conjugation of a hairpin pyrrole-imidazole polyamide to a quinone methide for control of dna...
TRANSCRIPT

Conjugation of a Hairpin Pyrrole-Imidazole Polyamide to a QuinoneMethide for Control of DNA Cross-Linking
Dalip Kumar, Willem F. Veldhuyzen, Qibing Zhou,† and Steven E. Rokita*Department of Chemistry and Biochemistry, University of Maryland,College Park, Maryland 20742. Received April 1, 2004; Revised Manuscript Received April 22, 2004
A series of quinone methide precursors designed for DNA cross-linking were prepared and conjugatedto a pyrrole-imidazole polyamide for selective association to the minor groove. Although reaction wasonly observed for DNA containing the predicted recognition sequence, yields of strand alkylation werelow. Interstrand cross-linking was more efficient than alkylation but still quite modest and equivalentto that generated by a comparable conjugate containing the N-mustard chlorambucil. Varying thelength of the linker connecting the polyamide and quinone methide derivative did not greatly affectthe yield of DNA cross-linking. Instead, intramolecular trapping of the quinone methide intermediateby nucleophiles of the attached polyamide appears to be the major determinant that limits its reactionwith DNA. Self-adducts of the quinone methide conjugate form readily and irreversibly as detectedby a combination of chromatography and mass spectroscopy. This result is unlike comparable self-adducts observed for oligonucleotide conjugates that form more slowly and remain reversible.Equivalent intramolecular alkylation of a polyamide by its attached chlorambucil mustard was notobserved under similar condition. The presence of DNA, however, did facilitate hydrolysis of thismustard conjugate.
INTRODUCTION
Distamycin continues to inspire development of newcompounds with a highly precise and predictable abilityto recognize an extended series of base pairs in duplexDNA since its original discovery over 40 years ago (1).This natural product is composed of three N-methylpyr-role rings attached through amide bonds to create amolecule that complements the bend of DNA’s minorgroove and offers hydrogen bonding to its preferred targetregion rich in A/T pairs (2). Such selectivity has sincebeen conferred to a number of reagents with covalentreactivity by conjugating the appropriate appendages todistamycin or related derivatives (3-5). Included in thisseries are a variety of alkylating agents. For example, adistamycin-benzoic acid N-mustard conjugate, tallimus-tine, was prepared and tested clinically as an anticancerdrug candidate (6, 7). Variations of tallimustine have alsobeen synthesized and examined under a range of biologi-cal conditions (8-11). Other alkylating and cross-linkingagents have similarly been attached to distamycin in-cluding a bis(hydroxylmethyl)pyrrole (12), a methyl alkylsulfate (13), mitomycin (14), a pyrrolobenzodiazepinederivative reminiscent of tomamycin (15), and a cyclo-propylpyrroloindole group related to CC-1065 and duo-carmycin (16).
Once distamycin and its conjugates were found to bindas a dimer in the minor groove (17), polyamides contain-ing the equivalents of a dimer structure were constructed(18). As predicted, these new hairpin-shaped derivativesbound tightly and precisely to chosen sequences of DNA.Inclusion of additional heterocyclic substituents allowedfor distinct recognition of each base pair (4, 19, 20).Further derivatization of such compounds with cyclopro-
pylpyrroloindole and chlorambucil appendages providedadditional efficiency and selectivity for alkylating andcross-linking DNA (Scheme 1) (21-23). Success with thepyrrole-imidazole polyamides created an extremely at-tractive system for directing the chemistry of quinonemethides to the minor groove of DNA.
Quinone methides are highly electrophilic and tran-sient intermediates that are implicated in alkylation ofDNA by drugs such as mitomycin (24) and tamoxifen (25),food additives such as butylated hydroxytoluene (BHT)(26), and certain natural products (27, 28). Our laboratoryhas prepared and studied a series of simple precursorsand their bioconjugates that form o-quinone methideintermediates. Activation of these precursors is easilycontrolled by addition of fluoride, a species that isotherwise rarely encountered under chemical and biologi-cal conditions (Scheme 2). The products formed byquinone methide reaction with strongly nucleophilicgroups in DNA tend to be unstable and readily reverseso that only adducts formed by the weakly nucleophilicexo amino groups of G and A are often isolated (nuc1 vsnuc2, respectively in Scheme 2) (29-34).
* To whom correspondence should be addressed. E-mail:[email protected].
† Current address: Department of Chemistry, Virginia Com-monwealth University, Richmond, VA 23284.
Scheme 1
915Bioconjugate Chem. 2004, 15, 915−922
10.1021/bc049941h CCC: $27.50 © 2004 American Chemical SocietyPublished on Web 06/04/2004

The tendency for an o-quinone methide to accumulateonly irreversible adducts can be overridden by attach-ment of a DNA ligand that is capable of maintaining closeproximity between a reactive nucleophile-electrophilepair. Reversibility of addition is not affected by thisstrategy. Rather, diffusion of the regenerated intermedi-ate is inhibited and consequently shielded from exposureto the weak nucleophiles forming irreversible adducts.This is best illustrated by the ability of a quinonemethide-acridine conjugate to act as a highly efficientand persistent cross-linker of duplex DNA through reac-tion primarily at G N7 in the major groove, a site thatwould otherwise form only an unstable and transientadduct (35). The ratio of monoalkylation to cross-linkingof this reagent is significantly smaller than that for anequivalent conjugate formed by chlorambucil. This desir-ably low ratio might also derive from the reversibility ofquinone methide addition if the conjugate is able tomigrate from sites allowing only monoalkylation to sitescapable of cross-linking. Certainly, an equivalent revers-ibility in product formation is responsible for convertingoligonucleotide-quinone methide self-adducts from inac-tive and undesirable side products to highly valuabletarget-promoted alkylating agents (36). A series of con-jugates between a pyrrole-imidazole polyamide and quino-ne methide precursors has now been prepared andexamined as described here to investigate whether asimilar synergy could be realized between a distamycin-based ligand, a quinone methide intermediate, and theminor groove of DNA.
MATERIALS AND METHODS
General. Solvents, chemicals, and reagents wereobtained in the highest purity commercially available andused without additional purification unless describedbelow. Chlorambucil, 4-(4′-methoxyphenyl)butyric acidand monomethyl adipoyl chloride were purchased fromICN Biomedicals, Aldrich Chemical Co. and Fluka Chemi-cal Corporation, respectively. Boc-â-alanine-Pam resinand (R)-2-Fmoc-4-Boc-diaminobutyric acid were pur-chased from Peptides International. Diisopropylethy-lamine (DIEA) and N,N-(dimethylamino)propylaminewere distilled from calcium hydride (CaH2) and storedover NaOH pellets. Fuming nitric acid was prepared bydistillation of a 50:50 solution of concentrated sulfuricacid and 70% nitric acid. Dimethylformamide (DMF) wasdistilled from P2O5 and stored over molecular sieves, and
acetonitrile was distilled from CaH2. Pyridine was freshlydistilled over NaOH pellets, and ethanol (EtOH) wasdried by distillation over magnesium and iodine (10:1w/w). Both methylene chloride (CH2Cl2) and toluene weredistilled from CaH2 prior to use. All aqueous solutionswere prepared with water purified to a resistivity of17.8-18.0 MΩ.
Silica gel for flash chromatography (230-400 mesh)was purchased from EM Sciences. Reverse phase C-18chromatography was performed using analytical (VarianMicrosorb-MV C-18, 300 C particle size, 250 mm × 4.6mm) and semipreparative (Alltech Econosphere C-18, 10um particle size, 250 mm × 10 mm) columns and a JascoPU-980 HPLC equipped with a Jasco MD-1510 UV-Visphotodiode array detector.
1H and 13C spectra were recorded on a Bruker 400 MHzspectrometer. All NMR chemical shifts (δ) are reportedin parts per million (ppm) and were determined relativeto the standard values of the solvent (Cambridge IsotopeLaboratories). Coupling constants (J) are reported inhertz (Hz). UV absorption spectra were obtained with aHewlett-Packard 8453 UV-Vis spectrophotometer. Lowresolution (LRMS) and high resolution (HRMS) massspectra were determined on a VG7070E instrument usingEI and FAB. Electrospray ionization mass spectrometrywas performed with a Finnigan LCQ system equippedwith a quadrupole ion trap.
Synthetic Procedures. N-Succinimidyl esters of 3-[4′-tert-butyldimethylsilyloxy-3′,5′-bis(acetoxymethyl)phenyl]-propionate (2), the related unsaturated derivative, andits conjugate 8 were prepared as described previously (35,37). The pyrrole-imidazole polyamide ImPy-â-ImPy-(R)γ-ImPy-â-ImPy-â-Dp and its chlorambucil conjugate 1 weresynthesized as first reported by Wurtz and Dervan (22)(Scheme 3), and all of the monomeric components neces-sary for this were prepared as described by Baird andDervan (38).
4-[4′-tert-Butyldimethylsilyloxy-3′,5′-bis(tert-bu-tyldimethylsilyloxymethyl)phenyl]butanoic Acid(10a). Formaldehyde (37%, 11.1 mL) was added to anaqueous solution of NaOH (25%, 7.7 mL) containingp-hydroxyphenylbutanoic acid 9a (2.0 g, 12 mmol) andmethanol (6 mL) at 0 °C. The resulting mixture washeated to 37 °C, stirred for 48 h, and then diluted withacetone (80 mL). The orange aqueous phase was isolated,diluted with methanol (10 mL), and again added toacetone (125 mL). The resulting white precipitate wasisolated by filtration, washed with acetone, and dried.This crude product (1.40 g, 4.93 mmol) was directlysilylated by addition of DMF (15 mL), tert-butyldimeth-ylsilyl chloride (TBS-Cl, 3.94 g, 26.1 mmol), and imidazole(4.00 g, 59.2 mmol). The reaction mixture was stirred atroom temperature for 20 h, diluted with water (100 mL),and extracted with ether (3 × 50 mL). The organic phaseswere combined, dried with MgSO4, and evaporated underreduced pressure. The residue was dissolved in methanol(40 mL), and potassium carbonate (2.0 g) was added inone portion. The solution was stirred for 40 min at roomtemperature, neutralized with 2 M HCl (150 mL), andextracted with ether (3 × 50 mL). The organic phase wasdried (MgSO4) and concentrated to yield a residue thatwas subject to silica gel flash chromatography usingEtOAc/hexanes (1:9). The desired product was isolatedas a colorless solid. Yield 56%; mp 78 °C. 1H NMR(CDCl3): δ 0.00 (s, 12H, Si(CH3)2), 0.07 (s, 6H, Si(CH3)2),0.92(s, 18H, C(CH3)3), 1.01 (s, 9H, C(CH3)3), 1.92-1.96(m, 2H, CH2), 2.36 (t, J ) 8.0 Hz, 2H, CH2), 2.63 (t, J )8.0 Hz, 2H, CH2), 4.68 (s, 4H, CH2), 7.16 (s, 2H, Ar-H).13C NMR (CDCl3): δ 0.00, 18.4, 18.8, 26.0, 26.1, 26.2,
Scheme 2
916 Bioconjugate Chem., Vol. 15, No. 4, 2004 Kumar et al.

33.3, 34.6, 60.6, 125.9, 131.5, 134.1, 146.2, 179.7. HRMS(FAB): calcd for C30H58O5Si3 (M)+ 582.3592, found582.2138.
4-[4′-tert-Butyldimethylsilyloxy-3′,5′-bis(acetoxy-methyl)phenyl]butanoic Acid (11a). Solid ferric chlo-ride (0.030 g, 0.19 mmol) was added to a stirred suspen-sion of 10a (0.33 g, 0.57 mmol) at 0 °C. The reactionmixture was maintained at this temperature and stirredfor 0.5 h before adding ice-cold water (100 mL). Thismixture was extracted with ether (3 × 10 mL). Theorganic layer was evaporated, and the residue wassuspended in water (50 mL). This was extracted withhexanes (3 × 10 mL). The organic phase was dried withMgSO4 and concentrated under reduced pressure. Theresulting residue was subjected to silica gel columnchromatography using EtOAc:hexanes (1:9), and thedesired product (viscous oil) was isolated in 81% yield.1H NMR (CDCl3): δ 0.17 (s, 6H, Si(CH3)2), 1.00 (s, 9H,C(CH3)3), 1.89 (m, 2H, CH2), 2.08 (s, 6H, COCH3), 2.35(t, 2H, CH2), 2.58 (t, 2H, CH2), 5.05 (s, 4H, CH2), 7.09 (s,2H, Ar-H). 13C NMR (CDCl3): δ 18.9, 21.0, 25.8, 26.2,33.3, 34.1, 61.9, 126.8, 130.3, 134.6, 149.6, 171.0. HRMS(FAB): calcd for C22H34O7SiNa (M + Na)+ 461.1972,found 461.1962.
N-Succinimidyl 4-[4′-tert-Butyldimethylsilyloxy-3′,5′-bis(acetoxymethyl)phenyl]butanoate (3). A mix-ture of 11a (0.050 g, 0.11 mmol), N-hydroxysuccinimide(0.048 g, 0.42 mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) (0.080 g, 0.42mmol) in DMF (5 mL) was stirred at room temperaturefor 12 h. The reaction mixture was poured into cold water(100 mL) and extracted with ether (3 × 10 mL). The
organic phase was dried with MgSO4, and solvent wasremoved at reduced pressure. The resulting residue wassubjected to preparative thin-layer chromatography usingEtOAc:hexanes (2:3) to afford the desired product as aviscous oil. Yield 67%. 1H NMR (CDCl3): δ 0.17 (s, 6H,Si(CH3)2), 1.00 (s, 9H, C(CH3)3), 2.01-2.05 (m, 2H, CH2),2.09 (s, 6H, COCH3), 2.60 (t, J ) 7.0 Hz, 2H, CH2), 2.09(t, J ) 7.0 Hz, 8.0 Hz, 2H, CH2), 2.83 (s, 4H, CH2), 5.06(s, 4H, CH2), 7.11(s, 2H, ArH). 13C NMR (CDCl3): δ -3.7,18.6, 21.0, 25.60, 25.88, 26.22, 30.23, 33.77, 61.90, 127.00,130.30, 134.00, 149.80, 168.40, 169.10, 171.0. HRMS(EI): calcd for C26H37NO9Si (M)+, 535.2238, found535.2227.
6-[4′-tert-Butyldimethylsilyloxy-3′,5′-bis(tert-bu-tyldimethylsilyloxymethyl)phenyl]hexanoic Acid(10b). This was prepared equivalently to that describedfor 10a starting from 9b (0.52 g, 2.5 mmol). Yield 44%;mp. 82 °C. 1H NMR (CDCl3): δ 0.06 (s, 12H, Si(CH3)2),0.13 (s, 6H, Si(CH3)2), 0.92 (s, 18H, C(CH3)3), 1.00 (s, 9H,C(CH3)3), 1.37-1.38 (m, 2H, CH2), 1.59-1.67 (m, 4H,CH2), 2.32 (t, J ) 8.0 Hz, 2H, CH2), 2.56 (t, J ) 8.0 Hz,2H, CH2), 4.67 (s, 4H, CH2), 7.13 (s, 2H, Ar-H). 13C NMR(CDCl3) δ 18.4, 18.72, 24.58, 25.94, 26.02, 28.67, 30.96,33.96, 35.21, 60.6, 125.8, 131.2, 135.3, 179.7. HRMS(FAB): calcd for C32H61O5Si3 (M - H)- 609.3827, found609.3826.
6-[4′-tert-Butyldimethylsilyloxy-3′,5′-bis(acetoxy-methyl)phenyl]hexanoic Acid (11b). This was pre-pared equivalently to that described for 11a starting from10b (025 g, 041 mmol). Yield 69% (viscous oil). 1H NMR(CDCl3): δ 0.18 (s, 6H, Si(CH3)2), 1.01 (s, 9H, C(CH3)3),1.38-1.42 (m, 2H, CH2), 1.57-1.71 (m, 4H, CH2), 2.06(s, 6H, OCOCH3), 2.44 (t, J ) 7.0, J ) 8.0 Hz, 2H, CH2),2.54 (t, J ) 8.0 Hz, 2H, CH2), 5.06 (s, 4H, CH2), 7.08(s, 2H, Ar-H). 13C NMR (CDCl3): δ -3.8, 18.6, 21.0, 22.1,23.9, 24.4, 25.8, 28.4, 30.9, 34.7, 35.0, 61.9, 126.6, 130.2,135.7, 149.4, 169.2, 170.9. HRMS (FAB): calcd ForC24H37O7Si (M - H)- 465.2309, found 465.2293.
N-Succinimidyl 6-[4′-tert-Butyldimethylsilyloxy-3′,5′-bis(acetoxymethyl)phenyl]hexanoate (4). Thiswas prepared equivalently to that described for 3 startingfrom 11b (0.10 g, 0.21 mmol). Yield 66% (liquid). 1H NMR(CDCl3): δ 0.16 (s, 6H, Si(CH3)2), 0.99 (s, 9H, C(CH3)3),1.43-1.45 (m, 2H, CH2), 1.61-1.63 (m, 2H, CH2), 1.74-1.77 (m, 2H, CH2), 2.08 (s, 6H, OCOCH3), 2.52-2.81(m, 4H, CH2), 2.82 (s, 4H, CH2), 5.05 (s, 4H, CH2), 7.08(s, 2H, Ar-H). 13C NMR (CDCl3): δ -3.8, 18.6, 21.0, 24.4,25.5, 25.8, 28.3, 30.8, 34.7, 61.9, 126.6, 130.2,135.7, 149.4, 168.5, 169.1, 170.9. HRMS (FAB): calcd forC28H41LiNO9Si (M + Li)+ 570.2711, found 570.2698.
Polyamide-Quinone Methide Conjugate (5). Asolution of N-succinimidyl-3-[4′-tert-butyldimethylsily-loxy-3′,5′-bis(acetoxymethyl)phenyl]propionate 2 (10 mg,19 µmol) in CH3CN (100 µL) was added to a solution ofImPy-â-ImPy-(R)γ-ImPy-â-ImPy-â-Dp (4 mg, 3 µmol) inDMF (320 µL). Reaction was initiated by addition ofDIEA (200 µL) and stirred for 10 min at room tempera-ture. The desired product was purified by preparativeHPLC using a solvent gradient of 25% CH3CN:0.1%(v/v) aq TFA to 50% CH3CN:0.1% (v/v) aq TFA over 40min (5 mL/min). Solvent was removed by lyophilizationto yield a white powder (0.3 mg, 6%). This yield wasmeasured by A306 based on the extinction coefficientof the polyamide-chlorambucil conjugate 1 (69 500M-1 cm-1) (22). LRMS (electrospray) m/z calcd forC83H111N26O18Si (M + H)+ 1787.8, found 1787.7.
Polyamide-Quinone Methide Conjugate (6). Thisproduct was prepared equivalently to that described forconjugate 5 starting from 3 (4 mg, 8 µmol). Yield 3%;
Scheme 3
Polyamide−Quinone Methide Conjugates Bioconjugate Chem., Vol. 15, No. 4, 2004 917

LRMS (electrospray) m/z calcd for C84H113N26O18Si (M +H)+ 1801.8, found 1801.7.
Polyamide-Quinone Methide Conjugate (7). Thisproduct was prepared equivalently to that described forconjugate 5 starting from 4 (4 mg, 8 µmol). Yield 2%;LRMS (electrospray) m/z calcd for C86H117N26O18Si (M +H)+ 1829.8, found 1829.5.
DNA Preparation and Analysis. Oligonucleotideswere synthesized by Invitrogen Life Technologies (Rock-ville, MD) and purified by gel electrophoresis understandard conditions (39). Oligonucleotides were labeledat their 5′ terminus with [γ-32P]-ATP (New EnglandBiolabs, Beverly, MA) as directed by the supplier. Typi-cally, a 5′-[32P] radiolabeled oligonucleotide (100 pmol,640 nCi), its unlabeled equivalent (200 pmol), and itscomplementary strand (330 pmol) were annealed in asolution of Tris (20 mM, pH 7) by placing the mixture(total volume 200 µL) in a microfuge tube. The tube wasplaced in a water bath (90° C), and the bath and the tubewere allowed to cool to room temperature over 3-4 h toafford a 2 µM solution of labeled duplex DNA.
DNA Alkylation and Cross-Linking by PolyamideConjugates. Typically, 10 µL of the DNA solutionsdescribed above (2 µM, 20 mM Tris, pH 7) were mixedwith water (2 µL) and a 15% aqueous acetonitrile solution(4 µL) of conjugate 1, 5, 6, 7, or 8 (15 µM) in a microfugetube. After addition of 4 µL of a salt solution (15 mMNaCl, 10 mM MgCl2, 5 mM CaCl2, ( 5 mM NaF), theresulting mixture (20 µL, 10 mM Tris, pH 7) of DNA(1 µM), reactant (3 µM), and salts (3 mM NaCl, 2 mMMgCl2, 1 mM CaCl2, ( 1 mM NaF) was incubated(37 °C) for 12 h. Aliquots (3 µL) were removed from thereaction mixtures and combined with a formamide load-ing solution (10 µL, 0.05% bromophenol blue and 0.05%xylene cyanol FF) and analyzed by 20% polyacrylamide(19:1 acrylamide:bisacrylamide) gel electrophoresis underdenaturing conditions (8 mM urea). DNA was detectedand analyzed by phosphoimagery.
RESULTS AND DISCUSSION
Design and Synthesis of the Quinone MethidePrecursors and Their Conjugates. A bis-functionalprecursor designed for cross-linking DNA through se-quential formation of two quinone methide intermediateshad previously been coupled to an acridine derivative anda triplex-forming oligonucleotide for delivery to the majorgroove (Scheme 2) (35, 40). This same precursor has nowalso been attached to a pyrrole-imidazole polyamide forits complementary delivery to the minor groove. This newconjugate additionally allowed for comparison to a relatedderivative prepared previously by Wurtz and Dervancontaining the N-mustard chlorambucil (1) (22) (Scheme3). This original conjugate was designed to bind in theminor groove of helical DNA containing 5′-(A/T)GC(A/T)GC(A/T)-3′ and cross-link neighboring sequences withits N-mustard attached to a central γ-aminobutyric acid(22). Synthesis of the polyamide was first accomplishedby manual solid-phase synthesis (22, 38) and repeatedin our laboratory in comparable yield. The N-mustardwas also coupled to this polyamide as described by Wurtzand Dervan (22).
Coupling between the available quinone methide pre-cursor 2 and the polyamide yielded a conjugate (5) thatcontained one less methylene unit in its linker than 1(Scheme 3). Consequently, a series of additional quinonemethide precursors were also prepared to more closelymimic the distance between the polyamide and itsattached N-mustard in 1. First, the alkyl chain connect-
ing the polyamide and quinone methide precursor wasextended by one carbon (6) to match the distance betweenthe polyamide and aromatic ring of chlorambucil. Second,a longer extension (7) was generated to approach moreclosely the 11-atom distance between the polyamide of 1and the site of nucleophilic addition by DNA on theN-mustard. In all cases, the linkers provided conforma-tional flexibility for sampling numerous sites of potentialreactivity. A contrasting analogue containing a conju-gated vinyl linker was additionally prepared (8) asdescribed previously (37) since this type of linkage haddemonstrated great effectiveness in other polyamideconjugates (16, 41, 42).
The activated esters 3 and 4 used for coupling to thepolyamide were constructed using the same strategydeveloped for the original bis-functional quinone methideprecursor 2 (35). The starting material 9a was formedby demethylation of 4-(4′-methoxyphenyl)butyric acid,and 9b was obtained from a Friedel-Craft acylation ofphenol by monomethyl adipoyl chloride and subsequentClemmensen reduction (43, 44). These materials werehydroxymethylated with formaldehyde under basic con-ditions and then silylated with tert-butyldimethylsilylchloride (TBS-Cl) under standard conditions to yield10a,b (Scheme 4) (35). The benzylic positions wereselectively substituted with acetate by treatment withacetic anhydride and FeCl3 (45), and finally the carboxylicacids were converted to their N-hydroxysuccinimideesters 3 and 4 in the presence of EDC. These activatedesters were condensed with the polyamide in a mixtureof CH3CN, DMF, and DIEA to yield the desired conju-gates capable of generating quinone methide intermedi-ates. Analysis by reverse phase (C-18) HPLC indicatedthat all polyamide amine was consumed in the presenceof the N-hydroxysuccinimide esters within 1 h and morethan 50% had already reacted within the first 10 min.This analysis also indicated that the conjugate formedin yields of up to 65%. However, product recovery afterreverse-phase chromatography was quite low and sug-gested problems with the final steps of isolation. Thismethod of purification still generated sufficient materialfor the studies below, and consequently these procedureswere not optimized. The molecular weights of eachconjugate were confirmed by electrospray mass spectros-copy.
Alkylation and Cross-Linking of a Target DNAUsing N-Mustard and Quinone Methide Conju-gates. The original N-mustard chlorambucil conjugate1 was targeted for selective reaction on either side of theTATA box of a HIV promoter and tested with a restrictionfragment of DNA that offered a range of sequencevariants (22). Indeed, the predicted sites were alkylated
Scheme 4
918 Bioconjugate Chem., Vol. 15, No. 4, 2004 Kumar et al.
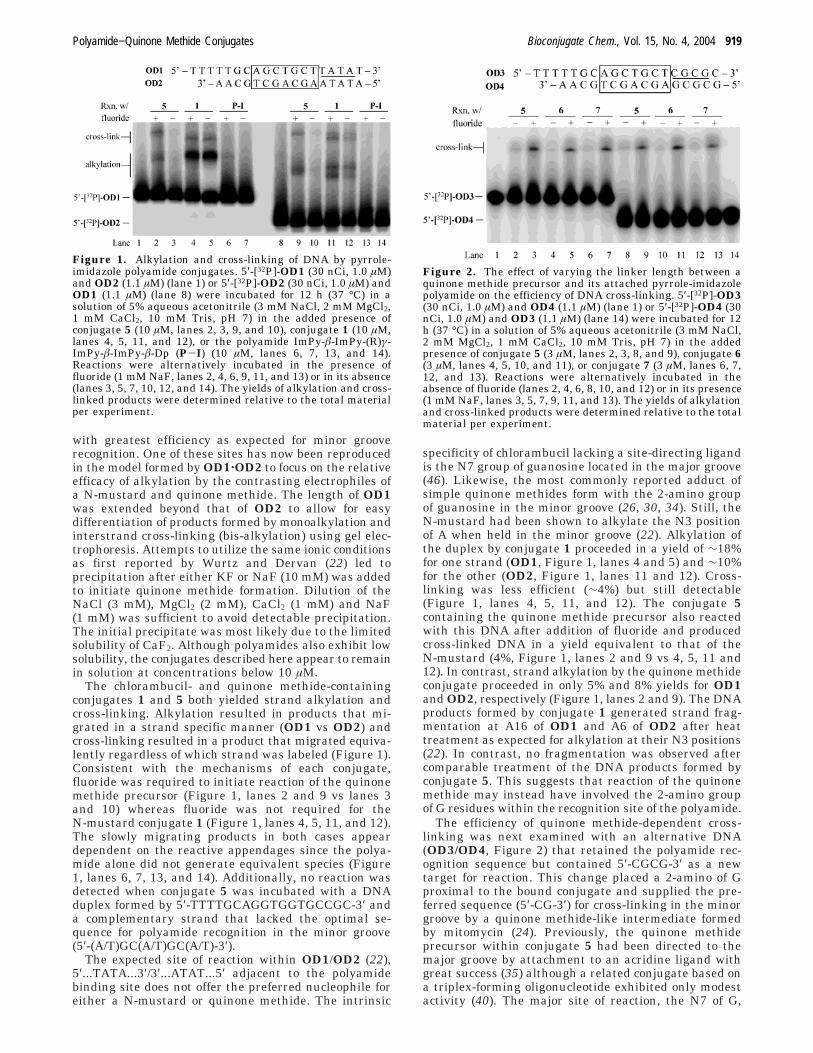
with greatest efficiency as expected for minor grooverecognition. One of these sites has now been reproducedin the model formed by OD1‚OD2 to focus on the relativeefficacy of alkylation by the contrasting electrophiles ofa N-mustard and quinone methide. The length of OD1was extended beyond that of OD2 to allow for easydifferentiation of products formed by monoalkylation andinterstrand cross-linking (bis-alkylation) using gel elec-trophoresis. Attempts to utilize the same ionic conditionsas first reported by Wurtz and Dervan (22) led toprecipitation after either KF or NaF (10 mM) was addedto initiate quinone methide formation. Dilution of theNaCl (3 mM), MgCl2 (2 mM), CaCl2 (1 mM) and NaF(1 mM) was sufficient to avoid detectable precipitation.The initial precipitate was most likely due to the limitedsolubility of CaF2. Although polyamides also exhibit lowsolubility, the conjugates described here appear to remainin solution at concentrations below 10 µM.
The chlorambucil- and quinone methide-containingconjugates 1 and 5 both yielded strand alkylation andcross-linking. Alkylation resulted in products that mi-grated in a strand specific manner (OD1 vs OD2) andcross-linking resulted in a product that migrated equiva-lently regardless of which strand was labeled (Figure 1).Consistent with the mechanisms of each conjugate,fluoride was required to initiate reaction of the quinonemethide precursor (Figure 1, lanes 2 and 9 vs lanes 3and 10) whereas fluoride was not required for theN-mustard conjugate 1 (Figure 1, lanes 4, 5, 11, and 12).The slowly migrating products in both cases appeardependent on the reactive appendages since the polya-mide alone did not generate equivalent species (Figure1, lanes 6, 7, 13, and 14). Additionally, no reaction wasdetected when conjugate 5 was incubated with a DNAduplex formed by 5′-TTTTGCAGGTGGTGCCGC-3′ anda complementary strand that lacked the optimal se-quence for polyamide recognition in the minor groove(5′-(A/T)GC(A/T)GC(A/T)-3′).
The expected site of reaction within OD1/OD2 (22),5′...TATA...3′/3′...ATAT...5′ adjacent to the polyamidebinding site does not offer the preferred nucleophile foreither a N-mustard or quinone methide. The intrinsic
specificity of chlorambucil lacking a site-directing ligandis the N7 group of guanosine located in the major groove(46). Likewise, the most commonly reported adduct ofsimple quinone methides form with the 2-amino groupof guanosine in the minor groove (26, 30, 34). Still, theN-mustard had been shown to alkylate the N3 positionof A when held in the minor groove (22). Alkylation ofthe duplex by conjugate 1 proceeded in a yield of ∼18%for one strand (OD1, Figure 1, lanes 4 and 5) and ∼10%for the other (OD2, Figure 1, lanes 11 and 12). Cross-linking was less efficient (∼4%) but still detectable(Figure 1, lanes 4, 5, 11, and 12). The conjugate 5containing the quinone methide precursor also reactedwith this DNA after addition of fluoride and producedcross-linked DNA in a yield equivalent to that of theN-mustard (4%, Figure 1, lanes 2 and 9 vs 4, 5, 11 and12). In contrast, strand alkylation by the quinone methideconjugate proceeded in only 5% and 8% yields for OD1and OD2, respectively (Figure 1, lanes 2 and 9). The DNAproducts formed by conjugate 1 generated strand frag-mentation at A16 of OD1 and A6 of OD2 after heattreatment as expected for alkylation at their N3 positions(22). In contrast, no fragmentation was observed aftercomparable treatment of the DNA products formed byconjugate 5. This suggests that reaction of the quinonemethide may instead have involved the 2-amino groupof G residues within the recognition site of the polyamide.
The efficiency of quinone methide-dependent cross-linking was next examined with an alternative DNA(OD3/OD4, Figure 2) that retained the polyamide rec-ognition sequence but contained 5′-CGCG-3′ as a newtarget for reaction. This change placed a 2-amino of Gproximal to the bound conjugate and supplied the pre-ferred sequence (5′-CG-3′) for cross-linking in the minorgroove by a quinone methide-like intermediate formedby mitomycin (24). Previously, the quinone methideprecursor within conjugate 5 had been directed to themajor groove by attachment to an acridine ligand withgreat success (35) although a related conjugate based ona triplex-forming oligonucleotide exhibited only modestactivity (40). The major site of reaction, the N7 of G,
Figure 1. Alkylation and cross-linking of DNA by pyrrole-imidazole polyamide conjugates. 5′-[32P]-OD1 (30 nCi, 1.0 µM)and OD2 (1.1 µM) (lane 1) or 5′-[32P]-OD2 (30 nCi, 1.0 µM) andOD1 (1.1 µM) (lane 8) were incubated for 12 h (37 °C) in asolution of 5% aqueous acetonitrile (3 mM NaCl, 2 mM MgCl2,1 mM CaCl2, 10 mM Tris, pH 7) in the added presence ofconjugate 5 (10 µM, lanes 2, 3, 9, and 10), conjugate 1 (10 µM,lanes 4, 5, 11, and 12), or the polyamide ImPy-â-ImPy-(R)γ-ImPy-â-ImPy-â-Dp (P-I) (10 µM, lanes 6, 7, 13, and 14).Reactions were alternatively incubated in the presence offluoride (1 mM NaF, lanes 2, 4, 6, 9, 11, and 13) or in its absence(lanes 3, 5, 7, 10, 12, and 14). The yields of alkylation and cross-linked products were determined relative to the total materialper experiment.
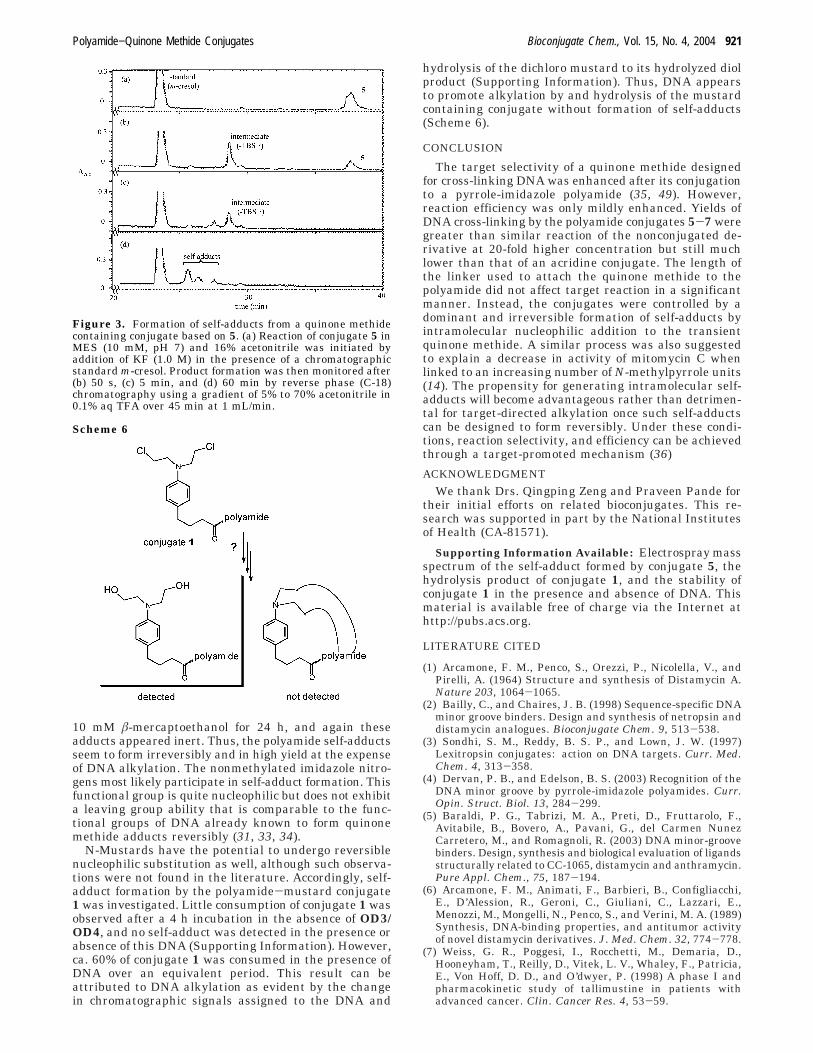
Figure 2. The effect of varying the linker length between aquinone methide precursor and its attached pyrrole-imidazolepolyamide on the efficiency of DNA cross-linking. 5′-[32P]-OD3(30 nCi, 1.0 µM) and OD4 (1.1 µM) (lane 1) or 5′-[32P]-OD4 (30nCi, 1.0 µM) and OD3 (1.1 µM) (lane 14) were incubated for 12h (37 °C) in a solution of 5% aqueous acetonitrile (3 mM NaCl,2 mM MgCl2, 1 mM CaCl2, 10 mM Tris, pH 7) in the addedpresence of conjugate 5 (3 µM, lanes 2, 3, 8, and 9), conjugate 6(3 µM, lanes 4, 5, 10, and 11), or conjugate 7 (3 µM, lanes 6, 7,12, and 13). Reactions were alternatively incubated in theabsence of fluoride (lanes 2, 4, 6, 8, 10, and 12) or in its presence(1 mM NaF, lanes 3, 5, 7, 9, 11, and 13). The yields of alkylationand cross-linked products were determined relative to the totalmaterial per experiment.
Polyamide−Quinone Methide Conjugates Bioconjugate Chem., Vol. 15, No. 4, 2004 919

forms a reversible adduct. Alternative delivery of thequinone methide to the minor groove was expected tosupport high levels of reaction due to the potentialadvantages of localizing the quinone methide proximalto the 2-amino group of guanosine that generates a stableadduct irreversibly (30, 31). Surprisingly, the quinonemethide conjugate 5 also cross-linked its new target OD3/OD4 with a low yield (∼5%) and did not generate mono-alkylation products detectably (Figure 2, lanes 3 and 9).
Many parameters could have impeded activity of thepolyamide conjugate including the length and flexibilityof the linker. While a long molecular spacer may allowaccess to many potential sites of reaction, the principleconformers of the spacer may not necessarily coincidewith those most reactive. In contrast, a short andrestricted spacer may stabilize a single conformer, butthis may or may not necessarily represent the mostreactive alignment. Certainly, the various linkers usedfor attaching N-mustards to ligands such as acridine (47)and triplex-forming oligonucleotides (40, 48) dramaticallyaffected target alkylation, and hence additional quinonemethide conjugates were prepared for more precisecomparison with the related polyamide-N-mustard con-jugate 1. The quinone methide conjugate 5 was extendedby one carbon unit to form conjugate 6 containing thesame four carbon link to the aromatic ring as that in theoriginal N-mustard conjugate 1. Another quinone me-thide conjugate 7 was prepared to mimic more closelythe 11 atom spacing between the sites of ligand attach-ment and DNA alkylation of 1. Surprisingly, neitherchange effected the apparent activity of the attachedquinone methide. Conjugates 6 and 7 both cross-linkedOD3/OD4 in low yield (3-6%) and no monoalkylatedderivatives were apparent in either case (Figure 2).
A short and rigid acrylamide linker has also been usedto join various reactive appendages to the termini ofdistamycin-like ligands (16, 41, 42). The successes achievedwith this design prompted construction of an equivalentquinone methide derivative (8) linked to the hairpinrather than terminal region of the polyamide as usedpreviously (37). However, incubation of conjugate 8 underconditions equivalent to those used for conjugates 1 and5-7 generated no observable alkylation or cross-linkingof OD3/OD4 (data not shown). This may reflect adecrease in the stability of the TBS-protected precursorrelative to its saturated derivative 5 (37). Alternatively,the modest activity of the pyrrole-imidazole polyamideconjugates 5-7 and even the lack of reaction of 8 mightinstead have originated from a common characteristicshared by all of these quinone methide derivatives.
Intramolecular Trapping of the Quinone MethideLimits Its Reaction with DNA. Quinone methideintermediates are highly unstable and have the potentialto react with a range of nucleophiles including thosepresent in the attached site-directing ligand. Such in-tramolecular trapping would typically suppress reactionwith the intended target, but the reversible nature of thequinone methide self-adducts formed with DNA allowssubsequent transfer of the reactive intermediate from anintra- to intermolecular position (36). This process ofreversible intramolecular trapping now serves a verybeneficial role in an appealing new strategy for targetpromoted alkylation. The nucleophilic groups of thepolyamide like those of DNA may also form intramolecu-lar adducts with the attached quinone methide. Whetherreaction with the polyamide suppresses or maintainstarget selective alkylation of DNA depends on the re-versibility of possible self-adducts. The lack of efficientcross-linking and alkylation of DNA by the conjugates
5-8, however, began to suggest that intramolecularadducts were forming and not capable of regeneratingthe quinone methide.
Reaction between the quinone methide of conjugate 5and its pyrrole-imidazole polyamide were consequentlyexamined by HPLC and electrospray mass spectrometryin a manner equivalent to that used to detect the self-adducts of an oligonucleotide-quinone methide conjugate(36). Within 12 h, conjugate 5 was completely consumedin the presence, but not absence, of 10 mM KF underneutral conditions, and several new products were de-tected by HPLC. Each exhibited a m/z of 1554 which isconsistent with loss of the silyl protecting group andsubstitution of both benzylic acetate groups with internalnucleophiles. No additional products were indicated bymass spectroscopy including no ions consistent with asingle intramolecular substitution and addition of onewater (m/z 1572), addition of two waters (m/z 1590), orother possible products (Scheme 5) (Supporting Informa-tion). HPLC-MS analysis was also repeated using veryhigh concentrations of fluoride (1 M KF) in order tomonitor the deprotected quinone methide precursor(- TBS) and its transformation to the self-adducts(Figure 3). Under these conditions, ∼60% of conjugate 5converted within 50 s to a transient intermediate that isconsistent with its deprotected derivative. Within 5 min,signals of the self-adducts appeared and by 60 min theprofile of self-adducts remained constant. Self-adductformation within the polyamide conjugate occurred muchmore rapidly than that observed for related oligonucle-otide conjugates (36). Both types of conjugates appear toform a diversity of self-adducts that likely result fromreaction with various nucleophiles along the attachedDNA ligand.
Generation of self-adducts of conjugate 5 would notnecessarily preclude subsequent reaction with a targetDNA as long as the adducts formed reversibly and thepolyamide self-adducts could still recognize the targetDNA. Conformational constraints created by intramo-lecular nucleophilic addition are likely to inhibit selectivebinding but affinity should be regained upon quinonemethide regeneration. Without such regeneration, nei-ther binding nor alkylation of DNA would likely bepossible. First, the stability of the polyamide self-adductsin the presence of a stoichiometric concentration of duplexDNA (0.3 µM, OD3/OD4) was monitored over 3 days. Nochange in the profile or quantity of self-adducts wasdetected by HPLC, and concurrently no alkylation orcross-linking of DNA was detected by gel electrophoresis.Next, the self-adducts were incubated in the presence of
Scheme 5
920 Bioconjugate Chem., Vol. 15, No. 4, 2004 Kumar et al.
10 mM â-mercaptoethanol for 24 h, and again theseadducts appeared inert. Thus, the polyamide self-adductsseem to form irreversibly and in high yield at the expenseof DNA alkylation. The nonmethylated imidazole nitro-gens most likely participate in self-adduct formation. Thisfunctional group is quite nucleophilic but does not exhibita leaving group ability that is comparable to the func-tional groups of DNA already known to form quinonemethide adducts reversibly (31, 33, 34).
N-Mustards have the potential to undergo reversiblenucleophilic substitution as well, although such observa-tions were not found in the literature. Accordingly, self-adduct formation by the polyamide-mustard conjugate1 was investigated. Little consumption of conjugate 1 wasobserved after a 4 h incubation in the absence of OD3/OD4, and no self-adduct was detected in the presence orabsence of this DNA (Supporting Information). However,ca. 60% of conjugate 1 was consumed in the presence ofDNA over an equivalent period. This result can beattributed to DNA alkylation as evident by the changein chromatographic signals assigned to the DNA and
hydrolysis of the dichloro mustard to its hydrolyzed diolproduct (Supporting Information). Thus, DNA appearsto promote alkylation by and hydrolysis of the mustardcontaining conjugate without formation of self-adducts(Scheme 6).
CONCLUSION
The target selectivity of a quinone methide designedfor cross-linking DNA was enhanced after its conjugationto a pyrrole-imidazole polyamide (35, 49). However,reaction efficiency was only mildly enhanced. Yields ofDNA cross-linking by the polyamide conjugates 5-7 weregreater than similar reaction of the nonconjugated de-rivative at 20-fold higher concentration but still muchlower than that of an acridine conjugate. The length ofthe linker used to attach the quinone methide to thepolyamide did not affect target reaction in a significantmanner. Instead, the conjugates were controlled by adominant and irreversible formation of self-adducts byintramolecular nucleophilic addition to the transientquinone methide. A similar process was also suggestedto explain a decrease in activity of mitomycin C whenlinked to an increasing number of N-methylpyrrole units(14). The propensity for generating intramolecular self-adducts will become advantageous rather than detrimen-tal for target-directed alkylation once such self-adductscan be designed to form reversibly. Under these condi-tions, reaction selectivity, and efficiency can be achievedthrough a target-promoted mechanism (36)
ACKNOWLEDGMENT
We thank Drs. Qingping Zeng and Praveen Pande fortheir initial efforts on related bioconjugates. This re-search was supported in part by the National Institutesof Health (CA-81571).
Supporting Information Available: Electrospray massspectrum of the self-adduct formed by conjugate 5, thehydrolysis product of conjugate 1, and the stability ofconjugate 1 in the presence and absence of DNA. Thismaterial is available free of charge via the Internet athttp://pubs.acs.org.
LITERATURE CITED
(1) Arcamone, F. M., Penco, S., Orezzi, P., Nicolella, V., andPirelli, A. (1964) Structure and synthesis of Distamycin A.Nature 203, 1064-1065.
(2) Bailly, C., and Chaires, J. B. (1998) Sequence-specific DNAminor groove binders. Design and synthesis of netropsin anddistamycin analogues. Bioconjugate Chem. 9, 513-538.
(3) Sondhi, S. M., Reddy, B. S. P., and Lown, J. W. (1997)Lexitropsin conjugates: action on DNA targets. Curr. Med.Chem. 4, 313-358.
(4) Dervan, P. B., and Edelson, B. S. (2003) Recognition of theDNA minor groove by pyrrole-imidazole polyamides. Curr.Opin. Struct. Biol. 13, 284-299.
(5) Baraldi, P. G., Tabrizi, M. A., Preti, D., Fruttarolo, F.,Avitabile, B., Bovero, A., Pavani, G., del Carmen NunezCarretero, M., and Romagnoli, R. (2003) DNA minor-groovebinders. Design, synthesis and biological evaluation of ligandsstructurally related to CC-1065, distamycin and anthramycin.Pure Appl. Chem., 75, 187-194.
(6) Arcamone, F. M., Animati, F., Barbieri, B., Configliacchi,E., D’Alession, R., Geroni, C., Giuliani, C., Lazzari, E.,Menozzi, M., Mongelli, N., Penco, S., and Verini, M. A. (1989)Synthesis, DNA-binding properties, and antitumor activityof novel distamycin derivatives. J. Med. Chem. 32, 774-778.
(7) Weiss, G. R., Poggesi, I., Rocchetti, M., Demaria, D.,Hooneyham, T., Reilly, D., Vitek, L. V., Whaley, F., Patricia,E., Von Hoff, D. D., and O’dwyer, P. (1998) A phase I andpharmacokinetic study of tallimustine in patients withadvanced cancer. Clin. Cancer Res. 4, 53-59.
Figure 3. Formation of self-adducts from a quinone methidecontaining conjugate based on 5. (a) Reaction of conjugate 5 inMES (10 mM, pH 7) and 16% acetonitrile was initiated byaddition of KF (1.0 M) in the presence of a chromatographicstandard m-cresol. Product formation was then monitored after(b) 50 s, (c) 5 min, and (d) 60 min by reverse phase (C-18)chromatography using a gradient of 5% to 70% acetonitrile in0.1% aq TFA over 45 min at 1 mL/min.
Scheme 6
Polyamide−Quinone Methide Conjugates Bioconjugate Chem., Vol. 15, No. 4, 2004 921

(8) Wyatt, M. D., Lee, M., and Hartley, J. A. (1997) Thesequence specificity of alkylation for a series of benzoic acidmustard and imidazole-containing distamycin analogues: theimportance of local sequence conformation. Nucleic Acids Res.25, 2359-2364.
(9) Brooks, N., McHugh, P. J., Lee, M., and Hartley, J. A. (2000)Alteration in the choice of DNA repair pathway with increas-ing sequence selective DNA alkylation in the minor groove.Chem. Biol. 7, 659-668.
(10) Baraldi, P. G., Romagnoli, R., Pavani, M. G., del CarmenNunez, M., Bingham, J. P., and Hartley, J. A. (2002) Benzoyland cinnamoyl nitrogen mustard derivatives of benzohetero-cyclic analogues of the tallimustine: synthesis and antitu-mour activity. Bioorg. Med. Chem. 10, 1611-1618.
(11) Wang, Y., Wright, S. C., and Larrick, J. W. (2003) Synthesisand preliminary cytotoxicity of nitrogen mustard derivativesof distamycin A. Bioorg. Med. Chem. Lett. 13, 459-461.
(12) Woo, J., Sigurdsson, S. T., and Hopkins, P. B. (1993) DNAinterstrand cross-linking reactions of pyrrole-derived bifunc-tional electrophiles: evidence of a common target site in DNA.J. Am. Chem. Soc. 115, 3407-3415.
(13) Encell, L., Shuker, D. E. G., Foiles, P. G., and Gold, B.(1996) The in vitro methylation of DNA by a minor groovebinding methyl sulfonate ester. Chem. Res. Toxicol. 9, 563-567.
(14) Paz, M. M., Das, A., and Tomasz, M. (1999) Mitomycin Clinked to DNA minor groove binding agents: synthesis,reductive activation, DNA binding and cross-linking proper-ties and in vitro antitumor activity. Bioorg. Med. Chem. 7,2713-2726.
(15) Damayanthi, Y., Reddy, B. S. P., and Lown, J. W. (1999)Design and synthesis of novel pyrrolo[2.1-c][1,4]benzodiazepine-lexitropsin conjugates. J. Org. Chem. 64, 290-292.
(16) Fregeau, N. L., Wang, Y., Pon, R. T., Wylie, W. A., andLown, J. W. (1995) Characterization of a CPI-lexitropsinconjugate-oligonucleotide covalent complex by 1H NMR andrestrained molecular dynamics simulation. J. Am. Chem. Soc.117, 8917-8925.
(17) Pelton, J. G., and Wemmer, D. E. (1989) Structuralcharacterization of a 2: 1 distamycin A-d(CGCAAATTGGC)complex by two-dimensional NMR. Proc. Natl. Acad. Sci.U.S.A. 86, 5723-5727.
(18) Dervan, P. B. (2001) Molecular recognition of DNA by smallmolecules. Bioorg. Med. Chem. 9, 2215-2235.
(19) Kielkopf, C. L., Baird, E. E., Dervan, P. B., and Rees, D.C. (1998) Structural basis for G-C recognition in the DNAminor groove. Nat. Struct. Biol. 5, 104-109.
(20) Kielkopf, C. L., White, S., Szewczyk, J. W., Turner, J. M.,Baird, E. E., Dervan, P. B., and Rees, D. C. (1998) A structuralbasis for recognition of A-T and T-A base pairs in the minorgroove of B-DNA. Science 282, 111-115.
(21) Tao, Z.-F., Fujiwara, T., Saito, I., and Sugiyama, H. (1999)Rational design of sequence-specific DNA alkylating agentsbased on Duocarmycin A and pyrrole-imidazole hairpinpolyamides. J. Am. Chem. Soc. 121, 4961-4967.
(22) Wurtz, N. R., and Dervan, P. B. (2000) Sequence specificalkylation of DNA by hairpin pyrrole-imidazole polyamideconjugates. Chem. Biol. 7, 153-161.
(23) Chang, A. Y., and Dervan, P. B. (2000) Strand selectivecleavage of DNA by diastereomers of hairpin polyamide-seco-CBI conjugates. J. Am. Chem. Soc. 122, 4865-4864.
(24) Tomasz, M. (1995) Mitomycin C: small, fast and deadly(but very selective). Chem. Biol. 2, 575-579.
(25) Fan, P. W., Zhang, F., and Bolton, J. L. (2000) 4-Hydroxy-lated metabolites of the antiestrogens tamoxifen andtoremifene are metabolized to unusually stable quinonemethides. Chem. Res. Toxicol. 13, 45-52.
(26) Lewis, M. A., Yoerg, D. G., Bolton, J. L., and Thompson,J. A. (1996) Alkylation of 2′-deoxynucleosides and DNA byquinone methides derived from 2,6-di-tert-butyl-4-methylphe-nol. Chem. Res. Toxicol. 9, 1368-1374.
(27) Morris, B. D., and Prinsep, M. R. (1998) Euthyroideones,novel brominated quinone methides from the Bryozoan Eu-thyroides episcopalis. J. Org. Chem. 63, 9545-9547.
(28) Zjawiony, J. K., Bartyzel, P., and Hamann, M. T. (1998)Chemistry of puupehenone: 1,6-conjugate addition to itsquinone methide system. J. Nat. Prod. 61, 1502-1508.
(29) Pande, P., Shearer, J., Yang, J., Greenberg, W. A., andRokita, S. E. (1999) Alkylation of nucleic acids by a modelquinone methide. J. Am. Chem. Soc. 121, 6773-6779.
(30) Veldhuyzen, W., Lam, Y.-f., and Rokita, S. E. (2001)2-Deoxyguanosine reacts with a model quinone methide atmultiple sites. Chem. Res. Toxicol. 14, 1345-1351.
(31) Veldhuyzen, W. F., Shallop, A. J., Jones, R. A., and Rokita,S. E. (2001) Thermodynamic versus kinetic products of DNAalkylation as modeled by reaction of deoxyadenosine. J. Am.Chem. Soc. 123, 11126-11132.
(32) Zhou, Q., and Turnbull, K. D. (2001) Quinone methidephosphodiester alkylation under aqueous conditions. J. Org.Chem. 66, 7072-7077.
(33) Freccero, M., Di Valentin, C., and Sarzi-AmadP, M. (2003)Modeling H-bonding and solvent effects in the alkylation ofpyrimidine bases by prototype quinone methide. A DFT study.J. Am. Chem. Soc. 125, 3544-3553.
(34) Freccero, M., Gandolfi, R., and Sarzi-AmadP, M. (2003)Selectivity of purine alkylation by a quinone methide. Kineticor thermodynamic control? J. Org. Chem. 68, 6411-6423.
(35) Veldhuyzen, W. F., Pande, P., and Rokita, S. E. (2003) Atransient product of DNA alkylation can be stabilized bybinding localization. J. Am. Chem. Soc. 125, 14005-14013.
(36) Zhou, Q., and Rokita, S. E. (2003) A general strategy fortarget-promoted alkylation in biological systems. Proc. Natl.Acad. Sci. U.S.A. 100, 15452-15457.
(37) Kumar, D., and Rokita, S. E. (2004) Synthesis of a hairpinpyrrole-imidazole polyamide conjugate containing a quinonemethide precursor and vinyl linking group. Tetrahedron Lett.45, 2887-2889.
(38) Baird, E. E., and Dervan, P. B. (1996) Solid-phase synthesisof polyamides containing imidazole and pyrrole amino acids.J. Am. Chem. Soc. 118, 6141-6146.
(39) Ellington, A., and Pollard, J. D. (1998) in Current Protocolsin Molecular Biology (Ausuble, F. M., Brent, R., Kingston,R. E., Moore, D. D., Seidman, J. G., Smith, J. A., and Struhl,K., Eds.) section 2.12, John Wiley and Sons, New York.
(40) Zhou, Q., Pande, P., Johnson, A. E., and Rokita, S. E. (2001)Sequence-specific delivery of a quinone methide intermediateto the major groove of DNA. Bioorg. Med. Chem. 9, 2347-2354.
(41) Tao, Z.-F., Saito, I., and Sugiyama, H. (2000) Highlycooperative DNA dialkylation by the homodimer of imidiazole-pyrrole diamide-CPI conjugate with vinyl linker. J. Am.Chem. Soc. 122, 1602-1608.
(42) Bando, T., Narita, A., Saito, I., and Sugiyama, H. (2002)Molecular design of a pyrrole-imidazole hairpin polyamidesfor effective DNA alkylation. Chem. Eur. J. 8, 4781-4790.
(43) Chu, L., Hutchins, J. E., Weber, A. E., Lo, J.-L., Yang, Y.-T., Cheng, K., Smith, R. G., Fisher, M. H., Wyvratt, M. J.,and Goulet, M. T. (2001) Initial structure-activity relation-ship of a novel class of nonpeptidyl GnRH receptor antago-nists: 2-arylindoles. Bioorg. Med. Chem. Lett. 11, 509-513.
(44) Aultz, D. E., and McFadden, A. R. (1977) Dibenz[b,e]-oxepinalkanoic acids as nonsteroidal antiinflammatory agents.3. ω-(6,11-dihydro-11-oxodibenz[b,e]oxepin-2-yl)alkanoic ac-ids. J. Med. Chem. 20, 1499-1501.
(45) Ganem, B., and Small, V. R. (1974) Ferric chloride in aceticanhydride. A mild and versatile reagent for the cleavage ofethers. J. Org. Chem. 39, 3728-3730.
(46) Kohn, K. W., Hartley, J. A., and Mattes, W. B. (1987)Mechanisms of DNA sequence selective alkylation of guanine-N7 positions by nitrogen mustards. Nucleic Acids Res. 15,10531-10549.
(47) Boritzki, T. J., Palmer, B. D., Coddington, J. M., andDenny, W. A. (1994) Identification of the major lesion fromthe reaction of an acridine-targeted aniline mustard withDNA as an adenine N1 adduct. Chem. Res. Toxicol. 7, 41-46.
(48) Reed, M. W., Lukhtanov, E. A., Gorn, V., Dutyavin, I., Gall,A., Wald, A., and Meyer, R. B. (1998) Synthesis and reactivityof aryl nitrogen mustard-oligodeoxyribonucleotide conjugates.Bioconjugate Chem. 9, 64-71.
(49) Zeng, Q., and Rokita, S. E. (1996) Tandem quinone methidegeneration for cross-linking DNA. J. Org. Chem. 61, 9080-9081.
BC049941H
922 Bioconjugate Chem., Vol. 15, No. 4, 2004 Kumar et al.