current organic chemistry chiral lewis acid catalyzed ene...
TRANSCRIPT

Current Organic Chemistry, 2000, 4, 305-342 305
Chiral Lewis Acid Catalyzed Ene-Reactions
Luiz Carlos Dias*
Instituto de Química - Universidade Estadual de Campinas - UNICAMP, CEP: 13083-970 - C.P. 6154 - Campinas - SP - Brazil
Abstract: This review covers recent progress in the use of chiral Lewis acidcatalysts in ene-reactions which involve carbonyl and imine compounds asenophiles. Chiral Lewis acid catalysts containing aluminum, titanium,ytterbium and copper are critically reviewed. Synthetic applications of recentsystems are specifically discussed.
Dedicated to the Brazilian Chemical Society
General Introduction mole-for-mole basis, many chemists are seeking newasymmetric catalysts, individual molecules each ofwhich mediate thousands of enantioselectiveconversions [4,9,10]. The strategy is to employ areagent that under normal circumstances does notreact with the substrate, but undergoes a selectivereaction under the influence of catalytic amounts of achiral compound [11,12,13].
Most of the molecules in the world are chiral, and awide range of biological and physical functions aregenerated through precise molecular recognition thatrequires strict matching of chirality [1]. Chirality is not aprerequisite for bioactivity but in bioactive moleculeswhere a stereogenic center is present greatdifferences in biological activities are usually observedfor both enantiomers as well as for the racemic mixture.At a molecular level, asymmetry dominates biologicalprocesses and a variety of functions responsible formetabolism and numerous biological responses occurbecause enzymes, receptors and other natural bindingsites recognize substrates with specific chirality [1].
One important criterion when considering anasymmetric synthesis is the degree ofenantioselectivity of a reaction. The pharmaceuticalindustry requires chiral products of greater than 99%ee, with less than 0.1% of the undesired enantiomer.
The design and development of efficient chiralcatalysts for enantioselective synthesis has becomeone of the most intense, dynamic and rapidly growingareas of organic chemical research [13,14]. Remarkableprogress in the development of catalytic asymmetricreactions has enabled the synthesis of various opticallyactive compounds with high optical purity [4].
Asymmetric synthesis is an important means bywhich enantiopure chiral molecules may be obtainedfor biological studies and sale, and the synthesis ofbiologically relevant natural and unnatural organicmolecules in optically pure form is of fundamentalimportance in medicinal chemistry and relateddisciplines [1,2,3]. Of particular importance is thedevelopment of asymmetric catalysts for the carbon-carbon bond forming reactions [4,5,6]. The use ofchiral catalysts is one of the most attractive methods forperforming asymmetric reactions, because comparedto the stoichiometric use of chiral auxiliaries, a smalleramount of a readily available chiral material is required.Therefore, a large quantity of the naturally andnonnaturally occurring chiral materials are directlyobtained with no need for further manipulation orremoval and recovery of the chiral auxiliary [7,8].Because stoichiometric reagents are costly to use on a
This review presents a comprehensive survey ofsome modern and highly selective methods for theenantioselective ene-reaction, in which asymmetricinduction is derived from the catalyst complex. Recentadvances in this area are turning chemist’s dreams intoreality at both academic and industrial levels.
Introduction to Ene-reaction
The ene-reaction is one of the most powerfulmethods for carbon-carbon bond construction insynthetic organic chemistry and it was first recognizedin 1943 by Alder and classified in his Nobel Lecture asan “indirect substitution addition” or “ene synthesis” in1950 [15,16]. The thermal and Lewis acid glyoxylateene-reaction was introduced more than 30 years ago
*Address correspondence to this author at the Instituto de Química -Universidade Estadual de Campinas - UNICAMP, CEP: 13083-970 -C.P. 6154 - Campinas - SP - Brazil; FAX: (019)-788-3023 - e-mail:[email protected]
1385-2728/00 $19.00+.00 © 2000 Bentham Science Publishers B.V.

306 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
by Klimova and Arbuzov et al. and has been studied byseveral groups [17-22]. The ene-reaction occursbetween an alkene having an allylic hydrogen (an"ene") and a compound containing an electron-deficient double bond (an "enophile") to form a σ-bondwith migration of the ene double bond and a 1,5-hydrogen shift (Scheme 1). It requires the energy foractivating the σ C-H and π X=Y bonds, while energy isgained on forming the σ Y-H and σ X-C bonds. Thethermal pericyclic ene-reaction can proceed through aconcerted six-electron pathway with a suprafacial threecomponent orbital interaction between the HOMO ofthe ene and the LUMO of the enophile (Scheme 1)[23-24].
(a) type I: reaction between all-carba-enecomponents with hetero-enophiles;
(b) type II: reaction between hetero ene componentand all-carba-enophile;
(c) type III: reaction occurring between hetero-enecomponents and hetero enophiles.
The ene-reaction is mechanistically related to theDiels-Alder cycloaddition, with enophiles orientedeither exo or endo with respect to the ene component,but requires a greater activation energy and highertemperatures. In the ene–reaction involving asuprafacial orbital interaction, the two electrons of theallylic σ C-H bond replace the two π electrons of thediene in the Diels-Alder. The ene process is favored byelectron withdrawing substituents on the enophiles, bystrain in the ene component, and by geometricalalignments that direct the components in favorablerelative positions. Whether the mechanism isconcerted or stepwise, positive charge is developed tosome extent at the ene component in Lewis acidpromoted reactions [23,24]. At this point, the Lewisacid promoted ene reactions differ from thermal enereactions where steric accessibility of the double bondand allylic hydrogen is the primary concern.
H
X
Y
X
YH
C
C
C
H
HOMO
CO R
H
+
ene enophile
1
2
3
bondformed
bondformed
LUMO
Ene-reactionMolecular Orbital interaction
ene: alkene, alkyne, allene, arene, carbon-heteroatom bondX=Y: C=C, C=O, C=N, C=S, O=O, N=N
32
1 allylictransposition
High demand within the pharmaceutical and fineindustries for efficient and economical methodologiesfor the asymmetric synthesis of both simple andcomplex molecules has resulted in new developmentsthrough the ene-reaction. An increasing emphasis hasbeen placed on developing catalyzed asymmetric enebond construction as a means of addressing the issuesof cost and operational simplicity inherent in industrialchemistry [25-28].
The use of Lewis acid catalysts has led toremarkable progress in the areas of both inter- andintramolecular ene reactions. When compared to theintermolecular reactions, intramolecular ene-reactionsare usually more facile, since these reactions takeadvantage of less negative activation entropy [29-32,37,42].
The carbonyl ene-reaction, particularly promoted bya stoichiometric to catalytic amount of Lewis acid hascurrently emerged as a useful method for theasymmetric synthesis of acyclic molecules [25-28]. Thisasymmetric reaction is a powerful tool for the synthesisof enantiomerically pure, complex molecules, althoughasymmetric catalysis of carbonyl-ene-reactions isdifficult to realize because the Lewis acid seems to berelatively far from the site of formation of the new chiralcenter. The selection of the central metals and thedesign of the chiral ligands are particularly important.
Scheme 1.
According to the nature of the reactants, the ene-reaction has been divided into two categories:
(1) all-carbon ene-reactions: takes place between anolefin bearing an allylic hydrogen atom (thecarba-ene) and an activated alkene or alkyne (thecarba-enophile) and,
(2) the hetero-ene-reaction: describes a reactionbetween an ene or enophile, either of whichcontains at least one heteroatom.

Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 307
One of the major problems associated with thecarbonyl-ene strategy is the limitation of the carbonylenophiles thus far developed. The synthetic potentialof the ene product heavily depends on thefunctionality of the carbonyl enophile employed. Thecarbonyl ene-reaction is severely substrate limited tohighly activated carbonyl components (e.g.glyoxylates, formaldehyde or chloral) or to highlyactivated ene components (e.g. 3-methylene-2,3-dihydrofuran). Usually, stoichiometric amounts ofpowerful Lewis acids are required owing to the lownucleophilicity of the olefin and tight binding of thehomoallylic alcohol product to the catalyst. Anotherproblem is that ene-reactions often suffer a seriousdrawback in terms of regiochemistry, for which stericaccessibility of hydrogen is an important factor.Reactions with unsymmetrical olefins usually give amixture of regioisomers.
induced by catalytic amounts of chiral Lewis acidcomplexes in the presence of molecular sieves 4A (MS4A) [36,37]. The authors described the first example ofan asymmetric ene-reaction between prochiral,halogenated aldehydes and alkenes catalyzed by chiralbinaphthol-derived aluminum complex (R)-5 (Scheme3, Table 1).
O
OAlMe
SiPh3
SiPh3
Me R1
R1 R
OHRCHO
The catalyst:
(R)-5
catalyst(20 mol%)
CH2Cl2-78 oC
MS 4A
4absolute configuration
established by conversion to the known
L-2-hydroxyisocaproic acid
+
Many natural products can be prepared at an earlystage of the synthetic scheme by taking advantage ofan asymmetric ene-reaction. Of special synthetic valueamong many carbonyl-ene variants is the glyoxylateene-reaction, which provides α-hydroxyesters ofbiological and synthetic importance.
Asymmetric Ene-Reactions
The first example of an asymmetric glyoxylate ene-reaction was described by Whitesell et al.in 1982 andconsisted of the use of a chiral glyoxylate andstoichiometric amounts of a Lewis acid [33,34].Addition of methylenecyclohexane 1 as the enecomponent to the chiral glyoxylate of 8-phenylmenthol2 in the presence of equivalent amounts of SnCl4afforded the corresponding ene product 3 with >99%diastereoselectivity in 94% yield (Scheme 2) [35].
Scheme 3.
Table 1. Addition of Alkenes to ProchiralHalogenated Aldehydes
entry R R1 catalyst (mol%) yield (%) ee (%)a
1 C6F5 Ph (R)-5 (20) 35 78
2 Cl3C Ph (R)-5 (20) 43 73
3 C6F5 SPh (R)-5 (20) 88 88
HORc*
O
O
ORc*
OH
O
Me
Ph
MeMe
O
+ -78 oC, 94%
>99 de
Rc*O- =
1
2
3
SnCl4 (100 mol%)
4 C6F5 SPh (R)-5 (10) 67 78
5 Cl3C SPh (R)-5 (20) 50 53
6 Cl3C Me (R)-5 (20) 79 78
a. Determined by HPLC analysis after conversion to the (-)-MTPA esters
The authors observed that the hindered 3,3'-bis-(triphenylsilyl)-substituent in chiral catalyst (R)-5 isessential to achieve good enantioselectivities since theuse of a catalyst derived from Me3Al and 3,3'-diphenylbinaphthol led to the racemic product 4 in lowyields. The best result in terms of yields andenantioselectivities was obtained in the ene-reactionbetween pentafluorobenzaldehyde and 2-(phenylthio)propene in the presence of 20 mol% ofcatalyst (R)-5 and MS 4A affording the corresponding
Scheme 2.
Chiral Aluminum Lewis Acid
In 1988, Yamamoto and coworkers provided the firstindication that asymmetry in ene-reactions could be

308 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
HOiPr
O
O
OH
O
OiPr
MePh
OH
OHPh
PhPh
OH
OH
O
O
OH
Ph
OH
Ph Ph
Ph
Ph
Me
OH
OH
+
(iPrO)2TiCl2 chiral diol
(10 mol% each)
MS 4A, -30 oC
4% ee (60%) 34% ee (72%)
44% ee (70%) 86% ee (82%)
1
6
7
8 9
10 11
ee's determined by lanthanide induced shift NMR measurement with (+)-Eu(DPPM)3 after conversion to the α-methoxy ester
Scheme 4.
product 4 in 88% yield and 88% ee (entry 3). With lessreactive methylene compounds, the observed yieldsand enantioselectivities in the catalyzed reaction weremodest and the stoichiometric reaction isrecommended.
Despite the good levels of asymmetric inductionobtained with this Al(III)-BINOL Lewis acid-basedcomplex, no Al(III)-derived Lewis acid catalyst systemthat affords higher levels of enantioselectivity in theene-reaction has been reported to date.
Chiral Titanium Lewis Acids
Titanium-based Lewis acid complexes haveundergone considerable development as vehicles foreffecting asymmetric catalytic ene-reactions.
Mikami and coworkers have reported an extremelyefficient asymmetric glyoxylate ene-reaction catalyzedby the titanium complexes (R)-12a/b , prepared in situfrom diisopropoxytitanium dihalide and optically activebinaphthol (BINOL) 1 1 in the presence of MS 4A(Schemes 4 and 5) [38]. Ene-reaction betweenmethylenecyclohexane 1 and isopropylglyoxylate 6 inthe presence of 10 mol% of a chiral catalyst derivedfrom a chiral diol and TiCl2(i-PrO)2 afforded thecorresponding ene-products (Scheme 4). The bestresult was obtained with the use of a chiral catalystderived from (R)-BINOL 1 1 and TiCl2(i-PrO)2 (86% eeand 82% yield). The discovery of the BINOL-derivedchiral catalyst was made after the screening of variouschiral catalysts derived from optically active diols [38].Tetra(alcoxy)titanium complexes provided for theexpedient synthesis of chiral catalyst structures byexploiting the facile substitution of monodentatealkoxide ligands for chelating optically active diols.Among the numerous chiral Ti(IV) complexes that areavailable from optically active chelating diols, Lewisacids derived from Binaphthol 1 1 (BINOL) haveundergone the largest degree of development ascatalysts for the ene-reaction (Scheme 4).
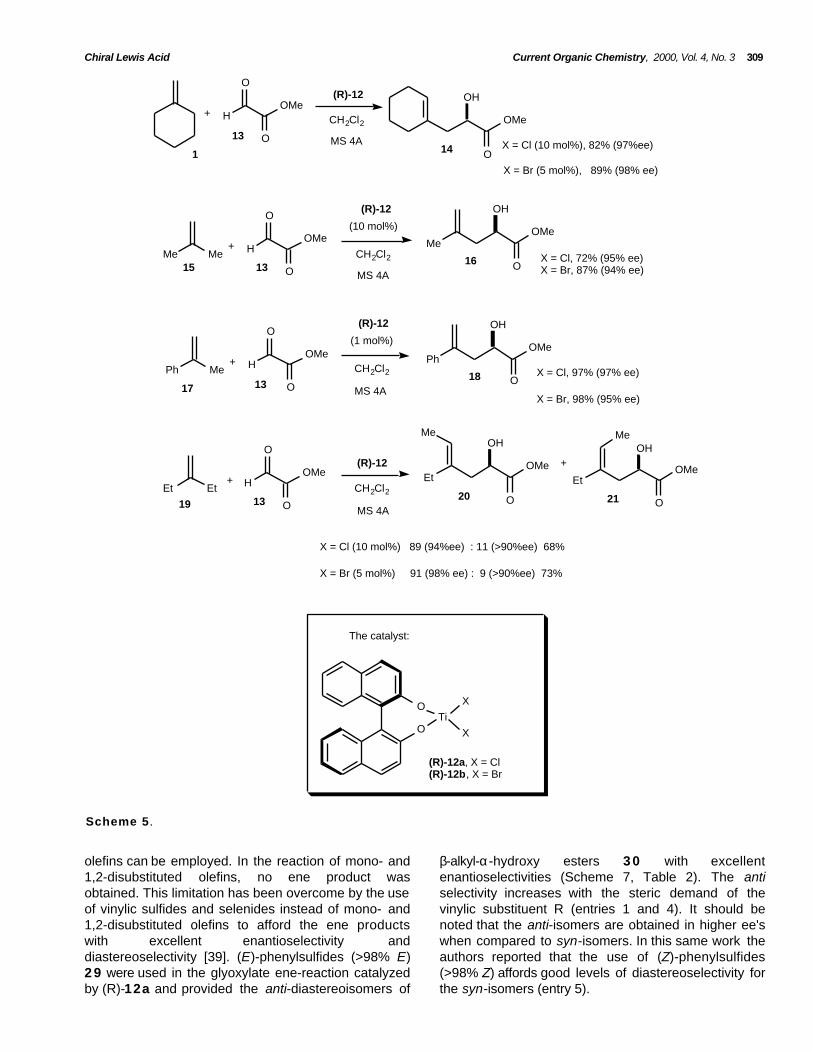
methylenecyclohexane 1 with methylglyoxylate 1 3 inthe presence of 10 mol% of catalyst (R)-12a affordedthe ene-product 14 in 83% yield and 97% ee. The useof 5 mol% of catalyst (R)-12b afforded the sameproduct in 89% yield and 98% ee. Especiallynoteworthy is the difference in asymmetric catalysisbetween the dibromo and dichloro catalysts. Thedibromide catalyst is superior to the dichloride in boththe reactivity and enantioselectivity for the glyoxylateene-reaction involving a methylene hydrogen shift inparticular (Scheme 5).
Both (R)- and (S)-BINOL 1 1 are commerciallyavailable in optically pure forms. It is important to pointout that the catalyst derived from (S)-BINOL 11 affordsthe (S)-alcohol and the catalyst derived from (R)-BINOL11 consistently affords the corresponding (R)-alcohol.
The high levels of enantiocontrol and rateacceleration observed with the BINOL catalysts areapparently due to the favorable influence of inherentC2-symmetry and the higher acidity of BINOLcompared to those of aliphatic diols. Remarkableenantioselectivities were observed with the use ofmethylglyoxylate 1 3 instead of isopropylglyoxylate 6(up to 99%, Scheme 5). The ene-reaction of
On the other hand, the dichloride catalyst is lower inreactivity but superior in enantioselectivity for certainglyoxylate ene-reactions involving a methyl hydrogenshift (Scheme 6).
However, due to limiting reactivity of the catalyst-glyoxylate complex, only nucleophilic 1,1-disubstituted
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 309
Me Me HOMe
O
O
CH2Cl2Me
OMe
O
OH
Ph Me HOMe
O
O
CH2Cl2Ph
OMe
O
OH
Et Et HOMe
O
O
CH2Cl2Et
OMe
O
OH
HOMe
O
O
CH2Cl2 OMe
O
OH
Me
O
OTi
X
X
EtOMe
O
OHMe
(10 mol%)
+
(R)-12
MS 4A
(1 mol%)
+
(R)-12
MS 4A
X = Cl, 72% (95% ee)X = Br, 87% (94% ee)
X = Cl, 97% (97% ee)
X = Br, 98% (95% ee)
+
(R)-12
MS 4A
X = Cl (10 mol%) 89 (94%ee) : 11 (>90%ee) 68%
X = Br (5 mol%) 91 (98% ee) : 9 (>90%ee) 73%
+
(R)-12
MS 4A X = Cl (10 mol%), 82% (97%ee)
X = Br (5 mol%), 89% (98% ee)
16
(R)-12a, X = Cl(R)-12b, X = Br
The catalyst:
+
17 13
1413
19 13
1
20 21
18
15 13
Scheme 5.
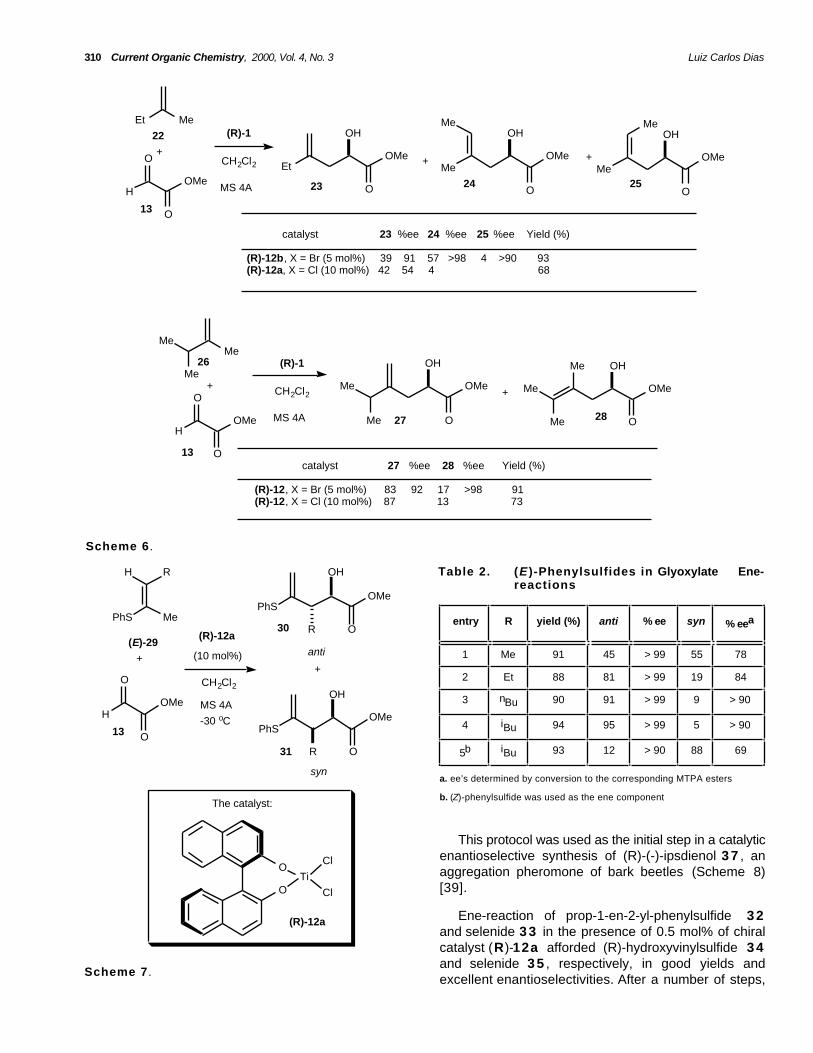
olefins can be employed. In the reaction of mono- and1,2-disubstituted olefins, no ene product wasobtained. This limitation has been overcome by the useof vinylic sulfides and selenides instead of mono- and1,2-disubstituted olefins to afford the ene productswith excellent enantioselectivity anddiastereoselectivity [39]. (E)-phenylsulfides (>98% E)2 9 were used in the glyoxylate ene-reaction catalyzedby (R)-12a and provided the anti-diastereoisomers of
β-alkyl-α-hydroxy esters 3 0 with excellentenantioselectivities (Scheme 7, Table 2). The antiselectivity increases with the steric demand of thevinylic substituent R (entries 1 and 4). It should benoted that the anti-isomers are obtained in higher ee'swhen compared to syn-isomers. In this same work theauthors reported that the use of (Z)-phenylsulfides(>98% Z) affords good levels of diastereoselectivity forthe syn-isomers (entry 5).
310 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
Et Me
HOMe
O
O
CH2Cl2 EtOMe
O
OH
MeOMe
O
OHMe
MeOMe
O
OHMe
Me
HOMe
O
O
CH2Cl2OMe
O
OH
OMe
Me
O
OHMe
Me
Me
Me
Me
Me
+
(R)-1
MS 4A
+ +
catalyst 23 %ee 24 %ee 25 %ee Yield (%)
(R)-12b, X = Br (5 mol%) 39 91 57 >98 4 >90 93(R)-12a, X = Cl (10 mol%) 42 54 4 68
+
(R)-1
MS 4A
+
catalyst 27 %ee 28 %ee Yield (%)
(R)-12, X = Br (5 mol%) 83 92 17 >98 91(R)-12, X = Cl (10 mol%) 87 13 73
22
23
13
24 25
27 28
13
26
Scheme 6.
O
OTi
Cl
Cl
PhS Me
HOMe
O
O
CH2Cl2
PhSOMe
O
OHH R
R
PhSOMe
O
OH
R
(R)-12a
The catalyst:
(10 mol%)+
(R)-12a
MS 4A
-30 oC
anti
syn
+
(E)-29
13
30
31
Table 2. (E )-Phenylsulfides in Glyoxylate Ene-reactions
entry R yield (%) anti % ee syn % eea
1 Me 91 45 > 99 55 78
2 Et 88 81 > 99 19 84
3 nBu 90 91 > 99 9 > 90
4 iBu 94 95 > 99 5 > 90
5b iBu 93 12 > 90 88 69
a. ee’s determined by conversion to the corresponding MTPA esters
b. (Z)-phenylsulfide was used as the ene component
This protocol was used as the initial step in a catalyticenantioselective synthesis of (R)-(-)-ipsdienol 3 7 , anaggregation pheromone of bark beetles (Scheme 8)[39].
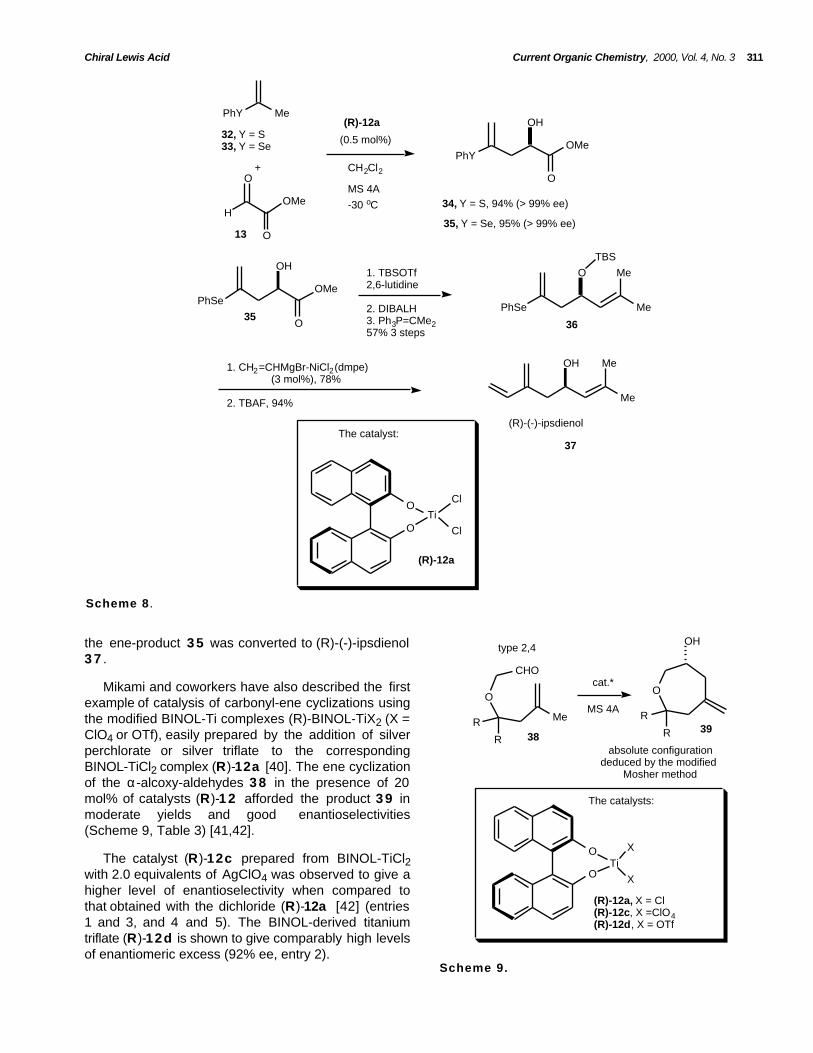
Ene-reaction of prop-1-en-2-yl-phenylsulfide 3 2and selenide 3 3 in the presence of 0.5 mol% of chiralcatalyst (R)-12a afforded (R)-hydroxyvinylsulfide 3 4and selenide 3 5 , respectively, in good yields andexcellent enantioselectivities. After a number of steps,
Scheme 7.
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 311
O
OTi
Cl
Cl
PhY Me
HOMe
O
O
CH2Cl2PhY
OMe
O
OH
PhSeOMe
O
OH
PhSe
O
OH Me
Me
Me
Me
TBS
(R)-12a
The catalyst:
(0.5 mol%)
+
(R)-12a
MS 4A
-30 oC 34, Y = S, 94% (> 99% ee)
35, Y = Se, 95% (> 99% ee)
32, Y = S33, Y = Se
(R)-(-)-ipsdienol
1. TBSOTf2,6-lutidine
2. DIBALH3. Ph3P=CMe257% 3 steps
1. CH2=CHMgBr-NiCl2(dmpe) (3 mol%), 78%
2. TBAF, 94%
13
3536
37
Scheme 8.
the ene-product 3 5 was converted to (R)-(-)-ipsdienol3 7 .
O
OTi
X
X
CHO
O
MeR
R
O
R
R
OH
The catalysts:
(R)-12a, X = Cl(R)-12c, X =ClO4(R)-12d, X = OTf
cat.*
MS 4A
type 2,4
absolute configurationdeduced by the modified
Mosher method
3839
Mikami and coworkers have also described the firstexample of catalysis of carbonyl-ene cyclizations usingthe modified BINOL-Ti complexes (R)-BINOL-TiX2 (X =ClO4 or OTf), easily prepared by the addition of silverperchlorate or silver triflate to the correspondingBINOL-TiCl2 complex (R)-12a [40]. The ene cyclizationof the α-alcoxy-aldehydes 3 8 in the presence of 20mol% of catalysts (R)-1 2 afforded the product 3 9 inmoderate yields and good enantioselectivities(Scheme 9, Table 3) [41,42].
The catalyst (R)-12c prepared from BINOL-TiCl2with 2.0 equivalents of AgClO4 was observed to give ahigher level of enantioselectivity when compared tothat obtained with the dichloride (R)-12a [42] (entries1 and 3, and 4 and 5). The BINOL-derived titaniumtriflate (R)-12d is shown to give comparably high levelsof enantiomeric excess (92% ee, entry 2).
Scheme 9.
312 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
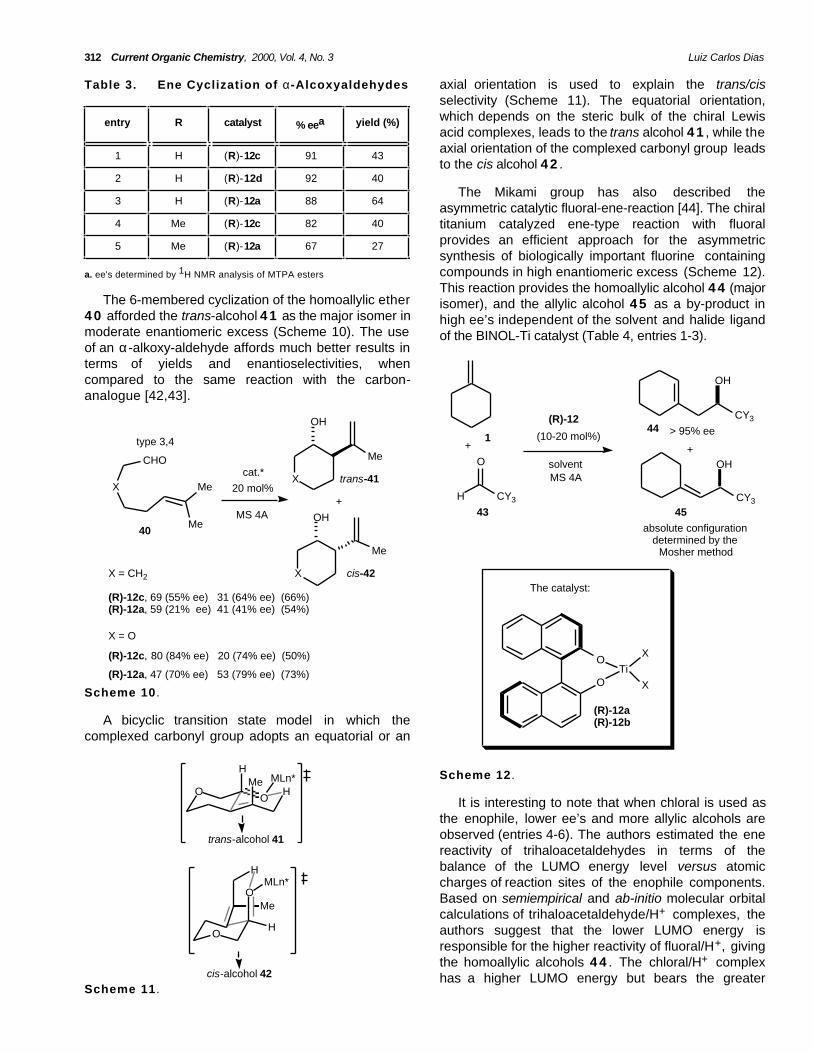
Table 3. Ene Cyclization of α-Alcoxyaldehydes axial orientation is used to explain the trans/cisselectivity (Scheme 11). The equatorial orientation,which depends on the steric bulk of the chiral Lewisacid complexes, leads to the trans alcohol 4 1 , while theaxial orientation of the complexed carbonyl group leadsto the cis alcohol 4 2 .
entry R catalyst % eea yield (%)
1 H (R)-12c 91 43
2 H (R)-12d 92 40The Mikami group has also described the
asymmetric catalytic fluoral-ene-reaction [44]. The chiraltitanium catalyzed ene-type reaction with fluoralprovides an efficient approach for the asymmetricsynthesis of biologically important fluorine containingcompounds in high enantiomeric excess (Scheme 12).This reaction provides the homoallylic alcohol 4 4 (majorisomer), and the allylic alcohol 4 5 as a by-product inhigh ee’s independent of the solvent and halide ligandof the BINOL-Ti catalyst (Table 4, entries 1-3).
3 H (R)-12a 88 64
4 Me (R)-12c 82 40
5 Me (R)-12a 67 27
a. ee's determined by 1H NMR analysis of MTPA esters
The 6-membered cyclization of the homoallylic ether4 0 afforded the trans-alcohol 4 1 as the major isomer inmoderate enantiomeric excess (Scheme 10). The useof an α-alkoxy-aldehyde affords much better results interms of yields and enantioselectivities, whencompared to the same reaction with the carbon-analogue [42,43].
O
OTi
X
X
H CY3
O solvent
CY3
OH
CY3
OH
(R)-12a(R)-12b
The catalyst:
+
(R)-12
MS 4A
(10-20 mol%)+
> 95% ee
absolute configuration determined by the
Mosher method
1
43
44
45
CHO
X Me
Me
X
OH
Me
X
OH
Me
cat.*
MS 4A
type 3,4
+
trans-41
(R)-12c, 80 (84% ee) 20 (74% ee) (50%)
(R)-12a, 47 (70% ee) 53 (79% ee) (73%)
20 mol%
X = O
(R)-12c, 69 (55% ee) 31 (64% ee) (66%)(R)-12a, 59 (21% ee) 41 (41% ee) (54%)
X = CH2
40
cis-42
Scheme 10 .
A bicyclic transition state model in which thecomplexed carbonyl group adopts an equatorial or an
O HO
HMe MLn*
O
O
H
MLn*H
Me
trans-alcohol 41
cis-alcohol 42
Scheme 11 .
Scheme 12 .
It is interesting to note that when chloral is used asthe enophile, lower ee’s and more allylic alcohols areobserved (entries 4-6). The authors estimated the enereactivity of trihaloacetaldehydes in terms of thebalance of the LUMO energy level versus atomiccharges of reaction sites of the enophile components.Based on semiempirical and ab-initio molecular orbitalcalculations of trihaloacetaldehyde/H+ complexes, theauthors suggest that the lower LUMO energy isresponsible for the higher reactivity of fluoral/H+, givingthe homoallylic alcohols 4 4 . The chloral/H+ complexhas a higher LUMO energy but bears the greater
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 313
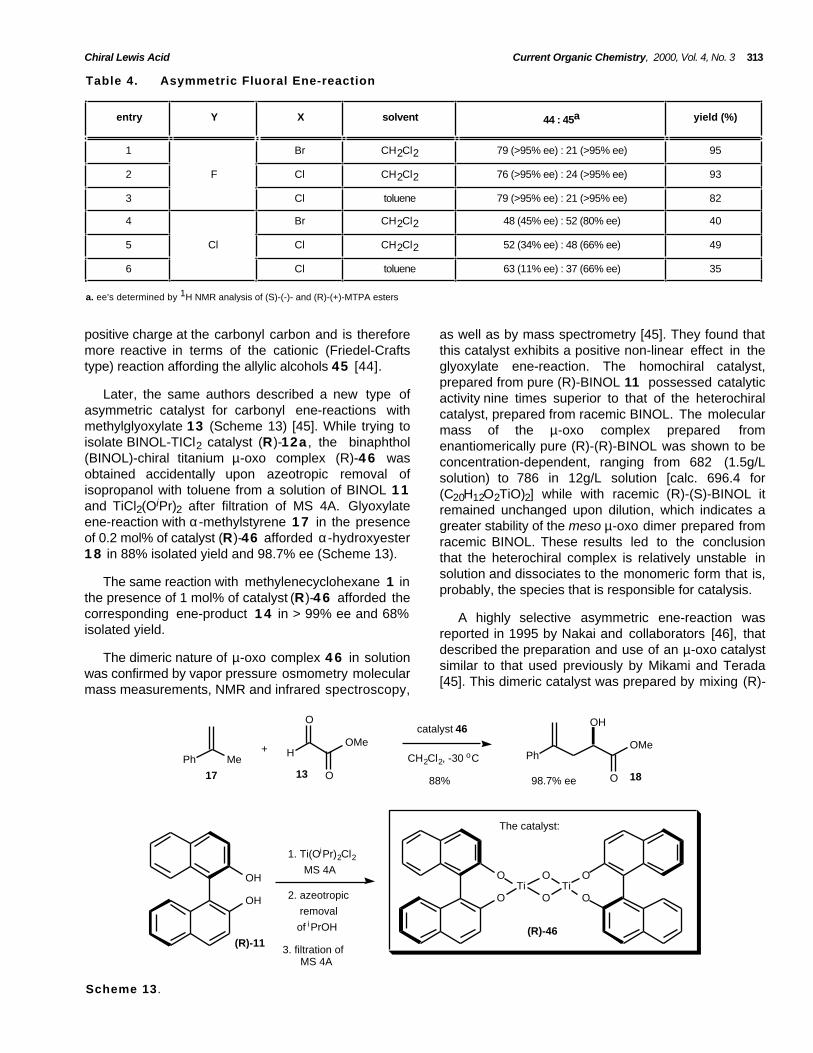
Table 4. Asymmetric Fluoral Ene-reaction
entry Y X solvent 44 : 45a yield (%)
1 Br CH2Cl2 79 (>95% ee) : 21 (>95% ee) 95
2 F Cl CH2Cl2 76 (>95% ee) : 24 (>95% ee) 93
3 Cl toluene 79 (>95% ee) : 21 (>95% ee) 82
4 Br CH2Cl2 48 (45% ee) : 52 (80% ee) 40
5 Cl Cl CH2Cl2 52 (34% ee) : 48 (66% ee) 49
6 Cl toluene 63 (11% ee) : 37 (66% ee) 35
a. ee's determined by 1H NMR analysis of (S)-(-)- and (R)-(+)-MTPA esters
positive charge at the carbonyl carbon and is thereforemore reactive in terms of the cationic (Friedel-Craftstype) reaction affording the allylic alcohols 45 [44].
as well as by mass spectrometry [45]. They found thatthis catalyst exhibits a positive non-linear effect in theglyoxylate ene-reaction. The homochiral catalyst,prepared from pure (R)-BINOL 11 possessed catalyticactivity nine times superior to that of the heterochiralcatalyst, prepared from racemic BINOL. The molecularmass of the µ-oxo complex prepared fromenantiomerically pure (R)-(R)-BINOL was shown to beconcentration-dependent, ranging from 682 (1.5g/Lsolution) to 786 in 12g/L solution [calc. 696.4 for(C20H12O2TiO)2] while with racemic (R)-(S)-BINOL itremained unchanged upon dilution, which indicates agreater stability of the meso µ-oxo dimer prepared fromracemic BINOL. These results led to the conclusionthat the heterochiral complex is relatively unstable insolution and dissociates to the monomeric form that is,probably, the species that is responsible for catalysis.
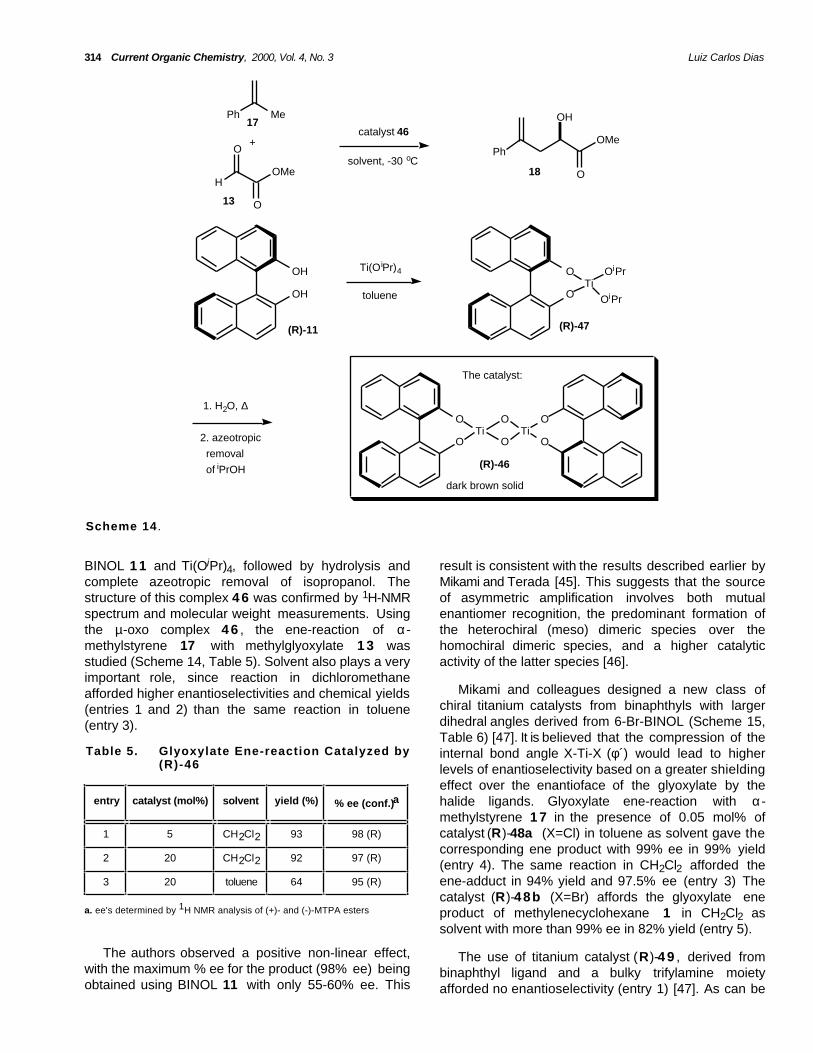
Later, the same authors described a new type ofasymmetric catalyst for carbonyl ene-reactions withmethylglyoxylate 13 (Scheme 13) [45]. While trying toisolate BINOL-TICl2 catalyst (R)-12a , the binaphthol(BINOL)-chiral titanium µ-oxo complex (R)-4 6 wasobtained accidentally upon azeotropic removal ofisopropanol with toluene from a solution of BINOL 1 1and TiCl2(OiPr)2 after filtration of MS 4A. Glyoxylateene-reaction with α-methylstyrene 1 7 in the presenceof 0.2 mol% of catalyst (R)-46 afforded α-hydroxyester1 8 in 88% isolated yield and 98.7% ee (Scheme 13).
The same reaction with methylenecyclohexane 1 inthe presence of 1 mol% of catalyst (R)-4 6 afforded thecorresponding ene-product 1 4 in > 99% ee and 68%isolated yield.
A highly selective asymmetric ene-reaction wasreported in 1995 by Nakai and collaborators [46], thatdescribed the preparation and use of an µ-oxo catalystsimilar to that used previously by Mikami and Terada[45]. This dimeric catalyst was prepared by mixing (R)-
The dimeric nature of µ-oxo complex 4 6 in solutionwas confirmed by vapor pressure osmometry molecularmass measurements, NMR and infrared spectroscopy,
Ph MeH
OMe
O
O
PhOMe
O
OH
OH
OH
O
OTi
O
OTi
O
O
+
catalyst 46
CH2Cl2, -30 oC
(R)-11
1. Ti(OiPr)2Cl2
MS 4A
2. azeotropic
removal
of i PrOH (R)-46
98.7% ee88%
The catalyst:
17 13 18
3. filtration of MS 4A
Scheme 13 .
314 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
Ph Me
HOMe
O
O
PhOMe
O
OH
OH
OH
O
OTi
OiPr
OiPr
O
OTi
O
OTi
O
O
+catalyst 46
solvent, -30 oC
The catalyst:
(R)-11
toluene
Ti(O iPr)4
(R)-47
1. H2O, ∆
2. azeotropic
removal
of iPrOH (R)-46
dark brown solid
17
13
18
Scheme 14 .
BINOL 1 1 and Ti(OiPr)4, followed by hydrolysis andcomplete azeotropic removal of isopropanol. Thestructure of this complex 4 6 was confirmed by 1H-NMRspectrum and molecular weight measurements. Usingthe µ-oxo complex 4 6 , the ene-reaction of α-methylstyrene 17 with methylglyoxylate 1 3 wasstudied (Scheme 14, Table 5). Solvent also plays a veryimportant role, since reaction in dichloromethaneafforded higher enantioselectivities and chemical yields(entries 1 and 2) than the same reaction in toluene(entry 3).
result is consistent with the results described earlier byMikami and Terada [45]. This suggests that the sourceof asymmetric amplification involves both mutualenantiomer recognition, the predominant formation ofthe heterochiral (meso) dimeric species over thehomochiral dimeric species, and a higher catalyticactivity of the latter species [46].
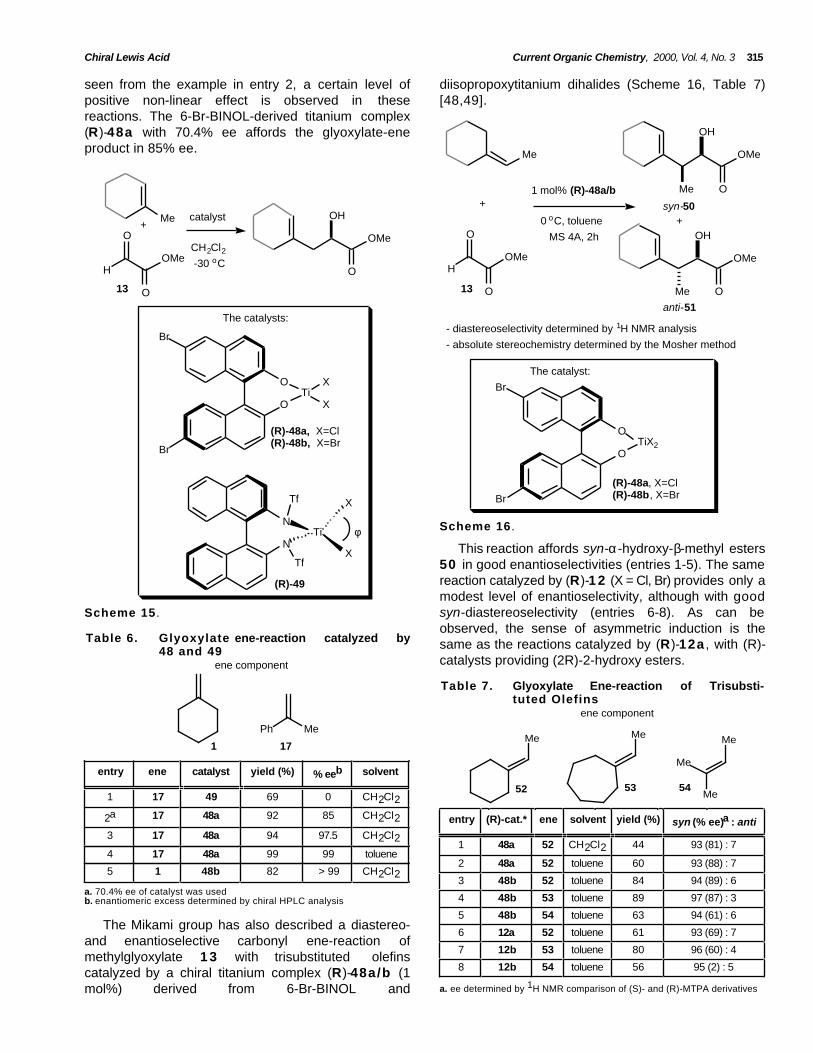
Mikami and colleagues designed a new class ofchiral titanium catalysts from binaphthyls with largerdihedral angles derived from 6-Br-BINOL (Scheme 15,Table 6) [47]. It is believed that the compression of theinternal bond angle X-Ti-X (φ´) would lead to higherlevels of enantioselectivity based on a greater shieldingeffect over the enantioface of the glyoxylate by thehalide ligands. Glyoxylate ene-reaction with α-methylstyrene 1 7 in the presence of 0.05 mol% ofcatalyst (R)-48a (X=Cl) in toluene as solvent gave thecorresponding ene product with 99% ee in 99% yield(entry 4). The same reaction in CH2Cl2 afforded theene-adduct in 94% yield and 97.5% ee (entry 3) Thecatalyst (R)-48b (X=Br) affords the glyoxylate eneproduct of methylenecyclohexane 1 in CH2Cl2 assolvent with more than 99% ee in 82% yield (entry 5).
Table 5. Glyoxylate Ene-reaction Catalyzed by(R)-46
entry catalyst (mol%) solvent yield (%) % ee (conf.)a
1 5 CH2Cl2 93 98 (R)
2 20 CH2Cl2 92 97 (R)
3 20 toluene 64 95 (R)
a. ee's determined by 1H NMR analysis of (+)- and (-)-MTPA esters
The authors observed a positive non-linear effect,with the maximum % ee for the product (98% ee) beingobtained using BINOL 11 with only 55-60% ee. This
The use of titanium catalyst (R)-4 9 , derived frombinaphthyl ligand and a bulky trifylamine moietyafforded no enantioselectivity (entry 1) [47]. As can be
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 315
seen from the example in entry 2, a certain level ofpositive non-linear effect is observed in thesereactions. The 6-Br-BINOL-derived titanium complex(R)-48a with 70.4% ee affords the glyoxylate-eneproduct in 85% ee.
diisopropoxytitanium dihalides (Scheme 16, Table 7)[48,49].
Me
HOMe
O
O Me
OH
O
OMe
Me
OH
O
OMe
Br
Br
O
OTiX2
+1 mol% (R)-48a/b
0 oC, toluene
MS 4A, 2h
+ syn-50
anti-51
13
(R)-48a, X=Cl(R)-48b, X=Br
The catalyst:
- diastereoselectivity determined by 1H NMR analysis
- absolute stereochemistry determined by the Mosher method
Me
HOMe
O
O
OMe
O
OH
O
O
N
NTi
X
X
TiX
X
Br
Br
Tf
Tf
+catalyst
CH2Cl2 -30 oC
(R)-48a, X=Cl(R)-48b, X=Br
(R)-49
The catalysts:
φ
13
Scheme 16 .
This reaction affords syn-α-hydroxy-β-methyl esters50 in good enantioselectivities (entries 1-5). The samereaction catalyzed by (R)-1 2 (X = Cl, Br) provides only amodest level of enantioselectivity, although with goodsyn-diastereoselectivity (entries 6-8). As can beobserved, the sense of asymmetric induction is thesame as the reactions catalyzed by (R)-12a , with (R)-catalysts providing (2R)-2-hydroxy esters.
Scheme 15 .
Table 6. Glyoxylate ene-reaction catalyzed by48 and 49
Ph Me
ene component
1 17
Table 7. Glyoxylate Ene-reaction of Trisubsti-tuted Olefins
Me Me Me
Me
Me
ene component
52 53 54
entry ene catalyst yield (%) % eeb solvent
1 17 49 69 0 CH2Cl2
2a 17 48a 92 85 CH2Cl2 entry (R)-cat.* ene solvent yield (%) syn (% ee)a : anti3 17 48a 94 97.5 CH2Cl2
1 48a 52 CH2Cl2 44 93 (81) : 74 17 48a 99 99 toluene
2 48a 52 toluene 60 93 (88) : 75 1 48b 82 > 99 CH2Cl2
3 48b 52 toluene 84 94 (89) : 6a. 70.4% ee of catalyst was used
4 48b 53 toluene 89 97 (87) : 3b. enantiomeric excess determined by chiral HPLC analysis
5 48b 54 toluene 63 94 (61) : 6The Mikami group has also described a diastereo-
and enantioselective carbonyl ene-reaction ofmethylglyoxylate 1 3 with trisubstituted olefinscatalyzed by a chiral titanium complex (R)-48a/b (1mol%) derived from 6-Br-BINOL and
6 12a 52 toluene 61 93 (69) : 7
7 12b 53 toluene 80 96 (60) : 4
8 12b 54 toluene 56 95 (2) : 5
a. ee determined by 1H NMR comparison of (S)- and (R)-MTPA derivatives
316 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
O
OTi
Cl
Cl
Me
HOMe
O
O
OMe
O
OH
R3Si
CH2Cl2
R3Si
MeOMe
O
OH
OMe
O
OTMS
R3Si
OMe
Me
O
OH
R3Si
OMe
Me
O
OTMS
R3Si
56
59
60
(R)-12a
(10 mol%)
+
-30 oC, 2h
(R)-12a
The catalyst:
+
+57
55a, R =Ph55b, R=Me
13
58
+
Scheme 17 .
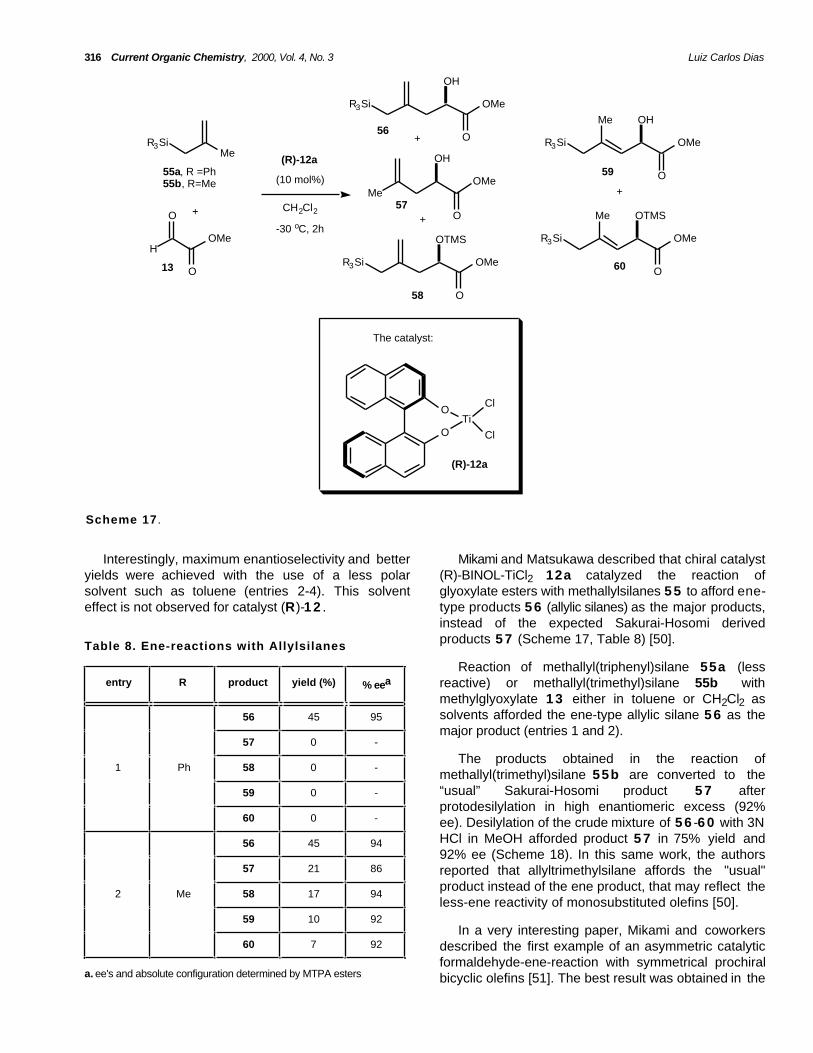
Interestingly, maximum enantioselectivity and betteryields were achieved with the use of a less polarsolvent such as toluene (entries 2-4). This solventeffect is not observed for catalyst (R)-1 2 .
Mikami and Matsukawa described that chiral catalyst(R)-BINOL-TiCl2 12a catalyzed the reaction ofglyoxylate esters with methallylsilanes 5 5 to afford ene-type products 5 6 (allylic silanes) as the major products,instead of the expected Sakurai-Hosomi derivedproducts 5 7 (Scheme 17, Table 8) [50].Table 8. Ene-reactions with Allylsilanes
entry R product yield (%) % eea
56 45 95
57 0 -
1 Ph 58 0 -
59 0 -
60 0 -
56 45 94
57 21 86
2 Me 58 17 94
59 10 92
60 7 92
a. ee's and absolute configuration determined by MTPA esters
Reaction of methallyl(triphenyl)silane 55a (lessreactive) or methallyl(trimethyl)silane 55b withmethylglyoxylate 1 3 either in toluene or CH2Cl2 assolvents afforded the ene-type allylic silane 5 6 as themajor product (entries 1 and 2).
The products obtained in the reaction ofmethallyl(trimethyl)silane 55b are converted to the“usual” Sakurai-Hosomi product 5 7 afterprotodesilylation in high enantiomeric excess (92%ee). Desilylation of the crude mixture of 5 6 -6 0 with 3NHCl in MeOH afforded product 5 7 in 75% yield and92% ee (Scheme 18). In this same work, the authorsreported that allyltrimethylsilane affords the "usual"product instead of the ene product, that may reflect theless-ene reactivity of monosubstituted olefins [50].
In a very interesting paper, Mikami and coworkersdescribed the first example of an asymmetric catalyticformaldehyde-ene-reaction with symmetrical prochiralbicyclic olefins [51]. The best result was obtained in the
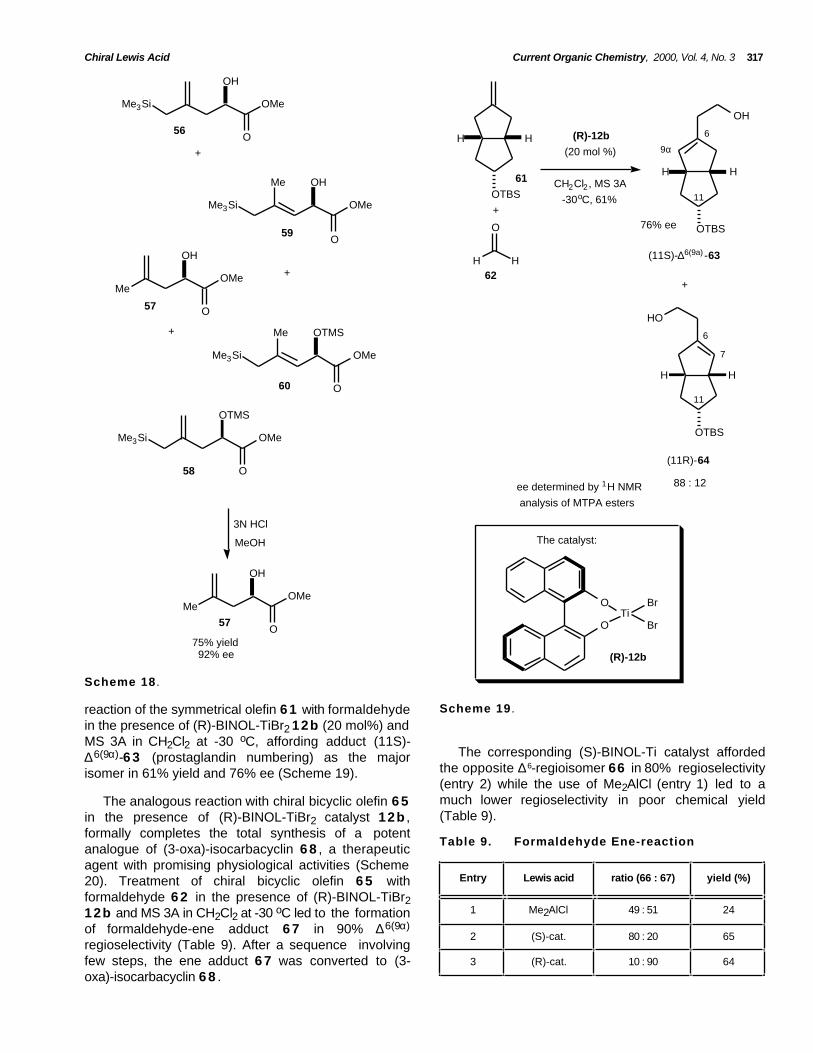
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 317
MeOH
MeOMe
O
OH
OMe
O
OH
Me3Si
MeOMe
O
OH
OMe
O
OTMS
Me3Si
OMe
Me
O
OH
Me3Si
OMe
Me
O
OTMS
Me3Si
3N HCl
75% yield 92% ee
+
+
56
57
58
59
60
57
+
Scheme 18 .
HH
OTBS
H H
O
HH
OTBS
OH
HH
OTBS
HO
O
OTi
Br
Br
+
(R)-12b
(20 mol %)
CH2Cl2, MS 3A
-30oC, 61%
76% ee
9α
6
11
(11S)-∆6(9a) -63
6
11
(11R)-64
7
+
88 : 12ee determined by 1H NMR
analysis of MTPA esters
61
62
(R)-12b
The catalyst:
Scheme 19 .reaction of the symmetrical olefin 6 1 with formaldehydein the presence of (R)-BINOL-TiBr2 12b (20 mol%) andMS 3A in CH2Cl2 at -30 oC, affording adduct (11S)-∆6(9α)-6 3 (prostaglandin numbering) as the majorisomer in 61% yield and 76% ee (Scheme 19).
The corresponding (S)-BINOL-Ti catalyst affordedthe opposite ∆6-regioisomer 66 in 80% regioselectivity(entry 2) while the use of Me2AlCl (entry 1) led to amuch lower regioselectivity in poor chemical yield(Table 9).
The analogous reaction with chiral bicyclic olefin 6 5in the presence of (R)-BINOL-TiBr2 catalyst 12b ,formally completes the total synthesis of a potentanalogue of (3-oxa)-isocarbacyclin 6 8 , a therapeuticagent with promising physiological activities (Scheme20). Treatment of chiral bicyclic olefin 6 5 withformaldehyde 6 2 in the presence of (R)-BINOL-TiBr212b and MS 3A in CH2Cl2 at -30 oC led to the formationof formaldehyde-ene adduct 6 7 in 90% ∆6(9α)
regioselectivity (Table 9). After a sequence involvingfew steps, the ene adduct 6 7 was converted to (3-oxa)-isocarbacyclin 6 8 .
Table 9. Formaldehyde Ene-reaction
Entry Lewis acid ratio (66 : 67) yield (%)
1 Me2AlCl 49 : 51 24
2 (S)-cat. 80 : 20 65
3 (R)-cat. 10 : 90 64
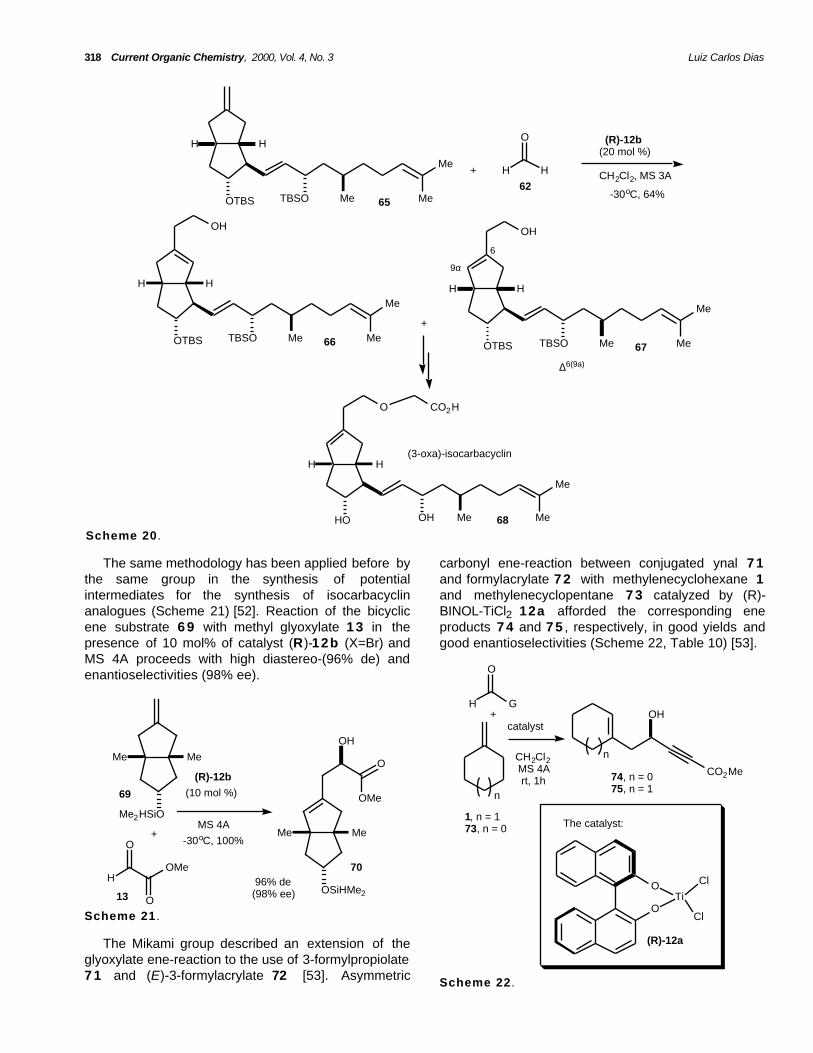
318 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
HH
OTBS
H H
O
MeTBSO Me
Me
HH
OTBS MeTBSO Me
Me
OH
HH
OTBS MeTBSO Me
Me
OH
HH
HO MeOH Me
Me
O CO2H
65
+
(R)-12b(20 mol %)
CH2Cl2, MS 3A
+
6
9α
∆6(9a)
(3-oxa)-isocarbacyclin
66 67
68
62-30oC, 64%
Scheme 20 .
The same methodology has been applied before bythe same group in the synthesis of potentialintermediates for the synthesis of isocarbacyclinanalogues (Scheme 21) [52]. Reaction of the bicyclicene substrate 6 9 with methyl glyoxylate 1 3 in thepresence of 10 mol% of catalyst (R)-12b (X=Br) andMS 4A proceeds with high diastereo-(96% de) andenantioselectivities (98% ee).
carbonyl ene-reaction between conjugated ynal 7 1and formylacrylate 7 2 with methylenecyclohexane 1and methylenecyclopentane 7 3 catalyzed by (R)-BINOL-TiCl2 12a afforded the corresponding eneproducts 7 4 and 7 5 , respectively, in good yields andgood enantioselectivities (Scheme 22, Table 10) [53].
O
O
H G
O
OH
CO2Me
Ti
Cl
Cl
1, n = 173, n = 0
+catalyst
74, n = 075, n = 1
n
nCH2Cl2 MS 4A rt, 1h
(R)-12a
The catalyst:
MeMe
Me2HSiO
HOMe
O
OMeMe
OSiHMe2
O
OMe
OH
+
(R)-12b
(10 mol %)
MS 4A
-30oC, 100%
96% de(98% ee)
69
13
70
Scheme 21 .
The Mikami group described an extension of theglyoxylate ene-reaction to the use of 3-formylpropiolate7 1 and (E)-3-formylacrylate 72 [53]. Asymmetric Scheme 22 .
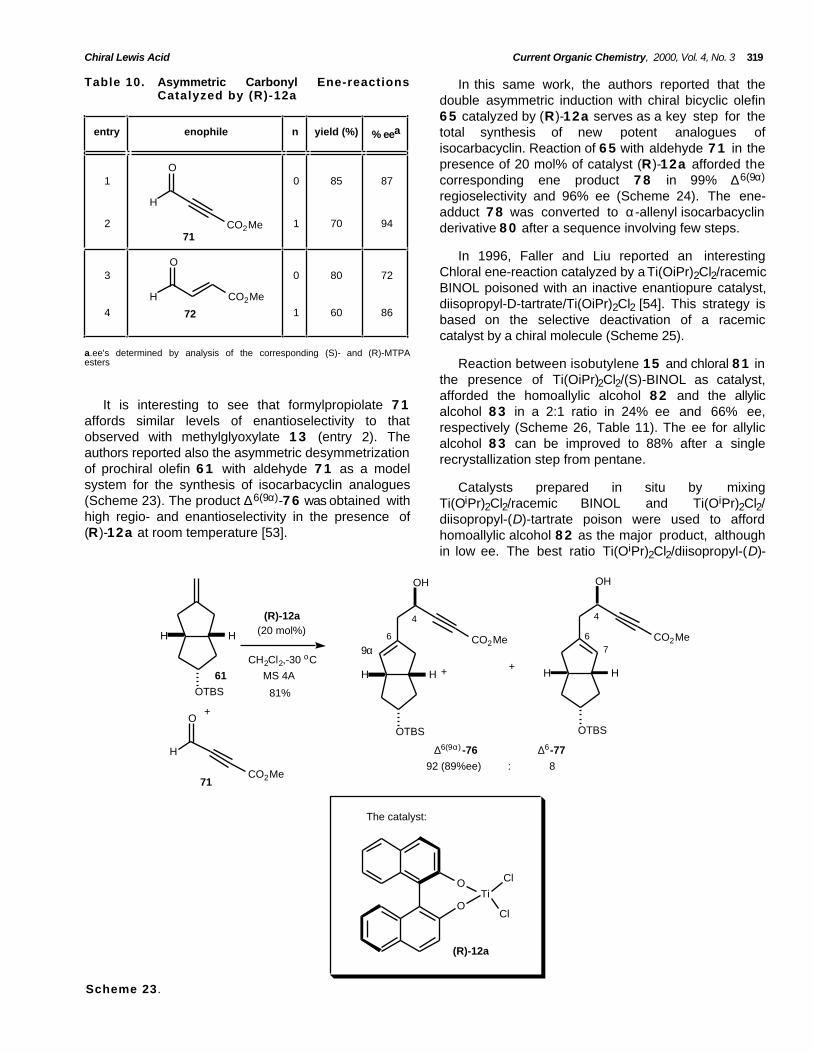
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 319
Table 10. Asymmetric Carbonyl Ene-reactionsCatalyzed by (R)-12a
In this same work, the authors reported that thedouble asymmetric induction with chiral bicyclic olefin6 5 catalyzed by (R)-12a serves as a key step for thetotal synthesis of new potent analogues ofisocarbacyclin. Reaction of 6 5 with aldehyde 7 1 in thepresence of 20 mol% of catalyst (R)-12a afforded thecorresponding ene product 7 8 in 99% ∆6(9α)
regioselectivity and 96% ee (Scheme 24). The ene-adduct 7 8 was converted to α-allenyl isocarbacyclinderivative 8 0 after a sequence involving few steps.
entry enophile n yield (%) % eea
1
2
H
O
CO2Me71
0
1
85
70
87
94
In 1996, Faller and Liu reported an interestingChloral ene-reaction catalyzed by a Ti(OiPr)2Cl2/racemicBINOL poisoned with an inactive enantiopure catalyst,diisopropyl-D-tartrate/Ti(OiPr)2Cl2 [54]. This strategy isbased on the selective deactivation of a racemiccatalyst by a chiral molecule (Scheme 25).
3
4
H
O
CO2Me
72
0
1
80
60
72
86
a.ee's determined by analysis of the corresponding (S)- and (R)-MTPAesters Reaction between isobutylene 15 and chloral 8 1 in
the presence of Ti(OiPr)2Cl2/(S)-BINOL as catalyst,afforded the homoallylic alcohol 8 2 and the allylicalcohol 8 3 in a 2:1 ratio in 24% ee and 66% ee,respectively (Scheme 26, Table 11). The ee for allylicalcohol 8 3 can be improved to 88% after a singlerecrystallization step from pentane.
It is interesting to see that formylpropiolate 7 1affords similar levels of enantioselectivity to thatobserved with methylglyoxylate 1 3 (entry 2). Theauthors reported also the asymmetric desymmetrizationof prochiral olefin 6 1 with aldehyde 7 1 as a modelsystem for the synthesis of isocarbacyclin analogues(Scheme 23). The product ∆6(9α)-7 6 was obtained withhigh regio- and enantioselectivity in the presence of(R)-12a at room temperature [53].
Catalysts prepared in situ by mixingTi(OiPr)2Cl2/racemic BINOL and Ti(OiPr)2Cl2/diisopropyl-(D)-tartrate poison were used to affordhomoallylic alcohol 8 2 as the major product, althoughin low ee. The best ratio Ti(OiPr)2Cl2/diisopropyl-(D)-
O
O
OH
CO2Me
Ti
Cl
Cl
H
O
CO2Me
HH
OTBS
HH
OTBS
OH
CO2Me
HH
OTBS
71
(R)-12a
CH2Cl2,-30 oC
MS 4A
(R)-12a
The catalyst:
+
+
6
9α
4 4
+
67
81%
∆6(9α) -76 ∆6-77
92 (89%ee) : 8
61
(20 mol%)
Scheme 23 .
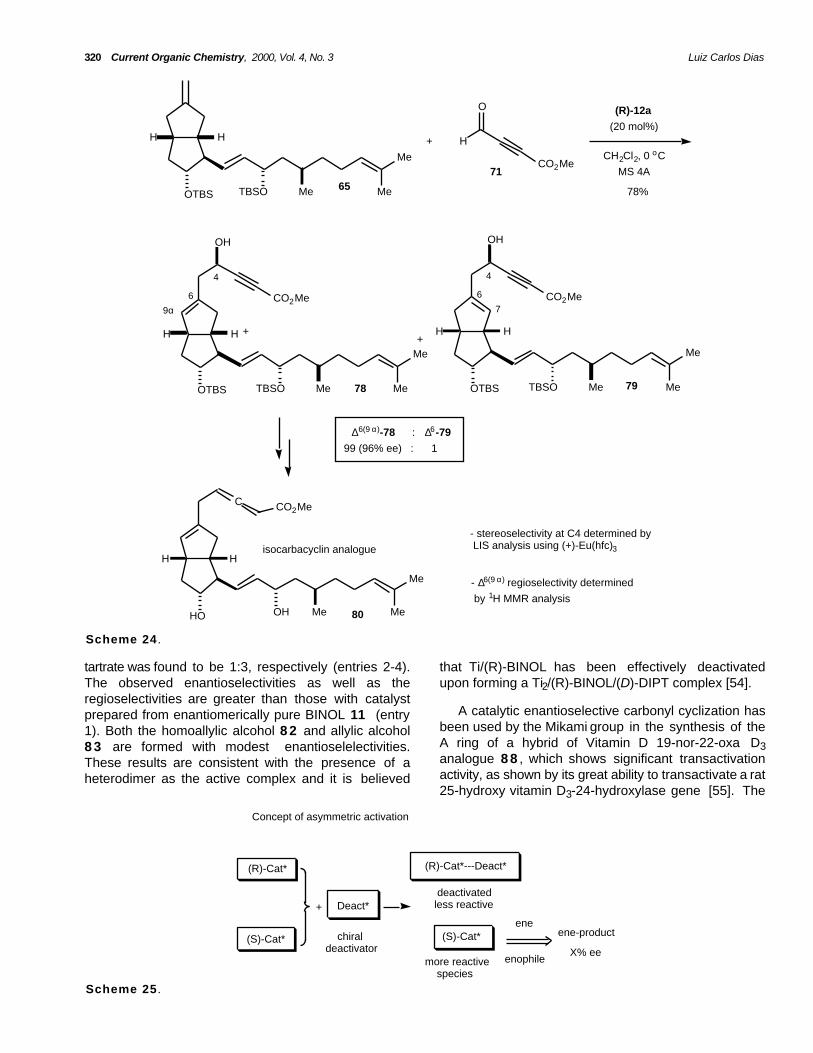
320 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
OH
CO2Me
H
O
CO2Me
HH
OTBS
OH
CO2Me
HH
OTBS
HH
OTBS MeTBSO Me
Me
HH
HO MeOH Me
Me
65
MeTBSO Me
Me
MeTBSO Me
Me
CO2MeC
71
(R)-12a
CH2Cl2, 0 oC
MS 4A
(20 mol%)
+
6
9α
4 4
+
6
7
78%
∆6(9 α)-78 : ∆6-79
99 (96% ee) : 1
isocarbacyclin analogue
80
7978
+
- stereoselectivity at C4 determined by LIS analysis using (+)-Eu(hfc)3
- ∆6(9 α) regioselectivity determined
by 1H MMR analysis
Scheme 24 .
tartrate was found to be 1:3, respectively (entries 2-4).The observed enantioselectivities as well as theregioselectivities are greater than those with catalystprepared from enantiomerically pure BINOL 11 (entry1). Both the homoallylic alcohol 8 2 and allylic alcohol8 3 are formed with modest enantioselelectivities.These results are consistent with the presence of aheterodimer as the active complex and it is believed
that Ti/(R)-BINOL has been effectively deactivatedupon forming a Ti2/(R)-BINOL/(D)-DIPT complex [54].
A catalytic enantioselective carbonyl cyclization hasbeen used by the Mikami group in the synthesis of theA ring of a hybrid of Vitamin D 19-nor-22-oxa D3analogue 8 8 , which shows significant transactivationactivity, as shown by its great ability to transactivate a rat25-hydroxy vitamin D3-24-hydroxylase gene [55]. The
(R)-Cat*
(S)-Cat*
+ Deact*
(R)-Cat*---Deact*
deactivatedless reactive
ene
enophile
ene-product
X% ee
(S)-Cat* chiral deactivator
more reactive species
Concept of asymmetric activation
Scheme 25 .
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 321
Me Me
H CCl3
O
Me CCl3
OH
Me CCl3
Me OH
+
catalyst (R)-12a
CH2Cl2 -20 oC
+
82
83
MS 4A
15
81
Scheme 26 .
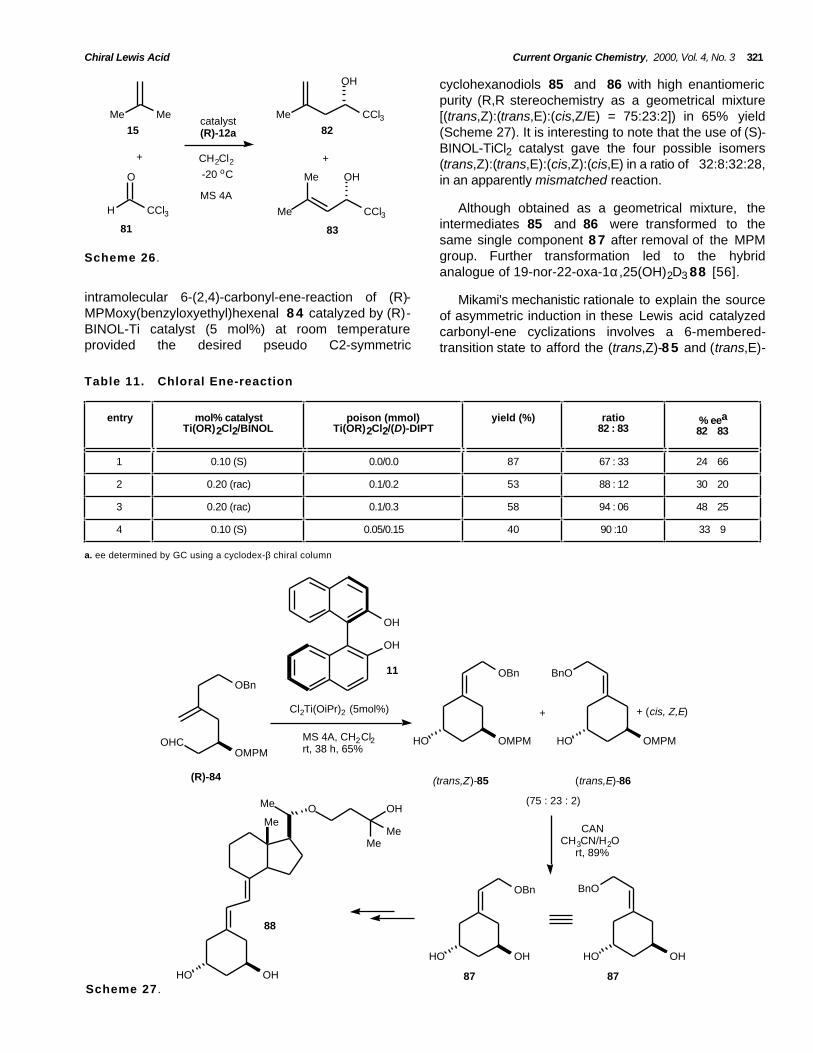
cyclohexanodiols 85 and 86 with high enantiomericpurity (R,R stereochemistry as a geometrical mixture[(trans,Z):(trans,E):(cis,Z/E) = 75:23:2]) in 65% yield(Scheme 27). It is interesting to note that the use of (S)-BINOL-TiCl2 catalyst gave the four possible isomers(trans,Z):(trans,E):(cis,Z):(cis,E) in a ratio of 32:8:32:28,in an apparently mismatched reaction.
Although obtained as a geometrical mixture, theintermediates 85 and 86 were transformed to thesame single component 8 7 after removal of the MPMgroup. Further transformation led to the hybridanalogue of 19-nor-22-oxa-1α,25(OH)2D3 88 [56].
intramolecular 6-(2,4)-carbonyl-ene-reaction of (R)-MPMoxy(benzyloxyethyl)hexenal 8 4 catalyzed by (R)-BINOL-Ti catalyst (5 mol%) at room temperatureprovided the desired pseudo C2-symmetric
Mikami's mechanistic rationale to explain the sourceof asymmetric induction in these Lewis acid catalyzedcarbonyl-ene cyclizations involves a 6-membered-transition state to afford the (trans,Z)-8 5 and (trans,E)-
Table 11. Chloral Ene-reaction
entry mol% catalystTi(OR)2Cl2/BINOL
poison (mmol)Ti(OR)2Cl2/(D)-DIPT
yield (%) ratio82 : 83
% eea82 83
1 0.10 (S) 0.0/0.0 87 67 : 33 24 66
2 0.20 (rac) 0.1/0.2 53 88 : 12 30 20
3 0.20 (rac) 0.1/0.3 58 94 : 06 48 25
4 0.10 (S) 0.05/0.15 40 90 :10 33 9
a. ee determined by GC using a cyclodex-β chiral column
OHC
OBn
OMPM
OH
OH
OBn
HO OMPM HO OMPM
BnO
OBn
HO OH HO OH
BnO
Me
OHHO
Me O OH
MeMe
87 87
(R)-84
Cl2Ti(OiPr)2 (5mol%)
MS 4A, CH2Cl2rt, 38 h, 65%
+
(trans,Z)-85 (trans,E)-86
(75 : 23 : 2)
CANCH3CN/H2O rt, 89%
+ (cis, Z,E)
88
11
Scheme 27 .
322 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
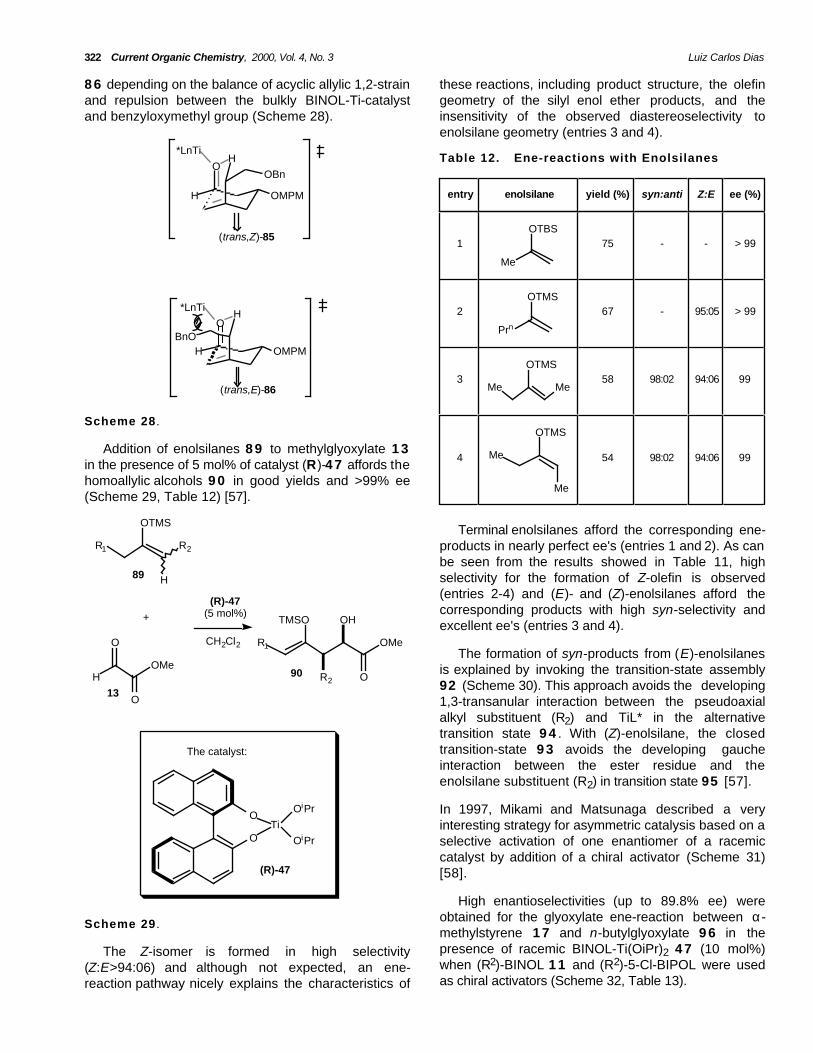
8 6 depending on the balance of acyclic allylic 1,2-strainand repulsion between the bulkly BINOL-Ti-catalystand benzyloxymethyl group (Scheme 28).
these reactions, including product structure, the olefingeometry of the silyl enol ether products, and theinsensitivity of the observed diastereoselectivity toenolsilane geometry (entries 3 and 4).
OMPM
O
H
H
OBn
*LnTi
OMPM
O
H
H*LnTi
BnO
(trans,Z)-85
(trans,E)-86
Table 12. Ene-reactions with Enolsilanes
entry enolsilane yield (%) syn:anti Z:E ee (%)
1
Me
OTBS75 - - > 99
2
Prn
OTMS67 - 95:05 > 99
3Me Me
OTMS58 98:02 94:06 99
Scheme 28 .
4 Me
OTMS
Me
54 98:02 94:06 99Addition of enolsilanes 8 9 to methylglyoxylate 1 3
in the presence of 5 mol% of catalyst (R)-4 7 affords thehomoallylic alcohols 9 0 in good yields and >99% ee(Scheme 29, Table 12) [57].
R1 R2
OTMS
H
H
O
O
OMe
R1
OH
R2
TMSO
O
OMe
O
OTi
OiPr
OiPr
+
(R)-47
The catalyst:
89
90
13
(R)-47(5 mol%)
CH2Cl2
Terminal enolsilanes afford the corresponding ene-products in nearly perfect ee's (entries 1 and 2). As canbe seen from the results showed in Table 11, highselectivity for the formation of Z-olefin is observed(entries 2-4) and (E)- and (Z)-enolsilanes afford thecorresponding products with high syn-selectivity andexcellent ee's (entries 3 and 4).
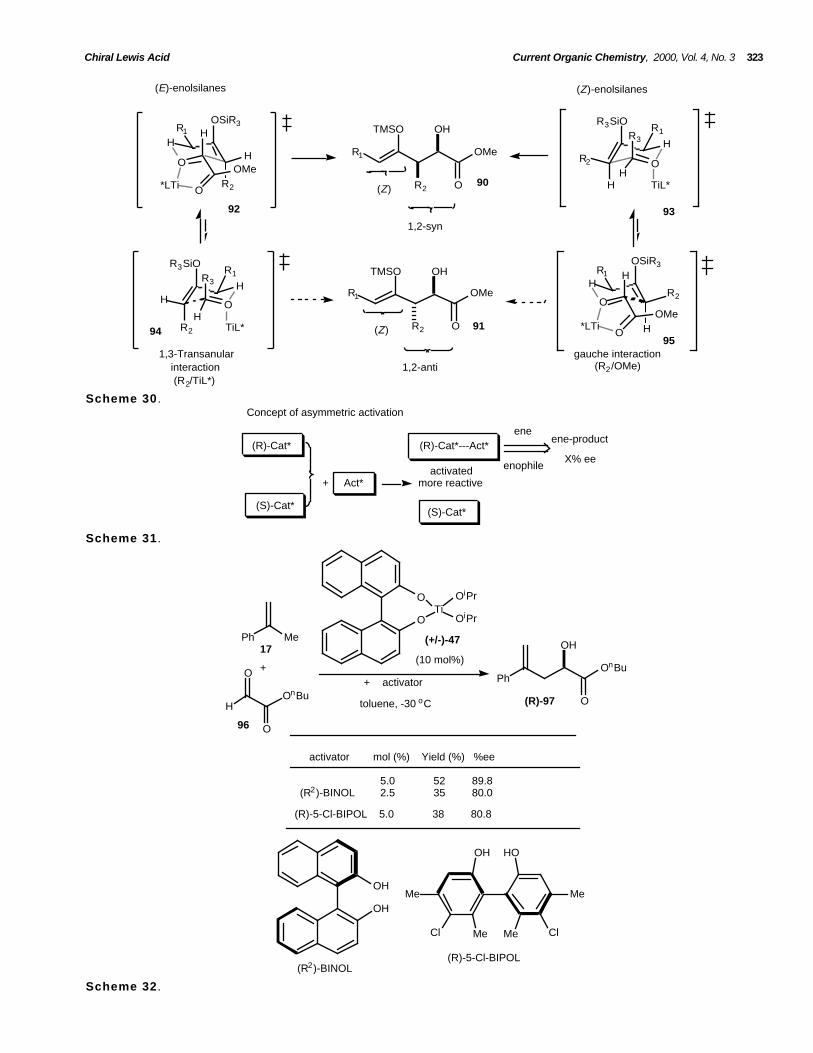
The formation of syn-products from (E)-enolsilanesis explained by invoking the transition-state assembly92 (Scheme 30). This approach avoids the developing1,3-transanular interaction between the pseudoaxialalkyl substituent (R2) and TiL* in the alternativetransition state 9 4 . With (Z)-enolsilane, the closedtransition-state 9 3 avoids the developing gaucheinteraction between the ester residue and theenolsilane substituent (R2) in transition state 95 [57].
In 1997, Mikami and Matsunaga described a veryinteresting strategy for asymmetric catalysis based on aselective activation of one enantiomer of a racemiccatalyst by addition of a chiral activator (Scheme 31)[58].
High enantioselectivities (up to 89.8% ee) wereobtained for the glyoxylate ene-reaction between α-methylstyrene 1 7 and n-butylglyoxylate 9 6 in thepresence of racemic BINOL-Ti(OiPr)2 4 7 (10 mol%)when (R2)-BINOL 1 1 and (R2)-5-Cl-BIPOL were usedas chiral activators (Scheme 32, Table 13).
Scheme 29 .
The Z-isomer is formed in high selectivity(Z:E>94:06) and although not expected, an ene-reaction pathway nicely explains the characteristics of
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 323
H
O
O*LTi
H
H
R2
OMe
OSiR3R1
H
OH
R3
H
R2
R3SiOR1
TiL*
R1
OH
R2
TMSO
O
OMe
R1
OH
R2
TMSO
O
OMe
H
OH
R3
R2
H
R3SiOR1
TiL*
H
O
O*LTi
H
R2
HOMe
OSiR3R1
(E)-enolsilanes (Z)-enolsilanes
1,3-Transanular interaction (R2/TiL*)
(Z)
1,2-syn
(Z)
1,2-antigauche interaction (R2/OMe)
92 93
90
9495
91
Scheme 30 .
(R)-Cat*
(S)-Cat*
+ Act* activatedmore reactive
ene
enophile
ene-product
X% ee
(S)-Cat*
Concept of asymmetric activation
(R)-Cat*---Act*
Scheme 31 .
Ph Me
HOnBu
O
O
PhOnBu
O
OH
O
OTi
OiPr
OiPr
OH
OH
OH HO
MeMe
Me Me ClCl
activator
toluene, -30 oC
(+/-)-47
(R)-97
(10 mol%)
(R2)-BINOL
activator mol (%) Yield (%) %ee
5.0 52 89.8 2.5 35 80.0
17
96
(R)-5-Cl-BIPOL 5.0 38 80.8
(R)-5-Cl-BIPOL(R2)-BINOL
++
Scheme 32 .
324 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
Ph Me
HOnBu
O
O
PhOnBu
O
OH
O
OTi
OiPr
OiPr
O
OTi
O
O
OiPr
OiPr
H
H
activator(10 mol%)
toluene, -30 oC
(R1)-47
(R)-97
(10 mol%)
17
96
The catalyst:
98
+
+
Scheme 33 .
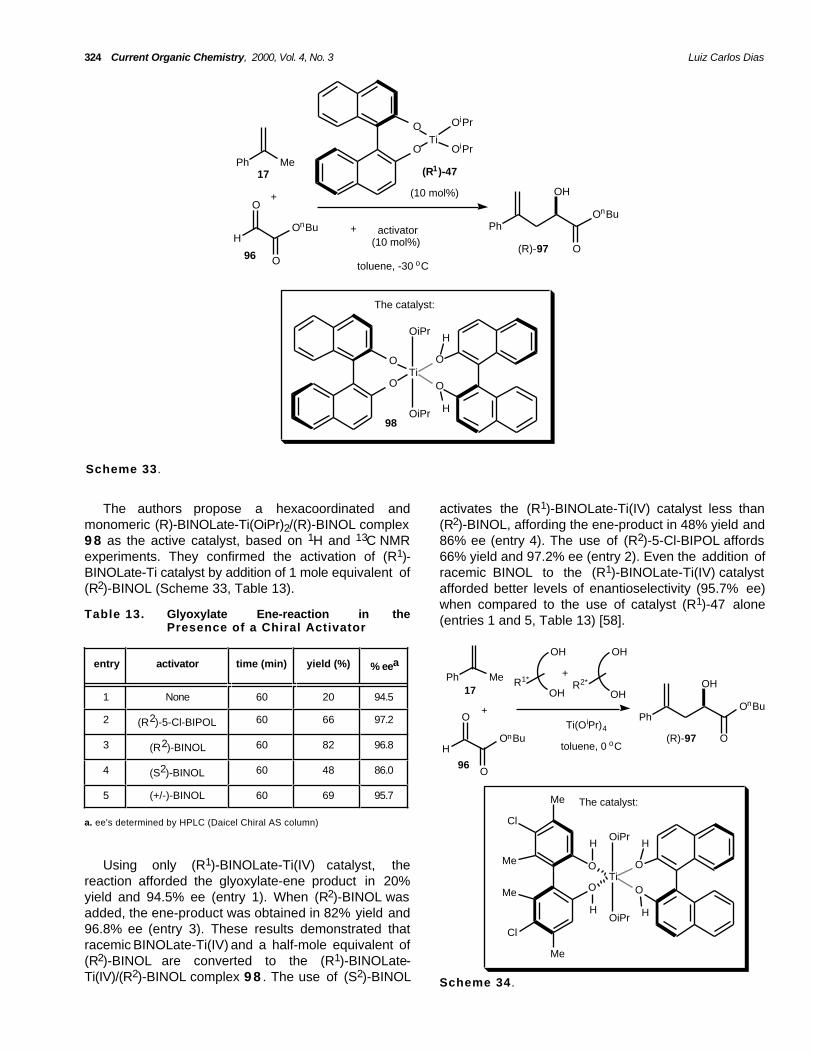
The authors propose a hexacoordinated andmonomeric (R)-BINOLate-Ti(OiPr)2/(R)-BINOL complex9 8 as the active catalyst, based on 1H and 13C NMRexperiments. They confirmed the activation of (R1)-BINOLate-Ti catalyst by addition of 1 mole equivalent of(R2)-BINOL (Scheme 33, Table 13).
activates the (R1)-BINOLate-Ti(IV) catalyst less than(R2)-BINOL, affording the ene-product in 48% yield and86% ee (entry 4). The use of (R2)-5-Cl-BIPOL affords66% yield and 97.2% ee (entry 2). Even the addition ofracemic BINOL to the (R1)-BINOLate-Ti(IV) catalystafforded better levels of enantioselectivity (95.7% ee)when compared to the use of catalyst (R1)-47 alone(entries 1 and 5, Table 13) [58].Table 13. Glyoxylate Ene-reaction in the
Presence of a Chiral Activator
Ph Me
HOnBu
O
O
PhOnBu
O
OH
OH
OH
O
OTi
O
O
OiPr
OiPr
H
H
Me
Cl
Me
Me
Cl
Me
H
H
OH
OHR2*
(R)-97
+
Ti(O iPr)4
17
96
The catalyst:
+
toluene, 0 oC
R1*
entry activator time (min) yield (%) % eea
1 None 60 20 94.5
2 (R2)-5-Cl-BIPOL 60 66 97.2
3 (R2)-BINOL 60 82 96.8
4 (S2)-BINOL 60 48 86.0
5 (+/-)-BINOL 60 69 95.7
a. ee's determined by HPLC (Daicel Chiral AS column)
Using only (R1)-BINOLate-Ti(IV) catalyst, thereaction afforded the glyoxylate-ene product in 20%yield and 94.5% ee (entry 1). When (R2)-BINOL wasadded, the ene-product was obtained in 82% yield and96.8% ee (entry 3). These results demonstrated thatracemic BINOLate-Ti(IV) and a half-mole equivalent of(R2)-BINOL are converted to the (R1)-BINOLate-Ti(IV)/(R2)-BINOL complex 9 8 . The use of (S2)-BINOL Scheme 34 .
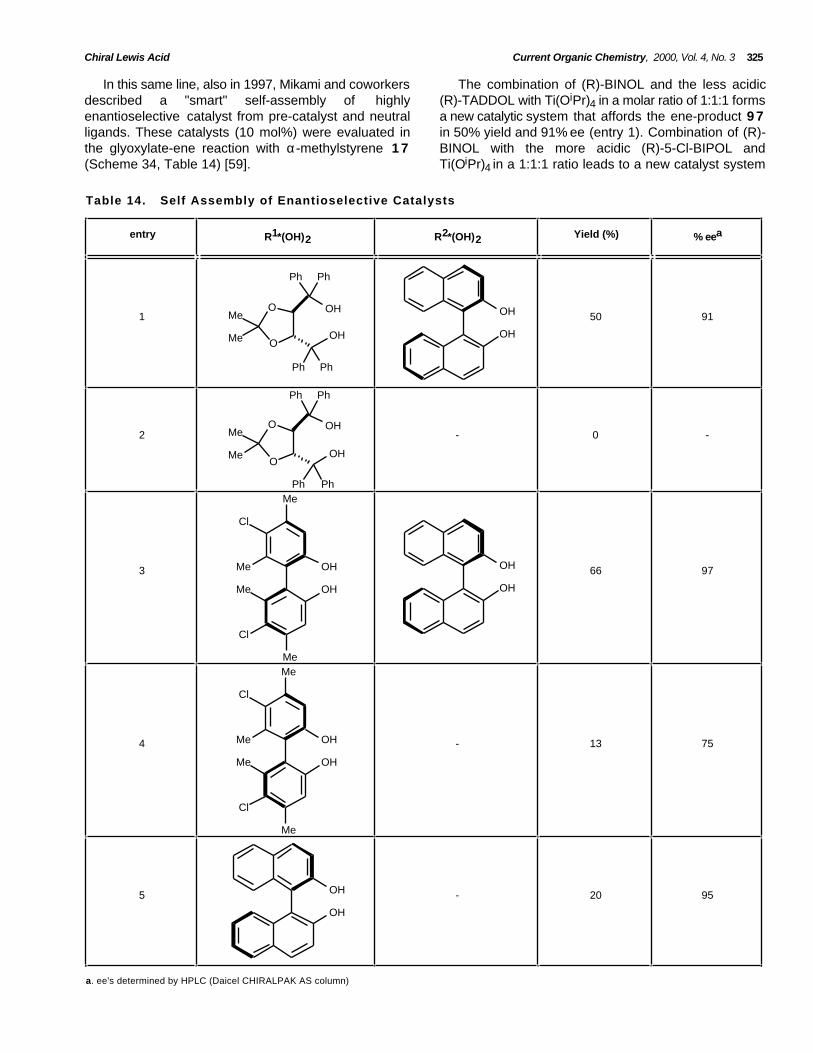
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 325
In this same line, also in 1997, Mikami and coworkersdescribed a "smart" self-assembly of highlyenantioselective catalyst from pre-catalyst and neutralligands. These catalysts (10 mol%) were evaluated inthe glyoxylate-ene reaction with α-methylstyrene 1 7(Scheme 34, Table 14) [59].
The combination of (R)-BINOL and the less acidic(R)-TADDOL with Ti(OiPr)4 in a molar ratio of 1:1:1 formsa new catalytic system that affords the ene-product 9 7in 50% yield and 91% ee (entry 1). Combination of (R)-BINOL with the more acidic (R)-5-Cl-BIPOL andTi(OiPr)4 in a 1:1:1 ratio leads to a new catalyst system
Table 14. Self Assembly of Enantioselective Catalysts
entry R1*(OH)2 R2*(OH)2 Yield (%) % eea
1O
O
OH
Ph
OH
Ph Ph
Ph
Me
Me
OH
OH
50 91
2O
O
OH
Ph
OH
Ph Ph
Ph
Me
Me
- 0 -
3
Me
Cl
Me OH
Me
OHMe
Cl
OH
OH
66 97
4
Me
Cl
Me OH
Me
OHMe
Cl
- 13 75
5 OH
OH
- 20 95
a. ee's determined by HPLC (Daicel CHIRALPAK AS column)
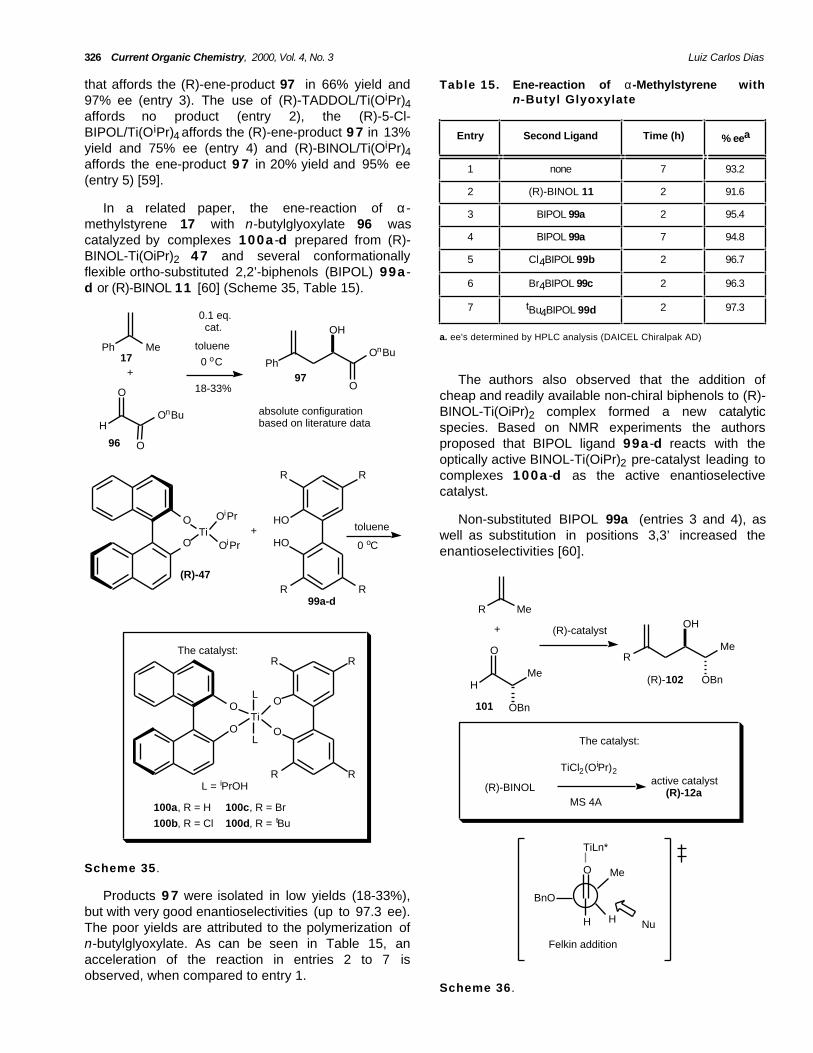
326 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
that affords the (R)-ene-product 97 in 66% yield and97% ee (entry 3). The use of (R)-TADDOL/Ti(OiPr)4affords no product (entry 2), the (R)-5-Cl-BIPOL/Ti(OiPr)4 affords the (R)-ene-product 9 7 in 13%yield and 75% ee (entry 4) and (R)-BINOL/Ti(OiPr)4affords the ene-product 9 7 in 20% yield and 95% ee(entry 5) [59].
Table 15. Ene-reaction of α-Methylstyrene withn-Butyl Glyoxylate
Entry Second Ligand Time (h) % eea
1 none 7 93.2
2 (R)-BINOL 11 2 91.6
In a related paper, the ene-reaction of α-methylstyrene 17 with n-butylglyoxylate 96 wascatalyzed by complexes 100a -d prepared from (R)-BINOL-Ti(OiPr)2 4 7 and several conformationallyflexible ortho-substituted 2,2’-biphenols (BIPOL) 99a -d or (R)-BINOL 11 [60] (Scheme 35, Table 15).
3 BIPOL 99a 2 95.4
4 BIPOL 99a 7 94.8
5 Cl4BIPOL 99b 2 96.7
6 Br4BIPOL 99c 2 96.3
7 tBu4BIPOL 99d 2 97.3
Ph Me
HOnBu
O
O
PhOnBu
O
OH
O
OTi
OiPr
OiPr
RR
HO
HO
R R
O
OTi
O
RR
R R
OL
L
+
0.1 eq. cat.
18-33%
The catalyst:
(R)-47
+
99a-d
100a, R = H 100c, R = Br
100b, R = Cl 100d, R = tBu
L = iPrOH
absolute configurationbased on literature data
17
96
97
toluene
0 oC
toluene
0 oC
a. ee's determined by HPLC analysis (DAICEL Chiralpak AD)
The authors also observed that the addition ofcheap and readily available non-chiral biphenols to (R)-BINOL-Ti(OiPr)2 complex formed a new catalyticspecies. Based on NMR experiments the authorsproposed that BIPOL ligand 99a -d reacts with theoptically active BINOL-Ti(OiPr)2 pre-catalyst leading tocomplexes 100a -d as the active enantioselectivecatalyst.
Non-substituted BIPOL 99a (entries 3 and 4), aswell as substitution in positions 3,3’ increased theenantioselectivities [60].
R Me
HMe
OR
Me
OBn
OH
OBn
O
H
BnO
Me
H
TiLn*
(R)-102
TiCl2(OiPr)2
MS 4A
+ (R)-catalyst
(R)-BINOLactive catalyst (R)-12a
The catalyst:
Nu
101
Felkin addition
Scheme 35 .
Products 9 7 were isolated in low yields (18-33%),but with very good enantioselectivities (up to 97.3 ee).The poor yields are attributed to the polymerization ofn-butylglyoxylate. As can be seen in Table 15, anacceleration of the reaction in entries 2 to 7 isobserved, when compared to entry 1.
Scheme 36 .

Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 327
Table 17. Role of Molecular Sieves (MS 4A)
entry MS 4A (mg) R = Me% yield % ee
R = Ph% yield % ee
1a 500 72 95 100 97
2b none 79 7 81 10
3c 500 - none 76 95 96 97
a. in situ preparation of the chiral catalyst;
b. absence of MS 4A;
c. absence of MS 4A after filtering of MS used for preparing the chiralcatalyst
In 1997, Mikami and coworkers described aninteresting double-stereodifferentiating study(Scheme 36). Carbonyl ene-reactions of (S)-benzyloxypropanal 1 0 1 catalyzed by (R)-BINOL-Ticomplex (R)-12a (matched pair) affords the anti-diastereoisomer 1 0 2 with good selectivities (entries 1,3, 4, Table 16) [61].
The (S)-catalyst (mismatched pair) provides the syn-isomer although in lower chemical yields andselectivities (entries 2 and 5). The use of achiraltitanium catalysts, like TiCl2(iPrO)2 and TiCl(iPrO)3 leadsto the corresponding anti-isomer in good yields (entries6 and 7).
the presence of MS 4A (entries 1 and 3). These resultsshowed that MS 4A is essential for the formation of thechiral catalyst but do not play an important role in theene-reaction. Analysis of 13C NMR spectra showed thehydroxy-carbon signal of BINOL at δ 153 ppm (s). Nochange was observed by mixing BINOL withTi(OiPr)2Cl2 in the absence of MS 4A. The addition ofMS 4A to a solution of BINOL and Ti(OiPr)2Cl2 lead to adownfield shift of the hydroxy-carbon signal (m, 160-163 ppm) indicating the formation of the BINOL-attached chiral catalyst [62]. It appears that MS 4Afacilitates the alcoxy-ligand exchange reactions in the insitu preparation step of the chiral catalyst BINOL-TiCl2.
Table 16. Diastereofacial Selectivity inCarbonyl-ene-reactions
Me Me1 73 15
ene component
entry ene catalyst yield (%) syn : antia
1 1 R 38 <1 : >99
2 1 S 10 84 : 16
3 73 R 55 <1 : >99
R Me
HOMe
O
O
ROMe
O
OH
O
OTi
Cl
Cl
+(0.1 mol%)
CH2Cl2 0 oC
The catalyst:
(R)-12a
(1 mmol)
13
catalyst4 15 R 24 <1 : >99
5 15 S 7 84 : 16
6 15 TiCl2(iPrO)2 60 87 : 13
7 15 TiCl(iPrO)3 48 91 : 09
a. Isomeric ratio determined by GC and/or HPLC analysis
The authors suggest that this reaction proceedthrough an open transition state with Felkin addition asdepicted in Scheme 36 [61].
The Role of Molecular Sieves
The use of molecular sieves is essential to obtainhigh levels of enantioselectivity in the asymmetriccatalytic glyoxylate-ene-reaction [38]. The authorsobserved no significant difference in rate and chemicalyield in these catalytic ene-reactions in the absenceand in the presence of MS 4A, but observed a lowoptical yield when the catalyst solution was prepared inthe absence of MS 4A (Scheme 37, Table 17).
Scheme 37 .
In 1997, Mikami and coworkers further elucidatedthe role of molecular sieves and proved that thedichlorotitanium complex (R)-12a is not the activetitanium catalyst based on 17O NMR and elementalanalysis [63]. The authors proposed a BINOL-Ti catalystcomposed of a µ3-oxo (Ti3O) as the active catalyst. Thedichlorotitanium complex (R)-12a , prepared fromBINOL dilithium salt 103 and TiCl4 in CH2Cl2 was usedas catalyst for the ene-reaction between α-methylstyrene 17 and n-butylglyoxylate 96 (Scheme
The authors observed that higher levels ofenantioselectivity are obtained using a catalyst solutionprepared in the presence of MS 4A. The use of acatalyst solution obtained by removal of the MS 4A byfiltration afforded the same levels of highenantioselectivity to that obtained for the reaction in
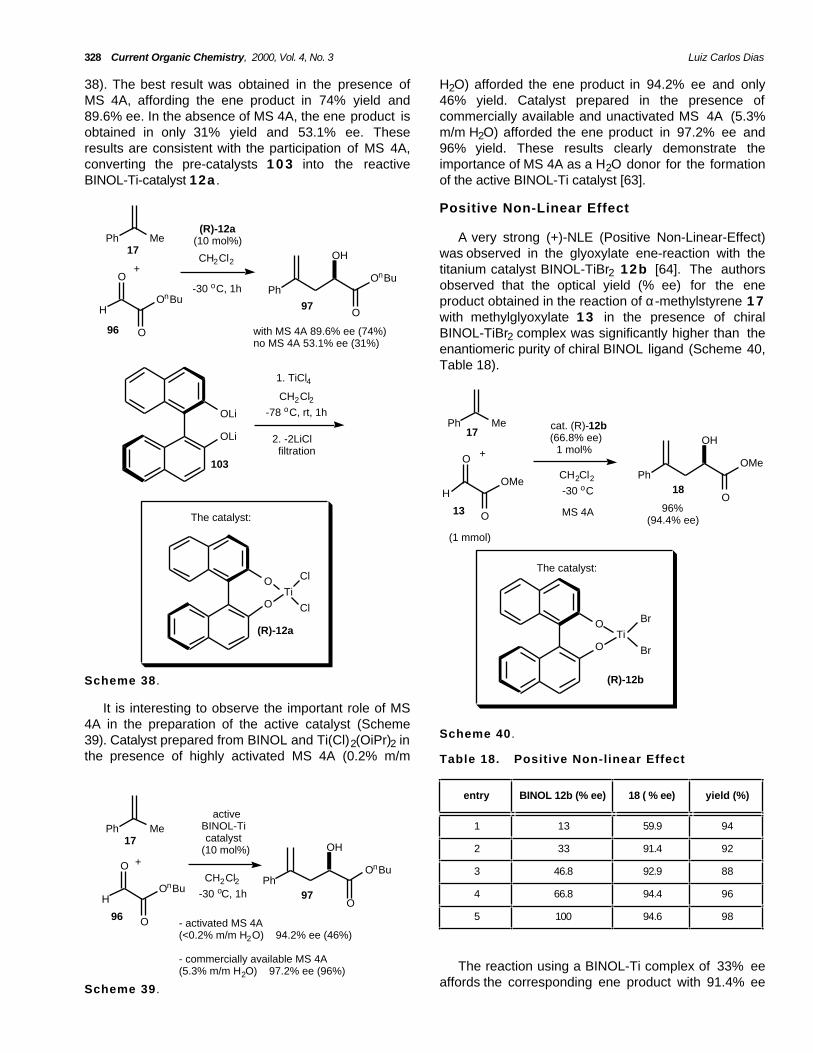
328 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
38). The best result was obtained in the presence ofMS 4A, affording the ene product in 74% yield and89.6% ee. In the absence of MS 4A, the ene product isobtained in only 31% yield and 53.1% ee. Theseresults are consistent with the participation of MS 4A,converting the pre-catalysts 1 0 3 into the reactiveBINOL-Ti-catalyst 12a .
H2O) afforded the ene product in 94.2% ee and only46% yield. Catalyst prepared in the presence ofcommercially available and unactivated MS 4A (5.3%m/m H2O) afforded the ene product in 97.2% ee and96% yield. These results clearly demonstrate theimportance of MS 4A as a H2O donor for the formationof the active BINOL-Ti catalyst [63].
Ph
HOnBu
O
O
Ph
OH
O
OnBu
OLi
OLi
O
OTi
Cl
Cl
Me
+
(R)-12a (10 mol%)
CH2Cl2
1. TiCl4
CH2Cl2
The catalyst:
with MS 4A 89.6% ee (74%)no MS 4A 53.1% ee (31%)
17
96
97
103
(R)-12a
-30 oC, 1h
-78 oC, rt, 1h
2. -2LiCl filtration
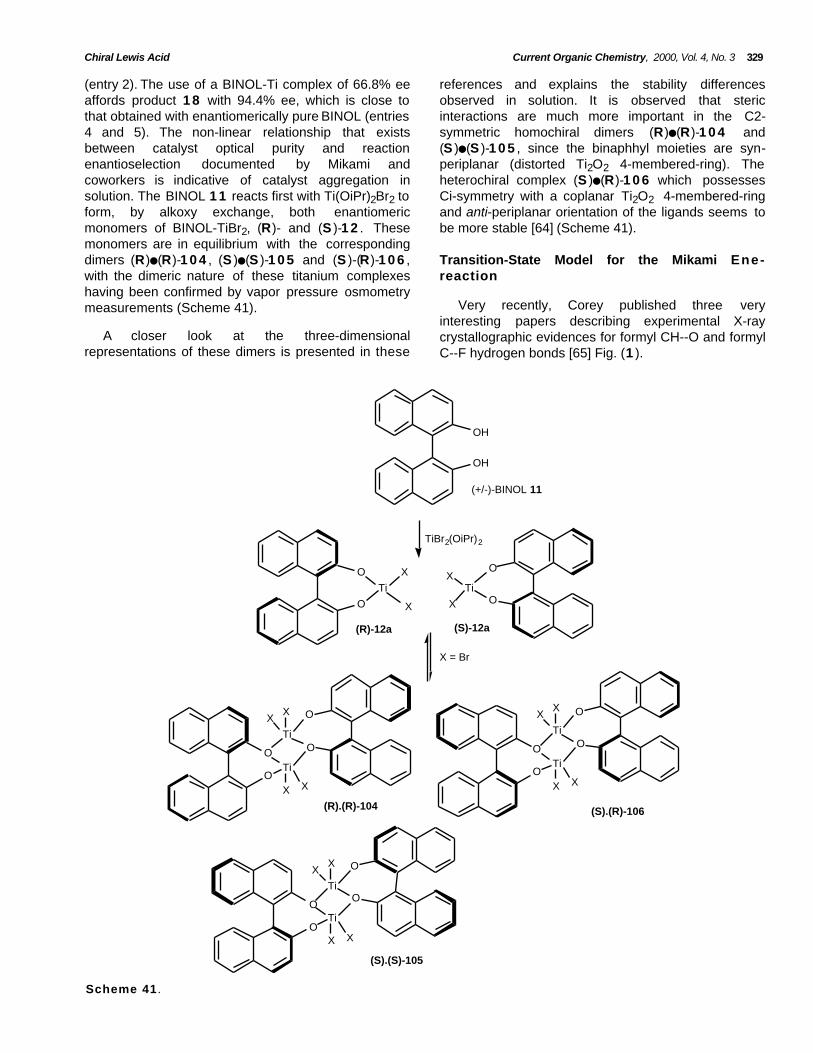
Positive Non-Linear Effect
A very strong (+)-NLE (Positive Non-Linear-Effect)was observed in the glyoxylate ene-reaction with thetitanium catalyst BINOL-TiBr2 12b [64]. The authorsobserved that the optical yield (% ee) for the eneproduct obtained in the reaction of α-methylstyrene 1 7with methylglyoxylate 1 3 in the presence of chiralBINOL-TiBr2 complex was significantly higher than theenantiomeric purity of chiral BINOL ligand (Scheme 40,Table 18).
Ph Me
HOMe
O
O
PhOMe
O
OH
O
OTi
Br
Br
+
cat. (R)-12b (66.8% ee) 1 mol%
CH2Cl2 -30 oC
The catalyst:
(R)-12b
(1 mmol)
17
13
18
MS 4A 96%(94.4% ee)
Scheme 38 .
It is interesting to observe the important role of MS4A in the preparation of the active catalyst (Scheme39). Catalyst prepared from BINOL and Ti(Cl)2(OiPr)2 inthe presence of highly activated MS 4A (0.2% m/m
Scheme 40 .
Table 18. Positive Non-linear Effect
entry BINOL 12b (% ee) 18 ( % ee) yield (%)
Ph
HOnBu
O
O
Ph
OH
O
OnBu
Me
+
active BINOL-Ti catalyst
(10 mol%)
- activated MS 4A(<0.2% m/m H2O) 94.2% ee (46%)
- commercially available MS 4A(5.3% m/m H2O) 97.2% ee (96%)
17
96
97
CH2Cl2-30 oC, 1h
Scheme 39 .
1 13 59.9 94
2 33 91.4 92
3 46.8 92.9 88
4 66.8 94.4 96
5 100 94.6 98
The reaction using a BINOL-Ti complex of 33% eeaffords the corresponding ene product with 91.4% ee
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 329
(entry 2). The use of a BINOL-Ti complex of 66.8% eeaffords product 1 8 with 94.4% ee, which is close tothat obtained with enantiomerically pure BINOL (entries4 and 5). The non-linear relationship that existsbetween catalyst optical purity and reactionenantioselection documented by Mikami andcoworkers is indicative of catalyst aggregation insolution. The BINOL 1 1 reacts first with Ti(OiPr)2Br2 toform, by alkoxy exchange, both enantiomericmonomers of BINOL-TiBr2, (R)- and (S )-1 2 . Thesemonomers are in equilibrium with the correspondingdimers (R)●(R)-1 0 4 , (S )●(S )-1 0 5 and (S )-(R)-1 0 6 ,with the dimeric nature of these titanium complexeshaving been confirmed by vapor pressure osmometrymeasurements (Scheme 41).
references and explains the stability differencesobserved in solution. It is observed that stericinteractions are much more important in the C2-symmetric homochiral dimers (R)●(R)-1 0 4 and(S )●(S )-1 0 5 , since the binaphhyl moieties are syn-periplanar (distorted Ti2O2 4-membered-ring). Theheterochiral complex (S )●(R)-1 0 6 which possessesCi-symmetry with a coplanar Ti2O2 4-membered-ringand anti-periplanar orientation of the ligands seems tobe more stable [64] (Scheme 41).
Transition-State Model for the Mikami Ene-reaction
Very recently, Corey published three veryinteresting papers describing experimental X-raycrystallographic evidences for formyl CH--O and formylC--F hydrogen bonds [65] Fig. (1 ).
A closer look at the three-dimensionalrepresentations of these dimers is presented in these
O
OTi
TiO
OXX
X X
O
OTi
TiO
OXX
X X
O
OTi
TiO
OXX
X X
O
O
Ti TiO
O
X
X X
X
OH
OH
(R).(R)-104 (S).(R)-106
(S).(S)-105
(R)-12a (S)-12a
(+/-)-BINOL 11
TiBr2(OiPr)2
X = Br
Scheme 41 .

330 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
H R
OB
F
F F
H R
OB
O
X Y
R
coordination enhances thepositive charge at formyl hydrogen
negative charge on boronincreases basicity of oxygen
Cl
Ti
OH
BINOL
B
Cl
Ti
Cl
BINOL
A C
O
Ti
O
Ti BINOLBINOL
Scheme 42 .
Based on the fact that both methylglyoxylate 1 3 (2-point-binding) and 3-methoxycarbonylpropynal 7 1 (1-point-binding) afford similar levels of enantioselectivity,the authors propose that bidentate coordination ofboth carbonyl groups of the glyoxylic esters is notessential. They propose that the aldehyde is activatedby complexation with the chiral catalyst (R)-BINOL-TiX2via the formyl lone electron pair syn to the formylhydrogen to form a pentacoordinated titanium structureFig. (3 )
Fig. (1). Formyl CH--F and CH--O hydrogen bonds. A trigonal bipyramidal geometry with the activatedaldehyde and one of the electronegative ligands in theapical position is proposed for this pentacoordinatedcomplex. The authors also propose formyl CH---Ohydrogen bonding with the closer and more accessibleoxygen lone pair of the BINOL generating structure107 Fig. (3 ).
In these papers, Corey describes the use of formylCH--O hydrogen bond as an additional factor thatcontributes to the high degree of enantioselectivitythat is observed in several enantioselective Lewis acidcatalyzed reactions. In the last paper of this series,Corey describes applications of this new kind ofhydrogen bond in determining transition-stategeometry in chiral Lewis-acid catalyzed aldol, carbonylallylation and Diels-Alder reactions [4]. The preferencefor this coplanar/eclipsed conformer derives from anattractive interaction between the formyl hydrogen(acidified by coordination of oxygen to the boron) andthe coplanar fluorine (more electron rich because of thenegative charge on boron).
O O
X
TiX O
OH
OCH3
107
aldehyde in apical position
CH--O hydrogen bond
An alternative explanation for this same fact comesfrom an interaction between the HOMO (oxygen lonepair) and LUMO (σ* B-F). This type of stabilizationcannot be ruled out although the energy of the HOMO(oxygen lone pair) is considerably lowered because ofthe positive charge on oxygen, and the energy of theLUMO (σ* B-F) is increased because of the negativecharge on boron Fig. (2 ).
Fig. (3). Complexation of aldehyde to BINOL-Ti catalyst.
Approach of the nucleophile from the top (re face)of the aldehyde is much more accessible thanapproach from the si face, shielded by the closenaphthol ring [66] Fig. (4 ).
H R
OB
F F
F
σ* B-F
O O
X
TiX O
OH
RH R
HOCH3
si face shielded
by the naphthol ring
Fig. (2). Molecular orbital interactions.
Also in 1997, Corey et al. presented a transition-state model for the Mikami enantioselelective ene-reaction based on the same arguments presentedabove [66]. Although the exact structure of theeffective Mikami catalyst is unknown, the authorsbelieve that any of the following species may functionas effective catalytic species (Scheme 42).
Fig. (40). Transition State for the Mikami ene-reaction.
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 331
Ytterbium Lewis Acids Copper Lewis Acids
Limited degrees of success have been achieved indeveloping optically active chiral catalysts derived fromlanthanides for the ene-reaction [67]. Chiral ytterbiumcomplex 108 generated from Yb(OTf)3 and (S)-6,6'-dibromo-binaphthol, produces a very modestasymmetric induction in the ene-reaction ofmethylglyoxylate 13 with α-methylstyrene 1 7(Scheme 43, Table 19). The best result in terms ofenantioselectivity (38% ee) for the derived α-hydroesters 1 8 was obtained using thisdibromoderivative (entry 2) [67].
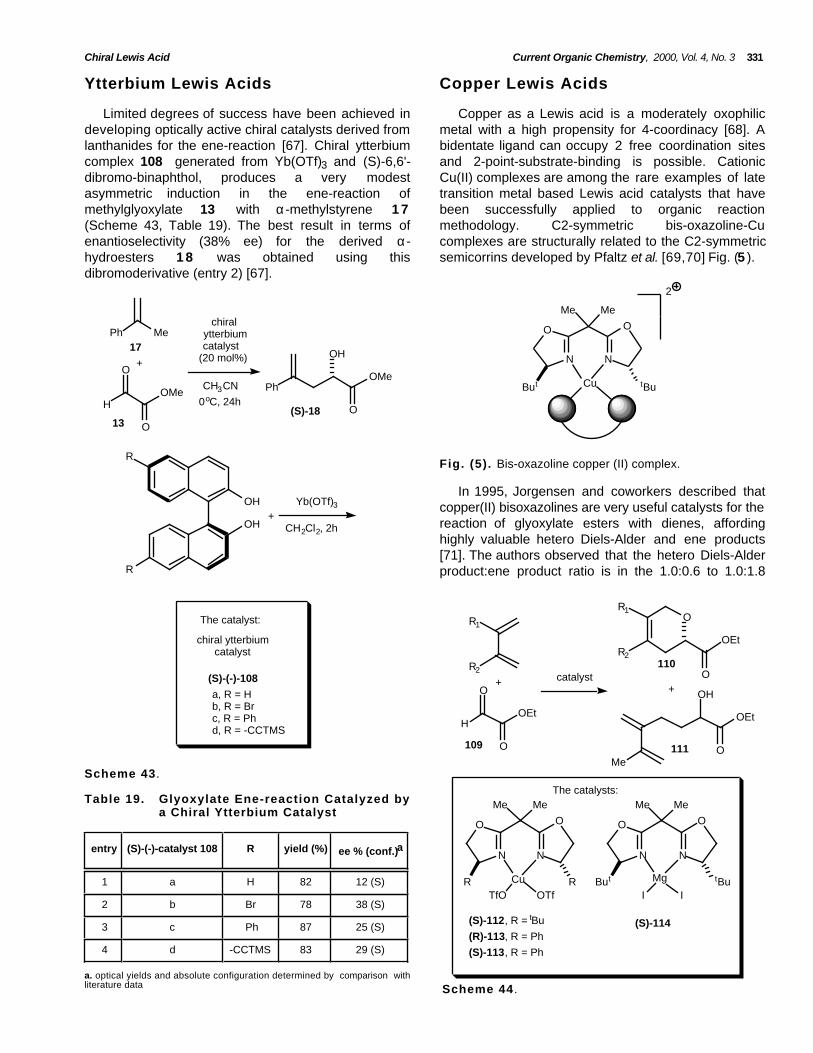
Copper as a Lewis acid is a moderately oxophilicmetal with a high propensity for 4-coordinacy [68]. Abidentate ligand can occupy 2 free coordination sitesand 2-point-substrate-binding is possible. CationicCu(II) complexes are among the rare examples of latetransition metal based Lewis acid catalysts that havebeen successfully applied to organic reactionmethodology. C2-symmetric bis-oxazoline-Cucomplexes are structurally related to the C2-symmetricsemicorrins developed by Pfaltz et al. [69,70] Fig. (5 ).
N
O O
N
Me Me
But tBuCu
2
Ph Me
HOMe
O
O
PhOMe
OH
O
OH
OH
R
Yb(OTf)3
R
+
chiral ytterbiumcatalyst
(20 mol%)
CH3CN
0oC, 24h
+
chiral ytterbium catalyst
CH2Cl2, 2h
The catalyst:
(S)-(-)-108
17
13(S)-18
a, R = Hb, R = Brc, R = Phd, R = -CCTMS
Fig. (5). Bis-oxazoline copper (II) complex.
In 1995, Jorgensen and coworkers described thatcopper(II) bisoxazolines are very useful catalysts for thereaction of glyoxylate esters with dienes, affordinghighly valuable hetero Diels-Alder and ene products[71]. The authors observed that the hetero Diels-Alderproduct:ene product ratio is in the 1.0:0.6 to 1.0:1.8
R1
R2
HOEt
O
O
OR1
R2
OEt
O
OEt
O
OH
Me
N
O O
N
Me Me
R RCu
TfO OTf
N
O O
N
Me Me
But tBuMg
I I
+ catalyst+
109
110
111
(S)-114(S)-112, R = tBu
(R)-113, R = Ph
(S)-113, R = Ph
The catalysts:
Scheme 44 .
Scheme 43 .
Table 19. Glyoxylate Ene-reaction Catalyzed bya Chiral Ytterbium Catalyst
entry (S)-(-)-catalyst 108 R yield (%) ee % (conf.)a
1 a H 82 12 (S)
2 b Br 78 38 (S)
3 c Ph 87 25 (S)
4 d -CCTMS 83 29 (S)
a. optical yields and absolute configuration determined by comparison withliterature data
332 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
Table 20. Glyoxylate Ene-reactions with Dienes
DA product (110) Ene product (111) ratio
entry R1 R2 catalyst yield (%) ee % (conf.) yield (%) ee % a 110:111
1 Me Me (S)-112 20 85 (S) 36 83 1.0:1.8
2 Me Me (R)-113 31 83 (S) 50 88 1.0:1.6
3 Me Me (S)-114 10 5 (S) 20 10 1.0:2.0
4 Me H (R)-113 33 80a 34 91 1.0:1.0
a. absolute configuration not assigned
range and is dependent on the chiral ligand attached tothe metal, the glyoxylate ester, and the reactiontemperature (Scheme 44, Table 20). These copper(II)bisoxazoline catalysts give much better hetero Diels-Alder selectivity when compared with the chiral BINOLand titanium complexes.
product ratio of 1.0:1.0. In the same reaction, the chiralBINOL titanium complex gives a 1.0:4.0 ratio [72]. Theauthors observed that using the tert-butyl-substitutedbisoxazoline ligand catalyst, the methyl glyoxylateesters gives the highest ee in the hetero Diels-Alderand the ene-reaction. They observed also that theabsolute stereochemistry in Diels-Alder products isdependent on the catalyst applied. The use of abisoxazoline ligand with a tert-butyl substituent at thechiral center gives the opposite stereochemistrycompared with a bisoxazoline ligand having a phenylsubstituent at the chiral center (entries 1 and 2). Theypropose that this result is explained by a geometricalchange at the copper atom, with a planar complex asthe intermediate when R=tBu, and a tetrahedral
As can be seen from the results in Table 19, thecopper catalysts (R)-1 1 3 and (S )-1 1 2 (entries 1 and 2)are much better catalysts than the correspondingmagnesium (II) iodide bisoxazoline complex (S )-1 1 4(entry 3) , both in terms of yields andenantioselectivities.
Using isoprene as the substrate and (R)-1 1 3 as thecatalyst afforded a hetero Diels-Alder product:ene
N
O O
N
Me Me
Ph PhCu
TfO OTf
CO2Me
SPh NHCbz
HOMe
O
O
MeO2CCO2Me
NHCbzSPhOH
HO2CCO2H
NH2NH2
R S
CO2Me
NHCbz115
(R)-113
+
catalyst(R)-113
42%117
The catalyst:
meso-DAP 118
94:06 diastereomeric excess determined at a later stage
unstable product
116
13
Scheme 45 .

Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 333
arrangement at the metal when the bisoxazoline ligandis phenyl. The authors attributed the turnover to achange in metal geometry from square planar totetrahedral.
1 1 3 , 119 and 120 are highly selective and effectiveenantioselective catalysts for glyoxylate ene-reactions(Scheme 46). These bis(oxazoline)-copper complexeswere initially used for asymmetric Diels-Alder reactionsand were reported to produce undesired ene sideproducts. These C2-symmetric copper-(II) complexesprovided excellent yields and enantioselectivitites inthe addition of a variety of olefins (including lessnucleophilic olefins) to glyoxylate esters (Table 21).
A very interesting synthetic application of thismethodology was used by Vederas and coworkers whoreported the use of bis(oxazoline)copper-(II) complex(R)-1 1 3 as a chiral catalyst for the ene-reaction of theN-Cbz-derivative of methyl (S)-4-(phenylthio)allylglycinate 1 1 6 and methylglyoxylate 1 3 affording1 1 7 in modest 42% yield and 88% de (S) (Scheme 45)[73]. This reaction was employed as a key step in thesynthesis of meso-diaminopimelic acid (meso-DAP)1 1 8 , a key constituent of bacterial peptidoglycan, anda potential target for development of new antibiotics.The alcohol 1 1 7 was transformed into the desired acid1 1 8 after a sequence involving few steps.
Ene-reaction of methylenecyclohexane 1 withethylglyoxylate 1 0 9 afforded (S )-1 2 1 in 97% ee (97%yield) in the presence of 10 mol% of (S)-catalyst 1 1 9(Table 21). The use of bis(aqua) catalyst (S )-1 2 0 (lessreactive) led essentially to the same result (entry 1). It isinteresting to note that the [Cu((S,S)-Ph-box)](OTf)2complex 1 1 3 affords the absolute stereochemistry(87% ee, 97% yield) of the resulting product oppositeto that produced by (S,S)-tBu-box catalysts 1 1 9 and1 2 0 , in perfect accordance with the results previouslyreported by the Jorgensen group and with thosereported by Vederas et al. [71,73].
Attempts to use N-(benzyloxy-carbonyl)-L-allylglycine methyl ester 1 1 5 in this reaction, in thepresence of several chiral catalysts failed because ofthe unreactive terminal olefin. To circumvent thisproblem, a sulfur substituent was temporally introducedat the terminus of the allylglycine residue. It is importantto point out that the use of chiral binaphthol-titaniumcomplexes as Lewis acids also failed and lead only torecovered starting materials [73].
The optimized Cu-(II)-bis-oxazoline catalyst systemhas been successfully applied to the ene-reaction ofunsymmetrical 1,1-disubstituted olefins (entries 3 and6), as well as with less nucleophilic monossubstitutedolefins (entry 4). The (S)-Ph-box-derived catalyst 1 1 3(10 mol%) mediates the addition of 2-methyl-1-heptene 1 2 8 to ethylglyoxylate 109 to afford (R)-129 in 91% ee, and 90:10 regioselectivity, the best eeobtained so far for this type of enophile (entry 6). Thisresult clearly demonstrate that this catalytic system candiscriminate between methyl and methylenehydrogens. The use of catalyst (S )-120 affords (S )-1 2 9 in 96% ee but with only 74:26 regioselectivity[74]. It is interesting to observe that bis(aqua) complex(R)-1 2 0 , readily prepared as a bench-stable solid, canbe used as catalyst with loadings as low as 0.1 mol%(entries 2, 5 and 6).
An extremely useful and highly enantioselectiveene-reaction was reported recently by Evans andcoworkers [74]. They described that the utilization ofthe bidentate bis(oxazolinyl)(box)-Cu(II) complexes
N
O O
N
Me Me
But tBuCu
N
O O
N
Me Me
But tBuCu
H2O OH2
N
O O
N
Me Me
Ph PhCu
(S)-119
2
2 SbF6
(S)-120
2
2 SbF6
(S)-113
2
2 OTf
The catalysts
blue solid
Scheme 46 .
The sense of asymmetric induction can berationalized by assuming that the reaction proceeds viathe intermediacy of the square planar catalyst-
N
O O
N
Me Me
But tBuCu
O O
OEtHNu
130
2
re face shielded by
the bulk tbutyl group
Scheme 47 .
334 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
Table 21. Glyoxylate Ene-reactions Catalyzed by Copper (II) Complexes
entry olefin product cat. (mol %) T (oC) yield (%) % ee (conf.)a
1
1
OEt
O
OH
121
120 (10)
119 (10)
113 (10)
0
0
0
97
97
99
97 (S)
97 (S)
87 (R)
2 Me Me15
MeOEt
O
OH122
120 (1)
113 (10)
0
0
83
92
96 (S)
92 (R)
3 MeBnO
123OEt
O
OH
OBn
124
120 (10)
113 (2)
25
25
62
88
98 (S)
92 (R)
4C3H7
125OEt
O
OHC3H7 126
119 (10) 25 96 98 (S)
5 Ph Me
17
OEt
O
OH
Ph
127
120 (1)
113 (10)
0
0
97
99
93 (S)
89 (R)
6 MeC4H9
128
OEt
O
OH
C4H9
129
120 (1)
113 (10)
25
25
89
81
96 (S)
91 (R)
a. absolute configuration assigned by conversion to MTPA estersb. ee's determined by GLC (Cyclodex-β) column or HPLC (Chiralcel OD-H column)
glyoxylate complex 130 (Scheme 47). The re face ofthe coordinated aldehyde is blocked by the tert-butylsubstituent and the approach of olefins occurs from theaccessible aldehyde si face.
reactions, as proposed earlier by Jorgensen et al. [75].According to the Evans group, Cu(II)-bis(oxazoline)complexes (S )-1 1 3 and (S )-1 1 9 catalyze the ene-reaction of methylenecyclohexane 1 andethylglyoxylate 1 0 9 with the enantiomeric excess ofthe product dependent on the oxazoline ringsubstituent (Scheme 48). α-hydroxy-ester 1 2 1 isobtained in 97% ee employing catalyst (S,S)-1 1 3 , andthe corresponding enantiomer is obtained in 87% eeusing complex (S )-1 1 9 . The use of complex (S )-1 3 1
More recently, Evans and coworkers at Harvardsuggested, based on structural and mechanisticstudies, that a change in geometry at the metal centeris not necessarily responsible for the reversal inenantioselectivity observed in glyoxylate ene-
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 335
N
O O
N
Me Me
But tBuCu
N
O O
N
Me Me
Ph PhCu
HOEt
O
O
OEt
O
OH
OEt
O
OH
N
O O
N
Me Me
Pri iPrCu
(S)-119(S)-113
2
2X 2X
2
a: X = OTf
b: X = SbF6
+
10 mol% (S)-119
CH2Cl2, 0 oC
(S):(R) = 6.5:93.5 (87% ee)
10 mol% (S)-113
CH2Cl2, 0 oC
(S):(R) = 98.5:1.5 (97% ee)
(S)-131
2
1
109
121121
2X
The catalysts
Scheme 48 .
afforded the corresponding ene product with only 36%ee (S). The authors believe that the intermediacy of adistorted square planar bis(oxazoline) Cu-(II)-substratecomplex is responsible for this reversal inenantioselectivity.
semiempirical calculations and crystallographictechniques to investigate this phenomenon (Scheme49). X-Ray crystal structures of the Cu[(S,S)-iPr-bis(oxazoline)](H2O)2(SbF6)2 and Cu[(S,S)-tBu-bis(oxazoline)](H2O)2(SbF6)2 complexes are alsopresented.
The Evans group employed doublestereodifferentiating experiments, EPR spectroscopy, In bis-aquo-complex 1 2 0 , the Cu-(II) center
presents a geometry distortion from square planarity,with the ligated water molecules distorted +33.3o awayfrom oxazoline substituents (Scheme 49). In the caseof phenyl-substituted complex 1 3 2 , the watermolecules tilt toward the oxazoline substituents by–9.3o. The authors observed also that these distortionsare independent of the nature of the counterion andthe absence of nonlinear effects in these glyoxylateene-reactions [75].
N
O O
N
Me Me
Ph PhCu
H2O OH2
N
O O
N
Me Me
But tBuCu
H2O OH2
(S)-132
2
2SbF6
(S)-120
2SbF6
O1-Cu-N1-C1 dihedral < +30.2o
O2-Cu-N2-C2 dihedral < +35.9o
O1-Cu-N1-C1 dihedral < - 11.3o
O2-Cu-N2-C2 dihedral < - 7.2o
2
Scheme 49 .
Ene-reactions with α-Tosyl-iminoEsters
Another highly enantioselective ene-reaction wasreported by the Jorgensen group in 1998 [76]. Theauthors described a highly enantioselective ene-reaction of tosyl- α-iminoesters with alkenes catalyzedby 0.1 mol % of chiral CuPF6-BINAP and CuClO4-BINAP complexes. This reaction afforded chiral α-aminoesters 1 3 4 , that can be used to prepare bothoptically active and biologically important natural andnon-natural α-aminoacids. The chiral phosphineligands (R)-BINAP and (R)-tol-BINAP in combinationwith copper (I) salts have been found to be the best in
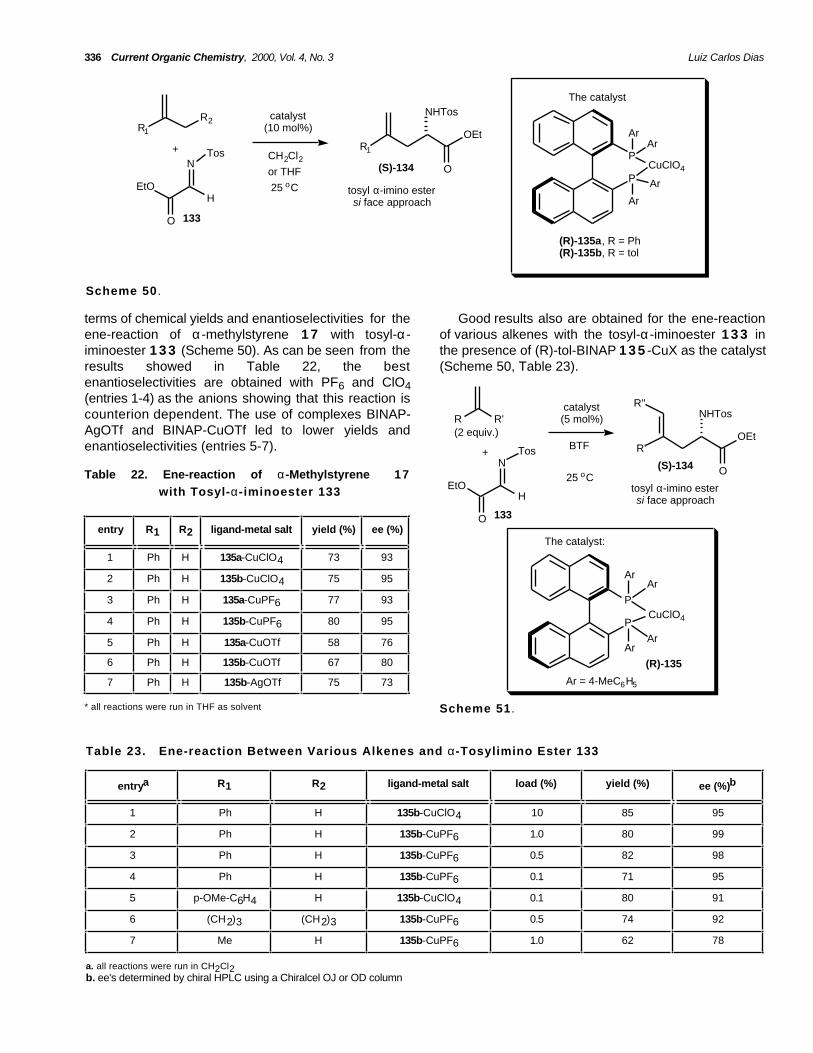
336 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
P
P
R1
R1
NHTos
N
H
Tos
R2
O
OEt
O
EtO
CuClO4
Ar
Ar
Ar
Ar
The catalyst
catalyst(10 mol%)
+
(R)-135a, R = Ph(R)-135b, R = tol
(S)-134
tosyl α-imino ester si face approach
133
CH2Cl2
or THF
25 oC
Scheme 50 .
terms of chemical yields and enantioselectivities for theene-reaction of α-methylstyrene 1 7 with tosyl-α-iminoester 1 3 3 (Scheme 50). As can be seen from theresults showed in Table 22, the bestenantioselectivities are obtained with PF6 and ClO4(entries 1-4) as the anions showing that this reaction iscounterion dependent. The use of complexes BINAP-AgOTf and BINAP-CuOTf led to lower yields andenantioselectivities (entries 5-7).
Good results also are obtained for the ene-reactionof various alkenes with the tosyl-α-iminoester 1 3 3 inthe presence of (R)-tol-BINAP 1 3 5 -CuX as the catalyst(Scheme 50, Table 23).
P
P
R R'
N
H
Tos
O
EtO
CuClO4
ArAr
ArAr
R'
NHTos
O
OEt
R''
The catalyst:
(R)-135
catalyst(5 mol%)
BTF
25 oC
+
Ar = 4-MeC6H5
tosyl α-imino ester si face approach
(2 equiv.)
(S)-134
133
Table 22. Ene-reaction of α-Methylstyrene 17with Tosyl-α-iminoester 133
entry R1 R2 ligand-metal salt yield (%) ee (%)
1 Ph H 135a-CuClO4 73 93
2 Ph H 135b-CuClO4 75 95
3 Ph H 135a-CuPF6 77 93
4 Ph H 135b-CuPF6 80 95
5 Ph H 135a-CuOTf 58 76
6 Ph H 135b-CuOTf 67 80
7 Ph H 135b-AgOTf 75 73
Scheme 51 .* all reactions were run in THF as solvent
Table 23. Ene-reaction Between Various Alkenes and α-Tosylimino Ester 133
entrya R1 R2 ligand-metal salt load (%) yield (%) ee (%)b
1 Ph H 135b-CuClO4 10 85 95
2 Ph H 135b-CuPF6 1.0 80 99
3 Ph H 135b-CuPF6 0.5 82 98
4 Ph H 135b-CuPF6 0.1 71 95
5 p-OMe-C6H4 H 135b-CuClO4 0.1 80 91
6 (CH2)3 (CH2)3 135b-CuPF6 0.5 74 92
7 Me H 135b-CuPF6 1.0 62 78
a. all reactions were run in CH2Cl2b. ee's determined by chiral HPLC using a Chiralcel OJ or OD column
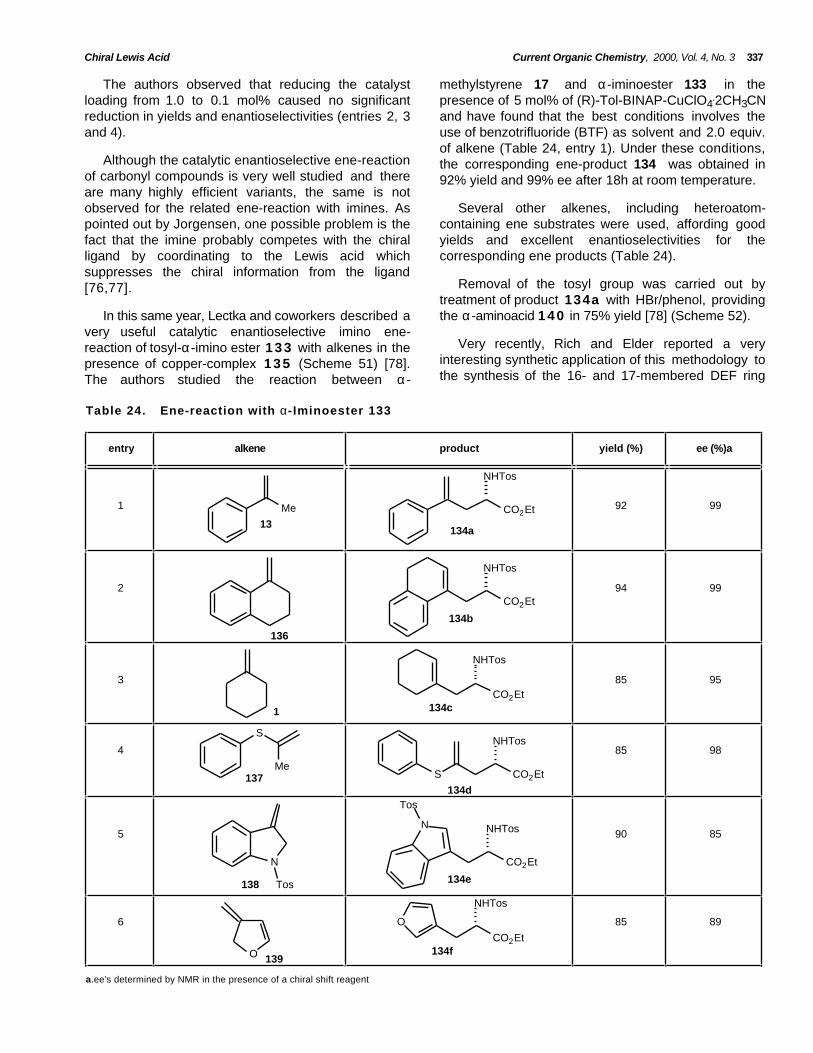
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 337
The authors observed that reducing the catalystloading from 1.0 to 0.1 mol% caused no significantreduction in yields and enantioselectivities (entries 2, 3and 4).
methylstyrene 17 and α-iminoester 133 in thepresence of 5 mol% of (R)-Tol-BINAP-CuClO4
.2CH3CNand have found that the best conditions involves theuse of benzotrifluoride (BTF) as solvent and 2.0 equiv.of alkene (Table 24, entry 1). Under these conditions,the corresponding ene-product 134 was obtained in92% yield and 99% ee after 18h at room temperature.
Although the catalytic enantioselective ene-reactionof carbonyl compounds is very well studied and thereare many highly efficient variants, the same is notobserved for the related ene-reaction with imines. Aspointed out by Jorgensen, one possible problem is thefact that the imine probably competes with the chiralligand by coordinating to the Lewis acid whichsuppresses the chiral information from the ligand[76,77].
Several other alkenes, including heteroatom-containing ene substrates were used, affording goodyields and excellent enantioselectivities for thecorresponding ene products (Table 24).
Removal of the tosyl group was carried out bytreatment of product 134a with HBr/phenol, providingthe α-aminoacid 1 4 0 in 75% yield [78] (Scheme 52).In this same year, Lectka and coworkers described a
very useful catalytic enantioselective imino ene-reaction of tosyl-α-imino ester 1 3 3 with alkenes in thepresence of copper-complex 1 3 5 (Scheme 51) [78].The authors studied the reaction between α-
Very recently, Rich and Elder reported a veryinteresting synthetic application of this methodology tothe synthesis of the 16- and 17-membered DEF ring
Table 24. Ene-reaction with α-Iminoester 133
entry alkene product yield (%) ee (%)a
1 Me
13CO2Et
NHTos
134a
92 99
2
136
NHTos
CO2Et
134b
94 99
3
1
NHTos
CO2Et134c
85 95
4
S
Me137 S CO2Et
NHTos
134d
85 98
5
N
Tos138
N
Tos
CO2Et
NHTos
134e
90 85
6
O 139
O
CO2Et
NHTos
134f
85 89
a.ee's determined by NMR in the presence of a chiral shift reagent

338 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
Ph
NHTos
O
OEt
Ph
NH2
O
OH
(S)-134a
1. PhOHHBr/AcOH
2. H2O
(S)-140Scheme 52 .
Compound 1 4 2 was converted to the 17-membered DEF ring system 1 4 3 of complestatin aftera few steps (Scheme 54). Acid catalyzed ringcontraction of this 17-membered ring (TFA, 50 oC)generated the 16-membered DEF ring system ofchloropeptin.
Conclusions
The progress in the catalytic asymmetric ene-reaction has been outstanding and impressive resultsthrough the use of titanium Lewis acids have beenobtained by Mikami et al. and Nakai et al. In particular,the asymmetric induction achieved by using chiralglyoxylate esters and the catalysis with BINOL-Lewisacid complexes are major achievements.
systems of chloropeptin and complestatin, two verypotent biologically active macrocyclic polypeptides[79]. Ene-reaction of tosyl α-imino ester 1 3 3 with 3-methyleneindoline 1 4 1 in the presence of (S)-tol-BINAP-CuClO4-2CH3CN in BTF as solvent afforded thefully protected 6-bromo-D-tryptophan 1 4 2 in 76% yieldand 94% ee (Scheme 53). The use of copper Lewis acids derived from bis-
oxazolines as described by Evans affords also fantasticresults in terms of yields and enantioselectivities for theglyoxylate ene-reaction.
NBr
Tos
P
P
N
H
Tos
O
EtO
CuClO4
ArAr
ArAr
N
Tos
CO2Et
NHTos
Br
The catalyst:
(S)-135
catalyst(5 mol%)
BTF
25 oC, 4h
+
Ar = 4-MeC6H5
76%, 94% ee
(R)-142
ee determined by Mosher ester analysis of the primary alchol obtained after ester reduction
141
133
With regard to the imino ene-reaction, the resultsdescribed recently by Jorgensen and Lectka are alsovery promising.
Despite these impressive recent advances, manyunsolved problems still remain and there are someother features of the catalytic ene-process that remainto be improved. These include limitations with regard toscope and frequent practical problems associated withcatalyst preparation and use, especially on large scale.Appropriate structural design of catalyst complexes toavoid the formation of oligomeric aggregation is also aworthwhile goal, since monomeric structures ofcatalysts would lead to enhanced catalytic activity.Reaction enantioselection is highly sensitive to minorvariations in catalyst preparation and, presumably, thesolution-state structure that is derived therefrom,resulting in nearly identical catalyst systems providingdifferent results, ranging from low to nearly perfectasymmetric induction. The control of the regio- andstereochemistry of the ene-reaction is still far fromdeveloped. It is therefore not surprising thatconsiderable attention has been focused on thedevelopment of metal catalyzed asymmetric variants ofthis reaction.
We predict that discoveries of even more practicalchiral Lewis acid catalysts, displaying substratetolerance and requiring lower catalyst loading, willcontinue to be a challenge for synthetic organicchemists. We expect to see more developments in thedirection of ene-reactions involving both rich- and poorenophile partners, which will need a new generation ofasymmetric catalysts. Further work on the design ofmore practical chiral Lewis acid catalysts and new
Scheme 53.
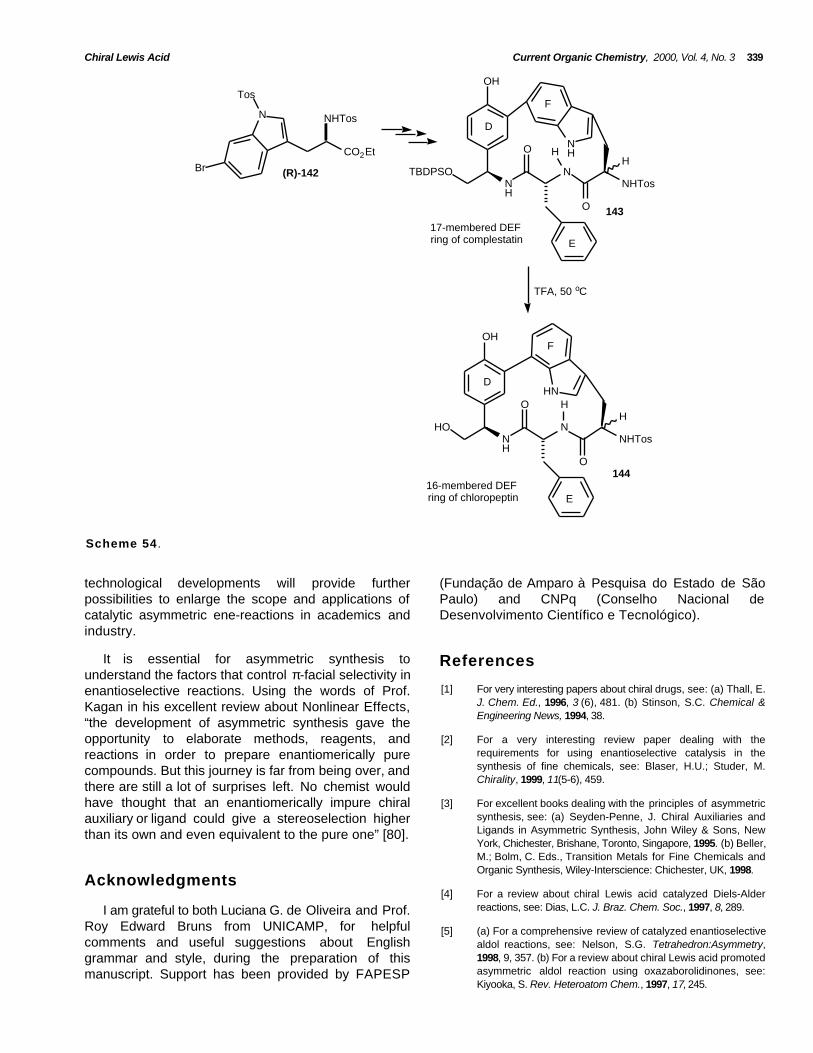
Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 339
N
Tos
CO2Et
NHTos
Br
OH
HONH
NNHTos
O
O
HHN
H
D
F
OH
TBDPSONH
NHTos
O
O
NH
H
D
F
N
H
(R)-142
E
E
17-membered DEF ring of complestatin
16-membered DEF ring of chloropeptin
143
144
TFA, 50 oC
Scheme 54 .
technological developments will provide furtherpossibilities to enlarge the scope and applications ofcatalytic asymmetric ene-reactions in academics andindustry.
(Fundação de Amparo à Pesquisa do Estado de SãoPaulo) and CNPq (Conselho Nacional deDesenvolvimento Científico e Tecnológico).
ReferencesIt is essential for asymmetric synthesis tounderstand the factors that control π-facial selectivity inenantioselective reactions. Using the words of Prof.Kagan in his excellent review about Nonlinear Effects,“the development of asymmetric synthesis gave theopportunity to elaborate methods, reagents, andreactions in order to prepare enantiomerically purecompounds. But this journey is far from being over, andthere are still a lot of surprises left. No chemist wouldhave thought that an enantiomerically impure chiralauxiliary or ligand could give a stereoselection higherthan its own and even equivalent to the pure one” [80].
[1] For very interesting papers about chiral drugs, see: (a) Thall, E.J. Chem. Ed., 1996, 3 (6), 481. (b) Stinson, S.C. Chemical &Engineering News, 1994, 38.
[2] For a very interesting review paper dealing with therequirements for using enantioselective catalysis in thesynthesis of fine chemicals, see: Blaser, H.U.; Studer, M.Chirality, 1999, 11(5-6), 459.
[3] For excellent books dealing with the principles of asymmetricsynthesis, see: (a) Seyden-Penne, J. Chiral Auxiliaries andLigands in Asymmetric Synthesis, John Wiley & Sons, NewYork, Chichester, Brishane, Toronto, Singapore, 1995. (b) Beller,M.; Bolm, C. Eds., Transition Metals for Fine Chemicals andOrganic Synthesis, Wiley-Interscience: Chichester, UK, 1998.
Acknowledgments[4] For a review about chiral Lewis acid catalyzed Diels-Alder
reactions, see: Dias, L.C. J. Braz. Chem. Soc., 1997, 8, 289.I am grateful to both Luciana G. de Oliveira and Prof.Roy Edward Bruns from UNICAMP, for helpfulcomments and useful suggestions about Englishgrammar and style, during the preparation of thismanuscript. Support has been provided by FAPESP
[5] (a) For a comprehensive review of catalyzed enantioselectivealdol reactions, see: Nelson, S.G. Tetrahedron:Asymmetry,1998, 9, 357. (b) For a review about chiral Lewis acid promotedasymmetric aldol reaction using oxazaborolidinones, see:Kiyooka, S. Rev. Heteroatom Chem., 1997, 17, 245.

340 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
[6] For an extensive review on selective reactions using allylicmetals, see: Yamamoto, Y.; Asao, N. Chem. Rev. , 1993, 93,2207.
[21] Whitesell, J.K.; Chen, H.-H.; Lawrence, R.M. J. Org. Chem.,1985, 50, 4663.
[22] Whitesell, J.K.; Lawrence, R.M; Chen, H.-H. J. Org. Chem.,1986, 51, 4779. [7] (a) For an excellent review on asymmetric synthesis with chiral
Lewis acid catalysts, see: Ishihara, K.; Yamamoto, H. Cat.Tech., 1997, 51. (b) For an excellent review on chiral Lewisacids in catalytic asymmetric reactions, see: Narasaka, K.Synthesis, 1991, 1.
[23] For mechanistic aspects of the ene-reaction, see: Paderes, G.D.;Jorgensen, W.L. J. Org. Chem., 1992, 57, 1904.
[24] For a paper about the mechanism of the carbonyl-ene reaction,see; Achmatowicz, O.; Florjanczyk, E.B. Tetrahedron, 1996, 52,8827.
[8] (a) For a very interesting paper about enantiomeric impurities inchiral catalysts, auxiliaries and synthons used inenantioselective synthesis, see: Armstrong, D.W.; Lee, J.T.;Chang, L.W. Tetrahedron:Asymmetry, 1998, 9, 143. (b) Reviewarticle about the use of chiral metal(salen) complexes inasymmetric catalysis: Sherrington, D.C. Chem. Soc. Rev.,1999, 28 (2), 85.
[25] For excellent reviews about asymmetric ene-reactions inorganic synthesis, see: (a) Mikami, K.; Shimizu, M. Chem.Rev., 1992, 92, 1021. (b) Borzilleri, R.M.; Weinreb, S.M.Synthesis, 1995, 347. (c) Mikami, K. Pure & Appl. Chem., 1996,68, 639.
[9] For an excellent review about stoichiometric asymmetricprocesses, see: Regan, A.C. J. Chem. Soc., Perkin Trans. 1,1999, 357.
[26] For reviews about general aspects of asymmetric carbonyl-ene-reactions, see: (a) Mikami, K.; Terada, M.; Narisawa, S.; Nakai,T. Synlett, 1992, 255. (b) Mikami, K. Advances in AsymmetricSynthesis, Vol. 1, pp. 1-44, JAI Press, Inc., 1995.[10] (a) For an excellent review about catalytic asymmetric process,
see: Wills, M. J. Chem. Soc. Perkin Trans. 1, 1998, 18, 3101. (b)enantioselective desymmetrization: Willis, M.C. J. Chem. Soc.,Perkin Trans. 1, 1999, 1765.
[27] For a short review on catalytic asymmetric carbonyl ene-reactions, see: Berrisford, D.J.; Bolm, C. Angew. Chem., Int. Ed.Engl., 1995, 34, 1717.
[11] (a) For a very interesting review on transition-metal-basedLewis acid catalysts, see: Bosnich, B. Aldrichimica Acta, 1998,31, 76. (b) For the first synthesis and structural characterizationof an enantiomerically pure planar-chiral Lewis acid complex,see: Tweddell, J.; Hoic, D.A.; Fu, G.C. J. Org. Chem., 1997, 62,8286. (b) For a very interesting review that highlights theprogress that has been made in the field of Lewis acid catalysisof carbon-carbon-bond forming reactions in aqueous solution,see: Engberts, J.B.F.N.; Feringa, B.L.; Keller, E.; Otto, S. Recl.Trav. Chim. Pays-Bas, 1996, 115, 457.
[28] For mechanistic studies of the metallo-ene-reaction, see:Gomez-Benjoa, E.; Cuerva, J.M.; Echavarren, A.M.; Martorell,G. Angew. Chem., Int. Ed. Engl., 1997, 36, 767.
[29] For a recent review about thermal and Lewis acid catalyzedintramolecular ene-reactions of allenylsilanes, see: Weinreb,S.M.; Smith, D.T.; Jin, J. Synthesis, 1998, 509.
[30] In 1997, Jenner described the effect of high-pressure kinetics ofLewis acid catalyzed cycloaddition and ene-reactions: Jenner, G.New J. Chem., 1997, 21, 1085.
[12] For a review dealing with recent developments in asymmetriccatalysis using synthetic polymers with main chain chirality,see: Pu, L. Tetrahedron:Asymmetry, 1998, 9, 1457.
[31] For solvent effects in asymmetric hetero Diels-Alder and ene-reactions, see: Johannsen, M.; Jorgensen, K.A. Tetrahedron,1996, 52, 7321.
[13] For an excellent review article about catalytic enantioselectiveconstruction of molecules with quaternary carbon stereocenters,see: Corey, E.J.; Guzman-Perez, A. Angew. Chem., Int. Ed.Engl., 1998, 37, 388.
[32] Excellent and comprehensive reviews of intramolecular ene-reactions: (a) Conia, J.M.; Perchec, P. Le Synthesis, 1975, 1. (b)Taber, D.F. Intramolecular Diels-Alder and Alder-EneReactions; Springer Verlag: Berlim, 1984.
[14] Recent review article about catalytic asymmetric additionreactions of carbonyls: Jorgensen, K.A.; Johannsen, M.; Yao, S.;Audrain, H.; Thorhauge, J. Acc. Chem. Res., 1999, 32, 605.
[33] Whitesell, J.K.; Bhattacharya, A.; Aguilar, D.A; Henke, K. J.Chem. Soc. Chem. Commun., 1982, 989.
[15] (a) Treibs, W.; Schmidt, H. Ber. Dtsch. Chem. Ges., 1927, 60,2335. (b) Grignard, V.; Doeuvre, J.C.R. Acad. Sci., 1930, 190,1164. (c) Ikeda, T.; Wakatsuki, K. J. Chem. Soc. Jpn., 1936, 57,425.
[34] Whitesell, J.K. Acc. Chem. Res., 1985, 18, 280
[35] Whitesell, J.K.; Bhattacharya, A.; Buchanan, C.M. Chen, H.H.;Deyo, D.; James, D.; Liu, C.-L.; Minton, M.A. Tetrahedron, 1986,42, 2993.
[16] (a) Alder, K.; Pascher, F.; Schmitz, A. Ber. Dtsch. Chem. Ges.,1943, 76, 27. (b) Nobel Lectures-Chemistry, 1942-1962;Elsevier: Amsterdam, pp. 253-305, 1964.
[36] Maruoka, K.; Hoshino, Y.; Shirasaka, T.; Yamamoto, H.Tetrahedron Lett., 1988, 29, 3967.
[17] (a) Klimova, E.I.; Arbuzov, Y.A. Dokl. Akad. Nauk. SSSR, 1965,167, 419. (b) Klimova, E.I.; Arbuzov, Y.A. Dokl. Akad. Nauk.SSSR, 1967, 173, 1332. (c) Klimova, E.I.; Arbuzov, Y.A. J. Org.Chem. USSR Engl., 1968, 4, 1726.
[37] For intramolecular ene-reactions of unsaturated aldehydescatalyzed by a chiral Lewis acid, see: (a) Sakane, S.; Maruoka,K.; Yamamoto, H. Tetrahedron Lett., 1985, 26, 5535. (b) Sakane,S.; Maruoka, K.; Yamamoto, H. Tetrahedron, 1986, 42, 2203. (c)another example of an intramolecular ene-reaction: Cossy, J.;Bouzide, A.; Pfau, M. J. Org. Chem., 1997, 62, 7106.[18] Achmatowicz, J.O.; Szechner, B. J. Org. Chem., 1972, 37, 964.
[19] Snider, B.B.; Straten, J.W.V. J. Org. Chem., 1979, 44, 3567. [38] (a) Mikami, K.; Terada, M.; Nakai, T. J. Am. Chem. Soc., 1989,111, 1940. (b) Mikami, K.; Terada, M.; Nakai, T. J. Am. Chem.Soc., 1990, 112, 3949.[20] Snider, B.B. Acc. Chem. Res., 1980, 13, 426.

Chiral Lewis Acid Current Organic Chemistry, 2000, Vol. 4, No. 3 341
[39] Terada, M.; Matsukawa, S.; Mikami, K. Chem. Commun., 1993,327.
[60] Chavarot, M.; Byrne, J.J.; Chavant, P.Y.; Pardillos-Guindet, J.;Vallée, Y. Tetrahedron:Asymmetry, 1998, 9, 3889.
[40] Mikami, K.; Terada, M.; Sawa, E.; Nakai, T. Tetrahedron Lett.,1991, 32, 6571.
[61] Mikami, K.; Matsukawa, S.; Sawa, E.; Harada, A.; Koga, N.Tetrahedron Lett., 1997, 38, 1951.
[41] In the ene-cyclizations, the carbon members where the tetherconnects the [1,5]-hydrogen shift system, are expressed in (m,n)fashion.
[62] (a) Dyer, A. An Introduction to Zeolite Molecular Sieves, Wiley,Chichester, 1988. (b) Davis, M.E. Acc. Chem. Res., 1993, 26,111.
[42] Mikami, K.; Sawa, E.; Terada, M. Tetahedron:Asymmetry, 1991,2, 1403.
[63] Terada, M.; Matsumoto, Y.; Nakamura, Y.; Mikami, K. Chem.Commun., 1997, 281.
[43] In 1990, Ziegler described an interesting synthesis oftrichothecene anguidine, in which one of the steps was anasymmetric ene-reaction conducted in the presence of chiralcatalysts to give relatively high diastereoselectivity but only lowlevels of enantioselectivity. Ziegler, F.E.; Sobolov, S.B. J. Am.Chem. Soc., 1990, 112, 2749.
[64] (a) Terada, M.; Mikami, K.; Nakai, T. J. Chem. Soc., Chem.Commun., 1990, 1623. (b) Mikami, K.; Terada, M. Tetrahedron,1992, 48, 5671.
[65] Corey, E.J.; Rohde, J.J.; Fisher, A.; Azimioara, M.D.Tetrahedron Lett., 1997, 38, 33. (b) Corey, E.J.; Rohde, J.J.Tetrahedron Lett., 1997, 38, 37. (c) Corey, E.J.; Barnes-Seeman,D.; Lee, T.W. Tetrahedron Lett., 1997, 38, 1699.[44] Mikami, K.; Yajima, T.; Terada, M.; Uchimaru, T. Tetrahedron
Lett., 1993, 34, 7591.[66] Corey, E.J.; Barnes-Seeman, D.; Lee, T.W.; Goodman, S.N.
Tetrahedron Lett., 1997, 38, 6513.[45] Terada, M.; Mikami, K. J. Chem. Soc., Chem. Commun., 1994,833.
[67] Qian, C.; Huang, T. Tetrahedron Lett., 1997, 38, 6721.[46] Kitamoto, D.; Imma, H.; Nakai, T. Tetrahedron Lett., 1995, 36,
1861. [68] (a) For a discussion on the coordination chemistry of Cu(II), see:Hathaway, B.J. In Comprehensive Coordination Chemistry,Wilkinson, G.; Gillard, R.D.; MaClevarty, J.A.; Eds.; PergamonPress: Oxford, Vol. 5, pp. 533, 1989. (b) A recent study ondivalent metal ion catalyst in Diels-Alder reactions in aqueousmedia concluded that Cu2+ is superior for bidentate metal iondienophilic substrates when compared to Zn2+, Ni2+, and Co2+:Otto, S.; Bertoncin, F.; Engberts, J.B.F.N. J. Am. Chem. Soc.,1996, 118, 7702.
[47] Mikami, K.; Motoyama, Y.; Terada, M. Inorganica Chimica Acta,1994, 222, 71.
[48] Terada, M.; Motoyama, Y.; Mikami, K. Tetrahedron Lett., 1994,35, 6693.
[49] It should be noted that the chiral titanium complexes derivedfrom binaphthol ditrifylamine (R)-49 afforded very low levels ofasymmetric induction (only 3% ee). [69] For a very interesting paper about chiral C2-symmetric
semicorrin and related ligands, see: Pfaltz, A. Synlett, 1999, 835.[50] Mikami, K.; Matsukawa, S. Tetrahedron Lett., 1994, 35, 3133,
[70] (a) For an improved procedure for the preparation of C2-symmetric chiral bis(oxazoline)-copper complexes, see: Evans,D.A.; Peterson, G.S.; Johnson, J.S.; Barnes, D.M.; Campos,K.R.; Woerpel, K.A. J. Org. Chem., 1998, 63, 4541. (b) For anexcellent review about the use of C2-symmetric chiralbis(oxazoline)-metal complexes in catalytic asymmetricsynthesis, see: Ghosh, A.K.; Mathiavanen, P.; Cappiello, J.Tetrahedron:Asymmetry, 1998, 9, 1.
[51] Mikami, K.; Yoshida, A. Synlett, 1995, 29.
[52] Mikami, K.; Narisawa, S.; Shimizu, M.; Terada, M. J. Am.Chem. Soc., 1992, 114, 9242.
[53] (a) Mikami, K.; Yoshida, A.; Matsumoto, Y. Tetrahedron Lett.,1996, 37, 8515. (b) see also: Mikami, K.; Yoshida, A. Angew.Chem., Int. Ed. Engl., 1997, 36, 858.
[71] Johannsen, M.; Jorgensen, K.A. J. Org. Chem., 1995, 60, 5757.[54] Faller, J.W.; Liu, X. Tetrahedron Lett., 1996, 37, 3449.
[72] Terada, M.; Mikami, K.; Takai, T. Tetrahedron Lett., 1991, 32,935.
[55] Mikami, K.; Osawa, A.; Isaka, A.; Sawa, E.; Shimizu, M.;Terada, M.; Kubodera, N.; Nakagawa, K.; Tsugawa, N.; Okano,T. Tetrahedron Lett., 1998, 39, 3359.
[73] Gao, Y.; Lane-Bell, P.; Vederas, J.C. J. Org. Chem., 1998, 63,2133.[56] A very similar approach for the synthesis of A ring has been
used by Courtney and coworkers: Courtney, L.F.; Lange, M.;Uskokovié, M.R.; Noukulich, P.M. Tetrahedron Lett., 1998, 39,3363.
[74] (a) Evans, D.A.; Burgey, C.S.; Paras, N.A.; Vojkovsky, T.;Tregay, S.W. J. Am. Chem. Soc., 1998, 120, 5824. (b) In arecent paper, Kanemasa and coworkers described thepreparation and use of transition-metal aqua complexes of 4,6-dibenzofurandiyl-2,2'-bis(4-phenyloxazoline). The cationic aquacomplexes prepared from their enantiopure ligand and varioustransition-metal (II) perchlorates are effective catalysts for theDiels-Alder reaction with excellent enantioselectivity andextreme chiral amplification, and high tolerance to water,alcohols, amines, and acids: Kanemasa, S.; Oderaotoshi, Y.;Sakaguchi, S.; Yamamoto, H.; Tanaka, J.; Wada, E.; Curran,D.P. J. Am. Chem. Soc., 1998, 120, 3074.
[57] (a) Mikami, K.; Matsukawa, S. J. Am.Chem. Soc., 1993, 115,7039. (b) Shoda, H.; Nakamura, T.; Tanino, K.; Kuwajima, I.Tetrahedron Lett. 1993, 34, 6281.
[58] (a) Mikami, K.; Matsukawa, S. Nature, 1997, 385, 613. (b)Review on chiral ligand acceleration: Berrisford, D.J.; Bolm, C.;Sharpless, K.B. Angew. Chem., Int. Ed. Engl., 1995, 34, 1059.
[59] Mikami, K.; Matsukawa, S.; Volk, T.; Terada, M. Angew. Chem.,Int. Ed. Engl., 1997, 36, 2768.
[75] Evans, D.A.; Johnson, J.S.; Burgey, C.S.; Campos, K.R.Tetrahedron Lett., 1999, 40, 2879.

342 Current Organic Chemistry, 2000, Vol. 4, No. 3 Luiz Carlos Dias
[76] Yao, S.; Fang, X.; Jorgensen, K.A. Chem. Commun., 1998, 2547. [79] Elder, A.M.; Rich, D.H. Org. Lett., 1999, 1(9), 1443.
[77] In this same paper, the authors have observed that the use of bis-(oxazolines) and different Lewis acids leads only to moderateee’s.
[80] For a very exciting review paper about nonlinear effects inasymmetric synthesis, see: Girard, C.; Kagan, H.B. Angew.Chem., Int. Ed. Engl., 1998, 37, 2922.
[78] Drury, W.J., III.; Ferraris, D.; Cox, C.; Young, B.; Lectka, T. J.Am. Chem. Soc., 1998, 120, 11006.