endotheliale mechanismen der gefäßdilatation bei der … · regel aus der summe der...
TRANSCRIPT

Aus dem Institut für Physiologie der Universität zu Lübeck
Direktor: Prof. Dr. med. Wolfgang Jelkmann
Endotheliale Mechanismen
der Gefäßdilatation
bei der funktionellen Hyperämie
im Skelettmuskel der Maus
Inauguraldissertation
zur Erlangung der Doktorwürde der
Universität zu Lübeck
- Aus der Medizinischen Fakultät -
Vorgelegt von
Malte Milkau
aus
Bad Oldesloe
Lübeck 2010

1. Berichterstatter: Prof. Dr. med. Cornelis de Wit
2. Berichterstatter: Prof. Dr. med. Uwe Wiegand
3. Berichterstatter: Prof. Dr. med. Axel Pries
Tag der mündlichen Prüfung: 17.02.2010
Zum Druck genehmigt: Lübeck, den 17.02.1010

INHALTSVERZEICHNIS
I
Inhaltsverzeichnis
1. Einleitung 1
1.1 Prinzipien der Durchblutungsregulation 1
1.2 Aufbau und Kontraktionsmechanismen der Blutgefä ße 3
1.3 Endothelabhängige Mechanismen der Vasodilatatio n 6
1.4 EDHF 8
1.5 Koordination des Gefäßverhaltens durch Gap junc tions 11
1.6 Funktionelle Hyperämie in der Skelettmuskulatur 14
1.7 Ziel der vorliegenden Arbeit 16
2. Material und Methoden 18
2.1 Versuchstiere 18
2.1.1 Allgemeines 18
2.1.2 Genotypisierung 19
2.2 Intravitalmikroskopie 22
2.2.1 Anästhesie und Präparation 22
2.2.2 Mikroskopie 26
2.2.3 Nervenstimulation 27
2.2.4 Prinzipieller Versuchsablauf 28
2.2.5 Stimulation mit vasoaktiven Substanzen 29
2.2.6 Untersuchungen zur funktionellen Hyperämie 30
2.2.7 Auswertung und Statistik 30
2.3 Textverarbeitung 32
3. Ergebnisse 33
3.1 Gefäßdilatationen durch vasoaktive Substanzen 33
3.1.1 Dilatationen nach Applikation von ACh und SNP in Wildtyptieren 33
3.1.2 Dilatationen nach Applikation von ACh und SNP in Cx40-defizienten
Tieren
34
3.2 Charakteristika der funktionellen Hyperämie 35
3.2.1 Abhängigkeit von Stimulationsdauer und -frequenz 35
3.2.2 Einfluss von Indomethacin und Nitro-L-Arginin 37
3.3 Rolle von K Ca bei der funktionellen Hyperämie 39

INHALTSVERZEICHNIS
II
3.3.1 Rolle von endothelialen KCa-Kanälen 39
3.3.2 Rolle des IKCa 43
3.3.3 Rolle des SKCa 44
3.4 Rolle von Connexinen bei der funktionellen Hype rämie 47
3.4.1 Rolle von Connexin 40 47
3.4.2 Austausch von Cx40 durch Cx45 50
4. Diskussion 53
4.1 Charakteristika der funktionellen Hyperämie 53
4.2 Bedeutung von K Ca für die funktionelle Hyperämie 55
4.3 Koordination der Gefäßdilatation durch Connexin e 57
4.4 Schlussfolgerungen und Ausblick 59
5. Zusammenfassung 63
6. Literatur 64
7. Anhang 78
7.1 Verwendete Substanzen 78
7.2 Lösungen und Puffer 80
7.3 Weitere Materialien 81
7.4 PCR 81
7.4.1 Verwendete Primer 81
7.4.2 Reaktionsansätze 82
7.4.3 Temperaturverläufe 82
8. Danksagung 83
9. Lebenslauf 85
10. Publikationen 87

ABKÜRZUNGSVERZEICHNIS
III
Abkürzungsverzeichnis
ACh Acetylcholin
Ado Adenosin
ATP Adenosintriphosphat
BKCa KCa mit großer (big) Leitfähigkeit
Bl6 siehe C57Bl/6
bp Basenpaare
C57Bl/6 Mausstamm mit definiertem genetischem Hintergrund
cAMP zyklisches Adenosinmonophosphat
cGMP zyklisches Guanosinmonophosphat
CNP C-Typ natriuretisches Peptid
CO2 Kohlendioxid
COX Cyclooxygenase
Cx Connexin
Cx40ko genetisch veränderter Mausstamm (siehe auch 2.1.1)
Cx40KI45 genetisch veränderter Mausstamm (siehe auch 2.1.1)
Cyp450 Cytochrom-P450-Epoxygenasen
DA Außendurchmesser
DI Innendurchmesser
dKCa-ko genetisch veränderter Mausstamm (siehe auch 2.1.1)
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
dNTPs Nukleotidlösung
EC Endothelzellen (endothelial cells)
EETs Epoxyeicosatriensäuren
EDHF Endothelium-Derived Hyperpolarizing Factor
EDTA Ethylendiamintetraessigsäure
eNOS endotheliale NO-Synthase
ER endoplasmatisches Retikulum
Fa. Firma
GJ Gap junctions
HZV Herzzeitvolumen
H2O2 Wasserstoffperoxid

ABKÜRZUNGSVERZEICHNIS
IV
I Stromstärke
IKCa KCa mit mittlerer (intermediate) Leitfähigkeit, auch KCa3.1
IKCa-ko genetisch veränderter Mausstamm (siehe auch 2.1.1)
Indo Indomethacin
i.p. intraperitoneal
IP3 Inositoltrisphosphat
i.v. intravenös
KCa Calcium-abhängiger Kaliumkanal
KCa2.3 siehe SKCa
KCa3.1 siehe IKCa
KG Körpergewicht
Kir Einwärts-Gleichrichter-Kaliumkanal (inward rectifier)
l Länge
L-NA Nitro-L-Arginin
LOX Lipoxygenase
M. Musculus
MEGJ myoendotheliale Gap junctions
MLCK Myosin Leichte Ketten (Chain) Kinase
MLCP Myosin Leichte Ketten (Chain) Phosphatase
MW Mittelwert
n Anzahl der Beobachtungen
NO Stickstoffmonoxid
η Viskosität
O2 Sauerstoff
∆P Druckdifferenz
PCR Polymerase-Kettenreaktion
PLA2 Phospholipase A2
PLC Phospholipase C
PGI2 Prostaglandin I2, Prostazyklin
PGs Prostaglandine
r Radius
R Strömungswiderstand
rcf Relative Zentrifugalkraft
SDS Sodiumdodecylsulfat

ABKÜRZUNGSVERZEICHNIS
V
SEM Standardfehler des Mittelwerts (standard error mean)
sGC lösliche (soluble) Guanylatzyklase
SKCa KCa mit kleiner (small) Leitfähigkeit, auch KCa2.3
SKCa-ko genetisch veränderter Mausstamm (siehe auch 2.1.1)
SMC Glatte Gefäßmuskelzellen (smooth muscle cells)
SNP Natriumnitroprussid (Sodiumnitroprusside)
tTA Tetracyclin Transaktivator
UCL1684 spezifischer Blocker des SKCa
V. Vena
VDCC Spannungsabhängiger Ca2+-Kanal (Voltage-Dependent
Ca2+-Channel), auch L-Typ-Calciumkanal

EINLEITUNG
1
1. Einleitung
1.1 Prinzipien der Durchblutungsregulation
Das Herz-Kreislaufsystem ist ein Organsystem, dessen Funktion lebensnotwendig
für jeden höheren Organismus ist. Mit dem Blut werden Sauerstoff (O2),
Energieträger, sonstige Nährstoffe und Signalstoffe zu den peripheren Geweben
befördert und Stoffwechselendprodukte und insbesondere Kohlendioxid (CO2)
abtransportiert. Außerdem ist das Kreislaufsystem unverzichtbar für die
Wärmeregulation und die Funktion des Immunsystems. Entscheidend für die
Erfüllung dieser Aufgaben ist, dass zu jeder Zeit ein bedarfsweise adaptierter
Blutfluss in den einzelnen Geweben und Organstromgebieten gewährleistet ist.
Betrachtet man das Gefäßsystem annähernd als ein Gebilde aus miteinander
kommunizierenden starren Rohren, in denen eine laminare Strömung einer
homogenen Flüssigkeit herrscht, so lassen sich einige physikalische Regeln
anwenden. Nach dem Ohmschen Gesetz wird die Gesamtstromstärke I
(Herzzeitvolumen, HZV) einerseits von der Perfusionsdruckdifferenz ∆P (arterieller
Blutdruck) und andererseits vom peripheren Gefäßwiderstand R bestimmt. Der
Gesamtwiderstand des Gefäßbettes berechnet sich nach der Kirchhoffschen
Regel aus der Summe der Einzelwiderstände der hintereinander liegenden
Gefäßabschnitte. Der Widerstand der einzelnen Abschnitte wiederum berechnet
sich nach dem Gesetz von Hagen-Poiseuille aus dem Kehrwert der vierten Potenz
des Radius (1/r4), der Länge (l) des betrachteten Gefäßabschnittes und der
Viskosität des Blutes (η).
I = R
∆P
Rges. = R1 + R2 + …
R = 8 • η • l
π • r4
Ohmsches Gesetz
Kirchhoffsche Regel (für Widerstände in Reihe)
Gesetz von Hagen-Poiseuille

EINLEITUNG
2
Die einzige in vivo nennenswert beeinflussbare Größe der drei letztgenannten ist
der Gefäßradius und auf Grund der oben bezeichneten Abhängigkeit hat bereits
eine geringe Gefäßerweiterung eine große Abnahme des Strömungswiderstandes
zur Folge.
Im Gefäßsystem des Menschen nimmt der Gesamtquerschnitt der Blutgefäße mit
abnehmender Gefäßgröße kontinuierlich zu. Auf der Ebene der Kapillaren ist ein
Gesamtquerschnitt erreicht, der 500- bis 800-mal dem Durchmesser der Aorta
ascendens entspricht. Kapillaren sind auf Grund ihres histologischen Aufbaus
(s.u.) nicht in der Lage, ihren Durchmesser aktiv zu verändern, daher sind die
ihnen vorgeschalteten Arteriolen mit einem regulierbaren Durchmesser von etwa
10 bis 100 µm die eigentlichen Widerstandsgefäße. Ihr Tonus und damit die
Gefäßweite hat den größten Anteil an der Regulation des Strömungswiderstandes.
Diese Arteriolen waren Objekt der Untersuchungen, die dieser Arbeit zu Grunde
liegen.
Da die Auswurfleistung des Herzens und damit das HZV begrenzt ist, ist es nicht
möglich, dass alle Organe gleichzeitig maximal perfundiert werden. Es muss
demzufolge eine differenzierte Durchblutungsregulation auf Ebene der einzelnen
Organe erfolgen. Dies wird dadurch erreicht, dass die Arteriolen (Widerstands-
gefäße) einzelner Organstromgebiete bedarfsadaptiert eine Widerstandsänderung
bewirken, so dass es bei gesteigertem Bedarf zu einer Mehrdurchblutung kommt
(funktionelle oder aktive Hyperämie). Diese Regulation basiert vor allem auf
lokalen Mechanismen. So können sowohl physikalische als auch biochemische
Faktoren den lokalen Gefäßtonus beeinflussen und so zu einer Konstriktion
(Verengung) oder einer Dilatation (Erweiterung) des Gefäßes führen. Ein Beispiel
für einen physikalischen Faktor ist die Wandschubspannung, die den
Kontraktionszustand eines Gefäßes beeinflussen kann. Außerdem ist das Gefäß
in der Lage, auf Änderungen des transmuralen Drucks zu reagieren (Bayliss-
Effekt). Ebenso kann ein verändertes extrazelluläres Milieu (zum Beispiel Anstieg
oder Abfall des Sauerstoffpartialdrucks oder des pH, Freisetzung von Metaboliten)
oder die Sekretion von vasoaktiven Substanzen den Kontraktionszustand eines
Gefäßes verändern.

EINLEITUNG
3
Im Gegensatz zu diesen lokalen Mechanismen dienen zentrale Mechanismen
weniger einer bedarfsorientierten Durchblutung einzelner Organe, sondern viel
mehr einer Regulation des systemischen Blutdrucks. Neurohumorale und
endokrine Einflüsse spielen hier eine Rolle. Das vegetative Nervensystem kann
durch sympathische konstriktorische Fasern den Gefäßtonus über adrenerge α1-
Rezeptoren beeinflussen. Der Parasympathikus beeinflusst den Gefäßtonus nur in
geringem Maße und in bestimmten Organsystemen (Geschlechtsorgane) und ist
an der Regulation des systemischen Blutdrucks nicht beteiligt. Weitere Effektoren
der zentralen Regulation sind Adrenalin, welches im Nebennierenmark produziert
wird, und Angiotensin II, welches im Gefäßlumen selbst aus Vorläufermolekülen
entsteht. Im Allgemeinen reagieren diese zentralen Regulationsmechanismen
entweder auf Zeichen einer Minderperfusion (z.B. in der Niere oder mediiert durch
bestimmte Barorezeptoren) oder auf eine erwartete Mehrleistung des Organismus
(z. B. Schreckreaktion mit Sympathikusaktivierung) und führen dann zu einer
Änderung des Gefäßtonus, um den systemischen Blutdruck im physiologischen
Bereich zu halten.
Der Anteil dieser verschiedenen lokalen und systemischen Mechanismen an der
Regulation des jeweiligen Gefäßtonus variiert in verschiedenen Geweben stark.
Die Versuche in dieser Arbeit beziehen sich auf die Durchblutung speziell des
Skelettmuskels und daher sollen die dort relevanten Mechanismen im Folgenden
noch näher betrachtet werden.
1.2 Aufbau und Kontraktionsmechanismen der
Blutgefäße
Das Herz-Kreislaufsystem besteht aus dem Herzen, das die treibende Kraft für
den Blutfluss generiert, sowie dem arteriellen und dem venösen Gefäßsystem.
Das arterielle System des Körperkreislaufs beginnt mit der Aorta und geht mit
abnehmender Größe der einzelnen Gefäße in die Arterien und die Arteriolen über
und führt dann zum kapillären System. Hier findet der Stoffaustausch mit den
Geweben statt. Den Kapillaren angeschlossen ist wiederum das venöse System
aus Venolen und größeren Venen, das das Blut zum Herzen zurückführt.

EINLEITUNG
4
Allen Gefäßen ist der grundlegende histologische Aufbau gemein [1]. Ihr Lumen ist
stets von einer einschichtigen Lage aus Endothelzellen ausgekleidet (Intima), die
die Grenzfläche zum fließenden Blut darstellen und vielfältige Aufgaben besitzen.
Außer ihrem Einfluss auf den lokalen Gefäßtonus (s.u.) nehmen sie auch wichtige
Funktionen im Zusammenhang mit der Immunabwehr und der Blutgerinnung wahr
und synthetisieren Bestandteile der extrazellulären Matrix der subendothelialen
Schicht. Die einzelnen Endothelzellen sind platte, polygonale Zellen, die mit ihrer
Längsachse parallel zum Blutstrom ausgerichtet sind. Sie sind sowohl durch Gap
junctions (s.u.), als auch durch Tight junctions miteinander verbunden und stellen
eine Diffusionsbarriere dar.
An das Endothel schließt sich die Media an, die vom Endothel durch eine dünne
Bindegewebsschicht und eine elastische Membran (Membrana elastica interna)
abgetrennt ist. Letztere ist von Öffnungen durchsetzt, um die Diffusion von Stoffen
zu erleichtern. Die Media besteht aus glatten Muskelzellen sowie Extra-
zellulärmatrix. Im Gegensatz zu den Endothelzellen sind die Gefäßmuskelzellen
mit ihrer Längsachse zirkulär um das Gefäß angeordnet. Die Dicke der Media ist
bei den großen Arterien am größten und nimmt mit abnehmender Gefäßgröße ab.
Auf der Ebene der Arteriolen und Venolen besteht sie aus einer Zelllage, in den
Kapillaren verschwindet sie völlig und es sind nur noch vereinzelte
Gefäßmuskelzellen aufzufinden. An die Media schließt sich die bindegewebige
Adventitia an, die das Gefäß im umliegenden Gewebe verankert.
Die Gefäßmuskelzellen der Arteriolen bestimmen mit ihrem Tonus den
Gefäßdurchmesser und damit den resultierenden Strömungswiderstand. Hierzu
besitzen sie einen kontraktilen Apparat aus Aktin- und Myosinfilamenten, der unter
ATP-Verbrauch zu einer Kontraktion der Muskelzelle führen kann [2]. Dieser
Apparat wird durch die Phosphorylierung und Dephosphorylierung der
regulatorischen leichten Kette des Myosins reguliert, ein höherer
Phosphorylierungsgrad führt zu einer gesteigerten Interaktion der kontraktilen
Elemente. Unter physiologischen Bedingungen herrscht ein Gleichgewicht
zwischen dem Enzym, das das Myosin phosphoryliert (Myosin Leichte Ketten
Kinase, MLCK) und dem Enzym, das es dephosphoryliert (Myosin Leichte Ketten
Phosphatase, MLCP). Ein entscheidender Faktor, der dieses Gleichgewicht

EINLEITUNG
5
beeinflusst, ist die intrazelluläre Calciumkonzentration, da die MLCK durch
Calcium aktiviert wird. Sie liegt in Ruhe bei etwa 10-7 mol/l und kann durch
spezielle Agonisten oder Membranpotentialänderungen verändert werden. Eine
Depolarisation der Zelle führt zu einer Öffnung von spannungsabhängigen
Calciumkanälen (VDCCs) und so zu einem Einstrom von Calcium [3]. Dies bewirkt
eine Aktivierung von Calciumkanälen im endoplasmatischen Retikulum (ER), was
den Calciumeinstrom ins Zellinnere noch verstärkt (Calicum-abhängige
Calciumfreisetzung). Umgekehrt führt eine Hyperpolarisation zum Schließen von
VDCCs und die Calciumkonzentration sinkt, da Ca2+ ständig in die intrazellulären
Speicher zurückgepumpt wird. Bei einem Anstieg des intrazellulären Calciums
kommt es zu einer Bindung von Calcium an das regulatorische Protein Calmodulin
und dieser Calcium-Calmodulin-Komplex bindet wiederum an die MLCK, so dass
ein aktiver Holoenzymkomplex entsteht. Dieser überträgt eine Phosphatgruppe
von ATP auf Myosin und ermöglicht so die Interaktion von Aktin und Myosin und
somit eine Kontraktion. Umgekehrt führt eine Hyperpolarisation über eine
Verminderung der intrazellulären Calciumkonzentration zu einer Erschlaffung der
Muskelzellen. Es gibt weiterhin Calcium-unabhängige Mechanismen, die den
Tonus der Gefäßmuskelzellen modulieren. So kann es durch die Hemmung der
MLCP über verschiedene Signalkaskaden zu einer Calcium-Sensitivierung
kommen, es wird dann bei gleichbleibender intrazellulärer Calciumkonzentration
ein höherer Kontraktionszustand erreicht. Analog dazu kann es durch eine
Aktivierung der MLCP durch intrazelluläre Botenstoffe wie cAMP oder cGMP zu
einer Calcium-Desensitivierung und einer Relaxation der Muskelzelle kommen.
Zusammengefasst sind die glatten Muskelzellen der Gefäßwand also durch einen
Ruhetonus charakterisiert, der durch verschiedene Einflussfaktoren in beide
Richtungen, hin zu einer Dilatation oder einer Konstriktion des Gefäßes,
beeinflusst werden kann, und der so den Gefäßwiderstand ändert und die lokale
Durchblutung reguliert.

EINLEITUNG
6
1.3 Endothelabhängige Mechanismen der Vasodilatatio n
Eine Änderung des Kontraktionszustandes eines Gefäßes kann erreicht werden,
indem ein physikalischer oder ein biochemischer Stimulus direkt auf die glatten
Muskelzellen der Gefäßwand einwirkt. Dies ist zum Beispiel beim Bayliss-Effekt
der Fall. Hier führt ein physikalischer Faktor, ein Anstieg des transmuralen Drucks,
zu einer Dehnung der Gefäßmuskelzellen und in der Folge zu einer Öffnung
mechanosensitiver Kationenkanäle und einem Einstrom von Calcium. Dies führt
zu einer Kontraktion des Gefäßes [4].
Andererseits kann der Kontraktionszustand eines Gefäßes auch durch Stimuli
beeinflusst werden, die nur mittelbar auf die Muskelzellen der Gefäßwand wirken
und primär einen Effekt auf das Endothel haben. Dieses kann daraufhin Autakoide
freisetzen, die in die Media diffundieren und dort den Gefäßtonus beeinflussen.
Ein physikalischer Stimulus, der auf diese Weise wirkt, ist die Schubspannung, die
das strömende Blut auf das Endothel ausübt [5]. Ein Anstieg des Blutflusses führt
zu einem Anstieg der Calciumkonzentration in den Endothelzellen [6] und so zu
einer Freisetzung vasodilatatorischer Mediatoren (flussinduzierte Dilatation),
insbesondere von Stickstoffmonoxid (NO) [7]. Ein typischer biochemischer
Mediator, der das Endothel aktiviert und so zu einer Gefäßdilatation führt und oft
als Testsubstanz der Endothelfunktion dient, ist Acetylcholin (ACh). Es aktiviert in
Endothelzellen über den muskarinischen M3-Rezeptor die Phospholipase C (PLC)
und diese spaltet wiederum Inositoltrisphosphat (IP3) aus Membranlipiden ab. IP3
wirkt dann auf Ionenkanäle im endoplasmatischen Retikulum und führt so zu
einem intrazellulären Calciumanstieg [8] und einer Hyperpolarisation der
Endothelzellen [9], was zu einer Freisetzung von Autakoiden führt, die eine
Gefäßdilatation bewirken.
Die klassischen Autakoide, die vom Endothel freigesetzt werden, sind NO und
Prostaglandin I2 (PGI2, Prostazyklin). NO wird im Endothel von der konstitutiv
exprimierten endothelialen NO-Synthase (eNOS) produziert, die ihrerseits von
einem Calcium-Calmodulin-Komplex bei Anstieg der intrazellulären Calcium-
konzentration aktiviert wird. Sie spaltet NO aus der Aminosäure L-Arginin ab und
es entsteht L-Citrullin. NO kann auf Grund seiner chemischen Eigenschaften in die
glatten Muskelzellen der Gefäßwand diffundieren und aktiviert dort die lösliche

EINLEITUNG
7
Guanylatzyklase (sGC), die cGMP produziert und so cGMP-abhängige
Proteinkinasen aktiviert [10-13]. Hierdurch wird einerseits ein Calcium-
unabhängiger Mechanismus der Relaxation der glatten Muskelzellen eingeleitet,
indem die Aktivität der MLCP gesteigert wird. Zusätzlich werden weitere
Zielproteine der cGMP-abhängigen Proteinkinasen aktiviert, die zu einer Senkung
des intrazellulären Calciums führen und so gleichzeitig eine Calcium-abhängige
Dilatation des Gefäßes bewirken [14].
Auch Prostazyklin wird im Endothel bei einem Anstieg des Calciums gebildet.
Durch eine Calciumerhöhung setzt die Phospholipase A2 (PLA2) Arachidonsäure
aus Membranlipiden frei. Die Cyclooxygenase (COX) bildet aus Arachidonsäure
Prostaglandin H2, welches daraufhin von der PGI2-Synthase zu Prostazyklin
umgesetzt wird. Dieses wirkt auf einen membranständigen G-Protein-gekoppelten
Prostaglandinrezeptor der Gefäßmuskelzellen und führt zur Synthese des
Botenstoffes cAMP durch die Adenylatzyklase. cAMP aktiviert nun analog zu
cGMP verschiedene Proteinkinasen und führt zu einer sowohl Calcium-
unabhängigen als auch einer Calcium-abhängigen Gefäßdilatation [15, 16]. Ein
weiterer Metabolisierungsweg der Arachidonsäure ist die Bildung von
Leukotrienen durch die Lipoxygenase (LOX), die allerdings keinen wesentlichen
Einfluss auf den Gefäßtonus haben.
Selbst nach Ausschaltung dieser beiden Signalwege durch eine pharmakologische
Blockade von eNOS und COX ist insbesondere in kleinen Blutgefäßen noch eine
Dilatation nach Stimulation des Endothels mit ACh zu beobachten. Da diese
Dilatation mit einer Hyperpolarisation der glatten Muskelzellen verbunden ist und
man bei der Entdeckung dieses Mechanismus von einem löslichen Faktor als
Mediator ausging, wurde dieser postulierte endotheliale Faktor als EDHF
(Endothelium-Derived Hyperpolarizing Factor) bezeichnet [17]. Auf Grund der
großen Bedeutung von EDHF für die vorliegende Arbeit soll hierauf im nächsten
Abschnitt noch näher eingegangen werden.
Die Bedeutung dieser drei endothelialen Mediatoren (NO, PGI2 und EDHF) variiert
in verschiedenen Organgebieten und in Gefäßen unterschiedlicher Größe. Es
konnte gezeigt werden, dass NO und Prostazyklin für die endothelabhängige
Dilatation vor allem in größeren Arterien verantwortlich sind, während in der
Mikrozirkulation die Bedeutung von EDHF in den Vordergrund tritt [18, 19].

EINLEITUNG
8
1.4 EDHF
Die genauen Mechanismen, die zu einer EDHF-Typ-Dilatation führen, sind bis
heute Gegenstand kontroverser Diskussionen. Eine Hyperpolarisation des
Endothels ist eine notwendige Voraussetzung für die Bildung von EDHF [9]. Sie
wird durch einen Kaliumausstrom erreicht, der durch die Öffnung von bestimmten
Ionenkanälen in der Zellmembran verursacht wird [20-22]. Hier sind Calcium-
abhängige Kaliumkanäle (KCa) beteiligt, die an Hand ihrer Leitfähigkeit in einen
Kanal mit geringer Leitfähigkeit (SKCa), einen mit mittlerer Leitfähigkeit (IKCa) und
einen Kanal mit großer Leitfähigkeit (BKCa) unterschieden werden. In
Endothelzellen werden SKCa und IKCa exprimiert, während BKCa auf glatten
Muskelzellen zu finden ist [23-27]. Ein Auslöser für die Öffnung dieser Kanäle ist
eine Erhöhung der intrazellulären Calciumkonzentration, die durch viele Faktoren
hervorgerufen wird, die das Endothel aktivieren [22]. EDHF ist nun dadurch
definiert, dass eine Hyperpolarisation der Gefäßmuskelzellen durch einen NO- und
Prostazyklin-unabhängigen Mechanismus vom Endothel erzeugt wird und so zu
einer Reduktion der dortigen Calciumkonzentration und einer Vasodilatation führt
[28].
Bisher sind verschiedene Mediatoren gefunden worden, die alle diese
Bedingungen erfüllen. Es gibt Hinweise darauf, dass verschiedene Epoxyeicosa-
triensäuren (EETs) in bestimmten Präparationen die Rolle eines EDHFs
einnehmen [29, 30]. EETs werden durch Cytochrom-P450-Epoxygenasen aus
Arachidonsäure synthetisiert. Der vermutete Mechanismus der Wirkung auf die
glatten Muskelzellen besteht in einer Öffnung von BKCa-Kanälen, einer daraus
folgenden Hyperpolarisation der Gefäßmuskelzellen und so einer Relaxation [31,
32], möglicherweise über einen G-Protein-gekoppelten Rezeptor [33].
Bei der Hyperpolarisation des Endothels kommt es zu einem Ausstrom von
Kaliumionen über SKCa- und IKCa-Kanäle und es ist denkbar, dass diese
Kaliumionen als EDHF wirken. Eine Erhöhung der extrazellulären Kalium-
konzentration kann sowohl Einwärts-Gleichrichter-Kaliumkanäle (Kir) als auch die
Na+-K+-ATPase der glatten Muskelzellen aktivieren, was zu einer
Hyperpolarisation der Gefäßmuskelzellen und einer Dilatation des Gefäßes führt
[34-38].

EINLEITUNG
9
Ebenso wie für EETs und Kalium konnte auch für Wasserstoffperoxid (H2O2)
gezeigt werden, dass es in bestimmten Präparationen wie ein EDHF wirkt [39-41].
Der genaue Mechanismus der endothelialen Bildung von H2O2 und des weiteren
Signalweges ist noch nicht geklärt, möglicherweise kommt es auch hier zu einer
Öffnung von BKCa-Kanälen. In der Literatur werden noch zwei weitere Substanzen
als mögliche Effektormoleküle einer EDHF-Dilatation diskutiert, das Endo-
cannabinoid Anandamid, das möglicherweise über Cannabinoidrezeptoren der
glatten Muskelzellen zu einer Hyperpolarisation führt [42], und das C-Typ
natriuretische Peptid (CNP), das über eine Erhöhung der cGMP-Konzentration in
den Gefäßmuskelzellen wirken soll [43].
Eventuell ist EDHF jedoch gar kein chemisch identifizierbarer Faktor, der vom
Endothel in die Media des Gefäßes diffundiert. In den letzten Jahren wurde die
Theorie entwickelt, dass es sich bei NO- und Prostazyklin-unabhängigen
Dilatationen auch um eine direkte Ladungsübertragung von den Endothel- in die
glatten Muskelzellen handeln könnte [44]. In einigen in vitro-Präparationen konnte
gezeigt werden, dass diese beiden Zellschichten in der Gefäßwand durch
myoendotheliale Gap junctions (MEGJ, siehe auch Abschnitt 1.5) verbunden sind
und dass die Blockade dieser Zellverbindungen EDHF-Dilatationen abschwächt
[45-48]. In vivo-Experimente zeigten allerdings Befunde auf, die gegen einen
reinen Ladungstransfer durch MEGJ im Sinne eines EDHF sprechen [9, 49]. Eine
weitere Möglichkeit, die Bedeutung von MEGJs für EDHF-Dilatationen zu
untersuchen, besteht darin, das Membranpotential in einer der beiden
Zellschichten zu verändern und zu messen, ob sich diese Änderungen auf die
jeweils andere Zellschicht ausbreiten. Auch in diesen Versuchen konnten in vitro
Hinweise auf MEGJ und eine direkte Kopplung gefunden werden [50, 51], was
sich jedoch in in vivo-Versuchen nicht verifizieren ließ [52, 53].
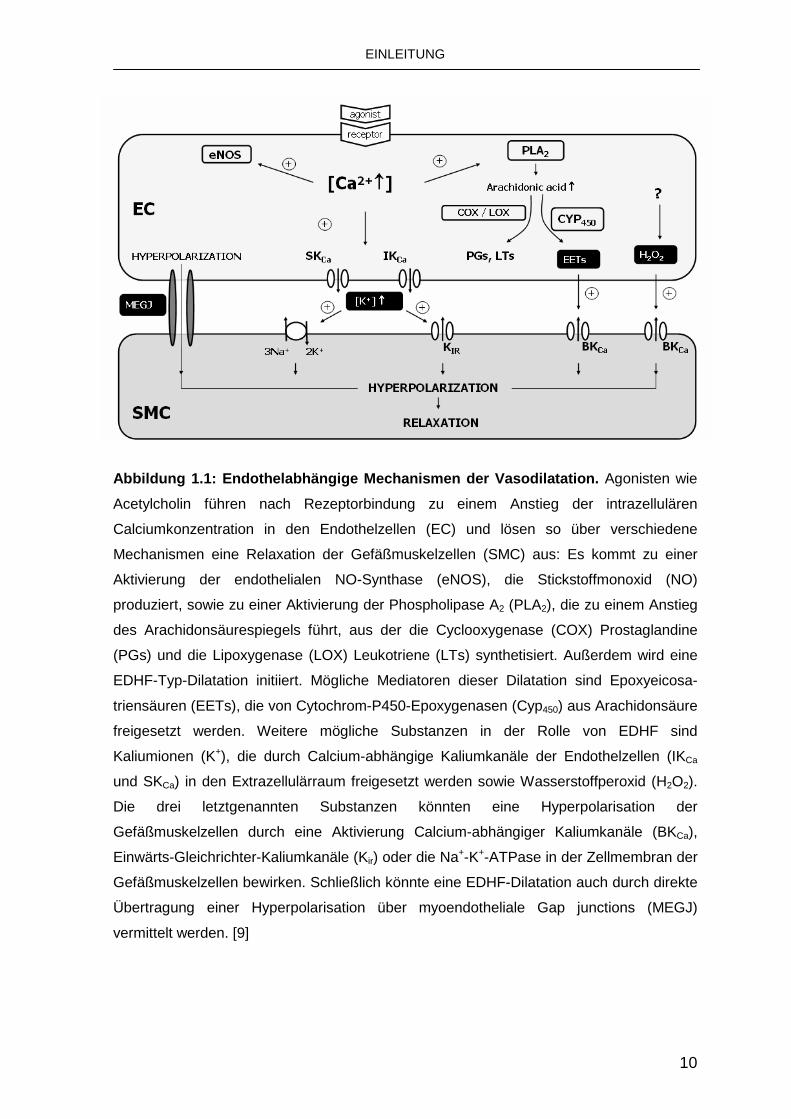
In Abb. 1.1 sind die endothelabhängigen Mechanismen einer Dilatation
schematisch zusammengefasst.
EINLEITUNG
10
Abbildung 1.1: Endothelabhängige Mechanismen der Va sodilatation. Agonisten wie
Acetylcholin führen nach Rezeptorbindung zu einem Anstieg der intrazellulären
Calciumkonzentration in den Endothelzellen (EC) und lösen so über verschiedene
Mechanismen eine Relaxation der Gefäßmuskelzellen (SMC) aus: Es kommt zu einer
Aktivierung der endothelialen NO-Synthase (eNOS), die Stickstoffmonoxid (NO)
produziert, sowie zu einer Aktivierung der Phospholipase A2 (PLA2), die zu einem Anstieg
des Arachidonsäurespiegels führt, aus der die Cyclooxygenase (COX) Prostaglandine
(PGs) und die Lipoxygenase (LOX) Leukotriene (LTs) synthetisiert. Außerdem wird eine
EDHF-Typ-Dilatation initiiert. Mögliche Mediatoren dieser Dilatation sind Epoxyeicosa-
triensäuren (EETs), die von Cytochrom-P450-Epoxygenasen (Cyp450) aus Arachidonsäure
freigesetzt werden. Weitere mögliche Substanzen in der Rolle von EDHF sind
Kaliumionen (K+), die durch Calcium-abhängige Kaliumkanäle der Endothelzellen (IKCa
und SKCa) in den Extrazellulärraum freigesetzt werden sowie Wasserstoffperoxid (H2O2).
Die drei letztgenannten Substanzen könnten eine Hyperpolarisation der
Gefäßmuskelzellen durch eine Aktivierung Calcium-abhängiger Kaliumkanäle (BKCa),
Einwärts-Gleichrichter-Kaliumkanäle (Kir) oder die Na+-K+-ATPase in der Zellmembran der
Gefäßmuskelzellen bewirken. Schließlich könnte eine EDHF-Dilatation auch durch direkte
Übertragung einer Hyperpolarisation über myoendotheliale Gap junctions (MEGJ)
vermittelt werden. [9]

EINLEITUNG
11
Zusammenfassend ist zu sagen, dass es bisher kein schlüssiges Konzept für die
Identität von EDHF gibt. Mehrere mögliche Substanzen sind identifiziert worden,
ohne dass eine Substanz hierbei im Vordergrund steht. Die oben dargestellten
Studienergebnisse lassen vermuten, dass es keinen uniformen EDHF gibt,
sondern dass die beteiligten Mediatoren in verschiedenen Gefäßgebieten und
Präparationen sowie möglicherweise auch speziesabhängig variieren.
1.5 Koordination des Gefäßverhaltens durch Gap
junctions
Da der Bedarf an Sauerstoff und Energieträgern in bestimmten Geweben mit der
Aktivität variiert, ist es notwendig, dass die Durchblutung diesem Bedarf
angepasst ist. Insbesondere in der Skelettmuskulatur gibt es eine große Differenz
zwischen Ruhedurchblutung und Blutfluss bei körperlicher Arbeit, die
Muskeldurchblutung kann um mehr als das 20-fache ansteigen [54].
Damit solch große Durchblutungssteigerungen möglich werden, muss das
Verhalten der Widerstandsgefäße über eine große Strecke koordiniert werden.
Wenn nur die Arteriolen unmittelbar im aktiven Gewebe dilatieren, wirken die
Abschnitte des Gefäßbaums, die weiter stromaufwärts lokalisiert sind,
flusslimitierend, und es kann nicht zu einer ausreichenden Blutflusssteigerung
kommen [55, 56]. Verdeutlichen lässt sich dies, indem man das Gefäßsystem
vereinfacht als ein Rohrsystem betrachtet (siehe Abschnitt 1.1). Entsprechend der
Kirchhoffschen Regel setzt sich der Gesamtströmungswiderstand aus der Summe
der Einzelwiderstände zusammen. Unter der Annahme, dass sich der
Gefäßwiderstand in der Skelettmuskulatur gleichmäßig auf intramuskuläre
Arteriolen und auf Gefäße außerhalb des Muskels verteilt, könnte selbst bei einem
Abfall des intramuskulären Gefäßwiderstandes auf vernachlässigbar kleine Werte
maximal eine Verdoppelung des Gesamtblutflusses erreicht werden, solange die
Gefäße außerhalb des Muskels ihre ursprünglichen Durchmesser beibehalten. Die
Dilatation muss sich also in stromaufwärts gelegene Gefäßabschnitte ausbreiten.
Eine solche Ausbreitung kann experimentell untersucht werden, indem ein Gefäß
strikt lokal mit einem Vasodilatator (z.B. Acetylcholin, ACh) stimuliert wird, so dass
es an der Stimulationsstelle zu einer Dilatation kommt. Diese Gefäßerweiterung

EINLEITUNG
12
wird dann nicht nur an der Stimulationsstelle beobachtet, sondern sie breitet sich
sehr schnell (< 1 s) entlang des Gefäßes aus, so dass es auch in einer Entfernung
von bis zu 20 Millimetern von der Stimulationsstelle zu einer koordinierten
Dilatation kommt. Diese Ausbreitung erfolgt sowohl nach stromaufwärts als auch
nach stromabwärts. Ein Transport von lokal applizierten vasoaktiven Substanzen
mit dem Blutstrom ist als Auslöser dieser koordinierten Dilatation auszuschließen,
da im Experiment in der Regel ein Gefäßabschnitt stromaufwärts von der
Stimulationsstelle betrachtet wird. Daher wird dieses Phänomen als ‚aufsteigende’
oder ‚fortgeleitete Dilatation’ bezeichnet [57]. Es konnte weiterhin gezeigt werden,
dass diese aufsteigenden Dilatationen nicht durch eine flussinduzierte Dilatation
hervorgerufen werden [58]. Auch die hohe Ausbreitungsgeschwindigkeit der
Fortleitung spricht gegen eine flussinduzierte Dilatation, da diese deutlich
langsamer entstehen und ca. 30 bis 150 s benötigen würde, um ihr Maximum zu
erreichen. Die Geschwindigkeit der Ausbreitung macht ein elektrisches Signal als
Vermittler der aufsteigenden Dilatation wahrscheinlich. Sie ist allerdings nicht
nerval vermittelt, da die Blockade schneller Natriumkanäle der Nerven durch
Tetrodotoxin keinen hemmenden Einfluss auf fortgeleitete Dilatationen hat [58].
Fortgeleitete Dilatationen lassen sich außer durch ACh auch durch Bradykinin und
Adenosin (Ado) auslösen [59, 60], nicht jedoch durch den NO-Donor
Natriumnitroprussid (SNP) [61]. SNP ruft zwar eine lokale Dilatation hervor, diese
breitet sich allerdings nicht entlang des Gefäßes aus. Die Tatsache, dass nicht
jede lokale Dilatation fortgeleitet wird, zeigt, dass es spezielle Mechanismen der
Fortleitung geben muss, die durch bestimmte Agonisten aktiviert werden (wie
ACh, Bradykinin, Ado), durch andere Substanzen (wie SNP) jedoch nicht. Zwei
der drei erstgenannten Substanzen (ACh und Bradykinin) haben gemeinsam, dass
sie primär auf das Endothel wirken und hier zu einer Hyperpolarisation und einem
Anstieg der intrazellulären Calciumkonzentration führen [8, 59], während SNP als
NO-Donor keine endotheliale Hyperpolarisation hervorruft, sondern direkt auf die
glatten Muskelzellen wirkt und hier zu einer Relaxation führt. Dies weist darauf hin,
dass eine endotheliale Hyperpolarisation an der Entstehung von fortgeleiteten
Dilatationen beteiligt ist. Die Rolle des Endothels kann zusätzlich dadurch belegt
werden, dass aufsteigende Dilatationen nach selektiver Zerstörung des Endothels
aufgehoben wurden [62, 63].

EINLEITUNG
13
Die endothelialen Autakoide NO und PGI2 sind allerdings weder bei der Auslösung
noch bei dem Prozess der Fortleitung beteiligt [64, 65]. Eine Schlüsselrolle für die
Auslösung fortgeleiteter Dilatationen scheint daher EDHF-Dilatationen
zuzukommen, und tatsächlich führt die Blockade von KCa-Kanälen an der
Stimulationsstelle zu einer Aufhebung der Gefäßdilatation sowohl an der
Stimulationsstelle als auch an entfernten Stellen [64, 66]. Ein weiterer Hinweis auf
die Beteiligung von EDHF ist eine endotheliale Hyperpolarisation auch an den
entfernten Stellen, wie sie mit der Bildung von EDHF assoziiert ist [67]. Diese
Befunde lassen insgesamt vermuten, dass sich eine endotheliale
Hyperpolarisation entlang des Gefäßes ausbreitet, die mit der Bildung von EDHF
assoziiert ist und so aufsteigende Dilatationen auslöst bzw. ermöglicht.
Membranpotentialänderungen können sich über mehrere Zellen ausbreiten, wenn
diese Zellen miteinander elektrisch gekoppelt sind. Dies ist in Gefäßen durch
spezielle Zellkontakte, Gap junctions (GJ), gegeben. GJ stellen niederohmige
Verbindungen zwischen einzelnen Zellen dar. Sie bestehen aus Connexin-
proteinen (Cx). Sechs dieser Connexine lagern sich zusammen und bilden einen
Halbkanal (Connexon) in der Zellmembran. Dieser Halbkanal dockt an einen
weiteren Halbkanal einer benachbarten Zelle an und so bilden sich Poren, die
gegen den Extrazellulärraum abgeschlossen sind. Sie ermöglichen den Durchtritt
von Ionen und Molekülen bis zu einer Größe von 1 kDa von einer Zelle in die
benachbarte und können zur elektrotonischen Ausbreitung einer Membran-
potentialänderung über mehrere Zellen führen [68, 69].
Vaskuläre Zellen können prinzipiell zwei Arten von GJ ausbilden: Homozelluläre
GJ, die jeweils entweder Endothelzellen oder glatte Muskelzellen untereinander
verbinden und so eine longitudinale Kommunikation ermöglichen, und
heterozelluläre GJ, die Endothelzellen mit den glatten Muskelzellen verbinden
(MEGJ, siehe Abschnitt 1.4) und eine transversale Kommunikation erlauben.
Connexine werden nach ihrem Molekulargewicht benannt und in vaskulären Zellen
werden von den zwanzig bekannten lediglich vier verschiedene Connexine
exprimiert: Cx37, Cx40, Cx43 und Cx45. Von diesen vier werden Cx37 und Cx40
hauptsächlich im Endothel der Widerstandsgefäße exprimiert. Cx43 erscheint in
geringen Mengen in den Endothelzellen größerer Gefäße sowie in den glatten

EINLEITUNG
14
Muskelzellen. Cx45 findet sich dagegen fast ausschließlich in den
Gefäßmuskelzellen [60, 62, 70-74].
Tatsächlich scheinen GJ und insbesondere Cx40 für die Weiterleitung der
Hyperpolarisation entlang des Endothels bei aufsteigenden Dilatationen eine
wesentliche Rolle zu spielen, da in genetisch veränderten Mäusen, die kein Cx40
exprimieren (Cx40 knock-out-Tiere, Cx40ko), fortgeleitete Dilatationen nach
Applikation von ACh deutlich abgeschwächt sind, während die lokale Antwort
intakt ist [75]. Auch der gentechnische Austausch von Cx40 durch Cx45 führt zu
einer deutlichen Reduktion der fortgeleiteten Dilatationen, Connexine sind also in
den Arteriolen nicht beliebig austauschbar [60].
Zusammenfassend lässt sich feststellen, dass aufsteigende Dilatationen auf einer
Hyperpolarisation beruhen, die in Zusammenhang mit einer lokalen EDHF-
Dilatation steht und sich über homozelluläre GJ im Endothel entlang des Gefäßes
ausbreitet und so das Gefäßverhalten koordiniert.
1.6 Funktionelle Hyperämie in der Skelettmuskulatur
Wie in Abschnitt 1.5 bereits erläutert, kann es in der Skelettmuskulatur zu einer
besonders ausgeprägten Durchblutungssteigerung kommen. Dies geschieht sehr
schnell (innerhalb 1 s) nach Beginn von Muskelkontraktionen [76]. Frühere
Studien konnten zeigen, dass es eine direkte Beziehung zwischen der Perfusion
und der Kontraktionsfrequenz eines Muskels [77], der Zunahme der
Laufgeschwindigkeit bei Ratten [78, 79] und der Sauerstoffaufnahme des Muskels
[80, 81] gibt.
Die Steigerung der Durchblutung eines bestimmten Organs wird durch eine
Dilatation der Widerstandsgefäße erreicht. Im Skelettmuskel könnte
möglicherweise zusätzlich zumindest ein gewisser Anteil der Durchblutungs-
zunahme durch eine Steigerung des Perfusionsdrucks erreicht werden, indem
Muskelkontraktionen eine Pumpfunktion auf die lokalen Venen ausüben und so
den Druck im venösen System absenken. Allerdings verhindert die
pharmakologisch herbeigeführte Unfähigkeit der Gefäße zu dilatieren die
Blutflusssteigerung bei Muskelkontraktionen [82-84]. Somit ist die Rolle der

EINLEITUNG
15
Muskelpumpe zu vernachlässigen und auch im Skelettmuskel ist eine Dilatation
der Widerstandsgefäße ausschlaggebend für die Mehrdurchblutung bei Arbeit.
Trotz der physiologisch wichtigen Rolle der aktiven Hyperämie ist es bis heute
unklar, welche Mechanismen die lokale Dilatation bei muskulärer Arbeit initiieren,
die sich dann entlang des Gefäßes ausbreitet. ACh, welches als Transmitter an
der motorischen Endplatte aus motorischen Nervenendigungen freigesetzt wird,
könnte von dort zu den benachbarten Widerstandsgefäßen diffundieren und zu
einer endothelvermittelten Dilatation führen [76, 85, 86]. Eine weit verbreitete und
viel untersuchte Hypothese ist metabolischer Natur, nämlich dass lokale
Mediatoren entsprechend der Arbeitsintensität, und damit dem Bedarf an
Sauerstoff und Energieträgern, freigesetzt werden und die Durchblutung steigern.
Eine andere Hypothese geht davon aus, dass die mechanische Verformung des
Gewebes durch Muskelkontraktionen selbst zu einer Gefäßdilatation führt [87-89].
Wahrscheinlich sind verschiedene Mechanismen involviert, die möglicherweise im
Zeitverlauf differenziert auftreten. So könnten spezielle Signalwege die schnelle
erste Phase der Dilatation unmittelbar nach den ersten Kontraktionen induzieren,
die dann durch andere (z. B. metabolische) Faktoren aufrechterhalten und an den
Bedarf angepasst wird [90]. Freigesetzte Mediatoren könnten von der arbeitenden
Skelettmuskulatur selbst, von den Erythrozyten im Gefäßlumen oder von den
Endothelzellen sezerniert werden und auf die glatten Gefäßmuskelzellen wirken.
Mögliche vasodilatatorische Metabolite sind unter anderem Laktat und Protonen,
die von arbeitenden Skelettmuskelzellen freigesetzt werden. Allerdings konnte für
beide Substanzen gezeigt werden, dass es keine direkte Korrelation zwischen
ihrer Konzentration und der funktionellen Hyperämie im Skelettmuskel gibt [91].
Die Konzentration von Adenosin steigt ebenfalls im arbeitenden Skelettmuskel an
und dieser Anstieg korreliert gut mit der Durchblutung [91, 92], jedoch führt die
Blockade von Adenosinrezeptoren zu keiner Beeinträchtigung der funktionellen
Hyperämie in der Skelettmuskulatur [93, 94]. Auch die Konzentration von
Kaliumionen steigt im arbeitenden Skelettmuskel an, da Kaliumionen bei der
Repolarisation des Aktionspotentials der Muskelzellen freigesetzt werden [95], und
dies könnte ebenfalls zu einer Gefäßdilatation im Rahmen der funktionellen
Hyperämie führen [96]. Es konnte außerdem gezeigt werden, dass eine lokale
Hypoxie durchblutungssteigernd wirkt [97], allerdings scheint dies kein

EINLEITUNG
16
wesentlicher Einflussfaktor bei der aktiven Hyperämie zu sein, da sowohl eine
Hyperoxie als auch eine verbesserte Sauerstoffversorgung bei Polyzythämie die
muskuläre Durchblutung nicht beeinflussen [98, 99]. Schließlich könnten auch die
endothelialen Autakoide NO und Prostazyklin sowie EDHF bei der Gefäßantwort
auf Muskelkontraktionen eine Rolle spielen. NO kann dabei nicht nur von den
Endothelzellen, sondern auch von den Skelettmuskelzellen produziert werden
[100, 101]. Außerdem kommt es in geringen Mengen gebunden an das
Hämoglobin in Erythrozyten vor und kann bei der Deoxygenierung freigesetzt
werden [102]. Nachdem die Bedeutung von NO für die Regulation des Ruhetonus
in Blutgefäßen entdeckt wurde, hoffte man, so auch die funktionelle Hyperämie
erklären zu können, allerdings ließ sich dies nicht oder zumindest nur partiell
bestätigen. Verschiedene Versuche, in denen unter anderem die NO-Produktion
gehemmt wurde, lieferten widersprüchliche Daten [103]. Bei Untersuchungen am
Menschen wurden sowohl Hinweise gegen [104-108] als auch für eine Beteiligung
von NO [109-113] erhoben. Ähnlich uneinheitliche Daten erbrachten Tierversuche
an Nagetieren und Hunden [114-120]. Die Datenlage bezüglich einer Rolle von
Prostazyklin ist ebenso widersprüchlich [121-124]. Beide Substanzen scheinen
also zumindest nicht in jeder Präparation und in jedem Versuchsaufbau zur
funktionellen Hyperämie beizutragen. Trotzdem scheint das Endothel eine
wichtige Funktion hierbei inne zu haben, da eine Zerstörung des Endothels der
zuführenden Blutgefäße im Tierversuch zu einer deutlich reduzierten
Blutflusssteigerung bei Muskelarbeit führte [125, 126]. Trotz zahlreicher
Untersuchungen gibt es also bis heute keine umfassende Aufklärung der
Mechanismen, die der funktionellen Hyperämie bei Skelettmuskelarbeit zu Grunde
liegen.
1.7 Ziel der vorliegenden Arbeit
Es gibt bis heute kein schlüssiges Konzept, wie die drastische
Durchblutungssteigerung bei muskulärer Arbeit und die hierfür notwendige
Dilatation der Widerstandsgefäße zu Stande kommt, obwohl viele beteiligte
Mechanismen gefunden wurden. Der Anteil dieser einzelnen Faktoren ist
allerdings jeweils nur gering und selbst die kombinierte Blockade verschiedener

EINLEITUNG
17
Mechanismen schwächte die Durchblutungssteigerung nur partiell ab. In den
letzten Jahren rückte das Endothel in den Fokus der Untersuchungen zur
funktionellen Hyperämie. Insbesondere EDHF-Typ-Dilatationen und hiermit
einhergehende Membranpotentialänderungen können zu der notwendigen
Koordination des Gefäßverhaltens führen.
Ziel der vorliegenden Arbeit war es, die Bedeutung der EDHF-Typ-Dilatation bei
der funktionellen Hyperämie im Skelettmuskel der Maus zu untersuchen. Hierzu
wurden Arteriolen im Cremastermuskel während Muskelkontraktionen mittels
Intravitalmikroskopie beobachtet. Da Calcium-abhängige Kaliumkanäle für die
Auslösung einer EDHF-Typ-Dilatation notwendig sind, wurde die funktionelle
Hyperämie in Tieren untersucht, die defizient für diese Kanäle waren. Eine weitere
Differenzierung der beteiligten Kaliumkanäle in ihre Subgruppen wurde durch die
Verwendung von Tieren mit einem selektiven knock-out einzelner Kaliumkanäle
sowie durch pharmakologische Intervention angestrebt. Ob die Ausbreitung einer
Membranpotentialänderung entlang des Endothels ebenso an der funktionellen
Hyperämie beteiligt ist, wurde schließlich an Tieren untersucht, die auf Grund
einer Defizienz für endotheliale Connexine eine gestörte Zellkopplung aufweisen.

MATERIAL & METHODEN
18
2. Material und Methoden
2.1 Versuchstiere
2.1.1 Allgemeines
Als Versuchstiere dienten Mäuse aus verschiedenen Mausstämmen. Die
untersuchten Tiere hatten zum Versuchszeitpunkt ein Gewicht zwischen 22,5 und
41 g und waren 3 bis 13 Monate alt. Die Mäuse wurden in der Tierhaltung des
Instituts für Physiologie der Universität gehalten und teils auch dort gezüchtet. Die
Tiere wurden maximal zu sechst in einem Standard-Makrolonkäfig (Typ 2)
gehalten und hatten Zugang zu Standardfutter und Trinkwasser. Alle Versuche
erfolgten nach den Richtlinien des Deutschen Tierschutzgesetzes und waren
durch das Ministerium für Umwelt, Naturschutz und Landwirtschaft des Landes
Schleswig-Holstein genehmigt (Aktenzeichen V312-72241.122-2, 02.06.2006).
Die Versuchstiere waren zum Teil genetisch modifiziert (Übersicht in Tab. 2.1).
Wt Wildtyptiere
Cx40ko Defizient für Connexin 40 (Cx40 knock-out)
Cx40KI45 Connexin 40-Gen durch Connexin 45-Gen ersetzt (Cx45 knock-in)
IKCa-ko Defizient für den IKCa-Kaliumkanal (IKCa knock-out)
dKCa-ko Defizient für den IKCa-Kaliumkanal und den SKCa-Kaliumkanal
(doppel-knock-out; induzierbarer knock-out, s.u.)
Tabelle 2.1: Versuchstiere
In Cx40ko-Tieren lag ein konventioneller knock-out für Cx40 vor [127], ebenso in
Cx40KI45-Tieren, allerdings war in diesen an Stelle der genetischen Information
für Cx40 das Gen für Cx45 eingefügt, so dass Letzteres exprimiert wurde [128]. In
IKCa-ko-Tieren lag wiederum ein konventioneller knock-out vor, so dass keine IKCa-
Kanäle exprimiert wurden [129]. dKCa-ko-Versuchstiere wurden durch Kreuzung
von IKCa-ko-Tieren mit SKCa-ko-Tieren erzeugt und von Prof. Dr. rer. nat. R. Köhler
zur Verfügung gestellt [130].

MATERIAL & METHODEN
19
Im Gegensatz zum konventionellen knock-out in IKCa-ko-Tieren handelte es sich
bei der genetischen Modifikation der SKCa-ko-Tiere um eine induzierbare
Genausschaltung. Hierbei war eine regulatorische Kassette aus verschiedenen
funktionellen Modulen zwischen dem Promotor des SKCa-Gens und dem
eigentlichen SKCa-Gen eingefügt. Auf den ursprünglichen Promotor folgte die
genetische Information für ein regulatorisches Protein, den Tetracyclin
Transaktivator (tTA), so dass dessen Expression vom SKCa-Promotor gesteuert
wurde. Zusätzlich war eine DNA-Sequenz eingefügt, die eine besonders effiziente
Transkription des tTA-Gens gewährleistet (adenovirus tripartite leader sequence).
Am Ende der eingefügten Genkassette befand sich wiederum ein Promotor (teto5
CMV), der in Anwesenheit von tTA zu einer Transkription des nachfolgenden
SKCa-Gens und so zu einer Expression des SKCa führte [131].
In derartig modifizierten dKCa-ko-Mäusen wird der IKCa nicht exprimiert, während
der SKCa überexprimiert wird, solange die Tiere kein Tetracyclin erhalten. Vor den
Experimenten wurde ihnen allerdings über mindestens 14 Tage statt des
Trinkwassers eine tetracyclinhaltige Lösung angeboten (Doxycyclin, 2 g/l).
Doxycyclin bindet an tTA und inaktiviert dieses Protein, so dass es den teto5-
Promoter nicht mehr aktiviert und die Transkription des SKCa-Gens nicht mehr
stattfindet. Zum Zeitpunkt der Versuche wurde in dKCa-ko-Tieren daher keiner
dieser beiden Kaliumkanäle exprimiert.
Cx40ko-Mäuse, Cx40KI45-Mäuse und entsprechende Wildtyptiere hatten einen
C57Bl/6-Hintergrund und waren 10-fach in diesen Hintergrund zurückgekreuzt.
IKCa- und dKCa-defiziente Tiere hatten einen SV129/C57Bl/6-Mischhintergrund und
als Kontrollen wurden Wildtyptiere mit einem entsprechenden genetischen
Hintergrund verwendet.
2.1.2 Genotypisierung
Zur Genotypisierung wurde den Mäusen im Alter von sechs Wochen ein etwa
einen Millimeter langes Stück der Schwanzspitze abgeschnitten und hieraus die
DNA extrahiert. Dafür wurden die Gewebsproben in Laird-Puffer für 24 Stunden in
einem Wasserbad zusammen mit Proteinase K bei 55 °C verdaut. Anschließend

MATERIAL & METHODEN
20
wurde die Proteinase durch 20-minütiges Erhitzen auf 95 °C inaktiviert und die
DNA mit eiskaltem Isopropanol gefällt, abzentrifugiert (13000 rcf, 15 Min.,
Microzentrifuge MC-13, Fa. Amicon, Witten) und mit Ethanol gewaschen.
Das DNA-Pellet wurde daraufhin in TE-Puffer aufgenommen und mit Hilfe der
Polymerase-Kettenreaktion (PCR) weiter untersucht. Die PCR ist eine Methode,
mit der eine sehr kleine Menge einer gesuchten DNA in großer Menge amplifiziert
werden kann, wenn die vor und hinter dem gesuchten DNA-Abschnitt liegenden
Basensequenzen bekannt sind. Dies erlaubt es, Primer zu verwenden, die zu
diesen Basensequenzen komplementär sind und so mit ihnen hybridisieren. Zur
Amplifikation wird die zu untersuchende Probe zusammen mit spezifischen
Primern, einer Nukleotidlösung (dNTPs) und einer thermostabilen DNA-
Polymerase (Taq-Polymerase) in einem Thermocycler zyklisch erwärmt und
wieder abgekühlt. Beim ersten Erwärmen werden die beiden Stränge der DNA-
Doppelhelix voneinander getrennt. Wenn der Ansatz nun wieder abgekühlt wird,
binden die Primer an die ihnen komplementären Abschnitte der DNA. Beim
anschließenden Erwärmen auf das Temperaturoptimum der Taq-Polymerase
synthetisiert diese beginnend an den Primern einen neuen DNA-Strang, der
komplementär zu der Matrize ist. Bei einem erneuten Temperaturzyklus wiederholt
sich dieser Vorgang. Die Vervielfältigung erfolgte in einem Thermocycler (iCycler,
Biorad, München). Die Reaktionsansätze, die verwendeten Primer und das
verwendete Programm sind im Anhang angegeben.
Die PCR-Produkte wurden anschließend mit einer Gelelektrophorese aufgetrennt
und identifiziert. Hierbei können DNA-Fragmente in einem Agarosegel durch ein
elektrisches Feld nach ihrer Größe separiert werden. Die Zugabe des DNA-
Interkalators Ethidiumbromid macht die DNA unter UV-Licht sichtbar. Wenn nun
die Größe der gesuchten DNA-Abschnitte bekannt ist, können die Banden
zugeordnet werden, die sich in der Elektrophorese darstellen. Die amplifizierte
DNA wurde in einem 0,5 % Agarosegel, das 0,2 µg/ml Ethidiumbromid enthielt, für
35 min bei 60 mA und 120 V in einer dafür gebauten Gelkammer aufgetrennt. Als
Negativkontrolle diente Wasser und als Referenzskala DNA Ladder Plus (100 bp).
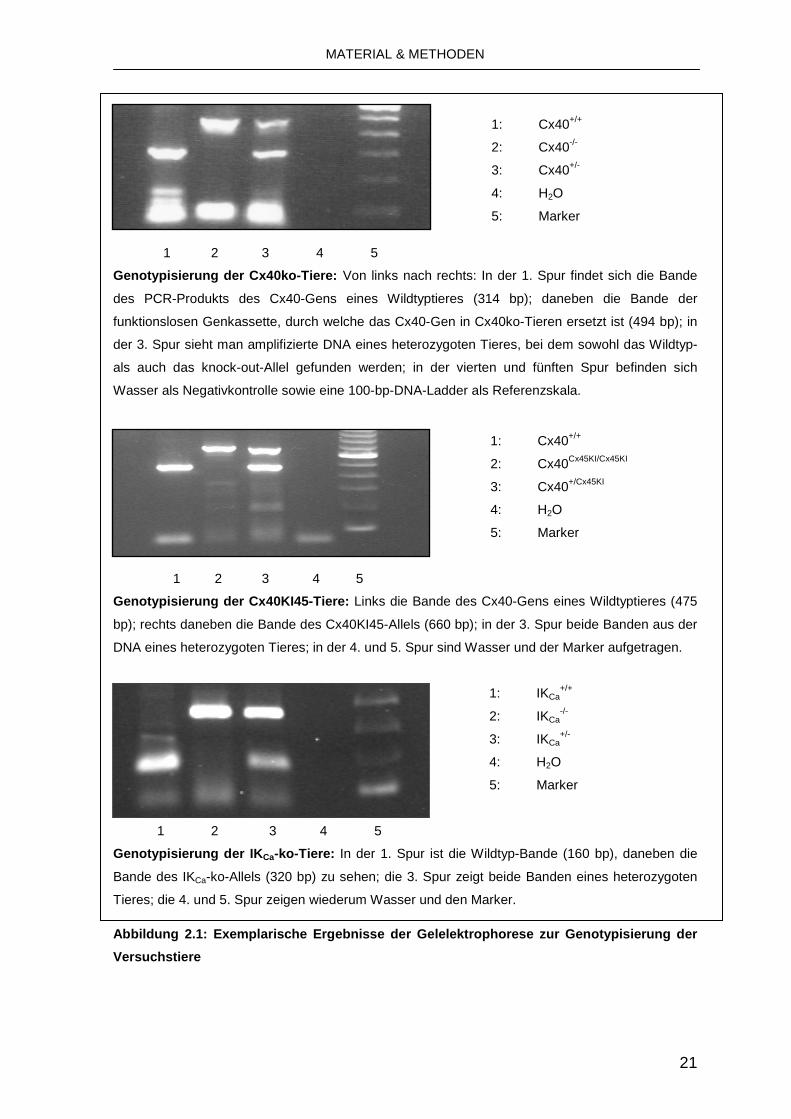
Die Länge der PCR-Produkte und exemplarische Ergebnisse sind in Abbildung 2.1
dargestellt.
MATERIAL & METHODEN
21
1: Cx40+/+
2: Cx40-/-
3: Cx40+/-
4: H2O
5: Marker
1 2 3 4 5
Genotypisierung der Cx40ko-Tiere: Von links nach rechts: In der 1. Spur findet sich die Bande
des PCR-Produkts des Cx40-Gens eines Wildtyptieres (314 bp); daneben die Bande der
funktionslosen Genkassette, durch welche das Cx40-Gen in Cx40ko-Tieren ersetzt ist (494 bp); in
der 3. Spur sieht man amplifizierte DNA eines heterozygoten Tieres, bei dem sowohl das Wildtyp-
als auch das knock-out-Allel gefunden werden; in der vierten und fünften Spur befinden sich
Wasser als Negativkontrolle sowie eine 100-bp-DNA-Ladder als Referenzskala.
1: Cx40+/+ 2: Cx40Cx45KI/Cx45KI
3: Cx40+/Cx45KI
4: H2O
5: Marker
1 2 3 4 5
Genotypisierung der Cx40KI45-Tiere: Links die Bande des Cx40-Gens eines Wildtyptieres (475
bp); rechts daneben die Bande des Cx40KI45-Allels (660 bp); in der 3. Spur beide Banden aus der
DNA eines heterozygoten Tieres; in der 4. und 5. Spur sind Wasser und der Marker aufgetragen.
1: IKCa+/+
2: IKCa-/-
3: IKCa+/-
4: H2O
5: Marker
1 2 3 4 5
Genotypisierung der IK Ca-ko-Tiere: In der 1. Spur ist die Wildtyp-Bande (160 bp), daneben die
Bande des IKCa-ko-Allels (320 bp) zu sehen; die 3. Spur zeigt beide Banden eines heterozygoten
Tieres; die 4. und 5. Spur zeigen wiederum Wasser und den Marker.
Abbildung 2.1: Exemplarische Ergebnisse der Gelelek trophorese zur Genotypisierung der
Versuchstiere

MATERIAL & METHODEN
22
Alle dKCa-ko-Versuchstiere wurden von der Arbeitsgruppe von Prof. Dr. rer. nat. R.
Köhler genotypisiert. Eine Kontrolle der erfolgten Genausschaltung wurde durch
die fehlende Dilatation nach Applikation von Acetylcholin bei knock-out-Tieren
ermöglicht (siehe Abschnitt 3.3.1).
2.2 Intravitalmikroskopie
2.2.1 Anästhesie und Präparation
Als Versuchstiere für die Intravitalmikroskopie wurden männliche Mäuse
verwendet. Sie wurden durch eine intraperitoneale Injektion eines Opiats
(Fentanyl, 0,05 mg/kg KG), eines Benzodiazepins (Midazolam, 5 mg/kg KG) und
eines zentralen α2-Agonisten (Medetomidin, 0,5 mg/kg KG) narkotisiert. Nach 15
Minuten war bei allen Mäusen das Toleranzstadium erreicht. Vor der eigentlichen
Präparation wurden der Hals und die Leistenregion rasiert.
Im ersten Schritt der Präparation wurde die Trachea freipräpariert, mit zwei Fäden
aufgespannt, inzidiert und mit einem Polyethylenkatheter (PE 50, DI: 0,5 mm, DA:
1 mm) intubiert, um die Spontanatmung der Maus zu erleichtern und das Tier im
Verlauf mechanisch beatmen zu können (Übersicht über die anatomischen
Verhältnisse in Abb. 2.2).
Danach wurde die Vena jugularis freipräpariert, aufgespannt und mit einer
Mikroschere inzidiert. In diese Öffnung wurde nun mit Hilfe eines
Operationsmikroskops (Typ 308 701, Fa. Wild GmbH, Heerbrugg, Schweiz; 6-
fache Vergrößerung) ein weiterer Polyethylenkatheter (PE 10, DI: 0,28 mm, DA:
0,61 mm) eingeführt, um ab diesem Zeitpunkt die Anästhetika mittels eines
Perfusors (Braun Perfusor secura ft, Fa. B. Braun, Melsungen) kontinuierlich
intravenös zu applizieren (Fentanyl 0,001 mg/h, Midazolam 0,1 mg/h,
Medetomidin 0,01 mg/h). Hierzu wurden diese in Ringerlösung verdünnt, die mit
einer Geschwindigkeit von 0,4 ml/h infundiert wurde. So wurden auch
Flüssigkeitsverluste durch die Präparation ausgeglichen. Die intravasale Lage des
Katheters wurde durch die Aspiration von Blut kontrolliert.

MATERIAL & METHODEN
23
Abbildung 2.2: Anatomie in der Halsregion der Maus (Schema)
Die Narkosetiefe wurde anhand des Kornealreflexes der Maus sowie durch die
Reaktion auf mechanische Reize kontrolliert und die Infusionsgeschwindigkeit der
Anästhetika bei Bedarf entsprechend korrigiert.
Anschließend wurde die Maus auf eine speziell angefertigte Unterlage überführt
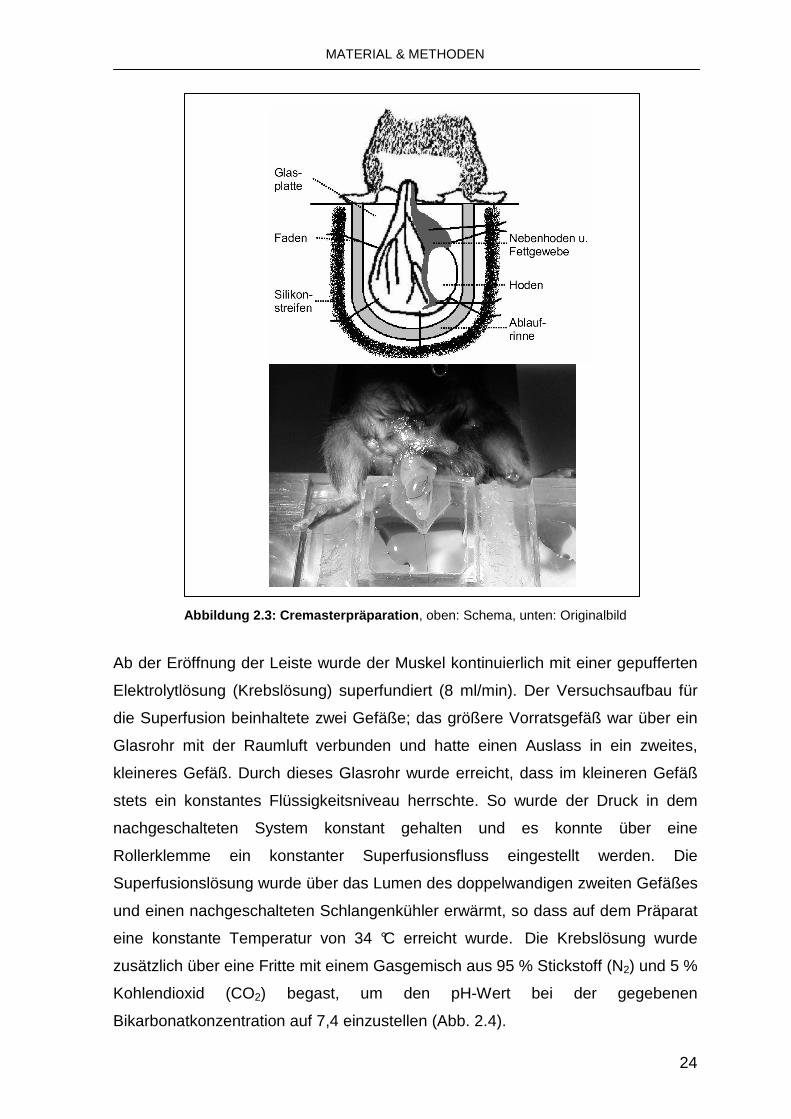
und die Leistenregion durch einen Schnitt eröffnet. Die Muskelpräparation erfolgte
nach der von Baez 1973 beschriebenen Methode [132] mit geringen
Modifikationen. Der Musculus cremaster wurde an seinem kaudalen Pol gefasst
und schonend von Bindegewebe befreit. Danach wurde er der Länge nach
eröffnet und mit mehreren Fäden über einem Deckglas aufgespannt
(Einzelknopfnähte mit atraumatischem monofilem Polypropylenfaden, 6/0
Prolene®). Das den Hoden und Nebenhoden mit dem M. cremaster verbindende
Bindegewebe wurde eingeschnitten und Hoden und Nebenhoden wurden in die
Bauchhöhle zurückgeschoben, um ein uneingeschränktes Blickfeld auf den M.
cremaster zu ermöglichen (Abb. 2.3).
Speicheldrüsen
V. jugularis
Trachea
MATERIAL & METHODEN
24
Abbildung 2.3: Cremasterpräparation , oben: Schema, unten: Originalbild
Ab der Eröffnung der Leiste wurde der Muskel kontinuierlich mit einer gepufferten
Elektrolytlösung (Krebslösung) superfundiert (8 ml/min). Der Versuchsaufbau für
die Superfusion beinhaltete zwei Gefäße; das größere Vorratsgefäß war über ein
Glasrohr mit der Raumluft verbunden und hatte einen Auslass in ein zweites,
kleineres Gefäß. Durch dieses Glasrohr wurde erreicht, dass im kleineren Gefäß
stets ein konstantes Flüssigkeitsniveau herrschte. So wurde der Druck in dem
nachgeschalteten System konstant gehalten und es konnte über eine
Rollerklemme ein konstanter Superfusionsfluss eingestellt werden. Die
Superfusionslösung wurde über das Lumen des doppelwandigen zweiten Gefäßes
und einen nachgeschalteten Schlangenkühler erwärmt, so dass auf dem Präparat
eine konstante Temperatur von 34 °C erreicht wurde. Die Krebslösung wurde
zusätzlich über eine Fritte mit einem Gasgemisch aus 95 % Stickstoff (N2) und 5 %
Kohlendioxid (CO2) begast, um den pH-Wert bei der gegebenen
Bikarbonatkonzentration auf 7,4 einzustellen (Abb. 2.4).

MATERIAL & METHODEN
25
Abbildung 2.4: Aufbau der Superfusionsvorrichtung (Schema)
Über eine peristaltische Pumpe (Fa. Ismatec, Wertheim-Mondfeld) konnten der
Superfusionslösung vasoaktive Substanzen zugeführt werden, die durch die
Superfusion gegenüber der ursprünglichen Konzentration 1:100 verdünnt wurden.
Im Folgenden wird als Konzentration immer die unmittelbar auf dem Präparat
herrschende Konzentration genannt.
Nach dem Umlagern der Maus auf den Mikroskopierplatz wurde sie über ein
mechanisches Beatmungsgerät (7025 Rodent Ventilator, Fa. Hugo Sachs
Elektronik, Freiburg) ventiliert (Frequenz 160 /min, Beatmungsdruck 2 cmH2O).
Die Effizienz der Beatmung konnte anhand der Thoraxexkursionen beurteilt
werden. Die Unterlage des Mikroskopiertisches war elektrisch beheizbar
(Elektrowerkstatt der Universität), so dass die ösophageal gemessene
Körpertemperatur der Maus bei 37 °C gehalten werden konnte. Nach Ende der
Präparation wurde eine 30-minütige Äquilibrierungsphase eingehalten, bevor das
eigentliche Versuchsprotokoll durchgeführt wurde. In dieser Zeit wurden
Narkosetiefe, Beatmung, Superfusionsfluss und Gefäßtonus im M. cremaster
regelmäßig kontrolliert. Nach Beendigung des Versuchsprotokolls wurden die
Versuchstiere mit einer letalen Dosis Pentobarbital i.v. getötet (0,3 ml, 48 mg).

MATERIAL & METHODEN
26
2.2.2 Mikroskopie
Die Mikrozirkulation im M. cremaster wurde mit einem Mikroskop (Nikon Eclipse
E600, Fa. Nikon, Tokyo, Japan) betrachtet (CFI Planfluor ECWD Objektiv: 20 x /
0,45, Fa. Nikon, Tokyo, Japan) und die mikroskopischen Bilder wurden mit einer
CCD-Kamera (Fa. PCO Computer Optics, Kelheim) auf einen Monitor übertragen
(600-fache Vergrößerung, siehe Abb. 2.5) und auf VHS-Kassette aufgezeichnet
(Videorecorder NV-FS2000, Fa. Panasonic, Osaka, Japan). Während der
Mikroskopie wurde der Muskel kontinuierlich weiter wie oben beschrieben mit
Krebslösung superfundiert.
Abbildung 2.5: Intravitalmikroskopie
(Beispielbild). Im oberen Bildabschnitt
eine Übersicht über einen Teil des
Gefäßbaumes im M. cremaster, unten
ein untersuchter Gefäßabschnitt als
Ausschnittsvergrößerung (links vor,
rechts nach Superfusion von 3 x 10-6 M
ACh). Der weiße Balken entspricht 20
µm.
L 32L 32L 32

MATERIAL & METHODEN
27
2.2.3 Nervenstimulation
Um Muskelkontraktionen auszulösen, wurde unter dem Mikroskop der den Muskel
versorgende Nerv aufgesucht. Als Stimulationselektrode diente eine
Platinelektrode (modifiziert nach Sarelius [133]). Diese bestand aus einem
Elektrodenhalter (Holder MEH7W15, Fa. WPI, Sarasota, USA), dessen Silberdraht
mit einem Platindraht (Durchmesser 25 µm) verbunden wurde. Um den Draht zu
stabilisieren, wurde eine ausgezogene Glaskapillare (Puller Model P2000, Fa.
Sutter Instruments, Novato, USA) über ihn gezogen, an deren Spitze er wenige
Millimeter herausragte. Die Öffnung der Kapillare wurde mit Silikon verschlossen.
Die Elektrode wurde mit Hilfe eines Mikromanipulators (Model M-152, Fa.
Narishige, Tokyo, Japan) unmittelbar an dem Nerven platziert. Als indifferente
Elektrode diente versilberter Kupferdraht (Durchmesser: 0,8 mm), der in die
Superfusionslösung eintauchte (siehe Abb. 2.6).
Zur Stimulation wurde eine Spannung angelegt, bei der es zu deutlich sichtbaren
Muskelkontraktionen kam (etwa 2,5 Volt, WPI Anapulse Stimulator 302-T, Fa.
WPI, Sarasota, USA). Die Stimulationsfrequenz betrug 10 oder 100 Hz. Beide
Stimulationsarten bewirkten makroskopisch deutlich sichtbare Muskel-
kontraktionen, bei Stimulation mit 10 Hz kam es zu Einzelzuckungen, bei 100 Hz
zu einer tetanischen Kontraktion. Der Muskel wurde für 1, 15 oder 60 Sekunden
stimuliert. Während bei der Stimulation über eine Sekunde kontinuierlich stimuliert
wurde, wurde bei den Stimulationen über 15 und 60 Sekunden ein
intermittierendes Stimulationsmuster aus 0,5 s Stimulation und 2,5 s Pause
verwendet (siehe Abb. 2.6). So konnte auch während der Stimulationszeit der
Gefäßdurchmesser ohne Beeinträchtigung durch Muskelkontraktionen betrachtet
werden. Die elektrischen Impulse wurden zur Kontrolle auf einem Oszilloskop (HM
205-3, Hameg Instruments, Mainhausen) dargestellt.

MATERIAL & METHODEN
28
Abbildung 2.6: Stimulation (Schema), oben: Aufbau, unten: zeitlicher Ablauf
2.2.4 Prinzipieller Versuchsablauf
Nach der Äquilibrierungsphase wurden die zu untersuchenden Gefäßabschnitte in
Arteriolen im M. cremaster ausgewählt. Es wurden Gefäße untersucht, die in der
zentralen Region des M. cremaster lagen, einen Ruhetonus aufwiesen und einen
Ruhedurchmesser von etwa 10 bis 20 µm hatten. Dann erfolgten die eigentlichen
Stimulationen (Superfusion mit vasoaktiven Substanzen bzw. elektrisch
herbeigeführte Muskelkontraktionen) und der Gefäßdurchmesser wurde
beobachtet.
Um die Dilatationsfähigkeit eines Gefäßes zu untersuchen, wurden Acetylcholin
(ACh) und Natriumnitroprussid (SNP) superfundiert. ACh wurde in Wasser gelöst
und auf die gewünschte Konzentration verdünnt. SNP wurde erst in
Natriumacetatlösung (1 mM) gelöst (auf 10 mM) und dann mit Wasser weiter auf
die gewünschte Konzentration verdünnt.

MATERIAL & METHODEN
29
In vielen Versuchen wurden außerdem Hemmstoffe bestimmter Enzyme
eingesetzt, um deren Einfluss auf den Gefäßdurchmesser auszuschalten. Hierbei
handelte es sich um Nitro-L-Arginin (L-NA, einen kompetitiven Hemmstoff der
endothelialen Stickstoffmonoxid-Synthase, eNOS) und Indomethacin (Indo, einen
kompetitiven Hemmstoff der Cyclooxygenase, COX). Sie wurden in 0,9 %iger
Natriumchloridlösung gelöst und dann kontinuierlich superfundiert (L-NA: 30 µM,
Indo: 3 µM). Mit beiden Substanzen wird nach einer Inkubationszeit von 30
Minuten eine effektive Hemmung beider Enzyme erreicht [134].
In anderen Versuchen wurde UCL 1684 eingesetzt, um den SKCa-Kaliumkanal
spezifisch zu blockieren [135-137]. Es wurde in 100 %iger DMSO-Lösung gelöst
(10 mM), mit Wasser verdünnt und dann kontinuierlich superfundiert (1 µM). Die
Experimente wurden nach einer Inkubationszeit von 20 Minuten begonnen, da
nach dieser Zeit eine suffiziente Blockade des SKCa zu erwarten ist.
Um den maximalen Durchmesser der untersuchten Gefäße zu bestimmen, wurde
das Präparat am Ende jedes Versuches mit supramaximalen Konzentrationen von
ACh, SNP und Adenosin superfundiert (jeweils 30 µM) und der resultierende
Durchmesser bestimmt.
2.2.5 Stimulationen mit vasoaktiven Substanzen
Um die Wirkung von ACh und SNP in verschiedenen Konzentrationen (10-7 bis
10-5 M) auf den Durchmesser der Arteriolen zu untersuchen, wurden pro
Versuchstier bis zu zehn Gefäßstellen unmittelbar vor und nach Zugabe des
jeweiligen Agonisten aufgesucht und es wurden Bildsequenzen der
Gefäßabschnitte aufgezeichnet. Das Aufsuchen von zehn verschiedenen
Gefäßabschnitten erfolgte in etwa einer Minute. Nachdem der Ruhedurchmesser
der Gefäße wieder annähernd erreicht war, wurde dieser Ablauf mit der
nächsthöheren Konzentration wiederholt.
MATERIAL & METHODEN
30
2.2.6 Untersuchungen zur funktionellen Hyperämie
Um die Gefäßreaktion auf muskuläre Arbeit zu untersuchen, wurde pro
Versuchstier eine Arteriole im Muskel betrachtet, die in der Nähe des stimulierten
motorischen Nerven und nicht unmittelbar benachbart zu einer Vene lag, um die
Diffusion von vasoaktiven Substanzen aus dem venösen Blut auszuschließen. Der
Durchmesser dieser Arteriole wurde sowohl unter Ruhebedingungen als auch bei
muskulärer Arbeit untersucht. Die muskuläre Arbeit wurde durch elektrische
Stimulation (siehe Abschnitt 2.2.3) nach dem Protokoll in Tab. 2.2 induziert.
Stimulation Dauer (s) Frequenz (Hz) Modus
1 1 10 Kontinuierlich
2 1 100 Kontinuierlich
3 15 10 Intermittierend
4 15 100 Intermittierend
5 60 10 Intermittierend
6 60 100 Intermittierend
Tabelle 2.2: Charakteristika der elektrischen Stimu lation zur Auslösung von Kontraktionen
Die Arteriolen wurden jeweils eine halbe Minute vor, während und zwei Minuten
nach der Stimulation aufgezeichnet. Die nächste Stimulation wurde erst
angewendet, nachdem die Gefäße ihren Ruhedurchmesser wieder erreicht hatten.
Zusätzlich wurde auch bei diesen Versuchen die durch Superfusion von ACh und
SNP (3 x 10-6 M) induzierte Durchmesseränderung untersucht.
2.2.7 Auswertung und Statistik
Die Bilder wurden zur Auswertung digitalisiert und die Auswertung erfolgte am
Computer mit handelsüblicher Laborsoftware (LABView, Fa. Texas Instruments,
Austin, USA), die im Institut für Physiologie an die Erfordernisse angepasst
worden war. Im Fall der Stimulation mit vasoaktiven Substanzen erfolgte eine
Durchmesserbestimmung der Gefäße zu einem Zeitpunkt unmittelbar vor bzw.
nach der Applikation der Agonisten. In Versuchen zur funktionellen Hyperämie

MATERIAL & METHODEN
31
erfolgte die Auswertung kontinuierlich über die Beobachtungszeit mit zwei
Messwerten pro Sekunde. In beiden Fällen wurden die Gefäßwände durch den
Auswertenden mit Hilfe der Maus bzw. der Cursortasten markiert und daraus der
Durchmesser errechnet (siehe Abb. 2.7). Die theoretisch erreichbare Auflösung
hierbei betrug 0,5 µm. Das Messsystem war zuvor mit einem Objektmikrometer
kalibriert worden.
Abbildung 2.7: Computergestützte
Auswertung des Gefäßdurch-
messers. Die Gefäßwände wurden
mit Hilfe der Cursortasten mit einem
Kasten markiert und hieraus der
Durchmesser errechnet.
Um Dilatationen in Gefäßen mit unterschiedlichem Ruhe- und Maximal-
durchmesser vergleichen zu können, wurde der Gefäßdurchmesser als Prozent
der maximal möglichen Dilatation angegeben und nach der folgenden Formel
berechnet:
% der maximalen Dilatation = [(D – D0) / (Dmax - D0)] x 100
D : Durchmesser des Gefäßes zum Beobachtungszeitpunkt
D0 : Ruhedurchmesser des Gefäßes unmittelbar vor der Stimulation
Dmax : Maximaler Durchmesser des Gefäßes
Bei einer Konstriktion des beobachteten Gefäßes erfolgte eine entsprechende
Berechnung nach der folgenden Formel:
% der maximalen Dilatation = (D / D0 x 100) – 100

MATERIAL & METHODEN
32
Der Tonus (Kontraktionszustand) eines Gefäßes wurde nach der folgenden
Formel berechnet und ist daher als dimensionslose Zahl angegeben:
Tonus = D0 / Dmax
Nach dieser Berechnung entspricht eine kleine Zahl einem hohen
Kontraktionszustand des Gefäßes.
Die statistische Auswertung erfolgte mit dem Programm Stata 8.0 (Fa. Stata Corp.
LP, College Station, USA), die graphische Darstellung mit dem Programm
SigmaPlot 2001 (Fa. SPSS Inc., Chicago, USA).
In die Analyse wurden nur Versuche einbezogen, bei denen das Protokoll
vollständig durchgeführt werden konnte. Die Ergebnisse sind im Folgenden als
Mittelwert ± Standardfehler (MW ± SEM) angegeben. Innerhalb einer Gruppe
wurden die Daten mittels des gepaarten t-tests verglichen, bei Vergleichen
zwischen verschiedenen Gruppen wurde eine Varianzanalyse durchgeführt. Bei
Mehrfachvergleichen wurde eine Korrektur nach Bonferoni angewendet.
Unterschiede wurden bei einer Irrtumswahrscheinlichkeit von 5 % (p ≤ 0,05) als
signifikant erachtet.
2.3 Textverarbeitung
Als Textverarbeitungsprogramm zur Verfassung der Dissertation diente Microsoft
Word 2002 (Fa. Microsoft, Seattle, USA), die Literaturverwaltung erfolgte mit
Endnote 9.0 (Fa. Thomson Scientific, Philadelphia, USA). Zum Erstellen einiger
Abbildungen dienten Microsoft Paint 5.1 und Microsoft Powerpoint 2002 (beides
Fa. Microsoft, Seattle, USA).

ERGEBNISSE
33
3. Ergebnisse
3.1 Gefäßdilatationen durch vasoaktive Substanzen
Zunächst wurde die Gefäßreaktion auf Acetylcholin (ACh) sowie auf
Natriumnitroprussid (SNP) in Wildtyp- und Cx40-defizienten Tieren analysiert. So
kann die Dilatationsfähigkeit der Gefäße beurteilt werden und es wird deutlich, ob
das Endothel in den knock-out-Mäusen trotz des veränderten Genotyps noch in
der Lage ist, eine suffiziente Gefäßdilatation hervorzurufen. Die Versuche wurden
in Anwesenheit von Indomethacin (Indo) und Nitro-L-Arginin (L-NA) wiederholt, um
den Effekt einzelner endothelialer Signalwege bei der Dilatation zu differenzieren.
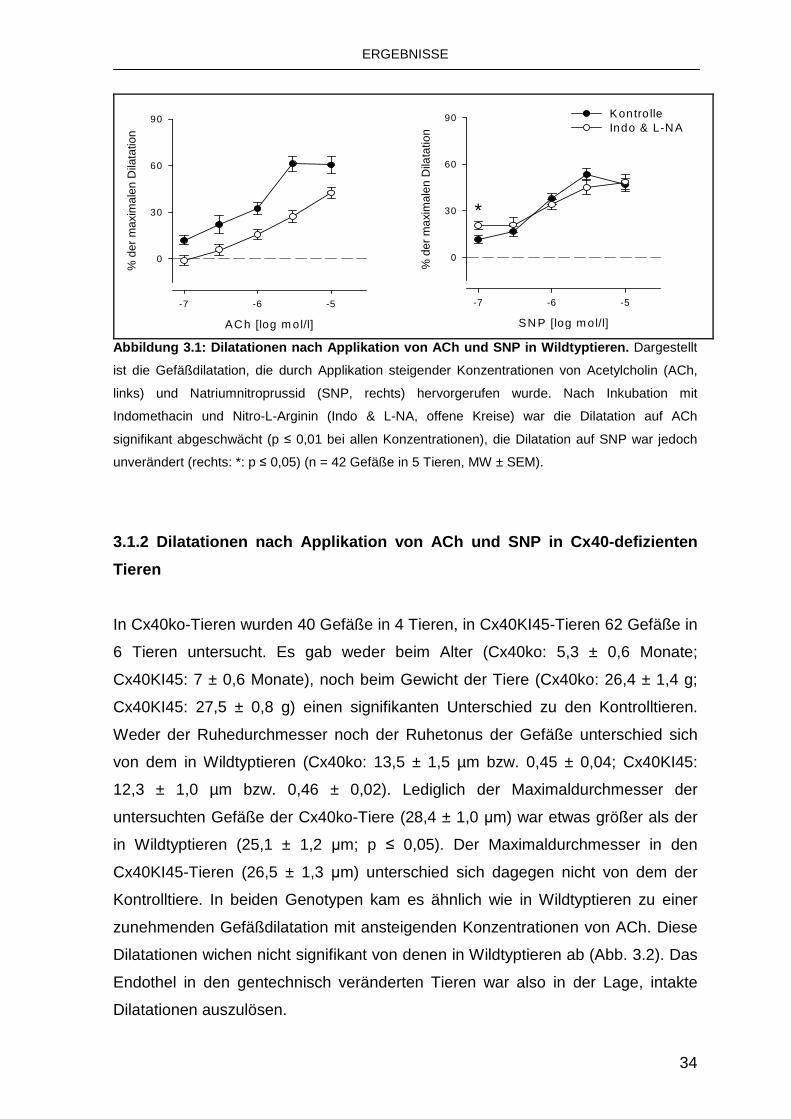
3.1.1 Dilatationen nach Applikation von ACh und SNP in Wildtyptieren
Es wurden 42 verschiedene Gefäße in 5 Tieren untersucht. Das Alter der Tiere
betrug zum Zeitpunkt der Versuche 6 ± 0,6 Monate, das Gewicht 28,3 ± 1,3 g. Der
Ruhedurchmesser der betrachteten Gefäße lag bei 12,3 ± 1,3 µm und der
Ruhetonus bei 0,47 ± 0,03. Der Maximaldurchmesser war 25,1 ± 1,2 µm. Die
Applikation ansteigender Konzentrationen von ACh führte zu einer
konzentrationsabhängigen Gefäßdilatation. Eine etwas geringer ausgeprägte
Dilatation wurde bei Gabe von SNP beobachtet (Abb. 3.1). Der Ruhedurchmesser
verringerte sich nach Inkubation mit Indo und L-NA signifikant auf 7,9 ± 0,9 µm (p
≤ 0,001) und somit war der Ruhetonus unter diesen Bedingungen ausgeprägter
(0,31 ± 0,02; p ≤ 0,001). Die Gefäßantwort auf ACh war nach dieser Behandlung
bei allen verwendeten Konzentrationen signifikant reduziert (p ≤ 0,01), es verblieb
jedoch eine deutlich ausgeprägte Dilatation. Im Gegensatz dazu war die
Gefäßdilatation auf SNP im Wesentlichen unverändert (Abb. 3.1). Dies zeigt, dass
ein geringer Anteil der ACh-vermittelten Dilatation im untersuchten Gefäßgebiet
durch NO und Prostaglandine vermittelt wird, die Dilatationsfähigkeit der
Gefäßmuskulatur bleibt jedoch nach Blockade dieser beiden Signalwege
unverändert.
ERGEBNISSE
34
* *
**
*
SN P [log m ol/l]
-7 -6 -5
% d
er m
axim
alen
Dila
tatio
n
0
30
60
90
*
ACh [log m ol/l]
-7 -6 -5
% d
er m
axim
alen
Dila
tatio
n
0
30
60
90 Kontro lleIndo & L-NA
Abbildung 3.1: Dilatationen nach Applikation von AC h und SNP in Wildtyptieren. Dargestellt
ist die Gefäßdilatation, die durch Applikation steigender Konzentrationen von Acetylcholin (ACh,
links) und Natriumnitroprussid (SNP, rechts) hervorgerufen wurde. Nach Inkubation mit
Indomethacin und Nitro-L-Arginin (Indo & L-NA, offene Kreise) war die Dilatation auf ACh
signifikant abgeschwächt (p ≤ 0,01 bei allen Konzentrationen), die Dilatation auf SNP war jedoch
unverändert (rechts: *: p ≤ 0,05) (n = 42 Gefäße in 5 Tieren, MW ± SEM).
3.1.2 Dilatationen nach Applikation von ACh und SNP in Cx40-defizienten
Tieren
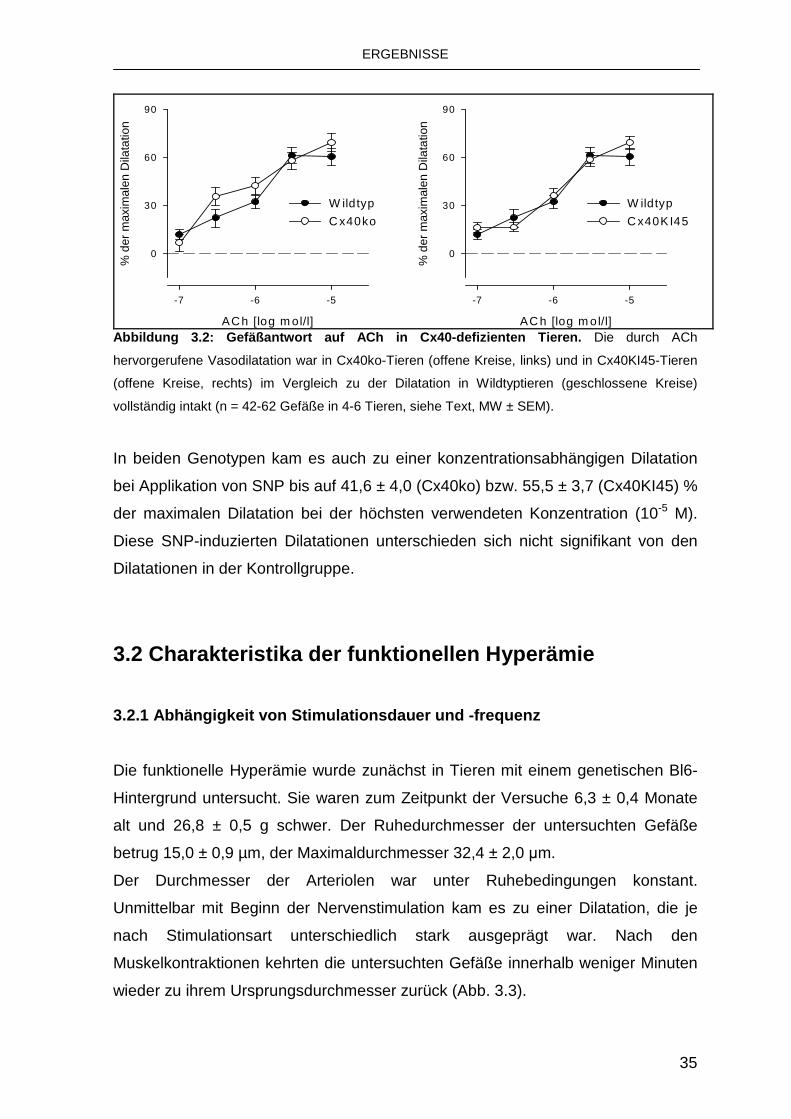
In Cx40ko-Tieren wurden 40 Gefäße in 4 Tieren, in Cx40KI45-Tieren 62 Gefäße in
6 Tieren untersucht. Es gab weder beim Alter (Cx40ko: 5,3 ± 0,6 Monate;
Cx40KI45: 7 ± 0,6 Monate), noch beim Gewicht der Tiere (Cx40ko: 26,4 ± 1,4 g;
Cx40KI45: 27,5 ± 0,8 g) einen signifikanten Unterschied zu den Kontrolltieren.
Weder der Ruhedurchmesser noch der Ruhetonus der Gefäße unterschied sich
von dem in Wildtyptieren (Cx40ko: 13,5 ± 1,5 µm bzw. 0,45 ± 0,04; Cx40KI45:
12,3 ± 1,0 µm bzw. 0,46 ± 0,02). Lediglich der Maximaldurchmesser der
untersuchten Gefäße der Cx40ko-Tiere (28,4 ± 1,0 µm) war etwas größer als der
in Wildtyptieren (25,1 ± 1,2 µm; p ≤ 0,05). Der Maximaldurchmesser in den
Cx40KI45-Tieren (26,5 ± 1,3 µm) unterschied sich dagegen nicht von dem der
Kontrolltiere. In beiden Genotypen kam es ähnlich wie in Wildtyptieren zu einer
zunehmenden Gefäßdilatation mit ansteigenden Konzentrationen von ACh. Diese
Dilatationen wichen nicht signifikant von denen in Wildtyptieren ab (Abb. 3.2). Das
Endothel in den gentechnisch veränderten Tieren war also in der Lage, intakte
Dilatationen auszulösen.
ERGEBNISSE
35
ACh [log m ol/l]
-7 -6 -5
% d
er m
axim
alen
Dila
tatio
n
0
30
60
90
AC h [log m ol/l]
-7 -6 -5
% d
er m
axim
alen
Dila
tatio
n
0
30
60
90
W ildtyp
C x40KI45
W ildtyp
C x40ko
Abbildung 3.2: Gefäßantwort auf ACh in Cx40-defizie nten Tieren. Die durch ACh
hervorgerufene Vasodilatation war in Cx40ko-Tieren (offene Kreise, links) und in Cx40KI45-Tieren
(offene Kreise, rechts) im Vergleich zu der Dilatation in Wildtyptieren (geschlossene Kreise)
vollständig intakt (n = 42-62 Gefäße in 4-6 Tieren, siehe Text, MW ± SEM).
In beiden Genotypen kam es auch zu einer konzentrationsabhängigen Dilatation
bei Applikation von SNP bis auf 41,6 ± 4,0 (Cx40ko) bzw. 55,5 ± 3,7 (Cx40KI45) %
der maximalen Dilatation bei der höchsten verwendeten Konzentration (10-5 M).
Diese SNP-induzierten Dilatationen unterschieden sich nicht signifikant von den
Dilatationen in der Kontrollgruppe.
3.2 Charakteristika der funktionellen Hyperämie
3.2.1 Abhängigkeit von Stimulationsdauer und -frequ enz
Die funktionelle Hyperämie wurde zunächst in Tieren mit einem genetischen Bl6-
Hintergrund untersucht. Sie waren zum Zeitpunkt der Versuche 6,3 ± 0,4 Monate
alt und 26,8 ± 0,5 g schwer. Der Ruhedurchmesser der untersuchten Gefäße
betrug 15,0 ± 0,9 µm, der Maximaldurchmesser 32,4 ± 2,0 µm.
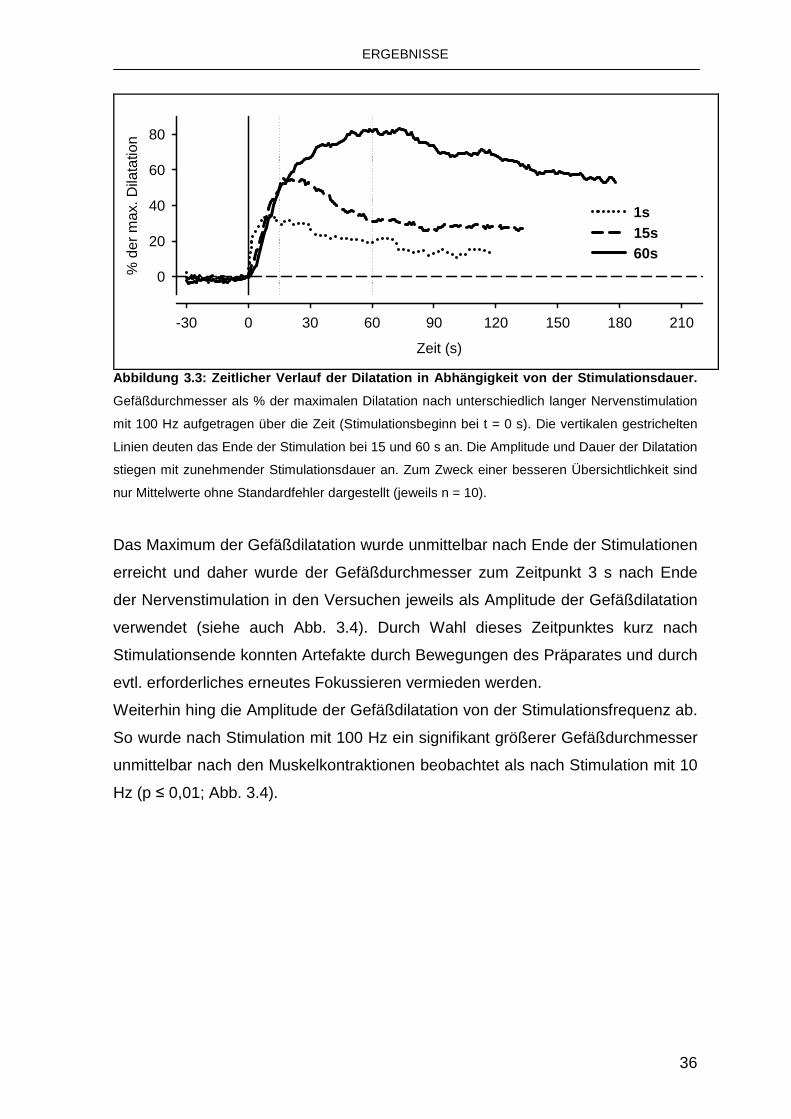
Der Durchmesser der Arteriolen war unter Ruhebedingungen konstant.
Unmittelbar mit Beginn der Nervenstimulation kam es zu einer Dilatation, die je
nach Stimulationsart unterschiedlich stark ausgeprägt war. Nach den
Muskelkontraktionen kehrten die untersuchten Gefäße innerhalb weniger Minuten
wieder zu ihrem Ursprungsdurchmesser zurück (Abb. 3.3).
ERGEBNISSE
36
Zeit (s)
-30 0 30 60 90 120 150 180 210
% d
er m
ax. D
ilata
tion
0
20
40
60
80
1s15s 60s
Abbildung 3.3: Zeitlicher Verlauf der Dilatation in Abhängigkeit von der Stimulationsdauer.
Gefäßdurchmesser als % der maximalen Dilatation nach unterschiedlich langer Nervenstimulation
mit 100 Hz aufgetragen über die Zeit (Stimulationsbeginn bei t = 0 s). Die vertikalen gestrichelten
Linien deuten das Ende der Stimulation bei 15 und 60 s an. Die Amplitude und Dauer der Dilatation
stiegen mit zunehmender Stimulationsdauer an. Zum Zweck einer besseren Übersichtlichkeit sind
nur Mittelwerte ohne Standardfehler dargestellt (jeweils n = 10).
Das Maximum der Gefäßdilatation wurde unmittelbar nach Ende der Stimulationen
erreicht und daher wurde der Gefäßdurchmesser zum Zeitpunkt 3 s nach Ende
der Nervenstimulation in den Versuchen jeweils als Amplitude der Gefäßdilatation
verwendet (siehe auch Abb. 3.4). Durch Wahl dieses Zeitpunktes kurz nach
Stimulationsende konnten Artefakte durch Bewegungen des Präparates und durch
evtl. erforderliches erneutes Fokussieren vermieden werden.
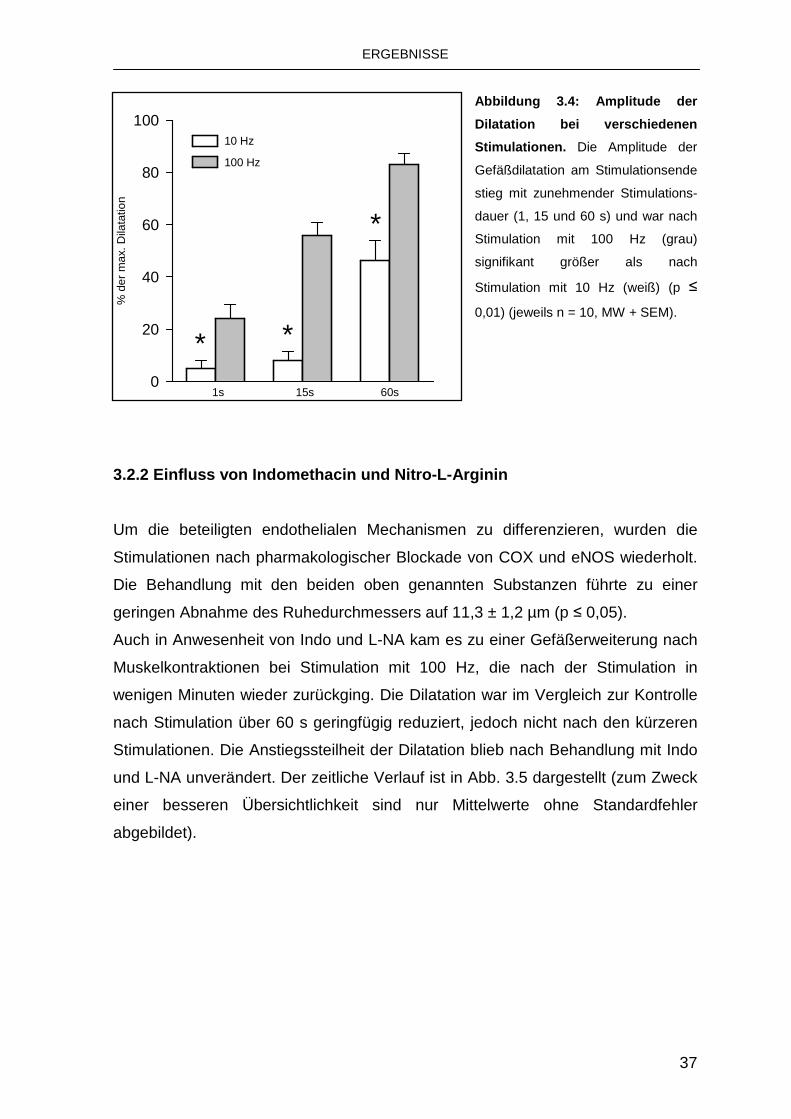
Weiterhin hing die Amplitude der Gefäßdilatation von der Stimulationsfrequenz ab.
So wurde nach Stimulation mit 100 Hz ein signifikant größerer Gefäßdurchmesser
unmittelbar nach den Muskelkontraktionen beobachtet als nach Stimulation mit 10
Hz (p ≤ 0,01; Abb. 3.4).
ERGEBNISSE
37
Abbildung 3.4: Amplitude der
Dilatation bei verschiedenen
Stimulationen. Die Amplitude der
Gefäßdilatation am Stimulationsende
stieg mit zunehmender Stimulations-
dauer (1, 15 und 60 s) und war nach
Stimulation mit 100 Hz (grau)
signifikant größer als nach
Stimulation mit 10 Hz (weiß) (p ≤
0,01) (jeweils n = 10, MW + SEM).
3.2.2 Einfluss von Indomethacin und Nitro- L-Arginin
Um die beteiligten endothelialen Mechanismen zu differenzieren, wurden die
Stimulationen nach pharmakologischer Blockade von COX und eNOS wiederholt.
Die Behandlung mit den beiden oben genannten Substanzen führte zu einer
geringen Abnahme des Ruhedurchmessers auf 11,3 ± 1,2 µm (p ≤ 0,05).
Auch in Anwesenheit von Indo und L-NA kam es zu einer Gefäßerweiterung nach
Muskelkontraktionen bei Stimulation mit 100 Hz, die nach der Stimulation in
wenigen Minuten wieder zurückging. Die Dilatation war im Vergleich zur Kontrolle
nach Stimulation über 60 s geringfügig reduziert, jedoch nicht nach den kürzeren
Stimulationen. Die Anstiegssteilheit der Dilatation blieb nach Behandlung mit Indo
und L-NA unverändert. Der zeitliche Verlauf ist in Abb. 3.5 dargestellt (zum Zweck
einer besseren Übersichtlichkeit sind nur Mittelwerte ohne Standardfehler
abgebildet).
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
1s
10 Hz
100 Hz
60s15s
*
* *
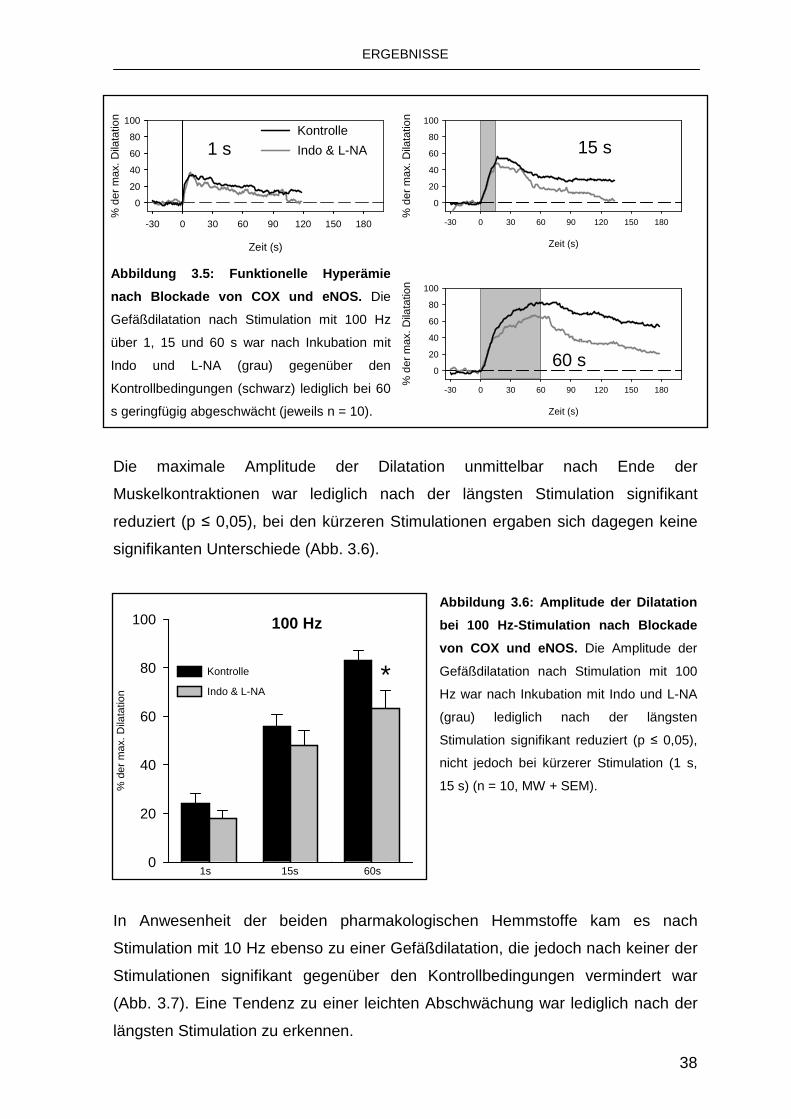
ERGEBNISSE
38
Die maximale Amplitude der Dilatation unmittelbar nach Ende der
Muskelkontraktionen war lediglich nach der längsten Stimulation signifikant
reduziert (p ≤ 0,05), bei den kürzeren Stimulationen ergaben sich dagegen keine
signifikanten Unterschiede (Abb. 3.6).
Abbildung 3.6: Amplitude der Dilatation
bei 100 Hz-Stimulation nach Blockade
von COX und eNOS. Die Amplitude der
Gefäßdilatation nach Stimulation mit 100
Hz war nach Inkubation mit Indo und L-NA
(grau) lediglich nach der längsten
Stimulation signifikant reduziert (p ≤ 0,05),
nicht jedoch bei kürzerer Stimulation (1 s,
15 s) (n = 10, MW + SEM).
In Anwesenheit der beiden pharmakologischen Hemmstoffe kam es nach
Stimulation mit 10 Hz ebenso zu einer Gefäßdilatation, die jedoch nach keiner der
Stimulationen signifikant gegenüber den Kontrollbedingungen vermindert war
(Abb. 3.7). Eine Tendenz zu einer leichten Abschwächung war lediglich nach der
längsten Stimulation zu erkennen.
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
15 s1 s
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
60 s
Indo & L-NA
Kontrolle
Abbildung 3.5: Funktionelle Hyperämie
nach Blockade von COX und eNOS. Die
Gefäßdilatation nach Stimulation mit 100 Hz
über 1, 15 und 60 s war nach Inkubation mit
Indo und L-NA (grau) gegenüber den
Kontrollbedingungen (schwarz) lediglich bei 60
s geringfügig abgeschwächt (jeweils n = 10).
1s 60s15s
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
*
100 Hz
Kontrolle
Indo & L-NA
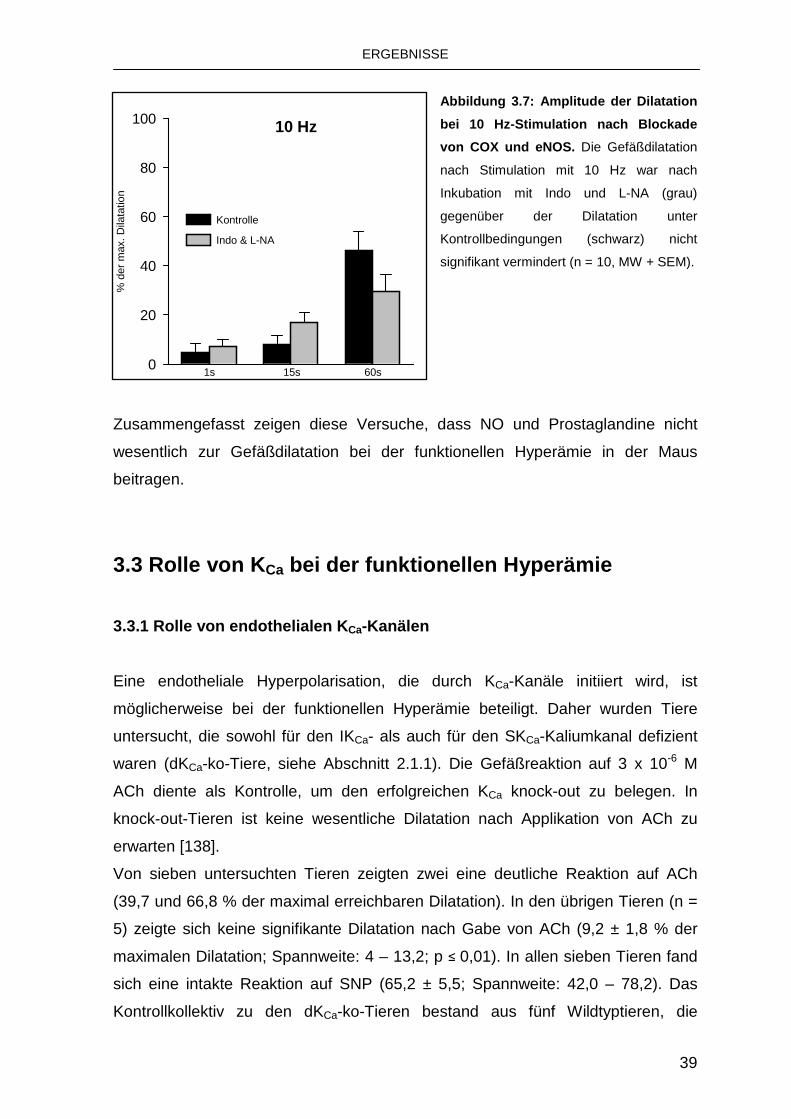
ERGEBNISSE
39
Abbildung 3.7: Amplitude der Dilatation
bei 10 Hz-Stimulation nach Blockade
von COX und eNOS. Die Gefäßdilatation
nach Stimulation mit 10 Hz war nach
Inkubation mit Indo und L-NA (grau)
gegenüber der Dilatation unter
Kontrollbedingungen (schwarz) nicht
signifikant vermindert (n = 10, MW + SEM).
Zusammengefasst zeigen diese Versuche, dass NO und Prostaglandine nicht
wesentlich zur Gefäßdilatation bei der funktionellen Hyperämie in der Maus
beitragen.
3.3 Rolle von K Ca bei der funktionellen Hyperämie
3.3.1 Rolle von endothelialen K Ca-Kanälen
Eine endotheliale Hyperpolarisation, die durch KCa-Kanäle initiiert wird, ist
möglicherweise bei der funktionellen Hyperämie beteiligt. Daher wurden Tiere
untersucht, die sowohl für den IKCa- als auch für den SKCa-Kaliumkanal defizient
waren (dKCa-ko-Tiere, siehe Abschnitt 2.1.1). Die Gefäßreaktion auf 3 x 10-6 M
ACh diente als Kontrolle, um den erfolgreichen KCa knock-out zu belegen. In
knock-out-Tieren ist keine wesentliche Dilatation nach Applikation von ACh zu
erwarten [138].
Von sieben untersuchten Tieren zeigten zwei eine deutliche Reaktion auf ACh
(39,7 und 66,8 % der maximal erreichbaren Dilatation). In den übrigen Tieren (n =
5) zeigte sich keine signifikante Dilatation nach Gabe von ACh (9,2 ± 1,8 % der
maximalen Dilatation; Spannweite: 4 – 13,2; p ≤ 0,01). In allen sieben Tieren fand
sich eine intakte Reaktion auf SNP (65,2 ± 5,5; Spannweite: 42,0 – 78,2). Das
Kontrollkollektiv zu den dKCa-ko-Tieren bestand aus fünf Wildtyptieren, die
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100 10 Hz
1s 60s15s
Kontrolle
Indo & L-NA
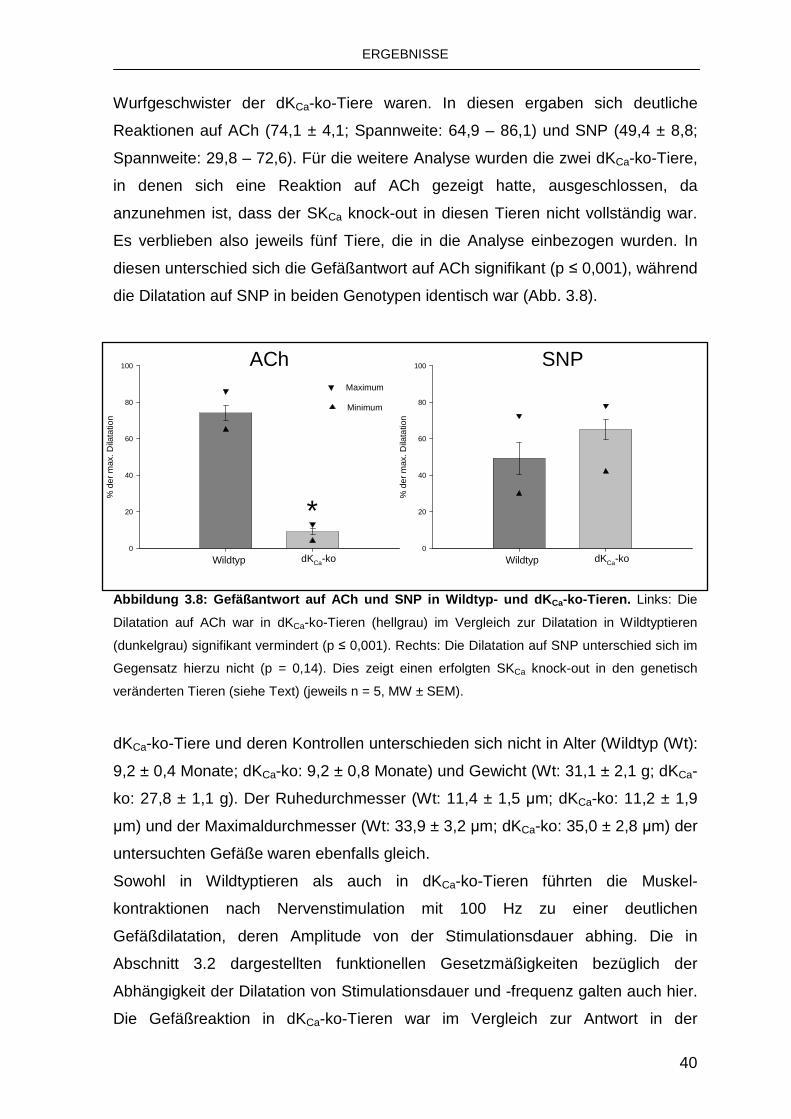
ERGEBNISSE
40
Wurfgeschwister der dKCa-ko-Tiere waren. In diesen ergaben sich deutliche
Reaktionen auf ACh (74,1 ± 4,1; Spannweite: 64,9 – 86,1) und SNP (49,4 ± 8,8;
Spannweite: 29,8 – 72,6). Für die weitere Analyse wurden die zwei dKCa-ko-Tiere,
in denen sich eine Reaktion auf ACh gezeigt hatte, ausgeschlossen, da
anzunehmen ist, dass der SKCa knock-out in diesen Tieren nicht vollständig war.
Es verblieben also jeweils fünf Tiere, die in die Analyse einbezogen wurden. In
diesen unterschied sich die Gefäßantwort auf ACh signifikant (p ≤ 0,001), während
die Dilatation auf SNP in beiden Genotypen identisch war (Abb. 3.8).
Abbildung 3.8: Gefäßantwort auf ACh und SNP in Wildtyp- und dK Ca-ko-Tieren. Links: Die
Dilatation auf ACh war in dKCa-ko-Tieren (hellgrau) im Vergleich zur Dilatation in Wildtyptieren
(dunkelgrau) signifikant vermindert (p ≤ 0,001). Rechts: Die Dilatation auf SNP unterschied sich im
Gegensatz hierzu nicht (p = 0,14). Dies zeigt einen erfolgten SKCa knock-out in den genetisch
veränderten Tieren (siehe Text) (jeweils n = 5, MW ± SEM).
dKCa-ko-Tiere und deren Kontrollen unterschieden sich nicht in Alter (Wildtyp (Wt):
9,2 ± 0,4 Monate; dKCa-ko: 9,2 ± 0,8 Monate) und Gewicht (Wt: 31,1 ± 2,1 g; dKCa-
ko: 27,8 ± 1,1 g). Der Ruhedurchmesser (Wt: 11,4 ± 1,5 µm; dKCa-ko: 11,2 ± 1,9
µm) und der Maximaldurchmesser (Wt: 33,9 ± 3,2 µm; dKCa-ko: 35,0 ± 2,8 µm) der
untersuchten Gefäße waren ebenfalls gleich.
Sowohl in Wildtyptieren als auch in dKCa-ko-Tieren führten die Muskel-
kontraktionen nach Nervenstimulation mit 100 Hz zu einer deutlichen
Gefäßdilatation, deren Amplitude von der Stimulationsdauer abhing. Die in
Abschnitt 3.2 dargestellten funktionellen Gesetzmäßigkeiten bezüglich der
Abhängigkeit der Dilatation von Stimulationsdauer und -frequenz galten auch hier.
Die Gefäßreaktion in dKCa-ko-Tieren war im Vergleich zur Antwort in der
ACh
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Minimum
Maximum
Wildtyp dKCa-ko
*
SNP
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Wildtyp dKCa-ko
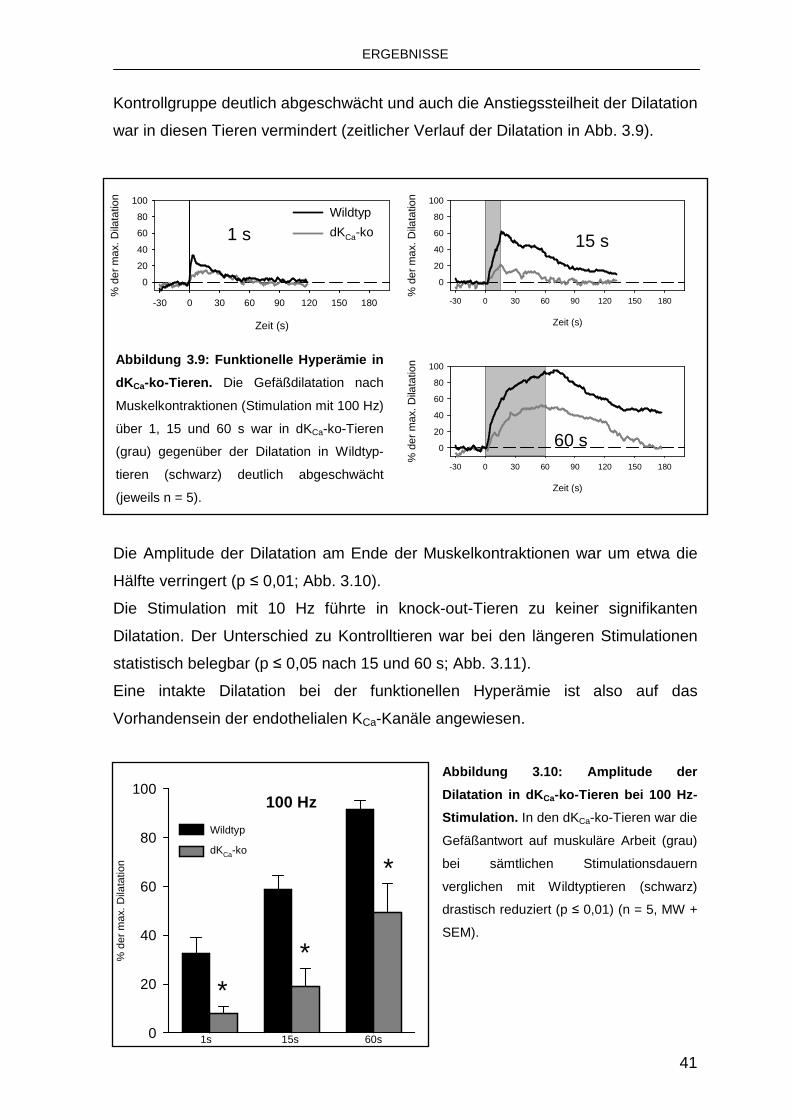
ERGEBNISSE
41
Kontrollgruppe deutlich abgeschwächt und auch die Anstiegssteilheit der Dilatation
war in diesen Tieren vermindert (zeitlicher Verlauf der Dilatation in Abb. 3.9).
Die Amplitude der Dilatation am Ende der Muskelkontraktionen war um etwa die
Hälfte verringert (p ≤ 0,01; Abb. 3.10).
Die Stimulation mit 10 Hz führte in knock-out-Tieren zu keiner signifikanten
Dilatation. Der Unterschied zu Kontrolltieren war bei den längeren Stimulationen
statistisch belegbar (p ≤ 0,05 nach 15 und 60 s; Abb. 3.11).
Eine intakte Dilatation bei der funktionellen Hyperämie ist also auf das
Vorhandensein der endothelialen KCa-Kanäle angewiesen.
Abbildung 3.10: Amplitude der
Dilatation in dK Ca-ko-Tieren bei 100 Hz-
Stimulation. In den dKCa-ko-Tieren war die
Gefäßantwort auf muskuläre Arbeit (grau)
bei sämtlichen Stimulationsdauern
verglichen mit Wildtyptieren (schwarz)
drastisch reduziert (p ≤ 0,01) (n = 5, MW +
SEM).
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
15 s1 s
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
60 s
dKCa-ko
Wildtyp
Abbildung 3.9: Funktionelle Hyperämie in
dKCa-ko-Tieren. Die Gefäßdilatation nach
Muskelkontraktionen (Stimulation mit 100 Hz)
über 1, 15 und 60 s war in dKCa-ko-Tieren
(grau) gegenüber der Dilatation in Wildtyp-
tieren (schwarz) deutlich abgeschwächt
(jeweils n = 5).
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100100 Hz
1s 60s15s
Wildtyp
dKCa-ko
**
*
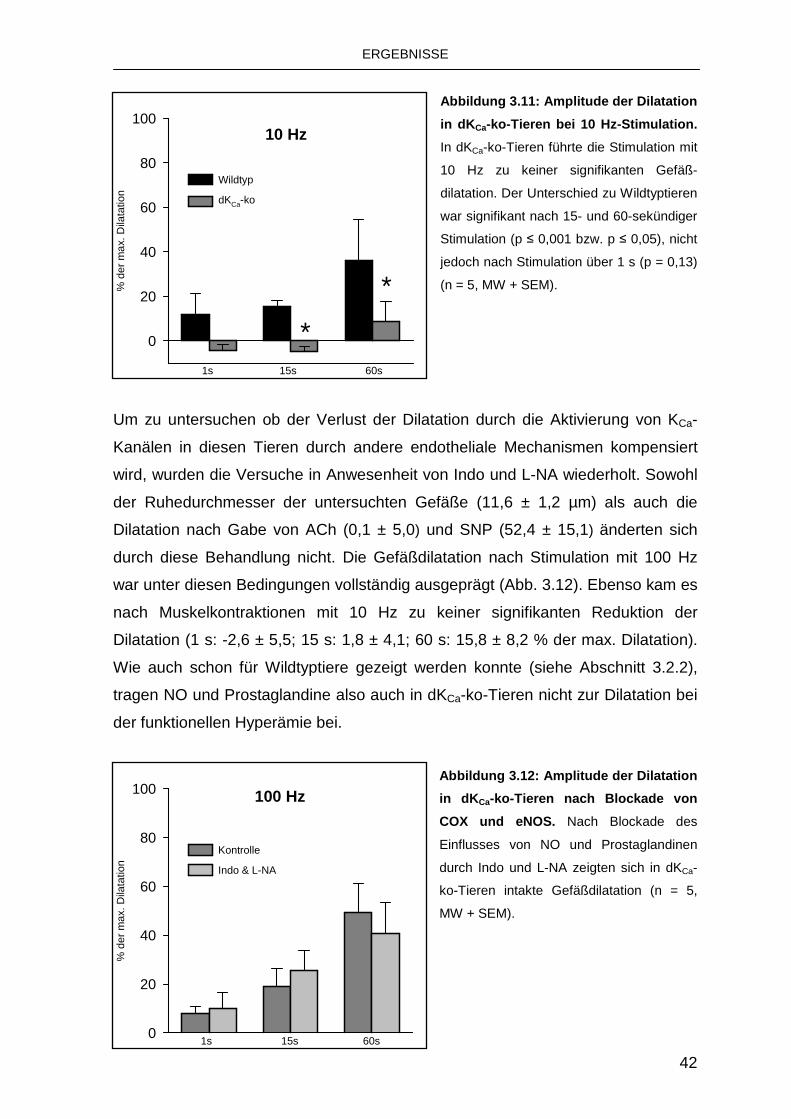
ERGEBNISSE
42
Abbildung 3.11: Amplitude der Dilatation
in dK Ca-ko-Tieren bei 10 Hz-Stimulation.
In dKCa-ko-Tieren führte die Stimulation mit
10 Hz zu keiner signifikanten Gefäß-
dilatation. Der Unterschied zu Wildtyptieren
war signifikant nach 15- und 60-sekündiger
Stimulation (p ≤ 0,001 bzw. p ≤ 0,05), nicht
jedoch nach Stimulation über 1 s (p = 0,13)
(n = 5, MW + SEM).
Um zu untersuchen ob der Verlust der Dilatation durch die Aktivierung von KCa-
Kanälen in diesen Tieren durch andere endotheliale Mechanismen kompensiert
wird, wurden die Versuche in Anwesenheit von Indo und L-NA wiederholt. Sowohl
der Ruhedurchmesser der untersuchten Gefäße (11,6 ± 1,2 µm) als auch die
Dilatation nach Gabe von ACh (0,1 ± 5,0) und SNP (52,4 ± 15,1) änderten sich
durch diese Behandlung nicht. Die Gefäßdilatation nach Stimulation mit 100 Hz
war unter diesen Bedingungen vollständig ausgeprägt (Abb. 3.12). Ebenso kam es
nach Muskelkontraktionen mit 10 Hz zu keiner signifikanten Reduktion der
Dilatation (1 s: -2,6 ± 5,5; 15 s: 1,8 ± 4,1; 60 s: 15,8 ± 8,2 % der max. Dilatation).
Wie auch schon für Wildtyptiere gezeigt werden konnte (siehe Abschnitt 3.2.2),
tragen NO und Prostaglandine also auch in dKCa-ko-Tieren nicht zur Dilatation bei
der funktionellen Hyperämie bei.
Abbildung 3.12: Amplitude der Dilatation
in dK Ca-ko-Tieren nach Blockade von
COX und eNOS. Nach Blockade des
Einflusses von NO und Prostaglandinen
durch Indo und L-NA zeigten sich in dKCa-
ko-Tieren intakte Gefäßdilatation (n = 5,
MW + SEM).
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
1s 60s15s
Wildtyp
dKCa-ko
*
*
10 Hz%
der
max
. Dila
tatio
n
0
20
40
60
80
100
1s 60s15s
Kontrolle
Indo & L-NA
100 Hz
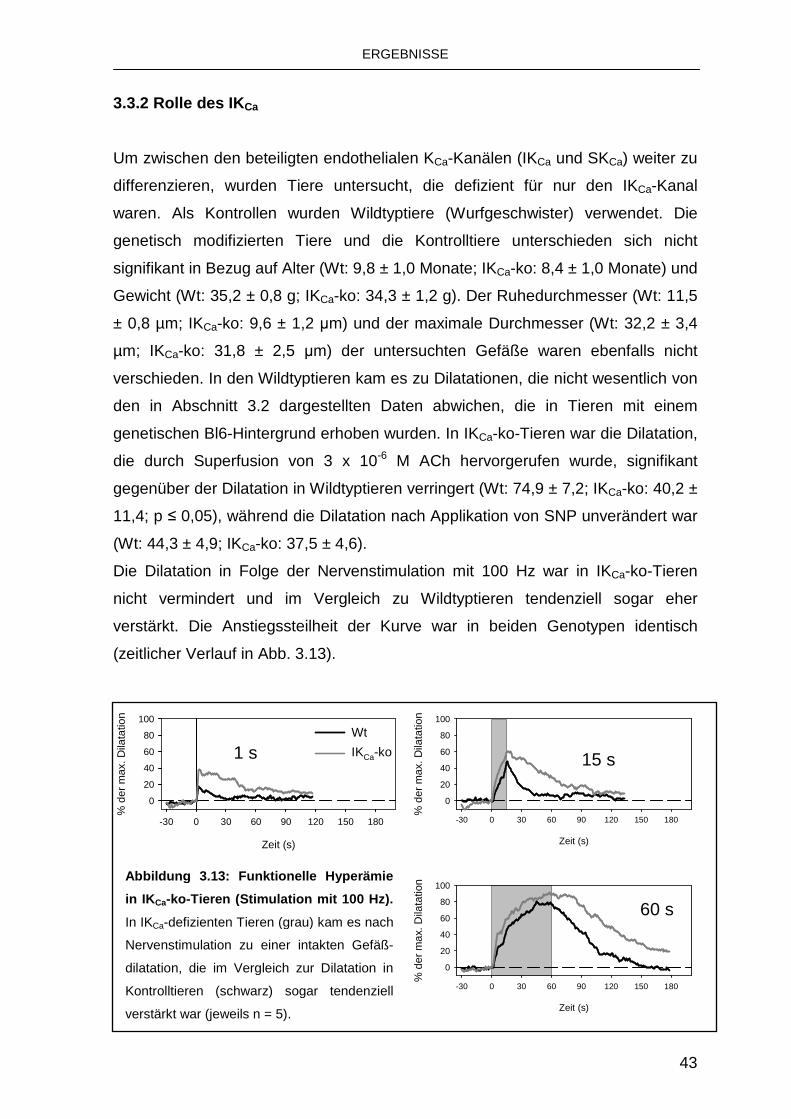
ERGEBNISSE
43
3.3.2 Rolle des IK Ca
Um zwischen den beteiligten endothelialen KCa-Kanälen (IKCa und SKCa) weiter zu
differenzieren, wurden Tiere untersucht, die defizient für nur den IKCa-Kanal
waren. Als Kontrollen wurden Wildtyptiere (Wurfgeschwister) verwendet. Die
genetisch modifizierten Tiere und die Kontrolltiere unterschieden sich nicht
signifikant in Bezug auf Alter (Wt: 9,8 ± 1,0 Monate; IKCa-ko: 8,4 ± 1,0 Monate) und
Gewicht (Wt: 35,2 ± 0,8 g; IKCa-ko: 34,3 ± 1,2 g). Der Ruhedurchmesser (Wt: 11,5
± 0,8 µm; IKCa-ko: 9,6 ± 1,2 µm) und der maximale Durchmesser (Wt: 32,2 ± 3,4
µm; IKCa-ko: 31,8 ± 2,5 µm) der untersuchten Gefäße waren ebenfalls nicht
verschieden. In den Wildtyptieren kam es zu Dilatationen, die nicht wesentlich von
den in Abschnitt 3.2 dargestellten Daten abwichen, die in Tieren mit einem
genetischen Bl6-Hintergrund erhoben wurden. In IKCa-ko-Tieren war die Dilatation,
die durch Superfusion von 3 x 10-6 M ACh hervorgerufen wurde, signifikant
gegenüber der Dilatation in Wildtyptieren verringert (Wt: 74,9 ± 7,2; IKCa-ko: 40,2 ±
11,4; p ≤ 0,05), während die Dilatation nach Applikation von SNP unverändert war
(Wt: 44,3 ± 4,9; IKCa-ko: 37,5 ± 4,6).
Die Dilatation in Folge der Nervenstimulation mit 100 Hz war in IKCa-ko-Tieren
nicht vermindert und im Vergleich zu Wildtyptieren tendenziell sogar eher
verstärkt. Die Anstiegssteilheit der Kurve war in beiden Genotypen identisch
(zeitlicher Verlauf in Abb. 3.13).
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
15 s1 s
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
60 s
IKCa-ko
Wt
Abbildung 3.13: Funktionelle Hyperämie
in IK Ca-ko-Tieren (Stimulation mit 100 Hz).
In IKCa-defizienten Tieren (grau) kam es nach
Nervenstimulation zu einer intakten Gefäß-
dilatation, die im Vergleich zur Dilatation in
Kontrolltieren (schwarz) sogar tendenziell
verstärkt war (jeweils n = 5).
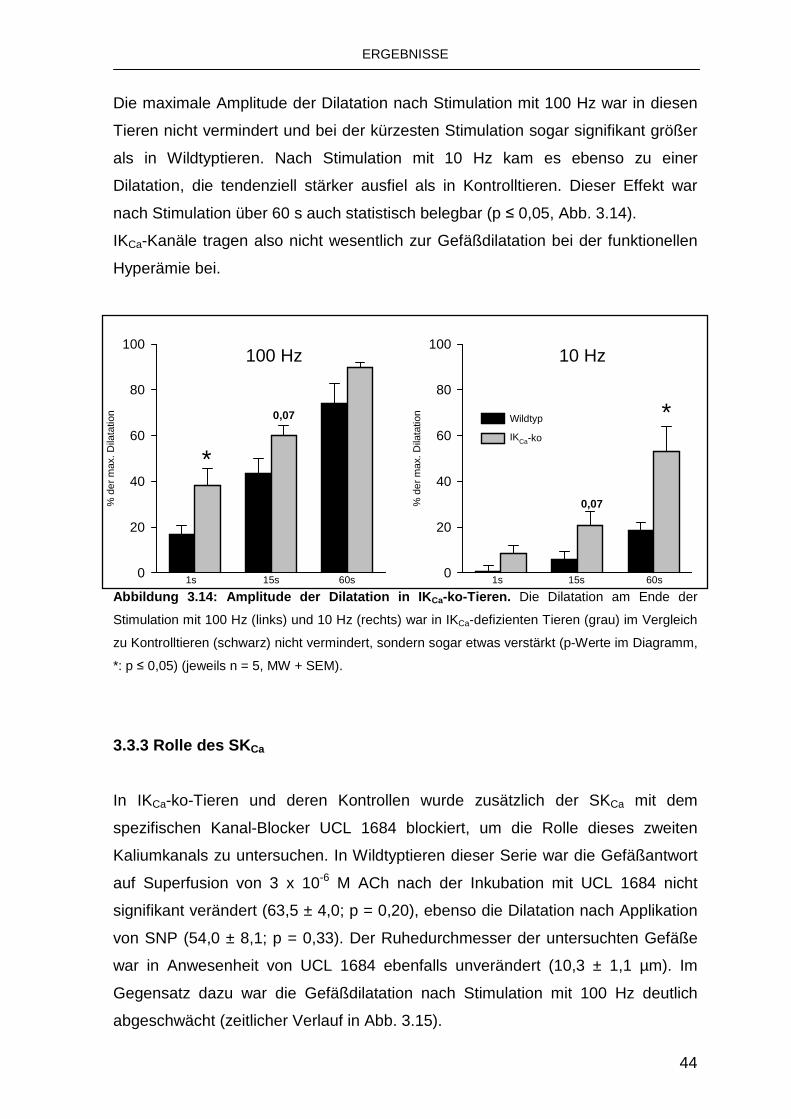
ERGEBNISSE
44
Die maximale Amplitude der Dilatation nach Stimulation mit 100 Hz war in diesen
Tieren nicht vermindert und bei der kürzesten Stimulation sogar signifikant größer
als in Wildtyptieren. Nach Stimulation mit 10 Hz kam es ebenso zu einer
Dilatation, die tendenziell stärker ausfiel als in Kontrolltieren. Dieser Effekt war
nach Stimulation über 60 s auch statistisch belegbar (p ≤ 0,05, Abb. 3.14).
IKCa-Kanäle tragen also nicht wesentlich zur Gefäßdilatation bei der funktionellen
Hyperämie bei.
Abbildung 3.14: Amplitude der Dilatation in IK Ca-ko-Tieren. Die Dilatation am Ende der
Stimulation mit 100 Hz (links) und 10 Hz (rechts) war in IKCa-defizienten Tieren (grau) im Vergleich
zu Kontrolltieren (schwarz) nicht vermindert, sondern sogar etwas verstärkt (p-Werte im Diagramm,
*: p ≤ 0,05) (jeweils n = 5, MW + SEM).
3.3.3 Rolle des SK Ca
In IKCa-ko-Tieren und deren Kontrollen wurde zusätzlich der SKCa mit dem
spezifischen Kanal-Blocker UCL 1684 blockiert, um die Rolle dieses zweiten
Kaliumkanals zu untersuchen. In Wildtyptieren dieser Serie war die Gefäßantwort
auf Superfusion von 3 x 10-6 M ACh nach der Inkubation mit UCL 1684 nicht
signifikant verändert (63,5 ± 4,0; p = 0,20), ebenso die Dilatation nach Applikation
von SNP (54,0 ± 8,1; p = 0,33). Der Ruhedurchmesser der untersuchten Gefäße
war in Anwesenheit von UCL 1684 ebenfalls unverändert (10,3 ± 1,1 µm). Im
Gegensatz dazu war die Gefäßdilatation nach Stimulation mit 100 Hz deutlich
abgeschwächt (zeitlicher Verlauf in Abb. 3.15).
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
1s 60s15s
10 Hz
*
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
1s 60s15s
100 Hz
Wildtyp
IKCa-ko
*
0,07
0,07
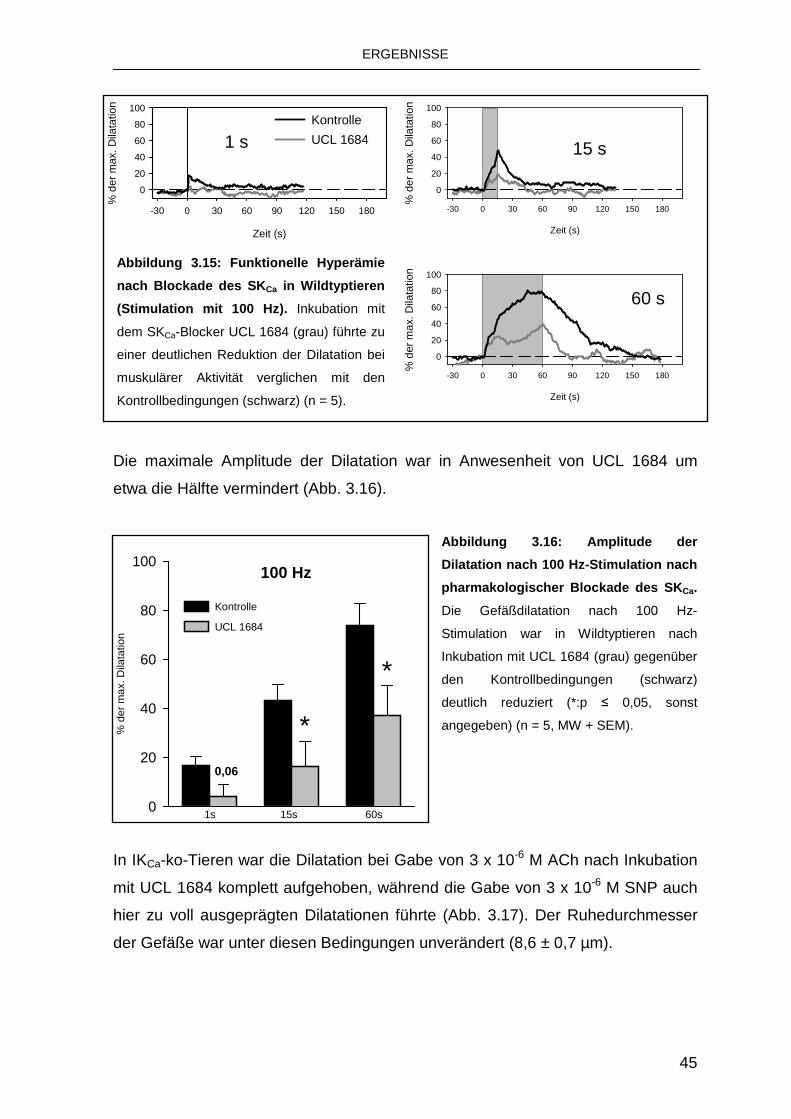
ERGEBNISSE
45
Die maximale Amplitude der Dilatation war in Anwesenheit von UCL 1684 um
etwa die Hälfte vermindert (Abb. 3.16).
Abbildung 3.16: Amplitude der
Dilatation nach 100 Hz-Stimulation nach
pharmakologischer Blockade des SK Ca.
Die Gefäßdilatation nach 100 Hz-
Stimulation war in Wildtyptieren nach
Inkubation mit UCL 1684 (grau) gegenüber
den Kontrollbedingungen (schwarz)
deutlich reduziert (*:p ≤ 0,05, sonst
angegeben) (n = 5, MW + SEM).
In IKCa-ko-Tieren war die Dilatation bei Gabe von 3 x 10-6 M ACh nach Inkubation
mit UCL 1684 komplett aufgehoben, während die Gabe von 3 x 10-6 M SNP auch
hier zu voll ausgeprägten Dilatationen führte (Abb. 3.17). Der Ruhedurchmesser
der Gefäße war unter diesen Bedingungen unverändert (8,6 ± 0,7 µm).
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
15 s1 s
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
60 s
UCL 1684
Kontrolle
Abbildung 3.15: Funktio nelle Hyperämie
nach Blockade des SK Ca in Wildtyptieren
(Stimulation mit 100 Hz). Inkubation mit
dem SKCa-Blocker UCL 1684 (grau) führte zu
einer deutlichen Reduktion der Dilatation bei
muskulärer Aktivität verglichen mit den
Kontrollbedingungen (schwarz) (n = 5).
1s 60s15s
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100100 Hz
Kontrolle
UCL 1684
*
0,06
*
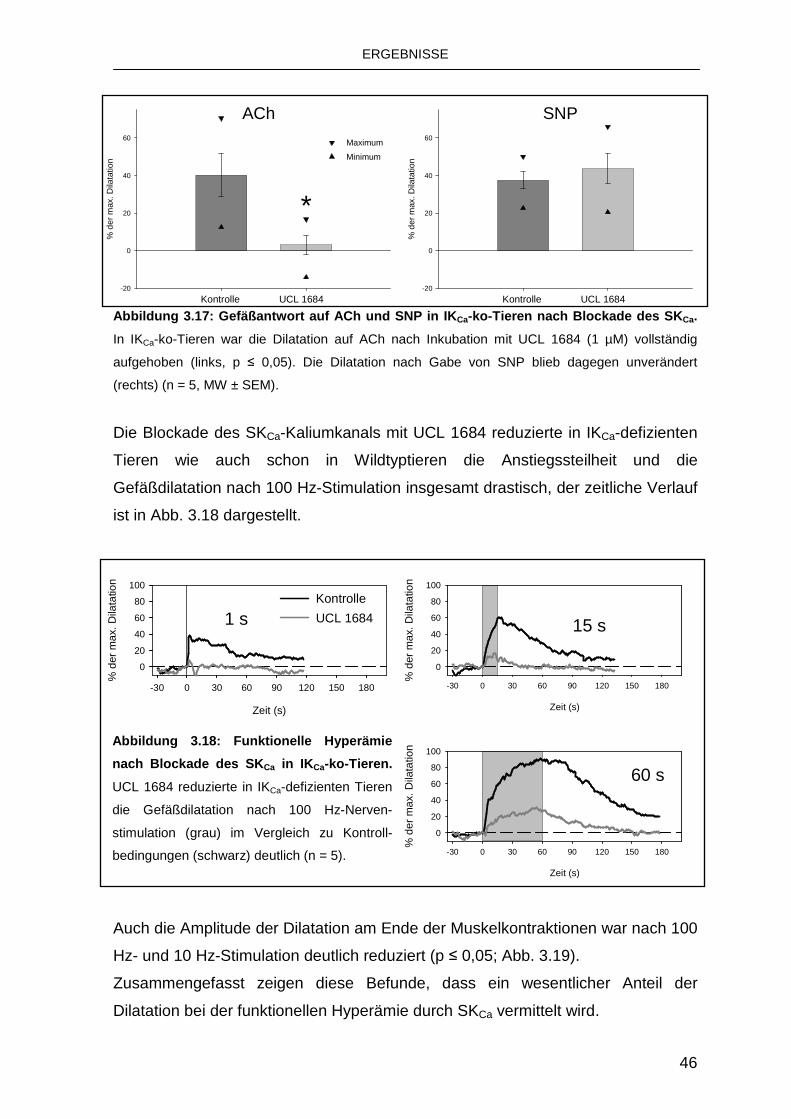
ERGEBNISSE
46
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
15 s1 s
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
60 s
UCL 1684
Kontrolle
Abbildung 3.17: Gefäßantwort auf ACh und SNP in IK Ca-ko-Tieren nach Blockade des SK Ca.
In IKCa-ko-Tieren war die Dilatation auf ACh nach Inkubation mit UCL 1684 (1 µM) vollständig
aufgehoben (links, p ≤ 0,05). Die Dilatation nach Gabe von SNP blieb dagegen unverändert
(rechts) (n = 5, MW ± SEM).
Die Blockade des SKCa-Kaliumkanals mit UCL 1684 reduzierte in IKCa-defizienten
Tieren wie auch schon in Wildtyptieren die Anstiegssteilheit und die
Gefäßdilatation nach 100 Hz-Stimulation insgesamt drastisch, der zeitliche Verlauf
ist in Abb. 3.18 dargestellt.
Auch die Amplitude der Dilatation am Ende der Muskelkontraktionen war nach 100
Hz- und 10 Hz-Stimulation deutlich reduziert (p ≤ 0,05; Abb. 3.19).
Zusammengefasst zeigen diese Befunde, dass ein wesentlicher Anteil der
Dilatation bei der funktionellen Hyperämie durch SKCa vermittelt wird.
SNP
% d
er m
ax. D
ilata
tion
-20
0
20
40
60
Kontrolle UCL 1684
ACh%
der
max
. Dila
tatio
n
-20
0
20
40
60
Kontrolle UCL 1684
*
Minimum
Maximum
Abbildung 3.18: Funktionelle Hyperämie
nach Blockade des SK Ca in IK Ca-ko-Tieren.
UCL 1684 reduzierte in IKCa-defizienten Tieren
die Gefäßdilatation nach 100 Hz-Nerven-
stimulation (grau) im Vergleich zu Kontroll-
bedingungen (schwarz) deutlich (n = 5).
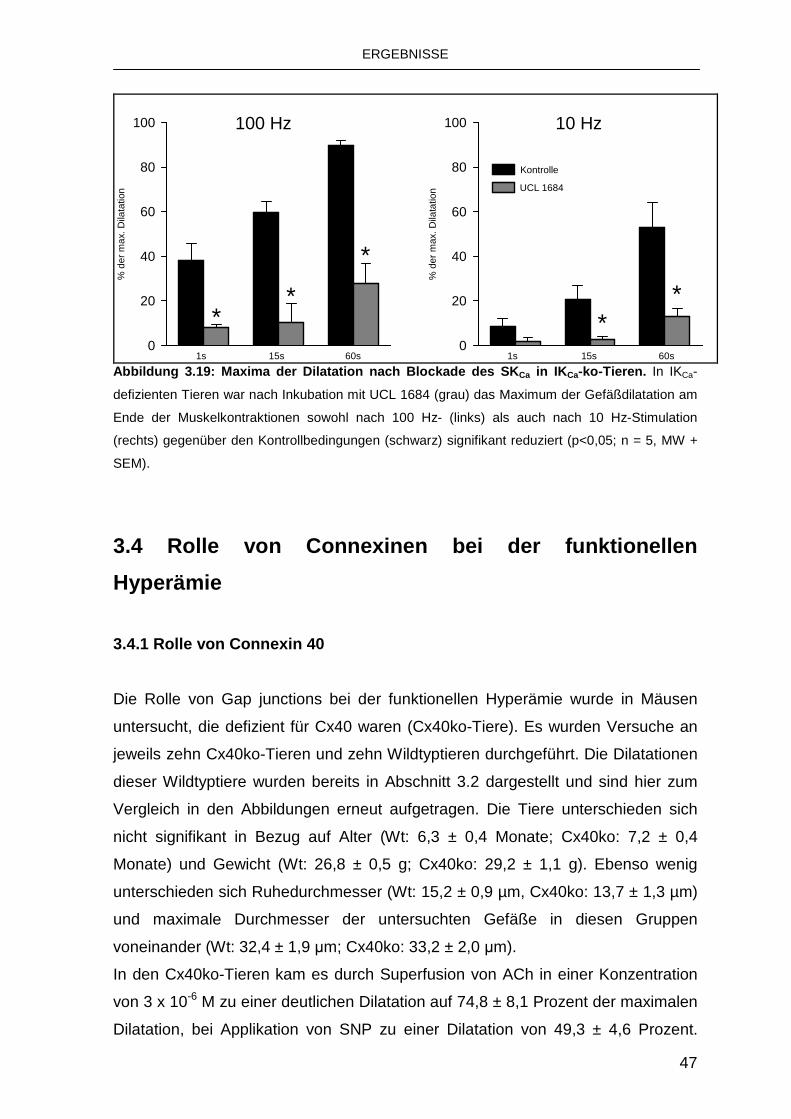
ERGEBNISSE
47
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
1s 60s15s
Kontrolle
UCL 1684
10 Hz
**
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
1s 60s15s
100 Hz
**
*
Abbildung 3.19: Maxima der Dilatation nach Blockade des SK Ca in IK Ca-ko-Tieren. In IKCa-
defizienten Tieren war nach Inkubation mit UCL 1684 (grau) das Maximum der Gefäßdilatation am
Ende der Muskelkontraktionen sowohl nach 100 Hz- (links) als auch nach 10 Hz-Stimulation
(rechts) gegenüber den Kontrollbedingungen (schwarz) signifikant reduziert (p<0,05; n = 5, MW +
SEM).
3.4 Rolle von Connexinen bei der funktionellen
Hyperämie
3.4.1 Rolle von Connexin 40
Die Rolle von Gap junctions bei der funktionellen Hyperämie wurde in Mäusen
untersucht, die defizient für Cx40 waren (Cx40ko-Tiere). Es wurden Versuche an
jeweils zehn Cx40ko-Tieren und zehn Wildtyptieren durchgeführt. Die Dilatationen
dieser Wildtyptiere wurden bereits in Abschnitt 3.2 dargestellt und sind hier zum
Vergleich in den Abbildungen erneut aufgetragen. Die Tiere unterschieden sich
nicht signifikant in Bezug auf Alter (Wt: 6,3 ± 0,4 Monate; Cx40ko: 7,2 ± 0,4
Monate) und Gewicht (Wt: 26,8 ± 0,5 g; Cx40ko: 29,2 ± 1,1 g). Ebenso wenig
unterschieden sich Ruhedurchmesser (Wt: 15,2 ± 0,9 µm, Cx40ko: 13,7 ± 1,3 µm)
und maximale Durchmesser der untersuchten Gefäße in diesen Gruppen
voneinander (Wt: 32,4 ± 1,9 µm; Cx40ko: 33,2 ± 2,0 µm).
In den Cx40ko-Tieren kam es durch Superfusion von ACh in einer Konzentration
von 3 x 10-6 M zu einer deutlichen Dilatation auf 74,8 ± 8,1 Prozent der maximalen
Dilatation, bei Applikation von SNP zu einer Dilatation von 49,3 ± 4,6 Prozent.
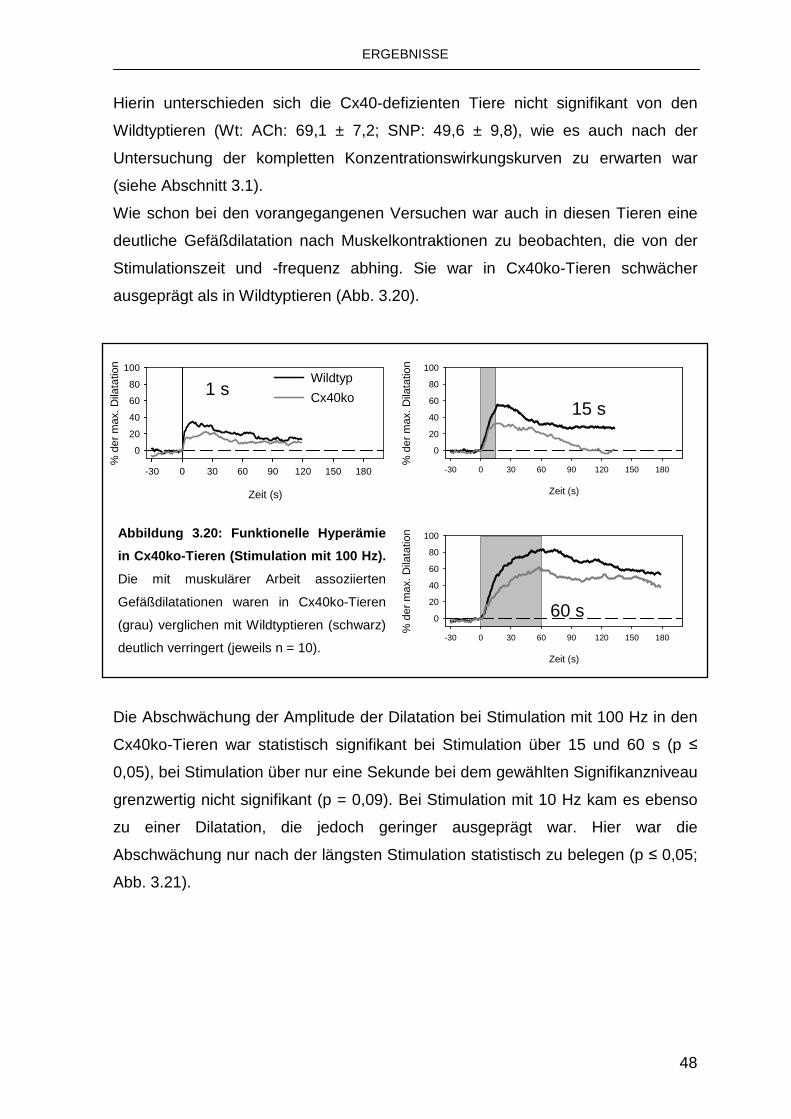
ERGEBNISSE
48
Hierin unterschieden sich die Cx40-defizienten Tiere nicht signifikant von den
Wildtyptieren (Wt: ACh: 69,1 ± 7,2; SNP: 49,6 ± 9,8), wie es auch nach der
Untersuchung der kompletten Konzentrationswirkungskurven zu erwarten war
(siehe Abschnitt 3.1).
Wie schon bei den vorangegangenen Versuchen war auch in diesen Tieren eine
deutliche Gefäßdilatation nach Muskelkontraktionen zu beobachten, die von der
Stimulationszeit und -frequenz abhing. Sie war in Cx40ko-Tieren schwächer
ausgeprägt als in Wildtyptieren (Abb. 3.20).
Die Abschwächung der Amplitude der Dilatation bei Stimulation mit 100 Hz in den
Cx40ko-Tieren war statistisch signifikant bei Stimulation über 15 und 60 s (p ≤
0,05), bei Stimulation über nur eine Sekunde bei dem gewählten Signifikanzniveau
grenzwertig nicht signifikant (p = 0,09). Bei Stimulation mit 10 Hz kam es ebenso
zu einer Dilatation, die jedoch geringer ausgeprägt war. Hier war die
Abschwächung nur nach der längsten Stimulation statistisch zu belegen (p ≤ 0,05;
Abb. 3.21).
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
15 s1 s
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
60 s
Cx40ko
Wildtyp
Abbildung 3.20: Funktionelle Hyperämie
in Cx40ko- Tieren (Stimulation mit 100 Hz).
Die mit muskulärer Arbeit assoziierten
Gefäßdilatationen waren in Cx40ko-Tieren
(grau) verglichen mit Wildtyptieren (schwarz)
deutlich verringert (jeweils n = 10).
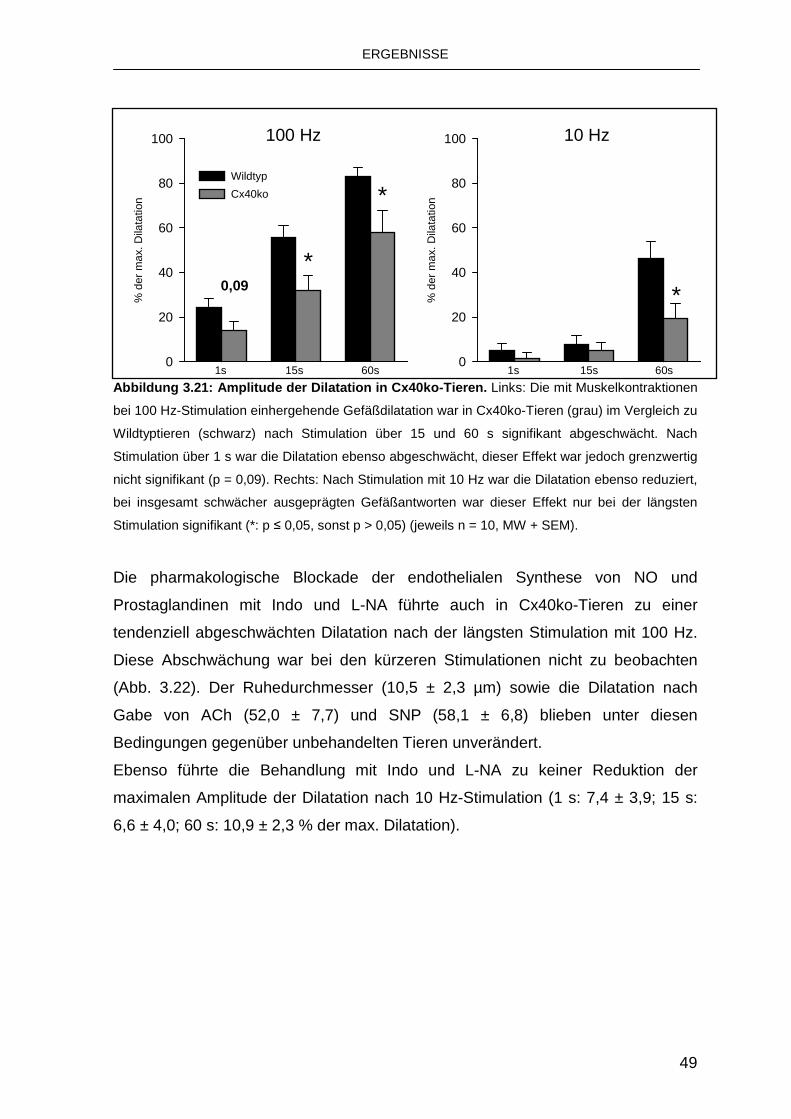
ERGEBNISSE
49
Abbildung 3.21: Amplitude der Dilatation in Cx40ko-Tieren. Links: Die mit Muskelkontraktionen
bei 100 Hz-Stimulation einhergehende Gefäßdilatation war in Cx40ko-Tieren (grau) im Vergleich zu
Wildtyptieren (schwarz) nach Stimulation über 15 und 60 s signifikant abgeschwächt. Nach
Stimulation über 1 s war die Dilatation ebenso abgeschwächt, dieser Effekt war jedoch grenzwertig
nicht signifikant (p = 0,09). Rechts: Nach Stimulation mit 10 Hz war die Dilatation ebenso reduziert,
bei insgesamt schwächer ausgeprägten Gefäßantworten war dieser Effekt nur bei der längsten
Stimulation signifikant (*: p ≤ 0,05, sonst p > 0,05) (jeweils n = 10, MW + SEM).
Die pharmakologische Blockade der endothelialen Synthese von NO und
Prostaglandinen mit Indo und L-NA führte auch in Cx40ko-Tieren zu einer
tendenziell abgeschwächten Dilatation nach der längsten Stimulation mit 100 Hz.
Diese Abschwächung war bei den kürzeren Stimulationen nicht zu beobachten
(Abb. 3.22). Der Ruhedurchmesser (10,5 ± 2,3 µm) sowie die Dilatation nach
Gabe von ACh (52,0 ± 7,7) und SNP (58,1 ± 6,8) blieben unter diesen
Bedingungen gegenüber unbehandelten Tieren unverändert.
Ebenso führte die Behandlung mit Indo und L-NA zu keiner Reduktion der
maximalen Amplitude der Dilatation nach 10 Hz-Stimulation (1 s: 7,4 ± 3,9; 15 s:
6,6 ± 4,0; 60 s: 10,9 ± 2,3 % der max. Dilatation).
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
*
1s 60s15s
*
1s 60s15s
Wildtyp
Cx40ko
*0,09
100 Hz 10 Hz
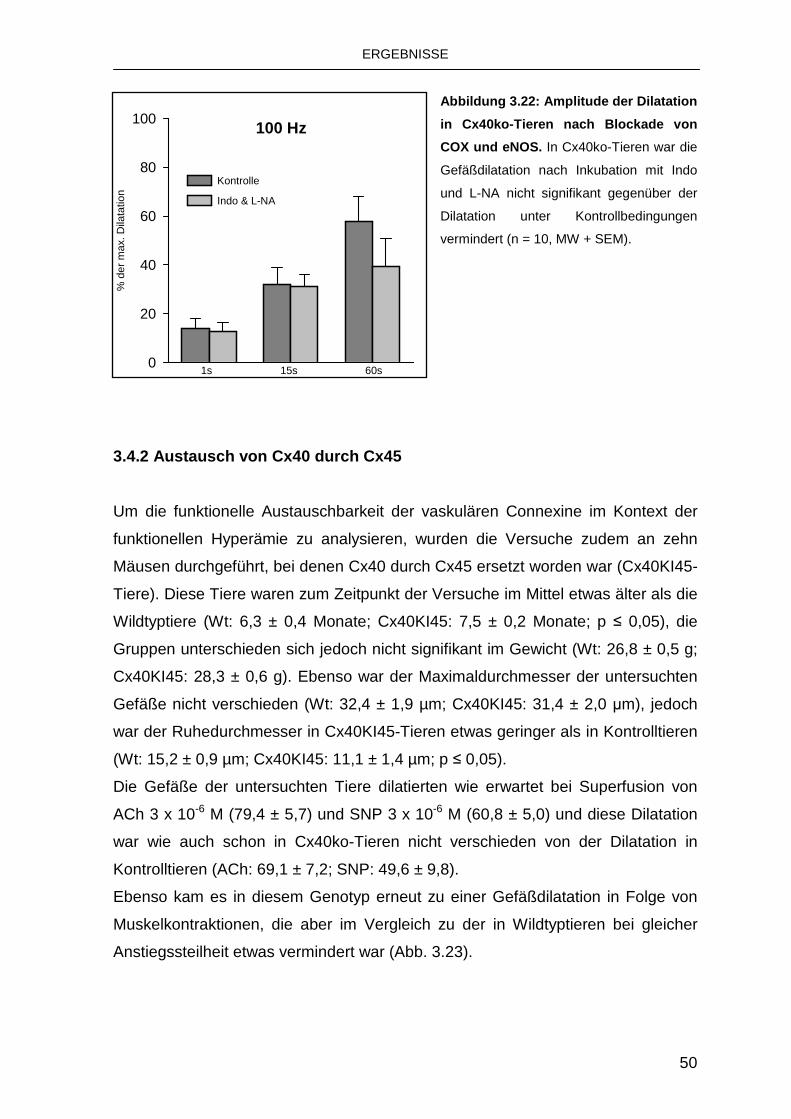
ERGEBNISSE
50
Abbildung 3.22: Amplitude der Dilatation
in Cx40ko-Tieren nach Blockade von
COX und eNOS. In Cx40ko-Tieren war die
Gefäßdilatation nach Inkubation mit Indo
und L-NA nicht signifikant gegenüber der
Dilatation unter Kontrollbedingungen
vermindert (n = 10, MW + SEM).
3.4.2 Austausch von Cx40 durch Cx45
Um die funktionelle Austauschbarkeit der vaskulären Connexine im Kontext der
funktionellen Hyperämie zu analysieren, wurden die Versuche zudem an zehn
Mäusen durchgeführt, bei denen Cx40 durch Cx45 ersetzt worden war (Cx40KI45-
Tiere). Diese Tiere waren zum Zeitpunkt der Versuche im Mittel etwas älter als die
Wildtyptiere (Wt: 6,3 ± 0,4 Monate; Cx40KI45: 7,5 ± 0,2 Monate; p ≤ 0,05), die
Gruppen unterschieden sich jedoch nicht signifikant im Gewicht (Wt: 26,8 ± 0,5 g;
Cx40KI45: 28,3 ± 0,6 g). Ebenso war der Maximaldurchmesser der untersuchten
Gefäße nicht verschieden (Wt: 32,4 ± 1,9 µm; Cx40KI45: 31,4 ± 2,0 µm), jedoch
war der Ruhedurchmesser in Cx40KI45-Tieren etwas geringer als in Kontrolltieren
(Wt: 15,2 ± 0,9 µm; Cx40KI45: 11,1 ± 1,4 µm; p ≤ 0,05).
Die Gefäße der untersuchten Tiere dilatierten wie erwartet bei Superfusion von
ACh 3 x 10-6 M (79,4 ± 5,7) und SNP 3 x 10-6 M (60,8 ± 5,0) und diese Dilatation
war wie auch schon in Cx40ko-Tieren nicht verschieden von der Dilatation in
Kontrolltieren (ACh: 69,1 ± 7,2; SNP: 49,6 ± 9,8).
Ebenso kam es in diesem Genotyp erneut zu einer Gefäßdilatation in Folge von
Muskelkontraktionen, die aber im Vergleich zu der in Wildtyptieren bei gleicher
Anstiegssteilheit etwas vermindert war (Abb. 3.23).
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
1s 60s15s
Kontrolle
Indo & L-NA
100 Hz
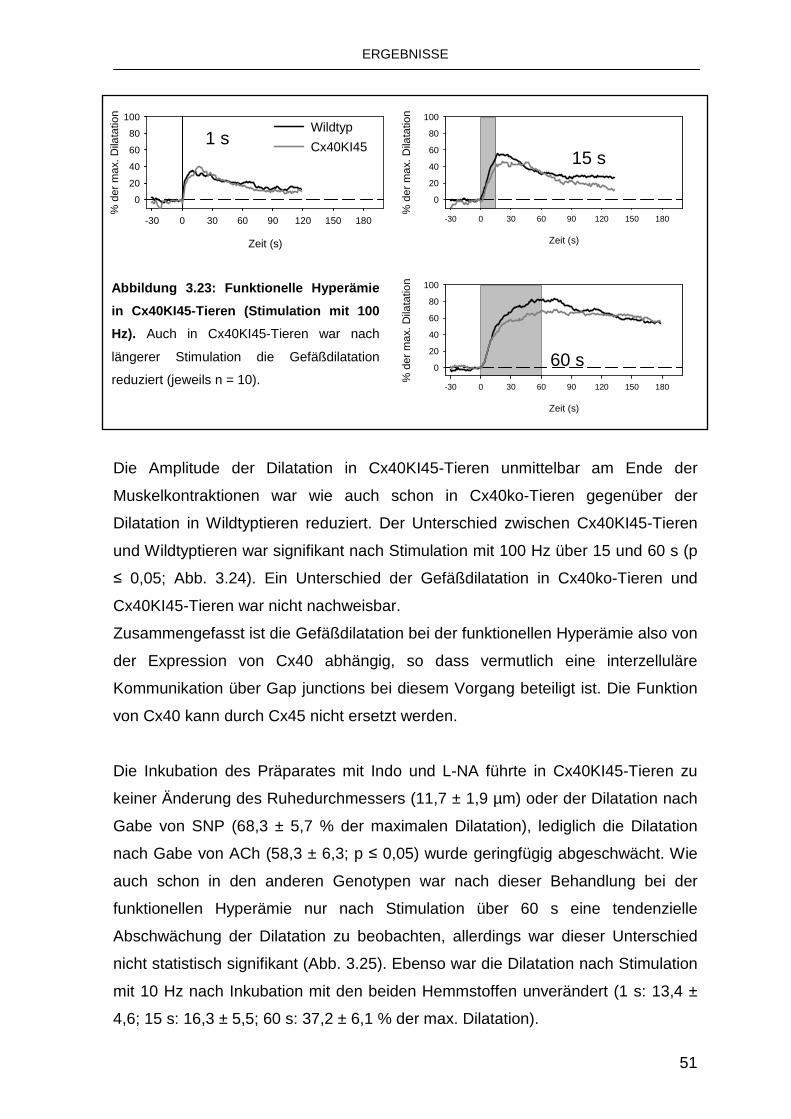
ERGEBNISSE
51
Die Amplitude der Dilatation in Cx40KI45-Tieren unmittelbar am Ende der
Muskelkontraktionen war wie auch schon in Cx40ko-Tieren gegenüber der
Dilatation in Wildtyptieren reduziert. Der Unterschied zwischen Cx40KI45-Tieren
und Wildtyptieren war signifikant nach Stimulation mit 100 Hz über 15 und 60 s (p
≤ 0,05; Abb. 3.24). Ein Unterschied der Gefäßdilatation in Cx40ko-Tieren und
Cx40KI45-Tieren war nicht nachweisbar.
Zusammengefasst ist die Gefäßdilatation bei der funktionellen Hyperämie also von
der Expression von Cx40 abhängig, so dass vermutlich eine interzelluläre
Kommunikation über Gap junctions bei diesem Vorgang beteiligt ist. Die Funktion
von Cx40 kann durch Cx45 nicht ersetzt werden.
Die Inkubation des Präparates mit Indo und L-NA führte in Cx40KI45-Tieren zu
keiner Änderung des Ruhedurchmessers (11,7 ± 1,9 µm) oder der Dilatation nach
Gabe von SNP (68,3 ± 5,7 % der maximalen Dilatation), lediglich die Dilatation
nach Gabe von ACh (58,3 ± 6,3; p ≤ 0,05) wurde geringfügig abgeschwächt. Wie
auch schon in den anderen Genotypen war nach dieser Behandlung bei der
funktionellen Hyperämie nur nach Stimulation über 60 s eine tendenzielle
Abschwächung der Dilatation zu beobachten, allerdings war dieser Unterschied
nicht statistisch signifikant (Abb. 3.25). Ebenso war die Dilatation nach Stimulation
mit 10 Hz nach Inkubation mit den beiden Hemmstoffen unverändert (1 s: 13,4 ±
4,6; 15 s: 16,3 ± 5,5; 60 s: 37,2 ± 6,1 % der max. Dilatation).
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
15 s1 s
Zeit (s)
-30 0 30 60 90 120 150 180
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
60 s
Cx40KI45
Wildtyp
Abbildung 3.23: Funktionelle Hyperämie
in Cx40KI45- Tieren (Stimulation mit 100
Hz). Auch in Cx40KI45-Tieren war nach
längerer Stimulation die Gefäßdilatation
reduziert (jeweils n = 10).
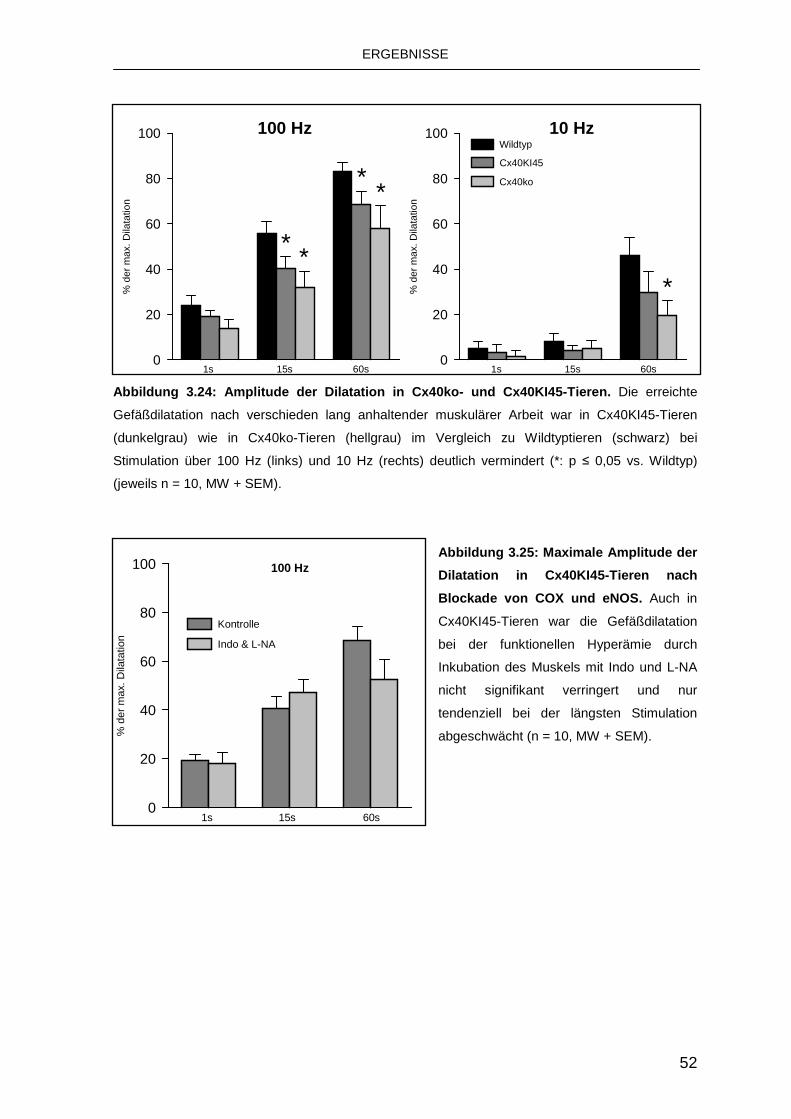
ERGEBNISSE
52
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100
*
**
**
1s 60s15s
10 Hz
1s 60s15s
Wildtyp
Cx40KI45
Cx40ko
100 Hz
Abbildung 3.24: Amplitude der Dilatation in Cx40ko- und Cx40KI45-Tieren. Die erreichte
Gefäßdilatation nach verschieden lang anhaltender muskulärer Arbeit war in Cx40KI45-Tieren
(dunkelgrau) wie in Cx40ko-Tieren (hellgrau) im Vergleich zu Wildtyptieren (schwarz) bei
Stimulation über 100 Hz (links) und 10 Hz (rechts) deutlich vermindert (*: p ≤ 0,05 vs. Wildtyp)
(jeweils n = 10, MW + SEM).
Abbildung 3.25: Maximale Amplitude der
Dilatation in Cx40KI45-Tieren nach
Blockade von COX und eNOS. Auch in
Cx40KI45-Tieren war die Gefäßdilatation
bei der funktionellen Hyperämie durch
Inkubation des Muskels mit Indo und L-NA
nicht signifikant verringert und nur
tendenziell bei der längsten Stimulation
abgeschwächt (n = 10, MW + SEM).
1s 60s15s
% d
er m
ax. D
ilata
tion
0
20
40
60
80
100 100 Hz
Kontrolle
Indo & L-NA

DISKUSSION
53
4. Diskussion
4.1 Charakteristika der funktionellen Hyperämie
Die funktionelle Hyperämie in arbeitendem Skelettmuskelgewebe ist ein basales
Prinzip in der Physiologie, dennoch sind deren Grundlagen noch immer nicht gut
verstanden. Das Ziel der vorliegenden Arbeit war, die zu Grunde liegenden
Signalmechanismen in den Blutgefäßen mit Hilfe von gentechnisch veränderten
Mäusen genauer zu charakterisieren. Zu diesem Zweck wurden zunächst einige
Parameter der Gefäßdilatation bestimmt, die durch Stimulation eines motorischen
Nerven in der versorgten Muskulatur hervorgerufen wurde.
Das Ausmaß der erreichten Gefäßdilatation hing sowohl von der Frequenz als
auch von der Dauer der Nervenstimulation ab. So konnte gezeigt werden, dass
Einzelkontraktionen (Stimulation mit 10 Hz) eine deutlich geringere Gefäßantwort
hervorrufen als eine tetanische Kontraktion (Stimulation mit 100 Hz). Dieses
entspricht dem physiologischen Bedarf und steht in Einklang mit den Ergebnissen
früherer Studien, die allerdings zum Teil in anderen Präparationen durchgeführt
wurden [77, 90, 139, 140]. Ebenso konnte gezeigt werden, dass die resultierende
Gefäßdilatation mit einer längeren Dauer der Muskelkontraktionen zunimmt, auch
dies bestätigt frühere Resultate (s.o.).
Obwohl bis heute kein schlüssiges Konzept existiert, wie es zu der ausgeprägten
Gefäßdilatation bei der funktionellen Hyperämie im Skelettmuskel kommt, wurde
schon früh postuliert, dass das Endothel als wichtiger Regulator des Gefäßtonus
eine entscheidende Rolle bei diesem Phänomen einnimmt. Hinweise hierauf
konnten auch experimentell gefunden werden, da die funktionelle Hyperämie nach
Zerstörung des Endothels deutlich reduziert war [125]. Daraufhin war es die
Hoffnung vieler Arbeitsgruppen, diese Funktion des Endothels mit der Freisetzung
von NO oder Prostaglandinen erklären zu können. Entsprechende Versuche
lieferten allerdings stets uneinheitliche und damit widersprüchliche Ergebnisse
(siehe Abschnitt 1.6). In der vorliegenden Arbeit wurde der Einfluss von NO sowie
Prostaglandinen auf die funktionelle Hyperämie in der Maus untersucht und es
zeigte sich in der verwendeten Präparation bei dem verwendeten Protokoll eine

DISKUSSION
54
kaum nachweisbare Abschwächung der Gefäßantwort. Lediglich nach der
längsten durchgeführten Stimulation (60 s) mit der höchsten Frequenz (100 Hz)
und damit einer tetanischen Muskelkontraktion wurde eine Abschwächung um
etwa 25 % gefunden. Bei allen anderen Stimulationsarten sowie bei den
Versuchen in Cx-defizienten Mäusen war die Gefäßdilatation nach Ausschaltung
von NO und Prostaglandinen voll ausgeprägt. Auch in KCa-defizienten Tieren, in
denen die Gefäßantwort unter Kontrollbedingungen bereits deutlich reduziert war,
kam es durch die pharmakologische Blockade von eNOS und COX zu keiner
weiteren signifikanten Abschwächung. Es lässt sich also ableiten, dass NO und
Prostaglandine in der untersuchten Präparation in der Maus nur unwesentlich zur
funktionellen Hyperämie beitragen und dass diese beiden endothelialen Autakoide
selbst nach dem Verlust anderer wichtigerer Funktionen des Endothels (z.B. in
KCa-defizienten Tieren) nicht in der Lage sind, die Gefäßdilatation im Sinne eines
back-up-Mechanismus aufrecht zu erhalten.
Naturgemäß stellt sich nach Betrachtung dieser Signalwege die Frage nach der
Bedeutung des dritten endothelialen Mechanismus, über den Gefäßdilatationen
hervorgerufen werden können, nämlich nach der Bedeutung von Dilatationen, die
durch den Endothelium-Derived Hyperpolarizing Factor (EDHF) vermittelt werden.
Diese Frage ist bis heute ungeklärt, da sie sich im Kontext der funktionellen
Hyperämie des Skelettmuskels schwer untersuchen lässt. Es ist noch umstritten,
welche chemische Substanz sich hinter EDHF verbirgt, was eine direkte
pharmakologische Blockade schwierig macht. Zusätzlich können viele der
Substanzen, die in Abschnitt 1.4 als mögliche Mediatoren einer EDHF-Dilatation
genannt wurden, auch aus den arbeitenden Skelettmuskelzellen stammen und
wären dann nicht sicher als EDHF zu charakterisieren. Darüber hinaus kann die
Dilatation, die nach Blockade von COX und eNOS nahezu unbeeinträchtigt
bestehen bleibt, nicht ohne Weiteres einem EDHF zugeschrieben werden, da
auch endothelunabhängige Mechanismen beteiligt sein könnten. Die Rolle des
Endothels und insbesondere der EDHF-Dilatation bei der funktionellen Hyperämie
im Skelettmuskel ist also bis heute nicht ausreichend bekannt. In der vorliegenden
Arbeit konnten neue Erkenntnisse gewonnen werden, die einige bisher ungeklärte
Aspekte der funktionellen Hyperämie aufzeigen und so zu einem besseren

DISKUSSION
55
Verständnis der Vorgänge bei der physiologischen Regulation der Widerstands-
gefäße in der Muskulatur führen.
4.2 Bedeutung von K Ca für die funktionelle Hyperämie
Ein Ereignis, das wesentlich an einer EDHF-Dilatation beteiligt ist, ist die Öffnung
von endothelialen KCa-Kanälen, die zu einer Hyperpolarisation der Endothelzelle
führt [20-22]. Im Endothel werden zwei verschiedene KCa exprimiert, nämlich IKCa
und SKCa (siehe Abschnitt 1.4). In der vorliegenden Arbeit konnte gezeigt werden,
dass in Tieren, die defizient für diese beiden Kanäle waren, die Gefäßdilatation im
Rahmen der funktionellen Hyperämie im Vergleich zur Gefäßantwort in
Wildtyptieren drastisch reduziert war. Diese Ergebnisse konnten in einer weiteren
Versuchsreihe bestätigt werden, in der in IKCa-defizienten Tieren der Kanal SKCa
pharmakologisch blockiert wurde. Dies zeigt, dass endotheliale KCa eine
herausragende Rolle bei der funktionellen Hyperämie einnehmen, und somit ergibt
sich ein deutlicher Hinweis darauf, dass EDHF-Dilatationen einen wesentlichen
Mechanismus der Gefäßdilatation im Rahmen der funktionellen Hyperämie
darstellen.
Interessanterweise war die Gefäßdilatation in IKCa-ko-Tieren vor der Blockade des
SKCa im Vergleich zu der in Wildtyptieren keineswegs reduziert, sondern
tendenziell sogar etwas verstärkt, obwohl diese Verstärkung nur bei einigen
Stimulationsmustern statistisch signifikant war (siehe Abb. 3.14). Dagegen war bei
Wildtyptieren nach Behandlung des untersuchten Muskels mit einem
pharmakologischen Blocker des SKCa (UCL 1684) bereits eine Reduktion der
Dilatation festzustellen, deren Ausmaß vergleichbar mit der Verminderung war, die
in Tieren mit einem doppelten knock-out für beide KCa-Kanäle beobachtet wurde.
Analog dazu konnte ebenso in IKCa-defizienten Tieren eine drastische
Verminderung der Gefäßdilatation durch eine zusätzliche Blockade des SKCa
erreicht werden. Folglich ist von den beiden endothelialen KCa-Kanälen SKCa der
Kanal, der für die Dilatation im Rahmen der funktionellen Hyperämie im
Skelettmuskel entscheidend ist, während IKCa im Gegensatz dazu kaum zur
Gefäßantwort beiträgt. Diese Resultate sind insbesondere daher von großer

DISKUSSION
56
Relevanz, da sich der Einfluss der einzelnen KCa auf die Gefäßdilatation nach
endothelialer Stimulation mit Acetylcholin völlig unterschiedlich darstellt. Hier hat
die Blockade des SKCa in Wildtyptieren keinen Effekt, während die Ausschaltung
des IKCa zu deutlich abgeschwächten Dilatationen führt. Lediglich in IKCa-
defizienten Tieren führt auch die Blockade des SKCa zu einer weiteren Abnahme
der Dilatation nach Stimulation des Endothels mit Acetylcholin [141-144]. Auch
diese Ergebnisse konnten in der vorliegenden Arbeit reproduziert werden.
Dieser Unterschied in der Aktivierung endothelialer KCa in Abhängigkeit von einem
spezifischen Stimulus, der die Dilatation auslöst, belegt, dass die beiden
exprimierten Kanäle nicht lediglich eine identische Funktion innehaben. Es muss
eine differenzierte Rolle der einzelnen Kanäle geben, die vom auslösenden
Stimulus abhängt. Denkbar wäre, dass die unterschiedlichen Aktivierungsmuster
der Kanäle durch ihre jeweilige spezielle Lokalisation in der Zellmembran bedingt
sind. So konnte kürzlich gezeigt werden, dass IKCa in der Umgebung von
myoendothelialen Gap junctions sowie in der Nähe von IP3-Rezeptoren des
endoplasmatischen Retikulums (ER) zu finden sind [145, 146]. Wahrscheinlich
führt eine Stimulation des Endothels mit Acetylcholin über das PLC-IP3-System zu
räumlich begrenzten Erhöhungen des intrazellulären Ca2+, die ebenfalls an den
IP3-Rezeptoren des ER lokalisiert sind (so genannte Ca2+-Pulsare) [145]. Die
Nähe dieser lokalen Ca2+-Erhöhungen zum IKCa legt nahe, dass dieser
Kaliumkanal durch sie aktiviert wird und eine wesentliche Rolle bei der
Acetylcholin-vermittelten Gefäßdilatation einnimmt. SKCa scheint dagegen eher im
Bereich der Endothelzellgrenzen und an homozellulären Gap junctions, die die
einzelnen Endothelzellen miteinander verbinden, lokalisiert zu sein [146, 147]. Es
ist nach wie vor ungeklärt, welches Signal zur Öffnung des SKCa führt, allerdings
könnte die Lage dieser Kanäle an mechanisch beanspruchten Zellgrenzen dafür
sprechen, dass eine Verformung des Gefäßes, wie sie auch bei
Muskelkontraktionen vorkommt, zu seiner Aktivierung führt. Ein weiterer Hinweis
auf diese mögliche mechanosensitive Funktion des SKCa ist kürzlich beschrieben
worden, da dieser Kanal auch für eine intakte EDHF-Dilatation im Rahmen einer
flussinduzierten Dilatation notwendig ist [130].

DISKUSSION
57
Die Ergebnisse der vorliegenden Arbeit zeigen also, dass endotheliale KCa-Kanäle
für die Gefäßdilatation bei der funktionellen Hyperämie in arbeitendem
Skelettmuskel von elementarer Bedeutung sind und weisen somit daraufhin, dass
eine EDHF-Dilatation beteiligt sein könnte. Im Gegensatz zu durch Acetylcholin
hervorgerufenen Dilatationen steht SKCa hierbei im Vordergrund, während IKCa
keine wesentliche Rolle einnimmt. Es muss daher eine differenzierte, Stimulus-
abhängige Aktivierung der einzelnen KCa geben, die möglicherweise mit der
Lokalisation der einzelnen Kanäle in der Zellmembran in Zusammenhang stehen
könnte.
4.3 Koordination der Gefäßdilatation durch Connexin e
Bei muskulärer Arbeit muss zusätzlich zu einer lokalen Dilatation der
intramuskulären Arteriolen auch eine Dilatation der vorgeschalteten Leitungs-
gefäße stattfinden, damit eine ausreichende Durchblutungssteigerung erreicht
werden kann (siehe Abschnitt 1.5). Ein Mechanismus, der wahrscheinlich zu
diesem Effekt beiträgt, ist die Fortleitung einer endothelialen Hyperpolarisation
entlang des Gefäßes, die zu einer aufsteigenden Dilatation führt und im
Experiment durch die lokale Gabe von Acetylcholin initiiert werden kann.
Tatsächlich konnte experimentell bestätigt werden, dass solche aufsteigenden
Dilatationen nicht nur durch pharmakologische Stimulation des Endothels, sondern
auch durch Kontraktionen einzelner Muskelfasern hervorgerufen werden [133,
148, 149], so dass eine Beteiligung dieser aufsteigenden Dilatationen an der
funktionellen Hyperämie in der Muskulatur zu erwarten ist.
In vorhergehenden Arbeiten konnte weiter gezeigt werden, dass aufsteigende
Dilatationen auf Cx40, ein Strukturprotein endothelialer Gap junctions, angewiesen
sind. Cx40-defiziente Tiere zeigen bei endothelialer Stimulation mit ACh
abgeschwächte fortgeleitete Dilatationen, erkennbar an einer reduzierten
Amplitude der Dilatation, die stromaufwärts von der Stimulationsstelle beobachtet
wird [75]. In der vorliegenden Arbeit konnte nun erstmals gezeigt werden, dass in
Cx40-defizienten Tieren auch die Gefäßdilatation im Rahmen der funktionellen
Hyperämie gegenüber Wildtyptieren abgeschwächt ist. Somit ist ein Cx40-

DISKUSSION
58
abhängiger Mechanismus bei der funktionellen Hyperämie beteiligt. Aus den
genannten Überlegungen ergibt sich, dass es sich hierbei wahrscheinlich um eine
Koordination des Gefäßverhaltens über eine gewisse Distanz zum Erreichen einer
maximal möglichen Durchblutungssteigerung handelt. Der beobachtete Effekt
beruht nicht auf einer generellen Einschränkung der endothelvermittelten
Dilatation in den genetisch modifizierten Tieren, denn die Dilatation nach globaler
Applikation von Acetylcholin auf den Cremastermuskel war in Wildtyptieren und
Cx40ko-Tieren identisch.
Weitere Versuche in Tieren, bei denen Cx40 durch ein anderes Connexin (Cx45)
ausgetauscht war, ergaben, dass auch die Dilatation in diesen Tieren gegenüber
Wildtyptieren deutlich reduziert ist. Ein wesentlicher Unterschied zwischen diesen
beiden Genotypen liegt darin, dass Cx40KI45-Tiere annähernd normotensiv sind
[150, 151], während Cx40ko-Tiere einen erhöhten Blutdruck aufweisen [152-154].
Diese Blutdruckveränderungen sind auf die veränderte Expression der Connexine
in juxtaglomerulären Renin-produzierenden Zellen der Niere zurückzuführen. Trotz
dieses Unterschieds im Blutdruck der Tiere ergab sich eine vergleichbare
Abschwächung der Dilatation in beiden Genotypen. Dies zeigt, dass die Reduktion
der funktionellen Hyperämie in Cx40ko-Tieren nicht durch die deutliche Hypertonie
dieser Tiere bedingt ist. Connexine müssen folglich eine Bedeutung für die
funktionelle Hyperämie haben, die über eine Blutdruckveränderung hinausgeht.
Cx40 hat also im Kontext der funktionellen Hyperämie spezielle Eigenschaften, die
nicht durch Cx45 übernommen werden können. Dies bestätigt frühere Versuche
zur Rolle von Connexinen bei fortgeleiteten Dilatationen in der Muskulatur. Nach
lokaler Stimulation mit Acetylcholin war Cx45 in Cx40-defizienten Mäusen
ebenfalls nicht in der Lage, den Verlust von Cx40 zu kompensieren, denn auch in
Cx40KI45-Tieren war die Amplitude der entfernten Dilatation reduziert [60]. Dies
ist jedoch nicht generell der Fall. In anderen Organen können unterschiedliche
Connexine wesentliche Funktionen natürlich exprimierter Connexine nach deren
Ausschaltung übernehmen. Dies wird zum einen dadurch belegt, dass Cx40KI45-
Tiere im Vergleich mit Cx40ko-Tieren einen deutlich reduzierten Blutdruck
aufweisen (s.o.), aber auch andere genetische Versuche, in denen Connexine
gegeneinander ausgetauscht wurden, zeigten ähnliche Ergebnisse. So ist Cx45 in

DISKUSSION
59
der Lage, die Funktion von Cx40 in der Reizleitung der Herzens nach einem
knock-out von Cx40 teilweise zu übernehmen [128]. Ein weiteres Beispiel für solch
einen funktionellen Ersatz findet man in der Organogenese. Cx43-defiziente Tiere
versterben kurz nach der Geburt an kardialen Fehlbildungen. Diese Funktion von
Cx43 in der Entwicklung des Herzens kann dadurch übernommen werden, dass
Cx32 oder Cx40 an gleicher Stelle im Genom der Maus eingefügt werden [155].
Cx40 und Cx45 unterscheiden sich zum Einen in ihrer Leitfähigkeit (etwa 200 pS
bei Cx40 und etwa 35 pS bei Cx45), zum Anderen aber auch in ihrem Gating und
ihrer Regulation [156-159]. Es ist noch nicht bekannt, welche spezielle Eigenschaft
von Cx40 für eine intakte Funktion vaskulärer Zellen notwendig ist.
Trotz der deutlichen Reduktion der Dilatation in Cx40-defizienten Tieren war nach
wie vor eine deutliche Gefäßantwort durch Muskelkontraktionen auslösbar. Dies
zeigt, dass es noch mindestens einen weiteren Mechanismus geben muss, der
diese ausgeprägte Flusssteigerung ermöglicht. Hier käme zum Einen die Diffusion
vasoaktiver Transmitter, die zum Beispiel aus den Skelettmuskelzellen stammen
könnten, oder auch ein rein mechanischer Stimulus durch die Kontraktionen in
Betracht [160]. Eine andere Möglichkeit wäre die Fortleitung eines weiteren
Signals, ähnlich der Hyperpolarisation der Endothelzellen, das sich in der glatten
Muskelschicht der Gefäße ausbreitet [49, 161, 162].
4.4 Schlussfolgerungen und Ausblick
In der vorliegenden Arbeit konnten neue Erkenntnisse gewonnen werden, die zu
einem besseren Verständnis der Gefäßregulation in der arbeitenden Skelett-
muskulatur führen. Es ergaben sich deutliche Hinweise darauf, dass EDHF-Typ-
Dilatationen wesentlich an der Gefäßdilatation beteiligt sind, die den notwendigen
Blutfluss für muskuläre Arbeit ermöglicht. Interessanterweise konnte zusätzlich
gezeigt werden, dass es eine differenzierte Aktivierung unterschiedlicher
beteiligter Kaliumkanäle geben muss, die von dem initialen Stimulus abhängt und
mit der Lokalisation der einzelnen Kaliumkanäle in der Zellmembran in
Zusammenhang stehen könnte. Zusätzlich konnte gezeigt werden, dass auch
endotheliales Connexin 40 eine elementare Voraussetzung für intakte

DISKUSSION
60
hyperämische Dilatationen darstellt. Insgesamt ergibt sich somit folgender
hypothetischer Mechanismus als mögliche Grundlage der funktionellen
Hyperämie:
Kontraktionen des Skelettmuskels führen zu einer Hyperpolarisation der
Endothelzellen der intramuskulären Gefäße. Diese kommt durch eine Öffnung von
SKCa-Kanälen zu Stande, die möglicherweise auf Grund ihrer Lokalisation an den
Zellgrenzen der Endothelzellen direkt durch eine mechanische Beanspruchung
aktiviert werden. Die Endothelzellhyperpolarisation initiiert eine lokale Dilatation
vom EDHF-Typ und führt so zu einem Widerstandsverlust der intramuskulären
Arteriolen. Zusätzlich breitet sich die Hyperpolarisation entlang des Gefäßes aus
und koordiniert das zelluläre Verhalten entlang des Gefäßbaumes. So wird erst die
ausgeprägte Durchblutungssteigerung, die für muskuläre Arbeit erforderlich ist,
ermöglicht. Diese Fortleitung erfolgt durch homozelluläre Gap junctions im
Endothel, die Cx40 als elementares Strukturprotein enthalten, dessen Funktion
nicht durch Cx45 ersetzt werden kann. Zusätzlich muss es weitere beteiligte
Mechanismen geben, da auch die Blockade obiger Signalwege keine komplette
Ausschaltung der hyperämischen Dilatation zur Folge hat. Allerdings scheinen
EDHF-Antworten für einen großen Anteil der Dilatation verantwortlich zu sein. Die
anderen endothelialen Autakoide (NO und Prostaglandine) tragen nur marginal zur
funktionellen Hyperämie bei.
Obwohl der beschriebene Mechanismus zu einem großen Teil auf den in dieser
Arbeit dargestellten Ergebnissen beruht, sind in der Zukunft noch weitere
Versuche erforderlich, um zentrale Aspekte zu bestätigen oder zu widerlegen.
Insbesondere das aktivierende Signal für die SKCa-Kanäle, die chemische Entität
von EDHF im Rahmen der funktionellen Hyperämie sowie weitere möglicherweise
beteiligte Signalwege und deren Anteil an der Gefäßantwort sind noch zu
entschlüsseln. Bereits in der Einleitung wurde erwähnt, dass sich die Rolle
verschiedener beteiligter Mechanismen der Dilatation wahrscheinlich im Laufe der
Gefäßantwort ändert. Unterschiedliche Signalwege könnten jeweils für die frühe
Phase der Dilatation und für die Aufrechterhaltung der Dilatation im weiteren
Verlauf verantwortlich sein. Diese verschiedenen zeitlichen Abläufe wurden in der
vorliegenden Arbeit mit dem gewählten Versuchsprotokoll nicht weiter

DISKUSSION
61
differenziert. Allerdings ist die Anstiegssteilheit der Dilatation bei SKCa-defizienten
Mäusen sowie nach Blockade des SKCa andeutungsweise reduziert. Dies spricht
für eine Beteiligung dieses Kanals an der ersten schnellen Phase der Dilatation. In
Versuchen nach Blockade von COX und eNOS sowie in Cx40-defizienten Tieren
kam es dagegen lediglich zu einer mehr oder weniger stark ausgeprägten
Verringerung der Amplitude der Dilatation, während die Anstiegssteilheit der Kurve
unverändert blieb. Dies könnte ein Hinweis darauf sein, dass die Signalwege, in
denen diese Proteine involviert sind, eher zur Aufrechterhaltung der Dilatation im
Verlauf beitragen.
Um die funktionelle Relevanz der untersuchten Mechanismen zu belegen, könnten
weitere Versuche durchgeführt werden, um die körperliche Leistungsfähigkeit (z.B.
die Ausdauerleistung) in den verschiedenen Genotypen zu quantifizieren und zu
vergleichen. In solchen Versuchen an wachen Tieren könnte auch ein potentieller
Einfluss von verwendeten Anästhetika ausgeschlossen werden. Bei einem
invasiven Versuchsaufbau, wie in dieser Arbeit verwendet, ist immer eine
Anästhesie notwendig, und diese könnte einen Einfluss auf die untersuchten
physiologischen Vorgänge haben. Barbiturate und inhalative Anästhetika
beeinflussen in einigen Präparationen EDHF-Dilatationen in der Mikrozirkulation
im Sinne einer Abschwächung [163, 164], dagegen gibt es für die in dieser Arbeit
verwendeten Substanzen keine derartigen Daten.
Insgesamt ist ein besseres Verständnis der funktionellen Hyperämie nicht nur von
akademischem Interesse, sondern auch wesentlich, um die Pathologie und
eventuell neue Therapiemöglichkeiten kardiovaskulärer Erkrankungen zu
verstehen bzw. zu entwickeln. Interessanterweise konnte kürzlich gezeigt werden,
dass endotheliale KCa auch im Menschen für eine EDHF-Dilatation unabdingbar
sind [165], und es wurde postuliert, dass Öffner dieser Kanäle möglicherweise als
Medikamente für Krankheiten eingesetzt werden können, in denen die
Endothelfunktion gestört ist [166-172]. Ebenso wie bei Kaliumkanälen ist auch
vorstellbar, dass Connexine in den nächsten Jahren zusätzliche klinische
Relevanz erhalten werden. Nicht nur im Tiermodell konnte der Einfluss von
Connexinen auf die Blutdruckregulation gezeigt werden [150, 152-154], auch im
Menschen gibt es Hinweise darauf, dass Connexine an der Blutdruckregulation

DISKUSSION
62
beteiligt sind [173, 174] und dass Polymorphismen in Connexingenen mit einem
höheren Risiko für arterielle Hypertonie einhergehen [175]. Außerdem spielen
Connexine eine Rolle bei Störungen der kardialen Reizleitung und bei
angeborenen Herzfehlern [176]. Zudem gibt es weitere seltene angeborene
Erkrankungen, die mit Mutationen in Connexingenen einhergehen [177], so dass
ein tieferes Verständnis der Physiologie dieser Proteinfamilie von großer klinischer
Bedeutung ist.

ZUSAMMENFASSUNG
63
5. Zusammenfassung
Die funktionelle Hyperämie, also die Mehrdurchblutung eines Gewebes bei einem
höheren Aktivitätszustand, ist ein basales Prinzip zur Regulation der Organ-
durchblutung. Dennoch sind die zu Grunde liegenden zellulären Mechanismen
noch unverstanden. Das Endothel mit seinem großen Einfluss auf den Gefäßtonus
könnte hieran beteiligt sein, z.B. über NO und Prostaglandine. Neben diesen
traditionellen Mediatoren rückt jüngst ein weiterer endothelialer hyperpolarisations-
abhängiger Mechanismus (EDHF) in den Vordergrund der Untersuchungen. Ziel
dieser Arbeit war es, die Bedeutung dieser EDHF-Typ-Dilatation für die
funktionelle Hyperämie zu untersuchen. Hierzu wurden Gefäßdilatationen nach
Muskelkontraktionen nach pharmakologischer oder genetischer Ausschaltung
endothelialer Kaliumkanäle, die essentiell für die Auslösung einer EDHF-Dilatation
sind, mittels Intravitalmikroskopie beobachtet. Zusätzlich wurde die Rolle einer
Koordination des zellulären Verhaltens in Tieren untersucht, denen bestimmte
Gap junction-Proteine (Connexine) fehlten.
In Mäusen, denen der endotheliale Calcium-abhängige Kaliumkanal SKCa fehlte,
war die aktivitätsinduzierte Dilatation etwa halbiert. Dagegen hatte das Fehlen des
zweiten Calcium-abhängigen Kaliumkanals IKCa keine Auswirkung auf die
Dilatation. Wurde dagegen in IKCa-defizienten oder Wildtyptieren SKCa
pharmakologisch blockiert, war wiederum eine deutliche Abschwächung der
Dilatation zu beobachten. Interessanterweise war die Dilatation, die durch die
klassische endothelstimulierende Substanz Acetylcholin induziert wurde, nicht von
SKCa sondern vornehmlich von IKCa abhängig. Die Daten zeigen, dass die
Aktivierung der endothelialen SKCa-Kanäle und eine folgende Hyperpolarisation
(EDHF-Typ) wesentlich an der Dilatation beteiligt sind. Weiterhin war die Dilatation
nach Muskelkontraktionen in Tieren, denen das Gap junction-Protein Connexin 40
fehlte, reduziert, und dies konnte nicht durch die Expression von Cx45 an Stelle
von Cx40 kompensiert werden. Dies lässt vermuten, dass eine lokal ausgelöste
EDHF-Typ-Dilatation sich entlang des Endothels in einer Cx40-abhängigen Weise
ausbreitet und so die Koordination des Gefäßverhaltens sichert.
Zusammengefasst scheint eine endotheliale Hyperpolarisation für die
Gefäßerweiterung bei der Mehrdurchblutung des arbeitenden Skelettmuskels und
die Koordination des zellulären Verhaltens entscheidend zu sein.

LITERATUR
64
6. Literatur
1. Lüllmann-Rauch R. Blutgefäße. Histologie. Verstehen - Lernen - Nachschlagen. 1. Ausgabe. Stuttgart: Georg Thieme Verlag, 2003:208 - 219
2. Linke WA und Pfitzer G. Kontraktionsmechanismen. In: Schmidt RF, Lang F und Thews G. Physiologie des Menschen mit Pathophysiologie. 29. Ausgabe. Heidelberg: Springer Medizin Verlag, 2005:116 - 143
3. Ledoux J, Werner ME, Brayden JE und Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006;21:69-78
4. Davis MJ, Meininger GA und Zawieja DC. Stretch-induced increases in intracellular calcium of isolated vascular smooth muscle cells. Am J Physiol 1992;263:H1292-9
5. Pohl U, Holtz J, Busse R und Bassenge E. Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension 1986;8:37-44
6. Ando J, Ohtsuka A, Katayama Y, Korenaga R, Ishikawa C und Kamiya A. Intracellular calcium response to directly applied mechanical shearing force in cultured vascular endothelial cells. Biorheology 1994;31:57-68
7. Lamontagne D, Pohl U und Busse R. Mechanical deformation of vessel wall and shear stress determine the basal release of endothelium-derived relaxing factor in the intact rabbit coronary vascular bed. Circ Res 1992;70:123-30
8. Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, Salama G, Segal SS und Kotlikoff MI. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ Res 2007;101:1300-9
9. de Wit C und Wolfle SE. EDHF and gap junctions: important regulators of vascular tone within the microcirculation. Curr Pharm Biotechnol 2007;8:11-25
10. Molina CR, Andresen JW, Rapoport RM, Waldman S und Murad F. Effect of in vivo nitroglycerin therapy on endothelium-dependent and independent vascular relaxation and cyclic GMP accumulation in rat aorta. J Cardiovasc Pharmacol 1987;10:371-8
11. Ignarro LJ, Harbison RG, Wood KS und Kadowitz PJ. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J Pharmacol Exp Ther 1986;237:893-900
12. Moncada S und Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol 2006:213-54

LITERATUR
65
13. Koeppen M, Feil R, Siegl D, Feil S, Hofmann F, Pohl U und de Wit C. cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension 2004;44:952-5
14. Lincoln TM, Dey N und Sellak H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol 2001;91:1421-30
15. Mitchell JA, Ali F, Bailey L, Moreno L und Harrington LS. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp Physiol 2008;93:141-7
16. Schroder H und Schror K. Prostacyclin-dependent cyclic AMP formation in endothelial cells. Naunyn Schmiedebergs Arch Pharmacol 1993;347:101-4
17. Vanhoutte PM. Endothelium-dependent hyperpolarizations: the history. Pharmacol Res 2004;49:503-8
18. Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, Takayanagi T, Nagao T, Egashira K, Fujishima M und Takeshita A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol 1996;28:703-11
19. Nishikawa Y, Stepp DW und Chilian WM. In vivo location and mechanism of EDHF-mediated vasodilation in canine coronary microcirculation. Am J Physiol 1999;277:H1252-9
20. Feletou M und Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol 2006;26:1215-25
21. Fitzgerald SM, Bashari H, Cox JA, Parkington HC und Evans RG. Contributions of endothelium-derived relaxing factors to control of hindlimb blood flow in the mouse in vivo. Am J Physiol Heart Circ Physiol 2007;293:H1072-82
22. Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM und Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci 2002;23:374-80
23. Bychkov R, Burnham MP, Richards GR, Edwards G, Weston AH, Feletou M und Vanhoutte PM. Characterization of a charybdotoxin-sensitive intermediate conductance Ca2+-activated K+ channel in porcine coronary endothelium: relevance to EDHF. Br J Pharmacol 2002;137:1346-54
24. Burnham MP, Bychkov R, Feletou M, Richards GR, Vanhoutte PM, Weston AH und Edwards G. Characterization of an apamin-sensitive small-conductance Ca(2+)-activated K(+) channel in porcine coronary artery endothelium: relevance to EDHF. Br J Pharmacol 2002;135:1133-43
25. Gauthier KM, Liu C, Popovic A, Albarwani S und Rusch NJ. Freshly isolated bovine coronary endothelial cells do not express the BK Ca channel gene. J Physiol 2002;545:829-36

LITERATUR
66
26. Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation 2005;12:113-27
27. Potocnik SJ, McSherry I, Ding H, Murphy TV, Kotecha N, Dora KA, Yuill KH, Triggle CR und Hill MA. Endothelium-dependent vasodilation in myogenically active mouse skeletal muscle arterioles: role of EDH and K(+) channels. Microcirculation 2009;16:377-90; 1 p following 390
28. Bolz SS, de Wit C und Pohl U. Endothelium-derived hyperpolarizing factor but not NO reduces smooth muscle Ca2+ during acetylcholine-induced dilation of microvessels. Br J Pharmacol 1999;128:124-34
29. Fisslthaler B, Fleming I und Busse R. EDHF: a cytochrome P450 metabolite in coronary arteries. Semin Perinatol 2000;24:15-9
30. Fleming I. Cytochrome p450 and vascular homeostasis. Circ Res 2001;89:753-62
31. Popp R, Bauersachs J, Hecker M, Fleming I und Busse R. A transferable, beta-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J Physiol 1996;497 ( Pt 3):699-709
32. Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR und Michelakis ED. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation 2003;107:769-76
33. Li PL und Campbell WB. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ Res 1997;80:877-84
34. Edwards G, Dora KA, Gardener MJ, Garland CJ und Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 1998;396:269-72
35. Edwards G, Gardener MJ, Feletou M, Brady G, Vanhoutte PM und Weston AH. Further investigation of endothelium-derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1-EBIO and ouabain. Br J Pharmacol 1999;128:1064-70
36. Edwards G und Weston AH. Potassium and potassium clouds in endothelium-dependent hyperpolarizations. Pharmacol Res 2004;49:535-41
37. de Clerck I, Boussery K, Pannier JL und Van De Voorde J. Potassium potently relaxes small rat skeletal muscle arteries. Med Sci Sports Exerc 2003;35:2005-12
38. Sobey CG und Faraci FM. Knockout blow for channel identity crisis: vasodilation to potassium is mediated via Kir2.1. Circ Res 2000;87:83-4
39. Matoba T, Shimokawa H, Kubota H, Morikawa K, Fujiki T, Kunihiro I, Mukai Y, Hirakawa Y und Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun 2002;290:909-13

LITERATUR
67
40. Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H und Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest 2000;106:1521-30
41. Marvar PJ, Hammer LW und Boegehold MA. Hydrogen peroxide-dependent arteriolar dilation in contracting muscle of rats fed normal and high salt diets. Microcirculation 2007;14:779-91
42. Randall MD, Alexander SP, Bennett T, Boyd EA, Fry JR, Gardiner SM, Kemp PA, McCulloch AI und Kendall DA. An endogenous cannabinoid as an endothelium-derived vasorelaxant. Biochem Biophys Res Commun 1996;229:114-20
43. Chauhan SD, Hobbs AJ und Ahluwalia A. C-type natriuretic peptide: new candidate for endothelium-derived hyperpolarising factor. Int J Biochem Cell Biol 2004;36:1878-81
44. Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol 2004;141:881-903
45. Chaytor AT, Bakker LM, Edwards DH und Griffith TM. Connexin-mimetic peptides dissociate electrotonic EDHF-type signalling via myoendothelial and smooth muscle gap junctions in the rabbit iliac artery. Br J Pharmacol 2005;144:108-14
46. Berman RS, Martin PE, Evans WH und Griffith TM. Relative contributions of NO and gap junctional communication to endothelium-dependent relaxations of rabbit resistance arteries vary with vessel size. Microvasc Res 2002;63:115-28
47. Ujiie H, Chaytor AT, Bakker LM und Griffith TM. Essential role of Gap junctions in NO- and prostanoid-independent relaxations evoked by acetylcholine in rabbit intracerebral arteries. Stroke 2003;34:544-50
48. Edwards G, Feletou M, Gardener MJ, Thollon C, Vanhoutte PM und Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br J Pharmacol 1999;128:1788-94
49. Welsh DG und Segal SS. Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol 1998;274:H178-86
50. Yamamoto Y, Klemm MF, Edwards FR und Suzuki H. Intercellular electrical communication among smooth muscle and endothelial cells in guinea-pig mesenteric arterioles. J Physiol 2001;535:181-95
51. Emerson GG und Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 2000;87:474-9
52. Siegl D, Koeppen M, Wolfle SE, Pohl U und de Wit C. Myoendothelial coupling is not prominent in arterioles within the mouse cremaster microcirculation in vivo. Circ Res 2005;97:781-8

LITERATUR
68
53. de Wit C, Boettcher M und Schmidt VJ. Signaling across myoendothelial gap junctions--fact or fiction? Cell Commun Adhes 2008;15:231-45
54. Joyner MJ und Wilkins BW. Exercise hyperaemia: is anything obligatory but the hyperaemia? J Physiol 2007;583:855-60
55. Lash JM. Contribution of arterial feed vessels to skeletal muscle functional hyperemia. J Appl Physiol 1994;76:1512-9
56. Segal SS. Integration of blood flow control to skeletal muscle: key role of feed arteries. Acta Physiol Scand 2000;168:511-8
57. Segal SS und Duling BR. Flow control among microvessels coordinated by intercellular conduction. Science 1986;234:868-70
58. Segal SS, Welsh DG und Kurjiaka DT. Spread of vasodilatation and vasoconstriction along feed arteries and arterioles of hamster skeletal muscle. J Physiol 1999;516 ( Pt 1):283-91
59. Duza T und Sarelius IH. Conducted dilations initiated by purines in arterioles are endothelium dependent and require endothelial Ca2+. Am J Physiol Heart Circ Physiol 2003;285:H26-37
60. Wolfle SE, Schmidt VJ, Hoepfl B, Gebert A, Alcolea S, Gros D und de Wit C. Connexin45 cannot replace the function of connexin40 in conducting endothelium-dependent dilations along arterioles. Circ Res 2007;101:1292-9
61. Chen Y und Rivers RJ. Nonvasomotor influence of sodium nitroprusside on arteriolar remote response to methacholine. J Vasc Res 2001;38:219-27
62. Looft-Wilson RC, Payne GW und Segal SS. Connexin expression and conducted vasodilation along arteriolar endothelium in mouse skeletal muscle. J Appl Physiol 2004;97:1152-8
63. Emerson GG und Segal SS. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res 2000;86:94-100
64. Hoepfl B, Rodenwaldt B, Pohl U und de Wit C. EDHF, but not NO or prostaglandins, is critical to evoke a conducted dilation upon ACh in hamster arterioles. Am J Physiol Heart Circ Physiol 2002;283:H996-H1004
65. Doyle MP und Duling BR. Acetylcholine induces conducted vasodilation by nitric oxide-dependent and -independent mechanisms. Am J Physiol 1997;272:H1364-71
66. Domeier TL und Segal SS. Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J Physiol 2007;579:175-86
67. Dora KA, Xia J und Duling BR. Endothelial cell signaling during conducted vasomotor responses. Am J Physiol Heart Circ Physiol 2003;285:H119-26

LITERATUR
69
68. Goodenough DA, Goliger JA und Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem 1996;65:475-502
69. Kumar NM und Gilula NB. The gap junction communication channel. Cell 1996;84:381-8
70. Haefliger JA, Nicod P und Meda P. Contribution of connexins to the function of the vascular wall. Cardiovasc Res 2004;62:345-56
71. Yeh HI, Rothery S, Dupont E, Coppen SR und Severs NJ. Individual gap junction plaques contain multiple connexins in arterial endothelium. Circ Res 1998;83:1248-63
72. Rummery NM und Hill CE. Vascular gap junctions and implications for hypertension. Clin Exp Pharmacol Physiol 2004;31:659-67
73. Griffith TM. Which connexins connect? Circ Res 2007;101:1219-21
74. Gabriels JE und Paul DL. Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed. Circ Res 1998;83:636-43
75. de Wit C, Roos F, Bolz SS, Kirchhoff S, Kruger O, Willecke K und Pohl U. Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ Res 2000;86:649-55
76. Naik JS, Valic Z, Buckwalter JB und Clifford PS. Rapid vasodilation in response to a brief tetanic muscle contraction. J Appl Physiol 1999;87:1741-6
77. Murrant CL. Stimulation characteristics that determine arteriolar dilation in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 2005;289:R505-R513
78. Armstrong RB und Laughlin MH. Rat muscle blood flows during high-speed locomotion. J Appl Physiol 1985;59:1322-8
79. Laughlin MH und Armstrong RB. Muscular blood flow distribution patterns as a function of running speed in rats. Am J Physiol 1982;243:H296-306
80. Bockman EL. Blood flow and oxygen consumption in active soleus and gracilis muscles in cats. Am J Physiol 1983;244:H546-51
81. Mohrman DE und Regal RR. Relation of blood flow to VO2, PO2, and PCO2 in dog gastrocnemius muscle. Am J Physiol 1988;255:H1004-10
82. Dobson JL und Gladden LB. Effect of rhythmic tetanic skeletal muscle contractions on peak muscle perfusion. J Appl Physiol 2003;94:11-9
83. Hamann JJ, Buckwalter JB und Clifford PS. Vasodilatation is obligatory for contraction-induced hyperaemia in canine skeletal muscle. J Physiol 2004;557:1013-20
84. Hamann JJ, Valic Z, Buckwalter JB und Clifford PS. Muscle pump does not enhance blood flow in exercising skeletal muscle. J Appl Physiol 2003;94:6-10

LITERATUR
70
85. Dyke CK, Dietz NM, Lennon RL, Warner DO und Joyner MJ. Forearm blood flow responses to handgripping after local neuromuscular blockade. J Appl Physiol 1998;84:754-8
86. Hellsten Y, Krustrup P, Iaia FM, Secher NH und Bangsbo J. Partial neuromuscular blockade in humans enhances muscle blood flow during exercise independently of muscle oxygen uptake and acetylcholine receptor blockade. Am J Physiol Regul Integr Comp Physiol 2009;296:R1106-12
87. Kirby BS, Carlson RE, Markwald RR, Voyles WF und Dinenno FA. Mechanical influences on skeletal muscle vascular tone in humans: insight into contraction-induced rapid vasodilatation. J Physiol 2007;583:861-74
88. Clifford PS. Skeletal muscle vasodilatation at the onset of exercise. J Physiol 2007;583:825-33
89. Koller A und Bagi Z. On the role of mechanosensitive mechanisms eliciting reactive hyperemia. Am J Physiol Heart Circ Physiol 2002;283:H2250-9
90. Armstrong ML, Dua AK und Murrant CL. Time course of vasodilation at the onset of repetitive skeletal muscle contractions. Am J Physiol Regul Integr Comp Physiol 2007;292:R505-15
91. Lott ME, Hogeman CS, Vickery L, Kunselman AR, Sinoway LI und MacLean DA. Effects of dynamic exercise on mean blood velocity and muscle interstitial metabolite responses in humans. Am J Physiol Heart Circ Physiol 2001;281:H1734-41
92. Hellsten Y, Maclean D, Radegran G, Saltin B und Bangsbo J. Adenosine concentrations in the interstitium of resting and contracting human skeletal muscle. Circulation 1998;98:6-8
93. Koch LG, Britton SL und Metting PJ. Adenosine is not essential for exercise hyperaemia in the hindlimb in conscious dogs. J Physiol 1990;429:63-75
94. Marshall JM. The roles of adenosine and related substances in exercise hyperaemia. J Physiol 2007;583:835-45
95. Hnik P, Holas M, Krekule I, Kuriz N, Mejsnar J, Smiesko V, Ujec E und Vyskocil F. Work-induced potassium changes in skeletal muscle and effluent venous blood assessed by liquid ion-exchanger microelectrodes. Pflugers Arch 1976;362:85-94
96. Armstrong ML, Dua AK und Murrant CL. Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 2007;581:841-52
97. Roach RC, Koskolou MD, Calbet JA und Saltin B. Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol 1999;276:H438-45

LITERATUR
71
98. Knight DR, Schaffartzik W, Poole DC, Hogan MC, Bebout DE und Wagner PD. Effects of hyperoxia on maximal leg O2 supply and utilization in men. J Appl Physiol 1993;75:2586-94
99. Frisbee JC, Murrant CL, Wilson BA und Barclay JK. Polycythemia decreases fatigue in tetanic contractions of canine skeletal muscle. Med Sci Sports Exerc 1999;31:1293-8
100. Silveira LR, Pereira-Da-Silva L, Juel C und Hellsten Y. Formation of hydrogen peroxide and nitric oxide in rat skeletal muscle cells during contractions. Free Radic Biol Med 2003;35:455-64
101. Frandsen U, Lopez-Figueroa M und Hellsten Y. Localization of nitric oxide synthase in human skeletal muscle. Biochem Biophys Res Commun 1996;227:88-93
102. McMahon TJ, Moon RE, Luschinger BP, Carraway MS, Stone AE, Stolp BW, Gow AJ, Pawloski JR, Watke P, Singel DJ, Piantadosi CA und Stamler JS. Nitric oxide in the human respiratory cycle. Nat Med 2002;8:711-7
103. Tschakovsky ME und Joyner MJ. Nitric oxide and muscle blood flow in exercise. Appl Physiol Nutr Metab 2008;33:151-61
104. Radegran G und Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol 1999;276:H1951-60
105. Shoemaker JK, Halliwill JR, Hughson RL und Joyner MJ. Contributions of acetylcholine and nitric oxide to forearm blood flow at exercise onset and recovery. Am J Physiol 1997;273:H2388-95
106. Frandsenn U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B und Hellsten Y. Exercise-induced hyperaemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with N(G)-nitro-L-arginine methyl ester in humans. J Physiol 2001;531:257-64
107. Saunders NR, Dinenno FA, Pyke KE, Rogers AM und Tschakovsky ME. Impact of combined NO and PG blockade on rapid vasodilation in a forearm mild-to-moderate exercise transition in humans. Am J Physiol Heart Circ Physiol 2005;288:H214-20
108. Saltin B. Exercise hyperaemia: magnitude and aspects on regulation in humans. J Physiol 2007;583:819-23
109. Mortensen SP, Nyberg M, Thaning P, Saltin B und Hellsten Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension 2009;53:993-9
110. Dyke CK, Proctor DN, Dietz NM und Joyner MJ. Role of nitric oxide in exercise hyperaemia during prolonged rhythmic handgripping in humans. J Physiol 1995;488 ( Pt 1):259-65

LITERATUR
72
111. Gilligan DM, Panza JA, Kilcoyne CM, Waclawiw MA, Casino PR und Quyyumi AA. Contribution of endothelium-derived nitric oxide to exercise-induced vasodilation. Circulation 1994;90:2853-8
112. Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M und Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol 2002;543:691-8
113. Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B und Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol 2007;581:853-61
114. Hussain SN, Stewart DJ, Ludemann JP und Magder S. Role of endothelium-derived relaxing factor in active hyperemia of the canine diaphragm. J Appl Physiol 1992;72:2393-401
115. Hester RL, Eraslan A und Saito Y. Differences in EDNO contribution to arteriolar diameters at rest and during functional dilation in striated muscle. Am J Physiol 1993;265:H146-51
116. Hirai T, Visneski MD, Kearns KJ, Zelis R und Musch TI. Effects of NO synthase inhibition on the muscular blood flow response to treadmill exercise in rats. J Appl Physiol 1994;77:1288-93
117. Maxwell AJ, Schauble E, Bernstein D und Cooke JP. Limb blood flow during exercise is dependent on nitric oxide. Circulation 1998;98:369-74
118. Musch TI, McAllister RM, Symons JD, Stebbins CL, Hirai T, Hageman KS und Poole DC. Effects of nitric oxide synthase inhibition on vascular conductance during high speed treadmill exercise in rats. Exp Physiol 2001;86:749-57
119. Ray CJ und Marshall JM. Nitric oxide (NO) does not contribute to the generation or action of adenosine during exercise hyperaemia in rat hindlimb. J Physiol 2009;587:1579-91
120. Lau KS, Grange RW, Isotani E, Sarelius IH, Kamm KE, Huang PL und Stull JT. nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiol Genomics 2000;2:21-7
121. Kilbom A und Wennmalm A. Endogenous prostaglandins as local regulators of blood flow in man: effect of indomethacin on reactive and functional hyperaemia. J Physiol 1976;257:109-21
122. Wilson JR und Kapoor SC. Contribution of prostaglandins to exercise-induced vasodilation in humans. Am J Physiol 1993;265:H171-5
123. Shoemaker JK, Naylor HL, Pozeg ZI und Hughson RL. Failure of prostaglandins to modulate the time course of blood flow during dynamic forearm exercise in humans. J Appl Physiol 1996;81:1516-21

LITERATUR
73
124. McKay MK, Gardner AL, Boyd D und Hester RL. Influence of venular prostaglandin release on arteriolar diameter during functional hyperemia. Hypertension 1998;31:213-7
125. Sagach VF, Kindybalyuk AM und Kovalenko TN. Functional hyperemia of skeletal muscle: role of endothelium. J Cardiovasc Pharmacol 1992;20 Suppl 12:S170-5
126. Duza T und Sarelius IH. Increase in endothelial cell Ca(2+) in response to mouse cremaster muscle contraction. J Physiol 2004;555:459-69
127. Kirchhoff S, Nelles E, Hagendorff A, Kruger O, Traub O und Willecke K. Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin40-deficient mice. Curr Biol 1998;8:299-302
128. Alcolea S, Jarry-Guichard T, de Bakker J, Gonzalez D, Lamers W, Coppen S, Barrio L, Jongsma H, Gros D und van Rijen H. Replacement of connexin40 by connexin45 in the mouse: impact on cardiac electrical conduction. Circ Res 2004;94:100-9
129. Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V, Bader M, de Wit C, Hoyer J und Kohler R. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res 2006;99:537-44
130. Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, Wulff H, de Wit C, Hoyer J und Kohler R. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 2009;119:2323-32
131. Bond CT, Sprengel R, Bissonnette JM, Kaufmann WA, Pribnow D, Neelands T, Storck T, Baetscher M, Jerecic J, Maylie J, Knaus HG, Seeburg PH und Adelman JP. Respiration and parturition affected by conditional overexpression of the Ca2+-activated K+ channel subunit, SK3. Science 2000;289:1942-6
132. Baez S. An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res 1973;5:384-94
133. Cohen KD, Berg BR und Sarelius IH. Remote arteriolar dilations in response to muscle contraction under capillaries. Am J Physiol Heart Circ Physiol 2000;278:H1916-23
134. de Wit C, von Bismarck P und Pohl U. Mediator role of prostaglandins in acetylcholine-induced vasodilation and control of resting vascular diameter in the hamster cremaster microcirculation in vivo. J Vasc Res 1993;30:272-8
135. Strobaek D, Jorgensen TD, Christophersen P, Ahring PK und Olesen SP. Pharmacological characterization of small-conductance Ca(2+)-activated K(+) channels stably expressed in HEK 293 cells. Br J Pharmacol 2000;129:991-9

LITERATUR
74
136. Rosa JC, Galanakis D, Ganellin CR, Dunn PM und Jenkinson DH. Bis-quinolinium cyclophanes: 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)- diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca(2+)-activated K+ channel. J Med Chem 1998;41:2-5
137. Liegeois JF, Mercier F, Graulich A, Graulich-Lorge F, Scuvee-Moreau J und Seutin V. Modulation of small conductance calcium-activated potassium (SK) channels: a new challenge in medicinal chemistry. Curr Med Chem 2003;10:625-47
138. Wolfle S, Kohler R, Hoyer J und de Wit C. IKCa is a major effector in EDHF-dilations in arterioles in vivo. Acta Physiol 2007;189, Supplement 653: O18-4
139. Dua AK, Dua N und Murrant CL. Skeletal muscle contraction-induced vasodilator complement production is dependent on stimulus and contraction frequency. Am J Physiol Heart Circ Physiol 2009;297:H433-42
140. VanTeeffelen JW und Segal SS. Rapid dilation of arterioles with single contraction of hamster skeletal muscle. Am J Physiol Heart Circ Physiol 2006;290:H119-27
141. Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, Hoyer J und Kohler R. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br J Pharmacol 2003;138:594-601
142. Hinton JM und Langton PD. Inhibition of EDHF by two new combinations of K+-channel inhibitors in rat isolated mesenteric arteries. Br J Pharmacol 2003;138:1031-5
143. Gluais P, Edwards G, Weston AH, Falck JR, Vanhoutte PM und Feletou M. Role of SK(Ca) and IK(Ca) in endothelium-dependent hyperpolarizations of the guinea-pig isolated carotid artery. Br J Pharmacol 2005;144:477-85
144. Wolfle SE, Schmidt VJ, Hoyer J, Kohler R und de Wit C. Prominent role of KCa3.1 in endothelium-derived hyperpolarizing factor-type dilations and conducted responses in the microcirculation in vivo. Cardiovasc Res 2009;82:476-83
145. Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI und Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A 2008;105:9627-32
146. Sandow SL, Neylon CB, Chen MX und Garland CJ. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (K(Ca)) and connexins: possible relationship to vasodilator function? J Anat 2006;209:689-98
147. Dora KA, Gallagher NT, McNeish A und Garland CJ. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res 2008;102:1247-55

LITERATUR
75
148. Berg BR, Cohen KD und Sarelius IH. Direct coupling between blood flow and metabolism at the capillary level in striated muscle. Am J Physiol 1997;272:H2693-700
149. Murrant CL und Sarelius IH. Local and remote arteriolar dilations initiated by skeletal muscle contraction. Am J Physiol Heart Circ Physiol 2000;279:H2285-94
150. Schweda F, Kurtz L, de Wit C, Janssen-Bienhold U, Kurtz A und Wagner C. Substitution of connexin40 with connexin45 prevents hyperreninemia and attenuates hypertension. Kidney Int 2009;75:482-9
151. Just A, Kurtz L, de Wit C, Wagner C, Kurtz A und Arendshorst WJ. Connexin 40 mediates the tubuloglomerular feedback contribution to renal blood flow autoregulation. J Am Soc Nephrol 2009;20:1577-85
152. Krattinger N, Capponi A, Mazzolai L, Aubert JF, Caille D, Nicod P, Waeber G, Meda P und Haefliger JA. Connexin40 regulates renin production and blood pressure. Kidney Int 2007;72:814-22
153. Spray DC. Hypertension in connexin40-null mice: a renin disorder. Kidney Int 2007;72:781-2
154. Wagner C, de Wit C, Kurtz L, Grunberger C, Kurtz A und Schweda F. Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ Res 2007;100:556-63
155. Plum A, Hallas G, Magin T, Dombrowski F, Hagendorff A, Schumacher B, Wolpert C, Kim J, Lamers WH, Evert M, Meda P, Traub O und Willecke K. Unique and shared functions of different connexins in mice. Curr Biol 2000;10:1083-91
156. van Veen TA, van Rijen HV und Jongsma HJ. Electrical conductance of mouse connexin45 gap junction channels is modulated by phosphorylation. Cardiovasc Res 2000;46:496-510
157. Beblo DA, Wang HZ, Beyer EC, Westphale EM und Veenstra RD. Unique conductance, gating, and selective permeability properties of gap junction channels formed by connexin40. Circ Res 1995;77:813-22
158. Bukauskas FF und Verselis VK. Gap junction channel gating. Biochim Biophys Acta 2004;1662:42-60
159. van Rijen HV, van Veen TA, Hermans MM und Jongsma HJ. Human connexin40 gap junction channels are modulated by cAMP. Cardiovasc Res 2000;45:941-51
160. Clifford PS, Kluess HA, Hamann JJ, Buckwalter JB und Jasperse JL. Mechanical compression elicits vasodilatation in rat skeletal muscle feed arteries. J Physiol 2006;572:561-7
161. Sandow SL, Looft-Wilson R, Doran B, Grayson TH, Segal SS und Hill CE. Expression of homocellular and heterocellular gap junctions in hamster arterioles and feed arteries. Cardiovasc Res 2003;60:643-53

LITERATUR
76
162. de Wit C. Different pathways with distinct properties conduct dilations in the microcirculation in vivo. Cardiovasc Res 2010;85:604-13
163. de Wit C, Esser N, Lehr HA, Bolz SS und Pohl U. Pentobarbital-sensitive EDHF comediates ACh-induced arteriolar dilation in the hamster microcirculation. Am J Physiol 1999;276:H1527-34
164. Loeb AL, Godeny I und Longnecker DE. Anesthetics alter relative contributions of NO and EDHF in rat cremaster muscle microcirculation. Am J Physiol 1997;273:H618-27
165. Gillham JC, Myers JE, Baker PN und Taggart MJ. Regulation of endothelial-dependent relaxation in human systemic arteries by SKCa and IKCa channels. Reprod Sci 2007;14:43-50
166. Grgic I, Kaistha BP, Hoyer J und Kohler R. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses--relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol 2009;157:509-26
167. Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, Kohler R und Wulff H. Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol 2009;75:281-95
168. Sheng JZ, Ella S, Davis MJ, Hill MA und Braun AP. Openers of SKCa and IKCa channels enhance agonist-evoked endothelial nitric oxide synthesis and arteriolar vasodilation. Faseb J 2009;23:1138-45
169. Kohler R, Kaistha BP und Wulff H. Vascular KCa-channels as therapeutic targets in hypertension and restenosis disease. Expert Opin Ther Targets 2010;14:143-55
170. Feletou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol 2009;156:545-62
171. Kohler R und Ruth P. Endothelial dysfunction and blood pressure alterations in K(+)-channel transgenic mice. Pflugers Arch 2010
172. Feletou M, Kohler R und Vanhoutte PM. Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep 2010;12:267-75
173. Hanner F, Sorensen CM, Holstein-Rathlou NH und Peti-Peterdi J. Connexins and the kidney. Am J Physiol Regul Integr Comp Physiol 2010
174. Kurtz L, Madsen K, Kurt B, Jensen BL, Walter S, Banas B, Wagner C und Kurtz A. High-Level Connexin Expression in the Human Juxtaglomerular Apparatus. Nephron Physiol 2010;116:p1-p8
175. Firouzi M, Kok B, Spiering W, Busjahn A, Bezzina CR, Ruijter JM, Koeleman BP, Schipper M, Groenewegen WA, Jongsma HJ und de Leeuw PW.

LITERATUR
77
Polymorphisms in human connexin40 gene promoter are associated with increased risk of hypertension in men. J Hypertens 2006;24:325-30
176. Gros D, Dupays L, Alcolea S, Meysen S, Miquerol L und Theveniau-Ruissy M. Genetically modified mice: tools to decode the functions of connexins in the heart-new models for cardiovascular research. Cardiovasc Res 2004;62:299-308
177. Laird DW. Life cycle of connexins in health and disease. Biochem J 2006;394:527-43

ANHANG
78
7. Anhang
7.1 Verwendete Substanzen
Substanz Hersteller
Acetylcholin Sigma-Aldrich, München
Adenosin Sigma-Aldrich, München
Agarose Invitrogen, Karlsruhe
Borsäure Sigma-Aldrich, München
Bromphenolblau Merck, Darmstadt
CaCl2 x 2 H2O Merck, Darmstadt
DMSO Sigma-Aldrich, München
DNA Ladder plus Invitrogen, Karlsruhe
dNTPs peqlab, Erlangen
Doxycyclin Sigma-Aldrich, München
EDTA Merck, Darmstadt
Ethanol Roth, Karlsruhe
Ethidiumbromid Roth, Karlsruhe
Fentanyl Curamed, Karlsruhe
Glycerol 87 % Sigma-Aldrich, München
Indomethacin Alpharma-isis, Langenfeld
Isopropanol Merck, Darmstadt
KCl Merck, Darmstadt
KH2PO4 Merck, Darmstadt
Nitro-L-Arginin Sigma-Aldrich, München
Medetomidin Pfizer, Karlsruhe
MgCl2 Invitrogen, Karlsruhe
MgSO4 x 7 H2O Merck, Darmstadt
Midazolam DeltaSelect, Dreieich
NaCl Merck, Darmstadt
NaHCO3 Merck, Darmstadt
Natriumacetat Merck, Darmstadt

ANHANG
79
Natriumnitroprussid Sigma-Aldrich, München
PCR-Puffer Invitrogen, Karlsruhe
Pentobarbital Merial, Halbergmoos
Primer zur PCR MWG-Biotech, Ebersberg
Proteinase K Invitrogen, Karlsruhe
Saccharose AppliChem, Darmstadt
SDS Porphyrin Systems, Lübeck
Taq-Polymerase Invitrogen, Karlsruhe
Tris-HCl (pH 8,0) Roth, Karlsruhe
Tris-Borat (pH 7,5) Sigma-Aldrich, München
UCL 1684 Tocris, Ellisville, USA
Gasgemisch 5 % CO2, 95 % N2 Linde, München

ANHANG
80
7.2 Lösungen und Puffer
Doxycyclinlösung TBE-Puffer
Doxycyclin 2 g/l Tris-Borat 89 mM
Saccharose 20 g/l Borsäure 89 mM
Trinkwasser EDTA 2 mM
Entmineralisiertes Wasser
Ladepuffer
Glycerol 87 % 3,45 ml TE-Puffer
Bromphenolblau 1 Spatelspitze Tris-HCl 10 mM
TBE-Puffer 6,55 ml EDTA 1 mM
Entmineralisiertes Wasser
Laird-Puffer
Tris-HCl 100 mM Superfusionslösung
SDS 0,2 % (Krebslösung)
EDTA 5 mM Na+ 138 mM
NaCl 200 mM K+ 5 mM
Entmineralisiertes Wasser Ca2+ 2,5 mM
Mg2+ 1,2 mM
Ringerlösung HCO3- 20 mM
Na+ 147 mM SO42- 1,2 mM
K+ 4 mM H2PO4- 1,2 mM
Ca2+ 2,5 mM Cl- 127,2 mM
Cl- 156 mM Entmineralisiertes Wasser
Entmineralisiertes Wasser

ANHANG
81
7.3 Weitere Materialien
Chirurgisches Nahtmaterial 6/0 (Prolene®) Ethicon, Hamburg
Platindraht (25 µm Durchmesser ) Chempur, Karlsruhe
Polyethylenkatheter PE 10 Schubert, München
Polyethylenkatheter PE 50 Schubert, München
7.4 PCR
7.4.1 Verwendete Primer
Tiere Primer Sequenz
IKCa-ko IKCa forward
IKCawt reverse
IKCako reverse
5’-CTTTGGATCCAGATGTTTCTTGGTGTTAAG-3’
5’-GCCACAGTGTGTCTGTGAGG-3’
5’-CGTGCAATCCATCTTGTTCA-3’
Cx40ko Cx40wt forward
Cx40wt reverse
Cx40ko forward
Cx40ko reverse
5'-GGGAGATGAGCAGGCCGACTTCCGGTGCG-3’
5'-GTAGGGTGCCCTGGAGGACAATCTTCCC-3'
5'-GGATCGGCCATTGAACAAGATGGATTGCAC-3'
5'-CTGATGCTCTTCGTCCAGATCATCCTGATCG-3'
Cx40KI45 Cx40 forward
Cx40wt reverse
Cx40cx45ki reverse
5’-AGGCAGGCTCATGTGGAGCT-3’
5’-AGGCTGAATGGTATCGCACC-3’
5’-TTCTTCCAGAGCCCGGTGCTG-3’
ANHANG
82
7.4.2 Reaktionsansätze
IKCa-ko Cx40ko bzw.
Cx40KI45
Probenvolumen 1,0 µl 3,5 µl
dNTPs je 25 mM 1,0 µl 0,4 µl
PCR Puffer 2,5 µl 2,5 µl
MgCl2 50 mM 0,63 µl 1,25 µl
Primermix 100 pmol/µl 3,0 µl 0,8 µl
Taq-Polymerase 5 U/µl 0,25 µl 0,25 µl
Entmineralisiertes Wasser 16,62 µl 16,3 µl
7.4.3 Temperaturverläufe
Cx40ko
Anzahl Temperatur Zeit
Einmalig 94 °C 1 min
38 Zyklen 94 °C
65 °C
72 °C
40 s
1 min
1 min
Einmalig 72 °C 7 min
IKCa-ko
Anzahl Temperatur Zeit
Einmalig 94 °C 3 min
10 Zyklen 94 °C
58 °C
72 °C
35 s
35 s
50 s
25 Zyklen 94 °C
58 °C
72 °C
35 s
35 s
50 s*
Einmalig 72 °C 10 min
* Verlängerung um 5 s mit jedem Zyklus
Cx40KI45
Anzahl Temperatur Zeit
Einmalig 94 °C 2,5 min
35 Zyklen 94 °C
68 °C
72 °C
40 s
30 s
40 s
Einmalig 72 °C 7 min

DANKSAGUNG
83
8. Danksagung
Mein ganz besonderer Dank gilt Herrn Prof. Dr. med. Cor de Wit für die
Überlassung dieses spannenden Themas und die fachlich hervorragende und
freundschaftliche Betreuung im Verlauf der Erstellung dieser Promotion. Durch
seine Kompetenz und allzeit vorhandene Diskussionsbereitschaft habe ich nicht
nur einen Einstieg in die wissenschaftliche Arbeitsweise bekommen, mir wurde
auch die Freude daran vermittelt. Sowohl bei der täglichen Arbeit als auch im
Rahmen verschiedener Kongressbesuche konnte Cor de Wit mich immer wieder
aufs Neue motivieren und half mir, auch über die eine oder andere Durststrecke
hinweg den roten Faden nicht zu verlieren.
Außerdem danke ich Herrn Prof. Dr. med. W. Jelkmann für die freundliche
Aufnahme im Institut für Physiologie und die Unterstützung, die mir für die
vorliegende Arbeit gewährt wurde. Ebenso danke ich allen weiteren Mitarbeitern
des Institutes, die für eine stets angenehme Arbeitsatmosphäre sorgten und mir
immer mit Rat und Tat zur Seite standen. Hierbei möchte ich ganz besonders Rita
Meuer hervorheben, die mir mit ihrer technischen Erfahrung nicht nur über allerlei
Hürden des Laboralltags hinweg geholfen hat, sondern mich auch oft mit einem
Becher Tee und ein paar netten Worten bei der Stange gehalten hat. Ebenso
wertvoll war für mich die Unterstützung der anderen Doktoranden der
Arbeitsgruppe, die mich zu Beginn der Arbeit geduldig in die Technik der
Intravitalmikroskopie einführten und auf deren Hilfe ich mich auch später jederzeit
verlassen konnte – vielen Dank insbesondere an Steffi Wölfle, Volker Schmidt und
Markus Böttcher.
Auch Herrn Prof. Dr. rer. nat. Ralf Köhler möchte ich an dieser Stelle nicht
unerwähnt lassen – vielen Dank für das Bereitstellen der genetisch veränderten
Mäuse, die für diese Arbeit von elementarer Bedeutung waren, sowie für die
konstruktive Mitarbeit an der gemeinsam herausgebrachten Veröffentlichung.

DANKSAGUNG
84
Dankbar bin ich auch dem Dekanat der Universität zu Lübeck sowie der
Studienstiftung des Deutschen Volkes e.V., die mir jeweils durch ihre finanzielle
Unterstützung erst ermöglicht haben, die nötige Zeit in den experimentellen Teil
dieser Arbeit zu investieren.
Schließlich möchte ich noch die Menschen erwähnen, die mir auch außerhalb des
Labors zu jeder Zeit den Rücken frei gehalten haben und mich bei allem
unterstützt haben. Hierbei sind in erster Linie meine Eltern zu nennen, die den
Grundstein für mein Studium und damit auch für diese Arbeit gelegt haben und mir
all dies erst ermöglichten. Mindestens ebenso wichtig war für mich die
Unterstützung meiner Freundin Heike, die jederzeit viel Geduld aufgebracht hat
und immer bereit war, sich auch unausgegorene und verworrene Gedankengänge
von mir anzuhören, die am nächsten Tag schon wieder Schnee von gestern
waren.
Zu guter Letzt danke ich noch all denen, die mir geholfen haben, möglichst viele
Tippfehler in dieser Arbeit noch zu entdecken und zu korrigieren, und die sich
dafür die Zeit genommen haben, sich in dieses komplexe Thema einzudenken –
vielen Dank noch einmal an meine Eltern und an meine Schwester Maren.

LEBENSLAUF
85
9. Lebenslauf

LEBENSLAUF
86

PUBLIKATIONEN
87
10. Publikationen
Originalarbeiten
Milkau M , Köhler R, de Wit C. “Crucial importance of the endothelial K+-
channel SK3 and connexin40 in arteriolar dilations during skeletal muscle
contraction.” FASEB J., September 1, 2010; 24(9): 3572 – 3579. Epub 28 Apr
2010.
Kongressbeiträge mit Abstractpublikationen
Vorträge
Milkau M , de Wit C. “Arteriolar dilations in response to exercise are attenuated
in Cx40-deficient mice.” Acta Physiol. 192 (Suppl. 663): 77, 2008.
Jahrestagung der Deutschen Physiologischen Gesellschaft, Köln, 2008.
Posterpräsentationen
Boettcher M, Milkau M , de Wit C. “EDHF-type dilations require Cx40 in small
vessels suggesting a role for myoendothelial gap junctions.” J.Vasc.Res. 43:
538, 2006. Jahrestagung der Gesellschaft für Mikrozirkulation und vaskuläre
Biologie, München, 2006.
Milkau M , de Wit C. “Arteriolar dilation in response to skeletal muscle
contraction is reduced in Cx40-deficient mice.” FASEB J. 22: 1144.2, 2008.
Experimental Biology, San Diego, USA, 2008.

PUBLIKATIONEN
88
de Wit C, Milkau M , Hoyer J, Köhler R, Schmidt V. “Ca2+-dependent K+-
channels (IK1, SK3) mediate arteriolar dilation in a distinct stimulus-dependent
fashion.” Acta Physiol. 195 (Suppl. 669): =212, 2009. Jahrestagung der
Deutschen Physiologischen Gesellschaft, Giessen, 2009.
de Wit C, Milkau M , Schmidt VJ, Wolfle SE, Hoyer J, Köhler R. “Ca2+-
dependent K+-channels (IK1, SK3) mediate arteriolar dilation in a distinct
stimulus-dependent fashion.“ FASEB J. 23: 948.22, 2009. Experimental
Biology, New Orleans, USA, 2009.
Kongressbeiträge ohne Abtractpublikationen
Vorträge
Milkau M , Köhler R, de Wit C. “Mechanisms of dilation during active hyperemia
in the skeletal muscle microcirculation.” Rostock workshop on smooth muscle
function, Rostock, 2007.
Milkau M , de Wit C. “Beteiligung von endothelialen KCa und Cx40 an der
arteriolären Dilatation bei Skelettmuskelarbeit.“ Ostseephysiologentagung,
Rostock, 2008.
Posterpräsentationen
Milkau M , de Wit C. “Durchblutungsregulation in der Muskulatur – Die Rolle
von Gap Junctions.“ 1. Lübecker Doktorandentag, Lübeck, 2007.
Milkau M , de Wit C. “Wie wird ein Muskel mit Sauerstoff versorgt? –
Mechanismen der Durchblutungssteigerung.“ 2. Lübecker Doktorandentag,
Lübeck, 2008. (Auszeichnung mit Posterpreis).