
The Clinical and Pathologic Constellation of Wegener Granulomatosis of the Orbit
Stephen R. Perry, MD,1 Jack Rootman, MD/,3 Valerie A. White, MD2,3
Purpose: Wegener granulomatosis (WG) may present as an orbital mass without obvious upper respiratory or systemic features. The authors examined the clinical and pathologic features of a series of cases of orbital WG to define the features of presentation and progression of this disorder.
Methods: Thirteen subjects with orbital presentations of WG were identified from the University of British Columbia Orbit Clinic index of diseases. Clinical features were correlated with the results of computed tomography in 12 cases and orbital biopsy in 11 cases. Antineutrophil cytoplasmic antibody (c-ANCA) testing was performed in five cases.
Results: The main ocular symptoms were decreased vision, redness, and ocular and facial pain, whereas the main signs were proptosis, scleritis, and lid inflammation. Progression was marked by an increased incidence of bilaterality and systemic features. Ear, nose, and throat features were discovered at presentation in 11 cases and became universal during the follow-up period. Initial antineutrophil cytoplasmic antibody test results were negative in five patients but became positive later in three patients. Orbital biopsy specimens typically had features of mixed inflammation, fat disruption, and small areas of necrosis. The combination of cyclophosphamide and oral steroids was highly effective in terminating disease episodes.
Conclusions: Orbital WG can be recognized by a constellation of clinical and radiologic findings with evidence of an often erosive, infiltrating, and restrictive fibrotic, inflammatory mass. Concurrent ear, nose, and throat or specific ocular findings such as scleritis with typical limbal infiltrate can occur. Biopsy results show mixed inflammation with evidence of necrosis that must not be regarded as a nonspecific finding. Ophthalmology 1997; 1 04:683-694
Over time, we have become impressed by the protean clinical and diagnostic features of orbital Wegener granulomatosis (WO). This disorder has a broad range of presentations and associations. It often is clinically and
Originally received: February 13, 1996. Revision accepted: December 12, 1996.
I Department of Ophthalmology, Kidderminster General Hospital, Kidderminster, W orcestershire, England.
2 Department of Ophthalmology, University of British Columbia and the Vancouver Hospital and Health Sciences Centre, Vancouver, British Columbia, Canada.
pathologically misdiagnosed or diagnosed late, which may lead to grave visual or systemic consequences. Advances in treatment over the past decade have greatly improved the prognosis, particularly when diagnosis is made early. These concerns prompted a retrospective review of our experience at the University of British Columbia Orbit Unit. Results suggest that a constellation of clinical, radiologic, and pathologic features may facilitate earlier recognition of this disease. Prompt initiation of effective, specific therapy leads to optimal outcome for
3 Department of Pathology, University of British Columbia and the Vancouver Hospital and Health Sciences Centre, Vancouver, British Columbia, Canada.
Presented at the American Society of Ophthalmic, Plastic and Reconstructive Surgery Annual Meeting, Chicago, Illinois, November 13, 1993; the Verhoeff Society Meeting, Rochester, Minnesota, May 12-
15, 1994, and the International Symposium on Eyelids, the Orbit and the Lacrimal System-XXVIlth International Congress of Ophthalmology, Toronto, Ontario, June 24-25, 1994.
Reprint requests to Jack Rootman, MD, Department of Ophthalmology, University of British Columbia, 2550 Willow St, Vancouver, British Columbia, Canada, V5Z 3N9.
683

Ophthalmology Volume 104, Number 4, April 1997
what initially may be a relatively benign manifestation of WG.
Materials and Methods
Thirteen subjects were identified from the index of diseases maintained by the University of British Columbia Orbit Unit. Data presented here include observations from
684
our clinic as well as information from distant referring centers that, for geographic reasons, shared in the followup care.
This retrospective study was made possible by formalized and meticulous recording of orbital disease l and by information available through systemic evaluations, orbital biopsy material (11 cases, 1 as an outside pathology consult), and computed tomography (CT) (12 cases). The diagnosis of WG was made from the observed overall

Perry et al . Wegener Granulomatosis of the Orbit
Figure 1. A, a 21-year-old man presented in January 1987 with a I-year history of recurrent, progressive, bilateral lid swelling associated with injection and proptosis. He also had noted some persistent nasal obstruction. Visual acuity was 20/20 in each eye with normal pupils, and he had markedly injected conjunctiva with chemosis and brawny lid edema. His proptosis measured 29 mm in the right eye and 30 mm in the left eye. He had symmetrical but moderate reduction of ocular movements. His general health was otherwise good. B,C, axial and coronal computed tomography scans showed diffuse bilateral infiltration replacing orbital fat, obliterating other structures, and causing proptosis, with swelling in his inferior and middle turbinates. He had a biopsy performed of both the orbit and nasal passages. The orbital biopsy specimen showed dense sclerosing inflammation often with angiocentric concentration of a mixed infiltrate of lymphocytes, plasma cells, polymorphonuclear leukocytes, giant cells, and follicles (D, original magnification, x4; E, original magnification, x25). Immunohistochemistry was consistent with a polyclonal inflammatory disorder, and a diagnosis of nonspecific sclerosing inflammation was made. Biopsy specimen of the nasal tissue showed granulation tissue with a mixture of both acute and granulomatous infiltration. There was no evidence of fibrinoid necrosis of vessels, but a few focal microabscesses were noted. Although there was suspicion of Wegener granulomatosis, a definitive diagnosis could not be made. The patient was treated with oral prednisone starting at 40 mg daily and decreasing to 5 mg weekly. This was followed by orbital radiation of 2000 cOy in 10 treatments. This led to only partial resolution of his problems, and it was not possible for him to stop receiving steroids. By September 1987, his visual acuity was 20/15 in each eye, his proptosis had reduced to 22 mm in each eye, and his nasal passages had cleared. Immunosuppression was considered, but he decided to continue receiving 10 mg of prednisone daily. He gradually stopped treatment over the next 10 months. F, The patient at 8 months poststeroid treatment with much improvement. His ocular situation remained stable until 1991, when a nasal obstruction developed. Repeat computed tomography scan showed reduced proprosis but persistent obliteration of all fat planes. He remained clinically stable until November 1992, when he was seen for nasal obstruction, shortness of breath, and sore throat. Systemic investigation at that point showed bilateral, ill-defined densities in the chest, and he had a subglottic inflammatory mass, of which a biopsy was done, the results of which showed only chronic inflammation. At this point, his c-ANCA test result was positive and a definitive diagnosis of Wegener granulomatosis was made. He began receiving Septra and corticosteroids. Within 6 weeks, he stopped taking steroids; his breathing had improved because of decreased obstruction of his airway and nasal passages and reduction of the pulmonary infiltrates. He was last seen in April 1995 and was completely stable while taking Septra with no respiratory or orbital symproms.
clinical pattern and its correlation with the orbital, radiologic, and histopathologic findings. Two patients presented with diagnostic systemic features evident at an early stage in the orbital process; thus, orbital biopsy was not obtained.
Results
Presentation to the Referring Specialist
The study group consisted of 5 males and 8 females comprising 10 white patients, 2 American Indian patients, and 1 Asian patient. The average age at ophthalmic presenta-
Table 1. Main Ocular Features at First Ophthalmic Presentation
Main Ocular Features
Proptosis Bilateral proptosis Proptosis with scleritis
Reduced visual acuity Bilateral reduced vision Reduced vision and scleritis Reduced vision with mass White eye and no mass
Scleritis Bilateral scleritis Scleritis with proptosis
Ocular pain Facial pain
Ocular injection Lid features
No.
7/13 3/7 1/7 7/13 4/7 4/7 2/7 1/7 6/13 3/6 1/6 9/13 6/9 9/13 4/13
tion was 44 years (range, 9 to 69 years), which is 2 years later than the first attributable symptoms (42 years). The average duration of follow-up was 30 months (range, 3 to 109 months).
The main features on first ophthalmic presentation are listed in Table 1. They differ from the disease encountered in follow-up in both site and severity of disease. The predominant initial reported symptoms were proptosis associated with ocular or facial pain and a red eye in which scleritis was diagnosed earlier. The level of visual disability at presentation was usually moderate; in only one case was it reduced below 20/200 (in one eye of a woman who suffered a chronic red eye for 1 year before presentation). Disease at presentation was bilateral in six cases (Fig 1).
In all but two cases, ear, nose, and throat symptoms were active during the 3 months before ophthalmic presentation (Table 2). Typical symptoms were plugged nose and ears, epistaxis or clear nasal discharge, pressure sensations in the ears or over the paranasal sinuses, draining ears, and hearing loss or tinnitus (Fig 2). Three of the patients had been treated symptomatically without a diagnosis being made: one with antibiotics for radiologic ethmoiditis, one with sinus curettage (no biopsy) and antibi-
Table 2. Overview of Disease Progression: 13 Patients
Unilateral Bilateral ENT Systemic Disease Disease Features Features
At presentation 7 6 11 5 During follow-up 2 11 13 8
ENT = ear, nose, and throat.
685

Ophthalmology Volume 104, Number 4, April 1997
Figure 2. A, computed tomography scan taken in 1987 of the inner ear showing bone erosion that was associated with drainage from the ear, hearing loss, and tinnitus. The patient had been referred with a 2-month history of progressive orbital cellulitis and infiltration treated as an infection. B,C, axial and coronal computed tomography scans. She had severe necrotizing scleritis and adjacent orbital infiltration on the left side, which was associated with clinically minimal scleritis on the right. There was an extensive choroidal and retinal detachment. Biopsy results of the orbit showed mixed inflammation with focal microabscesses consistent with features of Wegener granulomatosis. Systemic study results, however, were negative. The patient was treated with corticosteroids and cyclophosphamide, and both her ocular and ear, nose, and throat symptoms resolved, the left eye becoming phthisical. D, she was subsequently seen in June 1990 with some reported ear problems, but repeat computed tomography scans of the orbits and ears showed repair of the bone and no active disease. The patient's treatment consisted of prednisone 60 mg daily and cyclophosphamide 150 mg daily. The prednisone was reduced and discontinued over a 3-month period, and the patient stopped taking cyclophosphamide within 8 months.
otics, and one with local treatment for mastoiditis. Gum biopsy results before referral in one patient showed mixed inflammation suggesting WG, which was not pursued because the gum growths resolved without treatment. Two other patients with long histories of nasal discomfort had undergone surgery in the past without diagnosis (ethmoidectomy 16 years previously and nasal surgery 10 years previously).
Systemic symptoms were seen in five patients at presentation. Two patients had malaise and weight loss, and one patient experienced arthralgia. The final two had clinically active systemic disease: one with foot drop and chest crepitations associated with mild shadowing on X rays, and the other with multiple systemic problems including pneumonia, splenic infarct, congestive cardiac failure, and orbital inflammation with cranial nerve palsies. This latter was the only case not presenting primarily for ophthalmologic care.
686
Evolution
The disease progressed with an increased number and severity of signs and symptoms as well as the individual inflammatory and mass effects summarized in Tables 2 and 3. All patients eventually showed ear, nose, and throat features. In three cases, these were limited to mild hearing deficits associated with mastoiditis. Although 6 of 10 had unilateral disease at presentation, ocular features became bilateral in all but 2 patients.
The findings formed three relatively distinct patterns: diffuse orbital involvement, lacrimal involvement, or midline involvement associated with visual deterioration. The globe was always displaced away from the causative orbital mass. Lid swelling with brawny discoloration was particularly prominent in those with lacrimal gland involvement (Figs 3 and 4).
An orbital mass developed in all patients in the series,

Perry et al . Wegener Granulomatosis of the Orbit
Table 3. Main Categories of Ophthalmic Disease Encountered during Follow-up
Symptom or Sign
Orbital mass Proptosis Eyelid swelling and injection Optic neuropathy Scleritis Keratitis Ocular restriction Uveitis
No.
13/13 11/13 8/13 8/13 6/13 6/13 4/13 3/13
including those who presented initially with scleritis (6/ 13, 44%). Scleritis was usually described as nodular with necrosis and, by the time of consultation, was associated in all six patients with typical white, subepithelial, stromal, corneal infiltrates with a clear limbal zone (Figs SA and 5B).
Eight of the 13 patients lost visual acuity (total = 13 eyes) (Table 4). Nine eyes in five patients showed visual deterioration during ophthalmic evaluation; significantly, of those presenting with normal acuity, subsequent visual loss did not develop in any. Scleritis was the initial presentation in four of the eight patients with visual loss. In all eight, there was an associated orbital mass (midline, apical, or diffuse on CT scan) that caused proptosis in all but one affected eye. Optic neuropathy was diagnosed in all eight patients. Progressive loss was documented by swollen or pale disc, decreased vision, or an associated relative afferent pupillary defect. One patient had a fulminant course commencing with focal scleritis, progressive uveitis, retinal and choroidal detachments, and proptosis with restricted ocular movements. This developed into progressive, massive, necrotizing scleritis, panophthalmitis, and adjacent orbital infiltration. The final visual outcome of no light perception with phthisis was secondary to the destructive panophthalmitis and inflammatory orbital mass.
The Tempo of Evolution of Orbital Disease
The pattern of onset generally was subacute, evolving over weeks. Three patients had chronic disease: one with near continuous ocular inflammation for 2 years before referral and two with intermittent disease. One with intermittent disease had visual deterioration that persisted and contralateral proptosis that resolved 1 year previously. The other patient had suffered recurrent nasal and sinus problems over a 16-year period, and 10 years before ophthalmic presentation had been hospitalized for a self-limiting pneumonia with hemoptysis.
Disease activity commonly showed rapid acceleration after a relatively benign subacute onset, a pattern noted in 9 patients (with 10 episodes of acute activity). The ocular prodromal phase typically lasted 2 months, and all patients had at least 1 month of ocular or orbital disease before their accelerated episode. Subsequent deterioration
was dramatic (usiIally over a period as short as 1 week) and was manifested by increased pain and inflammatory signs; in 7 patients (10 eyes), visual acuity was reduced to less than 20/200. In five patients, these episodes were responsible for serious visual loss during the period of ophthalmic care (Table 4).
Four episodes of deterioration occurred on or soon after cessation of steroid therapy without combined immunosuppression. Two episodes occurred immediately after steroid reduction with already moderately active disease in which there was concern about a lack of response to immunosuppressive therapy. In one case, alternative immunosuppression (oral azathioprine 150 mg daily) had been substituted with a rapid reduction in oral steroids from 80 to 10 mg daily over a 2-week period. The other case involved abrupt cessation of steroid therapy (before referral) from 100 mg daily with institution of a highdose intravenous antibacterial regimen based on fears of missed infectious origin of the midline inflammatory mass. Another patient had finished an apparently effective course of systemic corticosteroids 2 months previously, then deteriorated.
Computed Tomography Features
Of the 12 patients who underwent CT scan, all showed either orbital mass or midline change. Eight had nasopharyngeal or sinus lesions, and six had evidence of midline bony erosion (Fig 6). Orbital masses were located as follows: seven presented with lacrimal gland involvement (Fig 7), four with midline disease, four with apical mass, and four with inferolateral disease. Four of the seven cases with lacrimal involvement showed no evidence of midline disease (Fig 8). Orbital masses tend to have infiltrative margins obscuring the adjacent fat planes. Two patients had bilateral diffuse orbital involvement (Fig 1).
Pathology
Biopsy material was available from II patients and showed several important and consistent features (Table 5), which together strongly suggest the diagnosis of orbital WG. All had features of fat disruption, usually consisting of focal areas of necrosis with free vacuoles, often evidenced by lipid-laden macrophages and giant cells with obliteration of normal fat spaces. Active or old fibrosis was seen in all specimens. In 10 of the 11 cases, there was a mixture of granulomatous and acute inflammation (Figs 10, 3B, 3C, 5B, 8B) with eosinophils or polymorphonuclear leukocytes or both and loosely formed granulomatous infiltrates. Nine of 11 cases had features of a smudgy necrosis (microabscesses), either in a stellate (Fig 3D) or a focal microscopic pattern or both. These areas were surrounded by mixed granulomatous and acute inflammatory infiltrates. Features of active necrotizing vasculitis (Fig 3B) were rare. This may reflect the size of the specimen, lack of vessels within the tissue, or direct local necrosis induced by polymorphonuclear leukocytes rather than ischemia. Three of 11 showed necrotizing vasculitis, and 3 had features that suggested ques-
687
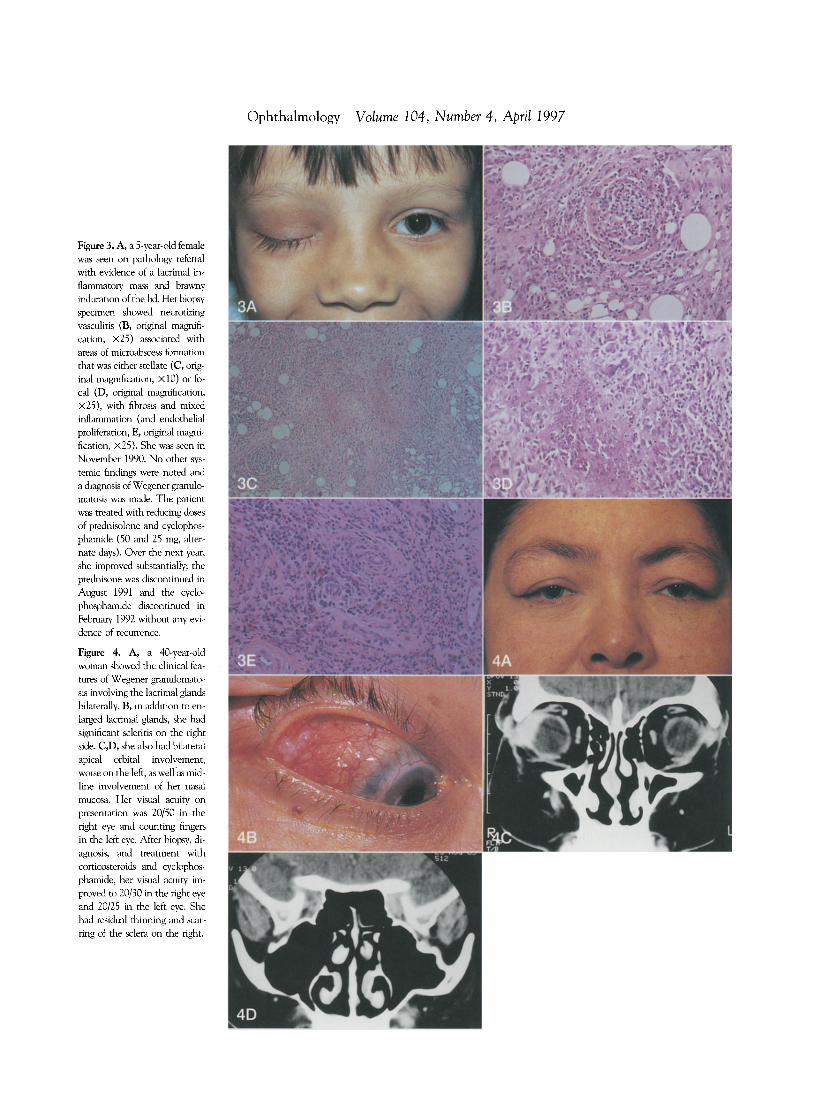
Figure 3. A, a 5-year-old female was seen on pathology referral with evidence of a lacrimal inflammatory mass and brawny induration of the lid. Her biopsy specimen showed necrotizing vasculitis (B, original magnification, x25) associated with areas of microabscess fonnation that was either stellate (C, original magnification, XI0) or focal (D, original magnification, x25), with fibrosis and mixed inflammation (and endothelial proliferation, E, original magnification, x25). She was seen in November 1990. No other systemic findings were noted and a diagnosis of Wegener granulomarosis was made. The patient was treated with reducing doses of prednisolone and cyclophosphamide (50 and 25 mg, alternate days). Over the next year, she improved substantially; the prednisone was discontinued in August 1991 and the cyclophosphamide discontinued in February 1992 without any evidence of recurrence.
Figure 4. A, a 40-year-old woman showed the clinical features of Wegener granulomatosis involving the lacrimal glands bilaterally. B, in addition to enlarged lacrimal glands, she had significant scleritis on the right side. C,D, she also had bilateral apical orbital involvement, worse on the left, as well' as midline involvement of her nasal mucosa. Her visual acuity on presentation was 20/50 in the right eye and counting fingers in the left eye. After biopsy, diagnosis, and treatment with corticosteroids and cyclophosphamide, her visual acuiry improved to 20/30 in the right eye and 20/25 in the left eye. She had residual thinning and scarring of the sclera on the right.
Ophthalmology Volume 104, Number 4, April 1997
Perry et al . Wegener Granulomatosis of the Orbit
tionable or old vasculitis with acellular obliteration of vessels (Fig 5B). There were features of activation of the endothelium (swelling and proliferation) in many (7/11) of the vessels studied (Fig 3E).
Outcomes of Various Treatment Regimens
The Effects of Oral Steroids. Steroids were used alone as initial therapy in 10 patients. Good control with remis-
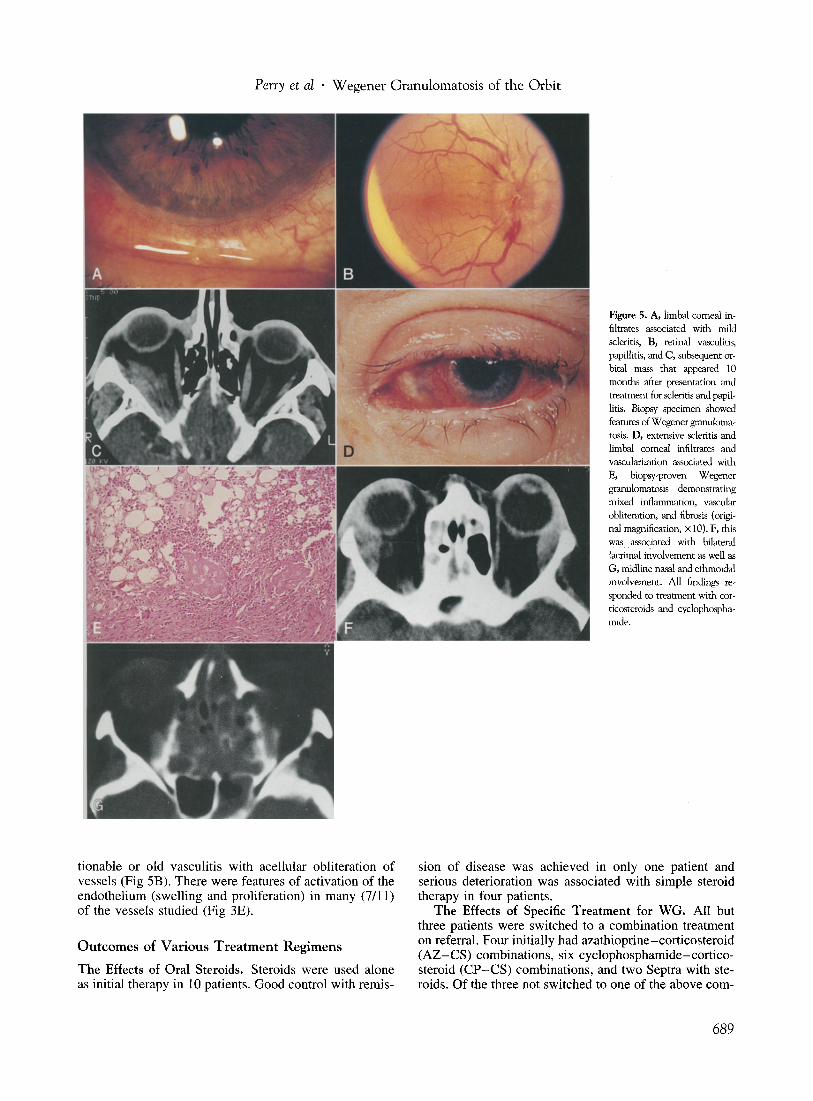
Figure 5. A, limbal corneal infiltrates associated with mild scleritis, B, retinal vasculitis, papillitis, and C, subsequent orbital mass that appeared 10 months after presentation and treatment for scleritis and papillitis. Biopsy specimen showed features of Wegener granulomatosis. D, extensive scleritis and limbal corneal infiltrates and vascularization associated with E, biopsy-proven Wegener granulomatosis demonstrating mixed inflammation, vascular obliterarion, and fibrosis (original magnification, x 10). F, this was associated with bilateral lacrimal involvement as well as G, midline nasal and ethmoidal involvement. All findings responded to treatment with corticosteroids and cyclophosphamide.
sion of disease was achieved in only one patient and serious deterioration was associated with simple steroid therapy in four patients.
The Effects of Specific Treatment for WG. All but three patients were switched to a combination treatment on referral. Four initially had azathioprine-corticosteroid (AZ-CS) combinations, six cyclophosphamide-corticosteroid (CP-CS) combinations, and two Septra with steroids. Of the three not switched to one of the above com-
689
Ophthalmology Volume 104, Number 4, April 1997
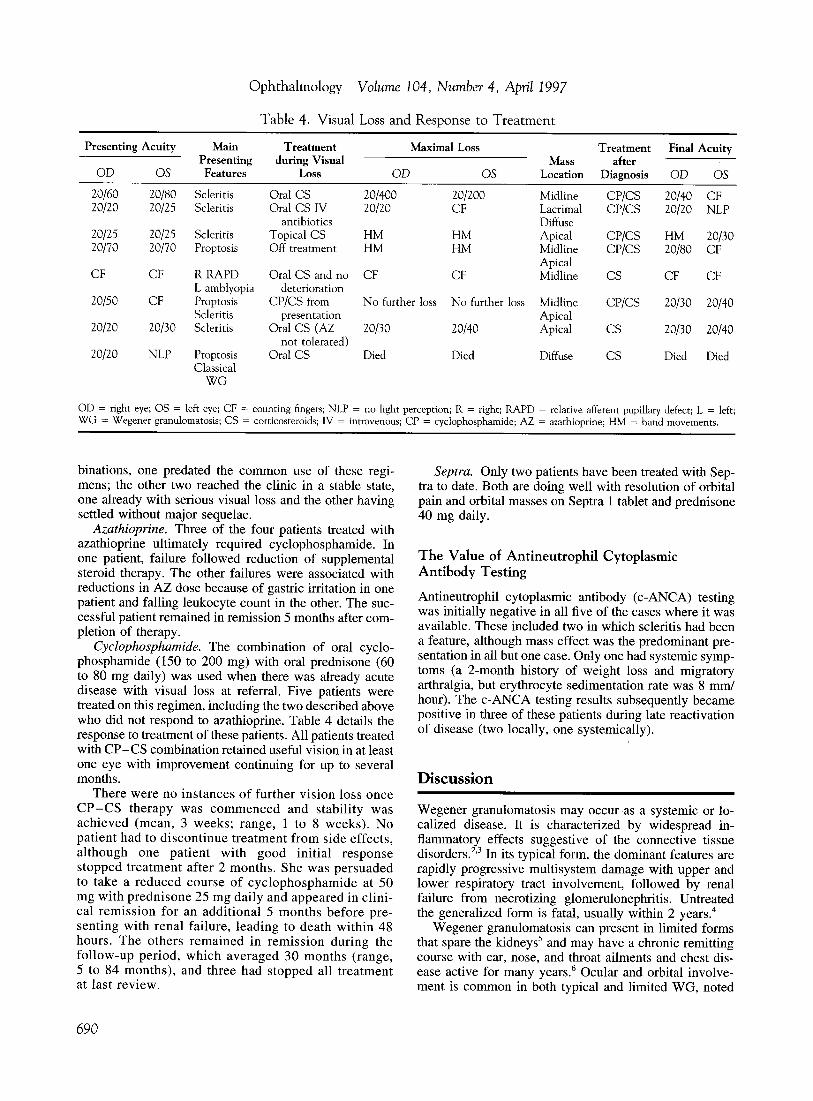
Table 4. Visual Loss and Response to Treatment
Presenting Acuity Main Treatment Maximal Loss Treatment Final Acuity Presenting during Visual Mass after
OD OS Features Loss OD OS Location Diagnosis OD OS
20/60 20/80 Scleritis Oral CS 20/400 20/200 Midline CP/CS 20/40 CF 20/20 20/25 Scleritis Oral CS IV 20/20 CF Lacrimal CP/CS 20/20 NLP
antibiotics Diffuse 20/25 20/25 Scleritis Topical CS HM HM Apical CP/CS HM 20/30 20/70 20/70 Proptosis Off treatment HM HM Midline CP/CS 20/80 CF
Apical CF CF RRAPD Oral CS and no CF CF Midline CS CF CF
L amblyopia deterioration 20/50 CF Proptosis CP/CS from No further loss No further loss Midline CP/CS 20/30 20/40
Scleritis presentation Apical 20/20 20/30 Scleritis Oral CS (AZ 20/30 20/40 Apical CS 20/30 20/40
not tolerated) 20/20 NLP Proptosis Oral CS Died Died Diffuse CS Died Died
Classical WG
00 = right eye; OS = left eye; CF = counting fingers; NLP = no light perception; R = right; RAPO = relative afferent pupillary defect; L = left; WG = Wegener granulomatosis; CS = corticosteroids; IV = intravenous; CP = cyclophosphamide; AZ = azathioprine; HM = hand movements.
binations, one predated the common use of these regimens; the other two reached the clinic in a stable state, one already with serious visual loss and the other having settled without major sequelae.
Azathioprine. Three of the four patients treated with azathioprine ultimately required cyclophosphamide. In one patient, failure followed reduction of supplemental steroid therapy. The other failures were associated with reductions in AZ dose because of gastric irritation in one patient and falling leukocyte count in the other. The successful patient remained in remission 5 months after completion of therapy.
Cyclophosphamide. The combination of oral cyclophosphamide (150 to 200 mg) with oral prednisone (60 to 80 mg daily) was used when there was already acute disease with visual loss at referral. Five patients were treated on this regimen, including the two described above who did not respond to azathioprine. Table 4 details the response to treatment of these patients. All patients treated with CP-CS combination retained useful vision in at least one eye with improvement continuing for up to several months.
There were no instances of further vision loss once CP-CS therapy was commenced and stability was achieved (mean, 3 weeks; range, 1 to 8 weeks). No patient had to discontinue treatment from side effects, although one patient with good initial response stopped treatment after 2 months. She was persuaded to take a reduced course of cyclophosphamide at 50 mg with prednisone 25 mg daily and appeared in clinical remission for an additional 5 months before presenting with renal failure, leading to death within 48 hours. The others remained in remission during the follow-up period, which averaged 30 months (range, 5 to 84 months), and three had stopped all treatment at last review.
690
Septra. Only two patients have been treated with Septra to date. Both are doing well with resolution of orbital pain and orbital masses on Septra 1 tablet and prednisone 40 mg daily.
The Value of Antineutrophil Cytoplasmic Antibody Testing
Antineutrophil cytoplasmic antibody (c-ANCA) testing was initially negative in all five of the cases where it was available. These included two in which scleritis had been a feature, although mass effect was the predominant presentation in all but one case. Only one had systemic symptoms (a 2-month history of weight loss and migratory arthralgia, but erythrocyte sedimentation rate was 8 mmJ hour) . The c-ANCA testing results subsequently became positive in three of these patients during late reactivation of disease (two locally, one systemically).
Discussion
Wegener granulomatosis may occur as a systemic or localized disease. It is characterized by widespread inflammatory effects suggestive of the connective tissue disorders?·3 In its typical form, the dominant features are rapidly progressive multisystem damage with upper and lower respiratory tract involvement, followed by renal failure from necrotizing glomerulonephritis. Untreated the generalized form is fatal, usually within 2 years.4
Wegener granulomatosis can present in limited forms that spare the kidneys5 and may have a chronic remitting course with ear, nose, and throat ailments and chest disease active for many years.6 Ocular and orbital involvement is common in both typical and limited WG, noted
Perry et al . Wegener Granulomatosis of the Orbit
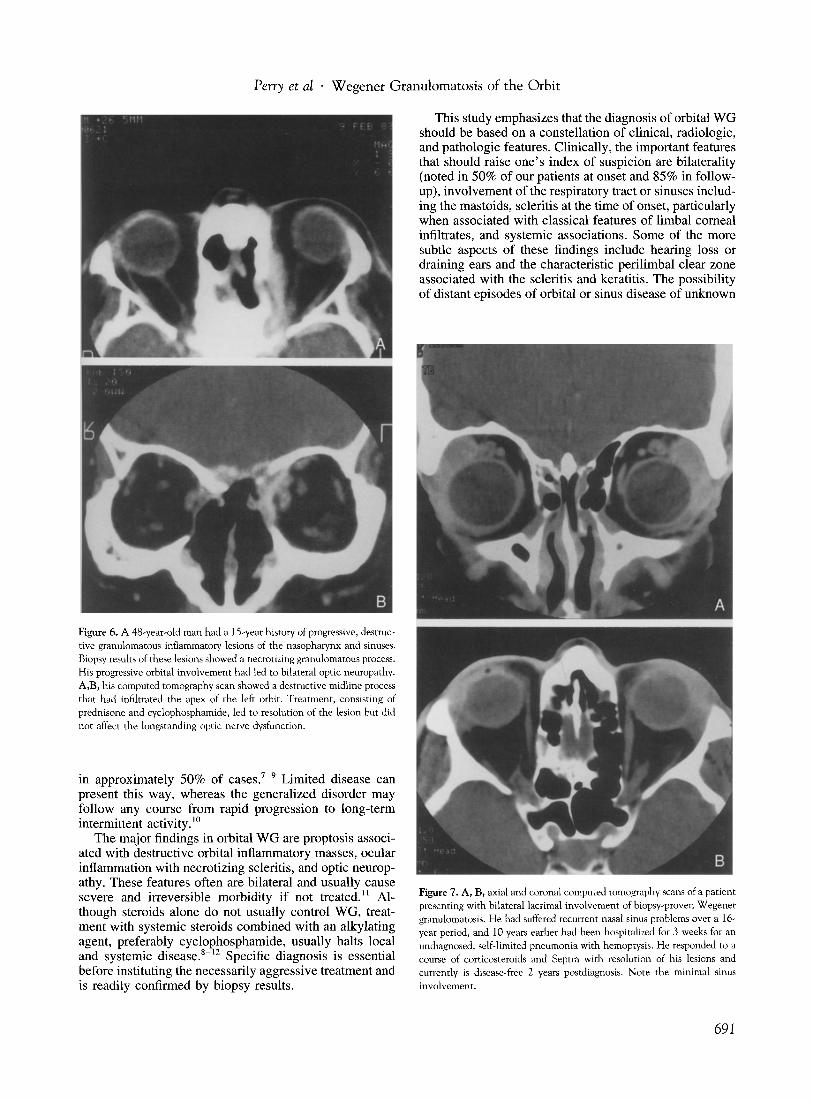
Figure 6. A 48-year-old man had a IS-year history of progressive, destructive granulomatous inflammatory lesions of the nasopharynx and sinuses. Biopsy results of these lesions showed a necrotizing granulomatous process. His progressive orbital involvement had led to bilateral optic neuropathy. A,B, his computed tomography scan showed a destructive midline process that had infiltrated the apex of the left orbit. Treatment, consisting of prednisone and cyclophosphamide, led to resolution of the lesion but did not affect the longstanding optic nerve dysfunction.
in approximately 50% of cases.7-
9 Limited disease can present this way, whereas the generalized disorder may follow any course from rapid progression to long-term intermittent activity. 10
The major findings in orbital WG are proptosis associated with destructive orbital inflammatory masses, ocular inflammation with necrotizing scleritis, and optic neuropathy. These features often are bilateral and usually cause severe and irreversible morbidity if not treated. I I Although steroids alone do not usually control WG, treatment with systemic steroids combined with an alkylating agent, preferably cyclophosphamide, usually halts local and systemic disease. 8
-12 Specific diagnosis is essential
before instituting the necessarily aggressive treatment and is readily confirmed by biopsy results.
This study emphasizes that the diagnosis of orbital WG should be based on a constellation of clinical, radiologic, and pathologic features. Clinically, the important features that should raise one's index of suspicion are bilaterality (noted in 50% of our patients at onset and 85% in followup), involvement of the respiratory tract or sinuses including the mastoids, scleritis at the time of onset, particularly when associated with classical features of limbal corneal infiltrates, and systemic associations. Some of the more subtle aspects of these findings include hearing loss or draining ears and the characteristic perilimbal clear zone associated with the scleritis and keratitis. The possibility of distant episodes of orbital or sinus disease of unknown
Figure 7. A, B, axial and coronal computed tomography scans of a patient presenting with bilateral lacrimal involvement of biopsy-proven Wegener granulomatosis. He had suffered recurrent nasal sinus problems over a 16-year period, and 10 years earlier had been hospitalized for 3 weeks for an undiagnosed, self-limited pneumonia with hemoptysis. He responded to a course of corticosteroids and Septra with resolution of his lesions and currently is disease-free 2 years postdiagnosis. Note the minimal sinus involvement.
691

Ophthalmology Volume 104, Number 4, April 1997
Figure 8. A 30-year-old woman presented with an I8-month history of a painless mass in the right upper lid. A, the computed tomography scan shows a lacrimal mass. B, biopsy results showed features consistent with Wegener granulomatosis, showing vascular occlusion, fibrinoid necrosis, and a mixed inflammatory infiltrate. She was treated successfully with steroids and azathioprine. No midline involvement has been noted in 2 years' follow-up.
cause must be recognized because this condition may progress in episodic fashion and may regress on its own.
This disorder ~ffects a wide age range from childhood to old age, and there was an average of 2 years of attributable symptoms before ophthalmic presentation. In our experience and in recent clinicopathologic consultations, clinicians frequently fail to recognize that this disease can occur in young people. Orbital inflammatory syndromes in childhood should include WG in the differential diagnosis, particularly when the sinuses, lacrimal gland, or bilateral orbits are involved.
It bears emphasis that WG involving the lacrimal gland can occur without necessarily having contiguous orbital or midline disease. Further, it may present as bilateral, diffuse orbital disease. Wegener granulomatosis can have an orbital presentation as a major feature before respiratory symptoms and signs are recognized.
The CT features suggestive of this disorder are involvement of the sinus structures, including mastoids, and a tendency for orbital masses to be associated with
692
infiltration and obliteration of adjacent fat planes. Midline disease is associated with bone erosion.
In this series, visual loss occurred only in those patients who presented with some reduced acuity, suggesting that the final process of major visual damage was already active at presentation and was largely attributable to optic neuropathy. This probably was because of early occult activity that affected the optic nerve even in those patients presenting with scleritis. This would fit with the eventual development of orbital masses in all of these patients.
Systemic features were usually minimal in those patients with limited disease, and major local destruction occurred with only mild nonspecific systemic features. However, limited presentation can be prodromal to systemic involvement.
Another important finding recognized in this and previous work7 is the potential for acute progression of chronic disease. This clearly was seen in 10 episodes of dramatic deterioration after relatively mild prodromal disease, which caused considerable morbidity during ophthalmic care. These episodes occurred with steroids as well as with no systemic therapy, particularly after reduction of steroid therapy. One event appeared to be triggered by orbital biopsy.
A number of investigations aid in early diagnosis, needed for effective treatment before serious deterioration occurs. The c-ANCA testing is useful when the results are positive13 and has helped in limited disease where the results are said to be positive in 60% to 70% of cases. 14
It is particularly useful in cases with scleritis in which studies of 17 patients all tested positive for c-ANCA 15,16
(although 1 initially tested negative but became positive later). However, in our series with orbital disease, five patients initially tested negative despite late positive conversion in three, reaffirming the diagnosis. Test sensitivities may vary, and perhaps our patients were tested earlier
Table 5. Pathologic Features of Wegener Granulomatosis
Pathologic Feature
Fat disruption/necrosis Fibrosis
Active (8) Old (3)
Mixed inflammation-granulomatous/PMNs Eosinophils Necrosis
Stellate (4) Micro (2) Both (3)
Vasculitis Necrotizing (3) Questionable (old) (3)
Other Activated vessels Giant cells
PMNs = polymorphonuclear neutrophils.
No.
11/11 11/11
10/11 10/11 9/11
3/11
7/11 5/11

Perry et al . Wegener Granulomatosis of the Orbit
in the disease process. Treatment should not be delayed by a negative test result in the presence of clinical and pathologic evidence that suggests this disorder.
The results of orbital biopsy were of particular interest in this series, which was biased toward patients with an obscure orbital inflammatory mass by the nature of the referral system. Orbital biopsy is difficult to interpree7.18
and should be studied in light of a constellation of clinical and histopathologic findings. These should include careful clinical study, c-ANCA testing,13,14,19 and CT scans of the orbit, paranasal sinuses, and mastoid sinuses,zo In our series, 11 patients underwent orbital biopsy because of the suggestive constellation of clinical features and the absence of other hard evidence. The histopathologic features in orbital biopsies differ from those of other sites. Necrotizing granulomas with vasculitis have been shown in only a third of orbital biopsies where confirmatory biopsy was available from other sites. 18
In this study, orbital biopsy was found invaluable, and a number of important observations should be emphasized. A mixed inflammation with lymphocytes often cuffing vessels, areas of fat necrosis and lipid-containing macrophages, mixed granulomatous stellate or focal microabscesses, and polymorphonuclear leukocytic and eosinophilic infiltrates with fibroplasia should not be left with a nonspecific title. Once stains for fungi and mycobacteria have ruled out infection, there are few other causes for this kind of mixed orbital inflammation. Hodgkin disease is very rare, and the former catchall diagnosis of pseudotumor is not acceptable. Acute idiopathic inflammation and sclerosing inflammation each have their typical appearance. Acute idiopathic inflammation is characterized by rapid onset over days or weeks and is associated with clinical inflammatory sigJ.1s. On investigation, it may present with diffuse orbital involvement or perineural involvement or it may localize to the lacrimal glands, peribulbar region, or extraocular muscles (as myositis). Biopsy specimens show largely lymphocytic infiltration without an acute component, necrosis, or fibrosis, nor is there any evidence of vascular infiltration or destruction. The acute idiopathic inflammations respond promptly to corticosteroids alone. Idiopathic sclerosing inflammation is characterized clinically by cicatricial infiltration, mass effect, and mild chronic inflammation. Evidence of cicatricial restriction of movement is frequent. Histologically, fibrosis is a characteristic finding and is associated with paucicellular inflammatory infiltrates consisting of plasma cells, lymphocytes, histiocytes, and occasional neutrophils within a dense collagenous stroma.2l ,22
In biopsy specimens from orbits with WG, the wide range of acute to chronic presentation is reflected in the relative predominance of cell types with the most chronic cases showing firm masses with large areas of desmoplasia in addition to the areas of active disease. The features described in the orbit (mixed inflammation, microscopic necrosis, and others) have been noted as characteristic of this disorder in other extravascular soft tissue sites, such as the mediastinum, retroperitoneum, and breast.23
The dramatic benefits of CP-CS combination therapy7,8,12 contrast with the partial response to AZ-CS and with the poor results of steroids alone. The presumptive use of the latter therapy reflects the difficulty of early diagnosis and unfortunately masks disease activity without bringing about remission or preventing dramatic deterioration. The one patient who settled on steroids may well have improved without treatment, as had other patients in this study who suffered self-limiting episodes attributable to WG before presentation.
The combination of Septra with steroids has been effective24 and, in the limited experience in this series, has eased symptoms dramatically within weeks of initiation in two patients with orbital masses, orbital pressure sensations, and nasal stuffiness. It appears to be a safe initial therapy where there is no early visual loss. Such loss appears to presage more serious deterioration, and a CPCS combination would seem an appropriate option.
Because this disorder may be of such serious consequence, we would encourage a nonrestrictive clinical and pathologic definition to make earlier diagnosis. The particular constellation of clinical signs and symptoms associated with typical and highly suggestive biopsy features should prompt diagnosis and activate treatment to prevent the sometimes catastrophic outcomes.
References
1. Rootman J. Diseases of the orbit: a multidisciplinary approach. Philadelphia: JB Lippincott, 1988; 85.
2. Goodman GC, Churg J. Wegener's granulomatosis. Pathology and review of the literature. Arch PathoI1954;58:533-53.
3. Fauci AS, Wolff SM. Wegener's Granulomatosis: studies in eighteen patients and a review of the literature. Medicine 1973;52:535-61.
4. Walton EW. Giant cell granuloma of the respiratory tract (Wegener's granulomatosis). Br Med J 1958;2:265-70.
5. Carrington CB, Liebow AA. Limited forms of angiitis and granulomatosis of Wegener's type. Am J Med 1966; 41:497-527.
6. Coppeto JR, Yamase H, Monteiro ML. Chronic ophthalmic Wegener's granulomatosis. J Clin Neuro-ophthalmol 1985;5:17-25.
7. Bullen CL, Lieseang TJ, McDonald TJ, DeRemee RA. Ocular complications of Wegener's granulomatosis. Ophthalmology 1983;90:279-90.
8. Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener's granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 1983;98:76-85.
9. Haynes BF, Fishman ML, Fauci AS, Wolff SM. The ocular manifestations of Wegener's granulomatosis. Fifteen years experience and review of the literature. Am J Med 1977;63: 131-41.
10. Coutu RE, Klein M, Lessell S, et al. Limited form ofWegener granulomatosis. Eye involvement as a major sign. JAMA 1975;233:868-71.
11. Spalton DJ, Graham EM, Page NG, Sanders MD. Ocular changes in limited forms of Wegener's granulomatosis. Br J Ophthalmol 1981;65:553-63.
12. Hollander D, Manning RT. The use of alkylating agents in
693

Ophthalmology Volume 104, Number 4, April 1997
the treatment of Wegener's granulomatosis. Ann Intern Med 1967;67:393-8.
13. Pulido JS, Goeken JA, Nerad JA, et al. Ocular manifestations of patients with circulating antineutrophil cytoplasmic antibodies. Arch Ophthalmol 1990; 108:845-50.
14. Nolle B, Specks U, Ludemann J, et al. Anticytoplasmic autoantibodies: their immunodiagnostic value in Wegener's granulomatosis. Ann Intern Med 1989;111:28-40.
15. Charles SJ, Meyer PA, Watson PG. Diagnosis and management of systemic Wegener's granulomatosis presenting with anterior ocular inflammatory disease. Br J Ophthalmol 1991; 75:201-7.
16. Soukiasian SH, Foster CS, Niles JL, Raizman MB. Diagnostic value of anti-neutrophil cytoplasmic antibodies in scleritis associated with Wegener's granulomatosis. Ophthalmology 1992; 99: 125-32.
17. Blodi FC, Gass JM. Inflammatory pseudotumour of the orbit. Br J Ophthalmol 1968;52:79-93.
18. Kalina PH, Lie JT, Campbell RJ, Garrity JA. Diagnostic value and limitations of orbital biopsy in Wegener's granulomatosis. Ophthalmology 1992;99:120-4.
694
19. Van der Woude FJ, Rasmussen N, Lobatto S, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener's granulomatosis. Lancet 1985; 1:425-9.
20. Simmons JT, Leavitt R, Kornblut AD, Fauci AS. CT of the paranasal sinuses and orbits in patients with Wegener's granulomatosis. Ear Nose Throat J 1987;66:134-40.
21. McCarthy JM, White VA, Harris G, et al. Idiopathic sclerosing inflammation of the orbit: immunohistologic analysis and comparison with retroperitoneal fibrosis. Mod Pathol 1993;6:581-7.
22. Rootman J, McCarthy M, White V, et al. Idiopathic sclerosing inflammation of the orbit. A distinct clinicopathologic entity. Ophthalmology 1994; 101:570-84.
23. Goulart RA, Mark EJ, Rosen S. Tumefactions as an extravascular manifestation of Wegener's granulomatosis. Am J Surg Pathol 1995; 19:145-53.
24. West BC, Todd JR, King JW. Wegener granulomatosis and trimethoprim-sulfamethoxazole: complete remission after a twenty-year course. Ann Intern Med 1987; 106:840-2.







