hipertension pulmonar - sitio de encuentro e … · hipertension pulmonar el presente artículo es...
TRANSCRIPT

HIPERTENSION PULMONAR
El presente artículo es una actualización al mes de octubre del 2006 del Capítulo del Dr. Carlos Lovesio, del Libro Medicina Intensiva, Dr. Carlos Lovesio, Editorial El Ateneo, Buenos Aires (2001)
CONCEPTO
La hipertensión pulmonar es una patología frecuente que puede complicar una amplia variedad de enfermedades cardíacas y respiratorias. También puede aparecer como una condición primaria o idiopática. La hipertensión pulmonar no es una enfermedad por si misma, sino una manifestación hemodinámica de una variedad de procesos patológicos.
La presión arterial sistólica pulmonar normal en sujetos sanos varía entre 18 y 30 mm Hg y la presión diastólica entre 4 y 12 mm Hg. El criterio de definición de hipertensión pulmonar de la World Health Organization es la presencia de una presión arterial sistólica pulmonar >40 mm Hg., que generalmente corresponde a una velocidad de regurgitación tricuspídea en el Doppler de >3,0 m/s. El criterio hemodinámico utilizado por el National Institutes of Health Registry on Primary Pulmonary Hypertension considera que existe hipertensión pulmonar cuando la presión media en la arteria pulmonar excede los valores de 25 mm Hg en el reposo y 30 mm Hg durante el ejercicio, con una presión capilar pulmonar o presión auricular izquierda por debajo de 15 mm Hg, y un volumen minuto cardiaco normal o reducido.
Algunos autores consideran que para definir la hipertensión pulmonar se debe hacer referencia a la resistencia vascular pulmonar (ver más adelante). Los valores obtenidos se expresan en Unidades Wood, considerándose valores por encima de 3 Wood como representativos de hipertensión pulmonar. Si el valor obtenido se multiplica por 80, se obtiene la determinación en dinas/seg.cm-5, correspondiendo a la hipertensión pulmonar valores por encima de 250.
Otra forma de evaluar el estado de la circulación pulmonar es utilizando el denominado gradiente transpulmonar, que es el resultado de restar la presión capilar pulmonar de la presión pulmonar media. Su valor normal es inferior a 6 mm Hg, considerándose de riesgo un valor superior a los 15 mm Hg.
La hipertensión pulmonar habitualmente es asintomática hasta que la condición alcanza sus períodos avanzados, admitiéndose que se requieren valores de presión arterial media en la arteria pulmonar mayores de 40 mm Hg para que se hagan evidentes los síntomas. Generalmente, estos se presentan como manifestaciones de la insuficiencia cardíaca derecha. La condición final se conoce como cor pulmonale, situación en la cual la hipertensión pulmonar severa ha alterado la función y estructura del ventrículo derecho, de modo que la dilatación y la falla ventricular derecha resultan en congestión hepática severa y edema periférico.

CLASIFICACIÓN ETIOPATOGÉNICA
La hipertensión pulmonar es habitualmente clasificada como primaria o secundaria. La hipertensión pulmonar secundaria responde a un proceso patológico conocido, tal como las enfermedades vasculares del colágeno o una cardiopatía. En la hipertensión pulmonar primaria, no se reconoce ninguna causa obvia. Esta clasificación tradicional en primaria y secundaria tiene una serie de limitaciones y no es útil en la práctica clínica. La Organización Mundial de la Salud (WHO) ha propuesto una nueva clasificación orientada al tratamiento (Tabla 1).
Tabla 1.- Clasificación de la hipertensión pulmonar de la Organización Mundial de la Salud (Modificación de Venecia, 2003)
1. Hipertensión arterial pulmonar
a. Hipertensión pulmonar idiopática
b. Hipertensión pulmonar familiar
c. Hipertensión pulmonar relacionada con las enfermedades vasculares del colágeno, shunts congénitos sistémico-pulmonares (Síndrome de Eisenmenger), hipertensión portal, infección por virus VIH, drogas o tóxicos (anorexígenos -fenfluramina y dexfenfluramina-), otros (enfermedades tiroideas, enfermedad por almacenamiento de glicógeno, enfermedad de Gaucher, telangiectasia hemorrágica hereditaria, hemoglobinopatías, desordenes mieloproliferativos, esplenomegalia, asplenia secundaria a esplenectomía quirúrgica)
d. Hipertensión pulmonar persistente del recién nacido
e. Asociada con compromiso significativo venoso o capilar: enfermedad pulmonar veno-oclusiva (PVOD), hemangiomatosis capilar pulmonar
2. Hipertensión pulmonar asociada con enfermedad cardiaca izquierda
Enfermedad cardiaca auricular o ventricular izquierda
Enfermedad valvular izquierda
3. Hipertensión pulmonar asociada con hipoxemia y/o enfermedad pulmonar
Enfermedad pulmonar obstructiva crónica
Enfermedad intersticial pulmonar
Desordenes respiratorios asociados con el sueño
Síndrome de hipoventilación alveolar

Exposición a elevadas alturas
Enfermedad pulmonar neonatal
Displasia capilar alveolar
4. Hipertensión pulmonar debida a enfermedad trombótica o embólica crónica
Obstrucción tromboembólica de las arterias pulmonares proximales
Obstrucción tromboembólica de las arterias pulmonares distales
Embolismo pulmonar por tumores, parásitos, cuerpos extraños
5. Misceláneos
Sarcoidosis, histiocitosis x, linfangiomatosis, compresión de los vasos pulmonares por adenopatías, tumores, mediastinitis fibrosante
CLASIFICACIÓN HEMODINÁMICA
Considerando que la hipertensión pulmonar es un diagnóstico hemodinámico, es conveniente desde el punto de vista pronóstico y terapéutico contar con una clasificación hemodinámica, tal como la que se expresa en la Tabla 2.
La presión arterial pulmonar está relacionada con el volumen minuto cardiaco (CO) y con la resistencia vascular pulmonar (RVP). Las siguientes ecuaciones ilustran las relaciones hemodinámicas entre la presión pulmonar arterial media (PAPM en mm Hg), el CO (en litros por minuto), la RVP (en dinas por segundo por cm-5), y las presiones capilar pulmonar (PCP) y capilar pulmonar media (PCPM).
1.- RVP = (PAPM - PCPM)/CO
2.- PAPM - PCP = RVP x CO
3.- PAPM = (RVP x CO) + PCPM
La RVP normal oscila entre 1 y 1,5 U Word o 80 a 120 dinas/seg/cm-5. Las ecuaciones precedentes son extremadamente útiles para definir las principales etiologías de la hipertensión pulmonar.

En la hipertensión venosa pulmonar, la presión arterial pulmonar puede estar aumentada mientras el CO y la RVP son normales (hipertensión pulmonar postcapilar). La hipertensión pulmonar también puede ser el resultado del aumento selectivo o no selectivo del CO o puede ser debida a un aumento de la resistencia en la circulación pulmonar. Los lugares de aumento de la resistencia en el lecho vascular pulmonar pueden ser los segmentos arteriolares y o los segmentos arteriales más proximales (hipertensión pulmonar precapilar). El aumento de la resistencia se puede observar en más de un segmento vascular pulmonar.
Tabla 2.- Perfil hemodinámico de diferentes tipos de hipertensión pulmonar.
1. Hipertensión pulmonar precapilar
Las presiones sistólica, diastólica y media de la arteria pulmonar son mayores que las normales; la presión pulmonar capilar media es normal. La resistencia vascular pulmonar está significativamente elevada; las presiones pulmonares arteriales sistólica y diastólica son significativamente mayores que la presión capilar pulmonar
2. Hipertensión pulmonar postcapilar
Las presiones sistólica, diastólica y media de la arteria pulmonar son mayores que las normales, la presión capilar pulmonar está elevada; la resistencia vascular pulmonar es normal; la presión pulmonar arterial de fin de diástole es igual o dentro de los 5 mm Hg de la presión capilar pulmonar media
3. Mixta
Las presiones sistólica, diastólica y media de la arteria pulmonar son mayores que las normales; la presión capilar pulmonar está elevada; la presión pulmonar arterial de fin de diástole es escasamente mayor que la presión capilar pulmonar media
4. Aumento selectivo o no selectivo del flujo sanguíneo pulmonar
Las presiones sistólica, diastólica y media de la arteria pulmonar pueden ser mayores que las normales; el flujo sanguíneo pulmonar está aumentado; la resistencia vascular pulmonar es normal o está ligeramente aumentada; la presión venosa pulmonar está escasamente aumentada o es normal
En la Tabla 3, por su parte, se indican las distintas condiciones clínicas que se encuentran habitualmente asociadas con las categorías hemodinámicas descriptas en la Tabla 2.

Tabla 3.- Condiciones clínicas asociadas con las categorías hemodinámicas de hipertensión pulmonar.
1. Hipertensión pulmonar precapilar
Hipertensión pulmonar primaria, hipertensión pulmonar asociada con enfermedades vasculares del colágeno, síndrome de Eisenmenger, enfermedad hepática (hipertensión portal), infecciones por el virus VIH, drogas anorexígenas, hipertensión pulmonar persistente del neonato, hipertensión pulmonar de las alturas, hipertensión pulmonar neurogénica, hipertensión pulmonar tromboembólica, estenosis de las ramas arteriales periféricas
2. Hipertensión pulmonar postcapilar
Fallo ventricular izquierdo diastólico o sistólico, estenosis o insuficiencia aórtica, insuficiencia mitral, obstrucción valvular mitral, enfermedad pulmonar veno-oclusiva
3. Mixta
Fallo ventricular izquierdo sistólico primario crónico, estenosis aórtica, insuficiencia valvular aórtica y mitral crónica, obstrucción mitral
4. Aumento selectivo o no selectivo del flujo sanguíneo pulmonar
Defectos septales auriculares, defectos septales ventriculares, ducto arterioso persistente, insuficiencia cardiaca de alto débito (hipertiroidismo), enfermedad hepática, anemia crónica
CLASIFICACIÓN FUNCIONAL
Los pacientes con hipertensión pulmonar pueden ser evaluados en forma subjetiva en función de su capacidad funcional. Esta evaluación es similar a la aceptada para la insuficiencia cardiaca congestiva por la New York Heart Association (NYHA), y en la actualidad se conoce como la clasificación de la World Health Organization (WHO)/NYHA (Tabla 4). La diferencia principal entre la clasificación funcional de la WHO y la clasificación de la New York Heart Association es la inclusión de los pacientes con síncope como clase funcional IV en la anterior.

Tabla 4.- Clasificación funcional de la hipertensión pulmonar (NYHA/WHO)
Clase I
Pacientes con hipertensión pulmonar pero sin limitaciones para la actividad física. La actividad física ordinaria no produce disnea ni fatiga, dolor torácico o síncope.
Clase II
Pacientes con hipertensión pulmonar que presentan una limitación ligera de la actividad física. Estos pacientes están confortables en reposo, pero la actividad física ordinaria produce disnea, fatiga o dolor torácico.
Clase III
Pacientes con hipertensión pulmonar que presentan una marcada limitación de la actividad física. Estos pacientes están confortables en reposo, pero la mínima actividad física produce disnea, fatiga, dolor torácico o casi-síncope.
Clase IV
Pacientes con hipertensión pulmonar que presentan una imposibilidad de realizar cualquier actividad física sin síntomas. Estos pacientes presentan signos de insuficiencia cardiaca derecha. La disnea y la fatiga se pueden presentar en el reposo, y cualquier actividad física las exacerba.
ANATOMÍA PATOLÓGICA
La única forma que presenta características anatomopatológicas propias es la hipertensión pulmonar primaria. En el pasado, la arteriopatía pulmonar plexogénica fue considerada el signo definitorio de la hipertensión pulmonar primaria, y las lesiones plexogénicas fueron consideradas de significación diagnóstica y patogénica. Varios estudios recientes han demostrado que la arteriopatía pulmonar primaria incluye un espectro de lesiones histopatológicas variando desde la clásica arteriopatía plexogénica a formas microtrombóticas no plexogénicas. Los tres tipos mayores de arteriopatías pulmonares involucran las arterias musculares como sigue: 1) hipertrofia aislada de la media; 2) hipertrofia de la media y fibroelastosis laminar concéntrica, que puede o no asociarse con lesiones plexiformes, lesiones angiomatoides, necrosis fibrinoide y arteritis necrotizante; y 3) hipertrofia medial y fibrosis no laminar excéntrica y concéntrica. La arteriopatía plexogénica pulmonar es una patente inespecífica de respuesta de la vasculatura pulmonar a la injuria hemodinámica.
La enfermedad pulmonar veno-oclusiva (PVOD), por su parte, es un síndrome clínico patológico responsable de un pequeño número de casos de hipertensión pulmonar. El rasgo característico de la PVOD es la oclusión extensa y difusa de las venas pulmonares por tejido fibroso, que puede ser blando y edematoso o denso y esclerótico. Las arteriolas pulmonares

exhiben una hipertrofia medial moderada a severa en la mitad de los casos, pero habitualmente no se evidencian lesiones de arteritis ni plexiformes.
FISIOPATOLOGÍA
Las distintas formas etiológicas citadas anteriormente presentan mecanismos fisiopatológicos variables según la causa.
La hipertensión pulmonar primaria es una enfermedad rara de etiología no clara. Si bien la mayoría de los casos son esporádicos, la enfermedad es familiar en el 6 al 10% de los casos. En estos casos, la misma se hereda como un trastorno autonómico dominante con penetrancia incompleta. En el 50% de los pacientes con hipertensión pulmonar familiar se han identificado mutaciones en el gen bone morphogenetic protein receptor II (BMPR2). Otras familias demuestran alteraciones en la misma región cromosómica donde reside este gen, el locus 2q32. Existe una tendencia inexplicable para desarrollar hipertensión pulmonar familiar en edades más precoces en las generaciones subsecuentes, fenómeno denominado anticipación genética. Una serie de factores de riesgo se han identificado en algunos pacientes con hipertensión pulmonar primaria. Es probable que muchos de estos pacientes sean susceptibles a estos factores de riesgo debido a su patente genética.
Varios mecanismos posibles han sido sugeridos para explicar la patogénesis de la hipertensión pulmonar primaria: disfunción de las células endoteliales del lecho arterial pulmonar; fenotipo anormal de las células musculares; respuesta de remodelación vascular anormal a los cambios hemodinámicos; respuesta anormal a la hipoxia de las células musculares del lecho pulmonar; anormalidades de los canales de potasio de la membrana que modulan la cinética del calcio; aumentados niveles de mediadores vasoconstrictores, tales como serotonina y endotelina 1; y reducida producción de vasodilatadores, tales como óxido nítrico y prostaciclina. Otros mediadores involucrados son la adrenomedulina, el péptido intestinal vasoactivo y el factor de crecimiento endotelial vascular.
Basado en experimentos en animales, se ha propuesto que el evento iniciador que en última instancia conduce a la hipertensión pulmonar severa plexogénica es la apoptosis de las células endoteliales en los sitios de bifurcación que también están expuestos a altas fuerzas de cizallamiento. Se ha propuesto que la susceptibilidad genética es probable que resida en el control del reemplazo apropiado de las células endoteliales muertas por apoptosis. La característica fundamental del endotelio vascular es la capacidad de mantener una disposición en monolámina; lo que probablemente ocurra en la hipertensión pulmonar severa es que se produzca la evolución a un fenotipo celular proliferativo, altamente resistente a la apoptosis.
Múltiples factores contribuyen al desarrollo de hipertensión pulmonar en el contexto de las enfermedades respiratorias. La hipoxia alveolar es un hallazgo común en muchas formas de enfermedades respiratorias, y produce una constricción selectiva de las arteriolas pulmonares. La vasoconstricción pulmonar hipóxica parece ser una propiedad intrínseca de las células musculares lisas pulmonares, siendo dependiente tanto de la disponibilidad de calcio extracelular como del estado de la membrana celular. La hipercarbia y la acidosis, que frecuentemente acompañan a las

enfermedades obstructivas crónicas del pulmón, potencian la respuesta vasopresora vascular a la hipoxia. Aunque la vasoconstricción hipóxica aguda puede ser reversible cuando se restaura la tensión normal de oxígeno, la hipoxia crónica produce un remodelamiento vascular, el cual solo responde parcial y lentamente a la corrección de los trastornos gasométricos.
La compresión mecánica de la vasculatura pulmonar también desempeña un rol en el desarrollo de hipertensión pulmonar en los pacientes con enfisema severo, y puede contribuir a deteriorar la dinámica vascular durante las descompensaciones agudas.
La policitemia que ocurre en los pacientes crónicamente hipoxémicos determina hiperviscosidad, lo que impide secundariamente el flujo sanguíneo a través de la circulación pulmonar. Una pérdida de la superficie seccional de la vasculatura como resultado de la destrucción de grandes volúmenes de parénquima pulmonar también contribuye a la elevación de la presión arterial pulmonar en condiciones tales como el enfisema bulloso avanzado y las enfermedades fibrosantes del pulmón.
El tromboembolismo puede resultan en una descompensación aguda pulmonar con aumento severo y transitorio de la presión arterial pulmonar, directamente como resultado de la obstrucción vascular por el trombo, o por la liberación de sustancias vasoactivas, o por agravamiento del deterioro gasométrico y potenciación de la vasoconstricción pulmonar hipóxica.
En los últimos años se ha descrito una forma de hipertensión pulmonar crónica asociada con el tromboembolismo, en la cual se genera un estrechamiento progresivo de la luz de las arterias pulmonares mayores con el consiguiente aumento de la presión. No todos los pacientes con esta patología tienen antecedentes claros de embolismo pulmonar. En un estudio prospectivo de 223 pacientes que se presentaron con embolismo pulmonar agudo, la incidencia de hipertensión pulmonar crónica tromboembólica sintomática fue del 3,1% al año y 3,8% a los dos años. Llamativamente, la enfermedad se ha constatado con mayor frecuencia en pacientes esplenectomizados.
En la esclerosis sistémica limitada, especialmente en la variante CREST, se produce una arteriopatía pulmonar. En la autopsia, hasta el 80% de los pacientes con el síndrome CREST tienen cambios histopatológicos consistentes con hipertensión arterial pulmonar; sin embargo, en vida, sólo el 10 al 15% de ellos tienen una hipertensión pulmonar clínicamente significativa. Se encuentran ocasionalmente hallazgos histológicos y evidencias clínicas de hipertensión pulmonar, en pacientes con lupus eritematoso sistémico, enfermedad mixta del tejido conectivo y artritis reumatoidea.
La oclusión trombótica in situ de la circulación pulmonar puede ocurrir en una serie de condiciones incluyendo la policitemia, anemia de células falciformes, eclampsia y displasia fibromuscular diseminada. En las enfermedades trombóticas, los vasos pequeños están afectados en forma difusa a través de todo el pulmón.
En los pacientes con SDRA se han descripto elevaciones de la presión arterial pulmonar. En adición a los factores previamente citados, la liberación de mediadores vasoactivos como resultado del proceso inflamatorio subyacente puede contribuir al proceso vascular pulmonar.

Se ha observado un aumento de la incidencia de hipertensión pulmonar en los individuos con infección por el virus de la inmunodeficiencia adquirida. La patogenia de la hipertensión pulmonar en estos casos no es clara, pero se ha sugerido como mecanismo posible el aumento de la liberación de endotelina 1 en respuesta a la infección viral. La ocurrencia de hipertensión pumonar es independiente del recuento de células CD4, pero parece estar relacionado con la duración de la infección por HIV.
Varios tipos de enfermedades hepáticas, incluyendo la cirrosis, hipertensión portal, trombosis de la vena porta y ausencia congénita de la vena porta, pueden asociarse con hipertensión pulmonar. Un mecanismo posible es el aumento de los niveles de péptidos vasoactivos intestinales que alcanzan el pulmón, produciendo una alteración de la hemodinamia pulmonar y una respuesta pulmonar vasodilatadora anormal.
Varias toxinas y drogas se han asociado con hipertensión pulmonar precapilar. Las drogas anorexígenas tales como la fenfluramina constituyen un riesgo bien documentado para el desarrollo de hipertensión pulmonar. Este riesgo aumenta con el empleo prolongado, por más de tres meses. El mecanismo de la hipertensión pulmonar secundario al consumo de drogas anorexígenas permanece desconocido, aunque se ha propuesto como mecanismo potencial el aumento de la liberación de serotonina y de sus efectos inhibitorios sobre los canales de potasio, conduciendo a un aumento del influjo de calcio citosólico. El uso de estimulantes del sistema nervioso central tales como la metanfetamina y cocaína se ha asociado con un riesgo aumentado de hipertensión arterial pulmonar.
En la insuficiencia ventricular izquierda y en las patologías valvulares del lado izquierdo del corazón, se produce un aumento de la presión pulmonar primariamente por elevación de la presión postcapilar, lo cual hace necesario un aumento de la presión en el circuito pulmonar para poder seguir manteniendo el flujo a través del pulmón. En esta situación, la diferencia entre la presión arterial diastólica pulmonar y la presión de enclavamiento pulmonar es relativamente pequeña (3 a 5 mm Hg), a pesar de la presencia de hipertensión pulmonar. Los cambios histopatológicos en el árbol arterial son relativamente modestos y potencialmente reversibles. En contraste, las condiciones en que las arterias y arteriolas pulmonares son el sitio primario de enfermedad se asocian con un aumento del gradiente de presión entre la presión diastólica y la presión de enclavamiento pulmonar, y las anormalidades vasculares varían entre proliferación intimal moderada y oclusión vascular.
En pacientes con defectos septales auriculares o ventriculares o ducto arterioso persistente se puede producir hipertensión pulmonar como consecuencia del aumento selectivo y no selectivo del flujo sanguíneo pulmonar. En general, la hipertensión pulmonar es menos frecuente y habitualmente menos severa en los shunts localizados antes de la válvula tricúspide (defectos auriculares septales).
En pacientes con estados de alto volumen minuto cardiaco, tales como la tirotoxicosis, anemia crónica y enfermedad hepática, se puede observar una moderada hipertensión pulmonar. En algunos pacientes con aumento del flujo sanguíneo pulmonar también se puede producir un aumento de la resistencia vascular pulmonar. Se ha sugerido que la vasodilatación pulmonar dependiente de flujo es anormal en estos pacientes, y se produce un aumento moderado en la resistencia vascular pulmonar. Con el desarrollo del complejo de Eisenmenger, se produce un

aumento marcado de la presión arterial pulmonar y de la resistencia vascular pulmonar y una reversión del shunt, pasando a ser de derecha a izquierda.
Es probable que la PVOD represente una patente común de injuria adquirida resultante de una multiplicidad de posibles insultos. Entre las causas involucradas se han citado infecciones, factores genéticos, exposición a drogas y tóxicos, diátesis trombótica y alteraciones autoinmunes.
Si bien se han identificado distintos mecanismos biopatológicos en las células y tejidos de pacientes con hipertensión pulmonar, las exactas interacciones entre estos mecanismos en la iniciación y progresión del proceso patológico no son bien conocidas. Las vías posibles (Fig. 1) incluyen la clásica interacción entre predisposición genética y factores de riesgo que pueden inducir cambios en diferentes tipos celulares (células musculares lisas, células endoteliales, células inflamatorias, plaquetas) y en la matriz extracelular de la microcirculación pulmonar. El imbalance entre factores trombogénicos, mitogénicos, proinflamatorios y vasoconstrictores en oposición a los mecanismos anticoagulantes, antimitóticos y vasodilatadores puede iniciar y perpetuar una serie de procesos tales como la vasoconstricción, proliferación, trombosis e inflamación en la circulación pulmonar. Estos mecanismos son responsables de la iniciación y progresión de los cambios obstructivos típicos de la hipertensión pulmonar. El aumento consecuente de la resistencia vascular pulmonar produce la sobrecarga ventricular derecha y eventualmente la falla ventricular y la muerte.

Predisposición genética Factores de riesgo
Fig. 1.- Potenciales mecanismos patogénicos y biopatológicos de la hipertensión arterial pulmonar.
CUADRO CLÍNICO
Los pacientes con hipertensión pulmonar habitualmente se presentan con síntomas inespecíficos. El síntoma más común es la disnea de esfuerzo, que es el síntoma inicial en el 60% de los pacientes y eventualmente se presenta en todos ellos. Con el aumento de la severidad de la hipertensión pulmonar, la tolerancia al ejercicio progresivamente declina, limitada por la disnea y la fatiga. La ortopnea puede ser experimentada por pacientes tanto con hipertensión pulmonar precapilar como postcapilar. La disnea paroxística nocturna, sin embargo, es exclusiva de la hipertensión venosa pulmonar.
Mutaciones BMPR-2Mutaciones ALK1Polimorfismo SHTTPolimorfismo ec-NOSPolimorfismo CPS
AnorexígenosInfección HIVAumento del flujo pulmonarHipertensión portalEnfermedades tejido conectivo
Daño vascular pulmonar
Cambios en la matriz, plaquetas y activaciónde células inflamatorias
Disfunción endotelialCambios en las vías del NO,
de la endotelina y de la prostaciclina
Disfunción de celulas musculares lisas
Vasoconstricción Proliferación
Trombosis Inflamación
Inicio y progresión de la enfermedad vascular pulmonar hipertensiva
FALLO VENTRICULAR DERECHO

El dolor torácico anginoso y el síncope aparecen en aproximadamente el 40% de los pacientes durante el curso de la enfermedad. Este último es particularmente importante debido a que implica una reducción marcada del volumen minuto cardíaco, siendo un signo de mal pronóstico. Del mismo modo, la distensión abdominal y los edemas se asocian con insuficiencia cardíaca derecha y son indicio de mal pronóstico.
El examen físico puede ser sugerente de la enfermedad causal. El hallazgo más consistente en la hipertensión arterial pulmonar, independiente de su causa, es el aumento de la intensidad del componente pulmonar del segundo ruido cardiaco (P2) en comparación con el componente aórtico del mismo ruido (A2). El examen del pulso yugular venoso puede demostrar la elevación de la presión venosa, mientras que la presencia de onda v se asocia con insuficiencia tricuspídea y falla cardíaca derecha. El examen pulmonar puede demostrar la presencia de EPOC o alteraciones de la caja torácica como enfermedades causales de la hipertensión pulmonar.
El examen cardíaco puede poner en evidencia los elementos propios del cor pulmonale: un segundo ruido aumentado de intensidad y desdoblado, un galope derecho o un clic de eyección pulmonar. En el electrocardiograma se puede hacer evidente la sobrecarga o hipertrofia de las cavidades derechas. La presencia de cianosis periférica se asocia con una reducción marcada del volumen minuto cardíaco, la existencia de un shunt de derecha a izquierda, o la presencia de patología pulmonar.
La presencia de fenómeno de Raynaud, artralgias o edema en las manos u otros síntomas de enfermedades del tejido conectivo alertan sobre la posibilidad de una hipertensión pulmonar relacionada con una enfermedad sistémica. Una historia de ronquidos o apnea provistos por un compañero del paciente exige la evaluación para alteraciones de la respiración asociadas con el sueño como causas potenciales o factor contributorio.
METODOLOGÍA DIAGNÓSTICA
El proceso diagnóstico de la hipertensión pulmonar requiere una serie de investigaciones destinadas a establecer el diagnóstico, reconocer la clase clínica de hipertensión pulmonar y el tipo, y evaluar el grado de deterioro funcional y hemodinámico. Con propósitos prácticos, es útil adoptar una evaluación secuencial que incluya cuatro estadios:
1. Sospecha clínica de hipertensión pulmonar
2. Detección de la hipertensión pulmonar
3. Identificación de la clase clínica de hipertensión pulmonar
4. Evaluación del grado de hipertensión arterial pulmonar (tipo, capacidad funcional, hemodinamia)
Historia clínica. La historia clínica del paciente se debe focalizar en síntomas obvios que se puedan asociar con una causa secundaria definida. Debido al componente genético reconocido

de la hipertensión pulmonar primaria, el interrogatorio sobre la presencia de síntomas o un diagnóstico establecido en otros miembros de la familia permite un reconocimiento precoz de la enfermedad. Se deben evaluar los antecedentes relativos a la presencia de enfermedades del tejido conjuntivo, exposición a tóxicos tales como supresores del apetito o agentes quimioterápicos (mitomicina-C, carmustina, etopósido, ciclofosfamida o bleomicina). Se debe explorar la exposición a VIH. Se debe requerir información sobre antecedentes relacionados con trombosis venosa o embolismo pulmonar, aunque la hipertensión pulmonar crónica tromboembólica puede ocurrir en ausencia de una historia reconocida de tromboembolismo.
Examen físico. En los pacientes con hipertensión pulmonar significativa, el examen físico puede mostrar los signos de hipertrofia ventricular derecha, así como de insuficiencia cardíaca derecha: elevación de la presión venosa yugular, hepatomegalia y edemas periféricos. En más del 90% de los pacientes con hipertensión pulmonar idiopática se reconoce un componente pulmonar del segundo ruido cardiaco acentuado a nivel del ápex, reflejando un aumento de la fuerza de cierre de la válvula pulmonar debido a la elevación de la presión pulmonar. Otros signos de aumento de la presión arterial pulmonar incluyen: 1) click de eyección sistólica precoz debido a la interrupción brusca de la apertura de la válvula pulmonar, 2) soplo de eyección medio sistólico causado por el flujo pulmonar transvalvular turbulento, 3) frémito palpable paraesternal izquierdo, 4) galope ventricular derecho S4, y 5) onda “a” prominente en el pulso yugular.
Signos físicos de enfermedad más avanzada incluyen un soplo diastólico de regurgitación pulmonar y un soplo holosistólico de regurgitación tricuspídea, galope ventricular derecho S3, marcada distensión de las venas yugulares y hepatomegalia pulsátil, edemas periféricos y ascitis como signos de fallo ventricular derecho. La presencia de baja presión arterial y extremidades frías constituyen signos ominosos ya que indican la presencia de una marcada reducción del volumen minuto cardiaco.
El examen físico puede brindar información respecto a la etiología. La cianosis sugiere un shunt derecha-izquierda, volumen minuto cardiaco severamente reducido o un deterioro marcado de la transferencia de gases a nivel pulmonar. La obesidad, cifoescoliosis o trastornos obstructivos respiratorios pueden identificar un síndrome de hipoventilación. Los signos cutáneos clásicos de la esclerodermia, así como la insuficiencia venosa periférica, exigen investigaciones especiales para su reconocimiento o descarte. Un raro hallazgo clínico que es virtualmente patognomónico de la hipertensión pulmonar crónica asociada con el tromboembolismo es un frote sobre los campos pulmonares periféricos, en particular sobre los lóbulos inferiores, que resulta del flujo sanguíneo turbulento en áreas parcialmente ocluidas.
Electrocardiograma. La hipertensión pulmonar se asocia con hipertrofia y dilatación de las cavidades derechas del corazón. Puesto que estos procesos producen alteraciones en el ECG, este examen simple puede sugerir la presencia de una hipertensión pulmonar hemodinámicamente significativa. La hipertrofia ventricular derecha y la desviación del eje a la derecha en el ECG se observan en el 87% y 79%, respectivamente, de los pacientes con hipertensión pulmonar idiopática. Los hallazgos ECG sugestivos de hipertensión pulmonar incluyen: 1) desviación a la derecha del eje eléctrico; 2) un onda R alta y una onda S pequeña con una relación R/S >1 en V1; 3) complejo qR en V1; 4) patente rSR´en V1; 5) onda S grande y R pequeña con relación R/S <1 en V5 o V6; o 6) patente S1, S2, S3. El aumento de la onda P es indicativo de agrandamiento

auricular derecho. A pesar de estos hallazgos, el ECG carece de suficiente sensibilidad (55%) y especificidad (70%) como método de elección para la detección de una hipertensión pulmonar significativa.
Ciertas características del ECG en pacientes con diagnóstico establecido de hipertensión pulmonar pueden tener valor pronóstico. Una onda P de amplitud ≥0,25 mV en DII se asocia con un aumento del riesgo de muerte de 2,8 veces a los seis años, y cada aumento adicional en un mm en DIII se corresponde con un aumento de 4,5 en dicho riesgo.
Radiografía de tórax. La radiografía de tórax es una parte importante de la evaluación, ayudando a identificar muchas causas de hipertensión pulmonar (Tabla 5). La presencia de anormalidades parenquimatosas, redistribución del flujo sanguíneo, agrandamiento cardíaco, y alteraciones estructurales de la pared torácica pueden contribuir al diagnóstico causal. En los pacientes ancianos, la radiografía de tórax es de limitado valor, excepto en aquellos con severa hipertensión pulmonar.
Los signos radiográficos generales que pueden ser tomados como sugestivos de hipertensión pulmonar son el aumento de la sombra de la arteria pulmonar principal y sus troncos, con una concomitante atenuación de la vasculatura pulmonar periférica (“pruning”). Estos hallazgos se presentan en la mayoría de los pacientes con hipertensión pulmonar idiopática, pero su ausencia no debe ser interpretada como excluyente del diagnóstico. Las dos dimensiones más útiles indicativas de hipertensión pulmonar son el tamaño de la rama descendente de la arteria pulmonar derecha y el índice hilio/torácico. En un adulto, una arteria descendente pulmonar derecha de más de 16 mm de ancho y un índice hilio/torácico mayor de 38% es indicativo de hipertensión pulmonar, aunque no permita cuantificar la magnitud de la misma.
Tabla 5.- Probabilidades diagnósticas de hipertensión pulmonar en función de los hallazgos radiológicos.
Condiciones en las cuales el examen radiográfico contribuye al diagnóstico:Enfermedad pulmonar obstructiva crónicaFibrosis pulmonarEnfermedad granulomatosa del pulmónDeformidades de la pared torácicaEnfermedad congénita del corazónEnfermedad valvular mitralEnfermedad cardíaca izquierda
Condiciones en las cuales el examen radiográfico no contribuye al diagnóstico:Hipertensión pulmonar primaria
Enfermedad embólica pulmonar recurrente
Gases en sangre. El examen de gases en sangre es de utilidad para la evaluación de los pacientes con sospecha de hipertensión pulmonar. La presencia de hipoxemia asociada a hipercapnia, si se excluye una alcalosis metabólica primaria, implica una severa alteración parenquimatosa pulmonar o un desorden neuromuscular. Debido a que la vasoconstricción hipóxica pulmonar solo ocurre cuando la PaO2 es menor de 60 mm Hg., la presencia de una

tensión parcial de oxígeno normal descarta a la hipoxemia como causa de hipertensión pulmonar. En este último caso, sin embargo, se debe evaluar la posible desaturación durante el sueño (síndrome de apnea de sueño) que puede cursar con hipertensión pulmonar y oxigenación normal durante la vigilia.
Estudios serológicos. Los exámenes específicos de sangre incluyen la determinación de anticuerpos contra el virus de la inmunodeficiencia adquirida, anticuerpos antinucleares, factor reumatoideo, tests de función hepática y tiroidea. La elevación de los anticuerpos antinucleares es común en pacientes con hipertensión pulmonar primaria y no necesariamente implica una enfermedad del colágeno asociada. No existe una patente específica de anticuerpos o títulos consistentemente asociados con hipertensión pulmonar primaria. En algunos casos, se puede reconocer una enfermedad del colágeno con manifestación exclusivamente pulmonar.
Ecocardiograma. El ecocardiograma es muy útil en presencia de hipertensión pulmonar moderada a severa, aunque puede no contribuir al diagnóstico en las formas leves de la enfermedad. Debido a la sobrecarga crónica de presión del ventrículo derecho (VD), la mayoría de los pacientes presentan agrandamiento de las cavidades derechas, hipertrofia del VD, y reducción global de la función sistólica ventricular izquierda. Esto se acompaña por un desplazamiento sistólico del septum interventricular (IVS) con aumento del grosor y una relación anormal IVS/pared ventricular izquierda posterior (>1) en respuesta a la sobrecarga de presión. El ventrículo izquierdo adquiere forma de D con reducción de los volúmenes diastólico y sistólico, pero con preservación de la función sistólica global (Fig. 2). Se han descrito derrame pericárdico y prolapso de la válvula mitral en asociación con la hipertensión pulmonar. En el caso particular de la hipertensión pulmonar aguda generada por el embolismo pulmonar, es característica la distensión no colapsable de la vena cava inferior (Fig. 3).
La presión arterial pulmonar sistólica (PAPs) se considera igual a la presión sistólica ventricular derecha (RVSP) en ausencia de estenosis de la válvula pulmonar u obstrucción del tracto de salida. Se puede obtener una estimación de la RVSP utilizando el ecocardiograma Doppler mediante el cálculo del gradiente de presión entre ventrículo derecho y aurícula derecha durante la sístole, aproximado por la ecuación modificada de Bernouilli como 4v2, en la cual v es la velocidad del jet tricuspídeo en metros por segundo. La RVSP se deriva adicionando la presión auricular derecha al gradiente (RVSP = 4v2 + RAP) (Fig. 4). El jet tricuspídeo es analizable en el 74% de los pacientes con hipertensión pulmonar. Al menos 10 estudios han informado una buena correlación entre la RVSP estimada por ecodoppler y por evaluación hemodinámica a través de la cateterización de las cavidades derechas. La presión diastólica pulmonar también puede ser estimada por ecodoppler, midiendo la velocidad de regurgitación a través de las válvulas pulmonares, y se correlaciona bien con las medidas invasivas (r = 0,92).
El ecocardiograma provee evidencia directa relativa a la función ventricular izquierda sistólica y diastólica y a la función y morfología valvular, lo cual permite reconocer las causas de hipertensión pulmonar producidas por un aumento de la presión venosa pulmonar. El ecocardiograma también permite el reconocimiento de posibles alteraciones cardiacas congénitas. Si la sospecha es elevada, el ecocardiograma transesofágico puede permitir una muy buen definición anatómica.

Fig. 2.- Ecocardiograma de paciente con hipertensión pulmonar secundaria a EPOC.
Fig. 4.- Ecocardiograma transtorácico. Severa hipertensión pulmonar por tromboembolismo crónico recidivante. Se observa la grosera dilatación de la aurícula derecha y del ventrículo derecho, la presencia de insuficiencia tricuspídea y una presión pulmonar estimada por eco de 59 mm Hg.
Fig. 3.- Distensión no colapsable de la vena cava inferior en paciente con hipertensión pulmonar.

Exámenes de función pulmonar. Los exámenes de función pulmonar permiten identificar las alteraciones primariamente pulmonares. Se recomienda incluir dentro de los mismos la espirometría, que establece la presencia de enfermedad obstructiva de la vía aérea; la determinación de los volúmenes pulmonares, que permite evaluar el componente restrictivo; y la medida de la capacidad de difusión de monóxido de carbono, que provee un índice de la superficie alvéolo capilar, sugiriendo el diagnóstico de enfermedad pulmonar intersticial o patología vascular obliterativa pulmonar. La hipertensión pulmonar aislada en la esclerodermia se asocia con una marcada disminución de la capacidad de difusión pulmonar para el monóxido de carbono (DLCO) en el momento del diagnóstico de la hipertensión.
Test de ejercicio. Los test cardiopulmonares durante el ejercicio son útiles en la evaluación de pacientes con causas no definidas de disnea, puesto que presentan una patente característica en presencia de enfermedad vascular pulmonar. Los test más comúnmente utilizados son la caminata de seis minutos; un ejercicio de ergometría estándar utilizando un protocolo de baja intensidad y graduado; un test de ejercicio cardiopulmonar con evaluación del intercambio gaseoso; un test de ejercicio en conjunción con una evaluación por ecodoppler de la presión arterial pulmonar; y un test de ejercicio en conjunción con la cateterización cardiaca derecha.
La determinación seriada de la clase funcional y de la capacidad al ejercicio permite establecer la severidad de la enfermedad, la respuesta a la terapéutica, y la progresión de la misma.
El test de la caminata de seis minutos (6MWT) es técnicamente simple y no costoso. Es predictivo de la sobrevida en la hipertensión pulmonar idiopática y se correlaciona en forma inversa con la severidad funcional de la clasificación NYHA. El 6MWT habitualmente se combina con el escore Borg para la evaluación del nivel subjetivo de disnea con el ejercicio. La reducción de la saturación arterial de oxígeno >10% durante la 6MWT aumenta el riesgo de mortalidad 2,9 veces en un periodo de seguimiento medio de 26 meses. El 6MWT es el end point primario tradicional para la gran mayoría de ensayos clínicos controlados realizados en pacientes con hipertensión pulmonar.
Centellograma de ventilación perfusión. La hipertensión pulmonar de origen tromboembólico es una condición potencialmente curable que debe ser considerada en todos los pacientes con hipertensión pulmonar inexplicable. El centellograma de ventilación/perfusión pulmonar en estos casos generalmente muestra uno o más defectos de perfusión segmentarios o mayores. Un centellograma V/Q normal prácticamente descarta el origen tromboembólico de la hipertensión pulmonar. Los defectos en parche, no segmentarios, son menos específicos, pero pueden asociarse con enfermedad tromboembólica. Aunque un centellograma negativo es altamente específico para descartar el tromboembolismo, se han descrito múltiples causas de centellogramas falsos positivos, incluyendo sarcoma de la arteria pulmonar, vasculitis de grandes vasos, compresión vascular extrínseca, enfermedad pulmonar veno-oclusiva, o hemangiomatosis capilar pulmonar. En pacientes con enfermedad pulmonar parenquimatosa, los defectos de perfusión se superponen a los defectos de ventilación.
Cateterización cardíaca derecha. En pacientes con sospecha de hipertensión pulmonar, la cateterización cardiaca derecha es requerida para confirmar la presencia de la hipertensión,

establecer el diagnóstico específico y determinar la severidad de la hipertensión. Además, la cateterización derecha se requiere para guiar la terapéutica.
En muchos pacientes, la cateterización derecha está indicada para confirmar y cuantificar el grado de hipertensión pulmonar sugerida por los métodos no invasivos. La hipertensión pulmonar postcapilar puede ser identificada por la presencia de una presión enclavada pulmonar elevada. La determinación de la saturación de oxígeno en las diversas cámaras y vasos permite identificar la presencia y localización de un shunt de izquierda a derecha.
Mediante la determinación de las presiones y del volumen minuto cardíaco, se puede establecer la magnitud de la resistencia vascular pulmonar (RVP).
La determinación de la resistencia vascular pulmonar es importante para establecer la utilidad de las terapéuticas vasodilatadoras (Ver tratamiento).
Por último, en el NIH Registry, que fue realizado antes de la era de las terapéuticas efectivas para la hipertensión pulmonar idiopática, los parámetros hemodinámicos demostraron ser predictivos de la sobrevida en pacientes con terapéutica convencional.
Angiografía pulmonar. Si la causa de la hipertensión pulmonar permanece sin diagnóstico, la angiografía puede contribuir a la detección de tromboembolismo pulmonar (Fig. 5). Al presente, la angiografía pulmonar continúa siendo el procedimiento diagnóstico de elección para la evaluación de pacientes con sospecha de hipertensión pulmonar por tromboembolismo crónico. En pacientes con hipertensión pulmonar por tromboembolismo recurrente crónico, la angiografía muestra múltiples defectos vasculares bajo la forma de oclusiones, estenosis, y redes intravasculares. En ocasiones se pueden encontrar grandes trombos centrales. En la hipertensión pulmonar primaria, la angiografia muestra una imagen característica de amputación de vasos periféricos (Fig. 6).
En pacientes con hipertensión pulmonar severa, la angiografía presenta riesgos. Se puede producir hipotensión arterial y bradicardia, e incluso paro cardíaco. Se recomienda en estos casos utilización técnicas de magnificación y substracción, administrando pequeños bolos de sustancia de contraste de baja osmolaridad para visualizar territorios seleccionados del árbol vascular.
Polisomnografía. Esta metodología permite medir varios parámetros fisiológicos en los pacientes durante el sueño, y permite explicar ciertos casos en los cuales existe una desproporción entre las características clínicas y la hipertensión pulmonar. Los pacientes con EPOC, insuficiencia cardíaca congestiva, y varios procesos neuromusculares, pueden presentar severos trastornos de la respiración durante el sueño. Las apneas centrales u obstructivas durante el sueño, pueden asociarse con episodios de hipoventilación e hipoxemia, que generan vasoconstricción pulmonar hipóxica y severa hipertensión pulmonar.

Fig. 5.- Angiografia pulmonar en Fig. 6.- Angiografia pulmonar en paciente con TEP masivo en arteria paciente con hipertensión pulmonar pulmonar derecha. primaria.
Tomografía helicoidal. La tomografía helicoidal es un método específico y sensible para
la detección de la enfermedad tromboembólica pulmonar aguda y crónica, y en algunos pacientes puede ser utilizada en lugar de la angiografía pulmonar (Fig. 7).
La dosis de contraste intravenoso utilizada es menor que para la angiografía pulmonar; y para los tromboémbolos localizados en las arterias pulmonares, la sensibilidad es probablemente igual a la de la angiografía. La tomografía también puede ser útil para sugerir diagnósticos alternativos, tales como sarcoma, vasculitis, enfermedades malignas y fibrosis mediastinal. La TAC también es útil para evaluar el parénquima pulmonar y determinar la extensión de los componentes obstructivos o restrictivos. Una patente en vidrio esmerilado en los lóbulos inferiores es sugestiva de enfermedad veno-oclusiva.
Fig. 7.- Tomografía helicoidal de tórax en paciente con trombo-embolismo pulmonar. Se observa la oclusión embólica de la rama izquierda de la arteria pulmonar.
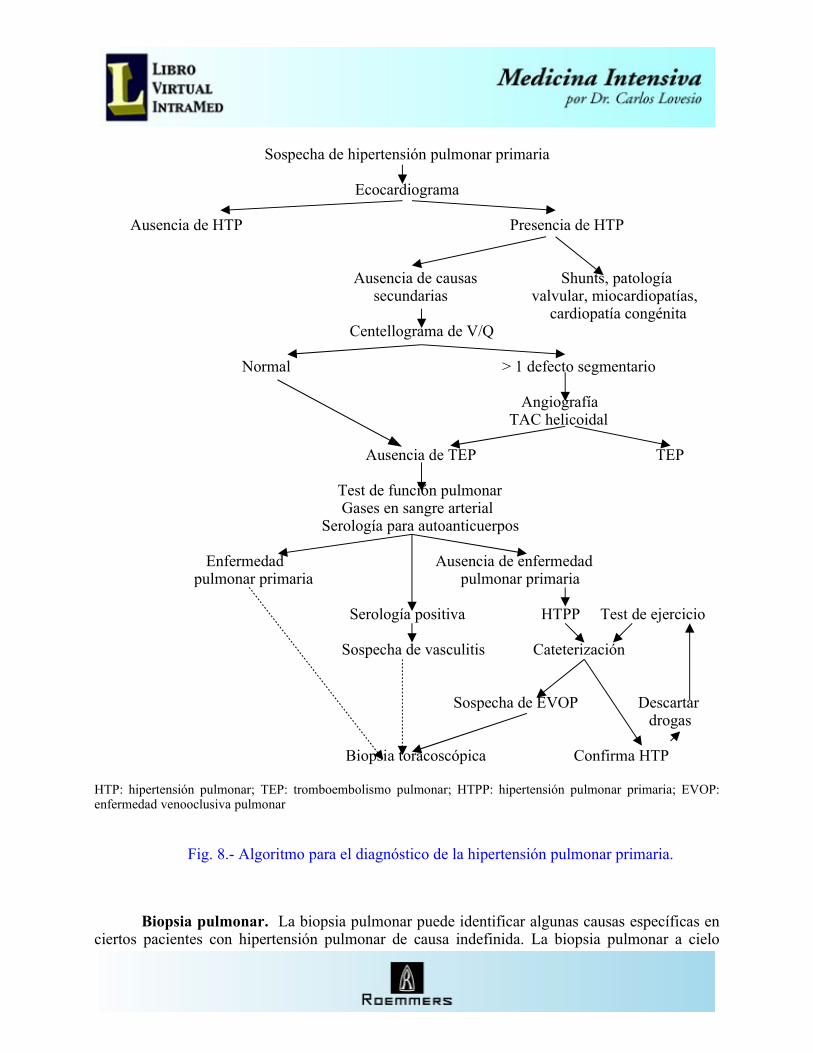
Sospecha de hipertensión pulmonar primaria
Ecocardiograma
Ausencia de HTP Presencia de HTP
Ausencia de causas Shunts, patología secundarias valvular, miocardiopatías,
cardiopatía congénita Centellograma de V/Q
Normal > 1 defecto segmentario
Angiografía TAC helicoidal Ausencia de TEP TEP
Test de función pulmonar Gases en sangre arterial Serología para autoanticuerpos
Enfermedad Ausencia de enfermedad pulmonar primaria pulmonar primaria
Serología positiva HTPP Test de ejercicio
Sospecha de vasculitis Cateterización
Sospecha de EVOP Descartar drogas
Biopsia toracoscópica Confirma HTP
HTP: hipertensión pulmonar; TEP: tromboembolismo pulmonar; HTPP: hipertensión pulmonar primaria; EVOP: enfermedad venooclusiva pulmonar
Fig. 8.- Algoritmo para el diagnóstico de la hipertensión pulmonar primaria.
Biopsia pulmonar. La biopsia pulmonar puede identificar algunas causas específicas en ciertos pacientes con hipertensión pulmonar de causa indefinida. La biopsia pulmonar a cielo

abierto o por toracoscopía involucra un riesgo sustancial de morbilidad y mortalidad en pacientes con hipertensión pulmonar. En adición, los hallazgos histopatológicos en las pequeñas arterias pulmonares son inespecíficos y pueden no diferenciar entre las diversas causas del padecimiento.
En raras ocasiones, sin embargo, la evaluación histopatológica puede establecer un diagnóstico de vasculitis activa, enfermedad pulmonar granulomatosa, enfermedad pulmonar veno-oclusiva, hemangiomatosis capilar pulmonar, enfermedad pulmonar intersticial o bronquiolitis. Sin embargo, puesto que la posibilidad de alterar el diagnóstico clínico en base a los hallazgos patológicos es baja y el riesgo es alto, no se recomienda la biopsia como método de rutina.
El National Institutes of Health de EE.UU, al establecer su registro de Hipertensión pulmonar primaria, definió un algoritmo que incluye los exámenes esenciales y los diagnósticos secundarios a excluir (Fig. 8). Utilizando esta metodología se puede establecer con certeza el diagnóstico, y eliminar las causas secundarias de hipertensión pulmonar.
PRONÓSTICO
Aunque la hipertensión pulmonar idiopática es percibida como una enfermedad progresiva, habitualmente con un mal pronóstico, la historia natural de la enfermedad es heterogénea, con algunos pacientes muriendo en meses del diagnóstico y otros viviendo por décadas. La evolución de pacientes con otras formas de hipertensión pulmonar ha sido menos descrita. En la última década, los avances en la terapéutica médica han cambiado el curso de la enfermedad, y han hecho que las decisiones terapéuticas sean más complicadas.
La sobrevida de los pacientes con hipertensión pulmonar primaria no tratados en el registro del NIH fue de 2,8 años a partir del diagnóstico, aunque como ya se adelantó, los nuevos tratamientos han mejorado este ominoso pronóstico (Tabla 6).
Tabla 6.- Sobrevida en pacientes tratados con hipertensión pulmonar.Años desde el diagnóstico
Grupos históricos sin tratamiento (%)
Grupos NYHA III y IV con terapia convencional
(%) (*)
Grupos NYHA III y IV en tratamiento con epoprosterenol (%)
1 68 77 872 52 733 48 41 634 305 34 27 54
* Warfarina, digoxina, diuréticos
Aunque las curvas de sobrevida han sido descritas especialmente para la hipertensión pulmonar idiopática, es claro que el diagnóstico de base asociado con la hipertensión pulmonar influencia el pronóstico. Las series iniciales sugirieron que el pronóstico de los pacientes con hipertensión pulmonar asociada con la esclerodermia era más desfavorable que el de los pacientes

con hipertensión pulmonar idiopática. La sobrevida en pacientes con hipertensión pulmonar asociada con el HIV parece similar a la de la población de hipertensos idiopáticos. En la enfermedad pulmonar obstructiva crónica y en el ARDS, la presencia de cor pulmonale contribuye significativamente a acortar la sobrevida. La sobrevida a tres años de los pacientes con obstrucción severa al flujo aéreo y un aumento en tres o cuatro veces la resistencia vascular pulmonar es de sólo el 10 al 15%.
Recientemente se reevaluaron los indicadores pronósticos en la HTP, constatándose que los más importantes son los síntomas, evaluados por la clase funcional, la respuesta al ejercicio y variables hemodinámicas. McLaughlin y col., en un informe sobre el pronóstico de la hipertensión pulmonar, establecieron una serie de parámetros para predecir un mal pronóstico, incluyendo: a) estado avanzado en la clasificación funcional NYHA-FC; b) baja performance en la caminata de seis metros; c) presencia de derrame pericárdico; d) elevada presión auricular derecha media; e) reducido índice cardiaco; f) elevada presión arterial pulmonar media; g) elevado índice RV (Tei) en ecocardiograma Doppler; h) hallazgos ECG de aumento de amplitud de la onda P en DII, patente qR en V1, y criterios de la WHO para hipertrofia ventricular izquierda; i) nivel elevado de péptido natriurético cerebral (>180 pg/mL); j) en pacientes con hipertensión pulmonar idiopática tratados con epoprostenol, persistencia de un estado III o IV de la clasificación NYHA luego de tres meses de terapéutica; k) en pacientes con hipertensión asociada con esclerodermia, reducción del DLCO (< 45% del predecible); l) valor pico VO2max
menor de 10,4 mL/kg/min.; y m) en pacientes pediátricos, el pronóstico se agrava con la menor edad.
En la hipertensión pulmonar primaria, los dos mecanismos más frecuentes de muerte son la falla ventricular derecha progresiva y la muerte súbita, siendo más frecuente la primera. Con la progresión de la falla ventricular derecha, el paciente se presenta con disnea progresiva, hipoxemia y una disminución creciente del volumen minuto cardíaco. La neumonía comúnmente es fatal debido a que la hipoxia alveolar produce mayor vasoconstricción pulmonar, con una incapacidad para mantener un adecuado volumen minuto cardíaco y el resultante final en shock cardiogénico. Cuando se produce acidemia e hipoxemia, también se pueden producir arritmias fatales.
Los mecanismos postulados para la muerte súbita en la hipertensión pulmonar primaria incluyen: bradiarritmias y taquiarritmias, embolismo pulmonar agudo, hemorragia pulmonar masiva e isquemia súbita del ventrículo derecho.
TRATAMIENTO
Como ya se adelantó, la hipertensión pulmonar puede responder a patologías específicas cardíacas o pulmonares, y mejorar substancialmente cuando las mismas son tratadas. Se admite que no existe tratamiento curativo para la hipertensión pulmonar primaria, excepto probablemente el transplante pulmonar, por lo que tampoco existe una propuesta terapéutica uniformemente aceptada y eficaz.

En la Tabla 7 se indican las diversas estrategias posibles de ser utilizadas, en función de las categorías hemodinámicas de hipertensión pulmonar.
Tabla 7.- Estrategias de tratamiento de la hipertensión pulmonar basadas en las categorías hemodinámicas de la enfermedad.
1. Hipertensión pulmonar postcapilar
Reducción de la presión capilar pulmonar: diuréticos, venodilatadores (nitratos) veno y arteriolo dilatadores mixtos (inhibidores de la enzima de conversión de angiotensina), terapia combinada
Disminución de la resistencia vascular pulmonar: vasodilatadores pulmonares (nitroglicerina, nitroprusiato, inhibidores de la enzima de conversión, prostaciclina, óxido nítrico inhalado, antagonistas de la endotelina)
Corrección de las causas primarias: tratamiento de las enfermedades valvulares del corazón, tratamiento de las miocardiopatías
2. Hipertensión pulmonar debida al aumento selectivo o no selectivo del flujo sanguíneo pulmonar
Reducción del flujo sanguíneo pulmonar: corrección de la causa primaria (reparo de los defectos septales cardiacos, tratamiento del hipertiroidismo), disminución de la relación entre resistencia vascular sistémica y pulmonar, empleo de vasodilatadores arteriolares (hidralazina, fentolamina)
3. Hipertensión pulmonar precapilar
Vasodilatadores: bloqueantes de los canales de calcio, infusión continua de prostaciclina, antagonistas de la endotelina
Tratamiento de la falla ventricular izquierda: digital, diuréticos, inhibidores de la enzima de conversión; dobutamina, dopamina o inhibidores de la fosfodiasterasa en perfusión intermitente, tratamiento de la causa primaria si existe
Terapia adyuvante: anticoagulación prolongada, oxígeno suplementario
Terapia quirúrgica: transplante pulmonar o cardiopulmonar, septostomía auricular, tromboendarterectomía pulmonar
Tratamientos en investigación: análogos de la prostaciclina (iloprost, baroprost, uniprost) antagonistas de la endotelina, óxido nítrico inhalado, donantes de óxido nítrico, terapia génica
Péptidos natriuréticos e inhibidores de la endopeptidasa

Inhibidores de la 5-fosfodiesterasa: sildenafil
Anticoagulación. Si bien no se ha comprobado que la anticoagulación pueda revertir los cambios patológicos de la hipertensión pulmonar idiópatica, su utilización está recomendada en estos pacientes, preferiblemente tan pronto como sea posible luego de realizar el diagnóstico, excepto que existan contraindicaciones específicas. El anticoagulante de elección es la warfarina, y la dosis debe ajustarse para mantener un INR de 2 a 3. Los pacientes con tromboembolismo pulmonar crónico deben ser tratados durante toda su vida. Algunos expertos extrapolan la evidencia que soporta el empleo de anticoagulación en pacientes con hipertensión pulmonar idiopática a otros pacientes con hipertensión pulmonar, pero este no es un concepto generalizado. En caso de utilizar anticoagulación en otras circunstancias, se debe tener en cuenta que los pacientes con esclerodermia tienen alto riesgo de sangrado gastrointestinal, y los pacientes con enfermedad cardiaca congénita tienen un riesgo aumentado de hemoptisis.
Oxígeno. La terapéutica con oxígeno es importante por múltiples razones. Debido a que la vasoconstricción pulmonar hipóxica puede contribuir a la hipertensión pulmonar, la reversión de la hipoxia es importante para reducir la posible sobrecarga ventricular derecha. Está demostrado que el oxígeno prolonga la vida en el cor pulmonale. La hiperventilación producida por la hipoxemia aumenta el trabajo respiratorio, por lo que el oxígeno suplementario puede ayudar a reducir este exceso de trabajo. Finalmente, la hipoxemia reduce la compliance ventricular y dificulta el lleno ventricular, lo que en la hipertensión pulmonar primaria se asocia con el riesgo potencial de disminuir aún más el volumen minuto cardíaco. La recomendación estándar es que los pacientes hipoxémicos con hipertensión pulmonar mantengan una saturación de oxígeno por encima del 90% durante el reposo, el ejercicio y durante el sueño. Esto puede ser difícil de lograr en pacientes con enfermedad pulmonar intrínseca concomitante, o shunt de derecha a izquierda intracardiaco. El empleo de oxígeno suplementario puede ser controvertido en pacientes con grandes shunts derecha a izquierda por enfermedad congénita cardiaca con fisiología de Eisenmenger, pero puede ayudar a disminuir la necesidad de flebotomía y potencialmente reducir la incidencia de disfunción neurológica y otras complicaciones.
Diuréticos. Los diuréticos están indicados en pacientes con evidencia de falla ventricular derecha. En el manejo a largo tiempo de pacientes con hipertensión pulmonar se considera importante mantener un volumen intravascular próximo a lo normal con diuréticos y restricción de sodio. Se debe tener en cuenta que la diuresis rápida y excesiva puede conducir a la hipotensión sistémica, insuficiencia renal y síncope. Se deben controlar estrechamente los niveles de electrolitos séricos y la función renal en pacientes en tratamiento con diuréticos.
Optimización del hematocrito. Los efectos adversos de una elevada viscosidad secundaria a una eritrocitosis severa (hematocrito ≅60%) superan a los beneficios potenciales del aumento de la capacidad de trasporte de oxígeno. La eritroferesis euvolémica o flebotomía para lograr un hematocrito de 50 a 55% parece tener efectos favorables inmediatos. No se han documentado efectos favorables adicionales disminuyendo el hematocrito por debajo de 50%. Las flebotomías se deben realizar en pequeños volúmenes (300 ml) en dos o tres días, con un intervalo de recuperación de 48 horas si se planea una cirugía electiva.

Digital y dobutamina. Puesto que la depresión de la contractilidad miocárdica parece ser uno de los eventos primarios en la progresión de la insuficiencia cardiaca derecha, los agentes inotrópicos se han considerado para el tratamiento de esta condición. La administración intravenosa por corto tiempo de digoxina en la hipertensión pulmonar idiopática produce un incremento modesto en el volumen minuto cardiaco y una reducción significativa de los niveles de norepinefrina circulantes; sin embargo, no hay datos sobre los efectos del tratamiento durante largo tiempo. De acuerdo a ello, el uso de la digital en pacientes con hipertensión pulmonar con fallo cardiaco derecho refractario se basa primariamente en el juicio del médico tratante. La digital puede ser utilizada en los pacientes con fibrilación auricular o aleteo auricular para disminuir la frecuencia cardiaca.
Los pacientes con hipertensión pulmonar en estadio terminal son tratados con dobutamina intravenosa en la mayoría de los centros de alta complejidad. Este tratamiento generalmente resulta en una mejoría clínica que puede persistir por un periodo variable de tiempo, en forma similar a lo que ocurre en pacientes con insuficiencia cardiaca izquierda avanzada.
Drogas vasodilatadoras. Las drogas vasodilatadoras tienen su indicación en el tratamiento de la hipertensión pulmonar primaria y en algunas formas de hipertensión secundaria, en base al concepto de que existe una vasoconstricción excesiva que puede iniciar las lesiones de remodelación que condicionan la irreversibilidad del cuadro. Debido a los riesgos potenciales del tratamiento, este tipo de drogas sólo deben ser empleadas en centros con experiencia en esta patología.
Si bien se han propuesto múltiples vasodilatadores para el tratamiento de la hipertensión pulmonar, en la actualidad las opciones son los bloqueantes de los canales de calcio, el óxido nítrico inhalado, las prostaglandinas y los antagonistas de los receptores de endotelina.
Debido a que los vasodilatadores tienen una serie de riesgos potenciales, se debe probar si los mismos son hemodinámicamente eficaces antes de iniciar un tratamiento prolongado. Específicamente, se debe realizar una cateterización derecha, y la administración aguda de vasodilatadores debe ser monitorizada para identificar los pacientes que tienen respuesta. El resultado ideal de un test de vasodilatadores en la hipertensión pulmonar primaria incluye: vasodilatación con una reducción en la resistencia vascular pulmonar de al menos un 20% y reducción en la presión de la arteria pulmonar en al menos 20%; incremento en el volumen minuto cardíaco resultante de la reducción de la postcarga ventricular derecha; con ausencia de un decremento significativo de la presión arterial sistémica. Recientemente, la Sociedad Europea de Cardiología formuló una definición de consenso de una respuesta positiva aguda a los vasodilatadores en pacientes con hipertensión pulmonar idiopática. La misma se define como un descenso de la presión arterial pulmonar media de al menos 10 mm Hg y un valor de presión arterial pulmonar final de no más de 40 mm Hg, con un aumento o al menos no cambio en el volumen minuto cardiaco. Algunos autores consideran que esta definición es poco sensible, ya que deja fuera de consideración a aquellos que responden con un descenso del 20% en la presión pulmonar, pero cuya presión pulmonar media continúa estando por encima de 40 mm Hg., y que eventualmente podrían responder a un tratamiento con vasodilatadores.

Las drogas utilizadas para la evaluación aguda son el epoprostenol intravenoso en dosis inicial de 1-2 ng/kg/min cada 5 a 15 minutos, hasta una dosis máxima de 12 ng/kg/min.; la adenosina intravenosa en dosis inicial de 50 µg/kg/min cada 2 minutos hasta una dosis máxima de 500 µg/kg/min.; la acetilcolina intravenosa en dosis de 1 mg/min durante 10 minutos, y el óxido nítrico inhalado, 10-80 ppm en aire durante cinco minutos. El peso de la evidencia favorece el empleo del epoprostenol IV o del óxido nítrico inhalado como agentes preferidos para el test de vasodilatadores, la adenosina IV puede ser utilizada si no se dispone de ninguno de los otros agentes. El rango de dosis de esta última es de 50-250 µg/kg/min, con incrementos de dosis de 50 µg/kg/min cada dos minutos.
Drogas bloqueantes de los canales de calcio. Los pacientes que responden a la acción de un vasodilatador de corta acción se deben someter a un ensayo de dosis convencionales por vía oral de un bloqueante de los canales de calcio. La indicación es en pacientes con hipertensión pulmonar con un índice cardiaco mayor de 2,1 l/min/m2 y o una saturación de oxígeno en sangre venosa mezclada por encima de 63%, y o una presión auricular derecha menor de 10 mm Hg. Todos los pacientes deben ser monitorizados hemodinámicamente durante la iniciación del tratamiento.
Las dosis de estas drogas que se han demostrado eficaces en la hipertensión pulmonar idiopática son relativamente elevadas (120-240 mg/día de nifedipina y 240-720 mg/día de diltiazem). Es recomendable, en pacientes vasoreactivos, iniciar el tratamiento con una dosis reducida (30 mg de nifedipina tres veces por día o 60 mg de dilitiazem tres veces por día) e ir aumentado la dosis en forma progresiva en las semanas siguientes hasta lograr el máximo régimen tolerado. Esta terapéutica se asocia con una mejoría significativa en la calidad de vida, regresión de la hipertrofia ventricular derecha y aumento de la sobrevida. Rich y colaboradores mostraron que la administración de 240 mg de nifedipina o 720 mg de diltiazem diarios en pacientes con hipertensión pulmonar primaria se asoció con una sobrevida a los cinco años del 95% en los respondedores, comparado con un 36% en los no respondedores. Desgraciadamente, sólo el 10-15% de pacientes con hipertensión pulmonar reúnen los criterios de un test positivo de vasoreactividad, y sólo la mitad de estos presentan una respuesta clínica y hemodinámica satisfactoria en el tratamiento de largo tiempo. Se acepta que sólo en estos casos se debe continuar un tratamiento con bloqueantes cálcicos exclusivamente.
Se debe tener presente que la administración de dosis elevadas de bloqueantes de los canales de calcio se asocia con efectos colaterales significativos, incluyendo hipotensión sistémica como resultado de vasodilatación periférica o de un efecto inotrópico negativo, exacerbación de la hipertensión pulmonar, hipoxemia, arritmias, y deterioro de la función ventricular derecha.
Prostaglandinas. Los mecanismos precisos de acción de las prostaciclinas en pacientes con hipertensión pulmonar son desconocidos y muy probablemente sean multifactoriales. Los mismos incluyen relajación de las células musculares lisas vasculares, inhibición de la agregación plaquetaria, normalización de las anormalidades de agregación, dispersión de las plaquetas agregadas, limitación de la injuria de células endoteliales, inhibición de la migración de células vasculares facilitando la remodelación de los cambios vasculares, mejoría del clearance

pulmonar de ET-1, efecto inotrópico directo, aumento de la utilización periférica de oxígeno por el músculo esquelético y mejoría hemodinámica durante el ejercicio.
Las prostaglandinas están contraindicadas en pacientes con enfermedad parenquimatosa pulmonar severa y disfunción cardiaca derecha. Los agentes disponibles para uso clínico son el epoprostenol, el iloprost, el beraprost y el treprostinil.
Los no respondedores pueden ser incluidos en un ensayo de epoprostenol (PGI2) intravenoso crónico. Esta técnica ha sido aprobada recientemente por la US Food and Drug Administration para el tratamiento de pacientes con hipertensión pulmonar primaria, siendo utilizada principalmente en pacientes refractarios a la terapia convencional. Los mismos se encuentran dentro de las clases III y IV de la clasificación de la NYHA, que incluye pacientes con saturación venosa de oxígeno (SvO2) <60% y en espera de trasplante pulmonar. En un estudio reciente (Sitbon y col.) la prostaciclina mejoró los parámetros hemodinámicos, la tolerancia al ejercicio, la calidad de vida y la sobrevida en pacientes incluidos dentro de los grupos precedentes, cuando se comparó con la terapéutica convencional. La falta de una respuesta hemodinámica favorable en forma aguda no es indicativa de un fracaso definitivo. En efecto, se han comprobado efectos beneficiosos a largo tiempo sin que exista una respuesta aguda. El mecanismo de esta respuesta no es conocido. En el estudio citado, el grupo que recibió epoprostenol tuvo una sobrevida al año del 85%, a los dos años del 70%, a los tres años del 63%, y a los cinco años del 55%. En el grupo control, la sobrevida fue del 58%, 43%, 33% y 28% para los mismos periodos de tiempo.
La dosis óptima de PGI2 en un paciente individual es variable. Se recomienda comenzar con un dosaje muy bajo de epoprostenol (1 a 2 ng/kg/min.), e incrementar la dosis gradualmente en 1 a 2 ng/kg/min., basado en los efectos colaterales y la tolerancia. Muchos pacientes alcanzan una dosis meseta, y pueden no requerir titulación en ascenso a partir de este punto. Mientras que muchos pacientes alcanzan la respuesta adecuada con dosis entre 25 y 40 ng/kg/min, el rango de dosis es amplio, con considerable variabilidad interindividual.
Debido a la corta vida media de la droga, es necesario colocar un catéter venoso a permanencia para su administración. La discontinuación abrupta del epoprostenol puede asociarse con un aumento por rebote en la presión arterial pulmonar que puede llevar a la falla ventricular derecha aguda e incluso a la muerte. Los efectos adversos habituales incluyen disconfort abdominal, dolor mandibular, diarrea, flashing, rash y dolor de pies. La sobredosis aguda se asocia con hipotensión sistémica, y la sobredosis prolongada puede producir un estado de circulación hiperdinámica con una insuficiencia cardiaca de alto débito. La mayor limitación de la infusión intravenosa continua de prostaciclina en humanos es la frecuencia de complicaciones asociadas con el sistema de administración, en particular infecciones, y el elevado costo, aproximadamente 100.000 U$S anual (2004). El manejo de pacientes en tratamiento crónico con epoprostenol rquiere una considerable infraestructura, incluyendo médicos y personal de enfermería con experiencia.
En la actualidad existen análogos de la prostaciclina que pueden ser administrados por vía no parenteral. El iloprost y el beroprost son dos análogos de la PGI2 que pueden ser administrados por inhalación y por vía oral, respectivamente. El iloprost es un análogo estable de la prostaciclina disponible para empleo intravenoso, oral y en aerosol. Tiene una vida media de

20 a 25 min. La terapéutica inhalante para la hipertensión pulmonar es un concepto atractivo que se ha aplicado en la práctica clínica. En la hipertensión pulmonar idiopática, la inhalación aguda de iloprost produce un efecto vasodilatador pulmonar más potente que la inhalación aguda de óxido nítrico. La dosis diaria recomendada es de 2,5 µg a 5 µg seis a nueve veces por día, con una dosis máxima de 45 µg/día. Para el empleo a largo tiempo, la relativa corta duración de acción del iloprost inhalado requiere de seis a nueve inhalaciones por día para obtener un beneficio clínico significativo. El iloprost inhalado ha sido aprobado en Europa para el tratamiento de la hipertensión pulmonar idiopática en clase funcional III y en EE.UU. para las clases funcionales III y IV.
El beraprost es el primer análogo de la prostaciclina químicamente estable y activo por vía oral. Es absorbido rápidamente en condiciones de ayuno; la concentración pico se alcanza luego de 30 minutos y la vida media es de 35 a 40 minutos luego de la administración oral. La dosis media es de 80 µg una vez por día. Estudios controlados contra placebo en Europa han reportado mejoría en la tolerancia al ejercicio con el beraprost en pacientes con hipertensión pulmonar primaria. El beraprost ha sido aprobado para su uso en Japón y Corea.
El treprostinil es un análogo de la PGI2, con una vida media de dos a cuatro horas, que puede ser administrado por vía subcutánea con una bomba de infusión. El mayor problema con esta droga es el dolor y el eritema que se producen en el sitio de inyección, lo que hace necesaria la frecuente rotación del mismo. La droga está disponible en una jeringa premezclada y prellenada, y por lo tanto no se requiere la preparación del medicamento en el momento de la administración. La dosis de trepostinil oscila entre 3 y 65 ng/kg/min, con una media de 22 ng/kg/min. Recientemente se ha introducido el treprostinil intravenoso, que es más conveniente que el epoprosterenol, ya que no requiere refrigeración y tienen una vida media más larga.
Se debe tener en cuenta, que en cualquier caso, estos tratamientos deben ser utilizados como un “puente” hasta disponer de un órgano para el trasplante.
Oxido nítrico inhalado. El oxido nítrico, un gas producido en los vasos sanguíneos y en otros órganos por la acción de la enzima óxido nítrico sintetasa sobre la L-arginina, juega un rol importante en la regulación del tono vasomotor en la circulación pulmonar. Una vez elaborado, el óxido nítrico activa la guanilato ciclasa citosólica, aumentando el nivel de GMPc, lo que resulta en una relajación de la musculatura lisa en el lecho pulmonar.
La inhalación de concentraciones relativamente bajas de óxido nítrico produce una vasodilatación pulmonar selectiva en las áreas ventiladas del pulmón en niños y adultos con hipertensión pulmonar crónica. Estos efectos son comparables a los producidos por la infusión de prostaciclina. Debido a los riesgos de la hipertensión pulmonar de rebote con el cese agudo de la terapéutica y a los efectos tóxicos potenciales, tales como la metahemoglobinemia y la inmunosupresión, el empleo a largo tiempo del ON inhalado es muy limitado. Las guías para el diagnóstico y el manejo de la hipertensión arterial pulmonar del American College of Chest Physicians proponen el uso del óxido nítrico inhalado exclusivamente para los tests de vasoreactividad.
Antagonistas de la endotelina. Como las endotelinas circulantes y la activación de los receptores de endotelina inducen vasoconstricción y aumento de la resistencia vascular pulmonar

y de la presión arterial pulmonar, se han evaluado antagonistas de la endotelina para el tratamiento crónico de la hipertensión pulmonar. Estudios experimentales han informado una reducción sustancial en la resistencia vascular y en la presión arterial pulmonar en las formas precapilar, postcapilar y mixta de hipertensión pulmonar.
El bosentan, un agente bloqueante no selectivo de la endotelina que puede ser utilizado por vía oral, se ha demostrado efectivo en los pacientes en clase III y IV de la WHO con hipertensión pulmonar idiopática o hipertensión pulmonar asociada con enfermedades del tejido conjuntivo, enfermedad congénita cardiaca con shunt derecha a izquierda, drogas y toxinas e infección por VIH. La droga ha sido aprobado por la Food and Drug Administration para el tratamiento de la hipertensión pulmonar primaria y la asociada con la esclerodermia. La droga también tendría efectos antiproliferativos sobre el músculo liso arterial pulmonar a través de un mecanismo que involucra la subregulación de receptores independientes del bloqueo de los receptores de endotelina.
El tratamiento con bosentan (inicial: 62,5 mg dos veces por día y a partir del segundo mes 125 mg dos veces por día) se asoció con una mejoría significativa en la clase clínica y un aumento en la distancia de caminata en seis minutos. Debido al riesgo de hepatotoxicidad, se requieren exámenes de función hepática al menos una vez por mes. El bosentan también puede producir anemia, edemas y teratogenicidad. La droga está contraindicada en el embarazo. La eficacia de esta droga se observa recién a los dos o tres meses de la iniciación de la terapéutica, por lo que no debe ser utilizada como droga única en pacientes con enfermedad avanzada clase IV que necesitan una respuesta más inmediata. En un estudio de Highland y colaboradores se comprobó que el bosentan es más costo-efectivo que el tratamiento con epoprostenol o treprostinil, y además presenta una mejoría neta en la calidad de vida.
El sitaxsentan es un compuesto de segunda generación en estudio en ensayos clínicos de fase IIb/III. Se considera que la droga tiene menos problemas de toxicidad hepática que el bosentan. Las dosis de 100 a 300 mg. producen una mejoría en al menos un nivel en la clasificación funcional de la NYHA en alrededor del 50% de los pacientes. No existen datos prospectivos que indiquen que el sitaxsentan reduzca la mortalidad. La droga se encuentra bajo revisión por la FDA.
Inhibidores de la 5-fosfodiesterasa. La inhibición de la degradación del óxido nítrico es una alternativa farmacológica para facilitar la vasodilatación pulmonar. La fosfodiesterasa-5 es la principal isoenzima responsable de la inhibición de la vasodilatación pulmonar inducida por el óxido nítrico. En teoría, la inhibición de esta isoenzima podría aumentar los niveles de AMPc y GMPc, mediadores de la vasodilatación, resultando en una disminución de la presión arterial pulmonar.
El inhibidor de la 5-fosfodiesterasa sildenafil, en dosis variable de 50 a 300 mg/día, atenúa la vasoconstricción pulmonar inducida por la hipoxia y potencialmente sería efectivo en el tratamiento de la hipertensión pulmonar. En adición a producir una mejoría en la hemodinamia cardiopulmonar, el sildenafil se ha asociado con una reducción de la masa ventricular derecha, pudiendo tener un rol en la prevención o reversión de la remodelación del ventrículo derecho secundaria a la hipertensión pulmonar. Los efectos colaterales del sildenafil incluyen enrojecimiento cutaneo, diarrea y quemazón retroesternal. Se ha comprobado que prolonga los

efectos vasodilatadores del ON inhalado y del iloprost aerosolizado. Recientemente se ha completado un ensayo multicéntrico con buenos resultados, habiendo sido aprobada la droga por la FDA para su empleo en la hipertensión pulmonar.
Aunque muchos autores establecen que existe una equivalencia entre el sildenafil, el epoprostenol y el bosentan, en la opinión de Archer y col., la eficacia, conveniencia y costo, sugieren una ventaja para el sildenafil como droga de primera línea para el tratamiento de pacientes en clase NYHA III.
En la Tabla 8 se indican los agentes disponibles y las dosis recomendadas para el tratamiento crónico de la hipertensión pulmonar primaria.
Tabla 8.- Drogas para el tratamiento crónico de la hipertensión pulmonar.
Nifedipina (oral) 30-240 mg/díaDiltiazem (oral) 120-720 mg/díaOxido nítrico (inhalado) 5-80 ppm (partes por millón)Epoprostenol infusión continua intravenosa 1-2 ng/kg/min hasta 20-40 ng/kg/minBeraprost (oral) 40-120 µg/día en 3 o 4 tomasIloprost nebulización 2,5 a 5,0 µg/6 a 9 veces por día (30 µg/día)Trepostinil infusión continua subcutánea 3 a 65 ng/kg/min (media: 22 ng/kg/min)Bosentan (oral)Sildenafil (oral)
125 mg dos veces por día50-100 mg una a tres veces por día
Tratamiento combinado. Al momento actual, son pocos los ensayos clínicos que han evaluado en forma prospectiva el empleo de terapéutica combinada en pacientes con hipertensión pulmonar. La práctica clínica común es adicionar una segunda terapéutica cuando existe una evidencia subjetiva y/o objetiva de deterioro clínico o progresión de la enfermedad durante la terapéutica inicial. Sin embargo, atacando más de una vía biológica crítica en la patogénesis de la hipertensión pulmonar en todos los pacientes, es probable que se obtengan mejores resultados clínicos y menor progresión de la enfermedad.
La eficacia y la seguridad de la combinación de bosentan y epoprostenol fue evaluada en un ensayo prospectivo en 33 pacientes con hipertensión pulmonar en clase III-IV de la WHO (BREATHE-2). Se demostró una mejoría en la hemodinamia, en la capacidad de ejercicio y en la clase funcional tanto en los enfermos que recibieron epoprostenol como en los que recibieron terapéutica combinada, sin diferencia estadísticamente significativa.
Varios estudios pequeños han evaluado los beneficios de combinar sildenafil oral y terapéutica con iloprostenol inhalado. Estos estudios demostraron una acción sinérgica del sildenafil oral y del iloprost inhalado en pacientes con hipertensión pulmonar, encontrándose en marcha dos estudios prospectivos destinados a comprobar los resultados precedentes.

La combinación de bosentan y sildenafil fue evaluada en nueve pacientes con hipertensión pulmonar idiopática. Al adicionar sildenafil, se comprobó una mejoría en las condiciones funcionales.
La mayor limitación de la polifarmacia de la hipertensión pulmonar es el costo del tratamiento. Todas las drogas aprobadas son costosas, y el costo del tratamiento puede hacerse exorbitante cuando se utilizan dos o más drogas en el mismo paciente.
Tromboendarterectomía pulmonar. El tratamiento de elección para la hipertensión pulmonar crónica de origen trombótico es la tromboendarterectomía pulmonar. Sólo los trombos organizados en los vasos proximales pueden ser abordados con esta técnica. La evaluación preoperatoria debe incluir una angiografía pulmonar completa para determinar el sitio y extensión de la trombosis. Cuando la cirugía tiene éxito, se comprueba una rápida y significativa mejoría hemodinámica. Estos pacientes deben permanecer anticoagulados por el resto de la vida, y en la mayoría de los casos se debe colocar un dispositivo de interrupción de la vena cava inferior.
Septostomía auricular con balón. La septostomía auricular con balón es un procedimiento invasivo que intenta la paliación de la HTP creando un defecto intraauricular para producir un shunt de derecha a izquierda. Creando este shunt se producen cambios hemodinámicos favorables, tales como la reducción de la presión auricular derecha, aumento del volumen de eyección ventricular izquierdo, mejoría de la capacidad de ejercicio, y prolongación de la sobrevida en comparación con controles históricos. El procedimiento es bien tolerado a pesar de que se produce una disminución en la saturación de oxígeno arterial, pero se asocia con un alto riesgo en pacientes críticos. Esta técnica debe ser utilizada en pacientes con hipertensión pulmonar severa refractaria al tratamiento con prostaglandinas, en especial si se asocia con síncope recurrente, y como un puente para el trasplante.

Hipertensión arterial pulmonar(Clase funcional NYHA III o IV)
Terapéutica convencional(anticoagulantes orales + oxígeno + diuréticos + digoxina)
Respuesta aguda a vasodilatadores
Si No
Bloqueantes de canales Clase III Clase IVde calcio
Respuesta sostenida Antagonistas de receptor de endotelina Prostaciclina intravenosa o análogos de prostaciclina o antagonistas de receptor
o prostaciclina intravenosa de endotelina Si No o análogos de prostaciclina
Falta de mejoría
Terapia o deterioro combinada?
Continuar con bloqueantes Sildenafil Septostomía atrial y/o de canales de calcio trasplante de pulmón
Fig. 9.- Algoritmo para el tratamiento de la hipertensión arterial pulmonar (Humbert M. y col.).
Trasplante pulmonar. En el momento actual, el trasplante pulmonar de cualquier tipo sólo debe ser considerado después del fracaso de la terapéutica médica, teniendo en cuenta el tiempo necesario para completar la evaluación pretrasplante y el tiempo que trascurre en lista de espera hasta la obtención de un órgano adecuado. La muerte en el periodo de espera constituye un problema particular en pacientes con mal estado funcional. En términos generales, los pacientes que continúan deteriorándose pese a la terapéutica con drogas vasodilatadoras, o que se deterioran luego de un intervalo de beneficio, deben ser considerados para trasplante pulmonar. El trasplante está indicado en la hipertensión pulmonar primaria cuando ofrece un beneficio en término de sobrevida, o cuando la calidad de vida del paciente es tan pobre que justifica su realización. Las características para derivar a un paciente con hipertensión pulmonar para evaluación para trasplante pulmonar incluyen: clase New York Heart Association III o IV; presión auricular derecha media >10 mm Hg, presión arterial pulmonar media >50 mm Hg, índice cardiaco <2,5 l/min/m2, y falta de respuesta a la administración intravenosa continua de prostaciclina.

La técnica de trasplante a utilizar puede ser la de pulmón-corazón en bloque, bipulmonar o unipulmonar. Si bien el método recomendado es el trasplante bipulmonar, la decisión respecto a cual procedimiento es el más apropiado en cada caso en particular debe ser dejada en manos de centros con experiencia en esta compleja patología. El número de trasplantes en estos pacientes ha sido pequeño; en Estados Unidos se han transplantado entre 49 y 66 pacientes por año con esta patología entre 1991 y 1995. En pacientes con hipertensión pulmonar idiopática, la sobrevida luego del trasplante es de 70%, 50%, 40% y 25% al año, tres años, cinco años y 10 años, respectivamente. Todos los pacientes con hipertensión pulmonar primaria deben ser tratados inicialmente con PGI2 y sólo aquellos pacientes que no responden a este tratamiento deben ser considerados para el trasplante.
En la Fig. 9 se presenta un algoritmo para el tratamiento de la hipertensión arterial pulmonar clase funcional III o IV de la clasificación NYHA.
BIBLIOGRAFIA
Archer S., Michelakis E.: An evidence-based approach to the management of pulmonary arterial hypertension. Curr Opin Cardiol 21:385-2006
Badesch D.: Pharmacologic therapy of pulmonary hypertension. 28th Educational and Scientific Symposium. SCCM, San Francisco, 1999
Badesch D., Abman S., Ahearn G.: Medical therapy for pulmonary arterial hypertension. ACCP Evidence-Based Clinical Practice Guidelines. Chest 126 (Suppl July) 35S-2004
Barst R., Rubin L., Long W.: A comparison of continous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 334:296-1996
Beaulieu Y., Marik P.: Bedside ultrasonography in the ICU. Part 1. Chest 128:881-2005
Bossone E., Bodini B., Mazza A.: Pulmonary arterial hypertension: the key role of echocardiography. Chest 127:1836-2005
British Cardiac Society Guidelines and Medical Practice Committee: Recommendations on the management of pulmonary hypertension in clinical practice. Heart 86(Suppl 1)i1-2001
Channick R., Williamson T.: Diagnosis and treatment of pulmonary arterial hypertension. Cardiol Clin 22:441-2004
Chatterjee K., De Marco T., Alpert J.: Pulmonary hypertension. Hemodynamic diagnosis and management. Arch Intern Med 162:1925-2002
Crisostomo M., Rubinstein I.: Pharmacotherapy of chronic pulmonary hypertension: a contemporary overview. Clin Pulm Med 5:191-1998
D’Alonzo G., Lodato R., Fuentes F.: Diagnostic approaches in pulmonary hypertension. J Crit Illness 2:17-1987

Dincer H., Presberg K.: Current management of pulmonary hypertension. Clin Pulmonary Med 11:40-2004
Doyle R., McCrory D., Channick R.: Surgical treatments/Interventions for pulmonary arterial hypertension. ACCP Evidence-Based Clinical Practice Guidelines. Chest 126: (Suppl 1): S63-2004
Executive Summary from World Symposium on Primary Pulmonary Hypertension 1998. World Health Organization Web site. Available at: http://www.who.int/ned/evd/pph.html
Farber H., Loscalzo J.: Pulmonary arterial hypertension.N Engl J Med 351:1655-2004
Fedullo P., Auger W., Channick R.: Chronic thromboembolic pulmonary hypertension. Clin Chest Med 16:353-1995
Fishman A.: Clinical classification of pulmonary hypertension. Clin Chest Med 22:385-2001
Galie N., Manes A., Ugoccioni L.: Primary pulmonary hypertension: insights into pathogenesis from epidemiology. Chest 114 (Suppl):184S-1998
Galie N., Manes A., Branzi A.: Medical therapy of pulmonary hypertension: the prostacyclins. Clin Chest Med 22:529-2001
Galie N., Torbicki A., Barst R.: Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J 25:2243-2004
Galie N., Ghofrani H., Torbicki A.: Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 353:2148-2005
Ghofrani H., Schermuly R., Rose F.: Sildenafil for long-term treatment of nonoperable chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 167:1139-2003
Griffiths M., Evans T.: Inhaled nitric oxide therapy in adults. N Engl J Med 353:2683-2005
Haj R., Cinco J., Mazer C.: Treatment of pulmonary hypertension with selective pulmonary vasodilators. Curr Opin Anaesthesiol 19:88-2006
Higenbottam T., Scott J., Spiegelhalter D.: The value of prostacyclin and heart-lung transplantation in severe pulmonary hypertension. Br Heart J 70:366-1993
Higenbottam T., Butt A., Dinh-Xaun A.: Treatment of pulmonary hypertension with the continuous infusion of a prostacyclin analogue, iloprost. Heart 79:175-1998
Higenbottam T., Laude L., Emery C.: Pulmonary hypertension as a result of drug therapy. Clin Chest Med 25:123-2004
Highland K., Strange C., Mazur J.: Treatment of pulmonary arterial hypertension: a preliminary decision analysis. Chest 124:2087-2003

Hillier S.: Recent advances in the treatment of pulmonary hypertension. Curr Opinion in Anaesth 16:331-2003
Hoeper M., Mayer E., Simonneau G.: Chronic thromboembolic pulmonary hypertension. Circulation 113:2011-2006
Humbert M., Sitbon O., Simonneau G.: Treatment of pulmonary arterial hypertension. N Engl J Med 351:1425-2004
Jones K., Higenbottan T.: A comparison of continous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 334:296-1996
Lilienfeld D., Rubin L.: Mortality from primary pulmonary hypertension in the United States 1979-1996. Chest 117:796-2000
Mandel J., Mark E., Hales C.: Pulmonary veno-occlusive disease. Am J Respir Crit Care Med 162:1964-2000
McGoon M., Gutterman D., Steen V.: Screening, early detection, and diagnosis of pulmonary arterial hypertension. ACCP Evidence-Based Clinical Practice Guidelines. Chest 126 (Suppl July): 14S-2004
McLaughlin V.: Classification and epidemiology of pulmonary hypertension. Cardiol Clin 22:327-2004
McLaughlin V., Presberg K., Doyle R.: Prognosis of pulmonary arterial hypertension. ACCP Evidence-Based Clinical Practice Guidelines. Chest 126 (Suppl July) 78S-2004
McLaughlin V., McGoon M.: Pulmonary arterial hypertension. Circulation 114:1417-2006
Mehta N., Khan I., Mehta R.: HIV-related pulmonary hypertension. Analytic review of 131 cases. Chest 118:1133-2000
Michelakis E., Weir K.: The pathobiology of pulmonary hypertension. Clin Chest Med 22:419-2001
Morley T., Zappasodi S., Belli A.: Pulmonary vasodilator therapy for chronic obstructive pulmonary disease and cor pulmonale: treatment with nifedipine, nitroglycerin, and oxygen. Chest 92:71-1987
Murali S.: Pulmonary arterial hypertension. Curr Opin Crit Care 12:228-2006
Naeije R., Vachiery J.: Medical therapy of pulmonary hypertension: conventional therapies. Clin Chest Med 22:517-2001
Neely C., Stein R., Matot I.: Calcium blockage in pulmonary hypertension and hypoxic vasconstriction. New Horizons 4:99-1996
Nunes H., Humbert M., Capron F.: Pulmonary hypertension associated with sarcoidosis: mechanisms, haemodynamics and prognosis. Thorax 61:68-2006

Olschewski H., Walmrath D., Schermuly R.: Aerosolized prostacyclin and iloprost in severe pulmonary hypertension. Ann Intern Med 124:820-1996
Olschewski H., Ghofrani H., Walmrath D.: Recovery from circulatory shock in severe primary pulmonary hypertension with aerosolization of iloprost. Intensive Care Med 24:631-1998
Olschewski H., Simonneau G., Galie N.: Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 347:322-2002
Opitz C., Wensel R., Winkler J.: Clinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J 26:1895-2005
Palevsky H., Fishman A.: The management of primary pulmonary hypertension. JAMA 265:1014-1991
Pepke Zaba J., Higenbottan T.: Inhaled nitric oxide as a cause of selective pulmonary vasodilatation in pulmonary hypertension. Lancet 338:1173-1991
Prielipp R.: Pulmonary hypertension and right heart failure. En 9th Critical Care Refresher Course. 34th Critical Care Congress, Phoenix, Arizona, EE.UU., January 2005
Ricciardi M., Bossone E., Bach D.: Echocardiographic predictors of an adverse response to a nifedipine trial in primary pulmonary hypertension. Chest 116:1218-1999
Rich S., Dantzker D., Ayres S.: Primary pulmonary hypertension: A national prospective study. Ann Intern Med 107:216-1987
Rich S., Kaufman E., Levy P.: The effect of high doses of calcium channel blockers on survival in primary pulmonary hypertension. N Engl J Med 327:76-1992
Rich S.: Medical treatment of primary pulmonary hypertension: a bridge to transplantation? Amer J Cardiol 75:63A-1995
Rich G.: Management of the patient with pulmonary hypetension and right ventricular failure. The American Society of Anesthesiologists, Inc. ASA Vol 33:2005
Rubin L.: Primary pulmonary hypertension: ACCP Consensus Statement. Chest 104:236-1993
Rubin L.: Primary pulmonary hypertension. N Engl J Med 336:111-1997
Rubin L., Badesch D., Barst R.: Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 346:896-2002
Rubin L.: Diagnosis and management of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 126: (Suppl 1):S7-2004
Rubin L., Badesh D.: Evaluation and management of the patient with pulmonary arterial hypertension. Ann Intern Med 143:282-2005
Salvi S.: α1-Adrenergic hypothesis for pulmonary hypertension. Chest 115:1708-1999

Schenk P., Petkov V., Madl C.: Aerosolized iloprost therapy could not replace long-term IV epoposterenol administration in severe pulmonary hypertension. Chest 119:296-2001
Schnader J., Janz T.: Management approaches to primary pulmonary hypertension. Clin Pulmonary Med 4:231-1997
Simonneau G., Barst R., Galie N.: Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary artery hypertension. Am J Respir Crit Care Med 165:800-2002
Sperling R., Creager M.: Nitric oxide and pulmonary hypertension. Coronary Artery Disease 10:287-1999
Tonz M., Segesser L., Schilling J.: Treatment of acute pulmonary hypertension with inhaled nitric oxide. Ann Thorac Surg 58:1031-1994
Voelkel N., Cool C.: Pathology of pulmonary hypertension. Cardiol Clin 22:343-2004
Williamson D., Wallman L., Jones R.: Hemodynamic effects of bosentan, an endothelin receptor antagonist, in patients with pulmonary hypertension. Circulation 102:411-2000
Zapol W.: Inhaling nitric oxide: A selective pulmonary vasodilator and bronchodilator. Chest 105 (Suppl) S87-1994