drepanocitosis finado
TRANSCRIPT

Drepanocitosis
ANEMIA DE CÉLULAS FALCIFORMES

Antecedentes James Herrick: Un estudiante de medicina de la isla de Grenada, primero en descubrir esta alteracion Linus Pauling: Demostró la movilidad electroforética anormal de la Hb S Vernon Ingram: Descubrió que la enfermedad se debía a la sustitución de un solo aminoácido en la
molécula de la hemoglobina Max Perutz; Perutz: Dicha estructura fue descifrada también elucidó las bases moleculares de su
función Janet Watson: Priorizo los síntomas que se manifestaban en los lactantes afectados sólo después que
la concentración de la hemoglobina fetal ha caído ayudó al entendimiento de la enfermedad.

BASES MOLECULAR
ES

Bases moleculares La drepanocitosis o A. F. se debe a la producción de una hemoglobina
mutante (Hb S), que es el resultado del reemplazo de la adenina por la timina en el codón del DNA (GTG en lugar de GAG) que codifica el ácido glutámico, en la posición 6 de la cadena α de la globina, lo que causa que este aminoácido sea sustituido por valina
La Hb S polimerice fácilmente protege al portador heterocigoto de sufrir el cuadro de paludismo endémico causado por Plasmodium falciparum.

GENÉTICA
Rasgo autosómico dominante. Formas de hemoglobinopatía S:
a. Forma heterocigota ó rasgo falciforme (HbAS)
b. Forma homocigota ó anemia falciforme (HbSS)
c. Forma doble heterocigota HbS-talasemia (HbS-Tal)
d. Forma doble heterocigota HbS-HbC (HbSC)
e. Otras formas de asociación de HbS con distintas hemoglobinopatías pueden tener una variabilidad clínica diversa.
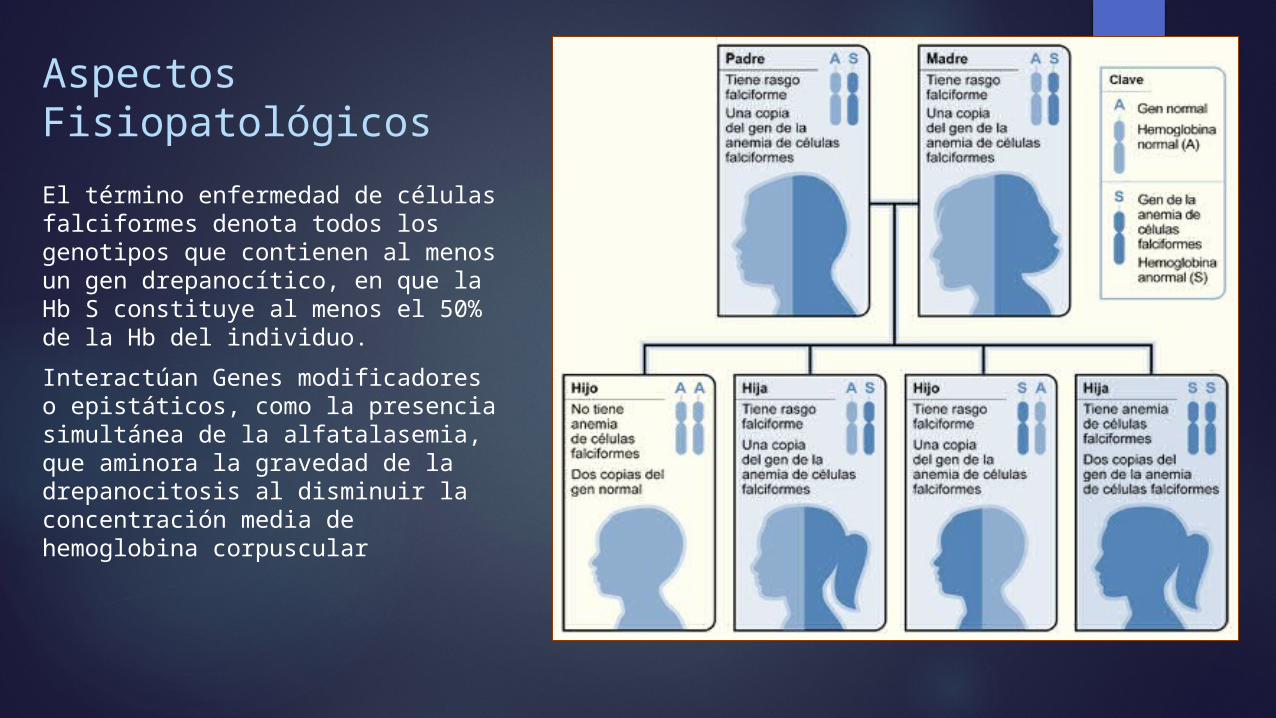
Aspectos Fisiopatológicos El término enfermedad de células falciformes denota todos los genotipos que contienen al menos un gen drepanocítico, en que la Hb S constituye al menos el 50% de la Hb del individuo. Interactúan Genes modificadores o epistáticos, como la presencia simultánea de la alfatalasemia, que aminora la gravedad de la drepanocitosis al disminuir la concentración media de hemoglobina corpuscular
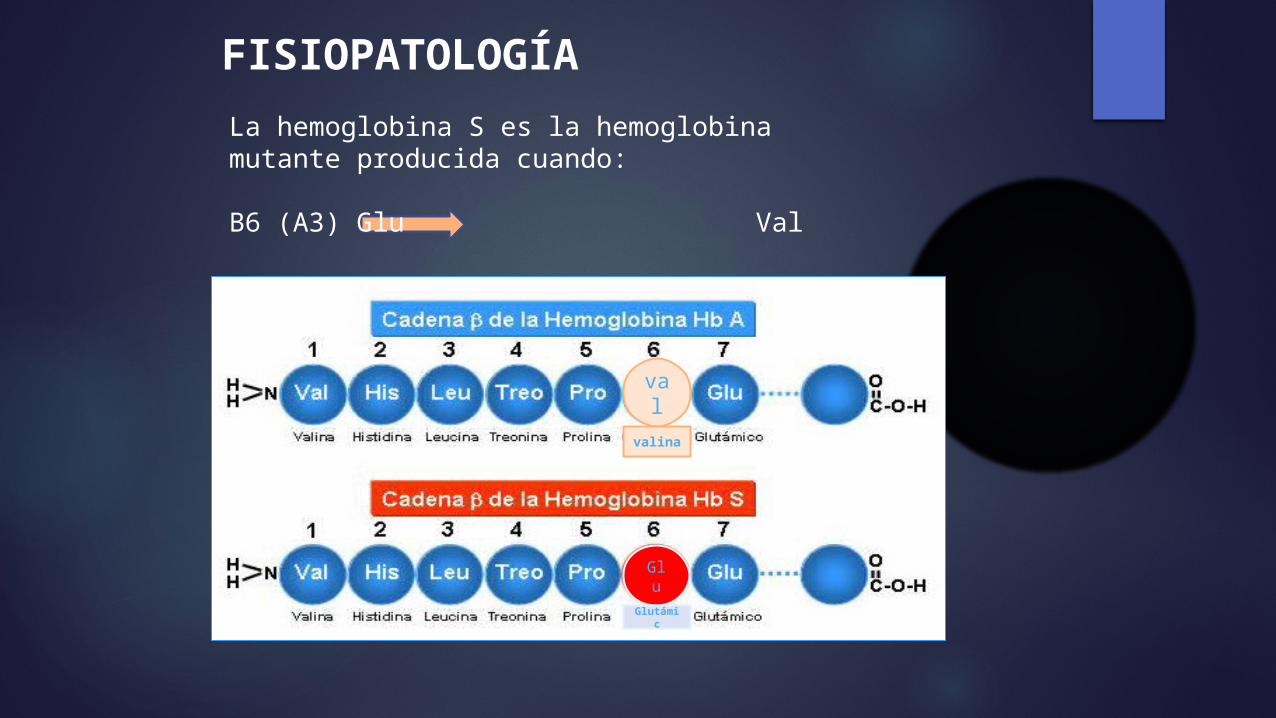
FISIOPATOLOGÍALa hemoglobina S es la hemoglobina mutante producida cuando:
Β6 (A3) Glu Val
val
valina
Glu
Glutámic
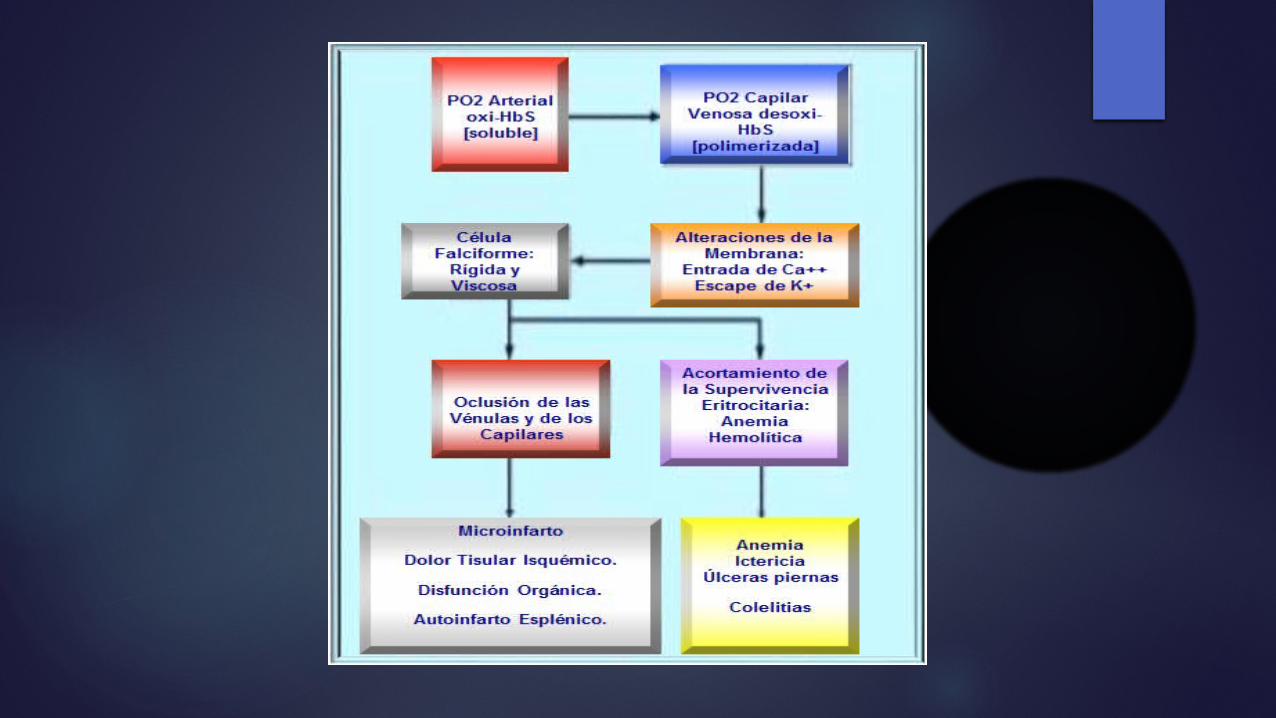
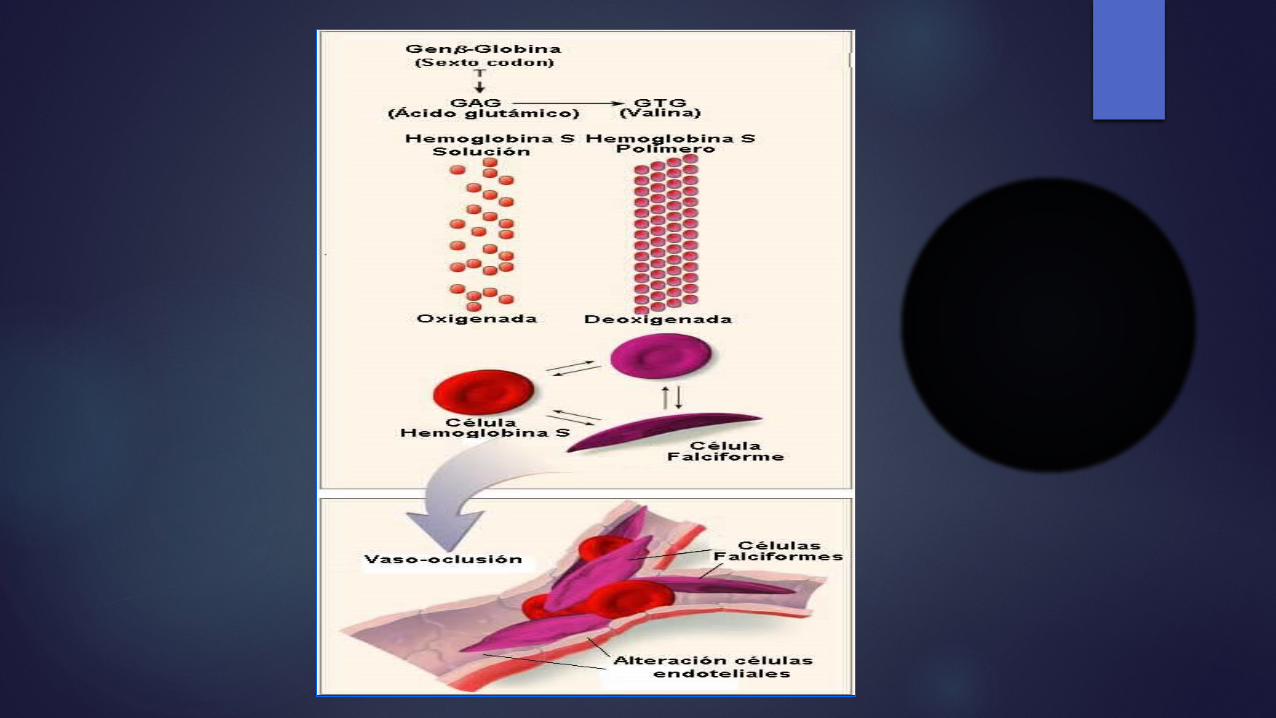
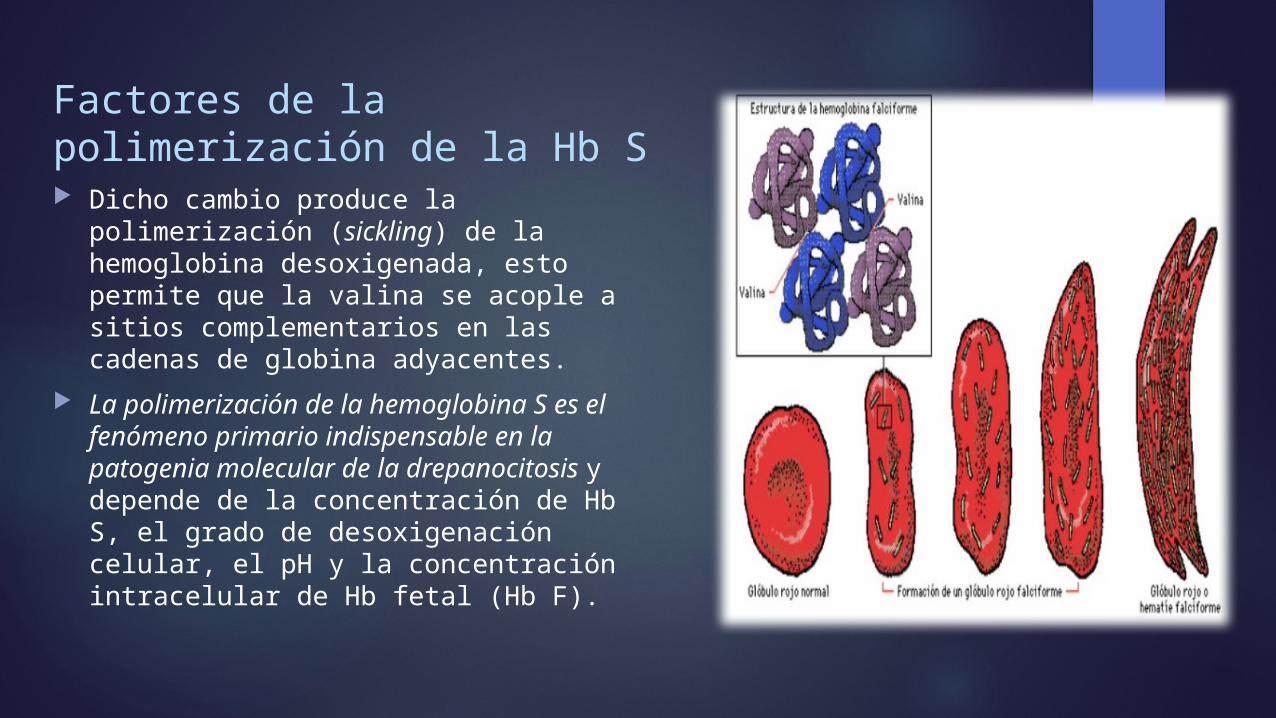
Factores de la polimerización de la Hb S Dicho cambio produce la polimerización
(sickling) de la hemoglobina desoxigenada, esto permite que la valina se acople a sitios complementarios en las cadenas de globina adyacentes.
La polimerización de la hemoglobina S es el fenómeno primario indispensable en la patogenia molecular de la drepanocitosis y depende de la concentración de Hb S, el grado de desoxigenación celular, el pH y la concentración intracelular de Hb fetal (Hb F).
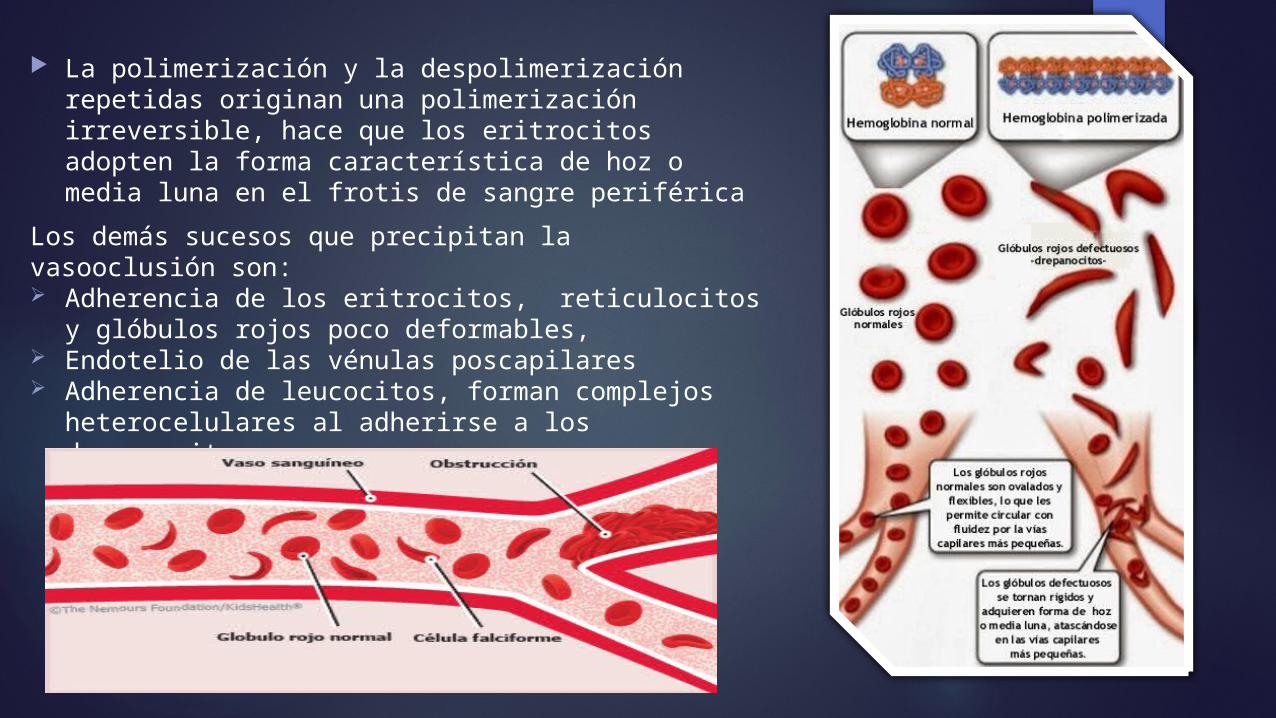
La polimerización y la despolimerización repetidas originan una polimerización irreversible, hace que los eritrocitos adopten la forma característica de hoz o media luna en el frotis de sangre periférica
Los demás sucesos que precipitan la vasooclusión son: Adherencia de los eritrocitos, reticulocitos y
glóbulos rojos poco deformables, Endotelio de las vénulas poscapilares Adherencia de leucocitos, forman complejos
heterocelulares al adherirse a los drepanocitos.
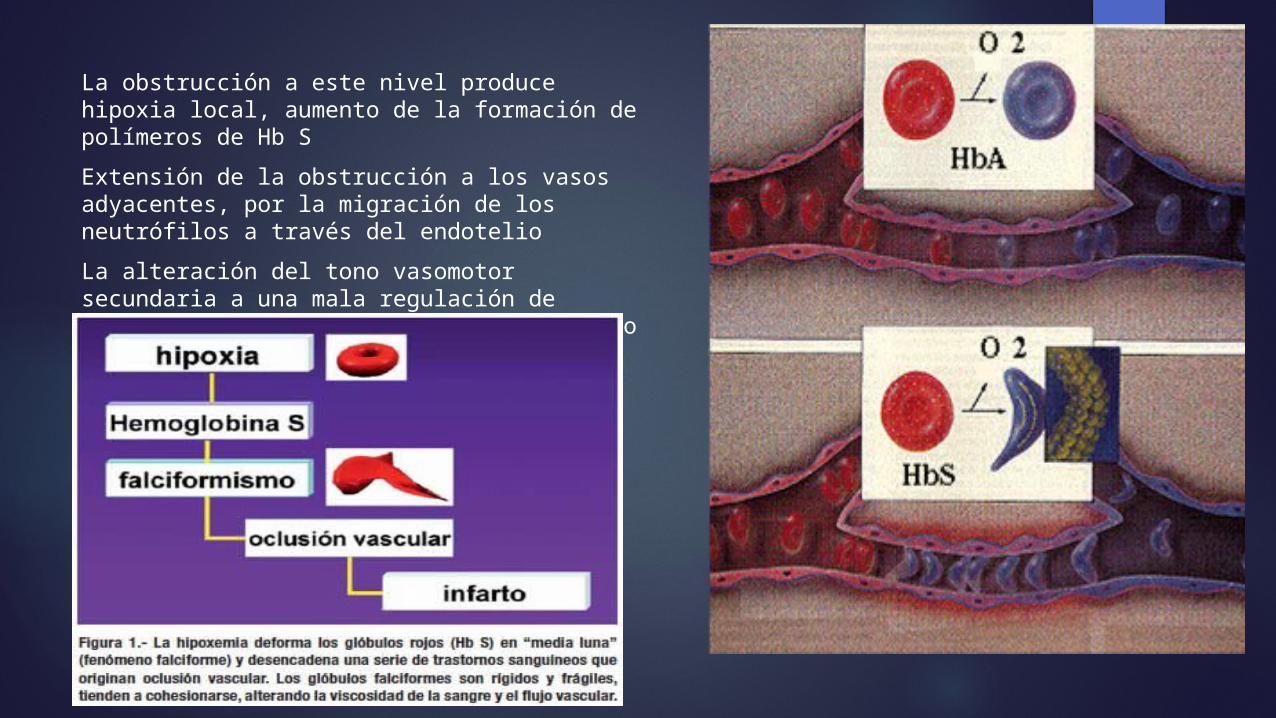
La obstrucción a este nivel produce hipoxia local, aumento de la formación de polímeros de Hb S Extensión de la obstrucción a los vasos adyacentes, por la migración de los neutrófilos a través del endotelio La alteración del tono vasomotor secundaria a una mala regulación de sustancias vasodilatadoras, como el óxido nítrico, contribuyen a este fenómeno.

Una regulación anormal de la homeostasia catiónica, que conduce a la formación de células drepanocíticas densas y a la polimerización irreversible de las mismas, secundaria al daño grave de la membrana eritrocítica, que lleva a una hemólisis acelerada.
La drepanocitosis constituye la presencia de una doble capa de lípidos disfuncional en el glóbulo rojo, con pérdida de la asimetría normal en los fosfolípidos de membrana, sobre todo la presencia de fosfatidilserina, normalmente del lado citoplásmico, en el lado externo de la misma.

Cuadro clínico y diagnóstico El neonato está protegido durante ocho a 10 semanas por el alto
porcentaje de Hb fetal presente. Es importante notar que 33% de los pacientes cursan asintomáticos. El cuadro clínico es el de un individuo estable por largos periodos, los
cuales se interrumpen por crisis de las siguientes clases: Hay un numeroso grupo adicional de manifestaciones clínicas, como el
síndrome torácico agudo, cuya repetición predispone a la hipertensión pulmonar; entre otras causas, están las infecciones diversas y la embolia grasa, posterior a la necrosis de la médula ósea.

El crecimiento de los niños afectados es más lento. Hay engrosamiento de los huesos a expensas de la cavidad medular, dactilitis y daño en la médula renal con pérdida de la capacidad de concentración.
En el ojo hay daño a la vasculatura retiniana que puede dar origen a infarto de la retina y desprendimiento posterior de ésta.
Las pacientes embarazadas con drepanocitosis resultan más afectadas por pielonefritis, infartos pulmonares, neumonía, hemorragia preparto, prematurez y muerte fetal.
DIAGNÓSTICO
HPLC (cromatografía líquida de alta presión)- Método inicial para RN ideal.
- Utiliza como muestra el papel secante del cribado metabolopatía neonatal.
- Útil para la cuantificación de HbS y F.
- Útil en el seguimiento de los tratamientos.
Prueba de la falciformación. Células drepanocíticas son obtenidas in vitro luego de adición de metabisulfito de sodio a la muestra.

DIAGNÓSTICO
Electroforesis de hemoglobina a pH alcalino y ácido (acetato- celulosa y agar citrato)
Precisa mayor muestra de sangre. Venopunción habitual. Sangre de cordón – cribado neonatal.

DIAGNÓSTICO Pacientes susceptibles de realización de estas técnicas:
- Población neonatal global
- Paciente sospechoso de cualquier edad:
a) APF de drepanocitosis
b) Individuo raza negra.
Sepsis+ meningitis+ neumococo
Dolor
ACVA
c) Anemia Síndrome torácico agudo
Priapismo
Hiperesplenismo agudo
Úlceras
- Anemia hemolítica aguda
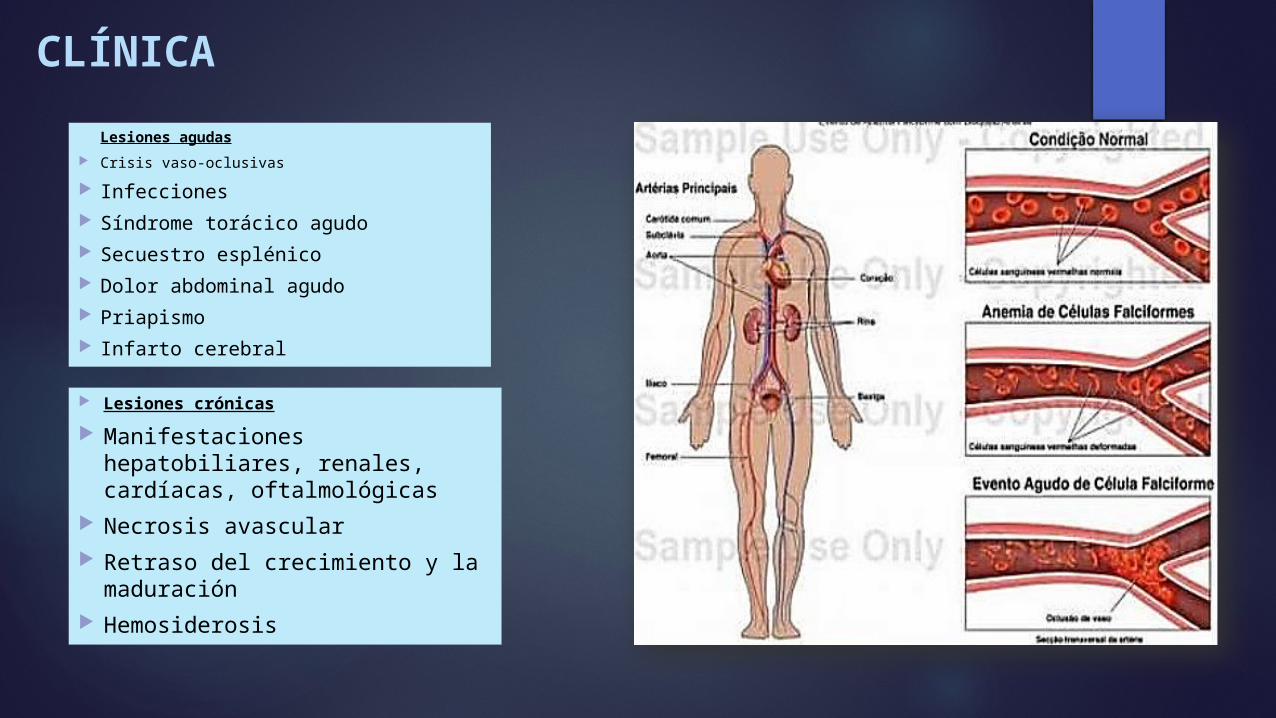
CLÍNICA
Lesiones agudas Crisis vaso-oclusivas
Infecciones Síndrome torácico agudo Secuestro esplénico Dolor abdominal agudo Priapismo Infarto cerebral
Lesiones crónicas
Manifestaciones hepatobiliares, renales, cardíacas, oftalmológicas
Necrosis avascular Retraso del crecimiento y la
maduración Hemosiderosis

SIGNOS Y SÍNTOMAS DE ALARMA
Temperatura ≥ 38,5ºC Dolor moderado o severo Síntomas respiratorios Dolor abdominal, distensión y/o
aumento agudo del bazo Signos o síntomas neurológicos Aumento de la palidez, fatiga o letargia Priapismo de >2-3 hs de persistencia

Crisis de infarto Es patognomónica y la más común. Provocada por la obstrucción de los vasos sanguíneos por
los drepanocitos rígidos, causa hipoxia y muerte hística; es más frecuente en huesos, tórax y abdomen.
Hay oclusión microvascular en la médula ósea, causando necrosis de la misma, sobre todo en los huesos largos, costillas, esternón, cuerpos vertebrales y pelvis.
El bazo es tan repetidamente afectado que a los seis u ocho años de edad el paciente se halla “autoesplenectomizado”, lo que se puede reflejar en la presencia de eritrocitos, conteniendo los cuerpos de Howell-Jolly en la sangre periférica

Crisis aplasia Con mucha frecuencia tiene su origen
en infecciones virales, principalmente la debida al parvovirus humano B19, que es citotóxico para los precursores eritroides, causando una reticulocitopenia de siete a 10 días de duración.

Crisis de secuestro esplénico Ocurre en la infancia temprana y se debe a un repentino atrapamiento masivo de
eritrocitos en el bazo. La cantidad de Hb es menor de 6 g/dl y su disminución es de 2 a 3 g o más con
respecto al valor basal, acompañada de esplenomegalia, reticulocitosis y en ocasiones por trombocitopenia.

Crisis hemolítica Se debe a un aumento en la tasa de hemólisis por diferentes razones.

Datos de laboratorio En el frotis de sangre periférica se observan drepanocitos (células en forma
de hoz o media luna), dianocitos y, si existe atrofia esplénica, cuerpos de Howell-Jolly, que corresponden a restos del núcleo.
El diagnóstico se basa en la inducción de drepanocitos por hipoxia o metabisulfito de sodio, o por ambos, y de manera más precisa mediante la electroforesis de hemoglobina a pH alcalino.
Esta técnica demuestra la presencia de Hb S, la cual se mueve lentamente,atrás de la Hb A.
El diagnóstico prenatal se basa en técnicas de secuenciación y amplificación del DNA, a partir de una muestra de las vellosidades coriónicas, obtenida por biopsia en el primer trimestre del embarazo y sometida a amplificación del DNA mediante la reacción en cadena de la polimerasa.
En el segundo trimestre del embarazo estudiando el líquido amniótico. También se puede hacer el diagnóstico en los eritrocitos fetales cuando se
encuentran circulando en la madre.

Tratamiento Consiste principalmente en evitar los factores que desencadenan las crisis,
como deshidratación, infecciones, anoxia, estasis circulatoria y exposición prolongada al frío.
Se deben administrar las vacunas para generar una respuesta inmune adecuada contra neumococos, meningococos y haemophilus inflienzae, bacterias que pueden causar mayores problemas debido a la asplenia funcional o anatómica.
La administración profiláctica de penicilina oral debe ser continua, una vez que el bazo deja de ser funcional.
La hidroxiurea es un citotóxico y citorreductor que actúa en la fase de síntesis (S) del ciclo celular, incrementa la concentración de hemoglobina F, regula la adhesividad del eritrocito y aumenta el óxido nítrico.
La hidroxiurea disminuye la frecuencia de crisis dolorosas, síndrome torácico agudo, número de hospitalizaciones y la necesidad de transfusión sanguínea.
No existe entonces un tratamiento específi co y la atención depende del cuadro clínico, aunque se ha utilizado el trasplante de médula ósea.
Bibliografía Librería Cochrane/ uptodate
Harrison Principios de medicina interna
http://www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf
Hematología 8ª Edición Manual CTO de Medicina y Cirugía. Editorial CTO
José Carlos Jaime Pérez/ Davis Gomez Almaguer. Hematología: La sangre y sus enfermedades 2ª Edición . Editorial Mc Graw Hill
Williams, Manual de Hematología, 8ª Edición, Editorial Mc Graw Hill, Abbadon_9117