encefalopatie farmaco-resistenti e trattamento · classificazione encefalopatie ... “epilessia...
TRANSCRIPT

Hospital meeting 27.5.2015
Dr. Alberto SpaliceDipartimento di Pediatria e NPISapienza Università di Roma
Encefalopatie farmaco-resistenti e trattamento

Definizioni
Encefalopatia epilettica
Sindrome elettroclinica nella quale l’attività epilettica critica ed intercritica (clinica ed elettroencefalografica)
determina un deterioramento progressivo delle funzioni cognitive e motorie.

Classificazione
Encefalopatie epilettiche età-correlate

Sindrome di Ohtahara (OS)
Esordio: primi tre mesi di vita, spesso entro le prime due settimaneTipo di crisi: prevalentemente spasmi tonici, crisi parziali, isolate o in clustersPattern EEG: suppression-burst continuoEziologia• Encefalopatia ipossico-ischemica• Malformazioni cerebrali (emimegalencefalia, agenesia del
corpo calloso, malformazioni corticali)• Errori congeniti del metabolismo• Forme monogeniche (STXBP1, ARX, SLC2A22 ..)

Sindrome di Ohtaharapattern EEG

Sindrome di Ohtahara
Evoluzione
Ø Sindrome di West (75%) a 3-6 mesi di vita
Ø Sindrome di Lennox-Gastaut
Terapia
Ø Valproato
Ø Benzodiazepine
Ø Dieta chetogena
Ø Corticosteroidoterapia
Ø Vigabatrin
Ø Zonisamide
Ø Immunoglobuline HD

Encefalopatia Epilettica Precoce (EME)
Esordio: primi tre mesi di vita, a volte nelle prime ore
Tipo di crisi:
mioclono focale, di solito al volto o alle estremità (dita, palpebre). Le mioclonie sono erratiche o frammentarie perchè possono spostarsi da un’area all’altra del corpo con un pattern asincrono o apparentemente casuale.
Crisi focali
Automatismi
spasmi tonici
Pattern EEG: suppression-burst prevalentemente nel sonno

Encefalopatia Epilettica Precoce (EME)
Eziologia
Atrofia corticale
Errori congeniti del metabolismo
Forme monogeniche (ErbB4)
Evoluzione
Sindrome di West (50%)
Terapia
Ø Fenobarbitale
Ø Valproato
Ø Benzodiazepine
Ø Corticosteroidoterapia
Ø Vigabatrin
Ø Zonisamide

OS vs EME
OS EME
Tipo principale di crisi Spasmi tonici Myoclonias
Altri tipi di crisi Crisi motorie focaliCrisi tonico-cloniche generalizzateEmiconvulsioni
Crisi motorie focaliSpasmi tonici
EEG Suppression-burst continuo SB discontinuo, non sempre presente all’esordio, spesso più distinto nel sonno
Evoluzione 75% progression nella WS12% progression nella LGS
Fino al 50% dei casi progressione nell’ipsaritmia, poi ritorno al suppression-burst
Eziologia Malformazioni cerebraliForme monogenicheMalattie metaboliche
Malattie metabolicheForme monogenicheMalformazioni cerebrali
Cause genetiche ARX, STXBP1, PKNP, SCL25A22, CDKL5
ErbB4, SCL25A22

SINDROME DI WEST

STORIA 1841 – W.J. West “Una forma particolare di convulsioni infantili”
(Lancet)
In precedenza Clarke: “Salaam convulsions”
1951 – Vazquez e Turner: descrizione di 10 casi di una nuova sindrome “Epilessia generalizzata in flessione”
1952 – Gibbs e Gibbs: introduzione del termine “ipsaritmia”
1958 – Sorel, Dusaucy-Bauloxe: efficacia dell’ACTH
1960 – Gastuat (9th Colloque de Marseille): Sindrome di West
1990, 1992 – Chiron, Gram: vigabatrin

EPIDEMIOLOGIA
Incidenza 1:2000
Forma più frequente di encefalopatia epilettica associata a ritardo mentale
Insorgenza: 4-6 mesi non oltre il 1° anno di vita
M>F

EZIOLOGIA
Sintomatica (85-90%)
precoce alterazione sviluppo
psicomotorio e presenza di segni
neurologici
Idiopatica / Criptogenetica (10-15%)
sviluppo neurologico normale, assenza di segni di encefalopatia

CAUSE
Infezioni pre e perinatalitoxoplasmacitomegalovirusrosoliainfezioni intracraniche batteriche o virali
Sindromi neurocutaneesclerosi tuberosaNF1Sturge-Weber
Malformazionidifetti di migrazione (agiria, pachigiria, lissencefalia)oloprosencefaliaSindrome di Aicardi

CAUSE
Malattie metabolicheü iperglicinemiaü malattie mitocondrialiü ipocalcemiaü ipomagnesemiaü ipoglicemiaü Iponatriemia
Malattie cromosomicheü sindrome di Downü sindrome X-fragile
Encefalopatia ipossico-ischemica
Malattie cerebrovascolari
Fome monogenicheü CDKL5ü ARXü STXBP1

Patogenesi
Ipotesi:
1) Disfunzione tronco-encefalica di neuroni serotoninergici in pz che rispondono ad ACTH c’è aumento metaboliti serotonina (Langlais 1991)
2) Anomalie immunologiche: HLA-DRw52, aumento cellule B attivate
3) Alterazioni asse encefalo-surrene: ridotti livelli di ACTH e aumento CRH nel liquor (Baram, 1992)
4) Iperespressione di sinapsi eccitatorie: attività parossistica continua declino cognitivo (Dulac, 1994)

SINTOMI
TRIADE SINTOMATOLOGICA
1) Spasmi
2) Ipsaritmia
3) arresto e/o regressione dello sviluppo psicomotorio

1) SPASMI
Brusche contrazioni simmetriche e bilaterali dei muscoli assiali
Spasmi in flessione grappoli di spasmi
Spasmi in estensione nell’addormentamento
Spasmi in flesso-estensione e al risveglio
Sintomi associati: grido, dispnea, arrossamento cutaneo, movimenti oculari abnormi, pianto.

1) SPASMI

2) IPSARITMIA
EEG intercritico associazione caotica di complessi punta-onda, polipunta-onda, onde lente.
EEG nel sonno andamento periodico (attività ipsaritmica intervallata a fasi di depressione elettrica.
EEG critico desincronizzazione dell’EEG nel sonno.

2) IPSARITMIA

2) IPSARITMIA

3) SVILUPPO PSICOMOTORIO 1/3 dei casi sviluppo psicomotorio normale
95% dei pz. arresto dello sviluppo o perdita delle acquisizioni dopo l’esordio degli spasmi
Altri problemi neurologici associati (diplegia spastica, quadriplegia o emiparesi)

DIAGNOSI
Clinica EEG delle 24 h VideoEEG Neuroradiologia (anomalie SNC) PET (rileva aree di ipometabolismo spesso
correlate con displasie corticali ed anomalie della sostanza bianca)

PROGNOSI
Spasmi ed ipsaritmia tendono a regredire prima dei 5 aa
55-60% dei pz possono sviluppare altri tipi di crisi o sindromi epilettiche evoluzione nella Sindrome di Lennox-Gastaut
Prognosi migliore nelle forme idiopatiche

TERAPIA ACTHefficace nel trattamento degli spasmi
non evidenze su dosaggio ottimale e durata del trattamento
dosaggio e durata: 1-2 UI/Kg a giorni alterni per 5 giorni a cicli per 2 4 settimane Vigabatrin
riduzione del numero di spasmi erisoluzione dell’ipsaritmia indicazione assoluta nella sclerosi tuberosa
effetti collaterali tossicità retinica Topiramato Prednisone Acido valproico (a dosi elevate) Nitrazepam Piridossina Zonisamide Lamotrigina

SINDROME DI LENNOX-GASTAUT

Descritta per la prima volta nel 1939 (Gibbs)
Incidenza: 5% dei pazienti epilettici (10% dei pazienti di età < 15 aa)
Età di esordio 2-8 anni
M>F

CAUSE
Criptogenetica (30%)
Sintomatica (70%) : - Cause pre/perinatali: prematurità, travaglio prolungato, distress respiratorio, sclerosi tuberosa, displasia corticale, bande eterotopiche - Cause postnatali: infezioni SNC, patologie metaboliche o degenerative del SNC, patologie cerebrovascolari
Circa 1/3 dei pazienti rappresentano un’evoluzione del quadro di spasmi infantili

PATOGENESI
Non è stato dimostrato alcun meccanismo patogenetico sottostante comunque i lobi frontali sembrano esercitare un ruolo predominante (l’età di esordio corrisponde con la maturazione dei lobi frontali)

CLINICA
Triade sintomatologica
1) crisi convulsive: tonico-assiali
atonicheassenze atipiche
2) EEG: PO lente diffuse, PPO fronto-temporali
3) ritardo mentale – turbe del comportamento

CLINICA
Crisi toniche: - durante addormentamento- assiali simmetriche o unilaterali- scatenate da rumori o da stimoli tattili
EEG: scariche di punte ritmiche a 10 c/s sulle regioni anteriori e sul vertice
Crisi atoniche: perdita di tono muscoli di nuca, tronco e arti
EEG: PO e PPO diffuse
Assenze atipiche: - esordio e termine graduale con perdita di coscienza incompleta
- spesso associate a mioclonie palpebrali
EEG: PO a 2-2,5 c/s diffuse

CLINICA

CLINICA
Stato di male nel 70-80% dei pazienti
Ritardo / arresto delle acquisizioni psicomotorie
Disturbi del comportamento (iperattività, aggressività, autismo)

EEG

PROGNOSI
Evoluzione cronicità con peggioramento dei disturbi intellettivi 15-20% dei pz miglioramento crisi
Fattori prognostici negativi:- Sindrome di West- esordio precoce (< 3 aa)- elevata frequenza delle crisi, con
numerosi stati di male- EEG attività di fondo costantemente
lenta senza fase di recupero; associazione di anomalie localizzate e PO lente diffuse

TERAPIA
CBZ e fenitoina: controllano crisi generalizzate tonico-cloniche e toniche
LTG e TPM: efficaci su drop attacks
Corticosteroidi: efficaci nelle forme idiopatiche e nello stato epilettico
VPA
Rufinamide
Acetazolamide

EPILESSIA MIOCLONICA SEVERA DELL’INFANZIA o SINDROME DI DRAVET

INTRODUZIONE
Charlotte Dravet 1978
Classificazione ILAE (1989) epilessie e sindromi indeterminate focali e generalizzate
Prevalenza 1/40000
Rapporto M/F 2/1
Elevata incidenza familiare di epilessia e/o CF predisposizione genetica

EZIOLOGIA
- No associazione con patologie cerebrali specifiche
- Atrofia cerebrale diffusa
- Microdisgenesie corticali e cerebellari
- Malformazioni midollo spinale

GENETICA
SCN1A (gene codificante per α Subunit Type 1 dei canali del sodio): mutato in varie forme di epilessia
- Generalized Epilepsy Febrle Seizure Plus (GEFS +)- Severe Myoclonic Epilepsy in Infancy (SMEI)- Intractable Childhood Epilepsy (with) Generalized Tonic – Clonic Convulsions (ICEGTC)
Farmaci attivi sui canali del sodio ? Importante traguardo da raggiungere: Identificazione della patologia molecolare
Terapia farmacologica specifica

CLINICA
- 1a crisi entro il 1o anno di vita • generalmente scatenata da febbre • durata variabile • crisi cloniche (generalizzate o parziali)
- crisi successive entro 6-8 settimane • temperatura corporea sempre più bassa crisi spontanee • durata sempre maggiore stato di male • crisi tonico-cloniche, emicloniche, assenze

CLINICA
2o-4o anno • crisi miocloniche pluriquotidiane scatenate da variazioni della luminosità
• crisi parziali complesse
• assenze atipiche
• progressivo ritardo psicomotorio
• deficit neurologici progressivi

EEG
INTERCRITICO
-Attività di fondo normale all’esordio
-PO e PPO • generalizzate e/o focali • rapide • isolate/brevi buffées • talvolta predominanza di lato • fotosensibilità

EEG

RMN • Atrofia cerebrale diffusa
• Displasie corticali e cerebellari • Raramente Megacisterna Magna

PROGNOSI
- Persistenza crisi - Progressivo ritardo psicomotorio linguaggio - Deficit neurologici progressivi
- Disturbi del comportamento
- Exitus: ~15,8%

DIAGNOSI DIFFERENZIALE
• Convulsioni Febbrili • Epilessia mioclonica benigna
• Sindrome di Lennox-Gastaut

TERAPIA
Benzodiazepine Acido Valproico
Fenobarbitale Sodio bromuroEtosuccimideTopiramato

MIDAZOLAM
Benzodiazepina ad azione ultrabreve
Somministrabile per via endovenosa, orale ed intranasale
Ha un'azione immediata di sedazione, blanda miorisoluzione ed amnesia anterograda
• Midazolam oromucosale (Buccolam)

Indicazioni
La prescrizione del Buccolam è a carico del SSN solo se:
Trattamento acuto delle crisi nelle epilessie dell’età pediatrica con elevato rischio di occorrenza di crisi prolungate o di stato di male epilettico
Forme idiopatiche:
Ø sindrome di Dravet,
Ø sindrome di Panayioutopulos
Trattamento acuto delle crisi nelle altre epilessie dell’età pediatrica, dopo un precedente episodio di crisi prolungata o stato di male epilettico:
Ø forme idiopatiche;
Ø forme sintomatiche a varia eziologia
- deve essere usato solo da genitori/persone che prestano assistenza a persone che abbiano ricevuto una diagnosi di epilessia,
- per i bambini di età compresa tra 3 e 6 mesi il trattamento deve essere eseguito in contesto ospedaliero in cui sia possibile il monitoraggio e siano disponibili presidi per la rianimazione.

Indicazioni
Attualmente prescrivibile anche a:
pazienti in età evolutiva con crisi convulsive febbrili prolungate
pazienti adulti con epilessia con crisi convulsive prolungate
a seguito del compimento del 18° anno ( come continuità terapeutica o inizio )

Midazolam oromucosale
Necessario piano terapeutico, valido 12 mesi
Dose varia a seconda dell’età:
Ø 6 mesi – 1 anno -> 2,5mg
Ø 1-5 anni -> 5mg
Ø 5-10 anni -> 7,5mg
Ø 10-18 anni -> 10mg

Come somministrare il farmaco

approssimativamente dopo 5 minuti. Circa l’80% delle crisi termina entro 10 minuti
Inizio dell’effetto terapeutico:
Durata dell’azione:
• il paziente può rimanere protetto da nuove crisi per circa 4 ore

Possibili effetti collaterali
• ipersensibilità acuta e glaucoma ad angolo stretto
• non è stata stabilita la sicurezza di Midazolam in gravidanza
• cura dentale: la soluzione ha un pH di 5,0-5,5 , che è leggermente acida ( come acido come l'aceto ) , è anche senza zucchero
Depressione respiratoria
Irrequietezza
Sonnolenza ( ore )
Perdita di memoriaControindicazioni e precauzioni

Epilessia Mioclono-Atonica di Doose
Esordio: 7 mesi- 8 anni (picco 1-5 anni); M>F
Eziologia
Sconosciuta
Forme monogeniche: SCN1A, SCN1B
Tipo di crisi: crisi miocloniche e crisi atoniche con frequenza plurigiornaliera, CF in 1/3 dei casi, drop attacks in 2/3 dei casi, più raramente GTCS e assenze atipiche
EEG ictale: P-O e PPO a 2-4 Hz, interictale: inizialmente normale, scariche di P-O a 3 c/s prevalenti nel sonno

Epilessia Mioclono-Atonica di Doose
Terapia
VPA
Etosuccimide
Benzodiazepine (CZP)
Lamotrigina
KD
Prognosi
epilessia: imprevedibile, in alcuni casi farmacoresistenza, in altri casi controllo delle crisi dopo circa 3 anni
Funzioni cognitive: prognosi variabile, correlata alla severità dell’epilessia

Sindrome di Landau-Kleffner(Sindrome afasia-epilessia o afasiaepilettica acquisita)
Esordio: 5-7 anni,
Eziologia: Sconosciuta
Clinica: afasia acquisita che si manifesta in bambini con normale sviluppo neurologico, che avevano già maturato capacità espressive.
Afasia: n più del 50% dei casi esordisce con apparente perdita delle capacità uditive e di comprensione (agnosia uditiva verbale) e successivamente con perdita del linguaggio (afasia percettiva ed espressiva).
Epilessia: costituisce il sintomo di esordio nel 40-50% dei casi, in alternativa compare in coincidenza con l’afasia. Le convulsioni sono parziali-complesse, ma possono anche essere generalizzate, tonico-cloniche, atoniche e tipo assenze

LKS
EEG: scariche di punte-onde lente in regione temporale o centro-temporale di solito sinistro che tendono ad essere attivate durante la fase di sonno lento e per buona parte del tracciato (più del 70%) tanto da assumere in alcuni casi caratteristiche riconducibili ad un quadro di ESES.

LKS
Terapia
Non esiste un trattamento terapeutico univoco.
VPA +/- BDZ
L’impiego dei cortocosteroidi è controverso
Prognosi: in genere benigne, risente della severità della crisi e del timing d’esordio (pre- o post- acquisizione del linguaggio)

Epilessia con punte-ondein sonno lento (CSWS o ESES)
Esordio: 5-7 anni di vita
Quadro elettro-clinico:
alterazione elettroencefalografica
deterioramento mentale: disturbi del linguaggio, disturbi motori e del comportamento
crisi convulsive polimorfe: parziali, generalizzate, assenze o mioclonie.
Il disturbo può essere presente sia nella forma completa che in quella senza segni clinici e con la sola alterazione elettroencefalografica
A volte evoluzione trifasica:
1. Epilessia isolata
2. Epilessia + regressione neuropsicologica
3. Disabilità intellettiva isolata
CSWS
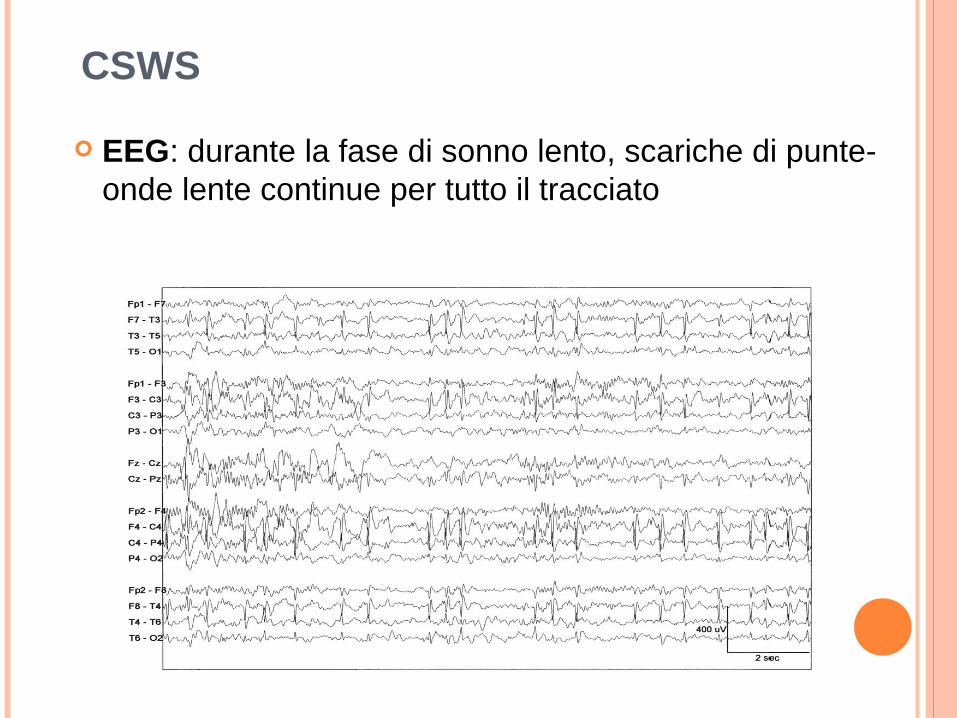
EEG: durante la fase di sonno lento, scariche di punte-onde lente continue per tutto il tracciato

CSWS
Terapia
Lamotrigina
Levetiracetam
VPA
Steroidi
BDZ
Prognosi: variabile, possibile remissione delle crisi con miglioramento delle funzioni motorie e cognitive e normalizzazione dell’EEG

GRAZIE DELL’ATTENZIONE