hepatic activation of the fam3c-hsf1-cam pathway...
TRANSCRIPT

Hepatic Activation of the FAM3C-HSF1-CaM PathwayAttenuates Hyperglycemia of Obese Diabetic MiceZhenzhen Chen,1 Liwei Ding,1 Weili Yang,1 Junpei Wang,2 Liming Chen,3 Yongsheng Chang,4 Bin Geng,5
Qinghua Cui,2 Youfei Guan,6 and Jichun Yang1
Diabetes 2017;66:1185–1197 | DOI: 10.2337/db16-0993
FAM3C is a member of the family with sequence similarity3 (FAM3) gene family, and this study determined its roleand mechanism in regulation of hepatic glucose/lipidmetabolism. In obese diabetic mice, FAM3C expressionwas reduced in the liver, and hepatic FAM3C restorationimproved insulin resistance, hyperglycemia, and fatty liver.FAM3C overexpression increased the expression ofheat shock factor 1 (HSF1), calmodulin (CaM), and phos-phorylated protein kinase B (Akt) and reduced that ofgluconeogenic and lipogenic genes in diabetic mouselivers with the suppression of gluconeogenesis and lipiddeposition. In cultured hepatocytes, FAM3C overex-pression upregulated HSF1 expression, which elevatedCaM protein level by inducing CALM1 transcription toactivate Akt in a Ca2+- and insulin-independent manner.Furthermore, FAM3C overexpression promoted nuclearexclusion of FOXO1 and repressed gluconeogenic geneexpression and gluconeogenesis in a CaM-dependentmanner in hepatocytes. Hepatic HSF1 overexpressionactivated the CaM-Akt pathway to repress gluconeo-genic and lipogenic gene expression and improve hy-perglycemia and fatty liver in obese diabetic mice. Inconclusion, the FAM3C-HSF1-CaM-Akt pathway playsimportant roles in regulating glucose and lipid metab-olism in hepatocytes independent of insulin and calcium.Restoring hepatic FAM3C expression is beneficial for themanagement of type 2 diabetes and fatty liver.
In the past decades, type 2 diabetes had become a seriouspublic health issue, affecting more than 300 million peopleworldwide in 2013 (1). Increased hepatic glucose productionas a result of insulin resistance had been accepted as beingcritical for the development of fasting hyperglycemia andtype 2 diabetes (2). Protein kinase B (Akt) is the key nodemolecule of insulin signaling, and Akt activation phosphor-ylates and inactivates FOXO1 and glycogen synthase kinase3 (GSK-3) to suppress gluconeogenesis and enhance glyco-gen synthesis in the liver (2–4). Akt also affects the activitiesof SREBP-1 and FOXO1 to modulate lipid metabolism (2,5).Under insulin-resistant status, activating Akt via insulin-independent pathway(s) represents a novel strategy forsuppressing hepatic gluconeogenesis and ameliorating hy-perglycemia (6,7).
The family with sequence similarity 3 (FAM3) genefamily contains four members, designated as FAM3A,FAM3B, FAM3C, and FAM3D (8). FAM3A plays importantroles in regulation of hepatic and glucose metabolism viaATP-P2 receptor–mediated activation of Akt pathways in-dependent of insulin (7). FAM3B (PANDER) exerts del-eterious effects on pancreatic b-cell function (9,10) andhepatic insulin signaling (11–14). To date, FAM3C, alsoknown as interleukin-like epithelial-mesenchymal transi-tion (EMT) inducer (ILEI), has been reported to be impor-tant in embryonic development, EMT, and the progression
1Department of Physiology and Pathophysiology, School of Basic Medical Sci-ences, Key Laboratory of Molecular Cardiovascular Science of the Ministry ofEducation, Center for Non-coding RNA Medicine, Peking University Health Sci-ence Center, Beijing, China2Department of Biomedical Informatics, School of Basic Medical Sciences, KeyLaboratory of Molecular Cardiovascular Science of the Ministry of Education,Center for Non-coding RNA Medicine, Peking University Health Science Center,Beijing, China3Department of Biophysics and Molecular Physiology, Key Laboratory of Mo-lecular Biophysics of Ministry of Education, Huazhong University of Science &Technology School of Life Science & Technology, Wuhan, China4National Laboratory of Medical Molecular Biology, Institute of Basic MedicalSciences, Chinese Academy of Medical Sciences and Peking Union MedicalCollege, Beijing, China
5Hypertension Center, Fuwai Hospital, Chinese Academy of Medical Sciencesand Peking Union Medical College, Beijing, China6Advanced Institute for Medical Sciences, Dalian Medical University, Dalian,China
Corresponding author: Jichun Yang, [email protected].
Received 15 August 2016 and accepted 18 February 2017.
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db16-0993/-/DC1.
Z.C. and L.D. contributed equally to this work.
© 2017 by the American Diabetes Association. Readers may use this article aslong as the work is properly cited, the use is educational and not for profit, and thework is not altered. More information is available at http://www.diabetesjournals.org/content/license.
Diabetes Volume 66, May 2017 1185
METABOLISM

of some cancers (15–17). In situ hybridization and North-ern blot analyses revealed that FAM3C was ubiquitouslyexpressed in all tissues (17). Cleaved FAM3C can be se-creted by cells, and the secreted FAM3C protein form isassociated with autophagy of tumors. Circulating FAM3Clevel is a biomarker for autophagy and some cancers(15,16,18). Secreted FAM3C protein is important formaintaining retina size and the function of the photo-receptor layer (19). Transgenic FAM3C overexpressionprotects mice against Alzheimer disease by destabilizingthe penultimate amyloid-b precursor (20). FAM3C alsoregulates osteogenic differentiation and bone metabo-lism (21). Whether and how FAM3C plays a role in theregulation of glucose and lipid metabolism remainsuncharacterized.
The current study revealed that FAM3C expression isdecreased in the livers of obese diabetic mice and thathepatic FAM3C restoration attenuates hyperglycemia, in-sulin resistance, and fatty liver. Mechanistically, FAM3Cupregulates HSF1 to directly induce CALM1 gene transcrip-tion, elevating calmodulin (CaM) protein level to activate thephosphatidyl inositol 3-kinase (PI3K)–Akt pathway and re-press gluconeogenic gene expression in an insulin- and Ca2+-independent manner in hepatocytes.
RESEARCH DESIGN AND METHODS
Experimental Animals and AntibodiesThe study used 8- to 10-week-old male C57BL/6 mice and8- to 10-week-old male db/db mice on a BKS background.The C57BL/6 mice were fed a 45% high-fat diet (HFD) ornormal diet (ND) (Medicience Ltd, Jiangsu, China) for12 weeks (6,7). All procedures involving experimental an-imals were approved by the Institutional Animal Care andUse Committee of Peking University Health Science Cen-ter, which complies with the Guide for the Care and Use ofLaboratory Animals.
Anti-FAM3C antibody was purchased from Abcam(Cambridge, U.K.). Anti-phosphorylated (p)Akt (phosphor-ylation at Ser473 or Thr308 site) and Akt antibodies werepurchased from Cell Signaling Technology, Inc. (Danvers,MA). If not indicated specifically, pAkt representedphosphorylation at Ser473 site. Other antibodies wereobtained from Santa Cruz Biotechnology, Inc. (Dallas,TX), Cell Signaling Technology, Inc., or other commercialcompanies.
Adenoviral Overexpression of FAM3C in Mouse LiversAdenovirus (Ad) expressing homo FAM3C was con-structed by SinoGenoMax Company (Beijing, China) usingthe homo FAM3C expression plasmid purchased fromOriGene (#RG201036). To overexpress FAM3C in thelivers of HFD and db/db mice, 1.0 3 109 plaque-formingunits of Ad-FAM3C or Ad-green fluorescent protein(GFP) were injected into mice via tail vein in 100 mLvolume (6,7). At the 4th and 7th day after virus injec-tion, oral glucose tolerance tests (OGTTs) were per-formed. On the 9th day, the mice were sacrificed for
experimental analysis. The serum was collected for mea-suring triglycerides (TG) and cholesterol (CHO) levels.The liver was collected for TG and CHO content measure-ment and other biochemical analyses.
OGTT, Insulin Tolerance Test, and Pyruvate ToleranceTest AssaysFor the OGTT, mice were fasted from 8 A.M. to 2 P.M. withfree access to drinking water in a cage with fresh bedding.Blood glucose was determined after 0 min, and mice wereorally administered glucose at the dose of 3 g/kg bodyweight. Blood glucose levels at 15, 30, 60, 90, and 120 minwere monitored. The pyruvate tolerance test (PTT) and in-sulin tolerance test (ITT) were performed on the 7th dayafter an injection of Ad-FAM3C or Ad-GFP, as detailed pre-viously (6,7). OGTT, ITT, and PTT were performed usingdifferent sets of mice.
Cell CultureHuman HepG2 cells were infected with 25 multiplicity ofinfection of Ad-GFP or Ad-FAM3C for 24 h. For insulinstimulation, infected cells were serum starved for 12 hand then stimulated with 10 nmol/L or 100 nmol/L insu-lin (Novo Nordisk) for 5 min. The cells were lysed for pro-tein analysis. Infected cells were treated with 50 mmol/LLY294002, 1 mmol/L wortmannin, 100 mmol/L chlorprom-azine (CPZ), and 100 mmol/L W-7 for 1 h before being lysedfor pAkt analysis. To knock down CALM1 expression, HepG2cells were transfected with the mixture of four sets of smallinterfering (si)RNAs against homo CALM1 mRNA (siRNAsequences are provided in Supplementary Table 1) for 6 h,and then the medium replaced with fresh DMEM/high-glucose medium. The cells were infected with Ad-GFPor Ad-FAM3C for 24 h. For depletion of extracellularCa2+, infected cells were treated with Ca2+-free DMEM/high-glucose medium plus 0.5 mmol/L EGTA in the ab-sence or presence of 100 mmol/L CPZ for 2 h before pAktanalysis.
Primary Mouse Hepatocyte CultureHepatocytes were isolated from mice by nonrecirculatingcollagenase perfusion through the portal vein, as pre-viously described (7). The isolated mouse hepatocyteswere plated on dishes coated with rat collagen type Iand cultured in RPMI 1640 containing 10% FBS at 37°Cin a 5% CO2 atmosphere.
Overexpression of CALM1 and HSF1 via PlasmidTransfection in HepG2 CellsHepG2 cells were plated in 6-well plates and transfectedwith 5 mg CALM1 or HSF1 plasmid using the ViaFecttransfection kit (Vigorous Biotechnology, Cat No T001).The transfected cells were incubated for 6 h, and then thetransfection medium was replaced with normal culturemedium. The cells were cultured for another 24 h, fol-lowed by treatment with CPZ or PI3K inhibitor for 1 hbefore being lysed for pAkt analysis. Plasmids expressinghomo CALM1 and HSF1 were purchased from OriGene(CALM1, Cat No SC115829; HSF1, Cat No RG200314).
1186 FAM3C-HSF1-CaM Attenuates Hyperglycemia Diabetes Volume 66, May 2017

siRNA-Mediated Silencing of FAM3C and HSF1in HepG2 CellsTo knock down FAM3C or HSF1 expression, HepG2 cellswere transfected with 50 nmol/L scrambled siRNA or50 nmol/L siRNAs mixtures against homo FAM3C mRNAor HSF1 (siRNA sequences are provided in SupplementaryTable 1). The expression levels of FAM3C, HSF1, CaM,and pAkt were analyzed 24 h after transfection.
Hydrodynamics-Based Plasmid Injection forOverexpressing HSF1 and CALM1 in C57BL/6Mouse LiversHydrodynamics-based transfection in animals by tailvein administration of naked plasmid DNA was de-tailed previously (22–24). Briefly, 8- to 10-week-old maleC57BL/6 mice were randomly divided into three groupsbased on OGTT, and 50 mg endotoxin-free pEGFP-C3,pHSF1, or pCALM1 dissolved in sterile saline (the volumeof saline was 10% the body weight) was injected into thetail vein in 7 s at room temperature. OGTTs were per-formed at 72 h after the plasmid injection. Mice weresacrificed 24 h later.
Real-Time PCR AssayTotal RNA of tissues and cells were extracted usingRNApure High-purity Total RNA Rapid Extraction Kit(BioTeke Corporation). The complementary DNA wassynthesized using RevertAid First Strand cDNA Synthe-sis Kit (Fermentas, K1622). Target gene mRNA levelwas normalized to that of b-actin in the same sample.Each sample was assayed in duplicate or triplicate in eachexperiment. Melting and amplification curves for eachPCR action were also analyzed for ensuring the specificityof the PCR amplification. The primer sequences for real-time PCR assays are listed in Supplementary Table 2.
ImmunoblottingCells, livers, and other tissues were lysed in fresh Rothlysis buffer on ice. The lysates were centrifuged at 4°C at12,000 rpm for 10 min, and the pellets were discarded.The protein concentration in the supernatant was deter-mined using bicinchoninic acid assay. Total proteins (80–120 mg) were separated by 10 or 12% SDS-PAGE and thentransferred to the Hybond-C Extra membrane (AmershamBiosciences). The Western blot assays were performed asdetailed previously (6,7).
Measurement of Intracellular Calcium LevelHepG2 cells seeded on coverslips were infected withAd-GFP or Ad-FAM3C for 24 h, loaded with 1 mmol/LFura-2–acetoxymethyl for 30 min, and then imaged underan Olympus IX71 fluorescence microscope. For depletionof extracellular calcium experiments, infected cells weretreated with calcium-free medium plus 0.5 mmol/L EGTAfor 2 h before the Fura-2–acetoxymethyl load. The emis-sion intensities under 340 nm and 380 nm illuminationwere recorded every 1 s, and the average ratio of theemission densities (F340-to-F380) in 300 s reflects thebasal intracellular free calcium level (6,7).
Glucose Production Assay in HepG2 CellsFor glucose production assay, cells were infected withAd-GFP or Ad-FAM3C for 24 h and then washed threetimes by PBS buffer solution. Cells were cultured in glucoseand phenol red-free DMEM medium, supplemented with20 mmol/L sodium lactate and 2 mmol/L sodium pyruvatefor 13 h. Insulin (100 nmol/L) was added to the cells foran additional 3 h. Glucose content in the medium wasdetermined using the Glucose Assay Kit (Sigma-Aldrich,GAGO-20) and normalized to total cellular protein content.
Chromatin ImmunoprecipitationThe protocol for chromatin immunoprecipitation (ChIP)experiments was detailed previously (25). HepG2 cellswere washed twice with PBS and cross-linked with 1%formaldehyde at room temperature for 10 min. The cellswere sonicated and centrifuged for 10 min at 4°C. Thesupernatants were immunocleared using 2 mg shearedsalmon sperm DNA, 20 mL preimmune serum, and pro-tein A-Sepharose for 2 h at 4°C or just diluted in dilu-tion buffer as input control. The sheared chromatin wasimmunoprecipitated with anti-HSF1 antibodies or IgGovernight at 4°C. The eluted immunoprecipitate and theinput control were digested with proteinase K. DNA wasextracted for PCR amplification with the specific primersspanning the putative HSF1 binding site in homo CALM1promoter (sense, 59-acaagcaagcaaagcccttt-39; antisense,59-agccagtaatgtgaacccca-39).
Cytosolic and Nuclear Distribution of FOXO1 ProteinCells were seeded on coverslips and infected with Ad-GFPor Ad-FAM3C as previously described (7). The cells wererinsed with PBS and then permeabilized with 0.2% TritonX-100/0.5% PBS for 10 min, followed by washing withPBS. The coverslips were blocked in 1% BSA for 30 minat 37°C and then incubated overnight with anti-FOXO1antibodies at 4°C. The coverslips were washed with PBS,followed by detecting with goat anti-rabbit Alexa Fluor594. After nuclear staining with DAPI, coverslips weremounted on glass slides using 70% glycerol in PBS.Mounted coverslips were imaged, and cells were visual-ized by fluorescence microscopy using a confocal laserscanning microscope.
Statistical AnalysisData are presented as mean 6 SEM. Statistical signifi-cance of differences between groups was analyzed by un-paired Student t test or by one-way ANOVA when morethan two groups were compared.
RESULTS
FAM3C mRNA and Protein Levels Were Reducedin the Livers of Diabetic MiceA previous study reported that FAM3C protein can besecreted after cleavage of the signal peptide (19). TwoFAM3C protein isoforms exist in cells; one is called thefull-length form and the other is called the secreted formwithout the signal peptide (20). Generally, two FAM3Cprotein isoforms (;26 and 22 kD) exist in most mouse
diabetes.diabetesjournals.org Chen and Associates 1187
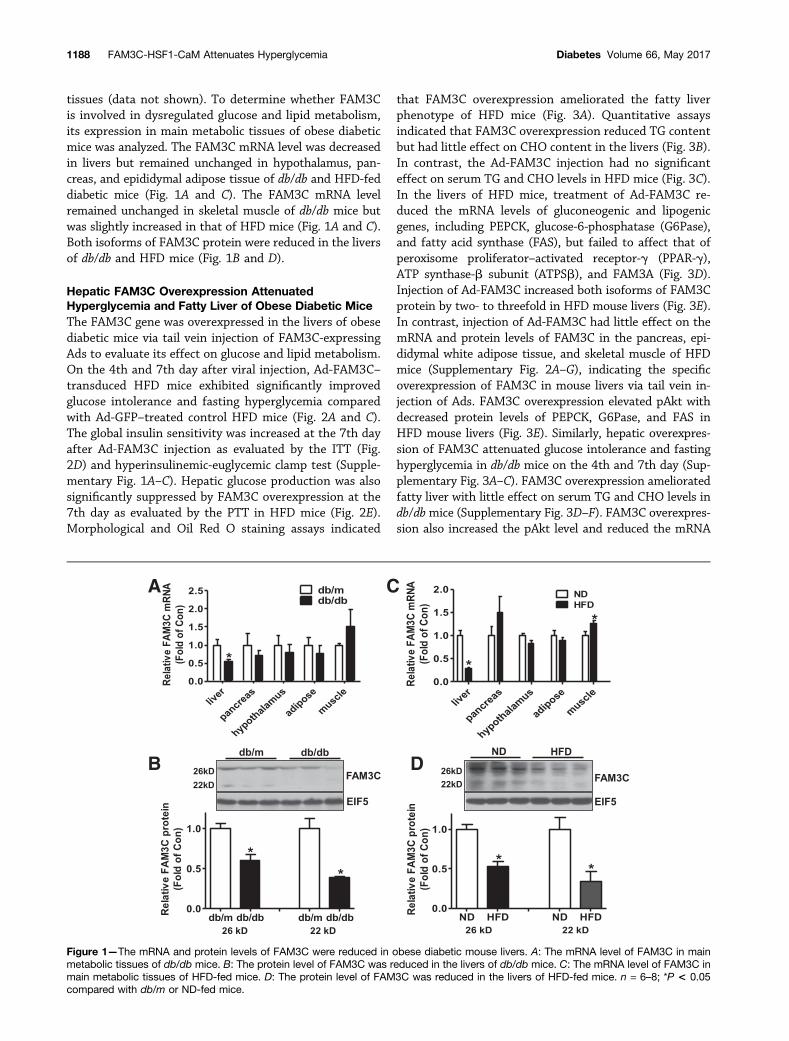
tissues (data not shown). To determine whether FAM3Cis involved in dysregulated glucose and lipid metabolism,its expression in main metabolic tissues of obese diabeticmice was analyzed. The FAM3C mRNA level was decreasedin livers but remained unchanged in hypothalamus, pan-creas, and epididymal adipose tissue of db/db and HFD-feddiabetic mice (Fig. 1A and C). The FAM3C mRNA levelremained unchanged in skeletal muscle of db/db mice butwas slightly increased in that of HFD mice (Fig. 1A and C).Both isoforms of FAM3C protein were reduced in the liversof db/db and HFD mice (Fig. 1B and D).
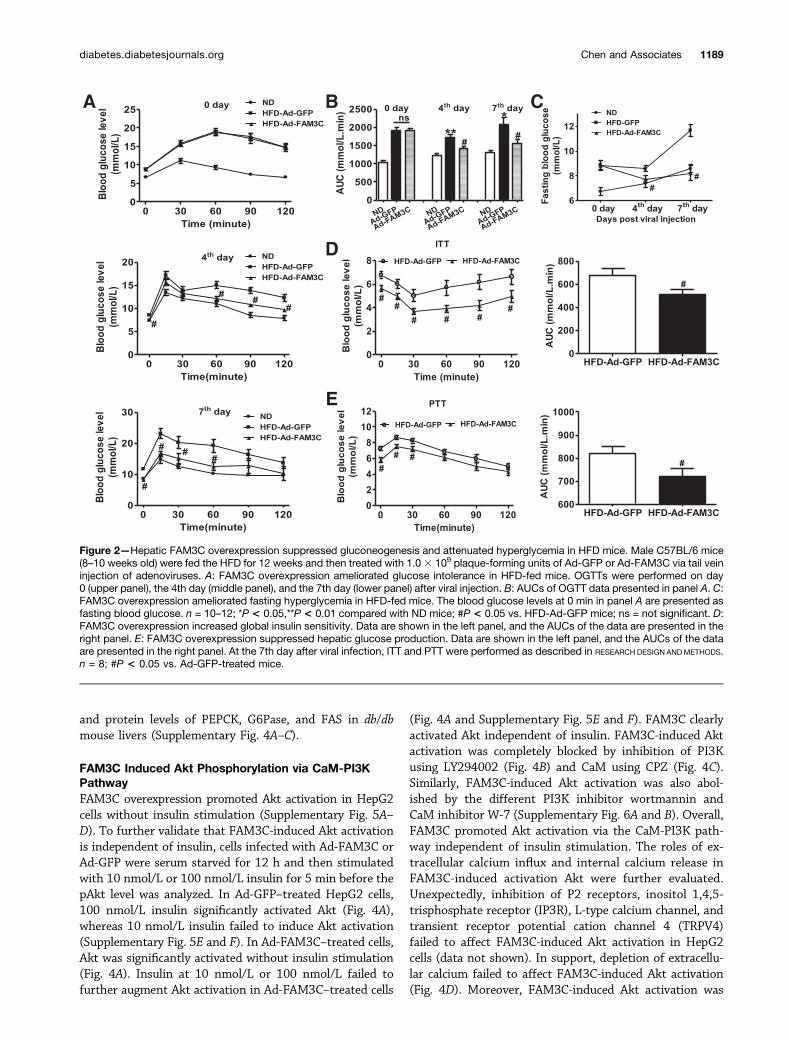
Hepatic FAM3C Overexpression AttenuatedHyperglycemia and Fatty Liver of Obese Diabetic MiceThe FAM3C gene was overexpressed in the livers of obesediabetic mice via tail vein injection of FAM3C-expressingAds to evaluate its effect on glucose and lipid metabolism.On the 4th and 7th day after viral injection, Ad-FAM3C–transduced HFD mice exhibited significantly improvedglucose intolerance and fasting hyperglycemia comparedwith Ad-GFP–treated control HFD mice (Fig. 2A and C).The global insulin sensitivity was increased at the 7th dayafter Ad-FAM3C injection as evaluated by the ITT (Fig.2D) and hyperinsulinemic-euglycemic clamp test (Supple-mentary Fig. 1A–C). Hepatic glucose production was alsosignificantly suppressed by FAM3C overexpression at the7th day as evaluated by the PTT in HFD mice (Fig. 2E).Morphological and Oil Red O staining assays indicated
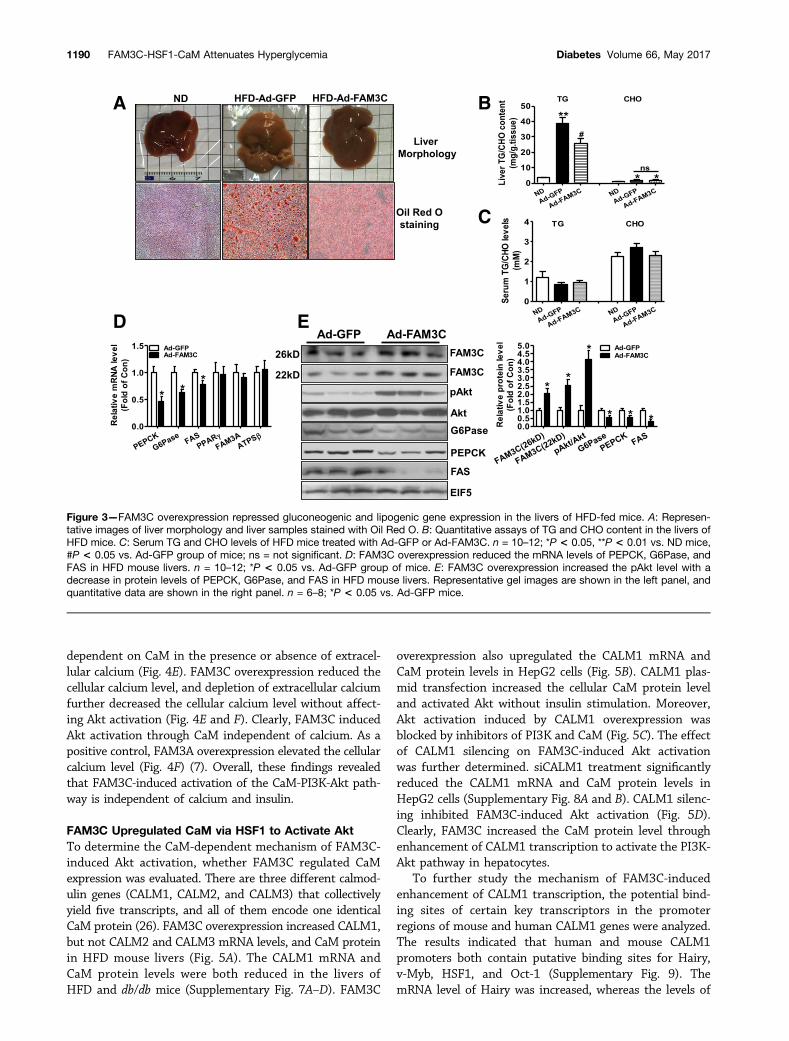
that FAM3C overexpression ameliorated the fatty liverphenotype of HFD mice (Fig. 3A). Quantitative assaysindicated that FAM3C overexpression reduced TG contentbut had little effect on CHO content in the livers (Fig. 3B).In contrast, the Ad-FAM3C injection had no significanteffect on serum TG and CHO levels in HFD mice (Fig. 3C).In the livers of HFD mice, treatment of Ad-FAM3C re-duced the mRNA levels of gluconeogenic and lipogenicgenes, including PEPCK, glucose-6-phosphatase (G6Pase),and fatty acid synthase (FAS), but failed to affect that ofperoxisome proliferator–activated receptor-g (PPAR-g),ATP synthase-b subunit (ATPSb), and FAM3A (Fig. 3D).Injection of Ad-FAM3C increased both isoforms of FAM3Cprotein by two- to threefold in HFD mouse livers (Fig. 3E).In contrast, injection of Ad-FAM3C had little effect on themRNA and protein levels of FAM3C in the pancreas, epi-didymal white adipose tissue, and skeletal muscle of HFDmice (Supplementary Fig. 2A–G), indicating the specificoverexpression of FAM3C in mouse livers via tail vein in-jection of Ads. FAM3C overexpression elevated pAkt withdecreased protein levels of PEPCK, G6Pase, and FAS inHFD mouse livers (Fig. 3E). Similarly, hepatic overexpres-sion of FAM3C attenuated glucose intolerance and fastinghyperglycemia in db/db mice on the 4th and 7th day (Sup-plementary Fig. 3A–C). FAM3C overexpression amelioratedfatty liver with little effect on serum TG and CHO levels indb/dbmice (Supplementary Fig. 3D–F). FAM3C overexpres-sion also increased the pAkt level and reduced the mRNA
Figure 1—The mRNA and protein levels of FAM3C were reduced in obese diabetic mouse livers. A: The mRNA level of FAM3C in mainmetabolic tissues of db/db mice. B: The protein level of FAM3C was reduced in the livers of db/db mice. C: The mRNA level of FAM3C inmain metabolic tissues of HFD-fed mice. D: The protein level of FAM3C was reduced in the livers of HFD-fed mice. n = 6–8; *P < 0.05compared with db/m or ND-fed mice.
1188 FAM3C-HSF1-CaM Attenuates Hyperglycemia Diabetes Volume 66, May 2017
and protein levels of PEPCK, G6Pase, and FAS in db/dbmouse livers (Supplementary Fig. 4A–C).
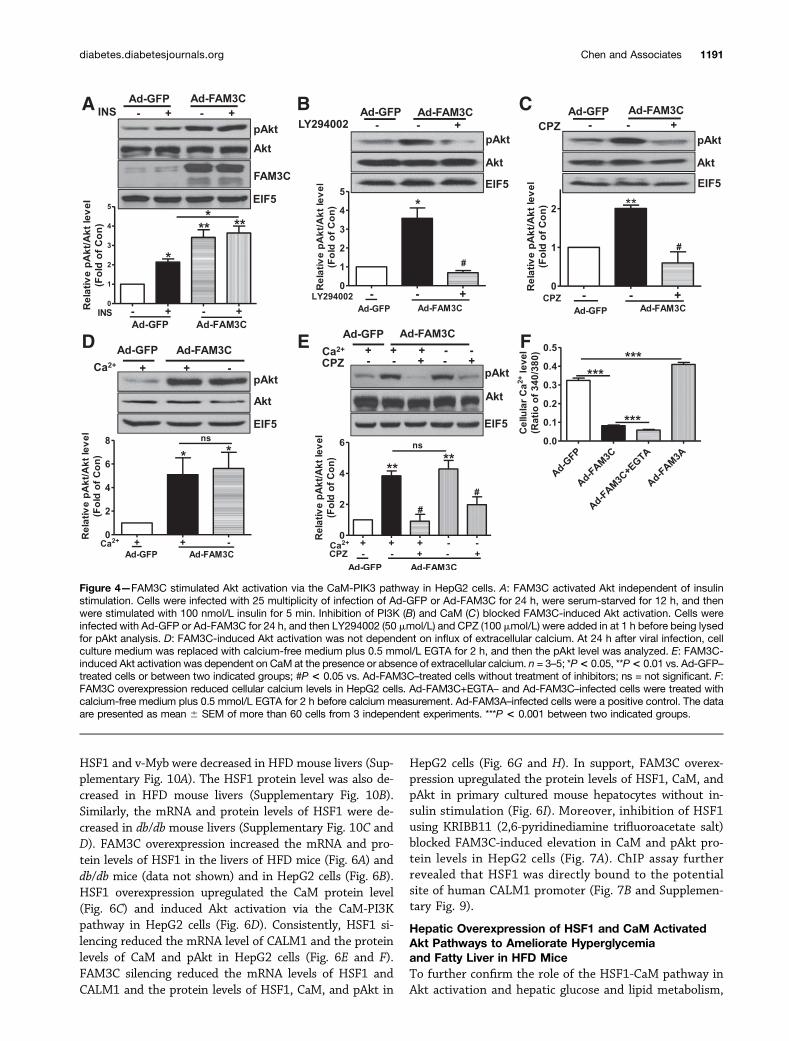
FAM3C Induced Akt Phosphorylation via CaM-PI3KPathwayFAM3C overexpression promoted Akt activation in HepG2cells without insulin stimulation (Supplementary Fig. 5A–D). To further validate that FAM3C-induced Akt activationis independent of insulin, cells infected with Ad-FAM3C orAd-GFP were serum starved for 12 h and then stimulatedwith 10 nmol/L or 100 nmol/L insulin for 5 min before thepAkt level was analyzed. In Ad-GFP–treated HepG2 cells,100 nmol/L insulin significantly activated Akt (Fig. 4A),whereas 10 nmol/L insulin failed to induce Akt activation(Supplementary Fig. 5E and F). In Ad-FAM3C–treated cells,Akt was significantly activated without insulin stimulation(Fig. 4A). Insulin at 10 nmol/L or 100 nmol/L failed tofurther augment Akt activation in Ad-FAM3C–treated cells
(Fig. 4A and Supplementary Fig. 5E and F). FAM3C clearlyactivated Akt independent of insulin. FAM3C-induced Aktactivation was completely blocked by inhibition of PI3Kusing LY294002 (Fig. 4B) and CaM using CPZ (Fig. 4C).Similarly, FAM3C-induced Akt activation was also abol-ished by the different PI3K inhibitor wortmannin andCaM inhibitor W-7 (Supplementary Fig. 6A and B). Overall,FAM3C promoted Akt activation via the CaM-PI3K path-way independent of insulin stimulation. The roles of ex-tracellular calcium influx and internal calcium release inFAM3C-induced activation Akt were further evaluated.Unexpectedly, inhibition of P2 receptors, inositol 1,4,5-trisphosphate receptor (IP3R), L-type calcium channel, andtransient receptor potential cation channel 4 (TRPV4)failed to affect FAM3C-induced Akt activation in HepG2cells (data not shown). In support, depletion of extracellu-lar calcium failed to affect FAM3C-induced Akt activation(Fig. 4D). Moreover, FAM3C-induced Akt activation was
Figure 2—Hepatic FAM3C overexpression suppressed gluconeogenesis and attenuated hyperglycemia in HFD mice. Male C57BL/6 mice(8–10 weeks old) were fed the HFD for 12 weeks and then treated with 1.03 109 plaque-forming units of Ad-GFP or Ad-FAM3C via tail veininjection of adenoviruses. A: FAM3C overexpression ameliorated glucose intolerance in HFD-fed mice. OGTTs were performed on day0 (upper panel), the 4th day (middle panel), and the 7th day (lower panel) after viral injection. B: AUCs of OGTT data presented in panel A. C:FAM3C overexpression ameliorated fasting hyperglycemia in HFD-fed mice. The blood glucose levels at 0 min in panel A are presented asfasting blood glucose. n = 10–12; *P < 0.05,**P < 0.01 compared with ND mice; #P < 0.05 vs. HFD-Ad-GFP mice; ns = not significant. D:FAM3C overexpression increased global insulin sensitivity. Data are shown in the left panel, and the AUCs of the data are presented in theright panel. E: FAM3C overexpression suppressed hepatic glucose production. Data are shown in the left panel, and the AUCs of the dataare presented in the right panel. At the 7th day after viral infection, ITT and PTT were performed as described in RESEARCH DESIGN ANDMETHODS.n = 8; #P < 0.05 vs. Ad-GFP-treated mice.
diabetes.diabetesjournals.org Chen and Associates 1189
dependent on CaM in the presence or absence of extracel-lular calcium (Fig. 4E). FAM3C overexpression reduced thecellular calcium level, and depletion of extracellular calciumfurther decreased the cellular calcium level without affect-ing Akt activation (Fig. 4E and F). Clearly, FAM3C inducedAkt activation through CaM independent of calcium. As apositive control, FAM3A overexpression elevated the cellularcalcium level (Fig. 4F) (7). Overall, these findings revealedthat FAM3C-induced activation of the CaM-PI3K-Akt path-way is independent of calcium and insulin.
FAM3C Upregulated CaM via HSF1 to Activate AktTo determine the CaM-dependent mechanism of FAM3C-induced Akt activation, whether FAM3C regulated CaMexpression was evaluated. There are three different calmod-ulin genes (CALM1, CALM2, and CALM3) that collectivelyyield five transcripts, and all of them encode one identicalCaM protein (26). FAM3C overexpression increased CALM1,but not CALM2 and CALM3 mRNA levels, and CaM proteinin HFD mouse livers (Fig. 5A). The CALM1 mRNA andCaM protein levels were both reduced in the livers ofHFD and db/db mice (Supplementary Fig. 7A–D). FAM3C
overexpression also upregulated the CALM1 mRNA andCaM protein levels in HepG2 cells (Fig. 5B). CALM1 plas-mid transfection increased the cellular CaM protein leveland activated Akt without insulin stimulation. Moreover,Akt activation induced by CALM1 overexpression wasblocked by inhibitors of PI3K and CaM (Fig. 5C). The effectof CALM1 silencing on FAM3C-induced Akt activationwas further determined. siCALM1 treatment significantlyreduced the CALM1 mRNA and CaM protein levels inHepG2 cells (Supplementary Fig. 8A and B). CALM1 silenc-ing inhibited FAM3C-induced Akt activation (Fig. 5D).Clearly, FAM3C increased the CaM protein level throughenhancement of CALM1 transcription to activate the PI3K-Akt pathway in hepatocytes.
To further study the mechanism of FAM3C-inducedenhancement of CALM1 transcription, the potential bind-ing sites of certain key transcriptors in the promoterregions of mouse and human CALM1 genes were analyzed.The results indicated that human and mouse CALM1promoters both contain putative binding sites for Hairy,v-Myb, HSF1, and Oct-1 (Supplementary Fig. 9). ThemRNA level of Hairy was increased, whereas the levels of
Figure 3—FAM3C overexpression repressed gluconeogenic and lipogenic gene expression in the livers of HFD-fed mice. A: Represen-tative images of liver morphology and liver samples stained with Oil Red O. B: Quantitative assays of TG and CHO content in the livers ofHFD mice. C: Serum TG and CHO levels of HFD mice treated with Ad-GFP or Ad-FAM3C. n = 10–12; *P < 0.05, **P < 0.01 vs. ND mice,#P < 0.05 vs. Ad-GFP group of mice; ns = not significant. D: FAM3C overexpression reduced the mRNA levels of PEPCK, G6Pase, andFAS in HFD mouse livers. n = 10–12; *P < 0.05 vs. Ad-GFP group of mice. E: FAM3C overexpression increased the pAkt level with adecrease in protein levels of PEPCK, G6Pase, and FAS in HFD mouse livers. Representative gel images are shown in the left panel, andquantitative data are shown in the right panel. n = 6–8; *P < 0.05 vs. Ad-GFP mice.
1190 FAM3C-HSF1-CaM Attenuates Hyperglycemia Diabetes Volume 66, May 2017
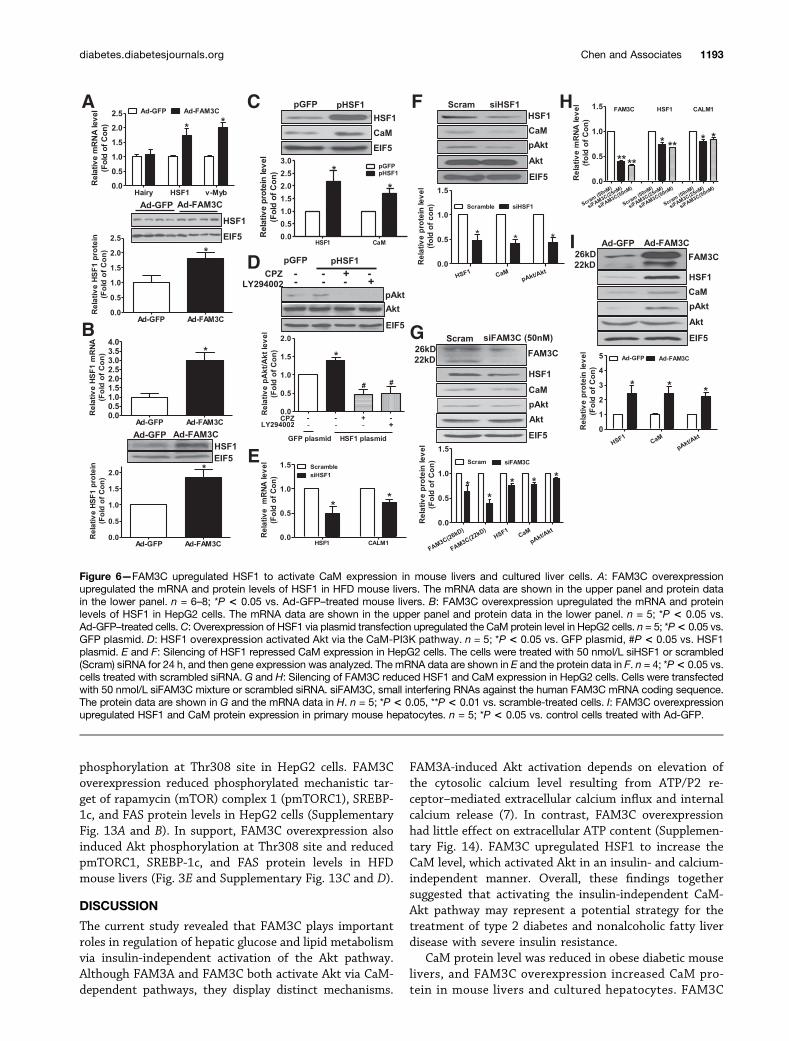
HSF1 and v-Myb were decreased in HFD mouse livers (Sup-plementary Fig. 10A). The HSF1 protein level was also de-creased in HFD mouse livers (Supplementary Fig. 10B).Similarly, the mRNA and protein levels of HSF1 were de-creased in db/db mouse livers (Supplementary Fig. 10C andD). FAM3C overexpression increased the mRNA and pro-tein levels of HSF1 in the livers of HFD mice (Fig. 6A) anddb/db mice (data not shown) and in HepG2 cells (Fig. 6B).HSF1 overexpression upregulated the CaM protein level(Fig. 6C) and induced Akt activation via the CaM-PI3Kpathway in HepG2 cells (Fig. 6D). Consistently, HSF1 si-lencing reduced the mRNA level of CALM1 and the proteinlevels of CaM and pAkt in HepG2 cells (Fig. 6E and F).FAM3C silencing reduced the mRNA levels of HSF1 andCALM1 and the protein levels of HSF1, CaM, and pAkt in
HepG2 cells (Fig. 6G and H). In support, FAM3C overex-pression upregulated the protein levels of HSF1, CaM, andpAkt in primary cultured mouse hepatocytes without in-sulin stimulation (Fig. 6I). Moreover, inhibition of HSF1using KRIBB11 (2,6-pyridinediamine trifluoroacetate salt)blocked FAM3C-induced elevation in CaM and pAkt pro-tein levels in HepG2 cells (Fig. 7A). ChIP assay furtherrevealed that HSF1 was directly bound to the potentialsite of human CALM1 promoter (Fig. 7B and Supplemen-tary Fig. 9).
Hepatic Overexpression of HSF1 and CaM ActivatedAkt Pathways to Ameliorate Hyperglycemiaand Fatty Liver in HFD MiceTo further confirm the role of the HSF1-CaM pathway inAkt activation and hepatic glucose and lipid metabolism,
Figure 4—FAM3C stimulated Akt activation via the CaM-PIK3 pathway in HepG2 cells. A: FAM3C activated Akt independent of insulinstimulation. Cells were infected with 25 multiplicity of infection of Ad-GFP or Ad-FAM3C for 24 h, were serum-starved for 12 h, and thenwere stimulated with 100 nmol/L insulin for 5 min. Inhibition of PI3K (B) and CaM (C ) blocked FAM3C-induced Akt activation. Cells wereinfected with Ad-GFP or Ad-FAM3C for 24 h, and then LY294002 (50 mmol/L) and CPZ (100 mmol/L) were added in at 1 h before being lysedfor pAkt analysis. D: FAM3C-induced Akt activation was not dependent on influx of extracellular calcium. At 24 h after viral infection, cellculture medium was replaced with calcium-free medium plus 0.5 mmol/L EGTA for 2 h, and then the pAkt level was analyzed. E: FAM3C-induced Akt activation was dependent on CaM at the presence or absence of extracellular calcium. n = 3–5; *P< 0.05, **P< 0.01 vs. Ad-GFP–treated cells or between two indicated groups; #P < 0.05 vs. Ad-FAM3C–treated cells without treatment of inhibitors; ns = not significant. F:FAM3C overexpression reduced cellular calcium levels in HepG2 cells. Ad-FAM3C+EGTA– and Ad-FAM3C–infected cells were treated withcalcium-free medium plus 0.5 mmol/L EGTA for 2 h before calcium measurement. Ad-FAM3A–infected cells were a positive control. The dataare presented as mean 6 SEM of more than 60 cells from 3 independent experiments. ***P < 0.001 between two indicated groups.
diabetes.diabetesjournals.org Chen and Associates 1191
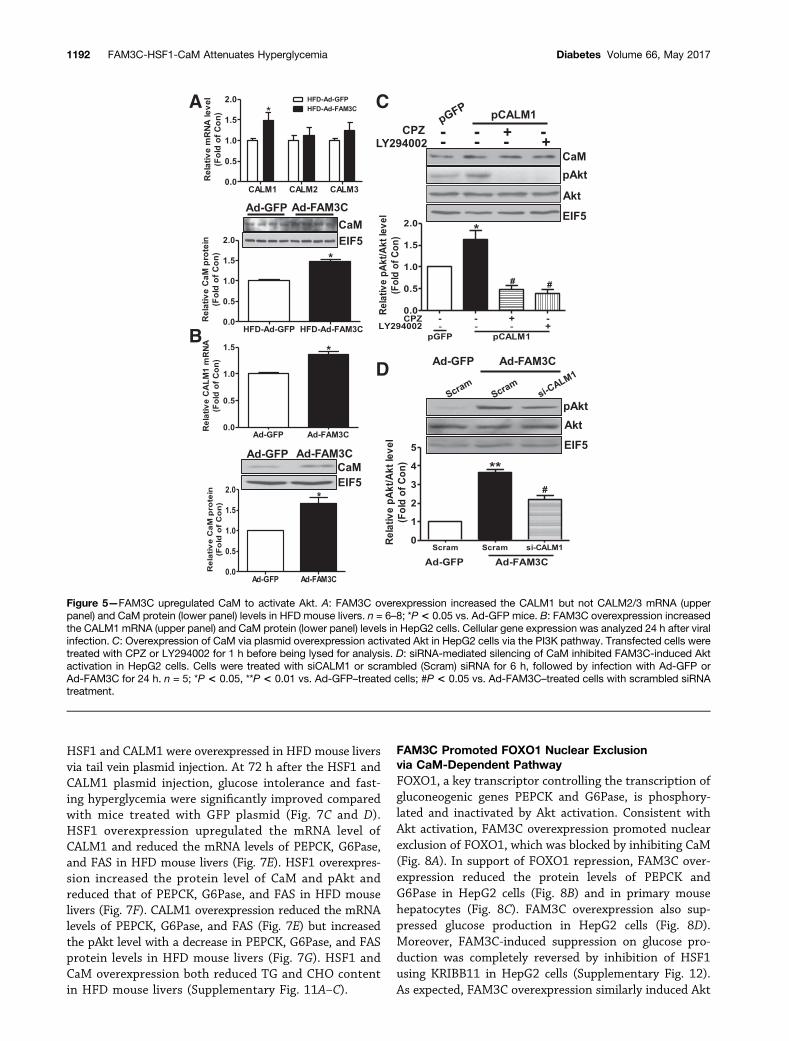
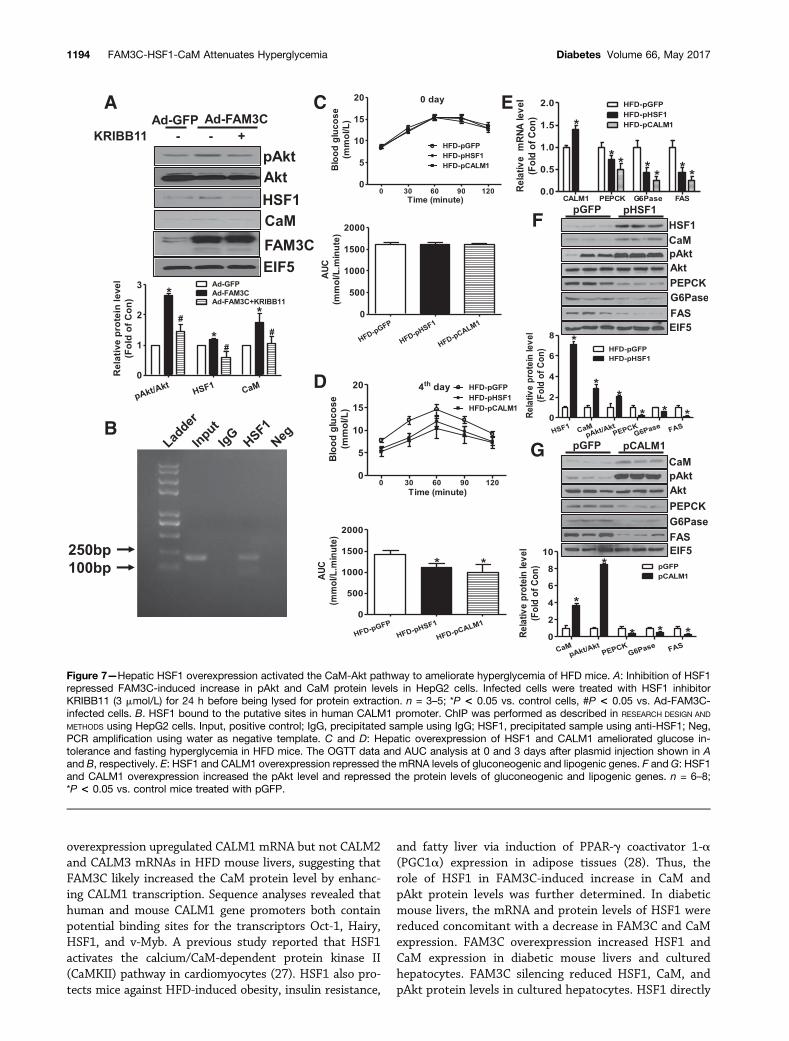
HSF1 and CALM1 were overexpressed in HFD mouse liversvia tail vein plasmid injection. At 72 h after the HSF1 andCALM1 plasmid injection, glucose intolerance and fast-ing hyperglycemia were significantly improved comparedwith mice treated with GFP plasmid (Fig. 7C and D).HSF1 overexpression upregulated the mRNA level ofCALM1 and reduced the mRNA levels of PEPCK, G6Pase,and FAS in HFD mouse livers (Fig. 7E). HSF1 overexpres-sion increased the protein level of CaM and pAkt andreduced that of PEPCK, G6Pase, and FAS in HFD mouselivers (Fig. 7F). CALM1 overexpression reduced the mRNAlevels of PEPCK, G6Pase, and FAS (Fig. 7E) but increasedthe pAkt level with a decrease in PEPCK, G6Pase, and FASprotein levels in HFD mouse livers (Fig. 7G). HSF1 andCaM overexpression both reduced TG and CHO contentin HFD mouse livers (Supplementary Fig. 11A–C).
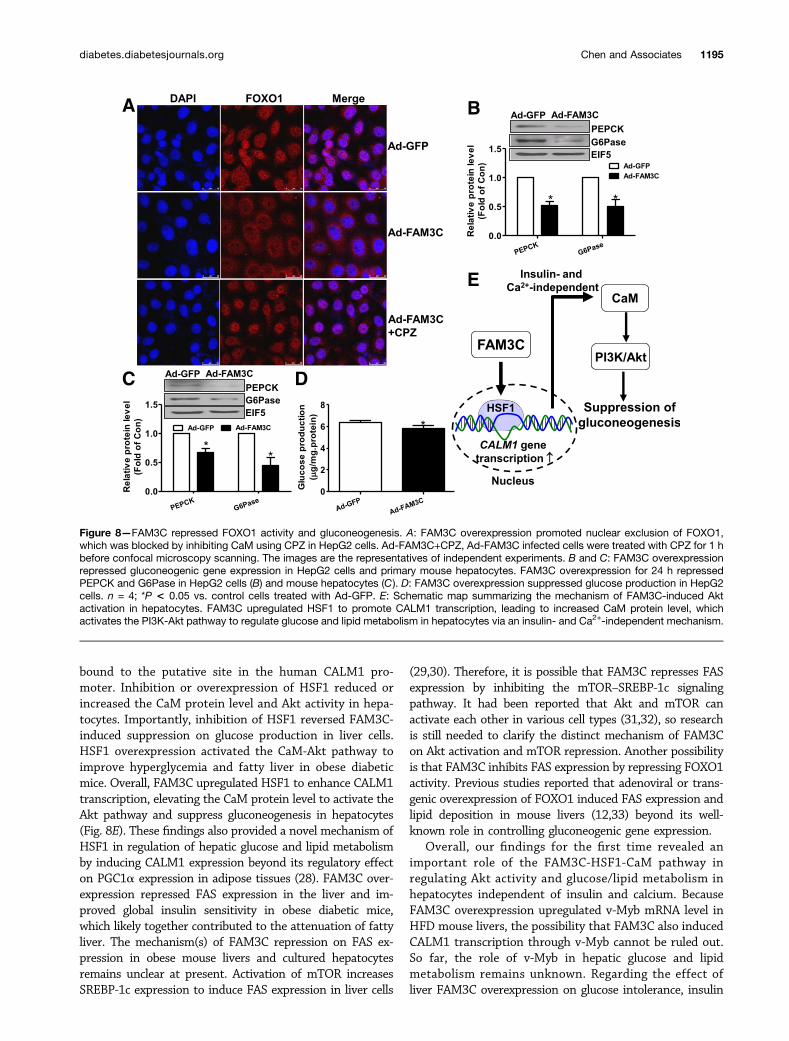
FAM3C Promoted FOXO1 Nuclear Exclusionvia CaM-Dependent PathwayFOXO1, a key transcriptor controlling the transcription ofgluconeogenic genes PEPCK and G6Pase, is phosphory-lated and inactivated by Akt activation. Consistent withAkt activation, FAM3C overexpression promoted nuclearexclusion of FOXO1, which was blocked by inhibiting CaM(Fig. 8A). In support of FOXO1 repression, FAM3C over-expression reduced the protein levels of PEPCK andG6Pase in HepG2 cells (Fig. 8B) and in primary mousehepatocytes (Fig. 8C). FAM3C overexpression also sup-pressed glucose production in HepG2 cells (Fig. 8D).Moreover, FAM3C-induced suppression on glucose pro-duction was completely reversed by inhibition of HSF1using KRIBB11 in HepG2 cells (Supplementary Fig. 12).As expected, FAM3C overexpression similarly induced Akt
Figure 5—FAM3C upregulated CaM to activate Akt. A: FAM3C overexpression increased the CALM1 but not CALM2/3 mRNA (upperpanel) and CaM protein (lower panel) levels in HFD mouse livers. n = 6–8; *P < 0.05 vs. Ad-GFP mice. B: FAM3C overexpression increasedthe CALM1 mRNA (upper panel) and CaM protein (lower panel) levels in HepG2 cells. Cellular gene expression was analyzed 24 h after viralinfection. C: Overexpression of CaM via plasmid overexpression activated Akt in HepG2 cells via the PI3K pathway. Transfected cells weretreated with CPZ or LY294002 for 1 h before being lysed for analysis. D: siRNA-mediated silencing of CaM inhibited FAM3C-induced Aktactivation in HepG2 cells. Cells were treated with siCALM1 or scrambled (Scram) siRNA for 6 h, followed by infection with Ad-GFP orAd-FAM3C for 24 h. n = 5; *P < 0.05, **P < 0.01 vs. Ad-GFP–treated cells; #P < 0.05 vs. Ad-FAM3C–treated cells with scrambled siRNAtreatment.
1192 FAM3C-HSF1-CaM Attenuates Hyperglycemia Diabetes Volume 66, May 2017
phosphorylation at Thr308 site in HepG2 cells. FAM3Coverexpression reduced phosphorylated mechanistic tar-get of rapamycin (mTOR) complex 1 (pmTORC1), SREBP-1c, and FAS protein levels in HepG2 cells (SupplementaryFig. 13A and B). In support, FAM3C overexpression alsoinduced Akt phosphorylation at Thr308 site and reducedpmTORC1, SREBP-1c, and FAS protein levels in HFDmouse livers (Fig. 3E and Supplementary Fig. 13C and D).
DISCUSSION
The current study revealed that FAM3C plays importantroles in regulation of hepatic glucose and lipid metabolismvia insulin-independent activation of the Akt pathway.Although FAM3A and FAM3C both activate Akt via CaM-dependent pathways, they display distinct mechanisms.
FAM3A-induced Akt activation depends on elevation ofthe cytosolic calcium level resulting from ATP/P2 re-ceptor–mediated extracellular calcium influx and internalcalcium release (7). In contrast, FAM3C overexpressionhad little effect on extracellular ATP content (Supplemen-tary Fig. 14). FAM3C upregulated HSF1 to increase theCaM level, which activated Akt in an insulin- and calcium-independent manner. Overall, these findings togethersuggested that activating the insulin-independent CaM-Akt pathway may represent a potential strategy for thetreatment of type 2 diabetes and nonalcoholic fatty liverdisease with severe insulin resistance.
CaM protein level was reduced in obese diabetic mouselivers, and FAM3C overexpression increased CaM pro-tein in mouse livers and cultured hepatocytes. FAM3C
Figure 6—FAM3C upregulated HSF1 to activate CaM expression in mouse livers and cultured liver cells. A: FAM3C overexpressionupregulated the mRNA and protein levels of HSF1 in HFD mouse livers. The mRNA data are shown in the upper panel and protein datain the lower panel. n = 6–8; *P < 0.05 vs. Ad-GFP–treated mouse livers. B: FAM3C overexpression upregulated the mRNA and proteinlevels of HSF1 in HepG2 cells. The mRNA data are shown in the upper panel and protein data in the lower panel. n = 5; *P < 0.05 vs.Ad-GFP–treated cells. C: Overexpression of HSF1 via plasmid transfection upregulated the CaM protein level in HepG2 cells. n = 5; *P< 0.05 vs.GFP plasmid. D: HSF1 overexpression activated Akt via the CaM-PI3K pathway. n = 5; *P < 0.05 vs. GFP plasmid, #P < 0.05 vs. HSF1plasmid. E and F: Silencing of HSF1 repressed CaM expression in HepG2 cells. The cells were treated with 50 nmol/L siHSF1 or scrambled(Scram) siRNA for 24 h, and then gene expression was analyzed. The mRNA data are shown in E and the protein data in F. n = 4; *P< 0.05 vs.cells treated with scrambled siRNA. G and H: Silencing of FAM3C reduced HSF1 and CaM expression in HepG2 cells. Cells were transfectedwith 50 nmol/L siFAM3C mixture or scrambled siRNA. siFAM3C, small interfering RNAs against the human FAM3C mRNA coding sequence.The protein data are shown in G and the mRNA data in H. n = 5; *P < 0.05, **P < 0.01 vs. scramble-treated cells. I: FAM3C overexpressionupregulated HSF1 and CaM protein expression in primary mouse hepatocytes. n = 5; *P < 0.05 vs. control cells treated with Ad-GFP.
diabetes.diabetesjournals.org Chen and Associates 1193
overexpression upregulated CALM1 mRNA but not CALM2and CALM3 mRNAs in HFD mouse livers, suggesting thatFAM3C likely increased the CaM protein level by enhanc-ing CALM1 transcription. Sequence analyses revealed thathuman and mouse CALM1 gene promoters both containpotential binding sites for the transcriptors Oct-1, Hairy,HSF1, and v-Myb. A previous study reported that HSF1activates the calcium/CaM-dependent protein kinase II(CaMKII) pathway in cardiomyocytes (27). HSF1 also pro-tects mice against HFD-induced obesity, insulin resistance,
and fatty liver via induction of PPAR-g coactivator 1-a(PGC1a) expression in adipose tissues (28). Thus, therole of HSF1 in FAM3C-induced increase in CaM andpAkt protein levels was further determined. In diabeticmouse livers, the mRNA and protein levels of HSF1 werereduced concomitant with a decrease in FAM3C and CaMexpression. FAM3C overexpression increased HSF1 andCaM expression in diabetic mouse livers and culturedhepatocytes. FAM3C silencing reduced HSF1, CaM, andpAkt protein levels in cultured hepatocytes. HSF1 directly
Figure 7—Hepatic HSF1 overexpression activated the CaM-Akt pathway to ameliorate hyperglycemia of HFD mice. A: Inhibition of HSF1repressed FAM3C-induced increase in pAkt and CaM protein levels in HepG2 cells. Infected cells were treated with HSF1 inhibitorKRIBB11 (3 mmol/L) for 24 h before being lysed for protein extraction. n = 3–5; *P < 0.05 vs. control cells, #P < 0.05 vs. Ad-FAM3C-infected cells. B. HSF1 bound to the putative sites in human CALM1 promoter. ChIP was performed as described in RESEARCH DESIGN AND
METHODS using HepG2 cells. Input, positive control; IgG, precipitated sample using IgG; HSF1, precipitated sample using anti-HSF1; Neg,PCR amplification using water as negative template. C and D: Hepatic overexpression of HSF1 and CALM1 ameliorated glucose in-tolerance and fasting hyperglycemia in HFD mice. The OGTT data and AUC analysis at 0 and 3 days after plasmid injection shown in Aand B, respectively. E: HSF1 and CALM1 overexpression repressed the mRNA levels of gluconeogenic and lipogenic genes. F and G: HSF1and CALM1 overexpression increased the pAkt level and repressed the protein levels of gluconeogenic and lipogenic genes. n = 6–8;*P < 0.05 vs. control mice treated with pGFP.
1194 FAM3C-HSF1-CaM Attenuates Hyperglycemia Diabetes Volume 66, May 2017
bound to the putative site in the human CALM1 pro-moter. Inhibition or overexpression of HSF1 reduced orincreased the CaM protein level and Akt activity in hepa-tocytes. Importantly, inhibition of HSF1 reversed FAM3C-induced suppression on glucose production in liver cells.HSF1 overexpression activated the CaM-Akt pathway toimprove hyperglycemia and fatty liver in obese diabeticmice. Overall, FAM3C upregulated HSF1 to enhance CALM1transcription, elevating the CaM protein level to activate theAkt pathway and suppress gluconeogenesis in hepatocytes(Fig. 8E). These findings also provided a novel mechanism ofHSF1 in regulation of hepatic glucose and lipid metabolismby inducing CALM1 expression beyond its regulatory effecton PGC1a expression in adipose tissues (28). FAM3C over-expression repressed FAS expression in the liver and im-proved global insulin sensitivity in obese diabetic mice,which likely together contributed to the attenuation of fattyliver. The mechanism(s) of FAM3C repression on FAS ex-pression in obese mouse livers and cultured hepatocytesremains unclear at present. Activation of mTOR increasesSREBP-1c expression to induce FAS expression in liver cells
(29,30). Therefore, it is possible that FAM3C represses FASexpression by inhibiting the mTOR–SREBP-1c signalingpathway. It had been reported that Akt and mTOR canactivate each other in various cell types (31,32), so researchis still needed to clarify the distinct mechanism of FAM3Con Akt activation and mTOR repression. Another possibilityis that FAM3C inhibits FAS expression by repressing FOXO1activity. Previous studies reported that adenoviral or trans-genic overexpression of FOXO1 induced FAS expression andlipid deposition in mouse livers (12,33) beyond its well-known role in controlling gluconeogenic gene expression.
Overall, our findings for the first time revealed animportant role of the FAM3C-HSF1-CaM pathway inregulating Akt activity and glucose/lipid metabolism inhepatocytes independent of insulin and calcium. BecauseFAM3C overexpression upregulated v-Myb mRNA level inHFD mouse livers, the possibility that FAM3C also inducedCALM1 transcription through v-Myb cannot be ruled out.So far, the role of v-Myb in hepatic glucose and lipidmetabolism remains unknown. Regarding the effect ofliver FAM3C overexpression on glucose intolerance, insulin
Figure 8—FAM3C repressed FOXO1 activity and gluconeogenesis. A: FAM3C overexpression promoted nuclear exclusion of FOXO1,which was blocked by inhibiting CaM using CPZ in HepG2 cells. Ad-FAM3C+CPZ, Ad-FAM3C infected cells were treated with CPZ for 1 hbefore confocal microscopy scanning. The images are the representatives of independent experiments. B and C: FAM3C overexpressionrepressed gluconeogenic gene expression in HepG2 cells and primary mouse hepatocytes. FAM3C overexpression for 24 h repressedPEPCK and G6Pase in HepG2 cells (B) and mouse hepatocytes (C). D: FAM3C overexpression suppressed glucose production in HepG2cells. n = 4; *P < 0.05 vs. control cells treated with Ad-GFP. E: Schematic map summarizing the mechanism of FAM3C-induced Aktactivation in hepatocytes. FAM3C upregulated HSF1 to promote CALM1 transcription, leading to increased CaM protein level, whichactivates the PI3K-Akt pathway to regulate glucose and lipid metabolism in hepatocytes via an insulin- and Ca2+-independent mechanism.
diabetes.diabetesjournals.org Chen and Associates 1195

resistance, and hepatic glucose production, it should also benoted that although Ad-FAM3C–treated mice had a lowerfasting blood glucose level, they exhibited comparable re-sponse to glucose, insulin, and pyruvate during OGTT,ITT, and PTT compared with Ad-GFP–treated mice. Thechanges of areas under the curve (AUCs) observed inthese tests may have been the result of the lower initialblood glucose level in FAM3C-overexpressing mice com-pared with control mice.
As reviewed by Berchtold and Villalobo (34), CaM gen-erally is bound and activated by increased cellular freecalcium to interact with target proteins and exert differ-ent functions. CaM also interacts with target proteins viaCa2+-independent mechanisms. CaM was previously re-ported to interact with insulin receptor substrate (IRS)1/2, PI3K, and Akt (35–37). The interaction betweenCaM and IRS proteins in rat soleus muscle is enhancedin insulin-resistant status induced by dexamethasone, andoverexpression of CaM in Chinese hamster ovary cellsinduces insulin resistance (35). Activation of CaMKII in-duces insulin resistance and Akt inhibition in the obesecondition (38); however, CaM also mediates Akt activa-tion in mouse mammary carcinoma cells (37). Silencing ofCaMKII inhibits H2O2-induced Akt activation in vascularsmooth muscle cells (39). Muscle-specific activation ofCaMKIV increases global insulin sensitivity in mice (40).FAM3A or ATPSb activates the CaM-PI3K-Akt pathway tosuppress gluconeogenic and lipogenic gene expression inliver cells (6,7). The current study further revealed thatFAM3C upregulated HSF1 to induce CALM1 transcrip-tion. An elevation in CaM protein level suppressed hepaticglucose production, improved insulin resistance and hy-perglycemia, and attenuated fatty liver in type 2 diabeticmice. FAM3C overexpression decreased cellular calcium inHepG2 cells. Depletion of extracellular calcium furtherreduced cellular calcium but failed to affect FAM3C-induced Akt activation in liver cells. Clearly, FAM3C acti-vates the CaM-Akt pathway independent of calcium. Insupport, direct CaM overexpression activated Akt to sup-press gluconeogenic and lipogenic gene expression inobese diabetic mouse livers. The FAM3C-HSF1-CaM-Aktsignaling axis represents a novel model of CaM actionindependent of calcium. Restoring hepatic FAM3C expres-sion to activate the HSF1-CaM-Akt pathway is beneficialfor type 2 diabetes with severe insulin resistance.
Regarding the role of FAM3C in promoting Akt acti-vation, several issues should be noted. Akt also playsimportant roles in cell proliferation and some cancers(41). The adverse effects of some anticancer drugs inhibitingAkt activity include global insulin resistance and increasedhepatic gluconeogenesis (42). Although mesenchymal trans-differentiation induced by transforming growth factor-b(TGF-b) is associated with upregulated Dab2 and FAM3Cexpression, the corelationship between FAM3C expres-sion and Akt activation remains unrevealed in the pre-vious studies (43,44). FAM3C repression by factors suchas fatty acids contributes to dysregulated glucose and lipid
metabolism, but long-term FAM3C overactivation by TGF-bmay trigger tumorigenesis in various cell types (15,43,44).It had been believed that dysregulated glucose metabolismis involved in various cancers. Gluconeogenesis is reducedin rat liver with Walker-256 tumor (45). Dexamethasoneattenuates hepatocellular carcinoma in mice with restoredgluconeogenesis in malignant hepatocytes (46). However,upregulation of PEPCK expression also promotes thegrowth of colon cancer (47). Metformin prevents or reducesthe risk of hepatocellular carcinoma in humans and animalswith diabetes with the suppression of hepatic gluconeogen-esis (48,49). Metformin also increases the pAkt level in di-abetic animal livers (50). FAM3C may represent a uniquemolecule determining the balance between dysregulated glu-coneogenesis and tumorigenesis. The upstream signals suchas TGF-b may play critical roles in determining whetherFAM3C promotes tumorigenesis. Actually, transgenic over-expression of FAM3C reduces brain amyloid-b depositionand ameliorates the memory deficit without triggeringtumorigenesis in a mouse Alzheimer disease model (20).
In summary, we presented novel data revealing thatthe insulin- and calcium-independent FAM3C-HSF1-CaM-Akt pathway plays important roles in regulating glucoseand lipid metabolism in the liver (Fig. 8E). Under obeseconditions, a decrease in hepatic FAM3C expression willlead to the inhibition of the HSF1-CaM-Akt pathway andcontribute to dysregulated glucose and lipid metabolism.Restoring the hepatic FAM3C-HSF1-CaM-Akt pathway isbeneficial for correcting dysregulated glucose and lipidmetabolism under severe insulin resistance.
Funding. This study was supported by grants from the Ministry of Scienceand Technology (2016YFC1304800) and the Natural Science Foundation of China(81471035/81670748/91339106/81322011/81422006/81390351).Duality of Interest. No potential conflicts of interest relevant to this articlewere reported.Author Contributions. Z.C. and L.D. wrote the manuscript. Z.C., L.D.,W.Y., and J.W. researched data and contributed to the discussion. Z.C., L.D.,Y.G., and J.Y. designed the study and revised and edited the manuscript. L.C.,Y.C., B.G., and Q.C. contributed to the discussion and reviewed and edited themanuscript. J.Y. is the guarantor of this work and, as such, had full access to allthe data in the study and takes responsibility for the integrity of the data and theaccuracy of the data analysis.
References1. Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE.Global estimates of diabetes prevalence for 2013 and projections for 2035. Di-abetes Res Clin Pract 2014;103:137–1492. Biddinger SB, Kahn CR. From mice to men: insights into the insulin re-sistance syndromes. Annu Rev Physiol 2006;68:123–1583. Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; regula-tion by AKT and 14-3-3 proteins. Biochim Biophys Acta 2011;1813:1938–19454. Matsuda S, Nakanishi A, Wada Y, Kitagishi Y. Roles of PI3K/AKT/PTENpathway as a target for pharmaceutical therapy. Open Med Chem J 2013;7:23–295. Sparks JD, Dong HH. FoxO1 and hepatic lipid metabolism. Curr Opin Lipidol2009;20:217–2266. Wang C, Chen Z, Li S, et al. Hepatic overexpression of ATP synthase b
subunit activates PI3K/Akt pathway to ameliorate hyperglycemia of diabetic mice.Diabetes 2014;63:947–959
1196 FAM3C-HSF1-CaM Attenuates Hyperglycemia Diabetes Volume 66, May 2017

7. Wang C, Chi Y, Li J, et al. FAM3A activates PI3K p110a/Akt signaling toameliorate hepatic gluconeogenesis and lipogenesis. Hepatology 2014;59:1779–17908. Zhu Y, Xu G, Patel A, et al. Cloning, expression, and initial characterizationof a novel cytokine-like gene family. Genomics 2002;80:144–1509. Cao X, Gao Z, Robert CE, et al. Pancreatic-derived factor (FAM3B), a novelislet cytokine, induces apoptosis of insulin-secreting beta-cells. Diabetes 2003;52:2296–230310. Yang J, Gao Z, Robert CE, et al. Structure-function studies of PANDER, anislet specific cytokine inducing cell death of insulin-secreting beta cells. Bio-chemistry 2005;44:11342–1135211. Yang J, Wang C, Li J, et al. PANDER binds to the liver cell membrane andinhibits insulin signaling in HepG2 cells. FEBS Lett 2009;583:3009–301512. Li J, Chi Y, Wang C, et al. Pancreatic-derived factor promotes lipogenesis inthe mouse liver: role of the Forkhead box 1 signaling pathway. Hepatology 2011;53:1906–191613. Moak SL, Dougan GC, MarElia CB, et al. Enhanced glucose tolerance inpancreatic-derived factor (PANDER) knockout C57BL/6 mice. Dis Model Mech2014;7:1307–131514. Cao X, Yang C, Lai F, et al. Elevated circulating level of a cytokine,pancreatic-derived factor, is associated with metabolic syndrome componentsin a Chinese population. J Diabetes Investig 2015;2015:581–58615. Waerner T, Alacakaptan M, Tamir I, et al. ILEI: a cytokine essential for EMT,tumor formation, and late events in metastasis in epithelial cells. Cancer Cell2006;10:227–23916. Grønborg M, Kristiansen TZ, Iwahori A, et al. Biomarker discovery frompancreatic cancer secretome using a differential proteomic approach. Mol CellProteomics 2006;5:157–17117. Pilipenko VV, Reece A, Choo DI, Greinwald JH Jr. Genomic organization andexpression analysis of the murine Fam3c gene. Gene 2004;335:159–16818. Kraya AA, Piao S, Xu X, et al. Identification of secreted proteins that reflectautophagy dynamics within tumor cells. Autophagy 2015;11:60–7419. Katahira T, Nakagiri S, Terada K, Furukawa T. Secreted factor FAM3C (ILEI)is involved in retinal laminar formation. Biochem Biophys Res Commun 2010;392:301–30620. Hasegawa H, Liu L, Tooyama I, Murayama S, Nishimura M. The FAM3superfamily member ILEI ameliorates Alzheimer’s disease-like pathology bydestabilizing the penultimate amyloid-b precursor. Nat Commun 2014;5:391721. Määttä JA, Bendre A, Laanti M, et al. Fam3c modulates osteogenic celldifferentiation and affects bone volume and cortical bone mineral density.Bonekey Rep 2016;5:78722. Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals bysystemic administration of plasmid DNA. Gene Ther 1999;6:1258–126623. Tsai WC, Hsu SD, Hsu CS, et al. MicroRNA-122 plays a critical role in liverhomeostasis and hepatocarcinogenesis. J Clin Invest 2012;122:2884–289724. Yang J, Chen S, Huang L, Michalopoulos GK, Liu Y. Sustained expression ofnaked plasmid DNA encoding hepatocyte growth factor in mice promotes liverand overall body growth. Hepatology 2001;33:848–85925. Zhou Y, Jia S, Wang C, et al. FAM3A is a target gene of peroxisome proliferator-activated receptor gamma. Biochim Biophys Acta 2013;1830:4160–417026. Nojima H. Structural organization of multiple rat calmodulin genes. J MolBiol 1989;208:269–28227. Peng W, Zhang Y, Zheng M, et al. Cardioprotection by CaMKII-deltaB ismediated by phosphorylation of heat shock factor 1 and subsequent expressionof inducible heat shock protein 70. Circ Res 2010;106:102–11028. Ma X, Xu L, Alberobello AT, et al. Celastrol protects against obesity andmetabolic dysfunction through activation of a HSF1-PGC1a transcriptional axis.Cell Metab 2015;22:695–70829. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepaticlipogenesis. Nat Rev Mol Cell Biol 2015;16:678–689
30. Latasa MJ, Griffin MJ, Moon YS, Kang C, Sul HS. Occupancy and function ofthe -150 sterol regulatory element and -65 E-box in nutritional regulation of thefatty acid synthase gene in living animals. Mol Cell Biol 2003;23:5896–590731. Fan W, Sun D, Liu J, et al. Adipose stromal cells amplify angiogenic sig-naling via the VEGF/mTOR/Akt pathway in a murine hindlimb ischemia model: a3D multimodality imaging study. PLoS One 2012;7:e4562132. Pellegrino R, Calvisi DF, Neumann O, et al. EEF1A2 inactivates p53 by wayof PI3K/AKT/mTOR-dependent stabilization of MDM4 in hepatocellular carcinoma.Hepatology 2014;59:1886–189933. Qu S, Altomonte J, Perdomo G, et al. Aberrant forkhead box O1 function isassociated with impaired hepatic metabolism. Endocrinology 2006;147:5641–565234. Berchtold MW, Villalobo A. The many faces of calmodulin in cell pro-liferation, programmed cell death, autophagy, and cancer. Biochim Biophys Acta2014;1843:398–43535. Li Z, Joyal JL, Sacks DB. Binding of IRS proteins to calmodulin is enhancedin insulin resistance. Biochemistry 2000;39:5089–509636. Fischer R, Julsgart J, Berchtold MW. High affinity calmodulin target se-quence in the signalling molecule PI 3-kinase. FEBS Lett 1998;425:175–17737. Deb TB, Coticchia CM, Dickson RB. Calmodulin-mediated activation of Aktregulates survival of c-Myc-overexpressing mouse mammary carcinoma cells.J Biol Chem 2004;279:38903–3891138. Ozcan L, Cristina de Souza J, Harari AA, Backs J, Olson EN, Tabas I. Ac-tivation of calcium/calmodulin-dependent protein kinase II in obesity mediatessuppression of hepatic insulin signaling. Cell Metab 2013;18:803–81539. Bouallegue A, Pandey NR, Srivastava AK. CaMKII knockdown attenuatesH2O2-induced phosphorylation of ERK1/2, PKB/Akt, and IGF-1R in vascularsmooth muscle cells. Free Radic Biol Med 2009;47:858–86640. Lee HY, Gattu AK, Camporez JP, et al. Muscle-specific activation of Ca(2+)/calmodulin-dependent protein kinase IV increases whole-body insulin action inmice. Diabetologia 2014;57:1232–124141. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and oppor-tunities. Nat Rev Drug Discov 2014;13:140–15642. Crouthamel MC, Kahana JA, Korenchuk S, et al. Mechanism and man-agement of AKT inhibitor-induced hyperglycemia. Clin Cancer Res 2009;15:217–22543. Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selectivetranslational induction of Dab2 and ILEI. Nat Cell Biol 2010;12:286–29344. Song Q, Sheng W, Zhang X, Jiao S, Li F. ILEI drives epithelial to mesen-chymal transition and metastatic progression in the lung cancer cell line A549.Tumour Biol 2014;35:1377–138245. Moreira CC, Cassolla P, Dornellas AP, et al. Changes in liver gluconeo-genesis during the development of Walker-256 tumour in rats. Int J Exp Pathol2013;94:47–5546. Ray K. Hepatocellular carcinoma: restoring gluconeogenesis: steroids couldtreat liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:69347. Montal ED, Dewi R, Bhalla K, et al. PEPCK coordinates the regulation ofcentral carbon metabolism to promote cancer cell growth. Mol Cell 2015;60:571–58348. Chen HP, Shieh JJ, Chang CC, et al. Metformin decreases hepatocellularcarcinoma risk in a dose-dependent manner: population-based and in vitrostudies. Gut 2013;62:606–61549. Pernicova I, Korbonits M. Metformin–mode of action and clinical implica-tions for diabetes and cancer. Nat Rev Endocrinol 2014;10:143–15650. Cleasby ME, Dzamko N, Hegarty BD, Cooney GJ, Kraegen EW, Ye JM.Metformin prevents the development of acute lipid-induced insulin resistance inthe rat through altered hepatic signaling mechanisms. Diabetes 2004;53:3258–3266
diabetes.diabetesjournals.org Chen and Associates 1197