
Anal. Chem. 1988. 60, 5-10 5
Two-Laser Photoionization Supersonic Jet Mass Spectrometry of Aromatic Molecules
James W. Hager and Stephen C. Wallace*
Department of Chemistry, University of Toronto, Toronto, Ontario M5S 1A1, Canada
We report the results of a comparative investigation of two- laser resonant photoionization mass spectrometry (MS) and conventional one-laser resonant photoionization MS of several aromatic molecules In a supersonic jet. The use of a sepa- rate, independently tunable laser to photoionize exclted-state molecules Is shown to provkle enhanced parent km production compared wlth the one-laser technique. By use of two lasers in the photoionization process, isomer discrimination can be simplified by selectively exploiting differences in ionization potentials and excited-state lifetimes. I n particular, we have Investigated the two-laser photoionization of phenanthrene, anthracene, pyrene, aniline, and several methylindole lsomers under supersonic expansion conditions. The added dimen- dons of employing two lasers in the ionization are discussed.
It is no longer impractical to use electronic absorption transitions of polyatomic molecules for selective identification of species in a multicomponent sample without extensive prior separation. The use of supersonic expansion to sharpen and simplify normally structureless absorption spectra has been shown to provide extreme spectral selectivity (1). Laser- photoionization mass spectrometry coupled with this cooling technology has been recently demonstrated to be a viable analytical method displaying excellent selectivity and limits of detection in the subpicogram range (2,3). Like other “low temperature” techniques, the cooling of the molecular internal degrees of freedom concentrates population in the lowest vibrational and rotational levels of the ground electronic state, significantly simplifying the resulting electronic spectra. This is of particular importance when one is dealing with large polyatomic molecules for which room temperature absorption spectra are often completely structureless. Supersonic cooling virtually eliminates vibrational hot bands and reduces rovi- bronic line widths to a fraction of an angstrom even for these large molecules (1). Since these simplified electronic spectra are extremely sensitive to very subtle changes in molecular structure, they can serve as “fingerprints” for individual an- alytes in multicomponent samples.
Conventional resonantly enhanced two-photon ionization (RBPI) is often used in conjunction with mass analyzers (2, 3) because of its high ionization efficiency for atoms and molecules of many different chemical classes and the op- portunity to obtain simultaneous mass and electronic spectral information. Furthermore, when low to moderate light in- tensities are used, R2PI can be a very soft ionization technique for a great many polyatomic molecules, leading to the exclusive production of parent ions (2 ,3 ) . There are, however, several drawbacks to R2PI-MS when performed with a single-laser system. In this paper, we examine the use of two-laser pho- toionization as a method of significantly broadening the ap- plicability of supersonic expansion laser photoionization mass spectrometry for analysis of polyatomic molecules.
One particular shortcoming of R2PI is that molecules for which twice the photon energy (2hv) does not reach the ion- ization potential (IP) are, in large part, inaccessible when using
singel-laser photoionization techniques. A particularly im- portant class of molecules for which this is generally observed is polycyclic aromatic hydrocarbons (PAH’s). For PAHs with three or more rings, the lowest energy regions of the first excited singlet state are significantly less than half way to the IP and require the absorption of at least three photons for ionization. One can, of course, tune the laser to higher energies and begin to observe ion production when 2hv > IP. Un- fortunately, the electronic transitions with the highest ab- sorption cross sections are located within a few nanometers of the band origin (4) . For wavelengths at which 2hv > IP, the So-S1 transitions are much weaker, the spectrum is con- siderably more congested, and, in general, less well studied. In order to exploit fully the spectral selectivity provided by supersonic cooling, one would ideally like to use the most intense electronic transitions characteristic of the analyte of interest. This is not possible when using one-laser pho- toionization as the ionizing source for the determination of this important class of environmental contaminants. Other species for which this can be an important consideration are aromatic molecules containing electron-withdrawing sub- stituents such as -NOz, -CN, and -COOH (5).
Spectral broadening due to power saturation effects is often observed with even moderate laser light intensities when using single-laser R2PI (6). This can significantly distort electronic spectra and reduce the selectivity of laser-based supersonic jet techniques. Power saturation arises because, for most aromatic molecules, the absorption cross section to the first excited state (al) is often quite large and is approximately 1 to 2 orders of magnitude larger than that from the excited state to the ground ionic state (a2) (7). Thus, one cannot, when using only a single laser, maximize ion production without saturating the So-S1 transition. Furthermore, with laser in- tensities near the So-S1 saturation limit, fragmentation can be extensive, thus leading to a degraded mass spectrum (6).
We have employed the technique of two-laser photoioni- zation MS coupled with a supersonic expansion in order to overcome the drawbacks of conventional RPPI-MS discussed above. In this technique, light from a low-intensity laser (hv,) selectively excites a given jet-cooled polyatomic molecule through a resonant absorption process. Subsequent absorption of light from a second tunable laser system (hv,) ionizes the excited-state molecule. One can examine the two-step pho- toionization scheme for two independently tunable lasers by using the rate equation approach of Letokhov (9) for the optically thin medium of a supersonic expansion. When a simple system with two discrete levels, “1” (the vibrationless level of the ground electronic state) and “2” (an intermediate energy vibronic state), and an ionization continuum is con- sidered, there are simple solutions to the rate equations for several limiting cases. For the case where the first step is saturated and the ionization step is not, the number of pho- toions is given by (9)
where az is the absorption cross section for the transition from level 2 to the ionization continuum, Iz is the intensity of the ionizing laser, rZ is the pulse width of the ionizing laser, and
Ni = U21272No
0003-2700/88/0360-0005$01.50/0 0 1987 American Chemical Society

6 ANALYTICAL CHEMISTRY, VOL. 60, NO. 1, JANUARY 1, 1988
No is the number of molecules in the irradiated volume. This equation describes the photoionization process for the con- ventional one-laser RBPI of most polyatomic molecules when the laser intensity is increased to maximize ion production. Since most often u2 < ul for aromatic molecules, the fiist step will saturate before the ionization step in the one-laser R2PI process. Unfortunately, the molecular parameter determining the photoionization efficiency is uz, the cross section for the ionization step, which is an unknown quantity for most polyatomics. In addition, saturation of the So-S1 transition in a two-step photoionization process results in power broadening (6), which distorts the electronic spectrum and thus reduces spectral selectivity. It is important to realize that saturation and power broadening can occur at low laser intensities when working with strongly absorbing molecules.
For saturation of the ionization step without saturating the excitation step to the intermediate level, the number of photoions produced is given by (9)
Thus, the photoionization efficiency is determined by the cross section for the excitation from level 1 to level 2, which is a much more easily obtainable value than that for absorption of the ionizing photon.
The maximum ionization efficiency is obtained when the intensity of the ionizing laser is 2u1/u2 time higher than that of the exciting radiation (9). Obviously, this condition can only be fulfilled by using two independent lasers. Thus, the ultimate sensitivity of two-laser photoionization can be sig- nificantly greater than that of the one-laser technique.
An independently wavelength tunable ionizing laser can be valuable in several other respects. First, the ionization cross section for most polyatomic molecules is not a constant function of energy above the adiabatic IP (8). When a polyatomic molecule is ionized from the vibrationless level of the first excited state, the ionization cross section is highest slightly above the threshold for ion production (8, IO). This phenomenon has been shown to arise from the fact that Franck-Condon factors for ionization from an excited elec- tronic state of an aromatic molecule favor transitions for the minimum change in vibrational state (8, IO). This leads to a high ionization onset near the threshold with a gradual decline in ion production a t higher energies. With only one laser system, one has little control over this energy-dependent ionization cross section.
Finally, the tunability of the ionizing laser introduces an additional source of selectivity compared with conventional one-color RBPI methods through accurate measurements of ionization potentials. Ionization thresholds are very sensitive to subtle structural factors and two-laser photoionization allows the determination of IP’s to within 0.5 meV (8). Thus, one can discriminate between isomeric species that have ionization potentials that differ by this small energy, in ad- dition to obtaining electronic spectral information.
In the present publication we present the results of an investigation of the two-color photoionization of a number of expansion-cooled aromatic molecules and compare the results with conventional R2PI-MS techniques.
EXPERIMENTAL SECTION A detailed description of the continuous supersonic jet quad-
rupole mass spectrometer apparatus used in the present study has been given elsewhere (8). Two independent laser systems were employed for the resonant two-step photoionization process. One of these consists of a Nd:YAG pumped dye laser (Quanta Ray DCR-1A and PDL-1) operated at 10 Hz. Light from this laser system was frequency doubled in KD*P (Quanta Ray WEX-1) to provide tunable ultraviolet radiation between 270 and 340 nm. Typical pulse energies in the UV were between 1 and 6 mJ, which could be precisely reduced by using Brewster plate neutral density
fiiters. The second system if a XeCl pumped dye laser (Lumonics 860-T and EPD-330), which was operated on several UV and visible dyes covering the 33M50-nm-wavelength region producing pulse energies of up to 15 mJ. Again, these energies could be reduced with neutral density filters. One of these laser systems was chosen as the excitation laser and was tuned to the wavelength range of the lowest excited-state transitions while the other laser provided the ionizing light. The excitation laser for the aniline and indole investigations reported here was the NdYAG pumped dye laser and the XeCl laser system served as the ionization laser. Due to the required wavelengths for the excitation and ionization, the roles of these lasers were reversed for the two-laser pho- toionization of the PAH’s described in the present study. In general, the exciting laser light intensity was maintained at <70 dlpulse. The ionizing laser, however, was operated at intensities at which saturation of this transition becomes significant. This allows one to operate in a regime where the ion signal is a function only of the intensity of the excitation laser and, in the present studies, is linearly dependent on this intensity.
The light from the two laser systems enters the supersonic expansion chamber in a coaxial, counterpropagating fashion and intersects the free jet expansion in the isentropic core. Neither laser beam was focused, although apertures were used to limit both spot sizes to approximately 7 mm.
The Nd:E‘AG laser was pulsed at 10 Hz by using the internal clock to fire the flashlamps. This clock was also used to trigger a fixed and a variable delay generator, which provided trigger pulses for the excimer thyratron and the NdYAG Q-switch circuit. This arrangement allows complete temporal overlap of the two laser pulses, limited only by the jitter in the Q-switch opening time (approximately 2 ns). This delay system also provided us with a variable delay for the arrival of the ionization laser pulse, with respect to the exciting laser pulse, by up to 1 ws. The temporal overlap of the two light pulses was monitored by an ITT biplanar photodiode (<1 ns rise time) and a 500-MHz transient digitizer (Tektronix 7912 AD).
For these investigations, we have employed a continuous, un- skimmed supersonic expansion with helium as the carrier gas at backing pressures of 1.6-2.0 atm and a 0.100-mm expansion orifice. The sample concentration in the expansion mixture was deter- mined by measuring the loss of sample at a given helium flow rate over a period of hours (11). The values obtained from this technique were found to be reproducible to within approximately 25%. All samples were obtained from Aldrich with stated purities of 99% or greater and used without further purification.
The spectra shown in this paper were obtained under computer control by using a Digital MINC-11 minicomputer. Data ac- quisition and power normalization were done as previously de- scribed (8).
RESULTS AND DISCUSSION Phenanthrene. One of the distinct advantages of two-color
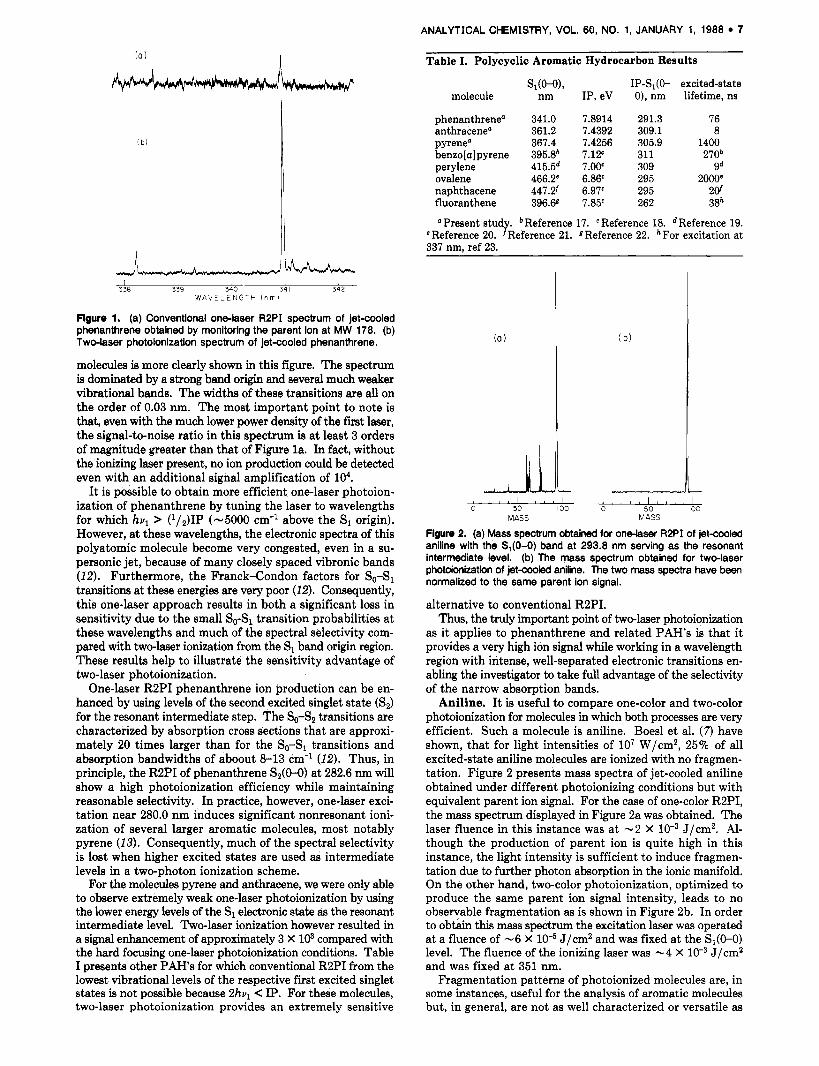
photoionization over one-color RBPI is the ability to ionize efficiently molecules for which 2hul does not reach the ioni- zation potential. Figure 1 displays two ionization spectra obtained under different laser excitation conditions for phenanthrene in a supersonic jet. For Figure la, only a single laser was used and operated a t approximately 6 mJ/pulse, focused to 0.5 mm, and tuned near the phenanthrene S1 band origin. Under these conditions, photoionization requires ab- sorption of two photons subsequent to electronic excitation since the IP is located approximately 0.5 eV above the 2hul energy of 7.3 eV. As one can see, the signal-to-noise ratio of this spectrum is poor, reflecting the very small three-photon ionization probability even when using these high laser in- tensities.
In Figure l b is the spectrum obtained by using a two-color ionization process. Here, the first laser was operated at ap- proximately 60 pJ/pulse and was collimated but unfocused. For this spectrum, the laser influences of the excitation and ionization lasers were -1.2 X and -1 X low2 J/cm2, respectively. The ionizing laser was tuned to a wavelength such that hul + hvq exceeds the adiabatic IP by about 10 meV. The spectral selectivity of supersonically cooling polyatomic
ANALYTICAL CHEMISTRY, VOL. 60, NO. 1, JANUARY 1, 1988 7
Table I. Polycyclic Aromatic Hydrocarbon Results
S,(O-o), IP-SI(O- excited-state molecule nm IP, eV 0), nm lifetime, ns
L L I 1 I 1 1
338 339 340 34 I 342 W A V E L E N G T H ( n m l
Figure 1. (a) Conventional one-laser R2PI spectrum of jet-cooled phenanthrene obtained by monitoring the parent ion at MW 178. (b) Two-laser photoionization spectrum of jet-cooled phenanthrene.
molecules is more clearly shown in this figure. The spectrum is dominated by a strong band origin and several much weaker vibrational bands. The widths of these transitions are all on the order of 0.03 nm. The most important point to note is that, even with the much lower power density of the first laser, the signal-to-noise ratio in this spectrum is at least 3 orders of magnitude greater than that of Figure la. In fact, without the ionizing laser present, no ion production could be detected even with an additional signal amplification of lo4.
It is possible to obtain more efficient one-laser photoion- ization of phenanthrene by tuning the laser to wavelengths for which hvl > (1/2)1P (-5000 cm-' above the S1 origin). However, a t these wavelengths, the electronic spectra of this polyatomic molecule become very congested, even in a su- personic jet, because of many closely spaced vibronic bands (12). Furthermore, the Franck-Condon factors for So-S1 transitions at these energies are very poor (12). Consequently, this one-laser approach results in both a significant loss in sensitivity due to the small So-S1 transition probabilities at these wavelengths and much of the spectral selectivity com- pared with two-laser ionization from the S1 band origin region. These results help to illustrate the sensitivity advantage of two-laser photoionization.
One-laser R2PI phenanthrene ion production can be en- hanced by using levels of the second excited singlet state (S,) for the resonant intermediate step. The So-S2 transitions are characterized by absorption cross sections that are approxi- mately 20 times larger than for the So-S1 transitions and absorption bandwidths of aboout 8-13 cm-' (12). Thus, in principle, the R2PI of phenanthrene S2(0-0) at 282.6 nm will show a high photoionization efficiency while maintaining reasonable selectivity. In practice, however, one-laser exci- tation near 280.0 nm induces significant nonresonant ioni- zation of several larger aromatic molecules, most notably pyrene (13). Consequently, much of the spectral selectivity is lost when higher excited states are used as intermediate levels in a two-photon ionization scheme.
For the molecules pyrene and anthracene, we were only able to observe extremely weak one-laser photoionization by using the lower energy levels of the S1 electronic state as the resonant intermediate level. Two-laser ionization however resulted in a signal enhancement of approximately 3 X 109 compared with the hard focusing one-laser photoionization conditions. Table I presents other PAHs for which conventional R2PI from the lowest vibrational levels of the respective first excited singlet states is not possible because 2hvl < IP. For these molecules, two-laser photoionization provides an extremely sensitive
phenanthrene" anthracene" pyrene" benzo[a] pyrene perylene ovalene naphthacene fluoranthene
341.0 361.2 367.4 395.8b 415.5d 466.2O 447.2f 396.68
7.8914 7.4392 7.4256 7.12c 7.0OC 6.86c 6.97c 7.85C
291.3 309.1 305.9 311 309 295 295 262
76 8
1400 270b
9d 20000
2 d 38h
" Present study. bReference 17. 'Reference 18. dReference 19. eReference 20. f Reference 21. Reference 22. hFor excitation at 337 nm, ref 23.
+-+---k MASS
( b )
-k--+-+ MASS
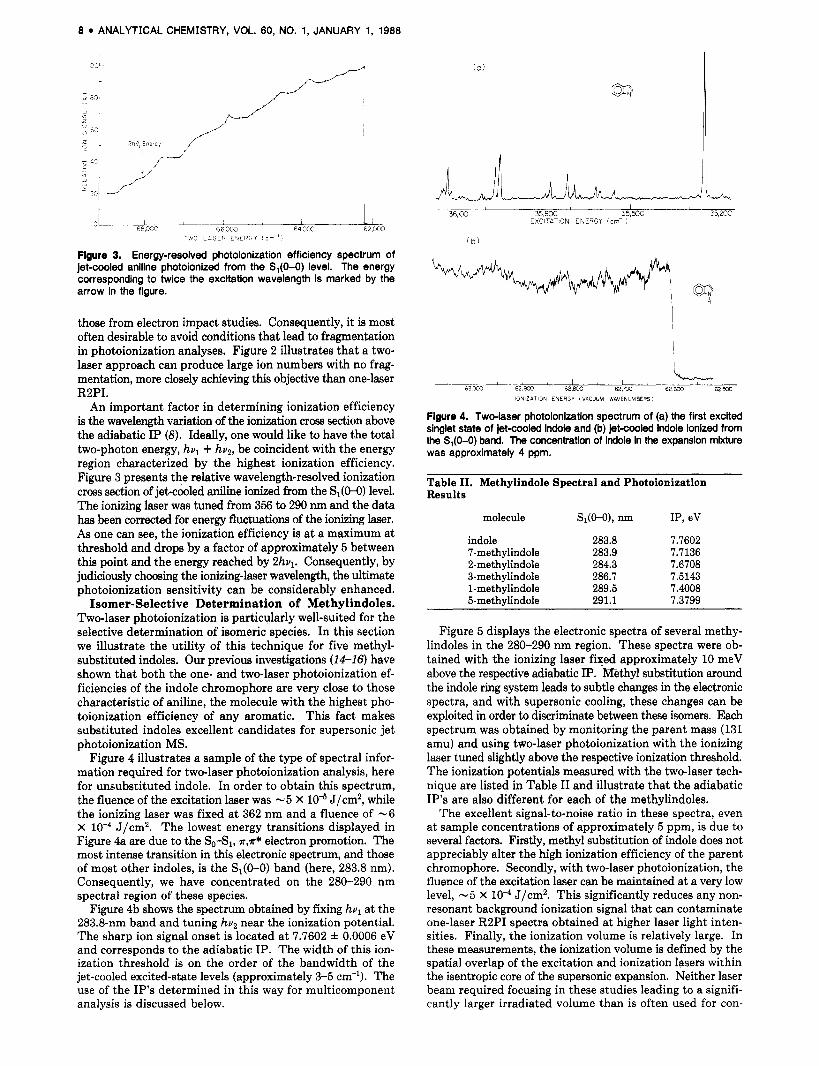
Figure 2. (a) Mass spectrum obtained for onalaser R2PI of jetcooled aniline with the S,(O-0) band at 293.8 nm serving as the resonant intermediate level. (b) The mass spectrum obtained for twdaser photoionkatlon of jetaoled aniline. The two mass spectra have been normalized to the same parent ion signal.
alternative to conventional R2PI. Thus, the truly important point of two-laser photoionization
as it applies to phenanthrene and related PAH's is that it provides a very high ion signal while working in a wavelength region with intense, well-separated electronic transitions en- abling the investigator to take full advantage of the selectivity of the narrow absorption bands.
Aniline. It is useful to compare one-color and two-color photoionization for molecules in which both processes are very efficient. Such a molecule is aniline. Boesl et al. (7) have shown, that for light intensities of lo7 W/cm2, 25% of all excited-state aniline molecules are ionized with no fragmen- tation. Figure 2 presents mass spectra of jet-cooled aniline obtained under different photoionizing conditions but with equivalent parent ion signal. For the case of one-color RBPI, the mass spectrum displayed in Figure 2a was obtained. The laser fluence in this instance was at -2 X J/cm2. Al- though the production of parent ion is quite high in this instance, the light intensity is sufficient to induce fragmen- tation due to further photon absorption in the ionic manifold. On the other hand, two-color photoionization, optimized to produce the same parent ion signal intensity, leads to no observable fragmentation as is shown in Figure 2b. In order to ob& this mass spectrum the excitation laser was operated at a fluence of -6 X J/cm2 and was fixed at the S1(O-O) level. The fluence of the ionizing laser was -4 X J/cm2 and was fixed at 351 nm.
Fragmentation patterns of photoionized molecules are, in some instances, useful for the analysis of aromatic molecules but, in general, are not as well characterized or versatile as
8 ANALYTICAL CHEMISTRY, VOL. 60, NO. 1, JANUARY 1, 1988
I
- c 7 / i4; .I
IES}
< .?L
- n:+ ir ', E n - r ~ r
~
~
.,I
I #I 660CS 640oC 62 OOO 68CCC
?L--
-@C L A S E S E ' Y E R S Y m
Figure 3. Energy-resolved photoionization efficiency spectrum of jet-cooled aniline photoionized from the S,(O-0) level. The energy corresponding to twice the excitation wavelength is marked by the arrow in the figure.
those from electron impact studies. Consequently, it is most often desirable to avoid conditions that lead to fragmentation in photoionization analyses. Figure 2 illustrates that a two- laser approach can produce large ion numbers with no frag- mentation, more closely achieving this objective than one-laser RZPI.
An important factor in determining ionization efficiency is the wavelength variation of the ionization cross section above the adiabatic IP (8). Ideally, one would like to have the total two-photon energy, hul + huZ, be coincident with the energy region characterized by the highest ionization efficiency. Figure 3 presents the relative wavelength-resolved ionization cross section of jet-cooled aniline ionized from the S1(M)) level. The ionizing laser was tuned from 356 to 290 nm and the data has been correded for energy fluctuations of the ionizing laser. As one can see, the ionization efficiency is a t a maximum at threshold and drops by a factor of approximately 5 between this point and the energy reached by 2hul. Consequently, by judiciously choosing the ionizing-laser wavelength, the ultimate photoionization sensitivity can be considerably enhanced.
Isomer-Selective Determination of Methylindoles. Two-laser photoionization is particularly well-suited for the selective determination of isomeric species. In this section we illustrate the utility of this technique for five methyl- substituted indoles. Our previous investigations (14-16) have shown that both the one- and two-laser photoionization ef- ficiencies of the indole chromophore are very close to those characteristic of aniline, the molecule with the highest pho- toionization efficiency of any aromatic. This fact makes substituted indoles excellent candidates for supersonic jet photoionization MS.
Figure 4 illustrates a sample of the type of spectral infor- mation required for two-laser photoionization analysis, here for unsubstituted indole. In order to obtain this spectrum, the fluence of the excitation laser was -5 X J/cm2, while the ionizing laser was fixed at 362 nm and a fluence of -6 X lo-* J/cm2. The lowest energy transitions displayed in Figure 4a are due to the So-Sl, *,a* electron promotion. The mast intense transition in this electronic spectrum, and those of most other indoles, is the S1(O-0) band (here, 283.8 nm). Consequently, we have concentrated on the 280-290 nm spectral region of these species.
Figure 4b shows the spectrum obtained by fixing hul at the 283.8-nm band and tuning hu2 near the ionization potential. The sharp ion signal onset is located at 7.7602 0.0006 eV and corresponds to the adiabatic IP. The width of this ion- ization threshold is on the order of the bandwidth of the jet-cooled excited-state levels (approximately 3-5 cm-'). The use of the IP's determined in this way for multicomponent analysis is discussed below.
( a )
%.d-" 1 ! ! ' & E
36,KX 35.800 35,500 EXCITAT ON ENERGY i cm- '
' b )
I ! 1 1 l
63wO 62 9xI 62 800 62.m 62 €CC 62 500 IONIZATION E N E R G Y (VACUUM WAVENUMBERSI
Figure 4. Twdaser photoionization spectrum of (a) the first excited singlet state of jet-cooled indole and (b) jet-cooied indole ionized from the S,(O-0) band. The concentration of indole in the expansion mixture was approximately 4 ppm.
Table 11. Methylindole Spectral and Photoionization Results
molecule Sl(M), nm IP, eV
indole 283.8 7.7602 7-methylindole 283.9 7.7136 2-methylindole 284.3 7.6708 3-methylindole 286.7 7.5143 1-methylindole 289.5 7.4008 5-methylindole 291.1 7.3799
Figure 5 displays the electronic spectra of several methy- lindoles in the 280-290 nm region. These spectra were ob- tained with the ionizing laser. fixed approximately 10 meV above the respective adiabatic IP. m t h y l substitution around the indole ring system leads to subtle changes in the electronic spectra, and with supersonic cooling, these changes can be exploited in order to discriminate between these isomers. Each spectrum was obtained by monitoring the parent mass (131 amu) and using two-laser photoionization with the ionizing laser tuned slightly above the respective ionization threshold. The ionization potentials measured with the two-laser tech- nique are listed in Table I1 and illustrate that the adiabatic IP's are also different for each of the methylindoles.
The excellent signal-to-noise ratio in these spectra, even at sample concentrations of approximately 5 ppm, is due to several factors. Firstly, methyl substitution of indole does not appreciably alter the high ionization efficiency of the parent chromophore. Secondly, with two-laser photoionization, the fluence of the excitation laser can be maintained at a very low level, -5 X J/cm2. This significantly reduces any non- resonant background ionization signal that can contaminate one-laser R2PI spectra obtained at higher laser light inten- sities. Finally, the ionization volume is relatively large. In these measurements, the ionization volume is defined by the spatial overlap of the excitation and ionization lasers within the isentropic core of the supersonic expansion. Neither laser beam required focusing in these studies leading to a signifi- cantly larger irradiated volume than is often used for con-

ANALYTICAL CHEMISTRY, VOL. 60, NO. 1, JANUARY 1, 1988 9
u, 2830 2840 2ffiQ
2 8 3 0 284 0 2850 2860 2870 W A V E L E N G T H l PI m I
1 1 I I 2870 288 0 2690 2910
2870 2880 2690 2430 W A V E L E N G T H l n m l
Figure 5. Two4aser photoionlzatlon spectrum of h e methyl-substltuted Indoles In a supersonic expanslon: (a) 7-methylindole ( - 5 ppm), (b) 2-methylindole (-5 pprn), (c) 3-methyllndole (-4 pprn), (d) 5- methyllndole (-2 ppm), and (e) 1-methylindole (-1 10 ppm).
ventional R2PI studies. Here, the ionization volume is ap- proximately 0.3 cm3 and is located in the high-density region of the expansion.
In Figure 6a is a two-laser photoionization spectrum of a 1:5 mixture of 2-methylindole and 7-methylindole with the ionizing laser fixed at an energy sufficient to induce ionization of both molecules (369 nm). One can see that, even though these are isomeric species with very similar electronic spectra, discrimination of one from another is readily accomplished with supersonic cooling. However, with the use of two-laser photoionization, much better discrimination is possible. This is illustrated in Figure 6b for the same methylindole mixture. In this instance, the detection method is identical with that for the spectrum in Figure 6a, but the ionization conditions have been altered. For the lower spectrum, the ionizing laser has been tuned to a wavelength (373 nm) that wil l ionize only 2-methylindole (hul + hv2 = 7.6900 eV). Thus, only molecules with IP < 7.6900 eV will be ionized via the two-laser process, which in this case corresponds to only 2-methylindole.
Although we have yet to interface our apparatus to a quantitative inlet system, such as a gas chromatograph, the limits of detection (LOD) can be estimated. On the basis of measured (16) absorption cross sections for resonant two-laser
I I I 283.5 284.0 284.5
(b ) i
I I I 283.5 284.0 2845
W A V E L E N G T H (nm)
Figure 8. Two-laser photoionization spectrum of a 1:5 mixture of P-methylindole and 7-methylindole under two sets of ionizing conditions: (a) ionizing laser tuned to 369 nm; (b) ionizing laser tuned to 373 nm.
ionization of indole and the approach of Boesl et al. (7), the LOD for the mass-resolved detection of the parent ion of the unsubstituted molecule is approximately 0.5 pg. The high two-step absorption cross sections of the indole chromophore are not expected to change significantly upon methyl sub- stitution leading to LOD’s of approximately the same order of magnitude for these derivatives.
The use of ionization potentials to select particular exci- tation spectra has numerous applications to the analysis of multicomponent samples containing isomeric substances. The methylindole illustration shows that discrimination of one isomer from another is a trivial matter, if one uses the two- laser photoionization technique. This will be of particular use when one isomer is present at much higher concentrations than the other. The species with the lower ionization potential can easily be selected and the electronic spectrum of this molecule can be stripped from the composite spectrum, al- lowing identification and quantification of the remaining component.
Time-Delayed Photoionization. An additional way in which two-laser photoionization can be employed in order to gain an advantage in isomer determination is by exploiting the differences in excited-state lifetimes by using a time delay between the excitation and ionization lasers. As an example, consider the photoionization of the isomers phenanthrene and anthracene with an excitation wavelength of 285 nm and an ionization wavelength sufficient to ionize both molecules. At this excitation wavelength, the spectra of both molecules are essentially structureless and consequently cannot be used for isomer selectivity. However, the use of two variably delayed lasers in the excitation-photoionization process allows one to discriminate very effectively against the isomer with the shorter excited-state lifetime. The measured excited-state decay times of phenanthrene and anthracene at this excitation wavelength are approximately 63 and 3 ns, respectively. Figure 7 displays the time-resolved ion yield of these two molecules as a function of delay between the arrival of the excitation and ionization laser pulses. The excitation laser was fixed at 285 nm and the ionization laser at 341 nm and only the parent masses at MW 178 were detected.

10 ANALYTICAL CHEMISTRY, VOL. 60, NO. 1, JANUARY 1, 1988
'. ".
r
0 20 40 60 80 cc 'L ?..& ~ , I I I 1
T I M E DELAY I n s )
Figwe 7. Ion yield as a fundon of delay between the excitation and ionization lasers for phenanthrene and anthracene in a supersonic expansion.
At delays in excess of 10 ns, phenanthrene ions are almost exclusively produced since >96% of the excited-state an- thracene population has relaxed prior to the arrival of the ionizing laser pulse, while only approximately 13% of the initially excited phenanthrene molecules has suffered the same fate after a 10-ns delay. For a 20-11s delay, -0.1% of the excited anthracene molecules are available for ionization and detection while -75% of the phenanthrene molecules that absorbed the first laser pulse remain in the excited state.
The two-laser approach to stepwise photoionization MS thus allows the investigator an extra dimension in the de- termination of isomers with differing excited-state lifetimes. Although the phenanthrene-anthracene system is charac- terized by widely differing lifetimes, this technique has the potential for discriminating between isomers with more similar excited-state decay times. Furthermore, time-delayed ioni- zation will be'particularly useful for analyses of multicom- ponent PAH/PCB samples. Contributions from molecules with short excited-state lifetimes, such as chlorinated aro- matics, to the photoionization spectra can easily be suppressed by delaying the arrival of the ionizing laser pulse by 10-100 ns. Delays on the order of 100 ns will lead to a significant enhancement of the contributions from PAH's such as ovalene (-2.0 ~ s ) , pyrene (-1.4 ps), benzopyrenes (-250 ns), and similar derivatives. At present, we are investigating time- delayed photoionization for the determination of these PAWS in real and simulated multicomponent samples.
In summary, two-laser photoionization provides several extra dimensions for resonant photoionization identification and analysis of species in multicomponent samples. The ability to tune the ionizing-laser wavelength independently with respect to the excitation laser provides a more efficient method of photoionization for a wide variety of aromatic molecules as well as allowing additional selectivity based on differences in ionization potentials and excited-state lifetimes. Thus, this technique offers several significant advantages over conventional one-laser R2PI.
Despite these advantages, the two-laser approach is not without drawbacks. This two-laser technique requires two independently tunable dye laser systems, which significantly increases experimental expense. More importantly, this method is much less effective than conventional one-laser
R2PI when the resonant intermediate level is characterized by a subnanosecond lifetime. If, for example, the excited-state lifetime of a particular analyte is dominated by fast nonra- diative processes, very little population will be available for ionization if the temporal overlap of the two laser pulses is not perfect. In certain instances, this difficulty can be ov- ercome by employing an ionizing wavelength sufficient to photoionize the analyte after the nonradiative process. This is similar to the approach taken by Smalley and co-workers for the study of intersystem crossing of jet-cooled aromatic (24,25) and N-heterocyclic molecules (26). Following initial excitation, an ArF excimer laser pulse at 193 nm (6.4 eV) was used to ionize the molecules in both the excited singlet and triplet manifolds with high efficiency. This approach promises to extend the analytical utility of two-laser ionization to a more diverse group of molecules.
ACKNOWLEDGMENT We are grateful to Gary Leach for experimental assistance. Registry NO. Phenanthrene, 85-01-8; anthracene, 120-12-7;
pyrene, 129-00-0; benzo[a]pyrene, 50-32-8; perylene, 198-55-0; ovalene, 190-26-1; naphthacene, 92-24-0; fluoranthene, 206-44-0; indole, 120-72-9; 7-methylindole, 933-67-5; 2-methylindole, 95-20-5; 3-methylindole, 83-34-1; 1-methylindole, 603-76-9; 5-methylindole,
LITERATURE CITED (1) Levy, D. H. A&. Chem. Phys. 1081, 47, 323. (2) Tembreull, R.; Sin, C. H.; Pang, H. M.; Lubman, D. M. Opt. News
1088, 12(10), 16. (3) Lubman, D. M. Anal. Chem. 1087, 59, 31A. (4) Hollas, J. M. Mgh Resolution Spectroscopy; Butterworths: London,
1982; Chapter 6. (5) Tembreull, R.; Sin, C. H.; Pang, H. M.; Lubman, 0. M. Anal. Chem.
1085, 57, 1186. (6) Johnson, P. M.; Otis, C. E. Annu. Rev. Phys. Chem. 1081, 32, 139. (7) Boesl, U.; Neusser, H. J.; Schlag, E. W. Chem. Phys. 1081, 55, 193. (8) Smith, M. A.; Hager, J. W.; Wallace, S. C. J. Chem. Phys. 1084, 80,
3097. (9) Letokhov, V. S. Nonlinear Laser Chemistry; Springer-Verlag: New
York, 1983; Chapters 3 and 4. (10) Duncan, M. A.; Deltr, T. G.; Smalley, R. E. J. Chem. Phys. 1081, 75,
21 18. (11) Tembreull, R.; Lubman, D. M. Anal. Chem. 1984, 56, 1962. (12) Ohta, N.; Baba, H. Mol. Phys. 1086, 59, 921. (13) Leach, G. MSc. Thesis, Unlversity of Toronto, 1986. (14) Hager, J. W.; Ivanco, M.; Smith, M. A,; Wallace, S. C. Chem. Phys.
1088. 105, 397. (15) Hager, J. W.; Demmer, D. R.; Wallace, S. C. J. Phys. Chem. 1087,
91, 1375. (16) Hager, J. W.; Wallace, S. C., unpublished work. (17) Greenblatt. G. D.; Nlssani, E.; Zaroura, E.; Haas, Y. J. Phys. Chem.
614-96-0.
1087, 91, 570. (18) Boschi, R.; Murrell, J. N.; Schmidt, W. Faraday Dlscuss. Chem. SOC.
1072, 54. 118. (19) Schwa*, S. A.; Topp. M. R. Chem. Phys. 1084, 86, 245. (20) Amkav, A.; Even, U.; Jortner, J. J. Chem. Phys. 1081. 74, 3745. (21) Amirav, A.; Even, U.; Jortner, J. J. Chem. Phys. I O S t , 75, 3770. (22) Chan. I. Y.; Dantus, M. J. Chem. Phys. 1085, 82, 4771. (23) Jandris, L. J.; Force, R. K.; Young, S. C. Appl. Spectrosc. 1085, 39,
266. (24) Duncan, M. A.; Dietz, T. G.; Smalley, R. E. J. Phys. Chem. 1981, 85,
7. (25) Dietz, T. G.; Duncan, M. A.; Smalley, R. E. J. Chem. Phys. 1082, 76,
1227. (26) Dietz, T. G.; Duncan, M. A.; Puiu, A. C.; Smailey, R. E. J. Phys.
Chem. 1082, 86, 4026.
RECEIVED for review April 23,1987. Accepted September 1, 1987. We acknowledge the financial support of the Natural Sciences and Engineering Research Council of Canada.







