gold speciation and transport in geological fluids: insights from experiments and physical-chemical...
TRANSCRIPT

Geological Society, London, Special Publications Online First
April 24, 2014; doi 10.1144/SP402.4, first publishedGeological Society, London, Special Publications
Alexandre V. Zotov and Kalin KouzmanovGleb S. Pokrovski, Nikolay N. Akinfiev, Anastassia Y. Borisova, modellinginsights from experiments and physical-chemical Gold speciation and transport in geological fluids:
serviceEmail alerting
new articles cite this article to receive free e-mail alerts whenhereclick
requestPermission
part of this article to seek permission to re-use all orhereclick
Subscribe
Collection London, Special Publications or the Lyell
to subscribe to Geological Society,hereclick
How to citeFirst and how to cite articles
for further information about Onlinehereclick
Notes
© The Geological Society of London 2014
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Gold speciation and transport in geological fluids: insights from
experiments and physical-chemical modelling
GLEB S. POKROVSKI1*, NIKOLAY N. AKINFIEV2, ANASTASSIA Y. BORISOVA1,3,
ALEXANDRE V. ZOTOV2 & KALIN KOUZMANOV4
1Geosciences Environnement Toulouse, GET (ex-LMTG), Universite de Toulouse,
CNRS-IRD-OMP, 14 Av. E. Belin, F-31400 Toulouse, France2Institute of Geology of Ore Deposits, Petrography, Mineralogy and Geochemistry Russian
Academy of Sciences, Staromonetny per. 35, 119017 Moscow, Russia3Geological Department, Moscow State University, Vorobevu Gory, 119899 Moscow, Russia
4Earth and Environmental Sciences, University of Geneva, rue des Maraıchers 13,
CH-1205 Geneva, Switzerland
*Corresponding author (e-mail: [email protected])
Abstract: This contribution provides an overview of available experimental, thermodynamic, andmolecular data on Au aqueous speciation, solubility, and partitioning in major types of geologicalfluids in the Earth’s crust, from low-temperature aqueous solution to supercritical hydrothermal-magmatic fluids, vapours, and silicate melts. Critical revisions of these data allow generation ofa set of thermodynamic properties of the AuOH, AuCl22 , AuHS, and Au(HS)2
2 complexes dominantin aqueous hydrothermal solutions; however, other complexes involving different sulphur forms,chloride, and alkali metals may operate in high-temperature sulphur-rich fluids, vapours, andmelts. The large affinity of Au for reduced sulphur is responsible for Au enrichment in S-richvapours and sulphide melts, which are important gold sources for hydrothermal deposits. Thermo-dynamic, speciation, and partitioning data, and their comparison with Au and S contents in naturalfluid inclusions from magmatic-hydrothermal gold deposits, provide new constraints on the majorphysical-chemical parameters (temperature, pressure, salinity, acidity, redox) and ubiquitous fluidcomponents (sulphur, carbon dioxide, arsenic) affecting Au concentration, transport, precipitation,and fractionation from other metals in the crust. The availability and speciation of sulphur and theirchanges with the fluid and melt evolution are the key factors controlling gold behaviour in mostgeological situations.
Since alchemists’ times, the reputation of gold hasbeen to be the most noble of the metals, that is, exist-ing as a native metal in the solid phase at all terres-trial conditions and being extremely unreactive incontact with most liquids and gases. However, theexistence of magmatic-hydrothermal gold oresunambiguously attests that gold may be concen-trated, transported, and deposited by geologicalfluids in quantities sufficient to form an economicdeposit, in which this metal is enriched by factorsof 1000–10 000 compared to its average concen-tration in the Earth’s crust (c. 1 ppb, Rudnick &Gao 2003). Detailed knowledge of aqueous chem-istry and solubility of Au in geological fluids overa wide range of temperatures and pressures is thusnecessary for understanding Au distribution andconcentration in the crust.
The solubility of gold has been the subjectof numerous alchemical, industrial, experimental,theoretical, and geologic studies for hundreds ofyears (e.g. see Williams-Jones et al. 2009 for a
historical overview). Despite the large amount ofinformation on Au geo(chemistry) gained so far,many questions remain about the physical-chemicaland geological processes and mechanisms thatcontrol Au mobilization and deposition, particularlyunder hydrothermal conditions at which most Auores form. An approach combining experimentaldata, physical-chemical modelling, and geologicalobservations is necessary to quantify the impactof these processes on the gold fate and distribu-tion. Probably the first quantitative account ofAu solubility in hydrothermal solutions was doneby Krauskopf (1951) who attempted to interpretthe few Au solubility experiments available at thattime (Ogryzlo 1935; Smith 1943; referencestherein) in terms of Au speciation and thermodyn-amics in solution. These works provided the foun-dation for our understanding of Au hydrothermalchemistry that is controlled by chemical enti-ties or species with sulphur, chloride, and oxygen,although the proposed stoichiometry of these
From: Garofalo, P. S., Ridley, J. R. (eds) Gold-Transporting Hydrothermal Fluids in the Earth’s Crust.Geological Society, London, Special Publications, 402, http://dx.doi.org/10.1144/SP402.4# The Geological Society of London 2014. Publishing disclaimer: www.geolsoc.org.uk/pub_ethics
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

species was tentative at that time. About twentyyears later, Helgeson & Garrels (1968) applied intheir paper an integrated approach combiningrare available experimental data, thermodynamicextrapolations of Au solubility, and considerationsof the budget of Au and accompanying elementsin gold veins based on geological observations.Their conclusions about Au transport in hydrother-mal fluids as aurous chloride species precipitatingin response to cooling were, however, criticized byBoyle (1969) who, based on field observations andgeochemistry of Au ores, argued that Au is primarilytransported as complexes with S, As, and Sb andis deposited in response to decompression andfluid-rock interaction rather than cooling.
The following 45 years of efforts of many otherore deposit geologists, experimentalists, and ther-modynamists studying Au in all its facets in hydro-thermal fluids, silicate melts, and minerals, have notsignificantly changed the state of this debate. Today,even if we know far better a substantial part ofpossible Au chemical species carrying this metal ingeological fluids, we have still rather limited data onAu contents and distribution in hydrothermal fluidsfrom direct analyses of fluid inclusions and in volca-nic gases and geothermal systems, and we alwayswonder why Au in its ores is associated with someparticular elements in particular phases (e.g.arsenian pyrite) or CO2-bearing fluids. With therapidly growing progress in experimental, spectro-scopic, analytical, and modelling techniques, ourknowledge of Au chemistry in hydrothermal fluids,magmatic vapours, silicate melts, and solid phases isexpanding rapidly, particularly in the last 10–15years. However, the application of these results togold ore deposits is always faced with the fascinat-ing complexity of natural processes responsible forAu mobilization, concentration, and deposition.
The present contribution is a humble attempt tosummarize what we have learned about gold ingeological fluids over the more than 50 years sincethe pioneering experimental and theoretical workscited above, and how this knowledge may help tobetter constrain our understanding of the impact ofdifferent factors leading to the formation of goldore deposits. In this paper, we first give an overviewof the available experimental and thermodynamicdata on Au aqueous speciation and solubility overa wide range of geological fluid conditions, fromlow-temperature aqueous solution to supercriticalhydrothermal fluids, volcanic gases, and silicatemelts. Next, on the basis of these data, we quantifythe effect on Au solubility of major parametersand components typical of geological fluids (temp-erature, pressure, salinity, acidity, redox, sulphur,CO2, arsenic). Then, we compare thermodyna-mic predictions with natural Au contents fromdirect analyses of fluid inclusions from porphyry
Cu–Au–Mo and associated deposits. The con-frontation between natural data and physical chem-istry provides useful contraints on understanding themechanisms and processes (cooling, decompres-sion, boiling, water-rock interactions, fluid mixing)controlling Au transport and deposition in differenttypes of deposits, which are discussed in a separatesection. Finally, we identify several remaining gapsin our knowledge of Au hydrothermal chemistryand discuss experimental, analytical, and modellingchallenges expected to help reduce these gaps in thenear future.
Overview of experimental and theoretical
data on Au speciation and solubility in
magmatic-hydrothermal fluids
General features of gold aqueous chemistry
Detailed knowledge of aqueous chemistry of Auover a wide range of temperature and pressure iscrucial for understanding Au distribution and con-centration by crustal fluids in various geologicalenvironments. This section briefly outlines themajor features of Au aqueous speciation and mol-ecular coordination.
Gold possesses the complete electronic structure[Xe] 5d106s1 and, like silver and copper, has a sin-gle s-electron outside a completed d-shell. Despitelarge similarities of outer-shell electronic configur-ations and ionization potentials, there are significantdifferences amongst these metals as to their redoxforms and affinities to different ligands. There isno simple explanation of these differences, butsome of them are likely to be due to relativisticeffects on the 6s-electrons of Au (Cotton & Wilkin-son 1988).
Although Au shows a wide range of oxidationstates, from I to VII, in synthetic chemical com-pounds, apart from Au0, the dominant oxidationstates of chemically-bound Au in natural fluidsand minerals are trivalent (auric gold, AuIII) andmonovalent (aurous gold, AuI). The former is,however, a strong oxidant and is stable only in oxi-dizing surface environments, whereas the latter isthe dominant Au form in the majority of hydro-thermal fluids and silicate melts (e.g. Pokrovskiet al. 2009a, b; references therein). Both in solidphase and aqueous solution, AuIII is almost alwayscoordinated with four ligands in a square geometry(Puddephatt 1978; Schwerdtfeger et al. 1992;Farges et al. 1993; see Table 1), whereas AuI sys-tematically forms linear two-coordinate structuresat naturally pertinent conditions (Schwerdtfegeret al. 1992; Tossell 1996), with only the few excep-tions of 3- and 4-coordinate AuI in some syntheticmetallo-organic compounds (e.g. Benfield et al.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from
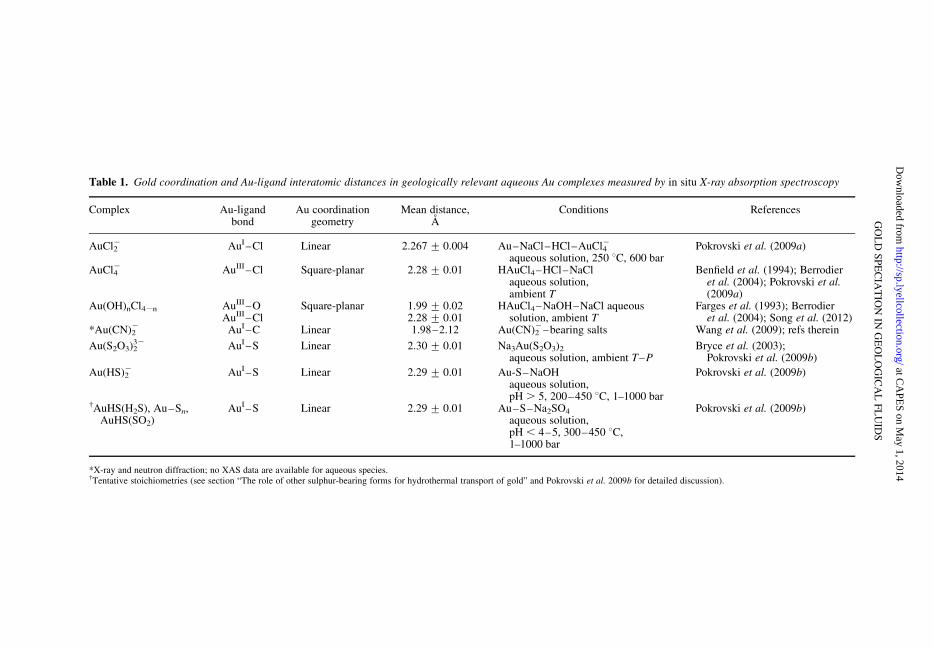
Table 1. Gold coordination and Au-ligand interatomic distances in geologically relevant aqueous Au complexes measured by in situ X-ray absorption spectroscopy
Complex Au-ligandbond
Au coordinationgeometry
Mean distance,A
Conditions References
AuCl22 AuI–Cl Linear 2.267 + 0.004 Au–NaCl–HCl–AuCl4
2
aqueous solution, 250 8C, 600 barPokrovski et al. (2009a)
AuCl42 AuIII–Cl Square-planar 2.28 + 0.01 HAuCl4–HCl–NaCl
aqueous solution,ambient T
Benfield et al. (1994); Berrodieret al. (2004); Pokrovski et al.(2009a)
Au(OH)nCl42n AuIII–OAuIII–Cl
Square-planar 1.99 + 0.022.28 + 0.01
HAuCl4–NaOH–NaCl aqueoussolution, ambient T
Farges et al. (1993); Berrodieret al. (2004); Song et al. (2012)
*Au(CN)22 AuI–C Linear 1.98–2.12 Au(CN)2
2–bearing salts Wang et al. (2009); refs therein
Au(S2O3)232 AuI–S Linear 2.30 + 0.01 Na3Au(S2O3)2
aqueous solution, ambient T–PBryce et al. (2003);
Pokrovski et al. (2009b)
Au(HS)22 AuI–S Linear 2.29 + 0.01 Au-S–NaOH
aqueous solution,pH . 5, 200–450 8C, 1–1000 bar
Pokrovski et al. (2009b)
†AuHS(H2S), Au–Sn,AuHS(SO2)
AuI–S Linear 2.29 + 0.01 Au–S–Na2SO4
aqueous solution,pH , 4–5, 300–450 8C,1–1000 bar
Pokrovski et al. (2009b)
*X-ray and neutron diffraction; no XAS data are available for aqueous species.†Tentative stoichiometries (see section “The role of other sulphur-bearing forms for hydrothermal transport of gold” and Pokrovski et al. 2009b for detailed discussion).
GO
LD
SP
EC
IAT
ION
ING
EO
LO
GIC
AL
FL
UID
S
at CA
PES on M
ay 1, 2014http://sp.lyellcollection.org/
Dow
nloaded from

1994; ICSD 2010; references therein). This rathersimple Au coordination contrasts with the largevariety of CuI, CuII, and AgI first-shell geometriesboth in solids and solution, from 2- to 6-coordinatesuch as linear, trigonal, distorted tetragonal, pyrami-dal or octahedral (e.g. Cotton & Wilkinson 1988;ICSD 2010; Pokrovsky et al. 2012; Pokrovskiet al. 2013a). Gold-ligand interatomic distances inthe main geologically relevant species in aqueoussolution have been recently determined usingdirect measurements by X-ray absorption spec-troscopy (XAS, Table 1). These distances arepretty constant for each ligand type (OH/H2O, Clor S) and almost independent of temperature andpressure, thus confirming the strong covalentnature of Au-ligand bonds and the constancy ofAu coordination over a wide T–P range. The stab-ility of main AuIII and AuI species with naturally-relevant ligands is discussed below.
Among natural ligands capable of complexingAuIII in aqueous solution, the Cl2 and OH2 ionsare the most common. Studies at room temperatureindicate that AuIII is strongly hydrolysed over awide pH range, but the identity and stability ofhydroxide species are poorly constrained becauseof low solubilities and formation of unstable solidphases (e.g. Au(OH)3) and Au colloids (Baes &Mesmer 1976; references therein). As a result, thethermodynamic properties of the Au3+ cation, whichis expected to form only in extremely acidic non-complexing media, are poorly known, and only
few theoretical estimates exist in the literature(Table 2).
The famous AuCl42 complex is responsible for
gold dissolution in aqua regia (i.e. a mixture ofconcentrated HCl and HNO3). Its structure andstability have been studied in detail at oxidizingnear-ambient conditions using potentiometry (Niko-laeva et al. 1972), Raman (Pan & Wood 1991; Pecket al. 1991; Murphy & Lagrange 1998), UV-visible(Usher et al. 2009), NMR (Sharps et al. 1993), andX-ray absorption spectroscopy (Farges et al. 1993;Berrodier et al. 2004; Pokrovski et al. 2009a; seeTable 1). This complex is dominant in acidicsolutions (pH ≤ �5) and, with an increase insolution pH, is progressively replaced by mixedAuCln(OH)2
42n species (where 0 ≤ n ≤ 4), withtotal gold solubility dropping in neutral-to-basicsolutions. On heating of acidic AuIII chloride sol-utions, gold is rapidly reduced to AuI even underhighly oxidizing conditions (Pokrovski et al.2009a). In natural aquatic systems, the presenceof reducing agents (e.g. sulphide, organic matter,ferrous iron) will further tend to reduce AuIII toAuI or Au0. Thus, the natural settings where AuIII
species may be important are limited to well-oxygenated, highly saline acidic groundwaters(Usher et al. 2009) and/or FeIII and MnIV oxyhy-droxide mineral surfaces (Berrodier et al. 2004).
The aurous cation, Au+, is one of the softestcations in solution, according to the soft-hard classi-fication of Pearson (1963) that suggests that large,
Table 2. Stability constants of main aqueous AuIII and AuI complexes at 25 8C and 1baraccording to available literature data
Reaction log K 25 8C,1 bar
References
Au3+ + 4Cl2 ¼ AuCl42 26.2 Nikolaeva et al. (1972)*
Au+ + 2CN2 ¼ Au(CN)22 38.7 Seward (1989)†
Au+ + 2SCN2 ¼ Au(SCN)22 17.0 Martell et al. (2004)‡
Au+ + OH2 ¼ AuOH0 10.2 Kissner et al. (1997)Au+ + Cl2 ¼ AuCl0 7.9 Akinfiev & Zotov (2010)
Au+ + 2Cl2 ¼ AuCl22 9.6 Akinfiev & Zotov (2010)
Au+ + 2Br2 ¼ AuBr22 12.4 Seward (1989)†
Au+ + 2I2 ¼ AuI22 19.0 Seward (1989)†
Au+ + HS2 ¼ Au(HS)0 24.6 Akinfiev & Zotov (2010)Au+ + 2HS2 ¼ Au(HS)2
2 30.2 Akinfiev & Zotov (2010)
Au+ + 2S2O322 ¼ Au(S2O3)2
32 26.0 Seward (1989)†
Au+ + 2SO322 ¼ Au(SO3)2
32 26.8 Webster (1986)†
Au(s) + H+ ¼ Au+ + 0.5 H2(g) 228.6 Shock et al. (1997)Au(s) + 3H+ ¼ Au3+ + 1.5 H2(g) 275.9 Shock et al. (1997)
*Recalculated to zero ionic strength using the AuCl22/AuCl4
2 standard potential (Nikolaeva et al. 1972), and Gibbsfree energies of Au3+ and AuCl2
2 from Shock et al. (1997) and Akinfiev & Zotov (2001), respectively.†Compiled/recalculated from older studies.‡Ionic strength ¼ 3.0.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

weakly charged, and easily polarizable cations,termed ‘soft’ (e.g. Au, Ag, Hg, Pt) form the stron-gest covalent complexes with ‘soft’ anions havingsimilar properties (e.g. HS2, CN2, S2O3
22, Br2,I2), whereas small, highly charged, and/or noteasily polarizable cations termed ‘hard’ (e.g. alka-line earth metals, rare earth elements, Al, Zr) prefer-entially form stable ionic-bond complexes with‘hard’ anions of similar properties (e.g. F2, CO3
22,SO4
22, OH2). Intermediate between these twoextremes are metals (e.g. Cu, Fe, Zn) and ligands(e.g. Cl2, SO3
22, NH3) called ‘borderline’; theytend to form between them the most stable com-plexes. Because of the higher charge and weakerpolarizability, AuIII is harder than AuI, whichexplains its preference to Cl2 (see above). It fol-lows from the soft-hard affinity rules that AuI
is unlikely to form stable complexes with hardligands like fluoride, carbonate, or sulphate.Indeed, no stability constants are known for suchspecies. The stability constants of AuIII and AuI
complexes with typical naturally-relevant ligandsare summarized in Table 2. The strongest AuI
inorganic complex, known so far in aqueous sol-ution, is dicyanide, Au(CN)2
2, which is widely usedfor leaching of Au from its ores (e.g. Muir 2011 for arecent review). This and other soft ligands, suchas S2O3
22, SCN2, and thiol (-SII) and amid (-NIII)functional groups of fulvic and humic acids, pro-duced by decomposition of plants and algae viamicroorganism activity, or by pyrite and sulphuroxidation, may contribute to the transport of AuI
in surface waters and mine drainage sites (e.g. Vlas-sopoulos et al. 1990). Some of these ligands, beingless toxic than CN2, are considered as alternativesto cyanide for selective extraction of Au fromhydrothermal ores (e.g. Adams 2005).
It should be noted that the chemical affinity ofa metal and a ligand themselves (which maybe quantified by the thermodynamic constant of acomplex formation reaction, Table 2) is not theonly property governing metal speciation innatural fluids. The availability (i.e. concentrationor activity) of the ligand is another major controllingfactor. Thus, in a dilute aqueous solution, devoid ofthe soft ligands discussed above, AuI prefers thehydroxide over the water ligand and is thus hydro-lysed by forming the neutral AuOH complex overa wide pH range (e.g. Vlassopoulos & Wood1990). Although hydroxide is rather hard, it isstrongly basic and thus a good donor for AuI,which may be explained by relativistic contractionof the 6 s orbital of AuI (Kissner et al. 1997).Thus, similarly to Au3+ (see above), the free Au+
cation is unstable in aqueous solution; as a result,its thermodynamic properties are poorly known(Table 2). Despite the stability constant values ofAuI–CN and AuI–I complexes are 9–10 orders of
magnitude higher than those of their AuI–SH andAuI–I v. AuI–Cl analogues (Table 2), the extremelylow concentrations of cyanide and iodide inmost natural waters, particularly under hydrother-mal conditions, render their complexes almostnegligible. Thus, the major carriers of gold in geo-logical fluids at elevated temperatures are hydrox-ide, chloride, and hydrogen sulphide (and possiblysome other sulphur forms such as polysulphides inS-rich fluids) as will be shown below.
Gold chloride, hydrogen sulphide, and
hydroxide species in aqueous solution and
dense supercritical fluid
Despite the almost 50-year work on gold in the earthscience community, experimental data on goldsolubility and aqueous speciation in the presenceof major natural ligands—chloride, hydroxide, andhydrogen sulphide—are not sufficiently system-atic and consistent. Thermodynamic reviews ofSverjensky et al. (1997) allowed generation of afirst set of thermodynamic properties of Au(HS)2
2
and AuI and AuIII chloride species based on rareexisting experiments. However, these data weresubject to large uncertainties because the parametersof these Au species were based on empirical corre-lations for the Au+ ion whose properties are notaccurate enough (see the section above). Anotherthermodynamic review published four years later(Akinfiev & Zotov 2001) allowed generalizing thestrategy of analysis of experimental informationavailable at that time on the stability constants ofAuI chloride, hydrogen sulphide, and hydroxidecomplexes. Recently published experimental dataon the stability of Au hydrogen sulphide complexesboth at low (Tagirov et al. 2006) and high tem-peratures (Stefansson & Seward 2004; Tagirovet al. 2005) have been considered in a more recentsummary (Akinfiev & Zotov 2010). In this section,after a systematic compilation of the availableexperimental data of Au speciation in hydrother-mal chloride- and sulphide-bearing solutions, wepresent a thermodynamic database for aqueousspecies in the Au–S2II–Cl–O–H system appli-cable over a wide range of temperatures (0–6008C) and pressures (1–3000 bars).
Historical background. To the best of our knowl-edge, the first laboratory investigations of gold solu-bility in model aqueous solutions at hydrothermalconditions in the presence of chloride and sulphidehave been those of Ogryzlo (1935) and Weissberg(1970). Although these works were lacking themodern-day accuracy of chemical analyses ofaqueous solutions, they have allowed establishingthe principal forms of Au in solution (hydroxide,
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

chloride, and sulphide complexes) and thus deter-mined the subsequent research directions in theexperimental geochemistry of Au in hydrothermalfluids. The following step in this research wasmarked by studies of Seward (1973) on Au hydro-gen sulphide species and Henley (1973) on chloridecomplexes under controlled oxygen (or hydrogen)fugacity, and by the potentiometric work of Niko-laeva et al. (1972) who reported the stabilities ofAuI and AuIII chloride species. These pioneeringworks were followed by a plethora of experimentalreports on Au solubility and speciation in hydro-thermal solutions, which are summarized in Table3 and briefly reviewed below for the three maintypes of Au aqueous species formed with hydroxide,chloride, and hydrogen sulphide.
AuI hydroxide complexes. The stability of the mono-hydroxide species AuOH0 has been investigated indetail over 20 years by the solubility method usingboth batch and flow-through hydrothermal reactors(Baranova et al. 1983; Zotov et al. 1985; Stefansson& Seward 2003a). The results are in good agreementwith one another, particularly at elevated tempera-tures (450–500 8C). Another solubility study (Bara-nova et al. 1977) reported the formation of thedihydroxide species, Au(OH)2
2, at very alkalineconditions (pH . 10), which are unlikely to occurin geological environments.
AuI chloride complexes. The thermodynamic prop-erties of AuCl2
2 in a wide temperature and pres-sure range were investigated by Zotov et al. (1989,1991) and Stefansson & Seward (2003b), usingquench-based batch-autoclaves and sampling-based flow-through reactors, respectively. Theagreement is good at 500 8C, the temperature atwhich chloride complexes began to be importantin natural systems (see below), but at and below400 8C the data of Stefansson & Seward (2003b)yield AuCl2
2 concentrations up to one order of mag-nitude lower than those of Zotov et al. (1989, 1991).Gammons & William-Jones (1995) employed adifferent method using Au–Ag alloys as redoxindicators and determined the stability constant ofAuCl2
2 at 3008C. Their value is in good agreementwith the Stefansson & Seward (2003b) study, andinterpolations between the previous studies at low(Nikolaeva et al. 1972) and high (Zotov et al.1989, 1991) temperatures. In contrast, there arelarge differences between those studies and theresults of Henley (1973) and Wood et al. (1987).The former showed unusually high solubilitiesand large data scatter (over two orders of magnitudein Au concentration); the latter used a complex mul-tiphase sulphide system at moderate temperatures(200–300 8C) at which it is difficult to demonstateequilibrium. Consequently, we do not recommend
these two studies for the thermodynamic analysisof AuCl2
2. More recently, Pokrovski et al. (2009a)used in situ X-ray absorption spectroscopy to estab-lish the structure of AuCl2
2 (Table 1) and demon-strate that this species is the dominant Au chloridecomplex in a wide T and salt concentration range,and that higher-order chloride species are negligibleat these conditions.
AuI hydrogen sulphide complexes. These Au speciesaccount for the major part of experimental studiesof Au at hydrothermal conditions. The basic infor-mation about the stoichiometry and stability ofthese complexes has been obtained by Seward(1973), Renders & Seward (1989), and Shenberger& Barnes (1989). These works found that thedominant Au species in H2S-bearing solutions areAuHS0 at acidic pH, Au(HS)2
2 at near-neutral pH,and Au2S2
22 or Au2(HS)2S22 at alkaline pH. In thenumerous later studies using different types of solu-bility methods (Hayashi & Ohmoto 1991; Zotov &Baranova 1995; Benning & Seward 1996; Gibertet al. 1998; Stefansson & Seward 2004; Tagirovet al. 2005, 2006), and, more recently, in situ spec-troscopic methods (Pokrovski et al. 2009b), thestability of Au(HS)2
2 at near-neutral pH, perti-nent to natural fluids, has been extended across awide T–P and compositional range. The numericalresults of Au solubility from these studies have beencompared by Pokrovski et al. (2009b) and reviewedby Akinfiev & Zotov (2010).
In contrast to all these studies whose results arein decent agreement, a few other solubility worksdiverge significantly from the main trend. Forexample, Baranova & Zotov (1998) reported unrea-listically high Au(HS)2
2 concentrations in equili-brium with the metal; in contrast, Pan & Wood(1994) found suspiciously low solubilities. Loucks& Mavrogenes (1999) investigated Au solubility athigher T–P than all existing studies (500–700 8C,and 1–4 kbar) and found Au concentrations up to1000 ppm, which were interpreted as a new spe-cies, AuHS(H2S)3
0. These data are, however, incon-sistent with more recent work (see section “The roleof other sulphur-bearing forms for hydrothermaltransport of gold” below). The results of Fleet &Knipe (2000), who measured Au solubility in flu-ids with extremely high H2S contents (up to H2Smole fractions of �0.3, equivalent to �20 mol/kgH2O) and interpreted it in terms of Au(HS)2
2, can-not be used for rigourous derivation of thermo-dynamic properties of aqueous Au complexes.Consequently, the four studies cited above werenot included in the following thermodynamic analy-sis. Gold species with sulphur ligands other thanH2S/HS2 will be discussed in section, “The roleof other sulphur-bearing forms for hydrothermaltransport of gold.”
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Table 3. Summary of published experimental studies on Au solubility and speciation in aqueous solution at hydrothermal conditions
Year Authors T, 8C P, bar System composition Main results
1935 Ogryzlo 200–300 svp Solubility of Au in HCl, NaCl, Na2CO3, andNa2S solutions
Observation of elevated Au solubility inalkaline sulphide solutions
1970 Weissberg 150–290 1000 Solubility of Au in alkaline solutions(0.25–0.9 m Na2S)
Elevated solubility growing with T andmNa2S
1972 Nikolaeva et al. 25–80 1 Potentiometry in KCl solutions (I ¼ 1) E8 of the reactionAuCl2
2 + e2 ¼ Auc + 2Cl2 at I ¼ 11972 Gadet & Pouradier 25 1 Potentiometry in Au–Cl–Br–NaOH solutions
using gold and calomel electrodes as afunction of pH
Equilibrium constants between AuX22,
AuXOH2, and Au(OH)22, where
X ¼ Cl or Br1973 Seward 175–250 1000 Solubility of Au in NaHS + NaCl solutions
(I ¼ 0.5) with PyPo buffer and pH208 �4–10First solubility study under fixed
f(O2) . AuHS0, Au(HS)22, and
Au2(HS)2S22 were suggested, and stabilityconstants of the two latter speciesdetermined between 175 and 2508C
1973 Henley 300–500 1000–2000 Solubility of Au in 0.5–2.0 m KCl solutions atMH and QMF buffers
Elevated Au solubility in chloride solutionsand its growth with increasing temperature
1977 Baranova et al. 25–200 svp Solubility of Au in H2O and NaOH with MtHebuffer
AuOH0 dominates below pH258 �12;Au(OH)2
2 appears at more alkaline pH1981 Belevantsev et al. 20 1 Solubility of Au2S(s) in sulphide solutions
at pH 6–12Stability constants of Au(HS)2
2,Au(HS)(OH)2, Au(HS)(H2S)0, andAu2(HS)2S22
1982 Nekrasov et al. 300 1000 Solubility of Au in Sb2S3–H2S(0.065 m)–HCl(0.1 m) solutions
Au solubility increases with increasing Sbconcentration; polynuclear Au–Sb speciesare suggested
1983 Baranova et al. 450 500 Solubility of Au in H2O at fixedlog f(O2) ¼ 217.3–27.8
AuOH0 is suggested
1984 Ryabchikov & Orlova 750 1500 Solubility of Au in acid chloride solutions inthe presence of granite melt and NNO buffer
Au solubility is up to 100s ppm; it increaseswith increasing Cl concentration
1985 Zotov et al. 300–500 500–1500 Au solubility in pure H2O, with Ni–NiO,Fe3O4–Fe2O3, and Cu–Cu2O buffers
Formation constants of the reactionAu + 0.5H2O + 0.25O2 ¼ AuOH0
1987 Wood et al. 200–350 svp Solubility of sulphide minerals (Fe, Sb, Zn, Pb,Ag, Bi, Mo) and Au in NaCl (0–5 m)solutions at PyPoMt buffer
AuCl22 and HAu(HS)2
0 are suggested
1989 Renders & Seward 25 1 Solubility of Au2S(s) in H2S–HS-solutions atpH 2–12 and total S 0.006–0.4 m
Stability constants of AuHS0, Au(HS)22, and
Au2S222
(Continued)
GO
LD
SP
EC
IAT
ION
ING
EO
LO
GIC
AL
FL
UID
S
at CA
PES on M
ay 1, 2014http://sp.lyellcollection.org/
Dow
nloaded from
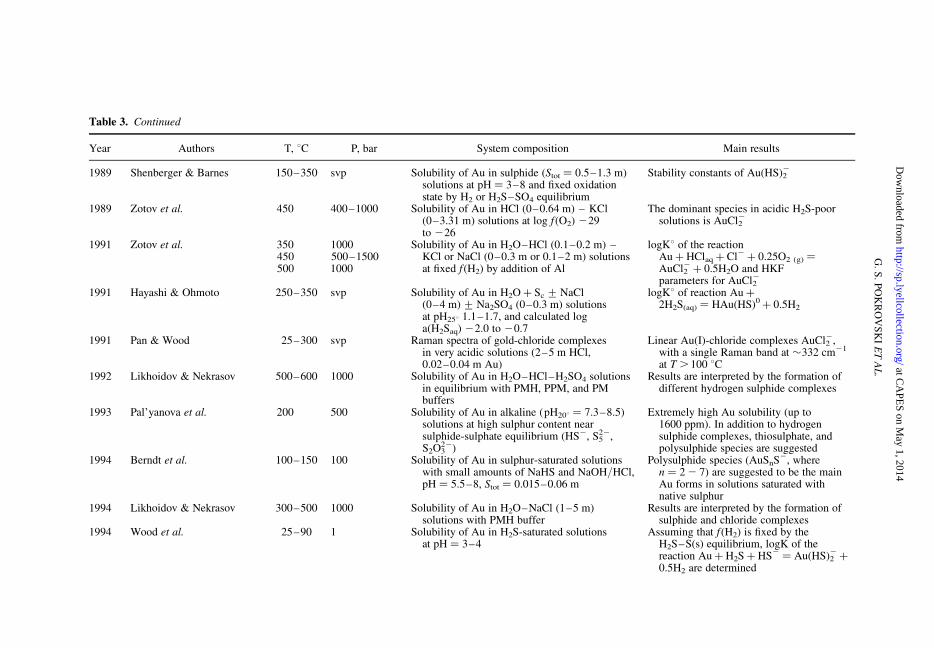
Table 3. Continued
Year Authors T, 8C P, bar System composition Main results
1989 Shenberger & Barnes 150–350 svp Solubility of Au in sulphide (Stot ¼ 0.5–1.3 m)solutions at pH ¼ 3–8 and fixed oxidationstate by H2 or H2S–SO4 equilibrium
Stability constants of Au(HS)22
1989 Zotov et al. 450 400–1000 Solubility of Au in HCl (0–0.64 m) – KCl(0–3.31 m) solutions at log f(O2) 229to 226
The dominant species in acidic H2S-poorsolutions is AuCl2
2
1991 Zotov et al. 350450500
1000500–15001000
Solubility of Au in H2O–HCl (0.1–0.2 m) –KCl or NaCl (0–0.3 m or 0.1–2 m) solutionsat fixed f(H2) by addition of Al
logK8 of the reactionAu + HClaq + Cl2 + 0.25O2 (g) ¼AuCl2
2 + 0.5H2O and HKFparameters for AuCl2
2
1991 Hayashi & Ohmoto 250–350 svp Solubility of Au in H2O + Sc + NaCl(0–4 m) + Na2SO4 (0–0.3 m) solutionsat pH258 1.1–1.7, and calculated loga(H2Saq) 22.0 to 20.7
logK8 of reaction Au +2H2S(aq) ¼ HAu(HS)0 + 0.5H2
1991 Pan & Wood 25–300 svp Raman spectra of gold-chloride complexesin very acidic solutions (2–5 m HCl,0.02–0.04 m Au)
Linear Au(I)-chloride complexes AuCl22,
with a single Raman band at �332 cm21
at T . 100 8C1992 Likhoidov & Nekrasov 500–600 1000 Solubility of Au in H2O–HCl–H2SO4 solutions
in equilibrium with PMH, PPM, and PMbuffers
Results are interpreted by the formation ofdifferent hydrogen sulphide complexes
1993 Pal’yanova et al. 200 500 Solubility of Au in alkaline (pH208 ¼ 7.3–8.5)solutions at high sulphur content nearsulphide-sulphate equilibrium (HS2, S5
22,S2O3
22)
Extremely high Au solubility (up to1600 ppm). In addition to hydrogensulphide complexes, thiosulphate, andpolysulphide species are suggested
1994 Berndt et al. 100–150 100 Solubility of Au in sulphur-saturated solutionswith small amounts of NaHS and NaOH/HCl,pH ¼ 5.5–8, Stot ¼ 0.015–0.06 m
Polysulphide species (AuSnS2, wheren ¼ 2 2 7) are suggested to be the mainAu forms in solutions saturated withnative sulphur
1994 Likhoidov & Nekrasov 300–500 1000 Solubility of Au in H2O–NaCl (1–5 m)solutions with PMH buffer
Results are interpreted by the formation ofsulphide and chloride complexes
1994 Wood et al. 25–90 1 Solubility of Au in H2S-saturated solutionsat pH ¼ 3–4
Assuming that f(H2) is fixed by theH2S–S(s) equilibrium, logK of thereaction Au + H2S + HS2 ¼ Au(HS)2
2 +0.5H2 are determined
G.
S.
PO
KR
OV
SK
IE
TA
L.
at CA
PES on M
ay 1, 2014http://sp.lyellcollection.org/
Dow
nloaded from

1994 Pan & Wood 200–350 svp Solubility of Au in H2O + NaOH(0.01–2 m) +Na2SO4(0.02–0.1 m) + H2S(0.3–2.1 m)solutions
logK of the reaction Au + H2S + HS2 ¼Au(HS)2
2 + 0.5H2
1995 Gammons & Williams-Jones 300 svp Solubility of Au–Ag alloys (XAu ¼ 0.3–1) andAgCl(s) in 3 m HCl
Equilibrium composition of Au–Ag alloysand logK of the reactionAu + 2Cl2 + H+ ¼ AuCl2
2 + 0.5H2
1995 Zotov & Baranova 23 & 80 1–500 Solubility of Au2S (23 8C) and AuAgS (80 8C)in near-neutral sulphide solutions
V8298(Au(HS)22)
1996 Benning & Seward 150–500 500–1500 Solubility of Au in aqueous solutions:(a) H2S(0.02–0.1 m Stot) + H2(0.035 bar),(b) H2S(0.01–0.1 m Stot) + H3PO4(0.001–0.14 m) + H2(0.035 bar), (c) H2S(0.02–0.11 m Stot) + NaOH(0.002–0.04 m) + H2(0.035 bar)
Stability constants of Au(HS)0 and Au(HS)22
1997 Gammons et al. 100–200 svp Disproportionation of AuCl22 into Au and
AuCl42 in HCl solutions (0.02–2 m)
logK of the reaction:3AuCl2
2 ¼ 2Au + AuCl42 + 2Cl2
1998 Baranova & Zotov 300–350 500 Solubility of Au in H2O, HCl (0.0002–0.2 m)and NaOH (0.05 m) with H2S(0.1 m) andH2(46–105 bar)
Stability constants of Au(HS) and Au(HS)22
1998 Gibert et al. 350–450 500 Solubility of Au in 0.5 m KCl solutions at QMFand PyPoMt or PyMtHe buffers
Reported stability constants of Au(HS)0 arein agreement with the data of Hayashi &Ohmoto (1991) when recalculated to theAuHS stoichiometry
1999 Loucks & Mavrogenes 550–725 1000–4000 Synthetic fluid inclusions method; solubility ofAu in H2O–KCl–HCl fluids at QMF andPPM buffers
Very high solubility of gold (up to1000 ppm). AuHS(H2S)3 is suggested andits HKF parameters determined
2000 Fleet & Knipe 100–400 1500 Solubility of Au in H2O–MgS solutions at veryhigh H2S concentrations (up to 20.3 m)
Stability constants of Au(HS)22
2000 Murphy et al. 25–250 svp, 850, 1500 Raman spectra of gold-chloride complexes Linear Au(I)-chloride complexes AuCl22,
with a single Raman band at �326 cm21
at T ¼ 250 8C2003a Stefansson & Seward 300–600 500–1500 Solubility of Au in H2O and NaOH (0–0.5 m)
solutions with 1028–1026 m H2, usinga flow-through reactor
logK of the reactionAu + H2O ¼ AuOH0 + 0.5H2
2003b Stefansson & Seward 300–600 500–1800 Solubility of Au in H2O + H2(4.1026–8 . 1024m) + HCl(0–1.4 m) +NaOH(0–0.3 m) + NaCl(0–1 m) solutionsusing in flow-through system
logK of the reactionAu + 2Cl2 + H+ ¼ AuCl2
2 + 0.5H2
(Continued)
GO
LD
SP
EC
IAT
ION
ING
EO
LO
GIC
AL
FL
UID
S
at CA
PES on M
ay 1, 2014http://sp.lyellcollection.org/
Dow
nloaded from

Table 3. Continued
Year Authors T, 8C P, bar System composition Main results
2004 Stefansson & Seward 100–500 500 Solubility of Au in H2O + H2 (1.6 . 1025–5.4 . 1024m) +
∑S(0.016–0.133 m)
+HCl(0–0.24 m) + NaOH(0–0.2 m)solutions in a flow-through system
Stability constants of Au(HS) and Au(HS)22
2004 Dadze & Kashirtseva 200–350 svp,300,1000
Solubility of Au in H2S-solutions:(a) thioacetamide (0.05–0.5 m) + NaOH(pHin ¼ 7.4–9) or HCl(pH258C ¼ 1.44),(b) thioacetamide (0.05–0.5 m) + NaOH(pH258C ¼ 7.4) + Na2SO4(0.05 m)
Stability constants of Au(HS) and Au(HS)22
2005 Tagirov et al. 350–500 500–1000 Solubility of Au in KCl(0.001–0.1 m) solutionsat QMF or QMA and PPM buffers
Stability constants of Au(HS) and Au(HS)22
2006 Tagirov et al. 25–250 svp Solubility of Au, Ag2S + Ag3AuS2 andAg3AuS2 + AgAuS (25 8C), and AgAuS(91–250 8C) in sulphide solutions (0.012–0.12 mS)
Stability constants of Au(HS)22
2009a Pokrovski et al. 25–500 600 Stability and structure of AuI and AuIII chloridecomplexes in Au–NaCl–HCl–AuCl4
2
aqueous solutions (pH , 2) using in situXAS spectroscopy
AuCl42 is unstable upon heating and is
rapidly transformed to Au(s) and AuCl22.
In the latter complex, Au is linearlycoordinated by 2Cl atoms at an averagedistance of 2.27 A.
2009b Pokrovski et al. 200–450 300–600 Au solubility and local structure in (a) S–NaOH, pH ¼ 5–8, (b) H2O–S(+H2SO4),pH ¼ 2–4, (c) H2O–S–Na2SO4 , pH ¼ 4–5aqueous solutions using in situ XASspectroscopy
First in situ measurement of Au–S speciesstructures at elevated T–P. In all systems,Au is linearly coordinated by 2S atoms at2.29 A. Au(HS)2
2 dominates at pH . 6,but at pH ,4–5, Au(HS)(H2S) and/orAu(HS)SO2 are tentatively suggested, andtheir stability constants reported.
svp, saturated vapour pressure of the aqueous solution; PPM, pyrite-pyrrhotite-magnetite; PMH, pyrite-magnetite-hematite; PM, pyrite-magnetite; MH, magnetite-hematite; QMF, quartz-muscovite-Kfeldspar; QMA, quartz-muscovite-andalusite; m, molality (number of moles of solute per 1 kg of water); I, ionic strength.
G.
S.
PO
KR
OV
SK
IE
TA
L.
at CA
PES on M
ay 1, 2014http://sp.lyellcollection.org/
Dow
nloaded from

Thermodynamic model. The thermodynamic prop-erties and stabilities of aqueous ions and complexesin hydrothermal fluids at elevated temperaturesand pressures are successfully described using theHelgeson–Kirkham–Flowers (HKF) equation ofstate (Helgeson et al. 1981; Tanger & Helgeson1988, Shock et al. 1989, 1997; Sverjensky et al.1997). The HKF model has been demonstrated tobe very versatile, being able to reproduce exper-imental stability constants of aqueous species andmineral solubilities in the liquid phase and densesupercritical fluid (density .�0.3–0.5 g/cm3) attemperatures from 0 8C to 1000 8C and pressuresat or above the vapour-liquid saturation curve(Psat) to 10 kbar (e.g. Sverjensky et al. 1997; Pok-rovski & Dubrovinsky 2011).
According to this model, any thermodynamicproperty of an aqueous species is regarded as asum of solvation and non-solvation (or structural)contributions (see Oelkers et al. 2009 for a recentreview). The former accounts for the interactionsof the species with the solvent molecules, whereasthe latter stems from the chemical bonding andmolecular structure of the species itself. The sol-vation term is treated using the electrostatic Born(1920) equation, in which the so-called Born par-ameter (v) of an ion is defined by its crystallo-graphic radius (Tanger & Helgeson 1988), whereasfor an aqueous complex, composed of differentions, empirical correlations between the entropyand the Born parameter are used (Shock & Helgeson1988). The non-solvation term is described by aseries of empirical equations with four adjustableparameters accounting for the species non-solvationvolume (a1, a2, a3, a4) and two adjustable par-ameters accounting for the species heat capacity(c1 and c2). To reduce the number of adjustableterms, the authors of the model proposed a seriesof empirical correlations amongst S, V, Cp at stan-dard state conditions (25 8C, 1 bar) and a1 – 4 andc1,2 coefficients, depending on the species chargeand based on a few aqueous species for which ther-modymanic properties were available in a wideT–P range. These correlations were updated withthe appearence of new experimental data, particu-larly for some neutral gases and complexes forwhich direct calorimetric (Cp) and volumetric(V ) measurements at elevated temperatures havebecome available in the last 15 years, allowing afar more precise refinement of v and a1 – 4 and c1,2
coefficients (e.g. Schulte et al. 2001; Perfetti et al.2008; Oelkers et al. 2009; references therein).These studies demonstrated that even neutral spe-cies have Born coefficients somewhat differentfrom zero, in contrast to the classical Born equa-tion that implies a zero v value for an unchargedspecies. However, these deviations from zero aresmall compared to v values for charged ions and,
for most mineral-fluid modelling purposes, maybe ignored.
Thus, to describe the thermodynamic propertiesof an aqueous component, one should specify itsstandard Gibbs free energy of formation andentropy at 25 8C, 1 bar (Df G8298 and S8298), andseven temperature- and pressure-independent par-ameters for the solvation part (Born parameter v),molar volume (a1, a2, a3, a4), and heat capacity(c1, c2). If data at different T and P are availablefor the stability constant of an aqueous complex(i.e. Gibbs free energy) or as direct measurementsof Cp and V for a neutral species, its HKF parametersmay be refined by adjusting them to match themeasured stability constants and using additionalcontraints from interparameter correlations. Forthis purpose, the OptimB computer code (Shvarov2010) has been used in the recent studies (Akinfiev& Zotov 2010; Tagirov et al. 2013), allowinguser-friendly and statistically robust HKF param-eter retrieval.
In the absence of reliable experimental high T–Pdata, the method of isocoulombic reactions may beapplied (Jackson & Helgeson 1985). It has beendemonstrated by a number of studies (e.g. Mesmeret al. 1988; Ruaya 1988) that, for an exchange reac-tion between aqueous species such as
AXmn + BYl
k = BXmn + AYl
k (1)
where A and B denote metals and X and Y ligandswith their corresponding charges (m, l) and stoi-chiometric coefficients (k, n), temperature andpressure dependences of the equilibrium constantof reaction (1) can be calculated, assuming nochange in the heat capacity (DrCp ≈ 0) and molarvolume (DrV ≈ 0) of the reaction. This approachwas successfully employed in recent papersreporting HKF parameters (Akinfiev & Zotov2001, 2010; Akinfiev & Tagirov 2006; Akinfievet al. 2006) and was used for some Au species asdetailed below.
An important issue that determines the deri-vation of HKF parameters from experimental Ausolubility studies is the thermodynamic propertiesof the major fluid constituents. These propertiesfor pure solid phases, pure gases (H2, O2, andH2S), ‘simple’ ions (Na+, Cl2, HS2, and OH2),and ion pairs (NaCl0 and NaOH0) were taken fromthe SUPCRT92 thermodynamic database (Johnsonet al. 1992) and its later extensions (Shock et al.1997; Sverjensky et al. 1997; http://geopig3.la.asu.edu:8080/GEOPIG/index.html) without anyrevision or adjustment, both because they are judgedto be reliable since based on a large experimentaldata set, and in order to maintain the internalconsistency. The stability constant of NaHS0 wasassumed to be equal to that of NaCl0, because
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

of similar ion radii and charges of Cl2 and HS2. TheHKF parameters of HCl0 were adopted fromTagirov et al. (1997). Thermodynamic propertiesof aqueous hydrogen sulphide H2S0(aq) and otherdissolved gaseous species were calculated usingthe equation of state (EoS) of Akinfiev & Diamond(2003) proposed for aqueous non-electrolytes. ThisEoS allows avoiding uncertainties inherent in theHKF model when predicting thermodynamic prop-erties of dissolved gases in water in near- and super-critical regions (Plyasunov & Shock 2001). It shouldbe noted that the HKF-based dataset for H2S0(aq)(Schulte et al. 2001) yields values of Gibbs freeenergy of aqueous H2S similar to those of theAkinfiev & Diamond (2003) model within 2 kJ/mol up to 500 8C. The model coefficients for thedissolved gases are provided in Table 4. A briefsummary of the optimization results from recentstudies (Akinfiev & Zotov 2001, 2010) for Auaqueous chloride, hydrogen sulphide, and hydroxidespecies is given below. The important feature ofthese compilations is that they used the originalexperimental solubility data points (where avail-able) rather than final stability constants reportedin the different experimental studies.
Optimization results and recommended thermodyn-amic properties of Au aqueous complexes
AuCl22. The available experimental studies
reporting the thermodynamic properties of the AuI
dichloride complex embrace a wide range of temp-eratures (25–500 8C) and pressures (1–1500 bar)(Nikolaeva et al. 1972; Gammons & Williams-Jones 1995; Zotov et al. 1995). Original experimen-tal data from these studies were recalculated toobtain Gibbs free energies of AuCl2
2 at the tempera-tures and pressures of these experiments (g8T,P).The obtained values of g8T,P were then used to
optimize values of DfG8298, S8298, C8p,298, and v ofthis complex by the OptimB computer code(Shvarov 2010). The value of the standard molalvolume of AuCl2
2 was estimated from the isocou-lombic reaction
Au(HS)−2 + AgCl−2 = AuCl−2 + Ag(HS)−2 (2)
and using the corresponding values of AgCl22 and
Ag(HS)22 from Akinfiev & Zotov (2001) and those
of Au(HS)22 as derived below. The stability con-
stants of AuCl22 reported by Stefansson & Seward
(2003b) are in good agreement with the present esti-mations above 450 8C, but by 1 log unit lower at400 8C. Figure 1 shows the optimization results interms of the difference in Gibbs energy values(g8T,P for AuCl2
2) between the model and experimen-tal datapoints for each original study as a functionof temperature.
AuHS0. A similar optimization procedure wasused to process available experimental data ofGibert et al. (1998), Baranova & Zotov (1998),Stefansson & Seward (2004), and Tagirov et al.(2005, 2006). The data of Hayashi & Ohmoto(1991) reporting Au solubility in sulphide- andsulphate-bearing solutions have been recalculatedto the AuHS stoichiometry. The data of Benning& Seward (1996) were excluded because they areinconsistent with more recent values reported bythe same group (Stefansson & Seward 2004), aswell as by Tagirov et al. (2005, 2006). The Gibbsfree energy value of AuHS0 at 25 8C and 1 bar hasbeen determined by Tagirov et al. (2006) to a highdegree of accuracy (+0.4 kJ/mol). The standardvalues of heat capacity and volume were estimatedfrom the isocoulombic reaction
AuCl−2 + AgHS0 = AuHS0 + AgCl−2 (3)
Table 4. Thermodynamic parameters of the Akinfiev & Diamond (2003) equation of state for gas-like neutral species inaqueous solution
Species Df G8g 298 j a b Df G8aq298 S8298 V8298 C8p, 298
cal . mol21 cm3. g21 cm3. K0.5. g21 cal . mol21 cal . mol21 K21 cm3. mol21 cal mol21 K21
H2 0 0.3090 28.4596 10.8301 4240 13.42 25.55 43.25O2 0 0.0600 29.7540 12.9411 3966 26.86 33.23 52.66H2S 28021 20.2017 212.8031 13.5396 26652 28.92 34.90 52.63CO2 294 254 20.085 28.8321 11.2684 292 256 28.47 32.71 50.35CH4 212 122 20.1131 211.8462 14.8615 28196c 21.55 37.18 59.63SO2 271 723 20.4234 214.1409 14.1959 271 840 37.79 38.51 56.32
The Gibbs function goaq(P, T) of an aqueous non-electrolyte at given temperature T(K) and pressure P(bar) in the framework of this EoS is expressed as
goaq(P,T) = go
g(T) − RT ln Nw + (1 − j)RT ln f o1 + RTj ln
RT
Mw
ro1
( )+ RTro
1 a + b103
T
( )0.5[ ]
,
where gog(T) stands for the Gibbs function of the corresponding species in the ideal gas state, Mw ¼ 18.0152 g mol21, Nw ¼ 1000/Mw mol,
f o1 and ro
1 are, respectively, the fugacity (in bars) and density (in g . cm23) of pure solvent (H2O) at given T and P, R ¼ 1.9872 cal . mol21 .
K21 and R ¼ 83.144 cm3 . bar . mol21 . K21 are the ideal gas constants, and j, a, and b denote empirical coefficients for each species in thetable. The data for aqueous species in the table (Df G8aq298, S8298, V8298, and C8p, 298) were computed using this EoS.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

and using the corresponding values of AuCl22
derived above and those of AgHS0 and AgCl22
from Akinfiev & Zotov (2001). It can be seen inFigure 1 that while the maximum deviations ing8T,P for AuHS do not exceed 1–2 kcal/mol, thetrends with increasing temperature of Gibert et al.’s
(1998) and Stefansson & Seward’s (2004) data aredifferent. The reasons of this remain unclear.
Au(HS)22. The available experimental data on
the stability of the AuI bis-hydrogen sulphidecomplex (Renders & Seward 1989; Shenberger &
Fig. 1. Differences in the Gibbs free energy of the indicated Au aqueous species between experimental values derivedfrom original studies (see section “Gold chloride, hydrogensulphide, and hydroxide species in aqueous solution anddense supercritical fluid” and Table 3) and calculated values using the recommended HKF parameters from Table 5(see text for details).
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Table 5. Standard thermodynamic properties (298.15 K and 1 bar) and HKF parameters of Au aqueous species recommended in this study
Species Df G8298* Df H8298* S8298† C8p,298
† V8298‡ a1
. 10§ a2. 1022* a3
‖ a4. 1024} c1
† c2. 1024} v . 1025*
AuCl22 236 795 246 664 47.16 226.4 68.6 11.4774 20.2425 22.2063 23.6158 27.0677 222.440 0.8623
AuHSaq 8344 13 193 50.86 1.8 56.5 9.4965 15.4057 20.3052 23.1459 238.1356 19.6524 0.0
Au(HS)22 3487 4703 77.46 3.3 75.1 12.3373 22.3421 3.0317 23.7026 253.6010 31.4030 0.7673
Au+ 39 000 45 182 16.50 0.58 31.4 6.1161 7.1520 2.9389 23.0747 7.7764 22.9158 0.1390
Au(OH)22 266 553 294 664 12.90 233.1 24.6 5.4533 5.5338 3.5749 23.0078 25.5091 29.7771 0.8465
AuClaq 23184 22077 41.73 215.1 53.1 9.0294 14.2654 0.1430 23.3687 22.6993 26.1162 0.0
AuOHaq 232 716 241 533 21.89 211.1 32.4 6.1937 7.3415 2.8644 23.0825 23.0370 23.9635 0.0
*cal . mol21.†cal . mol21K21.‡cm3 . mol21.§cal . mol21 . bar21.‖cal . K . mol21 . bar21.}cal . K . mol21.
G.
S.
PO
KR
OV
SK
IE
TA
L.
at CA
PES on M
ay 1, 2014http://sp.lyellcollection.org/
Dow
nloaded from

Barnes 1989; Berndt et al. 1994; Zotov & Baranova1995; Benning & Seward 1996; Baranova &Zotov 1998; Fleet & Knipe 2000) exhibit significantscatter; consequently, in the optimization we reliedon the recent data of Stefansson & Seward (2004)and Tagirov et al. (2005) reported in a wide T–Prange, together with low-temperature (25–150 8C)stability constants and the thermodynamic proper-ties of Au2S(s) provided by Tagirov et al. (2006).The value of V8298 for Au(HS)2
2 was adopted as amean of values reported by Benning & Seward(1996) and Baranova & Zotov (1998) based onpressure dependence of measured Au solubility,which is in agreement in both studies. At present,amongst all Au aqueous species, the stability ofAu(HS)2
2 appears to be the best-constrained in awide temperature (20–500 8C) and pressure (1–1000 bar) range (Fig. 1).
AuOH0. Experimental data on the hydroxidecomplex were reported by Zotov et al. (1995) in alimited T range (300–500 8C). Processing thesedata using the OptimB code yields Df G8298 andS8298 values, while C8p,298, and V8298 were estimatedusing the isocoulombic reaction
AuCl−2 + AgOH0 = AuOH0 + AgCl−2 (4)
with corresponding values of AgOH0 and AgCl22
from Akinfiev & Zotov (2001) and of AuCl22 as
derived above. Data of Stefansson & Seward(2003a), not used in the optimization, are in agree-ment with our predictions within better than0.5 log units in terms of AuOH0 concentration.
AuCl0, Au+, Au(OH)22. The thermodynamic
properties of these species are poorly known, andtheir formation constants have only been esti-mated at ambient conditions. The Df G8298 valueof Au(OH)2
2 was obtained from the estimate ofPeshchevitskii et al. (1976) for the reaction
AuCl−2 + 2OH− = Au OH( )−2 +2Cl− (5)
A value of Df G8298 ¼ 39.0 kcal/mol for Au+wasadopted from the SUPCRT92 database (Johnsonet al. 1992; Shock et al. 1997), while its Bornparameter was calculated using crystallographicion radius RAu+ ¼ 1.51 A (Huheey 1983). TheDf G8298 value of AuCl0 was taken from Sverjenskyet al. (1997). Other thermodynamic parameters forthese species were estimated using the followingisocoulombic reactions
AuCl−2 + Ag OH( )−2 = Au OH( )−2 +AgCl−2 (6)AuCl−2 + Ag+ = Au+ + AgCl−2 (7)
AuCl−2 + AgCl0 = AuCl0 + AgCl−2 (8)
and the corresponding S, Cp, and V values ofAg(OH)2
2, AgCl22, and Ag+ from Akinfiev &
Zotov (2001). The obtained thermodynamic pro-perties of Au aqueous complexes are reportedin Table 5, and the stability constants of the fourmain Au species (see reactions 10a to 10f below)are shown in Figure 2 as a function of temperatureat 1 kbar pressure.
The role of other sulphur-bearing forms in
hydrothermal transport of gold
Other hydrogen sulphide complexes. The major-ity of available solubility experiments in H2S-dominated solutions overviewed above suggest theformation of monohydrogen sulphide species of pre-sumed stoichiometry AuHS at acidic conditions,and of bis-hydrogen sulphide species, Au(HS)2
2
at neutral-to-basic pH. The latter species has beenrecently confirmed by in situ XAS measurementsof Au local structure and solubility in S–NaOH–H2O solutions (0.5–3 m S, pH �5–8) from 200to 450 8C (Pokrovski et al. 2009b). These exper-iments showed that Au is coordinated by two sul-phur atoms in a linear geometry at Au–S averagedistances of 2.29 + 0.01 A, and that the bulk solu-bility measured from the absorption edge height ofthe spectra (Figs 3 & 4) agrees within errors with
Fig. 2. Thermodynamic equilibrium constants of theformation reactions of the main Au chloride, hydroxide,and hydrogen sulphide complexes indicated in the figureas a function of temperature at 1000 bar according to therecommended HKF parameters in Table 5. Lines denotethe reaction constants between metallic gold and thecorresponding aqueous species from section “Majorparameters controlling Au transport and precipitation inhydrothermal fluids” (reactions 10).
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

the thermodynamic properties of Au(HS)22 recom-
mended above from solubility measurements inhydrothermal reactors.
However, at acid conditions (in systems such asS–H2O, S–Na2SO4–H2O, pH , 4–5), the XASstructural and solubility data of Pokrovski et al.(2009b) are inconsistent with the dominant forma-tion of AuHS0. Their data reveal linear S–Au–Sstructures with Au–S distances of 2.29 A, verysimilar to the experiments at neutral-to-basic pH,and solubilities of 1–2 orders of magnitude higherthat those calculated using the thermodynamic pro-perties of AuHS recommended above (Table 5).These findings were tentatively interpreted byPokrovski et al. (2009b) by the formation of theneutral AuHS(H2S)0 complex. Such species wasalso suggested by Hayashi & Ohmoto (1991)from solubility measurements in similar solutions.Wood et al. (1987) also assumed this species tobe dominant in acidic Cl-free solutions bufferedwith sulphide mineral assemblages. Other neutralAu species with H2S, such as tetrakis-hydrogensulphide, AuHS(H2S)3
0, have been suggested byLoucks & Mavrogenes (1999) in high-temperaturemetamorphic fluids on the basis of Au solubilityexperiments in FeS–FeS2-silicate assemblages
at temperatures 500–700 8C and pressures 2–4 kbars. However, this species was confirmedneither by XAS data of Pokrovski et al. (2009b)at lower T–P, nor by first-principles moleculardynamics simulations (MD) of Liu et al. (2011),who showed that this complex breaks down toAuHS(H2S) and two H2S molecules. In addition,the HKF parameters of the tetrakis-hydrogen sul-phide complexes suggested by Loucks & Mavro-genes (1999) yield calculated solubilities of 1–2orders of magnitude higher than those measuredby XAS at temperatures below 450 8C (Fig. 4; seePokrovski et al. 2009b for detailed discussion andcomparison).
However, three limitations do not yet allowdefinitive conclusions as for the dominant presenceof AuHS(H2S)0 in acidic S-rich (H2S . 0.1 m)solutions: (1) The scatter of datapoints in the solu-bility studies discussed above makes it difficult toobtain unambiguous derivation of complex stoi-chiometry. (2) The intrinsically poor sensitivity ofXAS to long distances, light neighbours (like H),and/or beyond-the-first shell neighbours aroundthe absorbing atom does not allow definitive con-straints on the complex stoichiometry beyondthe [S–Au–S] cluster (Pokrovski et al. 2009b).
Fig. 3. Local structure of AuI in sulphur-rich hydrothermal fluids revealed by in situ X-ray absorption spectroscopy(Pokrovski et al. 2009b). The left panel shows Fourier transform (FT) magnitudes of selected EXAFS spectra for neutraland acidic solutions of 0.5 m dissolved sulphur at 600 bar and indicated compositions and temperatures. Vertical dashedlines mark signals arising from the first S shell and from multiple scattering within the linear S–Au–S cluster. The rightpanel shows possible corresponding Au–S aqueous species whose structures and Au–S distances (in A) were calculatedusing first-principles quantum chemistry. While the structural results at pH . 5 are consistent with Au(HS)2
2, spectraat acidic pH do not detect the AuHS complex inferred from available solubility data. By contrast, they indicate theformation of species containing two sulphur atoms like bis-hydrogen sulphide, mixed HS–SO2, polysulphide, orthiosulphate complexes. The insufficient knowledge of sulphur speciation itself and low spectral resolution ofbeyond-the-first atomic shells do not allow distinguishing between those species (see Pokrovski et al. 2009b for details).
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

(3) Complex and poorly known sulphur speciationitself in the studied solutions does not allow exclu-sion of complexes with sulphur forms other thanhydrogen sulphide. The following section discussesthe possible role of such complexes.
Polysulphide and thiosulphate complexes. Theseligands often form as intermediate reaction productsin S-bearing solutions, in which sulphide andsulphate coexist, and during sulphur oxidation atlow-to-moderate temperatures where meta-stableequilibria may occur both in nature and experiments(e.g. Ohmoto & Lasaga 1982). Both types of ligandshave strong affinity for AuI (Table 2). The stabilityof Au(S2O3)2
32 is comparable to that for Au(HS)22
at ambient conditions (log10K for both species�26–30, see Table 2). According to Webster(1986), the thiosulphate complex may be responsi-ble for gold remobilization during low-temperatureoxidation of sulphide ores. For example, thiosul-phate concentrations in some geothermal springsat T ≤ 100 8C attain values comparable to thoseof H2S (Migdisov & Bychkov 1998; Xu et al.1998) and thus may bind Au and similar metals
(e.g. Ag). Thiosulphate is, however, unstable atacidic pH below 100 8C, breaking down to nativesulphur and sulphate (e.g. Kaasalainen & Stefansson2011).
Polysulphide Au complexes (AuSnS22, wheren ¼ 2–7) were reported to form at moderatetemperatures (100–150 8C) in Au solubility experi-ments in solutions saturated with elemental sul-phur (Berndt et al. 1994); however, because of thecomplexity of the system containing comparableconcentrations of different polysulphides and thio-sulphate, no preference could be given to any Auspecies. Other solubility studies at 200–300 8Cnear the sulphate-sulphide equilibrium also evokedpolysulphide and/or thiosulphate complexes toexplain the elevated Au solubilities (Pal’yanovaet al. 1993; Dadze & Kashirtseva 2004). These com-plexes were suggested to compete with hydrogensulphide for Au during its transport in near-surfaceepithermal and active geothermal systems. Unfor-tunately, a quantitative estimation of the impactof such ligands on Au transport by hydrothermalfluids is greatly hampered by the lack of data onthe stability and amounts of these sulphur forms at
Fig. 4. (a) Determination of the concentration of Au dissolved in the fluid phase can be done from the amplitude of theabsorption edge height (see Pokrovski et al. 2005b, 2006, 2009b for details). (b) and (c) Evolution of Au molality,measured from the spectrum absorption edge height as a function of time in the presence of an excess of native gold intwo experiments at neutral (b) and acidic (c) pH, shown in Figure 3 above. Each symbol corresponds to an XAS scan;arrows indicate temperature changes during the experiments; dashed horizontal lines (where present) denote thesteady-state Au concentration at each temperature. Grey error bars stand for the range of Au dissolved concentrations inequilibrium with metallic gold as calculated using the stability constants of Au(HS)2
2 and AuHS from availableliterature sources reviewed above. Double-end arrows in panel (c) represent the range of calculated Au concentrationsusing the stability constants of AuHS(H2S) derived from the batch-reactor solubility experiments of Hayashi & Ohmoto(1991) carried out in similar systems as those in the XAS experiments of Pokrovski et al. (2009b).
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

above-ambient temperatures and pressures. Recentin situ experiments indicate that such forms maybe stable in S-rich fluids at elevated temperatures.Pokrovski & Dubrovinsky (2011) and Pokrovski& Dubessy (2012), using in situ Raman spectros-copy in diamond-anvil and silica-capillary cells,showed that an unexpected sulphur species, the tri-sulphur radical ion S3
2, forms rapidly and reversiblyin sulphide-sulphate solutions between 250 8C and500 8C and pH ≥ 2–3 in a wide range of pressures,from Psat to 50 kbar. This trisulphur species, knownby chemists since the 1970s to exist in a variety ofionic non-aqueous liquids, silicate glasses, alkali-metal sulphide batteries, alkali chloride melts, andultramarine pigments (Chivers & Elder 2013),is now suggested to play a role in natural hydrother-mal fluids. In particular, by analogy with ‘classical’polysulphide forms (SnS22, see above), this speciesis expected to form stable complexes with AuI andthus may probably account, at least partly, for theAu elevated solubilities obtained by Hayashi &Ohmoto (1991), Loucks & Mavrogenes (1999) andPokrovski et al. (2009a). Detailed account of therole of this new sulphur form in Au transport bynatural hydrothermal fluids requires accurateknowledge of the S3
2 stability domain and speciallydesigned spectroscopic and solubility experimentson Au within this domain.
Sulphur dioxide and sulphites. Another importantsulphur ligand that might impact gold transport inhigh-T magmatic-hydrothermal fluids is sulphite(SO2, and its dissociation products, HSO3
2 andSO3
22). This sulphur form is particularly abundantat acidic pH and temperatures above 400 8C underoxidizing conditions typical of porphyry Cu–Audeposit formation (Kouzmanov & Pokrovski2012). Note that the stabilities of AuI sulphite andthiosulphate complexes at ambient conditions aresimilar (Table 2). Gold-sulphite or -SO2 speciesmay equally account for the observed Au twofoldS coordination and elevated solubilities comparedwith AuHS0 in Pokrovski et al.’s (2009b) exper-iments. Because metal-ligand interactions generallybecome ‘harder’ with increasing temperature,following the decrease of the solvent dielectric con-stant and thus reinforcement of coulombic attrac-tions (e.g. Crerar et al. 1985; Brimhall & Crerar1987), high-temperature conditions might be ben-eficial for Au complexing with SO2, which is a‘harder’ ligand than H2S. The formation of Au–SO2 species is thus expected to be favouredin magmatic fluids typical of active convergentmargins where SO2 often largely dominates overH2S (e.g. Borisova et al. 2006; references therein).The geological spatial and temporal links, widelydocumented in these areas between magmaticplutons and overlaying epithermal high sulphidation
Au–Cu deposits, are often interpreted by the trans-port of Au and Cu by oxidizing, acidic magmaticfluid or vapour phases enriched in SO2 (e.g. Heden-quist & Lowenstern 1994; Hedenquist et al. 1998;Heinrich 2007; Pokrovski et al. 2008a). No directdata exist on Au complexing by SO2 for magmaticfluids; however, Zajacz et al. (2010, 2011) reportedvariable Au metal solubility from a few ppm to100s ppm in aqueous SO2-dominated NaCl-bearingfluids at 1000 8C and 1.5 kbar from synthetic fluidinclusion experiments at oxidizing conditions (upto �NNO + 2.0). Although these Au concentrationsare lower than those measured in H2S-dominatedfluids (NNO 2 0.4) of similar total salt and sulphurcontent from those studies, sulphur speciation inthese systems is insufficiently constrained since itwas based on calculations using thermodynamicproperties of ideal gas species H2S(g) and SO2(g).Thus, as for other sulphur species of intermediatevalences, the major obstacle in quantifying thecontribution of SO2 in Au transport is the paucityof high T–P thermodynamic and PVTX data forSIV aqueous species and their partitioning betweenvapour, brine, and silicate melt. One of the majornear-future research challenges on magmatic-hydrothermal systems will be to assess the identityand amounts of the different complexes formed bysulphur in aqueous fluid, vapour phase, and silicatemelt, in particular using in situ spectroscopicmethods, both in model laboratory systems andnatural fluid/melt inclusions.
Vapour transport and vapour-liquid
partitioning of gold
One of the major last-decade advances in under-standing magmatic-hydrothermal systems, parti-cularly those hosting porphyry Au–Cu–Mo andassociated epithermal Au–Cu deposits, is the recog-nition that the vapour phase can transport significantquantities of metals including Au (Heinrich et al.1992, 1999, 2004; Audetat et al. 1998; Ulrichet al. 1999). This has motivated experimental andtheoretical research that allowed new constraintson the physical and chemical factors affecting thevapour-phase transport and vapour-brine partition-ing of Au and associated metals. These factors arediscussed in this section with a particular emphasison Au, whereas other metals are briefly mentionedfor comparison; they have been a subject of moredetailed recent reviews (e.g. Williams-Jones &Heinrich 2005; Simon & Ripley 2011; Kouzmanov& Pokrovski 2012).
Lessons from volcanic gases and natural fluidinclusions. Extensive work on volcanic gases andfumarole condensates and sublimates since the mid-20th century suggests selective vapour transport of
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

many metals (Symonds et al. 1987, Symonds &Reed 1993; Hedenquist et al. 1994; Taran et al.1995, 2001; Pokrovski et al. 2002a). Despite largevariations in a few cases, the majority of volcanicgas and fumarole vapour samples show meanmetal concentrations typically of a few ppm formany economic metals (Zn, Pb, Cu, Sn, Mo, Ag,As). By contrast with most metals and metalloids,for which large databases on their sublimationmineralogy and fumaroles contents are availablefrom many active volcanoes all over the world(e.g. Williams-Jones & Heinrich 2005; referencestherein), only a few studies report findings of Auas native metal or Cu–Ag–Au alloys in sublimatescollected on active volcanoes such as La Fossa,Italy (Filignati & Sbrana 1998), Colima, Mexico(Taran et al. 2000), and Kudryavy, Kurile Islands(Yudovskaya et al. 2006). The reported analysesof Au in fumarole vapour condensates are scarceand exhibit large variations, which reflect both thedifficulties of accurate high-temperature samplingand imperfections of chemical treatments andanalytical techniques for this poorly soluble tracemetal. For example, Au contents reported in vol-canic vapour condensates vary from �0.001 ppb(Hedenquist et al. 1994) to 5–20 ppb (Gemmel1987; Quisefit et al. 1989). More recent reports,from Colima and Kudryavy volcanos, however, sys-tematically show Au concentrations in condensatesless than 1 ppb (Taran et al. 2001; Yudovskayaet al. 2008; Chaplygin 2010), which are comparablewith Au contents in unaltered volcanic rocksfrom these areas (typically 0.1–1 ppb, Taran et al.2001) and are somewhat lower than the Au meancrustal abundance (1–2 ppb).
Gas phase transport and precipitation of manymetals and metalloids have been the subject ofextensive thermodynamic modelling based on gas-sublimate chemical equilibria in water-free systems(e.g. Symonds et al. 1987, 1992; Symonds & Reed1993; Tkachenko et al. 1999; Churakov et al.2000; Pokrovski et al. 2002a; references therein)and using chloride, sulphide, oxide/hydroxide, ornative metal gaseous species whose thermodynamicproperties are available in large databases (e.g.JANAF – Chase 1998; Ivtanthermo – Belov et al.1999; GASTHERM – Symonds & Reed 1993). Incontrast, limited modelling was devoted to Au,mostly because of the lack of data on its gaseousspecies. Gold gaseous species are not reported inthose databases, and the available data are limitedto Au, AuH, and AuS from the compilations ofPankratz et al. (1987), Pankratz (1982), and Barin(1995). To the best of our knowledge, the onlystudies that attempted to thermodynamically modelAu volcanic gas chemistry are those of Taran et al.(2000, 2001). These authors used the above ther-modynamic data to estimate Au transport and
precipitation by Colima volcanic gases and foundqualitative agreement between the Au distributionobserved in sampling silica tubes and their thermo-dynamic predictions. Other Au gas species, mostlyexplored by chemists in the 1970s, under HCl-rich highly oxidizing conditions include Au2Cl2(g)and Au2Cl6(g) (Hager & Hill 1970; Landsberg &Hoatson 1970; James & Hager 1978; Tagirovet al. 1993). However, the formation of such poly-meric species requires Au concentrations muchhigher than those in volcanic gases; it is also unli-kely that AuIII chloride complexes would be stablein natural volcanic systems where AuI is expectedto be the dominant gold redox state (see section‘General features of gold aqueous chemistry’). Itis clear that quantitative modelling of Au transportby volcanic gases awaits accurate thermodynamicdata for key Au gaseous species, such as hydroxides,chlorides, and sulphides.
The gold contents of the volcanic gas phaseboth measured and modeled thermodynamicallyare, however, 2–5 orders of magnitude lower thanthose measured over the last decade in vapour-likeinclusions from magmatic-hydrothermal deposits(Heinrich et al. 1999; Ulrich et al. 1999, 2001;Audetat & Pettke 2003; Kehayov et al. 2003;Baker et al. 2004; Williams-Jones & Heinrich 2005;Klemm et al. 2007, 2008; Seo et al. 2009, 2012).These inclusions, together with their high-salinityliquid counterparts, trapped in quartz and other min-erals during their growth from hydrothermal fluids,record vapour-liquid phase immiscibility eventsthat a saline metal- and volatile-bearing magmaticfluid undergoes upon its cooling and decompressionwhen rising to the surface (Hedenquist & Lowen-stern 1994; Heinrich et al. 1999; Kouzmanov &Pokrovski 2012; references therein). These eventsresult in the formation of assemblages of syngeneticinclusions of low-salinity aqueous vapour (typically,�10 wt% NaCl equiv, density �0.01–0.3 g/cm3) and hypersaline liquid (typically �20–80%NaCl equiv., density .0.5–0.6 g/cm3), producedby fluid unmixing. Metal partitioning betweenthese two phases is found to be in sharp contrast.Most base, alkali, and alkali earth metals (Na,K, Sr, Fe, Zn, Pb) are preferentially enriched inthe liquid, with vap/liq partitioning coefficientsKvap/liq typically less than 0.1, whereas Cu and Au(+As, +Mo) show a significantly stronger pre-ference for the vapour phase, with Kvapour/liquid
values from 0.1 to 100 (see compilation of Kouzma-nov & Pokrovski 2012 and Fig. 5). Among thesemetals, Au exhibits the highest K values, attainingalmost 100 in favour of the vapour phase (Fig. 5),with absolute Au concentrations as high as 10 ppm(see also section ‘Comparison of thermodynamicpredictions with natural data from porphyry Cu–Au–Mo and related deposits’ and Fig. 12 below).
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Such Au concentrations cannot be explainedby the available data on Au gas species in drysystems discussed above that yield ppb levels ofAu in H2S–HCl aqueous vapour saturated with themetal. The main difference between near-surfacevolcanic gases and hydrothermal vapours is thedensity, which increases by a factor of 100 fromthe surface (dvap ≈ 0.001 g/cm3 at ,10 bar) to afew km of depth (dvap ≈ 0.01–0.4 g/cm3 at 100–1000 bar). Vapour density thus appears to be themajor parameter affecting gold and other mineralssolubility in the vapour-phase domain, as will beshown below.
Solvation control on Au solubility in vapour. Recentsolubility studies of Au and CuCl, AgCl, SnO2, andMoO3 solids in unsaturated water vapour (i.e. atpressures below the vapour-liquid saturation curveof water or H2O-salt solution, Psat) have confirmedthat the dominant control on metal solubility iswater pressure (Williams-Jones & Heinrich 2005;Rempel et al. 2009; and references therein). Theyshow that at water pressures of a few hundred barsand temperatures of 200–400 8C, the solubility ofa solid oxide, chloride, or native metal is many
orders of magnitude higher, compared to thevolatility of this solid in a H2O-free system.More detailed overviews of Cu, Ag, Sn, and Mosolubility in these systems can be found elsewhere(Williams-Jones & Heinrich 2005; Kouzmanov &Pokrovski 2012; Pokrovski et al. 2013b); here, wefocus on the Au data.
Solubility experiments of native gold in under-saturated vapour of the HCl–H2O, H2S–S, andH2S–S–H2O systems were conducted by Archibaldet al. (2001) and Zezin et al. (2007, 2011) at temp-eratures 300–360 8C and pressures below Psat.These experiments showed enhanced volatility ofAu in the presence of water, which was explainedby the solid-gas reactions
Au(s) + HCl(gas) + mH2O(gas)
= AuCl · mH2O(gas) + 0.5H2(gas) (9a)Au(s) + H2S(gas) + mH2O(gas)
= AuS · mH2O(gas) + H2(gas) (9b)
where m is the apparent hydration number, whichdepends on the species identity, temperature, andpressure. With increasing pressure, in the chloridesystem it systematically increases from 3 to 5 and,in the sulphide system, from 0 to �2 in the T–Prange investigated. In H2O-poor experiments inthe presence of large amounts of hydrogen sul-phide gas (up to �100 bar H2S), Au solubility wasdescribed by the solvation reaction
Au(s) + (n + 1)H2S(gas)
= AuS · nH2S(gas) + H2(gas) (9c)
where n is close to 2 (Zezin et al. 2007).These studies reveal three fundamental controls
on vapour-phase transport of gold: (1) In theaqueous vapour phase, Au forms complexes withthe same ligands as in aqueous solution (chloride,sulphide, or water); however, Au ligation numberand Au redox state might be different (see below).(2) In contrast to the aqueous solution or densesupercritical fluid, in which charged Au speciesdominate (AuCl2
2, Au(HS)22, see section, ‘Gold
chloride, hydrogen sulphide, and hydroxide speciesin aqueous solution and dense supercritical fluid’),the major vapour species are likely to be uncharged(at least in unsaturated vapour and moderate PH2O
below the water critical point); this is in agreementwith the low dielectric constant of the vapourfavouring ion association. (3) As in aqueous sol-ution, the gold complexes in the vapour are solvatedby water molecules; the higher the pressure (orsolvent density), the more metal can be dissolvedin a vapour-solid system.
Fig. 5. Concentration ratios (equivalent to vapour-liquidpartition coefficient, K ¼ Cvapour/Cliquid) for sulphur,gold, and associated metals measured in coexistingvapour and hypersaline liquid inclusions from boilingassemblages in quartz from porphyry Cu–Au–Mo andrelated deposits, obtained by in situ laser ablation ICPMS(modified from Kouzmanov & Pokrovski 2012). Data arefrom Heinrich et al. (1999), Ulrich et al. (1999, 2001),Audetat & Pettke (2003), Kehayov et al. (2003), Bakeret al. (2004), Williams-Jones & Heinrich (2005), Klemmet al. (2007, 2008), and Seo et al. (2009, 2012). Note thatthe enhanced partitioning of Cu into the vapour phase (Kup to 100) is likely to be a post-entrapment artifact due toCu diffusion through quartz (see text and Lerchbaumer &Audetat 2012 for details).
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

The main limitation of this hydration/solvationapproach is a relatively narrow temperature andpressure range of available measurements andlarge changes in apparent hydration numbers withpressure for chloride species, which makes thepractical application difficult at the wide T–Prange of hydrothermal gold deposit conditions. Inaddition, gold coordination number and valencestate in S-bearing gaseous species suggested bythese studies from analyses of bulk Au concentra-tion dependence v. H2O and H2S fugacity remainpoorly constrained. For example, reactions (9b, c)require Au redox state to be +2 and coordina-tion number 3 in the species formed. This is incontradiction to both the dominant Au redox stateof +1 and typical coordination numbers of 2 influids and minerals at hydrothermal conditions(see above). Gold solubility, calculated by Zezinet al. (2011) using the stability constants of reac-tions (9a, b), appears, however, to be less than0.5 ppb Au in a typical hydrothermal vapour orliquid phase under porphyry-epithermal conditions(T � 350 8C, PH2O
� 100–400 bar, H2S concen-trations � few wt% as found in natural fluidinclusions, e.g. Seo et al. 2009). This calculatedAu concentration is many orders of magnitudelower than that measured in natural fluid inclusionsfrom porphyry-epithermal Au–Cu deposits (100–10 000 ppb; Seo et al. 2009; Kouzmanov & Pok-rovski 2012). These discrepancies imply that otherspecies control gold transport in natural liquid-likefluids and vapours at these conditions. More work,combining solubility with in situ spectroscopictechniques and molecular modelling, is needed tobetter understand the chemical state and molecularstructure of gold in hydrothermal vapour phases.
Fluid-density control on vapour-liquid partitioningof gold at hydrothermal conditions. Further insightinto the vapour transport of gold and associatedmetals (As, Cu, Ag, Zn) has recently been offeredby direct measurements of vapour-liquid partitioncoefficients of metals in model two-phase salt-watersystems analogous to brine- and vapour-like fluidinclusions from porphyry Cu–Au deposits at temp-eratures from 300 8C to 500 8C and pressures fromPsat to �500 bar (Pokrovski et al. 2005a, 2008a, b;Pokrovski 2010; references therein). These worksdemonstrate that the vapour-liquid distributionof elements obeys simple relationships involvingthe densities of the coexisting vapour and liquidphases. Figure 6 shows that, on a logarithmic scale,the partition coefficient of each metal (Kvapour/liquid,which is the ratio of metal mass concentrations inthe coexisting phases, Cvapour/Cliquid) is linearlyproportional to the ratio between the vapour andliquid densities (dvapour/dliquid), which are well-known in the H2O–NaCl system (e.g. Driesner &
Heinrich 2007). All lines tend to converge to thecritical point of the system where the concentra-tions are identical in both phases and the partitioncoefficient is, by definition, equal to one. Such‘ray diagrams’ have long been known for partition-ing of salts and acids between vapour and aqueoussolution (e.g. Styrikovich et al. 1955; Alvarezet al. 1994); they stem from classical thermodyn-amics and statistical mechanics showing that thehydration energy of a solute evolves linearly withthe solvent density (Mesmer et al. 1988; Andersonet al. 1991; Palmer et al. 2004). The relationshipsin Figure 6 confirm the validity of this model for avariety of metals and metalloids in a wide tempera-ture range. They support the findings in unsaturatedvapour systems (see above) and demonstrate thatwater-solute interaction (or hydration) is a keyfactor controlling metal vapour-phase solubility
Fig. 6. Vapour-liquid partition coefficients,log Kvapour/liquid ¼ log (mvapour/mliquid), where m is thenumber of moles of the element per 1 kg of fluid in thecorresponding phase, of selected metals and metalloidsat the two-phase equilibrium in the systemH2O + NaCl + KCl + HCl at temperatures between250 and 450 8C as a function of the vapour-to-liquiddensity ratio. Symbols stand for experimental datafrom the following sources: AsIII, CuI, AgI, andAuI–Pokrovski et al. (2005a); SbIII–Pokrovski et al.(2005a, 2008b); La–Shmulovich et al. (2002). Na, Fe,and Zn (omitted for clarity) plot close to Cu (Pokrovskiet al. 2005a). Limited data for MoVI (Rempel et al. 2009)and Pb (Pokrovski et al. 2008a) plot close to As and Cu,respectively (omitted for clarity). Lines represent theregression through the origin (i.e. critical point, c.p.) ofthe experimental data for each element using theequation log K ¼ n × log (dvapour/dliquid), where n is anempirical coefficient for each metal independent oftemperature and salinity (Pokrovski et al. 2005a).
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

and vapour-liquid partitioning. The following quali-tative trend of element volatility in a two-phasewater-salt system (in the order of decreasingKvapour/liquid values) may be established: B ≈AsIII . Si ≈ SbIII ≈ AuI . CuI ≈ FeII ≈ Na ≈K ≥ Zn . Ag ≈ Cd ≈ REE (Pokrovski 2010;Kouzmanov & Pokrovski 2012). Metalloids thatform neutral hydroxide species both in the vapourand liquid are distinctly more volatile than mostmetals forming charged chloride complexes in theliquid phase. Gold that forms predominantlyAuCl2
2 in the S-free liquid phase, according to avail-able thermodynamic data discussed above(section “Gold chloride, hydrogensulphide, andhydroxide species in aqueous solution and densesupercritical fluid”), appears to be the most volatilemetal investigated so far, as shown by the threeavailable experimental datapoints (Fig. 6) obtainedat 450 8C in the NaCl–HClH2O system in the pres-ence of the Fe2O3–Fe3O4 redox buffer (Pokrovskiet al. 2005a). The exact nature and composition ofAu species in the vapour phase remain unclear,and available data for AuCl . mH2O(gas) (Archibaldet al. 2001) do not allow reliable extrapolationsabove 350 8C. Whatever the exact species stoichi-ometry for Au and other metals in the vapourphase, it can be seen in Figure 6 that, contrary toAs (+B, +Mo, not shown) whose Kvapour/liquid
values approach one, gold and other metals areunable to enrich the vapour phase relative to thecoexisting liquid. All of them are concentrated inthe liquid by a factor of 10–1000 at conditionstypical of vapour-brine separation in porphyry andrelated systems at temperatures to at least 500 8C.
Vapour-liquid partitioning of gold at magmaticconditions. At magmatic temperatures (600–8008C), limited experimental data were obtained bythe method of synthetic fluid inclusions trapped inquartz for Au, Cu, Zn, Ag, and Fe in sulphur-freesystems, involving Na, K, and Fe chloride brines,silicate melts, and H2O–HCl vapour phases (seeSimon & Ripley 2011; Kouzmanov & Pokrovski2012 for reviews on different metals). Only twostudies explored Au solubility and partitioning inS-free vapour-brine-melt systems of NaCl–KCl–FeCl2–H2O-haplogranite compositions at 800 8C,1.0–1.5 kbar, and redox conditions buffered bythe Ni–NiO assemblage (Frank et al. 2002; Simonet al. 2005). Data on Au solubility in melts andfluid/melt partitioning will be discussed in section“Gold speciation and solubility in magmatic fluidsand fluid-melt partitioning.” The solubility of Auin the acidic brine phase was tentatively interpretedas AuHCl2
0, while hydroxide species (e.g. AuOH0)were suggested to form at near-neutral low-HClbrine compositions. A third study of the samegroup (Frank et al. 2011) explored a similar
system at the same T–P-redox conditions but in thepresence of iron sulphide assemblages to buffer sul-phur fugacity.
The Au vapour-brine partitioning coefficientsreported in these works are plotted in Figure 7.They also obey, despite the large data scatter, den-sity relationships similar to those establishedfor lower temperature hydrothermal conditions(shown by the red line labelled Au in this figure),although with absolute vapour-brine partition
Fig. 7. Vapour-liquid partition coefficientslog Kvapour/liquid ¼ log (mvapour/mliquid), where m is themass concentration of a metal per 1 kg of the vapour orliquid phase, of Au in two-phase model systemsH2O + NaCl + KCl + HCl + FeCl2 + sulphur +pyrrhotite measured at hydrothermal (350–500 8C) andmagmatic (800 8C) temperatures as a function of thevapour-to-liquid density ratio. Symbols stand forexperimental data from the following sources:S05–Simon et al. (2005), F11–Frank et al. (2011),P08–Pokrovski et al. (2008a). Vertical error barsdenote 1 standard deviation from the mean of multipleruns/analyses. For comparison, the straight lines throughthe critical point (c.p.) for the indicated elementsrepresent the density-model predictions in the S-freesystem based on data below 500 8C (Fig. 6) fromPokrovski et al. (2005a). At magmatic temperatures andsulphur content of a few wt%, the effect of sulphur on Auvapour-liquid partitioning is weak in front of the largeerrors that affect the data. In contrast, at hydrothermaltemperatures at acidic pH, Au partitions preferentiallyinto the vapour, Kvapour/liquid ≈ 1–10, whereas atneutral-to-basic pH, the effect of sulphur is muchweaker, consistent with the change of Au liquid-phasespeciation from neutral AuHS0 and/or AuHSH2S0 tonegatively charged Au(HS)2
2 at pH above �5 (seesection “Gold chloride, hydrogen sulphide, andhydroxide species in aqueous solution and densesupercritical fluid”).
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

coefficients �0.5 order of magnitude higher onaverage than at T , �500 8C for similar vapour/liquid density ratios (Fig. 6). Similar differences,not exceeding one unit of log Kvapour/liquid, wereobserved for Cu, Ag, Zn, and Fe. The increasingvolatility of Au and these metals at magmatic con-ditions compared to hydrothermal temperaturesmight be explained by possible formation ofhydroxide or hydrogen chloride species both invapour and brine (e.g. AuOH, AuHCl2, Simonet al. 2005).
Both vapour and liquid densities change inregular and predictable fashion that may be reason-ably approximated by the H2O–NaCl system(Driesner & Heinrich 2007) for natural vapour andbrine compositions dominated by Na, K, and Fechlorides. Consequently, the simple density trendsshown in Figures 6 and 7 provide an efficient prac-tical way for predicting vapour-liquid fractionationover the range of magmatic-hydrothermal condi-tions within an uncertainty less than one order ofmagnitude. Note that in S-poor water-salt systems,even at temperatures as high as 800 8C, Au andaccompanying metals (Ag, Cu, Fe, not shown)enrich the liquid phase compared to the vapour.
Effect of sulphur on Au vapour-liquid partitioning.The experimental data in S-free or S-poor systemsdiscussed above are in agreement with naturalcoexisting brine and vapour inclusions for allmetals and metalloids, except gold and copper,which show systematic enrichment in the vapourphase (Fig. 5). The most plausible explanation ofthis enrichment is the formation of volatile specieswith sulphur, the second most important ligandafter chloride (Heinrich et al. 1999). This hypoth-esis has recently been confirmed by experiments.The case of copper was discussed in detail in manyrecent papers (Pokrovski et al. 2008a; Frank et al.2011; Simon & Ripley 2011; Kouzmanov &Pokrovski 2012; Lerchbaumer & Audetat 2012;Pokrovski et al. 2013b) and will be only brieflysummarized here.
These experimental works showed that both athydrothermal (300–500 8C) and magmatic (600–800 8C) conditions, addition of reduced sulphur inthe system does increase Cu partitioning into thevapour, compared to a S-free system, but with absol-ute Kvapour/liquid values never exceeding one, con-trary to the natural data (Kvapour/liquid up to 100,Fig. 5). A plausible explanation of this systematicdiscrepancy was offered by recent measurementson natural and synthetic fluid inclusions in quartzreequilibrated with different fluid and melt compo-sitions. These data revealed rapid diffusion of theCu+ ion through the quartz leading to large post-entrapment changes in Cu concentrations in theinclusion (Li et al. 2009; Zajacz et al. 2009;
Lerchbaumer & Audetat 2012). Very recently, Seo& Heinrich (2013) analysed coeval vapour andbrine inclusions trapped in coexisting topaz,garnet, and quartz from Mole Granite (Australia)and showed that, in contrast to all other metals andsulphur whose concentrations are identical inquartz- and topaz/garnet-hosted inclusions, Cu con-centrations in vapour-like inclusions in quartz are1–2 orders of magnitude higher than in coevalinclusions in topaz and garnet. Since the structureof these silicate minerals does not allow rapid dif-fusion of cations compared to quartz, these findingsnicely confirm the experiments of Lerchbaumer &Audetat (2012). Seo & Heinrich (2013) developeda simple thermodynamic model according to whichthe diffusion occurs via the following exchangereaction between Cu+ and H+, which have elevateddiffusion coefficients in quartz but not in other sili-cate minerals: Cu+external + H+
inclusion ¼ Cu+inclusion +H+
external. Its Gibbs free energy is the driving forceof Cu+ diffusion into quartz to compensate for theoutward diffusion of H+ from the inclusion-hostedacidic fluid. This reaction is favourable becauseof the large acidity acquired in the quartz-hostedS-rich inclusion during cooling through SO2 dis-proportionation and precipitation of pyrite andchalcopyrite, whereas in the external fluid theacidity is rapidly neutralized by interaction withalkali aluminosilicate rocks (see below reactions10 and 11 and corresponding discussions). Theprocess continues until all reduced sulphur in theinclusion is consumed by precipitation with Cu+
coming from outside according to the reactionabove. This results in Cu/S ratios close to 2,similar to those in chalcopyrite in many naturalvapour inclusions (Seo et al. 2009; Kouzmanov &Pokrovski 2012). Thus, the elevated copper contentsin many vapour-like inclusions from porphyrydeposits (Fig. 5) might be a post-entrapment modi-fication, the extent of which depends on thethermal history of the host quartz and the chemicalevolution of the external fluid. In constrast, Auand base metals like Zn, Fe, or Pb are likely notbeing strongly affected by such diffusion processes.Seo & Heinrich (2013) also suggest that Au+ dif-fusion, if it does occur, is expected to be in oppo-site direction, that is, into the inclusion, so that Auconcentrations analysed in high-T vapour-likeinclusions from magmatic-hydrothermal depositsmight probably be correct or at least representminimal estimates. Other recent experiments inS-free systems also support these conclusions.Zajacz et al. (2009) imposed strong chemical gra-dients using concentrated HCl external solutionsto test post-entrapment modifications of fluid andmelt inclusions in quartz for a large number ofelements including alkaline and alkaline earthelements, Cu and Ag, and found that only cations
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

with a charge of +1 and a radius equal to or smal-ler than 1.0 A (Li+, Cu+, Na+, Ag+), diffusedthrough quartz, whereas those having higher elec-tric charges and/or larger radii (e.g. K, Rb, Cs) donot diffuse significantly, at least at the experimen-tal duration (,10 days) and temperature (500–720 8C). Because Au+ is similar in size to K+
(�1.37 A), it would not be expected to diffuse fastenough to create significant post-entrapment enrich-ment or depletion.
Direct measurements of gold vapour-liquid par-titioning in model sulphur-rich salt-water systems
are limited to two studies, to the best of ourknowledge. Pokrovski et al. (2008a) reportedKvapour/liquid(Au) values from 350 8C to 500 8Cand P ≤ 500 bar in the Au–NaCl–KCl–NaOHsystem in the presence of up to 2 wt% of nativesulphur or thioacetamide, using a hydrothermalreactor allowing accurate sampling of the vapourand liquid. Frank et al. (2011) measured vapour/brine and brine/melt partitioning of Au at 800 8Cand 1000–1500 bar in the system NaCl–KCl–FeC2–H2O-haplogranite in the presence of(Cu,Fe)S or pyrrhotite. Figure 7 shows that with
Fig. 8. (a) Solubility of native gold as a function of temperature at 1000 bar pressure in model aqueoussolutions of 10 wt% NaCl equivalent at pH 5 in equilibrium with the pyrite-magnetite-hematite (PMH), andpyrite-pyrrhotite-magnetite (PPM) oxygen and sulphur fugacity buffers, and at 0.1 wt% total dissolved sulphur andoxygen fugacity fixed by the magnetite-hematite buffer (MH). (b) H2S dissolved concentrations as a function oftemperature in the three model systems above. (c) Au solubility as a function of pressure at 400 8C, 10 wt% NaCl, pH 5,in equilibrium with pyrite-magnetite-hematite (�0.04 wt% S) and 0.1 wt%S with magnetite-hematite. (d) Au solubilityas a function of salinity at 400 8C, 1000 bar, pH 5, in equilibrium with pyrite-magnetite-hematite. Calculations wereperformed using the HCh computer code (Shvarov 2008). Thermodynamic data for Au aqueous complexes are from thisstudy (Table 5); those for minerals are from the SUPCRT database (Johnson et al. 1992), and those for sulphur speciesand other major fluid constituents and activity coefficients models are detailed in Pokrovski et al. (2009a, b). Thedominant Au aqueous species are indicated. The grey area in panel (c) shows the domain of low-density vapour-likefluids (density , 0.3 g/cm3), for which calculations are not reliable at present.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

the addition of 1–2 wt% of S to the water-saltsystem at acidic-to-neutral pH and temperatures350–500 8C, Au partition coefficients increase byup to 2 orders of magnitude, attaining values infavour of the vapour for Au (KAu . 1), whereasthe partitioning of Zn, Fe, Pb (not shown) isalmost unaffected. At magmatic conditions, theincrease in Au volatility is weaker, by a factor of2–3 on average, compared to S-poor compositions,but the scatter is large. Although the exact chemicalidentity and stoichiometry of Au complexes inthe vapour phase remain unconstrained, it is verylikely that gold forms uncharged complexes withreduced sulphur. The higher volatility of Au, com-pared to Cu and other base metals, is also consi-stent with the far greater stability of Au hydrogensulphide v. chlorides species in aqueous solutionand brine, as discussed above. Gold absoluteconcentrations measured in the vapour phase inequilibrium with metal both in hydrothermal andmagmatic experiments attain several tens of ppm,which is comparable with Au contents measuredin S-rich vapour-like fluid inclusions fromporphyry Au–Cu deposits (Kouzmanov & Pok-rovski 2012).
Another important factor controlling thevapour-liquid distribution of Au in S-rich systemsis the fluid acidity. Because a neutral species insolution generally has a far greater volatility thanits charged counterpart (Pokrovski et al. 2002a,2008a), Au is expected to be more volatile in S-richsystems under acidic conditions where AuHS0, andprobably AuHSH2S0, are more abundant in theliquid phase than Au(HS)22 (see above). It can beseen in Figure 7 that, at similar H2S concentrationsin the system (�0.5 wt% S), Au vapour-liquid par-titioning coefficients in the case where the liquidphase pH is below 5 are systematically higher thanthose at the liquid-phase pH ≥ 6, reflecting thechange of the dominant Au species in the liquid(e.g. see Fig. 9a below). Vapour-liquid unmixingof a neutral-to-basic fluid in which Au(HS)2
2 isdominant will thus not be favourable for Au frac-tionation into the vapour phase.
Gold speciation and solubility in magmatic
fluids and fluid-melt partitioning
Hydrothermal Au-bearing fluids are often sourc-ed from magmas (e.g. Hedenquist & Lowenstern1994). This section briefly summarizes the existingdata on Au contents, distribution, and partitioning inmagmatic systems containing aqueous fluids, sili-cate melts, and sulphide melts/minerals, based onavailable high-precision micro-analytical data bothfrom natural magmatic rocks and glasses andlaboratory experiments.
Natural systems. Table 6 summarizes the availabledata on gold concentrations measured in volcanicrocks, silicate glasses, fluids, and sulphide minerals.Most natural silicate melts contain gold at levelsof ,1–10 ppb, which is much lower than theppm-level Au concentrations obtained in laboratoryexperiments in similar systems saturated with met-allic gold (see below). The much lower natural Auconcentrations mean that most natural magmaticsystems are largely undersaturated with Au; this isconsistent with the absence of sensu stricto mag-matic gold deposits. By contrast, silicic melt andaqueous fluid inclusions from magmatic systemsspatially related to Cu–Zn massive sulphide oredeposits (Vikent’ev et al. 2012) are highly enrichedin gold, with tenors attaining several ppm, whichapproach the level of gold saturation in hydrousCl–S-bearing silicic systems (Table 6). Similarly,measured gold contents both in fluid and sulphidemelt inclusions in minerals from magmatic rockshosting porphyry-type Au–Cu deposits attainlevels of sub-ppm to ppm levels (Ulrich et al.1999; Halter et al. 2002, 2005), approaching theexperimental concentrations in the presence ofmetallic gold in hydrous S-bearing silicate melts(see below).
Gold concentrations in natural silicate glassesfrom evolved arc-related magmatic rocks in sub-duction zones display a dependence on SiO2
content, by showing maximum values, up to�10 ppb, at �60 wt% SiO2 or an mg# number of40 (mg# ¼ Mg/(Fetotal + Mg) × 100 mol%) in alarge range of rocks, from basalt to rhyolite (Mosset al. 2001; Sun et al. 2004; Jenner et al. 2010). Inthese arc-related magmas, the presence of magnetitecrystallizing below 60 wt% SiO2 marks the abruptdecrease of Au, Ag, and Cu contents to �1 ppb,which are close to those in MORB’s and primitivemantle (Sun et al. 2004; Jenner et al. 2010). Frac-tional crystallization of granitic rocks hostingporphyry Cu–Au–Mo deposits was also foundto lead to gold enrichment by a factor of 10–100in residual silicate melts (up to 100 ppb, Mustardet al. 2006). The redox state ( fO2
) and aluminumsaturation index (ASI ¼ Al/(Na + K + Ca) inmol%) of the melt are also important factors af-fecting Au contents. For example, Connors et al.(1993) and Sisson (2003) reported elevated Aucontents (5–35 ppb) in alkali mafic glasses andperalkaline felsic rocks (ASI , 1), with the highestAu concentration values (.30 ppb) being relatedto the most alkaline glasses containing nativegold inclusions. Borisova et al. (2006) also re-ported high gold concentrations (up to 22 ppb) inhydrous adakitic glasses of rhyolite compositionwith ASI . 1 under oxidizing conditions (SO2/H2S . 1). More recently, Ishimaru et al. (2011) dis-covered in mantle wedge-derived xenoliths a new
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

type of S-rich silicate melt or silicate-bearingaqueous fluid concentrating Au, PGE, and otherchalcophile metals (Cu, Ni). Their findings suggestthat metasomatic reactions of such a fluid/meltwith the mantle wedge resulted in Au, PGE, andvolatile enrichment of the upper mantle (Ishimaruet al., pers. comm.).
Experimental anhydrous systems. Gold concen-trations in dry basaltic melts vary from a few ppbto a few ppm and strongly depend on redoxconditions (Table 6). Gold valence state in thesemelts has been established to be AuI in the formof AuO0.5-like species, based on the dependenceof gold solubility on oxygen fugacity (Borisov &Palme 1996, 2000; Brenan et al. 2005). The
monovalent state of Au has also been confirmedby 197Au Mossbauer spectroscopy in synthetic gold-ruby silicate glasses (Wagner et al. 2000). At a con-stant redox potential, Au solubility in haplobasalticmelts was shown to slightly increase with increasingtemperature (Borisov & Palme 1996).
Experimental hydrous systems. Since 1994, thenumber of experimental studies on gold solubilityin melts, aqueous fluids, and sulphides in hydrousCl- and S-bearing systems is rapidly increasing.Most of these experiments were conducted at ele-vated pressures, approaching those of magma differ-entiation in the crust, and under buffered oxygenand sulphur fugacity using sulphide and oxide min-eral assemblages (Table 6). These measurements
Fig. 9. Solubility of native gold at 1000 bar and indicated temperatures in model 10 wt% NaCl equiv. aqueousfluids: (a) as a function of pH in equilibrium with the magnetite-hematite assemblage (MH) and 0.013 m H2S(�0.04 wt% S) in solution; (b) as a function of H2S dissolved concentration, pH 5, and oxygen fugacity fixed bymagnetite-hematite (MH); (c) as a function of oxygen fugacity (log f O2
, in bars) at 0.1 wt% total dissolved sulphur,together with (d) corresponding distribution of sulphur species in the fluid. Vertical dashed lines in panels (c) and(d) indicate the oxygen fugacity of the major mineral buffers. QFM, quartz-fayalite-magnetite; NNO, nickel-nickeloxide; MH, magnetite-hematite.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

report Au solubilities in silicate melts generallycomparable with those in anhydrous systems atsimilar T and fO2
conditions.In hydrous systems saturated with metallic gold,
two main phases concentrate gold at levels fromppm to wt%: an aqueous S/Cl-bearing fluid and aFe–Cu sulphide liquid. For example, Frank et al.(2002), Hanley et al. (2005), and Simon et al.(2005) demonstrated that Cl-bearing (NaCl–HCl)vapour and brine may dissolve up to �6000 ppmAu, depending on the HCl content in the system attemperatures from 800 8C to 1000 8C. Frank et al.(2002) suggested that gold speciation in silicatemelts at elevated HCl concentrations is dominatedby chloride complexes forming according to thereaction: AuO0.5
si-melt+HClfluid ¼ AuClsi-melt + 0.5H2Osi-melt. Similar Au chloride species (AuCl and/or AuCl . HCl) are likely to form in an aqueousfluid in equilibrium with this melt, yielding goldfluid/melt partitioning coefficients in favour ofthe aqueous fluid (Kfluid/Si-melt is between 8 and840 at 800 8C and 1 kbar; Frank et al. 2002; Simonet al. 2005, 2007b) mainly because of the far greaterHCl concentration in fluid compared to melt. Evenhigher Kfluid/Si-melt values for Au (1000–10 000)were reported by Hanley et al. (2005) for similarsystems at higher pressures (1.5 kbar) and byFrank et al. (2011) for S-bearing systems. Theexact gold speciation in the fluid and melt in theseexperiments remains, however, unconstrained.
Botcharnikov et al. (2010) inferred the formationof Au–Cl and Au–S complexes in Cl,S-bearinghydrous rhyodacite and andesite melts based ondependences of Au solubility v. Cl and S concen-trations. More recently, Botcharnikov et al. (2011)proposed Au–S–O-like and Au–Fe–S–O-likespecies in their experimental hydrous andesiticand basaltic melts based on evidence of maximumgold solubility attained during decomposition/dissolution of iron sulphides supplying highamounts of reduced sulphur to the melt. Similarly,Jego & Pichavant (2012) reported a gold solubi-lity increase in Fe-poor hydrous silicic melts atconditions of sulphide saturation (,NNO 2 1) upto the sulphide-sulphate transition (NNO + 0.5 toNNO + 2.0). Based on thermodynamic modellingand solubility dependences, these authors suggestedpolymeric Au2FeS2 species at redox conditionsclose to NNO 2 1 in S-bearing hydrous silicicmelts. However, in situ spectroscopic measure-ments of gold molecular environment are requiredto confirm this hypothesis. Zajacz et al. (2012)studied the solubility of Au in hydrous andesitemelts in the presence of salt and sulphur and demon-stated that it exhibits a redox dependence similarto that in aqueous solution (see Fig. 9c below) byreaching a maximum at redox close to the sulphide-sulphate transition (around NNO + 0.5 at 1000 8C).
They interpreted their data by the AuCl species inCl-bearing, S-free systems and by AuHS andmixed (K/Na)AuS complexes in S-bearing sys-tems. However, the interpretation of Au solubilityin such sulphur-rich systems may be complicatedby metallic gold nugget formation during the temp-erature rise and poorly known sulphur specia-tion, which may consist of species other thansulphide and sulphate, such as S3
2 and SO2 (Zajaczet al. 2012). Interestingly, similar alkali metalAu hydrogen sulphide complexes of the type(H2S)yAu(SH)(NaCl,KCl)x were hypothesized byZajacz et al. (2010) to explain the enhanced Ausolubility in aqueous S- and NaCl/KCl-bearingfluid phases at similar conditions (1000 8C,1.5 kbar, NNO 2 0.6, Stot up to �10 wt%, NaCl orKCl ≤ 0.7 m, Au � several 100s ppm). In additionto the effect of alkalis, their study also showedthat hydrogen sulphide complexes dominate overchlorides even at 1000 8C at S and Cl concentrationstypical of high-temperature vapours. Revealing theexact nature and structure of such new Au-carryingspecies requires in situ spectroscopic approachesboth for Au and S.
The majority of experiments clearly show thatAu is strongly concentrated in Fe/Cu sulphideliquids compared to silicate melts. For example,Bezmen et al. (1994) obtained Fe sulphide phasesenriched in gold up to �3 wt%, with Au partitioningcoefficients Ksulphide/Si-melt reaching 103–104.Similarly, Jugo et al. (1999), Bell et al. (2009),and Frank et al. (2011) reported in Cl–S–bearinggranite systems Au enrichments in sulphides from�0.7 to 6 wt% Au (depending on the sulphide),with corresponding Ksulphide/Si-melt values of1000–10 000. Li & Audetat (2012) reported sul-phide melt-mafic silicate melt partition coefficientsfor Au of the order of n × 102–n × 103 for muchhigher pressures (15–30 kbar), corresponding toupper mantle conditions. Simon & Ripley (2011)established an empirical dependence of the pyrrho-tite-silicic melt partition coefficients on fS2
for chal-cophile metals (Au, Cu, Ag). Jego & Pichavant(2012) also point to an important role of FeS in con-trolling gold solubility in S-bearing hydrous melts.Very recently, Zajacz et al. (2013) conducted sys-tematic measurements of Au, Cu, and Ag solubilityand partitioning in reduced sulphur magmas ofbasalt to rhyolite compositions and demonstratedthat pyrrhotite/silicate melt partitioning coefficientsfor Au (Ksulphide/Si-melt) increase by a factor of �5from basalt (K � 200) to rhyolite (K � 1100),which is primarily controlled by the FeO and FeSactivities in the silicate melt. Copper behaves simi-larly to gold with comparable partition coefficients,whereas Ag exhibits much lower K values (�50)independent of melt composition. These authorsconcluded that the Au (and Cu) budget is largely
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

G. S. POKROVSKI ET AL.
Table 6. Major published data on natural gold contents and experimental gold solubility and partitioningin silicate and sulphide melts and aqueous fluids
Location /Method
System T 8C, P kbar, fO2, fS2
Au in fluid, ppm{speciation}
Natural magmatic-volcanic systemsWorld Fresh silicic volcanic rocks
and glasses– –
Papua NewGuinea
Fresh volcanic glass and glassyrocks
– –
Argentina Magmatic rocks hostingporphyry Au–Cu deposit Bajo
de la Alumbrera
10008,≥3.5 kbar,
�6 wt% H2O
�2.0
Hawaii Anhydrous basalt, hawaiite,basanite
QFM20.3 + 1.3
–
Papua NewGuinea
Basalt to rhyodacite glasses QFM+2 –
Philippines Rhyolite/dacite melts(adakites) and aqueous fluid
inclusions
760–90082–10 kbar
–
Eastern Manusbasin
Arc-related basalt to rhyolite,and MORB from Atlantic and
Pacific ocean
– –
Ural Rhyodacite melt and aqueousfluid inclusions with 1–6 wt%
NaCl eq.
700–85085–9 kbar
�8
Experimental anhydrous systems saturated with metallic AuCAF Basalt and sulphides 1200 8C, 1 bar
log fO2¼ 29.2
–
CAF Haplobasalt 1300–14808, 1 bar, log fO2
from 211.5 to 20.7–
CAF Basalt & Fe–Ni liquid 12508,1 bar, IQF
–
CAF Fe-free haplobasalt 1200–15008, 1 bar,QFM22 to QFM+2
–
CAF Basalt – olivine 1260–13508, 1 bar QFM+0.6to QFM+5.4
–
Experimental hydrous systems saturated with metallic goldPCA MSS + Cu-rich sulphide
liquid + H2O + CO2–CH4–H2S–NaCl fluid
9008, 10 kbar 150–270
HPGA Mafic to intermediate melt &sulphides + NaCl + water
1200–13008; 100–400 MPa; variable fO2
–
CSPV Haplogranite + pyrrhotite (po)+ iss + water
8508, 1 kbarlog fO2
¼ 213.2 + 0.3,log fS2
¼ 21 + 0.7
–
CSPV Haplogranite + 70 wt% NaCl eq.aqueous brine with variable HCl
8008, 1 kbar, NNO[HCl]brine ¼ 3000–40 000 ppm
40–840{AuOH - at low HCl}{AuCl2H at high HCl}
CSPV Granite melt and 20–70 wt%NaCl eq. aqueous fluid
600–800 8C, 1.5 kbar, NNO,[HCl]brine ¼ 1.8 wt%
900–6000{AuCl, AuCl2H}
CSPV Haplogranite, aqueous NaClvapour + brine
800 8C, 1.1–1.45 kbar, NNO[HCl]vapour ¼ 670–1100 ppm
5–36 (vapour){AuOHvapour}
28–50 (brine){AuCl2H}
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Au in melt or glass, ppm{speciation}
Kfluid/melt Au in sulphide,ppm
Ksulphide/Si-melt References
0.0006–0.005(peralkaline)
≤0.0001–0.0005(peraluminous)�0.0005
(intermediate)
– – – Connors et al. (1993)
0.001–0.015 (maximumat �57 wt% SiO2)
– – – Moss et al. (2001)
bdl – ,0.1–4.3(FeS melt)
– Halter et al. (2002,2005)
0.001–0.035 – – – Sisson (2003)
0.0009–0.008 – – Sun et al. (2004)
0.00064–0.022 – – – Borisova et al. (2006)
�0.001 (Mg# ,40, SiO2
,60 wt%, magnetite),up to 0.007 (Mg# . 40,
no magnetite)
– – – Jenner et al. (2010)
�1.3(melt inclusions)
�6 – – Vikent’ev et al.(2012)
0.0004–0.0013 – 0.6–0.9 1200 + 900 Stone et al. (1990)
0.3–52(depending on fO2
){AuO0.5}
– – – Borisov & Palme(1996)
0.0007–0.013 – 0.8–172 1210 + 950 Crocket et al. (1997)
0.04–0.8 (QFM22)0.1–2.5 (QFM)
0.4–8.0 (QFM+2){Au1+}
– – – Borisov & Palme(2000)
0.9–12.3 – – – Brenan et al. (2005)
– – – – Ballhaus et al. (1994)
0.043–12 – 70–31 000 16 000 + 4000 Bezmen et al. (1994)
2.1–9.3 – 16 000–24 000(iss)
340–640 (po)
5700 + 2200(iss/Si-melt)
140 + 40(po/Si-melt)
Jugo et al. (1999)
�1{Au0 - at low HCl},{AuCl at high HCl}
40–840 (at HCl,10 000 ppm)
– – Frank et al. (2002)
0.2–1.2 2600–12 000 – – Hanley et al. (2005)
0.25–0.50 8–72 (vapour/melt)
56–100 (brine/melt)
– – Simon et al. (2005)
(Continued)
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

controlled by pyrrhotite in intermediate-to-felsicmagmas.
In summary, the main messages from theseresults are the following: (1) Gold is concentratedin magmatic aqueous Cl- and S-bearing fluidsand iron-copper sulphides at levels of 100–10 000 ppm. (2) Gold solubility both in hydrousand anhydrous silicate melts is several orders ofmagnitude lower (0.n to n ppm); it is generally
proportional to fO2, fS2
, and mCl, suggesting theformation of AuI oxygen, sulphide, or chloridespecies. (3) Au solubility in silicate melts reachesa maximum at the sulphide-sulphate redox tran-sition, similar to that in aqueous fluids. (4) Thepresence of iron sulphide is likely to be the mostimportant controlling factor of Au behaviour innatural silicate melts and the source of thiselement in degassing volatile aqueous phases.
Table 6. Continued
Location /Method
System T 8C, P kbar, fO2, fS2
Au in fluid, ppm{speciation}
CSPV Haplogranite, brine, vapour 8008, 1–1.45 kbar 4–41 (vapour)27–53 (brine)
CSPV Haplogranite andaqueous fluid
8008, 1.2 kbar; NNO �30 (fluid, S-free)�12 (fluid, S-bearing)
CSPV Haplogranite, sulphides(mss + iss + sulphide liquid),magnetite, NaCl–KCl–H2O
fluid
8008, 1.5 kbar; NNO,logfS2
¼ 25 4 0–
IHPV Cl–S-bearing hydrous system:rhyodacite to andesite
10508, 2 kbar; NNO –
RQ-TZMV Quartz and NaCl/KCl/LiCl/CaCl2–HCl–S–H2O fluid
10008, 1.5 kbar; NNO20.6 toNNO+1.9
1.9–4.3 (Na/KCl-free;up to 682 (Na/KCl-bearing)
AuCl, Na/KAuCl2, Na/KAu(HS)2
IHPV Basalt, andesite, FeS, CaSO4,H2O
10508, 2 kbar; QFM20.4 toQFM+3.3
–
CSPV Haplogranite, brine,vapour + sulphides
8008, 1 kbar; NNO 17–240 (brine),6–28 (vapour)
IHPV S-bearing hydrous(undersaturated) dacite
10008, 4 kbaraH2O
≤ 1log fS2
¼ 24 to +3
vapour phase not analysed
RQ-TZMV Andesite + H2O+ K/NaCl,FeCl2, S
10008, 2 kbarNNO20.5 to NNO+1.8
–
PCA Mafic silicate melt (SM) +monosulphide solid solution
(MSS) + sulphide liquid (SL)
1175–13008, 15–30 kbarQFM23.1 to QFM+1.0
–
RQ-MHC Basalt, dacite, andesite, rhyolite,H2SO4 aqueous solution
(5–9 wt%S), AuCuAg alloys
900–10308 2 kbarNNO20.8
log fS2� 23 to 22
–
IHPV, internally heated pressure vessel; CSPV, cold-seal pressure vessel; CAF, controlled atmosphere furnace; PCA, piston cylinderapparatus; HPGA, high pressure gas apparatus; RQ-TZMV, rapid quench titanium-zirconium-molybdenum vessel; RQ-MHC,rapid quench molybdenum-hafnium-carbide pressure vessel; T–P-fS2
, temperature-pressure-oxygen-sulphur fugacity; aH2O, water
activity; K, partitioning coefficient; Si-melt, silicate melt; iss, intermediate solid solution; mss, monosulphide solid solution;
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Factors of gold preconcentration and mobilizationin melts. These features of Au behaviour determinethe sources of Au for hydrothermal deposits and theore-forming potential of magmas. Simon & Ripley(2011) discussed several mechanisms which mayincrease ore-forming potential of a given magmaticsystem due to sulphide dissolution. Among themare the following: solidification/degassing fromthe early cumulus assemblage during magma
crystallization (Boudreau & Meurer 1999); degas-sing from magma saturated with an aqueous fluid;interaction with a hydrothermal fluid phase (e.g.Stavast et al. 2006); and magma oxidation (Bell &Simon 2011). An important mechanism leading toconcentration of Au, Cu, and Ag in magmatic Fe–Cu sulphide minerals is reduction of sulphate to sul-phide and oxidation of ferrous iron to ferric ironupon fractional crystallization of a FeII-bearing
Au in melt or glass, ppm{speciation}
Kfluid/melt Au in sulphide,ppm
Ksulphide/Si-melt References
0.5–0.9 (ASI ¼ 0.83–1.0) N.A. – – Simon et al. (2007a)
2.7 (ASI ¼ 1.02–1.13,S-free)
�0.6 (S-bearing)
15 + 2.5 (vapour/melt, S-free),
12 + 0.3(vapour/melt,
S-bearing)
– – Simon et al. (2007b)
0.02–0.65 – 9–286 (mss);107–7220 (iss);
200–64 000(Fe–Ni–Cu–S)
2300–10 000(mss/si-melt)
Bell et al. (2009)
0.2–2.5{Au–Clsi-melt, Au–Ssi-melt}
– – – Botcharnikov et al.(2010)
– – – – Zajacz et al. (2010)
up to 8{Au–S–Osi-melt , Au–Fe–
S–Osi-melt }
– 200–1400 100–300(sulphide/Si-melt)
Botcharnikov et al.(2011)
0.1–0.4 �1300 (brine/Si-melt),
120 (vapour/Si-melt)
300–500 1100–5000(iss/Si-melt)
Frank et al. (2011)
0.25–5{Au0si-melt, AuO0.5
si-melt} atNNO+1.5;
{Au2FeS2si-melt} at NNO 2 1
– – – Jego & Pichavant(2012)
Up to 5, increase withincreasing S and Cl; max
Au solubility is at NNO+0.5;{AuCl, AuHS,
Na/KAuS}
Zajacz et al. (2012)
3400 + 1200(MSS/SM)180 + 80
(SL/SM)
Li & Audetat (2012)
0.02–4.3{AuSNa si-melt}
– 20–600 180 + 20 (basalt)900 + 210
(dacite)1150 + 50
(rhyolite)
Zajacz et al. (2013)
(Fe–Ni) or Fe–Ni–Cu liquid, sulphide liquid; po, pyrrhotite; IQF, iron-silica phase-fayalite oxygen buffer; QFM, quartz-fayalite-magnetite oxygen buffer; NNO, nickel-nickel oxide oxygen buffer; ASI, aluminium saturation index; NaCl eq., salt content of thefluid expressed in wt% of NaCl equivalent; ‘ � ’ means approximate value; ‘–’ means not applicable or not analysed; bdl, belowdetection limit.
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

mafic silicate melt, leading to crystallization ofmagnetite and triggering the saturation in Cu-richsulphide phase. This sulphide phase (presumablyof bornite-like composition) efficiently scavengesAu and Ag from the silicate melt in arc-relatedmagmas (e.g. Jenner et al. 2010). The high contentsof chalcophile elements such as gold, copper, andsilver in magmatic iron sulphides make them animportant source of these metals for the degassingaqueous fluid at depth (e.g. Larocque et al. 2000).Crustal assimilation is also an important phenom-enon leading to magma saturation in volatiles andcontributing to ore-forming potential (Russellet al. 1995; Borisova et al. 2012a, 2013a). Mixingor hybridization of a sulphide-bearing basalticmagma with an hydrous sulphide-undersaturatedandesitic or dacitic magma may also be an impor-tant mechanism for supplying chalcophile metalsto aqueous S-bearing fluid due to dissolution ofthe basalt-hosted sulphides (e.g. Hattori & Keith2001; Halter et al. 2005; Borisova et al. 2012b).Gold-to-copper ratios in sulphide melt inclusionsfrom magmatic rocks hosting porphyry depositsare identical to those in the magmatic fluid andbulk ore, implying a magmatic sulphide sourcefor both Au and Cu (e.g. Ulrich et al. 1999; Halteret al. 2002). Based on experimental data in hydr-ous S-bearing systems, Botcharnikov et al. (2011)suggested that gold-rich basaltic-to-andesitic mag-mas can be generated in sulphide-saturated mantleat relatively oxidizing conditions corresponding tothe sulphide-sulphate boundary, which may occurabove subduction zones and mantle plumes. Theseauthors propose the two main controls on thegenesis of Au-enriched arc magmas: (1) metasoma-tism of the mantle wedge by Au-enriched slab-derived fluids; and (2) redox state and abundanceof sulphur in the magmatic source.
Major parameters controlling Au
transport and precipitation in
hydrothermal fluids
At the current state of our knowledge, quantitativemodelling of Au transport as a function of differentparameters of crustal fluids such as temperature,pressure, salinity, acidity, and redox conditionsare only possible for the aqueous liquid and densesupercritical fluid phase (density .0.3–0.4 g/cm3),for which large amounts of experimental data androbust thermodynamic models (e.g. HKF) are avail-able (see section “Gold chloride, hydrogen sulphide,and hydroxide species in aqueous solution anddense supercritical fluid”). This information allowsquantitative evaluation of the effect of these par-ameters on Au speciation and solubility, assumingthat the dominant aqueous gold species in the
majority of such fluids are those with the hydroxide,chloride, and hydrogen sulphide ligands (see section“Gold chloride, hydrogen sulphide, and hydroxidespecies in aqueous solution and dense supercriti-cal fluid”). The effect of other sulphur ligands(section “The role of other sulphur-bearing formsfor hydrothermal transport of gold”) that maypotentially be important in S-rich systems cannotbe evaluated at present owing to the extreme scarcityof the data and insufficient knowledge of sulphuraqueous speciation itself. Keeping these limitationsin mind, the dominant reactions controlling Ausolubility in a typical aqueous hydrothermal liquid,vapour, and supercritical fluid containing saltsand sulphur in the form of sulphate and sulphidebelow �600 8C and �5 kbar are assumed to be thefollowing:
Au(s) + 2H2S = Au(HS)−2 + 0.5H2 + H+,
in near-neutral and alkaline fluids (10a)
Au(s) + H2S = AuHS0 + 0.5H2, in acidic fluids
(10b)Au(s) + H+ + 2Cl− = AuCl−2 + 0.5H2, above
450−500 8C in acidic saline fluids
and brines (10c)
Au(s) + 2H2S = Au(HS)H2S0 + 0.5H2, tentative,
in acidic S-rich fluids and vapours
(10d)
Au(s) + H2O = AuOH0 + 0.5H2,
in Cl-poor and S-poor fluids (10e)
Au(s) + H+ + Cl− = AuCl0 + 0.5H2,
above 450−500 8C in acidic S-poor
vapours. (10f )
The Au(HS)H2S0 species (reaction 10d) suggestedfor S-rich (.1 wt% S) acidic conditions remainstentative because of other possible alternatives(polysulphide, sulphite) and the limited T–Pdomain of its reported stability constants (Pokrovskiet al. 2009b). Consequently, this complex wasnot considered systematically in the thermody-namic modelling below. Note, however, that itscontribution to Au solubility is relatively low at Scontents below 1 wt%, and its dependence on themajor parameters is very similar to that of AuHS0;as a result, neglecting Au(HS)H2S0 in solubilitycalculations below does not significantly affect themain trends. Gold hydroxide species (reaction 10e)appear to be minor compared to chlorides and sul-phides in most Au ore-deposit contexts at naturallypertinent Cl and S concentrations. However, at
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

some high-temperature magmatic sulphur-poorfluid compositions, this or mixed hydroxide-chlor-ide complexes might become more important (e.g.Simon et al. 2005), owing to the general increaseof the stability of ionic-like complexes with harderligands (OH2 and Cl2 v. HS2) with increasing tem-perature and decreasing the fluid dielectric con-stant (Brimhall & Crerar 1987). Gold monochloride(reaction 10f) is minor in fluids above 0.1 wt% saltat hydrothermal temperatures (,�500 8C), but itmay contribute at magmatic temperatures and inthe vapour phase in S-poor systems for the samereasons as those for AuOH above. Thus, reactions10a, b, and c are expected to adequately reflect themajor hydrothermal controls on gold behaviour, atleast in fluids containing less than 0.5–1.0 wt% S.
Figure 8 shows the solubility of native gold andthe range of predominance of its main species as afunction of the three key parameters—temperature,pressure, and salinity—at buffered or fixed sulphurconcentrations typical of the majority of Auore-forming environments. Figure 9 displays calcu-lated Au solubility as a function of acidity, sulphurcontent, and redox potential within the typicalrange of these parameters in the crust. Below, wediscuss the effect of each of these parameters onAu solubility. In addition, the effect of two ubiqui-tous components often associated with hydro-thermal gold deposits, carbon dioxide and arsenic,will be evaluated from a physical-chemical pointof view. Finally, possibilities of Au colloidal andhydrocarbon transport will be tackled. Solubilitycontrols on other metals usually accompanyinggold such as Cu, Ag, and Mo have recently beendiscussed elsewhere (Kouzmanov & Pokrovski2012; Pokrovski et al. 2013a) and thus will beonly briefly mentioned here for the sake of comple-teness (where necessary).
Temperature. Gold solubility in aqueous fluidscirculating in environments where sulphur andoxygen fugacity are buffered by common ironoxide and sulphide mineral assemblages (PPM ¼pyrite-pyrrhotite-magnetite, and PMH ¼ pyrite-magnetite-hematite) shows a pronounced increasewith increasing temperature, from ,0.001 ppmAu below 200 8C to �10 ppm Au above 500 8C.This increase is accompanied by progressivechanges of the dominant Au species in the fluidfrom AuHS0(+AuOH0) below 200 8C, to AuHS0 +Au(HS)2
2 between 250 8C and 450 8C, and toAuCl2
2 above 500 8C (Fig. 8a). These changes inAu solubility and speciation with temperaturereflect (1) the increase of H2S content in the fluidin equilibrium with the Fe-bearing sulphide min-erals (from �1025 at 150 8C to 0.5 mol/kg at500 8C, Fig. 8b), and (2) the increasing tendencyat elevated temperatures for stabilizing ionic-bond
complexes with harder ligands, such as chloridecompared to sulphide (e.g. Brimhall & Crerar1987). The large increase in solubility with tempera-ture in these systems is also typical for base metal(Cu, Fe, Zn, Pb) sulphide minerals but not for thesame reason as for Au. These metals form, overthe whole T range, almost exclusively chloridecomplexes whose stability largely increases withincreasing temperature (Barnes 1997; Kouzma-nov & Pokrovski 2012).
Gold solubility in Fe-poor fluids (i.e. in theabsence of pyrite) at a constant sulphur concen-tration (0.1 wt% S, which is typical of hydrothermalfluids) exhibits a particular temperature trend(Fig. 8a), not observed for any base metal. From150 to 350 8C, it shows elevated dissolved Auconcentrations of �1–10 ppm, with a weaklypronounced maximum at 200–250 8C. This is inagreement with the temperature evolution ofAu(HS)2
2 stability constants (Fig. 2), which showa maximum at these temperatures. Above 250 8C,Au dissolved concentrations decrease, show a weakminimum around 400 8C, and then increase againabove 450 8C due to the growing stability ofAuCl2
2. Thus, oxidized magmatic fluids of moderatesalinity, for example, those typical for porphyrysystems (Sillitoe 2010; Richards 2011), are able totransport significant amounts of Au above 450 8Cas chloride species, whereas Au transport in epi-thermal environments (≤350 8C) is strongly con-ditioned by the amount of ferrous iron in the fluidand rock, which is the main sink for hydrogen sul-phide in the form of pyrite (see below).
Pressure. A typical pressure effect on Au solubilityis shown in Figure 8c. Similar to other ore minerals,the pressure influence appears to be quite weak com-pared to that of temperature for liquid and super-critical fluids at densities above �0.5 g/cm3. Thisis explained by the low compressibility of densewater and a weak effect of pressure on the solventdielectric constant and density, which controlthe thermodynamic properties of aqueous species(Mesmer et al. 1988; Shock & Helgeson 1988).The minor increase of Au solubility with decreasingpressure apparent in Figure 8c is mostly due tothe increase in dissolved H2S concentration in thefluid (not shown) in the system in equilibrium withFe sulphides.
At pressures corresponding to the vapour-likedomain (shown by grey color in Fig. 8c), the avail-able experimental data and models do not allowreliable predictions of Au solubility. The preferen-tial partitioning of Au into vapour v. saline liquidrevealed by rare existing experiments (e.g. Pok-rovski et al. 2008a) and analyses of natural fluidinclusions (Kouzmanov & Pokrovski 2012; refer-ences therein) suggest that uncharged hydrogen
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

sulphide or chloride complexes might be stabilizedin the vapour phase with decreasing pressure (seesection “Vapour transport and vapour-liquid parti-tioning of gold”), but their stoichiometry and lig-ation number may be different from those of thenegatively charged Au bis-hydrogen sulphide anddichloride complexes dominant in dense aqueousfluid. However, even within our limited knowledgeof the low pressure domain, the effect of pressureitself in a single-phase fluid for the majority ofgeological situations in the crust is likely to be ofsecondary importance compared to that of tem-perature, sulphur amount, and redox potential. Anindirect pressure effect, generating vapour-liquidseparation or boiling, will be discussed in section“Fluid-phase controls on gold ore formation inhydrothermal systems.”
Salinity. At temperatures below 500 8C and near-neutral pH, the presence of salts (i.e. chloride) doesnot significantly influence Au solubility (Fig. 8d).The small effect of chloride is consistent with theweak stability of AuCl2
2 at such moderate tempera-tures and the predominance of hydrogen sulphidecomplexes, which are the solubility-controllingspecies. This Au speciation feature contrasts withthe pronounced solubility increase with salinityfor sulphide ore minerals of most base metals (Cu,Fe, Zn), which form chloride complexes in hydro-thermal fluids (Hemley & Hunt 1992; Hemleyet al. 1992; Yardley 2005; Kouzmanov & Pokrovski2012; references therein). The salinity effect on goldmay be more pronounced at acidic and S-poorconditions (pH , 4), but it is likely to be attenuatedby the general decrease of salinity in a cooling mag-matic fluid subjected to boiling and mixing withmeteoric waters. In contrast, in magmatic brinesabove 500 8C, chloride complexes are expected toplay a major role in gold transport, provided thatAu species with other sulphur-bearing ligands (e.g.SO2 and S3
2) turn out to be insignificant at suchconditions (see section “The role of other sulphur-bearing forms for hydrothermal transport of gold”).
Acidity. The fluid pH (by definition, pH is the nega-tive decimal logarithm of the activity of free pro-tons in solution, pH ¼ –log10aH+
) directly affectsthe concentrations of the charged complexesAuCl2
2 and Au(HS)22 in equilibrium with native
metal because their formation reactions involveprotons. The fundamental difference betweenthese two species is that the former consumes H+
(reaction 10c) and the latter produces H+ (reaction10a). Uncharged complexes (e.g. AuHS, Au(HS)H2S) are not affected by pH changes because noH+ is involved in their formation (e.g. reactions10b, d). It follows from reactions 10, a–f that, ina typical Cl- and S-bearing fluid, Au solubility
increases at both acidic and basic pH and passesthrough a minimum at near-neutral pH (Fig. 9a).With decreasing temperature and salt content, thisminimum shifts towards a more acidic pH. Again,the effect of pH is quite different for the majormetals (Fe, Cu, Zn) whose sulphide mineral solubi-lities decrease dramatically with increasing pHover the whole naturally relevant range (pH �3–9). For example, with all other parameters beingequal, a pH change from 4 to 5 yields a 100-folddecrease in pyrite and sphalerite solubility at 4008C and 10 wt% NaCl (Kouzmanov & Pokrovski2012, not shown), whereas Au solubility is almostunaffected (Fig. 9a).
Thus, at epithermal conditions (≤300 8C), fluidbuffering by alkali and alkali-earth aluminosilicaterocks or carbonates, yielding a fluid pH between 5and 7, may be favourable for Au transport asAu(HS)2
2, whereas at porphyry-deposit tempera-tures (≥400 8C), neutralization of the acidic fluidby interaction with such rocks will induce AuCl2
2
breakdown and gold precipitation. However, itshould be kept in mind that, in natural contexts,changes in fluid pH are often accompanied bychanges in both sulphur speciation itself and solubi-lities of major metal sulphides, so that the finaleffect of fluid-rock interactions on the fluid capacityto transport gold will depend on a given naturalcase (see section “Fluid-phase controls on gold oreformation in hydrothermal systems” below).
Reduced sulphur. The effect of H2S is straight-forward for all Au hydrogen sulphide complexes,by favouring their stability and thus gold solubi-lity (reactions 10a, b, and d). It can be seen inFigure 9b that, at near-neutral pH buffered by sili-cate rocks, fluids with H2S contents above1000 ppm S (≈ 0.03 mol/kg) are capable of trans-porting, over a wide temperature range, ppm-levelAu concentrations (which is �1000 times higherthan Au average tenors in the Earth’s crust). Atsulphur contents of several wt%, occurring influids from magmatic porphyry deposits and high-temperature metamorphic settings (Seo et al. 2009,2012; Tomkins 2010), Au(HS)2
2 and, potentially,other sulphide and/or polysulphide complexesmay transport 100s to 1000s ppm of Au with-out reaching saturation with the metal. The effectof reduced sulphur is fundamentally different forbase metals that are transported as chloride com-plexes but precipitate as sulphide minerals. Thus,high H2S concentrations are not favourable for Zn,Fe, Ag, and Mo mobility but are likely to be amajor condition for efficient Au transport.
One of the major controlling factors of Au be-haviour in geological fluids is, thus, the availabilityof reduced sulphur, which is often determinedby the capacity of an Fe-bearing rock and/or
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

dissolved ferrous iron to scavenge H2S from thefluid. A second major parameter is the speciationof sulphur itself in the fluid phase, which is highlysensitive to redox conditions, as will be shownbelow. A third issue, which cannot be quantifiedat present, is the potential existence of other H2S/HS2-bearing species of different stoichiometriesand/or of Au complexes with polysulphide (+sul-phite) ligands in S-rich fluids (see section “Therole of other sulphur-bearing forms for hydrother-mal transport of gold” above).
Redox potential. The effect of redox conditions(which are usually quantified in terms of oxygenfugacity, fO2
, and assuming the equilibriumbetween O2, H2, and H2O in the fluid at elevatedtemperature: H2 + 0.5O2 ¼ H2O) on Au solubilityis double, as shown in Figure 9c, d. First, in thedomain of predominance of H2S under reducingconditions, native Au solubility is proportional tofO2
, as can be seen in Figure 9c (or inversely pro-portional to fH2
, as shown by reactions 10a–f ). Attemperatures below 500 8C, this trend holds up torelatively high oxygen fugacities, corresponding tothe magnetite-hematite buffer (MH). At more oxi-dizing conditions, however, a second effect takesplace, which is the oxidation of H2S to sulphate orsulphur dioxide (depending on pH). This reinversesthe Au solubility trend v. fO2
because of the decreas-ing concentration of H2S in favour of the moreoxidized S species, which are not known to bindAu. At highly oxidizing conditions (above MH),where sulphate largely dominates, gold solubilityin salt-bearing fluids generally increases againowing to the formation of chloride species.Although such oxidizing conditions are rare innatural high-temperature settings, they may prob-ably occur in some iron oxide Cu–Au (IOCG)deposits (e.g. de Haller & Fontbote 2009). It fol-lows from the trends of Figure 9c, d that the maxi-mum capacity for Au transport occurs at redoxconditions slightly below those of MH, when thefluid H2S fraction is still high enough. Such con-ditions are typical for porphyry Cu–Au deposits(next two sections).
Interestingly, the effect of redox is very similarfor Au solubility in S- and Cl-bearing hydrous sili-cate melts in which Au concentrations in equili-brium with the metal reach maximum values at theH2S–SO4 transition and then decrease to more oxi-dizing conditions (Botcharnikov et al. 2011; Zajaczet al. 2012; see also section “Gold speciation andsolubility in magmatic fluids and fluid-melt parti-tioning” above).
SO2 breakdown. Another factor complicating Aubehaviour in the fluid phase, particularly in por-phyry systems, is SO2 breakdown on cooling. This
is one of the major phenomena that might leadto Au fractionation from Cu and other base metalsduring the porphyry-to-epithermal transition. Cool-ing of SO2-bearing magmatic fluid is accompaniedby large changes in sulphur speciation itself,which, in turn, affect both H2S concentration andacidity, according to the reaction of SO2 dispropor-tionation:
4SO2 + 4H2O = H2S + 3HSO−4 + 3H+ (11)
While this reaction does not cause significant redoxchanges because it is independent of oxygen fuga-city, it is pH-dependent, and thus its yield willdepend on the degree of H+ consumption duringfluid interactions with surrounding rocks. Cal-culations using thermodynamic properties foraqueous sulphur species from the SUPCRT database(Johnson et al. 1992; Sverjensky et al. 1997) showthat cooling of an SO2-bearing aqueous fluid in aclosed system from 600 8C to 200 8C yields onlypartial SO2 breakdown to sulphide and sulphate,accompanied by progressive pH changes fromneutral (pH �5 at 600 8C) to extremely acidic(pH , 2 below 250 8C). In contrast, where pH isbuffered by silicate rocks (e.g. albite, K-feldspar,muscovite, pH �5) during fluid ascent andcooling, reaction (11) is shifted to the right due toH+ consumption, resulting in �99% breakdown ofthe initial SO2 at temperatures below 400 8C.
The behaviour of Au and accompanying metalsin these two extreme cases is quite different. Thisis illustrated in Figure 10, where a model magmaticfluid, containing 10 wt% NaCl equiv., 1 wt% H2S,1 wt% SO2, 10 000 ppm Fe, 5000 ppm Cu, andan excess of metallic Au, is cooled numerically inequilibrium or not with the quartz + muscovite/andalusite + K-feldspar assemblage. The initialfluid composition corresponds to typical salt, sul-phur, and metal contents found in the ‘supercriticalfluid’ type of inclusions from major porphyry depos-its (Heinrich et al. 1999, 2004; Kouzmanov &Pokrovski 2012) and is similar to that adopted inprevious modelling studies (e.g. Heinrich 2005).
It can be seen in Figure 10a, b that, in the absenceof fluid neutralization by silicate rocks, reaction(11) causes acidification of the cooling fluid,which is favourable for Cu and Fe transport in theform of chloride complexes without significantloss, to temperatures �200–250 8C. In contrast,if the fluid is neutralized by interaction withalkali-alumino-silicate rocks (Fig. 10c, d), .90%of the initial Cu and Fe load will precipitatebetween 600 8C and 400 8C as magnetite andpyrite + chalcopyrite, which are the main Cu–Feminerals in high-temperature porphyry stages ofpotassic alteration (Sillitoe 2010; Kouzmanov &
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Pokrovski 2012). However, such a neutralized fluidwill be unable to carry significant amounts of Cuand Fe (and Pb, Zn, Ag) to lower-temperatureepithermal deposits (≤350 8C). The significanttransport of these metals to such settings by sulphur-rich fluids is, thus, only possible if the fluid does
not lose its acidity when it flows through the rock.It requires the rock to be already altered by anearlier portion of acidic fluid. Such an early acidicfluid prepares a favourable path for subsequentpulses of fluid rich in metals, both by reducing theneutralization capacity of the rock and creating
Fig. 10. Concentrations of principal sulphur forms (a, c) and Fe, Cu, and Au (b, d) as a function of temperature in amodel cooling magmatic fluid initially containing 10 wt% NaCl, 1 wt% H2S, 1 wt% SO2, 10 000 ppm Fe, 5000 ppm Cu,and an excess of metallic Au. Thermodynamic properties for S-species and Fe chloride complexes are taken from theSUPCRT database (Jonhson et al. 1992; Sverjensky et al. 1997), whereas those of Cu and Au complexes are,respectively, from Akinfiev & Zotov (2001, 2010) and Table 5 (this study). Panels (a) and (b) show the situation whenthe fluid cools down in a closed system and is not allowed to react with alkali silicate minerals. Note a dramaticacidification of such a fluid on cooling (shown by pH values on panel (b) due to the SO2 disproportionation reactiongenerating sulphuric acid (reaction 11). These acidic pH values are favourable for Fe and Cu transport as chloridecomplexes to temperatures as low as 300–250 8C; below these temperatures a significant part of Fe, Cu, and Sprecipitates as pyrite, chalcopyrite, covellite, and native sulphur. Upon cooling, the solubility of gold in the form ofAuCl2
2 decreases progressively over a wide T range, and its deposition is, thus, not focused to a narrow T window. Thecalculated Au solubility above 400 8C (.10 ppm) is higher than most natural gold concentrations in fluid inclusions,meaning that such SO2-bearing fluids are unsaturated with the metal and thus do not precipitate Au. Panels (c) and (d)show the same fluid, cooling in contact with an excess of the quartz + muscovite/andalusite + K-feldspar assemblagethat buffers the solution pH between 4.5 and 5.0 in the whole T range. Note a dramatic decrease of SO2 concentrationsbelow �500 8C, whereas elevated H2S contents (�0.1 mol/kg) are persisting down to 200 8C. These high H2Sconcentrations, coupled with near-neutral pH, are unfavourable for Cu and Fe solubility below 400–350 8C but arefavourable for massive Au transport (at �10 ppm level) to epithermal deposits. Note also a minimum of Au solubilityaround 450 8C, which corresponds to the high-T Au deposition with Cu in porphyry deposits. These two contrastingscenarios are likely to encompass the range of conditions natural fluids undergo.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

additional permeability due to alteration and partialsilicate mineral dissolution (Kouzmanov & Pok-rovski 2012).
The behaviour of Au upon the SO2 breakdown ismore complex, because of the change of dominantAu species from chloride (reaction 10c) to hydrogensulphide (reactions 10a, b, d) with the temperaturedecrease and the reaction (11) progress. Figure 10shows that, despite the large differences in Au spe-ciation and solubility-controlling factors betweenacidic (AuCl2
2 dominant, Fig. 10b) and neutral(Au(HS)2
2 dominant, Fig. 10d) fluids, with anexcess of sulphur over metals, a magmatic fluid,either buffered or not with rocks, will be able tocarry Au at concentrations above 1 ppm to fairlylow temperatures (�250 8C) without reaching sat-uration with the metal. Nevetheless, the potentialefficiency of Au precipitation is different foracidic and neutral fluids. In the case of transport asAuCl2
2, and when saturation with the metal isreached, a smooth and regular deposition wouldoccur over a wide T-range (Fig. 10b) and, thus, ona large spatial scale, which is not generally favour-able for creating ecomonic Au mineralization. Incontrast, the massive transport as Au(HS)2
2 mayoccur to temperatures as low as 150–200 8C (Fig.10d); such fluid would have a far greater potentialfor Au economic precipitation at such temperatures,when it gets diluted, oxidized, or loses its H2Sthrough interactions with Fe-bearing rocks withina narrow temperature and spatial window.
Effect of CO2. In addition to sulphur, carbon dioxide(CO2) is a very common volatile constituent offluids and vapours in the majority of gold deposits.It may attain more than 50 wt% in the fluid phasein carbonate-rich contexts such as greenstone-belt and Carlin-type gold, skarn-related gold, ormafic pegmatite Cu–Au and PGE deposits (e.g.Diamond 1990; Phillips & Evans 2004; Goldfarbet al. 2005; Vallance et al. 2009; Hanley &Gladney 2011). In porphyry Cu–Au deposits (e.g.Climax, Butte, Henderson, Bingham, El Salvador),it amounts to 10–20 wt% (�5–10 mol%) in deephigh-temperature parts corresponding to potassicalteration zones as indicated by analyses of high-temperature fluid inclusions (Rusk et al. 2008,2011). It is also detected in epithermal Audeposits at the level of a few wt% (Andre-Mayeret al. 2002).
Much more is known about CO2 in silicate melts.A detailed review of CO2 sources, contents, and be-haviour in magmas was provided by Lowenstern(2001) and Moore (2008). The two key propertiesof CO2 in magmatic systems distinguish this com-pound from water, chloride, and sulphur: (1) lowsolubility of CO2 in most types of silicate melts;and (2) absence of major mineral phases capable
of retaining CO2 in magmatic rocks. These pro-perties are responsible for early degassing of CO2
from magmas compared to water, salts, and sul-phur (Lowenstern 2001) and are likely to be thereason of the relatively modest CO2 concentrationsreported in fluids from intrusion-related Au depositssuch as Cu–Au porphyries (Rusk et al. 2008) ascompared to deposits in metamorphic or sedimen-tary carbonate-rich rock formations (e.g. Phillips& Evans 2004). Under hydrothermal conditions,CO2 may affect gold behaviour in different ways,both direct and indirect.
First, the presence of CO2 affects vapour-liquidphase relationships as compared to a CO2-freesystem. The PVTX properties of the H2O–NaCl–CO2 system are reasonably well constrained today,and physical-chemical models are available for pre-dicting densities of the vapour and liquid phasesin this system (e.g. Bowers & Helgeson 1983; Duanet al. 1995; Bakker 2009). These models are,however, not precise enough to quantify the exactT–P conditions of phase transition and compositionin the CO2–NaCl–H2O system, in contrast to theCO2-free NaCl–H2O system for which far moreaccurate models are now available (e.g. Driesner& Heinrich 2007; references therein). Nevertheless,existing data indicate that even moderate quantitiesof CO2 may significantly extend the vapour-liquidimmiscibility domain and increase the pressure ofphase separation. For example, the presence of10 wt% CO2 in supercritical aqueous fluid con-taining 10 wt% NaCl at 400 8C will increase thehomogenization pressure of the mixture (i.e. thepressure below which two phases, an aqueous vap-our and a saline liquid, coexist) from 270 bar (inCO2-free, 10 wt% NaCl–H2O system) to �500 bar(in the presence of CO2; Bakker 2009). This dif-ference corresponds to more than 2 km of depthunder hydrostatic pressure. In the evolution pathof the cooling and ascending magmatic fluid, thiswill allow an earlier (i.e. at greater depth) separationof the vapour and the corresponding Au fraction-ation into this phase in S-rich systems (section“Vapour transport and vapour-liquid partitioningof gold” above). A more detailed account of phaseseparation on Au behaviour in different geologicalsettings will be given in the section “Fluid-phasecontrols on gold ore formation in hydrothermalsystems”.
Second, because CO2 is a weak acid (log K of thedissociation reaction CO2 + H2O ¼ HCO3
2 + H+ is26 to 28 at hydrothermal conditions; Johnson et al.1992), its presence may influence the fluid pH, andthus, Au solubility. Boiling or phase separationlead to an increase in pH of the liquid phase dueto preferential partitioning of CO2 and other acidicvolatile components (HCl, H2S, SO2) into thevapour. As a numerical example illustrating this
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

phenomenon, boiling of a 10 wt% NaCl + 5 wt%CO2 aqueous solution at 350 8C, resulting inremoval of 90% of CO2 into the vapour phase,will increase the pH of the liquid by half an orderof magnitude (from 5.0 in initial solution to 5.5after boiling). If there is still a sufficient amount ofH2S remaining in the liquid, Au solubility willincrease accordingly (Fig. 9a). However, thisincrease may be attenuated by the departure ofH2S into the vapour (depending on the extent ofboiling), which will tend to decrease Au solubilityin the liquid phase (Fig. 9b). Thus, the final effectof boiling is difficult to generalize for Au. In high-pressure environments (.2 kbar) typical of Audeposits in metamorphic belts, where boiling doesnot occur, it was proposed that large amounts ofCO2 in the fluid may buffer pH at a value around 5and thus allow maintaining relatively high Auconcentrations in the form of Au(HS)2
2 (Phillips &Evans 2004). Such CO2 buffering control on pHmight, indeed, operate at low-to-moderate tempera-tures (≤350–300 8C), at which water-silicate rockreactions are slow. In contrast, in higher-temperature(≥400 8C) salt-rich fluids typical of porphyry orintrusion-related deposits, the effect of CO2 onfluid pH is likely to be attenuated by rapid fluidequilibration with silicate rocks, so that Au behav-iour will be little affected in the presence of CO2.
Third, CO2 anionic counterparts, bicarbonate(HCO3
2) and carbonate (CO322) ions, may poten-
tially act as direct ligands for ‘hard’ metals likeREE, Sn, Zr, U, Nb, and probably Fe (e.g. Seward& Barnes 1997; Wood & Samson 1998; Pokrovski2010; references therein). In contrast, base metals,Ag, and Au are not expected to be affected bycarbonate complexing both because of their gener-ally low chemical affinity to the ‘hard’ carbonateligand (section “General features of gold aqueouschemistry”) and the small abundance of carbonateions in acidic-to-neutral fluids typical of mostmagmatic-hydrothermal systems.
Fourth, when CO2 is present in large fractions,it may affect solvation phenomena in the fluidphase. Because most metal chloride and hydroxidecomplexes are solvated by water molecules bothin vapour and liquid or supercritical fluid phase(section “Vapour transport and vapour-liquid par-titioning of gold” above), lowering H2O activity(�mole fraction) in the presence of CO2 decreasesthe complex stability and thus, metal solubility.Alternatively, the effect of CO2 may be explainedusing the Born electrostatic equation, which usesthe dielectric constant of the fluid mixture (seesection “Gold chloride, hydrogen sulphide, andhydroxide species in aqueous solution and densesupercritical fluid”). Experimental measurementsavailable for a few oxide and chloride solids insupercritical H2O–CO2 mixtures attest to this
behaviour in a wide range of T–P–XCO2conditions
(Fig. 11). For example, quartz solubility in a 20 wt%CO2–H2O fluid at 600 8C and 2 kbar is 1.5 timeslower than in pure water at the same T–P(Walther & Orville 1983). A larger effect isobserved for ionic compounds, such as AgCl, forexample, whose solubility in the form of AgCl2
2 islowered by a factor of 3 in a 20 wt% CO2–H2Ofluid compared to pure H2O at 400 8C and 1 kbar(Akinfiev & Zotov 1999). A similar decrease ispredicted for Au solubility both as AuCl2
2 andAu(HS)2
2 species based on the Born equationof solvation (Fig. 11). By contrast, for neutraland less polar Au–S complexes such as AuHS0
or AuHS(H2S)0, CO2 would not have a strong influ-ence according to this electrostatic model (Fig. 11).
Fig. 11. Solubility of quartz, silver chloride, andmetallic gold in supercritical H2O–CO2 fluids atindicated temperatures, pressures, and pH as a functionof CO2 mole fraction, according to publishedexperimental measurements (Si and Ag) andthermodynamic predictions (Au, this study). Symbolsshow experimental data for quartz and AgCl(s) in theform of AgCl2
2 (NM00 – Newton & Manning 2000;WO83 – Walther & Orville 1983; AZ99–Akinfiev &Zotov 1999), whereas straight lines are their least-squarelinear fits through the data points. Curves for Au speciesrepresent calculated concentrations at indicated pH andsolution compositions and using the Born model for theGibbs free energy of solvation (Akinfiev & Zotov 1999)and Born coefficients (v) for Au species from Table 5:DGT,P ¼ Dv × (1/1mix 2 1/1water), where Dv is thechange in Born coefficient for the corresponding speciesformation reaction (10a-f), and 1water and 1mix are thedielectric constants of pure water and the H2O–CO2
mixture, respectively. Note the significant drop ofsolubility with increasing CO2 content for all speciesexcept AuHS whose v value is close to zero (reaction10b, Table 5).
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

However, this model does not take into accountpossible preferential solvation of Au–S non-polarspecies in a CO2 solvent (see Pokrovski et al.2008a, 2009b). Interestingly, the selective solva-tion capacities of supercritical CO2 are used inchemical engineering for synthesis and purificationof organometallic and organic compounds atmoderate temperatures (e.g. Erkey 2000). If suchsolvation occurs, Au solubility in the form ofuncharged sulphide complexes may increase withincreasing CO2 fraction in the fluid. A similarmechanism might influence the vapour-liquid parti-tioning of gold. The scarce data of Figure 7 inCO2-free vapour-liquid systems suggest that vola-tile Au-sulphur complexes may behave as regulargases and, thus, be enhanced in the presence ofCO2, both due to increase of the density contrastbetween liquid and vapour and specific solvationof neutral non-polar molecules like Au–H2S com-plexes by non-polar CO2 (Pokrovski et al. 2008a).This solvation effect of CO2 awaits, however, exper-imental confirmation for conditions relevant tohydrothermal gold deposits. Note that it may beefficient only at large CO2 fractions to change sig-nificantly the properties of the aqueous solvent(.10–20 mol% CO2), which are common in oro-genic gold deposits (e.g. Phillips & Evans 2004).In other types of hydrothermal Au deposits such asporphyry, epithermal, and Carlin, generally charac-terized by modest CO2 contents (,10 mol%), thesolvation effect on gold transport capacities byfluids is expected to be minor as compared to theother factors discussed above.
Role of arsenic and other metalloids (Sb, Se, Te).In many meso- and epithermal deposits (T ≤�300 8C), gold is found to be closely associatedwith arsenian pyrite, arsenopyrite, and sulphideminerals of Sb, Se, and Te (e.g. Boyle 1979; Cathe-lineau et al. 1989; Arehart et al. 1993; Cabri et al.2000). Both ‘visible’ native gold (i.e. particleslarger than �0.1 mm) in association with and ‘invis-ible’ gold incorporated into these minerals occur.The literature on arsenian pyrite over the last threedecades is full of discussions about the chemicaland redox state of both As and Au and Au–Aschemical and structural relationships in pyrite andarsenopyrite. Based on spectroscopic techniques(SIMS, TEM, Moessbauer, XAS, XPS) and interele-ment correlations, most studies agree that ‘invisible’gold occurs in two chemical states: the elementalform as nanometer-size particles of Au0, and chemi-cally bound gold with a still uncertain oxidationstate between Au3+ and Au12 (Marion et al. 1986;Arehart et al. 1993; Scaini et al. 1998; Simonet al. 1999; Cabri et al. 2000; Palenik et al. 2004;Reich et al. 2005; references therein). Accurateassessment of the Au oxidation state in As-pyrite
using interelement correlations is complicated bythe presence of many other elements in far greaterconcentrations (e.g. Cu, Sb, Ni, Co, Ag, Bi, Se,Te, Reich et al. 2005). The application of directspectroscopic techniques like XPS or XAS is ham-pered by their weak sensitivity to low Au concen-trations (,100–1000 ppm) and by the presenceof As, whose absorption edge energy is very closeto that of Au, resulting in peak overlap. Becauseof these limitations, the wide range of oxidationstates suggested for chemically bound Au shouldbe considered with care. The known gold chemistryboth in solid and solution implies that the onlystable Au oxidation state at hydrothermal conditionsis Au+ (see section “General features of gold aque-ous chemistry”). One of the most direct studiesof Au in As-pyrite published so far (Simon et al.1999), using near-edge absorption spectroscopy(XANES), reported the presence both of Au0 andAu+ in Au-rich (�1000 ppm) arsenian pyrites froma Carlin-type gold deposit. Chemically bound Au+
was found to have a nearest coordination numberof 2 and 4, based on comparison with Au-bearingstandard compounds with known valence andcoordination. However, the low signal-to-noiseratio of the EXAFS spectra from that study did notallow identification of the Au first-shell neighbours(S, As, or Fe).
As for arsenic, the majority of spectroscopic,analytical, and thermodynamic studies of arsenianpyrites indicate that As is likely to substitute forsulphur as As12, forming a Fe(As,S)2 solid solutionbetween pyrite and arsenopyrite (e.g. Fleet et al.1989; Pokrovski et al. 2002b; Reich et al. 2005;Blanchard et al. 2007; Liang et al. 2013; referencestherein). The only existing exception to these find-ings is an XPS study of Deditius et al. (2008),who reported the dominant presence of As3+,likely substituting for Fe2+, in pyrites from theYanacocha gold epithermal deposit. However,because of the high sensitivity of As-pyrites tosurface oxidation (e.g. Nesbitt et al. 1995; Lianget al. 2013), care is required in sample prepara-tion for the XPS technique that probes only first10 of nanometres of the sample surface. In addition,a coordination number of 6 by sulphur for As3+ inthe Fe2+ structural site (Deditius et al. 2008) con-tradicts the ubiquitous AsIII coordination numberof 3 and corresponding geometries (typically adistorted pyramid) in most solids and aqueous sol-ution (e.g. Borisova et al. 2010; Testemale et al.2011; references therein). Thus, in the absence ofadditional investigations, arsenic may be consi-dered most likely to exist as As12 in pyrite in themajority of Au deposits. From a thermodynamicpoint of view, As may be treated as a regular solidsolution FeS2–FeAsS (Blanchard et al. 2007) andusing the thermodynamic properties of pyrite and
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

arsenopyrite end-members (Pokrovski et al. 2002b;Perfetti et al. 2008).
The most intriguing feature of gold distributionin the majority of epithermal deposits is its positivecorrelation with As in pyrite. Total Au concenta-tions vary in a very large range from a few ppb to1000s ppm in As-pyrites and arsenopyrites. Basedon available SIMS, EMPA, XANES, and HRTEMstudies cited above, Reich et al. (2005) establishedan upper solubility limit of chemically bound Auin pyrites from different Carlin-type gold depositsformed between 150 and 250 8C: CAu ¼ 0.02 ×CAs + 4 × 1025 (mol%). Arsenian pyrites withAu/As ratios below 0.02 have the dominant Auform as Au+, whereas those with Au/As molalratios above 0.02 contain, in addition to Au+, sig-nificant fractions of Au0 nanoparticles. From basicthermodynamics postulating that stability domainsof most solid solutions extend with increasing tem-perature, it may be expected that far more goldas Au+ may be accommodated by arsenian pyriteat elevated temperatures and that part of the Aufound in native form in As-pyrite might be due toexsolution phenomena on cooling.
The fundamental reasons for the Au spatialassociation with As (and accompanying metalloidssuch as Sb, Se, Te) in sulpharsenides are subjectsof continuing debates. Different mechanisms havebeen proposed to account for the role of As andother metalloids in Au scavenging by pyrite andarsenopyrite. Earlier studies evoked Au transportby the fluid phase in the form of Au–As–S (+Sb,Se, Te) aqueous complexes and their precipitationwith ferrous iron (Boyle 1969; Boiron et al. 1989).However, such common precipitation due to theAu-metalloid species breakdown cannot explain themuch higher concentrations of As and Sb (wt%level) than Au (ppm level) in pyrites. In addition,although such complexes have been evoked inseveral studies (e.g. Seward 1973; Nekrasov et al.1982), they have not been convincingly demon-strated experimentally (see Wood & Samson 1998for discussion), and the available data reviewedhere strongly suggest that hydrogen sulphide andchloride complexes are sufficient to account forAu transport over a wide range of hydrothermalconditions.
Other studies suggested chemisorption of dis-solved Au as Au–S/As complexes at As-rich,Fe-deficient sites (Renders & Seward 1989; Mao1991; Fleet & Mumin 1997; Cepedal et al. 2008),electrochemically driven adsorption of negativelycharged Au(HS)2
2 complexes on semi-conductingAs-pyrite and arsenopyrite surfaces (Mironovet al. 1981; Moller & Kersten 1994), or Au3+ andAu+ precipitation on sulphide surfaces due to Aureduction (Maddox et al. 1998; Scaini et al. 1998).These hypotheses are, however, based mostly on
low temperature (,100 8C) experiments, some ofthem involving Au complexes which are unlikely athydrothermal conditions (e.g. AuCl4
2, see above).These limitations make it difficult to extrapolatethese results to the higher temperatures of Au-pyriteformation in nature (.150–200 8C). Different iso-morphic substitution models of Au in pyrite werealso invoked, including coupled substitutions ofAs3+ + Au+ for 2Fe2+, As3+ + Au3+ for 3Fe2+
(Deditius et al. 2008), or Cu+ + Au3+ for 2Fe2+
(Chouinard et al. 2005). However, at least someof them are inconsistent with the known As andAu crystal chemistry and redox states at hydrother-mal conditions, as discussed above.
Whatever the exact Au state in As-pyrite, nativeor chemically bound, an important aspect not expli-citly expressed in most studies is the role of commonprecipitation mechanisms of Au and associatedmetalloids. Thus, the association of native gold(Au0) with arsenopyrite and As-pyrite (As12) maysimply be due to the same reduction process in thehydrothermal fluid in which these elements are com-monly transported as Au1+ (chloride, sulphide,other species, see above) and As3+ (As(OH)3, Pok-rovski et al. 1996a; Perfetti et al. 2008; referencestherein). Such parallel reduction (Au1+ to Au0,and As3+ to As12) may, for example, occur duringfluid interaction with organic-rich sediments com-mon in Carlin-type gold deposits. Similarly, indeposits where gold is late compared to arseno-pyrite + pyrite (Heinrich & Eadington 1986;Genkin et al. 1998), native gold deposition ongrain boundaries and in cracks of pre-existingFe(As,S)2 or FeAsS may easily be explained byminor redissolution of arsenopyrite with formationof As(OH)3 and H2 according to the reaction like
FeAsS(s) + 3H2O + 2H+ + 2Cl−
= FeCl2 + As(OH)3 + H2S + 1.5H2 (12)
(or equivalent), a process that would act as a localredox trap for gold, as demonstated by thermo-dynamic calculations (Heinrich & Eadington 1986;Pokrovski et al. 2002b). Other factors such as fluidcooling or boiling are also favourable for As-pyrite/arsenopyrite precipitation together with Au,both in the native or chemically bound state,because of the breakdown of aqueous Au-hydrogensulphide complexes due to H2S scavenging by theseminerals (e.g. Velasquez et al. 2014). Similarcommon precipitation phenomena are likely to beresponsible for the close association of Au withother metalloids like Sb, Se, Te, or Bi, which aretransported as oxidized hydroxide species in thefluid phase (e.g. Sb(OH)3, Pokrovski et al. 2006;Bi(OH)3, Tooth et al. 2012), and usually precipi-tated in more reduced forms (antimonides, native
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

bismuth). These metalloids, if sufficiently abundant,may also form with Au individual solid phases (e.g.Au2Bi, AuSb2, AuTe2, AuAgTe2, Pb(Au,Te)S2,Ciobanu et al. 2006, 2009), alloys and solid sol-utions (e.g. Bi, Tooth et al. 2008), or melts (e.g.Ciobanu & Cook 2006; Wagner 2007), which mayscavenge Au from an undersaturated solution. Adeeper understanding of the gold-metalloids rela-tionships in hydrothermal systems awaits directexperimental studies at pertinent conditions anddevelopment of high-resolution (both in energyand space) in situ spectroscopic techniques.
Colloidal transport of gold. The chemically ‘soft’nature of gold (section “General features of goldaqueous chemistry”) allows easy delocalization ofouter-sphere electrons and establishment of bondsbetween gold atoms. This favours the formationof gold clusters and cages involving several Auatoms together with other ligands, leading to for-mation of gold nanoparticles and colloid-like aggre-gates, which may stay in solution for prolongedtimes. This property of gold was used for decoratingchina and glass articles using ‘liquid golds’, whichare gold multiatomic aggregates with thiolateligands bound around the Au cluster (Cotton &Wilkinson 1988), and is now widely employed innanotechnology (e.g. Cobley & Xia 2009).
In natural systems, the common observation ofcolloform gold with fine-grained quartz and amor-phous silica in epithermal deposits (Saunders1990, 2012; Herrington & Wilkinson 1993; Saun-ders & Schoenly 1995), and of nanoparticules ofgold with clays in weathering environments (e.g.Hough et al. 2008) incited researchers to suggestthat gold was transported in the form of colloids withsilica or clays. Such a transport might, indeed, besignificant in low-temperature dilute solutionswhere gold precipitation rates are slow and theabsence of electrolytes retards colloid coagulation.The affinity of both Au+ and Au3+ for aqueousor colloidal silica and the potential capability offorming stable Au–O–Si bonds may further beinferred using the analogy with other metals. It wasdemonstated by potentiometry and in situ XASmethods that aqueous silica significantly retardsFe, Al, and Ga hydrolysis and hydroxide precipi-tation by forming aqueous metal-silica polymericcomplexes in solution (e.g. Pokrovski et al. 1996b,2002c, 2003; references therein). Pokrovski et al.(2002c) showed that the values of stability constantsof metal-silicate complexes in solution are linearlycorrelated with those of the corresponding metalhydrolysis constants. Following this correlation,it is expected that both Au3+ and Au+ mightform stable silicate complexes. The exact stabilitydomains of such hypothetical complexes requireexperimental quantification. By analogy with the
low solubility of Au in silicate melts, these Au-silicate species in solution are expected to con-tribute to Au transport only in low-temperaturesulphur- and salt-poor Si-rich environments, prob-ably via specific adsorption and retention of Au oncolloidal silica particles.
At elevated temperatures and in salt- andsulphur-bearing hydrothermal fluids, such colloidsor Au–Si aggregates are unlikely to survive longenough to allow efficient Au transport, both due torapid crystallization kinetics and the presence ofchloride and sulphide ligands that are sufficient toaccount for Au transport (see above). The com-monly observed gold-quartz association in themajority of hydrothermal deposits is, thus, moreeasily explained by common depositional mechan-isms of both Au and Si upon fluid cooling (e.g.Helgeson & Garrels 1968) or boiling (Seward1989), rather than by transport as common Au–Sichemical entities.
Hydrocarbons as a potential medium for Au trans-port. The common association of gold with carbonseams and pyrobitumen in veins and hydrocarbonsin fluid inclusions in mining districts such as theCarlin deposits (Nevada) or Witwatersrand (SouthAfrica) tends to suggest that Au was transporteddirectly by hydrocarbons (see Williams-Joneset al. 2009 for an overview). This idea has been ver-ified by Williams-Jones & Migdisov (2007), whomeasured Au solubility in crude oil and found thatup to several 10s ppb of Au can be dissolvedat 250 8C. These original experiments point to apotentially significant capacity of hydrocarbonsfor transporting Au. The exact chemical speciationof Au in such media remains, however, unknown.Following the much higher affinity of Au+ (whichis a stable form in a strongly reducing hydrocarbonmedium) for sulphur compared to nitrogen or oxy-gen (section “General features of gold aqueouschemistry”), it may be expected that Au bindsthiol functional groups in oil. To what extent suchorganic transport may contribute to Au distribu-tion, compared to aqueous hydrothermal fluids,remains to be quantified. It should be noted thatAu solubility as Au(HS)2
2 species in a typical near-neutral aqueous solution at 200–250 8C and sul-phur content of 1000 ppm attains easily 100s to1000s ppb (e.g. Figs 8 & 9). This is at least 1–2orders of magnitude higher than the Au solubilityin oil from the available experiments cited above.Thus, Au-hydrocarbon transport and concentrationis likely to be restricted to specific petroleum-rich and water-poor environments, which mightoccur in some sedimentary basins. More syste-matic data on Au-organic matter interactionsat elevated temperatures are needed to resolvethis issue.
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Comparison of thermodynamic predictions
with natural data from porphyry
Cu–Au–Mo and related deposits
The gold solubility controls discussed above providea foundation for interpreting Au concentrationsin natural hydrothermal fluids and gold ore precipi-tation mechanisms in different geological settings.The rapidly growing database of gold and othermetals concentrations acquired over the last �15years, mainly using LA-ICPMS analyses of fluidinclusions from different types of hydrothermaldeposits (Yardley 2005; Kouzmanov & Pokrovski2012; this volume), together with the improvedknowledge of Au aqueous thermodynamics discus-sed above, allows a direct comparison of naturalgold contents in hydrothermal fluids with thermo-dynamic predictions. In this section, we focuson abundant fluid inclusion data on Au fromCu–Au–Mo porphyry and associated epithermaland skarn deposits. Detailed comparisons for othermetals are given elsewhere (Kouzmanov & Pok-rovski 2012).
Four genetic types of fluid inclusions may beidentified in porphyry and associated systems(Kouzmanov & Pokrovski 2012). These types reflectthe temperature-pressure evolution in the porphyryenvironment of a magmatic saline metal-bearingfluid, governed by the phase relationships of thewater-salt system: (a) supercritical fluid of moderatesalinity (�2–10 wt% NaCl) containing metalsand volatiles (CO2, H2S, SO2) exsolved from themagma; (b) aqueous low-density vapour and (c)hypersaline high-density liquid, both producedby unmixing of the supercritical fluid on coolingand decompression when it crosses the two-phasevapour-liquid boundary of the water-salt system;and (d) low-salinity aqueous solution generated bycondensation of the supercritical (a) or vapour-type(b) fluids above and their eventual mixing withmeteoric waters in the shallow epithermal environ-ment of porphyry systems. Thus, the characteris-tic parameters that distinguish among these typesare the temperature of homogenezation (Th) andsalinity (NaCl wt% equivalent) as derived frommicrothermometric measurements of inclusons. Inaddition, recent improvements of LA-ICPMS tech-niques allowed direct analyses of sulphur contentsin these fluid inclusions (Guillong et al. 2008;Seo et al. 2009, 2011, 2012; Catchpole et al. 2011,in press). This provides new constraints on theimpact of sulphur on Au behaviour based onthermodynamic calculations of Au speciation andsolubility.
Gold concentrations measured in the four typesof inclusions from porphyry and associated depositsare plotted v. Th, chlorinity (NaCl wt% equivalent),
and sulphur content (ppm) in Figure 12a–c. Notethat the temperature of homogenization providesan estimate of the minimum temperature ofentrapment. For coexisting liquid-vapour ‘boiling’assemblages of inclusions, a unique estimate oftemperature and pressure of entrapment is possibleusing the well-known PVTX properties of theH2O–NaCl system (e.g. Driesner & Heinrich2007); in all other cases, a correction is requiredfor the true temperature of entrapment. This correc-tion (towards higher temperatures) may be estimatedusing different geothermometers when available(trace element- or stable isotope-based). For thesupercritical and low-salinity types of fluid inclu-sions shown in Figure 12a, this correction usuallydoes not exceed 50–100 8C, implying that theentrapment temperature is usually higher that theobserved homogenization temperature. However,this should not change dramatically the main trendsin Au solubility discussed below. Another caveatwhen comparing Au contents in natural fluid inclu-sions with thermodynamic predictions of Au sol-ubility is that many natural fluids are probablyundersaturated with metallic gold because of thegenerally low abundance of this trace metal. As aresult, Au contents measured in natural fluid inclu-sions may not always be as sensitive to variationsof T, P, mS, or salinity as Au solubility itself(i.e. Au solution concentrations in equilibrium withthe metal). In addition, Au may also enter sulphideminerals as chemically bound or nano-particulateAu, whose solubility is different from that of bulkcrystalline Au. Nevetheless, the solubility trendsdiscussed below allow one to place rough limits onthe capacity of natural fluids to transport Au in agiven natural setting and to identify the most appar-ent trends.
The measured Au concentrations in Figure 12are compared with the calculated values as afunction of T, salinity, and H2S concentration inequilibrium with metallic gold and using the ther-modynamic properties of Au hydroxide, chloride,and hydrogen sulphide species recommendedabove (Table 5). The chosen redox and acidity con-ditions are typical of porphyry systems as inferredfrom mineral associations and wall-rock alterationpatterns. The assemblage magnetite-hematite waschosen as a proxy for the oxygen fugacity(Einaudi et al. 2003), and the pH was constrainedbetween 3 and 5 which corresponds to the differentextent of fluid buffering by alkali alumino-silicateassemblages common in porphyry deposits in awide temperature range (Meyer & Hemley 1967;Reed 1997). Acidic pH usually corresponds tofluids represented by the low-salinity aqueoustype of inclusions in high-sulphidation epithermaldeposits; they are produced by condensation ofthe magmatic HCl- and SO2-bearing vapour or
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

supercritical fluid phase generating acids on cooling(see section “Major parameters controlling Autransport and precipitation in hydrothermal fluids”above). Near-neutral pH values (close to 5) aretypical of high-temperature magmatic fluids, whichequilibrate with alkali-silicate rocks. Sulphur con-centrations in these calculations correspond tothose from fluid-inclusion analyses from porphyry(Seo et al. 2009, 2011, 2012) and high-to-intermedi-ate sulphidation epithermal (Catchpole et al. inpress) deposits; they typically range from 0.1 to1 wt% (Fig. 12c).
It can be seen in Figure 12 that analysed naturalgold concentrations span from �0.03 to �25 ppmfor the available dataset and are independent(within the data scatter) of the inclusion type, temp-erature, and salinity. The absence of clear tempera-ture dependence of natural Au concentrations agreeswith the calculated Au solubility trends with sulphurconcentrations at 0.1 wt% at pH 5 (Figs 8a & 12a).Higher sulphur contents, corresponding to anaverage value of the available analyses of fluidinclusions (0.7 wt%), yield Au solubilities at near-neutral pH higher than the majority of inclusions.More acidic pH (pH 3–4) and more reducingconditions (e.g. NNO buffer) would lead to lowerAu calculated solubilities than those shown by thecurves in this figure, better corresponding to thenatural analyses. Note that Au solubilities calcu-lated assuming sulphide buffering with the majorFe-bearing minerals (e.g. PPM buffer) yield Au con-centrations below 400–450 8C much lower than thenatural contents. This strongly suggests that mostepithermal fluids transporting Au (correspondingto the low-salinity aqueous type of inclusion inFig. 12a) were Fe-poor and exhibited no significantloss of sulphur during their interactions with rocks.At temperatures higher than 500 8C, most fluids areundersaturated with metallic gold, consistent withthe common lack of Au deposition above 500 8Cin porphyry systems.
The independence of Au natural contents of sal-inity (Fig. 12b), which contrasts with all base metals(see Kouzmanov & Pokrovski 2012), is consistentwith a weak fraction of Au chloride species at near-neutral H2S-rich fluids, particularly at temperaturesbelow 450–500 8C (Fig. 8d). The large majorityof Au fluid inclusion analyses are within the rangeof Au solubility calculated as a function of NaClwith S contents between 0.1 and 1 wt% at pH5. However, it should be noted that direct analyticaldata on sulphur contents are still very limited and atpresent only available for few porphyry and oneepithermal deposit. More data on S concentrationsare, thus, necessary for robust interpretion of Auconcentration trends.
Measured gold concentrations in all types ofinclusions where S contents are available do not
Fig. 12. Gold concentrations measured in four types offluid inclusions from porphyry and associated deposits(indicated by symbols) as a function of (a) temperatureof homogenization, (b) salinity, and (c) measuredsulphur concentrations, and their comparison withcalculated gold solubility (curves) at indicatedconditions of temperature, pH, sulphur content, salinity,and redox potential (redox buffers are defined in Figs 8 &9). Thermodynamic properties of Au species are fromTable 5, those of solid phases and major fluidconstituents and sulphur forms are from the SUPCRTdatabase. For data sources in panels (a) and (b), see thecaption of Figure 5; data in panel (c) are from Guillonget al. (2008), Seo et al. (2009, 2011, 2012), andCatchpole et al. (2011, in press).
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

show clear correlations with sulphur (Fig. 12c), butthe data scatter, together with simultaneous vari-ations of acidity and temperature, may obscure thetrends. Absolute sulphur concentrations measuredso far in fluid inclusions at temperatures above400 8C (.1000 ppm) are consistent with the solubi-lities of the major iron and copper sulphides (e.g.Fig. 8b). At lower temperatures, however, thesesolubilities are very low, and the elevated sulphurconcentrations, found in fluid inclusions of the‘aqueous solution’ type typical of epithermal depos-its, indicate that these fluids lack iron (and copper)compared to sulphur. This points to the originof such fluids from a condensed Fe-deficient butS-rich vapour phase, which is a widely acceptedmodern scenario of epithermal deposit formation(Heinrich 2005). This condensate should be acidic(pH , 3–4) to be able to transport Cu, Fe, Ag, Pb,and Zn, which are all present at elevated con-centrations in this fluid type (100s–1000s ppm,Kouzmanov & Pokrovski 2012). The high fluidacidity is consistent with the dissociation of vola-tiles such as HCl and the disproportionation ofSO2 to H2S and sulphuric acid on cooling (seesection “Major parameters controlling Au transportand precipitation in hydrothermal fluids”). Figure12c compares measured Au contents with calcu-lated Au metal solubilities as a function of sulphurconcentration at different T and pH reflecting theprogressive acidification of the magmatic fluid oncooling (e.g. Sillitoe 2010). It can be seen that thecalculations account reasonably well for the Aucontents within the typical pH, T, and redox poten-tial ranges of porphyry deposits, with only a fewexceptions for particularly Au-rich moderate-temperature ‘aqueous-type’ fluid inclusions, whichappear to be supersaturated with Au metal. Atpresent, it is difficult to quantitatively account forthese differences, which may be due to (1) uncer-tainties intrinsic to the determination of the trueentrapment temperature, (2) less acidic pH thanthose used in the calculations in Figure 12c,(3) colloidal Au transport (section “Major par-ameters controlling Au transport and precipitationin hydrothermal fluids”) and/or heterogeneousentrapment of Au nanoparticles (e.g. Pudack et al.2009; Kouzmanov et al. 2010), and (4) omissionof other Au species in calculations. The latterissue is particularly relevant in view of a recentdiscovery of polysulphide forms, such as S3
2, inS-rich acidic-to-neutral fluids, which mightaccount both for enhanced Au transport as directcomplexes with S3
2, and increased solubility ofother metal sulphide minerals in such solutionsdue to the consumption of H2S to form S3
2 (Pok-rovski 2011; Pokrovski & Dubrovinsky 2011).Additional experimental data on Au speciation inS-rich fluids coupled with systematic analyses of
S concentration and chemical state in natural fluidinclusions from porphyry-epithermal environmentswill confirm or reject this hypothesis.
Fluid-phase controls on gold ore
formation in hydrothermal systems
During their evolution in the crust, magmatic,hydrothermal, or metamorphic Au-bearing fluidsundergo five major processes causing gold andaccompanying metals redistribution and deposition:cooling, decompression, phase separation (orboiling), interaction with rocks, and mixing withexternal waters. All of them are interconnectedand may be overprinted on one another or act in par-allel. The improved knowledge of both Au naturalconcentrations and chemical speciation in theliquid and vapour phases may help to better estimatethe impact of each of these processes on gold pre-cipitation in mineral deposits in different geologicalenvironments. Below, we discuss major conse-quences of each of these processes on Au transportand deposition with examples of different golddeposit types.
Cooling
Temperature is believed to be the most direct factorthat influences mineral solubility and, thus, metaltransport and precipitation. Helgeson & Garrels(1968) were among the first to suggest, based onthermodynamic modelling, that cooling of anacidic saline fluid is an efficient mechanism of Audeposition with quartz and pyrite in veins. Sincethen, the effect of cooling was demonstrated forbase metals (Cu, Mo, Fe, Zn, Pb) in different geo-logical settings ranging from porphyry Cu–Auto VMS Zn–Pb deposits. For example, coolingof a single-source magmatic-hydrothermal fluidin a relatively small T interval (450–350 8C) waslikely the main cause of copper deposition in Cu–Au–Mo porphyry deposits such as Bingham (Land-twing et al. 2005), Bajo de la Alumbrera (Ulrichet al. 2001), and Butte (Rusk et al. 2004), andCu–Zn–Pb epithermal deposits such as Morococha(Catchpole et al. 2011, in press). These and othernatural observations are consistent with the gen-eral temperature dependence of metal sulphidemineral solubility that decreases dramatically withtemperature decrease in a saline fluid (Hemley &Hunt 1992; Hemley et al. 1992; Kouzmanov & Pok-rovski 2012).
The effect of T on gold deposition is, however,less straightforward than for other metals. As wasshown in the section “Major parameters controllingAu transport and precipitation in hydrothermalfluids,” gold behaviour in a cooling fluid is primarily
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

dependent on changes in reduced sulphur con-centration. With an excess of ferrous iron (andcopper) over sulphur in the fluid, pyrite + chalco-pyrite precipitation leading to sulphur removalfrom the fluid will cause Au precipitation. Thus,cooling of a magmatic saline fluid rich in Fe +Cu, such as those from porphyry systems, isexpected to deposit Au together with Cu and Fe sul-phides. This is likely to happen in high-temperatureparts of porphyry systems (Sillitoe 2010), as wellas in most Cu–Zn–Pb deposits on the seafloor(Candela 2004), where the rapid cooling of thefluid scavenges all H2S in the form of metal sul-phides, causing Au to precipitate. In contrast,vapour-like fluids produced by phase unmixingand enriched in H2S over Fe and Cu are expectedto carry Au in the form of volatile sulphide speciesdown to 150–200 8C without precipitation (e.g. Fig.8b). These conditions are likely to operate in high-sulphidation epithermal deposits spatially relatedto porphyry systems (e.g. Heinrich et al. 2004).
Cooling does not only cause metal sulphideprecipitation in porphyry systems; it is alsoaccompanied by large changes in sulphur speciationitself in the fluid phase, which consists of SO2 dis-proportionation into sulphate and sulphide (seereaction 11, Fig. 10). The extent of fluid pH buffer-ing by alkali silicate rocks will determine the ampli-tude of Cu, Fe, and Au transport and precipitationon cooling. In addition, the interplay between theinitial fluid redox, S, Cl, and major metal concen-trations may have different effects on Au behaviourin different deposits (e.g. Heinrich 2005). Thisuncertainty is superimposed with another majorunknown of Au aqueous chemistry, the existenceor not of dissolved complexes with sulphur formsother than H2S, such as SO2 or polysulphides(Pokrovski et al. 2008a, 2009b), which may beimportant in oxidizing environments typical of por-phyry deposits.
Decompression
The second change the cooling and ascending fluidundergoes is pressure decrease. Although, the mostobvious effect of decompression is phase separation(which will be discussed in the following subsec-tion), pressure decrease in a single-phase fluid mayalso affect Au v. Cu solubility. As was shown in thesection “Major parameters controlling Au transportand precipitation in hydrothermal fluids” pressurechanges affect Au solubility indirectly, mostly bychanging H2S concentration in the fluid (Fig. 8c).However, accurate quantification of this effect onAu solubilities is difficult at present because ofthe lack of experimental data, particularly for low-density vapours and supercritical fluids. Thus, onlya few qualitative observations can be done here.
Boyle (1969) argued, based on structural obser-vations, that pressure changes due to rock faultingand cracking are far more significant than arechanges in temperature for depositing gold andquartz, by saying that ‘if there is no dilatancy,there is no deposit.’ Recent thermo-mechanicalmodelling of the effect of earthquakes on gold-quartz vein formation (Weatherley & Henley 2013)suggests that faults opening during seismic eventsresult in a large fluid pressure drop owing to crea-tion of empty space, which, in turn, yields rapidand efficient quartz, gold, and other metals precipi-tation via so called ‘flash vapourization’ of the fluid.
Pressure was suggested to influence Cu/Auratios in porphyry deposits. Murakami et al.(2010) compiled Cu/Au ratios from 50 porphyryCu + Au + Mo deposits and noted that thesevalues have large variations, from 103 to 106, butincrease with increasing depth and pressure of oredeposition. This was explained by earlier precipi-tation of chalcopyrite compared to gold from acooling moderate-salinity fluid, whereas Au isstabilized in low-P, low-density fluids as sulphidecomplexes. Landtwing et al. (2010) suggested thatdeposit-scale variations of Cu/Au ratios in theBingham ore are due to different fluid paths, underslightly different pressure-density regimes, of acommon input fluid in the central part and the per-iphery of the porphyry stock. Thus, in the centralzone of the orebody, characterized by abundantfracturing and higher permeability (Gruen et al.2010), the magmatic fluid expands under hydro-static pressure and ore deposition occurs from avapour-like fluid enriched in S and Au but impover-ished in Cu, compared to the input magmatic fluid.In the deeper peripheral zones, by contrast, thisinput magmatic fluid cooled at higher pressureand, thus, underwent less contrasting phase separ-ation, leading to brine and vapour phases withcompositions closer to the original magmatic fluid.This resulted in Cu/Au ratios in precipitated oresimilar to those in the fluid. Similar Cu–Au zona-tions are observed in other porphyry Cu–Au depos-its (e.g. Batu Hijau, Indonesia, Arif & Baker 2004).However, it should be kept in mind that pressureevolution in porphyry deposits is easier to tracethan those of the other parameters and that thepressure and temperature decrease and associatedchanges (pH, sulphur speciation) occur all together;as a result, it is difficult at present to rigourouslyidentify the contribution of pressure itself on Audistribution.
Phase separation
The third key process, ultimately related to thetemperature and pressure decrease the fluid mayundergo when ascending to the surface, is boiling
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

or phase immiscibility. This phenomenon ismanifested in magmatic-hydrothermal systems bycoexisting liquid- and vapour-rich fluid inclusions.In the deep porphyry environment, phase separationresults in condensation of a hypersaline liquid fromthe dominant low-salinity vapour phase (Heinrich2007; Richards 2011), whereas in the epithermalenvironment, the phase separation occurs by boil-ing of the dilute aqueous liquid with formation ofvapour bubbles. The effect on gold transport anddeposition in these two cases is different.
As shown in the section “Vapour transport andvapour-liquid partitioning of gold” phase separationat high T–P results in a contrasting fractionationof metals, with As, S, and Au (and partly Cu)enriched in the vapour phase compared to Fe, Zn,Pb, Ag (Fig. 5). This enrichment was believed togenerate fluids forming Au–Cu epithermal depositsin near-surface environments (Heinrich 2005),when the vapour phase condenses into S-bearingaqueous solution transporting Au at relatively lowtemperatures (see above). Such conditions mayoccur in high-sulphidation Cu–Au (Hedenquistet al. 1993, 1998; Heinrich et al. 2004) and probablyCarlin-type Au (Muntean et al. 2011) deposits,spatially associated with magmatic intrusions,which may provide a source of vapour. However,alternative formation scenarios, for Carlin-type Audeposits, suggesting metamorphic breakdown ofsedimentary pyrite hosting Au and other metalsand subsequent concentration and deposition ofAu by hydrothermal processes involving rock sul-phidation, cannot be excluded (e.g. Large et al.2011; references therein). Whatever the source ofgold, sulphur plays a key role in the Au budget inthe brine and vapour phases. Note that the mass ofthe vapour produced during magmatic fluid evol-ution largely exceeds that of the brine in porphyrydeposits, with typical vapour/brine mass ratiosbetween 4 and 9 (e.g. Hedenquist et al. 1998; Land-twing et al. 2010; Lerchbaumer & Audetat 2012).Adopting a reasonable average value of 5 for thevapour-liquid partitioning coefficient of Au in aS-rich acidic porphyry environment (Figs 6 & 7;Pokrovski et al. 2008a), the vapour phase formedupon unmixing of the ascending magmatic fluidwill concentrate more than 90% of the totalAu. In contrast, adopting a recently recommendedKvapour/liquid value of 0.10–0.15 for Cu at thesame conditions (Lerchbaumer & Audetat 2012),the brine and vapour phases will likely carry com-parable copper amounts. Thus, these differences inAu and Cu distribution may also explain the Au/Cu zonations apparently caused by decompression,as discussed in the previous subsection.
In low-temperature/pressure epithermal envi-ronments (,350–300 8C), the density of thevapour phase is too low for significant Au
partitioning into the vapour phase (Figs 6 & 7).The main effect of liquid boiling under such con-ditions is removal of H2S and CO2 into thevapour. This leads to breakdown of Au-hydrogensulphide complexes and may result in gold precipi-tation in veins of bonanza zones (e.g. Saunders &Schoenly 1995; Simmons et al. 2005). Seward(1989) showed, using thermodynamic modelling,that, in active geothermal systems such asBroadlands-Ohaaki (New Zealand), boiling andconductive cooling of the deep Au(HS)2
2-bearingfluid at �290 8C in an open system, allowingremoval of the H2S-bearing vapour phase, inducesprecipitation of most of the Au over a �15 8C inter-val. The efficiency of the vapour removal stronglydepends on the permeability of the system.Focused fluid flow and efficient Au depositioninduced by boiling were suggested to be two keyfactors for the formation of large Au epithermaldeposits, based on studies of active geothermalsystems (e.g. Simmons & Browne 2007). Inaddition, boiling results in the liquid becomingmore alkaline (pH increase) due to a loss of CO2,which, in turn, may lead to precipitation of galena,sphalerite, chalcopyrite, pyrite, and argentite (seesection “Major parameters controlling Au transportand precipitation in hydrothermal fluids”for detailed discussion of the CO2 effect). Forexample, Velasquez et al. (2014) showed thatboiling of a CO2-bearing fluid at �250 8C inducedby faulting events in the El Callao mining dis-trict (Venezuela) was likely responsible for thecommon precipitation of Au and As-bearingpyrite. Boiling at temperatures below 300 8C islikely to be responsible for Au, Ag, and associatedbase metal sulphides deposition in low-sulphidationepithermal deposits (e.g. Andre-Mayer et al. 2002).
Fluid-rock interactions
The fourth event accompanying the evolution ofhydrothermal fluids is their interaction with wallrocks. This leads primarily to changes of pH and sul-phide content, which result in Au precipitation. Thiswas inferred from field observations more than 40years ago, by recognizing that slow fluid ‘diffusion’through the rock is one of the principal mechanismsof formation of most Au deposits (Boyle 1969).
In porphyry and related settings, fluid-rock inter-actions are expressed as alteration zonation cen-tred on the fluid source and propagating into thesurrounding rocks (Sillitoe 2010). Note that pH neu-tralization alone of an acidic S-rich fluid is notexpected to decrease Au solubility (Fig. 9a), but itleads to precipitation of major metals (Fe, Cu, Zn,Pb) as sulphides resulting in removal of reducedsulphur from the fluid and thus destabilization ofAu-hydrogen sulphide complexes. Skarn deposits
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

with a common metal zonation from proximalCu + Au to distal Zn + Pb zones, resulting from acombination of temperature decrease and neutraliz-ation of the acidic magmatic fluid by carbonaterocks, are a good example. Carbonate sedimentaryAu deposits in distal parts of porphyry systems (e.g.Cunningham et al. 2004) are another example offar-field flow of ore fluids accompanied by rock dis-solution of carbonates and the resulting pH rise.
Fluid interactions with an FeII-rich rock also actas a sink for hydrogen sulphide by precipitation ofpyrite + pyrrhotite. This efficient H2S consumptionfrom the fluid shifts reactions (10a, b, & c) tothe left, leading to gold precipitation. Mafic rocksrich in ferrous iron are, thus, an excellent sink forH2S. High Cu and Au grades are associated withmafic rocks, such as at the Oyu Tolgoi and Resol-utions deposits (see Sillitoe 2010). Wall-rocksulphidation is the primary control on metal depo-sition in deep and high-temperature orogenic goldsystems; in addition, with decreasing depth of for-mation, phase separation favoured by significantCO2 content may also contribute to Au precipitation(e.g. Mikucki 1998; references therein).
Fluid mixing
The fifth phenomenon is mixing of two fluidswith distinct temperature, acidity, or composition,which may induce gold ore formation in near-surface crustal settings such as seafloor hydrother-mal systems and Mississippi Valley-type deposits(Candela 2004). Mixing of gold-bearing hydro-thermal fluid with meteoric waters leads primarilyto dilution, oxidation, and additional cooling. Thesephenomena, together with boiling, discussed above,are likely to be responsible for Au deposition insome low-sulphidation epithermal deposits similarto meteoric-water-dominated active geothermalfields (e.g. Simmons et al. 2005). Mixing betweenfluids of contrasting acidities is more difficult toevaluate for Au. Such a mixing is expectedto have an indirect influence, for example, leadingto precipitation of Fe and other metal sulphideswith increasing pH and resulting in uptake ofreduced sulphur. For example, thermodynamicmodelling indicates that mixing of an acidic Au–Ag bearing fluid with meteoric alkaline waters wasthe main cause of Au–Ag sulphide deposition inthe epithermal Yunoe deposit, Russia (Pal’yanova& Savva 2009). Mixing between seawater and mag-matic fluid may also cause Au and Cu precipitationin IOCG deposits (e.g. de Haller & Fontbote 2009).By contrast, there is no robust evidence for incur-sion of groundwater into deep high-temperatureporphyry Cu–Au orebodies, and many studies(e.g. Hedenquist et al. 1998; Ulrich et al. 2001;Watanabe & Hedenquist 2001; Harris & Golding
2002; Seedorff & Einaudi 2004) show that mostporphyry fluids have a dominantly magmatic com-ponent. Similarly, there is no evidence of signifi-cant fluid mixing in deep orogenic Au deposits(Mikucki 1998).
Conclusions
This paper has presented an overview of goldchemical speciation, solubility, and partitioning inaqueous fluids and vapours over the wide range oftemperature and pressure conditions of the Earth’scrust. The data acquired using experimental andmodelling approaches show that aurous gold (Au+)is by far the most stable redox form of Au both insolids and fluids in the majority of hydrothermal-magmatic and metamorphic settings. The majorAu aqueous complexes in hydrothermal fluids arechloride and hydrogen sulphide species, AuCl2
2,Au(HS)2
2, and AuHS, for which robust thermodyn-amic data are available within the HKF model in awide T–P range. These data allow accurate pre-dictions of Au solubility and quantification of dif-ferent fluid parameters (e.g. temperature, pressure,salinity, redox, acidity, sulphur content) on Aumobility and precipitation in hydrothermal fluids.
In S-rich (.0.5–1.0 wt% S) hydrothermal andmetamorphic fluids, Au might also form solublecomplexes with polysulphides. Sulphide, chloride,and/or hydroxy-chloride forms of Au are alsoresponsible for its transport by the low-densityvapour phase, but their exact stoichiometry andstability remain poorly constrained. In magmaticfluids, Au may form species with alkali metals,chloride, and reduced sulphur. Gold is the mostvolatile economic metal in the H2O-salt-sulphurtwo-phase system, with vapour/liquid partition-ing coefficients (Kvapour/liquid) up to 10, likely dueto complexing with reduced sulphur. In silicatemelt-aqueous fluid systems, Au largely partitionsinto the aqueous fluid (Kfluid/Si-melt ≈ 1000) and/or into iron/copper sulphide melts relative to thesilicate melt (KFeS/Si-melt ≈ 1000–10 000). Thus,reduced sulphur appears to exert a key influenceon Au behaviour in melts, fluids, and vapours.
Comparison of thermodynamic predictions withnatural Au and S concentrations recently measuredin fluid inclusions from porphyry deposits in dif-ferent fluid types generated through the evolutionof magmatic fluid (single-phase fluid, vapour, brine,aqueous solution) further confirms the key roleof dissolved sulphur to account for the observednatural Au concentrations (typically between 0.1and 10 ppm). In most cases, elevated sulphurconcentrations (.0.1 wt%) are necessary to con-centrate and transport Au by the fluid phase inporphyry-epithermal settings. Changes in fluid
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

acidity and in the extent and rate of interactions withthe surrounding rocks during fluid ascent andcooling account reasonably well for the Au contentsobserved in natural fluid inclusions.
Although Au fractionation and deposition in thecrust may be caused by different overlapping pro-cesses, it is ultimately related to the behaviour ofsulphur. Thus, changes in sulphur availability, redoxstate, and speciation in the fluid during its evolutionare directly reflected on the gold fate. Amongst themain phenomena geological fluids undergo, cool-ing accompanied by decompression and water-rockinteractions is likely to be the major cause of Au(and associated metals) deposition and fraction-ation in most high-temperature porphyry contexts.Fluid immiscibility that occurs in these environ-ments may significantly impact the Au budget byconcentrating it in the sulphur-rich vapour phase,which is likely to provide a source of Au for shal-lower epithermal deposits. Fluid-rock interactionsuch as sulphidation and neutralization are likelyto be the main cause of Au deposition in orogenicand skarn deposits. Boiling and mixing with meteo-ric waters may contribute to Au deposition inshallow, low-temperature epithermal settings. Inmany cases, Au transport and deposition is alsoconditioned by the amount of more abundant basemetals (Fe, Cu, Zn) in the fluid, which competewith Au for sulphur. This competition may be criti-cal in magmatic-related deposits. In contrast, thedirect effect of other trace elements (As, Se, Te)on the Au transport and precipitation seems to beminor; their close association with Au in mostsettings may adequately be explained by commonsources and depositional processes. The effect ofCO2 on Au behaviour is to enhance vapour-brineseparation, which yields to changes in sulphurcontent and pH of the liquid phase in epithermallow-temperature environments, and direct Aupartitioning onto the vapour in high-temperaturemagmatic-related deposits.
Remaining gaps and future directions
This study has also highlighted that, in spite of sig-nificant progress in understanding the compositionand properties of Au-transporting geological fluidsat elevated temperatures and their thermodynamicmodelling, much remains to be done. This concernsanalytical, experimental, and theoretical fields ofresearch on gold whose major near-future chal-lenges are briefly outlined below.
Analytical
A number of analytical challenges for fluid inclu-sions, which are the only direct witnesses of
natural fluids, remain to be addressed. If Auconcentrations can now be routinely analysed inhydrothermal fluid inclusions at levels of a fewppm by modern LA-ICPMS machines (Seo et al.2009), the situation is different for sulphur, whichis the primary element controlling Au behaviour inmagmatic-hydrothermal systems. Sulphur remainsone of the most poorly quantified major fluid com-ponents. Only a few studies have measured Scontent in individual fluid inclusions using LA-ICPMS (Guillong et al. 2008; Seo et al. 2009,2011; Catchpole et al. 2011, in press). Such total sul-phur analyses should be coupled with in situ spec-troscopic determination of the abundances ofdifferent sulphur species using in situ micro-Ramanspectroscopy (e.g. Giuliani et al. 2003), and X-rayabsorption and emission spectroscopy (e.g. Metrichet al. 2009; Alonso Mori et al. 2010). New develop-ments of microanalytical techniques based onstandardless approaches for XAS (e.g. Cauzidet al. 2007) and LA-ICPMS (Borisova & Gouypers. comm.) should allow quantification of Auand associated metals in water-poor C–O–H fluidinclusions and gas bubbles. A better account of poss-ible fluid-inclusion post-entrapment modificationsdue to selective metal diffusion through the hostquartz (e.g. Zajacz et al. 2009; Lerchbaumer &Audetat 2012) is also essential for robust interpret-ation of Au, Cu, and Ag concentrations in the fluid.
In contrast with porphyry-style deposits forwhich a large dataset on fluid compositions has beenproduced for many metals including gold (e.g.Kouzmanov & Pokrovski 2012), the informationon Au content in fluids from shallow, low-to-moder-ate temperature environments like epithermal andCarlin-type deposits is fragmentary. In such set-tings, a recently developed method combining near-infrared microscopy with LA-ICPMS may allowdirect analyses of metal-precipitating fluids ininclusions hosted by opaque ore minerals such asenargite, wolframite, stibnite, sphalerite, tetrahe-drite, and pyrite (Kouzmanov et al. 2010). Thiswould allow verifying the broadly accepted para-digm that gangue (quartz, calcite) and associatedore (metal sulphide) minerals are cogenetic. Forexample, recent LA-ICP-MS analyses of sphalerite-hosted fluid inclusions from Mississippi Valley-typezinc-lead deposits indicate that sulphide ore min-erals precipitated from anomalously metal-richbatches of fluid compared to those precipitatingthe gangue minerals (Wilkinson et al. 2009).
Because Au is commonly associated with sul-phide minerals such as pyrite and chalcopyrite andenters their structure both as chemically boundand nano-particulate metal, quantification of con-tents and redox states of Au and associated tracemetals (e.g. As, Se, Te) in these minerals is requiredfor estimating Au budget in a deposit, understanding
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Au precipitation mechanisms, and developing effi-cient extraction procedures. Such in situ analysesshould be achieved via a combination of comp-lementary microanalytical and spectroscopic toolssuch as LA-ICPMS, SIMS, and XAS.
Experimental
The analytical issues discussed above shouldadvance in parallel with speciation and solubilityexperiments in model laboratory systems under con-trolled conditions that nature does not offer. If, involcanic vapours, H2S and SO2 are undoubtedlythe major sulphur species at 1 bar over a wide temp-erature range, sulphur speciation in dense aqueousfluids and silicate melts is quite different and rep-resent the major gap of our knowledge. The recentdiscovery of a polysulphide sulphur form in S-richfluids, the trisulphur ion S3
2, which is stable inaqueous solution above 250–300 8C over a widepressure range (Pokrovski & Dubrovinsky 2011;Pokrovski & Dubessy 2012; Jacquemet et al. inpress), might affect our interpretation of both sul-phur behaviour and its control of Au in S-richfluids typical of porphyry and orogenic deposits.In situ spectroscopic approaches (e.g. Raman) arenecessary to better constrain the stability domainof S3
2 and other intermediate sulphur forms (e.g.other polysulphides, SO2, and sulphites). Once it isachieved, specially designed solubility and spec-troscopy (e.g. XAS) experiments in systems wheresuch sulphur species are abundant should allowquantifying their effect on the solubility and trans-port of Au and other sulphur-loving metals (e.g.Cu, Mo, Pt).
Few experimental data are available on Au spe-ciation and partitioning in vapour-brine-silicatemelt systems (e.g. Lerchbaumer & Audetat 2012)and on Au solubility in the major sulphide mineralssuch as chalcopyrite, bornite, pyrhhotite, and pyriteat temperatures from 500 8C to 700 8C, typical ofporphyry Cu–Au deposits (e.g. Simon et al. 2000).This lack prohibits a quantitative account of Auvapour-phase transport and Au budget in such set-tings. Little is known about Au solubility and spe-ciation in deep metamorphic fluids (.2–4 kbar,.500 8C) enriched in sulphur produced by pyrite-to-pyrrhotite breakdown (e.g. Tomkins 2010).
The common association of gold with arsenicand other metalloids (Sb, Bi, Se, Te) in hydrother-mal deposits might indicate the existence of Aucomplexes with these elements (+sulphur) in thefluid phase. At present however, this hypothesisremains speculative owing to the absence of quanti-tative data on the stoichiometry and stability of suchspecies. Acquisition of such data would repre-sent a formidable challenge for experimentalistsboth because of the extreme toxicity of certain
metalloids (Se and Te) and poor knowledge oftheir own speciation in aqueous solution andgas phase. An additional issue here is the insuffi-ciently constrained thermodynamic properties ofAu-bearing As–Sb–Bi–Te–Se–S minerals (e.g.Au2Bi, AuSbS2, AuTe2, AuBiS2), which may hosta significant part of gold in epithermal deposits.The mechanisms and driving forces of Au scaven-ging by these chalcogenide minerals during precipi-tation would be another formidable task forcourageous experimentalists.
Another essential experimental need is to betterunderstand the effect of CO2 on gold transport.Although recent studies (e.g. Pokrovski et al.2008a; Hanley & Gladney 2011) evoke enhancedtransport of Cu, Au, Pd, and Ni by CO2-rich fluidsor vapours, yet there is no sound physical-chemicalinterpretation of this potentially important phenom-enon because of a lack of experimental data on oremetal solubility in CO2-rich fluids and vapours atmagmatic-hydrothermal conditions.
Recent experimental and fluid/melt inclusionstudies has pointed to hydrosilicate liquids as poten-tial transporting media for some metals, like Ta, Cr,or Cu, and alkalis (e.g. Kamenetsky & Kamenetsky2010; Smirnov et al. 2012; Borisova et al. 2012c;references therein). Such liquids, having gel-likeproperties, are composed mostly of silica, water, andNaOH; they may form at the magmatic-hydrother-mal transition during late evolutionary stages indeeply evolved granite and pegmatite magmas andplay a role in concentrating metals and affectingtheir distribution in hydrothermal-magmatic depos-its. To what extent such media may influence theAu budget in natural systems remains to be verifiedby further analytical and experimental work.
Modelling
A major theoretical challenge in ore-depositresearch on gold and accompanying metals will beinterpretation of experimental data of solubility/partitioning and metal speciation in the frameworkof physical-chemical equations of state, enablingpredictions over the wide range of conditions ofmagmatic-hydrothermal Au deposits, from aqueoussolution to hypersaline brine or low-density vapourphase. At present, accurate predictions for some Auspecies may only be done for the liquid and densesupercritical fluid phase (density .0.4 g/cm3)using the HKF equation of state (e.g. Akinfiev &Zotov 2010). Generation of a self-consistent data-set within this model based on recent experi-mental data is awaited for several Au species,potentially important at magmatic conditions (e.g.Au chlorides, hydroxides, alkali metal–chloride–sulphides, Zajacz et al. 2010). New models arecurrently under development attempting unified
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

descriptions of the vapour-liquid region (e.g. Akin-fiev & Diamond 2003), but they lack experimentaldata necessary for their parameterizing. Speciationand solubility data should be better integrated inuser-friendly databases and computer codes allow-ing calculations of thermodynamics and kineticsof fluid-rock interactions (e.g. Oelkers et al. 2009).
In the last decade, molecular modellingapproaches based on quantum chemistry and mol-ecular dynamics have given new insights intoatomic structure and hydration energy of ore metalcomplexes (e.g. Au, Cu, Ag), helping to interpretspectroscopic and solubility data and in makingchoices among possible speciation models todescribe experimental data (e.g. Sherman 2010;Liu et al. 2011; Pokrovski et al. 2009a, b, 2013a).Such approaches, yet in their beginning, areexpected to provide a direct link between the mol-ecular properties of the dissolved species and theirthermodynamic stability and solubility.
Another advance of the last decade is the growingapplication of physical hydrology approaches basedon heat distribution and fluid flow models andpermeability changes, which allowed integratedreactive transport models of fluid paths, pressureand temperature evolution, and three-dimensionalore distribution and shape (e.g. Driesner & Geiger2007; Ingebritsen & Appold 2012; Weis et al.2012). However, chemistry of fluid-rock inter-actions, mineral solubility, and chemical elementspeciation in the fluid phase are not yet quantitativelyand systematically accounted for in these models,in particular, for trace elements like gold. Integrationof chemistry and physics in the same conceptualmodel of ore deposit formation would, thus, beanother major computational challenge, which cancontribute both to our fundamental understandingof gold deposit formation and to improving goldore exploration and extraction.
This work was supported by the Agence Nationale de laRecherche (Grant SOUMET–ANR 2011 Blanc SIMI5-6/009-01 to G.S.P), University of Toulouse (GrantCO2MET to G.S.P.) and by the Russian Foundationfor Basic Research (grant 12-05-93107-CNRS_a). T. Pok-rovski and P. Pokrovski are acknowledged for their assist-ance in the manuscript preparation, and A. Borisov fordiscussions on Au solubility in melts. We thank P. Garo-falo for inviting us to write this contribution and Z.Zajacz and an anonymous reviewer for their thoughtfulcomments that greatly improved this paper.
References
Adams, M. D. (ed.) 2005. Advances in Gold Ore Proces-sing. Elsevier, Amsterdam.
Akinfiev, N. N. & Diamond, L. W. 2003. Thermodyn-amic description of aqueous nonelectrolytes at infinite
dilution over a wide range of state parameters. Geochi-mica et Cosmochimica Acta, 67, 613–627.
Akinfiev, N. N. & Tagirov, B. R. 2006. Effect of sel-enium on silver transport and precipitation by hydro-thermal solutions: thermodynamic description of theAg–Se–S–Cl–O–H system. Geology Ore Deposits,48, 402–413.
Akinfiev, N. N. & Zotov, A. V. 1999. Thermodynamicdescription of equilibria in mixed fluids (H2O-non-polar gas) over a wide range of temperature (25–700 8C) and pressure (1–5000 bars). Geochimica etCosmochimica Acta, 63, 2025–2041.
Akinfiev, N. N. & Zotov, A. V. 2001. Thermodynamicdescription of chloride, hydrosulphide, and hydroxidecomplexes of Ag(I), Cu(I), and Au(I) at temperaturesof 25–500 8C and pressures of 1–2000 bar. Geochem-istry International, 39, 990–1006.
Akinfiev, N. N. & Zotov, A. V. 2010. Thermodynamicdescription of aqueous species in the system Cu–Ag–Au–S–O–H at temperatures of 0–600C andpressures of 1–3000 bar. Geochemistry International,48, 714–720.
Akinfiev, N. N., Voronin, M. V., Zotov, A. V., Pro-
kof’ev, V. & Y. 2006. Experimental investigation ofthe stability of a chloroborate complex and thermodyn-amic description of aqueous species in the B–Na–Cl–O–H system up to 350 8C. Geochemistry Inter-national, 44, 867–878.
Alonso Mori, R., Paris, E., Giuli, G., Eeckhout, S. G.,Kavcic, M., Zitnik, M., Bucar, K., Pettersson,L. G. M. & Glatzel, P. 2010. Sulfur-metal orbitalhybridization in sulfur-bearing compounds studied byX-ray emission spectroscopy. Inorganic Chemistry,49, 6468–6473.
Alvarez, J., Corti, H. R., Fernandez-Prini, R. & Japas,M. L. 1994. Distribution of solutes between coexistingsteam and water. Geochimica et Cosmochimica Acta,58, 2789–2798.
Anderson, G. M., Castet, S., Schott, J. & Mesmer,R. E. 1991. The density model for estimation of ther-modynamic parameters of reactions at high temper-ature and pressure. Geochimica et CosmochimicaActa, 55, 1769–1779.
Andre-Mayer, A.-S., Leroy, J. L., Bailly, L., Chauvet,A., Marcoux, E., Grancea, L., Llosa, F. & Rosas, J.2002. Boiling and vertical mineralization zoning: acase study from the Apacheta low-sulfidation epither-mal gold-silver deposit, southern Peru. MineraliumDeposita, 37, 452–464.
Archibald, S. M., Migdisov, A. A. & Williams-Jones,A. E. 2001. The stability of Au-chloride complexesin water vapour at elevated temperatures and pressures.Geochimica et Cosmochimica Acta, 65, 4413–4423.
Arehart, G. B., Chryssoulis, S. L. & Kesler, S. E.1993. Gold and arsenic in iron sulfides fromsediment-hosted disseminated gold deposits: impli-cation for depositional processes. Economic Geology,88, 171–185.
Arif, J. & Baker, T. 2004. Gold paragenesis and chem-istry at Batu Hijau, Indonesia: implications for gold-rich porphyry copper deposits. Mineralium Deposita,39, 523–535.
Audetat, A. & Pettke, T. 2003. The magmatic–hydrothermal evolution of two barren granites: a
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

melt and fluid inclusion study of the Rito del Medioand Canada Pinabete plutons in northern NewMexico (USA). Geochimica et Cosmochimica Acta,67, 97–121.
Audetat, A., Gunther, D. & Heinrich, C. A. 1998. For-mation of a magmatic–hydrothermal ore deposit:insights with LA-ICP-MS analysis of fluid inclusions.Science, 279, 2091–2094.
Baes, C. F. Jr. & Mesmer, R. E. 1976. The Hydrolysisof Cations. Wiley, New York.
Bakker, R. J. 2009. Package FLUIDS. Part 3: correlationsbetween equations of state, thermodynamics and fluidinclusions. Geofluids, 9, 63–74.
Baker, T., Van Achterberg, E., Ryan, C. & Lang, J. R.2004. Composition and evolution of ore fluids in amagmatic–hydrothermal skarn deposit. Geology, 32,117–120.
Ballhaus, C., Ryan, C. G., Mernagh, T. P. & Green,D. H. 1994. The partitioning of Fe, Ni, Cu, Pt, Aubetween sulfide, metal, and fluid phases: a pilotstudy. Geochimica et Cosmochimica Acta, 58,811–826.
Baranova, N. N. & Zotov, A. V. 1998. Stability ofgold sulphide species (AuHSaq and Au(HS)2
2) at 300,350 8C and 500 bar: experimental study. Mineralogi-cal Magazine, 62A, 116–117.
Baranova, N. N., Barsukov, V. L., Dar’ina, T. G. &Bannykh, L. N. 1977. On the interaction of goldwith aqueous alkaline solutions at 25 and 250 8C. Geo-khimiya, 6, 877–884.
Baranova, N. N., Zotov, A. V., Bannykh, L. N.,Dar’ina, T. G. & Savel’ev, B. V. 1983. The effectof redox conditions on solubility of gold in water at450 8C and 500 atm. Geochemistry International, 20,117–122.
Barin, I. 1995. Thermochemical Data of Pure Sub-stances. 3rd edn. VHC, Weinheim (Federal Republicof Germany).
Barnes, H. L. (ed.) 1997. Geochemistry of HydrothermalOre Deposits. Wiley, New York.
Belevantsev, V. I., Peshchevitsky, B. I. & Shamovs-
kaya, G. L. 1981. Gold (I) sulfide complexes inaqueous solution. Izvestia Akademii Nauk SSSR, Sibirs-koe Otdelenie, Seria Khimia, 1, 81–87 (in Russian).
Bell, A. S. & Simon, A. 2011. Evidence for the alterationof the Fe3+/SFe of silicate melt caused by the degas-sing of chloride-bearing aqueous volatiles. Geology,39, 499–502.
Bell, A. S., Simon, A. & Guillong, M. 2009. Exper-imental constraints on Pt, Pd and Au partitioning andfractionation in silicate melt – sulfide – oxide –aqueous fluid systems at 800 8C, 150 MPa and variablesulfur fugacity. Geochimica et Cosmochimica Acta,73, 5778–5792.
Belov, G. V., Iorish, V. S. & Yungman, V. S. 1999.IVTANTHERMO for Windows – database on thermo-dynamic properties and related software. Calphad, 23,173–180.
Benfield, R. E., Filipponi, A., Bowron, D. T.,Newport, R. J. & Gurman, S. J. 1994. An x-rayabsorption study of gold coordination compounds:EXAFS refinements and double-electron excitationbackground. Journal of Physics: Condensed Matter,6, 8449–8468.
Benning, L. G. & Seward, T. M. 1996. Hydrosulphidecomplexing of Au(I) in hydrothermal solutions from150–400 8C and 500–1500 bar. Geochimica et Cos-mochimica Acta, 60, 1849–1871.
Berndt, M. E., Buttram, T., Earley, D. III & Seyfried,W. E. 1994. The stability of gold polysulfide com-plexes in aqueous sulfide solutions: 100 to 150 8Cand 100 bars. Geochimica et Cosmochimica Acta, 58,587–594.
Berrodier, I., Farges, F., Benedetti, M., Winterer,M., Brown, G. E. Jr. & Deveughele, M. 2004.Adsorption mechanisms of trivalent gold on iron-and aluminum-(oxy)hydroxides. Part 1: X-ray absorp-tion and raman scattering spectroscopic studies ofAu(III) adsorbed on ferrihydrite, goethite, and boeh-mite. Geochimica et Cosmochimica Acta, 68, 3019–3042.
Bezmen, N. I., Asif, M., Brugmann, G. E., Romanenko,I. M. & Naldrett, A. J. 1994. Distribution ofPd, Rh, Ru, Ir, Os, and Au beween sulfide and silicatemelts. Geochimica et Cosmochimica Acta, 58, 1251–1260.
Blanchard, M., Alfredsson, M., Brodholt, J.,Wright, K. & Catlow, C. R. A. 2007. Arsenic incor-poration into FeS2 pyrite and its influence on dissol-ution: a DFT study. Geochimica et CosmochimicaActa, 71, 624–630.
Boiron, M.-C., Cathelineau, M. & Trescases, J.-J.1989. Conditions of gold-bearing arsenopyritecrystallization in the Villeranges basin, Marche-Combrailles shear zone, France: a mineralogicaland fluid inclusion study. Economic Geology, 84,1340–1362.
Borisov, A. & Palme, H. 1996. Experimental determi-nation of the solubility of Au in silicate melts. Miner-alogy and Petrology, 56, 297–312.
Borisov, A. & Palme, H. 2000. Solubilities of noblemetals in Fe-containing silicate melts as derived fromexperiments in Fe-free systems. American Mineralo-gist, 85, 1665–1673.
Borisova, A. Y., Pichavant, M., Polve, M., Wieden-
beck, M., Freydier, R. & Caudaudap, F. 2006.Chemistry of silicic melts of the Mount Pinatubo,Philippines: implications for ore-forming potential ofadakitic magmatism. Geochimica et CosmochimicaActa, 70, 3702–3716.
Borisova, A. Y., Pokrovski, G. S., Pichavant, M.,Freydier, R. & Candaudap, F. 2010. Arsenicenrichment in hydrous peraluminous melts: insightsfrom LA-ICPMS and in situ X-ray Absorption Spec-troscopy. American Mineralogist, 95, 1095–1104.
Borisova, A. Y., Thomas, R., Salvi, S., Candaudap, F.,Lanzanova, A. & Chmeleff, J. 2012a. Tin andassociated metal and metalloid geochemistry by femto-second LA-ICP-QMS microanalysis of pegmatite-leucogranite melt and fluid inclusions: new evidencefrom melt-melt-fluid immiscibility. MineralogicalMagazine, 76, 91–133.
Borisova, A. Y., Toutain, J.-P., Stefansson, A., Gouy,S. & de Parseval, PH. 2012b. Processes controllingthe 2010 Eyjafjallajokull explosive eruption. Journalof Geophysical Research, 117, B05202.
Borisova, A. Y., Ceuleneer, G. et al. 2012c. A newview on the petrogenesis of the Oman ophiolite
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

chromitites from microanalyses of chromite-hostedinclusions. Journal of Petrology, 53, 2411–2440.
Borisova, A. Y., Martel, C. et al. 2013. Highly explo-sive 2010 Merapi eruption: Evidence for shallow-level crustal assimilation and hybrid fluid. Journalof Volcanology and Geothermal Reseach, 261,193–208, http://dx.doi.org/10.1016/j.volgeores.2012.11.002
Born, M. 1920. Volumen und Hydratationswarme derIonen. Zeitschrift fur Physik, 1, 45–48.
Botcharnikov, R. E., Linnen, R. L. & Holtz, F.2010. Solubility of Au in Cl- and S-bearing hydroussilicate melts. Geochimica et Cosmochimica Acta,74, 2396–2411.
Botcharnikov, R. E., Linnen, R. L., Wilke, M., Holtz,F., Jugo, P. J. & Berndt, J. 2011. High gold concen-trations in sulphide-bearing magma under oxidizingconditions. Nature Geoscience, 4, 112–115.
Boudreau, A. E. & Meurer, W. P. 1999. Chro-matographic separation of the platinum-groupelements, gold, base metals and sulfur during degas-sing of a compacting and solidifying crystal pile.Contributions to Mineralogy and Petrology, 134,174–185.
Bowers, T. S. & Helgeson, H. C. 1983. Calculation of thethermodynamic and geochemical consequences ofnonideal mixing in the system H2O–CO2–NaCl onphase relations in geologic systems: equation of statefor H2O–CO2–NaCl fluids at high pressures andtemperatures Geochimica et Cosmochimica Acta, 47,1247–1275.
Boyle, R. W. 1969. Hydrothermal transport and depo-sition of gold. Economic Geology, 64, 112–115.
Boyle, R. W. 1979. The geochemistry of gold and itsdeposits. Geological Survey of Canada, Bulletin,280, 584p.
Brenan, J. M., McDonough, W. F. & Ash, R. 2005.An experimental study of the solubility and partition-ing of iridium, osmium and gold between olivine andsilicate melt. Earth and Planetary Science Letters,237, 855–872.
Brimhall, G. H. & Crerar, D. A. 1987. Ore fluids: mag-matic to supergene. Reviews in Mineralogy andGeochemistry, 17, 235–321.
Bryce, R. A., Charnock, J. M., Pattrick, R. A. D. &Lennie, A. R. 2003. EXAFS and density functionalstudy of gold(I) thiosulphate complex in aqueous sol-ution. Journal of Physical Chemistry B, 107, 2516–2523.
Cabri, L. J., Newville, M., Gordon, R. A., Crozier,E. D., Sutton, S. R., McMahon, G. & Jiang, D.-T.2000. Chemical speciation of gold in arsenopyrite.Canadian Mineralogist, 38, 1265–1281.
Candela, P. A. 2004. Ores in the Earth’s crust. Treatiseon Geochemistry, 3, 411–431.
Catchpole, H., Kouzmanov, K., Fontbote, L.,Guillong, M. & Heinrich, C. A. 2011. Fluidevolution in zoned Cordilleran polymetallic veins –Insights from microthermometry and LA-ICP-MS offluid inclusions. Chemical Geology, 281, 293–304.
Catchpole, H., Kouzmanov, K., Putlitz, B., Font-
bote, L. & Seo, J. H. In press. Fluid evolution ofporphyry-related zoned base metal mineralization inthe Morococha district, Peru. Economic Geology.
Cathelineau, M., Boiron, M.-C., Holliger, P.,Marion, P. & Denis, M. 1989. Gold in arsenopyrites:crystal chemistry, location and state, physical andchemical conditions of deposition. EconomicGeology Monograph, 6, 328–341.
Cauzid, J., Philippot, P., Martinez-Criado, G.,Menez, B. & Laboure, S. 2007. Contrasting Cu-com-plexing behaviour in vapour and liquid fluid inclusionsfrom the Yankee Lode deposit, Mole Granite. Chemi-cal Geology, 246, 39–54.
Cepedal, A., Fuertes-Fuente, M., Martin-Izard, A.,Gonzalez-Nistal, S. & Barrero, M. 2008. Gold-bearing As-rich pyrite and arsenopyrite from the ElValle gold deposit, Asturias, northwestern Spain.Canadian Mineralogist, 46, 233–247.
Chaplygin, I. V. 2010. Behavior of chalcophile andhighly siderophile elements (Au, PGE, Re) in volcanicgases of Kudriavy volcano (Kuriles). In: 11th Inter-national Platinum Symposium, Ontario GeologicalSurvey, June 21–24, 2010, Miscellaneous release-Data, 269, Ontario.
Chase, M. W. Jr. 1998. NIST-JANAF thermo-chemical Tables. 4th edn. Journal of Physical andChemical Reference Data Monographs, MonographNo. 9, American Institute of Physics, New York.
Chivers, T. & Elder, P. J. W. 2013. Ubiquitous trisulfurradical ion: fundamentals and applications in materialscience, electrochemistry, analytical chemistry and geo-chemistry. Chemical Society Reviews, 42, 5996–6005.
Chouinard, A., Paquette, J. & Williams-Jones, A. E.2005. Crystallographic controls on trace-elementincorporation in auriferous pyrite from the Pascuaepithermal high-sulfidation deposit, Chile-Argentina.Canadian Mineralogist, 43, 951–963.
Churakov, S. V., Tkachenko, S. I., Korzinsky, M. A.,Bocharnikov, R. E. & Shmulovich, K. I. 2000.Evolution of composition of high-temperature fumaro-lic gases from Kudryavy volcano, Iturup, Kuril Islands:the thermodynamic modeling. Geochemistry Inter-national, 38, 436–451.
Ciobanu, C. L. & Cook, N. J. 2006. Gold scavengedby bismuth melts: an example from Alpine shearre-mobilizates in the Highis Massif, Romania. Mineral-ogy and Petrology, 87, 351–384.
Ciobanu, C. L., Cook, N. J. & Spry, P. G. 2006. Preface –Special Issue: telluride and selenide minerals in golddeposits – how and why? Mineralogy and Petrology,87, 163–169.
Ciobanu, C. L., Cook, N. J., Pring, A., Brugger, J.,Danyushevsky, L. V. & Shimizu, M. 2009. “Invis-ible gold” in bismuth chalcogenides. Geochimica etCosmochimica Acta, 73, 1970–1999.
Cobley, C. M. & Xia, Y. 2009. Gold and nanotechnology.Elements, 5, 309–313.
Connors, K. A., Noble, D. C., Bussey, S. D. & Weiss,S. I. 1993. Initial gold contents of silicic volcanicrocks: bearing on the behavior of gold in magmaticsystems. Geology, 21, 937–940.
Cotton, A. F. & Wilkinson, G. 1988. Advanced Inor-ganic Chemistry. 5th edn. Wiley, New York.
Crerar, D., Wood, S. & Brantley, S. 1985. Chemicalcontrols on solubility of ore-forming minerals inhydrothermal solutions. Canadian Mineralogist, 23,333–352.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Crocket, J. H., Fleet, M. E. & Stone, W. E. 1997.Implications of composition for experimental parti-tioning of platinum-group elements and gold betweensulfide liquid and basalt melt: the significance ofnickel content. Geochimica et Cosmochimica Acta,61, 4139–4149.
Cunningham, C. G., Austin, G. W., Naeser, C. W., Rye,R. O., Ballantyne, G. H., Stamm, R. G. & Barker,C. E. 2004. Formation of a paleothermal anomalyand disseminated gold deposits associated with theBingham Canyon porphyry Cu–Au–Mo system, Utah.Economic Geology, 99, 789–806.
Dadze, T. P. & Kashirtseva, G. A. 2004. Experimentalstudy of gold solubility in sulphide hydrothermalsolutions. In: Zharikov, V. A. & Fed’kin, V. V.(eds) Experimental Mineralogy, Some Results on theCentury’s Frontier. Moscow, Nauka, 1, 315–326 (inRussian).
de Haller, A. & Fontbote, L. 2009. The Raul-Condestable Iron Oxide Copper-Gold deposit, Cen-tral Coast of Peru: ore and related hydrothermalalteration, sulfur Isotopes, and thermodynamic con-straints. Economic Geology, 104, 365–384.
Deditius, A. P., Utsunomiya, S., Renock, D., Ewing, R.C., Ramana, C. V., Becker, U. & Kesler, S. E. 2008.A proposed new type of arsenian pyrite: composition,nanostructure and geological significance. Geochimicaet Cosmochimica Acta, 72, 2919–2933.
Diamond, L. W. 1990. Fluid inclusion evidence for P–V–T–X evolution of hydrothermal solutions in Late-Alpine gold-quartz veins at Brusson, Val d’Ayas,northwest Italian Alps. American Journal of Science,290, 912–958.
Driesner, T. & Geiger, S. 2007. Numerical simulation ofmultiphase fluid flow in hydrothermal systems. In:Liebscher, A. & Heinrich, C. A. (eds) Fluid-FluidInteractions. Reviews in Mineralogy and Geochemis-try, 65, 187–212.
Driesner, T. & Heinrich, C. A. 2007. The systemH2O–NaCl. Part I: correlation formulae for phaserelations in temperature–pressure–compositionspace from 0 to 1000 8C, 0 to 5000 bar, and 0 to 1XNaCl. Geochimica et Cosmochimica Acta, 71,4880–4901.
Duan, Z., Moller, N. & Weare, J. 1995. Equationof state for NaCl–H2O–CO2 system – prediction ofphase equilibria and volumetric properties. Geochi-mica et Cosmochimica Acta, 59, 2869–2882, http://www.geochem-model.org/programs.htm
Einaudi, M. T., Hedenquist, J. W. & Inan, E. E. 2003.Sulfidation state of fluids in active and extinct hydro-thermal systems: transitions from porphyry to epither-mal environments. Society of Economic GeologistsSpecial Publication, 10, 285–313.
Erkey, C. 2000. Supercritical carbon dioxide extraction ofmetals from aqueous solutions: a review. The Journalof Supercritical Fluids, 17, 259–287.
Farges, F., Sharps, J. A. & Brown, G. E. Jr. 1993. Localenvironment around gold(III) in aqueous chloride sol-utions: an EXAFS spectroscopy study. Geochimica etCosmochimica Acta, 57, 1243–1252.
Filignati, P. & Sbrana, A. 1998. Presence of native goldand tellurium in the active high-sulfidation hydrother-mal system of the La Fossa volcano (Vulcano, Italy).
Journal of Volcanology and Geothermal Research,86, 187–198.
Fleet, M. E. & Knipe, S. W. 2000. Solubility of native goldin H–O–S fluids at 100–400 8C and high H2S content.Journal of Solution Chemistry, 29, 1143–1157.
Fleet, M. E. & Mumin, A. H. 1997. Gold-bearingarsenian pyrite and marcasite and arsenopyrite fromCarlin Trend gold deposits and laboratory synthesis.American Mineralogist, 82, 182–193.
Fleet, M. E., MacLean, P. J. & Barbier, J. 1989.Oscillatory-zoned As-bearing pyrite from strata-boundand stratiform gold deposits: an indicator of ore fluidevolution. In: Keays, R. R., Ramsay, W. R. H. &Groves, D. I. (eds) The Geology of Gold Deposits:The Perspectives in 1988. Economic Geology Mono-graph, 6, 356–362.
Frank, M. R., Candela, P. A., Piccoli, P. M. &Glascock, M. D. 2002. Gold solubility, speciation,and partitioning as a function of HCl in the brine-silicate melt-mettalic gold system at 800 8C and100 MPa. Geochimica et Cosmochimica Acta, 66,3819–3732.
Frank, M. R., Simon, A. C., Pettke, T., Candela, P. A.& Piccoli, P. M. 2011. Gold and copper partitioningin magmatic-hydrothermal systems at 800 8C and100 MPa. Geochimica et Cosmochimica Acta, 75,2470–2482.
Gadet, M.-C. & Pouradier, J. 1972. Hydrolyse descomplexes de l’or (I). Comptes Rendus de l’Academiedes sciences, 275, serie C, 1061–1064.
Gammons, C. H. & Williams-Jones, A. E. 1995.The solubility of Au–Ag alloy + AgCl in HCl/NaCl solutions at 300 8C: new data on the stabil-ity of Au(I) chloride complexes in hydrothermalfluids. Geochimica et Cosmochimica Acta, 59,3453–3468.
Gammons, C. H., Yu, Y. & Williams-Jones, A. E. 1997.The disproportionation of gold(I) chloride complexesat 25 to 200 8C. Geochimica et Cosmochimica Acta,61, 1971–1983.
Gemmel, B. J. 1987. Geochemistry of metallic traceelements in fumarolic condensates from Nicaraguanand Costa Rican volcanoes. Journal of Volcanologyand Geothermal Research, 33, 161–181.
Genkin, A. D., Bortnikov, N. S. et al. 1998. A multidis-ciplinary study of invisible gold in arsenopyrite fromfour mesothermal gold deposits in Siberia, RussianFederation. Economic Geology, 93, 463–487.
Gibert, F., Pascal, M. L. & Pichavant, M. 1998. Goldsolubility and speciation in hydrothermal solutions:experimental study of the stability of hydrosulfidecomplex of gold (AuHS0) at 350 to 450 8C and500 bars. Geochimica et Cosmochimica Acta, 62,2931–2947.
Giuliani, G., Dubessy, J. et al. 2003. CO2–H2S–COS–S8–AlO(OH)-bearing fluid inclusions in ruby frommarble-hosted deposits in Luc Yen area, NorthVietnam. Chemical Geology, 194, 167–185.
Goldfarb, R., Baker, T., Dube, B., Groves, D. I., Hart,G. Jr. & Gosselin, P. 2005. Distribution, characterand genesis of gold deposits in metamorphic terranes.In: Economic Geology 100th anniversary volume.Society of Economic Geologists, Littleton, Colorado,USA, 407–450.
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Gruen, G., Heinrich, C. A. & Schroeder, K. 2010. TheBingham Canyon porphyry Cu–Mo–Au deposit. II.Vein geometry and ore shell formation by pressure-driven rock extension. Economic Geology, 105, 69–90.
Guillong, M., Latkoczy, C., Seo, J. H., Gunther, D. &Heinrich, C. A. 2008. Determination of sulfur in fluidinclusions by laser ablation ICP-MS. Journal ofAnalytical Atomic Spectrometry, 23, 1581–1589.
Hager, J. P. & Hill, R. B. 1970. Thermodynamic proper-ties of the vapor transport reactions in the Au–Clsystem by transpiration-mass spectrometric technique.Metallurgical and Materials Transactions B, 1,2723–2731.
Halter, W. E., Pettke, T. & Heinrich, C. A. 2002. Theorigin of Cu/Au ratios in porphyry-type ore deposits.Science, 296, 844–846.
Halter, W. E., Pettke, T. & Heinrich, C. A. 2005.Magma evolution and the formation of porphyryCu–Au ore fluids: evidence from silicate and sulfidemelt inclusions. Mineralium Deposita, 39, 845–863.
Hanley, J. J. & Gladney, E. R. 2011. The presence ofcarbonic-dominant volatiles during the crystallizationof sulfide-bearing mafic pegmatites in the NorthRoby zone, Lac des Iles Complex, Ontario. EconomicGeology, 106, 33–54.
Hanley, J. J., Pettke, T., Mungall, J. E. & Spooner,E. T. C. 2005. Investigations of the behavior and distri-bution of platinum and gold in silicate melt – brine mix-tures at 1.5 kbar, 600 to 800 8C using synthetic fluidinclusions methods: a laser ablation ICPMS pilot study.Geochimica et Cosmochimica Acta, 69, 2593–2611.
Harris, A. C. & Golding, S. D. 2002. New evidence ofmagmatic-fluid-related phyllic alteration: implicationsfor the genesis of porphyry Cu deposits. Geology, 30,335–338.
Hattori, K. H. & Keith, J. D. 2001. Contribution of maficmelt to porphyry copper mineralization: evidence fromMount Pinatubo, Phillipines, and Bingham Canyon,Utah, USA. Mineralium Deposita, 36, 799–806.
Hayashi, K. I. & Ohmoto, H. 1991. Solubility of goldin NaCl- and H2S-bearing aqueous solutions at 250–350 8C. Geochimica et Cosmochimica Acta, 55,2111–2126.
Hedenquist, J. W. & Lowenstern, J. B. 1994. The roleof magmas in the formation of hydrothermal ore depos-its. Nature, 370, 519–527.
Hedenquist, J. W., Simmons, S. F., Giggenbach, W. F.& Eldridge, C. S. 1993. White Island, New Zealand,volcanic-hydrothermal system represents the geo-chemical environment of high-sulfidation Cu and Auore deposition. Geology, 21, 731–734.
Hedenquist, J. W., Aoki, M. & Shinohara, H. 1994.Flux of volatiles and ore-forming metals from themagmatic-hydrothermal system of Satsuma Iwojimavolcano. Geology, 22, 585–588.
Hedenquist, J. W., Arribas, A. Jr. & Reynolds, T. J.1998. Evolution of an intrusion-centered hydro-thermal system: far Southeast-Lepanto porphyry andepithermal Cu–Au deposits, Philippines. EconomicGeology, 93, 373–404.
Heinrich, C. A. 2005. The physical and chemical evol-ution of low-salinity magmatic fluids at the porphyryto epithermal transition: a thermodynamic study.Mineralium Deposita, 39, 864–889.
Heinrich, C. A. 2007. Fluid – fluid interactions inmagmatic-hydrothermal ore formation. Reviews inMineralogy and Geochemistry, 65, 363–387.
Heinrich, C. A. & Eadington, P. J. 1986. Thermodyn-amic predictions of the hydrothermal chemistry ofarsenic and their significance for the parageneticsequence of some cassiterite-arsenopyrite-base metalsulfide deposits. Economic Geology, 81, 511–529.
Heinrich, C. A., Ryan, C. G., Mernagh, T. P. &Eadington, P. J. 1992. Segregation of ore metalsbetween magmatic brine and vapor – a fluid inclusionstudy using PIXE microanalysis. Economic Geology,87, 1566–1583.
Heinrich, C. A., Gunther, D., Audedat, A., Ulrich, T.& Frischknecht, R. 1999. Metal fractionationbetween magmatic brine and vapour, and the linkbetween porphyry-style and epithermal Cu–Au depos-its. Geology, 27, 755–758.
Heinrich, C. A., Driesner, T., Stefansson, A. &Seward, T. M. 2004. Magmatic vapor contractionand the transport of gold from porphyry to epithermalore deposits. Geology, 39, 761–764.
Helgeson, H. C. & Garrels, R. M. 1968. Hydrothermaltransport and deposition of gold. Economic Geology,63, 622–635.
Helgeson, H. C., Kirkham, D. H. & Flowers, G. C.1981. Theoretical prediction of the thermodynamic be-havior of aqueous electrolytes at high pressures andtemperatures: IV. Calculation of activity coefficients,osmotic coefficients, and apparent molal and standardand relative partial molal properties to 600 8C and5 kb. American Journal of Science, 291, 1249–1516.
Hemley, J. J. & Hunt, J. P. 1992. Hydrothermal ore-forming processes in the light of studies in rock-buffered systems: II. Some general geological appli-cations. Economic Geology, 87, 23–43.
Hemley, J. J., Cygan, G. L., Fein, J. B., Robinson, G. R.& D’angelo, W. M. 1992. Hydrothermal ore-formingprocesses in the light of studies in rock-bufferedsystems: I. Iron–copper–zinc–lead sulfide solubilityrelations. Economic Geology, 87, 1–22.
Henley, R. W. 1973. Solubility of gold in hydrothermalchloride solutions. Chemical Geology, 11, 73–87.
Herrington, R. J. & Wilkinson, J. J. 1993. Colloidalgold and silica in mesothermal vein systems. Geol-ogy, 21, 539–542.
Hough, R. M., Noble, R. R. P. et al. 2008. Naturallyoccurring gold nanoparticles and nanoplates. Geol-ogy, 36, 571–574.
Huheey, J. E. 1983. Inorganic Chemistry: Principles ofStructure and Reactivity. Harper and Row, New York.
ICSD 2010. Inorganic Crystal Structure Database, FIZKarlshure, http://www.fiz-karlsruhe.de/icsd.html
Ingebritsen, S. E. & Appold, M. S. 2012. The physicalhydrology of ore deposits. Economic Geology, 107,559–584.
Ishimaru, S., Arai, S., Borisova, A. Y. & Tamura, A.2011. Possible high-PGE-Au silicate melt/aqueousfluid in mantle wedge inferred from Ni metasomatismin Avacha peridotite xenoliths. In: Goldschmidt Con-ference Abstracts, Prague, Czech Republic. Mineralo-gical Magazine, 1087.
Jackson, K. J. & Helgeson, H. C. 1985. Chemical andthermodynamic constraints on the hydrothermal
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

transport and deposition of tin: I. Calculations of thesolubility of cassiterite at high pressures and tempera-tures. Geochimica et Cosmochimica Acta, 49, 1–22.
Jacquemet, N., Guillaume, D., Zwick, A. & Pok-
rovski, G. S. In press. In situ Raman spectroscopyidentification of the S32 ion in S-rich hydrothermalfluids from synthetic fluid inclusions. American Miner-alogist, http://dx.doi.org/10.2138/am.2014.4524
James, S. E. & Hager, J. P. 1978. High temperaturevaporization chemistry in the gold-chloride systemincluding formation of vapor complex species of goldand silver with copper and iron. Metallurgical Trans-actions, 9B, 501–508.
Jego, S. & Pichavant, M. 2012. Gold solubility in arcmagmas: experimental determination of the effect ofsulfur at 1000 8C and 0.4 GPa. Geochimica et Cosmo-chimica Acta, 84, 560–592.
Jenner, F. E., O’Neill, H. ST. C., Arculus, R. J. &Mavrogenes, J. A. 2010. The magnetite crisis in theevolution of arc-related magmas and the initial concen-tration of Au, Ag, and Cu. Journal of Petrology, 51,2445–2464.
Johnson, J. W., Oelkers, E. H. & Helgeson, H. C. 1992.SUPCRT92: a software package for calculating thestandard molal thermodynamic properties of minerals,gases, aqueous species, and reactions from 1 to5000 bar and 0 to 1000 8C. Computers & Geosciences,18, 899–947, available on line at: http://geopig.asu.edu/?q¼tools
Jugo, P. J., Candela, P. A. & Piccoli, P. M. 1999. Mag-matic sulfides and Au: Cu ratios in porphyry deposits:an experimental study of copper and gold partitioningat 850 8C, 100 MPa in a haplogranite melt-pyrrhotite-intermediate solid solution-gold metal assemblage, atgas saturation. Lithos, 46, 573–589.
Kaasalainen, H. & Stefansson, A. 2011. Sulfur specia-tion in natural hydrothermal waters, Iceland. Geochi-mica et Cosmochimica Acta, 75, 2777–2791.
Kamenetsky, V. S. & Kamenetsky, M. B. 2010. Mag-matic fluids immiscible with silicate melts: examplesfrom inclusions in phenocrysts and glasses, and impli-cations for magma evolution and metal transport.Geofluids, 10, 293–311.
Kehayov, R., Bogdanov, K., Fanger, L., von Quadt,A., Pettke, T. & Heinrich, C. A. 2003. The fluidchemical evolution of the Elatiste porphyry Cu–Au-PGE deposit, Bulgaria. In: Eliopoulos, D. G.(ed.) Mineral Exploration and Sustainable Develop-ment. Millpress, Rotterdam, 1173–1176.
Klemm, L. M., Pettke, T., Heinrich, C. A. & Campos, E.2007. Hydrothermal evolution of the El Tenientedeposit, Chile: porphyry Cu–Mo ore deposition fromlow-salinity magmatic fluids. Economic Geology,102, 1021–1045.
Klemm, L. M., Pettke, T. & Heinrich, C. A. 2008. Fluidand source magma evolution of the Questa porphyryMo deposit, New Mexico, USA. Mineralium Deposita,43, 533–552.
Kissner, R., Welti, G. & Geier, G. 1997. The hydrolysisof gold(I) in aqueous acetonitrile solutions. Journalof the Chemical Society, Dalton Transactions,1773–1777.
Kouzmanov, K. & Pokrovski, G. S. 2012. Hydro-thermal controls on metal distribution in porphyry
Cu(–Au–Mo) systems. Society of Economic Geol-ogists Special Publication, 16, 573–618.
Kouzmanov, K., Pettke, T. & Heinrich, C. A. 2010.Direct analysis of ore-precipitating fluids: combinedIR microscopy and LA-ICP-MS study of fluidinclusions in opaque ore minerals. EconomicGeology, 105, 351–373.
Krauskopf, K. B. 1951. The solubility of gold. EconomicGeology, 48, 858–870.
Landsberg, A. & Hoatson, C. L. 1970. The kinetics andequilibria of the gold-chloride system. Journal of theLess-Common Metals, 22, 327–339.
Landtwing, M. R., Pettke, T., Halter, W. E., Hein-
rich, C. A., Redmond, P. B., Einaudi, M. T. &Kunze, K. 2005. Copper deposition during quartz dis-solution by cooling magmatic-hydrothermal fluids:The Bingham porphyry. Earth and Planetary ScienceLetters, 235, 229–243.
Landtwing, M. R., Furrer, C., Redmond, P. B., Pettke,T., Guillong, M. & Heinrich, C. A. 2010. TheBingham Canyon porphyry Cu–Mo–Au deposit III.Zoned copper–gold ore deposition by magmaticvapor expansion. Economic Geology, 105, 91–118.
Large, R. R., Bull, S. W. & Maslennikov, V. V. 2011.A carbonaceous sedimentary source-rock model forCarlin-type and orogenic gold deposits. EconomicGeology, 106, 331–358.
Larocque, A. C. L., Stimac, J. A. & Huminicki,M. A. E. 2000. Evidence for open-system behaviorin immiscible Fe–S–O liquids in silicate magmas:implications for contributions of metals and sulfurto ore-forming fluids. Canadian Mineralogist, 38,1233–1249.
Lerchbaumer, L. & Audetat, A. 2012. High Cu concen-trations in vapor-type fluid inclusions: an artifact?Geochimica et Cosmochimica Acta, 88, 255–274.
Li, Y. & Audetat, A. 2012. Partitioning of V, Mn, Co, Ni,Cu, Zn, As, Mo, Ag, Sn, Sb, W, Au, Pb, and Bi betweensulfide phases and hydrous basanite melt at uppermantle conditions. Earth and Planetary ScienceLetters, 355–356, 327–340.
Li, Y., Audetat, A., Lerchbaumer, L. & Xiong, X. L.2009. Rapid Na, Cu exchange between synthetic fluidinclusions and external aqueous solutions: evidencefrom LA-ICP-MS analysis. Geofluids, 9, 321–329.
Liang, J.-L., Sun, W. D., Li, Y. L., Zhu, S. Y., Li, H., Liu,Y. L. & Zhai, W. 2013. An XPS study on the 1 valencestates of arsenic in arsenian pyrite: implications for Audeposition mechanism of Yang-shan Carlin-type golddeposit, western Qinling belt. Journal of Asian EarthSciences, 62, 363–372.
Likhoidov, G. G. & Nekrasov, I. Y. 1992. Solubility ofgold at 500 and 600 8C (total pressure ¼ 1 kbar) inthe presence of iron oxides and sulfides. Doklady Aka-demii Nauk SSSR, 323, 294–298 (in Russian).
Likhoidov, G. G. & Nekrasov, I. Y. 1994. Solubility ofgold in the system Na–Fe–S–Cl–H2O–O2 at 300–500 8C and total pressure of 1 kbar. Doklady AkademiiNauk SSSR, 336, 380–382 (in Russian).
Liu, X., Lu, X., Wang, R., Zhou, H. & Xu, S. 2011. Spe-ciation of gold in hydrosulphide-rich ore-formingfluids: insights from first-principles molecular dynam-ics simulations. Geochimica et Cosmochimica Acta,75, 185–194.
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Loucks, R. R. & Mavrogenes, J. A. 1999. Gold solubilityin supercritical hydrothermal brines measured in syn-thetic fluid inclusions. Science, 284, 2159–2163.
Lowenstern, J. B. 2001. Carbon dioxide in magmasand implications for hydrothermal systems. Minera-lium Deposita, 36, 490–502.
Maddox, L. M., Bancroft, G. M., Scaini, M. J. &Lorimer, J. W. 1998. Invisible gold: comparison ofAu deposition on pyrite and arsenopyrite. AmericanMineralogist, 83, 1240–1245.
Mao, S. H. 1991. Occurrence and distribution of invisiblegold in a Carlin-type gold deposit in China. AmericanMineralogist, 76, 1964–1972.
Marion, P. H., Regnard, J.-R. & Wagner, F. E. 1986.Etude de l’etat chimique de l’or dans les sulfures auri-fers par spectroscopie Mossbauer de 197Au. ComptesRendus Academie des Sciences (Paris) 302 II,571–574.
Martell, A. E., Smith, R. M. & Motekaitis, R. J. 2004.Critical: Critically Selected Stability Constants ofMetal Complexes Database. Version 8.0, NIST,Texas A & M University, College Station.
Mesmer, R. E., Marshall, W. L., Palmer, D. A., Simon-
son, J. M. & Holmes, H. F. 1988.Thermodynamics ofaqueous association and ionization reactions at hightemperatures and pressures. Journal of Solution Chem-istry, 17, 699–718.
Metrich, N., Berry, A. J., O’Neill, H. ST. C. & Susini,J. 2009. The oxidation state of sulfur in synthetic andnatural glasses determined by X-ray absorption spec-troscopy. Geochimica et Cosmochimica Acta, 73,2382–2399.
Meyer, C. & Hemley, J. J. 1967. Wall rock alteration. In:Barnes, H. L. (ed.) Geochemistry of Hydrothermal oreDeposits. Holt, Rinehart and Winston, New York,166–235.
Migdisov, A. A. & Bychkov, A. Y. 1998. The behav-iour of metals and sulphur during the formationof hydrothermal mercury-antimony-arsenic minerasi-zation, Uzon caldera, Kamchatka, Russia. Journalof Volcanology and Geothermal Research, 84,153–171.
Mikucki, E. J. 1998. Hydrothermal transport and deposi-tional processes in Archean lode-gold systems: areview. Ore Geology Reviews, 13, 307–321.
Mironov, A. G., Zhmodik, S. M. & Maksimova, E. A.1981. An experimental investigation of the sorptionof gold by pyrites with different thermoelectric proper-ties. Geochemistry International, 18, 153–160.
Moller, P. & Kersten, G. 1994. Electrochemicalaccumulation of visible gold on pyrite and arsenopyritesurfaces. Mineralium Deposita, 29, 404–413.
Moore, G. 2008. Interpreting H2O and CO2 contents inmelt inclusions: constraints from solubility exper-iments and modelling. Reviews in Mineralogy & Geo-chemistry, 69, 333–361.
Moss, R., Scott, S. D. & Binns, R. A. 2001. Gold contentof eastern Manus basin volcanic rocks: implications forenrichment in associated hydrothermal precipitates.Economic Geology, 96, 91–107.
Muir, D. M. 2011. A review of the selective leaching ofgold from oxidized copper-gold ores with ammonia-cyanide and new insights for plant control and oper-ation. Minerals Engineering, 24, 576–582.
Muntean, J. L., Cline, J. S., Simon, A. C. & Longo, A.A. 2011. Magmatic-hydrothermal origin of Nevada’sCarlin-type gold deposits. Nature Geoscience, 4,122–127.
Murakami, H., Seo, J. H. & Heinrich, C. A. 2010. Therelation between Cu/Au ratio and formation depth ofporphyry-style Cu–Au + Mo deposits. MineraliumDeposita, 45, 11–21.
Murphy, P. J. & LaGrange, M. S. 1998. Raman spec-troscopy of Au chloro-hydroxy speciation in fluids atambient T and P: a re-evaluation of the effects of pHand chloride concentration. Geochimica et Cosmochi-mica Acta, 62, 3515–3526.
Murphy, P. J., Stevens, G. & LaGrange, M. S. 2000.The effects of temperature and pressure on gold-chloride speciation in hydrothermal fluids: a Ramanspectroscopic study. Geochimica et CosmochimicaActa, 64, 479–494.
Mustard, R., Ulrich, T., Kamenetsky, V. S. &Mernagh, T. 2006. Gold and metal enrichment innatural granitic melts during fractional crystallization.Geology, 34, 85–88.
Nekrasov, I. Y., Konyushok, A. A. & Sorokin, V. I. 1982.Forms of gold(I) in antimony-bearing sulphide sol-utions. Doklady Academii nauk SSSR, 264, 207–210.
Nesbitt, H. W., Muir, L. J. & Pratt, A. R. 1995.Oxidation of arsenopyrite by air and air-saturated,distilled water, and implications for mechanism ofoxidation. Geochimica et Cosmochimica Acta, 59,1773–1786.
Newton, R. C. & Manning, C. E. 2000. Quartz solubilityin H2O–NaCl and H2O–CO2 solutions at deep crust-mantle pressures and temperatures: 2–15 kbar and500–900 8C. Geochimica et Cosmochimica Acta, 64,2993–3005.
Nikolaeva, N. M., Erenburg, A. M. & Antipina, V. A.1972. About the temperature dependence of the stan-dard potentials of halogenide complexes of gold.Izvestia Sibirskogo Otdeleniya AN SSSR, No. 9, SeriaKhimicheskih Nauk, 4, 126–128 (in Russian).
Oelkers, E. H., Benezeth, P. & Pokrovski, G. S. 2009.Thermodynamic databases for water-rock interaction.Reviews in Mineralogy and Geochemistry, 70, 1–46.
Ogryzlo, S. P. 1935. Hydrothermal experiments withgold. Economic Geology, 30, 400–424.
Ohmoto, H. & Lasaga, A. C. 1982. Kinetics of reactionsbetween aqueous sulfates and sulfides in hydrothermalsystems. Geochimica et Cosmochimica Acta, 46,1727–1745.
Pal’yanova, G. A. & Savva, N. E. 2009. Specific genesisof gold and silver sulphides at the Yunoe deposit(Magadan Region, Russia). Geology and Geophysics,50, 759–777 (in Russian).
Pal’yanova, G. A., Laptev Yu, V. & Kolonin, G. R.1993. An experimental study of gold mobility insulphur-saturated solutions in connection with con-ditions of gold-containing sulphide-rich assemblages.In: Hach-Ali, F., Torres-Ruiz, J. & Gervilla, F.(eds) Proceedings of II Biennial SGA Meeting“Current Research in Geology Applied to Ore Depos-its”. Granada, Spain, 527–530.
Palenik, C. S., Utsunomiya, S., Reich, M., Kesler,S. E., Wang, L. & Ewing, R. C. 2004. “Invisible”gold revealed: direct imaging of gold nanoparticles
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

in a Carlin-type deposit. American Mineralogist, 89,1359–1366.
Palmer, D. A., Simonson, J. M. & Jensen, J. P. 2004.Partitioning of electrolytes to steam and their solu-bilities in steam. In: Palmer, D. A., Fernandez-
Prini, R. & Harvey, A. H. (eds) Aqueous Systems atElevated Temperatures and Pressures. Elsevier,New York, 409–439.
Pan, P. & Wood, S. A. 1991. Gold-chloride complexesin very acidic aqueous solutions and at temperatures25–300 8C: a laser Raman spectroscopic study. Geo-chimica et Cosmochimica Acta, 55, 2365–2371.
Pan, P. & Wood, S. A. 1994. Solubility of Pt and Pd sul-fides and Au metal in aqueous bisulfide solutions. II.Results at 2008 to 350 8C and saturated vapor pressure.Mineralium Deposita, 29, 373–390.
Pankratz, L. B. 1982. Thermodynamic properties ofelements and oxides. Bureau of Mines, Bulletin, 672,57–58.
Pankratz, L. B., Mah, A. D. & Watson, S. W. 1987.Thermodynamic properties of sulfides. Bureau ofMines, Bulletin, 689, 29–30.
Pearson, R. G. 1963. Hard and soft acid and bases. Journalof the American Chemical Society, 85, 3533–3539.
Peck, J. A., Tait, C. D., Swanson, B. I. & Brown, G.E. Jr. 1991. Speciation of aqueous gold(III) chloridefrom ultraviolet/visible absorption and Raman/reson-ance Raman spectroscopies. Geochimica et Cosmo-chimica Acta, 55, 671–676.
Perfetti, E., Pokrovski, G. S., Ballerat-Busserolles,K., Majer, V. & Gibert, F. 2008. Densities andheat capacities of aqueous arsenious and arsenicacid solutions to 350 8C and 300 bar, and revisedthermodynamic properties of, and iron sulfarsenideminerals. Geochimica et Cosmochimica Acta, 72,713–731.
Peshchevitskii, B. I., Belevantsev, V. I. & Zemskov,S. V. 1976. New data on the chemistry of gold com-pounds in solutions. Izvestia Akademii Nauk SSSR,Sibirskoe Otdelenie, Seria Khimia, 4, 24–45 (inRussian).
Phillips, G. N. & Evans, K. A. 2004. Role of CO2 in theformation of gold deposits. Nature, 429, 860–863.
Plyasunov, A. V. & Shock, E. L. 2001. Correlation strat-egy for determining the parameters of the revisedHelgeson-Kirkham-Flowers model for aqueous none-lectrolytes. Geochimica et Cosmochimica Acta, 65,3879–3900.
Pokrovski, G. S. 2010. Enhanced vapor-phase transport oftin in hydrothermal systems or experimental artifacts?Journal of Volcanology and Geothermal Research,194, 63–66.
Pokrovski, G. S. 2011. A new form of sulfur at high temp-eratures and pressures: implications for metal trans-port in the lithosphere. SEG Newsletter, 86, 14–15.
Pokrovski, G. S. & Dubessy, J. 2012. In situ Raman spec-troscopy reveals new sulfur forms in high-temperaturegeological fluids. GeoRaman X, Nancy, France.Journal of Conference Abstracts, 77–78.
Pokrovski, G. S. & Dubrovinsky, L. S. 2011. The S32 ion
is stable in geological fluids at elevated temperaturesand pressures. Science, 331, 1052–1054.
Pokrovski, G. S., Gout, R., Zotov, A., Schott, J. &Harrichoury, J. C. 1996a. Thermodynamic
properties and stoichiometry of the arsenic(III) hydrox-ide complexes at hydrothermal conditions. Geochi-mica et Cosmochimica Acta, 60, 737–749.
Pokrovski, G. S., Schott, J., Harrichoury, J.-C. &Sergeyev, A. S. 1996b. The stability of aluminumsilicate complexes in acidic solutions from 25 to 1508C. Geochimica et Cosmochimica Acta, 60,2495–2501.
Pokrovski, G. S., Zakirov, I. V., Roux, J., Testemale,D., Hazemann, J.-L., Bychkov, A. Y. & Golikova,G. V. 2002a. Experimental study of arsenic speciationin vapor phase to 500 8C: Implications for As transportand fractionation in low-density crustal fluids and vol-canic gases. Geochimica et Cosmochimica Acta, 66,3453–3480.
Pokrovski, G. S., Kara, S. & Roux, J. 2002b. Stabilityand solubility of arsenopyrite, FeAsS, in crustalfluids. Geochimica et Cosmochimica Acta, 66, 2361–2378.
Pokrovski, G. S., Schott, J., Hazemann, J.-L., Farges,F. & Pokrovsky, O. S. 2002c. An X-ray AbsorptionFine Structure and Nuclear Magnetic Resonance spec-troscopy study of gallium-silica complexes in aqueoussolution. Geochimica et Cosmochimica Acta, 66,4203–4322.
Pokrovski, G. S., Schott, J., Farges, F. & Hazemann,J.-L. 2003. Iron(III)-silica interactions in aqueous sol-ution: insights from X-ray absorption fine structurespectroscopy. Geochimica et Cosmochimica Acta, 67,3559–3573.
Pokrovski, G. S., Roux, J. & Harrichoury, J.-C. 2005a.Fluid density control on vapor-liquid partitioningof metals in hydrothermal systems. Geology, 33,657–660.
Pokrovski, G. S., Roux, J., Hazemann, J.-L. & Teste-
male, D. 2005b. An X-ray absorption spectroscopystudy of argutite solubility and germanium aqueousspeciation in hydrothermal fluids to 500 8C and400 bar. Chemical Geology, 217, 127–145.
Pokrovski, G. S., Borisova, A. Y., Roux, J., Hazemann,J.-L., Petdang, A., Tella, M. & Testemale, D. 2006.Antimony speciation in saline hydrothermal fluids: acombined X-ray absorption fine structure and solubi-lity study. Geochimica et Cosmochimica Acta, 70,4196–4214.
Pokrovski, G. S., Borisova, A. Y. & Harrichoury, J.-C.2008a. The effect of sulfur on vapor-liquid fraction-ation of metals in hydrothermal systems. Earth andPlanetary Science Letters, 266, 345–362.
Pokrovski, G. S., Roux, J., Hazemann, J.-L., Borisova,A. Y., Gonchar, A. A. & Lemeshko, M. P. 2008b.In situ X-ray absorption spectroscopy measurementof vapor-brine fractionation of antimony at hydro-thermal conditions. Mineralogical Magazine, 72,667–681.
Pokrovski, G. S., Tagirov, B. R., Schott, J., Bazar-
kina, E. F., Hazemann, J.-L. & Proux, O. 2009a.An in situ X-ray absorption spectroscopy study of gold-chloride complexing in hydrothermal fluids. ChemicalGeology, 259, 17–29.
Pokrovski, G. S., Tagirov, B. R., Schott, J., Haze-
mann, J.-L. & Proux, O. 2009b. A new view ongold speciation in sulfur-bearing hydrothermal fluidsfrom in situ X-ray absorption spectroscopy and
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

quantum-chemical modelling. Geochimica et Cosmo-chimica Acta, 73, 5406–5427.
Pokrovsky, O. S., Pokrovski, G. S., Shirokova, L. S.,Gonzalez, A. G., Emnova, E. E. & Feurtet-Mazel,A. 2012. Chemical and structural status of copperassociated with oxygenic and anoxygenic phototrophsand heterotrophs: possible evolutionary consequences.Geobiology, 10, 130–149.
Pokrovski, G. S., Roux, J., Ferlat, G., Jonchiere, R.,Seitsonen, A. P., Vuilleumier, R. & Hazemann,J.-L. 2013a. Silver in saline hydrothermal fluids fromin situ X-ray absorption spectroscopy and first-principles molecular dynamics. Geochimica et Cosmo-chimica Acta, 106, 501–523.
Pokrovski, G. S., Borisova, A. Y. & Bychkov, A. Y.2013b. Speciation and transport of metals and metal-loids in geological vapors. Reviews in Mineralogy &Geochemistry, 76, 165–218.
Pudack, C., Halter, W. E., Heinrich, C. A. & Pettke,T. 2009. Evolution of magmatic vapor to gold-richepithermal liquid: the porphyry to epithermal transitionat Nevados de Famatina, northwest Argentina. Econ-omic Geology, 104, 449–477.
Puddephatt, R. J. 1978. The Chemistry of Gold. Elsevier,New York.
Quisefit, J. P., Toutain, J. P., Bergametti, G., Javoy,M., Cheynet, B. & Person, A. 1989. Evolutionv. cooling of gaseous volcanic emissions from Momo-tombo Volcano, Nicaragua: thermochemical modeland observations. Geochimica et Cosmochimica Acta,53, 2591–2608.
Reed, M. 1997. Hydrothermal alteration and its relation-ship to ore fluid composition. In: Barnes, H. L. (ed.)Geochemistry of Hydrothermal ore Deposits. JohnWiley & Sons, New York, 303–365.
Reich, M., Kesler, S. E., Utsunomiya, S., Palenik, C.S., Chryssoulis, S. L. & Ewing, R. C. 2005. Solubi-lity of gold in arsenian pyrite. Geochimica et Cosmo-chimica Acta, 69, 2781–2796.
Rempel, K. U., Williams-Jones, E. E. & Migdisov, A. A.2009. The partitioning of molybdenum (VI) betweenaqueous liquid and vapour at temperatures up to370 8C. Geochimica et Cosmochimica Acta, 73,3381–3392.
Renders, P. J. & Seward, T. M. 1989. The stability ofhydrosulphido and sulphido-complexes of Au(I) andAg(I) at 25 8C. Geochimica et Cosmochimica Acta,53, 245–253.
Richards, J. P. 2011. Magmatic to hydrothermal metalfluxes in convergent and collided margins. OreGeology Review, 40, 1–26.
Ruaya, J. R. 1988. Estimation of instability constants ofmetal chloride complexes in hydrothermal solutionsup to 300 8C. Geochimica et Cosmochimica Acta, 52,1983–1996.
Rudnick, R. L. & Gao, S. 2003. Composition of the con-tinental crust. Treatise on Geochemistry, 3, 1–64.
Rusk, B., Reed, M. H., Dilles, J. H. & Klemm, L. 2004.Compositions of magmatic-hydrothermal fluidsdetermined by LA-ICPMS of fluid inclusions fromthe porphyry copper-molybdenum deposit at Butte,Montana. Chemical Geology, 210, 173–199.
Rusk, B. G., Reed, M. H. & Dilles, J. H. 2008. Fluidinclusion evidence for magmatic-hydrothermal fluid
evolution in the porphyry copper-molybdenumdeposit at Butte, Montana. Economic Geology, 103,307–334.
Rusk, B., Emsbo, P., Hammersli, J., Hofstra, A., Hunt,A., Landis, G. & Rye, R. 2011. Origin and compo-sition of fluids that form giant porphyry Cu (Mo–Au)deposits. In: Proceedings of the 11th Biennial SGAMeeting. Antafogasta, Chile, 414–416.
Russell, J. K., Edwards, B. R. & Shyder, L. D. 1995.Volatile production possibilities during magmaticassimilation: heat and mass-balance constraints. In:Thompson, J. F. (ed.) Magmas, Fluids and Ore Depos-its. Mineralogical Association of Canada short CourseSeries, 23, 1–24.
Ryabchikov, I. D. & Orlova, G. P. 1984. Gold in mag-matic fluids. In: Tauson, L. V. (ed.) Physical ChemicalModels of Petrogenesis and ore Formation. Nauka,Siberian Brach, Novosibirsk, 103–111 (in Russian).
Saunders, J. A. 1990. Colloidal transport of gold andsilica in epithermal precious-metal deposits: evidencefrom the Sleeper deposit, Nevada. Geology, 18,757–760.
Saunders, J. A. 2012. Textural evidence of episodic intro-duction of metallic nanoparticles into bonanza epither-mal ores. Minerals, 2, 228–243.
Saunders, J. A. & Schoenly, P. A. 1995. Boiling, colloidnucleation and aggregation, and the genesis of bonanzaAu–Ag ores of the Sleeper deposit, Nevada. Minera-lium Deposita, 30, 199–210.
Scaini, M. J., Bancroft, G. M. & Knipe, S. W. 1998.Reactions of aqueous Au1+ sulphide species withpyrite as a function of pH and temperature. AmericanMineralogist, 83, 316–322.
Schulte, M. D., Shock, E. L. & Wood, R. H. 2001. Thetemperature dependence of the standard-state thermo-dynamic properties of aqueous nonelectrolytes. Geo-chimica et Cosmochimica Acta, 65, 3919– 3930.
Schwerdtfeger, P., Boyd, P. D. W., Brienne, S. &Burrell, A. 1992. Relativistic effects in gold chem-istry. 4. Gold (III) and gold (V) compounds. InorganicChemistry, 31, 3411–3422.
Seedorff, E. & Einaudi, M. T. 2004. Henderson por-phyry molybdenum system, Colorado: II. Decouplingof introduction and deposition of metals during geo-chemical evolution of hydrothermal fluids. EconomicGeology, 99, 39–72.
Seo, J. H., Guillong, M. & Heinrich, C. A. 2009.The role of sulfur in the formation of magmatic-hydrothermal copper-gold deposits. Earth and Plane-tary Science Letters, 282, 323–328.
Seo, J. H., Guillong, M., Aerts, M., Zajacz, Z. & Hein-
rich, C. A. 2011. Microanalysis of S, Cl, and Br influid inclusions by LA-ICP-MS. Chemical Geology,284, 35–44.
Seo, J. H., Guillong, M. & Heinrich, C. A. 2012. Sep-aration of molybdenum and copper in porphyry depos-its: the roles of sulfur, redox, and pH in ore mineraldeposition at Bingham Canyon. Economic Geology,107, 333–356.
Seo, J. H. & Heinrich, C. A. 2013. Selective copper dif-fusion into quartz-hosted vapor inclusions: Evidencefrom other host minerals, driving forces, and conse-quences for Cu-Au ore formation. Geochimica etCosmochimica Acta, 113, 60–69.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Seward, T. M. 1973. Thio complexes of gold and thetransport of gold in hydrothermal ore solutions. Geo-chimica et Cosmochimica Acta, 37, 379–399.
Seward, T. M. 1989. The hydrothermal chemistry of goldand its implications for ore formation: boiling and con-ductive cooling as examples. Economic GeologyMonograph, 6, 398–404.
Seward, T. M. & Barnes, H. L. 1997. Metal transport byhydrothermal ore fluids. In: Barnes, H. L. (ed.) Geo-chemistry of Hydrothermal ore Deposits. 3rd edn.Wiley and Sons, New York, 435–486.
Sharps, J. A., Brown, G. E. Jr. & Stebbins, J. F. 1993.Kinetics and mechanism of ligand exchange ofAu(III), Zn(II), and Cd(II) chlorides in aqueous sol-ution: an NMR study from 28–98 8C. Geochimica etCosmochimica Acta, 57, 721–731.
Shenberger, D. M. & Barnes, H. L. 1989. Solubility ofgold in aqueous sulfide solutions from 150 to 350 8C.Geochimica et Cosmochimica Acta, 53, 269–278.
Sherman, D. M. 2010. Metal complexation and ionassociation in hydrothermal fluids: insights fromquantum chemistry and molecular dynamics. Geo-fluids, 10, 41–57.
Shmulovich, K., Heinrich, W., Moller, P. & Dulski,P. 2002. Experimental determination of REE fraction-ation between liquid and vapour in the systems NaCl–H2O and CaCl2–H2O up to 450 8C. Contributions toMineralogy and Petrology, 44, 257–273.
Shock, E. L. & Helgeson, H. C. 1988. Calculation of thethermodynamic and transport properties of aqueousspecies at high pressures and temperatures: correlationalgorithms for ionic species and equation of state pre-dictions to 5 kb and 1000 8C. Geochimica et Cosmo-chimica Acta, 52, 2009–2036.
Shock, E. L., Helgeson, H. C. & Sverjensky, D. A.1989. Calculation of the thermodynamic properties ofaqueous species at high pressures and temperatures:standard partial molal properties of inorganic neutralspecies. Geochimica et Cosmochimica Acta, 53,2157–2183.
Shock, E. L., Sassani, D. C., Willis, M. & Sver-
jensky, D. A. 1997. Inorganic species in geologicfluids: correlations among standard molal thermodyn-amic properties of aqueous ions and hydroxide com-plexes. Geochimica et Cosmochimica Acta, 61,907–950.
Shvarov, Y. V. 2008. HCh: new potentialities for the ther-modynamic simulation of geochemical systemsoffered by windows. Geochemisty International, 46,834–839.
Shvarov, Y. V. 2010. OptimB: a program for calculationof the HKF parameters of an aqueous species using aset of values of its standard free energy at varioustemperatures and pressures. (http://www.geol.msu.ru/deps/geochems/soft/index_e.htm).
Sillitoe, R. H. 2010. Porphyry copper systems. EconomicGeology, 105, 3–41.
Simmons, S. F. & Browne, P. R. L. 2007. Hydrothermalminerals and precious metals in the Broadlands-Ohaakigeocthermal system: implications for understandinglow-sulfidation epithermal deposits. EconomicGeology, 95, 971–999.
Simmons, S. F., White, N. C. & John, D. A. 2005. Geo-logical characteristics of epithermal precious and
base metal deposits. Economic Geology, 100th Anni-versary Volume, 485–522.
Simon, A. C. & Ripley, E. M. 2011. The role of magmaticsulfur in the formation of ore deposits. In: Behrens, H.& Webster, J. D. (eds) Sulfur in Magmas and Melts.Reviews in Mineralogy and Geochemistry, 73,513–578.
Simon, G., Huang, H., Penner-Hahn, J. E., Kesler, S.E. & Kao, L.-S. 1999. Oxidation state of gold andarsenic in gold-bearing arsenian pyrite. AmericanMineralogist, 84, 1071–1079.
Simon, G., Kesler, S. E., Essene, E. J. & Chryssoulis,S. L. 2000. Gold in porphyry copper deposits: exper-imental determination of the distribution of gold inthe Cu–Fe–S system at 400 to 700 8C. EconomicGeology, 95, 259–270.
Simon, A. C., Frank, M. R., Pettke, T., Candela, P. A.,Piccoli, P. M. & Heinrich, C. A. 2005. Gold parti-tioning in melt-vapor-brine systems. Geochimica etCosmochimica Acta, 69, 3321–3335.
Simon, A. C., Frank, M. R., Pettke, T., Candela, P. A.,Piccoli, P. M., Heinrich, C. A. & Glascock, M.2007a. An evaluation of synthetic fluid inclusions forthe purpose of trapping equilibrated, cexisting, immis-cible fluid phases at magmatic conditions. AmericanMineralogist, 92, 124–138.
Simon, A. C., Pettke, T., Candela, P. A., Piccoli, P. M.& Heinrich, C. A. 2007b. The partitioning behavior ofAs and Au in S-free and S-bearing magmatic assem-blages. Geochimica et Cosmochimica Acta, 71,1764–1782.
Sisson, T. W. 2003. Native gold in a Hawaiian alkalimagma. Economic Geology, 98, 643–648.
Smirnov, S. Z., Thomas, V. G., Kamenetsky, V. S., Koz-
menko, O. A. & Large, R. R. 2012. Hydrosilicateliquids in the system Na2O–SiO2–H2O with NaF,NaCl and Ta: evaluation of their role in ore and mineralformation at high T and P. Petrology, 20, 271–285.
Smith, F. G. 1943. The alkali sulfide theory of gold depo-sition. Economic Geology, 38, 561–590.
Song, Z., Kenney, J. P. L., Fein, J. B. & Bunker, B. A.2012. An X-ray Absorption Fine Structure study of Auadsorbed onto the non-metabolizing cells of two soilbacterial species. Geochimica et Cosmochimica Acta,86, 103–117.
Stavast, W. J. A., Keith, J. D., Christiansen, E. H.,Dorais, M. J., Tingey, D., Larocque, A. & Evans,N. 2006. The fate of magmatic sulfides during intusionor eruption, Bingham and Tintic District, Utah. Econ-omic Geology, 101, 329–345.
Stefansson, A. & Seward, T. M. 2003a. The hydrolysisof gold(I) in aqueous solutions to 600 8C and 1500 bar.Geochimica et Cosmochimica Acta, 67, 1677–1688.
Stefansson, A. & Seward, T. M. 2003b. Stability ofchloridogold(I) complexes in aqueous solutions from300 to 600 8C and from 500 to 1800 bar. Geochimicaet Cosmochimica Acta, 67, 4559–4576.
Stefansson, A. & Seward, T. M. 2004. Gold(I) complex-ing in aqueous sulphide solutions to 500 8C at 500 bar.Geochimica et Cosmochimica Acta, 68, 4121–4143.
Stone, W. E., Crocket, J. H. & Fleet, M. E. 1990. Par-titioning of palladium, iridium, platinum, and goldbetween sulfide liquid and basalt melt at 1200 8C. Geo-chimica et Cosmochimica Acta, 54, 2341–2344.
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Styrikovich, M. A., Khaibullin, I. K. & Tshvirash-
vili, D. G. 1955. A study of salt solubility in high-pressure water steam. Doklady Akademii Nauk ofthe Union of Soviet Socialist Republics (USSR), 100,1123–1126.
Sverjensky, D. A., Shock, E. L. & Helgeson, H. C.1997. Prediction of the thermodynamic properties ofaqueous metal complexes to 1000 8C and 5 kb. Geo-chimica et Cosmochimica Acta, 61, 1359–1412.
Sun, W., Arculus, R. J., Kamenetsky, V. S. & Binns,R. A. 2004. Release of gold-bearing fluids in conver-gent margin magma prompted by magnetite crystalli-zation. Nature, 431, 975–978.
Symonds, R. B. & Reed, M. H. 1993. Calculation of multi-component chemical equilibria in gas-solid-liquidsystems: calculation methods, thermochemical data,and applications to studies of high-temperature volca-nic gases with examples from Mount St Helens. Amer-ican Journal of Science, 293, 758–864.
Symonds, R. B., Rose, W. I., Reed, M. H., Lichte, F. E. &Finnegan, D. L. 1987. Volatilization, transport andsublimation of metallic and non-metallic elements inhigh-temperature gases at Merapi Volcano, Indonesia.Geochimica et Cosmochimica Acta, 51, 2083–2101.
Symonds, R. B., Reed, M. H. & Rose, W. I. 1992. Origin,speciation, and fluxes of trace-element gases at Augus-tine volcano, Alaska: insights into magma degassingand fumarolic processes. Geochimica et Cosmochi-mica Acta, 56, 633–657.
Tagirov, V. K., Makarov, E. V. & Byukvin, V. A. 1993.Vapor composition and pressure over chlorides ofsilver and gold. Izvestiya Rossiskoi Akademii Nauk,Metally, 5, 67–71.
Tagirov, B. R., Zotov, A. V. & Akinfiev, N. N. 1997.Experimental study of the dissociation of HCl from350 to 500 8C and from 500 to 2500 bar: thermodyn-amic properties of HCl8(aq). Geochimica et Cosmochi-mica Acta, 61, 4267–4280.
Tagirov, B. R., Salvi, S., Schott, J. & Baranova, N. N.2005. Experimental study of gold-hydrosulphide com-plexing in aqueous solutions at 350–500 8C, 500 and1000 bars using mineral buffers. Geochimica et Cos-mochimica Acta, 69, 2119–2132.
Tagirov, B. R., Baranova, N. N., Zotov, A. V., Schott,J. & Bannykh, L. N. 2006. Experimental determi-nation of the stabilities Au2S(cr) at 25 8C andAu(HS)2
2 at 25–250 8C. Geochimica et CosmochimicaActa, 70, 3689–3701.
Tagirov, B. R., Baranova, N. N. et al. 2013. The specia-tion and transport of palladium in hydrothermal fluids:Experimental modelling and thermodynamic con-straints. Geochimica et Cosmochimica Acta, 117,348–373.
Tanger, J. C. & Helgeson, H. C. 1988. Calculation of thethermodynamic and transport properties of aqueousspecies at high pressures and temperatures: revisedequations of state for the standard partial molal proper-ties of ions and electrolytes. American Journal ofScience, 288, 19–98.
Taran, Y. A., Hedenquist, J. W., Korzhinsky, M. A.,Tkachenko, S. I. & Shmulovich, K. I. 1995.Geochemistry of magmatic gases from Kudryavyvolcano, Iturup, Kuril Islands. Geochimica et Cosmo-chimica Acta, 59, 1749–1761.
Taran, Y. A., Bernard, A., Gavilanes, J.-C. &Africano, F. 2000. Native gold in mineral pre-cipitates from high-temperature volcanic gases ofColima volcano, Mexico. Applied Geochemistry, 15,337–346.
Taran, Y. A., Bernard, A., Gavilanes, J.-C., Lunez-
heva, E., Cortes, A. & Armienta, M. A. 2001.Chemistry and mineralogy of high-temperature gasdischarges from Colima volcano, Mexico. Implicationsfor magmatic gas-atmosphere interaction.Journalof Volcanology and Geothermal Research, 108,245–264.
Testemale, D., Pokrovski, G. S. & Hazemann, J.-L.2011. Speciation of AsIII and AsV in hydrothermalfluids by in-situ X-ray absorption spectroscopy. Euro-pean Journal of Mineralogy, 23, 379–390.
Tkachenko, S. I., Porter, R. P., Korzinsky, M. A., van
Bergen, M. D., Shmulovich, K. I. & Schteinberg,G. S. 1999. Investigation of processes of ore andmineral formation from hogh-temperature fumarolegases at Kudryavy volcano, Iturup, Kurils Islands.Geokhimia, N88888 4, 410–422 (in Russian).
Tomkins, A. G. 2010. Windows of metamorphic sulfur lib-eration in the crust: implications for gold depositgenesis. Geochimica et Cosmochimica Acta, 74,3246–3259.
Tooth, B., Brugger, J., Ciobanu, C. & Liu, W. 2008.Modeling of gold scavenging by bismuth meltscoexisting with hydrothermal fluids. Geology, 36,815–818.
Tooth, B., Etschmann, B., Pokrovski, G. S., Teste-
male, D., Hazemann, J.-L., Grundler, P. &Brugger, J. 2012. Bismuth speciation in hydrothermalfluids: An X-ray absorption spectroscopy and solubi-lity study. Geochimica et Cosmochimica Acta, in press.
Tossell, J. A. 1996. The speciation of gold in aqueous sol-ution: A theoretical study. Geochimica et Cosmochi-mica Acta, 60, 17–29.
Ulrich, T., Gunther, D. & Heinrich, C. A. 1999. Goldconcentrations of magmatic brines and the metalbudget of porphyry copper deposits. Nature, 399,676–679.
Ulrich, T., Gunther, D. & Heinrich, C. A. 2001.Evolution of a porphyry Cu–Au deposit, based onLA-ICP-MS analysis of fluid inclusions, Bajo de laAlumbrera, Argentina. Economic Geology, 96,1743–1774 (correctly reprinted in 2002, 97, 1888–1920).
Usher, A., McPhail, D. C. & Brugger, J. 2009. A spec-trophotometric study of aqueous Au(III) halide-hydroxide complexes at 25–80 8C. Geochimica etCosmochimica Acta, 73, 3359–3380.
Vallance, J., Fontbote, L., Chiaradia, M., Mar-
kowski, A., Schmidt, S. & Vennemann, T. 2009.Magmatic-dominated fluid evolution in the JurassicNambija gold skarn deposits (southeastern Ecuador).Mineralium Deposita, 44, 389–413.
Velasquez, G., Beziat, D., Salvi, S., Borisova, A. Y.,Pokrovski, G. S. & de Parseval, P. 2014. Formationand deformation of pyrite and implications for goldmineralization at the El Callao Mining District, Vene-zuela. Economic Geology, 109, 457–486.
Vikent’ev, I. V., Borisova, A. Y., Karpukhina, V. S.,Naumov, V. B. & Ryabchikov, I. D. 2012. Direct
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

Data on the Ore Potential of Acid Magmas of theUzel’ginskoe Ore Field (Southern Urals, Russia).Doklady Earth Science, 443, 401–405.
Vlassopoulos, D. & Wood, S. A. 1990. Gold speciationin natural waters: I. Solubility and hydrolysis of goldin aqueous solution. Geochimica et CosmochimicaActa, 54, 3–12.
Vlassopoulos, D., Wood, S. A. & Mucci, A. 1990.Gold speciation in natural waters: II. The importanceof organic complexing – experiments with simpleorganic ligands. Geochimica et Cosmochimica Acta,54, 1575–1586.
Wagner, T. 2007. Thermodynamic modeling of Au–Bi–Te melt precipitation from high-temperaturehydrothermal fluids: Preliminary results. In: Proceed-ings of the Ninth Biennial SGA Meeting, Dublin,769–772.
Wagner, F. E., Haslbeck, S., Stievano, L., Calogero,S., Pankhurst, Q. A. & Martinek, K.-P. 2000.Before striking gold in gold-ruby glass. Nature, 407,691–692.
Walther, J. J. & Orville, P. M. 1983. The extraction-quench technique for determination of the thermodyn-amic properties of solute complexes: application toquartz solubility in fluid mixtures. American Mineral-ogist, 68, 731–741.
Wang, X.-B., Wang, Y.-L., Yang, J., Xing, X.-P., Li, J. &Wang, L.-S. 2009. Evidence of significant covalentbonding in Au(CN)2
2. American Journal of Science,131, 16 368–16 370.
Watanabe, Y. & Hedenquist, J. W. 2001. Mineralogicand stable isotope zonation at the surface over the ElSalvador porphyry copper deposit, Chile. EconomicGeology, 96, 1775–1797.
Weatherley, D. K. & Henley, R. W. 2013. Flash vapor-ization during earthquakes evidenced by gold deposits.Nature Geoscience, 6, 294–298.
Webster, J. G. 1986. The solubility of gold and silverin the system Au–Ag–S–O2–H2O at 25 8C and1 atm. Geochimica et Cosmochimica Acta, 50,1837–1845.
Weis, P., Driesner, T. & Heinrich, C. A. 2012. Porphyry-copper ore shells form at stable pressure-temperaturefronts within dynamic fluid plumes. Science, 338,1613–1615.
Weissberg, B. C. 1970. Solubility of gold in hydrothermalalkaline sulphide solutions. Economic Geology, 65,551–556.
Wilkinson, J. J., Stoffell, B., Wilkinson, C. C.,Jeffries, T. E. & Appold, M. S. 2009. Anomalouslymetal-rich fluids form hydrothermal ore deposits.Science, 323, 764–767.
Williams-Jones, A. E. & Heinrich, C. A. 2005. Vaportransport of metals and the formation of magmatic-hydrothermal ore deposits. Economic Geology, 100,1287–1312.
Williams-Jones, A. E. & Migdisov, A. A. 2007. Thesolubility of gold in crude oil: implications for oregenesis. In: Proceedings of the 9th Biennal SGAMeeting, Millpress, Dublin, 765–768.
Williams-Jones, A. E., Bowell, R. J. & Migdisov, A. A.2009. Gold in solution. Elements, 5, 281–287.
Wood, S. A. & Samson, I. M. 1998. Solubility of oreminerals and complexation of ore metals in
hydrothermal solutions. Reviews in EconomicGeology, 10, 33–80.
Wood, S., Crerar, D. A. & Borcsik, M. P. 1987. Solubi-lity of the assemblage pyrite-pyrrhotite-magnetite-sphalerite-galena-gold-stibnite-bismuthinite-argentite-molybdenite in H2O–NaCl–CO2 solutions from 200to 350 8C. Economic Geology, 82, 1864–1887.
Wood, S. A., Pan, P., Zhang, Y. & Mucci, A. 1994. Thesolubility of Pt and Pd sulfides and Au in bisulfide sol-utions. Mineralium Deposita, 29, 309–317.
Xu, Y., Schoonen, M. A. A., Nordstrom, D. K.,Cunningham, K. M. & Ball, J. W. 1998. Sulfurgeochemistry of hydrothermal waters in Yellowstonenational park: I. The origin of thiosulfate in hotspring waters. Geochimica et Cosmochimica Acta,62, 3729–3743.
Yardley, B. W. D. 2005. Metal concentrations in crustalfluids and their relationship to ore formation. EconomicGeology, 100, 613–632.
Yudovskaya, M. A., Distler, V. V., Chaplygin, I. V.,Mokhov, A. V., Trubkin, N. V. & Gorbacheva, S.A. 2006. Gaseous transport and deposition of gold inmagmatic fluid: evidence from the active Kudryavyvolcano, Kurile Islands. Mineralium Deposita, 40,828–848.
Yudovskaya, M. A., Tessalina, S., Distler, V. V., Cha-
plygin, I. V., Chugaev, A. V. & Dikov, Y. P. 2008.Behavior of highly-siderophile elements duringmagma degassing: a case at the Kudryavy volcano.Chemical Geology, 248, 318–341.
Zajacz, Z., Hanley, J. J., Heinrich, C. A., Halter,W. E. & Guillong, M. 2009. Diffusive reequilibrationof quartz-hosted silicate melt and fluid inclusions: areall metal concentrations unmodified? Geochimica etCosmochimica Acta, 73, 3013–3027.
Zajacz, Z., Seo, J. H., Candela, P. A., Piccoli, P. M. &Heinrich, C. A. 2010. Alkali metals control therelease of gold from volatile-rich magmas. Earth andPlanetary Science Letters, 297, 50–56.
Zajacz, Z., Seo, J. H., Candela, P. A., Piccoli, P. M. &Tossell, J. A. 2011. The solubility of copper in high-temperature magmatic vapors: a quest for the signifi-cance of various chloride and sulfide complexes. Geo-chimica et Cosmochimica Acta, 75, 2811–2827.
Zajacz, Z., Candela, P. A., Piccoli, P. M., Walle, M. &Sanchez-Valle, C. 2012. Gold and copper in volatilesaturated mafic to intermediate magmas: solubilities,partitioning, and implications for ore deposit forma-tion. Geochimica et Cosmochimica Acta, 91, 140–159.
Zajacz, Z., Candela, P. A., Piccoli, P. M., Sanchez-
Valle, C. & Walle, M. 2013. Solubility and partition-ing behaviour of Au, Cu, Ag and reduced S in magmas.Geochimica et Cosmochimica Acta, 112, 288–304.
Zezin, D. Y., Migdisov, A. A. & Williams-Jones, A. E.2007. The solubility of gold in hydrogen sulfide gas: anexperimental study. Geochimica et CosmochimicaActa, 71, 3070–3081.
Zezin, D. Y., Migdisov, A. A. & Williams-Jones, A. E.2011. The solubility of gold in H2O–H2S vapor at elev-ated temperatures and pressures. Geochimica et Cos-mochimica Acta, 75, 5140–5153.
Zotov, A. V. & Baranova, N. N. 1995. The solubilityof AuS2 and AuAgS in near-neutral sulphide solutionsat temperatures of 25 and 80 8C and pressures of
GOLD SPECIATION IN GEOLOGICAL FLUIDS
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from

1 to 500 bars. Proceedings of WRI-8, Vladivostok,773–776.
Zotov, A. V., Baranova, N. N., Dar’yna, T. G.,Bannykh, L. N. & Kolotov, V. P. 1985. The stabilityof AuOH0
sol in water at 300–500 8C and 500–1500 atm. Geochemistry International, 22, 156–161.
Zotov, A. V., Baranova, N. N., Dar’ina, T. G. &Bannykh, L. N. 1989. Gold (I) complexing in theKCl–HCl–H2O system at 450C and 500 atm. Geo-chemistry International, 26, 66–75.
Zotov, A. V., Baranova, N. N., Dar’ina, T. G. &Bannykh, L. N. 1991. The solubility of ib
aqueous chloride fluids at 350–500 8C and 500–1500 atm: thermodynamic properties of AuCl2,aq
2 upto 750 8C and 5000 atm. Geochemistry International,28, 63–71.
Zotov, A. V., Kudrin, A. V., Levin, K. A., Shikina, N.D. & Var’yash, L. N. 1995. Experimental studies ofthe solubility and complexing of selected ore elements(Au, Ag, Cu, Mo, As, Sb, Hg) in aqueous solutions.In: Shmulovich, K. I., Yardley, B. W. D. &Gonchar, G. G. (eds) Fluids in the Crust. Equilibriumand Transport Properties. Chapman & Hall, London,95–138.
G. S. POKROVSKI ET AL.
at CAPES on May 1, 2014http://sp.lyellcollection.org/Downloaded from