
Open Journal of Hematology, 2010, 1-4
Page 1 of 20 (Page number not for citation purposes)
Review Article
The Expanding Role of Proteasome-Based Therapy in the Treatment of Hematologic Malignancies
James J. Driscoll
2 Hematology-Oncology Service, Department of Medicine, Walter Reed Army Medical Center, Washington, DC, USA
,1 Roopa DeChowdhury,1 Jason Burris,1,2 Christina M. Annunziata1 1 Medical Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
Corresponding Author & Address: James J. DriscollMedical Oncology Branch, National Cancer Institute, 10 Center Drive, Building 10-Room 12N-226
Magnuson Cancer Center, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892 (Tel): 301-451-4401; (Fax): 301-480-6255; Email: [email protected] Published: 4th September, 2010 Accepted: 4th September, 2010 Received: 30th June, 2010 Revised: 11th August, 2010 Open Journal of Hematology, 2010, 1-4 © Driscoll et al.; licensee Ross Science Publishers ROSS Open Access articles will be distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided that the original work will always be cited properly. Keywords: Proteasome, Bortezomib, Multiple Myeloma, Hematologic Malignancies
ABSTRACT
The Ubiquitin (Ub)+Proteasome pathway is the major cellular pathway for the selective degradation of nuclear and cytosolic proteins. The proteasome is the catalytic core of the Ub+Proteasome pathway and has become an intriguing new target in drug development and cancer therapy for the treatment of hematologic malignancies. Successful pharmacologic inhibition of the proteasome with the boron-containing small molecule bortezomib led to US Food and Drug Administration (FDA) approval for the treatment of multiple myeloma (MM). That clinical success has propelled tremendous interest and application of proteasome inhibition to an increasing number of hematologic malignances. Inhibition of the proteasome results in the accumulation of multi-ubiquitinated proteins that are normally degraded through the tightly regulated Ub+Proteasome pathway. Such an accumulation leads to the stabilization of numerous cellular proteins that control the cell cycle, growth, proliferation and apoptosis. It is thought that the accumulation of multi-Ub~protein conjugates leads to apoptosis although there are numerous mechanisms proposed to explain how proteasome inhibition leads to cell death. However, not all patients respond to bortezomib-based therapy and moreover, those patients that do respond inevitably develop drug resistance. In addition, the mechanism of action for bortezomib remains incompletely characterized and, thus, newer proteasome inhibitors are needed and are in clinical development. The use of the proteasome inhibitor bortezomib has been expanded successfully from MM to other hematologic malignancies that include various lymphomas, Waldenström’s macroglobulinemia, Amyloidosis and Acute Myeloid Leukemia (AML). The proteasome is a validated therapeutic target and proteasome inhibitors modulate protein stability to effect tumor cell growth control and promote programmed cell death. Targeting the Ub+Proteasome pathway offers great promise in the treatment of hematologic and eventually solid tumor malignancies.
Open Journal of Hematology OPEN ACCESS
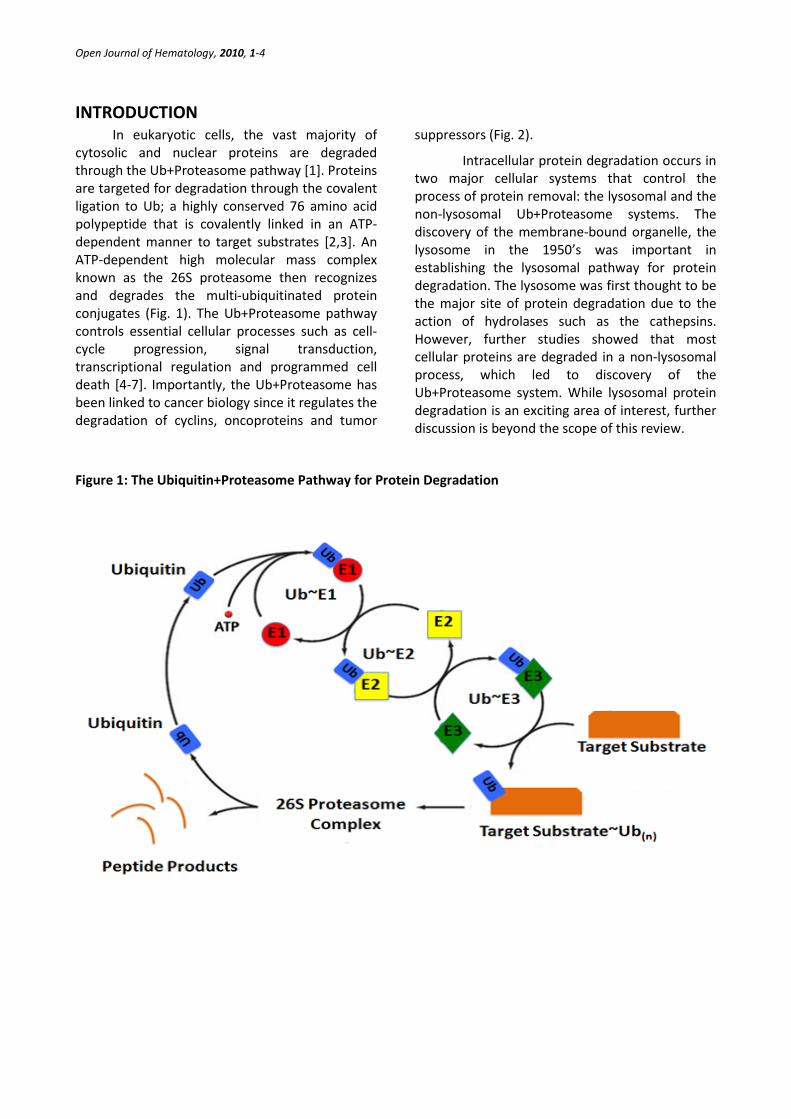
Open Journal of Hematology, 2010, 1-4
INTRODUCTION
In eukaryotic cells, the vast majority of cytosolic and nuclear proteins are degraded through the Ub+Proteasome pathway [1]. Proteins are targeted for degradation through the covalent ligation to Ub; a highly conserved 76 amino acid polypeptide that is covalently linked in an ATP-dependent manner to target substrates [2,3]. An ATP-dependent high molecular mass complex known as the 26S proteasome then recognizes and degrades the multi-ubiquitinated protein conjugates (Fig. 1). The Ub+Proteasome pathway controls essential cellular processes such as cell-cycle progression, signal transduction, transcriptional regulation and programmed cell death [4-7]. Importantly, the Ub+Proteasome has been linked to cancer biology since it regulates the degradation of cyclins, oncoproteins and tumor
suppressors (Fig. 2).
Intracellular protein degradation occurs in two major cellular systems that control the process of protein removal: the lysosomal and the non-lysosomal Ub+Proteasome systems. The discovery of the membrane-bound organelle, the lysosome in the 1950’s was important in establishing the lysosomal pathway for protein degradation. The lysosome was first thought to be the major site of protein degradation due to the action of hydrolases such as the cathepsins. However, further studies showed that most cellular proteins are degraded in a non-lysosomal process, which led to discovery of the Ub+Proteasome system. While lysosomal protein degradation is an exciting area of interest, further discussion is beyond the scope of this review.
Figure 1: The Ubiquitin+Proteasome Pathway for Protein Degradation

Open Journal of Hematology, 2010, 1-4
Page 3 of 20 (Page number not for citation purposes)
Figure 2: The Role of the Ubiquitin+Proteasome Pathway in Essential Cellular Processes
In eukaryotes, the 26S proteasome is a ~2.5-MDa structure that consists of over 30 different subunits and catalyzes the ATP-dependent degradation of nuclear and cytosolic proteins [8-12]. The 26S proteasome is a highly complex and mechanistically sophisticated proteolytic machine that recognizes, unfolds, translocates and cleaves multi-ubiquitinated proteins into peptides in a sequential “dis-assembly line” manner. It is reasonable that these multiple activities are enzymatically and physically linked to not only improve efficiency of proteolysis but also to prevent the escape of partially deubiquitinated or partially cleaved protein substrates (Fig. 3). This structure is found in both the cytoplasm and nucleus of all eukaryotic cells [9, 10, 16 and references therein]. The 26S proteasome consists of a barrel-shaped proteolytic core complex, known as the 20S proteasome that harbors the proteolytic activities and is capped at one or both ends by 19S regulatory complexes that fulfill multiple functions [11-17].
The 20S proteasome is a multicatalytic protease that exhibits various peptidase activities to function as the catalytic core the 26S
proteasome [21-30]. All peptidase activities for proteolytic cleavage of the protein substrate reside within the 20S structure. In mammalian tissues, the 20S proteasome is comprised of up to 14 different proteins, with each subunit represented twice. The ancestor of the eukaryotic 20S proteasome is the eubacterial and archaebacterial proteasome. The architectural characteristic of 20S proteasome is its composition of four seven-numbered rings, with two outer rings containing α subunits and two central rings composed of β subunits. It is apparent that the α subunits serve a structural function and the β subunits are responsible for the catalytic activity. These are classified as either α subunits or β subunits based on their similarities to the two subunits found in the 20S proteasome in the archaebacterium Thermoplasma acidophilum [21-24]. In the archaebacterial form the α and β subunits form four seven-membered rings that stack on top of each other to form a barrel-shaped structure [21-28]. Interestingly, during this process the number of active site β subunits has been reduced to three yielding six active site β subunits in the eukaryotic proteasome. In parallel, the

Open Journal of Hematology, 2010, 1-4
Page 4 of 20 (Page number not for citation purposes)
rather broad amino acid specificity of the archaeal β subunits has been narrowed down to three more distinct specificities, which are characterized as chymotrypsin-like, trypsin-like and peptidyl-glutamyl-peptide hydrolyzing activity, the latter
cleaving after acidic amino acids [21-24]. Numerous proteasome-interacting subunits have been described in various eukaryotic organisms [29].
Figure 3: Structural Models of the Fully Assembled 26S Proteasome
The 19S regulatory complexes function in the recognition, unfolding and translocation of multi-ubiquitinated proteins [30-36]. It is thought that these regulatory complexes facilitate entry of the unfolded Ub~conjugates into the 20S proteasome to promote degradation and also to prevent erroneous or unselective proteolysis [29-36]. Assembly of the 26S proteasome complex and the attachment of the 19S particles is chaperone-mediated and an elaborate multi-step process (Figure 3). The 19S regulatory particle of yeast, which is the most thoroughly studied version is composed of at least 17 different subunits. The 19S complex itself can be split into two different subcomplexes that are known as the base and the lid. Both subcomplexes are linked to each other through subunit Rpn10 (known as S5a in mammals) (Rpn, Regulatory Particle Non-ATPase) (10 and references therein). The base is composed
of a ring of six different ATPase subunits of the AAA-type (Rpt1 to Rpt6; Rpt, Regulatory Particle Triple “A” protein), which dock onto the α rings on both ends of the 20S core and two additional non-ATPase subunits, Rpn1 and Rpn2. The lid is composed of eight different subunits, Rpn3 and Rpn5 to Rpn11. Rpt5/S6a, is able to bind to multi-Ub chains, implying that this subunit forms part of the mechanism by which ubiquitinylated substrates are recognized by the 26S proteasome [37]. Upon binding to the α ring of the 20S complex, the ATPase ring appears to form a narrow pore to allow the substrate to then enter the catalytic core. The ATPases may function as an anti-chaperone to unfold the protein to allow entry into the catalytic complex [38-40]. The other four subunits of the base subcomplex are all non-ATPases. One of these non-ATPases is Rpn10/Pus1/S5a that contains a Ub-interacting

Open Journal of Hematology, 2010, 1-4
Page 5 of 20 (Page number not for citation purposes)
motif which binds to Ub chains [38,39]. Also, the non-ATPase subunit Uch2/UCH37 subunit is homologous to Ub hydrolases [34,35]. The other two non-ATPase subunits, Rpn1/Mts4/S2 and Rpn2/S1 may play a structural role and link the ATPase ring of the base sub-complex and the lid sub-complex [36-38]. The lid sub-complex consists of eight non-ATPase subunits. Rpn11/Pad1/S13 has been shown to have a novel metalloprotease domain and may play a role in Ub recycling by cleaving the Ub chain from the protein substrate [39]. The function of the other subunits is under investigation but importantly, the lid complex shows a remarkable conservation in its overall structure/subunit composition with two other protein complexes; the COP9/Signalosome and the eIF3 complex [41,42].
Selectivity in the Ub-pathway is mediated by E3 ubiquitin ligases that select proteins for degradation. E3 ligases function in concert with E2 Ub-conjugating (UBC) enzymes to elicit Ub
attachment to a lysine on the target protein through isopeptide bond formation. The E3 Ub ligase targets specific protein substrates for degradation by the proteasome. In general, the E3 Ub ligase is involved in polyubiquitination. Upon covalent linkage of a first Ub moiety to the substrate, a second is attached to the first, a third is attached to the second, and so forth to mark the substrate for proteasomal degradation. However, there are some ubiquitination events that are limited to mono-ubiquitination, in which only a single Ub is added by the Ub ligase to a substrate molecule. Mono-ubiquitinated proteins are generally not targeted to the proteasome for degradation, but may instead be altered in their subcellular localization, function or binding partners. Further complicating matters, different lysines on Ub can be targeted by an E3 to make conventional (K-48) or alternative, e.g., K-11, K23, chains.
Table I. Selected Proteasome Inhibitors as Therapeutic Agents in Oncology Inhibitor Compound Class Mechanism Status Naturally-Occurring Lactacystin β -lactone prodrug binds the β5 subunit pre-clinical
Aclacinomycin aklavinone inhibits the ChTL activity pre-clinical Eponemycin α'-β'-epoxyketone binds covalently the β5, β5i
and β1i catalytic subunits of the 20S proteasome and selectively inhibits the 3 major proteasome proteolytic activities at different rates
pre-clinical
Epoxomycin α'-β'-epoxyketone binds covalently to the β5i, β5, β2i and β2 catalytic subunits and inhibits the ChTL activity
pre-clinical
PR-39 cathelicidin highly basic arginine/ proline-rich peptide that binds α-7 subunit
pre-clinical
Synthetic Bortezomib boronyl dipeptide inhibits the ChTL activity FDA-approved for MM (1st line, Rel,
Ref) FDA-approved for Mantle Cell Lymphoma Early Phase- Refractory DLBCL Phase II-Waldenstrom’s Macroglobulinemia Phase II- Amyloidosis
Carfilzomib epoxyketone inhibits the ChTL activity Phase II Rel/ Ref MM Phase I Lymphomas Phase II Rel Solid Tumors

Open Journal of Hematology, 2010, 1-4
Page 6 of 20 (Page number not for citation purposes)
Salinosporamide (NPI-0052)
bicyclic β-lactone γ-lactam
inhibits the ChTL activity Phase I Rel/ Ref MM Phase I Adv Solid Tumors/ Rel/Ref Lymphomas
CEP-18770 boronyl peptide inhibits the ChTL activity Phase I MM MLN-9708 boronyl peptide inhibits the ChTL activity Pre-Clinical Hematologic and Solid
tumors Phase I Advanced Solid Tumors
TARGETING THE UB-PROTEASOME PATHWAY
Structurally, bortezomib is a modified dipeptidyl boronic acid derived from leucine and phenylalanine and is the first anti-neoplastic proteasome inhibitor. Bortezomib inhibits specifically the chymotrypsin-like activity, but this inhibition is sufficient to block all proteasomal catalytic activity [43, 44]. Proteasome inhibition induces apoptosis of cancer cells that are apparently more susceptible while simultaneously exerting a nontoxic effect on most normal cells. Disruption of the usually tightly regulated Ub+Proteasome system leads to the accumulation of high molecular weight Ub~protein conjugates that trigger apoptosis [45, 46]. Targeted protein degradation is a key mechanism for regulating numerous essential cellular signaling pathways that regulate proliferation, survival, and death. Furthermore, many proteasome substrates are found to be deregulated in tumor cells, including p53, cyclins, p21, p27, BRCA, IκB and NFκB. This makes modulation of proteasome activity an attractive strategy for cancer therapy and proteasome inhibitors have demonstrated antitumor activity alone or In combination with other therapies [47]. Numerous proteasome inhibitors, both naturally-occurring and synthesized are currently in pre-clinical development or early phase clinical trials (Table I).
MECHANISM OF PROTEASOME INHIBITOR INDUCED CELL DEATH
Bortezomib has shown cytotoxic activity against a variety of MM cell lines and in plasma cells from MM patients. A multitude of mechanisms have been proposed for the observed effect of bortezomib in MM cells. These putative mechanisms include promotion of apoptosis, inhibition of activation of nuclear factor-κB (NF-κB) in either or both the myeloma cells or the tumor microenvironment, reduction of adherence of
myeloma cells to the bone marrow stroma, blocked production and reduced intracellular signaling of interleukin-6 (IL-6), blocked production and efficacy of angiogenic factors, defective regulation of effectors of apoptosis such as B-cell lymphoma-2 (Bcl-2), overexpression, accumulation and functional alteration in tumor suppressor protein p53 levels and apoptotic capacity, accumulation of unfolded or multi-ubiquitinated immunoglobulin chains, the global accumulation of unfolded or misfolded proteins, the global accumulation of Ub~protein conjugates, the loss of free Ub and, finally, the induction of endoplasmic reticulum (ER) stress. It is noteworthy that bortezomib is effective in other non-MM cell lines that include lymphomas, leukemias and amyloidosis which do not synthesize abundant amounts of immunoglobulins. Furthermore, bortezomib is generally ineffective against most solid tumors and solid tumor cell lines although the NF-κB pathway is dysregulated in these same solid tumors. One attractive hypothesis is that proteasome inhibition prevents the clearance of misfolded proteins by the ER–associated degradation (ERAD) pathway, resulting in enhanced ER stress that leads to cell death.
The transcription factor NF-κB is a tightly regulated positive mediator of T- and B-cell development, proliferation, and survival and the controlled activity of NF-κB is required for the coordination of physiologic immune responses. NF-κB controls the expression of many genes that regulate apoptosis, cell proliferation and differentiation. Deregulation of NF-κB activation is a hallmark of several lymphoid malignancies and is directly linked to advanced disease. Constitutive NF-κB activation can promote continuous lymphocyte proliferation and survival and has recently been recognized as a critical factor in pathogenesis. Various molecular events lead to deregulation of NF-κB signaling in MM, Hodgkin’s disease and various T- and B-cell non-Hodgkin
Open Journal of Hematology, 2010, 1-4
Page 7 of 20 (Page number not for citation purposes)
lymphomas. The pivotal role of the NF-κB pathway in the inhibition of apoptosis, tumor promotion and progression, and the observation that NF-κB is constitutively activated in a large number of hematologic malignancies suggests that NF-κB inhibitors would be useful in cancer therapy. Proteasome inhibitors such as bortezomib may be
useful to block activation of the NF-κB pathway. However, it should be noted that NF-κB functions in activation of the innate and adaptive immune responses and that inhibitors of this pathway may also enhance the chemotherapy-induced apoptosis of normal hematopoietic progenitors.
Table II. Selected Bortezomib Regimens in Hematologic Malignancies
Hematologic Malignancy
Year Patient Population
Patients Regimen Response
Multiple Myeloma - SUMMIT 2003 Relapsed/
refractory 202 Bortezomib 1.3 mg/m2 on
D1, 4, 8, & 11 - 3 week cycle - Up to 8 cycles (24 weeks) - PO Dex (20 mg day of/after) for suboptimal response
35 % ORR - 27% CR/PR - 7 % MR
- CREST 2004 Relapsed/ refractory
54 Bortezomib 1-1.3 mg/m2 on D1, 4, 8, & 11 - 3 week cycle - Up to 8 cycles (24 weeks) - PO Dex (20 mg day of/after) for suboptimal response
37% CR/PR - 1 mg Bortezomib 50 % CR/PR - 1.3mg Bortezomib
- VISTA 2008 Initial Treatment
682 MP +/- Bortezomib 1.3mg/m2 - 9 6-week cycles - MP given on D1-4 - Bor - D1, 4, 8, 11, 22, 25, 29, & 32 – C1-4 - Bor - D1, 8, 22, & 29 – C5-9
>PR - 71% v 35% CR - 30% v 4%
Mantle Cell Lymphoma - PINNACLE 2006 Relapsed/
refractory 155 Bortezomib 1.3 mg/m2 on
D1, 4, 8, & 11 - 3 week cycle - Up to 1 yr
33-40% ORR - 8 % CR+CRu
- Update 2009 Relapsed/ refractory
155 (55 pts-still f/u)
Bortezomib 1.3 mg/m2 on D1, 4, 8, & 11 - 3 week cycle - Up to 17 cycles/4 cycles beyond CR
32% ORR - 8 % CR+Cru
2009 DLBCL Relapsed/ refractory
44 DA-EPOCH +Bortezomib 0.7-1.7 mg/m2 on D1 & 4 - 3-week cycles - Up to 6 cycles
42% ORR - 23% CR 83% v 13% ORR - ABC v. GCB
PTCL 2007 Relapsed/ refractory
12 Bortezomib 1.3 mg/m2 on D1, 4, 8, & 11 - 3 week cycle - Up to 6 cycles
67% ORR - 17% CR - 50% PR
Waldenstrom’s Macroglobulinemia
- Chen et al [84] 2007 Untreated/ 27 Bortezomib 1.3 mg/m2 on 26% ORR
Open Journal of Hematology, 2010, 1-4
Page 8 of 20 (Page number not for citation purposes)
Relapsed D1, 4, 8, & 11 - 3 week cycle - 2 cycles past CR/SD seen/PD/Tox
- No CR/all PR
- Treon et al [85] 2007 Relapsed/ refractory
27 Bortezomib 1.3 mg/m2 on D1, 4, 8, & 11 - 3 week cycle - Up to 8 cycles
85% ORR - No CRs - 48% major response - 37% minor response
- Treon et al [97] 2009 Untreated 23 Bortezomib 1.3mg/m2 & Dex 40mg on D1, 4, 8, & 11 + Rituximab 375mg/m2 on D11 - 4 3-week cycles - Followed by 4 additional cycles 12 weeks apart
96% ORR - 13% CR / 9% near CR - 13% VGPR / 48% PR - 13% MR
Primary Systemic (AL) Amyloidosis
- Kastritis et al 2007 Untreated/ Relapsed
18 Bortezomib 1.3mg/m2 on D1, 4, 8, & 11 + Dex 40mg on D1-4 - 3 week cycle - Up to 6 cycles
94% HR / 44% CHR
- Wechalekar et al [90] 2008 Relapsed/ refractory
20 Bortezomib 1.3mg/m2 on D1, 4, 8, & 11 +/- Dex - 3 week cycle
15% CHR / 65% PR
PROTEASOME INHIBITORS IN MULTIPLE MYELOMA Multiple myeloma (MM) is a B-cell disorder that produces malignant plasma cells and represents the second most common hematological malignancy [48-50]. The incidence and prevalence of MM are similar in the US and Europe with a total of ~40,000 cases diagnosed annually in both parts of the world. The course of MM is characterized by an asymptomatic or subclinical phase before diagnosis, a chronic phase lasting several years and an aggressive terminal phase. MM leads to progressive morbidity and eventual mortality by lowering resistance to infection and causing significant skeletal destruction with bone pain, pathological fractures, and hypercalcemia), anemia, renal failure, neurological complications and hyperviscosity.
The outcome of MM patients treated with conventional chemotherapy with or without high-dose therapy/autologous SCT has not been satisfactory, with median survivals ranging from 2 to 3 years for older patients and from 5 to 6 years for younger patients. Front-line approval of bortezomib was based upon the phase III VISTA
(VELCADE as Initial Standard Therapy in MM: Assessment with Melphalan and Prednisone) trial [51-55] (Table II). VISTA was a randomized, international, open-label trial compared VELCADE in combination with the current standard of care versus the standard of care alone in 682 previously untreated patients who were unsuitable for stem cell transplantation. VISTA demonstrated that patients receiving the standard of care plus VELCADE achieved a greater overall survival (OS) rate at 24 months (83%) compared to those who only received standard of care alone (70%). Furthermore, the CR rate in the VELCADE combination arm was 30% vs. 4% in the standard of care arm alone.
Bortezomib was initially approved as a third-line treatment of relapsed and refractory MM by the FDA under the accelerated approval program based upon results of the SUMMIT trial [53]. The SUMMIT trial studied 202 patients who had received at least two prior therapies and showed disease progression. Clinical remissions by SWOG criteria were observed in 17.6% of patients (95CI: 12%, 24%) and response lasted a median time of 1 year. In addition, the CREST trial in 54 subjects with relapsed MM showed similar

Open Journal of Hematology, 2010, 1-4
Page 9 of 20 (Page number not for citation purposes)
positive and promising responses [54-57].
In recent years, the availability of the novel effective immunomodulatory drugs (IMID’s), e.g., thalidomide and lenalidomide, in combination with bortezomib has resulted in a significant improvement in the long-term outcome. Moreover, novel drugs targeting new molecular pathways associated with Myelomagenesis and newly identified targets such as new proteaosme inhibitors, the aggresome and histone deacetylases (HDACs) that act through many different mechanisms of action have been developed and are currently being investigated in clinical trials to further improve the outcome of patients with MM [58-60] The treatment of MM patients with relapsed or refractory disease is even more challenging. Regimens containing IMID’s, particularly bortezomib in combination with IMID’s can be effective “rescue” regimens for patients refractory to primary treatment and were first approved for patients with relapsed or refractory MM. However, the duration of response is limited and all patients will eventually develop progressive disease. Among the next generation of novel drugs, the most promising are the IMID pomalidomide and the proteasome inhibitors carfilzomib (an epoxyketone), NPI-0052 (a bicyclic β-lactone γ-lactam) and a second-generation boron-based proteasome inhibitor MLN-9708. Furthermore, the HDAC inhibitors, particularly vorinostat (SAHA) and panobinostat (LBH 589) are gaining prominence in combination with bortezomib for relapsed/ refractory MM. Importantly, most of the new drugs that function as HDAC inhibitors have shown limited efficacy when used as monotherapy. A promising approach is the addition of a new agent, e.g., HDAC inhibitor as a third drug to a well established regimen such as bortezomib + dexamethasone to obtain an additive effect. The results the addition of panobinostat to either bortezomib + dexamethasone in myeloma cell lines, ex-vivo in freshly obtained myeloma cells and in vivo mouse models have been promising.
Panobinostat (PAN; LBH589) is a potent pan-deacetylase inhibitor (pan-DACi) proposed to also disrupt aggresome and HSP90 function and thus to promote cytotoxic misfolded protein aggregate formation and eventually cell death. Combination with bortezomib has shown synergistic cytotoxicity in preclinical studies.
Hence this combination is being evaluated in the PANORAMA program (PANobinostat ORAl in Multiple myelomA) Phase III (PANORAMA 1) and Phase II (PANORAMA 2) studies. PANORAMA 2 is a U.S.-based, multicenter, single-arm study to evaluate the efficacy of PAN + bortezomib+ DEX in pts with relapsed or bortezomib-refractory MM. In a phase Ib MM study, combination of oral PAN with bortezomib displayed a predictable and manageable safety profile with limited neurotoxicity and clinical efficacy seen with CR + PR + MR in 68% of pts, including 8 of 13 (62%) bortezomib-refractory pts. The combination of oral PAN and bortezomib has a predictable and manageable safety profile with promising activity in pts with relapsed, refractory MM, including MM refractory to prior bortezomib-based therapy.
BORTEZOMIB USE IN MANTLE CELL LYMPHOMA
Mantle cell lymphoma (MCL) is an aggressive lymphoma that is refractory to most current chemotherapy regimens has a poor outcome, especially when first-line treatment has failed [61, 62]. Several clinical studies have demonstrated that bortezomib has clinical effects on MCL and it appears that new therapeutic strategies such as radioactively labeled antibodies or molecular targeting agents, e.g. bortezomib, are warranted to further improve the clinical outcome of MCL. First line treatment of mantle cell lymphoma includes rituximab-based combination chemotherapy, e.g., hyperCVAD, CHOP, EPOCH [63, 64]. The PINNACLE trial, evaluated the effect of bortezomib in MCL patients that had failed at least one prior treatment. Results demonstrated a substantial response rate of 31% and disease progression was delayed by more than 6 months in >50% of MCL patients [65-68]. The PINNACLE study demonstrated a median time to progression (TTP) of 6.7 months, median time to next therapy (TTNT) of 7.4 months and median overall survival (OS) of 23.5 months. In responding patients, median TTP was 12.4 months, median duration of response (DOR) was 9.2 months, median TTNT was 14.3 months, and median OS was 35.4 months. The one-year survival rate was 69% overall and 91% in responding patients. Importantly, activity was seen in patients with refractory disease and toxicity was generally

Open Journal of Hematology, 2010, 1-4
Page 10 of 20 (Page number not for citation purposes)
manageable. Overall, the study indicated that bortezomib was associated with lengthy responses and notable survival in patients with relapsed or refractory MCL, with considerable TTP and TTNT in responding patients, to suggest a substantial clinical benefit and validated the use of bortezomib-based therapy in the treatment of relapsed and/or refractory MCL. Furthermore, the PINNACLE study was performed in a multicenter international setting and supports the accelerated addition of bortezomib as a treatment for relapsed MCL. Bortezomib was granted approval for treatment of relapsed or refractory MCL in 2006 represented an important step in second-line treatments.
BORTEZOMIB IN DIFFUSE LARGE B CELL LYMPHOMAS
Diffuse large-B-cell lymphoma (DLBCL) is an aggressive form of non-Hodgkin lymphoma (NHL) that accounts for 30% to 40% of the total incidence of NHL [69, 70]. Patients with DLBCL have been divided into 3 groups according to their gene profiling patterns: germinal-center B-cell–like DLBCL (GCB-DLBCL), activated B-cell–like DLBCL (ABC-DLBCL), and mediastinal or unclassified type 1 [71-73]. These subcategories are characterized by distinct differences in survival, responsiveness to various chemotherapies and dependence on signaling pathways, particularly the nuclear factor-kB (NF-κB) pathway. Aside from those patients eligible for allogeneic or autologous SCT, combination chemotherapy offers a potentially curative option for a subset of DLBCL patients. However, responses to cytotoxic chemotherapy, e.g, rituximab with cytoxan, hydroxyrubicin, oncovin, and prednisone, vary considerably depending on multiple factors, including disease stage and genetic profile, among others. In particular, patients with the ABC-DLBCL subtype, which is NF-κB–dependent, appear to have a significantly worse prognosis than the other subtypes. Collectively, these considerations have prompted the search for more effective treatment strategies in DLBCL.
In recent years considerable progress has been made in the treatment of patients with B-cell NHL [74-76]. Although responses can be achieved with combination chemotherapy regimens, a substantial proportion of patients are still not
cured. In recent years, the knowledge of the cellular and molecular biology of distinct types of B-cell NHL have led to the development of a new class of drugs that specifically targets unique disease-specific pathways. ABC DLBCL has a worse survival after upfront chemotherapy and is characterized by constitutive activation of the antiapoptotic nuclear factor–kappa B (NF-κB) pathway [78, 79]. Results suggest that bortezomib enhanced the activity of chemotherapy in ABC but not the GCB forms of DLBCL and provided a rational therapeutic approach based on the genetically distinct DLBCL subtypes [78].
A recent study demonstrated that the second-generation irreversible proteasome inhibitor carfilzomib was dramatically potentiated in combination with histone deacetylase inhibitors (HDACi’s) in DLBCL cells. Efficacy was seen in both the ABC- and GCB subtypes of DLBCL [80]. Although the proteasome inhibitor bortezomib has shown very limited single-agent activity in DLBCL, results of a recent study suggest that addition of bortezomib to standard chemotherapy improves outcomes in ABC-DLBCL. In addition, it has been proposed that synergistic interactions between HDAC and proteasome inhibitors in malignant hematopoietic cells reflect inhibition of HDACI-mediated NF-κB activation a phenomenon that may stem from RelA acetylation. However, it is noteworthy that HDACIs blocked NF-κB activation and promoted carfilzomib lethality in both ABC- and GC-type DLBCL cells, arguing against the possibility that this interaction is specific only for DLBCL cells addicted to the NF-κB pathway. In addition, there is evidence that proteasome inhibitor lethality may involve factors other than NF-κB inhibition; and in a recent study that used myeloma cells, bortezomib was unexpectedly shown to actually activate NF-κB. The initial rationale for the use of bortezomib in MM was the potential inhibition of NF-κB activity by blocking proteasomal degradation of IκBα. Bortezomib inhibits inducible NF-κB activity; however, its impact on constitutive NF-κB activity in MM cells is a point of discussion [81, 82]. Recent studies demonstrated that bortezomib-induced NF-κB activation was mediated via the canonical pathway. Moreover, other class of proteasome inhibitors also induced IκBα down-regulation associated with NF-κB activation. Bortezomib triggered phosphorylation of IKKβ and

Open Journal of Hematology, 2010, 1-4
Page 11 of 20 (Page number not for citation purposes)
its upstream receptor interacting protein2 (RIP2), whereas the IKKβ inhibitor MLN120B blocked bortezomib-induced I B downregulation and NF-κB activation, indicating a crucial role for RIP2/IKKβ signaling plays crucial role in bortezomib-induced NF-κB activation. Moreover, IKK inhibitors enhanced bortezomib-induced cytotoxicity. Our studies therefore suggest that bortezomib-induced cytotoxicity cannot be fully attributed to inhibition of canonical NF-κB activity in MM cells. Potentially, the high levels of immunoglobulin production and ER-Golgi protein transport would sensitize MM cells to proteotoxic stress, providing an attractive explanation for bortezomib's clinical activity and a potential means of identifying bortezomib-based combination approaches that will display even greater antitumor effects [82]. Consequently, the possibility that HDAC inhibitors potentiate the effect of carfilzomib through mechanisms unrelated to NF-κB cannot be excluded.
PERIPHERAL T-CELL LYMPHOMAS (PTCL) Peripheral T-cell lymphomas (PTCL’s) are a heterogeneous group of generally aggressive cancers that comprise ~15% of all NHL’s. There are multiple forms of aggressive PTCL’ and the current treatment approaches are similar to those used for B-cell lymphomas. These regimens have been used for PTCL patients with either localized or advanced stage disease and with autologous hematopoietic cell transplantation utilized in selected patients as well. Phase II clinical trials of bortezomib have demonstrated promising results in the treatment of patients with B-cell lymphomas [83]. NCCN guidelines now recommend bortezomib as second-line therapy for patients with relapsed or refractory PTCL in those patients that are not candidates for high-dose chemotherapy or autologous stem cell transplant. A multi-center phase II trial of bortezomib combined with a standard chemotherapy regimen in the treatment of previously untreated individuals with PTCL is ongoing.
WALDENSTRÖM’S MACROGLOBULINAEMIA
Waldenström’s macroglobulinemia is a rare type of slow-growing, NHL that results in the
overproduction of a monoclonal immunoglobulin (IgM) antibody by abnormal plasma cells. The malignant plasma cells multiply uncontrollably to produce high levels of IgM in the blood and eventually cause hyperviscosity. Treatment of symptoms that develop in WM patients may include plasmapheresis, chemotherapy and newly emerging biological therapies such as bortezomib. Bortezomib demonstrates potent activity against WM primary cells and cell lines [84, 85]. Interestingly, bortezomib has shown activity as a single agent as salvage therapy in WM patients. Moreover, ORR’s have reached 60-80% and major response rates have been observed in up to 50-60% of cases. A recent clinical study found that bortezomib was in fact an active agent in relapsed and refractory WM [86]. In this study, 27 patients with WM received up to eight cycles of bortezomib and all but one of the patients had relapsed or refractory disease. Following therapy, median serum IgM levels declined from 4.6 to 2.1 g/dL (p<0.0001) and the ORR was 85%. Common grade III/IV toxicities were sensory neuropathies (22.2%), leukopenia (18.5%), neutropenia (14.8%), dizziness (11.1%), and thrombocytopenia (7.4%).
In a phase II clinical study [86] evaluated the effectiveness and toxicity of single-agent bortezomib in WM. Symptomatic patients, untreated or previously treated, received bortezomib 1.3 mg/m2 intravenously days 1, 4, 8, and 11 on a 21-day cycle until two cycles past complete response (CR), stable disease (SD) attained, progression (PD), or unacceptable toxicity. A median of six cycles (range of 2 to 39) of bortezomib was administered. Twenty-one patients had a decrease in IgM of at least 25%, with 12 patients (44%) reaching at least 50% IgM reduction. Using both IgM and bi-dimensional criteria, responses included 7 PRs 26%, 19 SDs 70%, and 1 PD 4%. Total response rate was 26% and reductions in IgM levels were prompt with nodal responses lagging. The slower response in nodal disease may require prolonged or dose-adjusted schedule to allow prolonger treatment and avoid unwanted toxicities.
The recent WMCTG clinical trial 05-180 examined the activity of bortezomib, dexamethasone and rituximab in 23 symptomatic, untreated WM patients [87]. Patients received four cycles of induction with all three agents followed by four more cycles with each given

Open Journal of Hematology, 2010, 1-4
Page 12 of 20 (Page number not for citation purposes)
three months apart. The results demonstrated the bortezomib/ dexamethasone/ rituximab combination produced a durable response with high rates of response and CR. A Phase II trial of weekly bortezomib in combination with rituximab in relapsed/ refractory WM in 37 patients that had received one or more therapies demonstrated that this combination had significant activity and minimal neurological toxicity. It is clear that future studies are warranted and that bortezomib-based therapy holds promise as a leading therapeutic in the treatment of WM.
PRIMARY SYSTEMIC AMYLOIDOSIS
AL amyloidosis is the most common form of systemic amyloidosis in the U.S and is a rare disease that occurs in approximately eight of every 1,000,000 persons. It affects males and females equally and usually develops after the age of 40. At least 15 types of amyloidosis have been identified and each type is associated with deposits of a different protein. It is noteworthy that AL amyloidosis occurs in 5-15% of patients diagnosed with MM. Despite severe difficulties in the diagnosis and treatment of AL amyloidosis, patients with this disease can achieve long-term survival if properly managed. The optimal course of management requires early, accurate diagnosis, correct identification of the amyloid type, institution of prompt, appropriate and effective therapy and close follow-up with careful supportive care. Systemic involvement that affects the heart, kidneys and liver renders patients with worse outcomes and generally confers reduced response to various forms of chemotherapy. Bortezomib as treatment for systemic light chain amyloidosis has been reported in a phase II clinical study [90], a small case series and case reports [89, 91]. NCCN's Drug and Biologics Compendium lists systemic light chain amyloidosis as an indication for bortezomib. However, the NCCN guidelines indicate that this and other treatments for systemic light chain amyloidosis should be provided in the context of a clinical trial. Kastritis and colleagues [91] assessed the activity and feasibility of the combination of bortezomib and dexamethasone (BD) in patients with AL amyloidosis. A total of 18 patients were treated with BD including 7 that had relapsed or progressed after prior therapies. Eleven (61%) patients had 2 or more organs involved and the
kidneys and heart were affected in 14 and 15 patients, respectively. Among the evaluable patients, 94% had a hematological response and 44% had a hematological CR, including all 5 patients who had not responded to prior high dose dexamethasone-based treatment and 1 patient under dialysis. Five patients (28%) had a response in at least 1 affected organ. Hematological responses were rapid (median of 0.9 months) and median time to organ response was 4 months. The authors concluded that the combination of BD is feasible in patients with AL amyloidosis. Patients achieve a rapid hematological response and toxicity can be managed with close follow-up and dose adjustment. This treatment may be a valid option for patients with severe heart or kidney impairment.
Wechalekar et al [92] reported preliminary observations on the effectiveness of bortezomib in 20 patients with primary (AL) amyloidosis whose clonal disease was active despite prior treatment with a median of 3 lines of prior chemotherapy that included a thalidomide combination in all cases. Patients received a median of 3 (range of 1 to 6) cycles of bortezomib and 9 (45%) patients received concurrent dexamethasone. Three (15%) patients achieved complete hematological responses, and a further 13 (65%) achieved partial responses. Fifteen (75%) patients experienced some degree of toxicity, which in 8 (40%) cases resulted in discontinuation of bortezomib. Hence, bortezomib shows promise in the treatment of systemic AL amyloidosis.
It is mechanistically relevant that tumor cells that secrete amyloidogenic monoclonal proteins are similar to malignant plasma cells and demonstrate sensitivity to bortezomib. Since the cytotoxic effect of bortezomib on myeloma cells cannot be completely explained by the inhibition of the NF-κB pathway other mechanisms are definitely plausible. It is noteworthy that both amyloidogenic and plasma cells exhibit enhanced protein synthesis activity and protein secretory function. An attractive hypothesis is the accumulation Ub-conjugated proteins results in enhanced ER stress that then triggers apoptosis. Since normal cells may have less synthetic activity they may be less susceptible to the effects of proteasome inhibition. Hence, protein synthesis,

Open Journal of Hematology, 2010, 1-4
Page 13 of 20 (Page number not for citation purposes)
folding and ER transport may be linked to induction of ER stress by bortezomib-induced accumulation of ubiquitinated proteins. Cells with enhanced secretory activity may be further sensitized to proteasome inhibition. Mature plasma cells are terminally differentiated elements of the B lymphocytic lineage with a highly developed ER specialized in immunoglobulin secretion. ER quality control systems prevent mutated, misfolded or unfolded proteins from proceeding to the Golgi and the accumulation of misfolded proteins in the ER lumen initiates the unfolded protein response (UPR). Upon initiation of the UPR, cells then initiate steps to handle the accumulation of unfolded proteins, Protein translation is attenuated, transcription of genes that fulfill protein folding functions such ER resident chaperones and folding enzymes is altered and the ER-specific protein degradation (ERAD) system is activated. Entry of proteins into the ER and the stability of mRNA encoding secretory proteins is selectively inhibited as well. When these rescue mechanisms are not adequate to eliminate misfolded proteins from the ER, apoptotic pathways are then activated.
PROTEASOME INHIBITORS IN ACUTE MYELOID LEUKEMIA
Acute myeloid leukemia (AML) is an immunophenotypically heterogeneous group of diseases, with CD34+ cases being associated with drug resistance and poor outcome [93, 94]. Nearly 80% of patients with AML achieve a complete remission with induction chemotherapy. However, a high proportion of patients relapse, and eventually die of their disease. This is important, particularly due to the heterogeneity of AML, including a wide array of genetic lesions and immunophenotypic profiles. The CD34 antigen identifies early progenitor cells and, accordingly, AML can be divided into immature and mature forms (CD34+ and CD34–, respectively), the former subset associated with drug resistance and poorer outcome, as compared to the more mature CD34– cases. Moreover, at relapse, blast cells usually display a more immature phenotype, as a reflection of drug resistance and it has been suggested that the presence of an immature phenotype with age and cytogenetics represent important prognostic factors in AML.
A recent study demonstrated that bortezomib has marked in vitro activity in both AML cell lines and fresh blast cells obtained from AML patients [93]. Moreover, the antileukemic effect of bortezomib was seen in CD34+ and CD34– AML cells to suggest that proteasome inhibitors may in fact overcome the drug resistance associated with the immature CD34+ phenotype. Importantly, bortezomib was significantly more active than doxorubicin in the immature CD34+ cells, while there were no differences in its action on CD34– cells. Based upon these studies, a detailed analysis of the in vitro activity and mechanism of action of bortezomib on AML cells was performed [94]. Bortezomib was shown to be anti-proliferative and induced apoptosis in chronic phase (CP) CD34+ CML cells at clinically achievable concentrations. Moreover, bortezomib targeted primitive CML cells, with effects on CD34+38–, long-term culture-initiating (LTC-IC) and nonobese diabetic/severe combined immunodeficient (NOD/SCID) repopulating cells. Bortezomib was not selective for CML cells and induced apoptosis in normal CD34+38– cells and the effects against CML cells were seen when bortezomib was used alone or in combination with dasatinib. Bortezomib caused proteasome but not BCR-ABL inhibition and was effective in inhibiting proteasome activity and inducing apoptosis in cell lines expressing BCR-ABL mutations, including T315I. By targeting both TKI-insensitive stem and progenitor cells and TKI-resistant BCR-ABL mutations, bortezomib may offer a potential therapeutic option in certain leukemias.
CONCLUDING REMARKS The Ub+Proteasome pathway has been validated as a therapeutic target in oncology and revolutionized treatment of certain hematologic malignancies. The small molecule bortezomib was the first proteasome inhibitor to enter clinical use and received FDA approval for the treatment of patients with multiple myeloma, therefore validating inhibition of the proteasome as an anticancer target. Preclinical data also has demonstrated the synergistic effect of bortezomib with other chemotherapeutic agents and the ability to overcome drug resistance of targeted therapy in MM has since led to the development and investigation of more than 30 new

Open Journal of Hematology, 2010, 1-4
Page 14 of 20 (Page number not for citation purposes)
compounds in this disease and in the related plasma cell dyscrasias WM and Amyloidosis. The last decade has marked a new era in the treatment of diseases characterized by B cell disorders and monoclonal gammopathies and a paradigm shift has evolved to employ novel therapeutic agents that target the malignant clone and the surrounding bone marrow microenvironment. The finding that tumor cells are more sensitive to alteration of the Ub+Proteasome pathway and that inhibitors of the proteasome are useful in certain types of cancer therapy suggests that further investigation should provide new therapeutic strategies to attack proliferative disorders. Additionally, the combination of bortezomib with chemotherapeutic agents has yielded high response rates comparable or superior to those achieved in the SCT setting and more applicable to the elderly or frail patient populations. Together, these therapies should lead to higher response rates, more durable responses, less toxicity and prolonged survival for patients, making hematologic malignancies and certain plasma cell dyscrasias increasingly chronic and treatable diseases.
Inhibition of the proteasome peptidase activity prevents the degradation of all proteins normally degraded through the Ub+Proteasome pathway. The demonstrated efficacy of proteasome inhibition on cancer cells was at first surprising, because the proteasome is responsible for the degradation of most intracellular proteins and the initial expectation was that it would be equally toxic to normal cells. Recent evidence that pharmacological inhibition of the Ub+Proteasome pathway can be efficacious in the treatment of human cancers has set the stage for attempts to selectively inhibit the activities of disease-specific components within the pathway. Targeting the proteasome, while proven clinically efficacious, does not allow for this level of specificity. An increase in target specificity could increase the effectiveness of a therapeutic intervention while simultaneously diminishing unwanted side-effects and toxicities. Targeting of other components within the Ub+Proteasome pathway, e.g., E3 ligases, may provide the highest level of specificity and the preclinical successes of the MDM2 inhibitors are encouraging, although no E3 inhibitor has as of yet entered human clinical
trials. These important findings highlight the need for further research into the role of Ub+Proteasome system and protein homeostasis in cancer and other diseases. Since the greatest amount of specificity is present in the E3 Ub conjugation step, it is anticipated that drugs that target individual E3 ligases, possibly by blocking substrate binding, are likely to provide the greatest level of selectivity. However, E3’s are unconventional enzymes and the development of specific inhibitors represents a significant challenge. Specifically, it is likely that E3’s bind target substrates stoichiometrically and therefore do not function catalytically to select substrates for ubiquitination. Moreover, somewhat problematic is that each of these E3 ligases targets a finite number of substrates that are used in many cellular processes. Inhibition of E3 Ub ligase function globally is therefore likely to interrupt many diverse pathways while inhibition of a single E3 ligase could have a minimal effect. Pharmacologic targeting of E3 ligases could, in theory, provide an enhanced therapeutic index greater than targeting the proteasome because specific E3s ubiquitinate a small number of proteins, whereas the proteasome degrades all ubiquitinated proteins. A more detailed understanding of this system as well as the impact of targeting specific steps within this pathway has the potential to result in more disease-specific therapies with fewer off-target effects.
The Ub+Proteasome pathway is a highly complex, tightly regulated system that targets most cellular proteins for degradation. Recently, novel parallel pathways have been described that attach Ub-like molecules, e.g., Small Ubiquitin-like MOdifier (SUMO) and NEDD8 to target proteins [95, 96]. Identifying the underlying components and steps in these pathways is essential for the development of novel, mechanism-based drugs. The Ub+Proteasome, Sumoylation and Neddylation pathways are ideal candidates as targets as cytotoxic drugs that block key oncogenic steps in cancer and result in a proapoptotic cancer cell milieu. The therapeutic success of proteasome inhibitors in the treatment of hematological malignancies validates the proteasome as viable therapeutic target. Future efforts will investigate the therapeutic value of bortezomib in combination with other targeted and chemotherapeutic agents, the efficacy of

Open Journal of Hematology, 2010, 1-4
Page 15 of 20 (Page number not for citation purposes)
second-generation proteasome inhibitors and the identification of other components within the Ub+Proteasome pathway that may serve as novel targets. Finally, targeting components in the Sumoylation and Neddylation pathways will also be pursued to yield more selective therapeutics to develop mechanism-based drugs that modulate the Ub+Proteasome pathway.
LIST OF ABBREVIATIONS MM – multiple myeloma Ub – ubiquitin WM – Waldenström’s macroglobulinemia AML – acute myeloid leukemia SCT – stem cell transplantation CR – complete remission PR – partial remission SD – stable disease PD – progressive disease
ORR – overall response rate NF-κB – nuclear factor kappa-light-chain-enhancer of activated B cells IkB – inhibitor of NF-kappaB kinase FDA – Federal Drug Administration ATP – adenosine triphospahte MDa – megadalton kDa – kilodalton S – Svedberg unit Rpn – regulatory particle non-ATPase Rpt – regulatory particle triple “A” protein
CONFLICT OF INTEREST
The views expressed in this paper are those of the authors and not official views of the Department of Defense.
REFERENCES [1] Hershko A, Ciechanover A, Varshavsky A. Basic
Medical Research Award. The ubiquitin system. Nature Med. 2000; 6: 1073-1081.
[2] Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998; 67: 425-479.
[3] Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994; 79: 13-21.
[4] Ganoth D, Leshinsky E, Eytan E, Hershko A. A multicomponent system that degrades proteins conjugated to ubiquitin. Resolution of factors and evidence for ATP-dependent complex formation. J Biol Chem. 1988; 263: 12412-12419.
[5] Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991; 349: 132-138.
[6] Hoeller D, Dikic I. Targeting the ubiquitin in cancer therapy. Nature. 2009; 458: 438-444.
[7] Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol. 1995; 7: 215-223.
[8] Crews CM. Feeding the Machine: mechanisms of protein catalyzed degradation of ubiquitinated proteins. Curr Opin Chem Biol. 2003; 7: 534-539.
[9] Baumeister W, Walz J, Zühl F, Seemüller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998; 92: 367-380.
[10] Wolf DH, Hilt W. The proteasome: a proteolytic nanomachine of cell regulation and waste disposal. Biochim Biophys Acta. 2004; 1695: 19-31.
[11] Driscoll J, Goldberg AL. The proteasome (multicatalytic protease) is a component of the
1500-kDa proteolytic complex which degrades ubiquitin-conjugated proteins. J Biol Chem. 1990; 265: 4789-4792.
[12] Eytan E, Ganoth D, Armon T, Hershko A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc Natl Acad Sci USA. 1989; 86: 7751- 7755.
[13] Orlowski M, Wilk S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch Biochem Biophys. 2000; 383: 1-16.
[14] Hartmann-Petersen R, Gordon C. Proteins interacting with the 26S proteasome. Cell Mol Life Sci. 2004; 61: 1589-1595.
[15] Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997; 386: 463-71.
[16] Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999; 68: 1015-68.
[17] Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. A gated channel into the proteasome coreparticle. Nat Struct Biol. 2000; 7: 1062-1067.
[18] Pühler G, Weinkauf S, Bachmann L, Müller S, Engel A, Hegerl R, Baumeister W. Subunit stoichiometry and three-dimensional arrangement in proteasomes from Thermoplasma acidophilum. EMBO J. 1992; 11: 1607-16.

Open Journal of Hematology, 2010, 1-4
Page 16 of 20 (Page number not for citation purposes)
[19] Grziwa A, Baumeister W, Dahlmann B, Kopp F. Localization of subunits in proteasomes from Thermoplasma acidophilum by immunoelectron microscopy. FEBS Lett. 1991; 290: 186-190.
[20] Hegerl R, Pfeifer G, Pühler G, Dahlmann B, Baumeister W. The three-dimensional structure of proteasomes from Thermoplasma acidophilum as determined by electron microscopy using random conical tilting. FEBS Lett. 1991; 283: 117-121.
[21] Hilt W, Wolf DH. Stress-induced proteolysis in yeast. Mol Microbiol. 1992; 6: 2437-2442.
[22] Wenzel T, Baumeister W. Conformational constraints in protein degradation by the 20s proteasome. Nat Struct Biol. 1995; 2: 199-204.
[23] Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995; 268: 533-539.
[24] Driscoll J, Goldberg AL. Skeletal Muscle Proteasome can Degrade Proteins in an ATP-dependent Process that dose not Require Ubiquitin. Proc Natl Acad Sci USA. 1989; 88: 787-791.
[25] Seemüller E, Lupas A, Stock D, Löwe J, Huber R, Baumeister W. Proteasome from Thermoplasma acidophilum: a threonine protease. Science. 1995; 268: 579-582.
[26] Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem. 1997; 272: 25200-25209.
[27] Chen P, Hochstrasser M. Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell. 1996; 86: 961-972.
[28] Arendt CS, Hochstrasser M. Identification of the yeast 20S proteasome catalytic centers and subunit interactions required for active-site formation. Proc Natl Acad Sci USA. 1997; 94: 7156-7161.
[29] Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Ploegh H, Finley D. Multiple associated proteins regulate proteasome structure and function. Mol Cell. 2002; 10: 495-507.
[30] Braun BC, Glickman M, Kraft R, Dahlmann B, Kloetzel PM, Finley D, Schmidt M. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat Cell Biol. 1999; 1: 221-226.
[31] Gorbea C, Rechsteiner M. The mammalian regulatory complex of the 26S proteasome, in: Hilt W, Wolf DH. (Eds.), Proteasomes: The World of Regulatory Proteolysis, Landes Bioscience,
Georgetown/ Eurekah.com, Austin, USA, Georgetown/Austin, USA, 2000, pp. 91-128.
[32] Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994; 269: 7059-7061.
[33] Hofmann K, Falquet L. A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem Sci. 2001; 26: 347-350.
[34] Hölzl H, Kapelari B, Kellermann J, Seemüller E, Sümegi M, Udvardy A, Medalia O, Sperling J, Müller SA, Engel A, Baumeister W. The regulatory complex of Drosophila melanogaster 26S proteasomes. Subunit composition and localization of a deubiquitylating enzyme. J Cell Biol. 2000; 150: 119-130.
[35] Li T, Naqvi NI, Yang H, Teo TS. Identification of a 26S proteasome-associated UCH in fission yeast. Biochem Biophys Res Commun. 2000; 272: 270-275.
[36] Glickman MH, Rubin DM, Larsen CN, Schmidt M, Finley D, The regulatory particle of the yeast proteasome, in: Hilt W, Wolf DH. (Eds.), Proteasomes: The World of Regulatory Proteolysis, Landes Bioscience/Eurekah.com, Georgetown/Austin, USA, 2000, pp. 71-90.
[37] Lam YA, Lawson TG, Velayutham M, Zweier JL, Pickart CM. A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature. 2002; 416: 763-767.
[38] Wilkinson CR, Wallace M, Seeger M, Dubiel W, Gordon C. Mts4, a non-ATPase subunit of the 26 S protease in fission yeast is essential for mitosis and interacts directly with the ATPase subunit Mts2. J Biol Chem. 1997; 272: 25768-25777.
[39] Verma R, Aravind L, Oania R, McDonald WH, Yates JR 3rd, Koonin EV, Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002; 298: 611-615.
[40] Wilkinson CR, Ferrell K, Penney M, Wallace M, Dubiel W, Gordon C. Analysis of a gene encoding Rpn10 of the fission yeast proteasome reveals that the polyubiquitinbinding site of this subunit is essential when Rpn12/Mts3 activity is compromised. J Biol Chem. 2000; 275: 15182-15192.
[41] Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998; 94: 615-623.
[42] Kapelari B, Bech-Otschir D, Hegerl R, Schade R, Dumdey R, Dubiel W. Electron microscopy and subunit-subunit interaction studies reveal a first

Open Journal of Hematology, 2010, 1-4
Page 17 of 20 (Page number not for citation purposes)
architecture of COP9 signalosome. J Mol Biol. 2000; 300: 1169-1178.
[43] Elliott PJ, Ross JS. The proteasome: A new target for novel drug therapies. Am J Clin Pathol. 2001; 116: 637-646.
[44] Adams J. Proteasome inhibition in cancer: Development of PS-341. Semin Oncol. 2001; 28: 613-619.
[45] Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001; 61: 3071-3076.
[46] Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Treon SP, Munshi NC, Richardson PG, Hideshima T, Anderson KC. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci USA. 2002; 99: 14374-14379.
[47] Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009; 458: 438-44.
[48] International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003; 121: 749-57.
[49] Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment and survival. Cancer. 1975; 36: 842-854.
[50] Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Multiple Myeloma. Lancet. 2009; 374: 324-39.
[51] Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003; 8: 508-515.
[52] Mateos MV, Richardson PG, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Esseltine DL, Liu K, Cakana A, van de Velde H, San Miguel JF. Bortezomib Plus Melphalan and Prednisone Compared With Melphalan and Prednisone in Previously Untreated Multiple Myeloma: Updated Follow-Up and Impact of Subsequent Therapy in the Phase III VISTA Trial. J Clin Oncol. 2010; 28: 2259-66.
[53] Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein
DP, Anderson KC. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003; 348: 2609-2617.
[54] Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Bladé J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC; Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005; 352: 2487-2498.
[55] San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Mateos MV, Anderson KC, Esseltine DL, Liu K, Cakana A, van de Velde H, Richardson PG; VISTA Trial Investigators. Corrado C, Fantl D, Garcia J, Klein G, Riveros D, Hermann R, Hertzberg M, Horvath N, Marlton P, Spencer A, Drach J, Gisslinger H, Greil R, Gunsilius E, Linkisch W, Petzer A, Thaler J, Andre M, Beguin Y, Bron D, Demuynck H, De Prijck B, Delforge M, Doyen C, Ferrant A, Feremans W, Janssens A, Van de Velde A, Van Droogenbroeck J, Vermeulen P, Zachee P, Bahlis N, Belch A, Dolan S, Fox S, Lavoie A, Voralia M, Ai H, Hou J, Meng F, Shen Z, Zhao Y, Hajek R, Maisnar V, Koivunen E, Remes K, Sikiö A, Vanhatalo S, Attal M, Harousseau J, Hulin C, Michallet M, Salles G, Durk H, Engelhardt M, Gabor C, Goldschmidt H, Haenel M, Hess G, Knauf W, Schlag M, Welslau M, Zervas K, Zoumbos N, Borbényi Z, Fekete S, Illes A, Masszi T, Radvanyi G, Tarkovacs G, Ben-Yehuda D, Berrebi A, Nagler A, Rowe J, Boccadoro M, Cavo M, De Fabritiis P, Foa R, Lazzarino M, Liberati A, Morra E, Musto P, Pinto A, Kim K, Min C, Min Y, Suh C, Yoon S, Gibbons S, Hellmann A, Holowiecki J, Kloczko J, Kuliczkowski K, Komarnicki M, Robak T, Sulek K, Alexeeva J, Biakhov M, Domnikova N, Dunaev Y, Gaisarova G, Golenkov A, Loginov A, Patrin V, Pavlov V, Rossiev V, Rukavitsyn O, Savchenko V, Suvorov A, Yablokova V, Alegre A, Bladé J, De la Rubia J, Diaz-Mediavilla J, Hernandez J, Palomera L, Sureda A, Björkstrand B, Gruber A, Chao T, Huang S, Kuan-Der Lee C, Kuo C, Shih L, Cavet J, Kazmi M, Littlewood T, O'Dwyer M, Rule S, Russell N, Berdeja J, Bruetman D, Butler F, Callander N, Glass J, Gurtler J, Hanna W, Holladay C, Irwin D, Lewis R, Morrison V, Noga S, Roberts T, Schiller G. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008; 359: 906-917.

Open Journal of Hematology, 2010, 1-4
Page 18 of 20 (Page number not for citation purposes)
[56] Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Bladé J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC; Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005; 352: 2487-2498.
[57] Jagannath S, Richardson PG, Barlogie B, Berenson JR, Singhal S, Irwin D, Srkalovic G, Schenkein DP, Esseltine DL, Anderson KC; SUMMIT/CREST Investigators. Bortezomib in combination with dexamethasone for the treatment of patients with relapsed and/or refractory multiple myeloma with less than optimal response to bortezomib alone. Haematologica. 2006; 91: 929-934.
[58] Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, Berkers C, Nicholson B, Chao TH, Neuteboom ST, Richardson P, Palladino MA, Anderson KC. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005; 8: 407-419.
[59] Chauhan D, Singh A, Brahmandam M, Podar K, Hideshima T, Richardson P, Munshi N, Palladino MA, Anderson KC. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008; 111: 1654-1664.
[60] Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the Proteasome Inhibitor MLN9708 in Preclinical Models of Human Cancer. Can Res. 2010; 70: 1970-80.
[61] Lenz G, Dreyling M, Hiddemann W. Mantle cell lymphoma: Established therapeutic options and future directions. Ann Hematol. 2004; 83: 71-77.
[62] Schenkein D. Proteasome inhibitors in the treatment of B-cell malignancies. Clin Lymphoma. 2002; 3: 49-55.
[63] Richardson P. Clinical update: Proteasome inhibitors in hematologic malignancies. Cancer Treat Rev. 2003; 29: 33-39.
[64] O'Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, Straus D, Portlock C, Hamlin P, Choi E, Dumetrescu O, Esseltine D, Trehu E, Adams J, Schenkein D, Zelenetz AD. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin's lymphoma
and mantle cell lymphoma. J Clin Oncol. 2005; 23: 676-684.
[65] Goy A, Bernstein SH, Kahl BS, Djulbegovic B, M. J. Robertson MJ, Boral A, Shi H, Fisher RI. Bortezomib in relapsed or refractory mantle cell lymphoma (MCL): Results of the PINNACLE study. Journal of Clinical Oncology, 2006 ASCO Annual Meeting Proceedings Part 1. Vol 24, No. 18S (June 20 Supplement), 2006: 7512.
[66] Goy A, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Nasta S, O'Connor OA, Shi H, Boral AL, Fisher RI. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Ann Oncol. 2009; 20: 520-5.
[67] O’Connor OA. The emerging role of bortezomib in the treatment of indolent non-Hodgkin's and mantle cell lymphomas. Curr Treat Options Oncol. 2004; 5: 269-281.
[68] Bertoni F, Zucca E, Cotter FE. Molecular basis of mantle cell lymphoma. Br J Haematol. 2004; 124: 130-140.
[69] Armitage J, Coiffier B, Dalla-Favera R, Harris N, ed. Lippincott Williams & Wilkins 2004: 427-501.
[70] Gaynor ER, Fisher RI. Aggressive non-Hodgkin's lymphoma. In: Armitage JO, ed. Malignant Lymphoma. London: Arnold, 2000: 288-307.
[71] Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000; 403: 503-11.
[72] Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, Hurt EM, Zhao H, Averett L, Yang L, Wilson WH, Jaffe ES, Simon R, Klausner RD, Powell J, Duffey PL, Longo DL, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Montserrat E, López-Guillermo A, Grogan TM, Miller TP, LeBlanc M, Ott G, Kvaloy S, Delabie J, Holte H, Krajci P, Stokke T, Staudt LM; Lymphoma/Leukemia Molecular Profiling Project. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002; 346: 1937-47.
[73] Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, Fisher RI, Braziel RM, Rimsza LM, Grogan TM, Miller TP, LeBlanc M,

Open Journal of Hematology, 2010, 1-4
Page 19 of 20 (Page number not for citation purposes)
Greiner TC, Weisenburger DD, Lynch JC, Vose J, Armitage JO, Smeland EB, Kvaloy S, Holte H, Delabie J, Connors JM, Lansdorp PM, Ouyang Q, Lister TA, Davies AJ, Norton AJ, Muller-Hermelink HK, Ott G, Campo E, Montserrat E, Wilson WH, Jaffe ES, Simon R, Yang L, Powell J, Zhao H, Goldschmidt N, Chiorazzi M, Staudt LM. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004; 351: 2159-2169.
[74] Bea S, Zettl A, Wright G, Salaverria I, Jehn P, Moreno V, Burek C, Ott G, Puig X, Yang L, Lopez-Guillermo A, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Gascoyne RD, Connors JM, Grogan TM, Braziel R, Fisher RI, Smeland EB, Kvaloy S, Holte H, Delabie J, Simon R, Powell J, Wilson WH, Jaffe ES, Montserrat E, Muller-Hermelink HK, Staudt LM, Campo E, Rosenwald A; Lymphoma/Leukemia Molecular Profiling Project. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood. 2005; 106: 3183-3190.
[75] Goy A, Younes A, McLaughlin P, Pro B, Romaguera JE, Hagemeister F, Fayad L, Dang NH, Samaniego F, Wang M, Broglio K, Samuels B, Gilles F, Sarris AH, Hart S, Trehu E, Schenkein D, Cabanillas F, Rodriguez AM. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin's lymphoma. J Clin Oncol. 2005; 23: 667-675.
[76] Lenz G, Staudt LM. Aggressive Lymphomas. N Engl J Med. 2010; 362: 1417-1429.
[77] Dunleavy K, Wilson WH. Role of Molecular Subtype in Predicting Outcome of AIDS-Related Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2010; 28: e260-e260.
[78] Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, Grant N, Shovlin M, Jaffe ES, Janik JE, Staudt LM, Wilson WH. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood. 2009; 113: 6069-6076.
[79] Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001; 194: 1861-1874.
[80] Dasmahapatra G, Lembersky D, Kramer L, Fisher RI, Friedberg J, Dent P, Grant S. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood. 2010; 115: 4478-4487.
[81] Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, Mitsiades C, Munshi NC, Richardson PG, Carrasco RD, Anderson KC. Bortezomib induces canonical nuclear factor- B activation in multiple myeloma cells. Blood. 2009; 114: 1046-1052.
[82] McConkey DJ. Bortezomib paradigm shift in myeloma. Blood. 2009; 114: 931-932.
[83] Zinzani PL, Musuraca G, Tani M, Stefoni V, Marchi E, Fina M, Pellegrini C, Alinari L, Derenzini E, de Vivo A, Sabattini E, Pileri S, Baccarani M. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007; 25: 4293-4297.
[84] Vijay A, Gertz MA. Waldenström macroglobulinemia. Blood. 2007; 109: 5096-5103.
[85] Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, Castritis E, Bitsaktsis A, Pangalis GA. Treatment of relapsed or refractory Waldenstrom's macroglobulinemia with bortezomib. Haematologica. 2005; 90: 1655-1658.
[86] Chen CI, Kouroukis CT, White D, Voralia M, Stadtmauer E, Stewart AK, Wright JJ, Powers J, Walsh W, Eisenhauer E; National Cancer Institute of Canada Clinical Trials Group. National Cancer Institute of Canada Clinical Trials Group. Bortezomib is active in patients with untreated or relapsed Waldenstrom's macroglobulinemia: A phase II study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007; 25: 1570-1575.
[87] Treon SP, Hunter ZR, Matous J, Joyce RM, Mannion B, Advani R, Cook D, Songer J, Hill J, Kaden BR, Sharon D, Steiss R, Leleu X, Branagan AR, Badros A. Multicenter clinical trial of bortezomib in relapsed/refractory Waldenstrom's macroglobulinemia: Results of WMCTG Trial 03-248. Clin Cancer Res. 2007; 13: 3320-3325.
[88] Sitia R, Palladini G, Merlini G. Bortezomib in the treatment of AL amyloidosis: targeted therapy? Haematologica. 2007; 92: 1302-1305.
[89] Comenzo RL. Managing systemic light-chain amyloidosis. J Natl Compr Canc Netw. 2007; 5: 179-187.
[90] Kastritis E, Anagnostopoulos A, Roussou M, Toumanidis S, Pamboukas C, Migkou M, Tassidou A, Xilouri I, Delibasi S, Psimenou E, Mellou S, Terpos E, Nanas J, Dimopoulos MA. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007; 92: 1351-1358.
[91] Borde JP, Link R, Offensperger WB. Kappa-light chain amyloidosis of the liver, a rare cause of

Open Journal of Hematology, 2010, 1-4
Page 20 of 20 (Page number not for citation purposes)
liver enzyme elevation. Dtsch Med Wochenschr. 2008; 133: 1116-1120.
[92] Wechalekar AD, Lachmann HJ, Offer M, Hawkins PN, Gillmore JD. Efficacy of bortezomib in systemic AL amyloidosis with relapsed/refractory clonal disease. Haematologica. 2008; 93: 295-298.
[93] Colado E, Alvarez-Fernández S, Maiso P, Martín-Sánchez J, Vidriales MB, Garayoa M, Ocio EM, Montero JC, Pandiella A, San Miguel JF. The effect of the proteasome inhibitor bortezomib on acute myeloid leukemia cells and drug resistance associated with the CD34+ immature phenotype. Haematologica. 2008; 93: 57-66.
[94] Heaney NB, Pellicano F, Zhang B, Crawford L, Chu S, Kazmi SM, Allan EK, Jorgensen HG, Irvine AE, Bhatia R, Holyoake TL. Bortezomib induces apoptosis in primitive chronic myeloid leukemia cells including LTC-IC and NOD/SCID repopulating cells. Blood. 2010; 115: 2241-2250.
[95] Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A,
Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009; 458: 732-736.
[96] Driscoll JJ, Pelluru D, Lefkimmiatis K, Fulciniti M, Prabhala RH, Greipp PR, Barlogie B, Tai YT, Anderson KC, Shaughnessy JD Jr, Annunziata CM, Munshi NC. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 2010; 115: 2827-2834.
[97] Treon SP, Ioakimidis L, Soumerai JD, Patterson CJ, Sheehy P, Nelson M, Willen M, Matous J, Mattern J 2nd, Diener JG, Keogh GP, Myers TJ, Boral A, Birner A, Esseltine DL, Ghobrial IM. Primary therapy of Waldenström macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Onc. 2009; 27: 3830-3835.
Publish with ROSS Science Publishers and every scientist can easily read your work for free!
Your research papers will be: • available for free to the entire scientific community • peer reviewed and published immediately after
acceptance • cited in renowned open repositories upon
indexation of the journal • owned by yourself — author keep the copyright







