der medizinischen fakultät der friedrich-alexander ... · als dissertation genehmigt von der...
TRANSCRIPT

Etablierung und Anwendung von Echtzeit-PCR Verfahren
zur quantitativen Bestimmung der Parasitenbeladung
bei kutaner und viszeraler Leishmaniose in vitro und in vivo
Aus dem Mikrobiologischen Institut – Klinische Mikrobiologie, Immunologie und Hygiene
der Friedrich-Alexander-Universität Erlangen-Nürnberg Direktor: Prof. Dr. med. C. Bogdan
Der Medizinischen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur Erlangung des Doktorgrades Dr. med.
vorgelegt von
Theresia Julia Schulz
aus Plauen

Als Dissertation genehmigt von der
Medizinischen Fakultät der Friedrich-Alexander-Universität
Erlangen-Nürnberg
Vorsitzender des Promotionsorgans:
Gutachter:
Gutachter
Tag der mündlichen Prüfung:
Prof. Dr. Dr. h.c. J. Schüttler
Prof. Dr. C. Bogdan
Prof. Dr. S. Krappmann
23. November 2015

1. INHALTSVERZEICHNIS
1 INHALTSVERZEICHNIS 1 INHALTSVERZEICHNIS ................................................................................. 1
2 ZUSAMMENFASSUNG ................................................................................... 1
2.1 Deutsche Zusammenfassung .............................................................................. 1
2.1.1 Hintergrund und Ziele ................................................................................. 1
2.1.2 Methoden ..................................................................................................... 1
2.1.3 Ergebnisse und Beobachtungen .................................................................. 2
2.1.4 Praktische Schlussfolgerungen .................................................................... 2
2.2 English Abstract ................................................................................................. 3
2.2.1 Background ................................................................................................. 3
2.2.2 Methods ....................................................................................................... 3
2.2.3 Results ......................................................................................................... 3
2.2.4 Conclusion ................................................................................................... 4
3 EINLEITUNG .................................................................................................... 5
3.1 Leishmanien ....................................................................................................... 5
3.1.1 Klassifizierung und Epidemiologie ............................................................. 5
3.1.2 Übertragungszyklus ..................................................................................... 5
3.1.3 Durch Leishmania-Spezies verursachte Krankheitsbilder .......................... 6
3.1.4 Nachweismethoden ..................................................................................... 7
3.1.5 Aktuelle Therapieansätze ............................................................................ 8
3.1.6 Das Mausmodell der kutanen und viszeralen Leishmaniose .................... 10
3.2 Real-time PCR als diagnostisches Nachweisverfahren .................................... 11
3.2.1 Konventionelle bzw. Endpunkt-PCR ........................................................ 11
3.2.2 Reaktionsablauf und quantitative Auswertung der real-time PCR ........... 11
3.3 Fragestellung der vorliegenden Arbeit ............................................................. 15
4 MATERIAL & METHODEN .......................................................................... 17
4.1 Material ............................................................................................................ 17
4.1.1 Chemikalien .............................................................................................. 17
4.1.2 Oligonukleotide für Polymerase-Kettenreaktion (PCR) ........................... 18
4.1.3 Zytokine .................................................................................................... 19
4.1.4 Laborgeräte und Verbrauchsmaterialien ................................................... 19
4.1.5 Kits ............................................................................................................ 21

1. INHALTSVERZEICHNIS
4.1.6 Kulturmedien ............................................................................................. 22
4.1.7 Versuchstiere ............................................................................................. 23
4.1.8 Leishmanien .............................................................................................. 24
4.1.9 Klonierungsvektor/Plasmid ....................................................................... 24
4.2 Methoden .......................................................................................................... 24
4.2.1 Bestimmung der Zellzahl und –viabilität .................................................. 24
4.2.2 Gewinnung von Knochenmark aus Mäusen und Generierung von Knochenmarksmakrophagen (BM-MΦ) ................................................... 25
4.2.3 In vitro-Pulsinfektion von murinen Makrophagen (BM-MΦ) mit Leishmania major ...................................................................................... 26
4.2.4 Leishmanien-Erhaltungskultur .................................................................. 27
4.2.5 Experimentelles Modell der kutanen Leishmaniose ................................. 27
4.2.6 Experimentelles Modell der viszeralen Leishmaniose .............................. 27
4.2.7 Bestimmung der Erregerlast durch Grenzverdünnungsanalyse (Limiting Dilution Analysis) ...................................................................................... 28
4.2.8 DNA-Isolierung aus murinen Geweben und Zellkulturen ........................ 29
4.2.9 Bestimmung der DNA-Konzentration durch photometrische Messung ... 31
4.2.10 Quantitative real-time PCR ....................................................................... 31
4.2.11 Kinetoplasten-PCR .................................................................................... 37
4.2.12 Statistische Auswertung ............................................................................ 38
5 ERGEBNISSE .................................................................................................. 39
5.1 Etablierung und Evaluierung einer Leishmanien/Maus-Duplex-PCR zur quantitativen Bestimmung der Parasitenlast .................................................... 39
5.1.1 Verifizierung der geeigneten Primerkonzentration ................................... 40
5.1.2 Verifizierung der geeigneten Sondenkonzentration .................................. 45
5.1.3 Etablierung der Duplex Leishmanien/λ-PCR ............................................ 48
5.1.4 Etablierung einer Maus-PCR und Duplextest mit Leishmanien-PCR ...... 52
5.1.5 Generierung einer Standardkurve als Voraussetzung zur absoluten Quantifizierung .......................................................................................... 55
5.1.6 Vergleich der Sensitivität zwischen Kinetoplasten-PCR und 18S rDNA real-time PCR ............................................................................................ 58
5.1.7 Vergleich zwischen StepOne® Real-Time PCR System und 7900 HT Fast Real-Time PCR System .............................................................................. 60
5.2 Vergleich zwischen DiffQuik® und real-time PCR für in vitro-Pulsinfektionen muriner Knochenmarksmakrophagen .............................................................. 62
5.3 Vergleich zwischen Limiting Dilution Analysis und real-time PCR für in vivo-Infektionsexperimente ...................................................................................... 66

1. INHALTSVERZEICHNIS
5.3.1 Vergleich der Parasitenlast von BALB/c-bzw. C57BL/6-Mäusen mittels LDA und TaqMan nach subkutaner Infektion mit L. major ...................... 66
5.3.2 Vergleich der Parasitenlast von BALB/c-bzw. C57BL/6-Mäusen mittels LDA und TaqMan nach subkutaner Infektion mit L. infantum ................. 70
6 DISKUSSION .................................................................................................. 76
6.1 Etablierung eines real-time PCR-Systems zur Quantifizierung von Leishmanien ..................................................................................................... 76
6.1.1 Leishmanien-PCR ..................................................................................... 76
6.1.2 Duplex der Leishmanien-PCR mit λ-bzw. Maus-PCR ............................. 79
6.1.3 Vergleich zwischen konventioneller und real-time PCR .......................... 81
6.2 Leishmania real-time PCR im praktischen Gebrauch für in vitro/in vivo-Experimente...................................................................................................... 83
6.2.1 Anwendung der Leishmania real-time PCR bei in vitro-Infektionsexperimenten ............................................................................. 83
6.2.2 Anwendung der real-time PCR zur ex vivo-Analyse von Mausgewebe nach Infektion mit Leishmania .......................................................................... 85
7 LITERATURVERZEICHNIS.......................................................................... 92
8 ABKÜRZUNGSVERZEICHNIS .................................................................. 100

2. ZUSAMMENFASSUNG
1
2 ZUSAMMENFASSUNG
2.1 Deutsche Zusammenfassung
2.1.1 Hintergrund und Ziele
Bei der Leishmaniose handelt es sich um eine von Sandmücken übertragene Erkran-
kung. Einzellige Erreger der Gattung Leishmania rufen in Abhängigkeit von der jewei-
ligen Spezies eine kutane, mukokutane oder viszerale Verlaufsform hervor. Der Klima-
wandel wie auch die zunehmende Mobilität sind Risikofaktoren für deutlich höhere
Fallzahlen der Leishmaniose in Deutschland in naher Zukunft. Die Weiterentwicklung
von Nachweis- und Therapiemethoden nimmt damit einen wichtigen Stellenwert ein. In
der vorliegenden Arbeit sollte überprüft werden, ob eine für diesen Zweck entwickelte
real-time PCR die bisherigen Goldstandard-Methoden zur Quantifizierung der Parasi-
tenbeladung ersetzen könnte, welche sehr zeitaufwändig sind und experimentelle Erfah-
rung erfordern. Die neu etablierte PCR-Methode sollte auch auf ihre Anwendbarkeit in
Forschungsarbeiten am Mausmodell für kutane und viszerale Leishmaniose getestet
werden.
2.1.2 Methoden
Ein bereits publiziertes real-time PCR-Protokoll, basierend auf dem 18S rRNA-
Genabschnitt des Leishmaniengenoms, wurde zunächst evaluiert und bezüglich ver-
schiedener Parameter (Primerkonzentration, Sondenkonzentration etc.) optimiert. Um
die Parasitenlast in Leishmania-infizierten Zellen und Mausgewebe quantifizieren zu
können, wurde die Leishmania-spezifische PCR im Duplexverfahren mit einer PCR für
ein endogenes Mauskontrollgen etabliert. Anschließend wurde die Parasitenlastbestim-
mung mittels Leishmania-Duplex-PCR sowohl mit in vitro-infizierten Zellen als auch
mit Gewebe aus Leishmanien-infizierten Mäusen getestet und mit der jeweils herkömm-
lichen Diagnostik (in vitro-Infektion: mikroskopische Auszählung nach Zellfärbung mit
DiffQuik®-Lösung; ex vivo-Analyse: Grenzverdünnungsanalyse und anschließende
Kultur für 7-10 Tage) verglichen.

2. ZUSAMMENFASSUNG
2
2.1.3 Ergebnisse und Beobachtungen
In vitro mit Leishmania (L.) major infizierte murine Knochenmarkmakrophagen zeigten
in der mikroskopischen Auszählung erwartungsgemäß eine Abnahme der Infektionsrate,
wenn sie mit Immunstimulanzien inkubiert wurden, die Makrophagen zum Abtöten der
Parasiten aktivieren. Im Gegensatz dazu ergaben sich mit der in dieser Arbeit etablierten
Leishmania real-time PCR abweichende und zum Teil entgegengesetzte Ergebnisse,
sodass diese als Ersatz der bisherigen Untersuchungsmethode nicht geeignet erscheint.
Für in vivo-Untersuchungen wurden C57BL/6 und BALB/c- Mäuse mit L. major (kuta-
ne Leishmaniose) bzw. L. infantum (viszerale Leishmaniose) infiziert. In beiden Model-
len entwickelte sich der erwartete Infektionsverlauf. Das Ergebnis für die Beladung mit
Leishmanien gemessen mit Hilfe der konventionellen Grenzverdünnungsanalyse ent-
sprach den Daten aus der Literatur. Der Vergleich von real-time PCR und Grenzver-
dünnungsanalyse zeigte in beiden Modellen eine gute Übereinstimmung, sodass die
real-time PCR für ex vivo-Untersuchungen von Leishmanieninfektionen eine zuverläs-
sige und schnelle Quantifizierung zulässt.
2.1.4 Praktische Schlussfolgerungen
Ob die real-time PCR zum Nachweis der Leishmanienlast geeignet ist, hängt demnach
im Wesentlichen von den Versuchsbedingungen ab. Pulsinfektionsexperimente erstre-
cken sich meistens über wenige Tage; eine Abnahme von Parasiten kann hier in der
PCR nicht sicher detektiert werden. Hierfür ist wahrscheinlich die Parasiten-DNA, wel-
che nach dem Absterben der Erreger über eine längere Zeit in der Wirtszelle verbleibt,
verantwortlich. Bei ex vivo-Analysen von Geweben aus Leishmania-infizierten Mäusen,
bei denen sich die Infektion über längere Zeiträume ersteckte, zeigte sich eine gute Re-
produzierbarkeit und Korrelation zwischen den Ergebnissen der Duplex-PCR und der
konventionellen Methode. Damit wäre ein Einsatz des in dieser Arbeit evaluierten
Leishmania real-time PCR-Systems auch für Leishmaniose-Patienten denkbar und
könnte zum Beispiel zur Erfolgskontrolle einer Therapie eingesetzt werden.

2. ZUSAMMENFASSUNG
3
2.2 English Abstract
2.2.1 Background
Leishmaniasis is a disease transmitted by sand flies. Dependent on the species and strain
protozoan Leishmania parasites may cause a cutaneous, mucocutaneous or visceral in-
fection. Climate changes as well as the increasing mobility are risk factors for increas-
ing numbers of cases in Germany in the future. Advanced methods for parasite detection
and diagnosis are therefore of great relevance. This dissertation aimed to develop a real-
time PCR-based quantification method for Leishmania parasites which allows replace-
ment of the current time-consuming gold standard methods requiring significant exper-
imental know-how. In addition, the newly established PCR method should be evaluated
for its applicability for research studies in the murine model of cutaneous and visceral
leishmaniasis.
2.2.2 Methods
An already published real-time PCR protocol based on the 18S rRNA gene of the
Leishmania genome was initially evaluated and optimized for different parameters
(primer and probe concentrations etc.). To quantify the parasite burden in infected cells
and mouse tissue, a duplex PCR-system consisting of the Leishmania-specific PCR and
a PCR able to detect an endogenous mouse control gene was established. Afterwards
the duplex PCR-system was tested with in vitro infected cells as well as with tissue
samples from Leishmania-infected mice and compared to the respective conventional
diagnostic methods (in vitro infection: microscopic enumeration of the parasites after
cell staining with DiffQuik®; ex vivo: limiting dilution analysis followed by cultivation
for 7-10 days).
2.2.3 Results
In vitro, both the microscopically analyzed infection rate and the parasite load in L. ma-
jor-infected murine bone marrow-derived macrophages decreased when macrophages
get activated by stimulants known to induce parasite killing. The Leishmania real-time
PCR established in this dissertation, however, showed different or even opposite results.
Thus, the duplex-PCR method seems to be inappropriate to replace microscopic evalua-
tion of the parasite load in infected cells. For ex vivo examinations C57BL/6 and

2. ZUSAMMENFASSUNG
4
BALB/c mice were infected with L. major (cutaneous leishmaniasis) or L. infantum
(visceral leishmaniasis). In both models the expected course of infection developed. The
resulting Leishmania load measured with the conventional limiting dilution assay was
comparable to the results obtained with the real-time PCR indicating that the established
duplex-PCR method allows reliable and fast ex vivo quantification of parasite burden in
infected mouse tissues.
2.2.4 Conclusion
Whether real-time PCR is appropriate as a method for Leishmania quantification mainly
depends on the experimental setting that is used. In vitro experiments usually last only a
few days; here a decrease of the parasite load cannot be detected reliably. This is most
likely due to the detection of parasite DNA that will persist for some time in the host
cell after the death of the pathogens. In contrast, ex vivo analysis of tissues from
Leishmania-infected mice which had been infected for longer time periods showed a
good reproducibility and correlation of results by duplex-PCR and conventional evalua-
tion. Thus, the established Leishmania real-time PCR system might be also used for
quantification of parasite loads in human leishmaniasis patients in order to control for
the success of a therapeutic intervention.

3. EINLEITUNG
5
3 EINLEITUNG
3.1 Leishmanien
3.1.1 Klassifizierung und Epidemiologie
Einzellige Erreger der Gattung Leishmania sind begeißelte Flagellaten, die der Ordnung
der Kinetoplastida und Familie der Trypanosomatidae zugeordnet werden. Innerhalb der
Gattung sind über 20 verschiedene humanpathogene Spezies bekannt.
Die Leishmaniose ist endemisch in 88 Ländern der Welt und findet Verbreitung zwi-
schen dem 45. nördlichen und 30. südlichen Breitengrad [71]. Es wird unterschieden
zwischen der „Neu-Welt“-Leishmaniose mit Hauptendemiegebieten in Mittelamerika
sowie nördlichem und zentralem Südamerika sowie der „Alten Welt“; hier betroffen
sind weite Teile Asiens vom Nahen und Mittleren Osten bis Nordwestchina sowie die
Länder im Mittelmeerraum [42, 80]. Schätzungsweise 350 Mio. Menschen sind bedroht,
ca. 12 Mio. bereits infiziert; die Inzidenz beträgt rund 1,5 bis 2 Mio. Neuerkrankungen
pro Jahr [24].
Infolge des Klimawandels könnten sich die Endemiegebiete ausweiten und die Leish-
maniose an Bedeutung weiter zunehmen; so wurde beispielsweise das Auftreten von
Mücken der Gattung Phlebotomus, die als Vektoren für Leishmanien fungieren können,
in Süddeutschland beobachtet [73]. Die zunehmende Mobilität birgt außerdem die Ge-
fahr in sich, dass Leishmanien auch in Nicht-Endemiegebiete verschleppt werden. Das
Auftreten von autochthonen Leishmaniose-Fällen in Deutschland könnte dadurch eben-
falls erklärt werden [10, 53].
3.1.2 Übertragungszyklus
Die Übertragung von Leishmanien erfolgt durch weibliche Sandmücken (engl. sand
flies) der Gattung Phlebotomus, Lutzomyia und Psychodopygus. Durch eine Blutmahl-
zeit aufgenommen, entwickeln sich im Mitteldarm der Mücken über mehrere Zwischen-
stufen innerhalb einer Zeitspanne von 4 bis 25 Tagen [114] aus nicht-infektiösen pro-
zyklischen hochinfektiöse metazyklische Promastigoten (15 – 20 µm lang) mit einer ca.
20 µm langen Geißel im Bereich des vorderen Poles [90]. Nach Einwanderung in den
Proboscis der Mücke können bei einem erneuten Stich die Parasiten auf einen weiteren

3. EINLEITUNG
6
Wirtsorganismus übertragen werden [79], wo sie in der Haut sehr schnell von phagozy-
tierenden Zellen aufgenommen werden, insbesondere von Makrophagen (MΦ), Langer-
hanszellen, dendritischen Zellen und Granulozyten [85]. Im Phagosom dieser Zellen
findet die Transformation in die intrazelluläre, unbegeißelte und unbewegliche, amasti-
gote Form mit einem Durchmesser von 2 – 4 µm statt. Leishmania-Amastigote sind
gegenüber den äußeren Bedingungen im Phagosom und Phagolysosom unempfindlich
und können in diesem Zellkompartiment sehr gut überleben und sich sogar vermehren
[52]; nach starker Vermehrung ist die Freisetzung von Amastigoten durch Exozytose
oder Zelllyse und danach eine erneute Wirtszell-Infektion möglich [9]. Mücken können
beim Stich durch die Aufnahme freier Amastigoten oder Leishmania-beladener Phago-
zyten infiziert werden. Die infizierten Phagozyten zerfallen im Insektendarm, was zur
Freisetzung der Amastigoten führt [85], dadurch schließt sich der Infektionszyklus. Sel-
tener ist die Übertragung auch über die Plazenta, durch Bluttransfusionen, Geschlechts-
verkehr oder die gemeinsame Nutzung von Injektionsnadeln bei Drogenabhängigen
beobachtet worden [21, 68].
3.1.3 Durch Leishmania-Spezies verursachte Krankheitsbilder
Beim Menschen können drei große Krankheitsbilder voneinander abgegrenzt werden,
deren Ausprägung und Schweregrad einerseits von der/dem zugrundeliegenden Leish-
manien-Spezies/Stamm abhängig ist, zum anderen in starkem Zusammenhang mit dem
Immunstatus des Infizierten steht [67].
Die kutane Leishmaniose (CL), die überwiegend in Afghanistan, Syrien, Saudi-Arabien,
Brasilien, Peru, und im Iran vorkommt [25, 80], macht den größten Anteil der Leishma-
niosen aus. Sie ist gekennzeichnet durch das Auftreten einer Hautläsion an der Infekti-
onsstelle. Eine singulär auftretende Papel (auch als „Orientbeule“ bezeichnet) heilt nach
anfänglicher Schwellung und möglicher Ulzeration meist spontan innerhalb von 3 – 15
Monaten narbig ab [5]. Nach dem Ausheilen der Läsion kommt es meist zur Ausbildung
einer langanhaltenden Immunität [78], die Erreger persistieren dauerhaft [1]. Bei einer
Immunsuppression kann die Erkrankung erneut ausbrechen und in eine diffuse kutane
Leishmaniose übergehen, bis hin zum Befall innerer Organe (viszerale Leishmaniose)
mit letalem Ausgang [81, 114]. Erreger der kutanen Leishmaniose sind insbesondere L.
major, L. tropica, L. aethiopica und L. mexicana [80].

3. EINLEITUNG
7
Die mukokutane Leishmaniose, welche hauptsächlich in Lateinamerika Verbreitung
findet [97] und von den Spezies L. braziliensis, L. mexicana und L. peruviana hervorge-
rufen wird, ist gekennzeichnet durch rekurrierende und progredient verlaufenden Läsio-
nen in Bereich der Mund- und Nasenschleimhaut, welche nicht spontan abheilen [22].
Die viszerale Form der Leishmaniose (VL), auch Kala-Azar genannt, hat die schwer-
wiegendsten Auswirkungen auf den Wirtsorganismus und wird unter anderem durch L.
donovani und L. infantum / L. chagasi ausgelöst. Es kommt zum Befall innerer Organe
wie Milz, Leber und Knochenmark. Symptome sind Fieberschübe, Schleimhautblutun-
gen, Panzytopenie, Hepatosplenomegalie und ein zur Kachexie führender Gewichtsver-
lust [42], welcher unbehandelt fast immer letal endet. Verbreitung findet die viszerale
Leishmaniose vor allem in Bangladesch, Indien, Brasilien und dem Sudan.
3.1.4 Nachweismethoden
In Endemiegebieten treten eine Reihe von Krankheiten anderer Ätiologien auf, die im
klinischen Erscheinungsbild der Leishmaniose ähneln (z.B. Lepra, Hautkrebs, Tuberku-
lose bei der kutanen Form bzw. Malaria oder Schistosomiasis bei der viszeralen Leish-
maniose) [83], sodass eine Differentialdiagnose von großer Bedeutung ist.
Um die Diagnose einer Leishmaniose sicher zu stellen ist, ist der Nachweis des Erregers
unabdingbar. Aufgrund der weiten Verfügbarkeit und hoher Spezifität ist die Mikrosko-
pie in vielen Ländern der Goldstandard hierfür. Insbesondere in endemischen Gebieten
sind weiter ausgereifte Nachweistechniken zu teuer und nur selten verfügbar, sodass der
mikroskopische Nachweis das Mittel der Wahl ist. Mit Giemsa gefärbte Biopsieabstri-
che (CL) bzw. Punktate von Lymphknoten, Knochenmark oder Milz (VL) werden auf
das Vorliegen von Parasiten, vor allem aufgenommen in MΦ, untersucht. Problematisch
ist hierbei die geringe Anzahl an Amastigoten in chronischen Läsionen [6], was zu einer
Abnahme der Sensitivität führt.
Die kulturelle Anzucht auf geeigneten Nährböden/-medien zeichnet sich durch eine hö-
here Sensitivität aus. Allerdings ist die Kultivierung mit einer Reihe an Nachteilen ver-
bunden (siehe Kapitel 3.3).
Da das Ausmaß der klinischen Manifestation und auch der Therapieerfolg oft spezies-
abhängig sind [19], kann die Identifikation und Charakterisierung der Leishmanienspe-
zies sinnvoll sein, insbesondere bei der mukokutanen Verlaufsform. Dies ist z.B. mög-

3. EINLEITUNG
8
lich, wenn die Anzucht der Erreger mit der Multi-Lokus-Enzym-Elektrophorese
(MLEE) kombiniert wird [83].
Serologische Untersuchungen werden nicht routinemäßig durchgeführt. Aufgrund einer
eher geringen Menge an frei zirkulierenden Antikörpern, vor allem bei der CL [83], und
kreuzreagierenden Parasiten (z.B. Trypanosoma cruzi), können Sensitivität und Spezifi-
tät stark beeinträchtigt sein.
Trotz einer Vielzahl an erfolgreich evaluierten Methoden in der Leishmaniendiagnostik
geht die Tendenz immer mehr zur Molekulardiagnostik auf Basis der PCR (Polymerase
Chain Reaction) über. Vorteile gegenüber anderen etablierten Ansätzen wie kultureller
Anzucht ergeben sich im schnellen Vorliegen von Ergebnissen, die gleichzeitig eine
hohe sensible und spezifische Aussagekraft haben, dem DNA-Nachweis sowohl von
Amastigoten als auch von Promastigoten, sowie der Nutzbarkeit unterschiedlicher bio-
logischer Materialien [38]. Insbesondere, wenn eine niedrige Parasitenbeladung zu er-
warten ist, beispielsweise bei mukokutaner Leishmaniose [34] oder Proben aus Blut
[21] bzw. Konjunktivalsekret [102], ist die PCR anderen Methoden überlegen. Es exis-
tiert eine Vielzahl an PCR-Protokollen, die sich im technischen Ablauf (z.B. konventio-
nelle versus real-time PCR, siehe Kapitel 3.2), aber auch in den jeweiligen Zielgenen
voneinander unterscheiden. Hier seien stellvertretend die Mikrosatelliten-DNA [88],
SSU rRNA [107], das Tubulin-Gen [62], aber auch repetitive Sequenzen wie die Kine-
toplasten-DNA [23] genannt. Um vergleichbare Ergebnisse zu erhalten und die Durch-
führbarkeit in verschiedenen Zentren zu ermöglichen, ist es von großer Bedeutung,
PCR-basierte Protokolle zu standardisieren und zu optimieren.
Für weitere Fortschritte der Diagnostik in Endemiegebieten ist bei der Entwicklung
neuer bzw. Verbesserung bestehender Methoden auf eine einfache Durchführbarkeit
und möglichst niedrige Kosten zu achten.
3.1.5 Aktuelle Therapieansätze
Seit über 60 Jahren ist der Einsatz pentavalenter Antimonpräparate sowohl für die visze-
rale als auch kutane Leishmaniose erprobt und hat sich, insbesondere bei nicht vorbe-
handelten Patienten, mit einer Ansprechrate von ca. 95% bewährt [95]. Probleme erge-
ben sich aus der Toxizität bei systemischer Gabe und zunehmenden Resistenzraten, ins-
besondere bei chronisch Erkrankten [17]. Die Kombination mit Paromomycin, einem
Aminoglykosid-Antibiotikum, wird in gemeinsamer Gabe mit pentavalentem Antimon

3. EINLEITUNG
9
getestet und zeigt in Phase III-Studien eine erhöhte Effizienz, reduzierbare Behand-
lungsdauer und herabgesetzte Mortalität gegenüber der Einfachtherapie [49, 104].
Ebenso zeigt das auch als Mittel gegen Pilzinfektionen eingesetzte Amphotericin B,
welches durch Wechselwirkungen mit Sterinen die Permeabilität der Zellmembran be-
einflusst, gute Erfolge. Gerade in Gebieten mit hohen Resistenzen gegen Antimonprä-
parate, aber auch in Europa und den USA wird Amphotericin B als Medikament erster
Wahl eingesetzt [4]. Aufgrund von Nebenwirkungen (u.a. Nephrotoxizität [6]) ist der
Einsatz des weiterentwickelten, liposomalen Amphotericin B vorzuziehen: nach Endo-
zytose durch MΦ gelangt das Medikament in unmittelbare Nähe zum Erreger [42], die
Toxizität und Behandlungsdauer lassen sich reduzieren; schon durch einmalige Gabe
können über 90% der Patienten geheilt werden [103]. Nachteilig sind die hohen Kosten
des Medikaments, die einen flächendeckenden Einsatz in endemischen Gebieten derzeit
unmöglich machen [103]. Weitere Antipilzmittel, wie z.B. Azole, wirken ebenfalls
leishmanizid [105], denn sie blockieren die Synthese von Ergosterol, welches auch in
der Membran von Leishmanien nachweisbar ist. Sie dienen als Reservemedikation bei
Resistenz gegen Antimonverbindungen.
Miltefosin, ursprünglich für die Antitumortherapie entwickelt, zeigt in L. donovani un-
ter anderem Einflüsse auf den Etherlipidstoffwechsel, die Synthese von Membranankern
sowie die Signaltransduktion [63]. Hier stellt allerdings die lange Halbwertszeit (150-
200 Stunden) mit Erreichen von Medikamentenspiegeln unter dem therapeutischen Le-
vel ein hohes Risiko der Resistenzentwicklung dar [13].
Die kutane Leishmaniose heilt meist von selbst aus, insbesondere bei Immungesunden.
Eine Indikation zur Therapie ist jedoch gegeben, um die Selbstheilung zu beschleuni-
gen, die Narbenbildung zu reduzieren und eine Disseminierung des Erregers zu unter-
binden [71]. Insbesondere Infektionen mit L. tropica benötigen zur Ausheilung eine
große Zeitspanne (bis zu einem Jahr). Hier kann die zusätzliche Einleitung einer
Thermotherapie oder topischer Paromomycin-Behandlung sinnvoll sein [6]. Die wohl
beste Effizienz und niedrigste Rückfallrate zeigt klinischen Studien zufolge die intralä-
sionale Gabe von Antimonpräparaten [5].
Eine vorbeugende Impfung in Endemiegebieten könnte die Inzidenzraten drastisch sen-
ken, Versuche mit Lebendvakzinen sind wegen nicht tolerierbaren kutanen Narbenbil-
dungen gescheitert. Derzeitige Studien untersuchen unter anderem die Wirksamkeit von

3. EINLEITUNG
10
rekombinanten Antigenen, Speichelproteinen von Schmetterlingsmücken und genetisch
modifizierten Vakzinen als Bestandteil eines möglichen Impfstoffes [50, 54].
3.1.6 Das Mausmodell der kutanen und viszeralen Leishmaniose
Durch subkutane oder intradermale Injektion von L. major-Promastigoten wird in der
Maus eine kutane Leishmaniose ausgelöst, welche der kutanen Leishmaniose beim
Menschen ähnelt. Klinische Manifestation und Krankheitsverlauf weisen in gängigen
Inzuchtmausstämmen eine sehr unterschiedliche Ausprägung auf: die meisten Inzucht-
mausstämme wie C57BL/6-Mäuse bilden allenfalls lokale Läsionen an der Inokulati-
onsstelle und entwickeln langanhaltende Immunität, wohingegen es bei empfänglichen
Stämmen wie BALB/c Mäusen wegen fehlender Möglichkeiten der Parasitenkontrolle
zur generalisierten Leishmaniose kommt, welche letztlich letal verläuft [89]
Zur Induktion einer viszeralen Leishmaniose werden L. donovani oder L. infantum
Promastigoten intravenös in Mäuse injiziert. Im Verlauf der Infektion kommt es haupt-
sächlich zum Befall innerer Organe wie Milz und Leber. In der Milz findet ein langsa-
mer Anstieg der Parasitenlast statt und die Erreger können im Verlauf der Erkrankung
auf einem hohen Niveau persistieren [28]. Die Leber hingegen weist eine schnelle Ver-
mehrung der Parasiten auf, mit beginnender Immunantwort können die Erreger dann
aber kontrolliert werden und die Parasitenzahl im Lebergewebe wird im Verlauf der
Infektion reduziert. Bei der viszeralen Leishmaniose ist im Gegensatz zur L. major-
Infektion kein grundsätzlich abweichender Krankheitsverlauf zwischen BALB/c und
C57BL/6-Mäusen zu beobachten [46].
Sowohl im Mausmodell der kutanen als auch der viszeralen Leishmaniose wurden in
zahlreichen Studien wichtige Komponenten der Immunabwehr definiert, die für eine
erfolgreiche Kontrolle der Leishmanien notwendig sind. Unerlässlich ist das Vorhan-
densein des Zytokins Interferon-γ (IFN-γ), das von Natürlichen Killerzellen und T-
Helfer-Zellen produziert wird. Zusammen mit dem Zytokin Tumornekrosefaktor (TNF)
führt Interferon-γ zu einer potenten Aktivierung von infizierten Makrophagen, die, in-
folge der Expression der induzierbaren Stickstoffmonoxidsynthase und Produktion von
Stickstoffmonoxid, Leishmanien abtöten [70].

3. EINLEITUNG
11
3.2 Real-time PCR als diagnostisches Nachweisverfahren
3.2.1 Konventionelle bzw. Endpunkt-PCR
Herkömmliche PCR-Methoden funktionieren nach dem Prinzip, ein nachzuweisendes
Genprodukt, das zwischen zwei flankierenden Primern gelegen ist, zu vervielfältigen
und anschließend zu detektieren. Dies erfolgt durch die Aufspaltung eines DNA-
Doppelstranges bei ca. 94°C, Ausbildung eines komplementären Stranges mithilfe einer
hitzebeständigen DNA-Polymerase im Bereich zwischen den beiden Primern, im An-
schluss folgt die erneute Auftrennung des gebildeten Doppelstranges und Vorlage des
ursprünglichen und neu synthetisierten DNA-Abschnittes für die Bildung dazu kom-
plementärer Stränge im Folgezyklus usw.; durch die Abfolge vieler solcher Zyklen
nacheinander kommt es zur Anreicherung des nachzuweisenden Genproduktes. Anfangs
erfolgt die Vervielfältigung (Amplifikation) exponentiell, mit zunehmender Dauer die-
ses Prozesses werden Reagenzien verbraucht und damit kommt es zum Übergang in
einen linearen Produktzuwachs; letztlich erreicht die Menge des synthetisierten Gen-
produktes eine Plateauphase.
Bei der konventionellen PCR wird das amplifizierte Genprodukt erst nach Abschluss
der Reaktion nachgewiesen, indem das PCR-Produkt auf ein Agarose-Gel aufgetragen
und elektrophoretisch aufgetrennt wird. Die Laufgeschwindigkeit ist hierbei indirekt
proportional zum dekadischen Logarithmus der Moleküllänge [41]. Die Auswertung
erfolgt durch Fotografie des Geles im ultravioletten Licht; dabei wird der vorher zuge-
gebene Fluoreszenzmarker Ethidiumbromid sichtbar gemacht, welcher in die DNA
interkaliert. Eine untersuchte Probe wird als positiv beurteilt, wenn das PCR-Produkt
der erwarteten Größe entspricht, was durch den Vergleich mit einem parallel eingesetz-
ten DNA-Größenstandard mit DNA-Fragmenten bekannter Größen ermittelt werden
kann. Zur Überprüfung der Spezifität des PCR-Produktes kann es ggf. auch sequenziert
werden.
Diese Form der PCR wird auch als Endpunkt-PCR bezeichnet, weil die Detektion des
gesuchten Genproduktes erst im Anschluss an den Amplifikationsprozess erfolgt.
3.2.2 Reaktionsablauf und quantitative Auswertung der real-time PCR
Bei der real-time PCR wird das gebildete Produkt im Gegensatz zur konventionellen
PCR unmittelbar während der Amplifikation durch Messung eine Fluoreszenzsignals
3. EINLEITUNG
12
sichtbar gemacht, daher auch der Begriff „Echtzeit-PCR“ (engl. real-time). Das Ausmaß
der Fluoreszenzstärke ist dabei abhängig von der gebildeten Produktmenge [44] [40].
Die verwendeten Fluoreszenzfarbstoffe besitzen eine bestimmte Grundfluoreszenz, die
als Basislinie gesetzt wird. Ausgehend von dieser Hintergrundfluoreszenz wird ein
Schwellenwert (engl. Threshold) definiert, der der Standardabweichung der Basisfluo-
reszenz (gemessen zwischen dem 3. und 15. Zyklus) multipliziert mit 10 entspricht [37].
Eine Probe wird dann als positiv bewertet, wenn das Fluoreszenzsignal den Schwellen-
wert überschreitet, wobei der Threshold Cycle (CT) dem Zyklus entspricht, bei dem die-
ser Schwellenwert erstmals überstiegen wird. Je niedriger der CT-Wert ist, desto höher
war demzufolge die Ausgangsmenge des nachzuweisenden Genabschnittes. Verschiebt
sich der CT-Wert um einen Zyklus nach oben oder nach unten, so ist davon auszugehen,
dass eine halb so große bzw. doppelte Menge des interessierenden Gens in der einge-
setzten DNA vorlag. Der CT-Wert ist folglich die Basis für die Quantifizierung in der
real-time PCR [37].
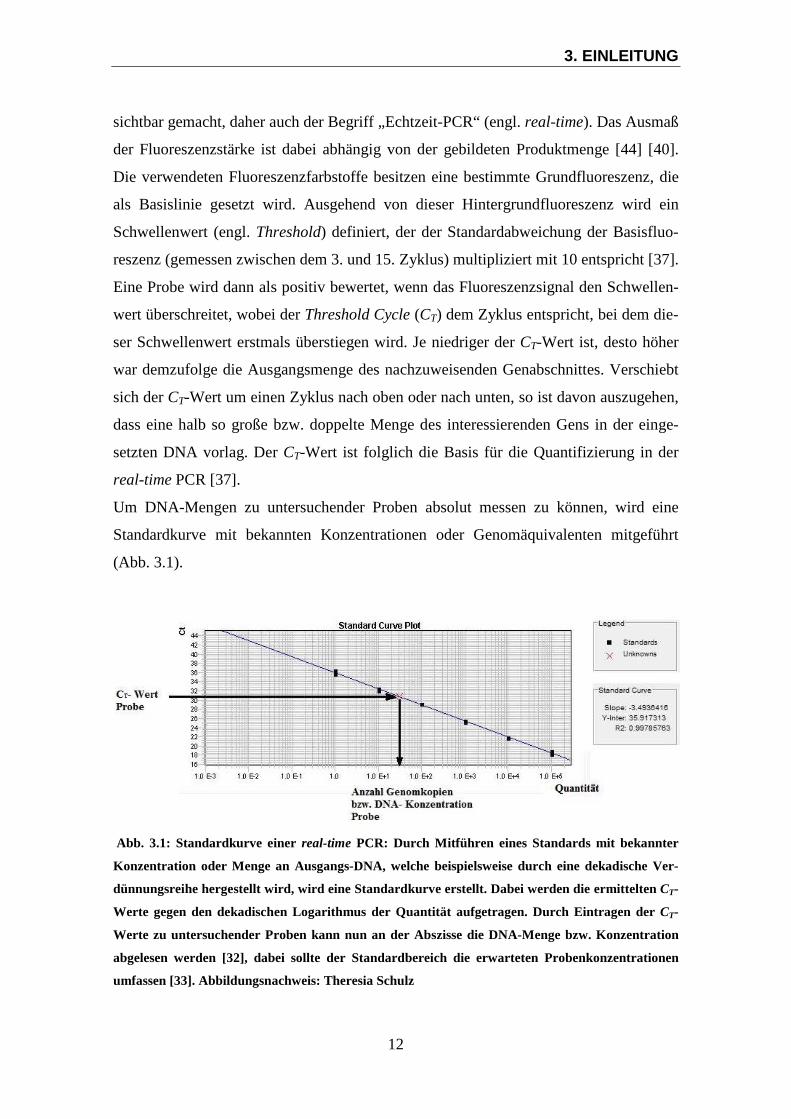
Um DNA-Mengen zu untersuchender Proben absolut messen zu können, wird eine
Standardkurve mit bekannten Konzentrationen oder Genomäquivalenten mitgeführt
(Abb. 3.1).
Abb. 3.1: Standardkurve einer real-time PCR: Durch Mitführen eines Standards mit bekannter
Konzentration oder Menge an Ausgangs-DNA, welche beispielsweise durch eine dekadische Ver-
dünnungsreihe hergestellt wird, wird eine Standardkurve erstellt. Dabei werden die ermittelten CT-
Werte gegen den dekadischen Logarithmus der Quantität aufgetragen. Durch Eintragen der CT-
Werte zu untersuchender Proben kann nun an der Abszisse die DNA-Menge bzw. Konzentration
abgelesen werden [32], dabei sollte der Standardbereich die erwarteten Probenkonzentrationen
umfassen [33]. Abbildungsnachweis: Theresia Schulz

3. EINLEITUNG
13
Als Fluoreszenzstoff wird heute meist SYBR Green® eingesetzt. Dieser Farbstoff bin-
det in doppelsträngiger DNA und zeigt nach Anregung eine etwa 1000-mal erhöhte
Fluoreszenz gegenüber dem ungebundenen Farbstoff [43, 44]. Vorteil ist die Möglich-
keit des universellen Einsatzes, da SYBR Green® nicht sequenzspezifisch bindet [14].
Allerdings besteht die Gefahr eines unspezifischen Fluoreszenzanstieges beim Vorlie-
gen von Artefakten, z.B. von Primerdimeren und unspezifischen PCR-Produkten [108].
Eine weitere Möglichkeit ist die Nutzung fluoreszierender Sonden (engl. probes). Diese
mit Farbstoffen markierten Oligonukleotide sind meist zwischen 20 und 30 Basenpaa-
ren lang und binden spezifisch an eine zwischen den Primern gelegene Sequenz. Erst
nach Bindung an die Zielsequenz kommt es zum Fluoreszenzanstieg. Es gibt eine Reihe
unterschiedlicher Sondensysteme, doch das Wirkprinzip ist allen gleich: der sogenannte
Fluoreszenz-Resonanz-Energie-Transfer (FRET) [15, 31]. Dies besagt, dass ein Fluo-
reszenzstoff nach Anregung mit Licht einer bestimmten Wellenlänge (A1) die aufge-
nommene Energie mit Licht einer anderen Wellenlänge wieder abstrahlt (E1, = Emissi-
on). Dabei hat jeder Fluoreszenzstoff ein eigenes Anregungs- und Emissionsspektrum.
Befindet sich ein Farbstoff 1 in ausreichender Nähe (max. 17-20 bp Abstand; bp = Ba-
senpaare) zu einem Farbstoff 2, dessen Anregungsspektrum dem Emissionsspektrum des
Ersteren entspricht (A2 = E1), so erfolgt bei Anregung keine Lichtabstrahlung von Farb-
stoff 1, sondern ein Energietransfer auf Farbstoff 2 [15]. Bei der quantitativen PCR kann
durch Messung der Emission von Farbstoff 1 oder 2 auf die räumliche Nähe beider Farb-
stoffe geschlossen werden [96]. In der vorliegenden Arbeit wurde die Emission von
Farbstoff 1 detektiert, der in diesem Fall als Reporterfarbstoff bezeichnet wird, während
Farbstoff 2 als Quencher fungiert. Die hier eingesetzten TaqMan-Sonden sind sogenann-
te Hydrolysesonden: durch die Exonukleaseaktivität der Taq-Polymerase werden die an
die Zielsequenz gebundenen Sonden aufgespalten, Reporter und Quencher werden
räumlich voneinander getrennt und es kommt zum Fluoreszenzanstieg bei E1 [37, 40,
45] (Abb. 3.2).
Mit dem Einsatz verschiedener Reporter- und Quencherfarbstoffe können mehrere
PCRs simultan in einem Ansatz erfolgen; diese Weiterentwicklung der real-time PCR
wird als Multiplex-PCR bezeichnet. Im vorliegenden Fall wurden die Fluoreszenzmar-
ker FAM und VIC als Reporter ausgewählt, sowie TAMRA und NFQ als Quencher.

NFQ ist zusätzlich an einen
mit der DNA-Helix im Bereich der kleinen Furche
stärkten Bindung der Sonde an die Zielsequenz und ermögl
zerer Sonden. Hierdurch steigen Sensitivität und Spezifität an, wodurch die benötigte
Sondenkonzentration und damit auch die Hintergrundfluoreszenz abnimmt
Abb. 3.2: Prinzip der TaqMan
sondengebunden; bei Anregung des Reporters kommt es zum Energietransfer (FRET) auf den
Quencher, welcher Licht einer bestimmten Wellenlänge
beide Farbstoffe in unmittelbarer Nähe zueinander befinde
ist. Bei Vorliegen von DNA mit der entsprechenden Zielsequenz kommt es im Rahmen der Ampl
fikation zur Anlagerung der Sonde an den komplementären Strang
Aktivität der Taq- Polymerase zur Spaltung der Sonde. Infolge der räumlichen Trennung
nicht mehr möglich und der Reporter strahlt Licht der Wellenlänge E
messbaren Fluoreszenzanstiegs bei
dungsnachweis: Theresia Schulz
3. EINLEITUNG
14
NFQ ist zusätzlich an einen Minor groove binder (MGB) gekoppelt; dieser hybridisiert
Helix im Bereich der kleinen Furche [56]. Der MGB führt zu einer ve
stärkten Bindung der Sonde an die Zielsequenz und ermöglicht so die Verwendung kü
Sonden. Hierdurch steigen Sensitivität und Spezifität an, wodurch die benötigte
Sondenkonzentration und damit auch die Hintergrundfluoreszenz abnimmt
TaqMan-Hydrolysesonden: Farbstoffe 1 (Reporter) und 2 (
sondengebunden; bei Anregung des Reporters kommt es zum Energietransfer (FRET) auf den
welcher Licht einer bestimmten Wellenlänge (E2) abstrahlt. Dies ist möglich,
beide Farbstoffe in unmittelbarer Nähe zueinander befinden, wie es bei der intakten Sonde der Fall
ist. Bei Vorliegen von DNA mit der entsprechenden Zielsequenz kommt es im Rahmen der Ampl
fikation zur Anlagerung der Sonde an den komplementären Strang (Annealing
Polymerase zur Spaltung der Sonde. Infolge der räumlichen Trennung
der Reporter strahlt Licht der Wellenlänge E1 ab. Die
messbaren Fluoreszenzanstiegs bei E1 dient als Grundlage der quantitativen Auswertung.
dungsnachweis: Theresia Schulz
3. EINLEITUNG
(MGB) gekoppelt; dieser hybridisiert
. Der MGB führt zu einer ver-
icht so die Verwendung kür-
Sonden. Hierdurch steigen Sensitivität und Spezifität an, wodurch die benötigte
Sondenkonzentration und damit auch die Hintergrundfluoreszenz abnimmt [57].
) und 2 (Quencher) sind
sondengebunden; bei Anregung des Reporters kommt es zum Energietransfer (FRET) auf den
ist möglich, wenn sich
bei der intakten Sonde der Fall
ist. Bei Vorliegen von DNA mit der entsprechenden Zielsequenz kommt es im Rahmen der Ampli-
Annealing) und durch die
Polymerase zur Spaltung der Sonde. Infolge der räumlichen Trennung ist FRET
. Die Höhe des jetzt
Grundlage der quantitativen Auswertung. Abbil-

3. EINLEITUNG
15
3.3 Fragestellung der vorliegenden Arbeit
Das bisher standardmäßig genutzte Verfahren zur Bestimmung der Parasitendichte mu-
riner Gewebe bei kutaner und viszeraler Leishmaniose in vivo ist die Grenzverdün-
nungsanalyse (Limiting Dilution Analysis, LD). Hierbei erfolgt das sequentielle Austit-
rieren von Zellsuspensionen über viele Verdünnungsstufen mit mindestens 12 Replika-
ten je Verdünnungsschritt. Nach Anzüchtung und Kultivierung wird durch die Auszäh-
lung positiver (Wachstum von Leishmanien) und negativer Replikate die Parasitenlast
statistisch ermittelt. Aufgrund des hohen Zeit- und Arbeitsaufwandes, der Anfälligkeit
gegen Pipettierfehler, z.B. Kreuzkontamination einzelner Verdünnungsstufen, Probleme
bei der statistischen Auswertung, die beispielsweise infolge des Überganges einer Ver-
dünnungsstufe mit 100% positiven Replikaten zu 100% negativen Replikaten in der
folgenden Verdünnungsstufe auftreten können, und einiger anderer Faktoren, hat diese
Methode etliche Nachteile. Eine Alternative könnte die Quantifizierung mittels real-
time PCR-Methode darstellen. Bisher veröffentlichte Arbeiten liefern klare Hinweise,
dass zwischen der Parasitenlast und dem PCR-Signal eine Korrelation besteht [64, 94].
Ziel 1: Die bereits beschriebene 18S rDNA-basierte real-time PCR [115] zum Nachweis
von Leishmanien-DNA soll etabliert und evaluiert werden (u.a. Ermittlung optimaler
Primer- und Sondenkonzentrationen, Testung der Spezifität und Sensitivität) und mittels
Amplifizierung von Proben unterschiedlicher Ausgangskonzentrationen an Leishma-
nien-DNA auf ihre quantitative Aussagekraft überprüft werden.
Die Quantifizierung infizierter muriner Organe bzw. Gewebe erfordert das gleichzeitige
mengenmäßige Erfassen eines Wirtszell-Genes, welches während der Infektion mög-
lichst nicht reguliert wird und als Bezugsgröße dient (Angabe als Leishmanien-DNA-
Äquivalente pro Wirtszell-Gen bzw. pro Wirtszelle). Dafür muss ein geeignetes Gen mit
bekannter Kopienzahl im murinen Genom definiert und ausgehend davon ein PCR-
System etabliert werden. Wünschenswert wäre ein Nebeneinander-Ablaufen beider
PCRs in einem Ansatz (sog. Duplex-PCR).
Ziel 2: Nach Etablierung eines geeigneten Leishmanien/Maus-PCR-Systems soll dieses
zunächst in in vitro-Bedingungen getestet werden. Hierbei soll analysiert werden, ob die
Parasitenlast in Zellkulturen von infizierten murinen Knochenmarksmakrophagen (BM-

3. EINLEITUNG
16
MΦ) mit Hilfe der real-time PCR verlässlich quantifiziert werden kann. Die dafür stan-
dardmäßig verwendete Methode ist die Fixierung und Anfärbung mittels DiffQuik®-
Färbereagenz, und die anschließende mikroskopische Ermittlung des prozentualen An-
teils infizierter Zellen und der Parasitenzahl in den infizierten Zellen, woraus sich Infek-
tionsrate und Parasitenlast bestimmen lassen. Es ist demnach zu prüfen, ob die Quanti-
fizierung über das PCR-Signal (ausgehend von einer DNA-Präparation aus entspre-
chenden Zellkulturen) mit den Resultaten aus mikroskopischer Auszählung nach Diff-
Quik®-Färbung übereinstimmt.
Ziel 3: Das etablierte Leishmanien/Maus-PCR-System soll schließlich für ex vivo-
Analysen von Geweben aus infizierten Mäusen (kutane Leishmaniose [L. major], visze-
rale Leishmaniose [L. infantum]) evaluiert werden. Damit verbunden ist die Fragestel-
lung, ob eine Quantifizierung der Parasitenlast durch real-time PCR unter Verwendung
von DNA aus Organen infizierter Mäuse mit den Ergebnissen der LD-Analyse korre-
liert.

4. MATERIAL & METHODEN
17
4 MATERIAL & METHODEN
4.1 Material
4.1.1 Chemikalien
2-Mercaptoethanol Sigma-Aldrich, Deisenhofen
2-Propanol Roth, Karlsruhe
Agarose ultra pure Invitrogen/Life Technologies, Karlsru-
he
Ammoniumchlorid Sigma-Aldrich, Deisenhofen
Brain-Heart-Infusion-Agar, pH 7,4 Charles River, Sulzfeld
DiffQuik® Färbereagenz Medion Diagnostics, Düdingen,
Schweiz
Dulbecco’s modified Eagle Medium
(DMEM)
Gibco/Life Technologies, Karlsruhe
dNTPs (20mM) = PCR Polymerization Mix GE-Healthcare, München
Ethanol Merck, Darmstadt
Ethidiumbromid Roth, Karlsruhe
Ethylenoxid (Anprolene®-Ampullen) Andersen Products, North Carolina
Fötales Kälberserum (FCS) Sigma-Aldrich, Deisenhofen
Humane DNA Bioline, Luckenwalde
LAL Wasser Lonza, Köln
Lambda-DNA New England Biolabs, Frankfurt/Main
MEM, nicht essentielle Aminosäuren, 100x Sigma-Aldrich, Deisenhofen
Phosphate buffered saline (PBS) Seromed-Biochrom, Berlin
pUC Mix MBI Fermentas, St. Leon-Rot
RPMI 1640 Gibco/Life Technologies, Karlsruhe
Hepes Gibco/Life Technologies, Karlsruhe
Penicillin-Streptomycin Gibco/Life Technologies, Karlsruhe
Schneider’s Insekten-Grundmedium Genaxxon BioScience, Ulm
TAE-Puffer, 50x Eppendorf, Hamburg

4. MATERIAL & METHODEN
18
Tetramethylammoniumchlorid (TE) Sigma-Aldrich, Deisenhofen
Taq Polymerase (5 U/µl) Invitrogen/Life Technologies, Karlsru-
he
TaqMan Gene Expression MasterMix 2x Applied Biosystems, Weiterstadt
Trypanblau Gibco/Life Technologies, Karlsruhe
Zap-Oglobin Beckman Coulter, Krefeld
4.1.2 Oligonukleotide für Polymerase-Kettenreaktion (PCR)
PCR/Zielgen Sequenz
TaqMan PCR
Leishmanien-
18S rDNA
[115]
Primer
Sonde
LEIS.U1
LEIS.L1
LEIS.P1
5’-AAGTGCTTTCCCATCGCAACT-3’
5’-GACGCACTAAACCCCTCCAA-3’
FAM-CGGTTCGGTGTGTGGCGCC-TAMRA
Leishmanien-
18S rDNA
(modifiziert)
[36]
Primer
Sonde
LEIS.U3
LEIS.L1
LEIS.P2
5’-TGCTTTCCCATCGCAACTTC-3’
5’-GACGCACTAAACCCCTCCAA-3’
FAM-TTCGGTGTGTGGCGCC-NFQ-MGB
Lambda D Primer LamD_F 5’-CCCTGCGGCTGGTAATGG-3’
[36] LamD_R 5’-TGAACACACCAGTGTAAGGGATGT-3’
Sonde LamD_P VIC-AAGGTTTCTTTGCTCGTCATA-NFQ-
MGB
Lambda C
[36]
Primer LamC_F
LamC_R
5’-CCGTTAACGATTTGCTGAACACA-3’
5’-GGCTGGTAATGGGTAAAGGTTTC-3’
Sonde LamC_P VIC-CAGTGTAAGGGATGTTTATGACGA-
NFQ-MGB

4. MATERIAL & METHODEN
19
Nidogen 3
[36]
Primer Nido.F3 5’-TCGGCTCAGTAGCGCCTT-3’
Nido.R3 5’-CCCAGGCCATCGGTTGT-3’
Nidogen 4
[36]
Sonde
Primer
Sonde
Nido.P3
Nido.F4
Nido.R3
Nido.P4
VIC-CCTGGCTGACTTGGA-NFQ-MGB
5’-ACCCAGCTTCGGCTCAGTAG-3’
5’-CCCAGGCCATCGGTTGT-3’
VIC-CCTTTCCTGGCTGACTT-NFQ-MGB
Kinetoplasten-
PCR
Primer Lei13A 5’-GTGGGGGAGGGGCGTTCT-3’
[86] Lei13B 5’-ATTTTACACCAACCCCCAGTT-3’
Primer/Sonden TaqMan-PCR
Applied Biosystems/Life Technologies,
Darmstadt
Primer/Sonden Kinetoplasten-PCR Sigma-Aldrich, Deisenhofen
4.1.3 Zytokine
Rekombinantes murines Interferon-γ (rmIFN-
γ)
Zur Verfügung gestellt von Dr. G.
Adolf, Ernst-Böhringer-Institut, Wien
Rekombinanter muriner Makrophagen-
kolonie-stimulierender Faktor (rmM-CSF)
R&D Systems, Wiesbaden-
Nordenstadt
Rekombinanter muriner Tumornekrosefaktor-
α (rmTNF-α)
R&D Systems, Wiesbaden-
Nordenstadt
4.1.4 Laborgeräte und Verbrauchsmaterialien
24-well Kulturplatten Cellstar® Greiner Bio-one, Frickenhausen
96-well Kulturplatten Nunc, Wiesbaden
Axioskop 40 Carl Zeiss, Jena
Axiovert 40C Mikroskop Carl Zeiss, Jena

4. MATERIAL & METHODEN
20
BioPhotometer Eppendorf, Hamburg
Brutschrank BBD 6220 Heraeus, Hanau
Brutschrank Heracell 240 Heraeus, Hanau
Chamber Slide LabTek® Thermo Fisher Scientific, Schwerte
Computer Optiplex 745 DELL, Frankfurt
Costar® Stipette® 5, 10, 25ml Sigma-Aldrich, Deisenhofen
Deckgläser Marienfeld, Lauda-Königshofen
Edelstahlsiebe VWR, Nürnberg
Elektrophoresekammer 40-1214 Mini L PeqLab, Erlangen
Feinwaage TE64 Sartorius, Göttingen
Gel Dokumentationssystem Gel iX Imager Intas, Göttingen
GeneAmp® PCR System 9700 Applied Biosystems/Life Technolo-
gies, Darmstadt
Injektionskanülen 20G/27G Becton Dickinson, Heidelberg
Injektionsspritzen 10ml Becton Dickinson, Heidelberg
Injektionsspritzen 20ml Codan, Lensahn
Kunststoffküvette UVette® Eppendorf, Hamburg
Magnetrührer RCT basic IKA, Staufen
MicroAmp® 48-well optical adhesive film Applied Biosystems/Life Technolo-
gies, Darmstadt
Fast optical 384-well reaction plate Applied Biosystems/Life Technolo-
gies, Darmstadt
MicroAmp® Fast optical 48-well reaction
plate
Applied Biosystems/Life Technolo-
gies, Darmstadt
Mikrowelle MW749 Clatronic, Kempen
Minishaker MS2 IKA, Staufen
Multipette® plus Eppendorf, Hamburg
Netzgerät E844 Consort, Turnhout, Belgien
Neubauer Zählkammer (0,02mm Tiefe) Brand, Wertheim
Neubauer Zählkammer (0,10mm Tiefe) Labor Optik, Bad Homburg
Optically Clear Adhesive Seal Sheets Thermo Fisher Scientific, Schwerte
PCR-Reaktionsgefäße 0,2 ml Eppendorf, Hamburg

4. MATERIAL & METHODEN
21
PCR-Reaktionsgefäße 1,5 ml SafeLock Eppendorf, Hamburg
PCR-Reaktionsgefäße 1,5 ml Standard Eppendorf, Hamburg
Petrischalen, steril verpackt Nunc, Wiesbaden
Pipetten 10, 20, 100, 1000µl Eppendorf, Hamburg
Pipetten 2, 10, 20, 200, 1000µl Gilson, Limburg
Pipettenspitzen 10 µl Sarstedt; Nümrecht
Pipettenspitzen 100 µl PeqLab, Erlangen
Pipettenspitzen 1000 µl Sarstedt, Nümrecht
Pipettierhilfe macro Brand, Wertheim
Pipettierhilfe Pipetboy acu Integra Biosciences, Fernwald
Plastikröhrchen (Falcons) 15ml Becton Dickinson, Heidelberg
Plastikröhrchen (Falcons) 50ml Becton Dickinson, Heidelberg
Plastiksiebe cell strainer Becton Dickinson, Heidelberg
Reagenzgläser 5ml (Round bottom tubes) Becton Dickinson, Heidelberg
Schnelltaster Kroeplin, Schlüchtern
StepOne® Real-Time PCR System Applied Biosystems/Life Technolo-
gies, Darmstadt
Sterilbank Hera Safe Heraeus, Hanau
TaqMan 7900 HT Fast Real-Time PCR Sys-
tem
Applied Biosystems/Life Technolo-
gies, Darmstadt
Taumler REAX3 Heidolph, Schwabach
Thermomixer compact Eppendorf, Hamburg
Tischzentrifuge 5417C Eppendorf, Hamburg
Tissue Lyser Qiagen, Hilden
Zellkulturschalen Nunc, Wiesbaden
Zentrifuge Multifuge 3 SR+ Heraeus, Hanau
4.1.5 Kits
High Pure PCR Template Preparation Kit
(100)
Roche Diagnostics, Mannheim

4. MATERIAL & METHODEN
22
4.1.6 Kulturmedien
Kaninchenblut-Schrägagar (Novy-MacNeal-Nicolle, NNN-Agar)
200 ml 1% (v/v) Brain-Heart-Infusion-Agar
50 ml frisches Kaninchenblut (Charles River, Sulzfeld)
50 ml PBS
100 IU/ml Penicillin G (30 000 IU)
0,1 mg/ml Streptomycin
Schneider’s Insektenzellmedium
Schneider´s Insekten-Grundmedium, ergänzt durch Zusatz von:
4 % (v/v) Aminosäure-Antibiotika-Pyruvat (AAP)-Lösung (25x)
10 mM Hepes
10 % (v/v) inaktiviertes FCS (56°C, 30 min)
2 % (v/v) Humanurin, steril
Aminosäure-Antibiotika-Pyruvat-Lösung enthält:
1.5 mg/ml Penicillin G (2500 U/ml)
3.25 mg/ml Streptomycinsulfat
25 mM Natriumpyruvat
50
50
6,8
13,8
mM
ml
mM
mM
L-Glutamin
DMEM, ohne Phenolrot
L-Asparagin
L-Arginin
in deionisiertem (Leitungswiderstand >18 MegaOhm) H2O (dH2O)

4. MATERIAL & METHODEN
23
Differenzierungsmedium für Knochenmarksmakrophagen
DMEM-Kulturmedium (incl. 4,5 g/l Glucose, L-Glutamin, Pyruvat), ergänzt durch Zu-
satz von:
10 % (v/v) inaktiviertes FCS
5 % (v/v) inaktiviertes Pferdeserum
50 µM 2-Mercaptoethanol
1 % (v/v) MEM nicht-essentielle Aminosäuren
15 % (v/v) Überstand der L929 Fibroblastenzelllinie (eigene Herstellung, enthält
m-CSF)
Zellkulturvollmedium
RPMI-1640 (incl. 300 mg/l L-Glutamin, 2000 mg/l NaHCO3),
ergänzt durch Zusatz von:
10 mM Hepes
50 µM 2-Mercaptoethanol
100 U/ml Penicillin G
100
5
mg/ml
% (v/v)
Streptomycin
inaktiviertes fötales Kälberserum (FCS)
Phosphatgepufferte Kochsalzlösung (PBS)
enthält:
0,2 g/l KCl
8,0 g/l NaCl
0,2 g/l KH2PO4
1,44 g/l Na2HPO4
in Aqua bidest.
4.1.7 Versuchstiere
Zur Gewinnung von murinen Organen wurden Mäuse der Inzuchtstämme
BALB/cAnNCrl (BALB/c) und C57BL/6NCrl (C57BL/6) im Alter von 6-12 Wochen
von Charles River Breeding Laboratories, Sulzfeld bezogen. Die Tiere wurden in Käfi-
gen mit Filterhaube gehalten und hatten freien Zugang zu Trinkwasser und Futter.

4. MATERIAL & METHODEN
24
Bei Infektionsexperimenten wurden regelmäßig der Allgemeinzustand und das Gewicht
der Tiere überprüft; die Fußdicke als Parameter der Läsionsentwicklung wurde mithilfe
eines Schnelltasters erfasst und dokumentiert.
4.1.8 Leishmanien
Für die in vitro-Infektion von BM-MΦ und für Mausinfektionsexperimente zum Modell
der kutanen Leishmaniose wurde der Stamm L. major MHOM/IL/81/FEBNI [98] einge-
setzt, der 1981 aus der Läsion einer israelischen Patientin isoliert wurde (Dr. F. Ebert,
Bernhard-Nocht-Institut für Tropenmedizin, Hamburg).
Für Mausinfektionsexperimente zur viszeralen Leishmaniose wurde der Stamm L. in-
fantum MHOM/00/98/LUB1 [10] eingesetzt. Dieser war aus dem Knochenmark eines
an viszeraler Leishmaniose erkrankten Kindes in Bayern isoliert worden.
4.1.9 Klonierungsvektor/Plasmid
Für quantitative Analysen des Leishmaniengehaltes zu untersuchender Proben wurde
von der Arbeitsgruppe ein Plasmid (pGEM-T Easy, Invitrogen/Life Technologies, Dar-
mstadt) zur Verfügung gestellt, in welches die 18S rRNA-Gensequenz aus dem Leish-
maniengenom [115] kloniert worden war (Karin Knoll, Ulrike Schleicher und Christian
Bogdan, unpublizierte Daten, siehe auch Kapitel 4.2.10).
4.2 Methoden
4.2.1 Bestimmung der Zellzahl und –viabilität
Zur Bestimmung der Zellzahl einer Zellsuspension wurde diese 1:10 in 0,08 % (v/v)
Trypanblau verdünnt.
Unter Zuhilfenahme einer Neubauer-Zählkammer kann die Zellzahl mikroskopisch be-
stimmt werden. Dazu wird ein Deckglas auf den Neubauer-Objektträger so aufgebracht,
dass „Newtonringe“ sichtbar werden. Damit ist die Kammer auf ein definiertes Volu-
men geeicht. Die Zählkammer kann nun mit der Trypanblau-gefärbten Zellsuspension
befüllt werden. Lebende und tote Zellen können voneinander unterschieden werden,
weil Trypanblau nur in solche Zellen eindringen kann, deren Zellmembran geschädigt
ist; und die Zellkerne blau anfärbt. Lebende Zellen mit intakter Membran hingegen
werden nicht angefärbt.

4. MATERIAL & METHODEN
25
Die Anzahl der Zellen in einer Suspension kann durch die folgende Formel ermittelt
werden:
Zellen/ml = n/c x f x k
Gesamtzellzahl = Zellen/ml x v
n = Summe der Zellen in den ausgezählten Großquadraten (bestehend aus 4 x 4 Unter-
quadraten)
c = Anzahl der ausgezählten Großquadrate
f = Verdünnungsfaktor in der Trypanblaulösung
k = Kammerfaktor der Neubauer-Zählkammer
(Zählkammer für Zellen: 1 x 104 , Zählkammer für Leishmanien: 5 x 104)
v = Gesamtvolumen der Suspension [in ml]
4.2.2 Gewinnung von Knochenmark aus Mäusen und Generierung von
Knochenmarksmakrophagen (BM-MΦ)
Nach Tötung von naiven Mäusen mittels Isofluran-Narkose und anschließender zervika-
ler Dislokation wurden diesen die Ober- und Unterschenkelknochen entnommen. Im
Anschluss erfolgte das Abtrennen der Epiphysen, Ausspülen der Knochenmarkszellen
mit Zellkulturvollmedium mit Hilfe einer 27G-Kanüle und einer Spritze und Zentrifuga-
tion bei 300 x g, 4°C für 10 min. Das entstandene Zellpellet wurde zur Generierung von
BM-MΦ verwendet. Entsprechend dem Protokoll von Schleicher und Bogdan [92] wur-
den einem Volumen von 50 ml MΦ-Differenzierungsmedium (enthält 5 ng/ml rmM-
CSF) 6 x 106 Knochenmarkszellen hinzugegeben. Die Differenzierung der Knochen-
markzellen zu Makrophagen (engl. bone marrow macrophages, Abk. BM-MΦ) erfolgt
in hydrophober Teflonfolie, die zuvor zu Schläuchen zusammengeschweißt und danach
mit Ethylenoxid (Anprolene®) sterilisiert wurde. Nach einer Inkubation für 7 bis 8 Tage
bei 37°C, 10% CO2, und 95% Luftfeuchtigkeit im Brutschrank können die reifen BM-
MΦ geerntet werden. Die hydrophobe Oberfläche erleichtert dabei das Ablösen der ad-
härenten Zellen.

4. MATERIAL & METHODEN
26
4.2.3 In vitro-Pulsinfektion von murinen Makrophagen (BM-MΦ) mit
Leishmania major
Für in vitro-Experimente wurden murine BM-MΦ von C57BL/6-Mäusen in 8-well
ChamberSlides mit promastigoten L. major für 24h infiziert. Pro well wurden 0,15x106
Makrophagen ausgesät; die gewählte Infektionsdosis (MOI= multiplicity of infection)
betrug 10 Leishmanien pro MΦ. Zusätzlich zu den Leishmanien wurden die MΦ mit
Zytokinen in unterschiedlicher Konzentration stimuliert. Danach wurden die Zellen
zweimal mit warmem PBS gewaschen. Leishmanien, welche nicht phagozytiert worden
sind, werden dadurch entfernt. Anschließend wurde frisches Kulturmedium (RPMI
1640 mit Zusatz von 5% FCS) zugegeben. Es erfolgte eine Kultivierung bei 37°C, 5%
CO2 und 95% Luftfeuchtigkeit im Zellinkubator.
Nach Ablauf von 24h erfolgte jeweils ein Wechsel des Mediums. Das Medium wurde
abgenommen, die Zellen zweimal mit PBS gewaschen und wieder mit frischem Medi-
um inkl. der Stimulanzien versetzt. Vor und nach dem Mediumwechsel erfolgte eine
mikroskopische Kontrolle der Zellen, um sicherzugehen, dass die MΦ nicht versehent-
lich durch den Waschvorgang entfernt wurden. Nach 48 bzw. 72h fand die Analyse der
Zellen mittels lichtmikroskopischer Auszählung statt. Dazu wurde das ChamberSlide
nach zweimaligem Waschen mit warmem PBS mittels DiffQuik® gefärbt. Neben dem
MΦ-Zellkern werden hier auch Kern und Kinetoplast der Leishmania-Amastigoten blau
angefärbt. So ist es möglich, Infektionsrate und Parasitenlast pro Zelle zu ermitteln. Pro
Kammer eines ChamberSlides wurden ca. 250 Zellen in mehreren Gesichtsfeldern aus-
gezählt, um die durchschnittliche Infektionsrate und Parasitenbeladung zu bestimmen.
Die durchschnittliche Anzahl der Parasiten pro MΦ, bezogen auf Gesamtzahl der MΦ,
lässt sich aus der Infektionsrate und der Parasitenzahl pro infiziertem Makrophagen wie
folgt berechnen:
Leishmanien pro MΦ = Infektionsrate [in %] x Ø Parasitenzahl pro infizierter MΦ
100
Parallel zu den verschiedenen Kulturansätzen im ChamberSlide wurden entsprechende
Kulturen und Stimulationen auch in der 24-well-Kulturplatte angesetzt. Pro well wurden
hier 1,5x106 Makrophagen ausgesät (MOI ebenfalls 10). Die Ansätze in der 24-well

4. MATERIAL & METHODEN
27
Platte dienten der Isolation von DNA, welche für TaqMan-Analysen eingesetzt werden
sollten. Das 24-well Format im Gegensatz zum ChamberSlide-Format wurde gewählt,
um die Ausbeute an DNA aus den einzelnen Stimulationsbedingungen zu erhöhen.
4.2.4 Leishmanien-Erhaltungskultur
Für die Weiterzucht der Leishmanien erfolgte abwechselnd die in vitro-Anzucht pro-
mastigoter Leishmanien bei 28°C, 5% CO2 und 95% Luftfeuchtigkeit über maximal 5
bis 8 Passagen auf Kaninchenblut-Schrägagarplatten, und die Infektion von BALB/c-
Mäusen [99]. Für die Anzucht größerer Mengen an Promastigoten wurden diese von der
Blutagarplatte in flüssiges Leishmanienmedium überführt, wo ein weiteres Wachstum
für 2 bis 4 Passagen erfolgte.
4.2.5 Experimentelles Modell der kutanen Leishmaniose
Zur Induktion einer kutanen Leishmaniose wurden die Versuchstiere der Inzuchtstämme
BALB/c und C57BL/6 subkutan (s.c.) mit L. major-Promastigoten, die aus einer statio-
när bewachsenen Blutagarkulturplatte entnommen und 2x mit PBS gewaschen worden
waren, infiziert. Dazu wurden jeweils in die Fußrückenhaut beider Hinterpfoten 3 x 106
L. major, resuspendiert in einem Volumen von 50 µl PBS, injiziert.
Der Infektionsverlauf wurde durch das regelmäßige Messen der Schwellung der Hinter-
pfoten dokumentiert und überwacht; hierzu erfolgte die Bestimmung der Fußdicke 2x
wöchentlich mit einem Schnelltaster. Der Allgemeinzustand der Tiere wurde täglich
kontrolliert.
4.2.6 Experimentelles Modell der viszeralen Leishmaniose
Für das experimentelle Modell der viszeralen Leishmaniose wurden die Versuchstiere
mit L. infantum-Promastigoten s.c. in beide Hinterpfoten infiziert. Die Infektion erfolgte
analog zum L. major-Modell, pro Fuß wurden jedoch nur 106 L.i., in 50 µl PBS, inji-
ziert.
Die Tiere entwickelten eine systemische Leishmaniose; eine Schwellung der infizierten
Haut war dagegen kaum zu beobachten. Die Fußdicke wurde jedoch regelmäßig (1x pro
Woche) überprüft, ebenso das Gewicht der Tiere. Der Allgemeinzustand der Tiere wur-
de täglich kontrolliert.

4. MATERIAL & METHODEN
28
4.2.7 Bestimmung der Erregerlast durch Grenzverdünnungsanalyse (Limi-
ting Dilution Analysis)
Die Erregerlast in den einzelnen Organen der infizierten Mäuse wurde jeweils durch
zwei verschiedene Methoden bestimmt: mithilfe der bereits etablierten Grenzverdün-
nungsanalyse (Limiting Dilution Analysis, LDA) sowie einer quantitativen real-time
PCR.
Es wurden folgende Organe untersucht: im Fall der kutanen Leishmaniose nach Infekti-
on mit L. major beide Hinterpfoten, die drainierenden poplitealen Lymphknoten und die
Milz; bei L. infantum-infizierten Mäusen zusätzlich Leber und Knochenmark von Ober-
und Unterschenkel.
Die Mäuse wurden nach Isofluran-Narkose durch zervikale Dislokation getötet. Danach
wurden die zu untersuchenden Organe entnommen, in sterilen Gefäßen gewogen und in
RPMI-Medium überführt.
Die weitere Aufbereitung erfolgte unter sterilen Bedingungen. Von allen Zellsuspensio-
nen der verschiedenen Organe wurden jeweils zwei identische Ansätze hergestellt, die
nach Abzentrifugation der Zellen mit den Leishmanien (10 min bei 4000U/min, 4°C)
auf unterschiedliche Art und Weise weiter verarbeitet wurden. Eines der Zellpellets
wurde für die LDA-Analyse in Leishmanienflüssigmedium resuspendiert, das andere
Zellpellet bei -20°C weggefroren, um daraus später DNA zu präparieren und die Parasi-
tenlast mittels TaqMan PCR zu evaluieren.
Die Hinterpfoten wurden nach Entfernung der Zehen mit einer Schere zerkleinert und
mithilfe eines Spritzenstempels durch ein Edelstahlsieb gerieben. Dieses wurde mehr-
mals mit PBS gespült. Das Zellpellet wurde auf zwei 50-ml Gefäße (Falcon-Röhrchen)
aufgeteilt, zentrifugiert (10 min bei 4000 U/min, 4°C) und die Überstände verworfen.
Ein Pellet wurde bei -20°C eingefroren, das zweite Pellet in 3 ml Schneider’s Medium
resuspendiert. Für die Parasitenbestimmung der Leber wurde ein kleines Stück abge-
trennt, gewogen (die gesamte Leber wurde ebenfalls gewogen) und durch ein Plastik-
sieb (cell strainer) gedrückt. Auch hier wurde mehrmals mit PBS gespült und die ent-
standene Suspension halbiert und zentrifugiert. Das Pellet für die Grenzverdünnungs-
analyse wurde in 3-10 ml Schneider’s Medium aufgenommen, das zweite Pellet einge-
froren. Milz und Lymphknoten wurden mit dem Stempel einer Injektionsspritze jeweils
durch ein Plastiksieb (cell strainer) gerieben, welches auf ein 50 ml Röhrchen (Falcon)

4. MATERIAL & METHODEN
29
gesetzt wurde. Stempel und Sieb wurden mehrmals mit PBS gespült und anschließend
zentrifugiert. Das entstandene Pellet der Milzzellen wurde zur Lyse der Erythrozyten in
5 ml Ammoniumchlorid (vorgewärmt auf 37°C) resuspendiert und 5 min bei Raumtem-
peratur belassen. Nach einer erneuten Zentrifugierung wurde das Pellet in PBS gelöst.
Es erfolgte eine mikroskopische Auszählung der Zellkonzentration (siehe: Bestimmung
der Zellzahl und –viabilität). Anschließend wurde das Volumen bestimmt, in dem sich
30 Mio. Zellen befinden, vom Gesamtvolumen entnommen und in ein neues Falcon-
Röhrchen gegeben. Beide Ansätze mit 30x106 Milzzellen wurden wiederum zentrifu-
giert, das Pellet für die LDA anschließend in 3 ml Schneider’s resuspendiert. Dadurch
ergibt sich eine Ausgangszahl von 107 Zellen/ml. Für Lymphknoten wurde analog ver-
fahren, allerdings entfällt hier die Zugabe von Ammoniumchlorid. Es wurden 3 Mio.
Zellen pro 3 ml Schneider’s eingestellt, sodass die Ausgangszahl 106 Zellen/ml beträgt.
Knochenmark wurde aus Ober- und Unterschenkelknochen gewonnen. Nach Abtren-
nung der Epiphysen wurden die Knochen mit PBS ausgespült. Nach Zentrifugierung
wurden auch hier die Zellen gezählt und anschließend zweimal 30 Mio. Zellen abzentri-
fugiert. Eines der Pellets wurde in 3 ml Schneider’s Medium resuspendiert, das andere
weggefroren.
Für die LDA wurden anschließend, ausgehend von der Ausgangssuspension, 16-24 Ver-
dünnungsstufen angelegt; je nach erwarteter Parasitenlast betrug der Verdünnungsfaktor
2, 3 oder 5. Hierbei wurden von jeder Verdünnungsstufe 12 Replikate zu je 100 µl in
96-well-Kulturplatten angelegt; diese wurden danach für 7-10 Tage im Brutschrank
kultiviert (bei 28°C, 5% CO2, 95% Luftfeuchtigkeit). Nach der Bebrütung erfolgte die
mikroskopische Kontrolle der Kulturplatten auf Leishmanienwachstum. Es wurde die
Anzahl an positiven (Wachstum erfolgt) und negativen (kein Wachstum erkennbar)
Wells für jede Verdünnungsstufe bestimmt, und mithilfe des Programmes Lcalc® 1.1
(Limiting dilution ananlysis software, StemCell Technologies Inc.) konnte nach den
Regeln der Poisson-Statistik unter Angabe der verwendeten Verdünnungsschritte, der
Zellzahl und des Organgewichtes die Erregerlast pro Organ bestimmt werden.
4.2.8 DNA-Isolierung aus murinen Geweben und Zellkulturen
Die Isolierung von DNA aus murinen Organgeweben und Makrophagenkulturen erfolg-
te mit dem High Pure PCR Template Preparation Kit der Firma Roche Diagnostics ent-
sprechend den Herstellerangaben. Das Aufreinigungsprinzip beruht auf Bindung der

4. MATERIAL & METHODEN
30
DNA an eine Silica-Membran; danach folgt ein Reinigungsprozess durch Waschpuffer
und anschließend die Elution der DNA von der Membran mithilfe eines Elutionspuffers.
Die zu untersuchende Probe enthielt 25-50 mg murines Organgewebe bzw. 104-106 Zel-
len von einer Zellkultur.
Nach Zugabe von 40 µl Proteinase K und 200 µl Tissue Lysis Buffer zum vorliegenden
Zellpellet bzw. zum Organstück erfolgt eine mehrstündige Inkubation, idealerweise
über Nacht, bei 55°C und 1100 U/min im Minishaker, um einen Verdau der Zell- und
Kernmembran sowie Freisetzung der nukleären DNA zu erreichen. Danach wird 200 µl
Binding Buffer zugegeben und für 10 min bei 72°C, 1400 U/min inkubiert. Zur Präzipi-
tation der DNA werden 100 µl reiner Isopropanol zugegeben und die gefällte DNA auf
die Silica-Membran-Isoliersäule aufgetragen. Es folgt eine einminütige Zentrifugation
bei 8700 U/min, wodurch die DNA an die Membran gebunden wird; der Durchfluss
wird anschließend verworfen. Die nächsten Waschschritte sind die Zugabe von je 500
µl Inhibitor Removal Buffer und Wash Buffer, nach deren Beigabe die Säule jeweils
wieder für eine Minute zentrifugiert wird. Nach der wiederholten Zugabe von 500 µl
Wash Buffer erfolgt eine 10-sekündige Zentrifugation bei 11000 U/min, um den restli-
chen gebundenen Alkohol aus der DNA zu entfernen. Im Anschluss wird das Säulchen
auf ein neues PCR-Reaktionsgefäß überführt und 100 µl Elution Buffer zugegeben, der
zuvor auf 70°C erhitzt wurde. Eine einminütige Zentrifugation bei 8700 U/min ergibt
als Durchsatz das Eluat mit der aufgereinigten DNA.
Um den Erfolg der DNA-Isolierung aus den verschiedenen Organen zu kontrollieren, und damit
falsch-negative PCR-Ergebnisse auszuschließen, wurden vor Beginn des Proteinase K-
Verdaus jeder Probe 10µl IPC Probenmix (=internal positive control) hinzugegeben.
Dieser setzt sich zusammen aus 4000 Kopien humaner DNA/µl sowie 15000 Kopien λ-
DNA/µl, welche dem λ-Phagen entstammt. In der quantitativen PCR wird die λ-DNA
ebenfalls amplifiziert. Ein positives Amplifikationsergebnis liefert einerseits die Bestä-
tigung, dass die DNA-Isolierung funktioniert hat, da die λ-DNA den Auf-
reinigungsprozess von Beginn an mit durchläuft; andererseits den Nachweis, dass die
erhaltene DNA frei von PCR-Inhibitoren ist. Somit erfüllt die λ – DNA den Zweck ei-
ner internen Positivkontrolle. Die Zugabe humaner DNA erfüllt beim Einsatz zellarmer
Proben die Funktion einer sogenannten „carrier-DNA“, um die Bildung einer ausrei-
chenden Menge an Präzipitat sicherzustellen.

4. MATERIAL & METHODEN
31
4.2.9 Bestimmung der DNA-Konzentration durch photometrische Messung
Im Anschluss erfolgte eine photometrische Konzentrationsbestimmung der aufgereinig-
ten DNA. Hierzu werden die DNA-Proben zunächst in TE-Puffer 1:50 vorverdünnt. Die
Messung erfolgt bei 260 nm (A260), da Nukleinsäuren bei dieser Wellenlänge ein Ab-
sorptionsmaximum besitzen. Dies lässt sich auf die in Nukleinsäuren enthaltenen Basen
und deren Spektraleigenschaften zurückführen. Für Doppelstrang-DNA bedeutet eine
optische Dichte von 1 eine Konzentration von ca. 50 µg/ml. Für Proteine liegt das Ab-
sorptionsmaximum bei 280 nm (A280). Eine Aussage über die Reinheit der Nukleinsäu-
relösung gibt der Quotient von A260 und A280 an; dieser sollte zwischen 1,6 und 2,0
liegen. Niedrigere Werte deuten auf eine Verunreinigung insbesondere durch Proteine
hin; Werte über 2,0 ergeben sich bei denaturierter DNA oder bei zusätzlichem Vorlie-
gen von RNA.
4.2.10 Quantitative real-time PCR
Für die real-time PCR wurden zwei verschiedene Messgeräte eingesetzt: das StepOne®
Real-Time PCR-System in Kombination mit der StepOne® Software v2.0 und das 7900
HT Fast Real-Time PCR-System mit der dazugehörigen Software SDS 2.3 zum Auswer-
ten der Daten.
Der Nachweis von Leishmanien-DNA erfolgte mithilfe des ribosomalen 18S rRNA-
Gens; das entsprechend der Publikation von Wortmann et al. [115] durch das Primer-
paar (Leis.U1/Leis.L1) und eine Sonde (Leis.P1) zu einem 62 bp langen Fragment amp-
lifiziert wurde. Zusätzlich wurde eine weitere, modifizierte Primer/Sonden-
Kombination getestet. Hierbei ist der Primer Leis.U3 gegenüber Leis.U1 um drei Ba-
senpaare im Leseraster Richtung 3´-Ende verschoben; die Sonde Leis.P2 enthält als
Quencherfarbstoff NFQ-MGB anstelle von TAMRA.
Ein zweites PCR-System dient der Messung des Gehaltes an Wirtszell-DNA, um den
Leishmaniengehalt einer Probe quantifizieren zu können. An die Publikation von Saidac
et al. [91] anlehnend wird das Single-Copy-Gen Nidogen nachgewiesen. Zwei verschie-
dene Primer/Sondensysteme wurden mittels des Programmes PrimerExpress 3.0 gene-
riert: Nido.3 enthält die Primer Nido.F3 und Nido.R3 sowie die Sonde Nido.P3, die
Modifikation Nido.4 enthält statt Nido.F3 den Primer Nido.F4 und die Sonde Nido.P4,
welche im DNA-Leseraster um einige bp Richtung 3‘-Ende verschoben sind. Die für die
Sonden verwendeten Farbstoffe sind VIC sowie NFQ-MGB.

4. MATERIAL & METHODEN
32
Das dritte verwendete PCR-System (λ-PCR) soll als Inhibitionskontrolle dienen: die
hier detektierte Zielsequenz entstammt der DNA des Bakteriophagen Lambda, welcher
aus infizierten E.coli-Zellen isoliert werden kann. Mithilfe der PrimerExpress-Software
wurden zwei alternative Primer-Sonden-Systeme im Bereich der Gene lamC und lamD
ausgesucht, weshalb die Primer/Sonden-Systeme entsprechend mit LambdaC bzw.
LambdaD bezeichnet wurden. Die Sonden sind analog zur Nidogen-PCR mit VIC bzw.
NFQ-MGB markiert. Zu Beginn der DNA-Präparation wurde allen Proben λ-DNA zu-
gegeben. Diese soll als interne Positivkontrolle für die Abwesenheit von PCR-
Inhibitoren und eine erfolgreiche DNA-Isolierung fungieren. Im Falle eines negativen
Ergebnisses der Leishmanien-PCR belegt die gleichzeitige Amplifikation von λ-DNA in
demselben Reaktionsansatz, dass die DNA-Isolierung funktioniert hat und die PCR-
Reaktion nicht inhibiert wurde. Damit ist sichergestellt, dass tatsächlich keine Leishma-
nien-DNA vorlag.
Für jede Probe erfolgten ein Dreifachansatz der Leishmanien/Maus-PCR sowie ein wei-
terer Ansatz der Leishmanien/λ-PCR. Die Dreifachmessung wurde zur Kontrolle der
Pipettiergenauigkeit durchgeführt. Als Ergebnis wird der jeweils der Mittelwert eines
Triplikates angegeben.
Die Leishmanien/Maus-PCR stellt eine Duplex-PCR dar, in der die Primer/Sonden-
Systeme zur Codierung sowohl von muriner als auch Leishmanien-DNA vorliegen. Die
Amplifikate beider PCRs können unabhängig voneinander durch das PCR-Gerät detek-
tiert werden, da zwei unterschiedliche sondengebundene Fluoreszenzmarker eingesetzt
werden. Eine gegenseitige Beeinflussung im Ablauf der PCR-Reaktion, z.B. durch feh-
lerhafte Anlagerung von Primern oder vorzeitiges Aufbrauchen freier Nukleotide, wur-
de durch entsprechende Testungen ausgeschlossen (siehe Kapitel 5.1).
Da die Validierung und Etablierung einer Leishmanien/Maus-Duplex-PCR ein wesent-
liches Ziel der vorliegenden Arbeit war, wird auf die Auswahl und Testung geeigneter
Primer- und Sondenkonzentrationen sowie sondenspezifischer Fluoreszenzmarker in
Kapitel 5.1 des Ergebnisteils näher eingegangen.
Der endgültige PCR-Ansatz (nach Abschluss der Etablierungsphase) setzt sich folgen-
dermaßen zusammen (Tab. 4.1):

4. MATERIAL & METHODEN
33
Reagenz Volumen [µl]
(ges.: 20 µl) Finale Konzentration
Master Mix 2x Konz. 10 1x Konz.
Prämix A
enthält:
Primer LEIS.U1/U3 [4 µM]
Sonde LEIS.P1[2 µM]
Primer Nido.F3/R3 [1,5 µM]
Sonde Nido.P3 [1 µM]
6
[2]
[2]
[2]
400 nM
200 nM
150 nM
100 nM
Proben-DNA 4
Tabelle 4.1: Konzentrations- und Mengenangabe eingesetzter Reagenzien im Ansatz der
Leishmanien/Maus-PCR; der Prämix A wurde zuvor in einer größeren Menge, ausreichend
für je 100 Ansätze, hergestellt und aliquotiert. Dabei enthalten 6µl des Prämixes jeweils 2µl
der Primer sowie 2µl Sonde der Leishmanien-PCR in den angegebenen Konzentrationen
sowie 2µl des Primer/Sonden-Gemisches für die Maus-PCR, welche bereits zuvor zusammen-
pipettiert worden waren (Volumenangabe der einzelnen Bestandteile deshalb in Klammern).
Analog dazu setzt sich der PCR-Ansatz für die Leishmanien/ λ-PCR zusammen, welche
ebenfalls als Duplex-PCR abläuft und somit ermöglicht, dass Leishmanien- und λ-DNA
in einem Ansatz zeitgleich detektierbar sind. Eingesetzte Volumina und die sich erge-
benden Konzentrationen des PCR-Ansatzes sind nachfolgend aufgeführt (Tab. 4.2).
Die Stammlösungen der Primer und Sonden wurden mit 1 mM Tris-HCl pH 8,5 (= 1:10
verdünnter Elutionspuffer des High Pure PCR Template Preparation Kits der Firma
Roche Diagnostics) verdünnt.

4. MATERIAL & METHODEN
34
Reagenz Volumen [µl]
(ges.: 20 µl) Finale Konzentration
Master Mix 2x Konz. 10 1x Konz.
Prämix A
enthält:
Primer LEIS.U1/U3 [4 µM]
Sonde LEIS.P1 [2 µM]
Primer LamD_F/R [1,5 µM]
Sonde LamD_P [1 µM]
6
[2]
[2]
[2]
400 nM
200 nM
150 nM
100 nM
Proben-DNA 4
Tabelle 4.2: Konzentrations- und Mengenangabe eingesetzter Reagenzien Ansatz der Leish-
manien/ λ-PCR; der Prämix B wurde zuvor in einer größeren Menge, ausreichend für je 100
Ansätze, hergestellt und aliquotiert. Dabei enthalten 6µl des Prämixes jeweils 2µl der Primer
sowie 2µl Sonde der Leishmanien-PCR in den angegebenen Konzentrationen sowie 2µl des
Primer/Sonden-Gemisches für die λ-PCR, welche bereits zuvor zusammenpipettiert worden
waren (Volumenangabe der einzelnen Bestandteile deshalb in Klammern).
Für die absolute Quantifizierung ist das Mitführen einer Standardkurve mit bekannten
Ausgangskonzentrationen für jeden Probenlauf erforderlich. Prinzipiell könnte für das
Erstellen einer Leishmanien-DNA Standardkurve Parasiten-DNA eingesetzt werden, die
aus Leishmania-Promastigotenkulturen mit bekannter Parasitenanzahl gewonnen wurde.
Problem hierbei ist jedoch, dass die Zahl und Konzentration an Leishmanien-
Genomäquivalenten ausgehend von einer mikroskopisch ausgezählten Leishmanienkul-
tur nur sehr ungenau ermittelt werden kann, da bei der DNA-Isolierung aus dem Leish-
mania-Promastigotenlysat von einem Verlust an DNA auszugehen ist, der nicht exakt
bestimmt werden kann und von Probe zu Probe schwanken kann. Des Weiteren gibt es
nur ungenaue Angaben zur Anzahl der 18S rRNA-Genkopien in Leishmania (laut Lite-
ratur ca. 200 Kopien pro Genom [61, 84, 109]). Um diese Schwierigkeiten zu vermei-
den, wird zur Erstellung der Leishmania-DNA-Standardkurve ein Plasmid verwendet
(pGEM-T Easy Vektor), in welches das Zielgen der Leishmanien-PCR (18S rDNA)
einkloniert worden war (unpublizierte Daten, Karin Knoll, Ulrike Schleicher, Christian

4. MATERIAL & METHODEN
35
Bogdan): mithilfe eines speziellen Lyseverfahrens erhält man nahezu reine Plasmid-
DNA, und da die Zahl der Basenpaare im Plasmid genau bekannt ist, kann nach photo-
metrischer Absorptionsmessung eine genaue Konzentrationsangabe erfolgen (in Plas-
mid-Kopien/µl). Für die Standardkurve von murinem Nidogen wird genomische DNA
aus der Maus eingesetzt, die aus Ohr-und Lebergewebe von naiven C57BL/6 Mäusen
präpariert worden war. Um eine bestehende Kontamination mit Leishmanien-DNA aus-
zuschließen, wurden alle hierfür verwendeten Gewebeproben zunächst mittels real-time
PCR untersucht; dabei wurden alle Gewebeproben mit positivem Amplifikationsergeb-
nis der Leishmanien-PCR von der weiteren Präparation für die Standardkurven-DNA
ausgeschlossen. Die Konzentration der beiden Standard-DNAs wird zunächst photomet-
risch bestimmt. Mithilfe der folgenden Formel:
1 µg von 1000 bp DNA = 1.52 x10-12 mol = 9.1 x 1011 Moleküle bzw. Kopien [77]
ergibt sich für 1 µg Plasmid-DNA (3076 bp, Summe aus 3015 bp des pGEM-T Easy
Plasmids und 61 bp des PCR-Produkts) eine Anzahl von 2,8x1011 Plasmid-Kopien.
Ausgehend von der DNA-Konzentration kann somit die Kopienzahl des Plasmids pro µl
berechnet werden. Die Quantifizierung von Leishmanien-DNA aus infizierten Mausge-
webeproben anhand der vorliegenden Standardkurve erfolgt demnach als Angabe von
Leishmania 18 rDNA-Kopien bezogen auf Menge an Maus-DNA.
Zur Erstellung der Standardkurve wurde eine dekadische Verdünnungsreihe über 6
Zehnerpotenzen pipettiert (Tab. 4.3).
Kopienzahl Leishmanien-Plasmid/µl Konzentration murine DNA [ng/µl]
105 102
104 101
103 100
102 10-1
101 10-2
100 10-3
Tabelle 4.3: Verdünnungsreihe Standardkurve

4. MATERIAL & METHODEN
36
Pro Verdünnungsstufe wurden 100 µl des Leishmanienplasmid/Mausgewebe-DNA-
Gemisches hergestellt. Die Verdünnung erfolgte in Elution Buffer (entnommen aus dem
High Pure PCR Template Preparation Kit, Roche Diagnostics). Beim Pipettieren der
Verdünnungsreihe war darauf zu achten, bei jeder Verdünnungsstufe die Pipettenspitzen
zu wechseln, um keine DNA zu „verschleppen“. Die Standardkurve wurde im doppelten
Ansatz angelegt. Pro Ansatz wurden in einem PCR-Lauf 4 µl der DNA eingesetzt, ent-
sprechend den zu untersuchenden Proben (siehe Tab. 4.1/4.2). Aus den sich ergebenden
CT-Werten dieser Ansätze mit bekannten Mengen wurde von der Software eine Stan-
dardkurve erstellt, die für den jeweiligen PCR-Lauf die Basis für die Quantifizierung
lieferte (siehe Kapitel 3.2.2 und Abb. 3.1).
Als Kontrolle wurde neben den Probentriplikaten bei jedem Probenlauf eine No templa-
te control im Dreifachansatz mitgeführt, in die anstelle der Proben-DNA Wasser zum
PCR-Mix hinzupipettiert wird. Ein negatives Amplifikationsergebnis weist darauf hin,
dass keine falsch positiven Ergebnisse zu erwarten sind, wie es beispielsweise bei Ver-
unreinigung der Primer/Sonden-bzw. des Mastermixes mit Leishmanien-DNA möglich
wäre.
Eine zusätzliche Validierung der Ergebnisse eines jeden PCR-Laufs lieferte die IPC
Wasser-Kontrolle (enthält 750 Kopien λ-DNA/µl), welche im Einfachansatz mitlief und
mit der Leishmanien/λ-PCR untersucht wurde. Sie enthält nur halb so viel λ-DNA wie
der IPC-Probenmix (1500 Kopien λ-DNA/µl; siehe Kapitel 4.2.8), welcher zu jeder
Probe vor der DNA-Präparation hinzugegeben wurde. Wenn man von einem etwa 50%-
igen DNA-Verlust beim Aufreinigungsprozess ausgeht, sollten die Konzentrationen an
λ-DNA in den Proben und in der IPC Wasser-Kontrolle einander entsprechen und auch
die Amplifikationsergebnisse (CT-Werte) ähnlich sein. Ist dies der Fall ist, kann davon
ausgegangen werden, dass die DNA-Isolation ohne größere Verluste stattgefunden hat.
Dies ist eine wichtige Voraussetzung für die Quantifizierung von Geweben mit geringer
Parasitenlast: würde im Aufreinigungsprozess nämlich zu viel DNA verloren gehen,
könnte dies zu falsch negativen Ergebnissen führen.
Der Ablauf und die Bedingungen für die verschiedenen quantitativen PCRs sind in Tab.
4.4 dargestellt.

4. MATERIAL & METHODEN
37
Phase Temperatur Zeit
1 50°C 2 min
2 95°C 10 min
3 95°C
15 sek 45x
60°C 1 min
Tabelle 4.4: Thermoprofil quantitative PCR
4.2.11 Kinetoplasten-PCR
Mit der hier verwendeten konventionellen PCR-Methode wird ein Abschnitt der Kine-
toplasten-Minicircle-DNA amplifiziert. Die Minicircle-DNA entstammt dem einzigen
Leishmanien-Mitochondrium, welches sich an der Flagellenbasis befindet. Die Ampli-
fikation wurde mit dem PCR-Gerät GeneAmp® PCR System 9700 durchgeführt.
PCR-Primer (13A ; 13B) entsprechen dem Protokoll von Rodgers [86], das entstehende
Amplifikat besitzt eine Länge von 120 bp.
Der PCR-Ansatz setzt sich wie folgt zusammen (Tab. 4.5):
Reagenz Volumen [µl]:
(ges.: 50 µl)
Finale
Konzentration
PCR-Polymerization Mix,
setzt sich zusammen aus:
Wasser
10x Taq Puffer
MgCl2 [50 mM]
dNTPs [20 mM]
Primer Lei13A [100 µM]
Primer Lei13B [100 µM]
37,55
5,0
1,5
0,25
0,25
0,25
1x
1,5 mM
100 µM
500 nM
500 nM
Taq Polymerase [5 U/µl] 0,2 0,02 U/µl
Proben-DNA 5
Tabelle 4.5: Konzentrations- und Mengenangabe eingesetzter Reagenzien je
PCR-Ansatz

4. MATERIAL & METHODEN
38
Bei jedem PCR-Lauf wurden zwei Positivkontrollen (genomische DNA von <0,1 bzw. <0,01
Leishmanien/µl nach mikroskopischer Auszählung) und eine Negativkontrolle (Wasser anstelle
der Proben-DNA) mitgeführt. Die Bedingungen der Kinetoplasten-PCR sind in Tab. 4.6.
dargestellt.
Temperatur Zeit
94°C 3 min
94°C 30 sek
55°C 30 sek 40x
72°C 30 sek
72°C 5 min
4°C ∞
Tabelle 4.6: Thermoprofil Kinetoplasten-PCR
Anschließend wurden pro PCR-Ansatz jeweils 10µl mit 5µl Blaumarker vermischt, auf
ein 2%iges Agarose-Gel in einer Elektrophoresekammer aufgetragen und das PCR-
Fragment durch Elektrophorese (45 min bei 120V) nach Größe aufgetrennt. Zur Be-
stimmung der PCR-Fragmentlänge wurde der DNA-Größenmarker pUC Mix mitge-
führt. Das in die DNA-Banden interkalierte Ethidiumbromid wurde durch UV-
Bestrahlung sichtbar gemacht und mit Hilfe des Gel-Dokumentationssystems Gel iX
Imager ausgewertet.
4.2.12 Statistische Auswertung
Für statistische Analysen wurde der t-Test für unabhängige Stichproben verwendet.
Signifikante Unterschiede mit einer Irrtumswahrscheinlichkeit von p<0,05 werden mit
* gekennzeichnet, wenn sich der Unterschied auf zwei verschiedene Messzeitpunkte
bezog. Beim Vergleich der beiden Mausstämme untereinander sind signifikante Unter-
schiede (p<0,05) mit einer Raute (#) deutlich gemacht.

5. ERGEBNISSE
39
5 ERGEBNISSE
5.1 Etablierung und Evaluierung einer Leishmanien/Maus-
Duplex-PCR zur quantitativen Bestimmung der Parasi-
tenlast
Die standardmäßig eingesetzte Methode zur Bestimmung der Parasitenlast muriner Ge-
webe bei kutaner und viszeraler Leishmaniose ist die Limiting Dilution Analyse, bei der
Verdünnungsreihen von den zu untersuchenden Gewebeproben angelegt werden und
aus dem Verhältnis der Leishmania-positiven und negativen Ergebnisse in den jeweili-
gen Verdünnungsstufen eine statistisch basierte Ermittlung der Erregerzahl erfolgt. Die
Quantifizierung bezieht sich auf lebende Leishmanien und erfolgt bei dieser Methode
indirekt. Bei in vitro-Infektionsexperimenten mit Makrophagenkulturen wird die Parasi-
tenbeladung meist durch mikroskopisches Auszählen der infizierten Zellen evaluiert.
Sowohl die LDA als auch die mikroskopische Analyse stellen eine zeitaufwändige Me-
thode dar und zeigen in Abhängigkeit vom jeweiligen Experimentator eine gewisse
Schwankungsbreite. Viele aufeinander folgende Arbeitsschritte erhöhen zudem die Feh-
leranfälligkeit.
In der vorliegenden Arbeit sollte deshalb ein PCR-System etabliert werden, welches –
basierend auf dem Prinzip der computerbasierten Echtzeitmessung – eine sehr präzise
und reproduzierbare Quantifizierung von Leishmanien ermöglichen würde und die bis-
herig eingesetzten Methoden ersetzen könnte.
Zur Messung von Leishmanien-DNA wurden Primer und Sonden entsprechend dem
Protokoll von Wortmann et al. [115] ausgewählt, die zum Nachweis des 18S rRNA-
Gens innerhalb des Leishmanien-Genoms geeignet sind. Laut Publikation ist das be-
schriebene PCR-System in der Lage, eine Vielzahl an Leishmanienspezies zu erfassen.
Zudem konnte zwischen geringen und hohen Konzentrationen an Leishmanien sehr gut
unterschieden werden, was eine unmittelbare Voraussetzung für eine Quantifizierung
darstellt. Zum Nachweis der Leishmania 18S rDNA wurden von Wortmann et al. die
beiden Primer LEIS.U1 und LEIS.L1 sowie die Sonde LEIS.P1 eingesetzt, welche mit

dem Reporter FAM und dem
die Lage des Primer/Sonden
Abb. 5.1: 18S rRNA-Gensequenz (hier: Spezies
der von Wortmann [115]
(gelb) und Primer LEIS.L1 (grün)
Im Zuge der Etablierung der Leishmanien
optimalen Amplifikationsb
leisten.
5.1.1 Verifizierung der
Zur Optimierung der von Wortmann
wurden einer der beiden Primer
genden als Wortmann modifiziert bezeichnet):
zu LEIS.P1 den Quencherfarbstoff NFQ
(minor groove binder) ermöglicht durch eine verstärkte DNA
Verkürzung der Sonde um drei Basenpaare
modifizierte Primer LEIS.U3 gegenüber LEIS.U1 um drei Basenpaare
5. ERGEBNISSE
40
FAM und dem Quencher TAMRA markiert ist. In Abb
die Lage des Primer/Sonden-Paares innerhalb der 18S rDNA-Sequenz dargestellt.
Gensequenz (hier: Spezies L. amazonensis) mit Lokalisationsa
eingesetzten Primer LEIS.U1 (grün markiert), Sonde LEIS.P1
LEIS.L1 (grün) .
Im Zuge der Etablierung der Leishmanien real-time PCR war es zunächst
Amplifikationsbedingungen auszutesten, um eine effiziente PCR zu gewäh
Verifizierung der geeigneten Primerkonzentration
Zur Optimierung der von Wortmann et al. beschriebenen Leishmania
einer der beiden Primer sowie die Sonde wie folgt leicht modifiziert (im Fo
genden als Wortmann modifiziert bezeichnet): die Sonde LEIS.P2 enthält im Gegensatz
farbstoff NFQ-MGB anstelle von TAMRA. Der Zusatz MGB
) ermöglicht durch eine verstärkte DNA-Bindungsfähigkeit eine
ng der Sonde um drei Basenpaare am 5´-Ende. Dem entsprechend wurde der
Primer LEIS.U3 gegenüber LEIS.U1 um drei Basenpaare
5. ERGEBNISSE
Abbildung 5.1 wird
Sequenz dargestellt.
Lokalisationsangabe
rkiert), Sonde LEIS.P1
zunächst nötig, die
auszutesten, um eine effiziente PCR zu gewähr-
18S rDNA-PCR
leicht modifiziert (im Fol-
nthält im Gegensatz
Der Zusatz MGB
Bindungsfähigkeit eine
sprechend wurde der
Primer LEIS.U3 gegenüber LEIS.U1 um drei Basenpaare im Leseraster

5. ERGEBNISSE
41
Richtung 3´-Ende verschoben. Mit dem modifizierten Primer sollte eine in manchen
Genbank-Einträgen vorhandene Basenfehlpaarung toleriert werden.
LEIS.U1 P1 L1 5´ GACAAGTGCTTTCCCATCGCAACT TCGGTTCGGTGTGT GGCGCCTTTGGAGGGGTTTAGTGCGTC CGG 3´
LEIS.U3 P2 L1 5´ GACAAG TGCTTTCCCATCGCAACTTC GGTTCGGTGTGTGGCGCC TTTGGAGGGGTTTAGTGCGTC CGG 3´
Abb. 5.2: Gegenübergestellt sind die Sequenzunterschiede der Primer- und Sondenpaare nach
Wortmann [115] (obere Zeile, Primer sind grün markiert und Sonde gelb) und Wortmann
modifiziert [36] (untere Zeile): die um drei bp verkürzte Sonde LEIS.P2 lässt eine Verschie-
bung von Primer LEIS.U3 gegenüber LEIS.U1 um ebenfalls 3 bp Richtung 3´-Ende zu.
Es wurden im Folgenden jeweils das ursprüngliche und modifizierte Primer/Sonden-
Gemisch getestet und miteinander verglichen, um die PCR mit möglichst hoher Sensiti-
vität sowie Spezifität auszuwählen.
Im Protokoll der von Wortmann publizierten Arbeit [115] wurden die Primer in einer
Endkonzentration von je 200 nM und die Sonde mit 20 nM eingesetzt. Da sich keine
Angaben zur analytischen Sensitivität finden ließen, wurden zur Optimierung des PCR-
Protokolls die Konzentrationen an Primern und Sonden neu evaluiert.
Zunächst erfolgte bei konstanter Sondenkonzentration (festgelegt auf 200 nM) die Er-
mittlung einer geeigneten Primerkonzentration. Dazu wurden in verschiedenen Ansät-
zen die Primer in Endkonzentrationen von 200, 300 und 400 nM eingesetzt. In Abbil-
dung 5.3 sind exemplarische Amplifikationskurven für unterschiedliche Primerkonzent-
rationen dargestellt (nach Originalprotokoll von Wortmann [115]). Dem Verlauf der
Kurven kann man entnehmen, dass die Primerkonzentration nur einen geringen Einfluss
auf das Ergebnis hat: die CT-Werte, welche als Basis für Detektion und Quantifizierung
anzusehen sind, liegen im gewählten Beispiel mit 36,83 (200 nM Primer), 35,96 (300
nM Primer) bzw. 35,57 (400 nM Primer) sehr nah beieinander. Vorausgesetzt ist eine
konstant belassene Konzentration an Leishmanien-DNA, im aufgezeigten Beispiel han-
delt es sich um eine 10-5 Verdünnung einer Leishmania-DNA-Lösung, die mikrosko-
pisch ermittelt 105 Leishmanien/µl enthielt.
Die verschiedenen Primerkonzentrationen wurden zusätzlich mit weiteren DNA-
Mischungen getestet, die neben Leishmanien-DNA auch humane DNA, Maus- oder λ-
DNA enthielt, um Kreuzreaktionen der Primer mit den verschiedenen DNA-Vorlagen
auszuschließen. Zum Einsatz kamen folgende Mischungen:

5. ERGEBNISSE
42
• 10-3-Verdünnung der Leishmania-DNA Stammlösung mit 105 L./µl
• 10-5-Verdünnung der Leishmania-DNA Stammlösung mit Zusatz von humaner
DNA 10 ng/µl
• 10-5-Verdünnung der Leishmania-DNA Stammlösung mit Zusatz von humaner
DNA 100 ng/µl
• 10-5-Verdünnung der Leishmania-DNA Stammlösung mit Zusatz von Maus-
DNA 10 ng/µl
• 10-5-Verdünnung der Leishmania-DNA Stammlösung mit Zusatz von Maus-
DNA 100 ng/µl
• 10-5-Verdünnung der Leishmania-DNA Stammlösung mit Zusatz von λ-DNA
1,5x103 Kop./µl
• 10-5-Verdünnung der Leishmania-DNA Stammlösung mit Zusatz von λ-DNA
1,5x106 Kop./µl
Der in Abb. 5.3 dargestellte Kurvenverlauf ließ sich mit allen DNA-Mischungen repro-
duzieren (Daten sind nicht gezeigt). Für die Primertestung des modifizierten Protokolls
ergaben sich keine wesentlichen Differenzen zu den beschriebenen Ergebnissen.

5. ERGEBNISSE
43
Abb. 5.3: Amplifikationskurven Leishmanien-PCR (hier: nach Original-Protokoll von
Wortmann et al. [115]) für unterschiedliche Primer-konzentrationen (200 nM - rot
dargestellt, 300 nM - schwarz, 400 nM - blau) bei konstanter Sondenkonzentration (200
nM). Eingesetzt wurde eine 10-5 Verdünnung einer Stammlösung, die mikroskopisch
ermittelt 105 Leishmanien/µl enthielt. Eines von acht ähnlichen Experimenten ist darge-
stellt; jeder Ansatz wurde als Einzelbestimmung durchgeführt.
Es zeigte sich, dass der Zusatz von humaner, Maus- und λ-DNA zur 10-5-Verdünnung
der L.-Stammlösung nicht zur Beeinträchtigung der Leishmanien-PCR führte: beim Zu-
satz unterschiedlicher Mengen „Fremd“-DNA ergaben sich bei konstanter Primerkon-
zentration von 400 nM CT-Werte zwischen 34,89 und 35,96 (es wurden je zwei Experi-
mente pro „Fremd“-DNA durchgeführt, für jeweils unterschiedliche Konzentrationen;
siehe oben). Im Vergleich zum Ansatz mit reiner Leishmanien-DNA (CT-Wert 35,57)
lagen die Abweichungen also bei weniger als einem Amplifikationszyklus.

5. ERGEBNISSE
44
Abb. 5.4: Amplifikationskurven Leishmanien-PCR nach Wortmann-Originalprotokoll [115] (Pri-
merkonzentration konst., 400 nM) für Ansätze mit reiner Leishmanien-DNA sowie dem Zusatz von
humaner bzw. Maus-DNA (finale Konzentration je 10 und 100 ng/µl) sowie λ-DNA (1500 und
150000 Kopien/µl). Eines von drei ähnlichen Experimenten (in Abhängigkeit von der Primerkon-
zentration) ist dargestellt; jeder Ansatz wurde als Einzelbestimmung durchgeführt.
Das Prinzip der quantitativen PCR beruht auf einer linearen, umgekehrt proportionalen
Beziehung zwischen dem CT-Wert und dem Logarithmus der eingesetzten Menge ent-
sprechend folgender Formel:
n = log x / log 2
n = Linksverschiebung des CT-Wertes (Angabe in PCR-Zyklen)
x = Eingesetzte Menge (Angabe als Faktor des Ausgangswertes)

5. ERGEBNISSE
45
Bei der 10-3-Verdünnung der Stammlösung mit 105 L./µl wurde ein CT-Wert von 28,78
ermittelt. Dies bedeutet, dass ein Fluoreszenzanstieg ca. 7 Zyklen früher detektiert wer-
den konnte als bei der 100fach stärker verdünnten Probe (10-5-Verdünnung; CT-Wert
35,57). Damit erfüllt sich die Erwartung, wonach entsprechend der o.g. Formel eine
doppelte bzw. hundertfache DNA-Menge zu einer Linksverschiebung des CT-Wertes
um einen bzw. rund 6,64 Zyklen führt. Da je Ansatz 4 µl Proben-DNA eingesetzt wer-
den, entspräche dies in der 10-5-Verdünnung der Stammlösung (105 L./µl) 4 Leishma-
nien im PCR-Ansatz. Aus dem konstant positiven Amplifikationsergebnis trotz geringer
DNA-Menge lässt sich ableiten, dass das PCR-Verfahren hoch sensitiv ist.
Bei künftig zu untersuchenden Proben ist mit eher niedrigen Mengen an Leishmanien-
DNA im Bereich der unteren Nachweisgrenze zu rechnen. Eine Konzentration von 400
nM Primer lässt im Vergleich zu 200 nM tendenziell auch höhere Sensitivität erwarten.
Außerdem traten keine falsch positiven Amplifikationsergebnisse auf, wenn Proben frei
von Leishmanien-DNA waren (getestet wurden humane, murine sowie λ-DNA; Daten
sind nicht gezeigt). Deshalb wurde die Konzentration auf 400 nM festgelegt.
5.1.2 Verifizierung der geeigneten Sondenkonzentration
Zur Bestimmung der optimalen Sondenkonzentration wurde die Primerkonzentration
konstant bei 400 nM belassen. Es wurden Ansätze mit 100 nM, 200 nM sowie 300 nM
Sonde getestet und gegenübergestellt.
Die hierbei ermittelten CT-Werte lagen für alle getesteten Sondenkonzentrationen nah
beieinander und zeigten auch beim Vergleich der beiden PCR-Systeme, Wortmann ori-
ginal versus Wortmann modifiziert, keine nennenswerten Unterschiede, wie in Tab. 5.1
an einer beispielhaften Probe erkennbar ist (hier: 10-5-Verdünnung der Stammlösung
mit 105 L./µl).

5. ERGEBNISSE
46
Für die 10-5-Verdünnung der Leishmanien-Stammlösung ergaben sich die in Abbildung
3.5 gezeigten Amplifikationskurven, welche für die Testung drei anderer Proben repro-
duziert werden konnten (Daten sind nicht gezeigt):
• 10-5-Verdünnung der Stammlösung mit Zusatz von humaner DNA 100 ng/µl
• 10-5-Verdünnung der Stammlösung mit Zusatz von Maus-DNA 100 ng/µl
• 10-5-Verdünnung der Stammlösung mit Zusatz von λ-DNA 1,5x106 Kop./µl
Wortmann [115] Wortmann modifiziert [36]
100 nM Sonde 35,95 35,61
200 nM Sonde 36,27 36,48
300 nM Sonde 36,42 36,96
Tabelle 5.1: Vergleich der CT-Werte der Leishmanien-PCR, durchgeführt nach dem Wortmann-
Originalprotokoll [115] oder dem modifizierten Protokoll [36] für eine 10-5 – Verdünnung einer
Stammlösung mit 105 Leishmanien/µl, in Abhängigkeit von der gewählten Sondenkonzentration.
Dargestellt ist sind Werte für eines von vier ähnlichen Experimenten; jeder Ansatz wurde als Ein-
zelbestimmung durchgeführt.

5. ERGEBNISSE
47
Abb. 5.5: Amplifikationskurven Leishmanien-PCR (nach Wortmann [115] original
und modifiziert [36]) für unterschiedliche Sondenkonzentrationen (100 nM - blaue
Kurven, 200 nM - grüne Kurven, 300 nM - orange bzw. violett) bei konstanter Primer-
konzentration (400 nM). Probe: 10-5- Verdünnung einer Stammlösung mit 105 Leish-
manien/µl. Dargestellt ist eines von vier ähnlichen Experimenten; jeder Ansatz wurde
als Einzelbestimmung durchgeführt.
Bei Betrachtung der Amplifikationskurven (Abb. 5.5) fiel jedoch auf, dass der Fluores-
zenzanstieg (entspricht ∆Rn der Y-Achse) bei einer Sondenkonzentration von 200 nM
fast doppelt so hoch war im Vergleich zu dem bei 100 nM. Eine weitere Erhöhung der
Sondenkonzentration auf 300 nM führte zwar zu einer weiteren Verstärkung des Fluo-
reszenzsignals, allerdings war der Effekt deutlich geringer ausgeprägt im Vergleich zum
Anstieg der Sondenkonzentration von 100 auf 200 nM. Basierend auf diesen Resultaten
wurde die Sondenkonzentration für alle nachfolgenden Untersuchungen auf 200 nM

5. ERGEBNISSE
48
festgelegt, da hier bei der gewählten Primerkonzentration von 400 nM ein deutliches
Fluoreszenzsignal zu detektieren war.
Wie bereits bei den Primertestungen (siehe 5.1.1) beschrieben konnte auch bei der Eva-
luation der Sondenkonzentration bestätigt werden, dass der Zusatz von humaner, Maus-
oder λ-DNA keinen nennenswerten Einfluss auf den CT-Wert der Leishmanien-PCR hat
(Daten sind nicht gezeigt). Für Ansätze ohne Leishmanien-DNA war das Amplifikati-
onsergebnis stets negativ, was Verunreinigungen der eingesetzten Lösungen und PCR-
Materialien ausschloss.
5.1.3 Etablierung der Duplex Leishmanien/λ-PCR
Ein nächstes Ziel bestand darin, zu überprüfen, ob die Leishmania 18S rDNA-PCR im
Duplexverfahren mit anderen PCRs eingesetzt werden kann, insbesondere mit einer
PCR zum Nachweis von λ-DNA und von Maus-DNA. Dies bietet mehrere Vorteile:
Pipettierungenauigkeiten getrennter Ansätze werden vereinheitlicht, außerdem verrin-
gert sich der Materialaufwand und Chemikalienverbrauch.
Wichtig ist hier, dass beide PCRs sich nicht gegenseitig beeinflussen. Um dies zu errei-
chen, müssen mehrere Bedingungen erfüllt sein:
• Sonden und Primer dürfen nur an der jeweiligen Ziel-DNA binden, sonst kann
es zum unspezifischen Fluoreszenzanstieg und damit zu falsch positiven Ampli-
fikationsergebnissen bzw. erniedrigten CT-Werten kommen.
• Es müssen genügend freie Nukleotide vorhanden sein, sonst besteht die Gefahr,
dass die Amplifikation mit verminderter Effizienz abläuft. Die Folge wäre eine
verminderte Sensitivität; geringe Mengen an Ziel-DNA könnten in einem sol-
chen Fall eventuell gar nicht mehr detektiert werden.
• Die PCR-Sonden müssen mit unterschiedlichen Reportern und Quenchern mar-
kiert sein, die vom PCR-Gerät als verschiedene Fluoreszenzsignale wahrge-
nommen werden können.
Die Leishmanien-Sonden LEIS.P1 (Wortmann Originalprotokoll [115]) bzw. LEIS.P2
(modifiziertes Protokoll [36]) enthalten FAM als Reporter. Für die Sonde der λ-PCR
wurde der Farbstoff VIC gewählt; als Quencher wurde NFQ-MGB festgelegt. Um ge-
genseitige Einflüsse zwischen beiden PCRs auszuschließen, wurden vorab Zielsequen-
zen, Primer und Sonden mit dem Programm PrimerExpress 3.0 verglichen. Diese Soft-
ware kann auf Grundlage der Sequenzdaten ermitteln, ob es zwischen den Primern und

5. ERGEBNISSE
49
Sonden der beiden PCRs zu Wechselwirkungen kommt, die durch lange Bereiche mit
komplementärer Basenpaarung oder durch Ausbildung von Sekundärstrukturen (z.B.
Primerdimere) bedingt sein können.
Entscheidend ist letztlich aber die Kompatibilität beider PCRs in der Praxis. Dazu wur-
de der Ablauf der PCR zwischen folgenden beiden Ansätzen verglichen: Leishmania
18S rDNA-PCR allein und Leishmania 18S rDNA-PCR im Duplex mit der λ-PCR. Als
Proben wurden analog zu den bisherigen Testreihen Leishmanien-DNA, reine λ-DNA,
Gemische aus Leishmanien- und λ-DNA, sowie reine Maus- bzw. humane DNA getes-
tet. Für die λ-PCR wurden zwei verschiedene Primer/Sonden-Systeme kreiert (im Be-
reich der Gene lamC bzw. lamD des λ-Phagen, deshalb mit Lambda C und Lambda D
bezeichnet) Für Primer und Sonde der λ-PCR wurden bewusst Konzentrationen ge-
wählt, die niedriger lagen als bei der Leishmanien-PCR (Primer 150 nM, Sonde 100
nM), um die Sensitivität der Leishmanien-PCR für Proben mit niedriger Parasitenlast zu
bewahren.
Insgesamt wurden also sechs verschiedene PCR-Mixe getestet: Leishmanien-PCR (nach
Wortmann [115]), Leishmanien-PCR plus Lambda C-PCR, Leishmanien-PCR plus
Lambda D-PCR, sowie die gleiche Konstellation mit dem modifizierten Leishmanien-
PCR Protokoll.
Abb. 5.6: Amplifikationskurven Leishmanien-PCR (Wortmann [115] original) - blau

5. ERGEBNISSE
50
dargestellt- im Vergleich zur Leishmanien/Lambda C-bzw. Leishmanien/Lambda D-
Duplex-PCR - grün bzw. violett; Probe: Leishmanien-DNA 10-5 Verdünnung einer
Stammlösung mit 105 Leishmanien/µl plus λ-DNA 1500 Kopien/µl. Dargestellt ist eines
von zwei ähnlichen Experimenten (in Abhängigkeit vom jeweiligen Leishmanien-PCR-
Protokoll); jeder Ansatz wurde als Einzelbestimmung durchgeführt.
In Abbildung 5.6 dargestellt sind Amplifikationskurven der Leishmanien-PCR (Original
Wortmann-Protokoll [115]), Leishmanien/Lambda C-Duplex sowie Leishma-
nien/Lambda D-Duplex für ein DNA-Gemisch aus Lambda-DNA (1500 Kopien/µl) und
der 10-5-Verdünnung der Stammlösung (105 L./µl- also rechnerisch 1 Parasit/µl).
Erkennbar ist, dass alle drei sich ergebenden Amplifikationskurven der Leishmanien-
PCR (als Monoplex-PCR sowie jeweils im Duplex mit Lambda C- bzw. Lambda D-
PCR) sehr nah beieinander liegen, was sowohl den Kurvenanstieg als auch das finale
Fluoreszenzniveau betrifft. Die Amplifikationskurven von Lambda C- bzw. Lambda D-
PCR sind nahezu identisch. Für das nach Wortmann modifizierte Protokoll [36] ließen
sich diese Ergebnisse reproduzieren.
Wie in Tab. 5.2 erkennbar ist, sind die CT-Werte der Duplex-PCRs nahezu identisch mit
der singulär durchgeführten Leishmanien-PCR. Dies gilt für beide PCR-Protokolle, ori-
ginal und modifiziert. Die Lambda-PCR hat also offenbar keinen Einfluss auf den Ab-
lauf der Leishmanien-PCR.
Wortmann [115] Wortmann modifiziert [36]
Leishmanien-PCR
(Monoplex) 36,14 36,93
Leishmanien/Lambda C-
PCR 36,71 37,15
Leishmanien/Lambda D-
PCR 36,44 36,23
Tabelle 5.2: Vergleich der CT-Werte Leishmanien-PCR für Wortmann-Originalprotoko ll [115]
und modifiziertes Protokoll [36] (nicht graphisch dargestellt) in Abhängigkeit vom PCR-Design
(Monoplex bzw. Duplex mit λ-PCR). Probe: Leishmanien-DNA 10-5 Verdünnung einer Stammlö-
sung mit 105 Leishmanien/µl plus λ-DNA 1500 Kopien/µl. Einzelnes Experiment; jeder Ansatz
wurde als Einzelbestimmung durchgeführt.

5. ERGEBNISSE
51
Umgekehrt hemmt auch das Vorhandensein von Leishmanien-DNA nicht die Lambda-
PCR: für reine λ-DNA (Probe: 1,5x103 Kop./µl) zeigt der CT-Wert der Lambda D-PCR
mit 25,90 keine nennenswerte Abweichung vom CT-Wert für ein Gemisch aus λ- und
Leishmanien-DNA (26,09; λ-DNA-Konzentration unverändert) (Lambda C-PCR: 27,90
bzw. 27,93).
Die CT-Werte für die λ-PCR sind niedriger, da die Ausgangsmenge an λ-DNA viel hö-
her ist. Im Vergleich zur Leishmanien-PCR ist der Fluoreszenzanstieg jedoch aufgrund
der geringeren Primer- und Sondenkonzentration niedriger.
In Abbildung 5.7 wird gezeigt, dass es bei der Duplex-PCR nicht zum unspezifischen
Anstieg der Leishmanien-PCR kommt, wenn nur λ-DNA als Probe im Ansatz vorhan-
den ist. Diese exemplarisch gezeigten Amplifikationskurven ergaben sich bei allen vier
getesteten Duplex-PCR-Mixen.
Abb. 5.7: Amplifikationskurven Leishmanien-PCR nach Wortmann original [115] -
blau dargestellt- im Duplex mit Lambda D-PCR - violett dargestellt; Probe: λ-DNA
1500 Kopien/µl. Dargestellt ist eines von vier ähnlichen Experimenten (in Abhängigkeit
von dem jeweiligen Duplex-PCR-Mix); jeder Ansatz wurde als Einzelbestimmung
durchgeführt.

5. ERGEBNISSE
52
Da bei der Leishmanien-PCR in Kombination mit der Lambda C-PCR hier leichte
Schwankungen in der Grundfluoreszenz auffielen (Daten sind nicht gezeigt), wurde die
Lambda D-PCR als interne Positivkontrolle aller folgenden Experimente festgelegt.
Die Ergebnisse für das modifizierte Leishmanien-Protokoll entsprachen denen des Ori-
ginalprotokolls, allerdings waren kleine Schwankungen der Basislinie erkennbar. In
diesem Punkt war die Original-PCR, wie von Wortmann [115] beschrieben, offenbar
weniger störanfällig.
5.1.4 Etablierung einer Maus-PCR und Duplextest mit Leishmanien-PCR
Da das Ziel der PCR-Etablierung die Quantifizierung von Leishmanien-DNA infizierter
muriner Gewebe bzw. Kulturen war, musste im nächsten Schritt eine weitere PCR zur
Detektion von Maus-DNA entwickelt werden. Um als Bezugsgröße verwendbar zu sein,
sollte als Zielgen ein DNA-Abschnitt des murinen Genoms mit bekannter Kopienzahl
gewählt werden.
In einer Publikation von Saidac et al. [91] wurde das murine Nidogen als Wirtszell-PCR
zur Quantifizierung von Borrelien-infiziertem Maus-Gewebe beschrieben. Dieses Sin-
gle-Copy-Gen kommt in jeder diploiden Mauszelle zweimal vor. Eine Angabe von
Leishmanien-DNA im Bezug zu einer bestimmten Menge an murinen Zellen wäre somit
möglich. Da Saidac et al. [91] nicht die TaqMan-Methode, sondern sogenannte Molecu-
lar Beacons einsetzten, mussten geeignete TaqMan-Primer- und Sonden für das murine
Nidogen als Zielsequenz getestet werden. Mithilfe des Programmes PrimerExpress 3.0
wurden auf Grundlage des beschriebenen Genabschnittes zwei geeignete Primer-
/Sondensysteme generiert. Die als Nido.3 und Nido.4 bezeichneten PCR-Systeme [36]
sind mit dem Reporter VIC und dem Quencher NFQ-MGB markiert und wurden im
Folgenden sowohl auf ihr Vermögen, murine DNA zuverlässig zu detektieren, als auch
auf ihre Kompatibilität im Duplex mit der Leishmanien-PCR überprüft.
Die Ergebnisse zeigten ein unbeeinträchtigtes Nebeneinander-Ablaufen der Leishma-
nien- und Maus-PCR-Systeme (Abb. 5.8). Im Vergleich zur Monoplex-Leishmanien-
PCR mit einem CT-Wert von 36,58 (Probe: Leishmanien-DNA 10-5 Verdünnung einer
Stammlösung mit 105 Leishmanien/µl plus Maus-DNA 100 ng/µl) lagen die CT-Werte
für die Leishmanien-PCR im Duplex mit Nido.3 bei 36,71 bzw. im Duplex mit Nido.4
bei 36,98. Des Weiteren wurden Proben mit niedriger konzentrierter Maus-DNA (0,1

5. ERGEBNISSE
53
ng/µl) von beiden Nidogen-PCRs zuverlässig amplifiziert; humane wie auch λ-DNA
wurde nicht amplifiziert (Daten sind nicht gezeigt).
Abb. 5.8: Amplifikationskurve Leishmanien-PCR (Wortmann Original [115]) - jeweils
hellblau dargestellt - im Vergleich zur Leishmanien/Nido.3- bzw. Leishmanien/Nido.4-
Duplex-PCR - Nido.3: hellgrün / Nido.4: dunkelgrün dargestellt; Probe: Leishmanien-
DNA 10-5-Verdünnung einer Stammlösung mit 105 Leishmanien/µl plus Maus-DNA
100 ng/µl. Dargestellt ist eines von zwei ähnlichen Experimenten (in Abhängigkeit vom
jeweiligen Leishmanien-PCR-Protokoll); jeder Ansatz wurde als Einzelbestimmung
durchgeführt.
Wie in Abbildung 5.8 erkennbar, zeigte sich für die Nido.4-PCR ein geringerer Anstieg
der Fluoreszenz im Vergleich zu Nido.3. Dies war durchgehend für alle getesteten Pro-
ben der Fall. Um zu überprüfen, ob höhere Sonden-bzw. Primerkonzentrationen der
Nido4.PCR dies kompensieren würden, wurden diese im nächsten PCR-Lauf jeweils
auf das Doppelte angehoben und mit den ursprünglichen Werten verglichen. Die Ampli-
fikationskurven ergaben keinen verbesserten PCR-Verlauf (Daten sind nicht gezeigt).
Deshalb wurde die Nido.3-PCR als Standard für die Maus-PCR festgelegt (auf den Zu-
satz Nido.3 wird deshalb im Folgenden verzichtet).

5. ERGEBNISSE
54
Mit der nach Wortmann modifizierten PCR [36] für Leishmania im Duplex mit der Ni-
dogen-spezifischen PCR zur Quantifizierung des Maus-DNA-Gehaltes ließen sich ähn-
liche Ergebnisse erzielen (Daten sind nicht gezeigt). Allerdings fielen hier erneut
Schwankungen der Basislinie bei der Leishmanien-PCR auf, insbesondere in den letzten
zehn PCR-Zyklen, wenn ein negatives Amplifikationsergebnis vorlag. Bei derartigen
Fluoreszenzschwankungen besteht die Gefahr, dass durch Überschreiten des Schwel-
lenwertes falsch positive Amplifikationsergebnisse zustande kommen. Aus diesem
Grund wurde das modifizierte Protokoll nicht weiter verwendet und stattdessen das Ori-
ginal-Protokoll als Leishmanien-PCR-Standard festgelegt (für die Leishmanien-PCR
wird deshalb im Folgenden auf den Zusatz „Original“ verzichtet).
Abbildung 5.9 zeigt die erfolgreiche Unterscheidung zwischen unterschiedlichen Men-
gen an Ausgangs-Maus-DNA: die CT-Werte der Nidogen-PCR liegen für 100 ng/µl
DNA bei 22, 48; für 10 ng/µl bei 26,37 sowie für 1 ng/µl bei 30,19. Der gleichmäßige
Abstand zwischen den CT-Werten deutet auf eine sehr präzise Quantifizierungsfähigkeit
der PCR hin.
Abb. 5.9: Amplifikationskurve Leishmanien-PCR - hellblau dargestellt - und Maus-
PCR - jeweils hellgrün dargestellt - bei gleichbleibender Konzentration an Leishma-
nien-DNA (Leishmanien-DNA 10-5-Verdünnung einer Stammlösung mit 105 Leishma-
nien/µl - rechnerisch 1 Parasit/µl) und Zusatz verschiedener Maus-DNA-Mengen (100
ng/µl; 10 ng/µl; 1 ng/µl). Einzelnes Experiment; jeder Ansatz wurde als Einzelbestim-
mung durchgeführt.

5. ERGEBNISSE
55
Im Nachfolgenden wurden diverse Positivproben von Patienten mit bekannter Leishma-
niose aus der diagnostischen Abteilung unseres Institutes sowie Organproben Leishma-
nien-infizierter Mäuse mit den beschriebenen PCR-Methoden getestet. Bei allen Proben
kam es zu einem positiven Amplifikationsergebnis der Leishmanien-PCR, für murine
Gewebe außerdem der Nidogen-PCR (Daten sind nicht gezeigt). Des Weiteren wurde
die DNA vier verschiedener Leishmanien-Spezies (L. major, L. infantum, L. brazilien-
sis, L. tropica) in der Leishmanien-PCR evaluiert und ebenfalls immer detektiert (Daten
sind nicht gezeigt). Zudem wurden Proben von drei verschiedenen, im Labor üblicher-
weise verwendeten Mausstämmen (C3H/HeJ, C57BL/6, BALB/c) überprüft, in denen
allesamt zuverlässig das Vorhandensein von Nidogen-DNA nachgewiesen werden
konnte.
5.1.5 Generierung einer Standardkurve als Voraussetzung zur absoluten
Quantifizierung
Um Proben mit unbekannten DNA-Mengen quantifizieren zu können, ist als Ver-
gleichswert das Mitführen von bekannten Standards erforderlich. Es eignet sich hierfür
eine Verdünnungsreihe. Aus den CT-Werten der einzelnen Verdünnungsstufen kann
eine Standardkurve erstellt werden. Die ermittelten CT-Werte der Proben werden in die-
se Kurve eingetragen, woraus wiederum die Ausgangsmenge an DNA ermittelt werden
kann.
Da für die Leishmanien-Quantifizierung ein Plasmid als Standard mitgeführt werden
sollte (siehe 4.2.10), wurde für dieses eine Verdünnungsreihe, ausgehend von 1010
Plasmid-Kopien/µl, bis hin zu 10-2 Kopien/µl, angefertigt.

5. ERGEBNISSE
56
Abb. 5.10: Amplifikationskurve der Leishmanien-Plasmid-Verdünnungsreihe (Zielkon-
zentration 1010 bis 10-2 Plasmid-Kopien/µl) in der Leishmanien-PCR; keine Amplifika-
tion (=Nulllinie) für 10 -1 und 10-2 Kopien/µl. Eines von zwei repräsentativen Experimen-
ten; jeder Ansatz wurde als Einzelbestimmung durchgeführt.
Abb. 5.11: Errechnete Standardkurve der Leishmanien-Plasmid-Verdünnungsreihe
(Zielkonzentration 1010 bis 10-2 Plasmid-Kopien/µl). CT-Werte sind gegen Konzentration
an Plasmid-DNA aufgetragen. Durch StepOne® Software errechnete Daten: Regressi-
onslinie: -3,392x (Konzentration Leishmanien-Plasmid) + 40,579; Regressionskoeffi-
zient R2=0,998. Eines von zwei repräsentativen Experimenten; jeder Ansatz wurde als
Einzelbestimmung durchgeführt.

5. ERGEBNISSE
57
In Abbildung 5.10 sowie 5.11 erkennt man die entstehenden Amplifikationskurven und
die daraus resultierende Regressionslinie der Standardkurve. Es zeigte sich eine äußerst
genaue und zuverlässige Quantifizierung über einen sehr großen dynamischen Bereich
(dynamic range) von insgesamt 10 Zehnerpotenzen. Bei 100 Kopien/µl, also 4 Kopien
im PCR-Ansatz (da 4 µl pro PCR eingesetzt wurden), fand eine Amplifikation statt (CT
41,12 ± 0,22; Mittelwert ± Standardabweichung aus zwei Verdünnungsreihen); ab der
10-1 -Verdünnung mit statistisch 0,4 Kopien wurde das Ergebnis negativ, wie es bei
korrekter Amplifizierung zu erwarten war. Dies zeigt, dass selbst kleinste Mengen der
Zielsequenz nachgewiesen werden, während Proben ohne entsprechende Ziel-DNA
zuverlässig zu negativen Ergebnissen führen. Das aus der Standardkurve (Abb. 5.11)
errechnete Bestimmtheitsmaß R² (= Regressionskoeffizient) untermalt mit einem Wert
von 0,998 die äußerst präzise Quantifizierung: dieser Koeffizient wird aus der Regressi-
onslinie der Standardkurve errechnet und zeigt die Größe der Übereinstimmung zwi-
schen der Regressionslinie und den individuell ermittelten CT-Werten auf; ein Wert von
1,00 entspräche dabei einer perfekten Übereinstimmung zwischen Regressionslinie und
den einzelnen Datenpunkten [75]. Zudem wird von der StepOne® Software die Effizi-
enz der PCR-Amplifikation berechnet, diese beträgt hier 97,145%. Sie ergibt sich aus
der Neigung der Standardkurve (in diesem Fall -3,392); diese würde im Idealfall (bei
einer Effizienz von 100%) den Wert 3,322 annehmen (siehe Formel in 5.1.1 zur Errech-
nung der CT-Wert-Abstände).
Für alle nachfolgenden PCRs wurde als Standard eine Verdünnungsreihe eingesetzt, die
Leishmanien- und Maus-DNA in einem Ansatz vereint. Ausgehend von 100 ng/µl
Maus-DNA (photometrisch ermittelt) und 105 Kopien des Leishmanien-Plasmids/µl
wurden sechs dekadische Verdünnungen erstellt (siehe 4.2.10), welche von nun an als
Bezugsgröße der Quantifizierung eingesetzt wurden; die Standardkurve wurde in jedem
PCR-Lauf als Duplikat mitgeführt.

5. ERGEBNISSE
58
5.1.6 Vergleich der Sensitivität zwischen Kinetoplasten-PCR und 18S
rDNA real-time PCR
Mithilfe der beschriebenen real-time PCR nach Wortmann [115] wird das 18S rRNA-
Gen nachgewiesen, welches Literaturangaben zufolge in ca. 160-200-facher Kopienzahl
vorliegt [61, 84, 109]. In einer konventionell durchgeführten PCR zum Nachweis von
Leishmanien wird häufig Kinetoplasten-Minicircles DNA amplifiziert [86] die in einer
viel höheren Kopienzahl (10000 bis 20000 pro Leishmanien-Genom) vorliegt [7, 65].
Es stellte sich daher die Frage, ob die konventionelle PCR in ihrer Sensitivität, insbe-
sondere bei niedriger Parasitenlast, der real-time PCR demnach nicht überlegen wäre. In
diesem Fall wäre die Kinetoplasten-PCR als komplementäre Methode bei fraglich nega-
tiven Ergebnissen in der real-time PCR denkbar.
Um die Sensitivität beider Methoden zu vergleichen, wurden Verdünnungsreihen von L.
major-DNA angefertigt (11 dekadische Verdünnungen von 5x104 bis 5x10-6 Leishma-
nien/µl). Diese wurden jeweils mit humaner bzw. muriner DNA versetzt (Konzentration
über die Verdünnungsstufen hinweg gleichbleibend), um Bedingungen „echter“ Proben
nachzuahmen, bei denen nie reine Leishmanien-DNA vorliegt. Erwartungsgemäß soll-
ten in der TaqMan-PCR bis zu Verdünnungsstufe 8 positive Amplifikationsergebnisse
vorliegen, denn dies entspräche mit 5x10-3 Leishmanien/µl bei einem eingesetzten Vo-
lumen von 4 µl einer Anzahl von 3,2 18S rRNA-Genkopien. Für die Kinetoplasten-PCR
wären theoretisch positive Ergebnisse bis Verdünnungsstufe 10 (5x10-5 Leishmanien/µl)
möglich: bei angenommener Kopienzahl der Minicircles-DNA von 10000 und einer
eingesetzten Menge von 5 µl DNA sollten dann 2,5 Kopien im Ansatz vorliegen.
In der Auswertung zeigte die TaqMan-PCR sichere Amplifikationsergebnisse bis zu
Verdünnungsstufe 7 bei Zusatz humaner DNA bzw. bis Verdünnung 8 bei Zusatz muri-
ner DNA. Die Erwartungen zur Sensitivität wurden demnach erfüllt. Für die Auswer-
tung der Kinetoplasten-PCR wurden die PCR-Ansätze nach erfolgter Amplifikation auf
Agarosegel (2%) aufgetragen. Danach wurde eine Spannungsquelle (120 V) für 45 min
angelegt und die Amplifikate mittels Ethidiumbromid-Färbung und Fotografie unter
UV-Licht sichtbar gemacht.

5. ERGEBNISSE
59
Abb. 5.12: Analyse der Kinetoplasten-PCR, aufgetragen auf Agarosegel 2%, Gelelektrophorese für
45 min bei 120V; anschließende Färbung mit Ethidiumbromid. Legende: pUC= Größenmarker;
H2O= Negativkontrolle; P1 = Positivkontrolle (<0,1 Leishmanien/µl); P2 = Positivkontrolle (<0,01
Leishmanien/µl); 1-11= Verdünnungen der Leishmanien-DNA - dekadische Verdünnungen von
5x104 bis 5x10-6 Leishmanien/µl, versetzt mit jeweils 50 ng/µl muriner DNA; weißer Pfeil gibt Lauf-
höhe der nachzuweisenden Bande an; Foto invers dargestellt. Eines von zwei repräsentativen Expe-
rimenten; jeder Ansatz wurde als Einzelbestimmung durchgeführt.
Wie in Abbildung 5.12 erkennbar ist, war bis zur Verdünnungsstufe 7 ein spezifisches
Leishmania-Kinetoplasten-PCR Amplifikat nachweisbar. Bei höheren Verdünnungen
war dies hingegen nicht mehr eindeutig erkennbar; stattdessen wurden unspezifische
Banden sichtbar. Analoge Ergebnisse wurden erzielt, wenn anstelle von muriner DNA
humane DNA zu der Verdünnungsreihe zugegeben wurde (Daten sind nicht gezeigt).
Die Kinetoplasten-PCR war im durchgeführten Experiment also nicht -wie angenom-
men- in ihrer Sensitivität der real-time PCR überlegen, sondern kam dieser maximal
gleich. Einen Vorteil des Einsatzes der Kinetoplasten-PCR bei Organen mit niedriger
Parasitenbeladung sollte es diesen Ergebnissen zufolge also nicht geben.

5. ERGEBNISSE
60
5.1.7 Vergleich zwischen StepOne® Real-Time PCR System und 7900 HT
Fast Real-Time PCR System
Alle bisherigen Erkenntnisse zu Etablierung und Evaluierung der verschiedenen real-
time PCR-Systeme wurden mit dem StepOne® Real-Time PCR System gewonnen. Da
dieses PCR-Gerät maximal 48 verschiedene PCR-Reaktionen parallel erfassen und ana-
lysieren kann, während das 7900 HT Fast Real-Time PCR System 384 simultane Ampli-
fikationen durchführen kann, sollte überprüft werden, ob die etablierte Leishmania 18S
rDNA–PCR und die murine Nidogen-PCR auf das real-time Gerät mit der größeren
Kapazität übertragbar war. Dies geschah insbesondere in Hinblick auf zukünftige Maus-
infektionsexperimente, welche eine große Menge an Proben erwarten ließen.
Um Ablauf der Amplifikation und Sensitivität beider Geräte vergleichen zu können,
sollten nun einige Proben mit dem 7900 HT Fast Real-Time PCR System amplifiziert
werden, die zuvor schon mittels StepOne® Real-Time PCR System quantifiziert worden
waren. Es bot sich an, die für den Vergleich mit der Kinetoplasten-PCR (siehe 5.1.6)
hergestellten Verdünnungsreihen (5x104 bis 5x10-6 Leishmanien/µl plus Zusatz muriner
bzw. humaner DNA) hier erneut zu verwenden, da damit auch die Sensitivität des neuen
Gerätes sehr gut evaluiert werden konnte. Es erfolgte eine absolute Quantifizierung der
einzelnen Verdünnungsstufen; die Basis hierfür bot das Mitführen der Standardkurve
(siehe 5.1.5). In Abbildung 5.13 ist der Vergleich der Resultate beider PCR-Geräte für
die einzelnen Verdünnungsstufen dargestellt (gezeigt sind 5x104 bis 5x10-2 Leishma-
nien/µl mit Zusatz muriner DNA).

5. ERGEBNISSE
61
Abb. 5.13: Vergleich der Auswertung einer dekadischen Leishmanien-Verdünnungsreihe (5x104 bis
5x10-2 Leishmanien/µl, versetzt mit jeweils 50 ng/µl muriner DNA) zwischen StepOne® Real-Time
PCR System (schwarze Balken) und 7900 HT Fast Real-Time PCR System (weiße Balken); angezeigt
sind die mithilfe der Standardkurve ermittelten Quantitäten (Angabe in Plasmid-Kopien/µl) für die
jeweils untersuchten Verdünnungsstufen (Angabe in Leishmanien/µl); - eines von zwei repräsenta-
tiven Experimenten, jeder Ansatz wurde als Einzelbestimmung durchgeführt.
Erkennbar ist, dass die ermittelten Quantitäten zwischen beiden TaqMan-Geräten ver-
gleichbar sind, ebenso wie die Sensitivität bei geringen DNA-Mengen. In einem zwei-
ten Experiment, dem die Verdünnungsreihe mit Zusatz humaner DNA zugrunde lag,
konnten diese Schlussfolgerungen bestätigt werden (Daten sind nicht gezeigt).
Die Bedingungen für einen Umstieg auf das 7900 HT Fast Real-Time PCR System wa-
ren demzufolge gewährleistet und die Vortestungen, bestehend aus Etablierung und
Evaluierung einer geeigneten real-time PCR, wurden als abgeschlossen betrachtet.

5. ERGEBNISSE
62
5.2 Vergleich zwischen DiffQuik® und real-time PCR für in
vitro-Pulsinfektionen muriner Knochenmarksmakropha-
gen
Nach Etablierung der Leishmanien/Maus-Duplex-PCR im real-time-Verfahren sollte
das PCR-System in einer in vitro-Anwendung getestet werden. Hierbei wurde unter-
sucht, ob die Bestimmung der Parasitenbeladung einer Leishmania-infizierten Makro-
phagenkultur mittels real-time PCR vergleichbare Ergebnisse liefert wie die mikrosko-
pische Ermittlung der Parasitenlast und Infektionsrate nach DiffQuik®-Färbung.
Im hier durchgeführten Versuch wurden Zellkulturen von C57BL/6 BM-MΦ mit
Leishmania-Promastigoten infiziert (MOI = 10) und danach entweder in Medium für
24-72h inkubiert oder mit IFN-γ ± TNF stimuliert, da diese Zytokinkombination be-
kanntermaßen die Expression von iNOS-Enzym und in der Folge die Produktion von
NO in den Makrophagen anregt, und dadurch die Parasiten in Abhängigkeit von der
freigesetzten NO-Menge abtötet [26, 48].
Für die mikroskopische Auswertung wurden die BM-MΦ in ChamberSlides ausgesät;
für die real-time PCR wurden 24-well-Plates verwendet, da eine größere Zellzahl benö-
tigt wurde, um ausreichend DNA für die anschließende PCR einsetzen zu können.
Nach mikroskopischer Auszählung wurden folgende Infektionsraten ermittelt (Abbil-
dung 5.14A):

5. ERGEBNISSE
63
Nach Ablauf der Pulsinfektion für 24h und anschließenden 48-stündigen Inkubation
sind ohne Zugabe von Stimulanzien ca. 50% aller Makrophagen infiziert (Medium + L.
major). Wie erwartet, reduzierte sich die Infektionsrate bei simultaner Zugabe von IFN-
γ und TNF-α auf unter 5%. In einem zweiten Experiment wurde zudem die Wirksam-
keit bei alleiniger Zugabe von IFN-γ (in unterschiedlich hohen Konzentrationen) bzw.
TNF-α untersucht: auch das Vorliegen von IFN-γ allein führte bei 20 ng/ml noch zu
einer leichten Abnahme der Infektionsrate, wohingegen bei den geringeren Konzentrati-
onen [2 bzw. 0,2 ng/ml] bzw. alleiniger Gabe von TNF-α kein Einfluss auf die Infekti-
onsrate mehr erkennbar war. Analoge Schlussfolgerungen lassen sich für die Angabe
der Parasitenlast pro infizierter Zelle ziehen (Abb. 5.14B).
Für eine bessere Vergleichbarkeit mit den TaqMan-Ergebnissen wurde auf Basis der
Infektionsrate und der Parasitenzahl pro infiziertem MΦ die Anzahl der Leishmanien in
Abb. 5.14: Infektion von C57BL/6 BM-MΦ mit L. major-Promastigoten (MOI=10; Pulsinfektion
für 24h, Gesamtdauer der Inkubation 72h); Zugabe von IFN-γ und TNF-α in unterschiedlich hohen
Dosierungen [Angabe in ng/ml]. Die ersten beiden Ansätze ohne Zugabe von Leishmanien (Nega-
tivkontrollen). Dargestellt sind Ergebnisse nach DiffQuik®-Färbung und anschließender mikro-
skopischer Auszählung; Mittelwert ± SD zweier unabhängiger Experimente für die ersten vier An-
sätze.
=nicht messbar
A: Angabe der MΦ-Infektionsraten [%]
B: Angabe von Leishmanien pro infizierte MΦ

5. ERGEBNISSE
64
der gesamten MΦ-Kultur, und davon ausgehend der Quotient Leishmanien/MΦ gesamt
errechnet, da mittels real-time PCR nur die Gesamtzahl aller MΦ (Menge der vorlie-
genden Maus-DNA) ermittelbar ist.
Zur Angleichung der Einheiten wurden die TaqMan-Ergebnisse (Angabe in Plasmid-
Kopien bezogen auf Maus-DNA) umgerechnet und der Einheit Leishmanien/MΦ ange-
glichen. Zunächst erhält man aus den TaqMan-Werten durch Kürzen der Einheiten die
Angabe (L.)-Plasmidkopien pro ng (Maus-DNA):
Einheitenangleichung aus TaqMan: � �������� = �� �������� ⇒ ������
(Leis= Angabe der Leishmanien in Kopien/µl; Nido= Angabe der Maus-DNA in ng/µl)
Danach war die Umrechnung von Plasmid-Kopien in Leishmanien erforderlich. In der
Literatur lassen sich Näherungswerte von 160-200 Kopien der 18S rDNA je Parasit fin-
den [61, 84, 109]. Der Einfachheit halber wurde von 200 Kopien ausgegangen. Mit der
Annahme, dass 200 ng murine DNA ca. 105 Nidogen-Kopien enthalten [91] (entspricht
beim Vorliegen des doppelten Chromosomensatzes 5x104 Zellen), ergibt sich eine Zell-
zahl von 250 MΦ pro ng DNA. Demzufolge können die TaqMan-Ergebnisse nach fol-
gender Formel in die Einheit Leishmanien/MΦ umgerechnet werden:
������ ÷ 250�Zellen$ ÷ 200�Plasmidkopien$ = . ∗ �012 3456789�54�:;
Annahme: 1 Parasit enthält 200 Kopien (der 18S rDNA); 1 ng (Maus-DNA) enthält
250 MΦ-Zellen (siehe oben)
Entsprechend diesen Umrechnungen sind die Parasitenbeladungen für beide Methoden
vergleichend in Abbildung 5.15 dargestellt.

5. ERGEBNISSE
65
Zunächst einmal lässt sich feststellen, dass die TaqMan-Ergebnisse glaubwürdige Werte
zwischen 0 und 10 Parasiten/MΦ liefern; auch sind die Werte der ersten beiden Ansät-
ze, denen keine Leishmanien hinzugefügt wurden, korrekterweise gleich null. Für reines
Medium ohne Zugabe von Stimulanzien wird laut TaqMan ein Wert von 0,5 L./MΦ
ermittelt, im Ansatz mit IFN-γ + TNF-α hingegen ist eine höhere Beladung (1,3 L./MΦ)
erkennbar. Dies steht im Gegensatz zu den DiffQuik®-Ergebnissen, denn mikrosko-
pisch betrachtet kommt es hier zu einer nahezu vollständigen Elimination der Parasiten.
Auch entspricht ein solches Ergebnis nicht den Erwartungen, da die Gabe der Stimulan-
zien ja zu einer Abtötung von Erregern führen soll. Die niedrigste Leishmanienbeladung
liegt laut TaqMan in den Ansätzen mit den gering konzentrierten Immunstimulanzien
vor, welche den DiffQuik®-Ergebnissen zufolge keine Abtötung bewirken, sondern in
Abb. 5.15: Parasitenlast nach Infektion von C57BL/6-BM-MΦ mit L. major-Promastigoten
(MOI=10; Pulsinfektion für 24h, Gesamtdauer der Inkubation 72h); Zugabe von IFN-γ und TNF-α
in unterschiedlich hohen Dosierungen [Angabe in ng/ml], Ergebnisse in Leishmanien/MΦ (gesamt).
Die ersten beiden Ansätze ohne Zugabe von Leishmanien (Negativkontrollen); Mittelwert ± SD
zwei unabhängiger Experimente für die ersten vier Ansätze.
Dargestellt sind Ergebnisse nach mikroskopischer Auswertung (DiffQuik®) bzw. TaqMan.
=nicht messbar

5. ERGEBNISSE
66
der Leishmanienbeladung pro MΦ sogar höhere Werte zeigen als bei den in Medium
gehaltenen Kulturen (z.B. Medium [2 Leishmanien/MΦ] versus TNF-α Stimulation [4
Leishmanien/MΦ]).
In der Zusammenschau führen diese Vergleiche zu der Schlussfolgerung, dass die Er-
gebnisse der real-time PCR nicht mit der mikroskopischen Auszählung korrelieren.
5.3 Vergleich zwischen Limiting Dilution Analysis und real-
time PCR für in vivo-Infektionsexperimente
5.3.1 Vergleich der Parasitenlast von BALB/c-bzw. C57BL/6-Mäusen mit-
tels LDA und TaqMan nach subkutaner Infektion mit L. major
Ein weiteres Ziel der vorliegenden Arbeit war die Evaluierung, ob die real-time PCR-
Methode für die Quantifizierung der Leishmanienlast in Organen von infizierten Mäu-
sen einsetzbar sei und sich mit den Ergebnissen aus der etablierten Limiting Dilution
Analysis (LDA) als vergleichbar erweisen würde.
In einem ersten Experiment wurden hierzu resistente C57BL/6 und suszeptible BALB/c
Mäuse mit 3x106 L. major-Promastigoten pro Hinterpfote infiziert.
Als Parameter für den klinischen Verlauf wurde die Schwellung der infizierten Fußhaut
gemessen (Abb. 5.16).

5. ERGEBNISSE
67
Wie in der Literatur bereits beschrieben [39], entwickelten BALB/c Mäuse eine pro-
gressive stetig fortschreitende Erkrankung, während C57BL/6 Mäuse nur vorüberge-
hend ein Anschwellen der infizierten Haut zeigten und nach mehreren Wochen klinisch
wieder völlig ausheilten.
Um über den Befall verschiedener Organe während der Akutphase der Infektion eine
Aussage zu gewinnen, wurde die Tiere 28 Tage nach Infektion (post infectionem, = p.i.)
getötet, die infizierte Fußhaut, die drainierenden poplitealen Lymphknoten sowie die
Milz zu Homogenaten bzw. Einzelzellsuspensionen aufgearbeitet und anschließend die
Leishmanienbeladung im Organ einmal mit Hilfe der real time PCR-Methode und in
Abb. 5.16: Verlauf der Fußschwellung bei BALB/c- und C57BL/6-Mäusen nach erfolgter Infektion
mit L. major; die Mäuse wurden s.c. mit 3 x 106 L. major-Promastigoten je Hinterpfote infiziert.
Dargestellt ist die prozentuale Zunahme der Fußdicke zum jeweiligen Zeitpunkt nach Infektion
(p.i.). Angabe von Mittelwert ± SD von anfänglich 12 Mäusen, davon vier BALB/c-Mäuse und acht
C57/BL6-Mäuse, von diesen wurden vier Mäuse mit den BALB/c-Mäusen an Tag 28 aus dem Ex-
periment genommen; vier weitere Mäuse wurden bis Tag 140 untersucht (Ende des Experiments);
einzelnes Experiment.

5. ERGEBNISSE
68
einem parallelen Ansatz mit Hilfe der LDA bestimmt. Um eine Aussage über Parasiten-
beladung in der Persistenzphase nach klinischer Ausheilung zu gewinnen, wurden
C57BL/6-Mäuse auch 140 Tage p.i. entsprechend obiger Angaben analysiert.
Pro Zeitpunkt und Mausstamm wurden je vier Mäuse untersucht, um ein repräsentatives
Ergebnis der Parasitenlastbestimmung zu erhalten.
Für den Vergleich der Parasitenbeladungen mussten zunächst einige Umrechnungen
erfolgen, denn die Einheiten der beiden Methoden sind verschieden: die LDA-
Ergebnisse sind in Leishmanien/Gramm Maus-Gewebe bzw. Leishmanien/Zellen ange-
geben; die Quantifizierung mittels Standardkurve (TaqMan) ergibt eine Angabe in
Leishmanien-Plasmidkopien pro ng Maus-DNA. Eine Umrechnung der TaqMan –Werte
in Leishmanien pro Mauszelle erfolgte analog dem in 3.2. beschriebenen Schema ent-
sprechend der Annahme, dass 200 ng murine DNA ca. 105 Nidogen-Kopien bzw. 5x 104
Zellen entspricht [91], woraus eine Zellzahl von 250 pro ng DNA folgt. Für die Um-
rechnung von Leishmanien-Kopien in Leishmanien wurde mit einem Näherungswert
von 200 Kopien je Parasit gerechnet [61, 84, 109]; da dies aber ein ungenauer Wert ist,
wurden die Ergebnisse hier zusätzlich in Plasmidkopien angegeben.
Für Organe, bei denen keine mikroskopische Zellzahl-Bestimmung möglich ist (hier:
Fuß, im folgenden Experiment außerdem Leber) sollte die Leishmanien-Zahl bezogen
auf 1g des jeweiligen Organgewebes angegeben werden. Hierfür wurde das im TaqMan
eingesetzte Organgewicht bestimmt und davon ausgehend die Anzahl der Leishmanien
für 1g des Gewebes berechnet. Somit konnte eine Umrechnung der TaqMan-Ergebnisse
und Angleichung an die LDA-Einheiten erfolgen.
In Abbildung 5.17 ist die Parasitenlast in der Hautläsion, dem Lymphknoten und der
Milz nach L. major-Infektion nach LD- und TaqMan-Analyse dargestellt. Die TaqMan-
Ergebnisse sind sowohl in Leishmanien als auch Leishmanien-Plasmidkopien pro Or-
ganzellen bzw. –gewicht gezeigt.

5. ERGEBNISSE
69
Abb. 5.17: Parasitenlast verschiedener Organe nach Infektion mit L. major. BALB/c- und C57BL/6-
Mäuse wurden beidseitig mit 3x106 L. major-Promastigoten subkutan infiziert. 28 (BALB/c und
C57BL/6 im Vergleich) bzw. 140 Tage (nur C57BL/6) nach der Infektion wurde die Parasitenbela-
dung in der Fußhaut, dem drainierenden Lymphknoten (LK) und der Milz mittels LD- und Taq-
Man-Analyse ermittelt. Gegenübergestellt sind die Messergebnisse von BALB/c - und C57BL/6 -
Mäusen. Die TaqMan-Ergebnisse sind jeweils in Leishmanien-Genkopien (Mittelreihe) und errech-
neter Leishmanienzahl (rechte Reihe, siehe Erklärungen im Text) angegeben und auf das Gewicht
des Gewebes (Gramm) bzw. Zellen des Zielorgans bezogen. Signifikante Unterschiede im t-Test
zwischen BALB/c- und C57BL/6-Mäusen (p<0,05) sind mit #, zwischen frühem und spätem Zeit-
punkt bei C57BL/6 mit * gekennzeichnet; Angabe von Mittelwert ± SD mit vier Mäusen je Gruppe
und Zeitpunkt; einzelnes Experiment.
Wie erwartet, wurde in Fuß, Lymphknoten sowie Milzgewebe von BALB/c-Mäusen
während der Akutphase der Infektion (d28 p.i.) eine signifikant erhöhte Leishmanienlast
im Vergleich zu C57BL/6-Mäusen bei der LD-Analyse ermittelt. Ein signifikanter Un-
terschied zwischen den beiden Mausstämmen zu diesem Infektionszeitpunkt konnte

5. ERGEBNISSE
70
auch mit Hilfe der TaqMan-Analyse erhoben werden. Ebenso zeigte sich für den
C57BL/6-Stamm in Fuß und Lymphknoten sowohl bei TaqMan als auch LDA eine sig-
nifikante Abnahme der Parasitenlast zum späten Infektionszeitpunkt (d140 p.i.), wäh-
rend dies bei der Milz für beide Methoden nicht zutraf. Im Milzgewebe ließen sich al-
lerdings insgesamt viel geringere Werte als in den anderen Organen feststellen. Insge-
samt zeigten die Bestimmung der Parasitenbeladung mittels LDA oder TaqMan über-
wiegend große Übereinstimmung, die in der LDA gewonnenen Aussagen zu signifikan-
ten Unterschieden zwischen den beiden Mausstämmen bzw. Zeitpunkten lassen sich im
TaqMan sogar zu 100% reproduzieren. Lediglich bei zwei Vergleichen ergaben sich
signifikante Unterschiede zwischen beiden Methoden: die Parasitenlast des Fußes in
BALB/c-Mäusen lag in der LD-Analyse um etwa zwei Zehnerpotenzen über den Taq-
Man-Werten. Für Lymphknoten ergaben sich in C57BL/6-Mäusen d28 p.i. um ca. eine
Zehnerpotenz erhöhte Werte in der TaqMan-Analyse. Für alle anderen Zeitpunkte sowie
Milzgewebe konnten keine signifikanten Abweichungen festgestellt werden. Damit
ergibt sich, dass in 77,8% der verglichenen Wertepaare keine signifikanten Abweichun-
gen zwischen den absoluten Werten beider Methoden auftreten.
5.3.2 Vergleich der Parasitenlast von BALB/c-bzw. C57BL/6-Mäusen mit-
tels LDA und TaqMan nach subkutaner Infektion mit L. infantum
In einem weiteren Experiment wurden BALB/c- und C57BL/6-Versuchstiere mit Pro-
mastigoten der Spezies L. infantum, welche beim Menschen eine viszerale Infektion
hervorrief [10], und nach intravenöser Injektion auch im Mausmodell eine viszerale
Leishmaniose auslöste (unpublizierte Daten, Ulrike Schleicher und Christian Bogdan),
subkutan in beide Hinterpfoten infiziert. Diese Injektionsart wurde bewusst gewählt, da
sie gegenüber einer i.v.-Injektion eher dem natürlichen Infektionsweg, nämlich einem
Stich durch die Sandmücke, entspricht. Da die allermeisten Daten aus der Literatur im
Mausmodell der viszeralen Leishmaniose nach intravenöser Injektion der Promastigoten
erhoben wurden [2, 35], sollte bei diesem Experiment auch evaluiert werden, ob eine
systemische Leishmaniose nach subkutaner Applikation der viszerotropen Erreger aus-
gelöst werden kann. Es wurde eine Infektionsdosis von 106 Parasiten je Fuß festgelegt.
Literaturdaten weisen darauf hin, dass es -im Gegensatz zur L. major-Infektion -im zeit-
lichen Verlauf einer L. infantum-Infektion keine signifikanten Unterschiede zwischen
BALB/c- und C57BL/6-Mäusen gibt [46, 55].

5. ERGEBNISSE
71
Zu untersuchende Organe waren neben Fußhaut, Lymphknoten und Milz auch das Kno-
chenmark und die Leber. Die Parasitenlast wurde an fünf Zeitpunkten (Tag 10, 28, 59,
118 und 174 nach Infektion) erfasst, wobei pro Zeitpunkt je zwei BALB/c- und zwei
C57BL/6-Mäuse untersucht wurden. Zur Vergleichbarkeit der Ergebnisse von LDA und
TaqMan erfolgte auch hier eine Anpassung der Einheiten, indem analog zu dem bei
5.3.1. beschriebenen Vorgehen die TaqMan-Ergebnisse in Leishmanien/Gramm (für
Fuß und Leber) bzw. Leishmanien/Zellen (für Lymphknoten, Milz und Knochenmark)
umgerechnet wurden.
Die Fußdicke der infizierten Mäuse wurde regelmäßig gemessen und dokumentiert, ihr
Verlauf ist in Abbildung 5.18 dargestellt.
Abb. 5.18: Verlauf der Fußschwellung bei BALB/c- und C57BL/6-Mäusen nach erfolgter L. infan-
tum-Infektion; die Mäuse wurden s.c. mit 106 L. infantum-Promastigoten je Hinterpfote infiziert
Dargestellt ist die prozentuale Zunahme der Fußdicke zum jeweiligen Zeitpunkt nach Infektion
(p.i.); Angabe von Mittelwert ± SD mit anfänglich zehn Mäusen je Gruppe, von diesen wurden je-
weils zwei Mäuse pro Zeitpunkt (Tag 10, 28, 59, 118 und 174 p.i.) aus dem Experiment genommen;
einzelnes Experiment.
Erkennbar ist eine nur mäßige Zunahme der Schwellung bei BALB/c-Mäusen (graue
Kurve), welche nach einem Maximum um Tag 21 wieder abnimmt und den Ausgangs-

5. ERGEBNISSE
72
wert erreicht. Bei C57BL/6-Mäusen (schwarze Kurve) ist ein stärkeres Anschwellen
(ca. 40% über Ausgangsniveau) erkennbar, welches ab der vierten Woche p.i. jedoch
genauso rasch wieder abebbt.
Die Ergebnisse der Parasitenlastbestimmung mittels LDA bzw. TaqMan sind in Abbil-
dung 5.19 dargestellt. Da die Werte der in Leishmanien umgerechneten Angaben (Taq-
Man) in 5.3.1 den LD-Ergebnissen sehr gut entsprachen, wurde hier auf die Angabe der
Leishmanien-Kopien (pro g bzw. Zellen) verzichtet.

5. ERGEBNISSE
73

5. ERGEBNISSE
74
Abb. 5.19: Parasitenlast verschiedener Organe nach Infektion mit L. infantum. BALB/c- und
C57BL/6-Mäuse wurden beidseitig mit 106 L. infantum-Promastigoten subkutan infiziert. An je-
weils fünf Zeitpunkten (Tag 10, 28, 59, 118 und 174 p.i) wurde die Parasitenbeladung in der Fuß-
haut, dem drainierenden Lymphknoten (LK), der Milz, der Leber und dem Knochenmark (KM)
mittels LD- und TaqMan-Analyse ermittelt. Gegenübergestellt sind die Messergebnisse von BALB/c
- bzw. C57BL/6 - Mäusen mittels LDA (linke Reihe) und TaqMan (rechte Reihe) in Leishmanien
pro Gramm Gewebe bzw. Zellen des Zielorgans. Signifikante Unterschiede im t-Test zwischen
BALB/c- und C57BL/6-Mäusen (p<0,05) in der LDA sind mit #, zwischen zwei benachbarten Zeit-
punkten mit * gekennzeichnet; Angabe von Mittelwert ± SD für vier (LDA) bzw. zwei (TaqMan)
Mäuse je Stamm und Zeitpunkt; einzelnes Experiment.
Eine Aussage zu Signifikanzen war nur für LDA-Werte möglich, da in einem zweiten
unabhängigen Experiment LD-Analysen für zwei weitere Mäuse pro Stamm und Zeit-
punkt durchgeführt wurden und somit eine ausreichende Zahl an Werten zur Durchfüh-
rung der statistischen Analyse gegeben war.
Die TaqMan-Ergebnisse wurden im Bezug auf Quantität und zeitlichen Verlauf mit den
LDA-Resultaten verglichen. Es ist erkennbar, dass die Kurven im zeitlichen Verlauf mit
Ausnahme der infizierten Haut in den meisten Fällen relativ gut einander entsprechen,
die absoluten Werte in der TaqMan-Messung aber durchgehend niedriger sind und zwi-
schen ein bis drei Zehnerpotenzen unter den LDA-Werten liegen.
Zu den einzelnen Organen lassen sich folgende Aussagen treffen:
Am Ort der Infektion in der Fußhaut stagnierte die Zahl der Parasiten bis d28 p.i. in
beiden Mausstämmen, danach kam es im weiteren Verlauf der Infektion zu einer Parasi-
tenabnahme, die in C57BL/6 Mäusen im Vergleich zu BALB/c Mäusen sehr viel deutli-
cher ausgeprägt war (LD-Analyse). Weder die Abnahme der Parasiten im zeitlichen
Verlauf noch der Unterschied zwischen C57BL/6 und BALB/c konnte in der TaqMan-
Analyse in ähnlicher Weise beobachtetet werden.
In den LD-Analysen kam es im drainierenden Lymphknoten von C57BL/6-Mäusen ab
d10 p.i. zu einer kontinuierlichen Abnahme der Parasitenbeladung um Faktor 10 im
weiteren Verlauf der Infektion. Im Gegensatz dazu fiel die Parasitenlast bei BALB/c-
Mäusen nur kurzfristig ab (d28 p.i.), erreichte aber zu späteren Zeitpunkten wieder das
Niveau von d10 p.i.. Diese Resultate finden sich auch bei den TaqMan-Analysen wie-
der, allerdings sind die Unterschiede im Vergleich zur LDA geringer ausgeprägt.

5. ERGEBNISSE
75
Im Gegensatz dazu wurde in der Milz in beiden Mausstämmen eine Zunahme der
Leishmanienzahl im zeitlichen Verlauf detektiert, die bei den C57BL/6-Tieren zwei log-
Stufen, bei den BALB/c-Mäusen sogar 3 log-Stufen umfasste (LDA). Ähnliche Ergeb-
nisse wurden auch mit der TaqMan-Methode erzielt. Eine Zunahme der Parasiten im
Verlauf der Infektion war mit beiden Methoden auch im Knochenmark von C57BL/6-
und BALB/c-Mäusen zu messen. Während der Anstieg der Parasitenzahl in C57BL/6-
Tieren bereits an d28 p.i. erkenntlich war, zeigte er sich in BALB/c-Mäusen erst am
letzten Messzeitpunkt (d174 p.i.). Jedoch ist bei beiden Methoden gleichermaßen eine
im Vergleich zu den anderen Organen niedrigere Parasitenlast erkennbar.
In der Leber wurde mit Hilfe der LD-Analysen für C57BL/6-Mäuse im zeitlichen Ver-
lauf ein kontinuierlicher Abfall auf ca. 102-103 Leishmanien/Gramm Lebergewebe fest-
gestellt. Auch bei BALB/c-Mäusen war im Gesamtverlauf eine Abnahme der Parasiten-
beladung erkennbar. In den TaqMan-Analysen hingegen war eine kontinuierliche Ab-
nahme der Parasitenzahl nicht zu beobachten. Die Parasitenbeladung war hier bei bei-
den Mausstämmen nur zwischenzeitlich reduziert, erreichte zum letzten Messzeitpunkt
aber wieder das Ausgangsniveau von d10 p.i..
In der Zusammenschau ergeben sich für den Infektionsverlauf folgende Schlussfolge-
rungen: i) die subkutane Injektion von viszerotropen L. infantum-Promastigoten in die
Fußhaut führt zu einer systemischen Leishmaniose. ii) In Analogie zum intravenösen
Infektionsmodell der viszeralen Leishmaniose kommt es auch nach s.c.-Infektion zur
kontinuierlichen Proliferation der Erreger in der Milz, während die Parasiten in der Le-
ber nach anfänglichem Wachstum kontrolliert werden können. iii) Eine starke Vermeh-
rung der Leishmanien am ursprünglichen Infektionsort (Fußhaut) und im drainierenden
Lymphknoten findet im Verlauf der Infektion nicht statt. iv) C57BL/6- und BALB/c-
Mäuse zeigen einen ähnlichen Infektionsverlauf.

6. DISKUSSION
76
6 DISKUSSION
Die herkömmliche Diagnostik der Leishmaniose und insbesondere die Quantifizierung
von Leishmanien-infizierten Geweben beruht auf der direkten Kultivierung oder dem
mikroskopischen Nachweis der Parasiten [116]. Die PCR-Methodik und insbesondere
die weiterentwickelte Form der real-time PCR zeigten bereits in experimentellen wie
klinischen Untersuchungen ihre Überlegenheit gegenüber diesen traditionellen Nach-
weismethoden, bezüglich Sensitivität [7, 106], Arbeitsaufwand und Schnelligkeit [30,
64, 116].
In der vorliegenden Arbeit wurde eine quantitative real-time PCR zur Detektion von
Leishmania-DNA und Wirtszell-DNA der Maus etabliert. Die Wirtszell-PCR diente
hierbei als Bezugsgröße zur Quantifizierung. Beide PCR-Systeme wurden auf ihre An-
wendung im Duplex-Verfahren überprüft.
Nach der Etablierung war es ein weiteres Ziel die Anwendbarkeit der PCR zur Quantifi-
zierung von Leishmania-Beladungen zu testen; einerseits für in vitro-Infektionsexperi-
mente mit murinen BM-MΦ, andererseits für ex vivo-Analysen zur Bestimmung der
Leishmanienbeladung in Gewebe von infizierten Mäusen. Zur Validierung der PCR
erfolgte jeweils der Vergleich mit den etablierten, standardmäßig eingesetzten Metho-
den.
6.1 Etablierung eines real-time PCR-Systems zur Quantifizie-
rung von Leishmanien
6.1.1 Leishmanien-PCR
Für die Detektion von Leishmanien-DNA mittels real-time PCR wurde das von Wort-
mann et al. [115] publizierte Primer/Sonden-System verwendet. Dieses lieferte Hinwei-
se auf hohe Spezifität und Sensitivität, sowie Differenzierung zwischen verschiedenen
DNA-Mengen: der CT-Wert, welcher bei der real-time PCR die Grundlage der Quantifi-
zierung bildet, wurde bei sehr hohen DNA-Konzentrationen bereits nach wenigen Amp-
lifikationszyklen erreicht; CT-Werte um 35-40 Zyklen deuteten auf geringe Mengen an
Leishmanien-DNA hin.

6. DISKUSSION
77
Zunächst wurden die Primer- und Sondenkonzentrationen neu evaluiert. Die Ergebnisse
experimenteller Untersuchungen ergaben optimale Amplifikations- und Detektionsbe-
dingungen bei Konzentrationen von jeweils 400 nm (Primer) und 200 nm (Sonde). Es
zeigte sich wie erwartet eine hohe Spezifität: Zusätze von Fremd-DNA hatten keinen
nennenswerten Einfluss auf die Amplifikation der Leishmanien-DNA. Bei Proben, die
das entsprechende Zielgen nicht enthielten, konnten keine falsch-positiven Ergebnisse
festgestellt werden. Dies bedeutet, dass die gewählte Gensequenz spezifisch für das
Leishmaniengenom ist [47, 115]. Die hohe Sensitivität zeigte sich darin, dass selbst
Proben mit rechnerisch weniger als zehn Genkopien im Ausgangsvolumen sicher als
positiv erkannt wurden. Auch für andere real-time PCR-Protokolle wurde eine entspre-
chende Sensitivität beschrieben [94, 116], was die Vorzüge dieser weiterentwickelten
PCR-Methode unterstreicht.
Die präzise Quantifizierungsfähigkeit umfasst eine weite Spanne an DNA-
Ausgangskonzentrationen (Minimum elf dekadische Potenzen). So kommt es unter Vo-
raussetzung optimaler Effizienz im PCR-Ablauf bei Einsatz der doppelten bzw. zehnfa-
chen DNA-Menge zu einer Linksverschiebung des CT-Wertes um einen Zyklus bzw.
rund 3,32 Zyklen. Mehrere dekadische Verdünnungsreihen zeigten in der Testung einen
konstant bleibenden CT-Abstand zwischen drei und vier Zyklen innerhalb der einzelnen
Verdünnungsstufen. Der extrem große Wertebereich der konstanten Quantifizierung
wurde mit einer Präparation des Leishmania 18S rDNA-Positiv-Kontrollplasmids aus E.
coli erreicht, welches für die Standardkurve hergestellt wurde. Die hohe Spannbreite
(dynamic range) der exakten Quantifizierung mit dem Leishmanien-Kontrollplasmid
sollte in der praktischen Anwendung, d.h. für Gewebeproben aus infizierten Mäusen,
ausreichend sein, um eine hohe Leishmanienlast in der Akutphase einer Infektion eben-
so sicher wie sehr niedrige Parasitenspiegel in der Persistenzphase erfassen zu können.
Verschiedene Leishmanienspezies (L. major, L. infantum, L. tropica, L. braziliensis)
zeigten in der Testung mit der von Wortmann beschriebenen PCR [115] allesamt ein
positives Amplifikationsresultat. Es wurde zusätzlich eine zweite, vom Original-
Protokoll leicht abweichende Leishmanien-PCR getestet. Differenzen in der Basenab-
folge bei einigen Leishmanienspezies sollten besser toleriert werden mithilfe einer ver-
änderten Sonde und einer daraus resultierenden abgewandelten Primersequenz. Trotz
der vermeintlichen Optimierung zeigte die Anwendung der modifizierten PCR eine er-

6. DISKUSSION
78
höhte Störanfälligkeit in Form von Fluoreszenzschwankungen bei negativen Proben.
Dieser entscheidende Nachteil im Vergleich zur Original-PCR führte zu der Entschei-
dung, das modifizierte PCR-System nicht weiter zu verwenden. In der Publikation von
Wortmann [115] wurden insgesamt 27 verschiedene Spezies überprüft und lieferten
ebenfalls durchweg positive Ergebnisse. Die befürchteten Basenfehlpaarungen führen in
der praktischen Testung also offenbar nicht zu Sensitivitätsverlusten. Die 18S rDNA-
PCR sollte somit universell zum Nachweis von Leishmaniosen geeignet sein [107].
Als Bezugsgröße zur Quantifizierung wurde eine Standardkurve aus Proben mit be-
kannten Konzentrationen erstellt. Mit dieser Methode kann die absolute DNA-Menge in
einer Probe ermittelt werden. Für die Generierung der Standardkurve wurde anstelle von
kompletten Organismen ein Plasmid mit der Zielsequenz der 18S rDNA verwendet, da
sich bei der Generierung des Standards ausgehend von einer Leishmanienkultur zwei
potentielle Fehlerquellen ergeben: die Parasitenzahl wird mikroskopisch ausgezählt und
kann keine zu 100% genauen Ergebnisse liefern; zudem geht bei der DNA-Extraktion
ein gewisser Anteil verloren geht, dessen Größe nicht bekannt ist. Der Fehler bei der
Plasmid-Verdünnungsreihe wurde als geringer eingeschätzt, weshalb diese als Referenz
für alle Experimente gewählt wurde.
Die Quantitäten der eingesetzten Proben wurden demzufolge in Plasmid-Kopien/µl er-
mittelt. Zum Vergleich der PCR mit anderen Methoden war aber eine Angabe in
Leishmanien erforderlich. Die Umrechnung gestaltete sich hierbei als schwierig, da die
ribosomalen Gene repetitive Sequenzen darstellen und die exakte Kopienzahl nicht be-
kannt ist; zudem könnten die Anzahl zwischen den einzelnen Spezies variieren. Litera-
turangaben sprechen von 160-200 Kopien pro Leishmaniengenom [61, 84, 109], biswei-
len wurden aber auch Werte unter 100 angegeben [47, 58]. Eine absolut genaue Um-
rechnung von Genkopien in Leishmanien ist demzufolge nicht möglich. Diese Schwie-
rigkeit könnte durch Verwendung eines anderen DNA-Abschnittes, mit bekannter Ko-
pienzahl, umgangen werden. Beispielsweise wurde ein real-time PCR-Protokoll basie-
rend auf dem DNA-Polymerase-Gen, einem Single-Copy-Gen, beschrieben [12]. Hier
wäre allerdings aufgrund der geringeren Kopienzahl ein Sensitivitätsverlust der PCR zu
befürchten, was bei Proben mit niedrigen Mengen an Leishmanien-DNA zu falsch nega-
tiven Ergebnissen führen könnte. Die beschriebene Problematik bezieht sich nur auf das
Ermitteln der genauen Parasitenzahl, was für den Vergleich der zu etablierenden PCR

6. DISKUSSION
79
mit anderen Nachweismethoden notwendig war. Für praktische Fragestellungen, wie
z.B. zeitliche Verläufe oder Vergleiche verschiedener Organgewebe, wäre die Umrech-
nung von Plasmid-Kopien in Leishmanien nicht unbedingt erforderlich, da sich bei-
spielsweise eine Verdopplung der Parasitenlast mit beiden Maßeinheiten gleichermaßen
feststellen ließe.
Für die Minicircle-Sequenz der Kinetoplasten-DNA wurden neben dem konventionellen
Schema auch real-time PCR-Protokolle beschrieben [69, 87], welche aufgrund der hö-
heren Kopienzahl (10000-20000 [86]) eventuell eine noch zuverlässigere Detektion sehr
geringer Leishmanienmengen (<zehn Parasiten im Ansatz) liefern könnten. Da die An-
gabe der Kopienzahlen aber noch ungenauer ist als bei der 18S rDNA, und selbst inner-
halb von Individuen einer Spezies stark schwanken kann, würde die Genauigkeit der
Quantifizierung abnehmen. Die hohe Variabilität der Zielsequenz verbunden mit der
geringen Menge von in öffentlichen Datenbanken zugänglichen Sequenzen würde hier
auch eine zuverlässige Methodenvalidierung erschweren, sodass der Einsatz solcher
PCR-Protokolle eher auf die Infektionsforschung beschränkt bleibt, wenn beispielswei-
se eine bereits bekannte Leishmanienspezies im Versuchstier nachgewiesen bzw. quan-
tifiziert werden soll. Für diagnostische Zwecke sind solche Protokolle weniger geeignet.
Mit der 18S rDNA-PCR nach Wortmann [115] wurde demnach eine geeignete Methode
für die vorliegenden Fragestellungen gewählt, die zudem einen guten Mittelweg zwi-
schen Sensitivität und präziser Quantifizierung gewährleistet.
6.1.2 Duplex der Leishmanien-PCR mit λ-bzw. Maus-PCR
Um bei einem negativen PCR-Ergebnis eine Amplifikationshemmung oder den Verlust
von DNA beim Aufreinigungsprozess ausschließen zu können, hat sich der Einsatz ei-
ner internen Inhibitionskontrolle bewährt [82]. Diese Kontrollreaktion läuft unter exakt
denselben Bedingungen ab und weist bei einem positiven Amplifikationsergebnis nach,
dass die DNA-Isolierung korrekt durchgeführt wurde und das gewonnene DNA-Eluat
keine PCR-Inhibitoren enthielt.
In der diagnostischen Abteilung unseres Instituts hat sich der Einsatz des Bakteriopha-
gen Lambda zu diesem Zweck bereits bewährt. Von Vorteil ist, dass der Lambdaphage
in der klinischen Diagnostik keine Rolle spielt, sodass die Zugabe von Lambda-DNA
keine störende Quelle der DNA-Kontamination darstellt. Zudem ist der Phage sehr gut
untersucht und seine komplette Genomsequenz bekannt. Zwei alternative Pri-

6. DISKUSSION
80
mer/Sonden-Systeme (Lambda C und Lambda D) waren auf Kompatibilität im Duplex
mit der Leishmanien-PCR überprüft worden. Für den genannten Zweck erwiesen sich
beide PCRs in der Testung als gut geeignet. Die Lambda C-PCR zeigte im Gegensatz zu
Lambda D größere Schwankungen in der Grundfluoreszenz. Die Gefahr hierbei wäre
ein falsch positives Ergebnis bei Überschreiten des CT-Wertes. Aus diesem Grund wur-
de die Lambda D-PCR als Standard-PCR für die interne Positivkontrolle festgelegt. Als
Alternative zum Lambdaphagen wäre prinzipiell auch die Verwendung eines murinen
Zielgens möglich (so bietet die später etablierte Nidogen-PCR auch gleichzeitig eine
interne Positivkontrolle), allerdings ist je nach Organ-und Zellsubstanz ein unterschied-
lich starkes Signal der Kontroll-PCR zu erwarten. Im Hinblick auf Untersuchungen von
Proben mit wenig Zellmaterial ist eine stets sichere Amplifikation des Kontrollgens
fraglich.
Zur Quantifizierung von Leishmania-Lasten in Mausexperimenten war es erforderlich,
in jeder Probe zusätzlich die Menge an muriner DNA zu messen, da diese als Bezugs-
größe zur Bestimmung der Parasitenbeladung dienen sollte. Hierzu wurde eine PCR
basierend auf dem Single-Copy-Gen Nidogen etabliert. Anhand der bekannten Kopien-
zahl (zwei pro Mauszelle) sollte so eine exakte Bestimmung der Zellzahl im murinen
Gewebe möglich sein. Auch hier war die PCR im Duplextest mit der Leishmanien-PCR
sehr gut kompatibel und zeigte ein gutes Quantifizierungspotential. Der Ablauf von
zwei PCRs in einem Ansatz (Duplex) führt nicht nur zu präziseren Ergebnissen (Weg-
fall von Schwankungen beim Pipettieren), sondern sorgt zusätzlich zu Einsparungen an
Material und Chemikalien. Zudem werden weniger Ansätze benötigt, sodass mehr Pro-
ben in einem PCR-Lauf untersucht werden können.
Zur Bestimmung der Parasitenbeladung in einer definierten Menge an Mausgewebe ist
eine absolute Quantifizierung bei jedem PCR-Lauf, und damit das Mitführen eines
Standards, erforderlich. Die Standardreihe wurde ebenfalls im Duplex angelegt und ent-
hielt sechs dekadische Verdünnungen (Leishmanien-DNA: 105-100 Plasmidkopien/µl;
genomische Maus-DNA: 102-10-3 ng/µl). Alternativ kann im real-time PCR-Verfahren
auch relativ nach der ∆CT- oder ∆ ∆CT-Methode quantifiziert werden. In diesem Fall
wird die Expression des/der untersuchten Gens/Gene auf ein nicht reguliertes House-
keeping-Gen (=endogene Kontrolle) normalisiert (∆CT-Methode) und danach ggf. auf
eine der untersuchten Proben kalibriert (∆ ∆CT-Methode) [12, 64]. Letztendlich kann

6. DISKUSSION
81
mit dieser Methode das Expressionslevel eines untersuchten Gens in verschiedenen
Proben sehr gut gegeneinander verglichen und relative Unterschiede aufgezeigt werden,
die Angabe einer absoluten Menge für das exprimierte Gen ist jedoch nicht möglich.
Genau diese Angabe war jedoch bei der Frage nach der Zahl der Leishmanien im infi-
zierten Gewebe notwendig. Aus diesem Grund kam zur Beantwortung der Fragestellung
nur eine real-time PCR mit absoluter Quantifizierung in Frage. Um die Ergebnisse ver-
schiedener PCR-Läufe vergleichbar zu machen, ist das Anlegen von Aliquots der Stan-
dardkurven-Chemikalien unbedingt erforderlich. Durch zu häufiges Auftauen und Ein-
frieren einer Stammlösung wäre nämlich ein allmählicher Verlust an DNA zu befürch-
ten, womit die Genauigkeit der Quantifizierung beeinträchtigt werden könnte.
Da die in dieser Arbeit etablierte 18S rDNA Leishmanien-PCR ein mehrfach abgesi-
chertes negatives Resultat mit humanen DNA-Proben (von Leishmania-negativen Pro-
banden) aufwies, ließe sich dieses PCR-Verfahren sehr wahrscheinlich auch recht gut in
einem Duplex-System zur Quantifizierung von Leishmanien in humanen Gewebe ein-
setzen. Die Schritte zur Etablierung und Verifizierung der Leishmania-Human-Duplex-
PCR könnten entsprechend dem hier beschriebenen Vorgehen übernommen werden.
Denkbar wäre beispielsweise ein Einsatz dieses PCR-Verfahrens bei der medikamentö-
sen Behandlung einer Leishmaniose, um die Wirksamkeit der gewählten Therapieform
sowie den zeitlichen Verlauf der Erkrankung nachzuvollziehen [116]. So konnte exemp-
larisch in einer Studie zu HIV-infizierten Patienten mit viszeraler Leishmaniose nach-
gewiesen werden, dass Rezidive assoziiert waren mit persistierend hohen Leishmanien-
DNA-Spiegeln im venösen Blut [11].
6.1.3 Vergleich zwischen konventioneller und real-time PCR
Die quantitative real-time PCR bietet gegenüber konventionellen PCR-Methoden eine
Reihe von Vorteilen: so werden in einem abgeschlossenen System (closed tube concept)
nacheinander Amplifikation, Detektion und Quantifizierung der Zielsequenz durchge-
führt. Dies sorgt für eine Verringerung des Arbeitsaufwandes; andererseits wird das
Kontaminationsrisiko [106, 110] vermindert (z.B. durch ein über den Elektrophorese-
puffer verschlepptes PCR-Produkt). Ein zusätzlicher Erregernachweis, welcher bei der
konventionellen Methode vonnöten ist (z.B. durch eine nested PCR), entfällt, da mittels
Hybridisierung der TaqMan-Sonden die Spezifität des Produktes bereits nachgewiesen
wird. Außerdem läuft die real-time PCR durch die Automatisierung um ein Vielfaches

6. DISKUSSION
82
schneller ab [30, 64]. Der Umgang mit kanzerogenen Farbstoffen wie Ethidiumbromid
fällt weg, da die Agarose-Gel-Auswertung nicht mehr nötig ist. Die Vorzüge der kon-
ventionellen PCR bleiben dennoch erhalten: beispielsweise die hohe Sensitivität oder
auch die Verwendbarkeit verschiedener Ausgangsmaterialien (Organ-oder Blutproben,
Zellkulturen usw.). Nachteilig hingegen ist der relativ hohe Preis für die benötigten re-
al-time-PCR-Geräte und Reagenzien (z.B. Sonden, PCR-Mix).
In der vorliegenden Arbeit galt es zu überprüfen, ob trotz optimierter real-time PCR-
Bedingungen die konventionelle Kinetoplasten-PCR der 18S rDNA-TaqMan PCR über-
legen sein könnte, da die nachzuweisende Sequenz der Kinetoplasten-PCR, die Mini-
circle-DNA, in viel höherer Kopienzahl vorliegt (10000-20000 [86] pro Leishmanien-
genom) als die 18S rDNA (160-200 Kopien [61, 84, 109]). Zur Untersuchung dieser
Fragestellung wurde ermittelt, bis zu welcher Verdünnungsstufe eine mit Leishmanien-
DNA versetzte Probe sicher als positiv erkannt werden konnte. Dabei zeigte sich, dass
die Kinetoplasten-PCR zwar an die Sensitivität der real-time PCR herankam, diese aber
nicht überbieten konnte. Daraus lässt sich schlussfolgern, dass die konventionelle PCR-
Methode der real-time PCR bei Verwendung der gleichen Zielstruktur deutlich unterle-
gen wäre. Die erhöhte Sensitivität und Spezifität bei der real-time PCR liegt unter ande-
rem darin begründet, dass neben den beiden Primern mit der Sonde noch ein zusätzli-
ches Oligonukleotid hybridisiert.
Für eine zuverlässige Quantifizierung mit der herkömmlichen PCR-Methode wäre zu-
dem ein vielfach größerer Arbeitsaufwand erforderlich [64]: so müsste von jeder Probe
eine Verdünnungsreihe hergestellt werden und diese nach dem PCR-Lauf mittels Aga-
rose-Gel-Auswertung auf das Vorliegen einer entsprechenden Bande in den jeweiligen
Verdünnungsstufen untersucht werden. All dies würde zu einem enormen Anstieg an
zeitlichem Aufwand und an Materialverbrauch führen, sodass die real-time PCR für
unsere Fragestellungen der herkömmlichen Methode in vielen Aspekten klar überlegen
ist [64].

6. DISKUSSION
83
6.2 Leishmania real-time PCR im praktischen Gebrauch für
in vitro/in vivo-Experimente
6.2.1 Anwendung der Leishmania real-time PCR bei in vitro-
Infektionsexperimenten
Nach Etablierung des Leishmanien/Maus-Duplex-PCR-Systems sollte dieses zunächst
für in vitro-Bedingungen getestet werden und mit der mikroskopischen Auswertung
nach DiffQuik®-Färbung verglichen werden. Dazu wurden murine BM-MΦ mit Leish-
manien-Promastigoten infiziert und unter Zugabe verschiedener Mengen an Immunsti-
mulanzien kultiviert, um unterschiedlich starke Abtötungen der Parasiten zu provozie-
ren. Es wurde überprüft, ob PCR-Signal und mikroskopische Auszählung bei der Quan-
tifizierung der Leishmanien miteinander korrelieren.
Die real-time PCR-basierte Methode böte gegenüber der Mikroskopie-Methode mehre-
re Vorteile: i) die Durchführung wäre schneller und weniger subjektiv durch den Expe-
rimentator beeinflussbar, ii) sie zeigt eine höhere Sensitivität [18, 83, 106, 116]. Nach-
teilig wäre hingegen, dass mittels PCR keine Bestimmung der Infektionsrate (Anteil
infizierter MΦ) möglich ist, sondern lediglich die Angabe von Promastigoten im Bezug
auf die Gesamtzahl der MΦ. Außerdem benötigt man eine höhere Anzahl an Zellen, um
ausreichend DNA für die PCR zu gewinnen. Bei Durchführung umfangreicher Experi-
mente müssten daher bei der PCR-Methode höhere Kosten für Verbrauchsmaterialien
und Chemikalien (z.B. Immunstimulanzien) einkalkuliert werden.
Zur mikroskopischen Auswertung von MΦ-Infektionsexperimenten gibt es zahlreiche
Publikationen, die der Validierung unserer Ergebnisse dienten. Der Vergleich der in
dieser Arbeit erzielten Resultate und den in der Literatur beschriebenen Daten zur mik-
roskopischen Leishmanienanalyse lieferte hierbei große Übereinstimmung. Wie publi-
ziert [8] führte die Zugabe von IFN-γ nur bei höheren Konzentrationen (20 U/ml; ent-
spricht 24 ng/ml) zu einer Senkung der Parasitenlast. In Analogie beobachteten wir bei
2 ng/ml (1,66 U/ml) nur eine minimale Reduktion der Parasitenbeladung, während die
Anzahl an intrazellulären Leishmanien bei 20 ng/ml IFN-γ (16,6 U/ml) um die Hälfte
verringert war. Durch Zugabe von IFN-γ plus TNF-α konnte die Kapazität der MΦ, in-
trazelluläre Pathogene wirksam abzutöten, wie beschrieben deutlich gesteigert werden

6. DISKUSSION
84
[8, 29, 100]. Im Gegensatz dazu stimulierte TNF allein MΦ nicht zum Abtöten von
Leishmanien [8].
Keine Übereinstimmung gab es beim Vergleich der mikroskopischen Auswertung mit
dem TaqMan-PCR Methode. Die höchsten Parasitenbeladungen wurden laut PCR in
den beiden Ansätzen gefunden, die den Literaturdaten und unserer DiffQuik®-Färbung
zufolge zur stärksten Abtötung der Promastigoten geführt hatten (Zugabe von TNF-α +
IFN-γ bzw. IFN-γ 20 ng/ml). Die niedrigsten Werte wurden im TaqMan-Verfahren für
die Proben gemessen, welche in der mikroskopischen Auswertung keinen abtötenden
Effekt erzielten (IFN-γ 2 bzw. 0,2 ng/ml) bzw. die Parasitenbeladung sogar erhöhten
(TNF-α ohne Zusatz von IFN-γ). Somit lässt sich schlussfolgern, dass die Ergebnisse
der mikroskopischen Auswertung nicht mit dem Signal der real-time PCR korrelieren.
Möglicherweise ist die Abtötung der Parasiten nach einem Zeitraum von wenigen Ta-
gen (hier: 72h) nicht detektierbar, weil die DNA abgestorbener Parasiten immer noch in
den Proben vorliegt und an die PCR-Primer bzw. Sonden bindet [12, 94]. So lieferten
beispielsweise Narbengewebe von ausgeheilter kutaner Leishmaniose in bis zu 80% ein
positives PCR-Resultat, obwohl die Erkrankung schon Jahre zurücklag und die Patien-
ten klinisch gesund waren [93]. Möglicherweise ist die freigesetzte DNA abgestorbener
Parasiten sogar besser detektierbar, da das Erbgut in den noch lebenden Organismen im
Zuge der DNA-Extraktion zu einem höheren Anteil mit dem restlichen Zellmaterial
ausgewaschen werden könnte. Dies würde auch erklären, warum die Ansätze mit der
stärksten Parasiten-Abtötung die höchsten PCR-Signale lieferten.
Um den Nachweis von Leishmania auf das Erbgut lebender Parasiten zu beschränken,
wäre es bei Experimenten dieser Art vorzuziehen, RNA anstelle von DNA zu messen
[59, 83]. Dies ist mit Hilfe der RT-(reverse transcription) real-time PCR möglich. Hier-
bei wird zunächst ein komplementärer DNA-Strang (cDNA) an die RNA synthetisiert,
welche dann als Matrize zur Amplifikation dient. Diese Methode ist zwar im Bezug auf
ihre Sensitivität der DNA-basierten real-time PCR sogar überlegen [106] und würde die
vorliegende Problematik beheben, aber sie könnte bei der Quantifizierung zu Problemen
führen, da von der vorliegenden RNA-Menge nur ungenaue Rückschlüsse auf die exak-
te Zellzahl der Parasiten erfolgen können.

6. DISKUSSION
85
In der Zusammenschau sind demnach die aktuellen PCR-Methoden und -Protokolle
keine adäquate Alternative zur herkömmlichen mikroskopischen Auswertung bei in
vitro-Pulsinfektionsexperimenten.
6.2.2 Anwendung der real-time PCR zur ex vivo-Analyse von Mausgewebe
nach Infektion mit Leishmania
Neben den in vitro-Infektionsexperimeneten mit Mφ wurde die real-time PCR zur Para-
sitenlastbestimmung auch in Leishmanien-infiziertem murinen Gewebe getestet und mit
der Standardmethode, der Limiting Dilution Analysis, verglichen. Da Infektionsverläufe
zumeist über einen längeren Zeitraum beobachtet werden, sollte eine Beeinträchtigung
der Ergebnisse durch Amplifikation von DNA abgestorbener Parasiten, die innerhalb
eines kurzen Zeitraumes akkumulieren, hier weniger zu Buche schlagen.
a. Infektion mit L. major
Nach subkutaner Infektion von L. major-Promastigoten in hoher Dosis entwickelten
suszeptible BALB/c- und resistente C57BL/6-Mäuse einen Krankheitsverlauf, der mit
Angaben in der Literatur sehr gut korrelierte. Die Fußschwellung als Parameter der lo-
kalen Infektion zeigte bei BALB/c-Mäusen beständig steigende Werte und nahm bis
zum letzten Messzeitpunkt an Tag 28 p.i. um ca. 85% zu [101, 111]. Ein Maximum der
Schwellung konnte bei C57BL/6 zwischen Tag 15 und 30 verzeichnet werden. Hier
nahm die Dicke um maximal 60% zu und sank anschließend wieder, nahezu bis auf
Ausgangswerte, ab [111, 112].
Zur Validierung der Parasitenlasten einzelner Organe wurden die Werte aus unserer LD-
Analyse zunächst mit publizierten Daten abgeglichen. Die Parasitenlast im Fuß stimmte
dabei sowohl an Tag 28 p.i. (BALB/c und C57BL/6) als auch Tag 140 p.i. (C57BL/6)
sehr gut Angaben aus der Literatur überein [20] [72]. Im poplitealen Lymphknoten und
der Milz lagen die Werte für die Parasitenbeladung im durchgeführten Experiment et-
was niedriger im Vergleich zu den veröffentlichten Ergebnissen anderer Labore (z.B.
BALB/c poplitealer Lymphknoten 4×103 nach 14 Tagen laut Literatur [111], eigenes
Experiment 102 Leishmanien/103 Zellen am Tag 28 p.i., C57BL/6 siehe [111] [112]).
Die Unterschiede zwischen unseren und Literaturdaten sind jedoch für alle Wertepaare
kleiner als zwei dekadische Potenzen und bekräftigen die Ergebnisse, nach denen für
BALB/c in allen Organen eine signifikant höhere Beladung mit Leishmanien festgestellt

6. DISKUSSION
86
wurde, während die Parasitenlast bei C57BL/6 in der Langzeitmessung auf einem nied-
rigen Niveau persistierte. Grundlegende Ergebnisse zum Mausmodell der kutanen
Leishmaniose nach L. major Infektion konnten also reproduziert werden.
Die real-time PCR-Ergebnisse zeigten eine sehr gute Übereinstimmung mit der An-
zucht-Methode, da sowohl die signifikanten Unterschiede zwischen beiden Stämmen
wie auch der zeitliche Verlauf bei C57BL/6 für alle Organe der LDA entsprachen. Le-
diglich in der Milz ergab sich nach TaqMan-Messung zum späteren Zeitpunkt ein An-
stieg, der so in der LDA nicht beobachtet wurde (Abb. 5.15). Es ist aber zu erwähnen,
dass in zwei von vier Mäusen keine Leishmanien-DNA mehr nachgewiesen werden
konnte, während in den anderen beiden Mäusen eine im Vergleich zur LD-Analyse sig-
nifikant erhöhte Leishmanienzahl detektiert wurde (maximal zwei Zehnerpotenzen Un-
terschied). Insgesamt war die Varianz zum Tag 140 p.i. in der PCR-Gruppe demnach
recht groß und es lässt sich keine abschließende eindeutige Aussage bzgl. eines Unter-
schiedes zur LD-Gruppe bilden. Hierzu wären weitere Wiederholungsexperimente nö-
tig, um die Fallzahlen zu erhöhen und so eine statistische Analyse auf eine breitere Ba-
sis zu stellen.
Unabhängig von dieser einzigen Diskrepanz lässt sich aber schlussfolgern, dass die re-
al-time PCR meist äquivalente Ergebnisse lieferte und geeignet ist, die LD-Analyse zu
ersetzen.
In einem zweiten Infektionsexperiment mit einer anderen Leishmanienspezies, L. infan-
tum, sollte überprüft werden, ob die gute Übereinstimmung beider Methoden reprodu-
zierbar ist.
b. Infektion mit L. infantum
In diesem abschließenden Versuch wurden C57BL/6- und BALB/c-Mäusen L. infan-
tum-Promastigoten eines viszerotropen Stammes [2] subkutan in beide Hinterpfoten
injiziert.
Zunächst wurde mit der Messung der Fußdicke der zeitliche Verlauf der lokalen Infek-
tion überprüft. Hierbei konnte festgestellt werden, dass die Kurve für C57BL/6 derjeni-
gen nach L. major-Infektion sehr ähnlich war mit einem Maximum vier Wochen nach
Infektion (Anstieg der Fußdicke um knapp 40%) und darauffolgender Abschwellung.
Bei BALB/c-Mäusen war fast gar keine Schwellung zu erkennen; die Zunahme betrug

6. DISKUSSION
87
maximal 15% nach 21 Tagen, woraufhin wieder die Ausgangswerte erreicht wurden.
Für die Parasitenlasten der untersuchten Organe wurden erneut die Ergebnisse der An-
zuchtmethode durch Abgleich mit publizierten Daten validiert, bevor sie mit TaqMan-
Messungen verglichen wurden. Für einige Werte wurde eine Einheiten-Umrechnung
vorgenommen, da sich in der Literatur meist Angaben bezogen aufs Gesamtorgan fin-
den lassen. Die Daten werden auch hinsichtlich der Injektionsarten der viszerotropen
Leishmanien (subkutan versus intravenös) diskutiert.
Leber: In unserem Experiment kam es im zeitlichen Verlauf bei beiden Stämme zu ab-
nehmenden Kurven um etwa zwei dekadische Potenzen; signifikante Unterschiede zwi-
schen C57BL/6 und BALB/c bestanden nicht.
Eine solche Abnahme der Parasitenbeladung in der Leber wurde sowohl für s.c. als auch
i.v.-Infektion mit L. infantum / L. chagasi bereits publiziert [2, 60, 76], und ist auch bei
i.v.-Infektion mit der Spezies L. donovani [27] beschrieben worden, welche ebenso eine
viszerale Verlaufsform auslöst. Für eine Infektionsdosis von 106 Parasiten sind in der
Literatur Werte um 105 Leishmanien pro Organ [2] in der frühen Infektionsphase be-
schrieben. Dies entspricht in etwa unseren Werten, ebenso wie der Abfall auf unter 104
im Verlauf [2]. In der Publikation von Oliveira et.al. [76] hingegen wurden selbst bei
höherer Infektionsdosis (107) nie Werte über 103 Parasiten ermittelt. Im Vergleich hier-
zu kommt es bei beiden Spezies nach intravenöser Infektion zu höheren Parasitenbela-
dungen gegenüber der subkutanen Infektion (bis zu 107 bei L. infantum) [2, 60, 76].
Beim zeitlichen Verlauf scheint außerdem die eingesetzte Infektionsdosis eine entschei-
dende Rolle zu spielen: so kommt es bei niedrigen Dosen (103 Leishmanien) zu einer
kontinuierlichen Abnahme der Parasitenlast; bei höheren Dosen (105) hingegen zu ei-
nem anfänglichen Anstieg auf ein Maximum um Tag 8-15 p.i. [16, 60], dem ein erneu-
ter Abfall der Parasitenbeladung folgt. Dieser sogenannte „Leberknick“ verschiebt sich
bei weiterer Dosissteigerung in der Zeitachse zunehmend nach hinten (Tag 45 p.i. bei
107 injizierten Parasiten [76]). Möglicherweise liefert eine begrenzte Kapazität des Im-
munsystems die Erklärung für dieses beobachtete Phänomen. Bereits publizierte Daten
bekräftigen unser Ergebnis, dass zwischen beiden Mausstämmen zu keinem Zeitpunkt
signifikante Differenzen ermittelt werden konnten [46].

6. DISKUSSION
88
Milz: In der Milz konnte bei beiden Mausstämmen im Infektionsverlauf eine stetige
Zunahme der Parasitenbeladung detektiert werden. Anfänglich gemessene Werte (pro g
Gewebe) lagen unter den Werten für Leber, am letzten Messzeitpunkt aber deutlich da-
rüber. Dieser grundsätzlich unterschiedliche Infektionsverlauf zwischen Leber- und
Milzgewebe wird auch in der Literatur wiederholt beschrieben, sowohl für L. infan-
tum/chagasi [2, 60, 76] als auch L. donovani, sowohl beim intravenösen wie auch sub-
kutanen Infektionsweg. Im Vergleich der absoluten Werte ist anfangs eine gute Über-
einstimmung mit Literaturdaten erkennbar (103 – 104 Parasiten/Organ nach einer Woche
[2, 76], jedoch war der Anstieg der Parasitenzahl in den hier beschriebenen Experimen-
ten rasanter (106 bis 107 Parasiten an Tag 59 im Vergleich zu 104 bis 105 [2, 76]). Die
Parasitenzahlen nach i.v.-Injektion waren im Vergleich zur durchgeführten s.c.-
Infektion in Abhängigkeit vom Zeitpunkt signifikant höher [2], gleich [60] oder leicht
erniedrigt [76]. In jedem Fall zeigten sich beim Vergleich der selbst erhobenen Daten
der s.c.-Infektion versus Literaturdaten der i.v.-Infektion mehr Unterschiede als bei der
Gegenüberstellung s.c.-Infektion eigener versus publizierter Daten.
Knochenmark: Für BALB/c war ab Tag 28 p.i. ein leichter Anstieg zu verzeichnen,
während bei C57BL/6 einem Maximum zum zweiten Messpunkt ein stetiges Absinken
der Parasitenlast folgte. Signifikante Unterschiede wurden weder im zeitlichen Verlauf
noch zwischen den beiden Stämmen gemessen. Literaturdaten zufolge kommt es zu
einem Anstieg der Parasitenlast zwischen Tag 7 und 14 p.i., auf den ein langsames Ab-
sinken folgt [16]. Diese Kurve konnten wir für C57BL/6-Mäuse gut nachvollziehen;
auch gibt es einen vergleichbaren Maximalwert von 103 Parasiten/Organ. Die Verlaufs-
kurve von BALB/c ähnelt mehr den von Oliveira et al. [76] beschriebenen Daten, wo-
nach es zu einem kontinuierlichen Anstieg der Parasitenlast kommt. Die in dieser Publi-
kation genannten Werte für L. chagasi stimmen mit unseren Angaben (zwischen 100
und 103 L./Organ) überein. Auffallend ist, dass im Knochenmark eine stets geringere
Parasitenlast als in den anderen Organen vorliegt; dies gilt sowohl für das vorliegende
Experiment als auch publizierte Daten [16, 60, 76].
Poplitealer drainierender Lymphknoten: Im gesamten Zeitraum des Experiments kam es
nur zu geringfügigen Schwankungen innerhalb einer Zehnerpotenz; ein geringes Absin-
ken der Parasitenlast (signifikant bei BALB/c) konnte an Tag 28 beobachtet werden. Es
wurden signifikant höhere Werte für BALB/c an den Tagen 118 und 174 gemessen, die

6. DISKUSSION
89
sich in absoluten Zahlen ausgedrückt aber nur auf ca. eine Zehnerpotenz bemessen. Da-
bei wurden - auf das Gesamtorgan bezogen - über den gesamten Infektionsverlauf Wer-
te zwischen 104 und 105 Leishmanien gemessen. Dies entspricht in etwa den Angaben
in der Literatur [2]. Die von Ahmed et al. publizierte Verlaufskurve für Lymphknoten
zeigt zudem den gleichen kleinen Knick nach unten etwa drei Wochen p.i., mit nachfol-
gendem Wiederanstieg auf Ausgangswerte. Für L. donovani (s.c.-Infektion) entsprach
die Verlaufskurve dem für L. infantum beschriebenen Verlauf, jedoch wurden trotz hö-
herer Infektionsdosis (5x106) um ca. zwei bis drei Zehnerpotenzen niedrigere Werte
gemessen [66].
Fuß: Hier ist eine langsame, aber signifikante Abnahme der Parasitenlast erkennbar, von
anfangs knapp 106 Parasiten/g Gewebe bis auf 104 (BALB/c) bzw. 102 (C57BL/6). Auch
in der Literatur werden sinkende Parasitenlasten für den Injektionsort bei s.c.-Infektion
beschrieben [2]; wobei der stärkste Abfall mit über 102 Parasiten/Organ schon etwas
früher (zwischen Woche 1 und 3 p.i.) als in unserem Experiment beobachtet wurde. Für
L. donovani (ebenfalls s.c.-Infektion) ist hingegen ein anfänglicher Anstieg bis Tag 14
beschrieben, auf den dann aber ein starker Abfall der Parasitenlast folgt [66]. Dabei
waren die Werte bei gleicher Infektionsdosis aber stets niedriger als nach L. infantum-
Infektion. Sowohl in unseren Beobachtungen wie auch in den publizierten Daten lässt
sich eine abnehmende Parasitenbeladung an der Injektionsstelle nach s.c.-Applikation
von Leishmania über den Verlauf der Infektion hinweg feststellen.
Zusammenfassend wurden in den hier durchgeführten Experimenten nach s.c.-Infektion
mit einer hohen Dosis von L. infantum-Promastigoten in den verschiedenen Organen für
den zeitlichen Verlauf der Parasitenlast stark voneinander abweichende Resultate er-
zielt, die den Daten aus der Literatur entsprechen [2, 76],. So stehen die sinkenden Wer-
te in Leber und Fuß im Kontrast zur Milz, in der ein stetiger Anstieg zu verzeichnen ist.
Die gleiche Dichotomie in der Parasitenbeladung zwischen den verschiedenen Organen
ist auch nach i.v.-Infektion von L. infantum oder L. donovani zu beobachten [113]. Un-
terschiede in der Immunantwort der Leber und der Milz werden als Ursache für den
differenten Verlauf diskutiert [87, 113].
Der Vergleich des generellen Krankheitsverlaufs mit dem i.v.-Infektionsweg führte zu
unterschiedlichen und sogar gegensätzlichen Beobachtungen: so wurden sowohl be-

6. DISKUSSION
90
schleunigte [51] als auch verlangsamte [76] Krankheitsverläufe in der Gegenüberstel-
lung mit der subkutanen Infektion festgestellt.
Zwischen den beiden Mausstämmen waren hingegen nur marginale Differenzen er-
kennbar, was die Daten aus der Literatur bestätigt [46, 55]. Die Tatsache, dass nach L.
infantum-Infektion bei gleicher oder sogar niedrigerer Dosis höhere Werte als für L.
donovani-Infektionen messbar waren [2, 16, 76], könnte auf das bereits bekannte,
schnellere Wachstum von L. donovani zurück zu führen sein, welches letztlich auch zu
einer beschleunigten Resolution in der Leber führt [113].
Der Verlauf der humanen viszeralen Leishmaniose entspricht einer disseminierten In-
fektion [42], welche nahezu immer letal endet. Die im Mausmodell eingesetzten Stäm-
me BALB/c und C57BL/6 spiegeln die Viszeralisation mit steigenden Parasitenbela-
dungen von Milz und Leber im Anfangsstadium der Infektion gut wider. Die Eindäm-
mung der Parasitenvermehrung und Selbst-Limitierung, die im Mausmodell z.B. in der
Leber zu beobachten ist, ist jedoch grundsätzlich verschieden vom Verlauf beim Men-
schen [55], sodass das der Verlauf der Erkrankung bei der viszeralen Leishmaniose zwi-
schen Maus und Mensch nur begrenzt vergleichbar ist.
Im Methodenvergleich zwischen LDA und TaqMan ergaben sich für die Infektionsver-
läufe in den verschiedenen Organen recht ähnliche Ergebnisse. So zeigte sich beispiels-
weise in der Milz ein Ansteigen der Werte, in den drainierenden Lymphknoten des Fu-
ßes eine konstante Parasitenlast. Einzelne Wertepaare wichen allerdings von den LD-
Resultaten ab: so zeigte sich ein Wiederanstieg in der Leber zum letzten Messzeitpunkt,
nachdem für die vorherigen Wertepaare ein Abfall erkennbar war, was wiederum den
Ergebnissen der Anzuchtmethode entsprach. Derartige Abweichungen sind aber mit
hoher Wahrscheinlichkeit auf die geringe Anzahl an Versuchstieren (zwei pro Messung)
zurückzuführen. Ein weiteres Beispiel liefern die Ergebnisse der letzten beiden Werte
für den Fuß: die wieder ansteigende Tendenz steht im Widerspruch mit dem beobachte-
ten Absinken der Parasitenlast in der LDA. Eine erhöhte Anzahl an Versuchstieren wäre
nötig, um diesen Effekt wirklich beurteilen und statistisch auswerten zu können.
Allerdings ist für alle Organe im Vergleich mit der LDA erkennbar, dass die Werte der
TaqMan-Messung gleichbleibend niedriger sind und um ca. ein bis zwei dekadische

6. DISKUSSION
91
Potenzen unter den LDA-Werten liegen. Dies steht im Kontrast zu den Ergebnissen des
vorher beschriebenen Infektionsexperiments mit L. major, wo im quantitativen Ver-
gleich der Ergebnisse diese Tendenz nicht auftrat. Der Effekt könnte demnach mit der
Leishmanienspezies zusammenhängen. Eine unterschiedliche Kopienzahl der 18S
rDNA je nach Spezies wäre durchaus denkbar. Diese Vermutung wird durch der Be-
obachtung bekräftigt, dass Probenmessungen von Leishmanienkulturen beider Spezies
(nach mikroskopischer Auszählung je 105 Parasiten/µl) in unseren Vortestungen unter-
schiedliche Quantitäten erbrachten: 1,06×107 Kopien/µl (L.m.) gegenüber 1,17×106 Ko-
pien/µl (L.i.) ergeben einen Unterschied von einer dekadischen Potenz. Bei Annahme
einer hundertprozentigen Effizienz der DNA-Aufreinigung und –Amplifikation, würde
dies Werte von 106 Kopien/Parasit für L. major, aber nur 11,7 Kopien für L. infantum
ergeben. Es bestehen also Hinweise, dass die Kopienzahl von 200 pro Organismus [61,
84, 109] in diesem Fall zu hoch gegriffen ist und nach unten korrigiert werden müsste.
Davon abgesehen bekräftigen die Ergebnisse der beiden in vivo-Experimente das große
Potential der real-time PCR-Methodik; durch weitere Entwicklung könnte hier ein der
Anzuchtmethode ebenbürtiges bzw. sogar überlegenes Verfahren etabliert werden, mit
dessen Hilfe man sehr genaue Parasitenlast-Quantifizierungen durchführen könnte. Eine
praktische Anwendung der Quantifizierung von Parasitenlasten wäre unter anderem bei
der Wirksamkeitsevaluierung verschiedener Medikamente denkbar [11]. So ist bei-
spielsweise bekannt, dass einige Leishmanienspezies auf Reservemedikamente wie Ke-
toconazol (L. mexicana) oder Fluconazol (L. major) sehr gut ansprechen [3, 74]. Der
Nachweis einer sinkenden Parasitenlast könnte bei der Erfolgsüberwachung einer sol-
chen alternativen Therapie nützlich sein, ebenso bei der Überprüfung der Wirkung neu
entwickelter Impfstoffe [106].

7. LITERATURVERZEICHNIS
92
7 LITERATURVERZEICHNIS
1. Aebischer T (1994). Recurrent cutaneous leishmaniasis: a role for persistent parasites? Parasitol Today 10:25-28
2. Ahmed S, Colmenares M, Soong L, Goldsmith-Pestana K, Munstermann L, Molina R, McMahon-Pratt D (2003). Intradermal infection model for pathogenesis and vaccine studies of murine visceral leishmaniasis. Infect Immun 71:401-410
3. Alrajhi AA, Ibrahim EA, De Vol EB, Khairat M, Faris RM, Maguire JH (2002). Fluconazole for the treatment of cutaneous leishmaniasis caused by Leishmania major. N Engl J Med 346:891-895
4. Alvar J, Croft S, Olliaro P (2006). Chemotherapy in the treatment and control of leishmaniasis. Adv Parasitol 61:223-274
5. Ameen M (2007). Cutaneous leishmaniasis: therapeutic strategies and future directions. Expert Opin Pharmacother 8:2689-2699
6. Ameen M (2010). Cutaneous leishmaniasis: advances in disease pathogenesis, diagnostics and therapeutics. Clin Exp Dermatol 35:699-705
7. Aviles H, Belli A, Armijos R, Monroy FP, Harris E (1999). PCR detection and identification of Leishmania parasites in clinical specimens in Ecuador: a comparison with classical diagnostic methods. J Parasitol 85:181-187
8. Bogdan C, Moll H, Solbach W, Rollinghoff M (1990). Tumor necrosis factor-alpha in combination with interferon-gamma, but not with interleukin 4 activates murine macrophages for elimination of Leishmania major amastigotes. Eur J Immunol 20:1131-1135
9. Bogdan C, Rollinghoff M (1998). The immune response to Leishmania: mechanisms of parasite control and evasion. Int J Parasitol 28:121-134
10. Bogdan C, Schonian G, Banuls AL, Hide M, Pratlong F, Lorenz E, Rollinghoff M, Mertens R (2001). Visceral leishmaniasis in a German child who had never entered a known endemic area: case report and review of the literature. Clin Infect Dis 32:302-306
11. Bossolasco S, Gaiera G, Olchini D, Gulletta M, Martello L, Bestetti A, Bossi L, Germagnoli L, Lazzarin A, Uberti-Foppa C, Cinque P (2003). Real-time PCR assay for clinical management of human immunodeficiency virus-infected patients with visceral leishmaniasis. J Clin Microbiol 41:5080-5084
12. Bretagne S, Durand R, Olivi M, Garin JF, Sulahian A, Rivollet D, Vidaud M, Deniau M (2001). Real-time PCR as a new tool for quantifying Leishmania infantum in liver in infected mice. Clin Diagn Lab Immunol 8:828-831
13. Bryceson A (2001). A policy for leishmaniasis with respect to the prevention and control of drug resistance. Trop Med Int Health 6:928-934
14. Bustin SA (2002). Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29:23-39
15. Cardullo RA, Agrawal S, Flores C, Zamecnik PC, Wolf DE (1988). Detection of nucleic acid hybridization by nonradiative fluorescence resonance energy transfer. Proc Natl Acad Sci U S A 85:8790-8794

7. LITERATURVERZEICHNIS
93
16. Carrion J, Nieto A, Iborra S, Iniesta V, Soto M, Folgueira C, Abanades DR, Requena JM, Alonso C (2006). Immunohistological features of visceral leishmaniasis in BALB/c mice. Parasite Immunol 28:173-183
17. Carter KC, Hutchison S, Henriquez FL, Legare D, Ouellette M, Roberts CW, Mullen AB (2006). Resistance of Leishmania donovani to sodium stibogluconate is related to the expression of host and parasite gamma-glutamylcysteine synthetase. Antimicrob Agents Chemother 50:88-95
18. Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, Alvar J, Boelaert M (2007). Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol 5:873-882
19. Choi CM, Lerner EA (2002). Leishmaniasis: recognition and management with a focus on the immunocompromised patient. Am J Clin Dermatol 3:91-105
20. Corware K, Harris D, Teo I, Rogers M, Naresh K, Muller I, Shaunak S (2011). Accelerated healing of cutaneous leishmaniasis in non-healing BALB/c mice using water soluble amphotericin B-polymethacrylic acid. Biomaterials 32:8029-8039
21. Cruz I, Morales MA, Noguer I, Rodriguez A, Alvar J (2002). Leishmania in discarded syringes from intravenous drug users. Lancet 359:1124-1125
22. Daneshbod Y, Oryan A, Davarmanesh M, Shirian S, Negahban S, Aledavood A, Davarpanah MA, Soleimanpoor H, Daneshbod K (2011). Clinical, histopathologic, and cytologic diagnosis of mucosal leishmaniasis and literature review. Arch Pathol Lab Med 135:478-482
23. de Bruijn MH, Barker DC (1992). Diagnosis of New World leishmaniasis: specific detection of species of the Leishmania braziliensis complex by amplification of kinetoplast DNA. Acta Trop 52:45-58
24. Desjeux P (1996). Leishmaniasis. Public health aspects and control. Clin Dermatol 14:417-423
25. Desjeux P (2004). Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis 27:305-318
26. Ding AH, Nathan CF, Stuehr DJ (1988). Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol 141:2407-2412
27. Engwerda CR, Murphy ML, Cotterell SE, Smelt SC, Kaye PM (1998). Neutralization of IL-12 demonstrates the existence of discrete organ-specific phases in the control of Leishmania donovani. Eur J Immunol 28:669-680
28. Engwerda CR, Kaye PM (2000). Organ-specific immune responses associated with infectious disease. Immunol Today 21:73-78
29. Esparza I, Mannel D, Ruppel A, Falk W, Krammer PH (1987). Interferon gamma and lymphotoxin or tumor necrosis factor act synergistically to induce macrophage killing of tumor cells and schistosomula of Schistosoma mansoni. J Exp Med 166:589-594
30. Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JD, Wengenack NL, Rosenblatt JE, Cockerill FR, 3rd, Smith TF (2006). Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev 19:165-256

7. LITERATURVERZEICHNIS
94
31. Förster VT (1948). Zwischenmolekulare Energiewanderung und Fluoreszenz. Anals of Physics (Leipzig) 2:55-75
32. Freeman WM, Walker SJ, Vrana KE (1999). Quantitative RT-PCR: pitfalls and potential. Biotechniques 26:112-122, 124-115
33. Fronhoffs S, Totzke G, Stier S, Wernert N, Rothe M, Bruning T, Koch B, Sachinidis A, Vetter H, Ko Y (2002). A method for the rapid construction of cRNA standard curves in quantitative real-time reverse transcription polymerase chain reaction. Mol Cell Probes 16:99-110
34. Garcia AL, Kindt A, Quispe-Tintaya KW, Bermudez H, Llanos A, Arevalo J, Banuls AL, De Doncker S, Le Ray D, Dujardin JC (2005). American tegumentary leishmaniasis: antigen-gene polymorphism, taxonomy and clinical pleomorphism. Infect Genet Evol 5:109-116
35. Garg R, Dube A (2006). Animal models for vaccine studies for visceral leishmaniasis. Indian J Med Res 123:439-454
36. Geißdörfer W unpublished data. 37. Gibson UE, Heid CA, Williams PM (1996). A novel method for real time
quantitative RT-PCR. Genome Res 6:995-1001 38. Goto H, Lindoso JA (2010). Current diagnosis and treatment of cutaneous
and mucocutaneous leishmaniasis. Expert Rev Anti Infect Ther 8:419-433 39. Handman E, Ceredig R, Mitchell GF (1979). Murine cutaneous leishmaniasis:
disease patterns in intact and nude mice of various genotypes and examination of some differences between normal and infected macrophages. Aust J Exp Biol Med Sci 57:9-29
40. Heid CA, Stevens J, Livak KJ, Williams PM (1996). Real time quantitative PCR. Genome Res 6:986-994
41. Helling RB, Goodman HM, Boyer HW (1974). Analysis of endonuclease R-EcoRI fragments of DNA from lambdoid bacteriophages and other viruses by agarose-gel electrophoresis. J Virol 14:1235-1244
42. Herwaldt BL (1999). Leishmaniasis. Lancet 354:1191-1199 43. Higuchi R, Dollinger G, Walsh PS, Griffith R (1992). Simultaneous
amplification and detection of specific DNA sequences. Biotechnology (N Y) 10:413-417
44. Higuchi R, Fockler C, Dollinger G, Watson R (1993). Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N Y) 11:1026-1030
45. Holland PM, Abramson RD, Watson R, Gelfand DH (1991). Detection of specific polymerase chain reaction product by utilizing the 5'----3' exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci U S A 88:7276-7280
46. Honore S, Garin YJ, Sulahian A, Gangneux JP, Derouin F (1998). Influence of the host and parasite strain in a mouse model of visceral Leishmania infantum infection. FEMS Immunol Med Microbiol 21:231-239
47. Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A,

7. LITERATURVERZEICHNIS
95
Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Muller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O'Neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ (2005). The genome of the kinetoplastid parasite, Leishmania major. Science 309:436-442
48. Iyengar R, Stuehr DJ, Marletta MA (1987). Macrophage synthesis of nitrite, nitrate, and N-nitrosamines: precursors and role of the respiratory burst. Proc Natl Acad Sci U S A 84:6369-6373
49. Jha TK, Olliaro P, Thakur CP, Kanyok TP, Singhania BL, Singh IJ, Singh NK, Akhoury S, Jha S (1998). Randomised controlled trial of aminosidine (paromomycin) v sodium stibogluconate for treating visceral leishmaniasis in North Bihar, India. BMJ 316:1200-1205
50. Kamhawi S, Belkaid Y, Modi G, Rowton E, Sacks D (2000). Protection against cutaneous leishmaniasis resulting from bites of uninfected sand flies. Science 290:1351-1354
51. Kaur S, Kaur T, Garg N, Mukherjee S, Raina P, Athokpam V (2008). Effect of dose and route of inoculation on the generation of CD4+ Th1/Th2 type of immune response in murine visceral leishmaniasis. Parasitol Res 103:1413-1419
52. Kaye P, Scott P (2011). Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol 9:604-615
53. Koehler K, Stechele M, Hetzel U, Domingo M, Schonian G, Zahner H, Burkhardt E (2002). Cutaneous leishmaniosis in a horse in southern Germany caused by Leishmania infantum. Vet Parasitol 109:9-17
54. Kubar J, Fragaki K (2005). Recombinant DNA-derived leishmania proteins: from the laboratory to the field. Lancet Infect Dis 5:107-114
55. Kumar R, Nylen S (2012). Immunobiology of visceral leishmaniasis. Front Immunol 3:251
56. Kumar S, Reed MW, Gamper HB, Jr., Gorn VV, Lukhtanov EA, Foti M, West J, Meyer RB, Jr., Schweitzer BI (1998). Solution structure of a highly stable DNA duplex conjugated to a minor groove binder. Nucleic Acids Res 26:831-838
57. Kutyavin IV, Afonina IA, Mills A, Gorn VV, Lukhtanov EA, Belousov ES, Singer MJ, Walburger DK, Lokhov SG, Gall AA, Dempcy R, Reed MW, Meyer RB, Hedgpeth J (2000). 3'-minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res 28:655-661
58. Lachaud L, Marchergui-Hammami S, Chabbert E, Dereure J, Dedet JP, Bastien P (2002). Comparison of six PCR methods using peripheral blood for detection of canine visceral leishmaniasis. J Clin Microbiol 40:210-215
59. Laskay T, Miko TL, Negesse Y, Solbach W, Rollinghoff M, Frommel D (1995). Detection of cutaneous Leishmania infection in paraffin-embedded skin

7. LITERATURVERZEICHNIS
96
biopsies using the polymerase chain reaction. Trans R Soc Trop Med Hyg 89:273-275
60. Leclercq V, Lebastard M, Belkaid Y, Louis J, Milon G (1996). The outcome of the parasitic process initiated by Leishmania infantum in laboratory mice: a tissue-dependent pattern controlled by the Lsh and MHC loci. J Immunol 157:4537-4545
61. Leon W, Fouts DL, Manning J (1978). Sequence arrangement of the 16S and 26S rRNA genes in the pathogenic haemoflagellate Leishmania donovani. Nucleic Acids Res 5:491-504
62. Luis L, Ramirez A, Aguilar CM, Eresh S, Barker DC, Mendoza-Leon A (1998). The genomic fingerprinting of the coding region of the beta-tubulin gene in Leishmania identification. Acta Trop 69:193-204
63. Lux H, Hart DT, Parker PJ, Klenner T (1996). Ether lipid metabolism, GPI anchor biosynthesis, and signal transduction are putative targets for anti-leishmanial alkyl phospholipid analogues. Adv Exp Med Biol 416:201-211
64. Mackay IM (2004). Real-time PCR in the microbiology laboratory. Clin Microbiol Infect 10:190-212
65. Marfurt J, Niederwieser I, Makia ND, Beck HP, Felger I (2003). Diagnostic genotyping of Old and New World Leishmania species by PCR-RFLP. Diagn Microbiol Infect Dis 46:115-124
66. Melby PC, Yang YZ, Cheng J, Zhao W (1998). Regional differences in the cellular immune response to experimental cutaneous or visceral infection with Leishmania donovani. Infect Immun 66:18-27
67. Molina R, Lohse JM, Pulido F, Laguna F, Lopez-Velez R, Alvar J (1999). Infection of sand flies by humans coinfected with Leishmania infantum and human immunodeficiency virus. Am J Trop Med Hyg 60:51-53
68. Morillas-Marquez F, Martin-Sanchez J, Acedo-Sanchez C, Pineda JA, Macias J, Sanjuan-Garcia J (2002). Leishmania infantum (Protozoa, kinetoplastida): transmission from infected patients to experimental animal under conditions that simulate needle-sharing. Exp Parasitol 100:71-74
69. Mortarino M, Franceschi A, Mancianti F, Bazzocchi C, Genchi C, Bandi C (2004). [Quantitative PCR in the diagnosis of Leishmania]. Parassitologia 46:163-167
70. Mougneau E, Bihl F, Glaichenhaus N (2011). Cell biology and immunology of Leishmania. Immunol Rev 240:286-296
71. Murray HW, Berman JD, Davies CR, Saravia NG (2005). Advances in leishmaniasis. Lancet 366:1561-1577
72. Nashleanas M, Kanaly S, Scott P (1998). Control of Leishmania major infection in mice lacking TNF receptors. J Immunol 160:5506-5513
73. Naucke TJ, Pesson B (2000). Presence of Phlebotomus (Transphlebotomus) mascittii Grassi, 1908 (Diptera : Psychodidae) in Germany. Parasitol Res 86:335-336
74. Navin TR, Arana BA, Arana FE, Berman JD, Chajon JF (1992). Placebo-controlled clinical trial of sodium stibogluconate (Pentostam) versus ketoconazole for treating cutaneous leishmaniasis in Guatemala. J Infect Dis 165:528-534
75. Nogva HK, Rudi K, Naterstad K, Holck A, Lillehaug D (2000). Application of 5'-nuclease PCR for quantitative detection of Listeria monocytogenes in

7. LITERATURVERZEICHNIS
97
pure cultures, water, skim milk, and unpasteurized whole milk. Appl Environ Microbiol 66:4266-4271
76. Oliveira DM, Costa MA, Chavez-Fumagalli MA, Valadares DG, Duarte MC, Costa LE, Martins VT, Gomes RF, Melo MN, Soto M, Tavares CA, Coelho EA (2012). Evaluation of parasitological and immunological parameters of Leishmania chagasi infection in BALB/c mice using different doses and routes of inoculation of parasites. Parasitol Res 110:1277-1285
77. Overbergh L, Giulietti A, Valckx D, Decallonne R, Bouillon R, Mathieu C (2003). The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J Biomol Tech 14:33-43
78. Peters N, Sacks D (2006). Immune privilege in sites of chronic infection: Leishmania and regulatory T cells. Immunol Rev 213:159-179
79. Pimenta PF, Turco SJ, McConville MJ, Lawyer PG, Perkins PV, Sacks DL (1992). Stage-specific adhesion of Leishmania promastigotes to the sandfly midgut. Science 256:1812-1815
80. Piscopo TV, Mallia AC (2006). Leishmaniasis. Postgrad Med J 82:649-657 81. Ready PD (2010). Leishmaniasis emergence in Europe. Euro Surveill
15:19505 82. Reiss RA, Rutz B (1999). Quality control PCR: a method for detecting
inhibitors of Taq DNA polymerase. Biotechniques 27:920-922, 924-926 83. Reithinger R, Dujardin JC (2007). Molecular diagnosis of leishmaniasis:
current status and future applications. J Clin Microbiol 45:21-25 84. Requena JM, Soto M, Quijada L, Alonso C (1997). Genes and chromosomes of
Leishmania infantum. Mem Inst Oswaldo Cruz 92:853-858 85. Rittig MG, Bogdan C (2000). Leishmania-host-cell interaction: complexities
and alternative views. Parasitol Today 16:292-297 86. Rodgers MR, Popper SJ, Wirth DF (1990). Amplification of kinetoplast DNA
as a tool in the detection and diagnosis of Leishmania. Exp Parasitol 71:267-275
87. Rolao N, Cortes S, Gomes-Pereira S, Campino L (2007). Leishmania infantum: mixed T-helper-1/T-helper-2 immune response in experimentally infected BALB/c mice. Exp Parasitol 115:270-276
88. Russell R, Iribar MP, Lambson B, Brewster S, Blackwell JM, Dye C, Ajioka JW (1999). Intra and inter-specific microsatellite variation in the Leishmania subgenus Viannia. Mol Biochem Parasitol 103:71-77
89. Sacks D, Noben-Trauth N (2002). The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol 2:845-858
90. Sacks DL, Pimenta PF, McConville MJ, Schneider P, Turco SJ (1995). Stage-specific binding of Leishmania donovani to the sand fly vector midgut is regulated by conformational changes in the abundant surface lipophosphoglycan. J Exp Med 181:685-697
91. Saidac DS, Marras SA, Parveen N (2009). Detection and quantification of Lyme spirochetes using sensitive and specific molecular beacon probes. BMC Microbiol 9:43
92. Schleicher U, Bogdan C (2009). Generation, culture and flow-cytometric characterization of primary mouse macrophages. Methods Mol Biol 531:203-224

7. LITERATURVERZEICHNIS
98
93. Schubach A, Haddad F, Oliveira-Neto MP, Degrave W, Pirmez C, Grimaldi G, Jr., Fernandes O (1998). Detection of Leishmania DNA by polymerase chain reaction in scars of treated human patients. J Infect Dis 178:911-914
94. Schulz A, Mellenthin K, Schonian G, Fleischer B, Drosten C (2003). Detection, differentiation, and quantitation of pathogenic leishmania organisms by a fluorescence resonance energy transfer-based real-time PCR assay. J Clin Microbiol 41:1529-1535
95. Seifert K (2011). Structures, targets and recent approaches in anti-leishmanial drug discovery and development. Open Med Chem J 5:31-39
96. Selvin PR, Hearst JE (1994). Luminescence energy transfer using a terbium chelate: improvements on fluorescence energy transfer. Proc Natl Acad Sci U S A 91:10024-10028
97. Silveira TG, Arraes SM, Bertolini DA, Teodoro U, Lonardoni MV, Roberto AC, Ramos M, Nerilo Sobrinho A, Ishikawa E, Shaw J (1999). [The laboratory diagnosis and epidemiology of cutaneous leishmaniasis in Parana State, southern Brazil]. Rev Soc Bras Med Trop 32:413-423
98. Solbach W, Forberg K, Kammerer E, Bogdan C, Rollinghoff M (1986). Suppressive effect of cyclosporin A on the development of Leishmania tropica-induced lesions in genetically susceptible BALB/c mice. J Immunol 137:702-707
99. Solbach W, Forberg K, Rollinghoff M (1986). Effect of T-lymphocyte suppression on the parasite burden in Leishmania major-infected, genetically susceptible BALB/c mice. Infect Immun 54:909-912
100. Stenger S, Solbach W, Rollinghoff M, Bogdan C (1991). Cytokine interactions in experimental cutaneous leishmaniasis. II. Endogenous tumor necrosis factor-alpha production by macrophages is induced by the synergistic action of interferon (IFN)-gamma and interleukin (IL) 4 and accounts for the antiparasitic effect mediated by IFN-gamma and IL 4. Eur J Immunol 21:1669-1675
101. Stenger S, Thuring H, Rollinghoff M, Bogdan C (1994). Tissue expression of inducible nitric oxide synthase is closely associated with resistance to Leishmania major. J Exp Med 180:783-793
102. Strauss-Ayali D, Jaffe CL, Burshtain O, Gonen L, Baneth G (2004). Polymerase chain reaction using noninvasively obtained samples, for the detection of Leishmania infantum DNA in dogs. J Infect Dis 189:1729-1733
103. Sundar S, Chatterjee M (2006). Visceral leishmaniasis - current therapeutic modalities. Indian J Med Res 123:345-352
104. Thakur CP, Kanyok TP, Pandey AK, Sinha GP, Zaniewski AE, Houlihan HH, Olliaro P (2000). A prospective randomized, comparative, open-label trial of the safety and efficacy of paromomycin (aminosidine) plus sodium stibogluconate versus sodium stibogluconate alone for the treatment of visceral leishmaniasis. Trans R Soc Trop Med Hyg 94:429-431
105. Urbina JA (1997). Lipid biosynthesis pathways as chemotherapeutic targets in kinetoplastid parasites. Parasitology 114 Suppl:S91-99
106. van der Meide W, Guerra J, Schoone G, Farenhorst M, Coelho L, Faber W, Peekel I, Schallig H (2008). Comparison between quantitative nucleic acid sequence-based amplification, real-time reverse transcriptase PCR, and

7. LITERATURVERZEICHNIS
99
real-time PCR for quantification of Leishmania parasites. J Clin Microbiol 46:73-78
107. van Eys GJ, Schoone GJ, Kroon NC, Ebeling SB (1992). Sequence analysis of small subunit ribosomal RNA genes and its use for detection and identification of Leishmania parasites. Mol Biochem Parasitol 51:133-142
108. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:RESEARCH0034
109. Villalba E, Ramirez JL (1982). Ribosomal DNA of Leishmania brasiliensis: number of ribosomal copies and gene isolation. J Protozool 29:438-441
110. Wall SJ, Edwards DR (2002). Quantitative reverse transcription-polymerase chain reaction (RT-PCR): a comparison of primer-dropping, competitive, and real-time RT-PCRs. Anal Biochem 300:269-273
111. Wilhelm P, Ritter U, Labbow S, Donhauser N, Rollinghoff M, Bogdan C, Korner H (2001). Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J Immunol 166:4012-4019
112. Wilhelm P, Wiede F, Meissner A, Donhauser N, Bogdan C, Korner H (2005). TNF but not Fas ligand provides protective anti-L. major immunity in C57BL/6 mice. Microbes Infect 7:1461-1468
113. Wilson ME, Jeronimo SM, Pearson RD (2005). Immunopathogenesis of infection with the visceralizing Leishmania species. Microb Pathog 38:147-160
114. World Health Organization (WHO) (2009) Leishmaniasis: background information. A brief history of the disease., p Available from: www.who.int/leishmaniasis/en/
115. Wortmann G, Sweeney C, Houng HS, Aronson N, Stiteler J, Jackson J, Ockenhouse C (2001). Rapid diagnosis of leishmaniasis by fluorogenic polymerase chain reaction. Am J Trop Med Hyg 65:583-587
116. Wortmann G, Hochberg L, Houng HH, Sweeney C, Zapor M, Aronson N, Weina P, Ockenhouse CF (2005). Rapid identification of Leishmania complexes by a real-time PCR assay. Am J Trop Med Hyg 73:999-1004

8. ABKÜRZUNGSVERZEICHNIS
100
8 ABKÜRZUNGSVERZEICHNIS °C Grad Celsius µm Mikrometer Abb. Abbildung MΦ Makrophage bidest. zweifach destilliert MGB Minor groove binder BM- MΦ aus Knochenmark generierte µm Mikrometer Makrophagen MgCl2 Magnesiumchlorid bp Basenpaare min Minute ca. zirka Mio. Million CL Kutane Leishmaniose mRNA messenger RNA CT Schwellenwertzyklus n Nano- (Threshold Cycle) NFQ Non fluorescent quencher DMEM Dulbecco’s modified Eagle’s nm Nanometer Medium ns nicht signifikant DNA Desoxyribonukleinsäure o.g. oben genannt dNTPs Desoxyribonukleosidtriphosphate p Ergebniswahrscheinlichkeit et al. und andere Autoren PBS Phosphate buffered saline FAM Carboxyfluorescein PCR Polymerase Chain Reaction FCS Fötales Kälberserum p.i. post infectionem g Gramm pUC DNA-Größenstandard ges. gesamt rDNA ribosomale DNA ggf. gegebenenfalls rmM- Rekombinanter muriner Makropha- h Stunde CSF genkolonie-stimulierender Faktor
HIV human immunodeficiency virus Rn normalized reporter value IFN Interferon RNA Ribonukleinsäure IPC Internal positive control RPMI Kultivierungsmedium
i.v. intravenös rRNA ribosomale RNA KM Knochenmark s.c. subkutan λ Lambda SD Standardabweichung l Liter sek Sekunde L. Leishmania SSU small subunit LDA Limiting dilution Analysis TAMRA Carboxytetramethylrhodamin L.i. Leishmania infantum TE Tetramethylammoniumchlorid LK Lymphknoten TAE Tris-Acetat-EDTA-Puffer L.m. Leishmania major TNF Tumor-Nekrose-Faktor
log logarithmisch U/min Umdrehungen pro Minute m Milli- VIC Trichlorophenylcarboxyfluorescein M Mol VL Viszerale Leishmaniose µ Mikro- v/v Volumen-Volumen-Verhältnis