Download - Validação de métodos analíticos - conceitos

Validação de métodos físico-quimicos
Agosto - 2013

Vanessa Rodrigues Lopes
Farmacêutica, graduada pela Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo – USP, São Paulo com especialização em
fármacos e medicamentos. Possui vários cursos de especialização nas áreas
de Validação Analítica, Estabilidade, Controle de Qualidade e Assuntos
Regulatórios por associações independentes. Experiência de 12 anos
adquirida nas empresas Bristol-Myers Squibb nas áreas de controle e
garantia de qualidade e Eurofarma na área de assuntos regulatórios onde
atua como especialista em assuntos regulatórios, como link entre as áreas
técnicas e regulatória na submissão de novos projetos, resposta à
exigências e treinamentos técnicos internos. Responsável pelos contatos
técnicos com a ANVISA para desenho de projetos e participação ativa nas
discussões de entidades para revisão da legislação técnica atual.

Tópicos
• Legislação envolvida no assunto;
• Importância da validação;
• Etapas prévias à validação;
• Parâmetros de validação e principais tendências;
• Reporte de validação;
• Mantendo “status” validado do método;
• Validação x verificação de métodos;
• Exercício – Montagem de protocolo de validação

Legislação e guias relevantes
• RDC 899/2003 – Validação de métodos analíticos;
• RDC 48/2009 – Alterações pós registro demedicamentos;
• RDC 27/2012 – Validação de métodos bioanalíticos
• RDC 31/2010 – Equivalência Farmacêutica – falasobre validação parcial;
• CP 11/2012 – Impurezas e produtos de degradação
• INMETRO - DOQ-CGCRE-008 – Orientações paravalidação analítica (rev. 03 – 2010);

Legislação e guias relevantes
• IUPAC: Harmonized guidelines for single laboratoryvalidation of methods of analysis – 2002
• FDA (draft): Analytical Procedures and MethodsValidation – 2000
• EURACHEM: The Fitness for Purpose of AnalyticalMethods (1998)
• ICHQ2(R1) – Validation of analytical procedures: Textand methodology

O que é validação?
• Ato documentado que atesta que qualquerprocedimento, processo, equipamento, material,atividade ou sistema realmente e consistentementeleva aos resultados esperados;
– RDC 17/2010 – Boas Práticas de Fabricação
• Process of defining an analytical requirement, andconfirming that the method under consideration hasperformance capabilities consistent with what theapplication requires.
– Eurachem: Fitness for purpose of analytical methods

Porque validar?• Demonstrar que o método é apropriado para a
finalidade pretendida, ou seja, a determinaçãoqualitativa, semi-quantitativa e/ou quantitativa defármacos e outras substâncias em produtosfarmacêuticos.
– RDC 899/2003 – Validação de métodos analíticos
• A validação é uma parte essencial de Boas Práticasde Fabricação (BPF), sendo um elemento da garantiada qualidade associado a um produto ou processoem particular.
– RDC 17/2010 – Boas Práticas de Fabricação

Quando validar?• Os métodos de controle de qualidade devem ser validados
antes de serem adotados na rotina, levando-se emconsideração as instalações e os equipamentos disponíveis.– Parágrafo único. Os métodos analíticos compendiais não requerem
validação, entretanto antes de sua implementação, devem existirevidências documentadas de sua adequabilidade nas condiçõesoperacionais do laboratório.
– RDC 17/2010 – Boas Práticas de Fabricação
• No caso de metodologia analítica descrita em farmacopéiasou formulários oficiais, devidamente reconhecidos pelaANVISA, a metodologia será considerada validada.
– RDC 899/2003 – Validação analítica


Quando revalidar?• A metodologia analítica deverá ser revalidada nas seguintes
circunstâncias:– mudanças na síntese da substância ativa,
– mudanças na composição do produto acabado,
– mudanças no procedimento analítico.
– Outras mudanças podem requerer validação dependendo da suanatureza.
– RDC 899/2003 – Validação de métodos analíticos
• Alterações de formulação (excipiente, sabor, cor), alteração deespecificações e métodos, inclusão de novo fabricante dofármaco ou alterações na sua rota de síntese, inclusão denova concentração ou forma farmacêutica do medicamento,
– RDC 48/2009 – Alterações pós registro

Responsáveis pela validação?
Responsável Técnico
assegurar a realização dos programas de validação
Responsável pelo Controle de Qualidade
assegurar que sejam feitas as validações necessárias, inclusive a validação dos métodos analíticos e calibração dos equipamentos de controle
Responsável pela Garantia da Qualidade
assegurar o correto cumprimento das atividades de validação
RDC 17/2010 – Boas Práticas de Fabricação
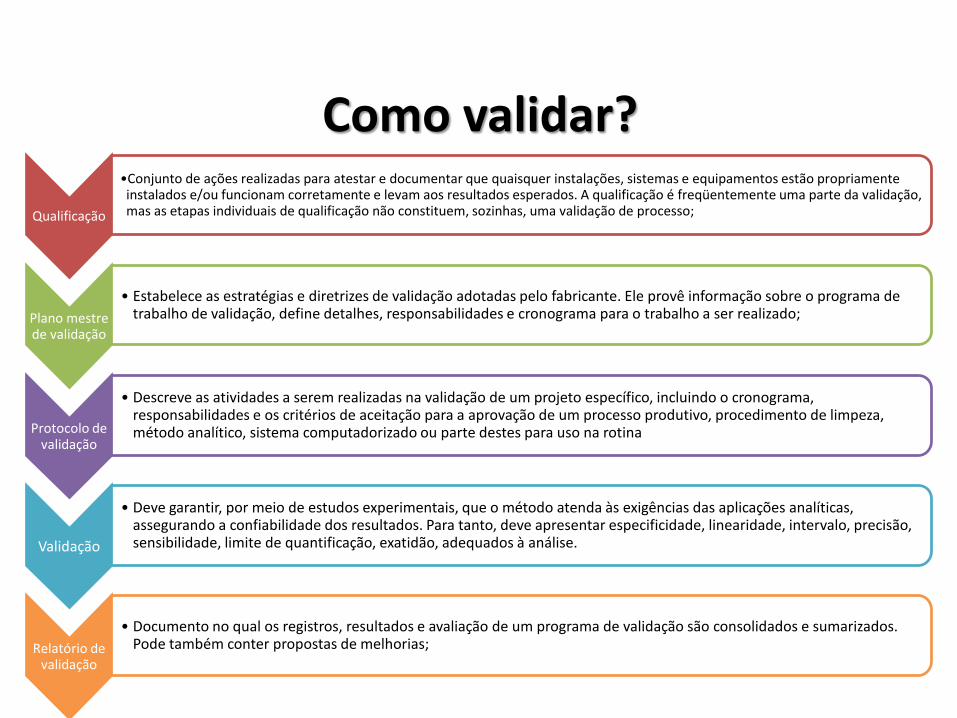
Como validar?
Qualificação
•Conjunto de ações realizadas para atestar e documentar que quaisquer instalações, sistemas e equipamentos estão propriamente instalados e/ou funcionam corretamente e levam aos resultados esperados. A qualificação é freqüentemente uma parte da validação, mas as etapas individuais de qualificação não constituem, sozinhas, uma validação de processo;
Plano mestre de validação
• Estabelece as estratégias e diretrizes de validação adotadas pelo fabricante. Ele provê informação sobre o programa de trabalho de validação, define detalhes, responsabilidades e cronograma para o trabalho a ser realizado;
Protocolo de validação
• Descreve as atividades a serem realizadas na validação de um projeto específico, incluindo o cronograma, responsabilidades e os critérios de aceitação para a aprovação de um processo produtivo, procedimento de limpeza, método analítico, sistema computadorizado ou parte destes para uso na rotina
Validação
• Deve garantir, por meio de estudos experimentais, que o método atenda às exigências das aplicações analíticas, assegurando a confiabilidade dos resultados. Para tanto, deve apresentar especificidade, linearidade, intervalo, precisão, sensibilidade, limite de quantificação, exatidão, adequados à análise.
Relatório de validação
• Documento no qual os registros, resultados e avaliação de um programa de validação são consolidados e sumarizados. Pode também conter propostas de melhorias;

Requisitos prévios para validaçãoPadrões de referência:
1.4. Deve-se utilizar substâncias de referência oficializadas pela FarmacopéiaBrasileira ou, na ausência destas, por outros códigos autorizados pela legislaçãovigente. No caso da inexistência dessas substâncias, será admitido o uso depadrões de trabalho, desde que a identidade e o teor sejam devidamentecomprovados (RDC 899/2003)
• padrão secundário (padrão de trabalho): padrão utilizado na rotinalaboratorial, cujo valor é estabelecido por comparação a um padrão dereferência (RDC 17/2010) – NÃO ACEITOS EM VALIDAÇÃO!!!
• padrão de referência: são exemplares de fármacos, impurezas, produtos dedegradação, reagentes, dentre outros, altamente caracterizados e da maiselevada pureza, cujo valor é aceito sem referência a outros padrões (RDC17/2010);
– Farmacopeico: Adquirido de um compêndio oficial reconhecido pela ANVISA (RDC37/2009);
– Caracterizado (primario): Análises para determinação absoluta da pureza eidentidade;
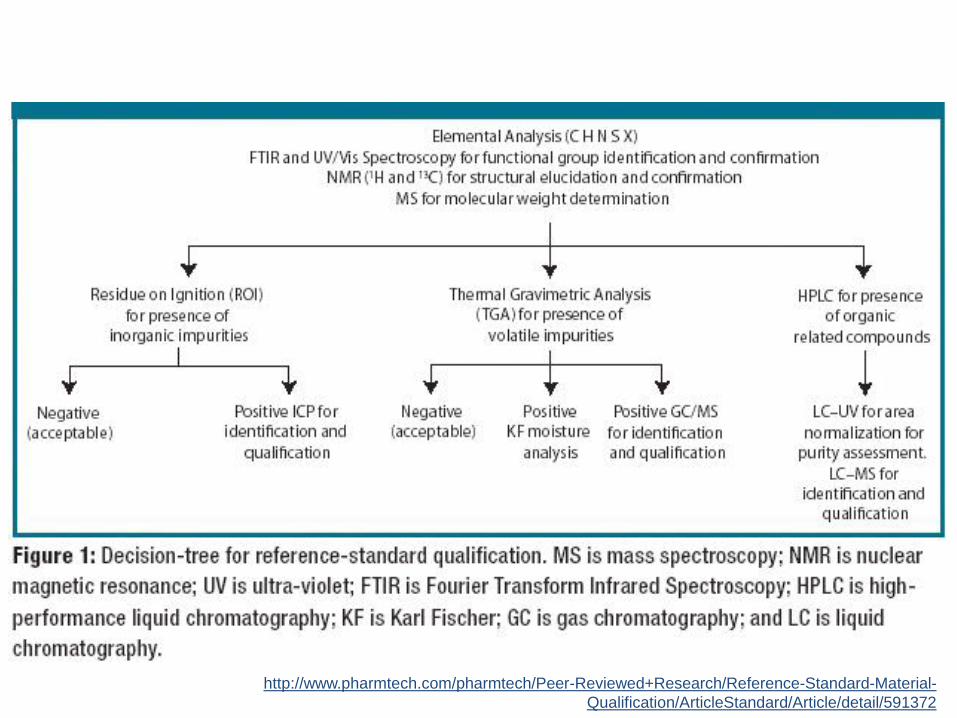
http://www.pharmtech.com/pharmtech/Peer-Reviewed+Research/Reference-Standard-Material-
Qualification/ArticleStandard/Article/detail/591372

Requisitos prévios para validaçãoQualificação e calibração de equipamentos:
1.9. Para a garantia da qualidade analítica dos resultados, todos osequipamentos utilizados na validação devem estar devidamentecalibrados e os analistas devem ser qualificados e adequadamentetreinados (RDC 899/2003);• qualificação: conjunto de ações realizadas para atestar e documentar que
quaisquer instalações, sistemas e equipamentos estão propriamente instaladose/ou funcionam corretamente e levam aos resultados esperados (RDC17/2010);
• calibração: conjunto de operações que estabelece, sob condiçõesespecificadas, a relação entre os valores indicados por um instrumento ousistema de medição ou valores representados por uma medida materializadaou um material de referência, e os valores correspondentes das grandezasestabelecidos por padrões (RDC 17/2010);

Requisitos prévios para validação
Treinamento dos analistas:
1.9. Para a garantia da qualidade analítica dos resultados,todos os equipamentos utilizados na validação devemestar devidamente calibrados e os analistas devem serqualificados e adequadamente treinados (RDC 899/2003);

Plano mestre de validação• Deve conter os elementos chave do programa de validação.
Deve ser conciso e claro, bem como conter, no mínimo:I. uma política de validação;
II. estrutura organizacional das atividades de validação;
III. sumário/relação das instalações, sistemas, equipamentos e processos que se encontram validados e dos que ainda deverão ser validados (situação atual e programação);
IV. modelos de documentos (ex: modelo de protocolo e de relatório) ou referência a eles;
V. planejamento e cronograma;
VI. controle de mudanças; e
VII. referências a outros documentos existentes.
– RDC 17/2010 – Boas práticas de fabricação

Protocolo de validação• Devem incluir, no mínimo, as seguintes informações:
I. objetivos do estudo;
II. local/planta onde será conduzido o estudo;
III. responsabilidades;
IV. descrição dos procedimentos a serem seguidos;
V. equipamentos a serem usados, padrões e critérios para produtos e processos relevantes;
VI. tipo de validação;
VII. processos e/ou parâmetros;
VIII. amostragem, testes e requisitos de monitoramento; e
IX. critérios de aceitação.
– RDC 17/2010 – Boas práticas de fabricação

Processos/Parâmetros da validação• No caso de metodologia analítica não descrita em
farmacopéias ou formulários oficiais, devidamentereconhecidos pela ANVISA, a metodologia será consideradavalidada, desde que sejam avaliados os parâmetrosrelacionados a seguir,– Especificidade e Seletividade
– Linearidade
– Intervalo
– Precisão
– Limite de detecção (sensibilidade)
– Limite de quantificação
– Exatidão
– Robustez
– RDC 899/2003 – Validação de métodos analíticos


Categoria Finalidade
ITestes quantitativos para a determinação do princípio ativo em produtos farmacêuticos ou matérias–primas
IITestes quantitativos ou ensaio limite para a determinação de impurezas e produtos de degradação em produtos farmacêuticos e matérias-primas
IIITestes de performance (por exemplo: dissolução, liberação do ativo, etc)
IV Testes de identificação
Categorias de testes
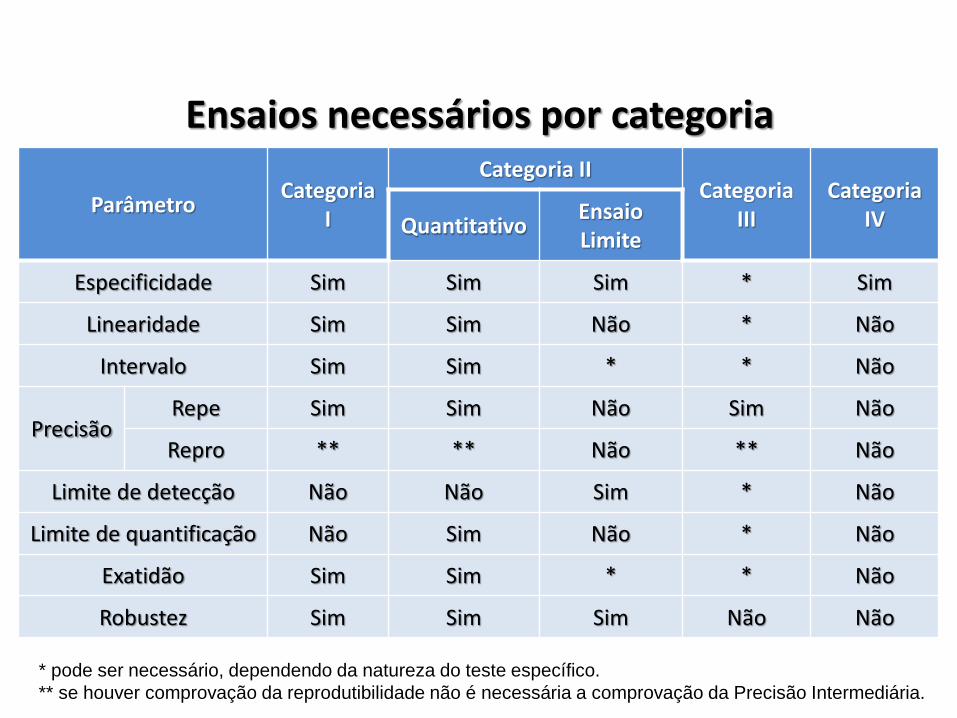
Ensaios necessários por categoria
ParâmetroCategoria
I
Categoria IICategoria
IIICategoria
IVQuantitativoEnsaio Limite
Especificidade Sim Sim Sim * Sim
Linearidade Sim Sim Não * Não
Intervalo Sim Sim * * Não
PrecisãoRepe Sim Sim Não Sim Não
Repro ** ** Não ** Não
Limite de detecção Não Não Sim * Não
Limite de quantificação Não Sim Não * Não
Exatidão Sim Sim * * Não
Robustez Sim Sim Sim Não Não
* pode ser necessário, dependendo da natureza do teste específico.
** se houver comprovação da reprodutibilidade não é necessária a comprovação da Precisão Intermediária.

EspecificidadeÉ a capacidade que o método possui de medir exatamente umcomposto em presença de outros componentes tais comoimpurezas, produtos de degradação e componentes da matriz
Contém compostos
semelhantes
Contém fármaco
Quantitativamente: Seleção entre o alvo e compostos semelhantes;
Qualitativamente: Ausência de interferência na presença de compostos
semelhantes (comprovar pureza cromatográfica do pico alvo)
• Impurezas “disponíveis”: Contaminar amostra
• Impurezas “indisponíveis”: Estudo de stress ou comparação de métodos

Especificidade – Planejamento do experimentoPreparação Objetivo
Padrões do alvo Avaliar a resposta básica do composto de interesse na técnica
Diluentes, fase móvel Avaliar ausência de resposta (ausência de picos no TR ou de resposta no comprimento de onda ou na técnica utilizada – titulação por exemplo)
Placebo branco Avaliar a ausência de resposta no TR ou comprimento de onda e localizar os picos relacionados ao placebo (HPLC/CG)
Placebo contaminado com padrão alvo
Avaliar o comportamento do alvo na matriz (se há alteração de resposta por exemplo ou se há interação que possa ser verificada –DAD)
Placebo contaminado com impurezas disponíveis (na concentração esperada)
Avaliar o comportamento das impurezas na matriz quando o alvo está ausente. Localizar os picos relacionados exclusivamente às impurezas disponíveis
Placebo contaminado com impurezas disponíveis (na concentração esperada) + alvo
Avaliar o comportamento do alvo na matriz e com a presença de impurezas
Amostras submetidas à stress
Avaliar a pureza dos picos ou ausência de resposta na presença de impurezas conhecidas e desconhecidas

Especificidade – Estudo de stress
• A empresa deverá apresentar estudos submetendo omedicamento às seguintes condições de estresse:
I. Aquecimento;
II. Umidade;
III. Solução ácida;
IV. Solução básica;
V. Solução oxidante;
VI. Exposição fotolítica; e
VII. Íons metálicos.
• Justificar tecnicamente a não utilização de qualquer uma dessascondições.
• Promover uma degradação de pequena extensão em torno de10-30% (dez a trinta por cento). Justificar degradação inferior;
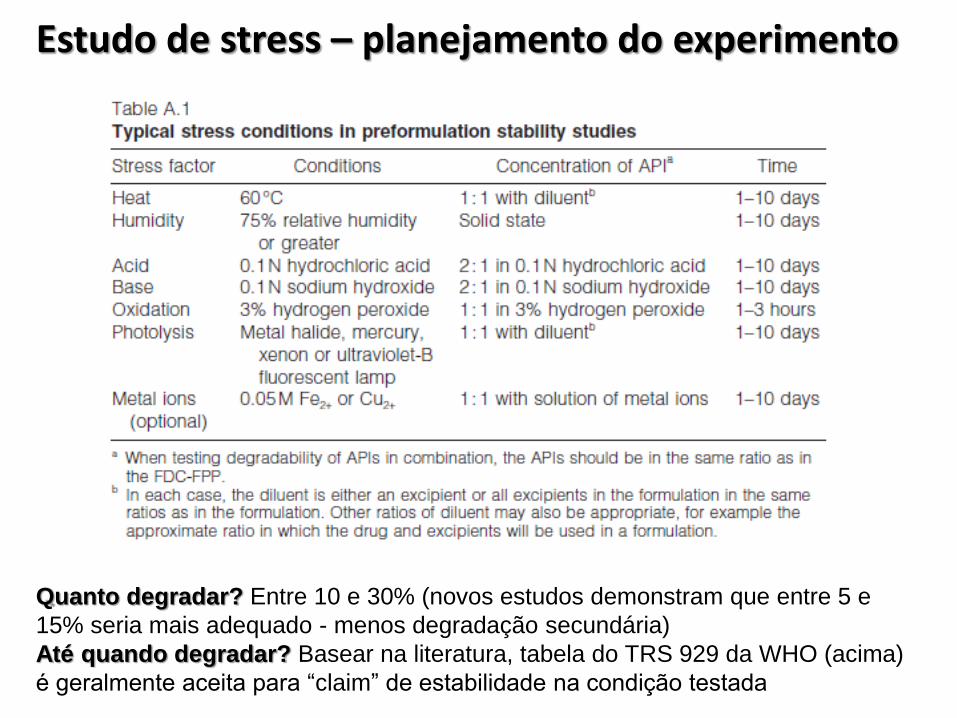
Estudo de stress – planejamento do experimento
Quanto degradar? Entre 10 e 30% (novos estudos demonstram que entre 5 e
15% seria mais adequado - menos degradação secundária)
Até quando degradar? Basear na literatura, tabela do TRS 929 da WHO (acima)
é geralmente aceita para “claim” de estabilidade na condição testada

Estudo de stress – Planejamento do experimentoPreparação Objetivo
Placebo degradado Avaliar e excluir os picos relativos exclusivamente à degradação do placebo
Padrão controle Padrão não degradado para calculo de recuperação após degradação
Padrão degradado Avaliar os picos relativos aos degradantes do padrão (overlay com placebo degradado para exclusão de picos do placebo). Avaliar também a característica indicativa de estabilidade
Amostra controle Avaliar os picos relativos à amostra (interação do ativo com os excipientes) e usada para calcular recuperação na degradação
Amostra degradada Avaliar os picos relativos à degradação do ativo na presença da matriz (placebo) que pode ser diferente da degradação do padrão sozinho (interação fármaco-excipiente)
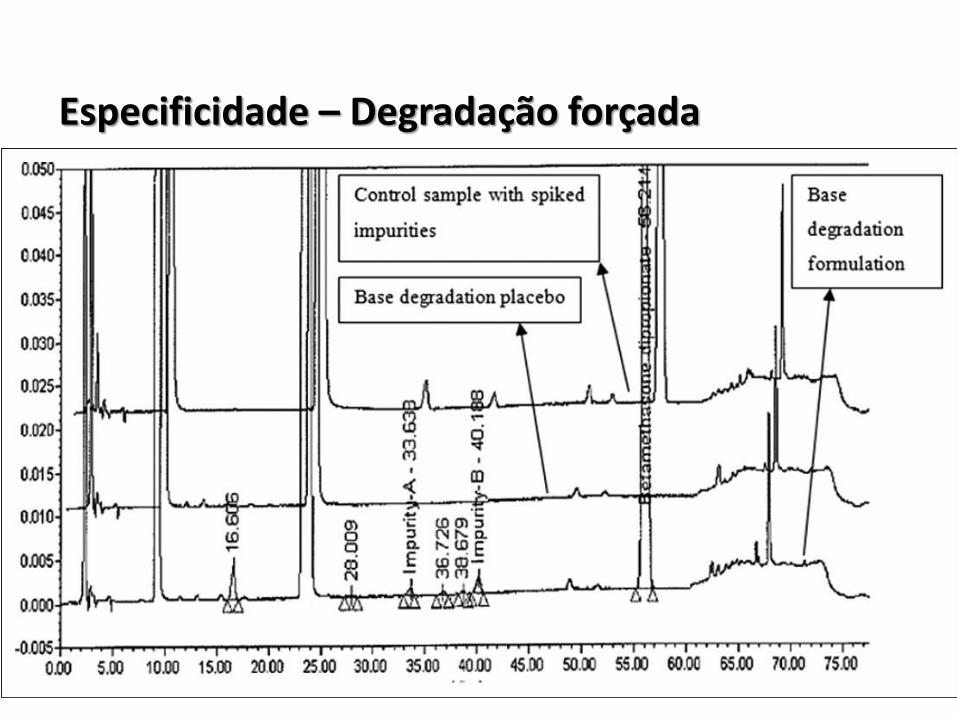
Especificidade – Degradação forçada


Especificidade – Principais exigências
• Ausência de degradação nas condições testadas ou degradação inferior a 10%;
• Degradação acima de 30%;
• Demonstrar condição indicativa de estabilidade do método;
• Demonstrar pureza de pico: Aplicável somente à métodos cromatográficos;
• Utilizar impurezas “disponíveis” para verificar se há interação (teor e produtos de degradação);
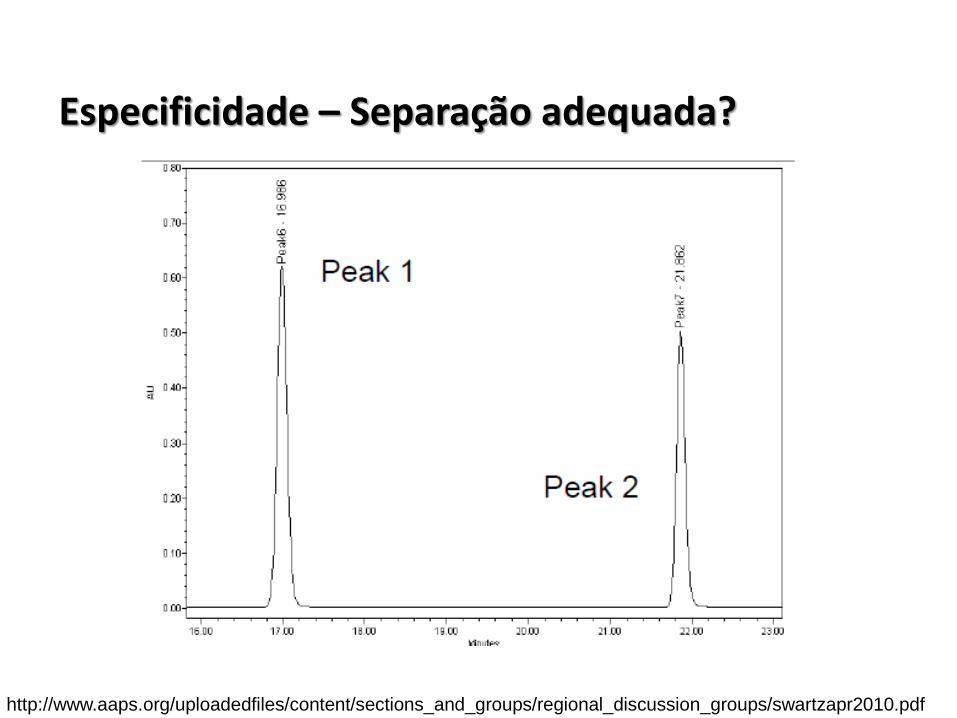
Especificidade – Separação adequada?
http://www.aaps.org/uploadedfiles/content/sections_and_groups/regional_discussion_groups/swartzapr2010.pdf

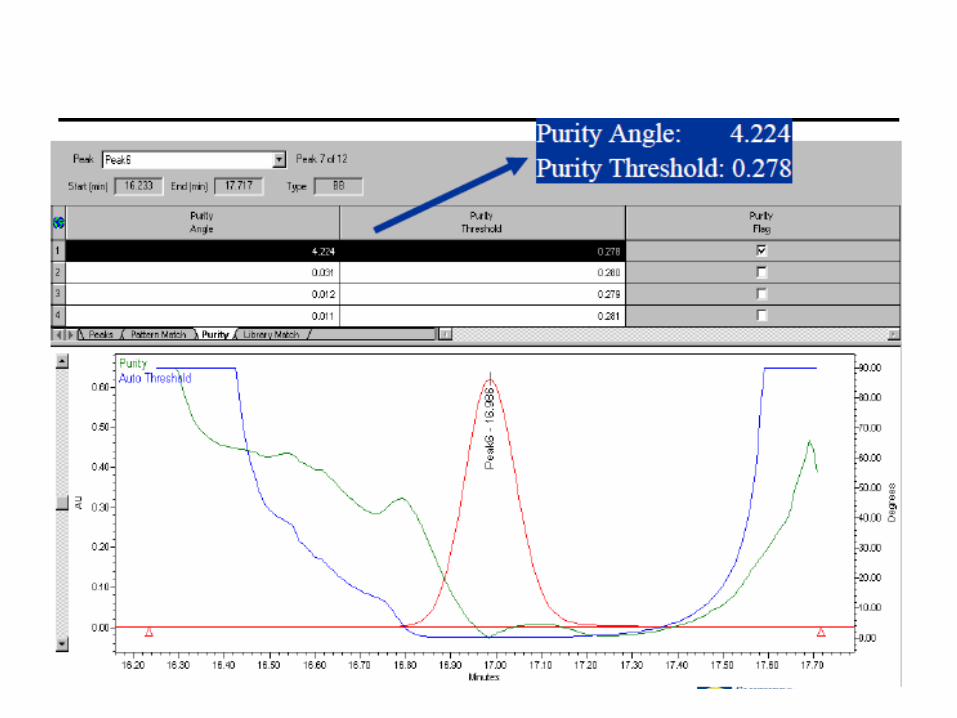

Especificidade – O que reportar?
• Cromatogramas de todas as soluções analisadas;
• “Overlay” de cromatogramas do placebo, placebo degradado, padrão, padrão degradado, amostra e amostras degradadas;
• Descrição de quais picos podem ser excluídos por pertencer exclusivamente à diluentes e/ou placebo/placebo degradado;
• Tabela demonstrando a pureza dos picos dos alvos da metodologia;
• Tabela constando a % de degradação nos estudos de stress e quantidade de impurezas verificadas.

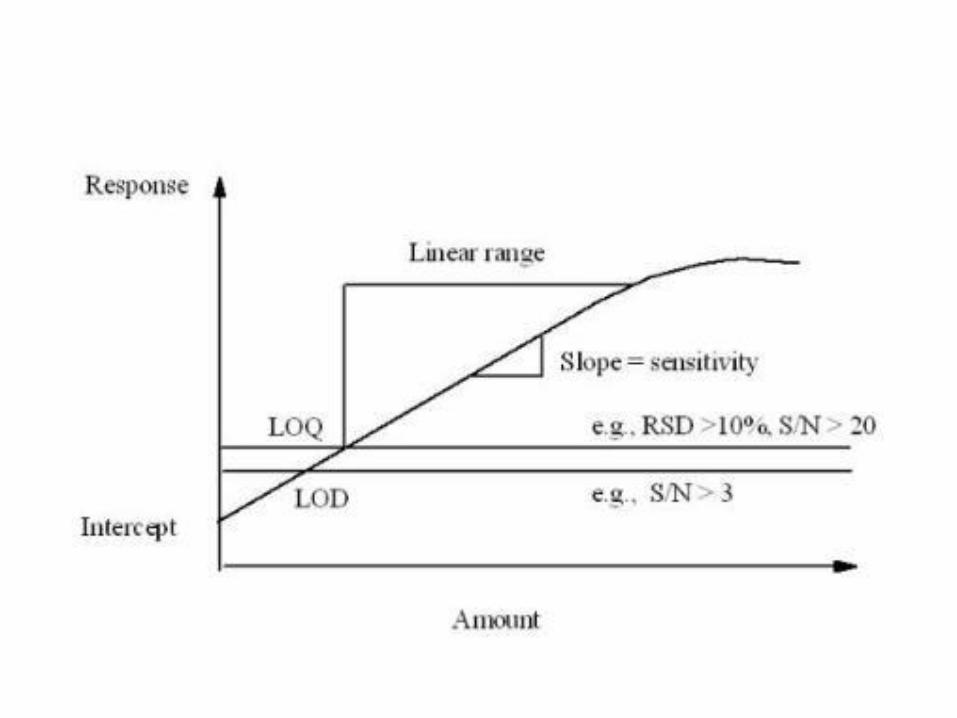
Limite de detecção – LD ou LoD
• Limite de detecção é a menor quantidade do analito presente em uma amostra que pode ser detectado, porém não necessariamente quantificado, sob as condições experimentais estabelecidas (RDC 899/2003);– O limite de detecção é estabelecido por meio da análise de soluções de
concentrações conhecidas e decrescentes do analito, até o menor nível detectável;
– Métodos não instrumentais: Pode ser feita visualmente, onde o limite de detecção é o menor valor de concentração capaz de produzir o efeito esperado (mudança de cor, turvação, etc);
– Métodos instrumentais: estimativa com base na relação de 3 vezes o ruído da linha de base. Pode ser determinado analisando 3 curvas contendo analito próximo ao limite de detecção ou de análise de amostras do branco;

Limite de quantificação – LQ ou LoQ
• É a menor quantidade do analito em uma amostra que pode ser determinada com precisão e exatidão aceitáveis sob as condições experimentais estabelecidas (RDC 899/2003);– Determinado, principalmente, para ensaios quantitativos de impurezas,
produtos de degradação em fármacos e produtos de degradação em formas farmacêuticas. Pode ser útil para validação de teor em métodos utilizados em validação de limpeza;
– O limite de quantificação é estabelecido por meio da análise de soluções contendo concentrações decrescentes do fármaco até o menor nível determinável com precisão e exatidão aceitáveis.
– Métodos instrumentais: estimativa com base na relação de 10 vezes o ruído da linha de base.
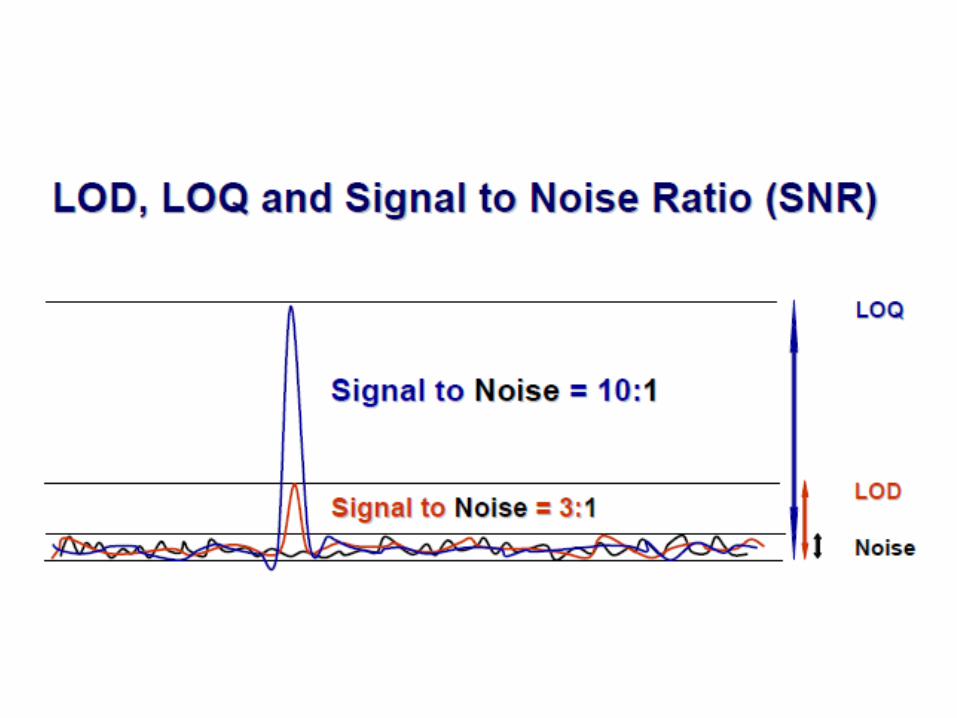

LD e LQ – O que reportar?
• Limite de detecção:– Tipo de abordagem utilizada (sinal ruído ou curvas);
– Valor de sinal ruído da linha de base com seu cromatograma;
– Cálculo utilizado para a determinação do LD
– Valor de LD na mesma unidade da especificação (usualmente em %)
• Limite de quantificação:– Tipo de abordagem utilizada (sinal ruído ou curvas);
– Valor de sinal ruído da linha de base com seu cromatograma;
– Cálculo utilizado para a determinação do LQ
– Verificação da precisão e exatidão no LQ
– Valor de LQ na mesma unidade da especificação (usualmente em %)

Linearidade
• É a capacidade de uma metodologia analítica dedemonstrar que os resultados obtidos são diretamenteproporcionais à concentração do analito na amostra,dentro de um intervalo especificado (RDC 899/2003).
– Análise de no mínimo 5 concentrações diferentes, conformetipo de validação – usualmente 3 replicatas de cadaconcentração;
– Coeficiente de correlação (mínimo 0,99), intersecção com oeixo Y, coeficiente angular, soma residual dos quadradosmínimos da regressão linear e desvio padrão relativo.
– Se não houver relação linear, realizar transformaçãomatemática.

Tipo de validação Faixa a ser validada
Determinação quantitativa do analito em matérias-primas ou em formas farmacêuticas
De 80% a 120% da concentração teórica do teste
Uniformidade de conteúdo De 70% a 130% da concentração teórica do teste
Determinação de impurezas Do nível de impureza esperado até 120% do limite máximo especificado. Quando apresentarem importância toxicológica ou efeitos farmacológicos inesperados, os limites de quantificação e detecção devem ser adequados às quantidades de impurezas a serem controladas
Dissolução De ±20% sobre o valor especificado para o intervalo. Caso a especificação para a dissolução envolva mais que um tempo, o alcance do método deve incluir –20% sobre o menor valor e +20% sobre o maior valor.
Concentrações conforme tipo de validação
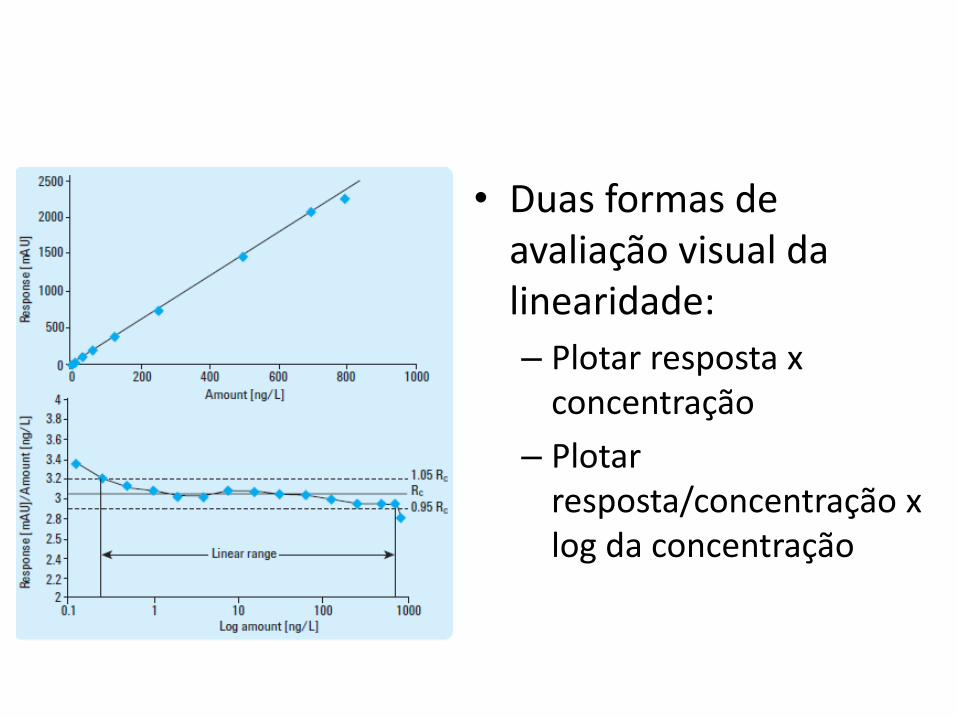
• Duas formas de avaliação visual da linearidade:
– Plotar resposta x concentração
– Plotar resposta/concentração x log da concentração

EXERCÍCIO

Exemplo – Escitalopram Tablets USP
• Citalopram é isômero óptico do escitalopram
• Padrão disponível: citalopram HBr
• Especificação: 90 a 110% do rotulado de escitalopram
• Concentração da amostra e padrão: 0,5 mg/mL de citalopram (0,62 mg/mL de citalopram HBr)
Linearidade – Escitalopram tablets – teor e UC
• Teor e uniformidade de conteúdo (UC) com mesmométodo
• Concentração alvo do método – 100%: 0,5 mg/mL decitalopram (usando 0,62 mg/mL de padrão decitalopram HBr)
• Faixa de validação para teor: 80 a 120%
• Faixa de validação para UC: 70 a 130%
• Faixa a ser validada: 70 a 130% da concentração alvo(considerando mesmo método para teor e UC)
• Considerar no mínimo 5 concentrações no intervalo
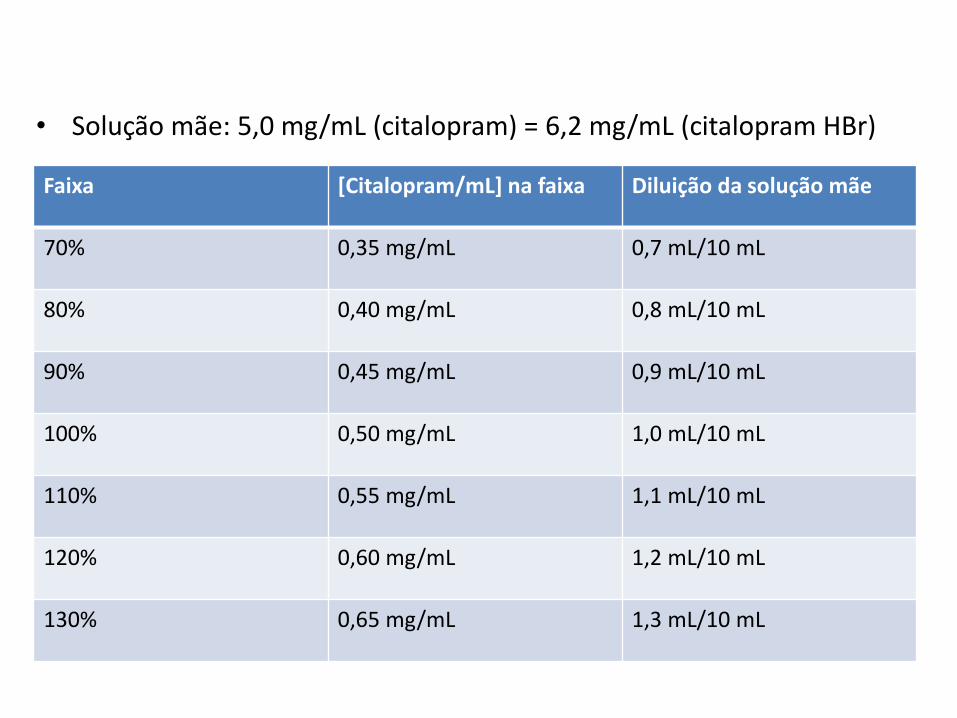
Faixa [Citalopram/mL] na faixa Diluição da solução mãe
70% 0,35 mg/mL 0,7 mL/10 mL
80% 0,40 mg/mL 0,8 mL/10 mL
90% 0,45 mg/mL 0,9 mL/10 mL
100% 0,50 mg/mL 1,0 mL/10 mL
110% 0,55 mg/mL 1,1 mL/10 mL
120% 0,60 mg/mL 1,2 mL/10 mL
130% 0,65 mg/mL 1,3 mL/10 mL
• Solução mãe: 5,0 mg/mL (citalopram) = 6,2 mg/mL (citalopram HBr)

Exemplo – Escitalopram Tablets USP
• Padrão disponível: Citalopram HBr
• Especificação: vide tabela
• Método não tem padrão das impurezas individuais: %área
• Concentração alvo do método: Especificação de cada impureza
• Faixa de linearidade: Do nível esperado (ausente) até 120% do máximo esperado (maior valor –0,5%) – LOQ até 0,6%
• Concentração do padrão: 0,5 mg/mL - 100%
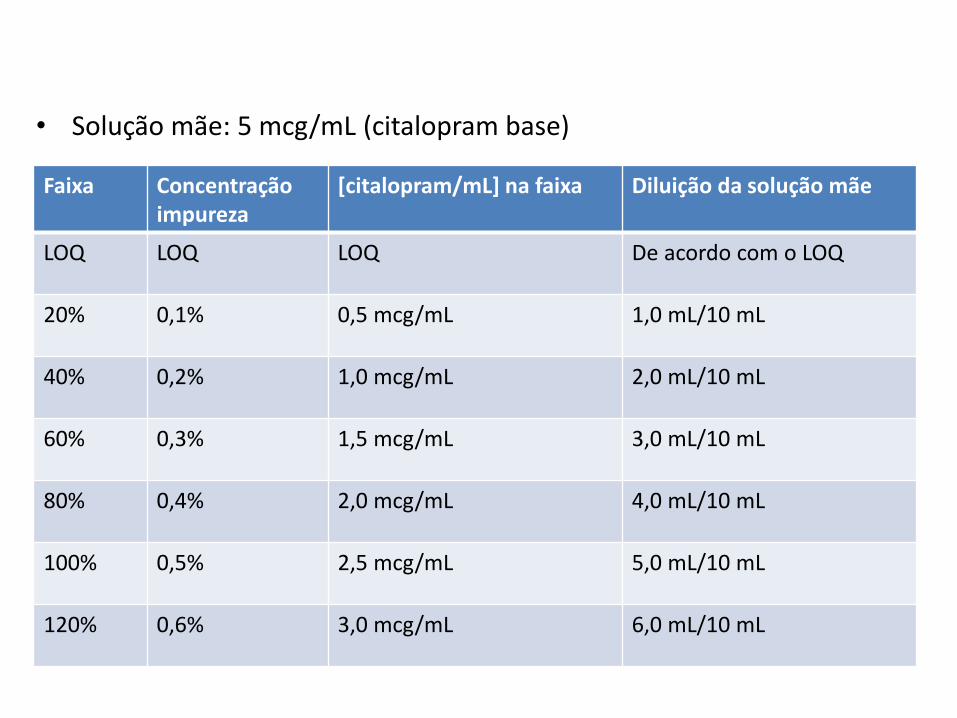
Faixa Concentraçãoimpureza
[citalopram/mL] na faixa Diluição da solução mãe
LOQ LOQ LOQ De acordo com o LOQ
20% 0,1% 0,5 mcg/mL 1,0 mL/10 mL
40% 0,2% 1,0 mcg/mL 2,0 mL/10 mL
60% 0,3% 1,5 mcg/mL 3,0 mL/10 mL
80% 0,4% 2,0 mcg/mL 4,0 mL/10 mL
100% 0,5% 2,5 mcg/mL 5,0 mL/10 mL
120% 0,6% 3,0 mcg/mL 6,0 mL/10 mL
• Solução mãe: 5 mcg/mL (citalopram base)

Exemplo – Escitalopram Tablets USP
• Padrão disponível: Escitalopram Oxalate
• Especificação: NLT 80% do rotulado de escitalopram
• Concentração da amostra e padrão: L/900 sendo L = 10 mg.
• Concentração alvo do método: 0,01 mg/mL (10 mg/900 mL) –100% (escitalopram base)
• Perfil de dissolução?
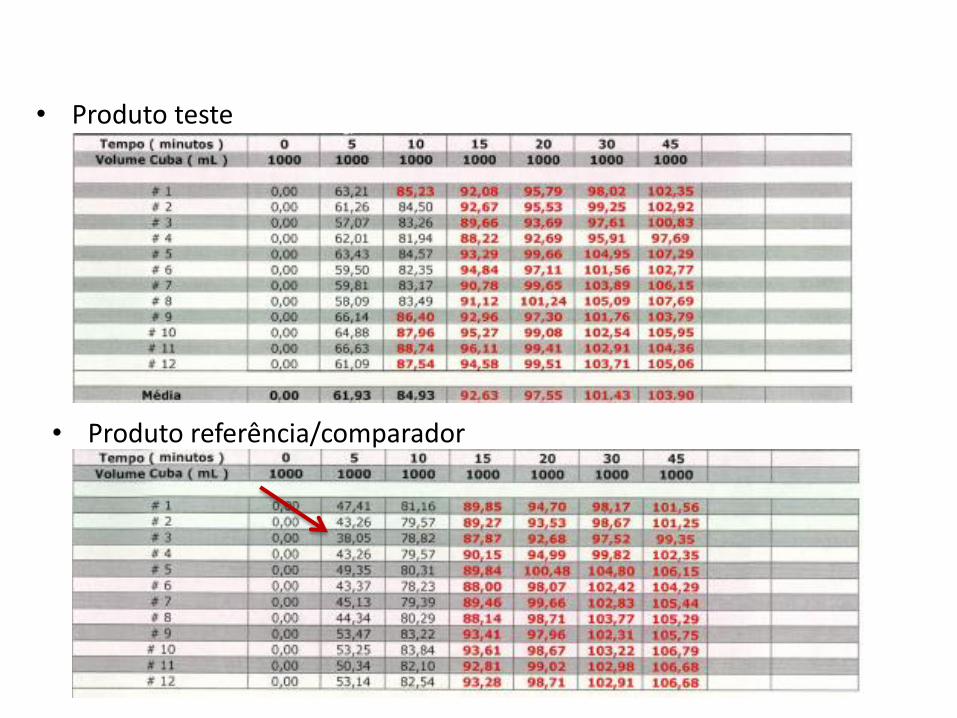
• Produto teste
• Produto referência/comparador

Linearidade – Escitalopram tablets – dissolução
• Concentração alvo do método – 100%: 0,01 mg/mL deescitalopram base livre
• Especificação de dissolução: minimo 80% dissolvido em30 minutos, ou seja, concentração mínima de 0,008mg/mL
• Perfil de dissolução – menor valor: 38,05%
• Faixa de validação para dissolução: -20% do menorvalor e +20% do maior valor (18% a 120%)
– 38% - 20% = 18%
– 100% + 20% = 120%
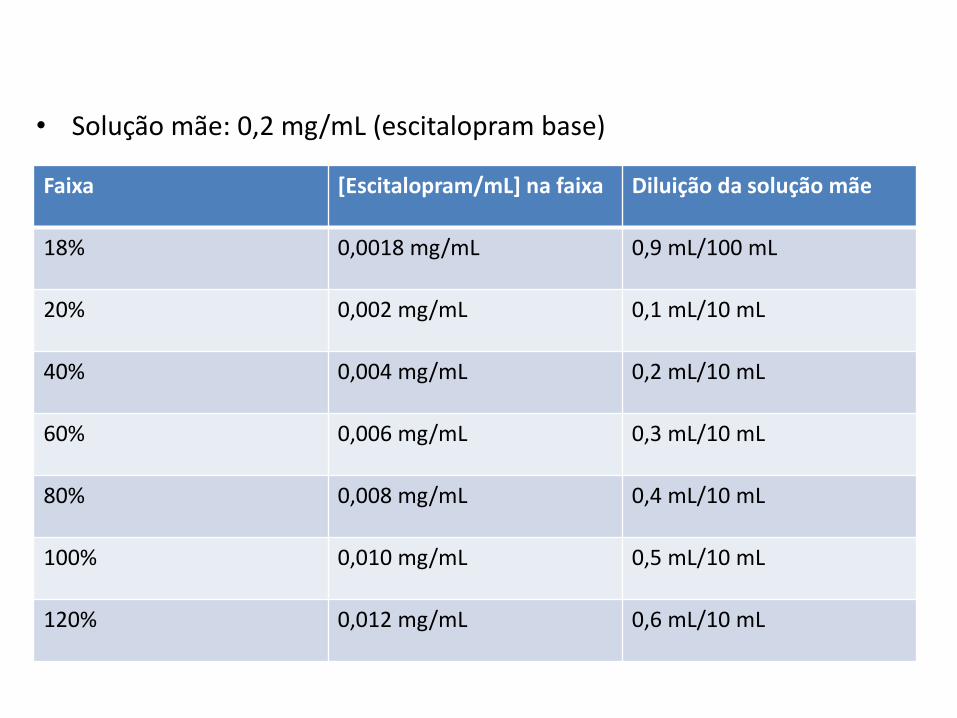
Faixa [Escitalopram/mL] na faixa Diluição da solução mãe
18% 0,0018 mg/mL 0,9 mL/100 mL
20% 0,002 mg/mL 0,1 mL/10 mL
40% 0,004 mg/mL 0,2 mL/10 mL
60% 0,006 mg/mL 0,3 mL/10 mL
80% 0,008 mg/mL 0,4 mL/10 mL
100% 0,010 mg/mL 0,5 mL/10 mL
120% 0,012 mg/mL 0,6 mL/10 mL
• Solução mãe: 0,2 mg/mL (escitalopram base)

Linearidade – O que pode acontecer?
• Concentração alvo do método – 100%: 0,01 mg/mL deescitalopram base livre
• Especificação de dissolução: minimo 80% dissolvido em30 minutos, ou seja, concentração mínima de 0,008mg/mL
• Perfil de dissolução – menor valor: 38,05%
• Faixa de validação para dissolução: -20% do menorvalor e +20% do maior valor (18% a 120%)
– 38% - 20% = 18%
– 100% + 20% = 120%
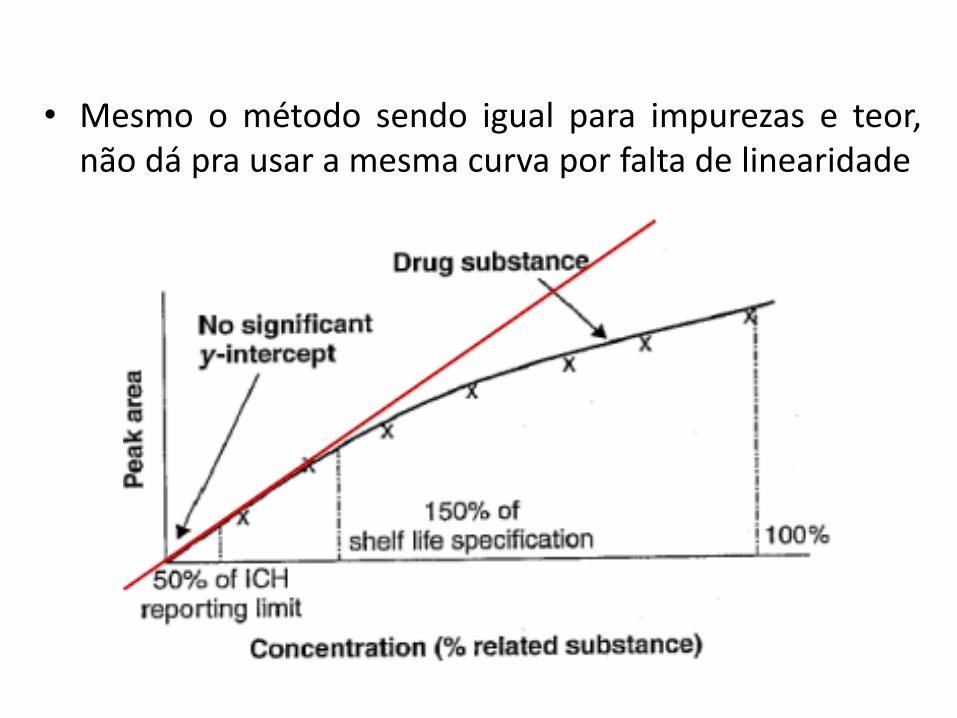
• Mesmo o método sendo igual para impurezas e teor,não dá pra usar a mesma curva por falta de linearidade
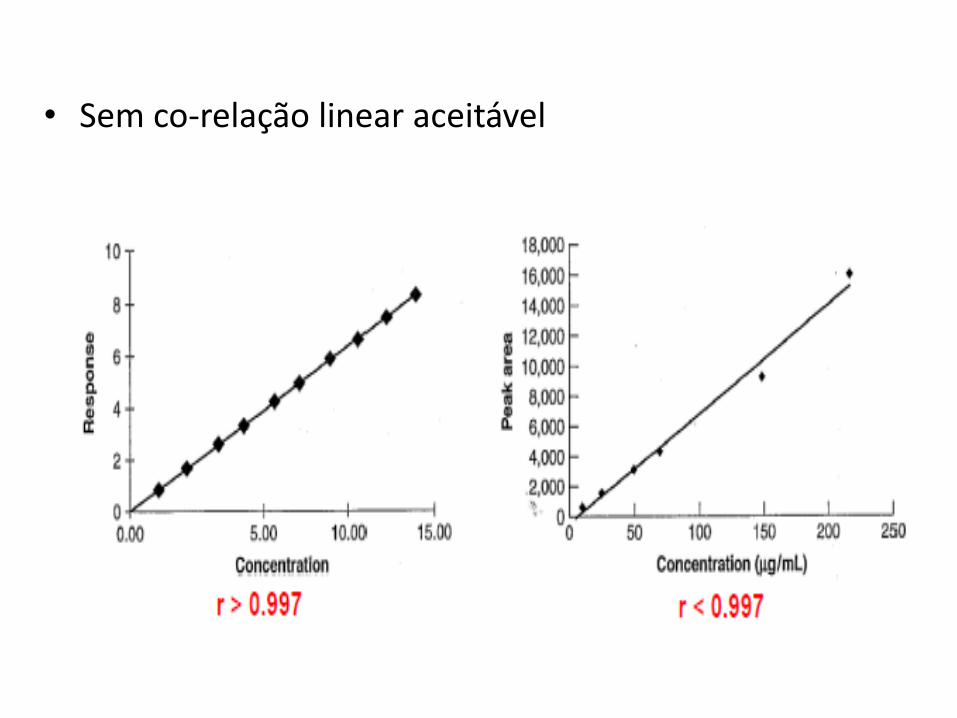
• Sem co-relação linear aceitável

• Pela ANVISA: Avaliação visual do gráfico dos resíduos – aparenta alguma co-relação?
• Normalmente os resíduos devem estar igualmente distribuidos acima e abaixo do zero, caso contrário aparentam co-relação;
• Calculo estatístico e minitabexistem para acessar não subjetivamente tal adequabilidade

Linearidade – O que reportar?
• Tabela contendo as concentrações x respostas obtidas analíticamente para no mínimo 5 concentrações;
• DPR verificado entre as replicatas de cada uma das concentrações (usualmente 3 replicatas);
• Grafico da linearidade para cada método
• Valor do coeficiente de co-relação (r);
• Gráfico de resíduos
• Equação da reta obtida e eventuais transformações matemáticas para obtenção de resposta linear (log por exemplo);
• Valor de y-intercepto obtido

Precisão
• A precisão é a avaliação da proximidade dos resultados obtidos em uma série de medidas de uma amostragem múltipla de uma mesma amostra.
– Repetibilidade (precisão intra-corrida): concordância entre os resultados dentro de um curto período de tempo com o mesmo analista e mesma instrumentação = no mínimo, 9 determinações no linear do método, ou seja, 3 concentrações na baixa, média e alta, com 3 réplicas cada ou mínimo de 6 determinações a 100% da concentração do teste(usualmente até 2%)
– Precisão do sistema – uma preparação avaliada 6 vezes;
– Precisão do método – 6 preparações avaliadas 1 vez;

Precisão
• Precisão intermediária (precisão inter-corridas): concordância entre os resultados do mesmo laboratório, mas obtidos em dias diferentes, com analistas diferentes e/ou equipamentos diferentes. Para a determinação da precisão intermediária recomenda-se um mínimo de 2 dias diferentes com analistas diferentes (usualmente até 5%)
• Reprodutibilidade (precisão inter-laboratorial): concordância entre os resultados obtidos em laboratórios diferentes. Estes dados não precisam ser apresentados para a concessão de registro (usualmente até 5%)– A precisão pode ser expressa como desvio padrão relativo (DPR) ou coeficiente de
variação (CV%), não podendo ser superior a 5%

Precisão do sistema
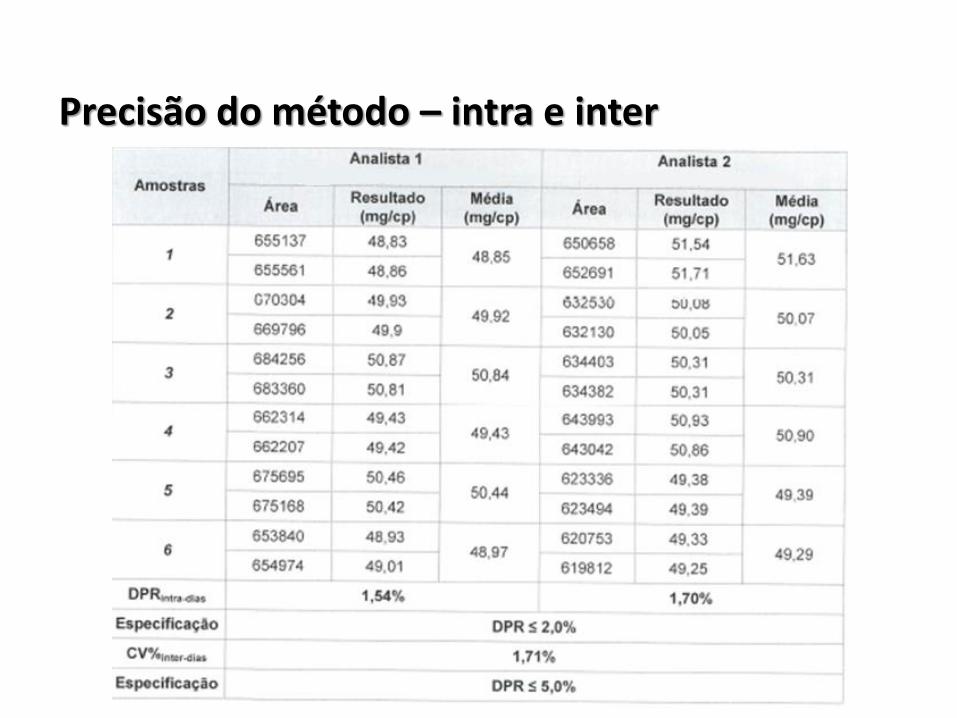
Precisão do método – intra e inter


Precisão – O que reportar?
• Tabela contendo os valores de avaliação da precisão do sistema e da variação encontrada;
• Tabela contendo os valores de avaliação da precisão do método por 1º analista usando 1º equipamento no 1º dia;
• Tabela contendo os valores de avaliação da precisão do método por 2º analista usando 2º equipamento no 2º dia;
• Tabela contendo a sumarização dos valores de precisão do método pelos 2 analistas;

Exatidão
• A exatidão de um método analítico é a proximidade dos resultados obtidos pelo método em estudo em relação ao valor verdadeiro.
• Fármaco
– Uso de metodologia analítica proposta na análise de uma substância de pureza conhecida (padrão de referência);
– Comparação dos resultados entre metodologias;
• Forma Farmacêutica
– Placebo contaminado com quantidade conhecida de fármaco ou da substância que se quer avaliar (no caso da indisponibilidade de amostras de certas impurezas e/ou produtos de degradação = comparação entre metodologias

Exatidão - Procedimento
• 9 determinações contemplando o intervalo linear do procedimento: – 3 concentrações na baixa – 3 replicatas;
– 3 concentrações na média – 3 replicatas;
– 3 concentrações na alta – 3 replicatas;
• Determinar a relação entre a concentração média determinada experimentalmente e a concentração teórica correspondente

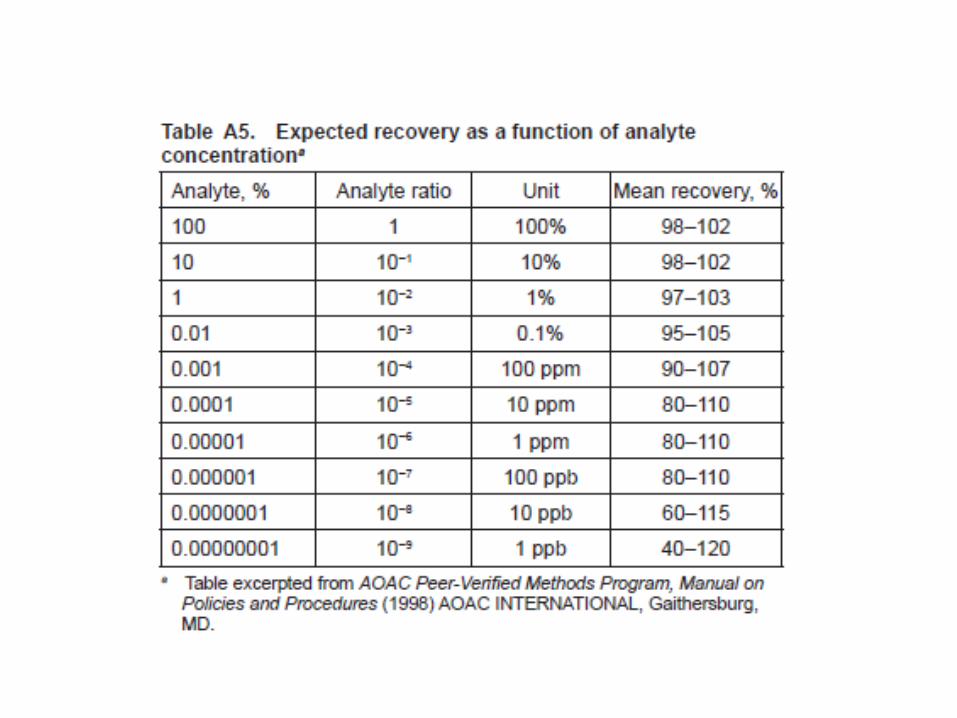
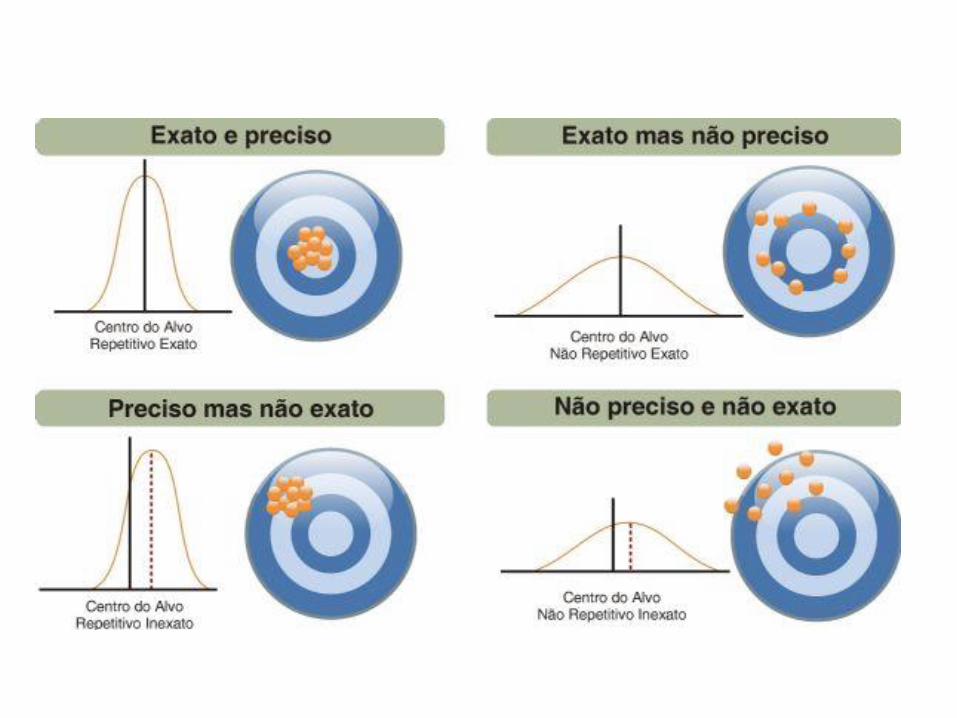

Robustez
• A robustez de um método analítico é a medida de sua capacidade em resistir a pequenas e deliberadas variações dos parâmetros analíticos. Indica sua confiança durante o uso normal (RDC 899/2003)– Durante o desenvolvimento da metodologia, deve-se considerar a avaliação da
robustez. Constatando-se a susceptibilidade do método à variações nas condições analíticas, estas deverão ser controladas e precauções devem ser incluídas no procedimento.
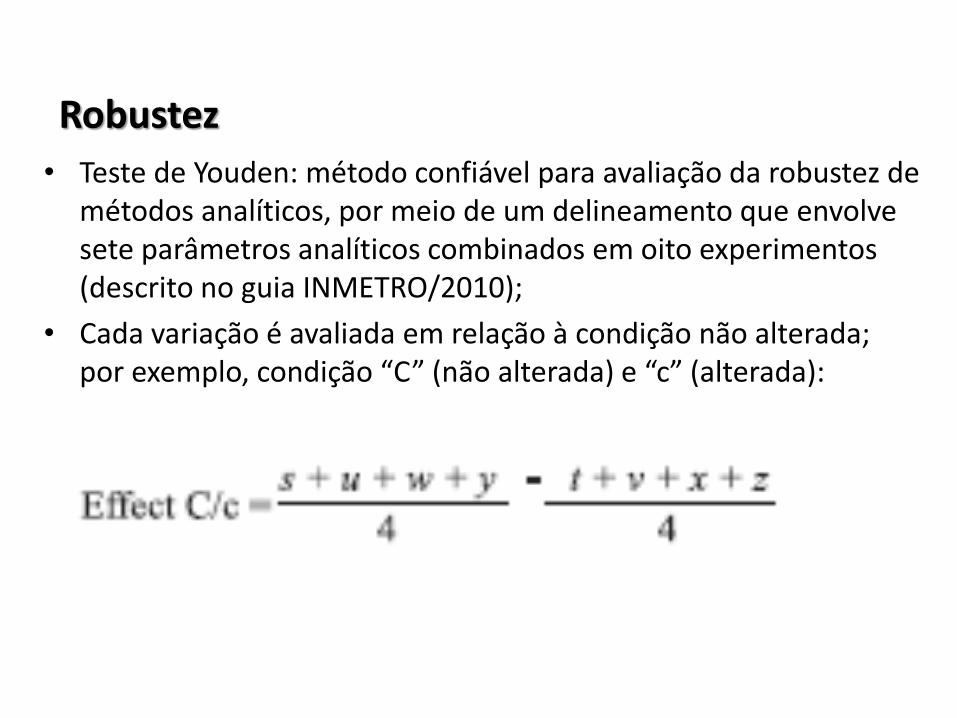
Robustez
• Teste de Youden: método confiável para avaliação da robustez de métodos analíticos, por meio de um delineamento que envolve sete parâmetros analíticos combinados em oito experimentos (descrito no guia INMETRO/2010);
• Cada variação é avaliada em relação à condição não alterada; por exemplo, condição “C” (não alterada) e “c” (alterada):


http://www.scielo.br/pdf/bjps/v45n2/v45n2a07.pdf

Robustez – O que reportar?
• Tabela com a avaliação dos resultados obtidos com a metodologia alterada x não alterada;
• Resultados do cálculo para matriz de Youden;
• Definição das especificações de alteração que devem ser consideradas significativas para cada parâmetro avaliado;
• Parâmetros que tiveram resultados significativamente diferentes da condição não alterada;
• Recomendações para inclusão no método analítico original – quais parâmetros devem ser estritamente monitorados;

Intervalo
• O intervalo especificado é a faixa entre os limites de quantificação superior e inferior de um método analítico. Normalmente é derivado do estudo de linearidade e depende da aplicação pretendida do método.
• É estabelecido pela confirmação de que o método apresenta exatidão, precisão e linearidade adequados quando aplicados a amostras contendo quantidades de substâncias dentro do intervalo especificado.

Estabilidade das soluções
• Não detalhada na RDC 899/2003;
• Necessária para garantir que o analito tem estabilidade no mínimo no tempo de análise;
• Importante para garantir as condições nas quais o analito é estável (refrigerado, ambiente) para estabelecer por exemplo,
– soluções-mãe de compostos pouco disponíveis ou caros,
– Tempo no qual as soluções podem ser mantidas para uma eventual investigação de resultados não conforme;

Relatório de validação
• Art. 490. Os relatórios devem refletir os protocolos seguidos e contemplar, no mínimo, o título, o objetivo do estudo, bem como fazer referência ao protocolo, detalhes de materiais, equipamentos, programas e ciclos utilizados e ainda, os procedimentos e métodos que foram utilizados.
– Art. 491. Os resultados devem ser avaliados, analisados e comparados com os critérios de aceitação previamente estabelecidos.
– Art. 492. Os Departamentos responsáveis pelos trabalhos de qualificação e validação devem aprovar o relatório completo.

Alterações permitidas em métodos já
validados
ANVISA não tem reconhecido e solicita precisão, especificidade e linearidade – Farmacopeia Mercosul em CP

Validação x verificação de métodos
• Não há guia da ANVISA. RDC 17/2010 é vaga.
• Capítulos da USP (farmacopeia americana) <1225> intitulado “validação de métodos compendiais” e <1226> “verificação de métodos compendiais”.
– Capítulo <1225> Descreve a validação de métodos analíticos com todos os parâmetros definidos. O resultado é um método validado para uma amostra específica. Este procedimento é recomendado para a validação de métodos desenvolvidos internamente.
– Capítulo <1226> Provê recomendações para a demonstração de que o laboratório tem a habilidade de utilizar o método de forma adequada.

Exemplos de parâmetros para
verificação de métodos
Não há guia da ANVISA, cada laboratório deve fornecer racional para escolha dos parâmetros de verificação

Referências• IUPAC - http://www.iupac.org/publications/pac/2002/pdf/7405x0835.pdf
• FDA -http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM122858.pdf
• ICH -http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf
• ISO 17025 - http://www.saba.org.ir/saba_content/media/image/2011/04/1821_orig.pdf
• INMETRO - http://www.inmetro.gov.br/Sidoq/Arquivos/CGCRE/DOQ/DOQ-CGCRE-8_03.pdf
• EURACHEM - http://www.eurachem.org/images/stories/Guides/pdf/valid.pdf
• AGILENT - http://www.chem.agilent.com/Library/primers/Public/5990-5140EN.pdf
• WHO – reference standards -http://www.who.int/medicines/areas/quality_safety/quality_assurance/GeneralGuidelinesEstablishmentMaintenanceDistributionChemicaReferenceSubstancesTRS943Annex3.pdf
• ANVISA – CP 11/2012 -http://portal.anvisa.gov.br/wps/wcm/connect/042a338049f229df96d7bfaa19e2217c/Consulta+P%C3%BAblica+n%C2%B0+11+GGMED.pdf?MOD=AJPERES
• AOAC – variabilidade - http://www.eoma.aoac.org/app_f.pdf
• Estatística - http://www.cprm.gov.br/publique/media/Andriotti_Tecnicas_estatisticas.pdf








