passant en solution, sera d’autant plus grande que la pluviométrie sera plus forte.
1. II. SCHOELLER, 1934, op. cit. 2. u L’influence du climat sur Ia composition chimique des eaux Routerrainea u, Bull. Soc. g6ol. Fr. (5). t. XI.
1941, p. 267-289.
75

Hydrologie des régbns arides
gtant donné que la pluviométrie diminue des régions tempérées aux régions tro- picales, pour augmenter de nouveau vers les régions équatoriales, on doit s’attendre à une augmentation de la teneur en chlorures, sulfates et résidu sec, en allant de la France au Sahara, avec de nouveau une diminution vers l’équateur. C’est bien ce qu’on observe.
L’évaporation exerce aussi une action particulièrement nette sur la composition chimique des eaux. Par évaporation la concentration des chlorures et des sulfates, et par là le résidu sec de l’eau contenue dans la couche superficielle du sol, augmentent. Cette eau plus ou moins renouvelée des profondeurs à la surface par capillarité amène une véritable ascension des sels, d’ailleurs fort bien constatée dans les régions tro- picales et subtropicales.
Lorsque l’évaporation devient très intense, les sels en dissolution peuvent passer à l’état de sursaturation et, par précipitation, former les croûtes calcaires, les carapaces gypseuses et m ê m e les croûtes salines, suivant le degré d’évaporation. C’est Caco3 qui se précipite le premier, car il est presque toujours très voisin de l’état de saturation, ainsi que l’ont démontré les recherches de l’auteur. Aussi le tuf calcaire se forme-t-il le premier du côté tempéré de la bordure des déserts, tandis que le gypse, qui demande pour précipiter une concentration plus poussée, donne des croûtes sur le côté désertique.
L a concentration est particulièrement nette après les pluies faibles, insuffisantes pour descendre jusqu’aux nappes et qui ne font qu’imprégner les couches superficielles. I1 se produit ainsi, par un jeu successif de descentes avec dilution et de remontées avec concentration de l’eau, une concentration progressive, si l’évaporation est par- ticulièrement intense. Seules les fortes pluies entrainent vers les nappes, en les diluant plus ou moins, ces eaux concentrées qui réapparaîtront aux sources. I1 est rare qu’elles s’acheminent directement des sources vers la mer, faute d’écoulement superficiel.
L’effet de l’évaporation s’ajoute donc à celui de la faiblesse de la pluviométrie. Aussi, dans Ies régions tropicales, aura-t-on des eaux salées, séléniteuses et à résidu sec élevé, tandis que dans les régions tempérées les eaux seront peu chargées.
L’élévation de température, déjà facteur primordial de l’évaporation, joue un rôle très important dans les phénomènes chimiques. Elle favorise en particulier les attaques des silicates et active les mises en solution. Enfin, concurremment avec l’humidité, elle préside à l’activité des micro-organismes du sol et à la combustion des matières organiques, avec production de gaz carbonique. Or ce gaz carbonique est le facteur essentiel de l’attaque des minéraux et des roches, silicates, carbonates.
L’auteur a examiné l’évolution des caractères chimiques des eaux depuis les régions tempérées jusqu’aux régions équatoriales. Voici les résultats concernant les divers éléments.
Résidu sec. Tout d’abord on constate une augmentation graduelle du r6sidu sec, des régions tempérées jusqu’au Sahara, puis une diminution dans les régions équatoriales.
Teneur en bicarbonates. Alors que pour tous les terrains la teneur en Ca, Mg, Na, C1, SO, varie nettement en fonction du climat, Ia teneur en bicarbonates reste fixée entre des limites très étroites, dans 75 % des cas, entre 2 et 8 milliéquivalents seulement. Mais malgré tout, pour tous les terrains, on constate une augmentation très graduelle de la teneur en bicarbonates des régions tempérées jusqu’aux régions des steppes (centre de la Tunisie), où il atteint son maximum, puis une diminution vers le désert (Sahara) et vers les régions équatoriales. I1 est très vraisemblable que l’augmentation de la teneur en bicarbonates vers les steppes se fasse dès les régions arctiques, car il enest ainsi en descendant des sommets des montagnes vers les plaines l. L a constance relative
1. H. SCAOELLEA, a Les variations de Ia teneur en gas carbonique des eaux souterraines, en fonction de l’altitude D, C. R. dead. Sc. Paris, t. 230, 1950, p. 560-561.
76

Géochimie des eaux souterraines
de la teneur en bicarbonates des eaux provient de la constance relative de la tension du CO, de l’atmosphère des sols proprement dits.
Dans les sols agricoles, la tension partielle du CO, varie généralement de 0,005 à 0,06, ce qui donne précisément des teneurs en HCO, de 2,s à 7 milliéquivalents à 150.
Les eaux d’infiltration ont dissous le CO, de cette atmosphère, permettant la mise en solution des bicarbonates. Or ce gaz carbonique des sols provient entièrement de l’activité biologique et de la décomposition de la matière organique dans le sol lui-même. Plus la végétation est abondante, plus la vie des micro-organismes dans le sol est intense, plus la production de CO, est grande, sa tension élevée et par là plus la teneur en bicarbonates est forte. Or l’abondance de la végétation et l’intensité de la vie des micro-organismes croissent
avec la température de l’air et la pluviométrie (bien entendu, jusqu’à certains optimums). L a teneur en bicarbonates des eaux est donc bien modifiée dans tel ou tel sens par les actions climatériques.
L’augmentation de la teneur en HCO,, du nord aux steppes, est due à un accroisse- ment de la température, avec humidité suffisante pour permettre une production de CO, importante.
L a diminution en direction du Sahara est provoquée, malgré l’augmentation continue de la température, par la décroissance de l’humidité du sol et par le manque de végé- tation. Au nord des steppes, le facteur de contrôle était la température, au sud c’est l’humidité du sol.
SO, et CI. Alors que dans les régions tempérées, les teneurs en rSO, et rC1 sont ex- trêmement faibles, le plus souvent nettement inférieures à la teneur en rHCO,, elles croissent rapidement vers le Sahara. Dès la région des steppes, chacun d‘eux dépasse rHCO,.
Mais le SO, croît plus lentement que le C1; c’est une conséquence des facteurs climatériques. L’augmentation de l’évaporation, qui va de pair avec la diminution de la pluviométrie, ne peut qu’amener une concentration des sels d’autant plus importante que ces sels sont plus solubles, la vitesse de dissolution étant proportion- nelle au déficit de saturation. L’augmentation plus rapide du C1 que du SO, est donc normale.
Mais cette évolution peut être troublée par des causes géologiques; abondance de gypse dans les sédiments des régions désertiques, abondance de chlorure de sodium dans certains terrains des régions tempérées.
Dans les régions équatoriales, nous retrouvons au contraire un taux extrêmement faible de SO, et de C1, souvent inférieur m ê m e B celui des eaux des régions tempérées. Nul doute que ce fait ne soit dû à la forte pluviométrie qui lessive intensément tous les terrains.
Ca, Mg, Na. Les modifications des anions entraînent celles des cations. C o m m e la variation du HCO, est négligeable en face de celles des ions SO, et C1, ce sont donc ces dernières qui entraîneront une variation des cations. L e Ca s’accroîtra ainsi des régions tempérées au désert, non en liaison avec HCO,,
mais avec SO,. L e M g subit le m ê m e sort que le Ca pour les mêmes raisons. Mais il a tendance
s’accroftre plus rapidement. On constate, en effet, que le rapport Mg-Ca est d’autant plus élevé que la concentration en sulfates est plus grande. L e sodium est, d’une manière générale, lié au chlore et subit par conséquent le m ê m e sort. Aussi voyons-nous dans les eaux de tous les terrains une augmentation graduelle de la teneur en C1, des régions tempérées aux régions désertiques.
Tous ces cations Ca, Mg, N a sont de nouveau en très faible quantité plus au sud, dans les régions équatoriales, par suite du lessivage intense dû à la pluviométrie.
77
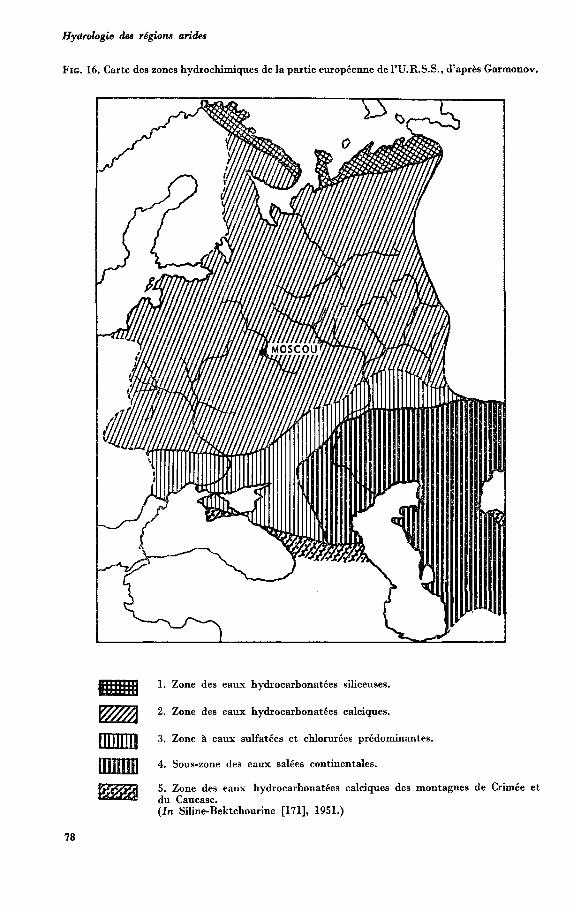
Hydrologie des régions arides
FIG. 16. Carte des zones hydrochimiques de la partie européenne de l'U.R.S.S., d'aprh Garmonov.
1. Zone des eaux hydrocarbonatées siliceuses.
2. Zone des eaux hydrocarbonatées calciques.
3. Zone
4. Sous-zone des eaux salées continentales.
5. Zone des eaux hydrocarbonatées calciques des montagnes de Crimée et du Caucase. (In Siline-Bektchourine [171], 1951.)
eaux sulfatées et chlorurées prédominantes.
ia

Géochimie des eaux souterraines
Une telle influence climatérique de la zonalité de la composition chimique des eaux souterraines apparaît dans la carte de Ia zonalité publiée par Garmonov l.
Reprenons le travail de Garmonov. D’après cet auteur, la nature des eaux des nappes phréatiques dépend de la zonalité des climats, de la zonalité de la couverture de terrain et des conditions d’altération. Les facteurs de la concentration des sels dans les eaux seraient : lo l’enlèvement des sels du sol; 20 le lessivage des sels contenus dans les terrains rocheux où les eaux des nappes phréatiques circulent; 30 l’enrichissement en sels par transport éolien; 40 l’évaporation des eaux souterraines à travers la surface du sol.
Garmonov a établi qyatre zones en Russie d’Europe (voir fig. 16). 1. Une zone des eaux phréatiques hydrocarbonatées siliceuses, coïncidant avec la
zone des sols de Toundra; elle occupe la côte nord de la presqu’île de Kola, la pres- qu’île de Kanin et le littoral plus à l’est, le cercle polaire en constituant sensiblement la limite sud.
2. Une zone des eaux hydrocarbonatées calciques, comprenant toute la partie centrale de la Russie. L a limite méridionale de cette zone passe approximativement par Mogilev, , Dniépropétrovsk, Kharkov, Penza, Oulianovsk et Taschly.
3. Une zone à eaux souterraines sulfatées et chlorurées prédominantes, se développant dans la partie méridionale de la Russie, au sud de la limite précédemment indiquée.
Mais on peut y distinguer une sous-zone continentale salée, c’est-à-dire essen- tiellement chlorurée, recouvrant la dépression au bord septentrional de la Caspienne, et la région à l’est de la Caspienne.
4. Une zone d’eaux hydrocarbonatées calciques, dans la partie montagneuse de la Crimée et du Caucase.
D’après les données de Priklonskii, il y a de nouveau une zone continentale salée, plus au sud, dans la partie basse de la Transcaucasie. En somme, on a en Russie la m ê m e zonalité que celle qui a été précédemment signalée :
eaux passant, du nord au sud, du type hydrocarbonaté au type sulfaté chloruré, puis au type chloruré. Les montagnes du Caucase et de la Crimée ramènent seulement au sud un Bot de type septentrional,
Naturellement, cette zonalité climatérique peut se trouver localement perturbée par des conditions géologiques. On voit donc qu’il y a une première zonalité climatérique, zonalité très large, indépendante de tout facteur géologique, et qui ne dépend que du degré d’aridité. Elle est sous l’influence de conditions tout à fait générales, qui tendent h effacer les autres causes. de zonalité. Elle est surtout caractérisée par les concentra- tions ou dilutions plus ou moins grandes des eaux. A l’intérieur de cette zonalité climatérique, on pourra trouver des zonalités géolo-
giques, dans lesquelles les variations de la composition chimique sont dues à la nature du terrain. Les causes géologiques de zonalités ne sont en effet que des causes parti- culieres amenant des perturbations dans la zonalité climatérique, de caractère beau- coup plus général. Enfin, on trouve partout une zonalité verticale, due au fait que les eaux, à mesure
qu’elles sont plus profondes, ont des vitesses de circulation de moins en moins grandes
LA COMPOSITION CHIMIQUE DES EAUX DANS LES DÉSERTS, D’APRBS KOUNINE
I1 n’est pas inutile de signaler ici les travaux soviétiques; ils apportent un certain nombre d’idées générales. Ainsi Kounine [ZO] nous donne quelques indications sur la formation des eaux
souterraines dans les déserts. Ces travaux confirment les conclusions formulées précé- demment et ont ainsi une valeur d‘illustration.
1. I. V. GAWONOV. u Zonalit6 des eaux des nappes libres dana la partie europdenne de l’U.R.S.S. v, Trauaux du Laboratoire dasprobldmes hydrogéologiques, /.cademie des sciences de l’U.R.S.S., t. III, 1948. In SILINE-BEKTCHOLRINE [171].
79

Hydrologie des régions arides
Kounine indique que, plus les eaux souterraines sont proches des zones d’alimen- tation, moins elles sont salées, et que la salure est d’autant plus forte que le trajet souterrain est plus long. Effectivement, c o m m e nous l’avions vu, la concentration des eaux souterraines dépend de la durée du contact eau-roche. I1 note que, moins les eaux sont salées, plus leur composition chimique est variée et qu’inversement, plus les eaux sont salées, plus leur composition est monotone et se rapproche d’une compo- sition standard. Ce n’est en s o m m e qu’un effet de la concentration qui aboutit finalement à une composition voisine de celle de l’eau de mer l. Pour les eaux souterraines salées et non pour les saumures, la minéralisation la plus
typique est NaC1, souvent CaCP. Les terrains traversés par l’eau ont une certaine inff uence sur la composition chimique
des eaux souterraines, en particulier au cous de la premihre étape de la minéralisation. Mais la principale influence sur la composition chimique des eaux souterraines est due à des facteurs indépendants de la composition des roches. Parmi ceux-ci figure l’éva- poration à l’intérieur du terrain, qui augmente graduellement la concentration et modifie la composition chimique.
Kounine apporte ainsi une confirmation éclatante à ce que nous avions indiqué.
LA FORMATION D E LA COMPOSITION CHIMIQUE DES E A U X SOUTERRAINES DANS LES R É G I O N S ARIDES, D’APRBS SILINE-BEKTCHOURINE
A. I. Siline-Bektchourine [173] a effectué une intéressante étude N Sur le problème de la formation de la composition cbimique des eaux des nappes libres dans les régions arides D.
I1 a réalisé des expériences de laboratoire sur des échantillons monolithes de terrain,pré- levés dans la basse plaine de la Caspienne, aux profondeurs de 2,5,4,5,6,5,8,5 et 10,8 m. Après avoir placé une partie de chaque échantdlon dans une sorte de perméamètre,
il a filtré à travers le premier, celui de 2’5 m de profondeur, de l’eau douce de la riviere Achtuba. L’Achtuba est une branche du cours inférieur de la Volga. Le filtrat résultant a été passé à travers l‘échantillon suivant de 4,5 m de profondeur; et le filtrat résultant à travers l’échantillon suivant, et ainsi de suite.
Cette opération a été répétée trois fois, toujours avec de l’eau de 1’Achtuba. L a composition chimique des extraits aqueux des sols était c o m m e indiqué dans
les deux tableaux suivants :
En pourcentage du poids sec du terrain.
Profandeur en mbtres
2.5 4.5 6.5 8.5 10.8
% HCO, . . . . . . . 0,050 SO,. . . . . . , . 0,918 Cl . . . . . . . . 0,041 Ca . . . . . . . . 0,278 Mg . . . . . . . . 0,054 Na . . . . . . . . 0,070
TOTAL. . . . . . 1,411 -
% 0,057 0,150 0,070 0,036 0,012 0,075
0,400 -
% 0,074 0,150 0,066 0,044 0,009 0,077
0,420 -
% 0,077 0,033 0,037 0,016 0,002 0,046
% 0,072 0,037 0,062 0,033 0,006 0,037
0,211 0,247
1. H. SCHOELLEH, 1914. op. cit.
80

Géochimie des eaux souterraines
En milliéqpuivalents/lires de la solution de l‘eau extraite.
Profondeur (en mhtres)
2,s 4.5 63 8 3 10,8
% % % HCO, . . . . . . . . 0,97 1,26 SO, . . . . . . . . 21,28 4,31 C1. . . . . . . . . 1,27 2,71 Ca . . . . . . . . 15,47 2,45 Mg . . . . . . . . 4,93 1,32 N a . . . . . . . . 3,12 4,51
TOTAL . . . . . . 47,04 16,56 - -
1,52 3,96 2,34 2,79 0,82 4,21
15,64 -
% 1,90 1,14 1,41 1,20 0,25 3,o - 8,90
% 1,56 1,02 2,31 2,15 0,66 2,08
9,78 -
Et voici la composition chimique de l’eau de l’Achtuba, exprimée en milliéquivalentsll :
HCOi so4 CI Ca M.5 N a Total
0,70 2,35 0,90 1,30 Néant 2,65 7,90
On voit que l’extrait aqueux est sulfaté-chloruré au sommet et chloruré-sulfaté en profondeur, et que jusqu’à 8,5 m de profondeur rNa>rCl, tandis qu’à 10 m, c’est l’inverse.
L’eau de 1’Achtuba est également sulfatée-chlorurée, avec rNa>rCl. L a première filtration à travers les sols de 2,5, 4 et 6,5 m montre tout d’abord un
accroissement très net de la concentration générale, en passant d‘un échantillon à un autre, puis une diminution de rSQJrC1, supérieur à 1 dans les deux premiers échantillons, inférieur dans les autres.
On constate de m b m e une modification du rapport rCliNa CI - N a
our ~ c1 négatif dans le
premier, positif et de plus en plus grand dans le deuxième puis le troisième. Les accroissements des éléments dans la solution ont été les suivants :
Entre 2,5 et 4,s m Entro 4,s et 6,5 m
HCO; . . . . . . . . . so, . . . . . . . . . c1. . . . . . . . . . Ca. . . . . . . . . . Mg . . . . . . . . . Na . . . . . . . . .
- 1,04 + 15,69 + 19,54 + 12,68 + 2944 + 19,07
- 1,80 + 3354 + 145,07 + 57,83 + 25,16 + 63,2
TOTbL . . . . . . . + 68,38 + 323.00
L a deuxième filtration a donne des eaux moins concentrées. Mais les mêmes phénomènes se produisent : lo baisse progressive du rapport rS04/rC1, supérieur à 1 jusqu’à 6,50 m
de profondeur et inférieur jusqu’à 8 et 10 m; 20 augmentation du rapport r --, CI- N a
c1 négatif jusqu’à 6,50 m et positif au-delà.
Dans les troisième et quatrième filtrations, on observe encore les memes processus, baisse générale de concentration à tous les niveaux, baisse de rSO,/rCl et augmentation
81

Hydrologie des rgwns arides
C1- Na CI
de r ___ vers la profondeur. Mais rSO,/rCl devient plus petit que 1 à des profondeurs
plus grandes, à 10 m dans la troisième filtration. Dans la quatrième, il reste supérieur à1,r- augmente vers la profondeur. Mais il ne devient positif que plus bas, à 10’5 m
dans la troisième filtration, et reste négatif au m ê m e niveau dans la quatrième filtration. En somme, les filtrations successives lessivent les terrains et, au fur et B mesure de
la filtration, donnent aux eaux des niveaux profonds les caractères des eaux des niveaux supérieurs.
C o m m e le montre A. I. Siline-Bektchourine, les eaux d’infihation se chargent en sels par dissolution, subissent des échanges de bases avec ce que j’appelle les per- mutolites. Bektchourine admet également des réactions de substitution, qui ne peuvent être considérées, en réalité, que s’il y a des précipitations. I1 vaudrait mieux se servir des produits de solubilité. L a diminution relative de la teneur en SO, m e paraît due à une mise en solution plus diftieile du S0,Ca que du NaCl. Et je pense que Ia diminution en valeur absolue du SO, P partir de la profondeur de 8,50 m dans la deuxième filtration et le ralentissement de son augmentation à partir de la m ê m e profondeur dans les troisième et quatrième filtrations sont plutôt dus à ce que le produit de solubilité [SO,] [Ca] a 6té atteint ou presque par suite de l’apport de Ca par les échanges de bases. Le produit de solubilité [SO,] [Ca] joue certainement un grand rôle. D’un autre côté, le fait que rC1- rNa, d’abord négatif vers la profondeur, s’accroît
de plus en plus et devient m ê m e positif, indique une m ê m e tendance qu’on peut mettre en corrélation avec l’augmentation de la concentration générale des eaux. En effet, on observe le m ê m e phénomène si l’on compare les eaux des diverses filtrations d’un m ê m e niveau. Lorsque la teneur en N a de la solution augmente, elle n’est plus en équi- libre avec celle de la permutolite. I1 y a alors échange de N a de la solution contre du Ca de la permutolitel. Les phénomènes ont donc été examinés d’une manière un peu différente de celle
de M. Bektehourine, sans faire intervenir des réactions chimiques, et en ne considérant que les ions en présence. Le travail de Siline-Bektchourine montre l’acquisition de la composition chimique
des eaux en trois étapes.
Première étape. 1. Dissolution de NaCl, CaSO,, MgCO, et autres sels par les eaux d’infiltration. 2. &change de bases, par exemple :
Cl-Na
c1
Ca [(HCO,),, SO,] + Na, -f Na, [(HCO,),, SO,] + Ca solution collorde solution collorde
rarement :
Ca [WCO,),, SO,] + Mg 2 Mg [(HCO,),, SO,] t- Ca solution collordc
3. Réaction de substitution : Na, (Mg) (HCO,), + CaSQ, = Na, (Mg) SO, + Ca(HCO,),
B ne considérer que s’il y a précipitation de CaCO,.
produire par augmentation de la teneur en Ca apportée par échange de bases. Le plus souvent, la précipitation de CaCO,, ou m ê m e celle de CaSO,, peut se
Deuxième étape. 1. Concentration des eaux. 1. H. SCHOELLER, a Relation entre la concentration en chlore de? calix souterraines et les €changes de bases aver Ics terrains
qui lea renferment n, C. R. Acod. SE. Pub, t. 232, 1951, p. 1432-1434.
82

Gwchimie des eaux souterraines
2. &change de bases : 2NaCl+ Ca (Mg) 2 Ca (Mg) C1, + 2Na solution collolde eolution colloide
3. Réaction de substitution : CaCI, + Na, (Mg) SO, = Na, (Mg) Cl, + CaSO,
Ici nous faisons la même remarque que précédemment.
Troisième étape. Mélange de l’eau d’infiltration avec l’eau de la nappe phréatique, avec réactions dites de substitution dues en réalité aux atteintes des produits de solubilité. I1 est bien évident que ce schéma ne s’applique qu’à des terrains et des eaux sem-
blables ii ceux qui ont été étudiés ici et qu’ailleurs les phénomènes suivent un autre ordre. Notamment l’échange N a solution contre Ca collo?de peut avoir lieu dans certains cas dans la première étape comme nous l’avons vu en Tunisie.
Néanmoins, ces trois étapes sont constantes : lo dissolution; 20 concentration; 30 mélange avec l’eau de la nappe phréatique.
QUELQUES CARACTBRES GÉNÉRAUX DES DÉSERTS ET SEMI-DÉSERTS D U POINT DE V U E GÉOCHIMIQUE [17, 291
Ainsi la composition chimique des eaux des déserts et semi-déserts dépend des condi- tions géographiques, liées elles-mêmes aux conditions hydrogéologiques. Dans les régions situées au pied de montagnes ou en bordure de bassins fermés, on rencontre des eaux douces; au contraire, dans les régions plus éloignées, la concentration en sels des eaux des nappes phréatiques s’accroît. Les eaux deviennent d’abord un peu, puis de plus en plus salées, jusqu’à être sursalées au centre des cuvettes.
L a composition chimique dépend également de la nature des roches et de la durée du trajet des eaux. Elle varie suivant l’alimentation des nappes. L’infiltration directe des pluies et des eaux superficielles donne des eaux douces qui peuvent gagner lesnappes. Mais quand l’évapotranspiration agit sur les eaux avant qu’elles descendent en profondeur, la teneur en sels augmente. On voit donc que l’hydrogéologie des déserts se caractérise par la variété de la compo-
sition chimique : eaux douces, eaux salées et eaux sursalées; on voit aussi que les lieux de formation et de présence des eaux douces, salées et sursalées sont souvent tout proches les uns des autres.
D e plus la zonalité suivant la profondeur n’est pas toujours respectée : on peut rencontrer en profondeur des eaux sous pression plus douces qu’en surface. Cela se constate dans les déserts russes aussi bien qu’au Sahara où la nappe d’eau douce, dite de l’Albien, se trouve sous les nappes salées du Miopliocène et du Quaternaire. L’étude des phénomènes hydrogéologiques actuels ne suffit pas à élucider la géo-
chimie des eaux souterraines dans les déserts et les semi-déserts. I1 faut prendre également en considération les oscillations des continents et les conditions paléo- géographiques. Dans les régions arides qui ont subi, au Quaternaire, des surélévations générales, les processus de lessivage et de dessalure des roches ont amené un abaissement de la teneur en sels non seulement des rivières et des bassins fermés, mais également des nappes. U n abaissement général provoque les phénomènes inverses : salure des roches, accumulation de sels dans les eaux souterraines.
Les conditions paléogéographiques ont certainement aussi leur importance : les périodes froides et pluvieuses auront favorisé l’apport d’eaux douces, et les périodes chaudes et sèches celui d’eaux salées. Ces eaux douces et ces eaux salées, en raison des temps géologiques que mettent souvent les eaux à parcourir les grandes nappes ont pu, dans certains cas, se maintenir dans les nappes profondes jusqu’à nos jours.
83

C H A P I T R E V
Les différents traceurs, la circulation microscopique de l’eau dans les terrains,
les traceurs radio-actifs
Les traceurs sont utilisés depuis longtemps en hydrologie en vue de suivre l’écoulement des eaux souterraines : 1. Pour savoir si un terrain est perméable. On regarde si le traceur pénètre dans le
terrain. 2. Pour rechercher une communication directe entre deux endroits. On jette le traceur
en amont dans la nappe ou le réseau d’eau et l’on examine son apparition en aval aux points où l’on pense qu’il doit arriver. I1 n’est donc pas question de temps. C’est une opération purement qualitative.
3. Pour rechercher la direction de l’écoulement. Dans une nappe, par exemple, on injecte le traceur dans un puits central et l’on repère son arrivée dans un des puits disposés en cercle autour du puits d’injection. C’est en somme une opération sem- blable à la précédente.
4. Pour mesurer la vitesse d’une circulation souterraine d’eau. I). s’agit alors de calculer le temps mis par le traceur pour aller d’un point à un autre. Cette opération quanti- tative n’est pas aussi simple qu’elle apparaît au premier abord, c o m m e nous le verrons plus loin.
5. Pour mesurer les débits. On verse dans le courant d‘eau en un point amont un débit donné d’eau renfermant une concentration donnée d’un traceur et l’on détermine en un point aval la concentration du traceur dans le courant.
LES CARACTÈRES D’UN TRACEUR IDÉAL
I1 est bien évident que n’importe quelle substance ne peut servir de traceur. Elle doit présenter certains caractères. Elle doit pouvoir être facilement déterminée quali- tativement, voire le plus souvent quantitativement, m ê m e lorsqu’elle se trouve en très faible concentration dans l’eau, car elle sera toujours très fortement dilu6e aux points de repérage.
Dans ses relations avec l’eau, le traceur doit être absent ou 2 la rigueur n’exister qu’en très petite quantité dans l’eau de la nappe ou des courants souterrains, pour qu’on puisse obtenir des différences utiles de concentration entre les points d’injection et les points de repérage.
I1 doit être facilement transporté par l’eau, c’est-à-dire soit se maintenir très aisé- ment en suspension, soit avoir une solubilité élevée.
I1 ne doit pas se décomposer ni se précipiter dans l’eau d’injection, ni dans l’eau de la nappe.
84

Les diflérents traceurs
I1 ne doit pas pouvoir se détruire sous l’influence de micro-organismes pendant son trajet souterrain.
Dans ses relations avec le terrain, il ne doit pas être absorbé, adsorbé ou retenu par le milieu poreux. I1 ne doit pas (( contaminer )) chimiquement le terrain trop longtemps, c’est-à-dire
être restitué trop tardivement par ce m ê m e terrain, ce qui écarterait les possibilités d’expériences successives, rapprochées les unes des autres. Enfìn il ne doit pas réagir chimiquement avec le milieu poreux. Du point de vue pratique, il doit pouvoir être obtenu facilement et à bas prix. 11
ne doit nécessiter que des opérations techniques simples, rapides et peu onéreuses. I1 ne doit présenter aucun danger dans les conditions normales d’emploi, au moment de l’injection, en raison de sa nocivité, car le traceur est obligatoirement utilisé en concentration élevée. Enfin il ne doit pas contaminer dangereusement l’eau étudiée, surtout au-delà d’un temps raisonnable.
LES DIFFÉRENTES SORTES DE TRACEURS NON RADIO-ACTIFS
Jusqu’à ces dernières années, on avait recours à des traceurs solides entraînés par l’eau, à des traceurs chimiques facilement solubles et facilement repérables et surtout à des traceurs colorants qui, à très grande dilution, sont encore visuellement distin- guables. Ils ont rendu bien des services. Ils ont chacun leurs qualités, mais aussi des défauts. Et maintenant l’avenir s’ouvre aux traceurs radio-actifs, à peu près tous détectables à des concentrations extrêmement faibles. Les uns ont une durée de vie faible, ce qui peut être extrêmement utile, tandis que d’autres ont une durée de vie très longue, ce qui est un avantage considérable dans l’étude des cheminements de très longue durée. Nous donnerons d’abord un aperçu des traceurs solides, chimiques et colorants, ce
qui permettra de mieux distinguer les qualités et les défauts des traceurs radio-actifs, car eux non plus ne sont pas parfaits.
Traceurs solides.
Nous pouvons distinguer trois sortes de traceurs solides : ceux qui flottent, ceux qui sont immergés et ceux qui sont en suspension.
Les traceurs qui flottent n’indiquent que la vitesse la plus élevée, celle du courant supérieur, et non la vitesse moyenne, qui importe le plus. En outre, ils ne peuvent être utilisés dans les nappes d’eau souterraines où ils sont retenus, ou m ê m e dans les terrains calcaires ordinaires. I1 faut qu’il y ait vraiment des chenaux de très grandes dimensions pour que le transport ne soit pas trop gêné.
Les traceurs immergés, c’est-à-dire ceux qui sont situés à une certaine profondeur dans la masse liquide et soutenus par un flotteur situé en surface, sont encore moins utilisables que les précédents en raison de leurs plus grandes dimensions.
Les traceurs en suspension sont plus facilement transportés par l’eau et peuvent ainsi servir en particulier dans les terrains largement fissurés. On leur demande de pouvoir se maintenir constamment en suspension, d‘être assez petits pour ne pas être arrêtés par les pores et chenaux. Leur emploi dépend donc essentiellement des dimensions de ceux-ci et de ceux-là. Et s’ils sont très finement divisés, ils ne doivent pas se fixer contre les parois des éléments constitutifs de l’aquifère. On a utilisé les traceurs solides les plus divers : balle d’avoine, son, grains d’amidon
facilement détectables par l’iode, levures (Saccharomyces cerevisiae, S. mycoderma), bactéries (B. violaceus, B. pyocyaneus, M. prodigiosus, M. aceti).
85

Hydrologie des régions arides
Traceurs chimiques solubles.
Les traceurs chimiques présentent cet avantage qu’il est facile d’en trouver de très solubles. Ils font ainsi vraiment corps avec l’eau qui les transporte. Et, lorsqu’ils ressortent des terrains en concentration suffisamment élevée, ils sont souvent aisément repérés par des réactions chimiques, ou m ê m e par des mesures de conductivité élec- trique.
Cependant ils présentent certains défauts. Leur diffusion, surtout si leur concentra- tion est élevée et si la vitesse de circulation de l’eau est extrêmement faible, est de nature à amener des perturbations. Mais ce n’est là, dans la plupart des cas, que leur moindre défaut.
Dans certains cas, on ne peut les injecter qu’à des concentrations élevées pour pouvoir les repérer à l’aval. Mais la densité de leur solution est alors beaucoup plus élevée que celle de l’eau, ce qui les fait descendre aux points les plus bas, empêche un mélange parfait et les place souvent hors circuit.
D’autre part, certains cations sont fortement adsorbés par l’argile ou la matière organique du terrain aquifère ou du toit ou du mur de celui-ci. C’est le cas des ions Na, K, Ca, Mg, Li, Ba, etc., suivant la composition de l’argile. Enfin, quelques-uns réagissent avec l’aquifère, en particulier avec l’argile. Une
concentration élevée de N a peut peptiser l’argile et diminuer la perméabilité de l’aquifère, modifiant ainsi la vitesse de circulation de l’eau. Les ions Ca agissent en sens contraire, en coagulant l’argile.
Les meilleurs traceurs seront donc les anions qui se fixent très diflicdement. Les traceurs chimiques les plus utilisés sont les chlorures (NaCl, CaCl,, LiC1,
NH,Cl), tous très solubles, facilement repérables par dosage classique du chlore OU par mesure de la conductivité électrique. Leur grand avantage est que l’ion C1 n’est pas adsorbé et ne réagit pas avec le terrain. Par conséquent il ne subit pas de retard trop important lors de son cheminement et se perd peu en route, d’où un excellent rendement. Mais l’écueil est la tentation de l’utiliser en concentration trop élevée au point d’injection : il risque alors de s’égarer, et de ne pas apparaître. D e plus, il est souvent nécessaire de l’employer eli très grande quantité ; l’opération est alors on6reuse. Ainsi pour marquer une eau de 10 mg de NaCl par litre, il faut en employer lo* fois plus que la fluorescéine reconnaissable à la dilution de 10-9. L à où 1 kg de fluorescéine est suffisant, il est nécessaire d’injecter 10 tonnes de NaCl. Autrement dit, là où 1,011 injecterait 50 litres d’eau ayant dissous de la fluorescéine, il faudrait injecter 38 m3 d’eau saturée en NaCl, avec les inconvénients précédemment relevés dus à la densité de la solution de NaCl. En outre, lorsqu’on a affaire à des eaux souterraines très chlorurées, comme dans
les pays arides, l’utilisation des chlorures comme traceurs devient encore plus difficile, car l’accroissement de la concentration en chlore aux points de détection est moins facile à saisir. Aussi y a-t-il avantage, dans ce cas, à employer le bichromate de sodium
(Crz0,Na,2H,0), dont la solubilité est de 2,380 kg à O0 et de 4,330 kg (à l’état anhydre) par litre d’eau. I1 peut être décelé à la dilution de 1 à 2 x en employant la diphé- nylcarbazide comme réactif1. Le nombre P de kilogrammes à employer dans les terrains calcaires (Ravier, Hours, Schneebeli [210]) est de :
dL P=3--’0.1V
a
J = débit en m3/s aux sources. L = longueur en mètres, à parcourir dans le sol.
i, F , GENET, Note sur des ddterminations de cheminement< souterrain3 p m la methode chimique m, Terre8 et eaux, suppl6- picnt scientifique, na 3, 19.54, p. 73-81.
86

Les diffërents traceurs
a = vitesse, en mètres/jour, du courant souterrain. V = réserves d’eau souterraine en milliers de mètres cubes.
Bien d’autres traceurs chimiques ont été utilisés : l’acide borique, le tétraborate de sodium, le bromoforme, les détergents commerciaux, le sucre, la dextrose, le phénol, ce dernier pouvant être décelé à l’odorat ou au goût, surtout si on le transforme en chlorophénol. Les sucres ont l’inconvénient d’être attaqués par les micro-organismes.
Traceurs colorants.
Les traceurs colorants présentent de très sérieux avantages. Leur solubilité est grande et leur détection peut se faire sur de très faibles concentrations de l’ordre de 10-9. Mais la plupart d’entre eux sont adsorbés par les matières argileuses ou les matières organiques. Certains réagissent avec le CO, dissous de l’eau, le pH, les substances ferrugineuses, les matières organiques. Aussi n’a-t-on pas enregistré uniquement des succès.
Généralement leur utilisation est à déconseiller dans les aquifères à porosité d’in- terstices, surtout si ceux-ci renferment des matières argileuses, ont des pores tres fins et ne laissent circuler l’eau qu’a très faible vitesse. Par contre ce sont des traceurs presque parfaits pour l’étude de la circulation de
l’eau dans les karst, à circulation rapide et à courants resserrés. Le traceur le plus connu est la fluorescéine (C,oH,oO,Na,). On utilise Bgalement
l’uranine qui n’est qu’une fluorescéine rendue plus soluble par l’addition de carbonate de soude.
L a fluorescéine est adsorbée par les matériaux argileux. Elle réagit avec les matières organiques, les oxydes ferriques. Elle est détruite par la ludere. Elle est affaiblie par le CO, dissous dans l’eau et l’acidité de l’eau. Mais, dans ce dernier cas, on peut la régénérer en alcalinisant l’eau. A l’œil nu elle est détectée à la concentration de 10-7, avec le fluorescope Trillat a la concentration de 2 x 10-9 et parfois de 1 x et avec le fluorescope électrique de Dienert à la concentration de 1 x 10-9. Ce dernier n’exige que 45 cm3 d’eau. I1 en faut 250 cm3 avec le fluorescope Trillat. On dissout dans l’eau la fluorescéine préalablement diluée dans de l’alcool additionné d’ammo- niaque (0’25 litre de NH, pour 5 litres d’alcool et pour 50 litres d’eau par kilogramme de fluorescéine). Le poids P de fluorescéine à employer est, d’après Ravier [210] :
P = 12 - + 0,02 v dL a
Oil P est le poids en kilogrammes de fluorescéine. k = 0’5 dans le cas de circulation en fissures ou en chenaux. k = 3 dans le cas de circulation en interstices. d est le débit des sources, en m3/s. L la longueur du trajet en mètres. V le volume de la réserve souterraine en milliers de mètres cubes. a la vitesse en mètres/jour du courant souterrain.
Si le cours d’eau ou bien l’injection d’eau pendant quarante-huit heures a un débit Dienert 1 utilisait les formules suivantes :
de plus de 5 l/s, on emploiera une quantité de fluorescéine : P = 2,5 x lO-’dL
Si le cours d’eau ou bien l’injection d’eau pendant douze heures a un débit de moins de 5 l/s, on emploiera :
1. F. DIENERT. Hydrologie agricole. Parie, Baillitc, 1932, 462 pages.
a7

Hydrologie des régions aridea
125 x 10-sd x L b
P =
où P est le poids en kilogrammes de fluorescéine. d le débit des sources, en m3/s. L la longueur du trajet, en mètres. b le débit moyen en m3/s dont on dispose pendant douze heures.
Bien d’autres colorants peuvent être utilisés. Dans le cas d’eaux alcalines, on emploie, outre la fluorescéine, l’éosine, l‘érythrosine,
le rouge congo. Avant de jeter les colorants dans la nappe on les dissout dane une petite quantité d’eau alcaline. Dans le cas d’eaux acides, on utilise le bleu de méthylène, le bleu d’aniline, le rouge ponceau. Ils sont alors, avant leur utilisation, dissous dans une eau faiblement acide.
Parmi les autres traceurs colorants notons la phénolphtaléine, la flavizine, le bleu carmin, le vert acide, le violet acide, la fuchsine acide.
Voici les quantités de colorants en grammes à employer par 10 mètres de longueur de trajet, selon Siline-Bektchourine [171].
Colorant Rochas Roches Roches Rochen argileuses sobleuses fissur6es karstiques
Fluoresc6ine . . Uranine . . . Éosine . . . . Erythrosine . . Rougecongo . . Bleu de méthylène. Bleu d’aniline . . Rouge ponceau . .
. . . . .
. . . . .
. . . . .
. . . . .
. . . . .
. . . . .
. . . . .
. . . . .
g
5-20 5-20 5-20 10-40 20-80 20-80 20-80 10-40
g
2-10 2-10 2-10 10-30 20-60 20-60 20-70 10-30
6
2-20 2-20 2-20 10-40 20-80 20-80 20-80 10-40
g
2-10 2-10 2-10 10-40 20-80 20-80 20-80 10-40
LA CIRCULATION D E L’EAU DANS LES TERRAINS
Les traceurs jetés en un point amont sont portés par l’eau jusqu’en un point aval où on les repère. Mais les filets liquides porteurs n’ont pas tous la m ê m e vitesse et ils ne suivent pas tous des chemins parallèles ou des chemins directs. Les diffhrentes portions du traceur arriveront donc au point aval en des temps variés, qu’il s’agit d’interpréter.
Diflérents trajets.
Les trajets sont nombreux. On peut ainsi distinguer : 1. Un trajet général qui dépend du vecteur caractérisant le m a x i m u m de gradient
du potentiel de vitesse, dérivant de la loi de Darcy. Mais, à l’intérieur de ce trajet général de vitesse moyenne, il y en a de plus rapides et de plus lents.
2. Des trajets irréguliers, plus rapides, dus à une perméabilité plus grande de certaines parties de l’aquifère, par suite de l’existence de pores plus larges et plus nombreux, de chenaux. Les cheminements plus faciles qui en résultent peuvent être parallèles ou parfois obliques au cheminement général.
3. Des trajets irréguliers plus lents, résultant d’une perméabilité plus faible, le long, ou en travers de certaines parties du circuit général.
88

Les diférents traceurs
4. Des trajets, les uns plus rapides, les autres plus lents, régulièrement disposés dans une roche homogène. Dans la m ê m e roche, par exemple un sable de granu- lométrie très hétérométrique, coexistent des interstices larges et des interstices étroits. Des filets liquides sont, sur certains trajets ou plus exactement certaines portions de trajet, les uns rapides, les autres lents. Mais c o m m e tous les pores communiquent entre eux, dans une roche homogène, les filets liquides de vitesse différente se mélangeront et les probabilités d’avoir en un point aval des filets ayant constamment cheminé plus rapidement que d’autres seront extrêmement faibles.
5. Des vitesses plus grandes et d’autres plus faibles dans un m ê m e conduit. Les filets liquides du centre des conduits se meuvent plus rapidement que ceux qui longent les parois. En mouvement turbulent, il y a brassage de sorte que toutes les molécules d’eau
possèdent à peu près la m ê m e vitesse de circulation moyenne lorsque le trajet est assez long. En mouvement laminaire, les filets liquides se meuvent parallèlement les uns
aux autres et dans un conduit ayant partout les mêmes sections en forme et en dimension, la distribution des vitesses est régulière.
Mais dans les roches, les sections des capillaires sont irrégulières le long du trajet des filets liquides. I1 y a donc ici également une possibilité de mélange entre filets liquides de vitesses différentes; d’autre part il est plus probable que dans le cas des roches à porosité d’interstices de trouver en aval des filets ayant constamment cheminé plus rapidement ou plus lentement ; cette probabilité reste d’ailleurs relativement faible, si le trajet est long.
On voit que la probabilité des points 4 et 5 est d’autant plus faible que la roche est à pores ou fissures plus fins et plus nombreux, tandis que, dans un massif calcaire à larges fissures très espacées, sans mouvements turbulents de l’eau, elle est beaucoup plus grande.
Trajet général.
C’est donc celui que définit la vitesse apparente de la loi de Darcy de l’écoulement laminaire de l’eau dans les terrains, ou, dans le cas de mouvement turbulent ou mixte, celui que définit la vitesse apparente
C’est la vitesse qu’on obtient en divisant le débit par la section de terrain. L a vitesse moyenne, plus grande que la vitesse apparente, est celle qu’on obtient en divisant le débit par la section du ou des conduits.
Ce sont ces vitesses apparentes ou moyennes qu’on cherche à obtenir à l’aide dea traceurs. Et il s’agit de voir comment on peut les obtenir à l’aide des différentes vitesses données par les traceurs.
Trajets détournés.
Ils apporteront des filets porteurs de traceurs, soit avant, soit après les filets porteurs des trajets directs, et pourront produire des pics indépendants du pic général. Mais on conçoit que les filets porteurs détournés puissent à l’arrivée interférer avec les filets porteurs directs. On ne peut établir aucune loi générale les concernant, sinon admettre leur existence
à partir des constatations faites aux points de repérage.
89
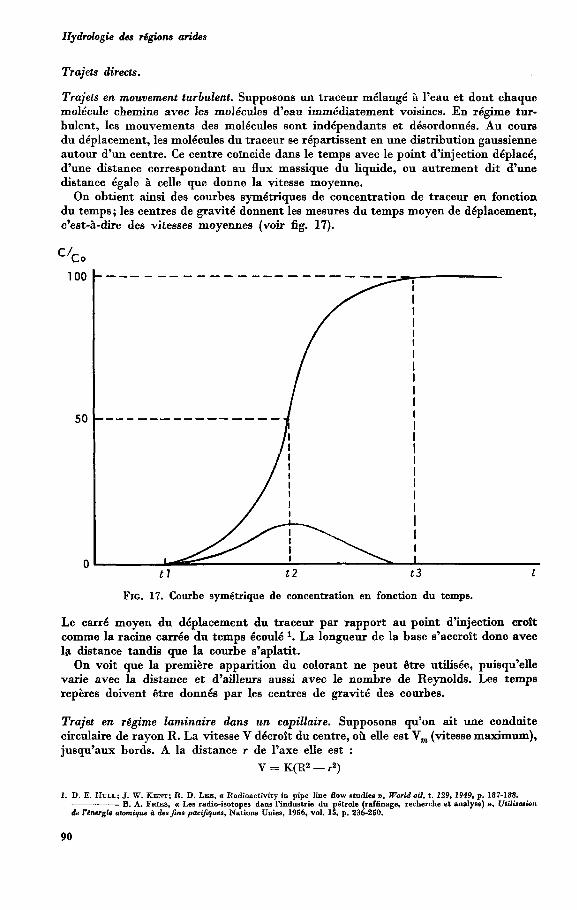
Hydrologie des régions arides
Trajets directs.
Trajets en mouvement turbulent. Supposons un traceur mélangé à l’eau et dont chaque molécule chemine avec les molécules d’eau immédiatement voisines. En régime tur- bulent, les mouvements des molécules sont indépendants et désordonnés. Au cours du déplacement, les molécules du traceur se répartissent en une distribution gaussienne autour d’un centre. Ce centre cohcide dans le temps avec le point d‘injection déplacé, d’une distance correspondant au flux massique du liquide, ou autrement dit d’une distance égale à celle que donne la vitesse moyenne. On obtient ainsi des courbes symétriques de concentration de traceur en fonction
du temps; les centres de gravité donnent les mesures du temps moyen de déplacement, c’est-à-dire des vitesses moyennes (voir fig. 17).
C’Co 1 O0
50
I I I I I I
I h II
/ I / i I I
I 1 I I I I I
tl t2 t3 t
FIG. 17. Courbe symétrique de concentration en fonction du temps.
L e carré moyen du déplacement du traceur par rapport au point d’injection croit c o m m e la racine carrée du temps écoulé l. L a longueur de la base s’accroft donc avec la distance tandis que la courbe s’aplatit. On voit que la première apparition du colorant ne peut être utilisée, puisqu’elle
varie avec la distance et d’ailleurs aussi avec le nombre de Reynolds. Les temps repères doivent être donnés par les centres de gravité des courbes.
Trajet en rdgime laminaire dans un capillaire. Supposons qu’on ait une conduite circulaire de rayon R. L a vitesse V décroît du centre, oh elle est V,,, (vitesse maximum), jusqu’aux bords. A la distance r de l’axe elle est :
V = K(R2 - r2)
1. D. E. HULL; J. W. KENT; R. D. LEE, a Radioactivity in pipe line flow studies n, World air, t. 129. 1949. p. 187-188. E. A. FRIES, u Les radio-isotopes dans l’industrie du p6trole (raffinage, recherche et analpe) n. UIilisorion
de I’lnergie afomque ¿ desfrns pacifiques, Nations Unies, 1956, vol. 15, p. 236-250.
90

Les digrents traceurs
avec
p étant la viscosité de l’eau. L a vitesse maximum est atteinte quand r = O :
V,,, = KR2
d’où
C o m m e la vitesse moyenne est : R2 U = K - 2
on voit que, dans une section, la vitesse des $lets les plus rapides est le double de la vitesse moyenne de l’eau dans le tube. Autrement dit, la vitesse des fìlets liquides amenant l’apparition première du colorant donnera le double de la vitesse moyenne effective de l’eau l.
Examinons maintenant la concentration moyenne du traceur au temps t, à une distance x du point de départ. Elle est [193] :
I
L a courbe de concentration du colorant à la distance x, en fonction du temps, présente toujours une queue excessivement longue. On constatera donc toujours une tendance à la présence d’un reste de nuage de traceur.
L a m ê m e équation montre que la médiane est égale aux deux tiers du temps théorique de (( détention 1) T, c’est-à-dire du temps moyen de transport du traceur, et que la moyenne harmonique est à 3/4 T. D’après Archibald, la meilleure mesure de la période de détention est le ‘?Oe centile. Mais aucune expérience n’a cependant été faite pour contrôler cette équation. I1 est à noter toutefois que, pour beaucoup d’auteurs, la médiane, qui correspond au centre de gravité, donnerait le temps de détention.
Trajet dans un sable de dimensions lat6rales illimit6es. Dans les cas précédents, il était question d’un acheminement dans un chenal bien limité latéralement. Ce serait celui de conduits aquifères, c o m m e il peut en exister dans le calcaire, mais plus rarement dans les roches à porosité d’interstices.
Dans ces dernières, il n’y a pas de limites latérales complètes pour les filets liquides, à part le toit et le m u r imperméables. I1 y a possibilité pour un filet liquide passant en un point donné de s’étaler en aval. Et c’est cet étalement qu’il y a lieu d’étudier avec Dane1 2.
Dans les conditions normales de circulation souterraine, le mouvement de l’eau dans les roches à porosité d’interstices s’effectue en régime laminaire. Mais alors que, dans un tube, il n’y a aucun mélange des filets liquides, il n’en va pas de m ê m e dans une roche à porosité d’interstices c o m m e les sables où tous les pores communiquent entre eux de six ou huit côtés à la fois (fig. 18 et 19).
1. P. DANEL, u La mesure des d6bits des eaux souterraines n, Actes du colloqua d’Ankara sur l’hydrologie de la zone aride. Unesco.
2. ID., ibad. 1953, p 104-112.
91

Hydrologie des régions arides
f c
I
18
19 20
FIG. 18. &coulement intergranulaire. FIG. 19. gcoulement intergrandaire. FIG. 20. Cône de diffusion ou cône de dispersion.
Un filet liquide allant de pore en pore aura donc tendance, en arrivant en face de chaque grain, à se subdiviser en deux, trois ou quatre autres filets qui le contourneront. Chacun de ces filets secondaires en fera autant en arrivant en face du grain aval et ainsi de suite. Mais le filet liquide voisin du premier en fera autant et tous les filets secondaires, tertiaires, etc., qui en résulteront, chemineront avec les filets secondaires, tertiaires, etc., issus du premier. On conçoit donc qu’il puisse se produire un mélange des filets liquides, les uns avec les autres.
Ainsi, au fur et à mesure du cheminement, les filets liquides tendront à s’écarter de plus en plus du chemin direct. I1 se produira un cône de dispersion (voir fig. 20). Si le filet liquide est porteur d’un traceur, ce traceur se répand donc graduellement
suivant un cône dont l’angle au sommet, variable suivant la nature des grains, est le plus souvent d’environ 60. L a répartition des concentrations dans chaque section affecte une forme de courbe en cloche, très voisine de la courbe de Gauss et, à l’intérieur du cône, la concentration diminue c o m m e l’inverse du carré de la distance au sommet. A une certaine distance du point d’injection, le diamètre du cône de dispersion
deviendra égal à l’épaisseur de la couche aquifère. Si le cône a bien un angle de 60, cette distance est sensiblement égale à dix fois l’épaisseur de l’aquifêre. Plus loin le cône s’aplatira de plus en plus. Et lorsque le diamètre latéral du cône aura un grand nombre de fois l’épaisseur de la couche, la concentration ne diminuera plus qu’inver- sement avec la distance.
92

Les différents traceurs
On peut imaginer que la circulation dans un sable se fait dans une série de capillaires plus ou moins parallèles et de sections différentes de rayons rl, r2, r3, etc., en série décroissante. L a vitesse maximum sera donc : V, = Kr; et la vitesse moyenne effec- tive sera
1 - MK(R! + Ri + Ri + ...)
4 2 u, = s = M(r: + r; + r: + ...)
q étant le débit de la section S du terrain.
L a vitesse moyenne effective sera donc inférieure à la vitesse moyenne du tube rl’ Ainsi Danel écrit : (( I1 semble que, dans un écoulement en milieu poreux, la vitesse
moyenne effective est en général inférieure ou au plus égale à la moitié de la vitesse maximum correspondant à l’apparition de la coloration. Expérimentalement on trouve que la vitesse moyenne effective est de la moitié au tiers de la vitesse maximum. D
Le temps d’apparition du maximum de concentration du traceur, après apparition du colorant, ne dépend que de la largeur de la vague de traceur au départ, c’est-à-dire de la quantité de solution de traceur et du temps qu’on a mis pour l’injecter au départ. On ne peut donc prendre ce maximum pour calculer la vitesse moyenne.
L’ADSORPTION ET LA RÉTENTION D E S TRACEURS
Les eaux renfermant les traceurs peuvent entrer en contact dans l’aquifère avec des substances ayant la propriéte de fixer des ions contenus dans l’eau; elles les échangent contre certains des leurs. L’adsorption peut être chimique à liaisons énergiques, ou simplement physique (adsorption de V a n der Waals à faibles liaisons entre l’adsorbant et l’adsorbé). Ces deux liaisons entre lesquelles il y a tous les intermédiaires peuvent d’ailleurs agir simultanément. Les substances contenues dans les terrains et SUS- ceptibles d’adsorber et d’échanger sont : lo les minéraux argileux (vermiculite, mont- morillonite spécialement et, à un degré moindre, illite, halloysite et kaolinite, ces minéraux étant rangés suivant l’ordre décroissant de capacité d’échange de bases) ; 20 les minéraux zéolithiques; 30 l’hydroxyde de fer; 40 les substances organiques, l’humus par exemple.
Nous appelons (( permutolites )) ces substances échangeuses. L e degré de fixation dépend de la nature des cations. Ceux-ci sont d’autant plus énergiquement fixés que l’ion est moins hydraté. Et les ions bivalents se fixent plus énergiquement que les ions monovalents à degré d’hydratation égal.
L a relation entre les ions A et B qui s’échangent entre eux, de l’eau au terrain et du terrain à l’eau, est de la forme :
[A] eau [BI eau ( [BI permut.
( [A] permut. & = k
Ainsi l’adsorption des cations est très aisée, en particulier celle de Ca, Mg, Na, K, NH,, Li, Rb, Sr, etc.
L’adsorption est d’autant plus facile que les terrains ont moins de cations précédem- ment fixés et que l’eau est plus riche en cations. Nous aurons à en tenir compte dans l’emploi des t r a “ radio-actifs cationiques, ce qui nous amènera à utiliser des entraîneurs.
93

Hydrologie des rdgwns arides
L‘adsorption sera faible dans les terrains purement siliceux, calcaires, dolomitiques, c o m m e il en existe beaucoup. Tandis que les adsorptions et les échanges seront d’autant plus importants qu’ils seront plus riches en permutolites : substances argileuses, orga- niques, hydroxyde ferrique, glauconie, etc.
Les anions et les éléments non ionisés peuvent subir une certaine adsorption physique et présenter des isothermes caractéristiques d’adsorption. Mais cette adsorption est de beaucoup plus faible que celle des cations.
Les anions CI-, Br-, I-, NO,, Cr,O,, SO,, le bore, ne subissent qu’une très faible réten- tion, pour ainsi dire négligeable. C’est la raison pour laquelle, lorsqu’on utilise un sel, l’anion et non le cation doit être choisi c o m m e traceur. Ainsi, dans le cas des chlorures, NaCl, CaCl,, LiC1, NH,Cl, le traceur tout indiqué est le chlore. I1 ne faut pas oublier que certaines substances peuvent réagir chimiquement, soit
avec les terrains, soit avec les substances contenues dans l’eau : SO, et Ba++ en présence l’un de l’autre précipitent. Le glycérophosphate de calcium réagit aussi chimiquement I1 en est de m ê m e du groupe CO, dissous, HCO;, CO,. Enfin il y a toujours lieu de te& compte des possibilités du dépassement des produits de solubilité.
L a rétention des traceurs adsorbable6 par les terrains crée un empoisonnement de ceux-ci, c’est-à-dire qu’après passage d’un traceur dans un terrain qui en a adsorbé une partie, ce terrain le restitue si l’on y fait passer une eau qui n’en contient pas ou dans laquelle la concentration en traceur [A] est
\ [A] permut. )+ [BI permut.
[A] eau < [BI eau
quand il s’agit d’échange. I1 se constitue ainsi une sorte d’empoisonnement du terrain, d’autant plus important
que l‘ion est fixé plus énergiquement et en plus grande quantité. Au cours de son cheminement dans les roches, la concentration du traceur diminue
graduellement le long de son parcours : lo par les phénomènes de dispersion latérale des divers filets liquides; 20 par le fait que des filets liquides circulent plus rapidement que d’autres; 30 par suite de la rétention du traceur par le terrain.
Nous avons vu qu’en raison de la dispersion latérale des divers filets liquides, la concentration du traceur diminue de l’amont à l‘aval. Si le terrain n’est pas limité 1 atéralement, la concentration décroît c o m m e l’inverse du carré de la distance comptée depuis l’origine. Si le flux chargé de traceur est limité par deux plans parallèles, ce qui est le cas des couches aquifères, la concentration diminue c o m m e l’inverse de la distance. Si le flux est limité latéralement de tous les côtés et si les filets liquides cheminent
tous avec la m ê m e vitesse, la concentration reste la m ê m e le long du trajet. Mais des filets liquides chargés de traceur avancent plus rapidement que d’autres.
Ils se mélangent alors avec des filets liquides en retard, non porteurs de traceur. Le front de liquide à une distance donnée du point de départ présente ainsi une certaine longueur dans le sens de la marche. L a concentration à l’extrémité amont du front est très faible et elle augmente progressivement vers l’extrémité aval où elle atteint sa valeur normale. Dans le cas d’un cheminement à filets tous parallèles les uns aux autres, c o m m e à l’intérieur d’un cylindre dans lequel à la section du point de départ la concen- tration est partout Co, la concentration C du colorant passe progressivement de la valeur O à la tête, à la valeur Co à la queue du front, C/Co va de O à 1.
Mais si, dans les mêmes conditions d’écoulement, il y a fixation des cations par le terrain, le front du traceur est retardé. En effet les cations sont fixés à une certaine distance du point de départ, diminuant progressivement la concentration en cations du traceur vers l’aval. Si un nouveau liquide dépourvu de traceur entre en contact avec le terrain ayant
fixé le traceur, c o m m e le liquide est sous-saturé vis-à-vis du terrain, une désorption de celui-ci se produit.
94

Les diffhents traceurs
L e traceur entrant plus loin en contact avec un terrain qui en est dépourvu, une nouvelle fixation a lieu et ainsi de suite, ce qui a pour effet de retarder le front. Nous avons donc à considérer avec Kaufmann et Orlob [206] les rapports entre
la vitesse du front du liquide lui-même et la vitesse U a du front retardé du traceur. Ce rapport est, d‘après Vermeulen et Hister :
1 - Ua -_ U ra.pb
I f - r[A].m
ra. pb r[A].m
DA = -
où ra
pb r[A] la concentration en milliéquivalents par centimètre cube de la phase liquide; m la porosité.
Ces équations (1) et (2) convenant à l’étude du débit constant à travers des colonnes de sol montrent que les traceurs ne peuvent être utilisés pour définir les variations de vitesse d’un liquide dans un milieu poreux. On peut étendre l’équation (1) en supposant qu’une lame de liquide de volume m.dx
parcourt lentement la distance dx à travers un milieu poreux, en transportant r[A].m.dx milliéquivalents de traceur. Le traceur se distribue entre l’eau et la roche. Si l’équilibre d’échange de fixation se fait instantanément, la lame de traceur parcourt la distance (Ua/TJ) dx de telle façon que :
est le nombre de milliéquivalents du traceur adsorbé par gramme de la phase solide ; la densité apparente du milieu poreux;
Ua Ule U U
r[A].m.dx = m.pb - dz.r[A].me - dx
Si la roche n’a aucune capacité d’adsorption, ra = O, d’où Ua = U; les deux se dé- placent ensemble. Si au contraire la roche a une capacité d’adsorption et si les phéno- mènes d’adsorption et de desorption sont instantanés, le front du traceur aura la m ê m e longueur et la m ê m e netteté que celui du liquide. I1 sera seulement retardé. Mais les réactions d’échange des ions entre le liquide et la roche demandent un
certain temps. Le front du traceur non seulement ne cheminera pas aussi vite que le front du liquide, mais il sera de plus en plus diffus.
LES MÉTHODES D’INJECTION DE s TRACEURS DANS L’AQUIPÈRE
I1 y a deux méthodes d’injection des traceurs : injection à débit continu, et injection massive de très courte durée.
Méthode d’injection continue, méthode par dilution.
Cette méthode consiste à verser un débit constant q de solution du traceur, à la concen- tration c, dans un courant dont on veut connaître le débit Q. On mesure en un point aval, oh le mélange est assuré, la concentration ce du courant,
Q + q. On a alors pour débit Q : e - eV
CV Q = q -
Et lorsque c, est trèe petit par rapport à c, l’équation précédente se simplifie :
95

Hydrotogie des régions arides
Mais il y a toujours lieu d’être très circonspect. Supposons qu’entre le point amont d’injection et le point aval s’ajoute un courant latéral, le débit mesuré sera celui du point aval. Si au contraire le courant se divise entre le point d’injection et le point de détection, le débit mesuré sera celui qui passe au point d’injection.
Cette méthode peut être appliquée aux courants circulant dans les massifs calcaires. Mais elle ne permet pas de calculer la vitesse lorsque les sections moyennes des écoule- ments sont inconnues entre les points amont et aval. Dans les nappes à perméabilité d‘interstices, il n’est possible de calculer ni les débits
ni les vitesses, car le débit Q déterminé ne peut être rapporté à une section quelconque de la nappe. C’est un débit partiel, de proportion inconnue. A fortiori on ne peut estimer la vitesse.
M6thode d’injection continue, mgthode de vitesse.
On injecte c o m m e précédemment au point amont, de manière continue, autant que possible avec un débit constant de solution du traceur; et l’on détermine le temps que met le front du traceur pour arriver au point d’observation. C o m m e la précédente, c’est une méthode onéreuse puisqu’elle nécessite une grande quantité de traceur.
1. Le terrain n’a aucune capacité d’adsorption. Les filets les plus rapides, porteurs de traceurs, arrivent au temps t, puis des filets de plus en plus lents, en m ê m e temps que les filets plus rapides, continuent à amener le traceur. Et, au bout d’un certain temps tg, tous les filets porteurs de traceur sont arrivés au point d’observation.
La courbe de concentration présente donc un front de concentration, qui croît de plus en plus rapidement, jusqu’à un temps t2, puis de plus en plus lentement jusqu’au temps tg à partir duquel la concentration reste constante.
C o m m e Dancl 1 l’a montré, la vitesse moyenne est au plus égale à la moitié de la vitesse maximum. Elle peut descendre au tiers de cette vitesse maximum. On se fonde sur ces valeurs pour déterminer la vitesse du courant. C o m m e nous l’avons vu, on ne peut utiliser le temps d’apparition du maximum d’intensité du traceur.
2. Le terrain a une capacité d’adsorption. Il faut alors en plus tenir compte des déca- lages dus aux phénomènes d’adsorption. Peu de travaux précis ont encore été faits sur ce sujet.
Deux cas sont à envisager :
Méth,ode d’injection par bougée.
Cette méthode consiste en une injection de très courte durée en un point amont. Puis l’on mesure le temps que le traceur met pour parvenir en aval.
Par suite de la différence de marche des divers flets liquides, la concentration en traceur à l’arrivée augmente jusqu’à une valeur maximum, car progressivement aux filets les plus rapides porteurs de traceur s’ajoutent les filets de plus en plus lents. Mais, par la suite, les filets les plus rapides amènent des eaux dépourvues de traceur de la partie amont de la bouffée; ces eaux se mélangent alors aux filets moins rapides; progressivement des filets de moins en moins rapides de la partie amont se comportent de la même manière. Si l’on construit un graphique de la concentration de l’eau au point d’observation
en fonction du temps, on obtient une courbe plus ou moins en cloche, mais avec une traîne. On a avantage à construire également une courbe cumulative de la concentra- tion en fonction du temps (voir fig. 21). Sur la courbe non cumulative on indique le temps t, d’apparition du traceur, la
hauteur ch du mode, c’est-à-dire la concentration maximum observée, le temps
1. P. DANEL, op. cit.
96

Les différents traceurs
t, correspondant à celui du mode, le temps t, de disparition du traceur, le temps moyen, t, = 1/2 (t, - te).
Sur la courbe cumulative on porte, outre les éléments précédents : le temps, t,, de la médiane, correspondant au centre de gravité, le temps, t,, du 10e centile, le temps t,, du 90e centile. On détermine l’écart D, interdécile, t,, - tlV Enfin on calcule le pourcentage de récupération du traceur, c’est-à-dire le pour-
centage du traceur retrouvé par rapport au traceur introduit. L a quantité de traceur retrouvé est donnée par la surface située sous la courbe cumulative. Ce pourcentage de récupération indique non seulement les pertes, mais aussi la dispersion des filets liquides à partir du point d’injection.
Ici aussi nous examinerons les deux cas : absence de phénomènes d’adsorption, adsorption notable du traceur.
Absence de phénomènes d’adsorption. En l’absence d’adsorption, la courbe de concentra- tion en fonction du temps affecte sensiblement une forme en cloche, à peu près symé- trique.
L a largeur de la courbe en cloche dépend essentiellement de la différence de vitesse des divers filets liquides circulant à l’intérieur de l’aquifère, de la différence à l’intérieur d’un m ê m e capillaire ou conduit aquifère et des différences dues aux diamètres divers des capillaires et des conduits. Cette largeur augmente avec la diversité des vitesses.
L a hauteur de la courbe en cloche, c’est-à-dire du pic, dépend tout d’abord des fac- teurs qui déterminent la courbe. C o m m e la surface sous la courbe représente la quantité de traceur écoulée, celle-ci reste la même, si à un allongement de la courbe correspond une diminution de la hauteur du pic.
Mais la hauteur de ce pic dépend aussi de la dispersion des filets liquides à partir du point d’injection. Si le milieu a de tiès grandes dimensions, de telle sorte que l’injection peut être considérée c o m m e ponctuelle, et que de ce point part un cône de dispersion régulier, la concentration le long d’une droite issue du point d’injection est inversement proportionnelle au carré de la distance. Dans une section perpendiculaire à l’axe du cône, la concentration est maximum sur cet axe et diminue graduellement de part et d’autre. Si le traceur remplit complètement toute l’épaisseur de l’aquifère, la diminution de
la concentration n’est plus que proportionnelle à la distance h partir de laquelle le traceur a rempli toute l’épaisseur de la couche aquifère.
Succession des pics. Les trajets détournés peuvent rendre dissymétrique la courbe, en augmentant la concentration de la traîne.
Dans d’autres cas, l’arrivée des trajets détournés peut produire des pics secondaires généralement situés en arrière du pic principal.
Mais on conçoit très bien qu’un trajet détourné passant par un conduit très conducteur puisse arriver au point d’observation avant le pic du mouvement général et m ê m e avant l’apparition du traceur du mouvement général. I1 importe surtout de connaître la vitesse moyenne de l’eau puisque c’est elle qui est
généralement l’objet de l’étude. Cette vitesse moyenne peut être évaluée dans un terrain d’après la porosité m et
le volume d’eau V qui s’est déplacé devant le front du traceur. Soit V, le volume du liquide contenu dans le terrain et qui sera déplacé devant
le front, c’est-à-dire entre le point d’injection et le point d’observation. Soit V le volume du liquide nécessaire pour déplacer V, on a alors :
Vf = s’ (1 - C) dV O CO
97

Hydrologie des régwns arides
V, peut être évalué par une intégration graphique de l’aire située au-dessus du front. Mais on remarque que les volumes peuvent être remplacés par les temps sur le
graphique car on peut passer des volumes V aux temps t en introduisant dans l’équation précédente les constantes m (porosité) et v (vitesse) :
t étant le temps moyen mis par le nuage pour arriver au point d’observation. C o m m e nous l’avons vu, d’après Danel, la vitesse moyenne serait ou supérieure ou égale à la moitié de la vitesse m a x i m u m correspondant à l’apparition du traceur, Et Danel trouva expérimentalement que cette vitesse moyenne est de la moitié à un tiers de la vitesse maximum.
D’après les expériences nombreuses faites en laboratoire sur des cylindres remplis de terre, la vitesse moyenne est donnée par la position du centre de gravité, c’est-à-dire par le temps avant lequel 50 yo du traceur est passé et après lequel 50 yo passera. D’après Archibald, au contraire, la meilleure mesure du temps théorique de détention est le 70,7e centile.
Pr6sence de phhomenes d’adsorption. Lorsqu’il y a des phénomènes d’adsorption et de désorption, de nombreuses incertitudes s’ajoutent. D’abord le temps d’apparition du traceur n’est plus le temps d’arrivée du ilet
liquide le plus rapide. Tout le front du traceur est décalé en arrière du front du liquide. Il se produit en effet un retard dû aux phénomènes d’adsorption et de désorption. Et ce retard est d‘autant plus grand que le traceur est plus fortement adsorbé et le trajet plus long. Aux diverses causes d’allongement du nuage du traceur qui jouent en dehors de tout
phénomène d’adsorption et de désorption, il faut ici en ajouter d’autres. Si les phé- nomènes d’adsorption et de désorption étaient instantanés, le front du nuage aurait toujours la m ê m e longueur sur tout son trajet. Mais les réactions d’échange demandent un certain temps. Aussi le front s’étend-il au fur et à mesure de sa progression. Si la vitesse de désorption est faible, il reste du traceur dans le terrain et la traîne du nuage devient très longue. La hauteur du pic, compte non tenu de tous les phénomènes en milieu non adsorbant,
diminue lorsque pour les causes précédentes le nuage s’allonge, mais elle dépend aussi des pertes définitives du traceur dans le terrain. Autrement dit le pic s’affaiblit d’autant plus que le milieu est plus adsorbant et le trajet plus long. On ne peut plus calculer la vitesse moyenne en utilisant par exemple l’apparition
du traceur, puisque cette apparition est retardée par rapport à l’arrivée des filets liquides les plus rapides. Et l’on ne peut calculer ce retard.
D e m ê m e le centre de gravité du traceur est retardé par rapport au centre de gravit6 des filets liquides, partis du point d’injection et arrivés au point d’observation. Ce retard ne peut lui-même être calculé.
Cela montre que s’il y a déjà des incertitudes avec les traceurs non adsorbables, ces incertitudes sont mille fois plus nombreuses avec les traceurs adsorbables, et que dès lors il y a lieu de rejeter ces derniers.
LES ENTRAINEURS
L a plupart des traceurs sont employés à des concentrations extrêmement faibles. Si donc il y a des phénomènes d’adsorption, la proportion de traceur adsorbé peut être très grande si le trajet est très long et la durée de contact avec le terrain très grande, de telle sorte que le traceur ne sera plus perceptible au point d’observation.
98

Les diffirents traceurs
Si au traceur radio-actif on ajoute son isotope non radio-actif, les phénomènes d’adsorption se porteront à la fois et indistinctement sur l’isotope radio-actif et l’isotope non radio-actif. La proportion de traceur radio-actif adsorbé sera donc considérablement diminuée. Ainsi au traceur 13lI on ajoutera l’iode ordinaire. I1 est donc souvent nécessaire d’ajouter un entraîneur, L a proportion de cet entraîneur sera indiquée plus loin.
UTILISATION DES TRACEURS RADIO-ACTIFS
En règle générale, l’emploi des traceurs radio-actifs ne s’impose que lorsque les traceurs chimiques ou colorants ont échoué, ou lorsqu’il est impossible de les utiliser. D e tonte façon, on a le plus souvent avantage à procéder, quand cela est possible, à un essai au traceur chimique ou colorant, avant d’utiliser le traceur radio-actif.
Avec les traceurs chimiques, on mesure les variations de concentration du corps ajouté. I1 faut donc prélever et analyser les échantillons ou dans certains cas mesurer la conductivité électrique, ce qui peut se faire sur place.
Avec les traceurs colorants, on mesure la variation de concentration du colorant sur échantillons.
Avec les traceurs radio-actifs, on détermine la variation de concentration du traceur par des mesures de la radio-activité, dans lesquelles il y a naturellement lieu de tenir compte de la variation de la radio-activité en fonction du temps. Rappelons que si l’activité au temps t = O est A,, elle devient, au temps t :
A = A,e -At
Si l’on désigne par T la période, c’est-à-dire le temps pendant lequel l’activité initiale a diminué de moitié, c’est-à-dire la demi-vie, on a :
t -0.69s~
A = .doe
0,693 T
h = -- étant la constante radio-active.
Mais la radio-activité est due à plusieurs radiations plus ou moins pénétrantes, à plus ou moins grande énergie de rayonnement, à plus ou moins grand pouvoir d’ioni- sation. Les rayonnements a, formés de noyaux d’hélium ayant une vitesse égale au ving-
tième de celle de la lumière, ont une grande énergie et un grand pouvoir d’ionisation. Mais ils sont peu p6nétrants, car ils sont arrêtés par une épaisseur de 3 à 8 c m d’air dans les conditions normales de pression et de température. Aussi, malgré leur grande éiiergie, ils ne peuvent guère être utilisés. Les rayonnements p, formés d’électrons, à vitesse voisine de celle de la lumière,
à pouvoir d’ionisation plus faible que celui des rayonnements a, sont par contre plus pénétrants. Ainsi il faut 0’47 mm d’épaisseur de plomb pour arrêter une Bnergie de 0,961 MeV et 0’92 mm pour arrêter une énergie de 2,2 MeV. Les épaissenrs d’eau d’arrêt sont les suivantes pour les diverses énergies 1.
1. CO~ISSARIAT A L’ENENGIE ATOMIQUE, Radio-Clémenrr artificiels préparés par le Commissariat à l’énergie alonrique, Liste no 4,, Gif-sur-1-vette, mars 1957, 137 pages.
99

Hydrologie des régions arides
Epaiabecr Énergie
nlm MeV
0,018 0,17 0,29 0,36 0,714 0,961 1,46 1,70 2,2
0,004 0,35 0,70 l,oo 2-6
66 8,O 10,l
4,1
I1 est à remarquer que l’absorption du rayonnement ß donne naissance à un rayonnement X d’autant plus grand que l’activité ß est importante.
Les rayonnements y, sans particules distinctes, semblables aux radiations lumineuses et aux rayons X, mais à longueur d’onde plus courte, ont un pouvoir d’ionisation semblable à celui du rayonnement ß. Mais ils sont beaucoup plus pénétrants. Ainsi la moitié est arrêtée par les épaisseurs suivantes de plomb : 0,89 M e V : 8 mm; 1,72 M e V : 12 mm; 2,76 M e V : 14 mm. La radio-activité se mesure par le taux de désintégration à la seconde. L’unité de
mesure, le millicurie, équivaut à 3 x lo’ désintégrations par seconde. Les appareils modernes de détection donnent des mesures précises de quelques
centaines de désintégrations par minute. I1 suffit donc d’une faible fraction de milli- curie pour mesurer la radio-activité. Toutes les substances radio-actives ne conviennent pas c o m m e traceurs. Elles doivent
en effet réunir un certain nombre de caractères : 1. Présenter une solubilité suffisante, pour être admise en concentration permettant
2. Présenter une faible adsorption, n’arrêtant pas les traceurs en route. 3. N e pas empoisonner les terrains. 4. Sensibilité de détection aux faibles concentrations, si possible au moyen d’appareils
5. Pouvoir être détectés, m ê m e dans les forages et les puits. 6. Avoir une demi-longueur de vie, adaptée au temps de parcours du trajet. 7. Pouvoir être obtenus facilement et à bas prix. 8. N e pas offrir de danger dans les conditions normales d’emploi. Les corps radio-actifs présentent en effet des dangers contre lesquels il faut se prémunir. En raison de leur faible pouvoir de pénétration, les rayonnements a sont les moins nocifs par exposition. Mais l’adsorption des corps émettant ces radiations est au contraire très dangereuse, car leur très grand pouvoir d‘ionisation entre alors en action.
Les rayonnements ß attaquent la peau et les tissus exposés. Les rayonnements y sont beaucoup plus dangereux, car leur grand pouvoir de péné-
tration leur permet d’atteindre les organes internes. I1 est donc bon de connaitre les doses de tolérance l.
1. Dans le cas d’exposition aux rayonnements de substances radio-actives situées à l’extérieur du corps humain, cela concernera spécialement les manipulations des traceurs radio-actifs, au cours de leur dilution. L a dose de rayonnement limite à laquelle l‘organisme humain peut être soumis sans inconvénient, pour un travail permanent, est de 0,3 roentgenIsemaine 2, ou 60 mr/8 h, mesuré dans l’au lorsqu’il
une détection au point d’observation.
portatifs.
1. COMMISSARIAT A ~)~NERCIE ATOMIQUE. Radio-QlPments artijÏeiels préparés par le Commissariat ù I’hergie atomique, Liste no 4,
2. Le roentgen est la quantité d e rayons X ou y telle que l’émission corpusculaire associée produise dans 1 cms d’air, a 00 et Gif-sur-Yvette, mare 1957, 137 pages.
760 mm de H g de pression, des ions partant l’unité électrostatique de charge de chaque signe.
100

Les différents. traceurs
s’agit de l’exposition de la totalité de l’organisme. L a dose de tolérance est plus élevée s’il s’agit d’une exposition partielle : 1,5 roentgen/semaine pour les mains.
L a dose approximative en roentgens/8 h à une distance de 1 m d’une source de C curies d’un émetteur de rayonnement y d’énergie E M e V est :
R = 4,4 EC 2. Dans le cas de contamination, c’est-à-dire d’adsorption, par les substances radio-
actives, le radio-élément est d’autant plus dangereux que sa période est plus longue, que le rayon qu’il émet est plus ionisant (m > ß > y) et qu’il se fixe plus sélecti- vement en un point du corps et s’en élimine plus difficilement. Sont peu toxiques : 24Na, 42K, 56Mn, ‘Wu, 76As, 85Kr; moyennement toxiques :
3H, 14C, 32P, 35S, W l , 59Fe, GoCo, %r, 1311, 137Cs, 140Ba, 19sAu; et très toxiques : 45Ca, 55Fe, 9lY, 9521, 144Ce, zlOBi. Dans l’eau de boisson, la concentration maximum d’activité tolérable est de
10-4 mc/l pour les émetteurs ß. Pour l’iode-131, émetteur ß y, se concentrant dans la thyrozde, la limite de tolérance n’est que de 10-5 mc/l seulement, tandis que pour le chrome-51, émetteur y pur, donc moins dangereux à absorber, elle est de 0,5 curie/m3. Celle du tritium est de 2 x 10-4 mc/cm3.
Le taux maximum d’iode-131 dans le corps, avec maximum dans la thyroïde, est de 0,3 microcurie, et celui du tritium est de 10 microcuries.
Digérents traceurs radio-actifs possibles.
I1 y a de nombreux traceurs possibles. Nous mentionnerons seulement ceux qu’on peut pbtenir le plus facilement à des prix raisonnables. Parmi ceux-ci nous distinguerons les émetteurs ß purs et les émetteurs ß et y.
Émetteurs p purs. Tritium (3H), période de 12,5 ans, énergie 0,019 MeV. Se trouve sous forme d‘hydro-
Carbone-14 (W), période de 5 700 ans. Energie 0,155 MeV. Phosphore-32 (32P), période de 14,3 jours. Énergie 1,712 MeV. P04H3 en solution
acide chlorhydrique de 5 à 10 mcjcm3 ou en solution neutre de phosphate de sodium à 2 mc/cm3.
Soufre-35 (35S), période 87,l jours. Énergie 0,166 MeV. Par exemple : SO,H, en solution aqueuse chlorhydrique 1 à 10 mc/cm3.
Calcium-45 (45Ca), 164 jours, 0,166 MeV. Cible : CaCO,; saturation, 530 PC. Strontium-90 (SoSr), 28 ans, 0,54 et 2,24 MeV. Cible : (N0J2Sr, 1 à 5 mc/cm3. Strontium-89 (asSr), 53 jours, 1,46 MeV. Cible : SrCO,; saturation, 99 [LC.
&metteurs p et y. Sodium-24 (24Na), 14,9 heures; ß : 1,39 MeV; y : 2,76 - 1,38 MeV. Cible : Nazco3 OU Fer-59 (59 Fe), 46,3 jours; ß : 0,46 - 1,56 - 0,27 MeV; y : 1,lO - 1,29 - 1,19. Cible : Cobalt-60 (‘Wo), 5,2 ans; ß : 0,32 M e V ; y : 1,33 - 1,17 MeV. Cible : CO OU CO,^,; lode-131 (131I), 8,05 jours; p : 0,61 - 0,34; y : 0,36 - 0,28 - 0,64 - 0,25 - 0,81 MeV. Thulium-170 (170Tm), 129 jours; ß : 0,97 - 0,98 MeV; y : 0,084 MeV. Cible : Tm2O3; Brome-@ (*2Br), 3 5 3 heures; ß : 0,465 MeV; y : 0,547 - 1,312 MeV. NH4Br, bromure
gène à 10 % de 3H, ou d’eau tritiée à 290 mc/cm3.
NaC1; saturation, 39 mc/g.
Fe; saturation, 3,7 mc.
saturation, 990 mc. Chlorure de cobalt 2 à 50 mc/mg.
INa : 10 à 50 mc/cm3.
saturation, 1,2 curies.
alcalin, 4 mc/mg.
10 1

Hydrologie des r6gions arides
Baryum-1@ (laoBa), 12 jours. On mesure les émissions y provenant du lanthane
Rubidium-86 (86Rb), 19,5 jours; ß : 1,822 - 0,716 M e V ; y : 1,076 MeV. Chlorure de Ruthenium-103 (losRu), 39,8 jours; ß : 0,217 - 0,698 M e V ; y : 0,498 MeV. Cible : Ru;
et qui sont plus pénétrantes que celles du baryum.
rubidium, saturation, 9,9 mc/g.
saturation, 2 mc/g.
Nous pouvons classer ces isotopes de la façon suivante :
Isotopes susceptibles d’être adsorbés ou de réagir avec l’eau ou avec le terrain. 24Na, 55Fe, 59Fe, 137Cs, l3lBa, S’JRb, 32P, 45Ca, goSr, 89Sr; Na, Fe, Cs, Ba, Rb, Ca, Sr ne pourront donc être utilisés dans les terrains argileux ou organiques qui fixeront les cations adsorbables.
Fe ne pourra non plus être employé dans les eaux où il risquera de précipiter par oxydation, B a précipitera par la présence de SO,, P ne sera pas non plus utilisable. Mais si les eaux souterraines sont très chargées en N a par exemple, ou en Ca, et si
elles sont en équilibre géochimique avec les terrains qu’eues traversent, la fixation de 24Na ou de 45Ca, suivant le cas, n’aura pas tendance à se produire. Et ces cations pourront servir d’entraîneurs. Si enfin les terrains ne contiennent aucun élément adsorbant, par exemple des
calcaires purs ou des sables ou des grès purement siliceux, les cations pourront servir de traceurs.
Isotopes non facilement adsorbables. a) Isotopes à très courte durée de vie (82Br, 35,9 heures). Il ne pourra donc servir qu’assez rarement en hydraulique souterraine, car les trajets d’un tel ordre de grandeur de durée à mesurer sont relativement rares.
b) Isotopes à courte durée de vie (1a1I, 8,05 jours). L a période est déjà plus intéres- sante, ce qui permet d’utiliser ce traceur dans un certain nombre de cas : sur de courtes longueurs pour des eaux à faible circulation de vitesse, et sur des longueurs déjà considérables pour les eaux ayant des vitesses importantes, c o m m e dans les massifs calcaires.
&ant donnée l’activité très élevée, il peut, sans entraîneur, être décelé avec une sensibilité élevée. Mais comme, malgré tout, l’iode subit une adsorption qui n’est pas toujours négli-
geable (surtout du fait de sa faible concentration), il y a intérêt à l’employer avec de l’iode non activé c o m m e entraîneur.
c) Isotopes à longue durée de vie (3H, 12,5 ans). Période tout à fait intéressante qui permet de l’employer dans le cas de vitesses faibles et de grandes longueurs de trajet.
minute et par gramme et son activité naturelle spécifique est extrêmement faible. Le grand avantage est que le tritium rentre dans la composition de l’eau et ne fait qu’un corps avec elle.
I1 a une activité d‘environ 2 x lOl6 désintégrations par
Méthodes d’emploi des traceurs radio-actifs.
Tout d’abord, avant d’utiliser un traceur radio-actif dans un aquifère, il y a lieu de connaître la façon dont se propagera le traceur dans le terrain et dans quelle pro- portion il peut être retenu par adsorption. On fera donc des essais de rendement sur échantillons en laboratoire, et on comparera les rendements à ceux de la fluorescéine et surtout à ceux du NaC1.
Pour cela on utilise un perméamètre à eau de grandes dimensions, en métal, de 1,5 m de hauteur et 0,90 m de diamètre environ (Kaufman et Orlob) [206, 207]), ou en verre de 1 m à 3 m de hauteur, 0,46 m de diamètre (Hours [204]).
102
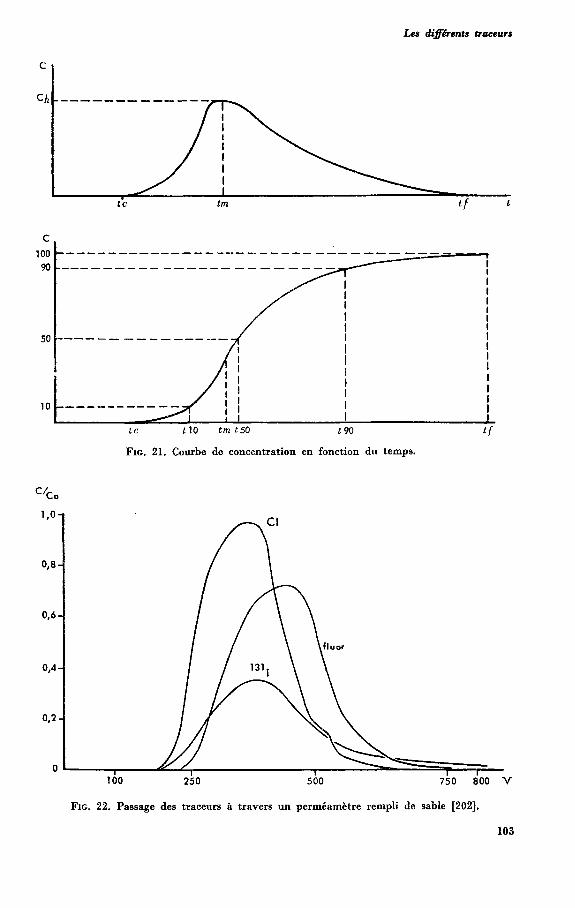
Les diffhents waceurs
C 1 O0 90
50
10
FIG. 21. Courbe de concentration en fonction du temps.
FIG. 22. Passage des traceurs B travers un permeametre rempli de sable [202].
103

Hydrologie des régions arides
L’eau entre dans le perméamètre par la partie supérieure et sort par le bas. Elle peut être maintenue à niveau constant dans le haut grâce à une vidange de trop-plein. On place un volume donné de terrain dans le perméamètre, en tâchant de lui redonner
ses conditions naturelles. Le terrain est ensuite imbibé d’eau jusqu’à saturation. Puis on déverse sur le haut de la colonne un volume V de solution de traceur à la concen- tration e,. Le temps du début de l’introduction du traceur est pris c o m m e origine des temps. Dès que la solution de traceur a été complètement absorbée par le terrain et adisparu
de la surface du sol, on fait arriver l’eau dans le perméamètre en la maintenant à charge constante, grâce au trop-plein. On a naturellement intérêt à utiliser la m ê m e eau que celle du courant souterrain dont on veut mesurer la vitesse. Pendant toute l’opération, on mesure les volumes d’eau qui s’écoulent, en prélevant des échantillons à intervalles réguliers, et on détermine la concentration e en traceur de chacun des échantillons. On porte alors sur un graphique, en ordonnées les rapports de concentra- tion c/c, et en abscisses les volumes d’eau cumulés correspondants (voir fig. 21, 22, 23). On construira un deuxième graphique, en portant en ordonnées les rapports c/e,
successifs cumulés (voir fig. 22).
FIG. 23. Passage des traceurs à travers un perméamètre rempli de limon sableux [202].
On calculera d’abord le rendement ~(VC) / C(V)c, à l’aide de la courbe cumulative. Si l’on opère avec des colonnes de longueurs différentes, on caractérisera la dimi- nution du rendement en fonction de la longueur du trajet (voir fig. 24). On pourra calculer le retard en prenant le chlorure de sodium c o m m e référence.
On considérera la concentration maximum e,, donnée par la hauteur du pic. I1 sera ainsi facile de comparer les différents traceurs entre eux.
I1 faut ensuite essayer de calculer la quantité de traceur radio-actif et la quantité d’entraîneur à employer. Nous citerons à ce sujet la méthode de Hours [204] : (( Le choix de la quantité
minimum d’entraîneur peut être basé sur le raisonnement suivant : pour utiliser au mieux le (( pouvoir marquant )) de nos millicuries, nous chercherons à rendre décelable,
104
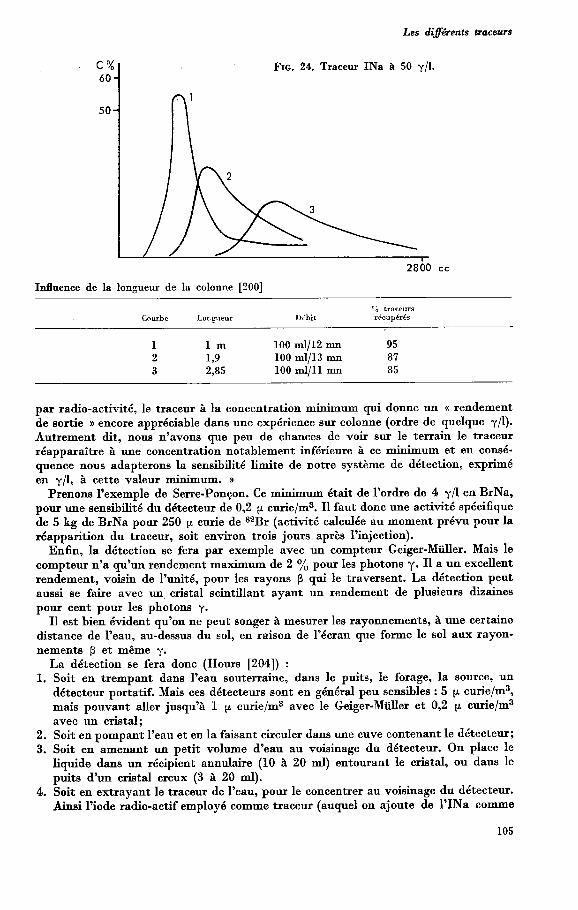
Les di.6rents traceurs
FIG. 24. Traceur INa à 50 y/l.
28ÒO cc Iduence de la longueur de la colonne [200]
o. tIneellTS Courbe Lougueur Dlbit r4cupéri.s
1 l m 100 d/12 m n 95 2 1,9 100 m1/13 m n 87 3 2,85 100 ml/ll m n 85
par radio-activité, le traceur à la concentration minimum qui donne un (( rendement de sortie )) encore appréciable dans une expérience sur colonne (ordre de quelque yo). Autrement dit, nous n’avons que peu de chances de voir sur le terrain le traceur réapparaître à une concentration notablement inférieure à ce minimum et en consé- quence nous adapterons la sensibilité limite de notre système de détection, exprimé en y/l, à cette valeur minimum. 1)
Prenons l’exemple de Serre-Ponçon. Ce minimum était de l’ordre de 4 y/1 en BrNa, pour une sensibilité du détecteur de 0’2 p curielm3. I1 faut donc une activité spécifique de 5 kg de BrNa pour 250 p curie de 82Br (activité calculée au moment prévu pour la rkapparition du traceur, soit environ trois jours après l’injection). Enfin, la détection se fera par exemple avec un compteur Geiger-Müller. Mais le
compteur n’a qu’un rendement m a x i m u m de 2 % pour les photons y. I1 a un excellent rendement, voisin de l’unité, pour les rayons ß qui le traversent. L a détection peut aussi se faire avec un cristal scintillant ayant un rendement de plusieurs dizaines pour cent pour les photons y. I1 est bien évident qu’on ne peut songer à mesurer les rayonnements, à une certaine
distance de l’eau, au-dessus du sol, en raison de l’écran que forme le sol aux rayon- nements ß et m ê m e y.
L a détection se fera donc (Hours [204]) : 1. Soit en trempant dans l’eau souterraine, dans le puits, le forage, la source, un
détecteur portatif. Mais ces détecteurs sont en général peu sensibles : 5 p curie/m3, mais pouvant aller jusqu’à 1 p curiejm3 avec le Geiger-Müller et 0’2 p curie/m3 avec un cristal;
2. Soit en pompant l’eau et en la faisant circuler dans une cuve contenant le détecteur; 3. Soit en amenant un petit volume d’eau au voisinage du détecteur. On place le
liquide dans un récipient annulaire (10 à 20 ml) entourant le cristal, ou dans le puits d’un cristal creux (3 à 20 ml).
4. Soit en extrayant le traceur de l’eau, pour le concentrer au voisinage du détecteur. Ainsi l’iode radio-actif employé c o m m e traceur (auquel on ajoute de 1’INa c o m m e
105

Hydrologie das rdgwns arides
entrdneur), après avoir ét6 oxydé avec du sulfate de nitrosyle, est rassedl6 dane du CC1,. L’iode est ensuite remis en solution aqueuse sou8 f o m e de INa-I0,Na par agitation avec une solution de soude. Et la solution est évaporée dana une coupelle que 1’011 place avec son résidu sous la fenêtre d’un compteur cloche [218].
Dif6rentes exphiences où ont 96 utilish les traceurs radio-actifs.
L a première expérience a été sans doute celle de Hess [203] sur la risere Susquehana, aux ]Etats-Unis d’Amérique; il a utilisé le radium. Mais le radium ne convient guère, car il peut être précipité par les ions SO, sous forme de RaSO,. D e plus les opérations sont trop onéreuses. Bien d’autres expériences ont été faites par la suite pour calculer les débits ou lee
vitesses d’écoulement des rivières. Citons, parmi tant d’autres, celles de Montem [208], Sons [214], Josendal, Sandford, Wilson [205], Simane [213], Harold A. Thomas Jr. (1956), Allen et Grindley [192]. On a également utilisé les traceurs radio-actifs pour l’étude de l’écoulement des
fluides dans les conduites, la recherche des fuites d’eau. Archibald [193], Hull, Kent et Lée (1949)’ Hull et Fries (1956)’ Puttman et Jefferson (1956)’ Seligman [212].
Relativement peu d’essais ont été effectués sur les nappes d’eau souterraines : Fox [201], Truesdale [216], Urbain, Lagrange, Hours, Geslin [218], Hours [204], Kaufman et Orlob [206, 2071, Brown, Parker et Smith [197], de Vessey et Czerny [220], etc. C’est peut-être l’étude des gisements de pétrole qui a le plus largement mis à contri-
bution les isotopes radio-actifs, en particulier pour détecter les communications directes existant entre les divers points d’un magasin : Comber et Tiratso (1950)’ Edwards et Holter [198], Russel [211], Watkins et Mardock [222], Flagg, Myers, Campbell, Terry, Mardock [199], Aebersold [190], Watkins et Dunning [221], etc. Nous ne donnerons ici que quelques exemples concernant les eaux souterraines.
Essai du Laboratoire de recherches de Californie [206, 2071. I1 s’agissait de comparer la vitesse de voyage de bactéries du groupe coliforme à la vitesse de déplacement du front d’eau d’égout dans un terrain. Les traceurs employés furent la fluorescéine, un chlorure, la dextrose et l’iode-131. Ils furent injectés au débit de 140 l/& par un puits de 12 pouces de diamètre dans une nappe captive située A 27,40 m de pro- fondeur. L’aquifère, de 1,34 m d’épaisseur moyenne, était composé de gravier et de sable ayant une taille effective de 0’56 mm, un coefficient d’uniformité de 6’9 et une porosité de 0’35. Vingt-trois puits de 6 pouces de diamètre furent placés jusqu’il 152 m de distance. I1 fallut employer de très grandes quantités de chlorure de sodium dont la densité provoqua des séparations de filets liquides et une arrivée sporadique du traceur aux points d’observation.
Les quantités déversées dans l’aquifère furent : a) de 170 litres de solution de fluorescéine sodique, à la concentration de 100 mg/kg, soit 17 grammes; de 1500 litres de solution de sucre ii la concentration de 6 g/kg, soit 9 kg; b) de 420 litres d’eau contenant 20 millicuries d’iode-131, pendant une durée de trois minutes.
Quant aux bactéries, elles furent introduites par une injection continue d’un mélange de 10 yo d’eau d’égout décantée et d’eau de recharge.
Le tableau suivant résume les constatations faites à trois puits d’observation situ& sur un m ê m e méridien.
106
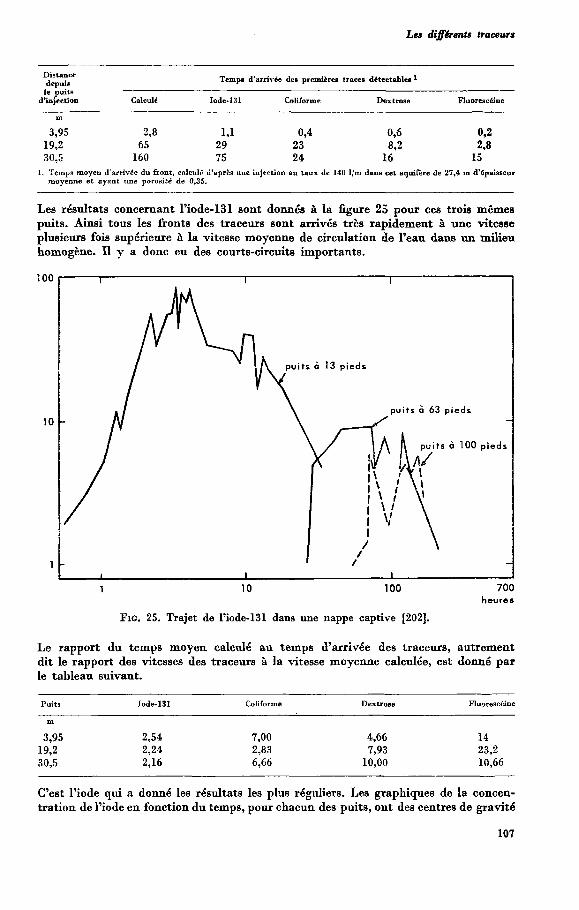
Lu diffhm traceurs
Temps d’arrivde des premihres tracen deteetables 1 Diatanoc depme le puitu -
d’injection Calculé Iode-131 Coliforme Dextrose F’luorescdine
3,95 2,8 1 3 1 0-4 19,2 65 29 23 30,5 160 75 24
1. Temps moyen d’arrivCe du front, calcul6 d‘aprha une injection au taux de 140 1/m dans cet aquifere de 27,4 m d’dpaisseur moyenne et ayant une porositd de 0,35.
Les résultats concernant l’iode-131 sont donnés 21 la figure 25 pour ces trois mêmes puits. Ainsi tous les fronts des traceurs sont arrivés tres rapidement à une vitesse plusieurs fois supérieure à la vitesse moyenne de circulation de l’eau dans un milieu homogène. I1 y a donc eu des courts-circuits importants.
FIG. 25. Trajet de l’iode-131 dans une nappe captive [202].
Le rapport du temps moyen calculé au temps d’arrivée des traceurs, autrement dit le rapport des vitesses des traceurs à la vitesse moyenne calculée, est donné par le tableau suivant.
Fluoreaekine Puits Iode- 131 Coliforme Dextrose ~~~
m
3,95 2,54 19,2 2,24 30,5 2,16
7,OO 2,83 6,66
4,66 7,93 10,oo
14 23,2 10,66
C’est l’iode qui a donné les résultats les plus réguliers. Les graphiques de la concen- tration de l’iode en fonction du temps, pour chacun des puits, ont des centres de gravité
107

Hydrologie des régions arides
peu éloignés du temps moyen calculé, sauf pour le premier puits où il semble y avoir un retard. On peut se demander pourquoi la marche des autres traceurs présente une telle irré-
gularité, c’est-à-dire pourquoi ces autres traceurs indiquent surtout un court-circuitage. Est-ce dû à la façon dont a été faite l’injection?
Pour détecter les très petites quantités d’iode radio-actif arrivant aux deux derniers puits, il fut nécessaire d’évaporer les échantillons, 20 cm3, et d’étudier le rayonnement ß avec un internal $ow counter. A u dernier puits l’activité de l’iode ne dépassait pas 6’5 coups/minute/cm3.
L a fluorescéine atteignit ce puits avec une concentration de 0,4 mg/kg au pic, suivi d’une traîne semblable à celle observée dans les perméamètres.
Essai de Serre-Ponçon, France [204]. I1 s’agissait de déterminer la possibilité d’une communication entre une galerie passant sous la Durance et un piézomètre foré de l’autre côté de la rivière à 100 m de distance, à travers des terrains rocheux et allu- vionnaires. Des quantités importantes de fluorescéine furent injectées dans le piézomètre. Mais
elles n’apparurent pas dans la galerie au cours de dix jours de pompage. Devant cet insuccès, on injecta, sous une pression de 30 m d’eau, 25 litres d’eau
ayant dissous 5 kg de bromure de sodium, marqués par 1 curie de brome-82. On rinça rapidement avec 500 litres d’eau.
L’eau de la galerie fut alors pompée à raison de 3’5 m3 à la minute et l’activité apparut dans la galerie vingt-quatre heures après l’injection. Le front du traceur était assez abrupt et la traîne très longue.
Essai ù Cauterets et Luz, France [218]. I1 s’agissait de savoir si les eaux thermales sulfurées sodifiées de Luz et de Saint-Sauveur étaient essentiellement alimentées par des eaux vadoses. Ce sont des eaux chaudes, ce qui implique un circuit profond, puisque la température moyenne annuelle de l’air est de 15” C environ. Les sources sortent au contact du granite et des schistes du Carbonifère inférieur.
A haute altitude, il y a des lacs situés au m ê m e contact. On suppose qu’ils alimentent le réseau aquifère.
Dans l’un de ces lacs, le lac du Labas, fut jeté 0’7 curie d’iode-131, sous forme d’iodure de sodium. L a longueur du trajet direct de l’eau a été évalué à 21 km. Des échantillons furent prélevés pendant treize jours de suite, de 2 litres au début,
de 15 litres à la fin, et ramenés à 100 cm3 environ. Les comptages ne révélèrent aucune tendance systématique à l’accroissement de
l’activité brute. L’expérience a montré que si l’origine de ces sources devait être vadose, le circuit était beaucoup plus lent qu’on ne l’avait pensé, ce qui n’a rien d’étonnant étant donné la longueur du trajet. Sinon, l’origine est ailleurs. Cette expérience ne fut d’ailleurs pas inutile, car elle permit d’étudier les mouvements de l’eau dans le lac.
Essais dans des terrainspétrolij’ères de l’Oklahoma (Nowata County) et du Kansas (Anderson County), &tats- Unis d’Amérique [222]. Ces essais sont particulièrement intéressants parce qu’ils indiquent les méthodes susceptibles d’être utilisées dans les études d’hy- drogéologie. Ils eurent pour but de connaître la direction et la vitesse de l’écoulement de l’eau et par conséquent d’estimer la perméabilité des terrains. Naturellement, avant tout essai, il faut rassembler toute la documentation possible
sur la lithologie et la structure du terrain perméable, toute la documentation sur les caractéristiques du puits d’injection, en particulier de ses propriétés hydrauliques. Si l’on a des échantillons du terrain, il y a lieu d’étudier au préalable l’absorption
des traceurs par ces terrains en utilisant la méthode du perméamètre, citée plus haut. Ensuite, aussi bien dans le puits d’injection que dans les puits d’observation où le
traceur peut apparaître, on procède à des logs de rayonnement et de neutrons, en se
108

Les diférents traceurs
servant d’appareils standard de mesure de rayonnement y et de chambres d’ionisation pour neutrons. Si les données préliminaires indiquent que l‘eau doit passer rapidement du puits
d’injection au puits d’observation, il peut être judicieux de procéder auparavant à un test avec un traceur colorant. On déterminera ainsi le temps approximatif du trajet, et par conséquent le moment où l’équipe de sondage à rayonnement y doit arriver pour détecter les premières apparitions, sans avoir à attendre trop longtemps.
D’ailleurs les apparitions positives de colorant confirmèrent l’existence de chenaux soupçonnés et permirent de mieux calculer les quantités de traceurs radio-actifs nécessaires.
Le taux d’injection fut habituellement de 3’8 litres en quinze minutes. L a radio- activité des fluides débités par tous les puits d’observation où le traceur pouvait apparaître fut continuellement mesurée à l’aide de tubes Geiger-Müller, sensibles au rayonnement y et descendus verticalement.
Des échantillons de liquide étaient pris à divers intervalles jusqu’au moment oh la radio-activité mesurée s’élevait nettement au-dessus de la radio-activité de fond. Après quoi les échantillons furent pris à intervalles fréquents.
Avant que le traceur n’atteignît le premier puits d’observation, l’appareil de carottage à rayonnement y fut placé devant la formation perméable. Puis, à l’arrivée du traceur radio-actif, on exécuta de nombreux carottages à rayons y,
ce qui permit de définir clairement les horizons à circulation plus facile de l’eau. Citons le test B opéré dans le Nowata County, Oklahoma, pour un projet d’injection
d’eau dans un gisement de pétrole contenu dans les sables de Bartlesville, à 155 m de profondeur. Les crépines des forages attaquaient 13’70 m de sable (la disposition des puits est indiquée à la fig. 26).
R-2
R-1
G-15
N 190-W
ti-14 H-16
G-17
I 165-P -
* F-16 F-14
FIG. 26. FIG. 27.
190-VET est le puits d’injection. Les productions respectives d’huile et d’eau de ces puits étaient :
Pairs Eau Huile Rapports
m3 llls
R 1,19 0,0476 2511 R-2 11,9 0,215 56/l 165-P 2,384 0,018 13011
109

Hydrologie des r€giona arides
Ces quantités exagérées d’eau par rapport à l’huile semblaient indiquer l’existence d’un écoulement par chenaux entre le puits d’injection et les puits de production. Les essais hydrauliques faits sur le puits d’injection 190-W, à l’aide d’unchor packer,
montrèrent que l’eau entrait dans les sables à la profondeur d’environ 158 m. Une solution de 226 g de fluorescéine dans environ 43 litres d’eau fut régulièrement
versée dans le puits d’injection 190-W. Le colorant apparut dans le puits R-2 environ onze heures après, c’est-à-dire après l’injection d’environ 1910 litres dans le puits 190-W et la production de 3 340 litres de pétrole et eau dans le puits R-2. Aucune fluorescence ne fut détectée dans l’eau des puits R-1 et 165-P. L a présence de colorant au puits R-2 confirma ainsi un écoulement préférentiel. Ensuite une solution contenant approximativement 87 millicuries d’iode-131 dans
environ 119 litres d’eau fut injectée dans le puits 190-W pendant quinze minutes. L e débit d’injection d‘eau dans le puits 190-W avait été augmenté depuis l’expérience faite avec le traceur colorant. Un accroissement de radio-activité fut constaté dans le puits R-2, deux heures
quarante-cinq après l’injection du traceur. Pendant ce temps 1 310 litres d’eau avaient été versés dans le puits 190-W. D’après la hauteur à laquelle était arrivé le liquide radio-actif dans le tubage au moment où le premier log fut fait, on calcula que le traceur avait atteint le puits R-2 en deux heures seulement. Un accroissement de radio-activité fut décelé à la surface dans le liquide produit par R-2, quatre heures et demie après l’injection. Pendant ce temps, 2 260 litres d’eau avaient été injectés. Les logs de rayonnement localisèrent nettement la zone d’entrée dans le puits R-2. Le début eut lieu quatre heures et demie et le pic sept heures après l’injection. Et
douze jours après l’eau avait toujours une radio-activité un peu plus grande que la radio-activité naturelle de fond.
L’intégration de l’activité observée dans l’eau du puits R-2 montra que 70 milli- curies, c’est-à-dire environ 80 yo de l’iode radio-actif, entrèrent dans le puits R-2. On observa un accroissement faible de radio-activité dans le puits R-1, environ
soixante-douze heures après l’injection du traceur. Mais le pic fut plus faible et la décroissance de la radio-activité plus rapide. Aucune radio-activité ne fut observée dans les quatre autres puits. Les données ainsi obtenues montrèrent que l’eau passe directement du puits 190-W
au puits R-2, à travers un ou deux chenaux, à une profondeur d’environ 158 m , ce qui semble impliquer l’existence d’une fracture continue, soit entre 190-W et R-1, sur une distance de 80 m, soit entre 190-W et R-1 viu R-2, sur une distance de 84 m environ. NOUS citerons encore l’essai de l’Anderson County (Kansas) dans les grès de Bartles-
ville. Le toit de la formation productive pétrolifère est à une profondeur allant de 236 à 241 m. L’épaisseur du sable est de 8 à 13 m aux environs immédiats. (Les puits sont disposés de la manière indiquée à la fig. 27.) 6-15 était le puits d’injection. Les puits d’exploitation H-16, F-16 et à un degré
moindre les pilits H-14 et F-14 donnaient une trop grande quantité d’eau. A lui seul le puits H-16 débitait plus d’eau qu’on n’en versait dans le puits 6-15, lequel ne pouvait donc être tenu pour seul responsable de la production excessive d’eau. Cependant les tests faits sur d’autres puits d’injection montrèrent que la tenue du puits 6-15 était anormale et pouvait bien tout de m ê m e être la cause majeure de la production relative élevée du puits H-16 et des puits voisins. L e faible débit relatif en eau du puits 6-17, situé presque en ligne droite entre le puits d’injection 6-15 et le puits H-16, permit de conclure à l’existence d’un chenal ou d’une zone de très grande per- méabilité entre les puits. On procéda à un premier test à l’aide de colorant pour confirmer l’existence d’une telle zone et établir la durée du trajet entre les puits.
Une solution de 226’8 g de fluorescéine fut dissoute dans environ 83 litres d’eau et injectée. On préleva des échantillons de liquide toutes les deux heures pendant
110

Les diff6rents traceurs
trois périodes de vingt-quatre heures et chaque jour la semaine suivante. L a première fluorescence fut détectée à la surface dans l’eau du puits H-16, cinquante-huit heures après l’injection du traceur, temps pendant lequel 19300 litres d’eau avaient été déversés dans le puits 6-15, et 31 100 litres de liquide avaient été produits dans le puits H-16. La fluorescence avait un pic d’intensité de 0’5 ppm et fut présente pendant au moins deux semaines.
L a première fluorescence de l’eau du puits F-16 fut observée après environ sept jours avec une concentration m a x i m u m de moins de 0,5 ppm; puis la fluorescence décrut rapidement. Aucune fluorescence ne fut observée dans l’eau des puits H-14, F-14 et
Des logs de rayonnement y furent faits sur les puits G-15. Puis environ 96 litres d’eau contenant 44 millicuries d’iode-131 furent mis dans le puits G-15, pendant une durée de quinze minutes. A ce moment l’injection d’eau y était de 10 500 litres par jour, tandis que lors de l’injection du colorant elle était de 8 O00 litres par jour.
Après injection on fit de nouveaux logs de rayonnement y, qui montrèrent un accroissement de rayonnement et par conséquent des zones d’entrée de l’eau, princi- palement entre 256 et 257 m de profondeur puis à un degré moindre à 252 m et à 255 m. Avant l’apparition du traceur dans les puits de production, des échantillons de liquide avaient été pris à des intervalles de temps pris au hasard, puis fréquem- ment et régulièrement. A aucun moment il n’y eut d’accroissement sensible de la radio-activité dans l’eau des puits F-14 et 6-17.
Dans le puits H-16, la première radio-activité apparut à la surface du puits trente- deux heures après l’injection du traceur. Or le calcul indiqua une durée de deux heures quarante-huit minutes de montée de l’eau depuis l’entrée dans le puits jusqu’au sommet du tubage. Le traceur atteignit donc le puits au bout de vingt-neuf heures. L a différence de temps (58 h - 32 h = 26 h) entre l’apparition de la fluorescéine et celle du traceur radio-actif dans l’eau du puits est peut-être due : lo au fait qu’on a injecté une plus grande quantité d’eau dans le cas du traceur radio-actif; 20 à la possibilité d’une adsorption partielle de la fluorescéine.
L a radio-activité fut constamment mesurée dans les puits F-16, H-16 et H-14. Au puits H-16, l’apparition eut donc lieu vingt-neuf heures après l’injection, puis il y eut un pic d’intensité au bout de quarante-trois heures suivi d’autres pics, l’un à soixante-quinze heures et l’autre à cent cinq heures. Or les logs de rayonnement y de ce puits y montrèrent trois niveaux d’entrée d’eau
à 258 m, 260 n et 262 m de profondeur. I1 n’y a pas de doute que chacun des trois pics est en relation avec chacune des trois venues d’eau. D e m ê m e la radio-activité de l’eau du puits F-16, mesurée en fonction du temps,
présenta trois pics, mais de moindre hauteur et allant en décroissant : le premier à cent huit heures, le second à cent trente-deux heures et le troisième à cent soixante-dix heures après l’injection. Or dans ce puits il y avait également trois entrées d’eau, bien indiquées par des accroissements d’activité sur les logs de rayonnement y : deux faibles à 240-241 m et à 244-245 m de profondeur, et un m a x i m u m à 262 m de profon- deur.
Dans le puits H-14, il y eut également trois pics, à cent trente-quatre, cent cinquante-huit et cent quatre-vingt-deux heures. Mais contrairement à ce qui s’est passé pour les autres forages la hauteur de ces pics est croissante. En outre, leur hauteur était beaucoup plus grande que celle des autres puits mais leur amplitude était plus faible. Ce fait doit être lié à des perméabilités plus grandes.
Essai du Wudi Raiyan, dans le désert de Libye [ZOO]. Des essais furent faits avec du chlorure de rubidium radio-actif pour montrer l’imperméabilité du fond du Wadi Raiyan. D’autres essais furent exécutés en figypte avec du chlorure de rubidium et montrèrent
111
6-17.
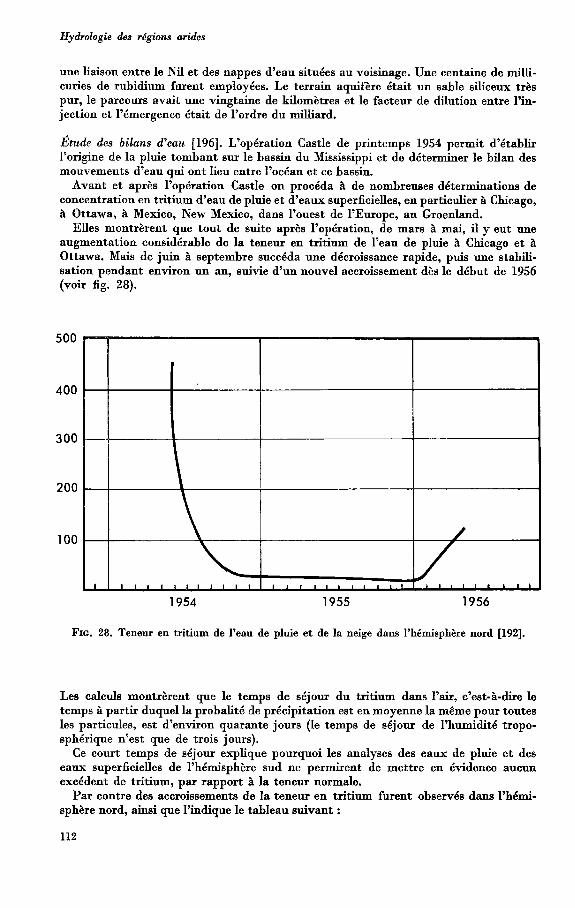
Hydrologie des régions arides
une liaison entre le Nil et des nappes d’eau situées au voisinage. Une centaine de milli- curies de rubidium furent employées. Le terrain aquifère était un sable siliceux très pur, le parcours avait une vingtaine de kilomètres et le facteur de dilution entre l’in- jection et l’émergence était de l’ordre du milliard.
& d e des bilans d’eau [196]. L’opération Castle de printemps 1954 permit d’établir l’origine de la pluie tombant sur le bassin du Mississippi et de déterminer le bilan des mouvements d’eau qui ont lieu entre l’océan et ce bassin. Avant et après l’opération Castle on procéda à de nombreuses déterminations de
concentration en tritium d’eau de pluie et d’eaux superficielles, en particulier à Chicago, à Ottawa, à Mexico, N e w Mexico, dans l’ouest de l’Europe, au Groenland.
Elles montrèrent que tout de suite après l’opération, de mars à mai, il y eut une augmentation considérable de la teneur en tritium de l’eau de pluie à Chicago et à Ottawa. Mais de juin à septembre succéda une décroissance rapide, puis une stabili- sation pendant environ un an, suivie d’un nouvel accroissement dès le début de 1956 (voir fig. 28).
500
400
300
200
1 O0
1954 1955 1956
FIG. 28. Teneur en tritium de l’eau de pluie et de la neige dans l’hémisphère nord [192].
Les calculs montrèrent que le temps de séjour du tritium dans l’air, c’est-à-dire le temps à partir duquel la probalité de précipitation est en moyenne la m ê m e pour toutes les particules, est d’environ quarante jours (le temps de séjour de l’humidité tropo- sphérique n’est que de trois jours).
Ce court temps de séjour explique pourquoi les analyses des eaux de pluie et des eaux superficielles de l’hémisphère sud ne permirent de mettre en évidence aucun excédent de tritium, par rapport à la teneur normale. Par contre des accroissements de la teneur en tritium furent observés dans l’hémi-
sphère nord, ainsi que l’indique le tableau suivant :
112

Les différents traceurs
Accroissement (T/H x lola)
Atomes de T par c m 2
Lac Michigan . . . . . . . . 5 4 (187 20) x 107
LacTahoe(Californie) . . . . . . 7 i90 x 107
Atlantique-Nord . . . . . . . 2 100 x 107 Pacifique-Nord . . . . . . . . 2 100 x 107
Lac de Zurich . . . . . . . . 26 67 x lo7
LacCrater(0regon) . . . . . . 6 1800 X lo7
L a teneur normale en tritium est de 1O-l’ à rapportée à un atome d’hydrogène. On calcula également la quantité de tritium qui s’est précipitée par centimètre
carré à la suite de l’opération Castle. Celle-ci est indiquée dans la colonne de droite du tableau précédent. On peut admettre qu’il est ainsi tombé en moyenne sur l’hémi- sphère nord 200 X lo7 atomes par centimètre carré (lo7 atomes de tritium donnent une désintégration par minute). I1 en résulta un remarquable accroissement de la teneur en tritium des eaux des rivières et des fleuves de l’hémisphère nord. Ainsi la teneur en tritium de l’eau du Mississippi passa de 4’7 (4,5) à 44 x 10-18 T/H, soit une augmen- tation de 39 x 10-l8 T/H. C’est-à-dire que 7,7 m d’eau se melangèrent avec l’eau de pluie transportant le tritium originaire de l’opération Castle. Après la période de décroissance du tritium, il y eut une constance de la teneur en
tritium de l’eau de la rivière pendant une période d’un an. Cela suppose un mélange complet dès les premiers mois. Les pluies ne se sont pas enrichies plus que l’eau des rivières en tritium à la suite des nouvelles évaporations du cycle de l’eau, tout au contraire. C’est donc que le mélange des eaux tritiées avec les eaux continentales n’est pas essentiellement superficiel et que de nouveaux mélanges pourraient ulté- rieurement continuer à se produire.
T/H) de l’eau du Mississippi, la pluie et la neige tombées à Chicago avaient une teneur régulière de 21 x 10-18 T/H et l’eau de l’océan une teneur de 2,5 x 10-lS T/H. On en déduisit que le tiers des pluies en moyenne est de l’eau réévaporée en provenance des continents et les deux tiers en provenance des océans. Pour procéder à ce calcul on avait soustrait 5 unités du tritium de l’eau de la rivière et 7 de l’eau de pluie afin de tenir compte du tritium du rayonnement cosmique lors de l’utilisation de la bombe. Ainsi sur les 770 mm de la pluie annuelle tombant sur le bassin du Mississippi,
520 proviennent des océans et 250 de la réévaporation des eaux continentales. On compte c o m m e run-off 280 mm. I1 y a donc 770 mm - 280 mm = 490 mm
d’évapotranspiration dans le bassin du Mississippi, représentant le tiers de l’humidité de l’air donnant naissance aux pluies. Les deux autres tiers provenant de l’océan correspondent donc a 980 mm, soit 1 m. Or il en retombe, nous l’avons vu, 520 mm sous forme de pluie. I1 en résulte que les (1 O00 mm - 520 mm) = 480 mm restants ainsi que les (490 mm - 250 mm) = 240 mm d’eaux du sol sont réévaporés.
Ainsi les vents amènent à l’océan moins d’humidité des eaux du sol de la vallée nord du Mississippi (240 mm par an) que le Mississippi n’en transporte (280 mm). On peut en conclure qu’il y a un équivalent de 8 m d‘eau du sol qui se mélange en
quelques mois avec la pluie dans la vallée du Mississippi. Environ 520 mm d’eau océanique se précipitent en pluie par an, 490 mm d’eau se réévaporent, 280 mm d’eau retournent aux océans par les rivieres, 240 mm d’eau du sol retournent aux océans par évaporation.
Pendant ia période de constance de la teneur en tritium (44 x
L a figure 29 montre le cycle des eaux qui en résulte. D’autres observations ont été faites sur des sources thermales, les Steamboat Springs
du Nevada et les geysers du Yellowstone Park (Wyoming).
113
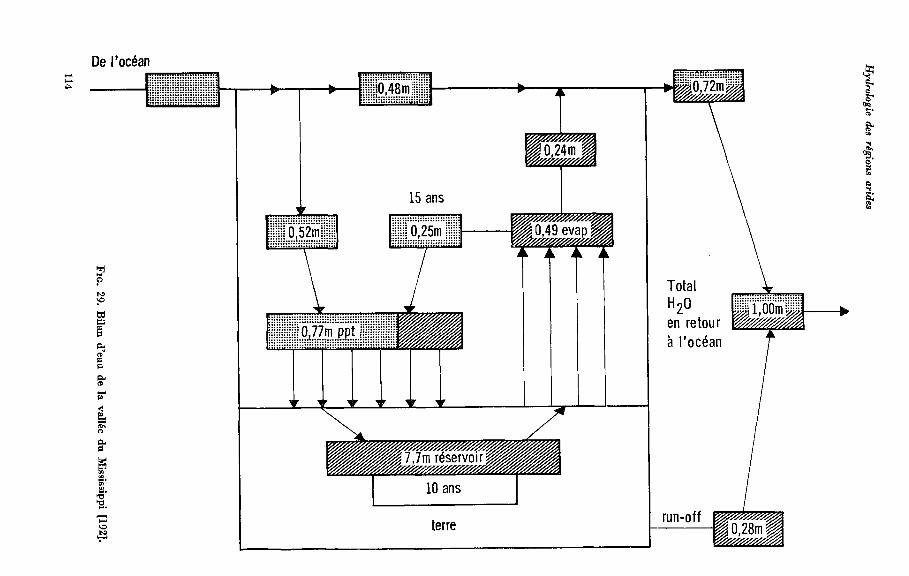
Hydrologie des régions arides
........... ........... ........... ........... ........... ........... ........... ........... ........... ...........
..... ........... ........... ........... ........... ........... ........... ...........
c
u O 114
FIG. 29. Bilan d'eau de la vallée du Mississippi [192].

Les différents traceurs
On savait déjà, d’après les études récentes de H. Craig1 basées sur les rapporta D/H, 018/01a7 que ces eaux devaient très probablement avoir une origine météorique.
Les dosages du tritium montrèrent que de petites quantités de tritium apportées par la pluie étaient apparues très rapidement dans presque toutes ces sources; et que très vraisemblablement le mélange complet n’était pas terminé. L’âge de l’eau de ces sources doit donc être de l’ordre de cinquante ans.
1. H. CRAIG; G. BOATO; D. E. WJDTE, a Nuclear processea in geologic settings. Second oonference, September 8-10, 1955 w. Na6. Aead. Se. (1956). publication no 400.
115

B I B L I O G R A P H I E
BILANS ET RESSOURCES
1. BAKER, D., (( Safe yield of ground water reservoirs n, Association internationale d’hydrologie scienti$que, Assemblée de Bruxelles, 1951, t. 2, p. 160. (Publication no 33.)
2. BAUMANN, P., Groundwater phenomena related to basin recharge ))9 Proceedings of the American Society of Civil Engineers, 1955, t. 81, no 806, 25 pages.
3. BERKALOFF, E., (( fitude du bilan d’eau Ain Ketena D, Bulletin économique et social de la Tunisie, no 44, 1950.
4. BOGOMOLOV, G.V., (( Classification des ressources d’eau souterraines et évaluation de leurs réserves n, Association internationale d’hydrologie scientijique, Symposia Darcy, 1956, t. 2, p. 262-271. (Publication no 41.)
5. -; PLOTNIKOV, N.A., (( Classifying the underground water resources and appraising their reserves, )) Congrès géologique international de Mexico, 1956, t. 2, p. 48.
6. -; -, (( Classification des ressources des eaux souterraines et leur représentation sur des cartes D, Abstracts of the reports at the XIth General Assembly of the International Asso- ciation of Scientijic Hydrology, Moscou, Akad. Nauk S.S.S.R., 1957, p. 45-41.
Coefficients d’infiltration, coe%cients d’évaporation. Étude d’un cas précis dans les calcaires gréseux (Pliocène et Quaternaire de la côte du Maroc) D, Association inter- nationale d’hydrologie scienti$que, Assemblée de Bruxelles, 1951, t. 4, p. 12-15. (Publication no 35.)
8. CHEBOTAREV, I. I., (( Principles of the estimation of subterranean waters )), Wat. &: Wat. Engng, 1951.
9. CLODIUS, S., ((Neue Zahlen zum Schema des Wasser-Kreislaufes D, Bohrtechnik - Brunnenbau, 1955, t. 6, no 1, p. 21-23.
10. COLAS, R., (( L’alimentation en eau des régions arides D, Société des ingénieurs civils de France, 1956, p. 396-412 (Mémoire 109, no 5).
11. COLLINS, B. W., (( Ground water in the hydrologic cycle, with special reference to Canter- bury, New Zealand )), Report Seventh Science Congress, Christchurch, New Zealand, Royal Society of New Zealand, 1953, p. 127-139.
12. DANEL, P., (( La réserve des débits des eaux souterraines D, Tech. mod., Construction, 1952, p. 245.
13. DROUHIN, G., (( Incidences de l’utilisation des eaux souterraines sur l’équilibre hydrolo- gique D, Tech. de l’eau, no 122, 1957, p. 15-22.
14. DUBIEF, J., Essai sur l’hydrographie superJicielle au Sahara, Birmandreis, Alger, Direction du Service de la colonisation et de l’hydraulique, Direction des études scientifiques, vol. 1, 1953, p. 457, 41 fig.
7. BOLELLI, E.,
116

Bibliographie
15. FOURMARIER, P., (( Les réserves aquifères en Belgique D, Tech. de l‘eau, 1952, p. 25-27. 16. GARMANOV, I.; KAMENSKI, G., N La formation des eaux souterraines et leur répartition
par zones, dans les régions arides de l’U.R.S.S. I), Congrès géologique international de Mexico, 1956, t. 20, p. 50.
17. GUIRSKI, N.K., Quelques particularités de la dynamique des eaux souterraines des déserts et des semi-déserts, 1958, 23 p.. 7 fig. (Communication privée.)
18. KOCH, P., Contribution cl l’étude de la corrélation entre les précipitations et les niveaux de nappe dans le bassin amont de la Seine. (La houille blanche, t, 10, numéro spécial B.), 1955.
19. KOUDELINE, B.I., N Principles of the regional estimate of natural resources of underground waters and problems of water balance n, Abstracts of the reports at the XIth General Assembly of the International Association of Scientific Hydrology, Moscou, Akad. Nauk S.S.S.R.,
Principes nouveaux pour distinguer ce qui est dû a l’écoulement souterrain sur l’hydrographie des fleuves D, Bulletin de l’Association internationale d’hydrologie scienti- fique, no 7, 1957, p. 25-35.
21. -, (( Importance des structures géologiques pour les calculs du bilan d’eau de multiples années n, Bulletin de l’Association internationale d’hydrologie scientifique, no 7, 1957, p. 36-40.
22. KOUNINE, V.N., (( Conditions of the formation of underground waters in the desert )I,
Abstracts of the reports at the Xlth General Assembly of the International Association of Scientific Hydrology, Moscou, Akad. Nauk S.S.S.R., 1957, p. 54-57.
23. MAMADEVAN, C., (( Hydrology of arid and sub-arid regions in India D, Congrès géologique international de Mexico, 1956, t, 20, p. 23.
24. MANIL, G., (( Un aspect du problème du bilan de l’eau dans le sol. Les données de Penman sur l‘évaporation en conditions naturelles D, Bulletin de l’Institut agronomique de Gembloux, 1954, t. 22, no 3-4, p. 255-271.
25. PENMAN, H., (( The water balance of catchment areas n, Association internationale d’hydro- logie scientifique, Assemblée de Bruxelles, 1951, t. 3, p. 434-443. (Publication no 34.)
26. -, Components in the water balance of a catchment area D, Quarterly J. R. met. Soc., 1955, t. 81, no 348, p. 280-284.
27. SCHOELLER, H., N Condensations occultes, en particulier dans les affleurements de terrains calcaires ou gréseux de l’Afrique du Nord D, Colloques internationaux du Centre national de la recherche scienti$que, Alger, marB 1951; Paris, 1953, no 35, p. 353-363.
Certaines lois de formation des eaux souterraines dans les zones arides de la terre D, Congres géologique international de Mexico,
29. -; -, Quelques règles de la formation des eaux souterraines dans les régions arides du globe, Moscou, Académie des sciences de l’U.R.S.S., Université d’État de Moscou, Institut de recherches géologiques, 1958, 23 p. (Communication privée.)
30. TIXERONT, J., (( Les ressources en eau dans les régions arides D, Ann. Ponts Chauss. (France), 1956, t. 126, no 3, p. 965-997.
31. -; BERKALOFF, E., Recherches sur le bilan d’eau en Tunisie N, Association internationale d’hydrologie seienti$que, Assemblée de Rome, 1954, t. 3, p. 553-558. (Publication no 38.)
32. -; -; CAINE, A.; MAUDUECH, E., Bilan d’eau des massifs calcaires en Tunisie. Cas des captages de Tunis et de Bizerte D, Association internationale d’hydrologie scienti- $que, Assemblée de Rome, 1954, t. 3, p. 553-558. (Publication no 38.)
33. TROXELL, H.C., (( The influence of ground water storage on the run-off in the San Bernardino and Eastern San Gabriel Mountains of northern California )I, Trans. Amer. geophys. Un., 1953, vol. 34, no 4, p. 552-562.
34. TUINZARD, H., (( Influence of the atmospheric pressure on the head of artesian water and phreatic water D, Association internationale d’hydrologie scienti$que, Assemblée de Rome, 1954, t. 2, p. 32-37. (Publication no 37.)
1957, p. 52-65. 20. -,
28. SILINE-BEKTCHOURINE, A.I. ; PLOTNIKOV, N.A.,
1956, t. 20, p. 56-57.
117

Hydrologie des régions arides
35. TURC, L., u Nouvelle formule pour le calcul du bilan de l’eau en fonction de valeur moyennes annuelles des précipitations et de la température D, C.R. Acad. Sci. France, 1951, t. 233, no 11, p. 633-635.
36. -, L., (( Relations entre les précipitations, l’évaporation et l’écoulement D, Sols africains, 1953, vol. 3, no 1, p. 138-172.
37. -, (( Le bilan de l’eau des sols, relations entre les précipitations et l’écoulement )), Ann. agron., série A, 1954, vol. 5, no 4, p. 491-596.
38. -, (( Calcul du bilan de l’eau. ]Evaluation en fonction des précipitations et des tempé- ratures D, Association internationale d’hydrologie scientijique, Assemblée de Rome, 1954, t. 3, p. 188-202. (Publication no 38.)
Hydrology of range lands as affected by the presence of brush vegeta- tion )), Association internationale d’hydrologie scientifique, Assemblée de Bruxelles, 1951, t. 3, p. 226-234. (Publication no 34.)
40. SOCIÉTÉ HYDROTECHNIQUE DE FRANCE, ALGER, (( Pluie, évaporation, filtration et écoule- ment. Comptes rendus des troisièmes journées de l’hydraulique )), La houille blanche, 1955.
39. VEIHMEYER, F.J.,
FLUCTUATIONS DES NAPPES
41. ACHTEN, M.A., u Fluctuations des nappes aquifères n, Tech. de l’eau, 1952, no 6, p. 31-39. 42. BOGARDI, J., (( Der Einfluss des Niederschlags und der Temperatur auf die Veränderungen
des Grundwasserspiegels D, Acta tech. Acad. Sci. Hungar., 1952, vol. 5, no 2, p. 219-245. 43. COLLINS, B.W., (( Fluctuations of ground water levels in New Zealand and their significance ))*
Association internationale d’hydrologie scientijique, Assemblée de Rome, 1954, t. 2, p. 25-31. (Publication no 37.)
44. CUSHMAN, R.L.; HALPENY, L.C., (( Effect on western drought on the water resources of Safford Valley, Arizona )I, Trans. Amer. geophys. Un., 1955, vol. 36, no 1, p. 87-94.
45. DENNER, J., (( La variation du niveau des eaux souterraines pour une longue série d’années, Gewäserkunde Tagung in Freiburg i. B. n, Gas und Wasserfach, 1955, t.-96, no 18, p. 611- 613.
46. -, (( 86 jährige Grundwasserganglinie von Berlin (Ein Ergebniss des ältesten exiestier- enden Grundwasserdienstes n, 2. dtsch. geol. Ges., 1954, t. 106, p. 70-74.
47. DOLLÉ, M., (( Oscillations de la surface piézométrique du réseau aquifère du Turonien supérieur D, Association internationale d’hydrologie scientijìque, Assemblée de Bruxelles, 1951, t. 3, p. 136-142. (Publication no 33.)
48. -, (( Oscillations de la surface piézométrique du réseau aquifère du Turonien supérieur n, Tech. de l’eau, 1952, no 68, p. 7-12.
49. FERRIS, J.G., (( Cyclic fluctuations of water level as a basis for determining aquifer trans- missibility )), Association internationale d’hydrologie scientifique, Assemblée de Bruxelles, 1951, t. 2, p. 213. (publication no 33.)
50. GRUNDY, F., (( The ground water depletion curve, its construction uses )), Association inter- nationale d’hydrologie scientijìque, Ãssemblée de Bruxelles, 1951, t. 2, p. 213. (Publication no 33.)
51. MÜGGE, R., (( Aufzeichnung von Luftdruck und Erbebenwellen mit Hilfe von Brunnen- spiegeln )), Association internationale d’hydrologie scientifique, Assemblée de Rome, 1954, t. 2, p. 49-52. (Publication no 37.)
52. NEMETH, E., (( Recherche graphique sur la corrélation entre la fluctuation de la nappe phréatique et celle de la température n9 Association internationale d’hydrologie scientijique, Assemblée de Rome, 1954, t. 2, p. 598-601. (Publication no 37.)
53. PAAVEL, V., (( Die Veränderlichkeit der Niederschlagshöhen und die Bemessung der Grund- wasser-fassungsanlagen )), Gas und Wusserfach, 1955, vol. 96, no 18, p. 599-601.
54. RAFFO, J.M., (( Variaciones de la napa freatica en relación con la precipitación, la presión atmosférica y la temperatura )I, Association internationale d’hydrologie scientijique, Assem- blée de Rome, 1954, t. 2, p. 99-112. (Publication no 37.)
118

Bi bbgrtaphie
55. THOMAS, H.E., (( Fluctuations of ground water levels D, Association internationale d'hydro- logie scientifique, Assemblée de Bruxelles, 1951, t. 2, p. 143-146. (Publication no 33.)
56. TISON, G., (i Nouvelles recherches sur les fluctuations des nappes aquifères n, Association internationale d'hydrologie scientifique, Assemblée de Rome, 195ii t. 2, p. 38-48. (Publication no 37.)
57. -, (( Fluctuations des nappes aquifères de types divers et particulièrement des nappes d'alluvions )), Association internationale d'hydrologie scienti$que, Symposia Darcy, 1956, t. 2, p. 210-221. (Publication no 41.)
58. TISON, L.J., (( Au sujet des fluctuations des nappes aquifères étendues n, Association inter- nationale d'hydrologie scientifique, Assemblée de Bruxelles, 1951, t. 2, p. 195-201. (Publica- tion no 33.)
59. -, (( Une étude analytique-des fluctuations des nappes aquifères est-elle possible? )), Ann. Association Ingénieurs Ecole spéciale de Gand, 1952, no 3, p. 89-99.
60. WAITE, H.A.; THOMAS, H.E., Effect of current drought upon water supplies in Cedar Valley, Utah D, Trans. Amer. geophys. Un., 1955, vol. 36, no 5, p. 805-812.
61. WERNER, P.W.; SANDGUIST, K.J., (( On the ground water recession curve for large water sheds n, Association internationale d'hydrologie scientifique, Assemblée de Bruxelles, 1951, t. 2, p. 202-212. (Publication no 33.)
RECHERCHES ET EXPLOITATION DES NAPPES DANS LES RÉGIONS ARIDES
62. CHEBOTAREV, I.I., (c Yield capacity of the aquifer D, Wat. & Wat. Engng., 1954, p. 355-357. 63. CLAIR, A., K lhude hydrologique du chott Chergui D, Terres et eaux, supplément scienti-
fique, 1956, no 7, p. 3-34. 64. H o m , R.H.L.; WILKE, H.R.; BLOODGOOD, D.E., x Application of air photo interpretation
in the location of ground water D, J. Amer. ?Vat. Wks. Ass., 1956, vol. 48, no 11, p. 138- 139.
65. LOEHNBERG, A., i( Water supply and drainage in semi arid countries )), Trans. Amer. geophys. Un., 1957, vol. 38, no 4, p. 501-510.
66. LOWY, H., (i Geophysical prospection of underground water in the desert by means of electromagnetic interference fringes N, Proc. Instn. Radio Engrs., 1956, vol. 44, no 8, p. 1062.
67. SCHOELLER, H., (( Zone et rayon d'appel, débits spécifiques des forages, des puits. Calcul des constantes des couches aquifères et de la longueur du front d'emprunt )I, I.U.G.G. Newsletter, no 13, Londres, International Union of Geodesy and Geophysics, 1956, p. 143- 160.
68. -, (i Méthode de détermination du rayon d'appel des forages et du coefficient de Darcy. Application B l'étude de la nappe des sables paléocènes de l'Aquitaine I), Association inter- nationale d'hydrologie scientifique, Symposia Darcy, 1956, t. 2, p. 67-75. (Publication no 41.)
PERMÉABILITÉ ET D&BITS DES PUITS ET FORAGES EN RÉGIME PERMANENT
69. BINDEMAN, N.N., (Méthodes de détermination de la conductibilité des roches par pompage
70. -, (Méthodes graphiques de détermination du débit des puits), Gostroizdat, 1951. 71. BORELI, M., (( Free surface flow toward partially penetrating wells D, Trans. Amer. geophys.
72. HANSEN, V.E., (i Unconfined ground water flow to multiple wells D, Proc. Amer. Soc. civ.
73. HANTUSH, M.S.; JACOB, C.E., E( Plane potential flow of ground water with linear leakage x,
74. -; -, (( Steady three dimensional flow to a well, in a two layered aquifer n, Trans.
et déversement), Ougletchizdat, 1951.
Un., 1955, vol. 36, no 4, p. 664-672.
Engrs., vol. 78, no 142, 18 pages.
Trans. Amer. geophys. Un., 1954, vol. 35, no 6, p. 917-936.
Amer. geophys. Un., 1955, vol. 36, no 2, p. 286-292.
119

Hydrologie des régions arides
75. INESON, J., (( Darcy’s law and the evaluation of peheability )), Association internationale d’hydrologie scientifique, Symposia Darcy, 1956, t. 2, p. 165-172. (Publication no 41.)
76. KREPS, H., (( Ein neues Verfahren zur Ermittlung der Grundwassergeschwindigkeit aus einem Pumpenversuch D, Die Wassenuirtschaf, 1954, t. 44, no 9, p. 228-232.
77. LELLI, M., (( S d a relazione fra la reciproca influenza di due pozzi freatici e la potenza della falda d‘acqua sotteranea attraversata )). G. Gen. civ., 1950, t. 88, no 7-8, p. 423-426.
78. NAHRGANG, G., Zur Theorie des volkommenen und unvolkommenen Brunnens, Berlin, Göt- tingen, Heidelberg ; Springer, 1954, 43 pages.
79. -, (( L’hypothèse de Dupuit-Thiem pour le calcul d’un puits et l’écoulement réel au voisinage d’un puits vertical à surface libre D, Association internationale d’hydrologie scien- tifique, Symposia Darcy, 1956, t. 2, p. 173-183. (Publication no 41.)
80. REMSON, I.; LANG, S.M., (( A pumping test method for the determination of specific yield D, Trans. Amer. geophys. Un., 1955, vol. 36, no 2, p. 321-355.
81. SCHMIDT, H. J., (( Abweichungen eines Ergiebigkeitsgezetzes bei einem Brunnen mit freiem Grundwasserspiegels D, Gas und Wasserfach, 1949, t. 90, no 23, p. 622.
82. SCHOELLER, H., (( Méthode de détermination du rayon d’appel des forages et du coefficient de Darcy. Application à l’étude de l’épuisement de la nappe des sables paléocènes #Aqui- taine )), Association internationale d’hydrologie scientijique, Symposia Darq, 1956, t. 2, p. 67-75. (Publication no 41.).
83. TCHARNYI, I.A., (( Démonstration rigoureuse de la formule de Dupuit dans l’écoulement sans pression )), Dokl. Akad. Nauk S.S.S.R., 1951, t. 79, no 6.
84. VIBERT, A., (< Sur une démonstration rigoureuse des formules de Dupuit ))* Génie civ., 1954, no 1, p. 10-12.
85. -, (( Puits captants et galeries drainantes D, Génie civ., 1950, t. 70, no 17, p. 329-333. 86. WEN HSIUNG LI; BOCE, P.; BENTON, S.F., (( A new formula for flow into partially pene-
trating wells in aquifers 11, Trans. Amer. geophys. Un., vol. 35, no 5, p. 805-812.
PEWÉABILITÉ ET DÉBITS DES FORAGES ET PUITS EN RÉGIME NON PERMANENT
87. BOULTON, N.S., (( The drawdown of the water table under non-steady conditions near a pumped well in an unconfined formation )), Proc. Instn. civ. Engrs., part. 3, vol. 3, no 2,
88. -, (( Unsteady radial flow to a pumped well allowing for delayed yield from storage )), Association internationale d’hydrologie scientifique, Assemblée de Rome, 1954, t. 2, p. 472- 477. (Publication no 37.)
89. COOPER, H.H.; JACOB, C.E. x A generalized graphical method for evaluating formation constants and summarizing well field history D, Trans. Amer. geophys. Un., 1946, vol. 27,
90. GELIS, E. DE, Qéments d’hydraulique souterraine, Rabat, Ministère de la production indus- trielle et des mines, Service géologique, 1956, 84 pages. (Notes et mémoires, no 138.)
91. GOLDSCHMIDT, M.J.; JACOBS, M., ((Underground water in the Haifa-Acco sand dunes and its replenishment )), Association internationale d’hydrologie scientifique, Symposia Darcy, 1956, t. 2, no 41, p. 39-56.
92. HANTUSH, M.S.; JACOB C.E., (( Non-steady Green’s functions for an infinite strip of leaky aquifer D, Trans. Amer. geophys. Un., 1955, vol. 36, no 1, p. 101-112.
93. -; ~ , (I Non-steady radial flow in infinite leaky aquifer D, Trans. Amer. geophys. Un., 1955, vol. 36, no 1, p. 95-100.
94. HANTUSH, M.S., (( Analysis of data from pumping tests in leaky aquifers )), Trans. Amer. geophys. Un., 1956, vol. 37, no 6, p. 735-737.
95. INESON, J., (( The drawdown of the water table under non-steady conditions near a pum-. ped well in an unconfined formation D, Proc. Instn. civ. Engrs., 1955, part. 3, vol. 4, no 1, p. 218.
p. 564-579.
p. 526-534.
120

Bibliographie
96. JACOB, C.E.; LOHMAN, S.W., (( Non-steady flow to a well of constant drawdown in an extensive aquifer I), Trans. Amer. geophys. Un., 1952, vol. 33, no 4, p. 559-570.
97. NELSON, W.B.; THOMAS, H.E., (( Pumping from wells on the floor of the Sevier Desert, Utah D, Trans. Amer. geophys. Un., 1953, vol. 34, no 1, p. 74-85.
98. REMSON, I.; VAN HYLCKAMA, T.E.A., (( Nomographs for the rapid analysis of aquifer tests I), J. Amer. Wat. Wks. Ass., vol. 48, no 5, p. 511-516.
99. RORABAUGH, NI., Graphical and theoretical analysis of step drawdown test of artesian well, 1953, numéro spécial 362, 23 pages (Proc. Amer. Soc. civ. Engrs.).
100. SCHNEEBELI, G.; HUARD DE LA M ~ R R E , P., (( Nouvelles méthodes de calcul pratique des écoulements de filtration non permanents à surface libre )), La houille blanche, no 3, 1953.
101. -, Sur l'hydraulique des puits D, Association internationale d'hydrologie scientijique Symposia Darcy, 1956, t. 2, p. 10-27. (Publication no 41.)
102. THEIS, C.V.; BROWN, R.H., Use of slide rule in solving ground water problems involving application of the non equilibrium formula, tirage à part sans indication, 2 pages. (U.S. Geological Survey.)
103. UBELL, K., (( Unsteady flow of ground water caused by well drawdown D, Association internationale d'hydrologie scientiJique, Symposia Darcy, 1956, t. 2, p. 129-132. (Publication no 41.).
104. VEN TE CHOW, (( O n the determination of transmissibility and storage coefficients from pumping test data I), Trans. Amer. geophys. Un., 1952, vol. 33, p. 397-405.
GÉOCHIMIE DES EAUX SOUTERRAINES
105. ARNDT, R.H., (( Radioactivity of rivers and lakes in parts of Garland and Hot Springs Countries, Arkansas D, Econ. Geol., 1953, vol. 48, p. 551-567.
106. BEHNE, W., (( Untersuchungen zur Geochemie des Chlor und Brom D, Geochimica et Cosmo- chimica Acta, 1953, vol. 3, no 4, p. 186-214.
107. BOCHERT, H., (( Zur Geochimie des Kohlenstoffs D, Geochimica et Cosmochimica Acta, 1951, vol. 2, no 1, p. 62-75.
108. BUYDENS, R., (( Le comportement des fluorures dans l'eau D, Bull. Acad. Méd. Belg., 1954, t. 19, no 5, p. 217-242.
109. -, (( L'importance du fluor et de l'iode dans les eaux d'infiltration I), Bulletin du Centre belge d'étude et de documentation des eaux, 1951, no 13, p. 145-150.
110. CARVALHO, A.H. DE, (( Preuve analytique indirecte de l'existence de l'anion silicate dans les eaux naturelles D, XVe Congrès de chimie analytique, Lisbonne, 1956, p. 234-235.
111. CAZAUX, P.; CANELLAS, J.; THOMASSIN, R., (( Contribution à la connaissance de la consti- tution chimique des eaux sulfurées )), Ann. Inst. Hydrol. 1954, t. 25, p. 47-74.
112. CHEBOTAREV, I.I., (( Reporting, interpretation and utilization of water analyses )), Wut. & Wut. Engrg., 1952, p. 132-137.
113. -, (( Les probGmes de salinité dans les régions arides D, Vat. & Wut. Engrg., 1955, t. 59, no 707, p. 10-19.
114. -, (( Metamorphism of natural waters in the crust of weathering D, Geochimica et Cosmo- chimica Acta, 1955, vol, 8, no 1-2, p. 22-48; no 3, p. 137-170.
115. -, (( Geochemical metabolism in natural waters n, Congres géologique international de Mexico, 1956, t. 20, p. 210.
116. -, (( Geochemical types of waters in arid regions D, Congrès géologique international de Mexico, 1956, t. 20, p. 48-49.
117. CHILINGAR, G.V., Durov's classification of natural waters and chemical composition of atmospheric precipitations in U.S.S.R. A review )), Trans. Amer. geophys. Un., 1956, vol. 37, no 2, p. 193-196.
118. -, (( C1 and SO4 content of atmospheric precipitation in U.S.S.R. i), Trans. Amer. geophys. Un., 1956, vol. 37, no 4, p. 410-412.
121

Hydrologie des régions arides
119. CHILINGAR, G.V., Soviet’s methods of reporting and displaying results of chemical analyses of natural waters and methods of recognizing oil-field waters. A summary D, Trans. Amer. geophys. Un., 1957, vol. 38, no 2, p. 219-221.
120. DANSGAARD, W., (( The Ola abundance in fresh water n, Geochimica et Cosmochimica Acta,
121. -, (( The Ola abundance in natural waters D, Nature, 1954, vol. 174, no 4422, p. 241-260. 122. DAUVILLIEH, A., (( Sur le cycle du sel n, C. R. Acad. Sci. France, 1956, t. 242, no 1, p. 47-
50. 123. DAVIS, R.; SCHAEFFER, O.A., (( Chlorine-36 in nature D, Ann. N.Y. Acad. Sci., 1955, t. 62,
no 5, 17 pages. 124. DOLE, M.; LANE, G.A.; RUDD, P.; ZANKELIES, D.A., (( Isotopic composition of atmospheric
oxygen and nitrogen D, Geochimica et Cosmochimica Acta, 1954, vol. 6, p. 65-78. 125. DOWNES, R.G., (( Cyclic salt as a dominant factor in the genesis of soils in South-Eastern
Australia )), Austr. Agric. Res., 1954, t. 5, no 3, p. 448-464. 126. DUBILER, A.S., Sur le Probleme de la composition chimique des eaux souterraines de la
région précaspienne nord-occidentale )I, Trud. Lab. gidrogeol. Probl., 1955, t. 12, p. 120- 138.
127. DUROV, S.A., (( Sur la question de l’origine de la composition de l’eau des Karsts )), J. chim. Ukr., 1956, t. 22, no 1, p. 106-111.
128. DYE, J.F., (( Calculation of effect of temperature on pH, free carbon dioxide and the three forms of alkalinity D, J. Amer. Wut. Wks. Ass., 1952, vol. 44, no 4, p. 356-372.
129. EPSTEIN, L.; MAYEDA, T., (( Variation of Ola content of waters from natural sources n, Geochimica et Cosmochimica Acta, 1953, vol. 2, no 3, p. 213-225.
130. FIGUEROL, (( Variaciones quimicas de una pizarra situada en la pared de un horno D, 3*& Reunión internacional sobre reactividad de los sólidos, Madrid, Resumenes, 1956, p. 45.
131. FRIEDMAN, L; UREY, H.C., (( Deuterium content of natural waters D, Bull. geol. Soc. Amer., 1952, vol. 63, no 12, part. 2, p. 1252.
132. -; -, (( Deuterium content of natural waters P, Ann. mineralogist, 1953, vol. 38, no 3-4, p. 338.
133. GEORGES, W.O.; HASTINGS, W.W., (( Nitrate in the ground water of Texas n, Trans. Amer. geophys. Un., 1951, vol. 32, no 3, p. 450-456.
134. GIUGHIARELLI, M.G., (( Ricerche sul tenore di F nelle acque di alimentazione della Puglia e Lucania >), Boll. Soc. ital. Biol. sper., 1952, t. 28, no 6, p. 1182-1183.
135. GORBOV, A.F., (( Sur la zonalité de la composition chimique des eaux dans la steppe de Kulundiusk )), Dokl. Akad. Nauk. S.S.S.R., 1951, vol. 81, no 5, p. 871-874.
136. GUYOT, J., (( Étude géochimique des eaux dans la région de Biskra D, Terres et eaux, supplé- ment scientifique, 1955, no 6, p. 3-15.
137. HANYA, T., (( Relation entre la nature chimique des eaux douces et la nature géologique du milieu n, J. chem. Soc. Japan, Pure chem. sect., 1953, vol. 74, no 5, p. 365-367; no 6, p. 450-452; no 7, p. 522-525.
138. HASTINGS, W. W., (( Report of Committee on the chemistry of natural waters, 1951-1952 D, Trans. Amer. geophys. Un., 1951, vol. 32, no 5, p. 769.
139. (( Report of Research committee on chemistry of natural waters 1951-1952 D, Trans. Amer. geophys. Un., 1953, vol. 34, no 3, p. 484-487.
140. HERTTAN, A., (( La configuration chimique des eanx souterraines I), Bulletin du Centre belge d’études et de documentation des eaux, 1955, no 27, p. 37-45.
141. IWASAKI, I.; TAREETANI, I.; KATSURA, T.; TACHIBANA, K., (( Recherches géochimiques sur les dépôts des sources chaudes I.Si0, dans la source chaude de Shiroike n, J. chem. Soc. Japan, Pure chem. sect., 1953, vol. 74, no 10, p. 857-859.
142. KELLER, G., (( Grundwasserversalaungen im saxonischcn Faltungsfeld Niedersachsens D, Geotektonisches Symposium zu Ehren von Hans Stille, Stuttgart, 1956.
1954, vol. 6, p. 241-260.
122

Bibliographie
143. KOBAYASHI, S., (( Distribution du fluor dans les eaux terrestres, sous divers états n, Bull. chem. Soc. Japan, 1954, t. 27, no 6, p. 314-317.
144. KORCENSHTEJN, V.N., (( Traits hydrochimiques caractéristiques de l’horizon aquifère Khadoumsky de l’élévation de Stavropol n, Dokl. Akad. Nauk. S.S.S.R., 1955, vol. 104, no 5, p. 771-774.
145. KORITNIG, S., (( Ein Beitrag zur Geochemie des Fluor )I, Geochimica et Cosmochimica Acta,
146. LA MOREAUX, P. E., (( Fluoride in ground water of Alabama D, Trans. Amer. Inst. min., (metall.) Engrs., 1951, vol. 187, p. 886-888.
147. LOVE, S. K., (( Radio-activité naturelle de l’eau )I, J. indust. Engng. Chem., 1951, vol. 43, no 7, p. 1541-1544.
148. MARGAT, J.; MARTIN, R., (( Essai sur l’évaporation et les variations de concentration des eaux souterraines dans une nappe phréatique d’un pays désertique D, Association inter- nationale d’hydrologie scientifique, Assemblée de Rome, 1954, t. 2, p. 72-84. (Publication no 37.)
149. MUCHEMBLE, G., (( Observations sur les eaux souterraines radio-actives du nord de la France et la radio-activité des roches encaissantes D, Ann. Inst. Hydrol., 1952, t. 23, no 72,
150. NORING, P., (( Chemische und physikalische Eerscheinungen bein einfiltriertem Grund- wasser D, Association internationale d’hydrologie scientijìque, Assemblée de Rome, 1954, t. 2, p. 113-117. (Publication no 37.)
151. OAIRA, S., u Distribution of heavy water in natural waters I), J. Earth Sci., Nagoya Univ., Japan, 1953, vol. 1, p. 42-61.
152. OVCEINNIKOV, A. M., (( Sur les principes de la classification hydrogéochimique des eaux souterraines D, Bull. Soc. Nat. Moscou, Section géologie, 1953, t. 28, no 2, p. 86-87.
153. RICHTER, W.; FLATHE, H., (( Die Versalzung von Küsteunahen Grundwassern dargestelt an einem Teil der deutschen Nordseekiiste n, Association internationale d’hydrologie scien- tifique, Assemblée de Rome, 1954, t. 2, p. 118-130. (Publication no 37.)
154. RIDDER, M. DE, Fluor in drinkwater D, Bulletin du Centre belge d’études et de documentation des eaux, 1950, no 7, p. 421-423.
155. RONA, E.; UREY, W. D., (( Radioactivity of ocean sediments. Radium and uranium content of ocean and river waters I), Amer. J. Sci., 1952, vol. 250, no 4, p. 241-262.
156. SAITO, N., (( On the existence of radiocolloids in natural waters D, Association internationale d’hydrologie scientifique, Assemblée de Bruxelles, 1951, t. 3, p. 512-516. (Publication no 34.)
157. SAJDAKOVSKIJ, S. Z., (( Sur la formation des eaux hydrosulfurées dans l’extrémité SW de la plate-forme russe )), Dokl. Akad. Nauk. S.S.S.R., 1955, vol. 103, no 2, p. 303-304.
158. -; THILCHUE, V. G.; CVIK, S. M., (( Sur le problème des conditions de formation des eaux souterraines du type chloruro-alcalino calcique D, Dokl. Akad. Nauk S.S.S.R., 1952, vol. 80, no 5, p. 791-792.
159. SARUHASHI, K., (( O n the equilibrium concentration ratio of carbonic acid substances dissolved in natural waters. A study of the metabolism in natural waters D, Papers Meteorol. Geophys. Japan, 1955, vol. 6, no 1, p. 38-55. &de sur les transformations dans les eaux naturelles. Sur le rapport de la concentration
21 l’équilibre des substances dérivées de l’acide carbonique dissoutes dans les eaux natu- relles )), J. Chem. Soc. Japan, Pure chem. Sect., 1955, t. 76, no 111, p. 1294-1308.
161. SAVCHENKO, P. S., (( Teneur en iode des eaux souterraines dans la région de la rivi6re Severnyj Donetz n, Dokl. Akad. Nauk S.S.S.R., 1954, vol. 99, no 2.
162. SCHNEIDER, H., (( Chemie des Grundwasser n, in Die Wasserschiessung, Deutsch Verein von Gas und Wasserfachmännern, Essen, 1952, p. 53-71.
163. SCHOELLER, H., (( Relation entre la concentration en chlore des eaux souterraines et les échanges de bases avec les terrains qui les renferment n, C. R. Acad. Sci., France, 1951, t. 132,
1951, vol. I, p. 80-116.
p. 29-64.
160.
p. 1432-1434.
123

Hydrologie des régions arides
164. SCHOELLER, H., (( Condensations occultes, en particulier dans les affleurements de terrains calcaires ou gréseux de l'Afrique du Nord n, Colloque international no 35 b Alger du Centre national de la recherche scientifique, 1951, p. 353-363.
165. -, (( Contribution à l'étude du fluor des eaux souterraines D, Ann. Inst. Hydrol., 1952, t. 23, no 72, p. 2-19.
166. -, (( Essai sur la qualité chimique de l'eau destinée à l'alimentation de l'homme dans les pays arides D, Terres et eaux, Alger, Supplément scientifique, 1955, no 24, p. 4-11.
167. -, (( La solubilité du fer dans les eaux souterraines D, Ann. Inst. Hydrol., 1955, t. 26, no 78, p. 1-37.
168. -, Géochimie des eaux souterraines ; application aux eaux de gisement de pétrole, Pans, Technip, 1956, 213 pages; et Rev. Inst. franç. Pétrole, 1955, vol. II, p. 181-213; 219-246;
169. SCHWILLE, E., Chlorure et nitrate dans les eaux souterraines de la Hesse rhénane et de la Rhénanie n, Gas und Wasserfacht, 1953, t. 94, no 14, p. 410-414.
170. -, (( Ionenumtausch und der Chemismus von Grund- und Mineralwässern n, 2. der geolo- gischen Gesellschaft, 1954, t. 106, p. 16-22.
171. SILINE-BEKTCHOURINE, A. I., Hydrogéologie spéciale, 1 vol., Moscou, Gossendarctvennoe iz datelctvo geologitcheshoi 4iteratouri, 1951, 394 pages.
172. -, Zonalité hydrochimique des eaux souterraines du synclinal attenant ä la Caspienne )), Izvest. Akad. Nauk S.S.S.R.; ser. Geol., 1952, no 4, p. 27-40.
173. -, K Sur les questions de la formation de la composition chimique des eaux Souterraines dans les régions arides )), M. G. Ou outhchenie zapouski, bip, 176, geologuia, Izv, 1956, p. 175- 193.
174. -, (( Types of hydrochemical maps in hydrogeology )i, Abstracts of the reports at the XI general assembly of the International Union of Geodesy and Geophysics. The international Association of Scientific Hydrology. Moscou, Akad. Nauk S.S.S.R., 1957, p. 69-70.
175. SPIRO, N. S., GRAMBERG, I. S., VOOH, C. I., (( Sur la classification générique des eaux natu- relles N, Dokl. Akad. Nauk, S.S.S.R., 1953, t. 93, no 3, p. 531-534.
176. STANKO, M., (( Radioactivity of waters issuing from sedimentary rocks N, Econ. Geol., 1952, vol. 47, no 5, p. 543-547.
177. STRAUB, J.; KOVACS, E., ((Antagonisme fluor-iode )), Népegészséggigy, 1956, t. 37; p. 162-164. 178. SUGAWARA, K.; TOCRIKUVO, L., s The determination of argon in natural waters, with
special reference to the metabolism of oxygen and nitrogen )), J. Earth Sci. Nagoya Uniu., Japan, 1955, t. 3, p. 77-84.
179. TAGEEVA, N. W., (t Sur certains types géochimiques d'eaux souterraines )), Izvest. Akad.
180. -, TCKHOMIROVA, M. M., (( Sur la géochimie des eaux naturelles du type chlorures de M g et de N a )), DOM. Akad. Nauk S.S.S.R., 1954, t. 96, no 1, p. 121-124.
181. TAKAHISA, H. ; FUMIO, S., (( Étude statistique sur les caractères géochimiques des eaux douces au Japon D, Geochimica et Cosmochimica Acta, 1956, vol. 9, no 5, p. 249-255.
182. TAMRAZJAN, G. P., (( Sur les lois de variation de la composition chimique des eaux de la série Makop, dans les limites du Caucase D, Dokl. Akad. Nauk S.S.S.R., 1954, t. 96, no 6,
183. VINCK, (( La salinité des eaux souterraines dans le Schleswig-Holstein. Origine, étendue, signification et suppression n, Gewässer kunde-Tagung in Freiburg im B., Gas und Wasser- fach, 1955, t. 96, no 18, p. 611-613.
184. VOLKER, A.; MONTSMA, E. O.;(( Détermination des saliilités des eaux à grandes profondeurs dans le sous-sol du Zuiderzee par prospection géophysique D, Association internationale d'hydrologie scientifique, Assemblée de Rome, 1954, t. 2, p. 151-161. (Publication no 37).
185. VORONKOV, P. P.; SOKOLOVA, O. K., (( Influence du lieu de prélèvement des eaux sur la quantité d'ions en solution D, Dokl. Akad. Nauk S.S.S.R., 1951, t. 81, no 4, p. 561-564.
507-552; 671-719; 823-874.
Nauk, S.S.S.R., SC. Geol., 1954, no 1, p. 69-76. 5
p. 1229-1232.
124

Bibliographie
186. VORONKOV, P. P.; SOKOLOVA, O. K., (( Particularité de formation de la composition chi- mique des eaux superficielles dans les différentes zones géographiques n, Dokl. Akad. Nauk S.S.S.R., 1954, t. 94, no 2, p. 293-296.
187. WAGNER, R., (( Zum Chemismus tiefer Grundwasser in einem Teil Nord-Westdeutschlands D, Association internationale d'hydrologie scientijque, Assemblée de Rome, 1954, t. 2, p. 131-137. (Publication no 37.)
188. WEDEMEYER, F. W., (c L'eau de source contenant des nitrates provoque la m6thémoglo- binémiose chez les jeunes enfants I), Archiv für Kinderheilkunde, 1956, t. 152, p. 267-275.
189. ZELLER, E. J.; WRAY, J. L., (( Factors influencing precipitation of calcium carbonate I), Bull. Amer. Ass. Petrol. Geol., 1956, vol. 40, no 1, p. 140-152.
TRACEURS RADIO-ACTIFS
190. AEBERSOLD, P. C., (I Le rôle des isotopes en technologie et dans l'industrie )), UtiEisation de l'énergie atomique à des fcns pacijiques, Nations Unies, 1956, vol. 14, p. 3-15.
191. ALBERTS, A. A. ; COCANOVER, R. D. ; BUNDRANT, C.O., (I Application of radio-isotopes to subsurface surveys D, AIME, Petroleum Branch, Fall Meeting, San Antonio, oct. 1954.
192. ALLEN, F. €1.; GRINDLEY, J., (( Radioactive tracers in the Thames Estuary n, The Dock and Harbour Authority, janv. 1957, vol. 37, no 435, p. 302-306.
193. ARCHIBALD, R. S., II Radiotracers in flow tests n, J. Boston Soc. civ. Engrs., 1950, vol. 37, no 1, p. 49-116.
194. BAVEL, C. H. M. VAN; HOOD, E. E.; UNDERWOOD, N., (I Vertical resolution in the neutron method for measuring soil moisture n, Trans. Amer. geophys. Un., 1954, vol. 35, no 4,
95. BEATTY, K. O.; FERREL, J. K.; RICHARDSON, (( Les radio-isotopes dans l'étude de Ia dyna- mique des fluides I), Utilisation de l'énergie atomique à des jîns pacifiques, Nations Unies,
Continental water balance, groundwater inventory and storage times, surface ocean mixing rates and world-wide water circulation patterns from cosmic-ray and bomb tritium N, Geochimica et Cosmochimica Acta, 1957, vol. 12, p. 277-296.
197. BROWN, R. E.; PARKER, H. M.; SMITH, J. M., (( Décharge terrestre des déchets liquides )), Utilisation de Z'énergie atomique à desJins pacifcques, Nations Unies, 1956, vol. 9, p. 763-769.
198. EDWARDS, J. BI.; KOLTER, L. E., n Radio-isotopes for water-input proíiles, in water flood operations D, Oil Gas J., 29 sept. 1954, p. 53-60.
199. FLAGG, A. H.; MYERS, J. P.; CAMPBELL, J. L. P.; TERRY, J. M.; MARDOCK, E. S., (( Radio- active tracers in oil production problems )), J. Petrol. Tech. ; AIME, Petroleum Transactions,
200. FOX, C. S., (( L'emploi des isotopes radio-actifs pour contrôler le mouvement des eaux
201. -, CI Radioactive isotopes tracer underground waters )I, Public Works, janv. 1952,
202. GUERON, J., (I Emploi des radio-éléments en hydrologie )), lm Congrès international de
203. HESS, V. F., (( O n the use of a radioactive tracer method in measurements of water n,
204. HOURS, R., II Les traceurs radio-actifs en hydrologie )), Mémoires et travaux de la Société
205. JOSENDAL, V. A.; SANDFORD, B. B.; WILSON, J. W., (( Improved multiphase flow studies
206. KAUFMAN, W. J.; ORLOB, G. T., (( A n evaluation of ground water tracers D, Trans. Amer.
p. 595-600.
1956, vol. 15, p. 230-235. 196. BEGGEMAN, F.; LIBBY, W .F.,
1955, VOL 204, p. 1-6.
souterraines D, Ingegneria, 1951, p. 305-307.
p. 57-58.
spéléologie, 1953, vol. 2, p. 301-306.
Trans. Amer. geophys. Un., 1943, vol. 2, p. 587-594.
hydrotechnique de France, 1955, vol. 1, p. 14-24.
employing radioactive tracers )), J. Petrol. Tech., 1952, vol. 4, p. 65-76.
geophys, Un., 1956, vol. 37, no 3, p. 297-306.
125

Hydrologie des régions arides
207. KAUFMAN, W. J.; ORLOB, G. T., (i Measuring ground water movement with radio-active and chemical tracers D, J. Amer. Wut. Wks. Ass., 1956, vol. 48, no 5, p. 559-572.
208. MONTENS, A., (( Die Verwendung von radioaktiven Isotopen bei Strömungs- und Geschwin- digkeits Messungen D, Gas und Wasserfach, 1952, vol. 93, no 14, p. 411-416.
209. (( Radioactive Isotopes in Petroleum Research o, Petroleum, 1952, vol. 15, p. 61-65. 210. RAVIER; HOURS, R.; SCHNEEBELI, G., (( Emploi de traceurs en hydrologie D, Association
211. RUSSEL, M., (i Radioactive iodine used as a tracer n, World Oil, 1954, vol. 138, p. 266-275, 212. SELIGMAN, H., (( Progrès récents dans les utilisations industrielles des isotopes radio-actifs 1).
Utilisation de l’énergie atomique ù des jìns paci$ques, Nations Unies, 1956, vol. 14, p. 16-18. 213. SIMANE, C., (( L’emploi des radio-isotopes en Tchécoslovaquie D, Utilisation de l’énergie
atomique ù des fins paci$ques, Nations Unies, 1956, vol. 14, p. 65-68. 214. SONS, E., (( Die Messung von Fliesszeiten in Wasserlaufen mit Hilfe von radioaktiven
Stoffen D, Die Wasserwirtschaft, 1952, vol. 42, no 10, p. 313-317. 215. STANLEY, D. R., (( Sand hltration studied with radiotracers 11, Amer. Soc. civ. Engrs.,
1955, vol. 81, p. 592. 216. TRUESDALE, G. A., (( Use of radioactive isotopes in tracing sewage flown, Atomics and atomic
technology, 1954, vol. 5, p. 304-312. 217. URBAIN, P., (( Sur l’état actuel des recherches hydrologiques par la méthode des traceurs
radio-actifs D, Association internationale d’hydrologie scienti$que, Assemblée de Rome, 1954, t. 2, p. 238-247. (Publication no 37.)
218. -; LAGRANGE, R.; HOURS R.; GESLIN, M., (( Sur l’emploi des traceurs radio-actifs sur le terrain en géologie et hydrogéologie I), Ann. Inst. Hydrol., 1954, vol. 25, no 76, p. 7-26.
219. U.S. NATIONAL BUREAU OF STANDARDS, Maximum permissible amounts of radio-isotopes in the human body, and maximum permissible concentrations in air and water, 1953. (Hand- book no 52).
220. VESSEY, E. de ; CZERNY, Gyoeo, (( A study of underground flow of radioactive isotopes and tracing ions D, Hidrologiai Kölony, 1957, vol. 37, p. 44-56.
221. WATKINS, J. W.; DUNNING, H. N., ((Les isotopes radio-actifs dans la recherche appliquée à la production du pétrole D, Utilisation de l’énergie atomique Ù des jìns pacijìques, Nations Unies, 1956, vol. 15, p. 37-44.
222. -; MARDOCH, E. S., (( Use of radioactive iodine as a tracer in water flooding operations )I, J. Petrol. Tech., AIME, 1954, vol. 6, p. 117-124.
française pour l’étude des eaux, 1955, no 40, p. 1-2.
126

I-. ORERTHUR. RENNES-PARIS. - D6pBt legal imprimeur no 5404.