epigenetics and epigenomics - profs.sci.univr.itprofs.sci.univr.it/delledonne/insegnamenti/genomi 4...
TRANSCRIPT

Epigenetics and epigenomics
In eukaryotic cells, the DNA is packaged within the nucleus in a structure called chromatin. This structure is a highly dynamic nucleoprotein complex that plays a central role in regulating how and when DNA is copied and transcribed into RNA. Thus chromatin states vary from cell type to cell type and along chromosomes. Typically, a multi-cellular organism will be characterized by one genome, but by as many epigenomes as there are cell types.
Epigenetics encompasses all processes that lead to heritable changes in gene expression (during development or across generations) without changes in the DNA sequence itself. In eukaryotes, chromatin is at the heart of most epigenetic processes. The epigenome refers to these states at the whole genome level. EPIGENETICA: Variazione del profilo trascrizionale indipendente dalla sequenza di DNA ed ereditabile attraverso le generazioni cellulari

Perché interessarcene?
• Cancro: modifiche epigenetiche possono spiegare come i fattori ambientali scatenino il cancro
• Cambiamenti Prenatali: modifiche molecolari al DNA fetali e materno prima della nascita possono rendere il nascituro suscettibile al diabete di tipo 2
• Disordini Mentali: cambiamenti epigenetici sono stati correlati a malattie come Alzheimer, schizofrenia, autismo
• Malattie Croniche: condizioni croniche come il lupus eritematoso sistemico o l’asma hanno una componente ambientale che può causare cambiamenti epigenetici che a loro volta possono interessare la progressione della malattia
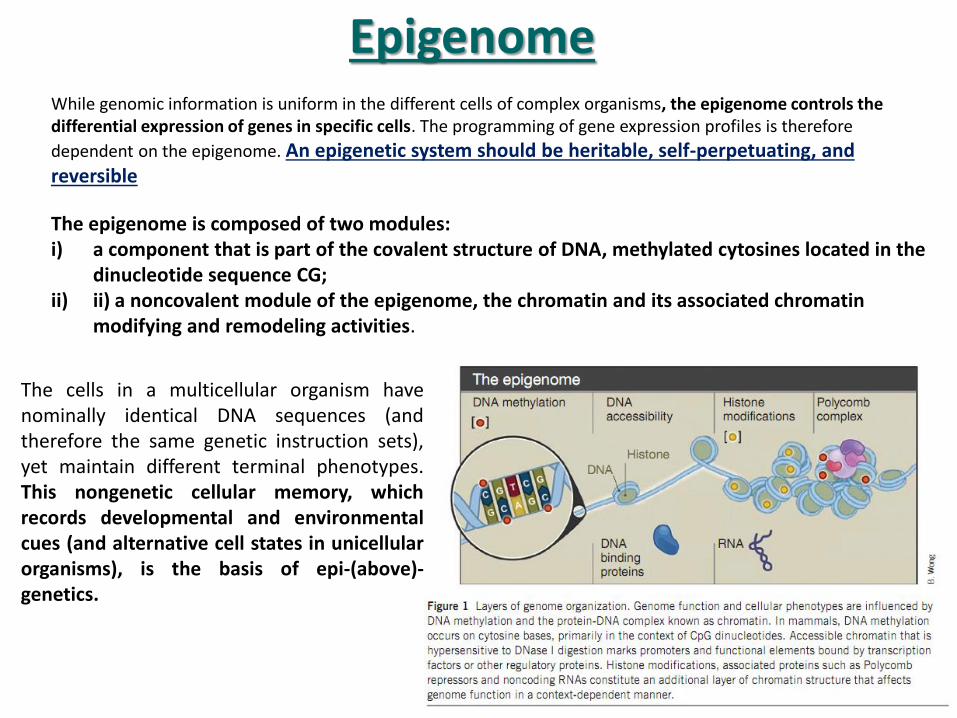
The cells in a multicellular organism have nominally identical DNA sequences (and therefore the same genetic instruction sets), yet maintain different terminal phenotypes. This nongenetic cellular memory, which records developmental and environmental cues (and alternative cell states in unicellular organisms), is the basis of epi-(above)-genetics.
While genomic information is uniform in the different cells of complex organisms, the epigenome controls the differential expression of genes in specific cells. The programming of gene expression profiles is therefore
dependent on the epigenome. An epigenetic system should be heritable, self-perpetuating, and reversible
The epigenome is composed of two modules: i) a component that is part of the covalent structure of DNA, methylated cytosines located in the
dinucleotide sequence CG; ii) ii) a noncovalent module of the epigenome, the chromatin and its associated chromatin
modifying and remodeling activities.
Epigenome

MECCANISMI
• Modifiche dello stato della cromatina
• Metilazione delle citosine

CROMATINA: VANTAGGI
• Impacchettare il DNA
• Ripartire correttamente il materiale genetico tra le cellule figlie
• Regolare l’espressione genica in modo tessuto-specifico
• Trasmettere lo stato di attività della cromatina all’interno di una linea differenziata

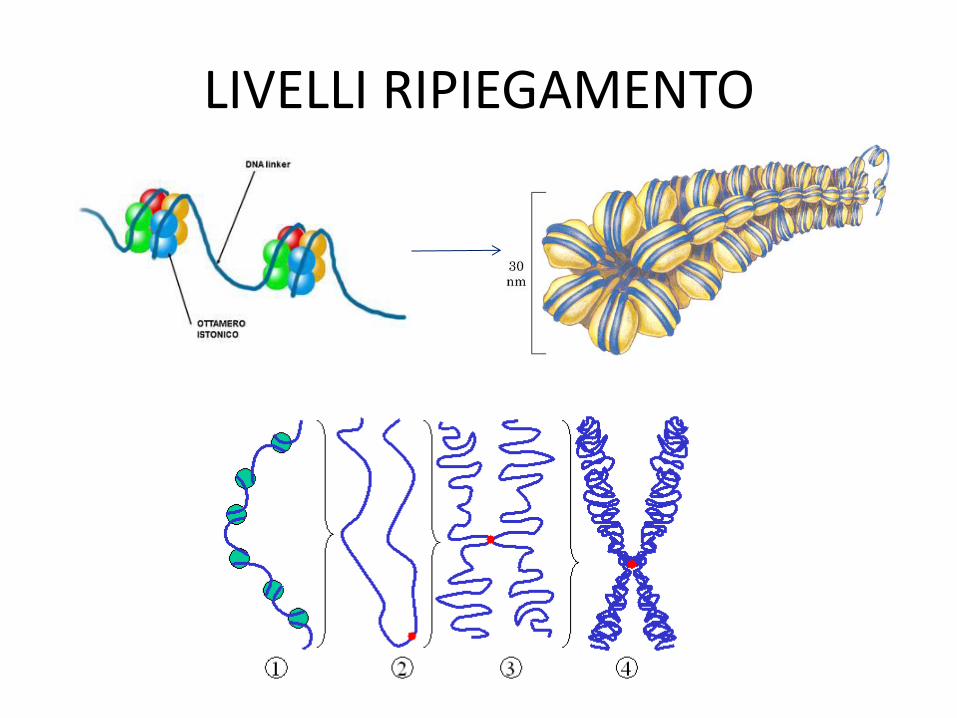
LIVELLI RIPIEGAMENTO
1. Fibra 10nm (collana di perle): core di proteine istoniche alle quali si legano 147 nucleotidi (nucleosoma) + tratto di DNA linker tra 2 nucleosomi
2. Fibra 30nm: interazione tra le code N-terminali degli istoni del core per ulteriore ripiegamento a solenoide
3. Interazione della fibra 30nm con l’impalcatura presente del nucleo genera anse e permette un’ulteriore compattazione
4. Compattazione finale per dare la tipica struttura dei cromosomi metafasici
I legami nel nucleosoma sono tra le proteine e lo scheletro del DNA, quindi sequenza-indipendenti

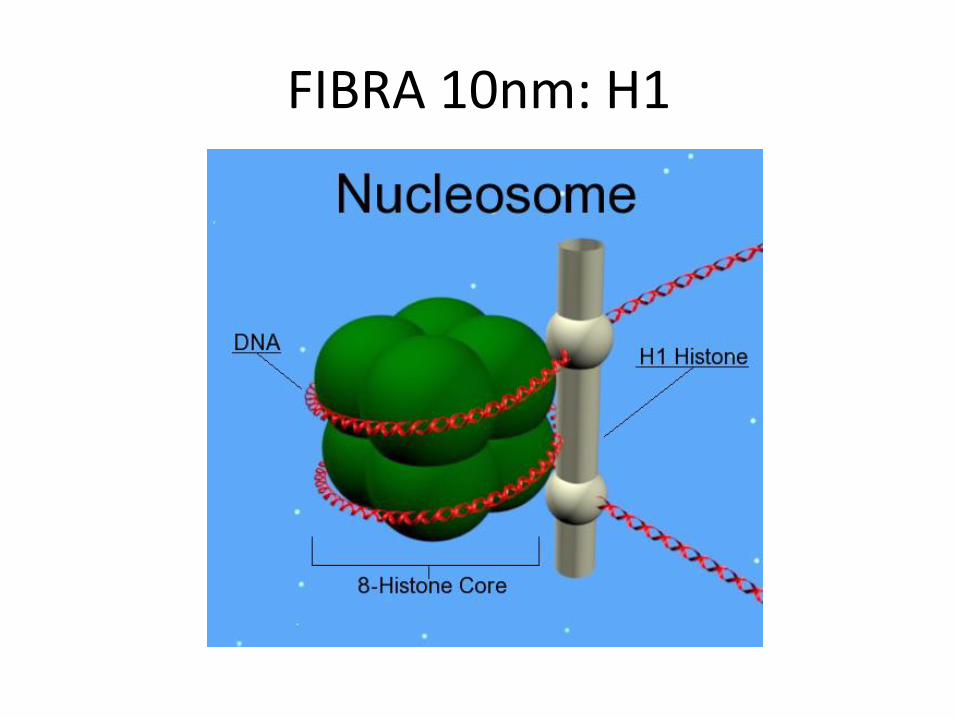
FIBRA 10nm
• Istoni H2A, H2B, H3, H4 formano il core del nucleosoma
– Un tetramero centrale, formato da 2 H3 + 2 H4 e 2 dimeri laterali, formati da H2A + H2B
– Estremamente conservati!
• Istone H1:
– Tiene vicine le estremità del DNA in entrata e uscita dal nucleosoma
– Maggiore variabilità genetica rispetto agli altri istoni
FIBRA 10nm: H1
LIVELLI RIPIEGAMENTO

CROMATINA
• Anse di cromatina maggiormente presenti verso il centro del nucleo sembrano essere silenziate
• Anse di cromatina maggiormente esposte verso la membrana nucleare sembrano essere più trascrizionalmente attive
• Sono possibili temporanei svolgimenti del DNA dal core istonico (i nucleosomi respirano)

Modifiche della cromatina
• Modifiche covalenti a carico degli istoni
– Acetilazione (influenza la condensazione della cromatina), fosforilazione, monoubiquitinazione. Sono modifiche reversibili
– Metilazione; è parzialmente reversibile e pare ci sia corrispondenza tra metilazione degli istoni e del DNA
• Rimodellamento degli istoni
– Complessi proteici ATP-dipendenti
– Omologie con elicasi, ma non hanno attività elicasica
– Deformano temporaneamente il DNA
Le modifiche degli istoni corrispondono a segnali per il reclutamento o l’esclusione di proteine della cromatina
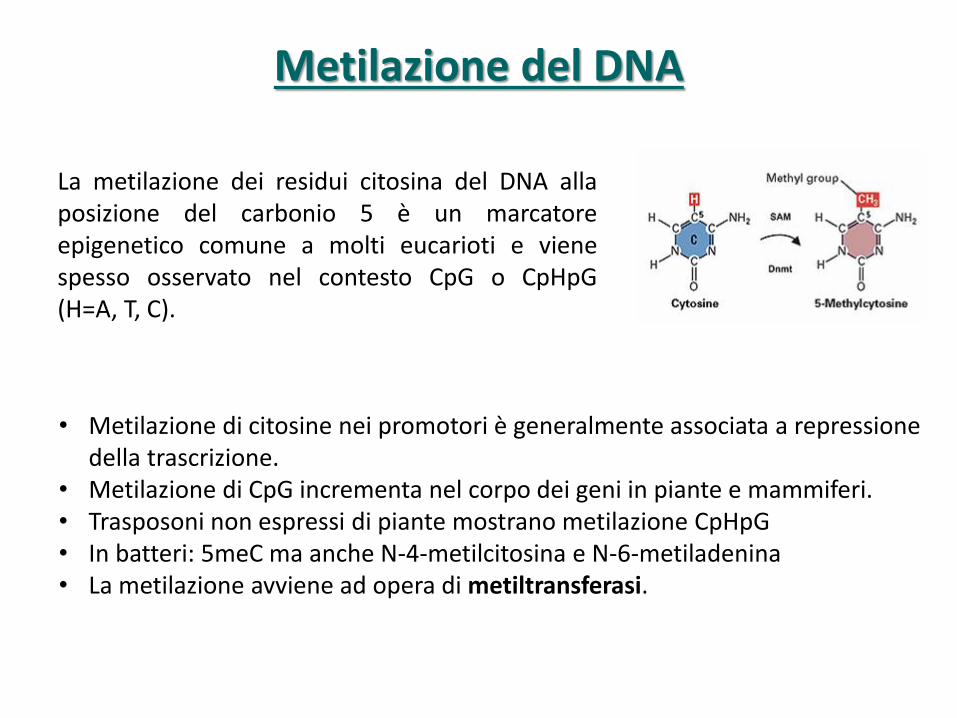
Metilazione del DNA
La metilazione dei residui citosina del DNA alla posizione del carbonio 5 è un marcatore epigenetico comune a molti eucarioti e viene spesso osservato nel contesto CpG o CpHpG (H=A, T, C).
• Metilazione di citosine nei promotori è generalmente associata a repressione della trascrizione.
• Metilazione di CpG incrementa nel corpo dei geni in piante e mammiferi. • Trasposoni non espressi di piante mostrano metilazione CpHpG • In batteri: 5meC ma anche N-4-metilcitosina e N-6-metiladenina • La metilazione avviene ad opera di metiltransferasi.

METILAZIONE DEL DNA
CGCGCGCGCG TATAAA
Isola CpG TATAbox Gene
Metilazione isola CpG
Silenziamento del gene

1. Accessibilità della cromatina: sequenziamento di siti di taglio della DNAsi I nella cromatina nucleare
2. Modificazioni istoniche: dati ottenuti tramite sequenziamento di DNA arricchito tramite immunoprecipitazione della cromatina con anticorpi specifici per le modificazioni istoniche
3. Espressione dell’RNA: sequenziamento degli sRNA o di lncRNA
4. Metilazione del DNA: dati ottenuti tramite sequenziamento di DNA trattato con bisolfito
Metodologie di analisi dell’epigenoma
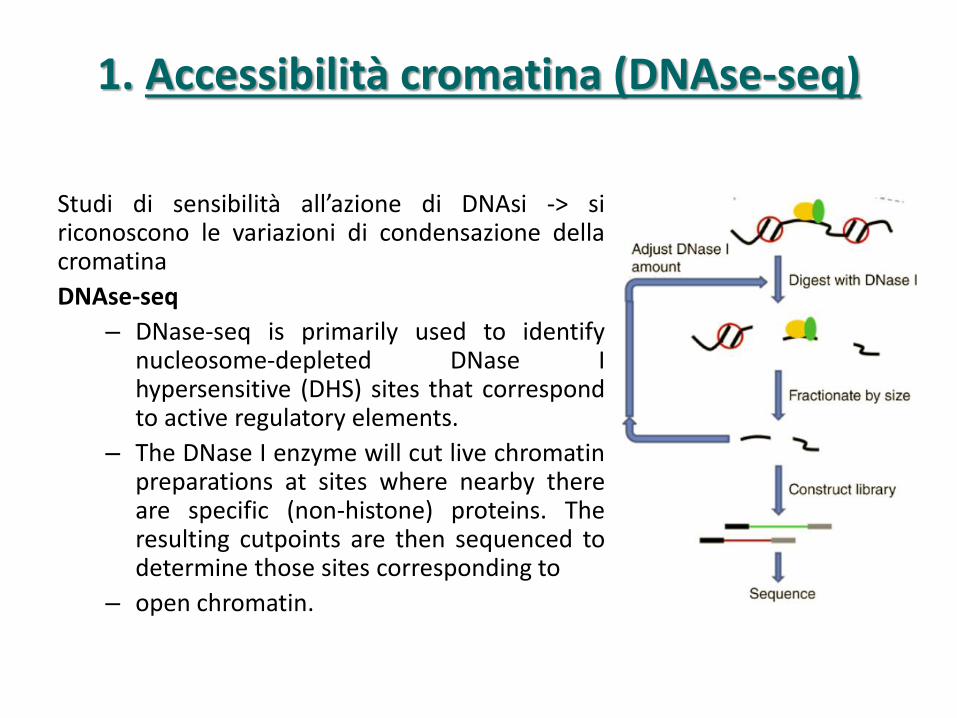
1. Accessibilità cromatina (DNAse-seq)
Studi di sensibilità all’azione di DNAsi -> si riconoscono le variazioni di condensazione della cromatina
DNAse-seq
– DNase-seq is primarily used to identify nucleosome-depleted DNase I hypersensitive (DHS) sites that correspond to active regulatory elements.
– The DNase I enzyme will cut live chromatin preparations at sites where nearby there are specific (non-histone) proteins. The resulting cutpoints are then sequenced to determine those sites corresponding to
– open chromatin.
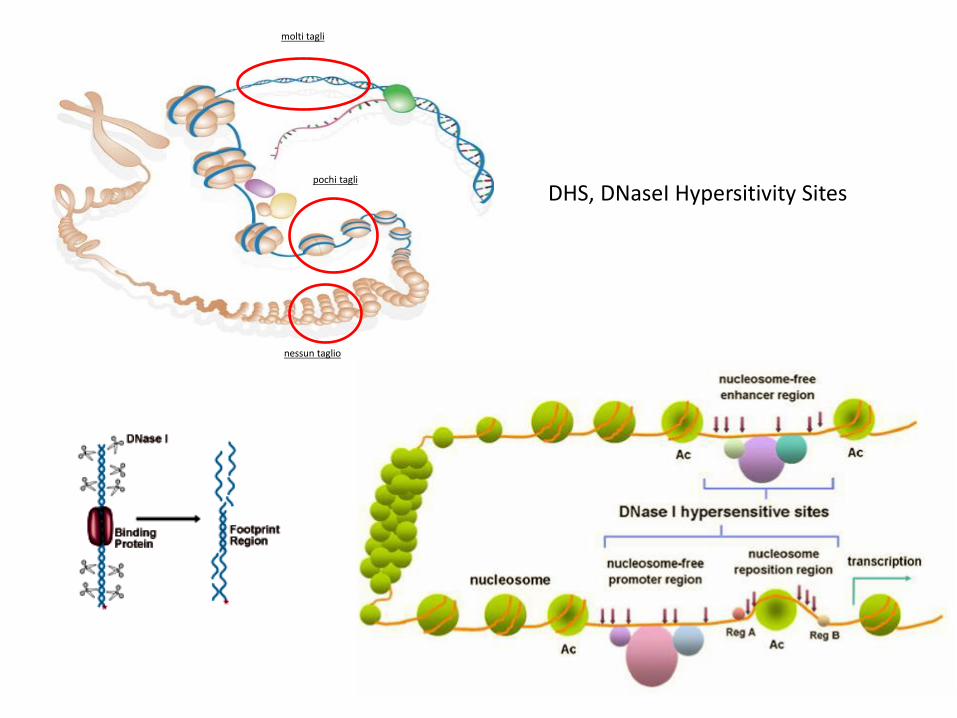
molti tagli
pochi tagli
nessun taglio
DHS, DNaseI Hypersitivity Sites

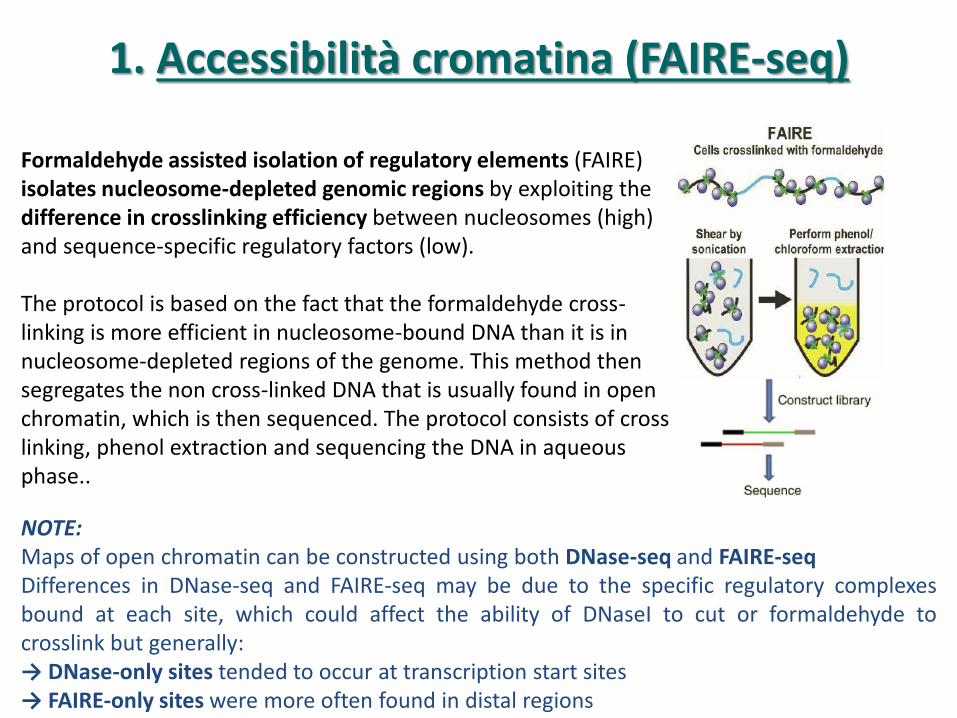
1. Accessibilità cromatina (FAIRE-seq)
Formaldehyde assisted isolation of regulatory elements (FAIRE) isolates nucleosome-depleted genomic regions by exploiting the difference in crosslinking efficiency between nucleosomes (high) and sequence-specific regulatory factors (low). The protocol is based on the fact that the formaldehyde cross-linking is more efficient in nucleosome-bound DNA than it is in nucleosome-depleted regions of the genome. This method then segregates the non cross-linked DNA that is usually found in open chromatin, which is then sequenced. The protocol consists of cross linking, phenol extraction and sequencing the DNA in aqueous phase..
NOTE: Maps of open chromatin can be constructed using both DNase-seq and FAIRE-seq Differences in DNase-seq and FAIRE-seq may be due to the specific regulatory complexes bound at each site, which could affect the ability of DNaseI to cut or formaldehyde to crosslink but generally: → DNase-only sites tended to occur at transcription start sites → FAIRE-only sites were more often found in distal regions

2. Modificazioni istoniche
• Immunoprecipitazione (ChIP-Seq): distinguere lo stato di acetilazione: – Reagente che instaura legami crociati proteine-DNA
(formaldeide)
– Estrazione (delicata!) cromatina
– Rottura meccanica del DNA
– Uso di anticorpi contro istoni acetilati
– Rottura legami istoni-DNA
– Sequenziamento DNA: la sequenza rivela quali regioni del genoma erano associate a istoni acetilati
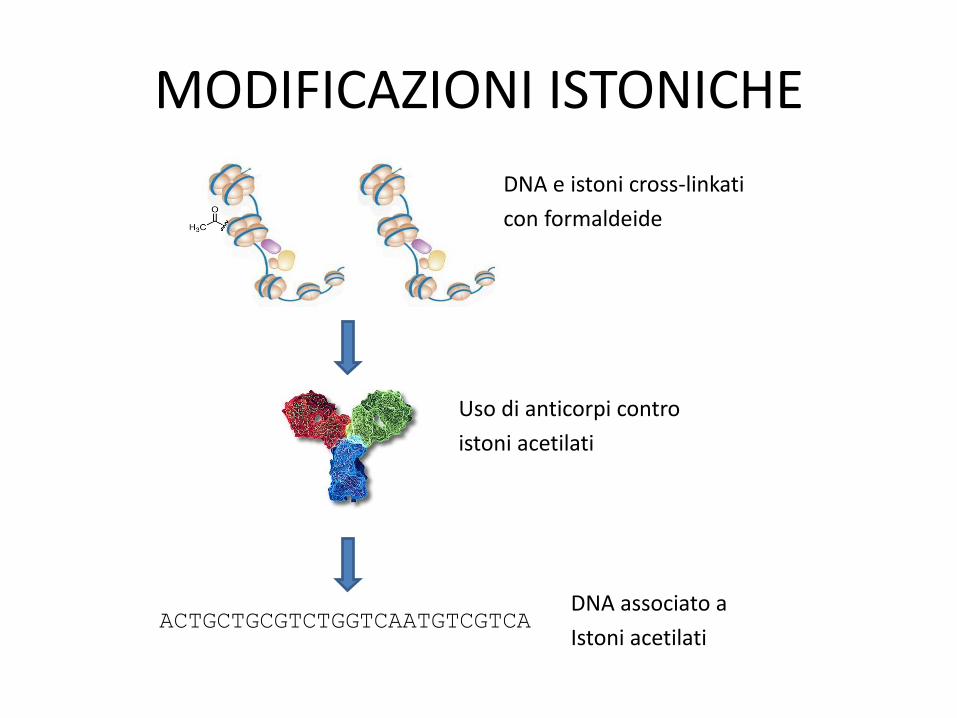
MODIFICAZIONI ISTONICHE
ACTGCTGCGTCTGGTCAATGTCGTCA
DNA e istoni cross-linkati
con formaldeide
Uso di anticorpi contro
istoni acetilati
DNA associato a
Istoni acetilati
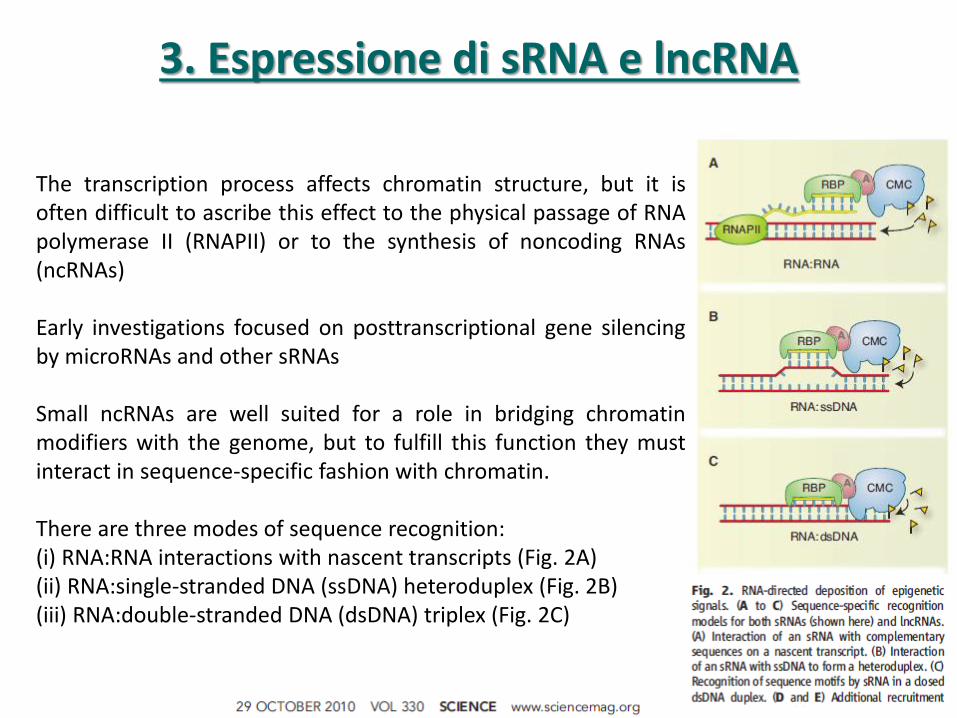
3. Espressione di sRNA e lncRNA
The transcription process affects chromatin structure, but it is often difficult to ascribe this effect to the physical passage of RNA polymerase II (RNAPII) or to the synthesis of noncoding RNAs (ncRNAs) Early investigations focused on posttranscriptional gene silencing by microRNAs and other sRNAs Small ncRNAs are well suited for a role in bridging chromatin modifiers with the genome, but to fulfill this function they must interact in sequence-specific fashion with chromatin. There are three modes of sequence recognition: (i) RNA:RNA interactions with nascent transcripts (Fig. 2A) (ii) RNA:single-stranded DNA (ssDNA) heteroduplex (Fig. 2B) (iii) RNA:double-stranded DNA (dsDNA) triplex (Fig. 2C)

• Il DNA viene metilato soprattutto a livello di citosine nelle ripetizioni dei dimeri CG (isole CpG)
• Le isole CpG si trovano soprattutto in prossimità del promotori o al 5’ di un gene
• Se metilate, silenziano il gene a cui sono associate
4. Metilazione del DNA

La metilazione delle citosine viene rilevata utilizzando delle tecniche basate su un qualche pretrattamento del DNA:
a) Digestione con endonucleasi sensibili a metilazione b) Arricchimento per affinità c) Trattamento con bisolfito
NOTA: Tecniche di biologia molecolare standard come PCR e clonaggio cancellano l’informazione sulla posizione delle citosine metilate nel DNA.
Rilevazione delle citosine metilate

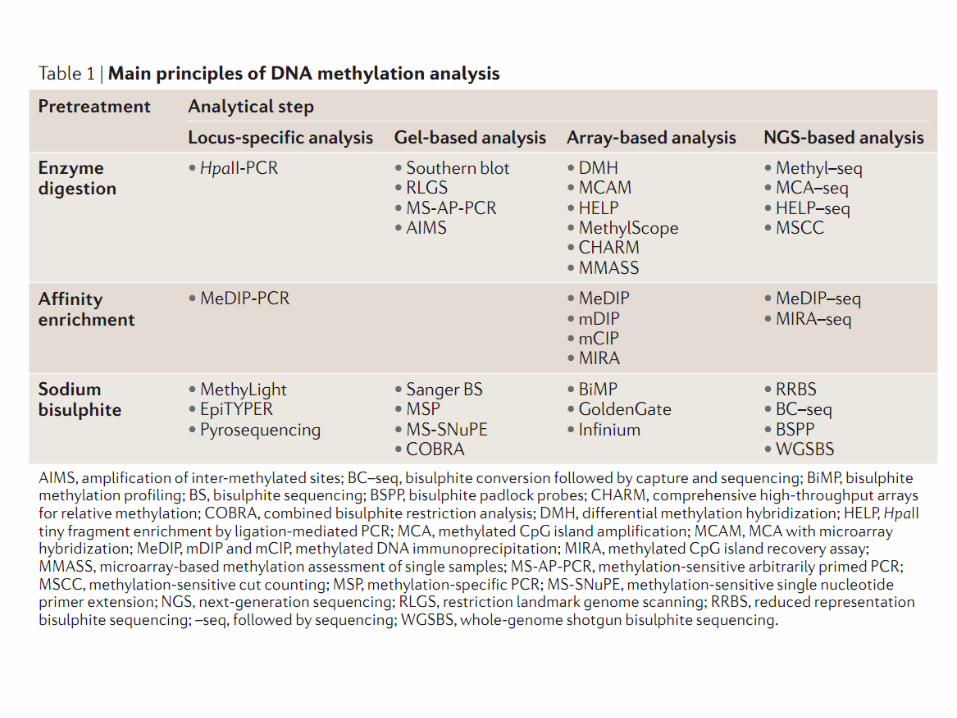

4a) Analisi del DNA metilato mediante digestione con endunucleasi
• Ad ogni enzima di restrizione sequenza specifico corrisponde una DNA metiltransferasi che protegge il DNA endogeno metilando delle basi sul sito di riconoscimento. Quindi il profilo di taglio di questi enzimi è indice dello stato di metilazione: dove non taglia, il DNA è metilato
• Nella tecnica restriction landmark genome scanning (RLGS) differenze in metilazione vengono rilevate come differenze nei profili di restrizione generati tramite digestione con un enzima di restrizione metilazione sensibile separata tramite elettroforesi bidimensionale.
• Le tecniche basate su elettroforesi sono molto laboriose e tendono ad essere abbandonate in favore di altre tecnologie
• Altre tecniche sono basate sui profili differenziali ottenuti da digestione con enzimi di restrizione metilazione-sensibili e successiva ibridazione ad un array specifico per isole CpG oppure sequenziamento NGS
HpaII MspI
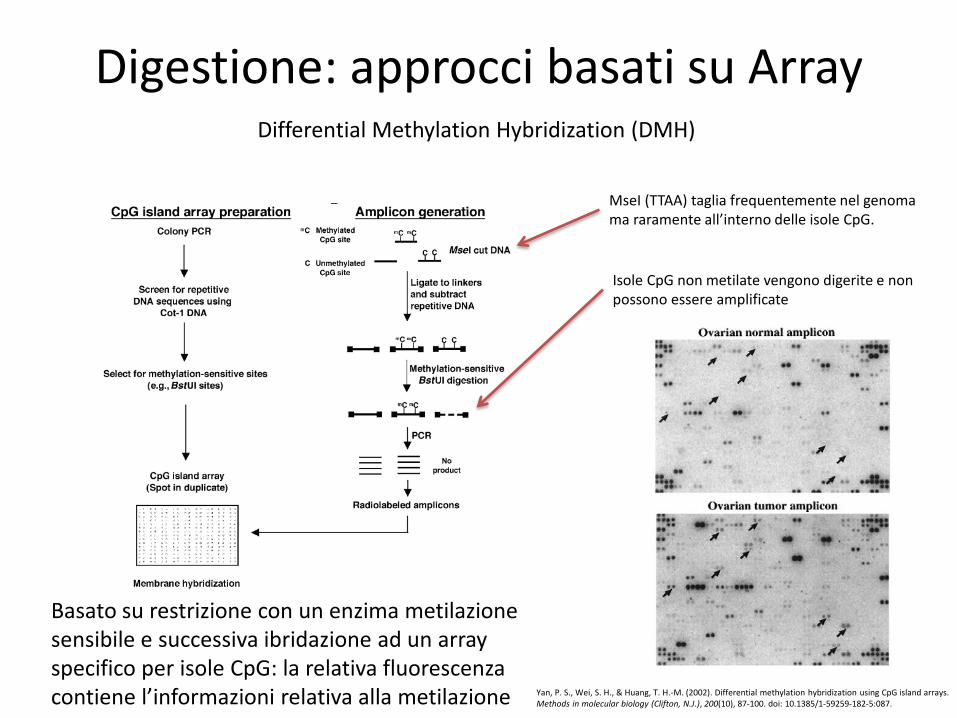
Digestione: approcci basati su Array Differential Methylation Hybridization (DMH)
Basato su restrizione con un enzima metilazione sensibile e successiva ibridazione ad un array specifico per isole CpG: la relativa fluorescenza contiene l’informazioni relativa alla metilazione
MseI (TTAA) taglia frequentemente nel genoma ma raramente all’interno delle isole CpG.
Isole CpG non metilate vengono digerite e non possono essere amplificate
Yan, P. S., Wei, S. H., & Huang, T. H.-M. (2002). Differential methylation hybridization using CpG island arrays. Methods in molecular biology (Clifton, N.J.), 200(10), 87-100. doi: 10.1385/1-59259-182-5:087.
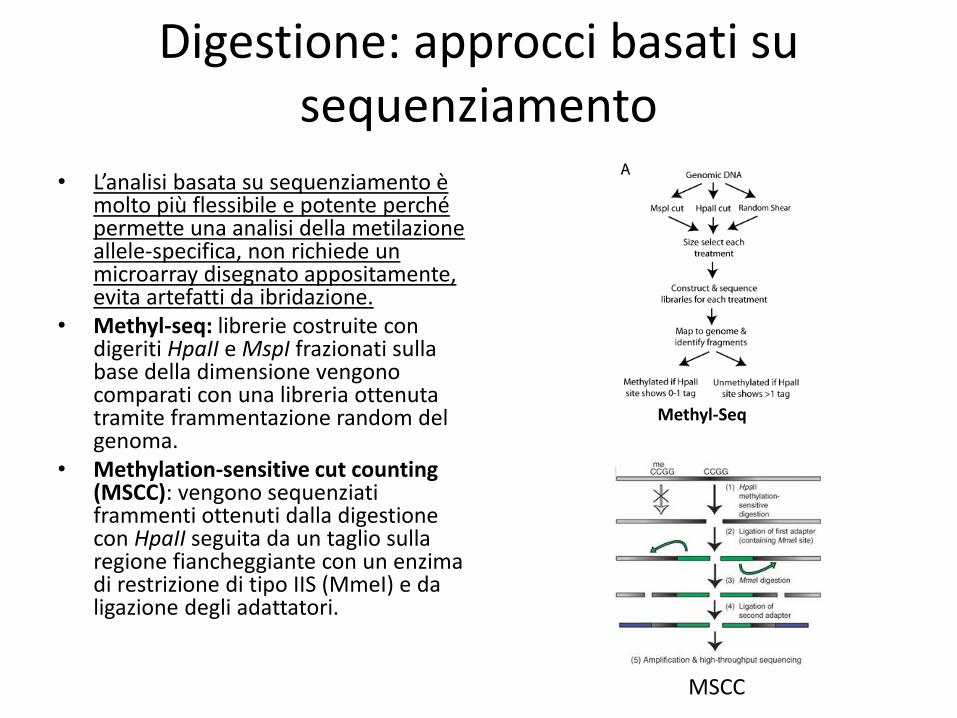
Digestione: approcci basati su sequenziamento
• L’analisi basata su sequenziamento è molto più flessibile e potente perché permette una analisi della metilazione allele-specifica, non richiede un microarray disegnato appositamente, evita artefatti da ibridazione.
• Methyl-seq: librerie costruite con digeriti HpaII e MspI frazionati sulla base della dimensione vengono comparati con una libreria ottenuta tramite frammentazione random del genoma.
• Methylation-sensitive cut counting (MSCC): vengono sequenziati frammenti ottenuti dalla digestione con HpaII seguita da un taglio sulla regione fiancheggiante con un enzima di restrizione di tipo IIS (MmeI) e da ligazione degli adattatori.
Methyl-Seq
MSCC
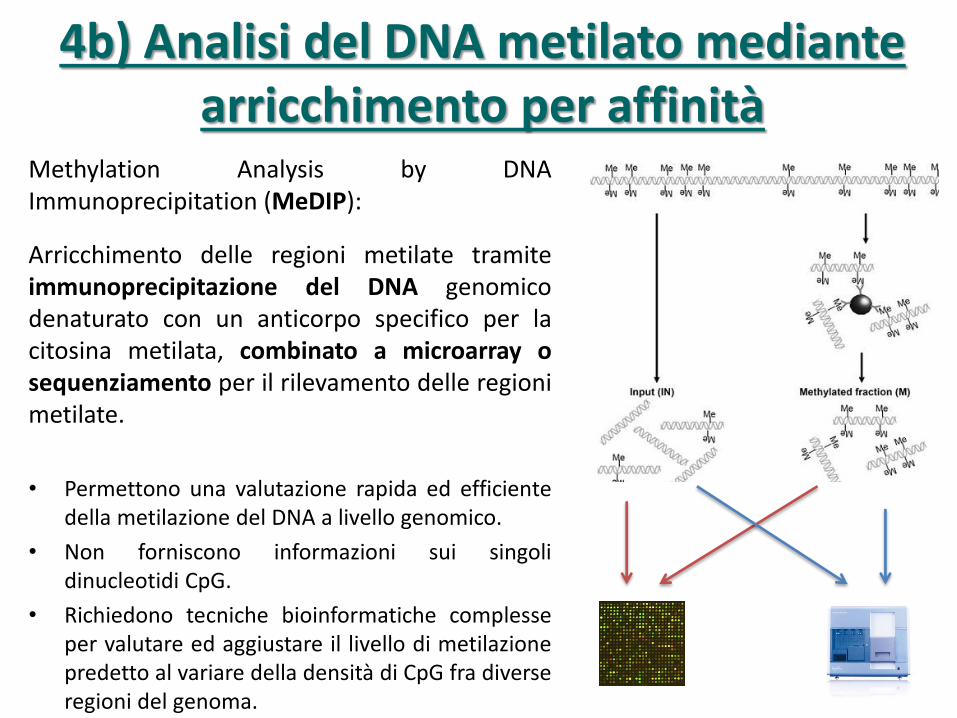
Methylation Analysis by DNA Immunoprecipitation (MeDIP):
Arricchimento delle regioni metilate tramite immunoprecipitazione del DNA genomico denaturato con un anticorpo specifico per la citosina metilata, combinato a microarray o sequenziamento per il rilevamento delle regioni metilate.
• Permettono una valutazione rapida ed efficiente della metilazione del DNA a livello genomico.
• Non forniscono informazioni sui singoli dinucleotidi CpG.
• Richiedono tecniche bioinformatiche complesse per valutare ed aggiustare il livello di metilazione predetto al variare della densità di CpG fra diverse regioni del genoma.
4b) Analisi del DNA metilato mediante arricchimento per affinità

• Metodi basati su array: – Uso 2 pool, uno arricchito e uno non arricchito,
marcati con differente fluorofori
– Ibrido
– La fluorescenza indica quali regioni del genoma sono metilate
• Metodi basati su sequenziamento – Uso un pool arricchito
– Sequenzio

• Il sodio bisolfito deammina chimicamente molto più rapidamente i residui di citosine non metilate rispetto alle citosine metilate causando una conversione da C a U
• Produce informazione sulla metilazione del DNA con la risoluzione di una singola base.
4c) Analisi del DNA metilato mediante trattamento con bisolfito
“Una differenza epigenetica viene trasformata in una differenza genetica”
Il DNA trattato con bisolfito può essere analizzato mediante:
a) Analisi microarray con sonde basate sulla versione convertita dal bisolfito
b) Analisi SNP (di fatto, la conversione C->T può essere monitorata come se fosse un qualsiasi SNP
c) Sequenziamento
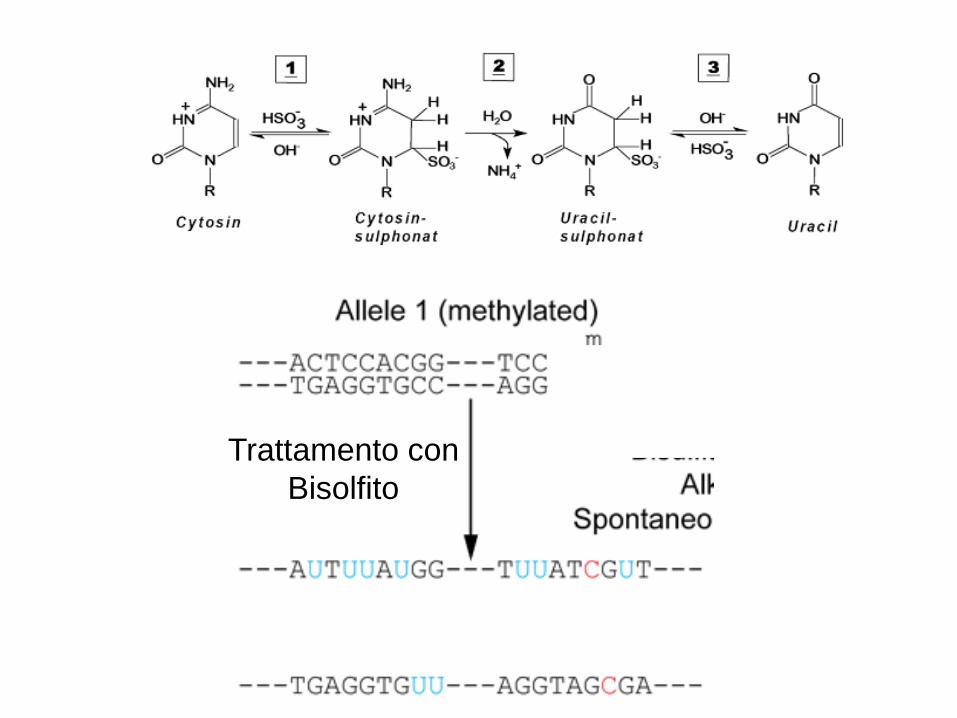
Trattamento con
Bisolfito

Bisolfito e microarray
• La tecnica del sodio bisolfito non è facilmente adattabile all’analisi array: – La conversione delle C ad U riduce il numero di basi a
3, diminuendo notevolmente la complessità delle sequenze e quindi risultando in una diminuita specificità di ibridazione.
– Richiede array dedicati basati sulla versione convertita dal bisolfito del genoma.
– In genomi complessi richiede l’amplificazione di regioni individuali del genoma prima dell’ibridazione ad un array ad oligo dedicato.

Bisolfito e GoldenGate BeadArray Basato su primer extension metilazione specifica in multiplex di DNA convertito con bisolfito fino a 1536 differenti loci CpG. Per ogni sito si utilizzano dei primer specifici per le sequenze metilate e non-metilate. I primer per i due differenti stati di metilazione sono marcati con due differenti fluorofori e i prodotti vengono ibridati al bead array.
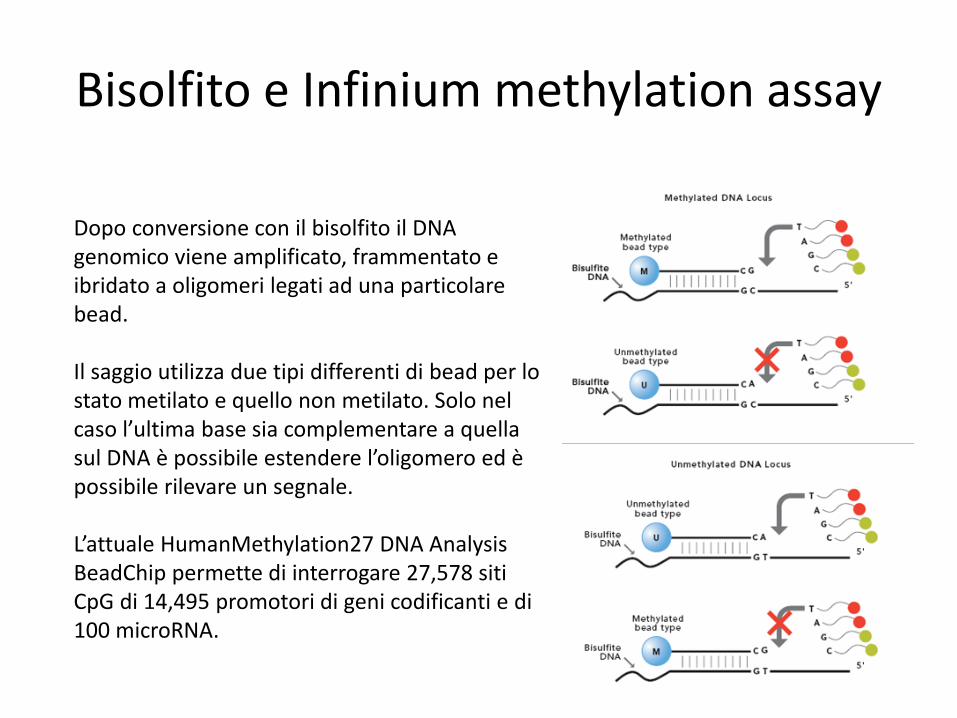
Bisolfito e Infinium methylation assay
Dopo conversione con il bisolfito il DNA genomico viene amplificato, frammentato e ibridato a oligomeri legati ad una particolare bead. Il saggio utilizza due tipi differenti di bead per lo stato metilato e quello non metilato. Solo nel caso l’ultima base sia complementare a quella sul DNA è possibile estendere l’oligomero ed è possibile rilevare un segnale. L’attuale HumanMethylation27 DNA Analysis BeadChip permette di interrogare 27,578 siti CpG di 14,495 promotori di geni codificanti e di 100 microRNA.
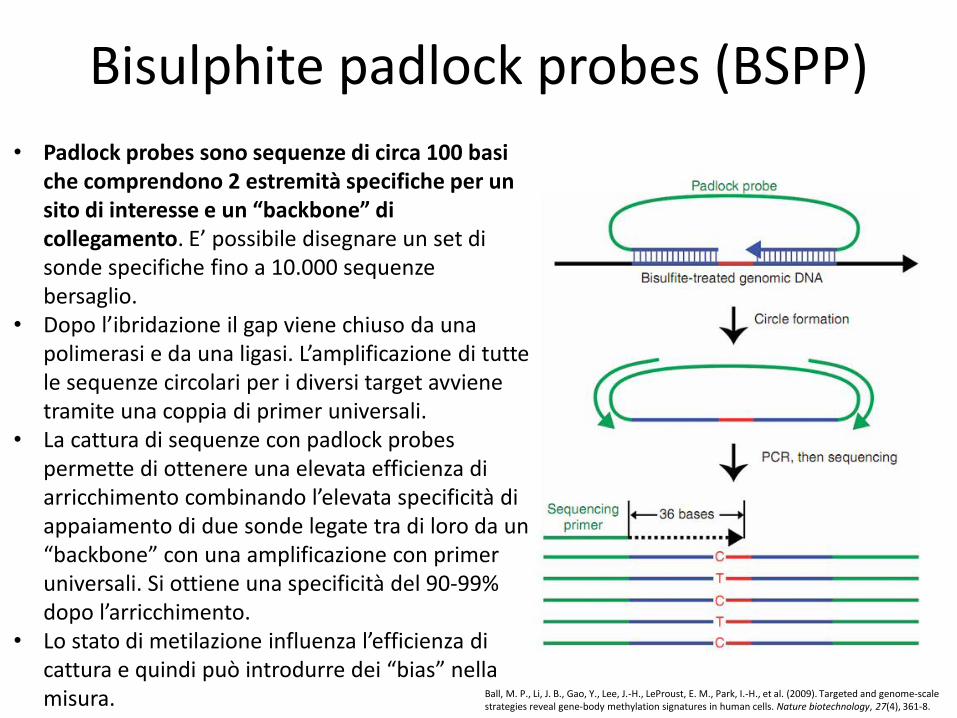
Bisulphite padlock probes (BSPP)
Ball, M. P., Li, J. B., Gao, Y., Lee, J.-H., LeProust, E. M., Park, I.-H., et al. (2009). Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nature biotechnology, 27(4), 361-8.
• Padlock probes sono sequenze di circa 100 basi che comprendono 2 estremità specifiche per un sito di interesse e un “backbone” di collegamento. E’ possibile disegnare un set di sonde specifiche fino a 10.000 sequenze bersaglio.
• Dopo l’ibridazione il gap viene chiuso da una polimerasi e da una ligasi. L’amplificazione di tutte le sequenze circolari per i diversi target avviene tramite una coppia di primer universali.
• La cattura di sequenze con padlock probes permette di ottenere una elevata efficienza di arricchimento combinando l’elevata specificità di appaiamento di due sonde legate tra di loro da un “backbone” con una amplificazione con primer universali. Si ottiene una specificità del 90-99% dopo l’arricchimento.
• Lo stato di metilazione influenza l’efficienza di cattura e quindi può introdurre dei “bias” nella misura.

Bisolfito e Whole-genome shotgun bisulfite sequencing (WGSBS)
• DNA convertito con bisolfito è particolarmente adatto ad approcci basati su sequenziamento.
• Consente l’analisi dello stato di metilazione del genoma con una risoluzione di una singola base.
• Il problema principale è rappresentato dalla bassa complessità del
DNA convertito con il bisolfito che può impedire l’assegnamento delle read a posizioni univoche sul genoma.
• L’utilizzo di read di lunghezza maggiore e strategie basate su
sequenziamento paired-end facilita l’allineamento univoco delle sequenze: – Read più lunghe: sequenze lunghe sono generalmente più complesse di
sequenze corte – Paired-end read: oltre all’informazione della sequenza, ho un vincolo in
più per allineare, cioè la distanza tra read accoppiate

Analisi di DNA arricchito per regioni specifiche del genoma
• E’ possibile eseguire l’analisi su campioni arricchiti su specifiche regioni (complessità) del genoma utilizzando la cattura tramite array, la cattura tramite padlock o tramite selezione ibrida in soluzione.
• Cattura prima della conversione con bisolfito: – usa un array genomico standard – lo stato di metilazione non influenza l’efficienza di
cattura – Poco efficiente verso le regioni ricche in CG. Richiede
quantità notevoli di DNA genomico (non è possibile amplificare prima della cattura perché verrebbe persa la metilazione)
• Cattura dopo la conversione a bisolfito: – Dopo la conversione e prima della cattura è possibile
amplificare il DNA tramite una PCR mediata da adattatori (BC-seq).
– Richiede permutazioni multiple degli oligonucleotidi di cattura
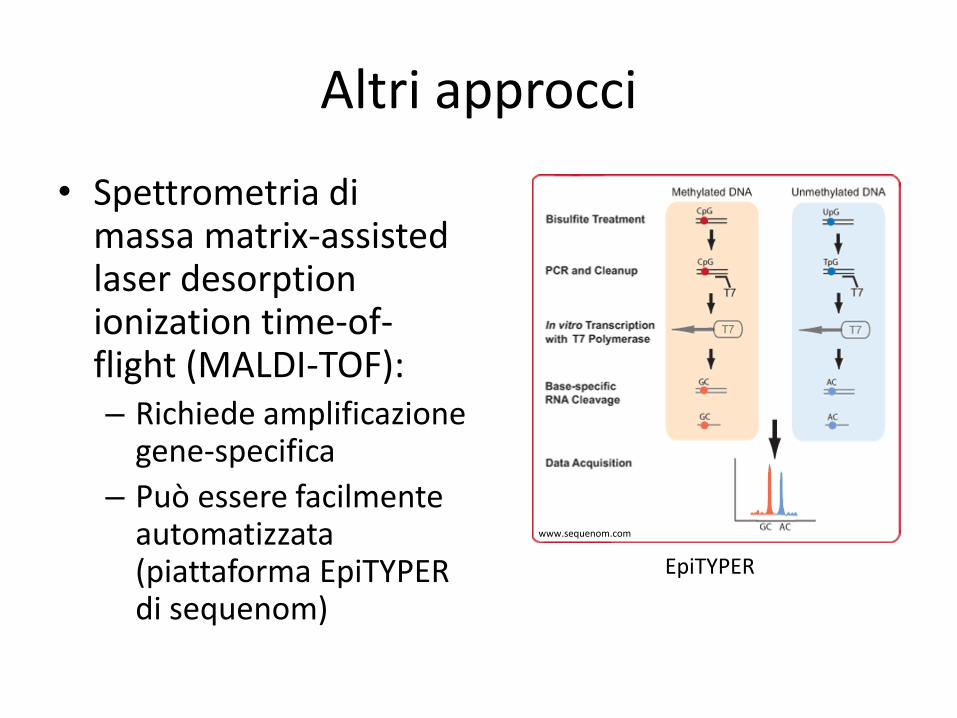
Altri approcci
• Spettrometria di massa matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF): – Richiede amplificazione
gene-specifica
– Può essere facilmente automatizzata (piattaforma EpiTYPER di sequenom)
www.sequenom.com
EpiTYPER

Scelta della metodica
Dipende da diversi fattori, come numero di campioni, quantità e qualità dei campioni, risoluzione e copertura desiderate.


The prevailing assumption is that mammalian DNA methylation is located almost exclusively in the CG context. However, in particular in embryonic stem cells, a handful of studies have detected non-CG methylation (mCHG and mCHH, where H= A, C or T) that comprises almost 25% of all cytosines at which DNA methylation is identified There are widespread differences in the composition and patterning of cytosine methylation between human embryonic stem cells and fetal fibroblasts (fibroblasts are the most common cells of connective tissue in animals). Nearly one-quarter of all methylation identified in embryonic stem cells are in a non-CG context, suggesting that embryonic stem cells may use different methylation mechanisms to affect gene regulation. Methylation in non-CG contexts show enrichment in gene bodies and depletion in protein binding sites and enhancers. Non-CG methylation disappear upon induced differentiation of the embryonic stem cells, and is restored in induced pluripotent stem cells. There are hundreds of differentially methylated regions proximal to genes involved in pluripotency and differentiation, and widespread reduced methylation levels in fibroblasts is associated with lower transcriptional activity.
Human DNA methylome in stem cells and fibroblasts


Frammentazione random
Immagine modificata da: Lister, R., & Ecker, J. R. (2009). Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome research, 19(6), 959-66. doi: 10.1101/gr.083451.108.
Mappa della metilazione a risoluzione di singola base in 2 linee cellulari
Analisi MethylC-Seq su 2 linee cellulari (2 repliche biologiche ciascuna):
Cellule staminali embrionali umane
H1
Fibroblasti del polmone fetale
IMR90
Miliardi di read 1.16 1.18
Resa totale in sequenza (Gbp)
87.5 91.0
Profondità media (per strand)
14.2 X 14.8 X
In ciascuna linea cellulare più dell’86% del genoma è coperto da almeno una read per un totale del 94% delle citosine del genoma.

Validazione della metilazione tramite bisulphite-PCR di siti specifici
• H9, una seconda linea cellulare embrionale umana ha mostrato metilazione non-CG in posizioni conservate
• IMR90 induced pluripotent stem cells (iPS) mostrano un ripristino della metilazione non-CG
• Cellule H1 indotte a differenziare con BMP4 (bone morphogenetic protein 4) perdono la metilazione non-CG come le IMR90
La metilazione non-CG è caratteristica di uno stato di cellula staminale embrionale. Essa non è dovuta a semplici differenze genetiche tra le due linee cellulari
In the H1 stem cells we detected abundant DNA methylation in non-CG contexts (mCHG and mCHH, where H=A, C or T)
DNA methylation sequence context is displayed according to the key and the percentage methylation at each position is represented by the fill of each circle. Non-CG methylated positions indicated by an asterisk are unique to that cell type and ‘14’ indicates a mCHH that is shifted 4 bases downstream in H9 cells. iPS, induced pluripotent stem cell.
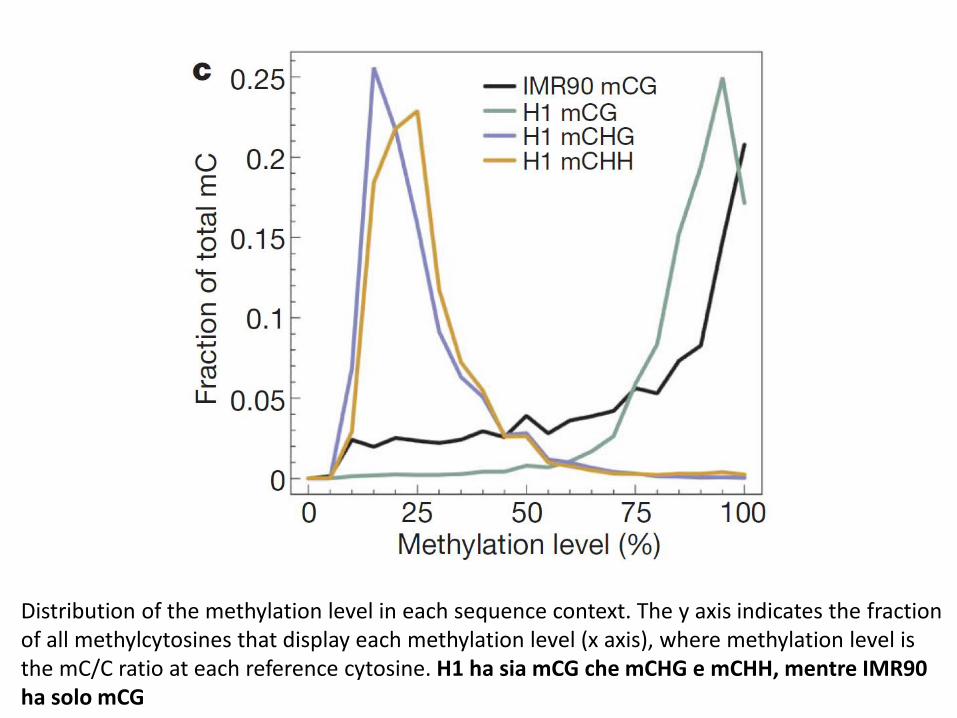
Distribution of the methylation level in each sequence context. The y axis indicates the fraction of all methylcytosines that display each methylation level (x axis), where methylation level is the mC/C ratio at each reference cytosine. H1 ha sia mCG che mCHG e mCHH, mentre IMR90 ha solo mCG
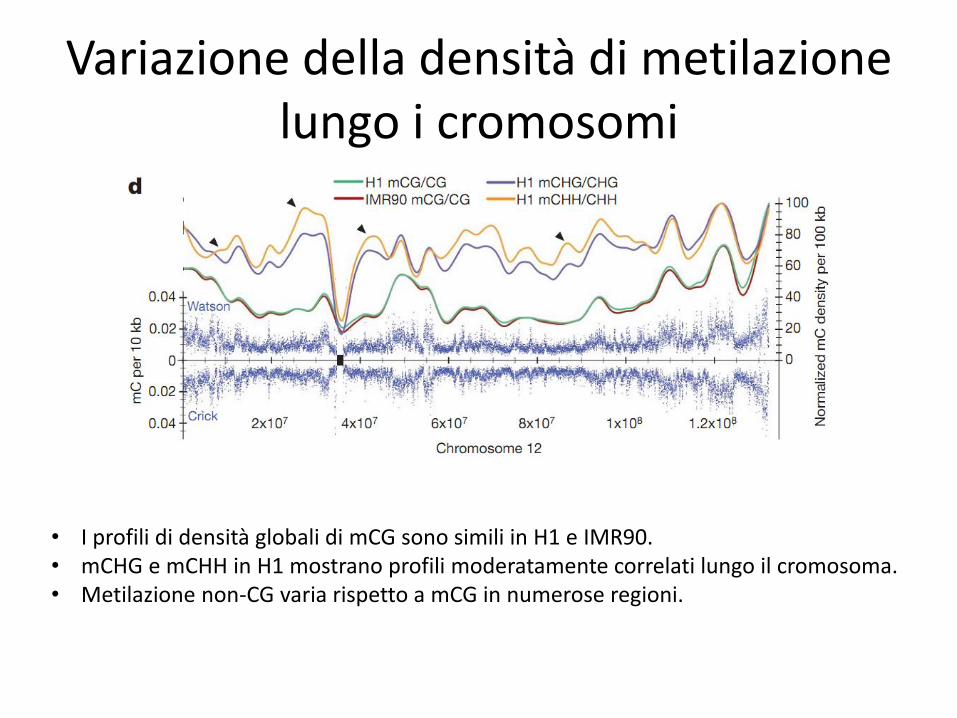
Variazione della densità di metilazione lungo i cromosomi
• I profili di densità globali di mCG sono simili in H1 e IMR90. • mCHG e mCHH in H1 mostrano profili moderatamente correlati lungo il cromosoma. • Metilazione non-CG varia rispetto a mCG in numerose regioni.
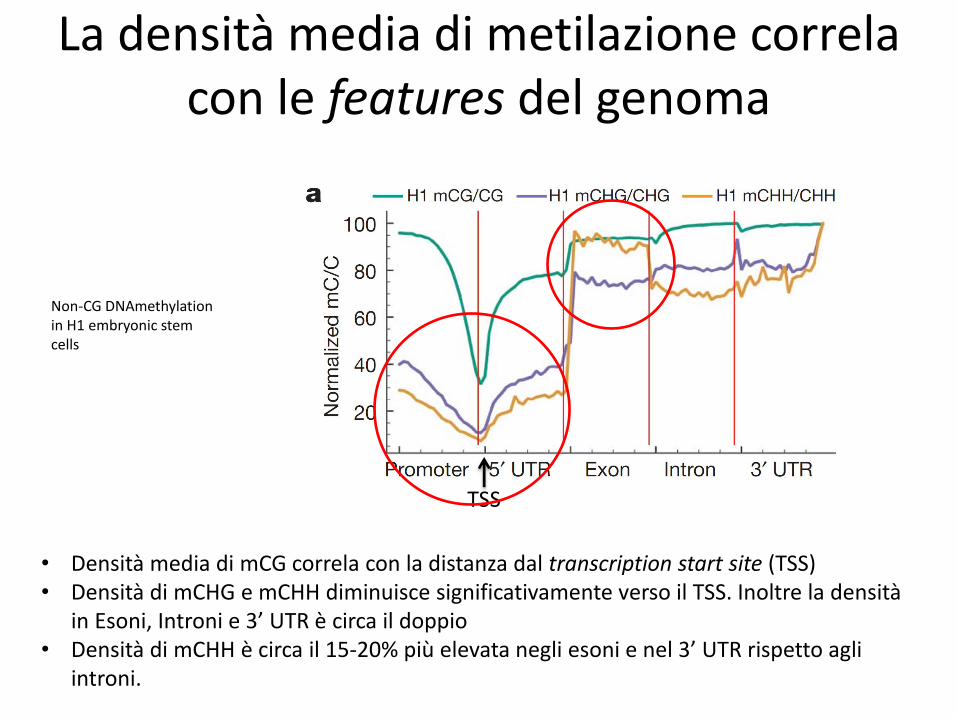
La densità media di metilazione correla con le features del genoma
• Densità media di mCG correla con la distanza dal transcription start site (TSS) • Densità di mCHG e mCHH diminuisce significativamente verso il TSS. Inoltre la densità
in Esoni, Introni e 3’ UTR è circa il doppio • Densità di mCHH è circa il 15-20% più elevata negli esoni e nel 3’ UTR rispetto agli
introni.
TSS
Non-CG DNAmethylation in H1 embryonic stem cells
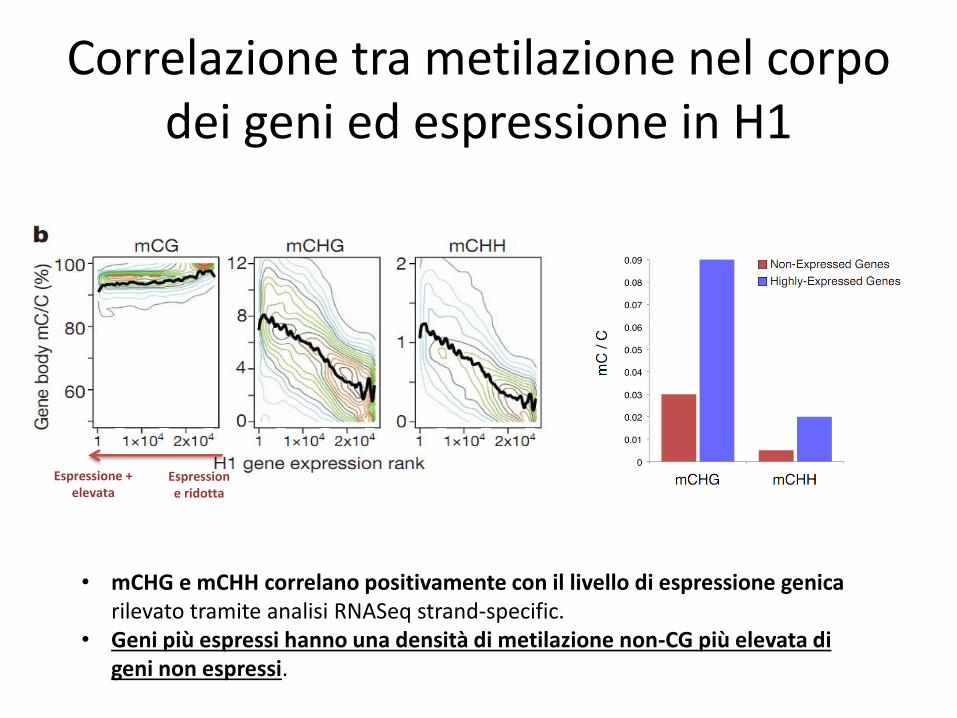
Correlazione tra metilazione nel corpo dei geni ed espressione in H1
Espressione + elevata
Espressione ridotta
• mCHG e mCHH correlano positivamente con il livello di espressione genica rilevato tramite analisi RNASeq strand-specific.
• Geni più espressi hanno una densità di metilazione non-CG più elevata di geni non espressi.
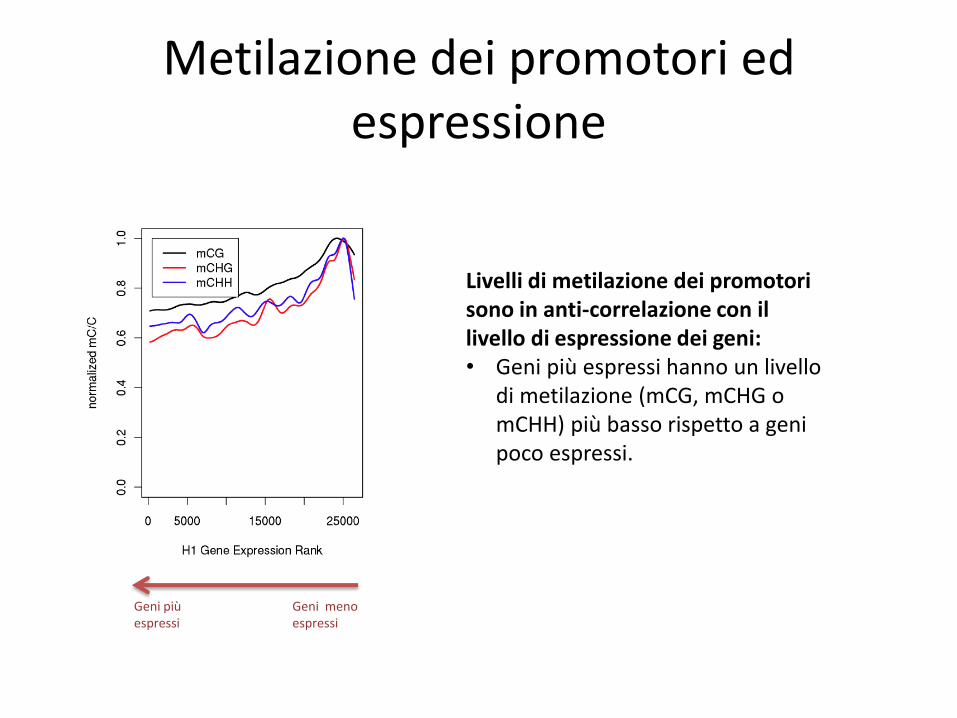
Metilazione dei promotori ed espressione
Geni più espressi
Geni meno espressi
Livelli di metilazione dei promotori sono in anti-correlazione con il livello di espressione dei geni: • Geni più espressi hanno un livello
di metilazione (mCG, mCHG o mCHH) più basso rispetto a geni poco espressi.

Using Arabidopsis thaliana, the workhorse of modern plant genetics, it has been determined how often and where in the genome epigenetic modifications occur -- and how often they disappear again. To determine the rate and distribution of methylation changes in the genome, ten Arabidopsis lines coming from the same stock, but that had been propagated independently for 30 generations by self-fertilization, have been searched for differences in the methylation pattern in comparison to the common ancestor. For each line, about 14 million cytosines were investigated. On average, every plant had almost 3 million methylated cytosines. The vast majority of these were the same in all lines, but about 6 percent had changed since the lines had become separated. At these positions, at least one of the individuals was different, with either methylation gained or lost relative to the ancestor. Each of the lines had about 30,000 such epimutations, which was 1,000 times more than DNA mutations. With 30,000 epimutations after 30 generations, the geneticists had expected that 1,000 epimutations occurred in each generation. But when parents were compared with their immediate offspring, it was found that the epimutation rate was three to four times as high. Therefore, many epimutations are apparently not stable and return to their original state after a few generations. New epimutations are often not maintained over the long term. A new epimutation thus must have a strong evolutionary advantage so that it can become established before being lost again. Because reverse mutations do not necessarily happen in the next generation, it is still possible that epigenetic differences contribute to inheritance of traits between parents and their children or grandparents and their grandchildren. Another difference to ordinary mutations is that epimutations do not occur randomly, but often at the same places in the genome. While genes were disproportionately often affected, methylation of mobile DNA elements, transposons, was very stable. This appears to make sense, since it was already known that artificially induced loss of methylation has a much greater effect on the activity of transposons than of regular genes.
Epigenetic Changes over several generations
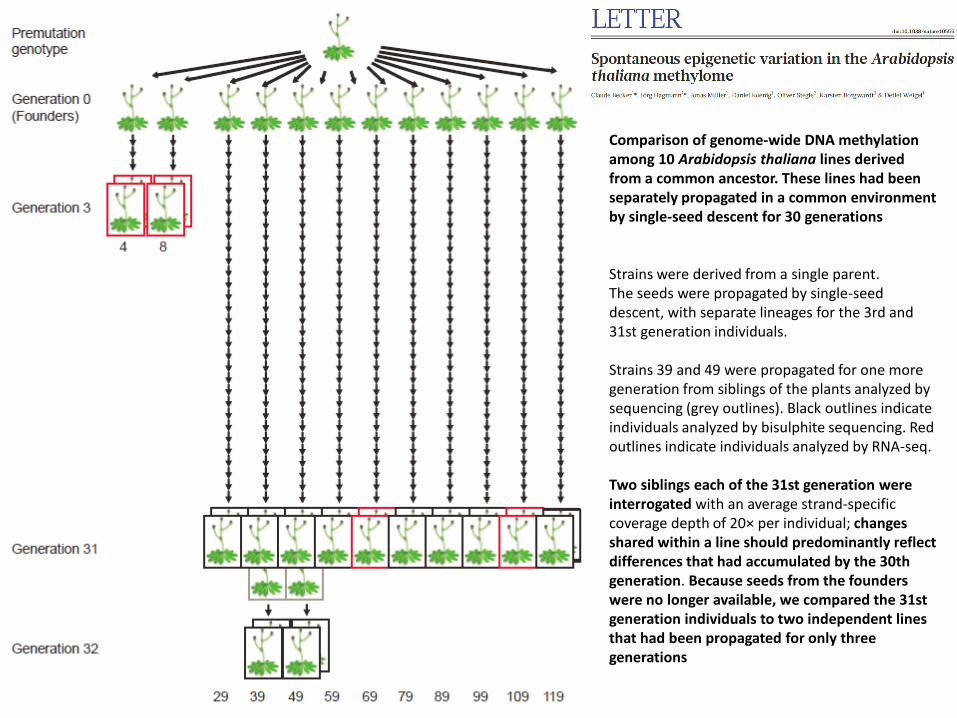
Comparison of genome-wide DNA methylation among 10 Arabidopsis thaliana lines derived from a common ancestor. These lines had been separately propagated in a common environment by single-seed descent for 30 generations Strains were derived from a single parent. The seeds were propagated by single-seed descent, with separate lineages for the 3rd and 31st generation individuals. Strains 39 and 49 were propagated for one more generation from siblings of the plants analyzed by sequencing (grey outlines). Black outlines indicate individuals analyzed by bisulphite sequencing. Red outlines indicate individuals analyzed by RNA-seq. Two siblings each of the 31st generation were interrogated with an average strand-specific coverage depth of 20× per individual; changes shared within a line should predominantly reflect differences that had accumulated by the 30th generation. Because seeds from the founders were no longer available, we compared the 31st generation individuals to two independent lines that had been propagated for only three generations
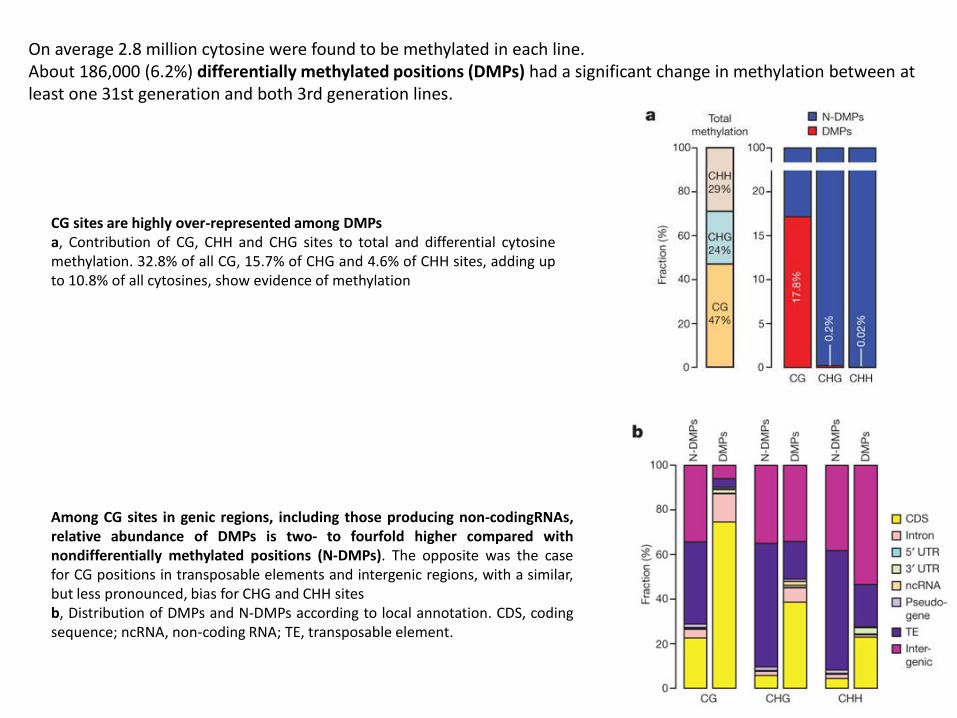
On average 2.8 million cytosine were found to be methylated in each line. About 186,000 (6.2%) differentially methylated positions (DMPs) had a significant change in methylation between at least one 31st generation and both 3rd generation lines.
CG sites are highly over-represented among DMPs a, Contribution of CG, CHH and CHG sites to total and differential cytosine methylation. 32.8% of all CG, 15.7% of CHG and 4.6% of CHH sites, adding up to 10.8% of all cytosines, show evidence of methylation
Among CG sites in genic regions, including those producing non-codingRNAs, relative abundance of DMPs is two- to fourfold higher compared with nondifferentially methylated positions (N-DMPs). The opposite was the case for CG positions in transposable elements and intergenic regions, with a similar, but less pronounced, bias for CHG and CHH sites b, Distribution of DMPs and N-DMPs according to local annotation. CDS, coding sequence; ncRNA, non-coding RNA; TE, transposable element.
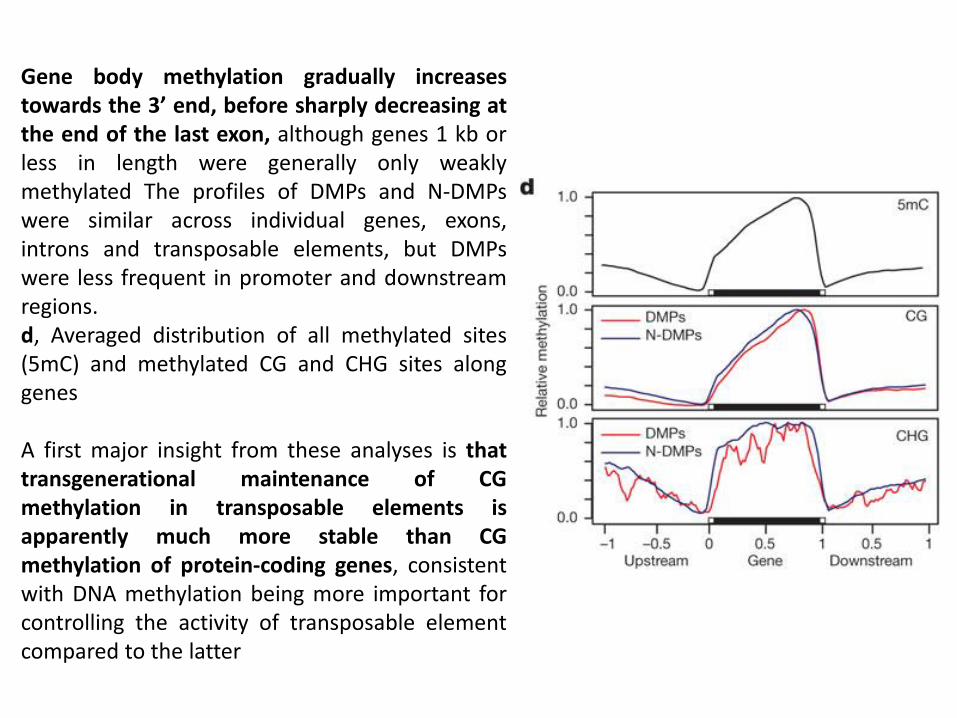
Gene body methylation gradually increases towards the 3’ end, before sharply decreasing at the end of the last exon, although genes 1 kb or less in length were generally only weakly methylated The profiles of DMPs and N-DMPs were similar across individual genes, exons, introns and transposable elements, but DMPs were less frequent in promoter and downstream regions. d, Averaged distribution of all methylated sites (5mC) and methylated CG and CHG sites along genes A first major insight from these analyses is that transgenerational maintenance of CG methylation in transposable elements is apparently much more stable than CG methylation of protein-coding genes, consistent with DNA methylation being more important for controlling the activity of transposable element compared to the latter
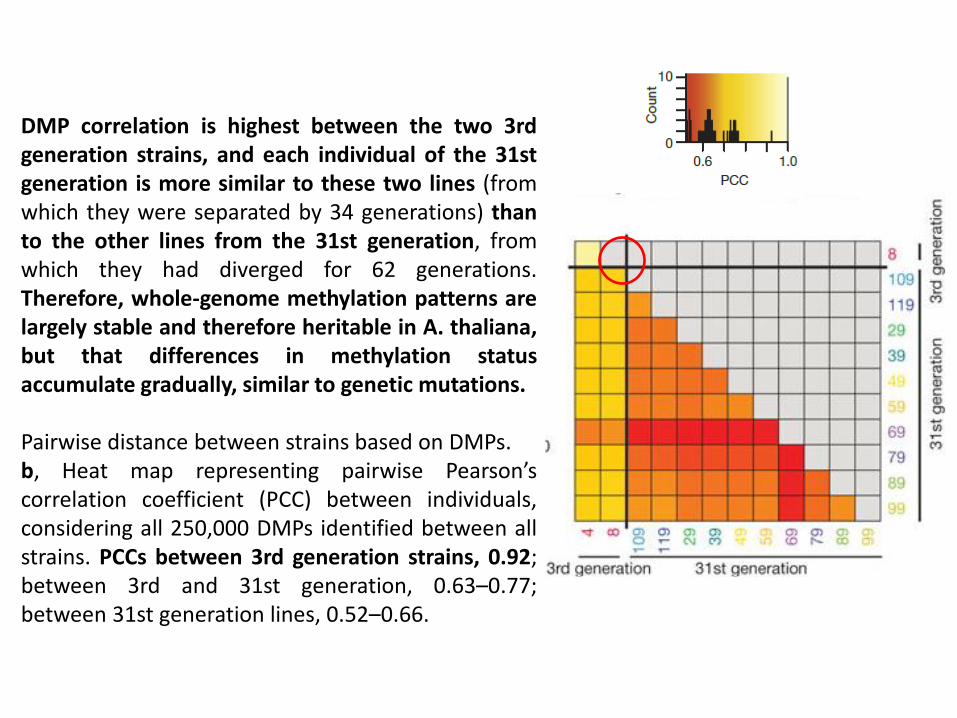
DMP correlation is highest between the two 3rd generation strains, and each individual of the 31st generation is more similar to these two lines (from which they were separated by 34 generations) than to the other lines from the 31st generation, from which they had diverged for 62 generations. Therefore, whole-genome methylation patterns are largely stable and therefore heritable in A. thaliana, but that differences in methylation status accumulate gradually, similar to genetic mutations. Pairwise distance between strains based on DMPs. b, Heat map representing pairwise Pearson’s correlation coefficient (PCC) between individuals, considering all 250,000 DMPs identified between all strains. PCCs between 3rd generation strains, 0.92; between 3rd and 31st generation, 0.63–0.77; between 31st generation lines, 0.52–0.66.

Compared to genetic mutations, the frequency of epimutations at single cytosine residues was many orders of magnitude higher, with an average of close to 30,000 DMPs in the analysed sequence space, compared with less than 30 DNA sequence mutations per strain. Thirty-two per cent of DMPs between generations 3 and 31 occurred more than once, and 13% more than twice. If DMPs arose randomly, the expected rate of recurrent events would be less than 1%. This indicates that certain positions are particularly prone to increases or decreases in methylation rate. The analysis of 32nd generation of lines 39 and 49 revealed how many DMPs emerge from one generation to the next. These individuals were progeny of siblings of the individuals interrogated in the 31st generation, and shared changes in the 32nd generation should reflect differences that arose between the 30th and 31st generation. We found on average over 3,300 between-generation DMPs. This is in the same range as DMPs between siblings (on average, about 5,000), but more than we would have expected from the 30,000 that had accumulated between the 3rd and each of the 31st generation lines. One explanation is that frequent transgenerational changes in methylation status occur at a limited number of sites, and that only a fraction of new DMPs is maintained over the longer term. This is corroborated by the observation that more than two-thirds of DMPs distinguishing the 32nd from the 31st generation in lines 39 and 49 had already been found in other 31st generation individuals. Perhaps the most important finding is that the number of epimutations does not increase linearly with time, indicating that many are not stably inherited over the long term. However, a DMR that had become demethylated after 31 generations, was found re-methylated in the following generation. This suggests that DNA methylation in specific regions of the genome can fluctuate over relatively short timescales. Such sites can be considered as going through recurrent cycles of forward and reverse epimutation, which is very different from what is found at the level of the genome sequence, where reverse mutations are exceedingly rare.

RNA interference (RNAi) is an evolutionarily conserved mechanism that uses short antisense RNAs that are generated by ‘dicing’ (chopping) dsRNA precursors to target corresponding mRNAs for cleavage. Recent developments have revealed that there is also extensive involvement of RNAi-related processes in regulation at the genome level. Indeed, dsRNA and proteins of the RNAi machinery can direct epigenetic alterations to homologous DNA sequences to induce transcriptional gene silencing or, in extreme cases, DNA elimination. Furthermore, in some organisms RNAi silences unpaired DNA regions during meiosis. These mechanisms facilitate the directed silencing of specific genomic regions.
RNA interference (RNAi)

microRNA
I microRNA sono corte molecole di RNA non codificante di ~22 nucleotidi
sono regolatori chiave dell’espressione genica a livello post-trascrizionale
taglio dei trascritti bersaglio
repressione della traduzione
Deadenilazione dell’mRNA e sua degradazione
sono in genere altamente conservati
hanno ruoli fondamentali in molti processi biologici, tra cui:
Sviluppo
Differenziazione cellulare
Proliferazione cellulare
Apoptosi
Risposta agli stress
Oncogenesi
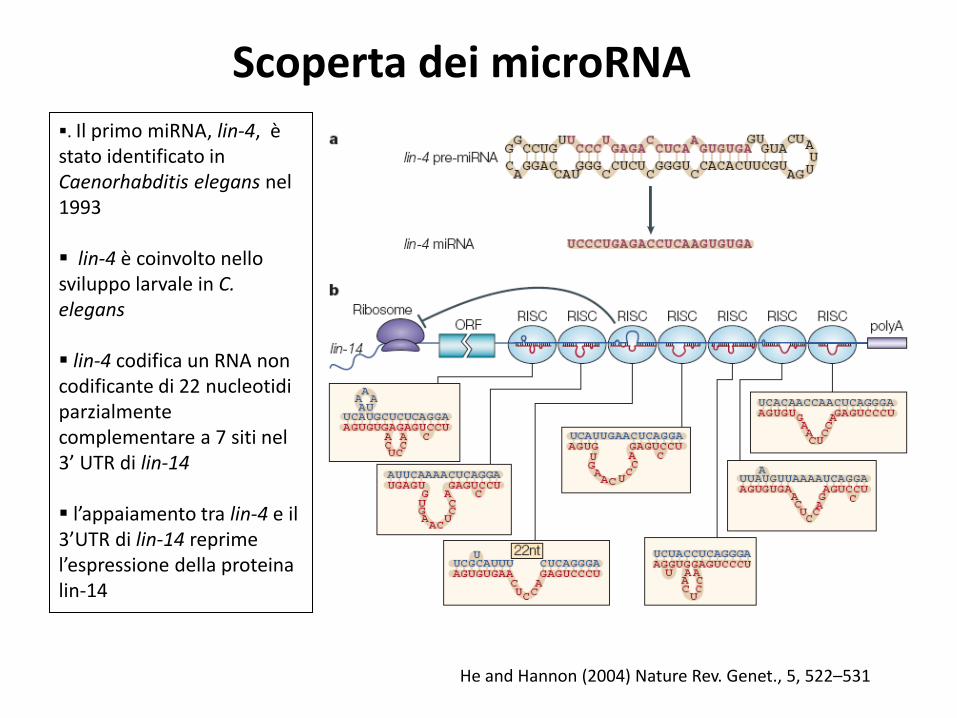
Scoperta dei microRNA
. Il primo miRNA, lin-4, è stato identificato in Caenorhabditis elegans nel 1993
lin-4 è coinvolto nello sviluppo larvale in C. elegans lin-4 codifica un RNA non codificante di 22 nucleotidi parzialmente complementare a 7 siti nel 3’ UTR di lin-14 l’appaiamento tra lin-4 e il 3’UTR di lin-14 reprime l’espressione della proteina lin-14
He and Hannon (2004) Nature Rev. Genet., 5, 522–531
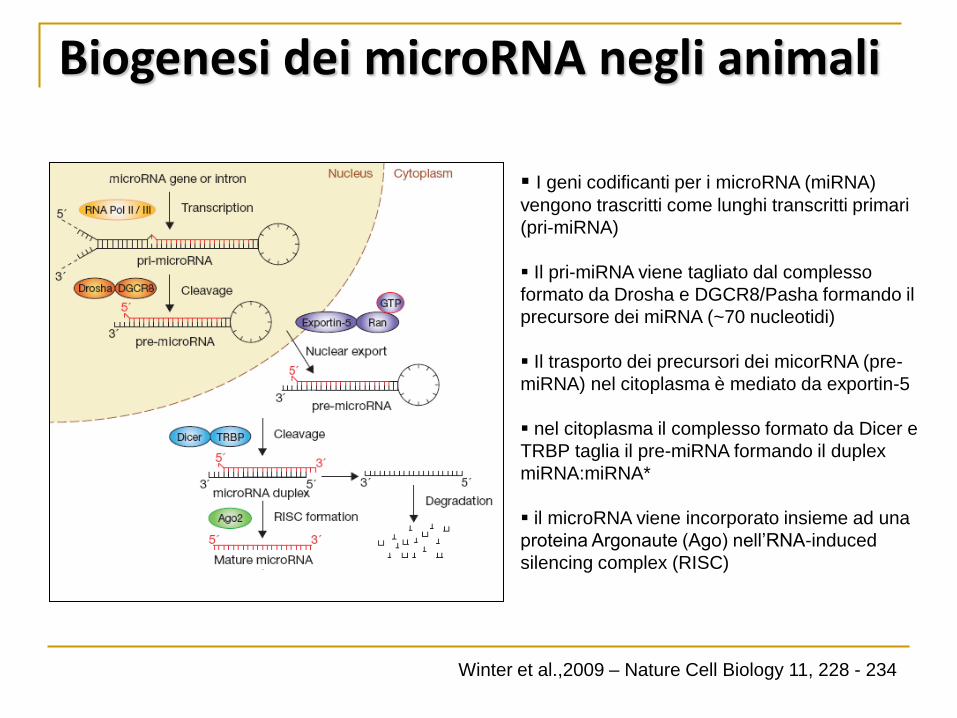
Biogenesi dei microRNA negli animali
I geni codificanti per i microRNA (miRNA)
vengono trascritti come lunghi transcritti primari
(pri-miRNA)
Il pri-miRNA viene tagliato dal complesso
formato da Drosha e DGCR8/Pasha formando il
precursore dei miRNA (~70 nucleotidi)
Il trasporto dei precursori dei micorRNA (pre-
miRNA) nel citoplasma è mediato da exportin-5
nel citoplasma il complesso formato da Dicer e
TRBP taglia il pre-miRNA formando il duplex
miRNA:miRNA*
il microRNA viene incorporato insieme ad una
proteina Argonaute (Ago) nell’RNA-induced
silencing complex (RISC)
Winter et al.,2009 – Nature Cell Biology 11, 228 - 234

Biogenesi dei microRNA nelle piante
• non sono stati individuati omologhi di Drosha e dei suoi cofattori nelle
piante
il processamento da pri-miRNA a pre-miRNA è mediato dalla proteina
nucleare Dicer-like 1 (DCL1) in associazione con HYPONASTIC
LEAVES 1 (HYL1) e SERRATE (SE).
DCL1 e HYL1 sono anche responsabili della formazione del duplex
miRNA:miRNA*
Hua Enhancer (HEN1) media la metilazione dei miRNA di pianta
Hasty media il trasporto del miRNA nel citoplasma
i miRNA:miRNA* duplex si associano alla proteina Ago
il miRNA* viene degradato mentre l’altro filamento rimane nel
complesso RISC
Wahid et al., 2010 – Biochimica et Biophysica Acta 1803 (11), 1231-1243
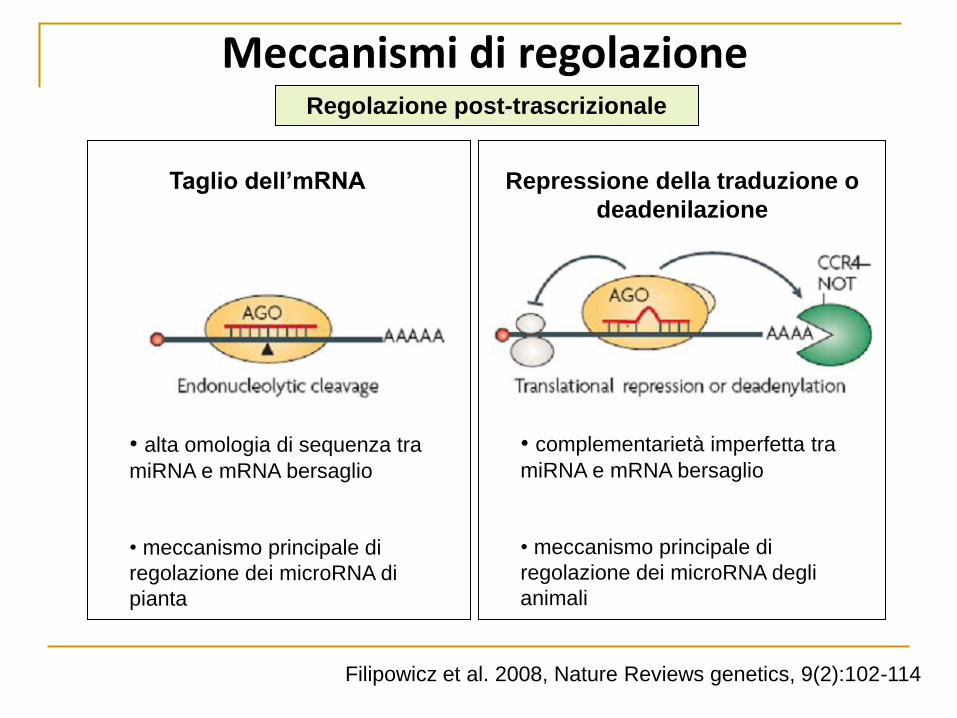
Meccanismi di regolazione Regolazione post-trascrizionale
Taglio dell’mRNA Repressione della traduzione o
deadenilazione
• alta omologia di sequenza tra
miRNA e mRNA bersaglio
• meccanismo principale di
regolazione dei microRNA di
pianta
• complementarietà imperfetta tra
miRNA e mRNA bersaglio
• meccanismo principale di
regolazione dei microRNA degli
animali
Filipowicz et al. 2008, Nature Reviews genetics, 9(2):102-114
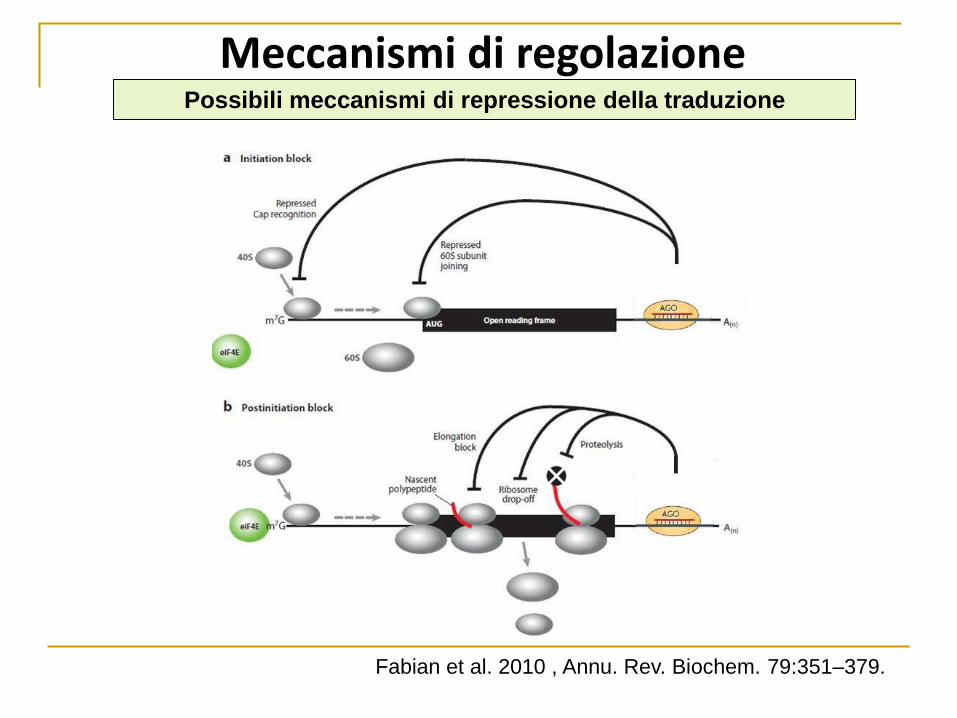
Meccanismi di regolazione Possibili meccanismi di repressione della traduzione
Fabian et al. 2010 , Annu. Rev. Biochem. 79:351–379.
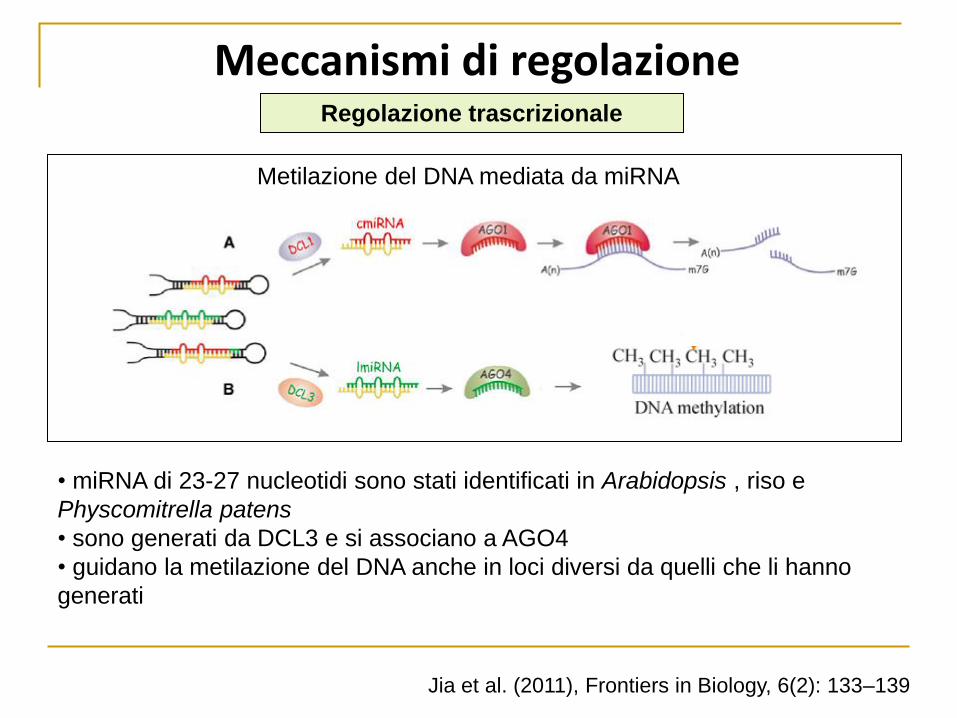
Meccanismi di regolazione
Metilazione del DNA mediata da miRNA
• miRNA di 23-27 nucleotidi sono stati identificati in Arabidopsis , riso e
Physcomitrella patens
• sono generati da DCL3 e si associano a AGO4
• guidano la metilazione del DNA anche in loci diversi da quelli che li hanno
generati
Jia et al. (2011), Frontiers in Biology, 6(2): 133–139
Regolazione trascrizionale
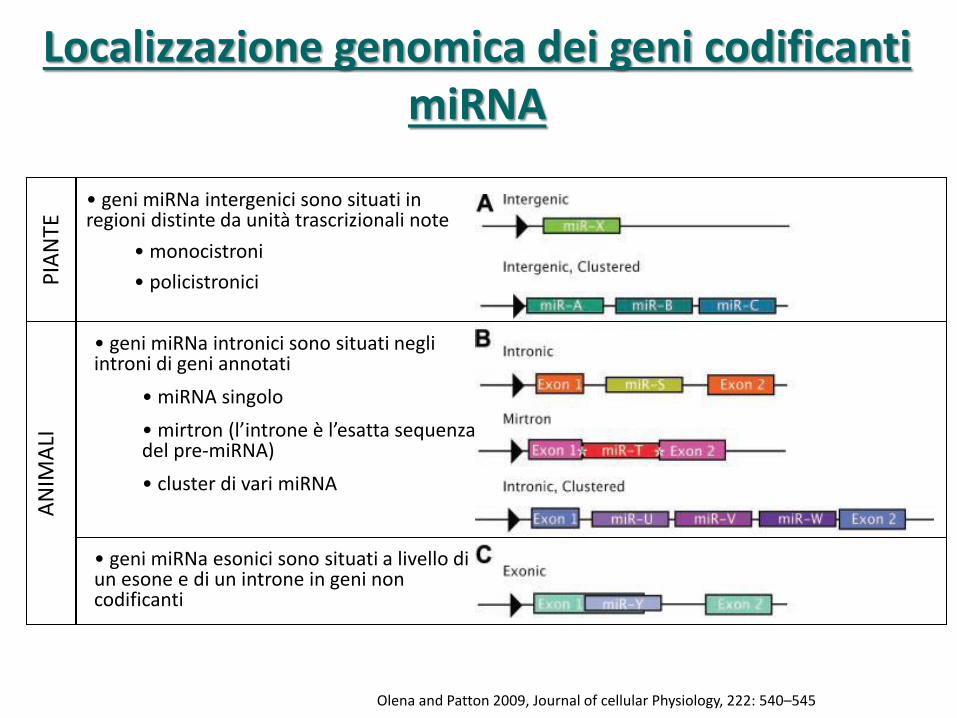
Olena and Patton 2009, Journal of cellular Physiology, 222: 540–545
• geni miRNa intergenici sono situati in regioni distinte da unità trascrizionali note
• monocistroni
• policistronici
• geni miRNa intronici sono situati negli introni di geni annotati
• miRNA singolo
• mirtron (l’introne è l’esatta sequenza del pre-miRNA)
• cluster di vari miRNA
• geni miRNa esonici sono situati a livello di un esone e di un introne in geni non codificanti
PIA
NTE
A
NIM
ALI
Localizzazione genomica dei geni codificanti
miRNA

Analisi del profilo di espressione dei miRNA: problematiche
I miRNA maturi sono molecole corte; miRNAs presentano un contento in GC etereogeneo ampio range di Tm; I microRNA maturi non hanno il poly(A); la sequenza target è presente anche nel trascritto primario(primiRNA) e nel precursore (pre-miRNA); miRNAs appartenenti alla stessa famiglia possono differire anche di un singolo nucleotide(e.g., la famiglia Let-7).

Tecniche di analisi del profilo di espressione dei miRNA
Real Time PCR
microarray
Sequenziamento massivo

Real time PCR
retrotrascrizione (RT):
Primer lineari Primer stem-loop
rilevamento del prodotto amplificato attraverso molecole fluorescenti:
SYBR Green I Sonde Taqman
Vantaggi: alta sensibilità e specificità richiede basse quantità di materiale Svantaggi: la dimensione limitata e l’etereogeneo contenuto in GC dei microRNA ostacola il disegno dei primer
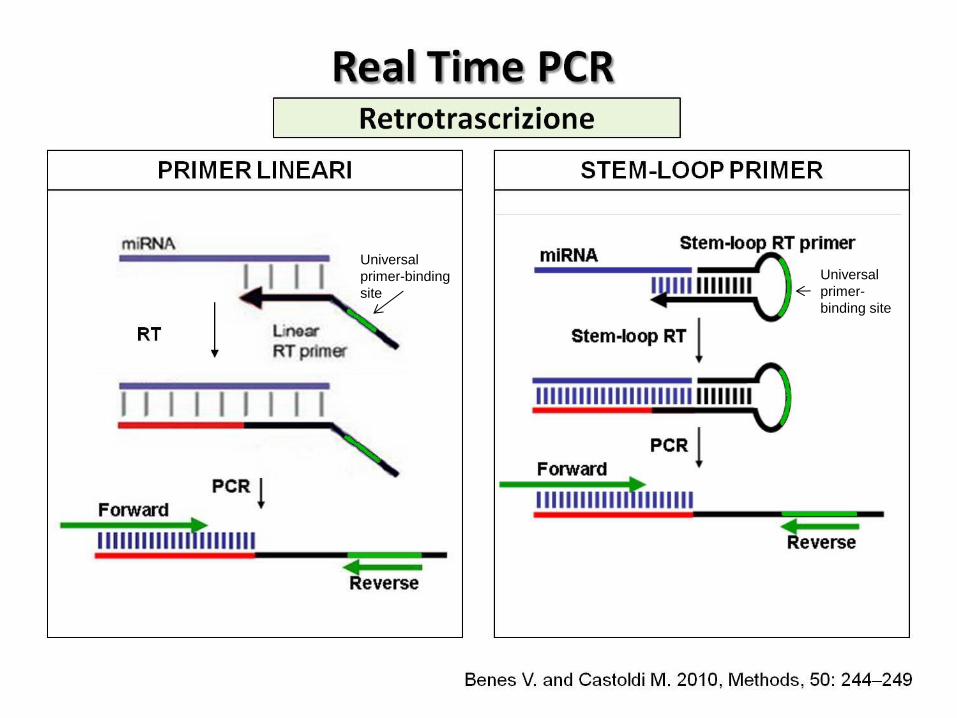
Universal
primer-binding
site
Universal
primer-
binding site
Retrotrascrizione
STEM-LOOP PRIMER PRIMER LINEARI
Benes V. and Castoldi M. 2010, Methods, 50: 244–249
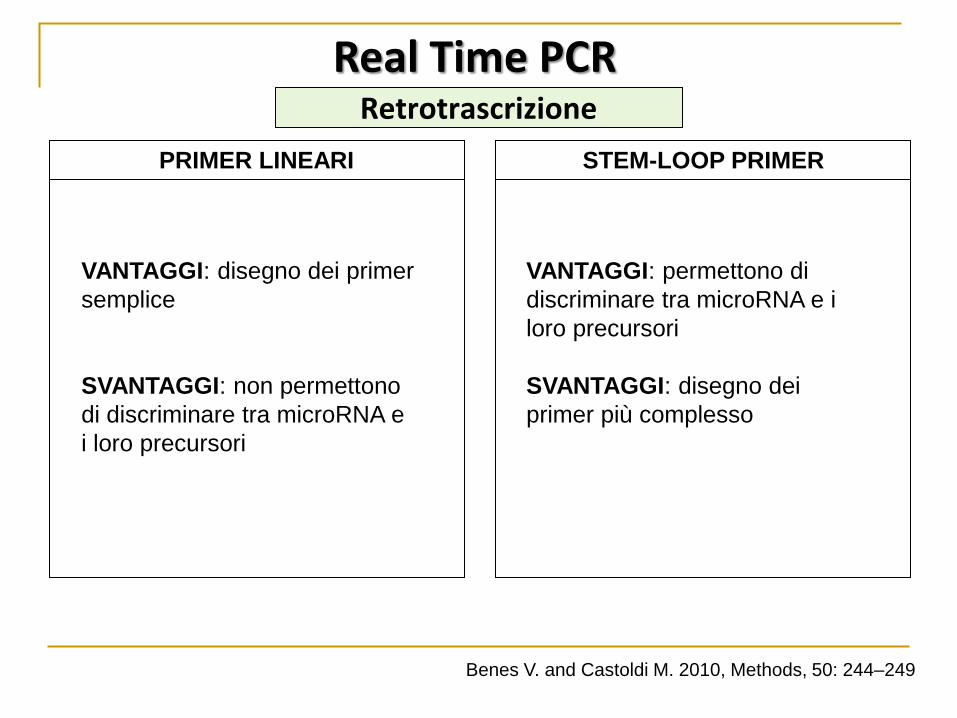
Real Time PCR
VANTAGGI: disegno dei primer
semplice
SVANTAGGI: non permettono
di discriminare tra microRNA e
i loro precursori
VANTAGGI: permettono di
discriminare tra microRNA e i
loro precursori
SVANTAGGI: disegno dei
primer più complesso
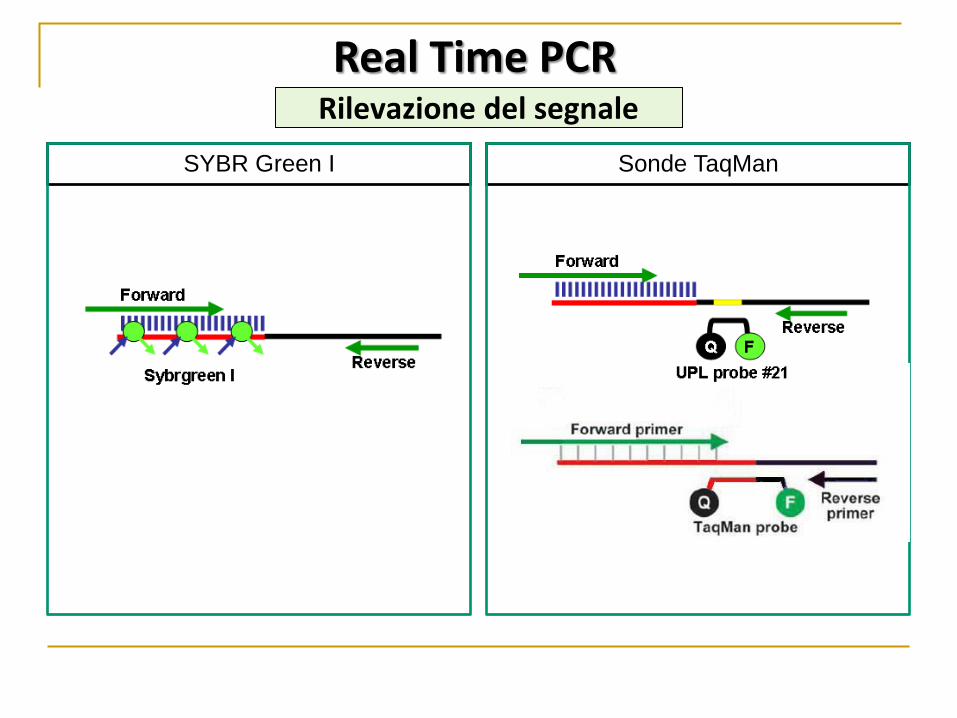
SYBR Green I Sonde TaqMan
Real Time PCR Rilevazione del segnale

SYBR Green I Sonde TaqMan
Real Time PCR Rilevazione del segnale
non permette di discriminare tra
differenti prodotti di PCR in quanto
si lega a tutti i dsDNA presenti
nella reazione, inclusi prodotti non
specifici come dimeri di primer.
Maggiore specificità: anche se
nella reazione sono presenti
dimeri di primer o altri prodotti di
amplificazione non specifici,
questi non genereranno
fluorescenza.
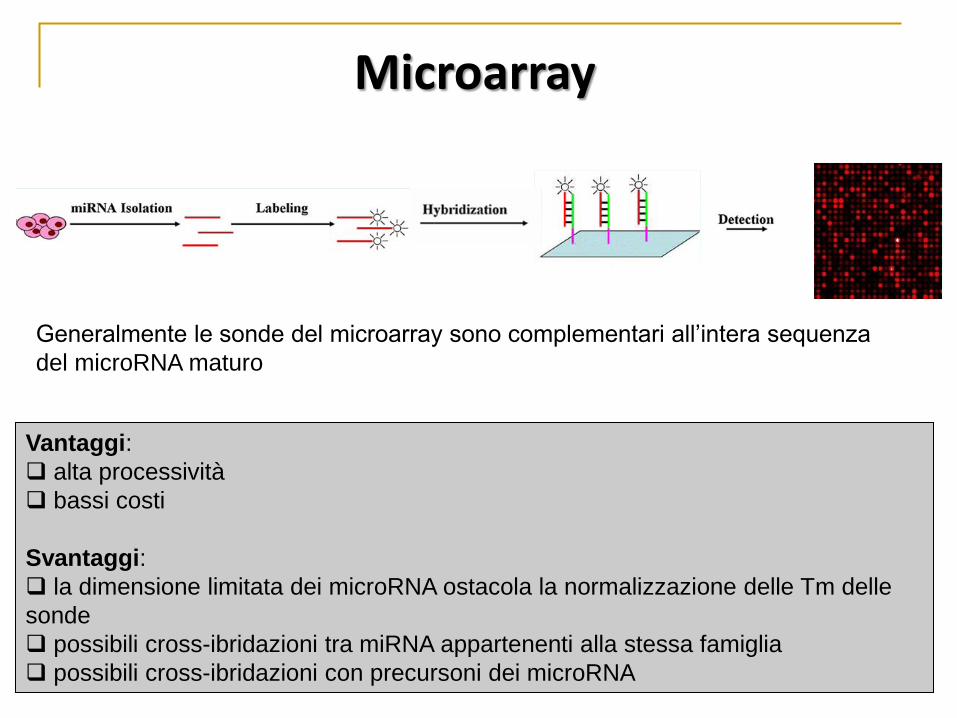
Vantaggi:
alta processività
bassi costi
Svantaggi:
la dimensione limitata dei microRNA ostacola la normalizzazione delle Tm delle
sonde
possibili cross-ibridazioni tra miRNA appartenenti alla stessa famiglia
possibili cross-ibridazioni con precursoni dei microRNA
Generalmente le sonde del microarray sono complementari all’intera sequenza
del microRNA maturo
Microarray
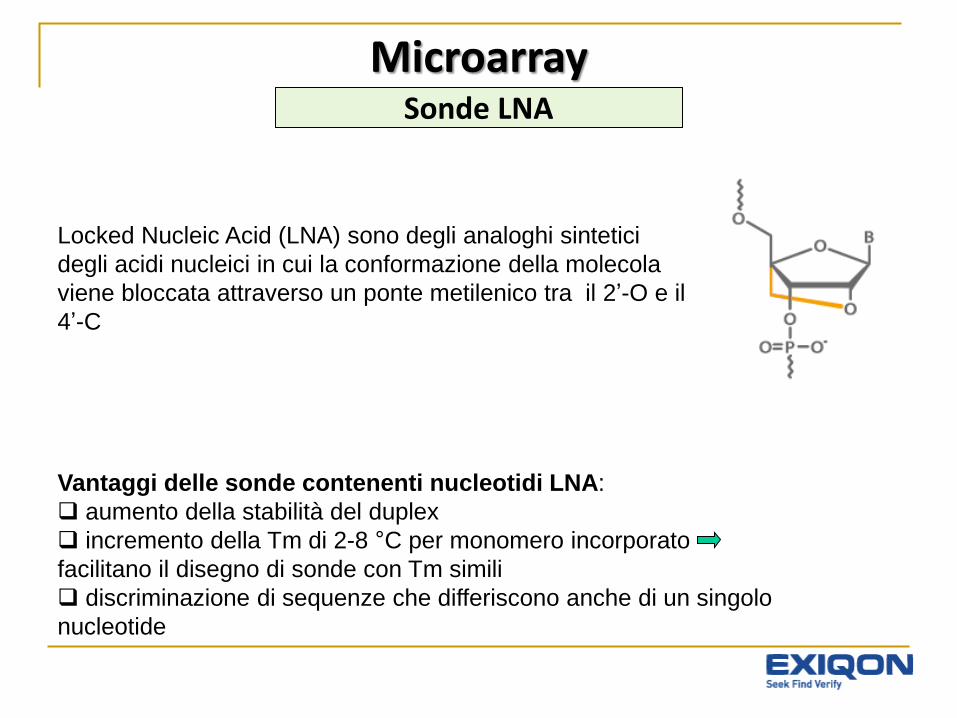
Vantaggi delle sonde contenenti nucleotidi LNA:
aumento della stabilità del duplex
incremento della Tm di 2-8 °C per monomero incorporato
facilitano il disegno di sonde con Tm simili
discriminazione di sequenze che differiscono anche di un singolo
nucleotide
Locked Nucleic Acid (LNA) sono degli analoghi sintetici
degli acidi nucleici in cui la conformazione della molecola
viene bloccata attraverso un ponte metilenico tra il 2’-O e il
4’-C
Microarray Sonde LNA
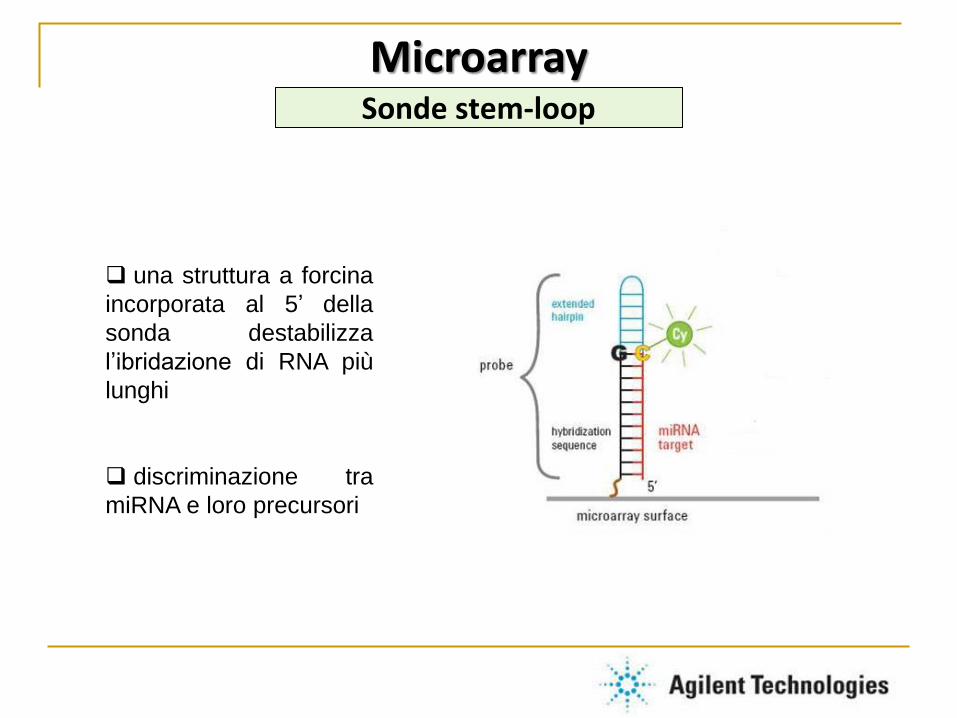
una struttura a forcina
incorporata al 5’ della
sonda destabilizza
l’ibridazione di RNA più
lunghi
discriminazione tra
miRNA e loro precursori
Microarray Sonde stem-loop
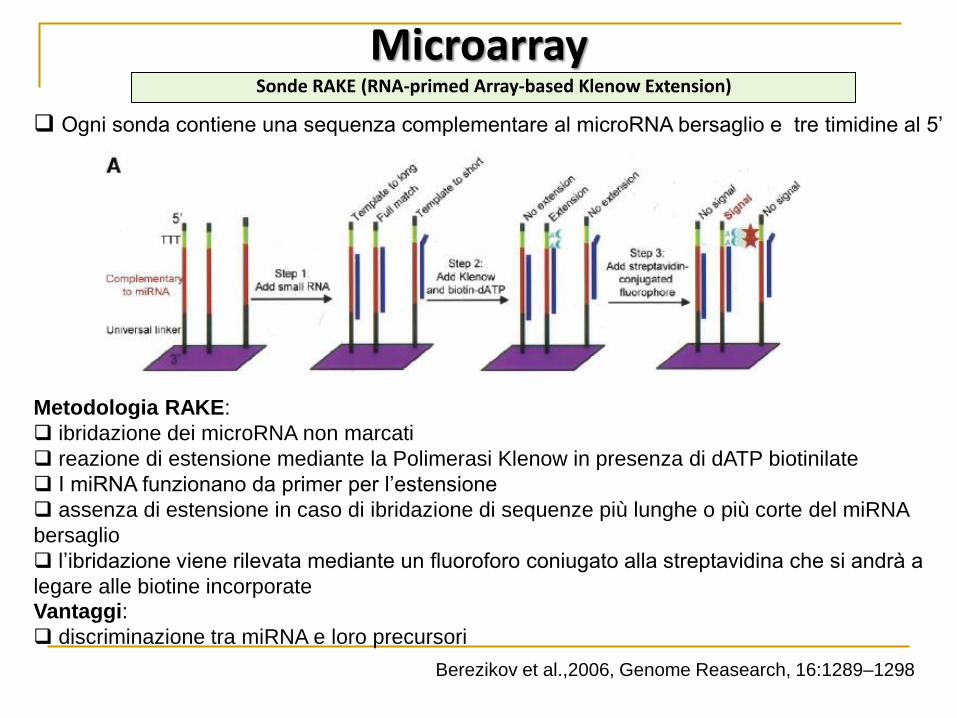
Vantaggi:
discriminazione tra miRNA e loro precursori
Berezikov et al.,2006, Genome Reasearch, 16:1289–1298
Metodologia RAKE:
ibridazione dei microRNA non marcati
reazione di estensione mediante la Polimerasi Klenow in presenza di dATP biotinilate
I miRNA funzionano da primer per l’estensione
assenza di estensione in caso di ibridazione di sequenze più lunghe o più corte del miRNA
bersaglio
l’ibridazione viene rilevata mediante un fluoroforo coniugato alla streptavidina che si andrà a
legare alle biotine incorporate
Ogni sonda contiene una sequenza complementare al microRNA bersaglio e tre timidine al 5’
Microarray Sonde RAKE (RNA-primed Array-based Klenow Extension)
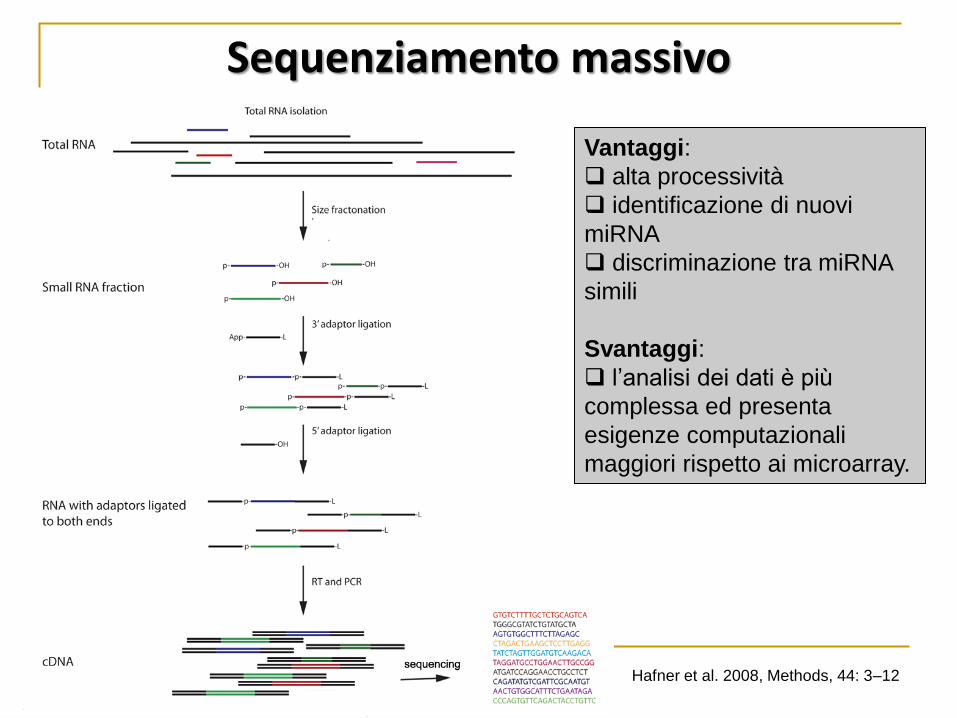
Vantaggi:
alta processività
identificazione di nuovi
miRNA
discriminazione tra miRNA
simili
Svantaggi:
l’analisi dei dati è più
complessa ed presenta
esigenze computazionali
maggiori rispetto ai microarray.
Hafner et al. 2008, Methods, 44: 3–12
Sequenziamento massivo

sequenze
Allineamento con i precursori
presenti in miRBase (banca dati
pubblica di microRNA)
Le sequenze che non allineano con
miRNA noti e che mappano su regioni
del genoma che possono formare
strutture a forcina possono
rappresentare nuovi miRNA
Sequenziamento massivo Analisi di espressione e identificazione di nuovi miRNA