enfermedades neuromusculares - mic · neurológicas •neurona motora superior –lesiones con...
TRANSCRIPT

Enfermedades NeuromuscularesDr. René Gutiérrez Jiménez
Asesor: Dr. Aurelio Martínez Lozano

Miller: Miller's Anesthesia, 6th ed. 2005
Condiciones que afectan algún
componente de la unidad motora:
neurona motora nervios periféricos
placa neural, músculos

Neurológicas
• Neurona motora superior
– Lesiones con parálisis espástica con o sin atrofia
– Esclerosis lateral primaria, traumatismos, enfermedades neurovasculares
• Neurona motora inferior
– Lesiones con parálisis flácida
– Atrofia muscular espinal
– Poliomielitis

Neurológicas
• Nervio periférico
– Lesiones inclusive en raíces nerviosas
– Neuropatía diabética
– Hernia de disco
– Trauma
– Compresión
• Unión mioneural
– Defecto en receptores o neurotransmisores
– Miastenia gravis

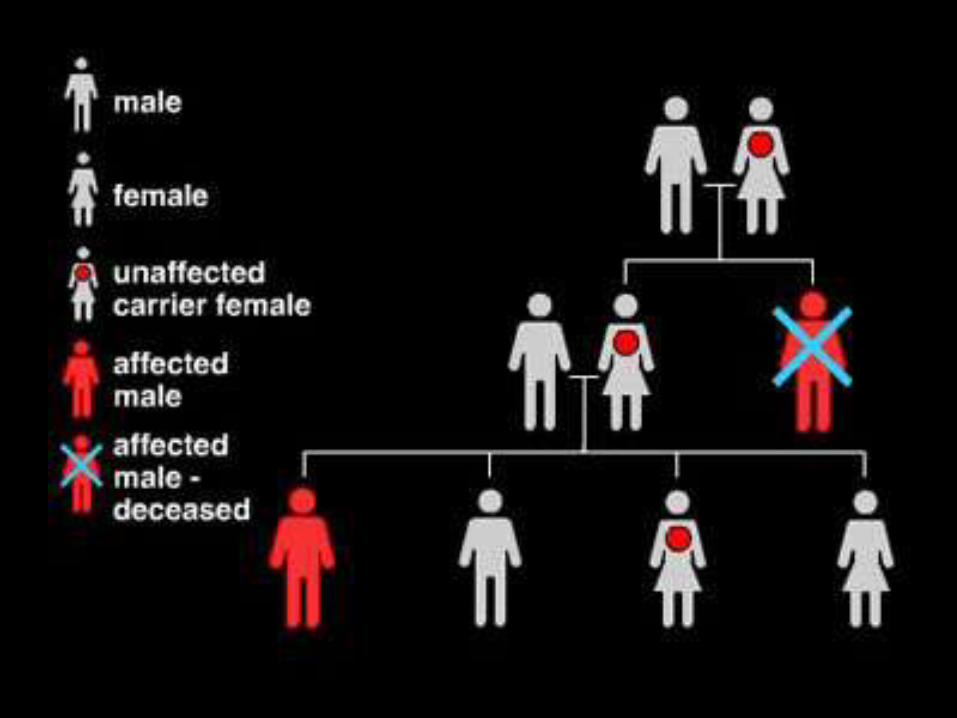
Distrofias musculares
Tachdjian's Pediatric Orthopaedics: by John A. Herring
Grupo de enfermedades determinadas
geneticamente progresivas del sistema
musculo esquelético
Cambios patológicos en las fibras
musculares sin anormalidades en la
inervación del mismo y en los nervios
periféricos

Características esenciales
• Miopatía definida clínicamente sin lesion
en nervios o sentidos
• Afectan toda la musculatura
• Es progresiva
• Cambios histológicos incluyen
degeneración muscular
• Todas son hereditarias

Historia
Edward Meryon (1852)
• Cuatro casos de atrofia progresiva y debilidad
muscular
Guillaume-Benjamin-Amand Duchenne (1868) • Describe la parálisis muscular mieloesclerótica
– Debilidad progresiva
– Afección únicamente muscular

Sir William Richard Gowers (1879)
• Signo de Gowers

Clasificación de Walton
• Distrofias musculares puras.
• Recesiva ligada al x.
– Distrofia muscular de Duchenne
– Distrofia muscular de Becker
• Autonómica recesiva:
– Cintura escapular y pélvica
– Fascioescapular infantil
• Autosómica dominante:
– Congénita
– Fascioescapulohumeral.
– Escapuloperonea.
– Distal.
– Ocular.
– Distrofias musculares con miotonía

Exploración física
• Historia clínica (enfermedad progresiva)
• Posición, marcha
• Inspección de piel, facies y ojos
• Fuerza muscular
• Evaluación neurológica

Auxiliares diagnósticos
• Laboratorio
– CPK
– Distrofina (Duchenne)
• Electromiografía
• Estudios de neuroconducción
• Biopsia
• Resonancia Magnetica
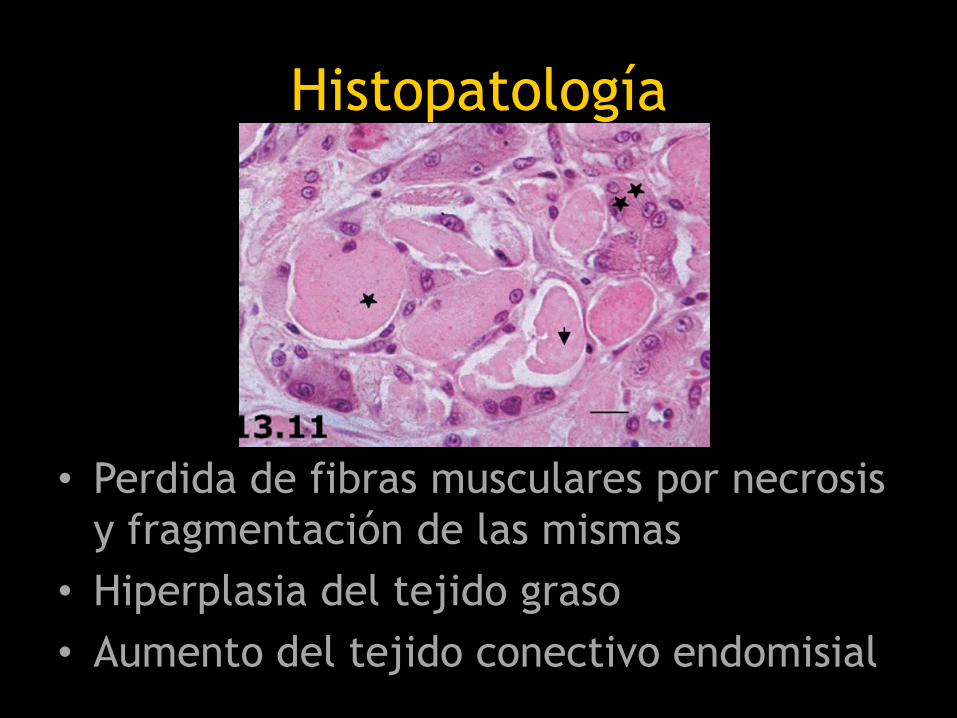
Histopatología
• Perdida de fibras musculares por necrosis
y fragmentación de las mismas
• Hiperplasia del tejido graso
• Aumento del tejido conectivo endomisial

Distrofia muscular de Duchenne
• Distrofia muscular mas común
• 2 a 4 años de edad
• Ausencia de distrofina
• 1 - 3,500 niños
• Hombres
• Síndrome de Turner gen X0
• Translocaciones

Etiología
• Mutación del gen xp21 del cromosoma X
• La distrofina es necesaria para la
estabilidad del citoesqueleto
• Esto causa degeneración muscular
progresiva y perdida funcional

Etiología de la degeneración
muscular
• Mecánica
• Calcio
• Vascular
• Regulación genética
• Glucolisación
• Inflamatoria

Cuadro clínico
• Manifestación clínica de los 3 a los 6 anos
• Signo de Gower (15 meses)
• Dificultad en la marcha (de puntas)
• Pie equino
• Extensores de la cadera
• Realineación del cuerpo
• Aumento de lordosis
• Recurvatum
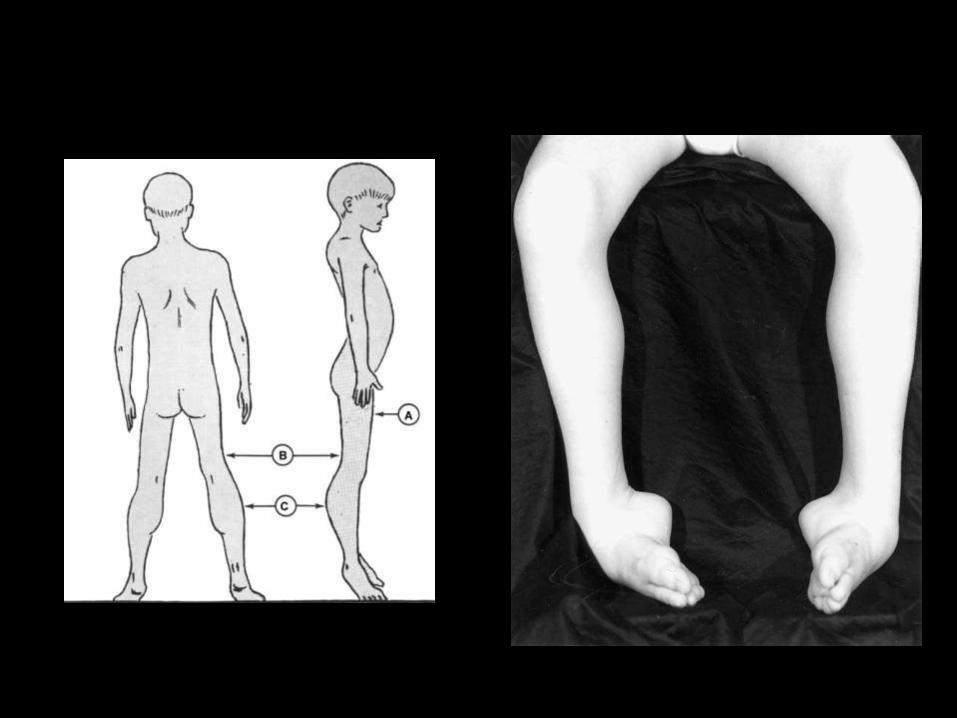

Cuadro clínico
• Seudohipertrofia
• Afección simétrica
• La debilidad tiene distribución proximal– Glúteo mayor– Abductores– Psoas ilíaco. Músculos del abdomen– Por ultimo músculos intercostales
• Signo de Meryon

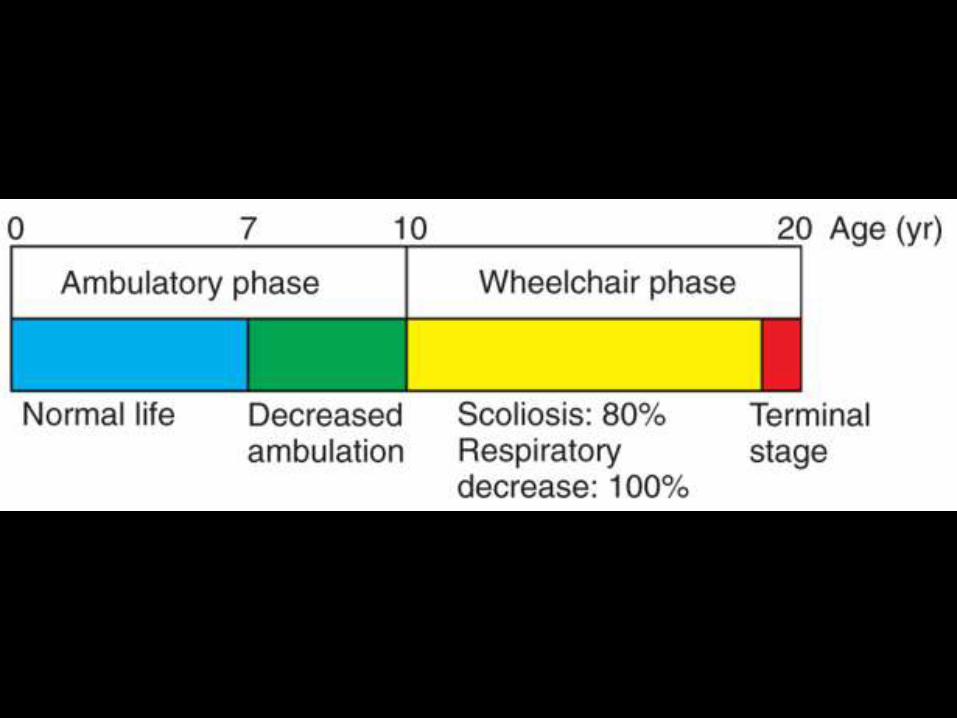
Cuadro clínico
• Trastornos de personalidad.
• Deterioro intelectual.
• Para los 10-15 años el niño tiende a estar en silla de ruedas.

Tratamiento médico
• Terapia física
• Apoyo psicológico y nutricional
• Prednisona y Deflazacort

• Prolonga la deambulación por 2 años
• Retrasa el avance de la capacidad vital y
postpone la ventilación mecánica
• Mejora la función cardiaca, disminuye la
escoliosis y la cirugía de columna

Efectos secundarios
• Obesidad
• Hiperglucemia
• Hipertensión
• Osteoporosis
• Fracturas vertebrales
• Cataratas (5ª)

Intervenciones ortopédicas en el
manejo de la distrofia de
Duchenne
Duchenne Muscular Dystrophy : Advances in Therapeutics
Neurological Disease and Therapy ; V. 79Author: Chamberlain, Jeffrey S.; Rando, Thomas A. Publication: New York Informa Healthcare, 2006.

Concepto
Los principios de la rehabilitación
ortopédica en la distrofia muscular de
Duchenne se basa en el entendimiento de
la evolución natural de la misma según
los patrones de debilidad contractura y
deformidad y estadificar el tratamiento
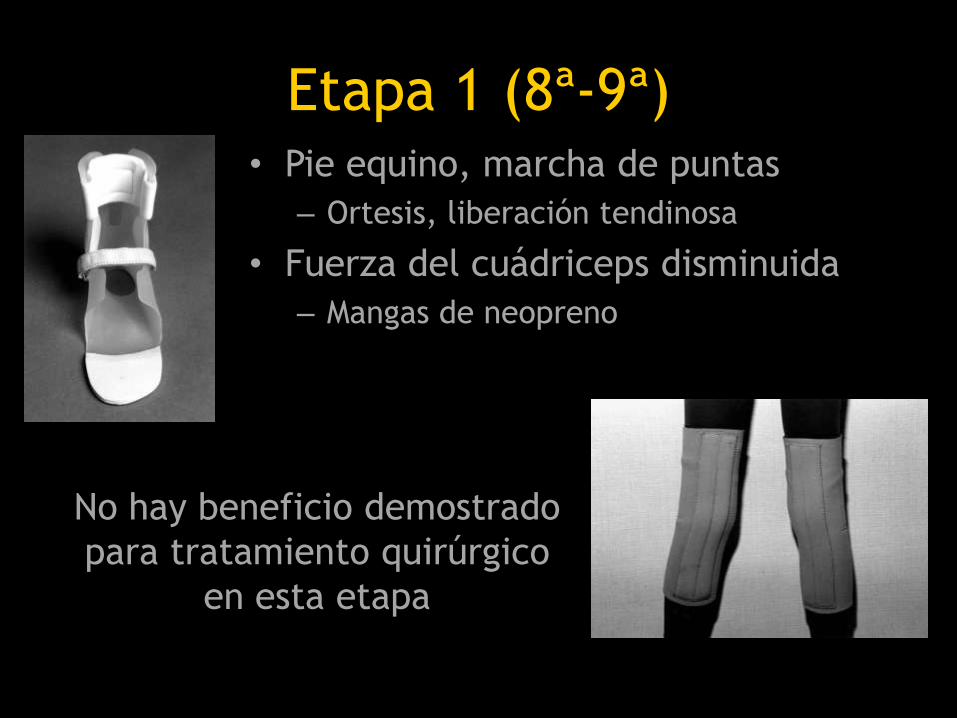
Etapa 1 (8ª-9ª)• Pie equino, marcha de puntas
– Ortesis, liberación tendinosa
• Fuerza del cuádriceps disminuida
– Mangas de neopreno
No hay beneficio demostrado
para tratamiento quirúrgico
en esta etapa

Fracturas
• Pacientes con densidad mineral baja
• Fémur 50%, tibia 25%
• Tratamiento conservador
• Posición funcional

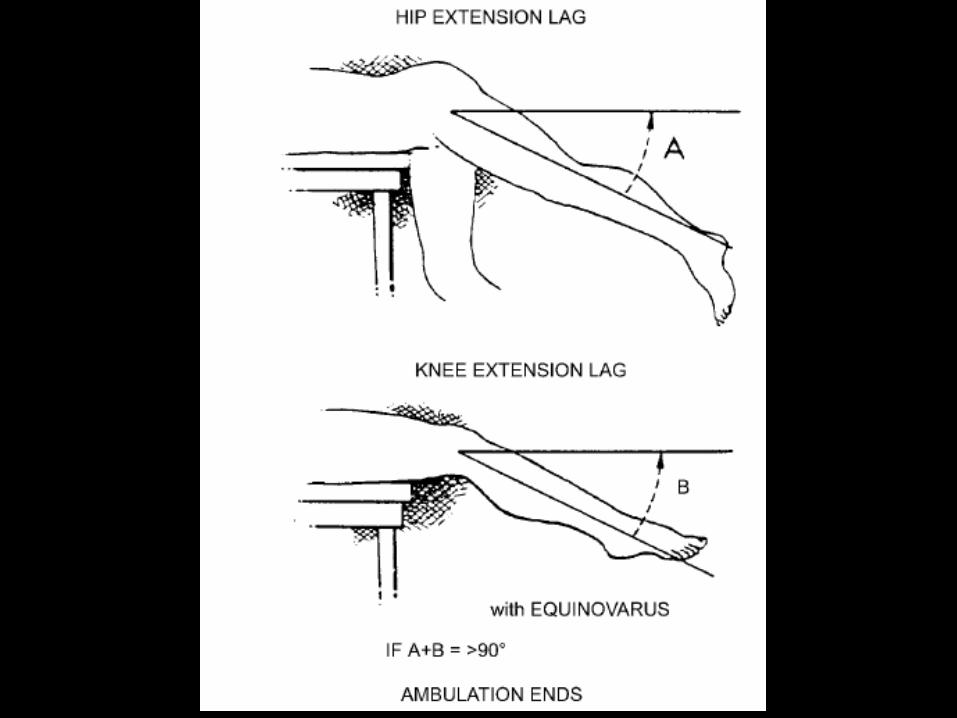
Etapa 2 (9ª-12ª)
• Pacientes dejan de caminar
• Se relaciona con la posición del paciente
• Evaluación de la extensión de cadera y
rodilla
• Tratamiento con ortesis
• Tratamiento quirúrgico

Tratamiento Medico
• Realizar ejercicios de estiramiento
• La inactividad es perjudicial
• El reposo duradero en cama ocasionará
perdida importante de la potencia
muscular.
• Control de peso.
• Apoyo psicológico (motivar a caminar).

Ortesis
– Deben utilizarse al final de la ambulación independiente.
– Ortesis suprarotulianas: • se usarán antes de que el niño quede limitado a
la silla de ruedas.
• Amplian el periodo de ambulación.
• Mejoría funcional.
• Evitar la formación de contracturas al deambular.
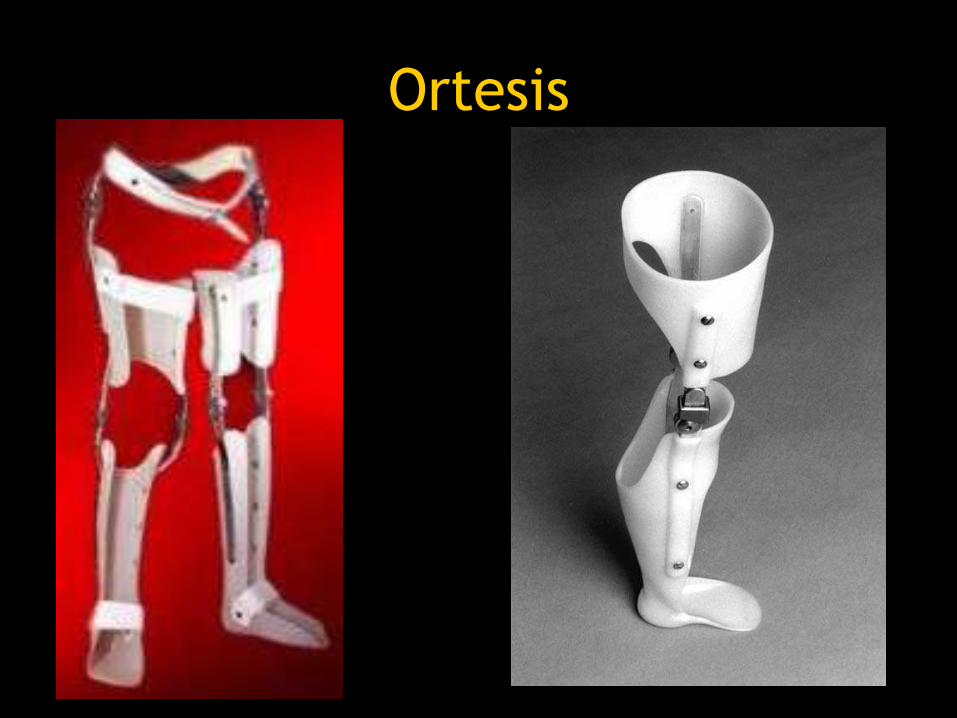
Ortesis

Clasificación de Shapiro y Specht
• Abordaje temprano extensivo
ambulatorio
• Abordaje ambulatorio moderado
• Abordaje ambulatorio mínimo
• Abordaje de rehabilitación
• Abordaje paliativo
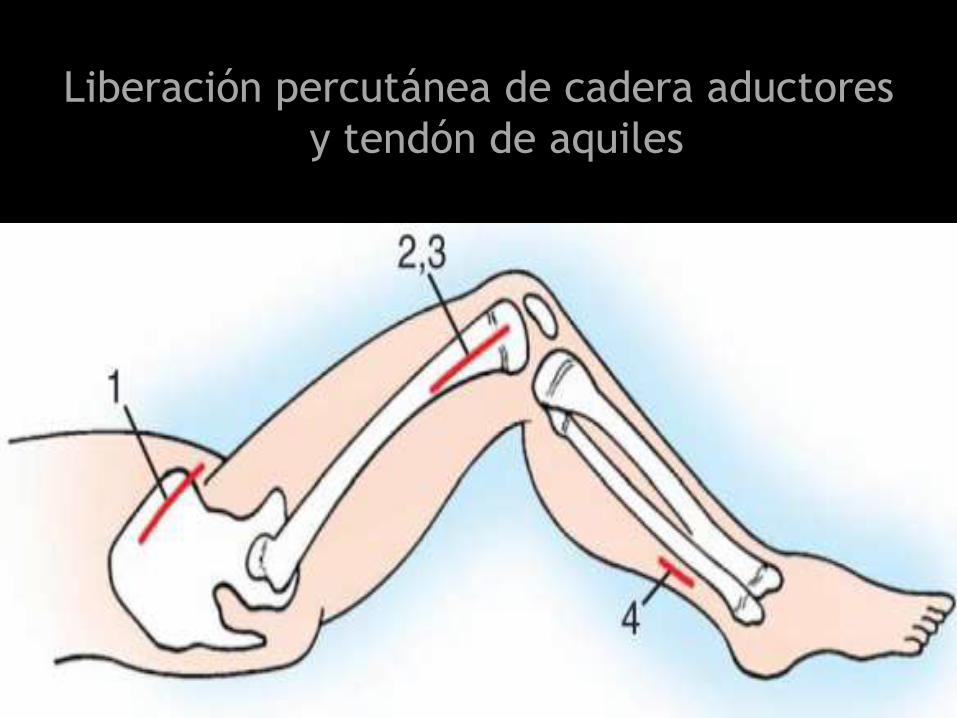
Liberación percutánea de cadera aductores
y tendón de aquiles
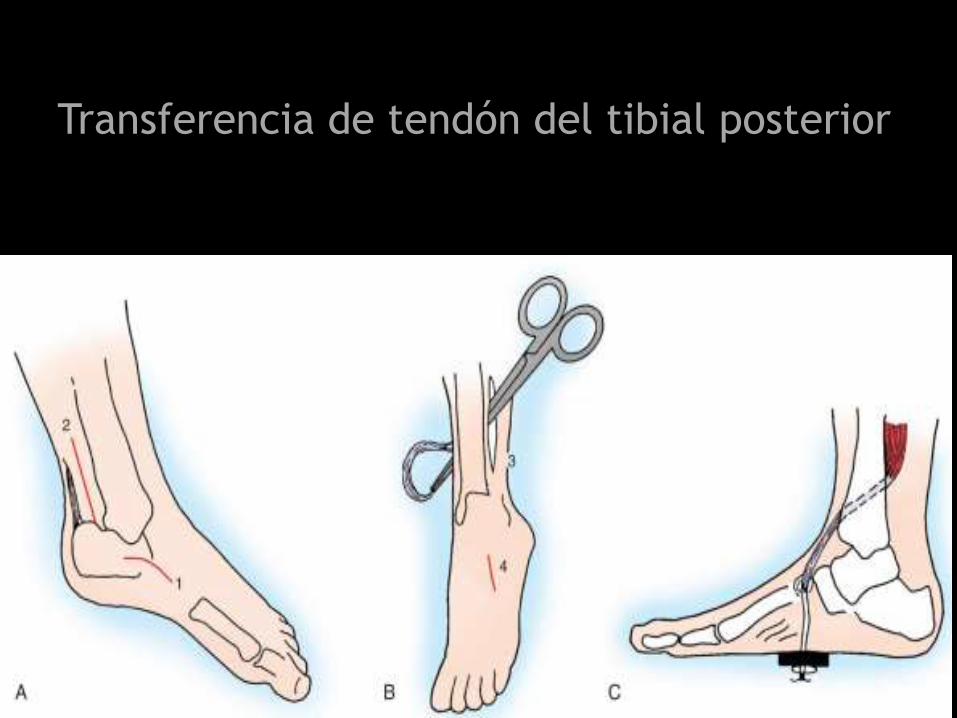
Transferencia de tendón del tibial posterior
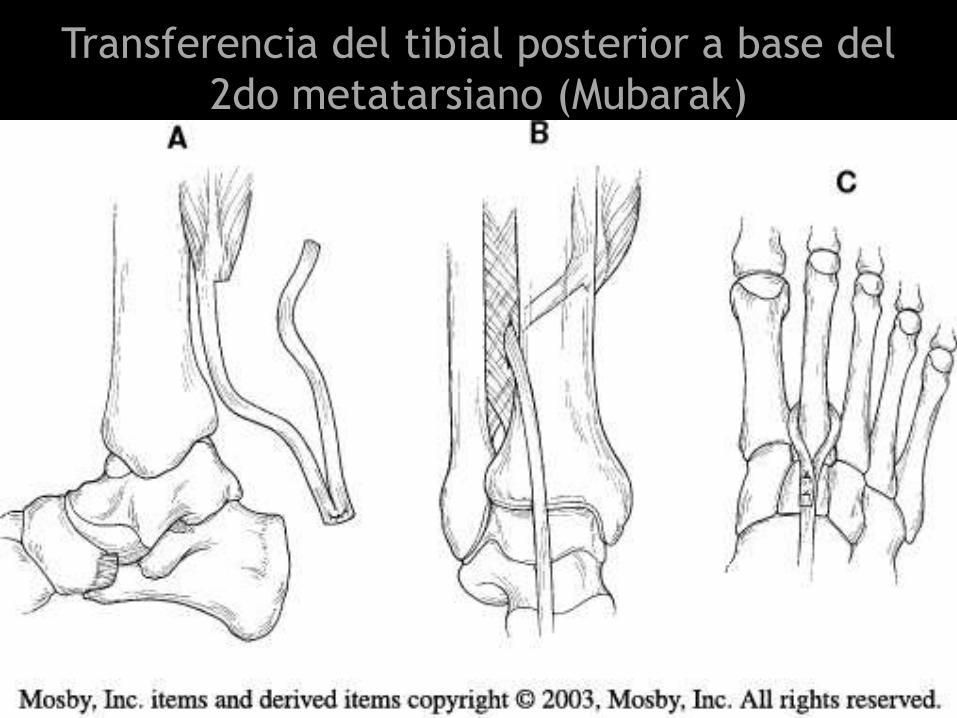
Transferencia del tibial posterior a base del
2do metatarsiano (Mubarak)

Etapa 3 (no ambulatorio)
• Liberación tendinosa no recomendada
• Pie equino varo severo
– Osteotomía del tarso
• Pacientes que no pueden usar zapatos
– Tendotomías
• Ortesis
• Compresiones neuropatías

Silla de ruedas
– Se utiliza cuando el niño no puede seguir con la ambulación independiente.
– Debe contar con respaldo para evitar la hiperextensión del cuello.
– Contar con apoyo adecuado para evitar escoliosis.
Síndrome piriforme

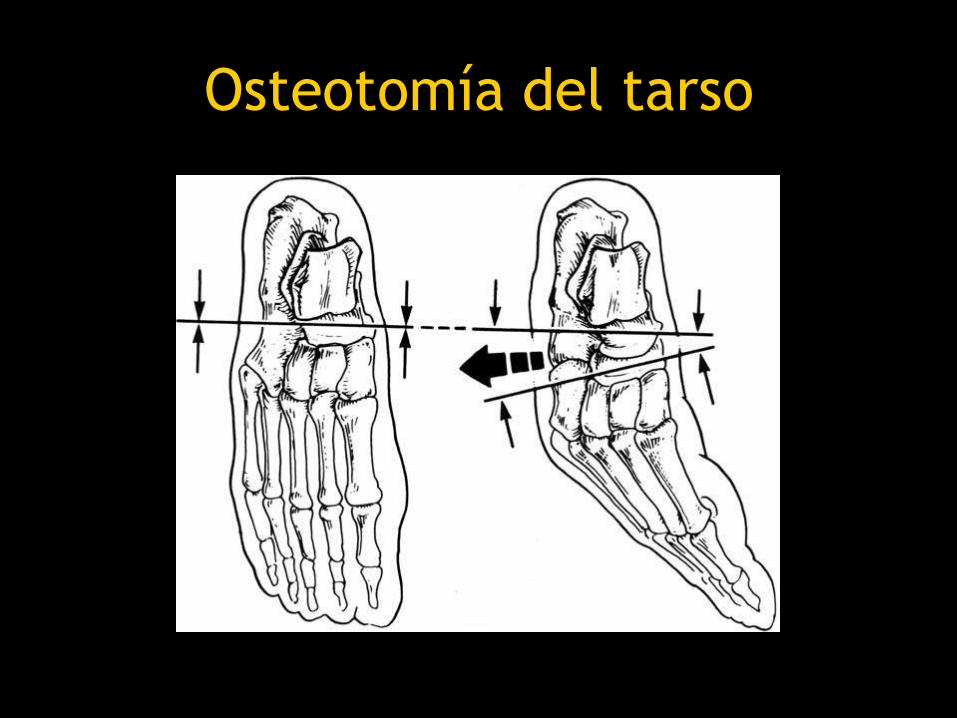
Osteotomía del tarso
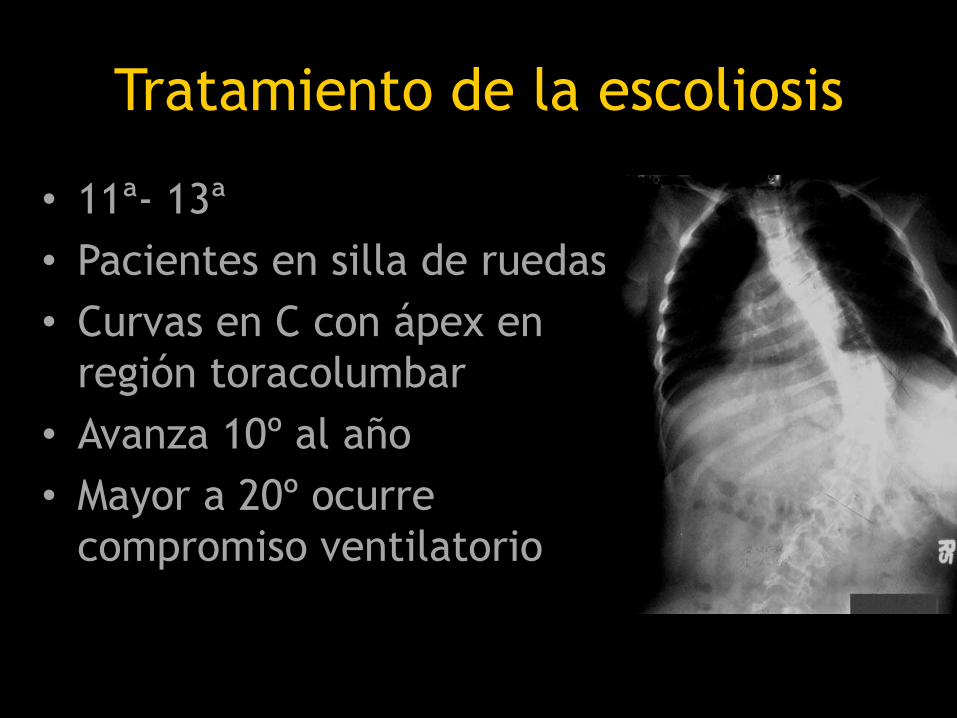
Tratamiento de la escoliosis
• 11ª- 13ª
• Pacientes en silla de ruedas
• Curvas en C con ápex en
región toracolumbar
• Avanza 10º al año
• Mayor a 20º ocurre
compromiso ventilatorio

Tratamiento de la escoliosis
• Entre mas se retrase el tratamiento
mayor el compromiso pulmonar
• Capacidad vital -30% = intubación
• Indicaciones quirúrgicas
– Capacidad vital mayor a 30%
– Corrección mayor a 30º

Tratamiento de la escoliosis
• Fusión posterior
• Nivel de artrodesis se discute
• Nivel superior
– Una arriba de la vertebra inicial
• Nivel inferior
– L5 si la oblicuidad pélvica es menor a 15º
• Influyen en el paciente psicológicamente

Distrofia muscular de Becker
• Ligado a X
• Aparición tardía (7a)
• Prevalencia de 2.3 en 100,000
• Desarrollo de la enfermedad mas lento
• Pacientes pueden caminar a veces hasta
los 40-50 años
• Cardiopatías

Emery-Dreifuss
• Ligada a X (xq28)
• Manifestaciones clínicas a los 5a-15a
• Progresión lenta
• Contractura codos y tobillos a edad temprana
• Cardiomiopatías como causa de muerte
• Ausencia de parálisis facial y miotonías
• Tratamiento sintomático


Distrofia de los músculos de la
cintura escapular y pélvica
• Autosómico recesivo
• Niveles de distrofina normales
• Síntomas iniciales afectan la cintura
pélvica y escapular
• Cualquier sexo

Fascioescapular infantil
• Rápido avance
• Lordosis lumbar progresiva
• Segunda década dejan de caminar
• Perdida de la audición
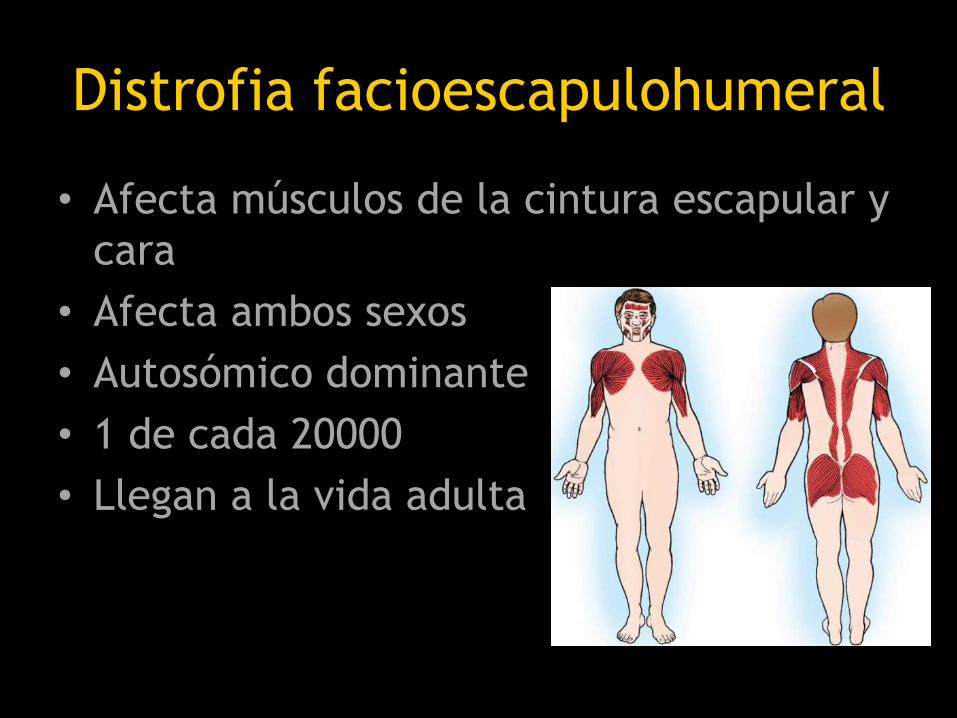
Distrofia facioescapulohumeral
• Afecta músculos de la cintura escapular y
cara
• Afecta ambos sexos
• Autosómico dominante
• 1 de cada 20000
• Llegan a la vida adulta

Miotonía de Thomsen
Estado de retraso de la relajación de los
músculos después de contracción voluntaria
o estimulación mecánica o eléctrica
Debilidad muscular que se presenta en la
primera y segunda década de la vida
Goetz: Textbook of Clinical Neurology, 3rd ed 2007 Elsevier

Miotonía de Thomsen
• Neonatal y en la infancia
• Autosómico dominante
• Sin preferencia de genero
• Miotonía general
• Dificultad para la deambulación
• Hipertrofia presente
• Sin afección endocrina o cardiaca

Miotonía de Thomsen
Orthopaedic Manifestations of Congenital Myotonic Dystrophy During
childood and Adolescence Federico Canavese, MD and Michael D.
Sussman, MD J Pediatr Orthop & Volume 29, Number 2, March 2009
• Gran debilidad muscular
• Mejora en la primera década
• 1 en 100,000
• Cardiopatías en la segunda década
• No hay contractura en miembros
superiores

Miotonía de thomsen
• Tratamiento
– Sulfato de quinina, procainamida para
disminuir la miotonía
– Calentamiento previo a movilización
• Pronostico
– El cuadro permanece estático

Distrofia miotónica
• Enfermedad progresiva
• Adolescencia y adultez
• 1-80000
• Autosómico dominante

Distrofia miotónica
• Fascie miotónica
• Ptosis
• CK elevada
• Cambios histológicos de distrofia
• La musculatura mas afectada es la de la
cara, manos y lengua
• Asociado a retardo mental

Distrofia miotónica
• No existe tratamiento
• Al termino de 20 años los pacientes
muestran incapacidad grave y la muerte

Miositis osificante
• Calcificación heterotópica y del tejido
muscular
• Patogenesis importante lesion traumática
• Mas común en adolescentes
• Asociado a maltrato infantil

Miositis osificante
• Tipo 1
– Traumática única
• Tipo 2
– Traumática múltiple
• Tipo3
– Asociada a desordenes neurológicos

Miositis osificante
• Zona externa
• Zona periférica
• Zona adyacente
• Zona central
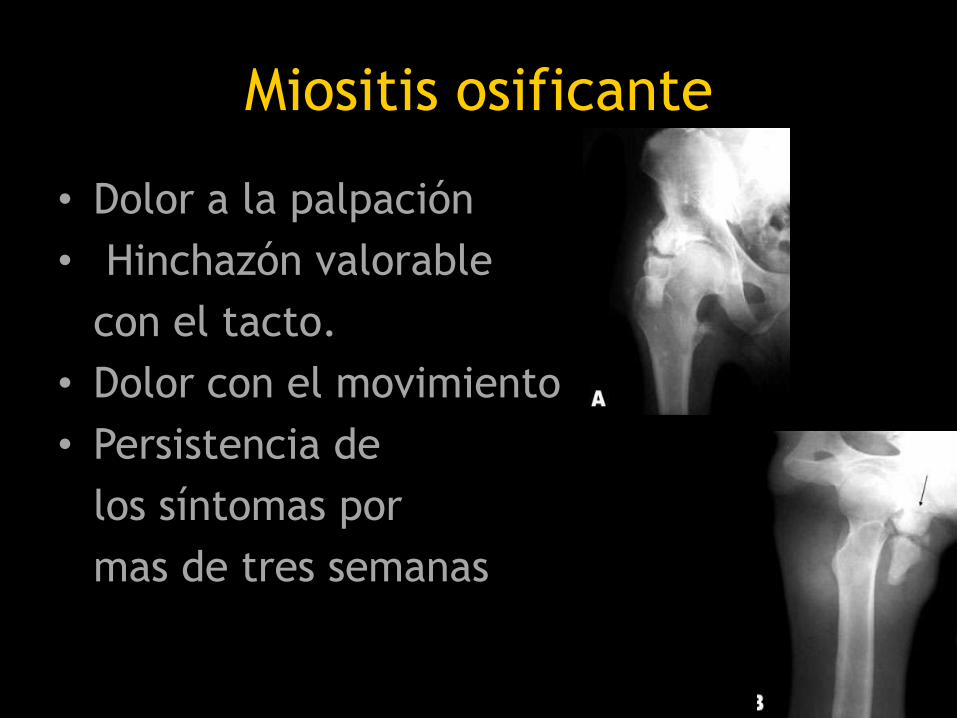
Miositis osificante
• Dolor a la palpación
• Hinchazón valorable
con el tacto.
• Dolor con el movimiento
• Persistencia de
los síntomas por
mas de tres semanas

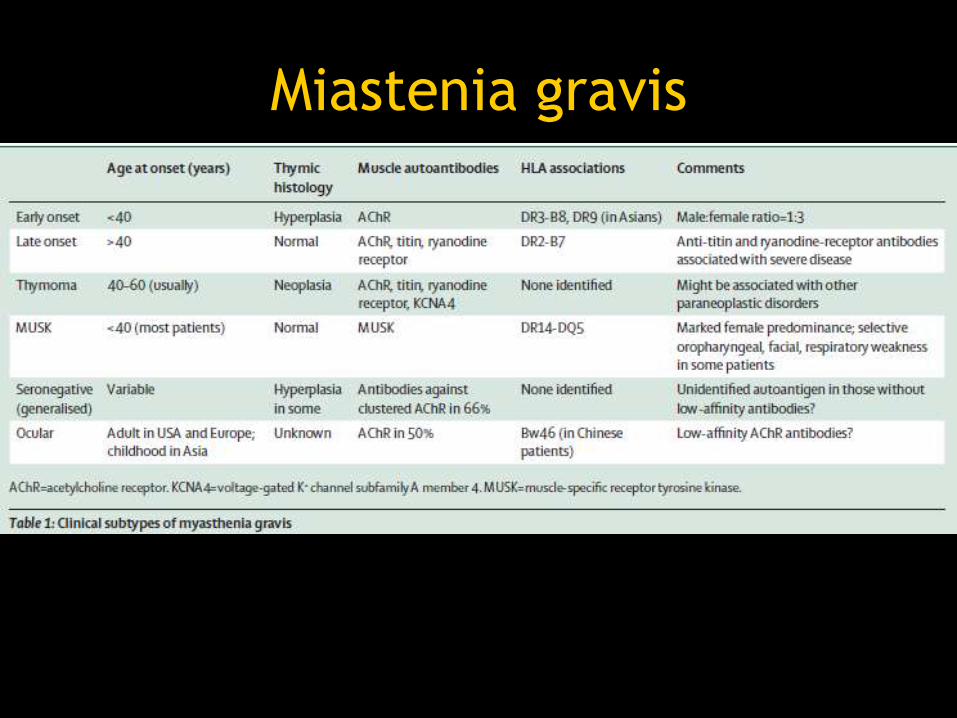
Miastenia gravis
• Enfermedad autoinmune de la unión
neuromuscular
• Afecta el receptor de acetilcolina
• Predisposición genética
• 20-100,000
• Mujeres 3:1 hombres (<40)

Miastenia Gravis
• Fatiga excesiva
fluctuante
• Debilidad
generalizada (2a)
• Síntomas iniciales
disfagia, disartria
(15%)
Miastenia gravis
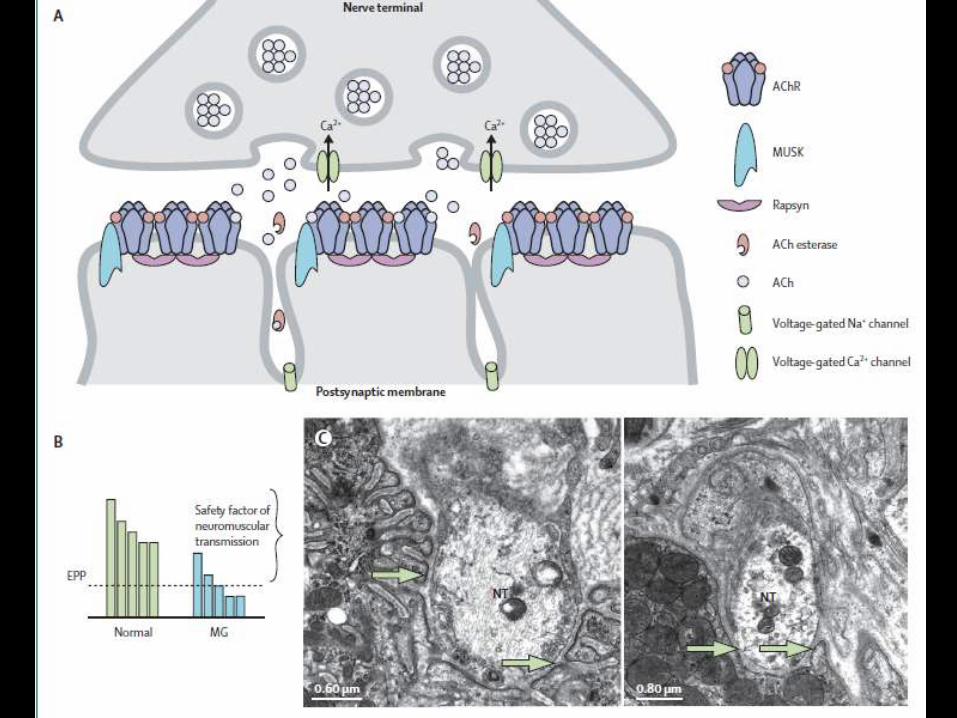
Miastenia gravis

Principios del tratamiento
• Individualizado según el subtipo
• Recobrar vida normal minimizando los
efectos secundarios de la terapia
• Infecciones son asociadas a
inmunosupresores

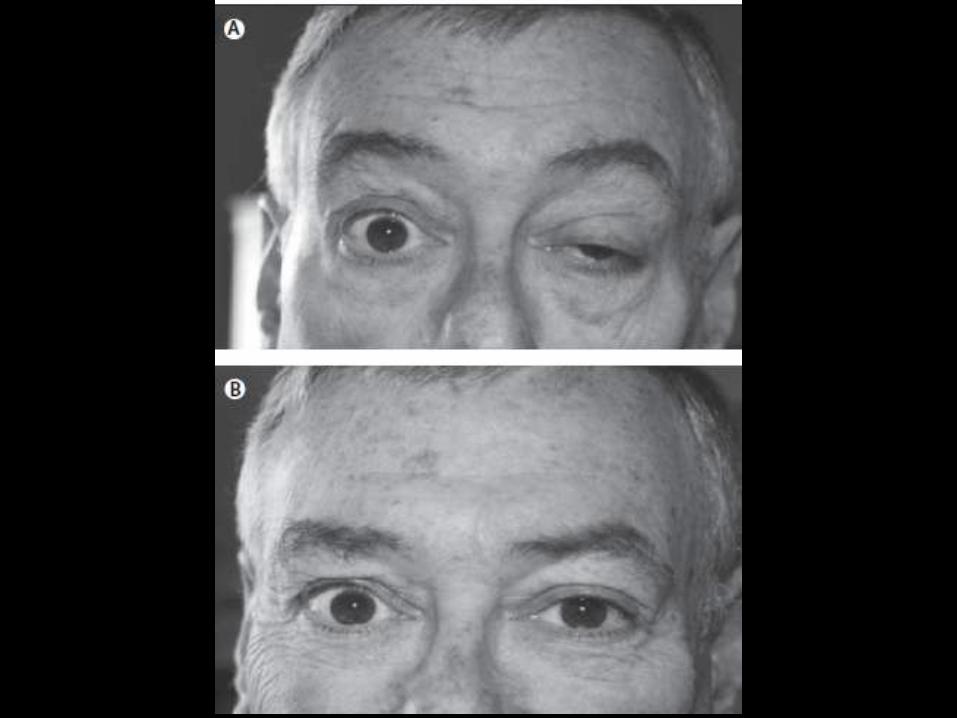

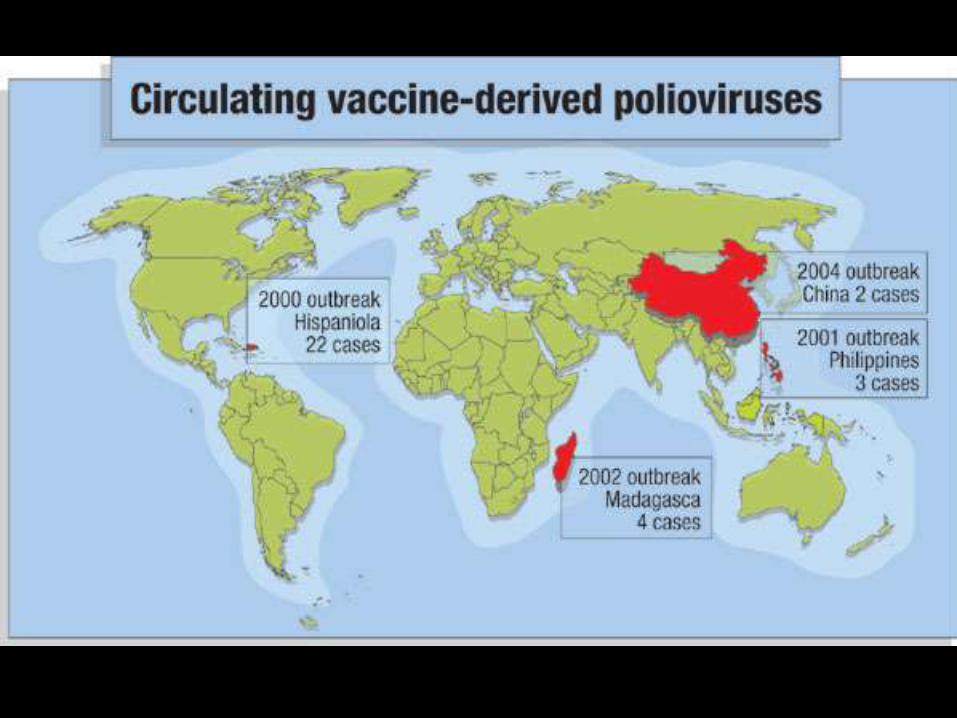
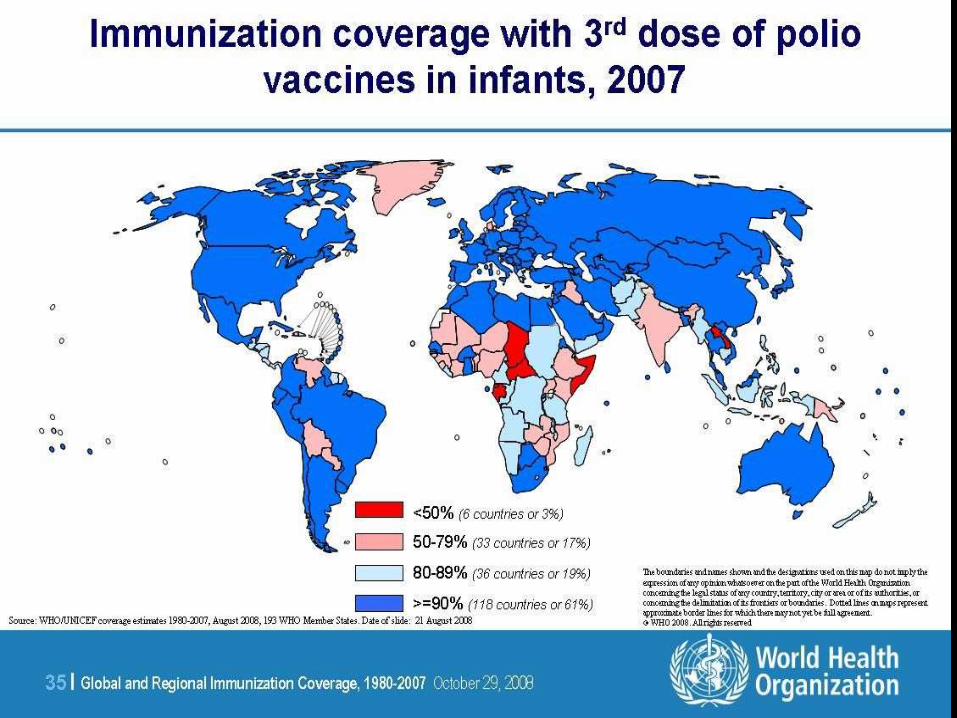
Poliomielitis
• Enfermedad infecciosa aguda que ataca
las astas anteriores de el cordón espinal
• Brunhilde (1), Lansing (2), and Leon (3)
• 1% forma paralitica
• Vacuna de la polio
• Vía hematógena
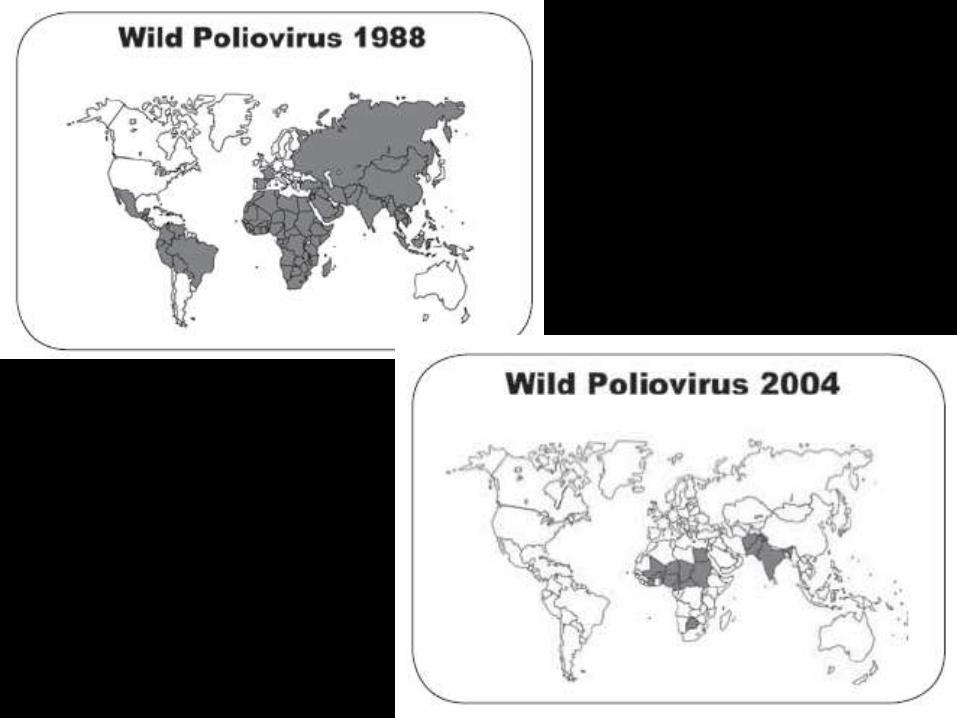

Poliomielitis
• La lesion es por producción de toxinas,
isquemia, edema, hemorragia
• Atrofia de neuronas motoras
• El grado de parálisis depende de la
cantidad de neuronas motoras lesionadas
• La recuperación de la función muscular
es mas notable a los 3 a 6 meses.

Etapas de la enfermedad
• Etapa aguda
– Malestar general, encefalomielitis
– Meningismo
– Dolor intenso a la movilización
– Tratamiento para prevención de la deformidad
– Perdida de la elasticidad
– Parálisis respiratoria

Etapas de la enfermedad
• Etapa convaleciente
– 2 días posterior a desaparición de la fiebre
– Disminuye la progresión de la parálisis
– Evaluación continua de la progresión
– 30% perdida de la fuerza en 3 meses se
considera parálisis permanente

Etapas de la enfermedad
• Etapa crónica
– 2 años después
– Contractura articular
ocurre por la fuerza de
la musculatura
dominante
– La edad influye en las
deformidades oseas

Tratamiento
• Etapa crónica
– Corregir deformidades.
– Evitar deformidades.
– Ejercicios de estiramiento pasivo.
– Ejercicios activos de hipertrofia.

Tratamiento

Tratamiento
• Etapa convaleciente
– Restauración y conservación de arco de movimiento normal
– Prevención de deformidades
– Alcanzar el mejor estado funcional

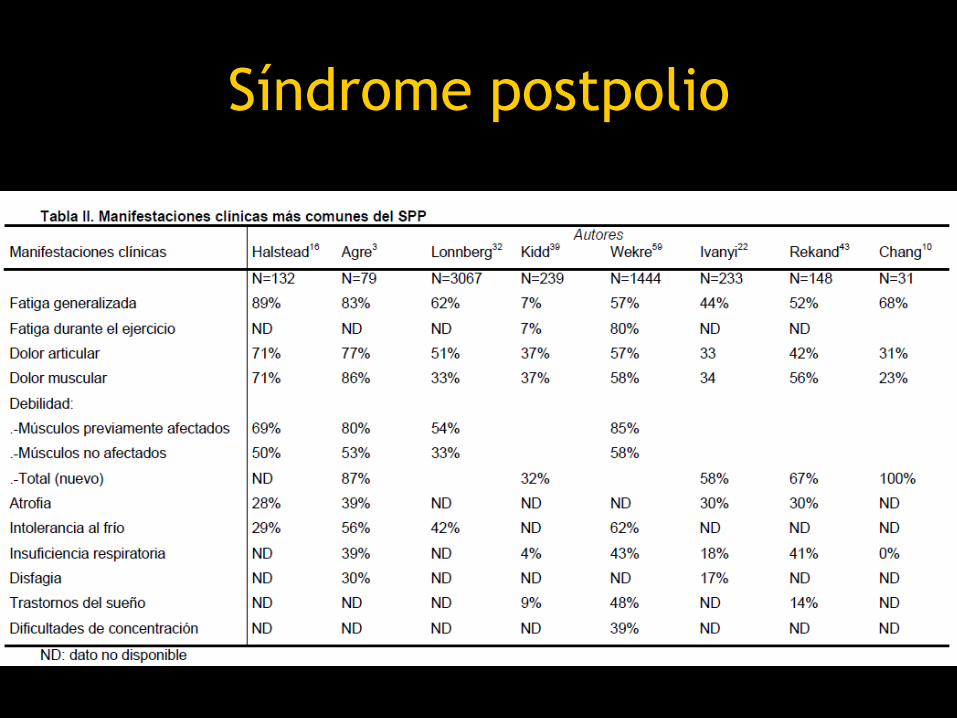
Síndrome postpolio
• Fatiga progresiva, debilidad muscular y
dolor y que aparece varias décadas
después de un episodio de poliomielitis
paralítica
• Se considera un síndrome neurológico
específico secundario a denervación
• Edad promedio 35a
Síndrome postpolio

Diagnostico
1.Antecedentes de poliomielitis paralítica
2. Intervalo de estabilidad clínico-funcional al menos 20 años.
3. Aparición, gradual o abrupta, de debilidad muscular no atribuible a la falta de uso
4. Signos electromiográficos compatibles con denervación aguda y reinervación cró-nica.
5. Exclusión de otros procesos neurológicos

Tratamiento
• Cambio del estilo de vida
• Sintomático
• Rehabilitación
• Ortesis
• Tratamiento quirúrgico
no indicado
