gout and hyperuricemia in chronic kidney disease · pdf filewhat is clinically significant?...
TRANSCRIPT

WHAT IS CLINICALLY SIGNIFICANT?
Gout and Hyperuricemia in Chronic Kidney Disease
› Background
› Pathogenic Relationships
› Kidney Manifestations
› Role of Lowering Serum Uric Acid
› Conclusion

ETIOLOGIES OF HYPERURICEMIA AND GOUTBecause humans lack uricase, they cannot convert the uric acid generated during purine metabolism into a soluble form. This can lead to an increased risk for hyperuricemia and monosodium uric acid crys-tallization in joints and tissues, a hallmark of gout. Hyperuricemia can be caused by the overproduction of uric acid, but is more often the result of insufficient kidney uric acid excretion. Because a family history is often seen in primary gout, much research is focused on genes that cause hyperuricemia, notably those that regulate renal uric acid transport, such as human urate transporter 1 (URAT1).11 Genetic polymorphisms in anion transporters such as URAT-1 and SLC2A9, which encodes for GLUT9, can cause hyperuricemia by decreasing proximal tubular uric acid clearance.12-14 Other non-modifiable risk factors for gout include male gender, increasing age, and menopause.11,15
Approximately 15% of uric acid clearance occurs via the gastrointestinal tract, and therefore small bowel disease can contribute to increased serum uric acid.16 A variety of medications can increase serum uric acid, including loop and thiazide diuretics. High intake of meat, shellfish, alcohol, and fructose also can cause hyperuricemia,17,18 while obesity increases the risk for its development by three-fold.19 For many people, these causes of hyperuricemia will not lead to gout, but for those who are susceptible, these factors may trigger gout attacks.18
EPIDEMIOLOGY AND PATHOGENIC RELATIONSHIPS GOUT AND CKDGout has been steadily increasing worldwide, and is now the most common type of inflammatory arthro- pathy.11 In the United States alone, its prevalence more than doubled between the 1960s and the 1990s,20,21 and it is now estimated at 3.9% of U.S. adults (8.3 million adults — 6.1 million men and 2.2 million women).20,22 Hyperuricemia is also common, with a prevalence of 6-8% in healthy adults, and a prevalence of 1 in 3 adults who have uncontrolled hypertension and several cardiovascular risk factors.23 Concomitant-ly, the prevalence of CKD has been increasing, with estimates at 14% of adults in the United States,24 and 8-16% globally.25 The parallel rise in gout and CKD (Figure 2) has led to investigations to study their rela-tionship, including a retrospective cohort study based on 54 years of follow-up data from the Framingham Heart Study, which found that the risk of develop-ing gout in CKD doubled compared to subjects not having CKD and gout at baseline (HR=2.09; 95% CI 1.41 to 3.08). This difference remained significant after adjusting for other known gout risk factors.1,26
2
Uric acid and chronic renal disease 47
Figure 1. The potential interrelationships of uric acid, xanthine oxidase activity, and clinical endpoints of cardiovascular and renal diseaseAdapted from: Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol 2005;25:43-9.
BACKGROUNDIn chronic kidney disease (CKD), gout is a concomitant illness that increases health risk and treatment burden. The direct clinical impact of gout is also amplified by the comorbidities it shares with CKD — hypertension, diabe-tes, and features of the metabolic syndrome. Ironically, CKD may often lead to gout, as observational studies have found CKD to be the third most common independent risk factor for gout after obesity and hypertension.1,2 CKD is associated with decreased excretion of uric acid and resultant hyperuricemia, a major risk factor for gout. Other mechanisms may be implicated in CKD since variations in serum uric acid do not account for most of the risk for developing gout.1,3
Conversely, there is evidence that gout and associated hyperuricemia may independently impair kidney function.4,5 Hyperuricemia in the presence of gout, as well as the use of gout medications that are potentially harmful to the kid-neys, are initiation factors for developing CKD, and progres-sion factors for worsening CKD.6
Observational data also suggest that hyperuricemia, even in the absence of gout, may independently worsen CKD,7,8 possibly via a pathogenic role in hypertension (Figure 1), and diabetic nephropathy, the two leading causes of CKD.9,10 This bulletin highlights the clinical significance of both gout and hyperuricemia by discussing their etiologies, epidemiol-ogy, and relationship to CKD.
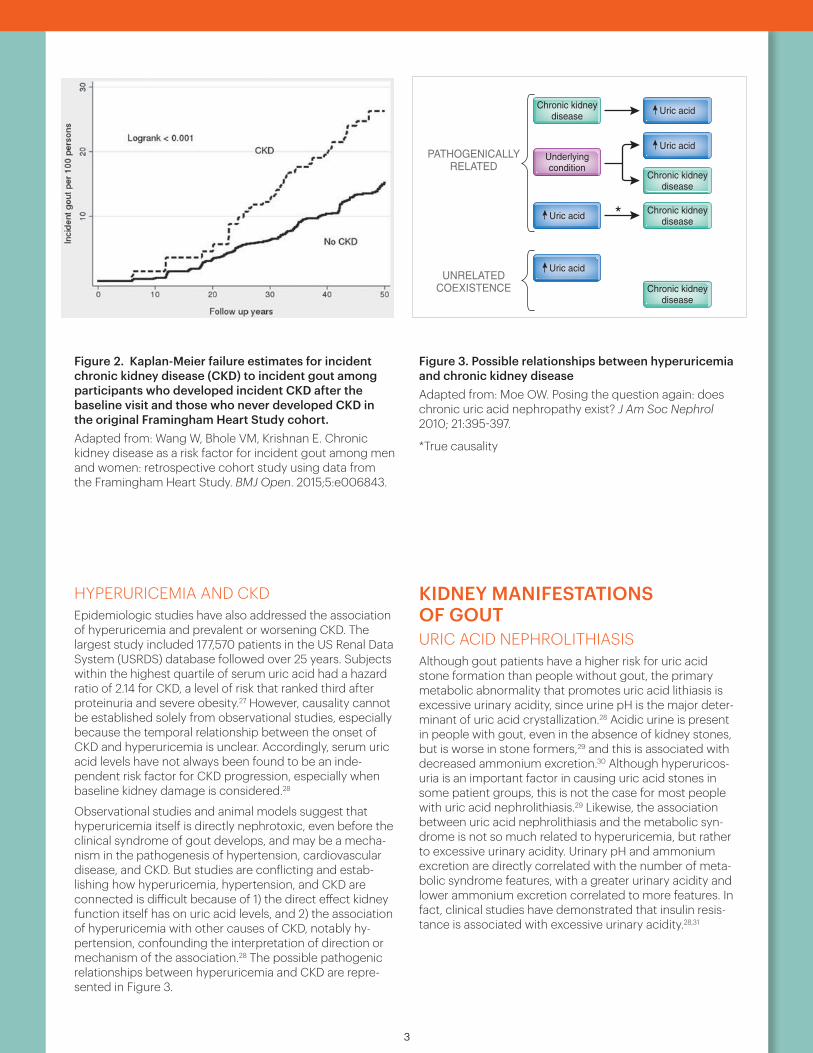
Figure 2. Kaplan-Meier failure estimates for incident chronic kidney disease (CKD) to incident gout among participants who developed incident CKD after the baseline visit and those who never developed CKD in the original Framingham Heart Study cohort. Adapted from: Wang W, Bhole VM, Krishnan E. Chronic kidney disease as a risk factor for incident gout among men and women: retrospective cohort study using data from the Framingham Heart Study. BMJ Open. 2015;5:e006843.
HYPERURICEMIA AND CKDEpidemiologic studies have also addressed the association of hyperuricemia and prevalent or worsening CKD. The largest study included 177,570 patients in the US Renal Data System (USRDS) database followed over 25 years. Subjects within the highest quartile of serum uric acid had a hazard ratio of 2.14 for CKD, a level of risk that ranked third after proteinuria and severe obesity.27 However, causality cannot be established solely from observational studies, especially because the temporal relationship between the onset of CKD and hyperuricemia is unclear. Accordingly, serum uric acid levels have not always been found to be an inde-pendent risk factor for CKD progression, especially when baseline kidney damage is considered.28
Observational studies and animal models suggest that hyperuricemia itself is directly nephrotoxic, even before the clinical syndrome of gout develops, and may be a mecha-nism in the pathogenesis of hypertension, cardiovascular disease, and CKD. But studies are conflicting and estab-lishing how hyperuricemia, hypertension, and CKD are connected is difficult because of 1) the direct effect kidney function itself has on uric acid levels, and 2) the association of hyperuricemia with other causes of CKD, notably hy-pertension, confounding the interpretation of direction or mechanism of the association.28 The possible pathogenic relationships between hyperuricemia and CKD are repre-sented in Figure 3.
Figure 3. Possible relationships between hyperuricemia and chronic kidney diseaseAdapted from: Moe OW. Posing the question again: does chronic uric acid nephropathy exist? J Am Soc Nephrol 2010; 21:395-397.
KIDNEY MANIFESTATIONS OF GOUT URIC ACID NEPHROLITHIASIS Although gout patients have a higher risk for uric acid stone formation than people without gout, the primary metabolic abnormality that promotes uric acid lithiasis is excessive urinary acidity, since urine pH is the major deter-minant of uric acid crystallization.28 Acidic urine is present in people with gout, even in the absence of kidney stones, but is worse in stone formers,29 and this is associated with decreased ammonium excretion.30 Although hyperuricos-uria is an important factor in causing uric acid stones in some patient groups, this is not the case for most people with uric acid nephrolithiasis.29 Likewise, the association between uric acid nephrolithiasis and the metabolic syn-drome is not so much related to hyperuricemia, but rather to excessive urinary acidity. Urinary pH and ammonium excretion are directly correlated with the number of meta-bolic syndrome features, with a greater urinary acidity and lower ammonium excretion correlated to more features. In fact, clinical studies have demonstrated that insulin resis-tance is associated with excessive urinary acidity.28,31
3
with CKD in general; the incidence rate of gout was 4.26(95% CI 3.39 to 5.35) per 1000 person-years amongthose with CKD in general and 2.43 (95% CI 2.18 to2.71) among those without. The Kaplan-Meier failureestimates for this analysis are shown in figure 2.In unadjusted Cox regression models, CKD in general
was also associated with an increased risk of gout with anHR of 1.47 (95% CI 1.14 to 1.90); in the multivariableCox models identical to those used in the main analyses,CKD in general was associated with HR of 1.61 (95% CI1.17 to 2.21) to incident gout (see online supplementarytable S1).Moreover, we performed a cross-sectional analysis for
influence of SUA on the relationship between CKD andgout. As aforementioned, since SUA data are only avail-able in five visits (visits 1–4 and 13), we chose to performa logistic regression on visit 4 because it was the last visitof the continuous four visits. In the multivariable logisticregression, SUA was included in an additional testbesides other covariates as aforementioned; in this add-itional test, the ORs for CKD (as shown in online supple-mentary table S2) and SUA (data not shown) were 1.82(1.13 to 2.93) and 3.35 (2.73 to 4.11) for men, 0.81 (0.44to 1.46) and 1.97 (1.56 to 2.49) for women, and 1.27(0.89 to 1.82) and 2.66 (2.30 to 3.09) overall.Comparisons have been made between unadjusted andSUA adjusted ORs as well as multivariable ORs with or
without SUA in the model. In both comparisons, theresults did not differ materially, as shown in online sup-plementary table S2.Probabilistic sensitivity analyses for the impact of mis-
classification, unmeasured confounding and selectionbias on the estimated association between CKD and goutare shown in online supplementary figure S1. The fre-quency distribution of bias and error adjusted ORs areshown in online supplementary figure S1A, where themedian bias-adjusted relative risk was 10.47 with 95%simulation ranging from 1.83 and 248. Changes in themagnitude adjusted OR in response to changes in mag-nitude and direction of bias are shown in online supple-mentary figure S1B.
DISCUSSIONOur analysis of data collected on the original FHS parti-cipants indicates that CKD is an independent risk factorfor gout. The analysis suggested that the HR of CKD togout is approximately 2.0 (non-prevalent CKD) toapproximately 1.5 (CKD in general). This result issimilar to another study using MRFIT data,8 where theadjusted HR for gout among those with CKD was 1.61(1.60 to 1.61).In this prospective study based on 54 years of follow-up
data from FHS participants, the risk of gout associatedwith CKD was doubled (HR=2.09; 95% CI 1.41 to 3.08)in participants free from CKD and gout at baseline.These associations persisted in both sexes, as well as inour additional analyses including those with CKD atbaseline, and were independent of other known riskfactors of gout, including age, BMI, alcohol use,smoking, hypertension and diabetes. Overall, thesefindings provide prospective evidence that individualswith CKD are at an increased future risk of gout inde-pendent of other known risk factors. Our sensitivity ana-lyses indicated that even after assuming extreme andunlikely degrees of misclassification and bias, the relativerisk estimate is about 1.8, suggesting that the true rela-tive risk might be stronger.Historically, the impact of CKD on gout has been
debated,1 25 26 as different studies observed differentresults. One factor that was often involved in the debatesis SUA. However, not all patients with hyperuricaemiadevelop gout, and the reasons remain unclear.27 28
Furthermore, it is difficult to ascertain if hyperuricaemiapreceded CKD or whether the reverse is true.25 To testthe influence of SUA with the relationship between
Table 2 Incidence rates for gout according to chronic kidney disease (CKD) status in the original Framingham Heart Studycohort
Men WomenIncident gout Rate (95% CI) Incident gout Rate (95% CI)
No CKD 204 3.60 (3.13 to 4.12) 121 1.57 (1.32 to 1.88)CKD 27 10.24 (7.02 to 14.93) 19 4.62 (2.95 to 7.24)
Figure 1 Kaplan-Meier failure estimates for incident chronickidney disease (CKD) to incident gout among participantswho developed incident CKD after the baseline visit and thosewho never developed CKD in the original Framingham HeartStudy cohort.
4 Wang W, et al. BMJ Open 2015;5:e006843. doi:10.1136/bmjopen-2014-006843
Open Access
Of foremost importance is whetherthere is a biologic basis for this entity.Human histopathologic data shows in-terstitial inflammation and fibrosis coex-isting with crystalline uric acid deposits,which are interpreted by some as “smok-ing guns.”6 A scatter of physiology stud-ies also showed abnormal indices of renaland endothelial function in the backdropof asymptomatic hyperuricemia. Noneof these provide proof and most of themare challenged by studies showing con-trary results. The most convincing datato date are derived from rodent models.Heinig and Johnson7 eloquently summa-rized this body of literature recently. In-duction of hyperuricemia elevates BPand produces renal microvascular, glo-merular, and tubulointerstitial lesionsthat are without doubt detrimental to theorganism. The mechanisms by whichthese lesions transpire are not yet knowneven with these models in hand. Based onthe rodent database, one will have toagree with the compelling view espousedby Henin and Johnson that the data arevery close to satisfying Koch’s criteria foruric acid nephropathy.
In contradistinction, the human evi-dence supporting a biologic mechanismof uric acid–induced injury in the kidneyis rather weak in comparison to the ro-dent data. One needs to exercise height-ened caution to extrapolate data gath-ered from species such as rodents
endowed with uricase and evolved withextremely low plasma and tissue levels ofuric acid, which are then rendered se-verely hyperuricemic experimentally.Similar findings in higher primates withrelative high normal plasma uric acidlevels would be substantially more per-suasive. The current status of the benchdata is convincing for the rodent modelbut still uncertain for humans. For hu-mans, one needs to turn to clinical datathat are more challenging to unravel.
Because one is equipped almost ex-clusively with epidemiologic data in hu-mans, one can apply some of Hill’sguidelines to attempt to gain more in-sight into causality. The strength of asso-ciation is variable from study to studyranging from none to weak8 –10; spacedoes not permit exhaustive citation of allstudies. A recent article in this journalprovided one of the strongest associa-tions to date. Obermayr et al.11 prospec-tively followed more than 21,000 pa-tients for a median of 7 yr with differentserum uric acid levels but the same base-line estimated GFR. They found mild hy-peruricemia (7.0 to 8.9 mg/dl) nearlydoubled the risk for incident kidney dis-ease, and more severe hyperuricemia(�9.0 mg/dl) tripled the risk. The sig-nificant odds ratios survived albeit di-minished with adjustments for baselineestimated GFR, gender, age, drugs po-tentially altering serum uric acid levels,
and components of the metabolic syn-drome. This is one of the strongest asso-ciations that survived multiple adjust-ments by robust statistical models anddoes support, although not prove, apathogenic role for uric acid.
Dose-response in humans is difficultto establish, because one cannot possibly“dose” the uric acid level. However, inthe study by Obermayr et al.,11 a modelwas constructed with the prospectivedata that displayed a curvilinear relation-ship between risk and the serum uric acidas a continuous variable. Consistency offinding is one of the weakest links in ourcurrent database. As pointed out earlier,more than its weight of negative associa-tions balances each positive study. Forthis discussion, specificity of the associa-tion as proposed by Hill is less relevantand need not be fulfilled because hyper-uricemia can lead to multiple conse-quences, and chronic kidney disease canresult from hyperuricemia alone or inconjunction with many other predispo-sitions. Temporal relation can be power-ful and revealing in population-baseddata. The occurrence of a risk factor be-fore the disease of interest lends supportto a causal relationship. This is particu-larly important because reduction ofGFR begets hyperuricemia, a fact that in-cites little debate. The study of Obermayret al. showed that hyperuricemia pre-cedes reduction in glomerular filtration.The amelioration of disease after elimi-nation or reduction of the risk factor alsostrongly supports causality. Unfortu-nately, we do not have the luxury of suchdata. Although the animal data thatemerged in the last decade are compel-ling, definitive proof of uric acid assum-ing a causative role in chronic kidney dis-ease is still not very satisfactory after allthese years. With that background, a fewgeneral points are noteworthy.
For the reasons stated in the begin-ning of this commentary, the motivationdriving one to pose this question willcontinue until a definitive affirmative ornegative answer is secured. The effect ofuric acid per se on progression of renaldisease in humans is unlikely to be large.The impact, if it exists, is unlikely to be ofthe same magnitude as hypertensive or
Uric acid
Uric acid
PATHOGENICALLYRELATED
Chronic kidneydisease
Underlyingcondition
*
UNRELATEDCOEXISTENCE Chronic kidney
disease
Uric acid
Chronic kidneydisease
Chronic kidneydisease
Uric acid
Figure 1. Possible relationships between hyperuricemia and chronic kidney disease.Asterisk indicates true causality.
CLINICAL COMMENTARY www.jasn.org
396 Journal of the American Society of Nephrology J Am Soc Nephrol 21: 395–397, 2010
*True causality

4
KIDNEY MANIFESTATIONS OF HYPERURICEMIAURATE NEPHROPATHY Urate nephropathy is defined as urate crystal deposition in the physiologic pH of the renal medulla, rather than the tubular lumen obstruction caused by uric acid lithiasis in tumor lysis syndrome or uricase knockout mice. Patholog-ical features observed in the kidneys of gout patients are those mainly associated with hypertensive kidney disease: advanced arteriosclerosis, glomerulosclerosis, and inter-stitial fibrosis. (Figure 4) Urate crystal deposition is present, but the focal nature of that feature appears inconsistent with the diffuse nature of the kidney disease.32 And possibly analogous to the fact that most people with hyperuricemia do not develop gout, 86% of postmortem cases with renal medullary urate deposits had not developed gout in life.28,33
Figure 4. Urate nephropathyAdapted from: Taylor WJ, Grainger R. In: Terkeltaub R, ed. Gout and Other Crystal Arthropathies: 1st ed. Philadelphia, PA: Elsevier Saunders; 2012:105-120.
OTHER DISEASESLess common kidney manifestations of hyperuricemia in-clude: familial juvenile hyperuricemic nephropathy (FJHN), an autosomal dominant disease leading to end-stage renal disease (ESRD) at an early age, reduced fractional excretion of uric acid, renal interstitial uric acid deposits, and sporad-ic gouty arthritis; complete or partial hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency (Lesch-Nyhan and Kelley-Seegmiller syndromes, respectively); acute uric acid-related nephropathy, a condition that is not usually associated with gout, but with a sudden, massive uric acid load caused by chemotherapy-induced tumor lysis.28
LOWERING SERUM URIC ACID TO TREAT GOUT IN THE SETTING OF CKDThe 2012 American College of Rheumatology (ACR) Guidelines emphasizes both non-pharmacologic and phar-macologic approaches for managing gout and lowering serum uric acid levels. The recommended serum uric acid level should be low enough to effectively improve and maintain the signs and symptoms of gout, a goal most often associated with a level of <6 mg/dL. The two first-line options for urate lowering therapy (ULT) are the xanthine oxidase inhibitors, febuxostat and allopurinol. Febuxostat does not require renal dose adjustment in mild to moderate CKD. Because insufficient evidence exists on its safety in CKD stage 4 or worse (eGFR of 0-29 mL/min/1.73 m2), no recommendation was issued for febuxostat in this setting. In CKD, the starting dose for allopurinol should not be more than 100 mg/day in moderate to severe CKD, followed by gradual upward titration of the maintenance dose, which can exceed 300 mg/day. Anti-inflammatory prophylaxis of acute gouty arthritis also involves the continuation of es-tablished pharmacologic ULT, without interruption, during an acute gout attack.17
The ACR recommends prophylaxis when initiating ULT be-cause the rapid decrease in serum urate can often trigger gout flares. First line options include low-dose colchicine (0.6 mg orally once or twice daily, with lower doses for moderate-to-severe CKD (eGFR of 15-59 mL/min/1.73 m2) and potential drug-drug interactions). Although low-dose non-steroidal anti-inflammatory drugs (NSAIDs) such as naproxen, 250 mg orally twice daily is recommended by the ACR. NSAIDs should generally be avoided in CKD. A stronger level of evidence exists for using colchicine.34
LOWERING SERUM URIC ACID TO TREAT CKD In view of the pathogenic connection between CKD and hyperuricemia, researchers have been evaluating how lowering serum uric acid levels might reduce the progres-sion of CKD. Data is based mostly on observational stud-ies and small clinical trials,35-37 and a significant problem with comparing their data, aside from methodology and confounding disease states and treatments, is the lack of uniformity in defining hyperuricemia. Levy, et al conducted a large retrospective review of 111,992 patients defined as having hyperuricemia at a level of > 7 mg/dL, and found that patients treated with uric acid lowering therapy (one of the following: allopurinol, febuxostat, or probenecid), who achieved a serum uric acid < 6 mg/dL, showed a 37% reduction in outcome events.38 In a recent small prospec-tive, randomized, open-label study with 21 patients receiv-ing febuxostat and 19 receiving conventional treatment, the authors reported renoprotective effects from febuxostat in hyperuricemic patients with CKD.39
At a single center, post hoc analysis of 113 patients who had completed a 2-year, single-blind, randomized controlled trial of allopurinol (56 patients received allopurinol and 57 were in the control group), showed improved estimated glomerular filtration rate and reduced cardiovascular risk.40
Chapter 9 — Clinical Features of Gout 111
cases, this is idiopathic. Acidic urine is observed in people with gout, even in the absence of renal stones, but is worse in stone formers,52 and this is associated with a deficit of ammo-nium excretion.55
Although hyperuricosuria is a very important factor in leading to uric acid stones in some patient groups, this is not present in most people with uric acid nephrolithiasis. Hyperuricosuria may occur in rare metabolic conditions such as Lesch-Nyhan syndrome, Kelley-Seegmiller syndrome, some glycogen storage diseases, or rare cases of URAT1 (SLC22A12)56 or GLUT9 (SLC2A9)57 mutations associated with severe hypouricemia (see Chapter 4).
Recently, an association between the metabolic syn-drome and uric acid nephrolithiasis has been identified. This association appears not to be due to the known asso-ciation of hyperuricemia and the metabolic syndrome but rather an association between an unduly acidic urinary pH and the metabolic syndrome. The principal mechanisms of urinary acidity in uric acid nephrolithiasis are increased net acid excretion and impaired buffering caused by inadequate ammonium excretion. Urinary pH and ammonium excre-tion are directly correlated with the number of metabolic syndrome features, with more features being associated with a more acidic urine and lower ammonium excretion. In stud-ies using the hyperinsulinemic euglycemic clamp technique, insulin resistance was shown to be associated with excessive urinary acidity.58
Urate NephropathyThe extent to which hyperuricemia causes rather than is caused by renal impairment has long been a subject for dis-cussion and is reviewed in Chapter 19. It is important to clar-ify terminology. Urate interstitial nephropathy is urate crystal deposition in the physiologic pH of renal medulla, as opposed to tubular lumen obstruction due to uric acid urolithiasis seen in tumor lysis syndrome or uricase knockout mice.
With specific respect to urate nephropathy (“gouty nephropathy”), Sydenham9 describes “morbific matter” in what may be an early account of gouty nephopathy:
The viscera in time are so much injured, from the stagna-tion of the morbific matter therein, that the organs of secre-tion no longer perform their functions, whence the blood, overcharged with vitiated humours, stagnates, and the gouty matter ceases to be thrown upon the extremities as formerly, so that at length death frees him from his misery.
In the middle of the 20th century, it was well recognized that patients with gout could frequently develop renal impair-ment.59 In that era prior to the widespread introduction of effective urate-lowering therapy or antihypertensive therapy, renal failure in patients with primary gout was not rare. For example, a case series of approximately 300 patients with mainly untreated gout from the 1950s observed significant renal disease in 18% to 25% of cases, and renal histologic changes, in 1960, were observed in the large majority of peo-ple with gout.59 However, whether these observations were due to hyperuricemia directly or to hypertension that is often coexistent with gout is difficult to determine. Pathologically, the kidneys of people with gout are characterized mainly by changes typically associated with hypertensive renal disease: advanced arteriosclerosis, glomerulosclerosis, and intersti-tial fibrosis (Figs. 9-10 and 9-11). Urate crystal deposition is observed, but the focal nature of that feature seems incon-sistent with the diffuse nature of the renal disease.60 Further-more, perhaps analogous to the fact that most people with hyperuricemia do not develop gout, 86% of postmortem cases with renal medullary urate deposits did not have gout in life.61
In people with elevated serum urate, it has been postulated that hyperuricemia itself is directly nephrotoxic, even prior to the development of the clinical syndrome of gout, and may be a mechanism for the increased rate of hypertension, car-diovascular disease and renal impairment that is observed
Figure 9-10 Urate nephropathy. There is chronic tubulointerstitial nephri-tis. Note the tubule distended by a collection of needle-like spaces, origi-nally containing urate deposits, associated with multinucleated giant cells (PAS stain, magnification ×40). (© 2011 American College of Rheumatology.
Available online at Rheumatology Image Bank: http://images.rheumatology
.org/viewphoto.php?albumId=75676&;imageId=2862276.)
Figure 9-11 This is chronic urate nephropathy with pale yellowish tan tophaceous deposits in the medulla. (Image © Edward C. Klatt, MD, Savan-
nah, Georgia, USA. All rights reserved. Available at http://library.med.utah.edu/
WebPath/RENAHTML/RENAL121.html.)
Figure 4

5
The larger Preventing Early Renal Function Loss (PERL) Allopurinol study, an international, multi-center, stratified, double-blind, placebo-controlled, parallel-group random-ized clinical trial, is currently investigating the effect of allopurinol in delaying the progression of CKD in type 1 diabetes. The reason for creating this trial stems from growing evidence that hyperuricemia may have a role in the pathogenesis of diabetic nephropathy and the pro-gression of CKD observed in type 1 diabetes. This evidence includes epidemiologic data,41 and prospective data from trials such as Reduction of Endpoints in Non-Insulin Depen-dent Diabetes Mellitus with the Angiotensin II Antagonist Losartan (RENAAL), in which lowering serum uric acid through the use of losartan accounted for 20% of the reno-protection provided by this drug.41,42 Mounting evidence over the last decade provides increasing support for the hypothesis that hyperuricemia leads to vasoconstriction and vascular remodeling that results in hypertension and contributes to the progression of CKD in at risk individuals.10
CONCLUSIONGout and hyperuricemia are clinically significant in the setting of CKD, especially when drugs used for their management can further impair kidney function. The established role of CKD as an independent risk factor for gout may therefore warrant screening for CKD when gout is first diagnosed.23 The role of hyperuricemia as an independent risk factor for CKD, however, is still being debated. Large randomized controlled trials can provide definitive answers about its relationship to CKD, and how its treatment might forestall CKD progression in populations such as those with hypertension and diabetic nephropathy.
Until the completion of large randomized controlled trials that support safety and efficacy, the ACR does not recommend serum uric acid lowering therapy for asymptomatic hyperuricemia.17 The dangers of inappropriately treating asymptomatic hyperuricemia are well documented,43-45 especially in the elderly.41
As emphasized by both the ACR and the National Kidney Foundation, lifestyle and dietary modifications that ameliorate the features of gout and hyperuricemia, along with appropriate pharmacologic treatments for gout and uric acid nephrolithiasis, are the proven strategies for reducing the risk of developing or worsening CKD.17,46
DISCLAIMER
Information contained in this National Kidney Foundation educational resource is based upon current data available at the time of publication. Information is intended to help clinicians become aware of new scientific findings and developments. This clinical bulletin is not intended to set out a preferred standard of care and should not be construed as one. Neither should the information be interpreted as prescribing an exclusive course of management.

1. Krishnan E. Chronic kidney disease and risk of incident gout among middle-aged men: a seven-year prospective observational study. Arthritis Rheum. 2013;65:3271-3278.
2. Juraschek SP, Kovell LC, Miller ER, et al. Association of kidney disease with prevalent gout in the United States in 1988-1994 and 2007-2010. Semin Arthritis Rheum. 2013;42:551-561.
3. Roddy E, Choi HK. Epidemiology of gout. Rhem Dis Clin N Am. 2014;40:155-175.
4. Jing J, Kielstein JT, Schultheiss UT, et al. Prevalence and correlates of gout in a large cohort of patients with chronic kidney disease. Nephrol Dial Transplant. 2015;30:613-621.
5. Yu KH, Kuo CF, Luo SF, et al. Risk of end-stage renal disease associated with gout: a nationwide population study. Arthritis Res Ther. 2012;14:R83.
6. Badve SV, Brown F, Hawley CM, et al. Challenges of conducting a trial of uric-acid-lowering therapy in CKD. Nat Rev Nephrol. 2011;7:295-300.
7. Obermayr RP, Temmi C, Gutjahr G, et al. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol. 2008;19:2407-2413.
8. Feig DI. Uric acid: a novel mediator and marker of risk in chronic kidney disease? Curr Opin Nephrol Hypertens. 2009;18:526-530.
9. Bellomo G. Uric acid and chronic kidney disease: a time to act? World J Nephrol. 2013;2:17-25.
10. Feig DI. Serum uric acid and the risk of hypertension and chronic kidney disease. Curr Opin Rheumatol. 2014;26:176-185.
11. Roddy E, Doherty M. Epidemiology of gout. Arthritis Res Ther. 2010;12:223.
12. Graessler J, Graessler A, Nuger S, et al. Association of the human urate transporter 1 with reduced renal uric acid excretion and hyperuricemia in a German Caucasian population. Arthritis Rheum. 2006;54:292-300.
13. McArdle PF, Parsa A, Chang YP, et al. Association of a common nonsynonymous variant in GLUT9 with serum uric acid levels in old order Amish. Arthritis Rheum. 2008;58:2874-2881.
14. Parsa A, Brown E, Weir MR, et al. Genotype-based changes in serum uric acid affect blood pressure. Kidney Int. 2012;81:502-507.
15. Hak AE, Curhan GC, Rodstein F, et al. Menopause, post-menopausal hormone use, and risk of incident gout. Ann Rheum Dis. 2010;69:1305-1309.
16. Cannella AC, Mikuls TR. Understanding treatments for gout. Am J Manag Care. 2005;11:S451-S458.
17. Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431-1446.
18. Choi HK. A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheum. 2010;22:165-172.
19. Hwang LC, Tsai CH, Chen TH. Overweight and obesity-related metabolic disorders in hospital employees. J Formos Med Assoc. 2006;105:56-63.
20. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63:3136-3141.
21. Lawrence RC, Felson DT, Helmick CG, et al. The National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: part II. Arthritis Rheum. 2008;58:26-35.
22. Centers for Disease Control and Prevention (CDC). National Center for Health Statistics (NCHS). National Health and Nutrition Examination Survey Questionnaire (NHANES) 2007-2008. Hyattsville, MD: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2009. Available at: http://wwwn.cdc.gov/nchs/nhanes/search/nhanes07_08.aspx. Accessed May 10, 2014.
23. Juraschek SP, Kovell LC, Miller ER, et al. Dose-response association of uncontrolled blood pressure and cardiovascular disease risk factors with hyperuricmemia and gout. PLoS ONE; 2013. 8:Article ID e56546.
24. U.S. Renal Data System (USRDS). USRDS 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2013.
25. Jha V, Garcia-Garcia G, Iseki K, et al. Chronic kidney disease: global dimension and perspectives. Global kidney disease 3. Available at: www.thelancet.com. Published online May 31, 2013. http://dx.doi.org/10.1016/50140-6736(13)60687-X.
26. Wang W, Bhole VM, Krishnan E. Chronic kidney disease as a risk factor for incident gout among men and women: retrospective cohort study using data from the Framingham Heart Study. BMJ Open. 2015;5:e006843.
27. Hsu CY, Iribarren C, McCulloch CE, et al. Risk factors for end-stage renal disease: 25-year follow-up. Arch Intern Med. 2009;169:342-350.
28. Taylor WJ, Grainger R. In: Terkeltaub R, ed. Gout and Other Crystal Arthropathies: 1st ed. Philadelphia, PA: Elsevier Saunders; 2012:105-120.
29. Cameron MA, Sakhaee K. Uric acid nephrolithiasis. Urol Clin North Am. 2007;34:335-346.
30. Gibson T, Highton J, Potter C, et al. Renal impairment and gout. Ann Rheum Dis. 1980;39:417-423.
31. Sakhaee K, Maalouf NM. Metabolic syndrome and uric acid nephrolithiasis. Semin Nephrol. 2008;28:174-180.
32. Feig DI, Kang D, Johenson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2009;359:1811-1821.
33. Linnane JW, Burry AF, Emmerson BT. Urate deposits in the renal medulla. Nephron. 1981;29:216-222.
34. Khanna D, Khanna PP, Fitzgerald JD. 2012 American College of Rheumatology Guidelines for Management of Gout. Part 2: Therapy and Antiinflammatory Prophylaxis of Acute Gouty Arthritis. Arthritis Care Res. 2012; 64:1447-1461.
35. Fairbanks LD, Cameron JS, Venkat-Raman G, et al. Early treatment with allopurinol in familial juvenile hyperuricaemic nephropathy (FJHN) ameliorates the long-term progression of renal disease. QJM. 2002;95:597-607.
36. Siu YP, Leung KT, Tong MK, et al. The use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47:51-59.
37. Kanbay M, Ozkara A, Selcoki Y, et al. Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearance, and proteinuria in patients with normal renal functions. Int Urol Nephrol. 2007;39:1227-1233.
38. Levy GD, Rashid N, Niu F, et al. Effect of Urate-lowering therapies on renal disease progression in patients with hyperuricemia. J Rheumatol. 2014;41:955-962.
39. Tanaka K, Nakayama M, Kanno M, et al. Renoprotective effects of febuxostat in hyperuricemic patients with chronic kidney disease: a parallel-group, randomized, controlled trial. Clin Exp Nephrol. Published online: 13 February 2015.
40. Goicoechea M, Garcia de Vinuesa S, Verdalles U, et al. Allopurinol and progression of CKD and cardiovascular events: long-terms follow-up of a randomized clinical trial. Am J Kidney Dis. 2015;65:543-549.
41. Maahs DM, Caramori ML, Cherney DZI, et al. Uric acid lowering to prevent kidney function loss in diabetes: the preventing early renal function loss (PERL) allopurinol study. Curr Diab Rep. 2013;13:550-559.
42. Miao Y, Ottenbros SA, Laverman GD, et al. Effect of a reduction in uric acid on renal outcomes during losartan treatment: a post hoc analysis of the reduction of endpoints in non-insulin-dependent diabetes mellitus with the Angiotensin II Antagonist Losartan Trial. Hypertension. 2011;58:2-7.
43. Carnovale C, Venegoni M, Clementi E. Allopurinol overuse in asymptomatic hyperuricemia: a teachable moment. JAMA Internal Medicine. July 2014;174:1031-1032.
44. Kang Y, Kim MJ, Jang HN, et al. Rhabdomyolysis associated with initiation of febuxostat therapy for hyperuricaemia in a patient with chronic kidney disease. J Clin Pharm Therapeutics. 2014;39:328-330.
45. Pasina L, Brucato AL, Djade CD. Inappropriate prescription of allopurinol and febuxostat and risk of adverse events in the elderly: results from the REPOSI registry. Eur J Clin Pharmacol. 2014;70:1495-1503.
46. A Clinical Update on Gout: Optimizing Care for Patients with Chronic Kidney Disease. 2014, National Kidney Foundation, Inc. www.kidney.org/professionals.
30 East 33rd StreetNew York, NY 10016 800.622.9010 www.kidney.org
© 2015 National Kidney Foundation, Inc. 02-10-6972_JBF
Sponsored by
Takeda Pharmaceuticals, USATakeda did not contribute to the development of the content for this resource. The content does not necessarily reflect the views, opinions, or commercial interests of Takeda.