cooperative single-atom active centers for attenuating
TRANSCRIPT
doi.org/10.26434/chemrxiv.14377016.v2
Cooperative Single-Atom Active Centers for Attenuating Linear ScalingEffect in Nitrogen Reduction ReactionKe Ye, Min Hu, Qin-Kun Li, Yi Luo, Jun Jiang, Guozhen Zhang
Submitted date: 07/04/2021 • Posted date: 08/04/2021Licence: CC BY-NC-ND 4.0Citation information: Ye, Ke; Hu, Min; Li, Qin-Kun; Luo, Yi; Jiang, Jun; Zhang, Guozhen (2021): CooperativeSingle-Atom Active Centers for Attenuating Linear Scaling Effect in Nitrogen Reduction Reaction. ChemRxiv.Preprint. https://doi.org/10.26434/chemrxiv.14377016.v2
We elucidate how the cooperation of two active centers can attenuate the linear scaling effect in NRR, throughthe first-principle study on 39 SACs comprised of two adjacent (~4 Å apart) four N-coordinated metal centers(MN4 duo) embedded in graphene.
File list (2)
download fileview on ChemRxivNRR_manuscript_new.pdf (1.28 MiB)
download fileview on ChemRxivNRR SI_new_v2.docx (1.97 MiB)

1
Cooperative single-atom active centers for attenuating linear scaling
effect in nitrogen reduction reaction
Ke Yea†, Min Hua†, Qin-Kun Lib, Yi Luoa, Jun Jianga, Guozhen Zhanga*
a Hefei National Laboratory for Physical Sciences at the Microscale, Chinese Academy of Sciences Center for Excellence in
Nanoscience, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui
230026, China.
b Department of Materials Science and NanoEngineering, Rice University, Houston, Texas, 77005, United States.
KEYWORDS: density-functional calculation; single-atom catalysis; scaling relations; nitrogen reduction reaction.
ABSTRACT: Cooperative effects of adjacent active centers are critical for single-atom catalysts (SACs) as active site density
matters. Yet how it affects scaling relationships in many important reactions like nitrogen reduction reaction (NRR) is underexplored.
Herein we elucidate how the cooperation of two active centers can attenuate the linear scaling effect in NRR, through the first-
principle study on 39 SACs comprised of two adjacent (~4 Å apart) four N-coordinated metal centers (MN4 duo) embedded in
graphene. Bridge-on adsorption of dinitrogen-containing species appreciably tilts the balance of adsorption of N2H and NH2 towards
N2H and thus substantially loosens the restraint of scaling relations in NRR, achieving low onset potential (V) and direct N≡N
cleavage (Mo, Re) at room temperature, respectively. The potential of MN4 duo in NRR casts new insight into circumventing
limitations of scaling relations in heterogeneous catalysis.
Heterogeneous catalysis lies at the heart of the chemical
industry.1 The ideal scenario for heterogeneous catalysis, as
illustrated by the Sabatier principle, seeks a delicate balance
between the activation of reactants and the desorption of
products.2 Yet for most reactions involving multiple
intermediates, scaling relationships between adsorption
strengths of different intermediates make such a balance
difficult to achieve, limiting optimization of catalysts.3
Breaking scaling relations is crucial for heterogeneous catalyst
design. Nørskov and co-workers have suggested using multiple
active centers to achieve it based on the first-principle
calculations4, which is supported by two recent experimental
studies.5-6 However, the ambiguity, and complexity of
structures of active centers in heterogeneous catalysis make it
difficult to gain a straightforward and precise understanding on
how catalysts could work around the restraint of scaling
relations posed to reactions. A simpler catalyst model with a
well-defined structure of the active center would be helpful to
decipher it.
The emerging single-atom catalysts (SACs) combining the
merits of both homogeneous and heterogeneous catalysts have
demonstrated excellent performances in a number of different
reactions.7-11 Because SACs possess simple and unified active
centers with well-defined composition and coordination
environments, they are deemed an ideal type of catalysts model
to provide mechanistic insight into heterogeneous catalysis.
Particularly, the advent of high-density SACs due to the rapid
advance of synthetic technologies12-18 and precise control of
inter-site distance19-21 would facilitate the study of cooperative
effects of multiple active centers. The synergetic effects
between adjacent active sites on heterogeneous catalysis have
drawn increasing attention in experimental work.5, 22-26 In
parallel, the computational approach plays an indispensable role
in dissecting mechanisms and structure-property relationships
associated with SACs.27 Previously, we have conducted a first-
principle study on cooperative communications between two
neighboring active centers, and impacts of active site density on
oxygen reduction reaction (ORR) and nitrogen reduction
reaction (NRR) respectively.28-30, which has prompted ourself
to continue the exploration of cooperative single-atom active
centers.
In the present work, we employed high-density SACs as
model systems to systematically investigate impacts of multiple
active centers on scaling relationships between adsorption
strengths of different reaction intermediates in electrocatalytic
nitrogen reduction reaction (eNRR).4, 31 eNRR has been inspired
by enzymatic catalysis of nitrogenase at mild conditions and
regarded as a promising solution of energy-saving nitrogen

2
fixation.31 Unfortunately, it is hampered by the challenge of
realizing optimal activation energy (Ea) of N2 and adsorption
energy (ΔEads) of nitrogen-containing intermediate species
simultaneously due to the scaling relations that couple favorable
(unfavorable) Ea and unfavorable (favorable) ΔEads together.
For eNRR, based on the Bell-Evans-Polanyi principle, the
strong coupling between adsorption strength of *NH2 and *N2H
(* denotes adsorbed species) is in the center of scaling relations
that undermine catalytic activities.4 To surpass this limitation,
it requires a decoupling mechanism to allow them to be
optimized independently.
Scheme 1. Various adsorption configurations of N2 on (a)
MN4/G and (b) MN4 duo/G.
Well-established eNRR mechanisms, however, resolve
around associative pathways (Figure S1)32 assuming single
active center, which adsorbs dinitrogen via either end-on or
side-on manners (Scheme 1). Meanwhile, alternative adsorption
mode of N2 via ensemble active centers33 has largely been
underexplored. Previously, we found an alternative reaction
path based on cooperative bridge-on adsorption of N2 (Scheme
1) by two closely arranged (~6 Å) Mo-N-C sites.29 This
discovery naturally raised the question that whether the bridge-
on manner might initiate a game-changing strategy of NRR.34 It
then motivated us to explore in this work how the scaling
relations evolve from single active center to two cooperative
active centers as the bridge-on adsorption pattern of N2 and
other nitrogen species are adopted.
Figure 1. Top view of MN4/G and MN4 duo/G models used in
this work, with the annotation of the philosophy of building
them.
A single metal center coordinated with four nitrogen atoms
(shorted as MN4) constitutes the essential moiety of many
excellent molecular catalysts derived from metal porphyrins
and metal phthalocyanines.35-37 The marriage of MN4 active
center and two-dimensional carbon support (shorted as MN4/C)
benefits from the ease of synthesis13-18, which has generated
quite a few promising SACs, including NRR catalysts38-43. As
shown in Figure 1, we placed two adjacent MN4 within
graphene (shorted as MN4 duo/G) as a prototype model of high-
density MN4 SACs to investigate how the cooperation of single-
atom centers will impact scaling relations in NRR. Note that our
MN4 duo model is different from other dual-atom M-N-C
models44-48 (Figure S2) in two aspects: (1) our model consists
of two well-separated MN4 centers while others either have a
metal-metal bond (essentially dimer) or share coordinating
atoms; (2) our model focuses on cooperation between two
individual active centers while others emphasize one active
center with two interconnecting metal atoms.
The results and discussion are organized into two parts. In the
first part, we surveyed the scaling relationship between
adsorption strength of *NH2 and *N2H in NRR occurring on 39
different MN4/G and MN4 duo/G (Figure S3), represented by
their respective Gibbs adsorption free energies (∆G*NH2 and
∆G*N2H), and showed how it appreciably shifts as the
adsorption mode of *N2 and *N2H varies from end-on to side-
on to bridge-on. Equivalently, the significant changes of scaling
relations for bridge-on mode were demonstrated in the form of
a favorable shift of the volcano plot. In the second part, under
the guidance of altered scaling relations, we investigated
several potential NRR catalysts, and achieved low onset
potential (V) and direct N≡N cleavage (Mo, Re) at room
temperature (energy barrier < 0.85 eV), respectively. Our
results demonstrate the importance of cooperation of
monodispersed active centers, which casts new insight into the
rational design of SACs for NRR at mild conditions.
RESULTS AND DISCUSSION
After building two different sets of MN4-type SACs: MN4/G
and MN4 duo/G (M=transition metals and main group IIIA-VA
metals), we first examined their stabilities in terms of
thermodynamic stability represented by formation energy (Ef)
and electrochemical stability represented by dissolution
potential (Udiss). The majority of them are found to be
thermodynamically stable (See Figure S4, Table S1, and
associated text in Supporting Information). Since we used the
atomic energy in bulk metal as the reference state of the metal,
the assignment of “unstable” in the sense of formation energy
does not mean it would decompose or undergo aggregation. We
expected a large energy barrier for metal migration from one
site to another will prevent them from clustering.29 Further, as
we explore the scaling relationship of adsorption of different
intermediates of NRR, these seemly “unstable” MN4 duo
models are all valuable data points that can help sketch the
whole picture we need.
All possible adsorption patterns of N2 as illustrated in
Scheme 1 were investigated for all MN4 and MN4 duo models.
The main group metals of interest could not effectively activate

3
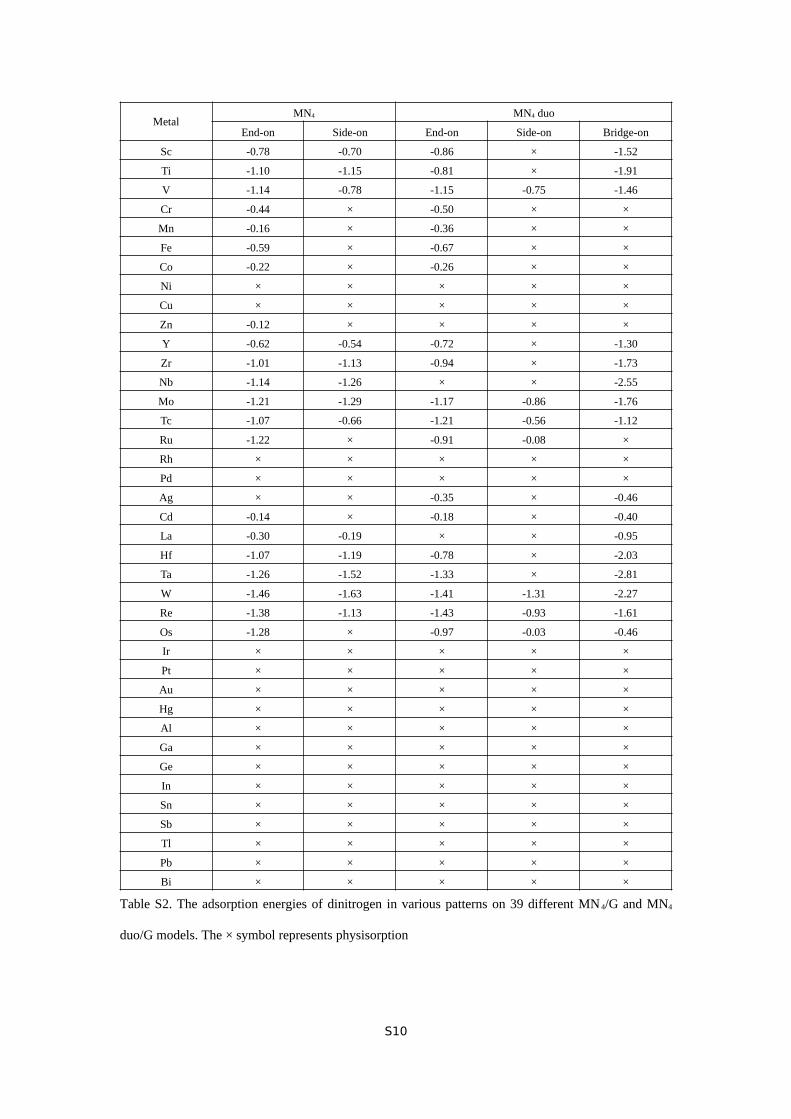
adsorbed N2, because they invoke physisorption or very weak
chemisorption. Transition metals develop five different
scenarios for the adsorption of N2 (Table S2). In the rest of
presentation, only MN4 and MN4 duo enabling substantial
chemisorption of N2 will be examined in the study of scaling
relations of NRR intermediates.
For associative NRR pathways, the first (*N2 + H+ + e- →
*N2H) and sixth (*NH2 + H+ + e- → *NH3) reductive
hydrogenation steps are the most likely potential-limiting
steps.34 Their respective free energy changes (∆G1 and ∆G6) are
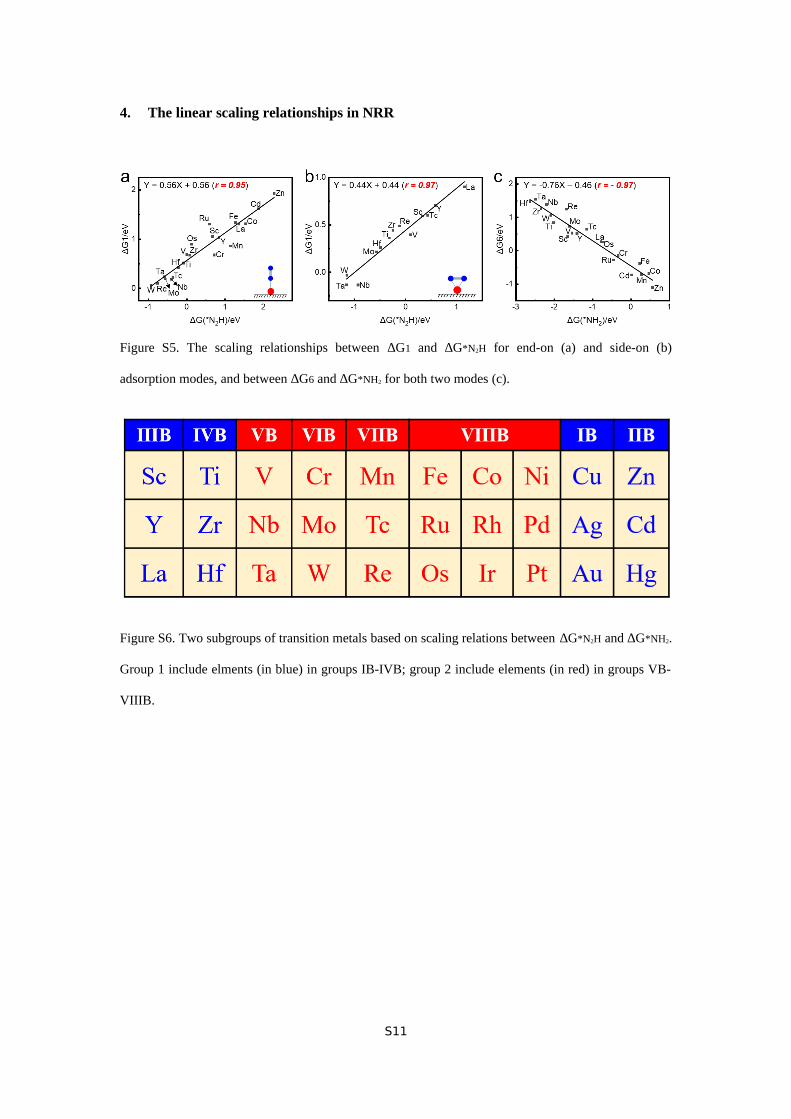
coupled through an inversely proportional relationship. Since
∆G1 and ∆G6 scale well with ∆G*N2H for better ∆G*NH2
respectively (Figure S5), the key scaling relations in NRR
eventually fall onto ∆G*N2H and ∆G*NH2.
Figure 2. (a) The scaling relationship between ∆G*NH2 and
∆G*NH2 for end-on adsorption mode on MN4/G. (b) The
differential charge density profiles of end-on *N2H and
*NH2 by VN4/G and YN4/G with an isosurface value of
5×10−3 e Å−3. Yellow and blue bubbles represent charge
accumulation and depletion, respectively. (c) Projected
crystal orbital Hamilton population (pCOHP) between the
metal centers (V and Y) and the nitrogen adatom. The values
of integrated COHP (ICOHP) are shown in red bold italics.
On MN4/G, as expected, the adsorption of NH2 scales well
with N2H in end-on mode (Figure 2a), in line with their scaling
relationship established on pure metals surfaces.4 The transition
metals can be divided into two groups based on two respective
fitting lines. Group 1 (labels in blue) includes the elements in
groups IB-IVB, while group 2 (labels in red) contains those in
groups VB-VIIIB. (Figure S6) Note that the balance between
adsorption of N2H and NH2 shifts towards N2H from group 1 to
group 2, which can be interpreted by the amount of charge
transfer from metal to nitrogen species (Figure 2b) based on
Bader charge profile and the bonding strength between them
(Figure 2c) using the integrated crystal orbital Hamilton
population (ICOHP)49-52 values (more negative value indicates
stronger bonding). Take V and Y for example, more negative
charges transfer from V to N2H than to NH2, while less from Y
to N2H than NH2; the M-N bonding interaction is appreciably
stronger in MN4-N2H than in MN4-NH2 for V, while it’s weaker
in the former than the latter for Y. The same distinction between
group 1 and 2 can be found on all types of adsorption mode on
both MN4/G and MN4 duo/G (Figure S7). Since preferable
enhancing the adsorption of N2H over NH2 is favorable to
mitigate the uphill energetics of activating the adsorbed N2 and
producing NH3 from NH2, we then focus on the study of
transition metals in group 2 in the rest of work.
From MN4/G to MN4 duo/G, the scaling relationships
between ∆G*N2H and ∆G*NH2 in end-on and side-on adsorption
modes do not change significantly (Figure 3), as only one metal
center bonds with adsorbates. However, bridge-on adsorption
of N2H on MN4 duo/G appreciably alters the landscape of
scaling relations (Figure 3a). As Figure 3a shows, bridge-on
mode appreciably reduces the slope and shifts up the intercept
of scaling line relative to end-on mode while side-on mode
reduces slope at the cost of shifting down the intercept. Note
that smaller slope and higher intercept values enable
strengthening adsorption of N2H much more than NH2, which
facilitates dinitrogen activation without impeding the final
reductive hydrogenation producing ammonia. Again, the
strengthening of N2H in bridge-on mode relative to end-on and
side-on is demonstrated using Bader charge and ICOHP profiles
(Figure 3b). Compared to end-on and side-on modes where only
one metal center bond with N2H, bridge-on mode has two
collaborative metal centers binding to N2H, which increases
electrons transferred to anti-bonding orbitals of N2H and overall
metal-nitrogen bonding strength.
In addition, ∆G1 barely scales with ∆G*N2H (Figure 3c) for
bridge-on mode, which is in sharp contrast to the well scaling
between the two in end-on and side-on modes. Here the internal
energy part plays a dominant role while the entropy factor has
negligible contribution (Table S3). The change of dependence
of ∆G1 on ∆G*N2H can be explained by the change of binding
strength from *N2 to *N2H. The integral of partial density of
states (pDOS) of nitrogen atoms from -3 eV to 0 eV (Fermi
level is reset to 0) is an indicator of effective number of
electrons involved in the bonding interaction between nitrogen
and metal atoms. Thus, the difference of this integral between
*N2 and *N2H roughly reflects the difference of their binding
strength to metal centers. Clearly, bridge-on mode has the
smallest difference while end-on and side-on bear comparable
changes (Figure 3d). As two cooperative metal centers
accommodate dinitrogen-containing species together, they not
only bind them stronger but also mitigate the uphill energetics
of first dinitrogen hydrogenation. Consequently, in bridge-on
associated NRR pathways, ∆G1 and ∆G6 are decoupled (Figure
S8), creating room for substantial improvement of overall
catalytic activity, as seen in the volcano plots of NRR occurring
on MN4 duo (Figure 4).
Based on the volcano plot associated with bridge-on
adsorption mode (Figure 4c), we have identified four selected
MN4 duo/G models (M = Mo, Re, V, Os) as potential NRR
catalysts. Although Tc appears to be the closest element to the
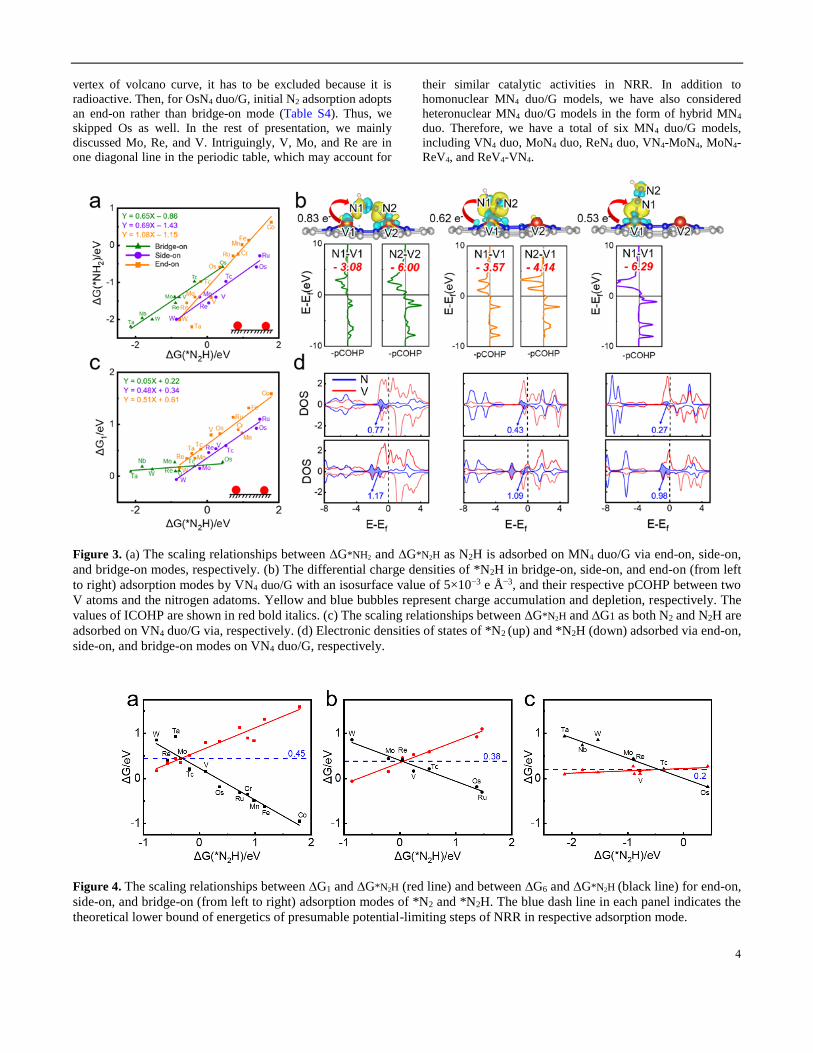
4
vertex of volcano curve, it has to be excluded because it is
radioactive. Then, for OsN4 duo/G, initial N2 adsorption adopts
an end-on rather than bridge-on mode (Table S4). Thus, we
skipped Os as well. In the rest of presentation, we mainly
discussed Mo, Re, and V. Intriguingly, V, Mo, and Re are in
one diagonal line in the periodic table, which may account for
their similar catalytic activities in NRR. In addition to
homonuclear MN4 duo/G models, we have also considered
heteronuclear MN4 duo/G models in the form of hybrid MN4
duo. Therefore, we have a total of six MN4 duo/G models,
including VN4 duo, MoN4 duo, ReN4 duo, VN4-MoN4, MoN4-
ReV4, and ReV4-VN4.
Figure 3. (a) The scaling relationships between ∆G*NH2 and ∆G*N2H as N2H is adsorbed on MN4 duo/G via end-on, side-on,
and bridge-on modes, respectively. (b) The differential charge densities of *N2H in bridge-on, side-on, and end-on (from left
to right) adsorption modes by VN4 duo/G with an isosurface value of 5×10−3 e Å−3, and their respective pCOHP between two
V atoms and the nitrogen adatoms. Yellow and blue bubbles represent charge accumulation and depletion, respectively. The
values of ICOHP are shown in red bold italics. (c) The scaling relationships between ∆G*N2H and ∆G1 as both N2 and N2H are
adsorbed on VN4 duo/G via, respectively. (d) Electronic densities of states of *N2 (up) and *N2H (down) adsorbed via end-on,
side-on, and bridge-on modes on VN4 duo/G, respectively.
Figure 4. The scaling relationships between ∆G1 and ∆G*N2H (red line) and between ∆G6 and ∆G*N2H (black line) for end-on,
side-on, and bridge-on (from left to right) adsorption modes of *N2 and *N2H. The blue dash line in each panel indicates the
theoretical lower bound of energetics of presumable potential-limiting steps of NRR in respective adsorption mode.

5
Using these models, we calculated the full NRR pathways
starting from bridge-on adsorption and obtained the onset
potential of NRR that signifies the activity of the catalyst of
interest. We first examined the adsorption energies of bridge-
on adsorbed N2, associated charge redistribution, and geometric
changes of reactant-active center complexes. As shown in Table
S5, the computed binding energies are between -1.46 and -1.82
eV for bridge-on adsorption, accompanied by substantial charge
transfer (0.59 ~ 0.93 |e| by Bader charge analysis) from both two
MN4 sites to N2. The corresponding charge distribution is also
demonstrated by the differential charge densities of N2-MN4
duo binding complexes (Figure S9), which illustrates electron
gain in the antibonding orbital of N2. As a result, the adsorbed
N2 is activated, with an appreciable stretching of N-N bond
from 1.11 Å (free N2 in the gas phase) to 1.18 ~ 1.23 Å.
Figure 5. The reaction profile of the bridge-on adsorption initiated0020030NRR pathway on VN4 duo/G model. Roman
numbers denote all reaction species along the pathway.
Then we investigated the N≡N bond dissociation following
the bridge-on adsorption. The transition state structures are
showing in Figure S10. Remarkably, 3 out of 6 selected models,
including MoN4 duo, ReN4 duo, and MoN4-ReN4, enable direct
N≡N bond breaking with a reaction barrier below 0.91 eV
(Table S6), a well-documented threshold for chemical reactions
available at room temperature.53 This unusual phenomenon
suggests a dissociative NRR mechanism, in sharp contrast to
common N≡N bond dissociation aided by reductive
hydrogenation seen in associative NRR pathways. Intriguingly,
this pathway will avoid the formation of *N2H, one of two key
intermediate species in NRR, suggesting the possibility of
circumventing the well-established scaling relationship for
NRR by direct N≡N bond dissociation through the joint effort
of two adjacent active centers.
To understand it, we adopted the N-N distance as the reaction
coordinate to understand the trend of energy barriers of N-N
dissociation. Intriguingly, the barriers are positively correlated
with the distances (Figure S11-a), because a smaller difference
of N-N distance between the transition state (TS) and initial
state will result in a smaller barrier for N-N bond dissociation.
This is typical for an early TS, whose structure is close to the
corresponding initial state. Then we examined the relationship
between the stabilities of all six models and the corresponding
energy barrier of TS and found that the lower stability of the
catalyst results in a smaller energy barrier (Figure S11-b). Since
the ReN4 duo is the least stable one among these six models, it
has the lowest direct N2 dissociation barrier. Similarly, MoN4
duo and MoN4-ReN4 enables direct N2 dissociation at room
temperature for their relative lability. The direct N≡N breaking
aided by bridge-on adsorption may open a new possibility of
breaking scaling relationships applied on NRR, which will be
investigated in the future. In contrast, as the stabilities increase,
which are the cases of VN4 duo, VN4-MoN4, and VN4-ReN4,
the dissociative pathway is unfavorable at room temperature;
thus, NRR will proceed via associative mechanisms in which
N≡N breaking takes place after one or two steps of reductive
hydrogenation. Their bridge-on adsorption initiated NRR
pathways is similar to the one occurring on MoN3 duo

6
embedded in graphene.29 The balance between stability and
lability of these catalysts may be a key factor for determining
the preferred reaction pathways of NRR occurring on them.
The full reaction coordinates of NRR with all six of these
models are collected in Figure S12 through Figure S17. Based
on the Gibbs free energy data therein, we found that VN4 duo/G
possesses the lowest limiting potential (0.18 V) and thus bears
the highest theoretical activity. The reaction coordinates are
shown in Figure 5. For the strong adsorption, the ΔG*N2→*N2H
for bridge-on *N2 is 0.11eV and the N−N distance is stretched
to 1.31Å. The second reductive hydrogenation (*NNH →
*HNNH) is endothermic (ΔG*N2H→*HNNH = 0.16 eV), with the
N−N distance of 1.37 Å. Intriguingly, this bridge-on *HNNH
resembles the bridging “diazene” species found in
nitrogenase.54-55 Then *HNNH dissociates and produces two
*NH, each of which is attached to one V atom. The energy
barrier (0.78 eV) for this N≡N bond breaking can be easily
overcome at room temperature. Then the two *NH alternately
undergo reductive hydrogenation steps, producing two *NH2.
Both reductive hydrogenation of *NH2 and desorption of NH3
from *NH3 are thermodynamically uphill. The electrochemical
step can be promoted only by applying an external potential. At
U= −0.17 V, ΔG*NH2→*NH3 becomes zero, and the uphill
energetics is substantially mitigated (Figure 5). This remarkably
low limiting potential benefits from the cooperation of two
adjacent VN4 sites that help breaking the scaling relationship of
NRR by properly balancing the adsorption energies of *N2H
and *NH2. For the endergonic (∼0.7 eV) desorption of
ammonia, we expect that the solvation of ammonia can mitigate
the unfavorable energetics because it will stabilize the desorbed
ammonia.4, 39, 43, 56-57 Thus, NH3 desorption may not be a main
obstacle in NRR and, hence, is not considered in detail here.
CONCLUSION
In this work, we have conducted a systematic first-principle
study on impacts of bridge-on N2 adsorption by two adjacent
MN4 sites on scaling relationships within electrocatalytic NRR
and found the key to attenuate the scaling relations is to create
inhomogeneous adsorption between dinitrogen-containing
species and nitrogen-containing species.
We confirmed that the scaling relationship between
adsorption of N2H and NH2 hold for NRR occurring on single
active center, regardless of MN4 or MN4 duo. Since the room of
improvement of NRR activity associated with one active center
is limited, we have to go beyond conventional end-on and side-
on adsorption modes. Intriguingly, the side-on fashion benefits
from different metal-nitrogen bonding patterns for *N2H and
*NH2, hinting a path for the evolution of NRR catalysts.43-44, 58
The bridge-on adsorption mode can be viewed as a two-center
version of “side-on”. It further widens the gap of metal-nitrogen
bonding strengths between *N2H and *NH2, enabling us to
decouple the optimization of (*N2 + e- + H+ → *N2H) and
(*NH2 + e- + H+ → *NH3). Thus, it shows the potential of
making substantial progress towards breaking scaling relations
in NRR. Importantly, the bridge-on pattern relies on the
proximity of two metal active sites, which could well explain
other reported promising NRR catalytic systems invoking
bridge-on like activation of N2 by multiple active centers.59-61
Based on the survey, we then identified VN4 duo and ReN4
duo as promising NRR catalysts through associative
mechanisms and dissociative mechanisms, respectively. For
VN4 duo, the remarkably low onset potential (0.18 eV) indicates
the level of excellent activity of NRR that can be achieved by
high-density SACs. For ReN4 duo, the most appealing feature
is the low energy barrier (0.52 eV) of direct N≡N bond
dissociation enabling a dissociative mechanism for NRR at
room temperature, which for sure will inspire more mechanistic
study on dissociative pathways with multiple concerted active
centers. We also expect it motivates more exploration on how
the delicate balance between stability and lability of active
centers would affect the dynamics of N2 activation process.
In summary, through a comprehensive understanding of
scaling relationships for NRR in MN4-type single-atom
catalysts, we found cooperation of two adjacent single-atom
active centers can attenuate scaling relation effect and facilitate
nitrogen fixation. Although our computational models are based
on ideal reaction conditions, we expect our work casting new
light on NRR mechanisms with collective active centers will be
helpful for the rational design of high-density SACs and
prompting mechanistic study on breaking scaling relationships
existing in other reactions (e.g. CO2 reduction62 and oxygen
reduction reaction) aided by cooperative multiple active
centers. We will also continue the investigation of cooperative
single-atom active centers under more realistic reaction
conditions in the future work.
COMPUTATIONAL DETAILS
All spin-polarized density functional theory (DFT)
calculations were performed using the Perdew-Burke-
Ernzerhof (PBE)63 functional in conjunction with plane-wave
projected augmented wave (PAW)64 method as implemented in
Vienna ab initio simulation program (VASP)65-66. The kinetic
cutoff energy for the plane-wave basis set was set to be 480 eV.
The Gaussian smearing method was adopted with a width of 0.1
eV to describe partial occupancies of each orbital. The first
Brillouin zone was sampled by a Monkhorst–Pack scheme with
a 3 × 3 × 1 k-point grid. To avoid the interaction between two
periodic units, a vacuum space exceeds 15 Å was employed.
Structures were fully relaxed until the forces were converged to
less than 0.02 eV Å-1. The Grimme’s D3 dispersion correction
scheme was used to describe the van der Waals interaction. The
solvent effect on scaling relations are not taken into account as
a previous study showed that no significant change that can alter
the scaling relations has been found as solvent effect was
incorporated.67 More details regarding adopted computational
models, calculation of various energy values and selection of
energetic descriptors are given in the Supporting Information.
ASSOCIATED CONTENT
Supporting Information

7
Computational detail, geometrical structures and stability
validation of MN4 and MN4 duo models, the adsorption energies of
N2, differential charge densities, various scaling relationships and
Gibbs free energy diagrams for NRR occurring on selected MN4
duo models.
AUTHOR INFORMATION
Corresponding Author
*Guozhen Zhang, Email: [email protected]
ORCID
Guozhen Zhang: 0000-0003-0125-9666
Jun Jiang: 0000-0002-6116-5605
Author Contributions
†K.Y. and M.H. contributed equally to this work.
Note
The authors declare no competing financial support.
ACKNOWLEDGMENTS
This work was financially supported by the Ministry of Science and
Technology of the People’s Republi0063 of China (No.
2018YFA0208702, 2018YFA0208603, and 2017YFA0303500)
and the National Natural Science Foundation of China (21703221,
21790351, and 21633006). The Supercomputing Center of
University of Science and Technology of China is acknowledged
for the computing resource.
REFERENCES
(1) Nørskov, J. K.; Studt, F.; Abild-Pedersen, F.; Bligaard, T.
Fundamental Concepts in Heterogeneous Catalysis; John Wiley & Sons,
Inc., 2014.
(2) Medford, A. J.; Vojvodic, A.; Hummelshøj, J. S.; Voss, J.; Abild-
Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J. K. From the
Sabatier Principle to a Predictive Theory of Transition-Metal
Heterogeneous Catalysis. J. Catal. 2015, 328, 36-42.
(3) Zhao, Z.-J.; Liu, S.; Zha, S.; Cheng, D.; Studt, F.; Henkelman, G.; Gong,
J. Theory-Guided Design of Catalytic Materials Using Scaling
Relationships and Reactivity Descriptors. Nat. Rev. Mater. 2019, 4, 792-804.
(4) Montoya, J. H.; Tsai, C.; Vojvodic, A.; Norskov, J. K. The Challenge
of Electrochemical Ammonia Synthesis: A New Perspective on the Role of
Nitrogen Scaling Relations. Chemsuschem 2015, 8, 2180-6.
(5) Wang, P.; Chang, F.; Gao, W.; Guo, J.; Wu, G.; He, T.; Chen, P.
Breaking Scaling Relations to Achieve Low-Temperature Ammonia
Synthesis through Lih-Mediated Nitrogen Transfer and Hydrogenation. Nat.
Chem. 2017, 9, 64-70.
(6) Mao, C.; Wang, J.; Zou, Y.; Qi, G.; Yang Loh, J. Y.; Zhang, T.; Xia, M.;
Xu, J.; Deng, F.; Ghoussoub, M.; Kherani, N. P.; Wang, L.; Shang, H.; Li,
M.; Li, J.; Liu, X.; Ai, Z.; Ozin, G. A.; Zhao, J.; Zhang, L. Hydrogen
Spillover to Oxygen Vacancy of TiO2-XHy/Fe: Breaking the Scaling
Relationship of Ammonia Synthesis. J. Am. Chem. Soc. 2020, 142, 17403-
17412.
(7) Qiao, B.; Wang, A.; Yang, X.; Allard, L. F.; Jiang, Z.; Cui, Y.; Liu, J.;
Li, J.; Zhang, T. Single-Atom Catalysis of Co Oxidation Using Pt1/FeOx.
Nat. Chem. 2011, 3, 634-41.
(8) Cui, X.; Li, W.; Ryabchuk, P.; Junge, K.; Beller, M. Bridging
Homogeneous and Heterogeneous Catalysis by Heterogeneous Single-
Metal-Site Catalysts. Nat. Catal. 2018, 1, 385-397.
(9) Wang, A.; Li, J.; Zhang, T. Heterogeneous Single-Atom Catalysis. Nat.
Rev. Chem. 2018, 2, 65-81.
(10) Mitchell, S.; Perez-Ramirez, J. Single Atom Catalysis: A Decade of
Stunning Progress and the Promise for a Bright Future. Nat. Commun. 2020,
11, 4302.
(11) Zhuo, H. Y.; Zhang, X.; Liang, J. X.; Yu, Q.; Xiao, H.; Li, J.
Theoretical Understandings of Graphene-Based Metal Single-Atom
Catalysts: Stability and Catalytic Performance. Chem. Rev. 2020, 120,
12315-12341.
(12) Wan, X.; Liu, X.; Li, Y.; Yu, R.; Zheng, L.; Yan, W.; Wang, H.; Xu,
M.; Shui, J. Fe–N–C Electrocatalyst with Dense Active Sites and Efficient
Mass Transport for High-Performance Proton Exchange Membrane Fuel
Cells. Nat. Catal. 2019, 2, 259-268.
(13) Yin, P.; Yao, T.; Wu, Y.; Zheng, L.; Lin, Y.; Liu, W.; Ju, H.; Zhu, J.;
Hong, X.; Deng, Z.; Zhou, G.; Wei, S.; Li, Y. Single Cobalt Atoms with
Precise N-Coordination as Superior Oxygen Reduction Reaction Catalysts.
Angew. Chem. Int. Ed. 2016, 55, 10800-5.
(14) Chen, Y.; Ji, S.; Wang, Y.; Dong, J.; Chen, W.; Li, Z.; Shen, R.; Zheng,
L.; Zhuang, Z.; Wang, D.; Li, Y. Isolated Single Iron Atoms Anchored on
N-Doped Porous Carbon as an Efficient Electrocatalyst for the Oxygen
Reduction Reaction. Angew. Chem. Int. Ed. 2017, 56, 6937-6941.
(15) Wu, J.; Zhou, H.; Li, Q.; Chen, M.; Wan, J.; Zhang, N.; Xiong, L.; Li,
S.; Xia, B. Y.; Feng, G. Densely Populated Isolated Single Co-N Site for
Efficient Oxygen Electrocatalysis. Adv. Energy Mater. 2019, 9, 1900149.
(16) Bauer, G.; Ongari, D.; Tiana, D.; Gaumann, P.; Rohrbach, T.; Pareras,
G.; Tarik, M.; Smit, B.; Ranocchiari, M. Metal-Organic Frameworks as
Kinetic Modulators for Branched Selectivity in Hydroformylation. Nat.
Commun. 2020, 11, 1059.
(17) Liu, K.; Zhao, X.; Ren, G.; Yang, T.; Ren, Y.; Lee, A. F.; Su, Y.; Pan,
X.; Zhang, J.; Chen, Z.; Yang, J.; Liu, X.; Zhou, T.; Xi, W.; Luo, J.; Zeng,
C.; Matsumoto, H.; Liu, W.; Jiang, Q.; Wilson, K.; Wang, A.; Qiao, B.; Li,
W.; Zhang, T. Strong Metal-Support Interaction Promoted Scalable
Production of Thermally Stable Single-Atom Catalysts. Nat. Commun.
2020, 11, 1263.
(18) Xiong, Y.; Sun, W.; Xin, P.; Chen, W.; Zheng, X.; Yan, W.; Zheng, L.;

8
Dong, J.; Zhang, J.; Wang, D.; Li, Y. Gram-Scale Synthesis of High-
Loading Single-Atomic-Site Fe Catalysts for Effective Epoxidation of
Styrene. Adv. Mater. 2020, e2000896.
(19) Wu, J.; Xiong, L.; Zhao, B.; Liu, M.; Huang, L. Densely Populated
Single Atom Catalysts. Small Methods 2019, 4, 1900540.
(20) He, Z.; He, K.; Robertson, A. W.; Kirkland, A. I.; Kim, D.; Ihm, J.;
Yoon, E.; Lee, G. D.; Warner, J. H. Atomic Structure and Dynamics of
Metal Dopant Pairs in Graphene. Nano Lett. 2014, 14, 3766-72.
(21) Lin, Y. C.; Teng, P. Y.; Yeh, C. H.; Koshino, M.; Chiu, P. W.; Suenaga,
K. Structural and Chemical Dynamics of Pyridinic-Nitrogen Defects in
Graphene. Nano Lett. 2015, 15, 7408-13.
(22) Li, H.; Wang, L.; Dai, Y.; Pu, Z.; Lao, Z.; Chen, Y.; Wang, M.; Zheng,
X.; Zhu, J.; Zhang, W.; Si, R.; Ma, C.; Zeng, J. Synergetic Interaction
between Neighbouring Platinum Monomers in CO2 Hydrogenation. Nat.
Nanotechnol. 2018, 13, 411-417.
(23) Zou, N.; Zhou, X.; Chen, G.; Andoy, N. M.; Jung, W.; Liu, G.; Chen,
P. Cooperative Communication within and between Single Nanocatalysts.
Nat. Chem. 2018, 10, 607-614.
(24) Jiao, J.; Lin, R.; Liu, S.; Cheong, W. C.; Zhang, C.; Chen, Z.; Pan, Y.;
Tang, J.; Wu, K.; Hung, S. F.; Chen, H. M.; Zheng, L.; Lu, Q.; Yang, X.; Xu,
B.; Xiao, H.; Li, J.; Wang, D.; Peng, Q.; Chen, C.; Li, Y. Copper Atom-Pair
Catalyst Anchored on Alloy Nanowires for Selective and Efficient
Electrochemical Reduction of CO2. Nat. Chem. 2019, 11, 222-228.
(25) Fu, J.; Dong, J.; Si, R.; Sun, K.; Zhang, J.; Li, M.; Yu, N.; Zhang, B.;
Humphrey, M. G.; Fu, Q.; Huang, J. Synergistic Effects for Enhanced
Catalysis in a Dual Single-Atom Catalyst. ACS Catal. 2021, 11, 1952-1961.
(26) Tang, Y.; Wei, Y.; Wang, Z.; Zhang, S.; Li, Y.; Nguyen, L.; Li, Y.;
Zhou, Y.; Shen, W.; Tao, F. F.; Hu, P. Synergy of Single-Atom Ni1 and Ru1
Sites on Ceo2 for Dry Reforming of Ch4. J. Am. Chem. Soc. 2019, 141,
7283-7293.
(27) Zhang, X.; Chen, A.; Chen, L.; Zhou, Z. 2d Materials Bridging
Experiments and Computations for Electro/Photocatalysis. Adv. Energy
Mater. 2021.
(28) Li, Q.-K.; Li, X.-F.; Zhang, G.; Jiang, J. Cooperative Spin Transition
of Monodispersed FeN3 Sites within Graphene Induced by CO Adsorption.
J. Am. Chem. Soc. 2018, 140, 15149-15152.
(29) Ye, K.; Hu, M.; Li, Q. K.; Han, Y.; Luo, Y.; Jiang, J.; Zhang, G.
Cooperative Nitrogen Activation and Ammonia Synthesis on Densely
Monodispersed Mo-N-C Sites. J. Phys. Chem. Lett. 2020, 11, 3962-3968.
(30) Han, Y.; Li, Q. K.; Ye, K.; Luo, Y.; Jiang, J.; Zhang, G. Impact of
Active Site Density on Oxygen Reduction Reactions Using Monodispersed
Fe-N-C Single-Atom Catalysts. ACS Appl. Mater. Interfaces 2020, 12,
15271-15278.
(31) Chen, J. G.; Crooks, R. M.; Seefeldt, L. C.; Bren, K. L.; Bullock, R.
M.; Darensbourg, M. Y.; Holland, P. L.; Hoffman, B.; Janik, M. J.; Jones,
A. K.; Kanatzidis, M. G.; King, P.; Lancaster, K. M.; Lymar, S. V.; Pfromm,
P.; Schneider, W. F.; Schrock, R. R. Beyond Fossil Fuel-Driven Nitrogen
Transformations. Science 2018, 360.
(32) Honkala, K.; Hellman, A.; Remediakis, I.; Logadottir, A.; Carlsson,
A.; Dahl, S.; Christensen, C. H.; Nørskov, J. K. Ammonia Synthesis from
First-Principles Calculations. Science 2005, 307, 555-558.
(33) Jeong, H.; Lee, G.; Kim, B. S.; Bae, J.; Han, J. W.; Lee, H. Fully
Dispersed Rh Ensemble Catalyst to Enhance Low-Temperature Activity. J.
Am. Chem. Soc. 2018, 140, 9558-9565.
(34) Qing, G.; Ghazfar, R.; Jackowski, S. T.; Habibzadeh, F.; Ashtiani, M.
M.; Chen, C. P.; Smith, M. R., 3rd; Hamann, T. W. Recent Advances and
Challenges of Electrocatalytic N2 Reduction to Ammonia. Chem. Rev. 2020,
120, 5437-5516.
(35) Wang, Y.; Yuan, H.; Li, Y.; Chen, Z. Two-Dimensional Iron-
Phthalocyanine (Fe-Pc) Monolayer as a Promising Single-Atom-Catalyst
for Oxygen Reduction Reaction: A Computational Study. Nanoscale 2015,
7, 11633-41.
(36) Melville, O. A.; Lessard, B. H.; Bender, T. P. Phthalocyanine-Based
Organic Thin-Film Transistors: A Review of Recent Advances. ACS Appl.
Mater. Interfaces 2015, 7, 13105-18.
(37) Maitarad, P.; Namuangruk, S.; Zhang, D.; Shi, L.; Li, H.; Huang, L.;
Boekfa, B.; Ehara, M. Metal-Porphyrin: A Potential Catalyst for Direct
Decomposition of N2O by Theoretical Reaction Mechanism Investigation.
Environ. Sci. Technol. 2014, 48, 7101-10.
(38) Li, X. F.; Li, Q. K.; Cheng, J.; Liu, L.; Yan, Q.; Wu, Y.; Zhang, X. H.;
Wang, Z. Y.; Qiu, Q.; Luo, Y. Conversion of Dinitrogen to Ammonia by
Fen3-Embedded Graphene. J. Am. Chem. Soc. 2016, 138, 8706-9.
(39) Zhao, J.; Chen, Z. Single Mo Atom Supported on Defective Boron
Nitride Monolayer as an Efficient Electrocatalyst for Nitrogen Fixation: A
Computational Study. J. Am. Chem. Soc. 2017, 139, 12480-12487.
(40) Han, L.; Liu, X.; Chen, J.; Lin, R.; Liu, H.; Lu, F.; Bak, S.; Liang, Z.;
Zhao, S.; Stavitski, E.; Luo, J.; Adzic, R. R.; Xin, H. L. Atomically
Dispersed Molybdenum Catalysts for Efficient Ambient Nitrogen Fixation.
Angew. Chem. Int. Ed. 2019, 58, 2321-2325.
(41) Tao, H.; Choi, C.; Ding, L.-X.; Jiang, Z.; Han, Z.; Jia, M.; Fan, Q.;
Gao, Y.; Wang, H.; Robertson, A. W.; Hong, S.; Jung, Y.; Liu, S.; Sun, Z.
Nitrogen Fixation by Ru Single-Atom Electrocatalytic Reduction. Chem
2019, 5, 204-214.
(42) Zhao, W.; Zhang, L.; Luo, Q.; Hu, Z.; Zhang, W.; Smith, S.; Yang, J.
Single Mo1(Cr1) Atom on Nitrogen-Doped Graphene Enables Highly
Selective Electroreduction of Nitrogen into Ammonia. ACS Catal. 2019, 9,
3419-3425.
(43) Choi, C.; Back, S.; Kim, N.-Y.; Lim, J.; Kim, Y.-H.; Jung, Y.
Suppression of Hydrogen Evolution Reaction in Electrochemical N2
Reduction Using Single-Atom Catalysts: A Computational Guideline. ACS
Catal. 2018, 8, 7517-7525.
(44) Guo, X.; Gu, J.; Lin, S.; Zhang, S.; Chen, Z.; Huang, S. Tackling the
Activity and Selectivity Challenges of Electrocatalysts toward the Nitrogen
Reduction Reaction Via Atomically Dispersed Biatom Catalysts. J. Am.

9
Chem. Soc. 2020, 142, 5709-5721.
(45) Wang, X.; Qiu, S.; Feng, J.; Tong, Y.; Zhou, F.; Li, Q.; Song, L.; Chen,
S.; Wu, K. H.; Su, P.; Ye, S.; Hou, F.; Dou, S. X.; Liu, H. K.; Max Lu, G.
Q.; Sun, C.; Liu, J.; Liang, J. Confined Fe-Cu Clusters as Sub-Nanometer
Reactors for Efficiently Regulating the Electrochemical Nitrogen
Reduction Reaction. Adv. Mater. 2020, 32, e2004382.
(46) He, T.; Puente Santiago, A. R.; Du, A. Atomically Embedded
Asymmetrical Dual-Metal Dimers on N-Doped Graphene for Ultra-
Efficient Nitrogen Reduction Reaction. J. Catal. 2020, 388, 77-83.
(47) Deng, T.; Cen, C.; Shen, H.; Wang, S.; Guo, J.; Cai, S.; Deng, M.
Atom-Pair Catalysts Supported by N-Doped Graphene for the Nitrogen
Reduction Reaction: D-Band Center-Based Descriptor. J. Phys. Chem. Lett.
2020, 11, 6320-6329.
(48) Lv, X.; Wei, W.; Huang, B.; Dai, Y.; Frauenheim, T. High-Throughput
Screening of Synergistic Transition Metal Dual-Atom Catalysts for
Efficient Nitrogen Fixation. Nano Lett. 2021, 21, 1871-1878.
(49) Deringer, V. L.; Tchougreeff, A. L.; Dronskowski, R. Crystal Orbital
Hamilton Population (COHP) Analysis as Projected from Plane-Wave
Basis Sets. J. Phys. Chem. A 2011, 115, 5461-6.
(50) Maintz, S.; Deringer, V. L.; Tchougreeff, A. L.; Dronskowski, R.
Analytic Projection from Plane-Wave and Paw Wavefunctions and
Application to Chemical-Bonding Analysis in Solids. J. Comput. Chem.
2013, 34, 2557-67.
(51) Maintz, S.; Deringer, V. L.; Tchougreeff, A. L.; Dronskowski, R.
Lobster: A Tool to Extract Chemical Bonding from Plane-Wave Based Dft.
J. Comput. Chem. 2016, 37, 1030-5.
(52) Nelson, R.; Ertural, C.; George, J.; Deringer, V. L.; Hautier, G.;
Dronskowski, R. Lobster: Local Orbital Projections, Atomic Charges, and
Chemical-Bonding Analysis from Projector-Augmented-Wave-Based
Density-Functional Theory. J. Comput. Chem. 2020, 41, 1931-1940.
(53) Young, D. C., A Practical Guide for Applying Techniques to Real
World Problems, John Wiley & Sons, Inc., New York, 1st edn, 2001.
(54) Saouma, C. T.; Kinney, R. A.; Hoffman, B. M.; Peters, J. C.,
Transformation of an [Fe(η2-N2H3)]+ Species to Pi-Delocalized [Fe2(μ-
N2H2)]2+/+ complexes. Angew. Chem. Int. Ed. 2011, 50, 3446-9.
(55) Hoffman, B. M.; Lukoyanov, D.; Yang, Z. Y.; Dean, D. R.; Seefeldt,
L. C. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage.
Chem. Rev. 2014, 114, 4041-62.
(56) Peterson, A. A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov,
J. K. How Copper Catalyzes the Electroreduction of Carbon Dioxide into
Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311-1315.
(57) Skulason, E.; Bligaard, T.; Gudmundsdóttir, S.; Studt, F.; Rossmeisl,
J.; Abild-Pedersen, F.; Vegge, T.; Jonsson, H.; Nørskov, J. K. A Theoretical
Evaluation of Possible Transition Metal Electro-Catalysts for N2 Reduction.
Phys. Chem. Chem. Phys. 2012, 14, 1235-1245.
(58) Ling, C.; Ouyang, Y.; Li, Q.; Bai, X.; Mao, X.; Du, A.; Wang, J. A
General Two‐Step Strategy–Based High‐Throughput Screening of Single
Atom Catalysts for Nitrogen Fixation. Small Methods 2018, 3, 1800376.
(59) Wang, S.; Shi, L.; Bai, X.; Li, Q.; Ling, C.; Wang, J. Highly Efficient
Photo-/Electrocatalytic Reduction of Nitrogen into Ammonia by Dual-
Metal Sites. ACS Cent. Sci. 2020, 6, 1762-1771.
(60) Qiu, W.; Xie, X. Y.; Qiu, J.; Fang, W. H.; Liang, R.; Ren, X.; Ji, X.;
Cui, G.; Asiri, A. M.; Cui, G.; Tang, B.; Sun, X. High-Performance
Artificial Nitrogen Fixation at Ambient Conditions Using a Metal-Free
Electrocatalyst. Nat. Commun. 2018, 9, 3485.
(61) Ma, X. L.; Liu, J. C.; Xiao, H.; Li, J. Surface Single-Cluster Catalyst
for N2-to-NH3 Thermal Conversion. J. Am. Chem. Soc. 2018, 140, 46-49.
(62) Ouyang, Y.; Shi, L.; Bai, X.; Li, Q.; Wang, J. Breaking Scaling
Relations for Efficient CO2 Electrochemical Reduction through Dual-Atom
Catalysts. Chem. Sci. 2020, 11, 1807-1813.
(63) Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient
Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865.
(64) Blöchl, P. E. Projector Augmented-Wave Method. Phys. Rev. B 1994,
50, 17953.
(65) Furthmüller, G. K. J., Efficient Iterative Schemes for Ab Initio Total-
Energy Calculations Using a Plane-Wave Basis Set. Phy. Rev. B 1996, 54,
11169-11186.
(66) Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the
Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758-1775.
(67) Park, J.; Roling, L. T. Elucidating Energy Scaling between Atomic
and Molecular Adsorbates in the Presence of Solvent. AIChE Journal 2020,
66:e17036.
TOC graphics

10

download fileview on ChemRxivNRR_manuscript_new.pdf (1.28 MiB)

Supporting Information
Cooperative single-atom active centers for attenuating linear
scaling effect in nitrogen reduction reaction
Ke Ye[a]†, Min Hu[a]†, Qin-Kun Li[b], Yi Luo[a], Jun Jiang[a], Guozhen Zhang[a]*
a. Hefei National Laboratory for Physical Sciences at the Microscale, Chinese Academy of Sciences Center for Excellence in
Nanoscience, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026,
China
b.Department of Materials Science and NanoEngineering, Rice University, Houston, Texas, 77005, United States
Email: [email protected]
S1
CONTENT
1. Computation detail.........................................................................................4
2. The descriptors used for NRR.........................................................................4
Figure S1. Three main pathways for associative mechanisms of NRR.........4
3. The definition and stability test of MN4/G and MN4 duo/G models.................5
Figure S2. Representation of various dual-atom M-N-C models...................5
Figure S3. Representation of (a) MN4/G and (b) MN4 duo/G models and (c)
the scope of metals investigated in this work.............................................6
Table S1. Atomic radius(R) of 39 metal elements, computed formation
energy(Ef), computed dissolution potential(Udiss), and the distance from
metal atom to graphene layer(d) of metal atoms in all 39 different MN4/G
models and MN4 duo/G models....................................................................8
Figure S4. Computed formation energy(Ef) and dissolution potential(Udiss)
of metal atoms in all 39 different (a) MN4/G models and (b) MN4 duo/G
models, respectively....................................................................................9
Table S2. The adsorption energies of dinitrogen in various patterns on 39
different MN4/G and MN4 duo/G models. The × symbol represents
physisorption.............................................................................................10
4. The linear scaling relationships in NRR........................................................11
Figure S5. The scaling relationships between ∆G1 and ∆G*N2H for end-on
(a) and side-on (b) adsorption modes, and between ∆G6 and ∆G*NH2 for
both two modes (c)....................................................................................11
Figure S6. Two subgroups of transition metals based on scaling relations
between ∆G*N2H and ∆G*NH2. Group 1 include elments (in blue) in groups
IB-IVB; group 2 include elements (in red) in groups VB-VIIIB.....................11
Figure S7. The scaling relationships between ∆G*N2H and ∆G*NH2 for (a)
end-on adsorption mode on MN4/G; (b) side-on adsorption mode on MN4/G;
(c) end-on adsorption mode on MN4 duo/G; (d) side-on adsorption mode
on MN4 duo/G; (e) bridge-on adsorption mode on MN4 duo/G. Blue and red
dots represent metal atoms from subgroup 1 and subgroup 2,
respectively................................................................................................12
Table S3. The entropy factor (TS) of *N2 and *N2H on MN4 duo/G via bridge-
on mode.....................................................................................................12
Figure S8. The relationship between ∆G1 and ∆G6 for bridge-on adsorption
initiated NRR occurring on MN4 duo/G.......................................................13
5. The catalytic activity related to the bridge-on adsorption model................13
Table S4. The adsorption energies of dinitrogen adsorbed on six selected
MN4 duo/G via end-on, side-on, and bridge-on manners...........................13
Table S5. The computed binding energies(ΔE*N2) of bridge-on manner on
six selected MN4 duo/G, the bond length of adsorbed dinitrogen, and the
amount of Bader charge transferred from MN4 duo/G to adsorbed
S2

dinitrogen...................................................................................................14
Figure S9. Differential charge densities of chemisorbed N2 on the selected
MN4 duo/G models. The charge depletion and accumulation were depicted
in blue and yellow, respectively. The iso-surface value is 0.005 e/Å3........14
Figure S10. The transition state (TS) for the N-N bond dissociation of
bridge-on *N2. The N-N distance and imaginary frequency in each of TS
are given in Å and cm-1, respectively.........................................................15
Table S6. The energy barriers and N-N distances of transition states for N-
N bond breaking for bridge-on adsorbed N2 on selected MN4 duo/G models.
...................................................................................................................15
Figure S11. Calculated energy barriers for direct N-N bond breaking as a
function of the formation energy of MN4 duo and a function of the N-N
distance in transition states, respectively..................................................16
Figure S12. Gibbs free energy diagram for NRR on VN4 duo/G models.....16
Figure S13. Gibbs free energy diagram for NRR on MoN4 duo/G models.. .16
Figure S14. Gibbs free energy diagram for NRR on ReN4 duo/G models....17
Figure S15. Gibbs free energy diagram for NRR on MoN4-ReN4 duo/G
models.......................................................................................................17
Figure S16. Gibbs free energy diagram for NRR on MoN4-VN4 duo/G
models.......................................................................................................18
Figure S17. Gibbs free energy diagram for NRR on ReN4-VN4 duo/G models.
...................................................................................................................18
6. Reference.....................................................................................................19
S3

1. Computation detail
The Gibbs free energy change (ΔG) of every elemental step was calculated by using the
computational hydrogen electrode (CHE) model proposed by Nørskov and co-workers1-3, which uses
one-half of the chemical potential of hydrogen as the chemical potential of the proton-electron pair.
According to this method, ΔG can be determined as follows:
ΔG = ΔE + ΔZPE - TΔS + ΔGU + ΔGpH
where ΔE is the electronic energy difference directly obtained from DFT calculations, ΔZPE is the
change in zero-point energies, T is the temperature (T = 300 K), and ΔS is the entropy change. ΔG U is
the free energy contribution related to the applied electrode potential U, ΔG pH is the pH-dependent
correction of free energy of solvated protons. ΔGpH is computed as 2.303 × kBT × pH (or equivalently,
0.059 × pH), in which kB is the Boltzmann constant and the value of pH is set to be zero. The ZPE of
adsorbed species was extracted from the harmonic vibrational frequency calculations. For simplicity,
only the adsorbate vibrational modes were calculated explicitly, while the catalyst sheet was fixed
based on the assumption that vibrations of the sheets are negligible, as adopted in previous theoretical
studies.4 The entropies of adsorbed species were neglected. The entropies and vibrational frequencies
of molecules in the gas phase were taken from the NIST database.5 The onset potential (UOnset) is
determined by the potential-limiting step which has the most positive ∆G (∆Gmax) as computed by UOnset
= − ∆Gmax/e. The solvent effect on scaling relations are not taken into account as a previous study
showed that no significant change that can alter the scaling relations has been found as the solvent
effect was incorporated.6
2. The descriptors used for NRR
Figure S1. Three main pathways for associative mechanisms of NRR.
The associative mechanisms of NRR has three different types of forms —— distal, alternating,
and enzymatic, all of which consist of six consecutive reductive hydrogenation steps (Figure S1). These
S4

three pathways have an NH3 moiety formed before the complete breaking of the N-N bond. The
complexity of these series of reactions makes it tedious to identify the effectiveness of a potential NRR
catalyst based on the exploration of full reaction pathways. To accelerate the evaluation of catalysts’
activities, we employed the energetic descriptors proposed by Ling et al.7 They chose the Gibbs free
energy changes (∆G) of the first and last reduction steps in NRR as descriptors, i.e. ∆G*N2→*N2H for
*N2 + H+ + e- → *N2H, and ∆G*NH2→*NH3 for *NH2 + H+ + e- → *NH3, for these two uphill steps are
the most likely potential-limiting steps of associative mechanisms of NRR.
3. The definition and stability test of MN4/G and MN4 duo/G models
Figure S2. Representation of various dual-atom M-N-C models.
S5

Figure S3. Representation of (a) MN4/G and (b) MN4 duo/G models and (c) the scope of metals
investigated in this work.
Single MN4/G consists of one metal atom coordinated with four pyridinic nitrogen atoms in the
graphene matrix. MN4 duo/G consists of two adjacent metal atoms coordinated respectively with four
pyridinic nitrogen atoms in the graphene matrix. We first built the single MN4/G model by anchoring
one metal atom onto a nitrogen-doped double vacancy (DV) of graphene (Figure S3a), which has been
a well-established molecular scaffold to stabilize single active site under electrochemical conditions.8-10
Then we built MN4 duo/G by placing two MN4 moieties adjacent to each other, in which two metal
atoms are separated by ~ 4 Å (Figure S3b). We examined a total of 39 metal elements, as collected in
Figure S2c, to get a comprehensive overview of the scale relationships in NRR.
The stabilities of all 39 different MN4/G models and 39 MN4 duo/G models have been evaluated
from two dimensions: formation energy (Ef) for thermodynamic stability and dissolution potential
(Udiss) for electrochemical stability, which are defined as:
Ef = EMN4/G – EN4/G –E(metal, bulk) (1)
Udiss = U diss。 (metal, bulk) – Ef/ne (2)
Where E(M, bulk) is the average energy of one metal atom in its most stable bulk structure, EMN4/G and
EN4/G are the total energies of MN4/G and MN4 duo/G models and graphene substrate, U diss。 (metal,
bulk) and n are the standard dissolution potential of bulk metal and the number of electrons involved in
the dissolution, respectively. Based on the above definition, systems with Ef < 0 eV are considered to
be thermodynamically stable, while materials with Udiss > 0 V vs SHE are regarded as
electrochemically stable. The exact values of Ef and Udiss are listed in Table S1. We note that most of
the experimentally synthesized SACs11 are thermodynamically and electrochemically stable according
S6

to the above evaluation criteria. As Figure S4a shows, these models are unevenly distributed into four
different zones: 18 out of 39 metals are stable from both thermodynamic and electrochemical points of
view, 12 are thermodynamically stable yet electrochemically unstable, 4 are electrochemically stable
yet thermodynamically unstable, and 5 are unstable in either dimension. Likewise, their corresponding
MN4 duo models are also divided into four different categories (Figure S4b).
The geometries of various MN4/G models also drew our attention. As Table S1 shows, FeN4,
CoN4, and NiN4 bear almost co-planar configurations, all other MN4/G contain a metal center sitting
above the N4/G plane ranging from 0.05 Å (MnN4 and AlN4) to 2.77 Å (HgN4). And for most of the
metals, the deviation from the plane of nitrogen-doped graphene is no less in MN4 duo@G than
MN4/G. Since the DV of N-doped graphene has a fixed cavity, the out-of-plane distance for the metal
center may be determined by its radii and the metal-nitrogen bonding strength (Table S1).
S7
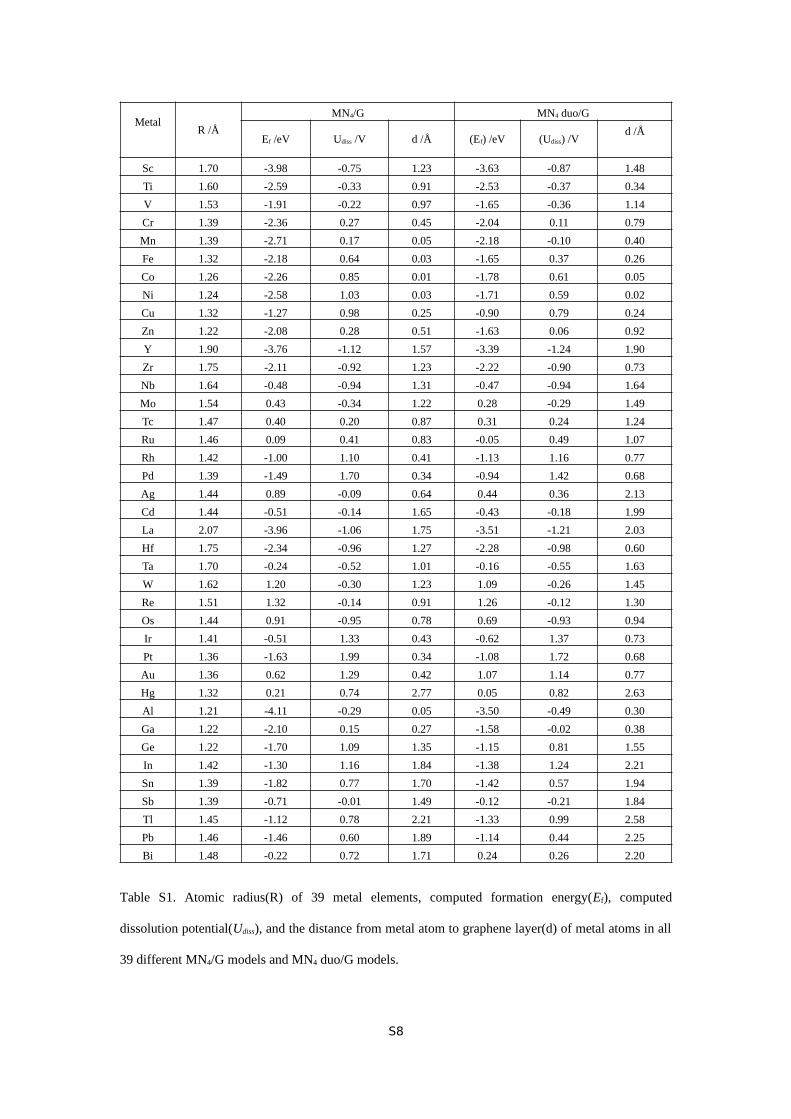
MetalR /Å
MN4/G MN4 duo/G
Ef /eV Udiss /V d /Å (Ef) /eV (Udiss) /Vd /Å
Sc 1.70 -3.98 -0.75 1.23 -3.63 -0.87 1.48
Ti 1.60 -2.59 -0.33 0.91 -2.53 -0.37 0.34
V 1.53 -1.91 -0.22 0.97 -1.65 -0.36 1.14
Cr 1.39 -2.36 0.27 0.45 -2.04 0.11 0.79
Mn 1.39 -2.71 0.17 0.05 -2.18 -0.10 0.40
Fe 1.32 -2.18 0.64 0.03 -1.65 0.37 0.26
Co 1.26 -2.26 0.85 0.01 -1.78 0.61 0.05
Ni 1.24 -2.58 1.03 0.03 -1.71 0.59 0.02
Cu 1.32 -1.27 0.98 0.25 -0.90 0.79 0.24
Zn 1.22 -2.08 0.28 0.51 -1.63 0.06 0.92
Y 1.90 -3.76 -1.12 1.57 -3.39 -1.24 1.90
Zr 1.75 -2.11 -0.92 1.23 -2.22 -0.90 0.73
Nb 1.64 -0.48 -0.94 1.31 -0.47 -0.94 1.64
Mo 1.54 0.43 -0.34 1.22 0.28 -0.29 1.49
Tc 1.47 0.40 0.20 0.87 0.31 0.24 1.24
Ru 1.46 0.09 0.41 0.83 -0.05 0.49 1.07
Rh 1.42 -1.00 1.10 0.41 -1.13 1.16 0.77
Pd 1.39 -1.49 1.70 0.34 -0.94 1.42 0.68
Ag 1.44 0.89 -0.09 0.64 0.44 0.36 2.13
Cd 1.44 -0.51 -0.14 1.65 -0.43 -0.18 1.99
La 2.07 -3.96 -1.06 1.75 -3.51 -1.21 2.03
Hf 1.75 -2.34 -0.96 1.27 -2.28 -0.98 0.60
Ta 1.70 -0.24 -0.52 1.01 -0.16 -0.55 1.63
W 1.62 1.20 -0.30 1.23 1.09 -0.26 1.45
Re 1.51 1.32 -0.14 0.91 1.26 -0.12 1.30
Os 1.44 0.91 -0.95 0.78 0.69 -0.93 0.94
Ir 1.41 -0.51 1.33 0.43 -0.62 1.37 0.73
Pt 1.36 -1.63 1.99 0.34 -1.08 1.72 0.68
Au 1.36 0.62 1.29 0.42 1.07 1.14 0.77
Hg 1.32 0.21 0.74 2.77 0.05 0.82 2.63
Al 1.21 -4.11 -0.29 0.05 -3.50 -0.49 0.30
Ga 1.22 -2.10 0.15 0.27 -1.58 -0.02 0.38
Ge 1.22 -1.70 1.09 1.35 -1.15 0.81 1.55
In 1.42 -1.30 1.16 1.84 -1.38 1.24 2.21
Sn 1.39 -1.82 0.77 1.70 -1.42 0.57 1.94
Sb 1.39 -0.71 -0.01 1.49 -0.12 -0.21 1.84
Tl 1.45 -1.12 0.78 2.21 -1.33 0.99 2.58
Pb 1.46 -1.46 0.60 1.89 -1.14 0.44 2.25
Bi 1.48 -0.22 0.72 1.71 0.24 0.26 2.20
Table S1. Atomic radius(R) of 39 metal elements, computed formation energy(Ef), computed
dissolution potential(Udiss), and the distance from metal atom to graphene layer(d) of metal atoms in all
39 different MN4/G models and MN4 duo/G models.
S8
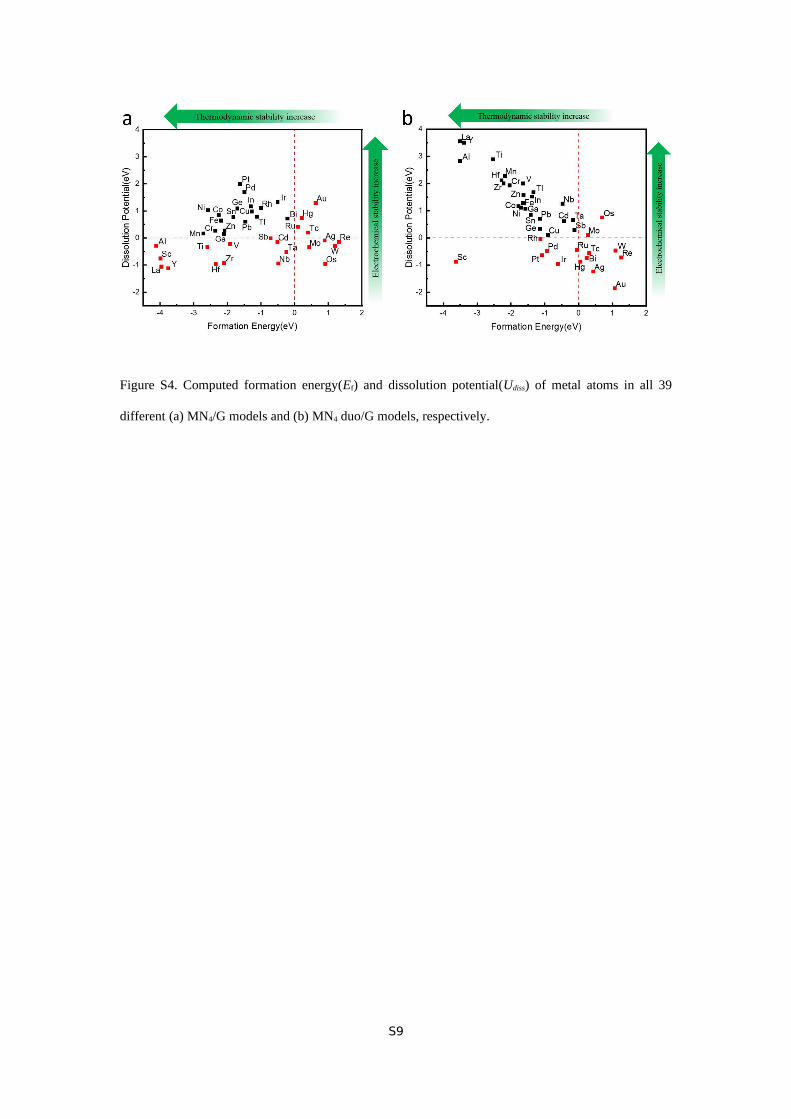
Figure S4. Computed formation energy(Ef) and dissolution potential(Udiss) of metal atoms in all 39
different (a) MN4/G models and (b) MN4 duo/G models, respectively.
S9
MetalMN4 MN4 duo
End-on Side-on End-on Side-on Bridge-on
Sc -0.78 -0.70 -0.86 × -1.52
Ti -1.10 -1.15 -0.81 × -1.91
V -1.14 -0.78 -1.15 -0.75 -1.46
Cr -0.44 × -0.50 × ×
Mn -0.16 × -0.36 × ×
Fe -0.59 × -0.67 × ×
Co -0.22 × -0.26 × ×
Ni × × × × ×
Cu × × × × ×
Zn -0.12 × × × ×
Y -0.62 -0.54 -0.72 × -1.30
Zr -1.01 -1.13 -0.94 × -1.73
Nb -1.14 -1.26 × × -2.55
Mo -1.21 -1.29 -1.17 -0.86 -1.76
Tc -1.07 -0.66 -1.21 -0.56 -1.12
Ru -1.22 × -0.91 -0.08 ×
Rh × × × × ×
Pd × × × × ×
Ag × × -0.35 × -0.46
Cd -0.14 × -0.18 × -0.40
La -0.30 -0.19 × × -0.95
Hf -1.07 -1.19 -0.78 × -2.03
Ta -1.26 -1.52 -1.33 × -2.81
W -1.46 -1.63 -1.41 -1.31 -2.27
Re -1.38 -1.13 -1.43 -0.93 -1.61
Os -1.28 × -0.97 -0.03 -0.46
Ir × × × × ×
Pt × × × × ×
Au × × × × ×
Hg × × × × ×
Al × × × × ×
Ga × × × × ×
Ge × × × × ×
In × × × × ×
Sn × × × × ×
Sb × × × × ×
Tl × × × × ×
Pb × × × × ×
Bi × × × × ×
Table S2. The adsorption energies of dinitrogen in various patterns on 39 different MN 4/G and MN4
duo/G models. The × symbol represents physisorption
S10
4. The linear scaling relationships in NRR
Figure S5. The scaling relationships between ∆G1 and ∆G*N2H for end-on (a) and side-on (b)
adsorption modes, and between ∆G6 and ∆G*NH2 for both two modes (c).
Figure S6. Two subgroups of transition metals based on scaling relations between ∆G*N2H and ∆G*NH2.
Group 1 include elments (in blue) in groups IB-IVB; group 2 include elements (in red) in groups VB-
VIIIB.
S11
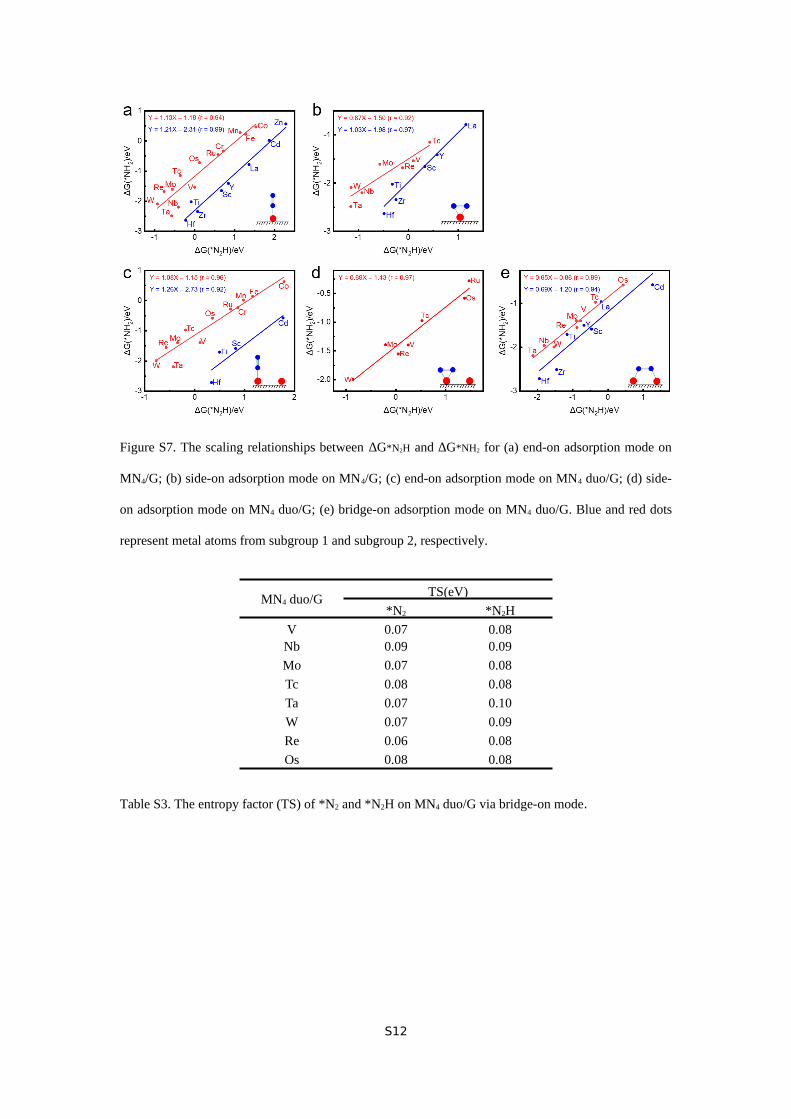
Figure S7. The scaling relationships between ∆G*N2H and ∆G*NH2 for (a) end-on adsorption mode on
MN4/G; (b) side-on adsorption mode on MN4/G; (c) end-on adsorption mode on MN4 duo/G; (d) side-
on adsorption mode on MN4 duo/G; (e) bridge-on adsorption mode on MN4 duo/G. Blue and red dots
represent metal atoms from subgroup 1 and subgroup 2, respectively.
MN4 duo/GTS(eV)
*N2 *N2H
V 0.07 0.08Nb 0.09 0.09
Mo 0.07 0.08
Tc 0.08 0.08
Ta 0.07 0.10
W 0.07 0.09
Re 0.06 0.08
Os 0.08 0.08
Table S3. The entropy factor (TS) of *N2 and *N2H on MN4 duo/G via bridge-on mode.
S12
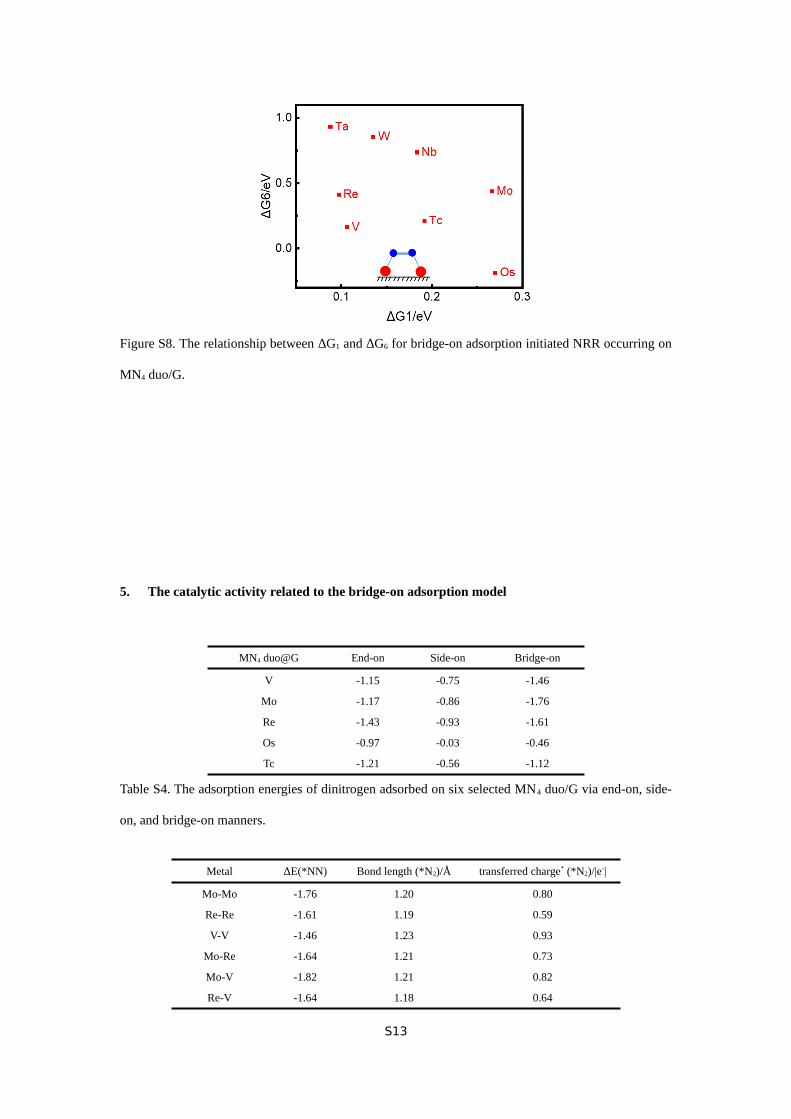
Figure S8. The relationship between ∆G1 and ∆G6 for bridge-on adsorption initiated NRR occurring on
MN4 duo/G.
5. The catalytic activity related to the bridge-on adsorption model
MN4 duo@G End-on Side-on Bridge-on
V -1.15 -0.75 -1.46
Mo -1.17 -0.86 -1.76
Re -1.43 -0.93 -1.61
Os -0.97 -0.03 -0.46
Tc -1.21 -0.56 -1.12
Table S4. The adsorption energies of dinitrogen adsorbed on six selected MN4 duo/G via end-on, side-
on, and bridge-on manners.
Metal ΔE(*NN) Bond length (*N2)/Å transferred charge* (*N2)/|e-|
Mo-Mo -1.76 1.20 0.80
Re-Re -1.61 1.19 0.59
V-V -1.46 1.23 0.93
Mo-Re -1.64 1.21 0.73
Mo-V -1.82 1.21 0.82
Re-V -1.64 1.18 0.64
S13
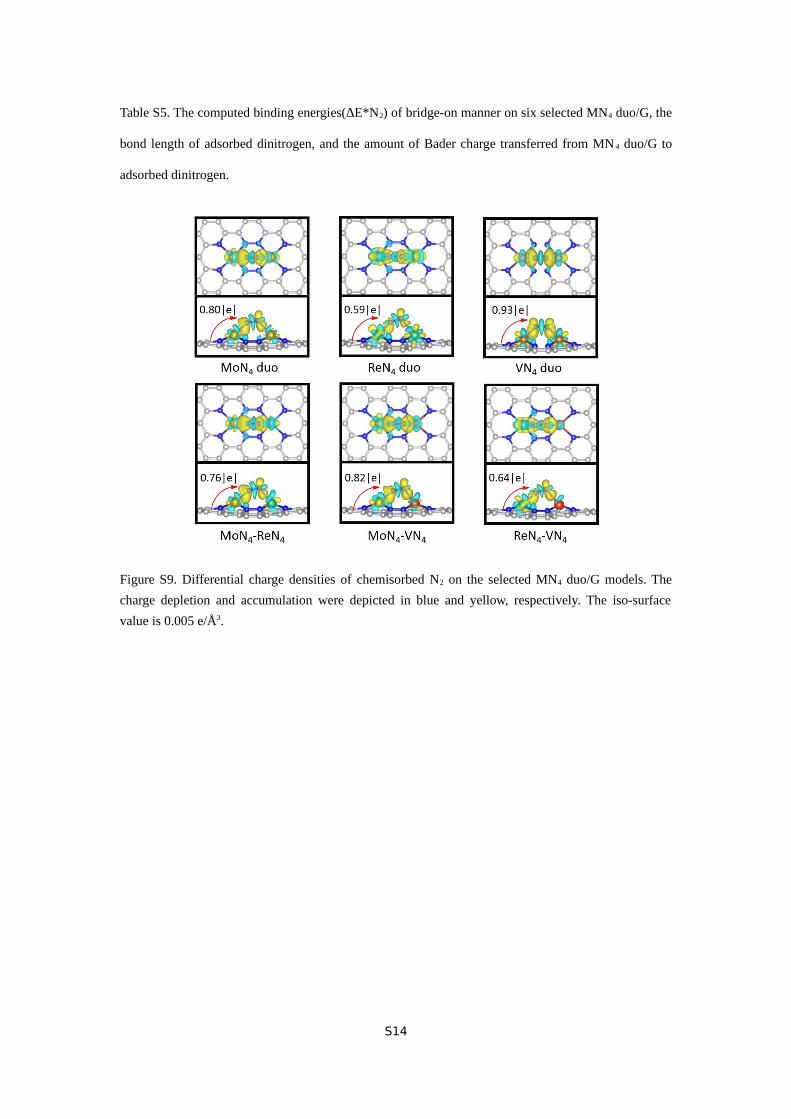
Table S5. The computed binding energies(ΔE*N2) of bridge-on manner on six selected MN4 duo/G, the
bond length of adsorbed dinitrogen, and the amount of Bader charge transferred from MN 4 duo/G to
adsorbed dinitrogen.
Figure S9. Differential charge densities of chemisorbed N2 on the selected MN4 duo/G models. The
charge depletion and accumulation were depicted in blue and yellow, respectively. The iso-surface
value is 0.005 e/Å3.
S14
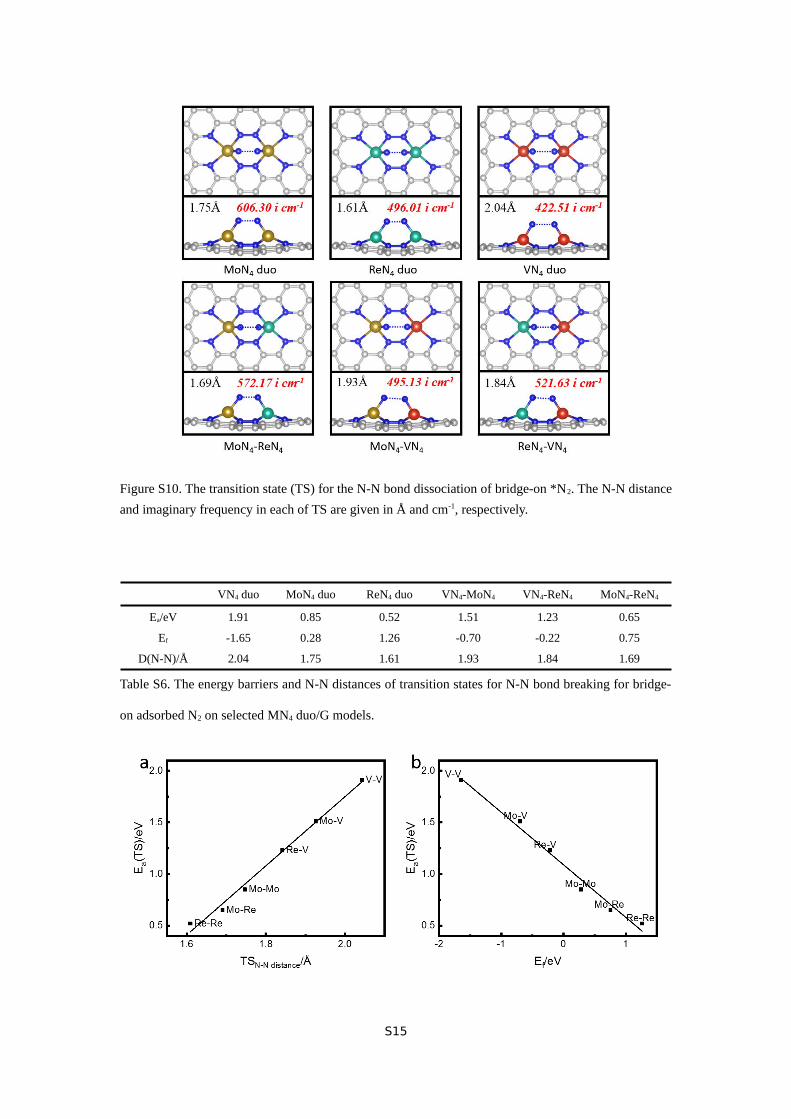
Figure S10. The transition state (TS) for the N-N bond dissociation of bridge-on *N2. The N-N distance
and imaginary frequency in each of TS are given in Å and cm-1, respectively.
VN4 duo MoN4 duo ReN4 duo VN4-MoN4 VN4-ReN4 MoN4-ReN4
Ea/eV 1.91 0.85 0.52 1.51 1.23 0.65
Ef -1.65 0.28 1.26 -0.70 -0.22 0.75
D(N-N)/Å 2.04 1.75 1.61 1.93 1.84 1.69
Table S6. The energy barriers and N-N distances of transition states for N-N bond breaking for bridge-
on adsorbed N2 on selected MN4 duo/G models.
S15
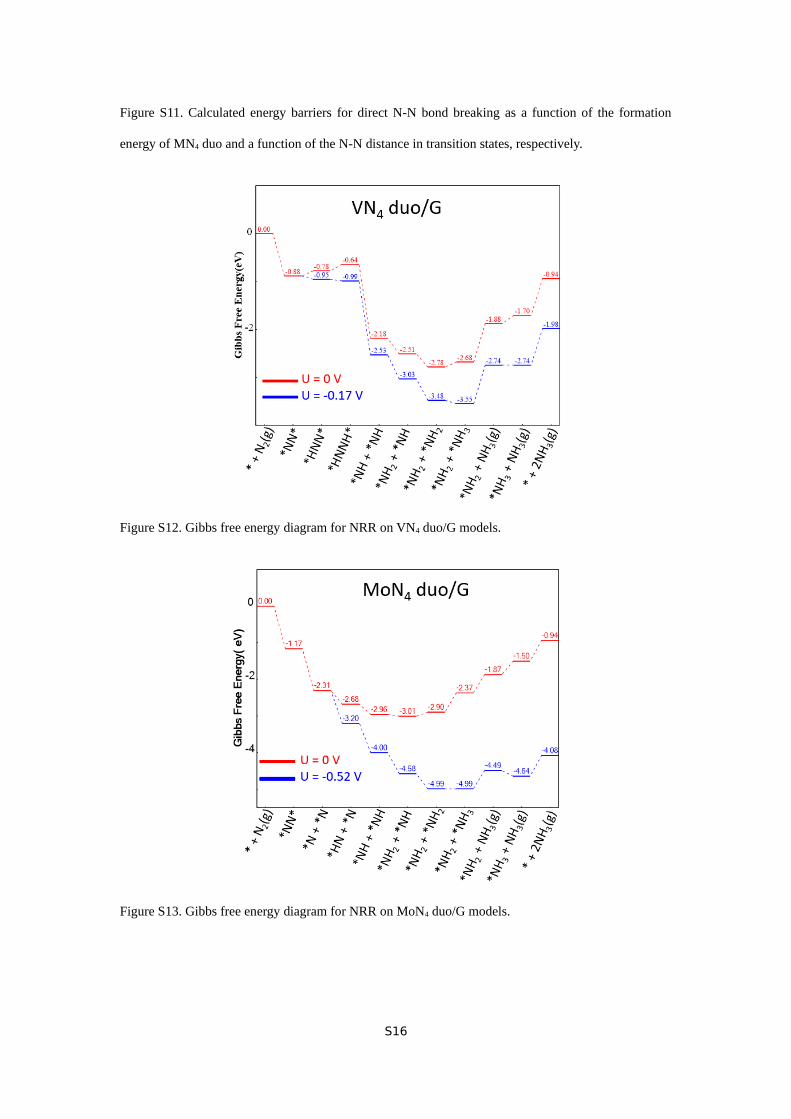
Figure S11. Calculated energy barriers for direct N-N bond breaking as a function of the formation
energy of MN4 duo and a function of the N-N distance in transition states, respectively.
Figure S12. Gibbs free energy diagram for NRR on VN4 duo/G models.
Figure S13. Gibbs free energy diagram for NRR on MoN4 duo/G models.
S16
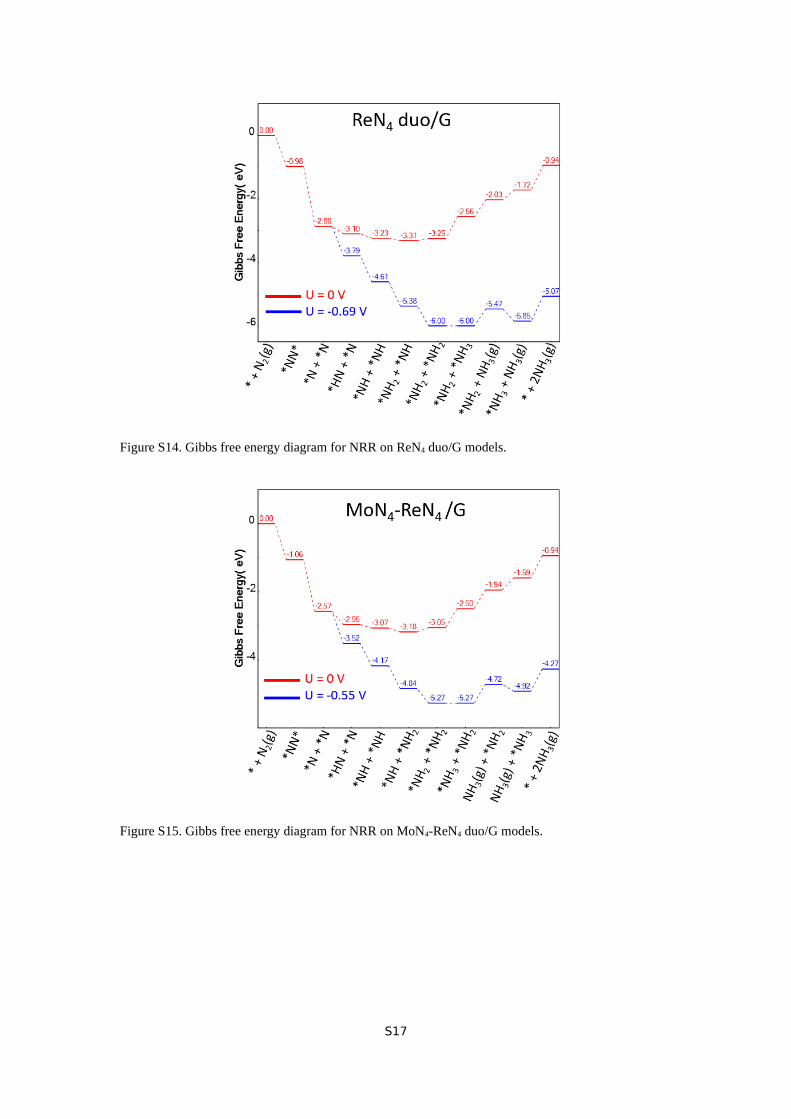
Figure S14. Gibbs free energy diagram for NRR on ReN4 duo/G models.
Figure S15. Gibbs free energy diagram for NRR on MoN4-ReN4 duo/G models.
S17
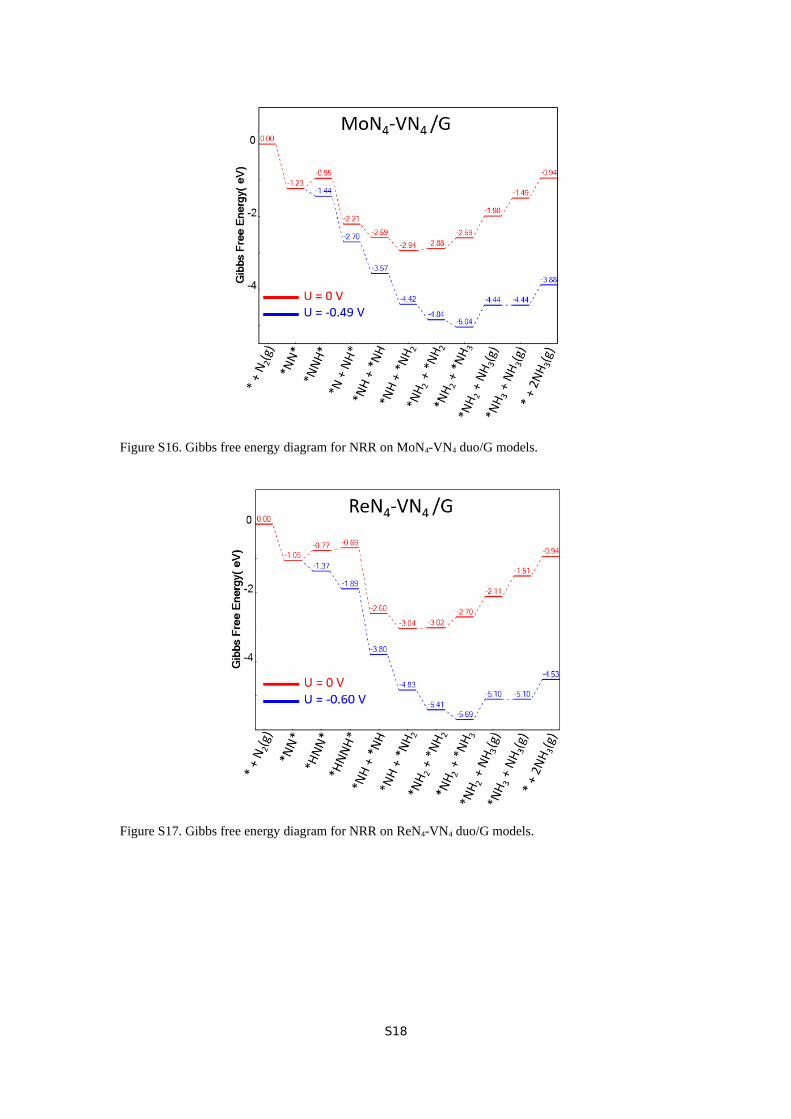
Figure S16. Gibbs free energy diagram for NRR on MoN4-VN4 duo/G models.
Figure S17. Gibbs free energy diagram for NRR on ReN4-VN4 duo/G models.
S18

6. Reference
(1) Nørskov, J. K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J. R.; Bligaard, T.; Jonsson, H.
Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108,
17886-17892.
(2) Rossmeisl, J.; Logadottir, A.; Nørskov, J. K. Electrolysis of Water on (Oxidized) Metal Surfaces.
Chem. Phys. 2005, 319, 178-184.
(3) Peterson, A. A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J. K. How Copper Catalyzes
the Electroreduction of Carbon Dioxide into Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311-
1315.
(4) Zhao, J.; Chen, Z. Single Mo Atom Supported on Defective Boron Nitride Monolayer as an
Efficient Electrocatalyst for Nitrogen Fixation: A Computational Study. J. Am. Chem. Soc. 2017, 139,
12480-12487.
(5) Computational Chemistry Comparison and Benchmark Database. http://cccbdb.nist.gov/.
(6) Park, J.; Roling, L. T. Elucidating Energy Scaling between Atomic and Molecular Adsorbates in the
Presence of Solvent. AIChE Journal 2020, 66: e17036.
(7) Ling, C.; Ouyang, Y.; Li, Q.; Bai, X.; Mao, X.; Du, A.; Wang, J. A General Two‐Step Strategy–
Based High‐Throughput Screening of Single Atom Catalysts for Nitrogen Fixation. Small Methods
2018, 3, 1800376.
(8) Liu, X.; Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S. Z. Building up a Picture of the Electrocatalytic
Nitrogen Reduction Activity of Transition Metal Single-Atom Catalysts. J. Am. Chem. Soc. 2019, 141,
9664-9672.
(9) Guo, X.; Gu, J.; Lin, S.; Zhang, S.; Chen, Z.; Huang, S. Tackling the Activity and Selectivity
Challenges of Electrocatalysts toward the Nitrogen Reduction Reaction Via Atomically Dispersed
Biatom Catalysts. J. Am. Chem. Soc. 2020, 142, 5709-5721.
(10) Wang, Y.; Tang, Y. J.; Zhou, K. Self-Adjusting Activity Induced by Intrinsic Reaction
Intermediate in Fe-N-C Single-Atom Catalysts. J. Am. Chem. Soc. 2019, 141, 14115-14119.
(11) Xiong, Y.; Sun, W.; Xin, P.; Chen, W.; Zheng, X.; Yan, W.; Zheng, L.; Dong, J.; Zhang, J.; Wang,
D.; Li, Y. Gram-Scale Synthesis of High-Loading Single-Atomic-Site Fe Catalysts for Effective
Epoxidation of Styrene. Adv. Mater. 2020, e2000896.
S19

download fileview on ChemRxivNRR SI_new_v2.docx (1.97 MiB)