conceptos básicos de farmacocinética clínica€¦ · web view · 2008-12-16introducciÓn. el...
TRANSCRIPT

Curso de Farmacología Clínica Año 2008
FARMACOCINÉTICA CLÍNICA
INTRODUCCIÓNEl concepto de farmacocinéticaLos conocimientos descriptivos y teóricos de la farmacología clínica, han experimentado
durante la pasada década un avance extraordinario. Al profesional veterinario que trabaja en
una determinada área de la ciencia médica, le resulta difícil mantenerse al corriente de las
publicaciones sobre los últimos avances de su especialidad. De allí que para mantenerse
actualizado sea imperioso que tenga acceso a libros y artículos de revisión que profundicen en
temáticas concretas.
El conocimiento de la disposición de una molécula de medicamento en los distintos fluidos
corporales, órganos y tejidos, constituye el primer paso para comprender la capacidad de éste
para llegar al sitio de acción, concentrarse en cantidad suficiente y por el tiempo necesario para
poder ejercer su efecto antes de ser eliminado del organismo.
La disposición de la molécula de un medicamento dentro del organismo es equivalente a un
“viaje” que ésta realiza a través del mismo. Las vías de administración, serían el equivalente de
las diferentes vías de acceso a un circuito turístico, y de la misma manera, unas permitirían que
las moléculas del medicamento ingresaran al organismo de forma más rápida o más fácil que
otras.
Este concepto de “viaje” o “periplo” del medicamento, es el concepto básico que debemos
retener para comprender el sentido del término “farmacocinética”, ya que el mismo supone la
aplicación de los principios de la cinética o movimiento al vocablo griego “φαρμαχον”
(pharmakon) que se utilizaba en la antigüedad para designar tanto a venenos como
medicamentos.
Tanto el término como el concepto de farmacocinética, fueron empleados por primera vez en
Alemania en el año 1905, por el profesor F. H. Dost, quien definió a la farmacocinética como:
“La ciencia del análisis cuantitativo entre organismo y medicamento”.
En esta definición se hace explícito el primer aspecto importante de la farmacocinética, que es
el estudio de la relación entre organismo y medicamento, ya que considerando los procesos a
los que éste es sometido (absorción, distribución, biotransformación y eliminación), la
farmacocinética puede ser también definida como la ciencia que estudia “lo que el organismo le
hace al medicamento”.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
1

Curso de Farmacología Clínica Año 2008
El segundo aspecto lo constituye la cuantificación de los procesos anteriormente mencionados,
es decir, la cuantificación de la magnitud y la velocidad a la cual se producen los procesos de
absorción, distribución, biotransformación y eliminación.
A manera de síntesis, podemos decir que el objetivo de la farmacocinética es estudiar la
evolución temporal de las concentraciones y cantidades de los medicamentos y sus metabolitos
en los fluidos biológicos y excretas, y construir modelos adecuados para interpretar los datos
obtenidos. Para lograr esto último se emplea una representación matemática o modelo de una
parte o la totalidad del organismo para poder de esa manera:
a- Reducir los datos experimentales (concentración del medicamento en fluidos biológicos
o tejidos) a cifras o parámetros que puedan tener una significación biológica.
b- Emplear esta información para formular predicciones acerca de los resultados de futuros
ensayos como ser; diferentes dosis o intervalos entre dosis.
De lo anterior, se deduce que el objetivo primordial de los estudios farmacocinéticos, es el
generar información para optimizar el empleo clínico de los fármacos en la consecución del
efecto terapéutico deseado.
Farmacocinética clínicaLa farmacocinética clínica, es la rama de la farmacología que se ocupa del estudio científico de
los medicamentos en el hombre y los animales, y sus objetivos son:
a- Mejorar la atención al paciente en el sentido de promover la utilización más segura y
eficaz de los medicamentos.
b- Incrementar los conocimientos de la terapia farmacológica a través de la investigación.
c- Transmitir los conocimientos adquiridos a través de la enseñanza.
Para poder diseñar u optimizar los regímenes terapéuticos, se deben conocer en detalle los
factores fisiológicos y patológicos que afectan la disposición de los fármacos (absorción,
distribución, metabolismo y excreción) en un organismo viviente.
En lo que respecta a nuestra profesión, es innegable el gran desafío, que de por si representa,
la variabilidad de los mencionados procesos respecto del factor especie. Como un ejemplo
representativo de lo mencionado, podemos mencionar a la especie bovina, la que durante sus
primeros meses de vida se comporta como un monogástrico para luego presentar la fisiología
digestiva de un poligástrico, lo que no solo modifica la posibilidad de la vía oral como una
opción para administrar un medicamento, sino también la variación del pH de la orina (ácida en
el neonato y alcalina en el adulto), lo que modifica diametralmente el patrón de eliminación y
reabsorción de fármacos.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
2

Curso de Farmacología Clínica Año 2008
Otro un ejemplo representativo es la deficiente capacidad del gato para formar conjugados con
el ácido glucurónico, lo cual modifica considerablemente el patrón de eliminación y reabsorción
de ciertos fármacos en esta especie.
Como consecuencia de lo explicado anteriormente se deduce que no siempre es posible aplicar
los mismos esquemas posológicos ni emplear el mismo arsenal terapéutico en diferentes
especies, como por ejemplo perros y gatos, ni tampoco en un individuo de una determinada
especie a lo largo de sus diferentes etapas de vida.
En términos más simplistas, la farmacocinética clínica es la disciplina de las ciencias sanitarias
que se basa en la aplicación de los principios farmacocinéticos para el tratamiento seguro y
eficaz de los pacientes mediante:
a- Programación inicial de los regímenes de dosificación, vías de administración, formas de
dosificación (en algunos casos individualizada), basándose en los conocimientos
generales que al presente se disponen acerca de la farmacocinética de cada
medicamento, el conocimiento de la fisiopatología de la enfermedad, el fin que se
persigue con la medicación y el análisis de variables tales como; especie, edad, sexo,
estado corporal (obesidad, emaciación), influencia farmacogenética, función renal y
función hepática entre otras.
b- Perfeccionamiento y ajuste de los regímenes de dosificación empleando como base el
monitoreo de las concentraciones séricas del medicamento u en otros fluidos corporales.
En otros casos el ajuste de la posología puede realizarse mediante medición directa de
la respuesta clínica, o mediante la realización de evaluaciones indirectas de la misma
como por ejemplo; las basadas en la modificación de ciertos parámetros bioquímicos.
VARIABILIDAD DE LA RESPUESTA A LOS FÁRMACOSEs bien conocido en estadística, que mediciones repetidas de una misma variable nunca
proporcionan resultados idénticos. Esto es totalmente válido para las denominadas ciencias
exactas como para las ciencias biológicas. Una de las causas de esta variabilidad la constituye
los errores de medición. Existen dos clases de error en la medición; los errores sistemáticos y
los errores aleatorios o al azar.
Los errores sistemáticos están asociados a la exactitud del método empleado para la
realización de la misma. Los errores de este tipo nos proporcionan resultados o valores
demasiado altos o demasiado bajos y la medición realizada carece de exactitud, y por lo tanto
no pueden analizarse estadísticamente. Por otra parte, los errores aleatorios o al azar, son
aquellos en los que los valores de las variables difieren del valor verdadero por azar.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
3

Curso de Farmacología Clínica Año 2008
Es una observación común, que el material biológico por su propia naturaleza es variable. De
manera que en las variables biológicas de los animales, es algo absolutamente normal hallar
diferencias. Se puede comprobar que el tipo y magnitud de los fenómenos biológicos que
pueden medirse, varían no solo de una especie a otra, sino que dentro de la misma especie lo
hacen de un individuo a otro y dentro del mismo individuo a lo largo del tiempo.
Para un clínico, la principal preocupación es la variabilidad de la respuesta clínica de un
individuo a los fármacos, siendo esta variación en la respuesta de tipo cualitativo o cuantitativo.
La variabilidad de tipo cualitativa, es aquella en donde la administración de un fármaco del
que se espera una respuesta clínica determinada es seguida por una respuesta discontinua de
la población, en donde en unos individuos se observa respuesta y otros individuos no
responden al tratamiento.
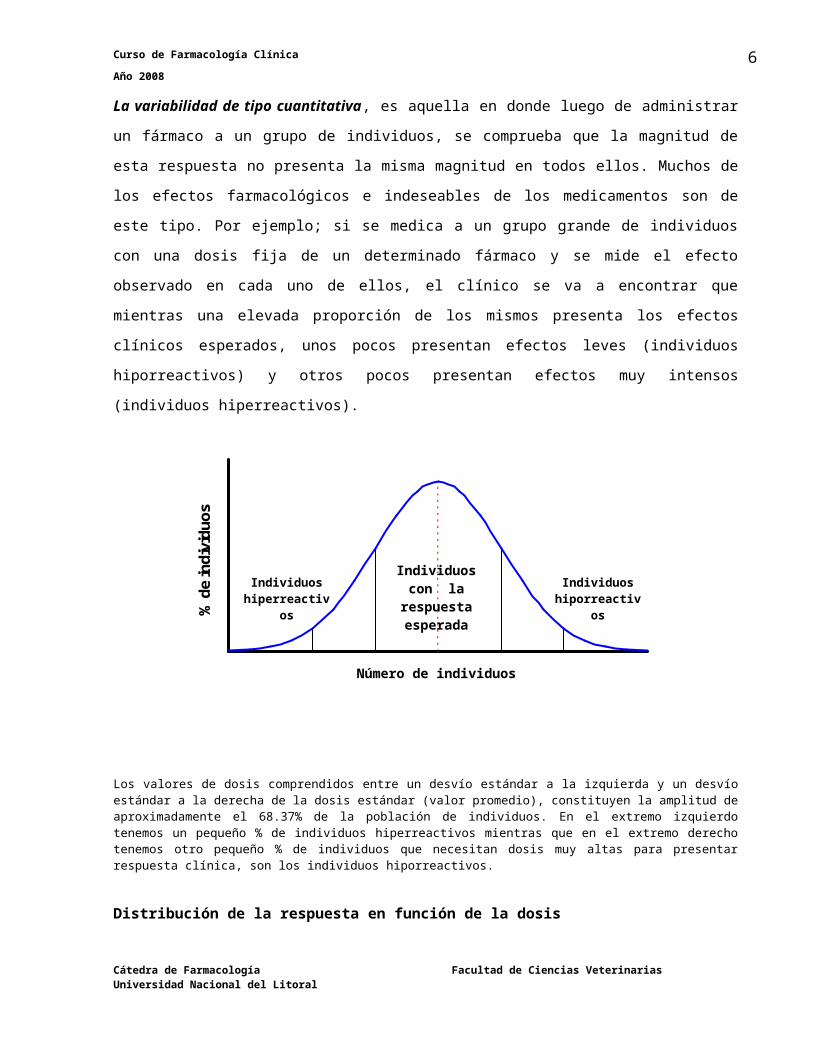
La variabilidad de tipo cuantitativa, es aquella en donde luego de administrar un fármaco a
un grupo de individuos, se comprueba que la magnitud de esta respuesta no presenta la misma
magnitud en todos ellos. Muchos de los efectos farmacológicos e indeseables de los
medicamentos son de este tipo. Por ejemplo; si se medica a un grupo grande de individuos con
una dosis fija de un determinado fármaco y se mide el efecto observado en cada uno de ellos, el
clínico se va a encontrar que mientras una elevada proporción de los mismos presenta los
efectos clínicos esperados, unos pocos presentan efectos leves (individuos hiporreactivos) y
otros pocos presentan efectos muy intensos (individuos hiperreactivos).
Los valores de dosis comprendidos entre un desvío estándar a la izquierda y un desvío estándar a la derecha de la dosis estándar (valor promedio), constituyen la amplitud de aproximadamente el 68.37% de la población de individuos. En el extremo izquierdo tenemos un pequeño % de individuos hiperreactivos mientras que en el extremo derecho tenemos otro pequeño % de individuos que necesitan dosis muy altas para presentar respuesta clínica, son los individuos hiporreactivos.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
4
Dosis
% d
e in
divi
duos
Individuos con la respuesta
esperada
Número de individuos
Individuos hiperreactivos
Individuos hiporreactivos

Curso de Farmacología Clínica Año 2008
Distribución de la respuesta en función de la dosisSi realizáramos un experimento y tomásemos un grupo muy grande de individuos y los
medicáramos con un determinado fármaco, haciendo variar la dosis desde valores muy bajos
hasta valores muy altos, hasta obtener la respuesta clínica esperada, entonces encontraremos
que las dosis empleadas son muy variables.
A la vez, tendremos pocos individuos que alcanzarán la respuesta clínica con una dosis mínima
(hiperreactivos), un grupo grande de individuos que alcanzan la respuesta clínica con una dosis
estándar, y un grupo de pocos individuos que necesitarán dosis mayores para alcanzar la
misma respuesta clínica que los anteriores (hiporreactivos). Estamos entonces ante una
distribución de dosis que puede ser bien definida con una distribución normal un valor promedio
y una varianza. Si bien la proporción en la que estos dos extremos se presentan en la clínica
diaria es baja, hay que tener presente que en biología como en cualquier otro orden de la vida
lo poco probable no quiere decir imposible, y el clínico debe estar precavido ante la posible falta
de eficacia o respuesta excesiva (que puede llegar a ser tóxica o letal en el caso de los agentes
anestésicos) para tomar las medidas pertinentes al caso.
Análisis de la variabilidad de la respuesta farmacológicaSon dos los factores que determinan que a una misma dosis dos individuos reaccionen o que
alcancen una respuesta clínica distinta, estos son el factor farmacodinámico y el
farmacocinética.
Factor farmacodinámicoEste factor se origina tanto por la diferente afinidad que puede presentar una droga a los
receptores específicos de la misma, o del diferente grado de actividad intrínseca que puede
desencadenarse en células de distintos individuos. La distribución de la respuesta
farmacológica está también relacionada con los niveles de concentración plasmática, que según
el principio básico de la farmacología, reflejan los niveles de concentración de fármaco que se
hallan en el sitio de acción del mismo o también llamado biofase.
De esta manera, bajas concentraciones plasmáticas en individuos en los cuales los receptores
específicos tengan gran afinidad por el fármaco y/o mayor actividad intrínseca reaccionarán
alcanzando el máximo efecto clínico en presencia de bajas concentraciones plasmáticas,
mientras que lo contrario ocurrirá con individuos con receptores con baja afinidad y/o baja
actividad intrínseca, en los cuales para alcanzar el máximo efecto terapéutico se necesitará
alcanzar concentraciones plasmáticas elevadas..
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
5
Curso de Farmacología Clínica Año 2008
A diferencia de la primera curva, en esta se muestra que para lograr un mismo efecto se necesitan distintas concentraciones plasmáticas en diferentes grupos de pacientes. La concentración plasmática es la mejor medida práctica de que se dispone para evaluar en forma indirecta la concentración de un fármaco en el sitio de acción (recordar el concepto de sistema linear), por lo que las diferencias entre individuos en este caso pueden ser atribuidas a diferencias en la sensibilidad de los receptores, por lo que estaríamos hablando de diferencias de tipo farmacodinámico.
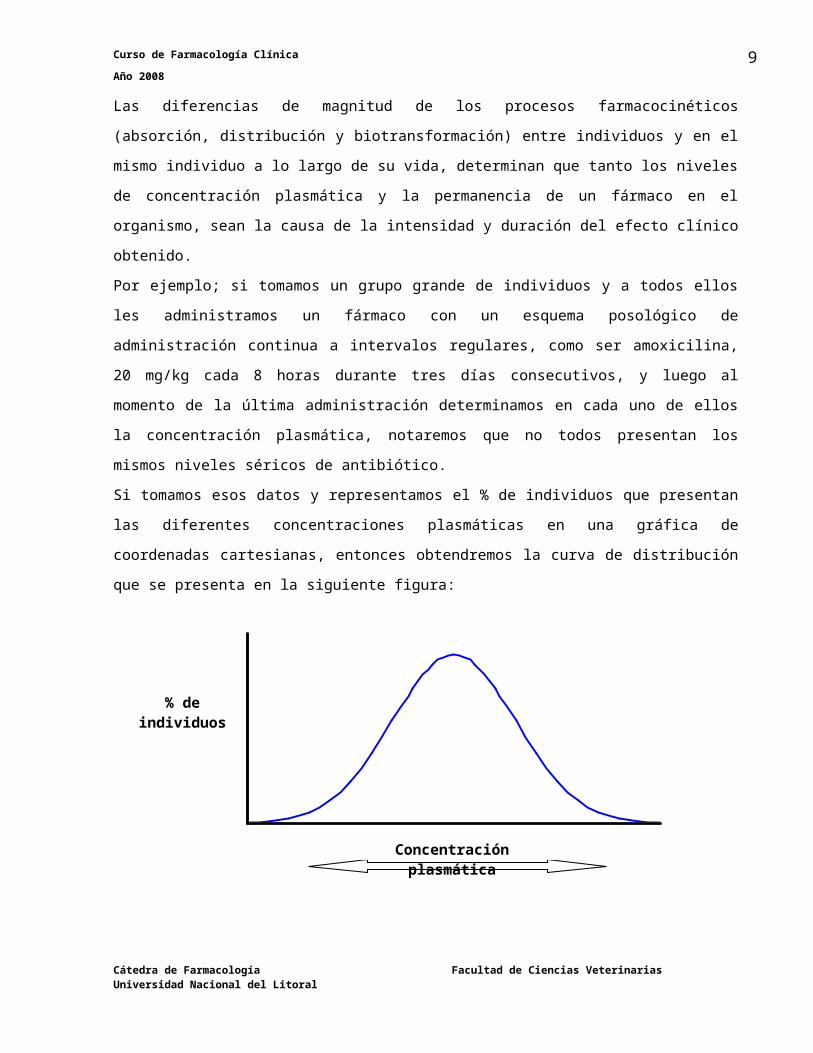
Factores farmacocinéticosLas diferencias de magnitud de los procesos farmacocinéticos (absorción, distribución y
biotransformación) entre individuos y en el mismo individuo a lo largo de su vida, determinan
que tanto los niveles de concentración plasmática y la permanencia de un fármaco en el
organismo, sean la causa de la intensidad y duración del efecto clínico obtenido.
Por ejemplo; si tomamos un grupo grande de individuos y a todos ellos les administramos un
fármaco con un esquema posológico de administración continua a intervalos regulares, como
ser amoxicilina, 20 mg/kg cada 8 horas durante tres días consecutivos, y luego al momento de
la última administración determinamos en cada uno de ellos la concentración plasmática,
notaremos que no todos presentan los mismos niveles séricos de antibiótico.
Si tomamos esos datos y representamos el % de individuos que presentan las diferentes
concentraciones plasmáticas en una gráfica de coordenadas cartesianas, entonces
obtendremos la curva de distribución que se presenta en la siguiente figura:
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
6
Dosis
% d
e in
divi
duos% de
individuos que
manifiestan efecto clínico
máximoIndividuos
hiperreactivos
Individuos con la respuesta
esperadaIndividuos
hiporreactivos
Concentración plasmática

Curso de Farmacología Clínica Año 2008
La variación absoluta de las concentraciones plasmáticas respecto del valor promedio, observada entre individuos que reciben una idéntica dosis, es diferente según el fármaco que se emplee.
Si transformamos los valores absolutos de concentración plasmática en porcentaje de variación
respecto de la media (valor/promedio x 100), notaremos que en farmacología clínica la
diferencia de concentraciones plasmáticas entre individuos que se considera aceptable es
aquella que fluctúa entre un 2 y un 30% del valor promedio observado.
VENTANA O AMPLITUD TERAPÉUTICAUno de los objetivos de la farmacocinética clínica, es el ajuste de los regímenes posológicos
(dosis, intervalo entre dosis, vía de administración), con el objeto de mantener las
concentraciones plasmáticas dentro de límites que garanticen la eficacia del fármaco y eviten la
toxicidad del mismo. Ventana terapéutica, es entonces la amplitud o límites de
concentraciones séricas (máximo y mínimo) dentro de las cuales existe una elevada
probabilidad de obtener la respuesta clínica deseada con una baja probabilidad de efectos
tóxicos.
Conceptos erróneos de margen terapéuticoCuando en farmacología clínica se habla de margen o ventana terapéutica, se asumen algunos
supuestos básicos que no siempre son ciertos. Es conveniente entonces tomar conocimiento de
ciertas limitaciones respecto del alcance del concepto “ventana terapéutica”. El primer supuesto
básico que se asume es que el margen terapéutico ha sido investigado y definido para cada
fármaco.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
7
Dosis
% d
e in
divi
duos% de
individuos
Concentración plasmática

Curso de Farmacología Clínica Año 2008
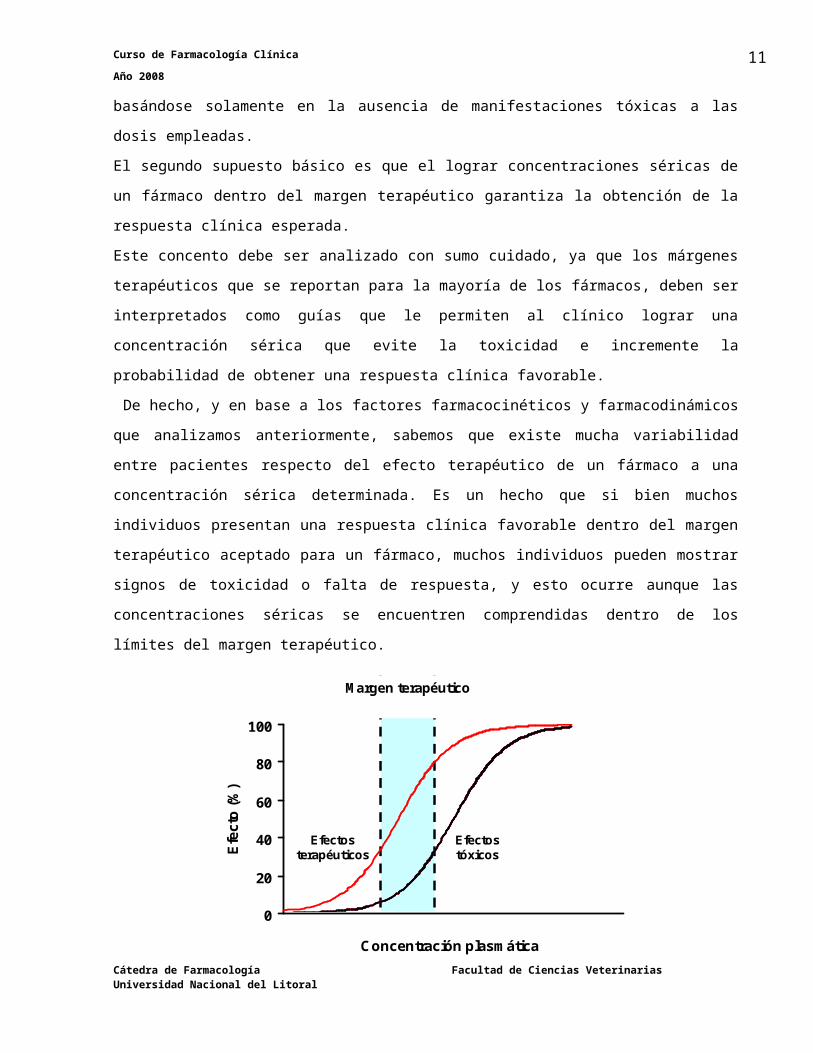
Este supuesto básico es una verdad a medias, ya que el “margen terapéutico” para muchos
fármacos solamente está basado en datos anecdóticos acerca de la eficacia clínica del mismo o
mas bien basándose solamente en la ausencia de manifestaciones tóxicas a las dosis
empleadas.
El segundo supuesto básico es que el lograr concentraciones séricas de un fármaco dentro del
margen terapéutico garantiza la obtención de la respuesta clínica esperada.
Este concento debe ser analizado con sumo cuidado, ya que los márgenes terapéuticos que se
reportan para la mayoría de los fármacos, deben ser interpretados como guías que le permiten
al clínico lograr una concentración sérica que evite la toxicidad e incremente la probabilidad de
obtener una respuesta clínica favorable.
De hecho, y en base a los factores farmacocinéticos y farmacodinámicos que analizamos
anteriormente, sabemos que existe mucha variabilidad entre pacientes respecto del efecto
terapéutico de un fármaco a una concentración sérica determinada. Es un hecho que si bien
muchos individuos presentan una respuesta clínica favorable dentro del margen terapéutico
aceptado para un fármaco, muchos individuos pueden mostrar signos de toxicidad o falta de
respuesta, y esto ocurre aunque las concentraciones séricas se encuentren comprendidas
dentro de los límites del margen terapéutico.
El segmento en la abcisa que se halla entre las dos líneas de puntos, es el intervalo entre concentraciones plasmáticas que garantizan la mayor eficacia terapéutica con un mínimo riesgo de efectos tóxicos en una población de individuos. El punto en donde la línea de puntos de la izquierda corta al eje de las abcisas representa la mínima concentración plasmática que es capaz de producir el mínimo % de efectos clínicos (línea sigmoidea izquierda). El punto en donde la línea de puntos de la derecha corta al eje de las abcisas representa la mínima concentración plasmática que es capaz de producir el mínimo % de efectos tóxicos (línea sigmoidea derecha).
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
8
Margen terapéutico
0
20
40
60
80
100
Concentración plasmática
Efec
to (%
)
Efectos tóxicos
Efectos terapéuticos
Curso de Farmacología Clínica Año 2008
Para muchos fármacos se ha determinado un margen o ventana o límites (mínimo y máximo) de
concentraciones plasmáticas, que en la mayoría de los individuos de una población permiten
obtener el efecto terapéutico deseado con un mínimo riesgo de efectos indeseables dosis-
dependientes.
En la siguiente figura se presenta la evolución de las concentraciones plasmáticas de un
individuo dentro de la ventana terapéutica.
La franja sombreada representa el margen o la ventana terapéutica, los días marcados en el eje de las abcisas representan los momentos en los cuales se administra el fármaco por vía intravenosa.
Este ejemplo corresponde a un esquema terapéutico de administraciones repetidas a intervalos
de tiempo regulares. Las administraciones se realizan a intervalos de un día que coinciden con
el tiempo de semivida del fármaco. Nótese que la concentración plasmática asciende
abruptamente luego de la administración de la dosis diaria, y que el estado de equilibrio
estacionario se alcanza luego de la quinta administración, que en este caso equivale a cinco
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
9
Tiempo (días)
Con
cent
raci
ón p
lasm
átic
a(μ
g/m
l)

Curso de Farmacología Clínica Año 2008
semividas (t1/2el x 5). También se puede apreciar que la amplitud de concentraciones plasmáticas
tiene un valor máximo (Cmax) que en razón de evitar los efectos tóxicos no se debe sobrepasar
y una concentración plasmática mínima (Cmin), por debajo de la cual el efecto terapéutico no se
logra.
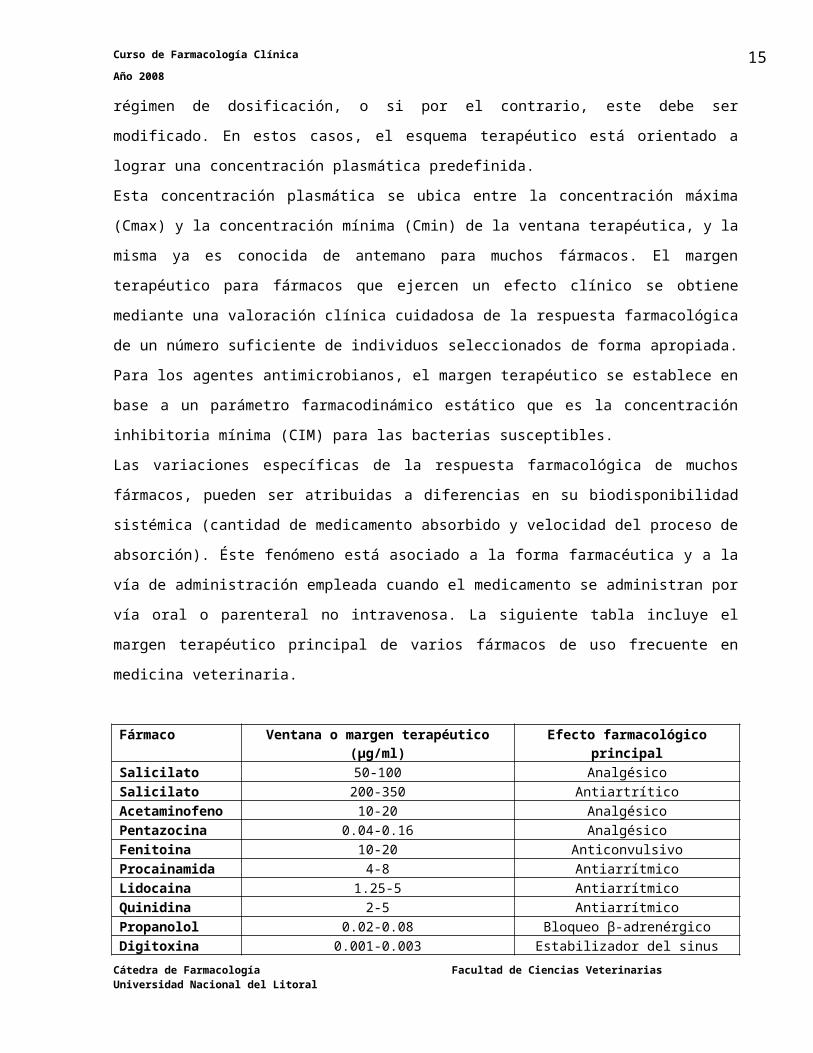
En la siguiente tabla presentamos algunos ejemplos de ventana terapéutica y algunos efectos
adversos dosis-dependientes de dos fármacos conocidos; teofilina y fenitoina (difenilhidantoina).
μg/ml Teofilina μg/ml Fenitoína50 convulsiones 50 alteraciones mentales40 arritmias 40 ataxia30 nerviosismo, 30 nistagmo
nauseas y vómitos 20margen terapéutico15
margen terapéutico 105 Sin protección contra
las convulsiones0 Sin mejoría en la función respiratoria 0
La teofilina es un fármaco que se emplea en terapéutica como broncodilatador y la velocidad
con la cual se metaboliza varía mucho entre un individuo y otro. La fenitoína es un fármaco
antiepiléptico que se emplea para tratamientos de por vida, y a diferencia del primero presenta
un metabolismo saturable, por lo que pequeñas variaciones en la dosis pueden producir
grandes variaciones en los niveles séricos. En ambos casos, en pacientes que se hallan en
estado crítico, es necesario determinar los niveles séricos para realizar el ajuste de la posología
a fin de evitar tanto la falta de efecto terapéutico como la aparición de toxicidad.
FUNDAMENTOS DE LA FARMACOCINÉTICA CLÍNICALa utilidad de la farmacocinética en la práctica clínica, reside en que permite calcular las dosis y
el intervalo de dosificación de un fármaco a fin de poder lograr las concentraciones plasmáticas
que se desean alcanzar para un individuo, en particular para lograr el efecto clínico deseado.
Para muchos fármacos como por ejemplo los antihipertensivos o los cardiotónicos como la
digoxina, no es necesario efectuar cálculos farmacocinéticos, porque para manipular el plan de
administración (dosis e intervalo) a fin de obtener el efecto terapéutico buscado se toma como
referencia la magnitud de un efecto farmacológico observable que en el primer caso es la
presión arterial y en el segundo caso es el efecto de toxicidad extracardíaca.
En cambio para otro tipo de fármacos como pueden ser los antiepilépticos o los antibióticos
aminoglucósidos, el método anterior no da resultado. Esto se debe porque el efecto terapéutico
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
10
Curso de Farmacología Clínica Año 2008
es difícil de evaluar debido a que no disponemos de parámetros clínicos o efectos terapéuticos
observables y no se puede saber si continuar con el régimen de dosificación, o si por el
contrario, este debe ser modificado. En estos casos, el esquema terapéutico está orientado a
lograr una concentración plasmática predefinida.
Esta concentración plasmática se ubica entre la concentración máxima (Cmax) y la
concentración mínima (Cmin) de la ventana terapéutica, y la misma ya es conocida de
antemano para muchos fármacos. El margen terapéutico para fármacos que ejercen un efecto
clínico se obtiene mediante una valoración clínica cuidadosa de la respuesta farmacológica de
un número suficiente de individuos seleccionados de forma apropiada. Para los agentes
antimicrobianos, el margen terapéutico se establece en base a un parámetro farmacodinámico
estático que es la concentración inhibitoria mínima (CIM) para las bacterias susceptibles.
Las variaciones específicas de la respuesta farmacológica de muchos fármacos, pueden ser
atribuidas a diferencias en su biodisponibilidad sistémica (cantidad de medicamento absorbido y
velocidad del proceso de absorción). Éste fenómeno está asociado a la forma farmacéutica y a
la vía de administración empleada cuando el medicamento se administran por vía oral o
parenteral no intravenosa. La siguiente tabla incluye el margen terapéutico principal de varios
fármacos de uso frecuente en medicina veterinaria.
Fármaco Ventana o margen terapéutico (μg/ml) Efecto farmacológico principalSalicilato 50-100 AnalgésicoSalicilato 200-350 AntiartríticoAcetaminofeno 10-20 AnalgésicoPentazocina 0.04-0.16 AnalgésicoFenitoina 10-20 AnticonvulsivoProcainamida 4-8 AntiarrítmicoLidocaina 1.25-5 AntiarrítmicoQuinidina 2-5 AntiarrítmicoPropanolol 0.02-0.08 Bloqueo β-adrenérgicoDigitoxina 0.001-0.003 Estabilizador del sinusDigoxina 0.001-0.003 Inductor del ritmo y antiarrítmicoSulfamidas 50-150 BacteriostáticoTrimetoprim 2-6 BacteriostáticoTetraciclinas 1.5-4 BacteriostáticoEritromicina 2-8 BacteriostáticoKanamicina 4-32 BactericidaGentamicina 4-16 BactericidaPenicilinas 0.1-25 Bactericida
Estudios de farmacocinética clínica
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
11

Curso de Farmacología Clínica Año 2008
Los estudios de farmacocinética clínica se basan en la aceptación de los siguientes supuestos
básicos:
a- La concentración del fármaco en el sitio de acción es proporcional a su concentración
plasmática.
b- La intensidad de los efectos de los fármacos está relacionada con su concentración en
el sitio de acción.
c- La duración del efecto está relacionada con el tiempo en que la molécula del fármaco
está presente en el sitio de acción.
Obviamente, se presentan situaciones en las que no es fácil interpretar o directamente no existe
una correlación lineal entre concentración plasmática de un fármaco y su efecto como por
ejemplo:
a- Moléculas que actúan en forma irreversible como los organofosforados.
b- Cuando se desarrolla tolerancia farmacodinámica.
c- Cuando se realiza una terapia combinada con fármacos sinérgicos y antagonistas.
d- Cuando el fármaco presenta metabolitos activos (como el diazepan que se metaboliza a
nordiazepan o en caso del albendazole que se metaboliza a albendazole sulfóxido
(ricobendazole).
e- Cuando la acción del fármaco no es rápidamente reversible como ocurre con la
reserpina o los agentes antineoplásicos.
VARIACIÓN TEMPORAL DE LOS EFECTOS DE LOS FÁRMACOSLa administración de un fármaco a un paciente, produce en éste efectos en relación a la dosis
administrada y también en relación al tiempo transcurrido desde su administración. Los factores
que influyen sobre el curso temporal de los efectos son:
La vía de administraciónEmpleando la vía intravenosa, se obtienen los efectos de manera más rápida que empleando la
vía intramuscular u oral. Esto es así, porque al administrase el fármaco directamente dentro de
la circulación general, se evita el tiempo que demanda el proceso de absorción, al tiempo que
logramos concentraciones elevadas y eficaces de manera casi instantánea.
Características del fármacoa- En el caso más simple, el efecto está directamente relacionado con las concentraciones
plasmáticas obtenidas como lo que ocurre con la teofilina.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
12

Curso de Farmacología Clínica Año 2008
b- Otros casos como el enalapril (antihipertensivo), las concentraciones plasmáticas y el
efecto difieren debido a que con las dosis comúnmente empleadas, se logran
concentraciones plasmáticas mucho mas elevadas que las necesarias para ejercer su
acción (inhibición de la convertasa de angiotensina). Además, la actividad de la
convertasa, se recupera más lentamente de lo que podría esperarse en relación a la
caída de las concentraciones plasmáticas.
c- En el caso de la warfarina (anticoagulane oral), como su mecanismo de acción es inhibir
la activación de la vitamina K, es necesario esperar a que disminuyan las
concentraciones del los factores de coagulación activos para observar los efectos
anticoagulantes.
d- Otro caso, lo constituyen ciertos fármacos cuyo efecto es acumulativo, como lo es el
caso de la toxicidad renal por acción de la gentamicina, que es mayor si se administra
como infusión constante que si se la administra en forma intermitente.
METODOLOGÍA DE LA FARMACOCINÉTICAPara describir el destino de los fármacos en el organismo, la farmacocinética recurre al uso de
modelos que implican suposiciones que no necesariamente se corresponden con realidades
fisiológicas. En el modelo que se presenta a continuación, se han representado varios
compartimientos o lugares hipotéticos en donde las moléculas del fármaco y sus metabolitos se
distribuyen.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
13

Curso de Farmacología Clínica Año 2008
Si examinamos la representación gráfica del modelo distinguiremos:
a- Un compartimiento central; la sangre o el plasma.
b- Un compartimiento de tejidos activos o sitio de acción.
c- Un compartimiento de tejidos inactivos o de depósito.
d- Sitios de metabolización y excreción.
Se entiende por compartimiento a un lugar imaginario, en donde las moléculas del fármaco ya
sea en su forma libre o no unida a proteínas plasmáticas o tisulares ingresa y sale. Se asume
que el fármaco libre es el único que se distribuye en el interior de un compartimiento de manera
uniforme y homogénea.
Los compartimientos para un fármaco, no necesariamente son los mismos que para sus
metabolitos, sean estos activos o inactivos. Sin embargo, el modelo que se describe en la figura
es demasiado complejo para ser abordado en este texto, por lo que, a los fines del aprendizaje,
desarrollaremos el análisis de un modelo constituido por un solo compartimiento.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
14
Curso de Farmacología Clínica Año 2008
No obstante, los fármacos que habitualmente se emplean en terapéutica veterinaria se
comportan como si el organismo estuviera constituido por dos compartimientos; un
compartimiento central y un compartimiento periférico, por lo que también desarrollaremos
algunos aspectos de un modelo de dos compartimientos.
MODELO DE UN COMPARTIMIENTOEste es el modelo más simple y más antiguo con el que se puede representar a un organismo y
considera que hay un ingreso de fármaco y que este se distribuye de manera instantánea, por lo
que la concentración de este sería idéntica en cualquier sitio del mismo en donde esta se
midiese.
En consecuencia, midiendo la concentración plasmática y conociendo la dosis administrada,
podemos calcular el volumen teórico del compartimiento. También se asume la existencia de
una sola vía de salida del fármaco ya sea por metabolismo, excreción o ambos procesos a la
vez.
Modelo en estado de equilibrioEn este modelo, el estado de equilibrio o estado estable se alcanza cuando la velocidad de
ingreso del fármaco al sistema es igual a la velocidad de salida.
El ejemplo más simple de esta condición es el caso de la infusión intravenosa continua de un
fármaco, donde el estado de equilibrio estacionario se alcanza luego de administrar el fármaco
durante un período equivalente al de cinco semividas del mismo.
En esta condición tenemos que:
a- La velocidad de entrada del fármaco es igual a la dosis administrada en un intervalo de
tiempo determinado (Ej. mg/min). En caso de una administración intermitente; por
ejemplo cada cuatro horas, la dosis administrada se denomina dosis de mantenimiento
(DM) la cual si la dividimos por el intervalo entre administraciones (T) nos daría como
resultado la velocidad de entrada.
b- La velocidad de salida del fármaco (eliminación) sería igual a la cantidad total de
fármaco en el organismo (M) que se expresaría como masa multiplicada por la constante
de eliminación que se expresa en unidades de tiempo recíproco (tiempo -1).
Si representamos todo lo que mencionamos anteriormente en un esquema tendremos que:
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
15

Curso de Farmacología Clínica Año 2008
El cubo representaría al compartimiento, el cual considerando que un organismo viviente está
compuesto de aproximadamente un 80% de agua, éste tendría un volumen teórico o Vd, que
significa volumen de distribución.
Teniendo en cuenta que los fluidos corporales son los que disuelven la dosis del fármaco,
entonces; Cp es la concentración plasmática resultante de la disolución del fármaco ingresado
en el mencionado volumen teórico.
Una representación gráfica de las concentraciones plasmáticas en función del tiempo según
este modelo considerando un esquema posológico de perfusión intravenosa (A) y un esquema
posológico de administración extravascular intermitente (B) se presentan a continuación.
En la figura A se representan las concentraciones plasmáticas estables que se alcanzan durante la administración de un fármaco por perfusión intravenosa continua. En la figura B, se presenta la fluctuación de las concentraciones plasmáticas, cuando el fármaco es administrado a idénticas dosis a intervalos de tiempo regulares. Ambas representaciones corresponden simulaciones realizadas con un modelo de un compartimiento.
Administración intravenosa rápida (bolo intravenoso)
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
16
Tiempo
Con
cent
raci
ón
plas
mát
ica
Tiempo
Con
cent
raci
ón
plas
mát
ica
A B
Velocidad de salida
kel x M (eliminación)
Velocidad de entrada
DM/T (dosificación)
Cp
Vd

Curso de Farmacología Clínica Año 2008
En el modelo de un solo compartimiento, la administración de un fármaco en forma rápida por
vía intravenosa (bolo intravenoso), produce una elevada concentración plasmática inicial (C0).
Como el ingreso del fármaco no es continuo, si medimos las concentraciones del mismo a
diferentes tiempos posadministración, obtendremos la siguiente curva que describe la evolución
temporal de las concentraciones plasmáticas en el organismo.
Gráfica que describe la evolución temporal de concentraciones plasmáticas en función del tiempo luego de la administración de un fármaco por vía intravenosa, considerando un modelo de un compartimiento.
En la figura, puede observarse que las concentraciones plasmáticas del fármaco van
disminuyendo a medida que el tiempo transcurre desde el momento de su administración. El
descenso de las concentraciones plasmáticas se debe a la eliminación del fármaco por parte de
los mecanismos depuradores del organismo (metabolización y excreción). La curva puede ser
descripta mediante una función exponencial cuya ecuación es la que se presenta a
continuación.
Cp = C0. e –kel . t
Donde C0 es la concentración plasmática estimada al tiempo mismo de finalizada la
administración intravenosa (este valor asume que la distribución es instantánea), e es la base
de los logaritmos naturales (2.71828) y kel es la constante de eliminación de primer orden. Esta
constante se interpreta como la fracción de fármaco que es eliminada por unidad de tiempo
transcurrido y t es el tiempo expresado en su correspondiente unidad (minutos, horas o días).
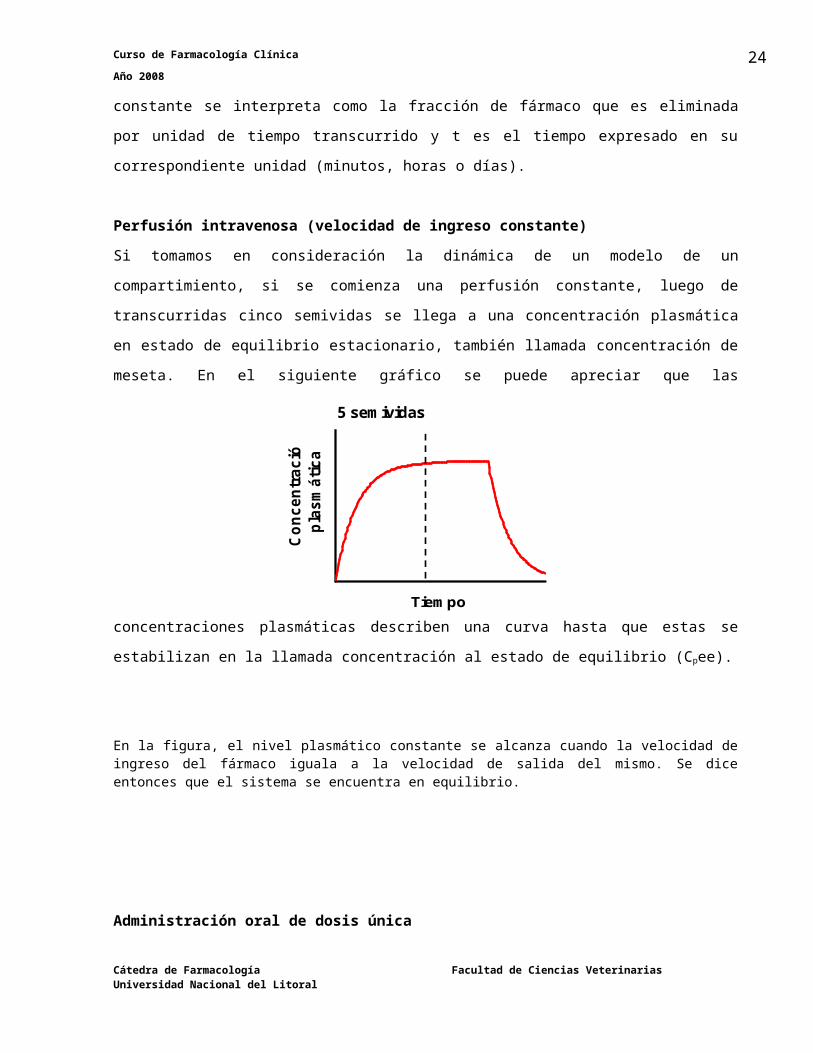
Perfusión intravenosa (velocidad de ingreso constante)
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
17
Con
cent
raci
ón
plas
mát
ica
Tiempo

Curso de Farmacología Clínica Año 2008
Si tomamos en consideración la dinámica de un modelo de un compartimiento, si se comienza
una perfusión constante, luego de transcurridas cinco semividas se llega a una concentración
plasmática en estado de equilibrio estacionario, también llamada concentración de meseta. En
el siguiente gráfico se puede apreciar que las concentraciones plasmáticas describen una curva
hasta que estas se estabilizan en la llamada concentración al estado de equilibrio (Cpee).
En la figura, el nivel plasmático constante se alcanza cuando la velocidad de ingreso del fármaco iguala a la velocidad de salida del mismo. Se dice entonces que el sistema se encuentra en equilibrio.
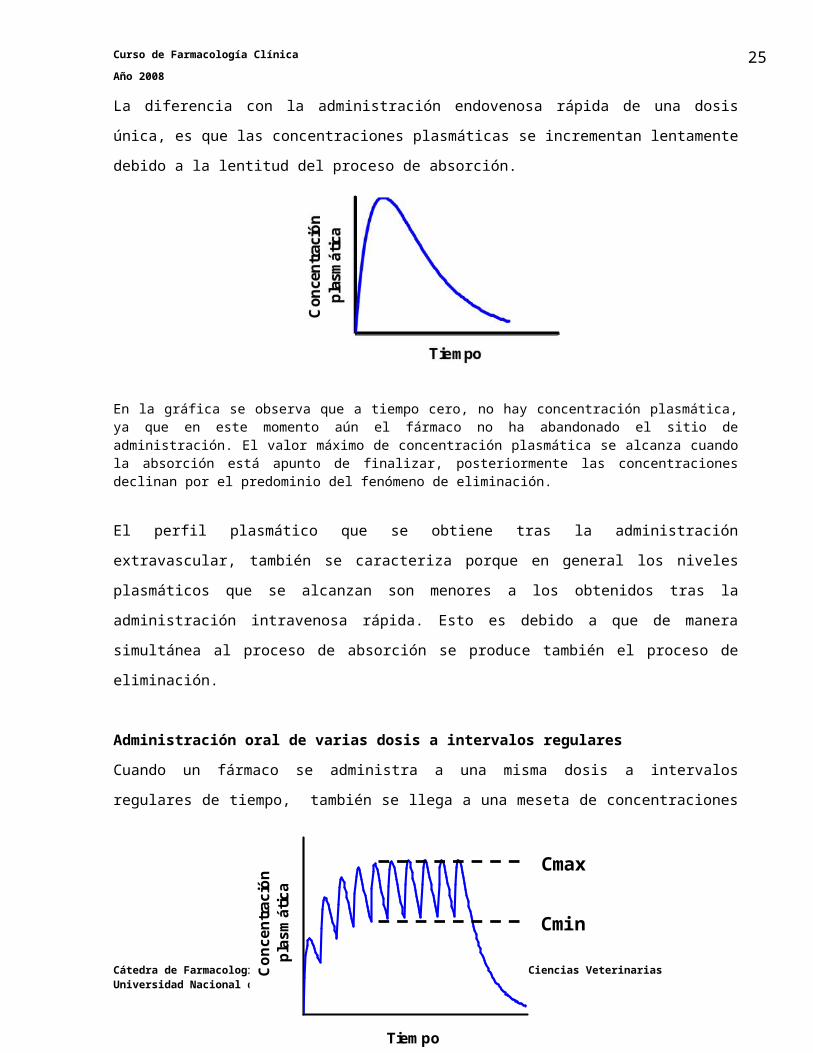
Administración oral de dosis únicaLa diferencia con la administración endovenosa rápida de una dosis única, es que las
concentraciones plasmáticas se incrementan lentamente debido a la lentitud del proceso de
absorción.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
18
Tiempo
Con
cent
raci
ón
plas
mát
ica
5 semividas
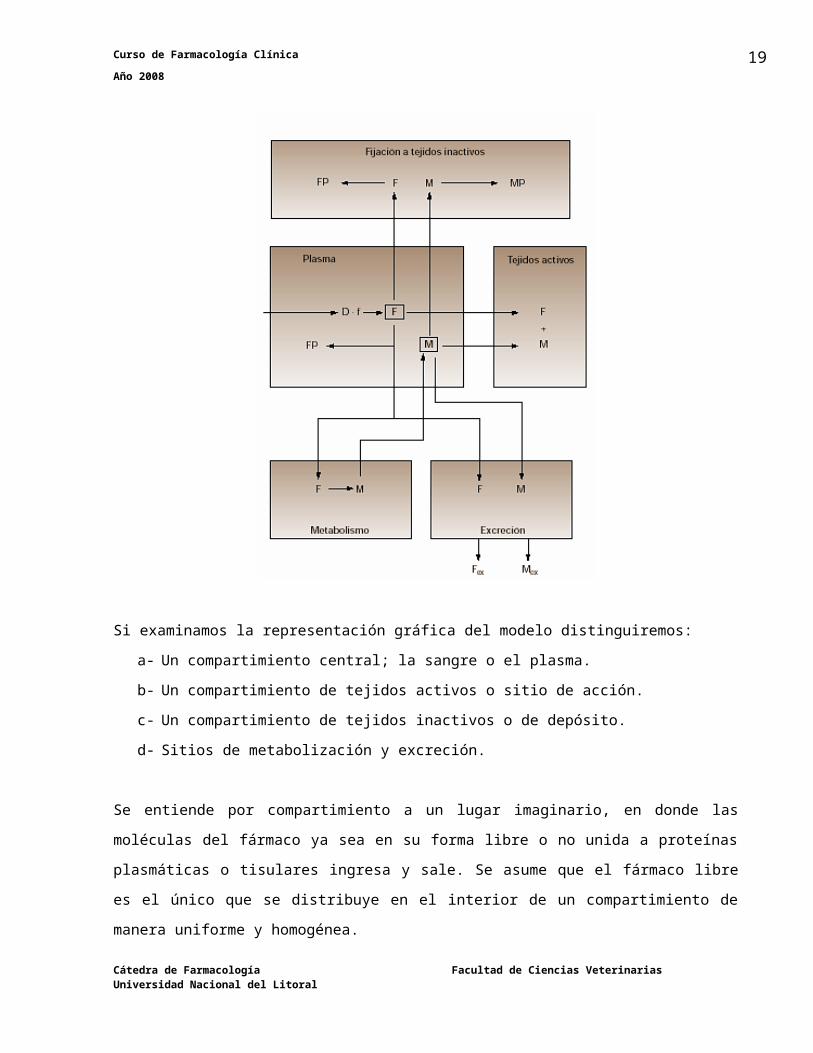
Curso de Farmacología Clínica Año 2008
En la gráfica se observa que a tiempo cero, no hay concentración plasmática, ya que en este momento aún el fármaco no ha abandonado el sitio de administración. El valor máximo de concentración plasmática se alcanza cuando la absorción está apunto de finalizar, posteriormente las concentraciones declinan por el predominio del fenómeno de eliminación.
El perfil plasmático que se obtiene tras la administración extravascular, también se caracteriza
porque en general los niveles plasmáticos que se alcanzan son menores a los obtenidos tras la
administración intravenosa rápida. Esto es debido a que de manera simultánea al proceso de
absorción se produce también el proceso de eliminación.
Administración oral de varias dosis a intervalos regularesCuando un fármaco se administra a una misma dosis a intervalos regulares de tiempo, también
se llega a una meseta de concentraciones plasmáticas que se caracteriza por la presencia de
picos (Cmax) y valles (Cmin) de concentración plasmática.
MODELO DE DOS COMPARTIMIENTOSEste modelo es más complejo. Consta de un compartimiento central y un compartimiento
periférico. Los fármacos ingresan al compartimiento central y luego desde allí se distribuyen al
compartimiento periférico. Un esquema de este modelo se presenta a continuación.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
19
Tiempo
Con
cent
raci
ón
plas
mát
ica
Cmax
Cmin
k12
Velocidad de salida k10 x M
(eliminación)
Velocidad de entrada
DM/T (dosificación)
C2
Vp
C1
Vc
k21

Curso de Farmacología Clínica Año 2008
En este esquema Vc representa el volumen del compartimiento central y C1 la concentración de
medicamento en este compartimiento, Vp representa el volumen del compartimiento central y C2
la concentración de fármaco en el mismo. Los procesos de distribución y redistribución se hallan
representados por las constantes de velocidad de primer orden de pasaje desde el
compartimiento central al periférico (k12) y del compartimiento periférico al central (k21). El
fármaco es eliminado desde el compartimiento central por un procesote orden uno (k10) y la
velocidad de eliminación viene expresada como el producto entre k10 y la masa de fármaco
eliminada (M).
En este modelo, la administración de la dosis de un fármaco en forma de bolo intravenoso
produce de manera inmediata elevadas concentraciones del fármaco como resultado de su casi
instantánea y homogénea distribución en el compartimiento central (plasma y tejidos con mayor
irrigación). Esta elevada concentración inicial C0, es seguida por una caída de las
concentraciones plasmáticas como resultado del predominio del proceso de distribución, hasta
que luego de alcanzarse el estado de equilibrio entre los compartimientos central y periférico la
curva de disposición plasmática es la resultante del predominio del proceso de eliminación.
La curva de concentraciones plasmáticas resultante en escala natural difiere muy poco de la
obtenida con un modelo de un compartimiento, por lo que las fases de distribución y de
eliminación se hacen evidentes cuando los datos de concentración plasmática en función del
tiempo se representan en una gráfica semilogarítmica.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
20
C1
C2
Tiempo
ln C
once
ntra
ción
pl
asm
átic
a
Fase de distribución o α
Fase de eliminación o β

Curso de Farmacología Clínica Año 2008
En la gráfica, se puede apreciar una fase de distribución o también llamada fase α y una fase de eliminación o fase β. Este modelo es el que mejor explica y describe la disposición de la mayoría de los fármacos en el organismo.
La curva de concentraciones plasmáticas resultante puede ser descripta por la siguiente ecuación biexponencial:
Cp = C1. e –α.t + C2. e –β.t
Donde α es la constante de distribución, β es la constante de eliminación aparente y C1 y C2 son
las concentraciones plasmáticas que se estiman extrapolando a tiempo cero las pendientes de
las fases de distribución y eliminación respectivamente y e es la base de los logaritmos
naturales.
TIPOS DE CINÉTICA DE LOS MEDICAMENTOSTal como la definición del término “farmacocinética” lo expresa, los fármacos están sometidos a
procesos de absorción, distribución y eliminación (metabolismo y excreción).
Por lo tanto los estudios farmacocinéticos pueden abarcar estos tres campos:
a- cinética de absorción
b- cinética de distribución
c- cinética de eliminación
CINÉTICA DE ABSORCIÓNLa cinética del proceso de absorción de los fármacos se desarrollará con mayor detalle cuando
se trate el tema de biodisponibilidad y bioequivalencia.
Mencionaremos aquí que el proceso de absorción de un fármaco desde el tracto gastrointestinal
sigue por lo general una cinética de primer orden. No obstante, es importante aclarar que en la
especie rumiante, los pre-estómagos actúan como un sistema de liberación sostenida de los
fármacos hacia el tracto digestivo posterior simulando una cinética de absorción de orden cero,
Un fenómeno similar resulta de la inyección parenteral de formas medicamentosas (implantes,
soluciones de depósito) que liberan el principio activo a la circulación sistémica a velocidad
constante (cinética de orden cero).
CINÉTICA DE DISTRIBUCIÓNPor no tener una aplicación práctica en clínica, solo mencionaremos que estos estudios se
refieren al paso de los fármacos entre los distintos compartimientos. En el caso de la
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
21

Curso de Farmacología Clínica Año 2008
administración de la dosis única de un fármaco en forma de bolo intravenoso, durante la fase α
se manifiesta el predominio de la difusión del fármaco desde el compartimiento central al
periférico. Esto da como resultado que las concentraciones plasmáticas decrezcan al tiempo
que se incrementan los niveles de fármaco en el compartimiento periférico.
En la gráfica el perfil sombreado representa la evolución de las concentraciones de fármaco en el compartimiento periférico, nótese su similitud con el perfil observado tras la administración extravascular de una dosis única en un modelo de un compartimiento.
Nótese que al tiempo que en la curva de concentraciones plasmáticas reproduce la inflexión
entre la fase α y β, se alcanza máximo nivel de fármaco en el compartimiento periférico.
Posteriormente, luego de haberse alcanzado el estado de pseudo-equilibrio, las pendientes
finales de las concentraciones plasmáticas y las tisulares muestran una idéntica pendiente, lo
que indica la estrecha relación entre los procesos de distribución, redistribución y eliminación
durante la fase terminal de disposición plasmática de un fármaco.
El concepto de distribución de un fármaco, y el tiempo que este requiere para lograr
concentraciones en el compartimiento periférico es de suma importancia, ya que la velocidad de
ingreso a este depende de factores tales como tamaño de la molécula, grado de ionización,
liposolubilidad y flujo sanguíneo. Esto debe ser tenido en cuenta cuando el sitio de acción del
fármaco se halla en el compartimiento periférico, ya que habrá un período de espera entre la
administración y el comienzo de los efectos que será variable para cada fármaco.
CINÉTICA DE ELIMINACIÓN
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
22
Niveles de fármaco en el compartimiento periférico
Tiempo
Con
cent
raci
ón

Curso de Farmacología Clínica Año 2008
En farmacocinética, la velocidad se expresa en unidades de masa (μg, mg, etc.) por unidad de
tiempo (min, h, etc.). Existen dos tipos de cinética de eliminación, la cinética lineal (orden uno)
y la cinética no lineal (orden cero).
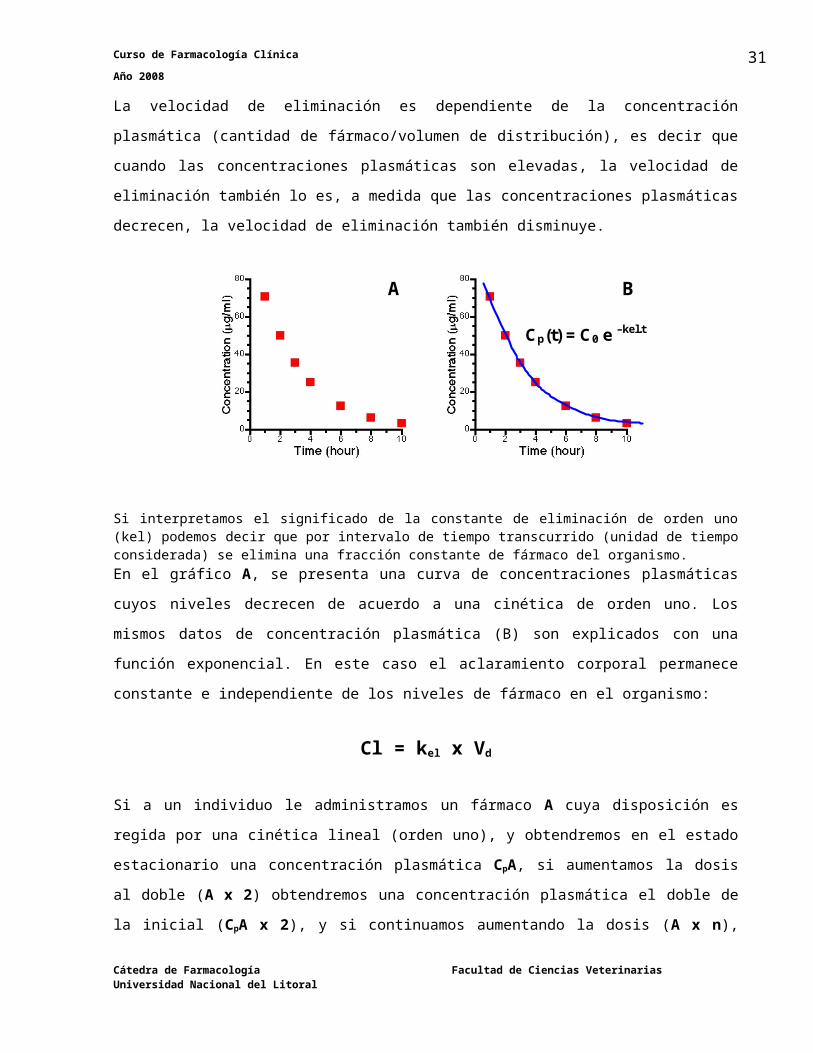
Cinética lineal (orden uno)La velocidad de eliminación es dependiente de la concentración plasmática (cantidad de
fármaco/volumen de distribución), es decir que cuando las concentraciones plasmáticas son
elevadas, la velocidad de eliminación también lo es, a medida que las concentraciones
plasmáticas decrecen, la velocidad de eliminación también disminuye.
Si interpretamos el significado de la constante de eliminación de orden uno (kel) podemos decir que por intervalo de tiempo transcurrido (unidad de tiempo considerada) se elimina una fracción constante de fármaco del organismo. En el gráfico A, se presenta una curva de concentraciones plasmáticas cuyos niveles decrecen
de acuerdo a una cinética de orden uno. Los mismos datos de concentración plasmática (B) son
explicados con una función exponencial. En este caso el aclaramiento corporal permanece
constante e independiente de los niveles de fármaco en el organismo:
Cl = kel x Vd
Si a un individuo le administramos un fármaco A cuya disposición es regida por una cinética
lineal (orden uno), y obtendremos en el estado estacionario una concentración plasmática CpA,
si aumentamos la dosis al doble (A x 2) obtendremos una concentración plasmática el doble de
la inicial (CpA x 2), y si continuamos aumentando la dosis (A x n), obtendremos un aumento de
la concentración plasmática proporcional a la dosis administrada (CpA x n).
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
23
Cp(t) = C0 e –kel.t
A B
02468
1012
0 50 100 150Dosis (mg)
mg/
ml
Curso de Farmacología Clínica Año 2008
Cinética no lineal (orden cero)En este tipo de cinética, la velocidad de eliminación es independiente de la cantidad de fármaco
presente en el organismo. Este fenómeno se explica por la saturación de los sistemas
biológicos encargados de eliminar el fármaco del organismo. En este caso la cantidad de
fármaco que se elimina por intervalo de tiempo es constante y su valor está definido por la
máxima capacidad de los sistemas de eliminación.
Si administramos un fármaco por la vía intravenosa, y se miden las concentraciones
plasmáticas del fármaco a intervalos de tiempo regulares, la gráfica resultante a escala normal
(A) es una recta que puede ser explicada por una función de primer grado o también llamada
ecuación de la recta (B).
En este tipo de cinética, la interpretación de la constante de eliminación de orden cero (k0) es
muy diferente a la de un proceso de orden uno, ya que el valor de la constante de orden cero
hace referencia exacta a la cantidad de fármaco (masa) que es eliminada por intervalo de
tiempo transcurrido.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
24
0
20
40
60
80
100
0 5 10 15 20Time (hour)
0
20
40
60
80
100
0 5 10 15 20Time (hour)
Con
cent
raci
ón (μ
g/m
l)
Con
cent
raci
ón (μ
g/m
l) A B
Cp(t) =C0 – kel.t
Curso de Farmacología Clínica Año 2008
Otra diferencia con el proceso anterior es que el aclaramiento no se comporta como una
constante sino que este varía en función de la concentración plasmática:
Cl = Vm / (Km + Cp)
Si a un paciente le administramos un fármaco B cuya disposición plasmática está regida por un
proceso de orden cero, obtendremos una concentración plasmática CpB1. Si continuamos
incrementando las dosis, las concentraciones plasmáticas irán aumentando en forma
proporcional a la dosis hasta un punto en el que los sistemas de eliminación se saturen. A partir
de ese momento las concentraciones plasmáticas se incrementarán en forma desproporcionada
respecto de la dosis administrada. Al incrementarse las concentraciones plasmáticas se
incrementan los tiempos de semivida de eliminación. Ej: alcohol y difenilhidantoína.
PARÁMETROS FARMACOCINÉTICOSConstante de eliminaciónLa constante de eliminación (kel) representa la fracción (o porcentaje) de la cantidad total de
fármaco en el organismo que es eliminada en unidad de tiempo. Debe tenerse presente que el
proceso de orden uno es un proceso exponencial, por lo que kel puede ser interpretada de una
forma general como una fracción de fármaco que se elimina en un intervalo infinitesimal de
tiempo. La kel en un modelo de dos compartimientos es un parámetro farmacocinético híbrido
que representa la suma de los procesos de distribución (k12), redistribución (k21) y de eliminación
desde el compartimiento central (k10).
Cálculo de kel
En un modelo de un compartimiento, la evolución de la concentración plasmática en función del
tiempo se describe mediante la siguiente función exponencial:
kel.t0p e C(t)C
Tomando logaritmos naturales:
.t k- C ln(t)C ln el0p
reordenando:
(t)C lnC ln.t k p0el
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
25

Curso de Farmacología Clínica Año 2008
t(t)C
Cln
t(t)lnClnC
k p
0
p0el
Cuando la concentración plasmática disminuye a la mitad de la concentración estimada a
tiempo cero (C0), el tiempo trascurrido se denomina semivida o vida media (t1/2). Si operamos
con la ecuación anterior para el momento en que el cociente entre las dos diferencias es 2
entonces tenemos que:
1/21/2el t
0.693t
2 lnk
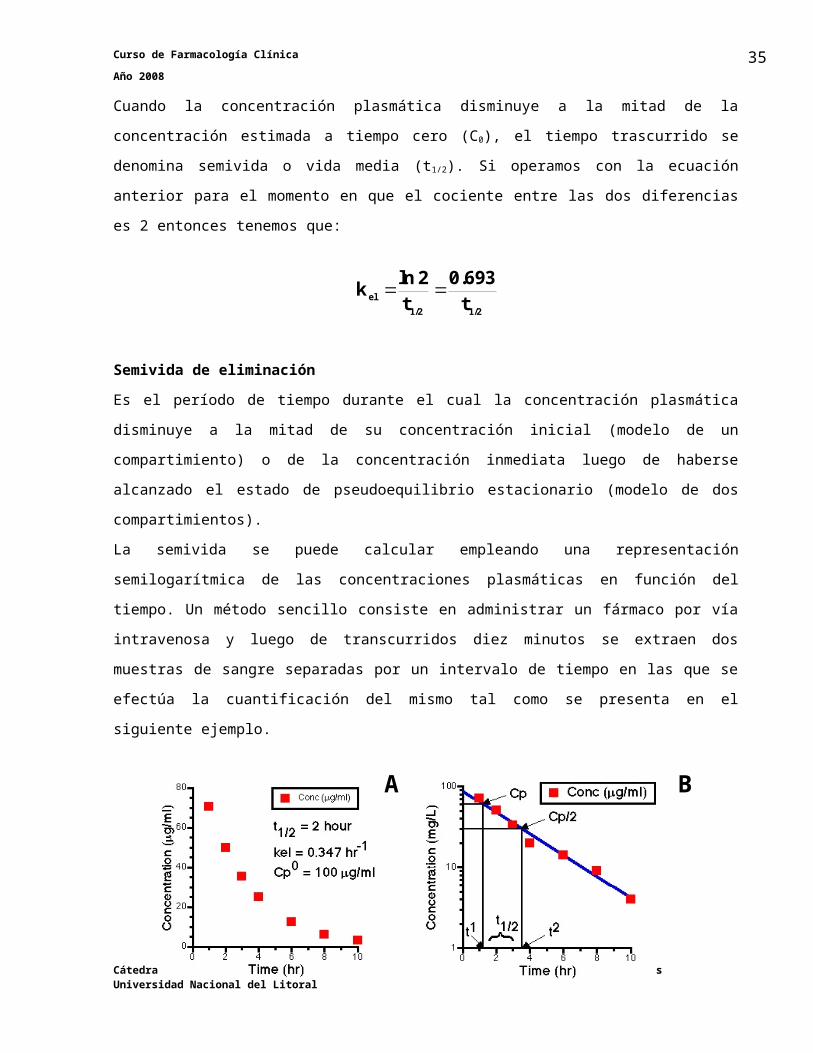
Semivida de eliminaciónEs el período de tiempo durante el cual la concentración plasmática disminuye a la mitad de su
concentración inicial (modelo de un compartimiento) o de la concentración inmediata luego de
haberse alcanzado el estado de pseudoequilibrio estacionario (modelo de dos
compartimientos).
La semivida se puede calcular empleando una representación semilogarítmica de las
concentraciones plasmáticas en función del tiempo. Un método sencillo consiste en administrar
un fármaco por vía intravenosa y luego de transcurridos diez minutos se extraen dos muestras
de sangre separadas por un intervalo de tiempo en las que se efectúa la cuantificación del
mismo tal como se presenta en el siguiente ejemplo.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
26
A B

Curso de Farmacología Clínica Año 2008
Si administramos un fármaco por vía intravenosa a un paciente a una determinada dosis y
determinamos las concentraciones plasmáticas de la misma a las 2 y 8 horas post
administración obtendremos una curva de concentraciones plasmáticas en función del tiempo
que se reprendan en escala original (A) y en escala semilogarítmica (B).
Los valores de concentración plasmática obtenidos son:
Cp a las dos horas de la inyección: 50 μg/ml
Cp a las ocho horas de la inyección: 6.25 μg/ml
El intervalo de tiempo entre las dos concentraciones es estimado como:
8 horas – 2 horas = 6 horas
Cálculo matemático de la semividaSe emplea la ecuación para el cálculo de kel
0.347 6
2.079 66.2550ln
k el
Se reordena la ecuación realizando pasaje de términos y tenemos que:
2 0.3470.963
kel0.693 t 1/2
En este caso la semivida estimada es de dos horas.
Semivida en una cinética no linealEn una cinética de eliminación de orden cero, al permanecer constante la velocidad de
eliminación, si aumenta la concentración plasmática, la semivida de eliminación se prolonga.
Por ejemplo; en humanos el ácido acetil salicílico que se hidroliza rápidamente a ácido salicílico
y luego se elimina conjugado con glicina, ácido glucurónico o como ácido salicílico libre. Si se
administran 500 mg del fármaco, la semivida del mismo es de 2 horas. Si la dosis se incrementa
a un gramo, la semivida se prolonga a cuatro horas. En casos de intoxicación se han
determinado semividas de 20 horas.
RELACIÓN DE LA SEMIVIDA CON OTROS PARÁMETROS FARMACOCINÉTICOSCon el volumen de distribución y el aclaramiento corporal o clearanceLa relación con este parámetro se presenta en la siguiente ecuación:
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
27

Curso de Farmacología Clínica Año 2008
a- Para dos fármacos con el mismo valor de aclaramiento corporal o clearance, la semivida
es mayor para el que presente el volumen de distribución mayor.
b- Si el volumen de distribución se incrementa a causa de algún proceso patológico la
semivida se prolonga y viceversa.
c- Si el clearance disminuye la semivida se prolonga y viceversa.
VOLUMEN DE DISTRIBUCIÓNEl volumen de distribución no es una entidad real, es solamente un concepto, ya que es el
factor de proporcionalidad que relaciona la concentración plasmática y la cantidad de fármaco
presente en el organismo.
donde M(t) es la masa de fármaco presente en el organismo a un tiempo determinado y Cp(t) es
la concentración plasmática observada a ese mismo tiempo.
Podemos también definirlo como el volumen hipotético en el cual el fármaco debería estar
disuelto para hallarse en la misma concentración que la observada en plasma.
El volumen de distribución se expresa en litros o ml por kg de peso. El valor del volumen de
distribución para la mayoría de fármacos se halla reportado en la literatura clásica.
El volumen de distribución en farmacocinética no es un volumen filológico, ya que la mayoría de
los fármacos presentan un volumen de distribución mucho mayor que el volumen de líquidos
presente en un organismo viviente. Sin embargo, algunos fármacos presentan un volumen de
distribución que puede ser interpretado casi como fisiológico como por ejemplo:
a- manitol, bloqueantes neuromusculares o ciertos antibióticos como los aminoglucósidos
que tienen un volumen de distribución similar al del líquido extracelular (0.2 litros/kg).
b- antipirina, alcohol y cafeína, que tienen un volumen de distribución similar al agua
corporal total (0.6 litros/kg).
c- heparina y azul de evans, que tienen un volumen de distribución similar al del plasma
(0.05 litros/kg).
Es importante considera que el volumen de distribución para un mismo fármaco puede variar
con la condición del paciente. Por ejemplo, en un cuadro urémico el volumen de distribución de
la digoxina disminuye a la mitad y en el neonato este es mayor que en los adultos. Otro caso
interesante lo constituyen las sulfamidas, cuyo volumen de distribución se incrementa en los
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
28

Curso de Farmacología Clínica Año 2008
cuadros de hipertermia. En el siguiente cuadro se describe la relación entre le valor del volumen
de distribución y el grado de distribución. Los valores aquí representados pueden ser
interpretados como una aproximación del grado de distribución.
Interpretación de los valores del volumen de distribución (Vss)
Fármaco fuertemente unidoa proteínas del plasma
0.06 litros/kgv. plasma/kg
Fármacos que atraviesanel endotelio capilar
0.2 litros/kg
v. plasma + v. liquido extracelular/kgFármacos que difundenatravés de membranas celulares
0.6 litros/kgv. plasma + v. extracelular + v. intracelular = agua total del organismo
Fármacos que se fijana ciertos tejidos
+ de 1 litro/kgconcepto puramente matemático
Una de las principales limitaciones del volumen de distribución es que los elevados valores
indican que el fármaco se halla fuera del espacio vascular pero no especifica donde ni de que
manera (forma libre o fijada a proteínas tisulares).
La magnitud del volumen de distribución están determinadas por dos propiedades físico
químicas; la unión a proteínas y el coeficiente de partición.
Unión a proteínasLas proteínas son macromoléculas capaces de atraer y unirse a pequeñas moléculas. Las
proteínas plasmáticas circulan dentro del organismo mientras que otro tipo de proteínas
(tisulares) son estacionarias. Los fármacos pueden unirse tanto a las proteínas circulantes como
a las tisulares con diferentes consecuencias. Una unión elevada de fármaco a las proteínas
plasmáticas (ibuprofeno y fármacos similares) darán como consecuencia volúmenes de
distribución pequeños a consecuencia de la retención del fármaco dentro del compartimiento
central. Por otra parte, fármacos con una elevada unión a proteínas tisulares como es el caso
de la digoxina tendrán volúmenes de distribución elevados. En un punto intermedio tenemos
fármacos que se unen extensivamente tanto a proteínas plasmáticas y tisulares. En plasma los
fármacos que se comportan como ácidos tienden a unirse a la albúmina, mientras que en el
caso de los compuestos básicos, estos presentan gran afinidad por las alfa glicoproteínas
ácidas.
Coeficiente de partición
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
29

Curso de Farmacología Clínica Año 2008
La fracción libre de los fármacos (forma no unida) se distribuye en el organismo dependiendo de
su afinidad por las estructuras lipídicas mas que por su afinidad por permanecer en el espacio
vascular acuoso. La capacidad de un fármaco de difundir fuera del espacio vascular se evalúa
mediante el coeficiente de partición octanol/agua (o/w). Por ejemplo; un fármaco con un
coeficiente de partición mayor a la unidad, tenderá a acumularse en los tejidos grasos, mientras
que un fármaco con coeficiente de partición con un valor menor a la unidad presentará afinidad
por permanecer disuelto en la fase acuosa.
Lo explicado hasta aquí demuestra que el volumen de distribución está lejos de ser un volumen
fisiológico y que tampoco puede ser interpretado como un indicador de la capacidad de un
fármaco de difundir fuera del espacio vascular.
Cálculo del volumen de distribución Los cálculos que se presentan aquí, se realizan considerando una disposición asociada a un
modelo de un compartimiento.
a- Calculando la concentración plasmática a tiempo cero.En un ensayo de farmacocinética, el único dato concreto de que disponemos es el de la dosis
exacta de fármaco administrado por la vía intravenosa.
De manera análoga a la estimación de kel, la regresión lineal de los datos log transformados
permite describir los datos de concentración plasmática con la ecuación de la recta donde el
punto de intersección en el eje de las ordenadas es el logaritmo natural de la concentración
estimada a tiempo cero (lnC0). Si tomamos entonces el antilogaritmo de lnC0 (C0) y hallamos el
cociente entre la dosis administrada y este obtendremos el volumen de distribución tal como
representa a continuación.
0d C
DosisV
b- Utilizando los datos de clearance y de la constante de eliminación.El valor del Vd que comúnmente se reporta en la literatura es el estimado mediante el cociente
entre el clearance y la constante de eliminación según la siguiente ecuación:
eld k
ClV
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
30
Curso de Farmacología Clínica Año 2008
Este valor se denomina volumen de área y es el que se emplea en la clínica para realizar los
cálculos de dosificación individual.
CLEARANCE O ACLARAMIENTO CORPORALEn fisiología se utiliza la determinación del clearance de la creatinina, una substancia endógena
con ciertas características farmacocinéticas que la hacen ideal para este propósito. La
creatinina se elimina por vía urinaria en un 100%, porque no sufre biotransformación alguna y
su concentración plasmática es muy constante porque se elimina a la misma velocidad con la
que se va produciendo.
La creatinina ingresa a la circulación general a velocidad constante mediante un proceso de
orden cero y se elimina por un proceso de orden uno, de allí el estado de equilibrio estacionario
del sistema y las concentraciones plasmáticas en estado de meseta.
Para determinar el clearance de creatinina (Clcr) se obtiene una muestra de orina de 3 horas o
de 12 horas, donde se cuantifica la creatinina.
Así se obtiene el valor de creatinina en miligramos excretada por minuto. También se cuantifica
la creatinina en plasma (Cpcr), la cual se supone constante durante las horas de recolección de
orina. Si representamos lo explicado con un modelo de un compartimento tenemos que:
Considerando que la producción de creatinina es igual a la excreción, entonces la concentración
plasmática de creatinina no varía (modelo en estado de equilibrio). Entonces la fórmula para
determinar el clearance de creatinina es la siguiente:
/minutomililitroscrC
minuto por eliminada cantidadClcrp
En farmacología, para determinar el clearance de los fármacos tenemos dos problemas:
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
31
Velocidad de salida
kel x M (eliminación)
Velocidad de entrada
DM/T (dosificación)
Cp
Vd
CpcrExcreción de creatininamg/minuto
Producción de creatininamg/minuto

Curso de Farmacología Clínica Año 2008
a- No podemos cuantificar directamente la cantidad eliminada, ya que la eliminación
comprende la excreción por todas las vías, más la transformación a metabolito.
b- A diferencia de la creatinina cuyos niveles séricos se mantienen constantes, la
concentración plasmática del fármaco es variable en el tiempo.
En consecuencia, para hallar el clearance de un fármaco debemos recurrir a :
a- Utilizando una infusión continuaCuando se alcanza el estado de equilibrio estacionario de un fármaco mediante una infusión
intravenosa continua, entonces podemos calcular la velocidad de infusión como el cociente
entre la dosis administrada y el tiempo de infusión:
eeC CltD
p
donde t es el tiempo de la infusión y Cpee es la concentración plasmática al estado de equilibrio
estacionario. Por lo tanto conociendo la velocidad de infusión y la concentración al estado de
equilibrio estacionario podemos calcular el clearance como:
eeCtD
Clp
b- Utilizando el cálculo del área bajo la curva de concentración plasmáticaEn un gráfico de la evolución de las concentraciones plasmáticas en función del tiempo, el área
delimitada por estas se conoce como área bajo la curva de concentración plasmática en función
del tiempo o ABC. En la siguiente figura el ABC se representa sombreada.
Al calcular el ABC estamos calculando el valor de la integral entre tiempo 0 e infinito de la
concentración plasmática multiplicada por la diferencial del tiempo.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
32

Curso de Farmacología Clínica Año 2008
Si reemplazamos el término Cp por la ecuación exponencial que describe la curva de
disposición plasmática tenemos que:
De acuerdo con lo que hemos visto al tratar el problema de la extrapolación, C0 es igual a la
dosis dividida por el volumen de distribución, con lo cual la ecuación anterior queda como:
.VdkD ABC
el
Reagrupando obtenemos que:
ABCD Cl
Experimentalmente, si administramos una dosis D a un individuo y luego determinamos varias
concentraciones plasmáticas podemos determinar el ABC por medio de la regla trapezoidal.
Como conocemos la dosis entonces podemos calcular el clearance.
c- Utilizando la fórmula Cl = kel x VdEsta fórmula puede explicarse utilizando las ecuaciones del estado de equilibrio estacionario y
la definición de clearance que es la relación entre la velocidad de eliminación (kel x M) y una
concentración de referencia (Cpee).
eeCMkCl
p
el
Reagrupando tenemos que:
M k eeC Cl elp
Multiplicando y dividiendo por el volumen de distribución tenemos que:
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
33

Curso de Farmacología Clínica Año 2008
VdMVdk eeC Cl elp
Pero M/Vd es la Cpee y entonces nos queda
.Vdk Cl el
Eliminando Cpee de ambos miembros tenemos
.VdkCl el
SIGNIFICADO DEL CLEARANCEEl clearance de un fármaco puede interpretarse como:
a- El volumen de plasma que se depura completamente de un fármaco por unidad de
tiempo.
b- La fracción del volumen de distribución que se depura del fármaco por unidad de tiempo.
c- La constante de proporcionalidad entre la velocidad de eliminación y la concentración
plasmática.
El clearance es un concepto biológicamente significativo e importante. Su magnitud es
independiente del modelo farmacocinético (uno, dos o mas compartimientos). Dado que es un
parámetro independiente, los valores de clearance que aparecen en las tablas se calculan
mediante el método del ABC.
El clearance total de un fármaco puede expresarse como a suma de los clearance de varios
órganos, siendo los más importantes el hígado y el riñón.
Cl = ClH + ClR +ClOTROS
Donde ClH es el clearance hepático, el ClR es el clearance renal y ClOTROS es el clearance debido
a otros órganos.
CLEARANCE HEPÁTICO
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
34
Curso de Farmacología Clínica Año 2008
El valor máximo del clearance hepático es el volumen de sangre arterial que arriba a este
órgano por unidad de tiempo y que representa el 30% del débito cardíaco, el cual varía según la
especie en función de su peso corporal según se presenta en la siguiente tabla.
Pesocorporal
Kg0.2 3 10 50 70 100 500
Débitocardíaco
ml/kg/min244 146 116 86 80 75 55
ClearanceHepático máximo
ml/kg/min73 44 35 26 24 23 17
Por ejemplo, en el hombre el clearance hepático máximo es de aproximadamente 1500 ml por
minuto, que es el flujo sanguíneo hepático. Un fármaco con este clearance sería totalmente
extraído del hígado en un solo pasaje.
Los fármacos que se eliminan por metabolismo hepático pueden clasificarse como:
a- Fármacos de alto clearance (alta extracción) con un valor mayor a 1000 ml por minuto
como la morfina, el verapamil, propanolol y lidocaína. Para estos fármacos el valor del
clearance está determinado por el flujo hepático.
b- Fármacos con bajo clearance hepático. Para estos fármacos, el clearance no depende
de flujo sanguíneo hepático sino de la capacidad intrínseca del órgano para metabolizar
el fármaco.
CLEARANCE RENALAl igual que en el caso del clearance hepático, el valor máximo del clearance renal es el valor
del débito sanguíneo de este órgano, que representa el 20% del débito cardíaco y que también
varía según la especie en función del peso corporal según la siguiente tabla.
Pesocorporal
Kg0.2 3 10 50 70 100 500
Débito
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
35

Curso de Farmacología Clínica Año 2008
cardíacoml/kg/min
244 146 116 86 80 75 55
ClearanceRenal máximo
ml/kg/min49 30 23 17 16 15 11
Para que un fármaco se elimina completamente de la sangre en un solo pasaje a través de los
riñones debe ser secretada activamente por los túmulos renales.
Cátedra de Farmacología Facultad de Ciencias Veterinarias Universidad Nacional del Litoral
36