case histories
DESCRIPTION
Case Histories. Case 1: Positive Newborn Metabolic Screen. History : NMS test result shows elevated Phenylalanine (0.75 umole/l; normalTRANSCRIPT

Case HistoriesCase Histories

Case 1: Positive Newborn Metabolic
ScreenHistoryHistory: : NMS test result shows elevated Phenylalanine NMS test result shows elevated Phenylalanine
(0.75 umole/l; normal <0.125)(0.75 umole/l; normal <0.125) Term pregnancyTerm pregnancy
Normal P/L/DNormal P/L/D BWt 3.1 kg, BWt 3.1 kg, Normal neonatal courseNormal neonatal course
Questions:1. Describe briefly what your initial
counselling to parents would be.2. What investigations would you under take
to confirm diagnosis?

Results of investigations
Results Plasma
PHE=1.2umole/l; tyrosine = 0.05 umole/l
Urine organic acids increased PPA,PLA,PAA
Questions1. What other
tests need to be done to be sure this baby needs diet treatment?
2. What is the basis of the diet treatment?
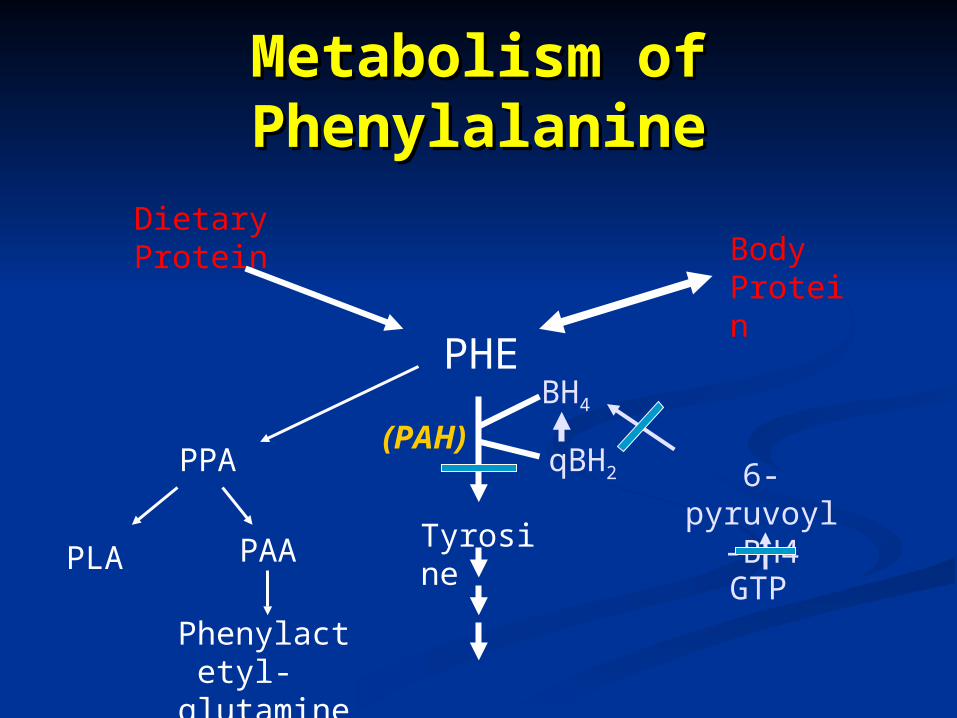
Metabolism of Metabolism of PhenylalaninePhenylalanine
PHE
Dietary Protein Body
Protein
Tyrosine
PPA
PLA PAA
Phenylactetyl- glutamine
BH4
qBH2
(PAH)6-pyruvoyl-
BH4
GTP
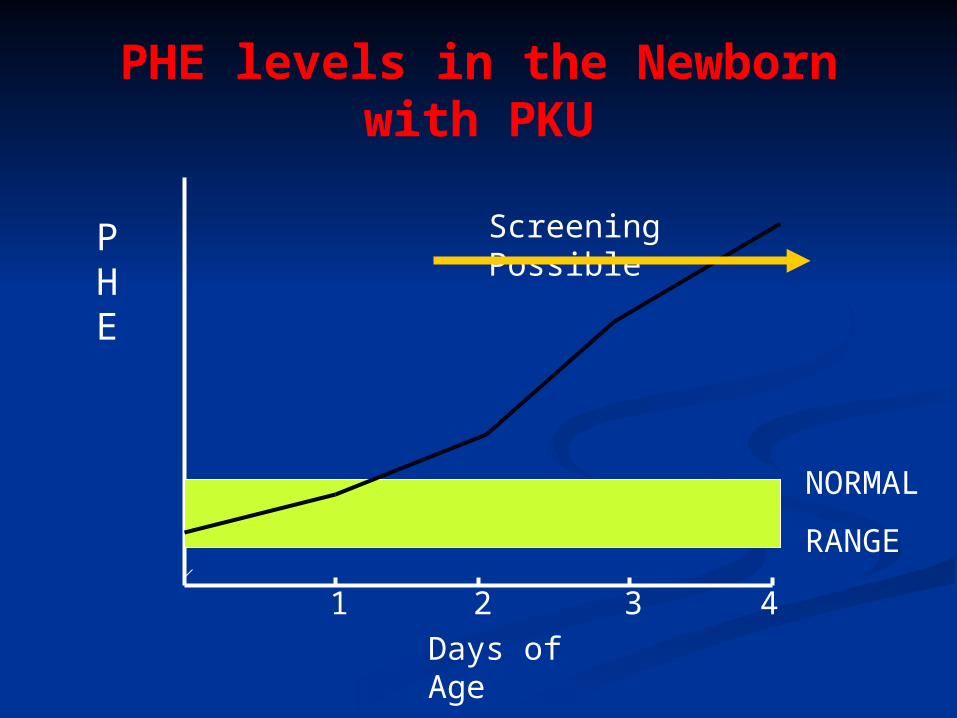
PHE levels in the Newborn with PKU
PHE
Days of Age
1 2 3 4
NORMAL
RANGE
Screening Possible

Untreated Phenylketonuria
Signs / Symptoms Signs / Symptoms mental retardationmental retardation hypopigmentation eczema-like rasheczema-like rash autistic-like behaviorautistic-like behavior autosomal recessiveautosomal recessive high bloodhigh blood
phenylalanine levelsphenylalanine levels

PKU: Diagnostic work-up
Confirm that PHE level is elevated Rule out biopterin deficiency
disorders Urine pterin levels Dihydrobiopterin reductase activity Biopterin load test (optional) If present start DOPA/carbiDOPA/5HTP
If BH4 disorder not diagnsosed & PHE above 0.4 mM/l, start low PHE diet

Natural Foods
Contains some normal nutrients
and all those being restricted
Nutritional Treatment of PKU
Diet has two components:
Must meet all nutritional needs + limit intake of restricted nutrients to amts sufficient for growth
Medical Formula
Contains all nutrients
except those being restricted
+
Case 2: Hepatomegaly with Abnormal Liver Pathology
History: The pathologist in your hospital calls to discuss
an abnormal liver biopsy result 8 month old boy with (R) abdominal mass
extending down into the iliac fossa Seen by Oncology re: ? Tumor; taken to OR
for open biopsy
QuestionWhat do you see in the following biopsy?
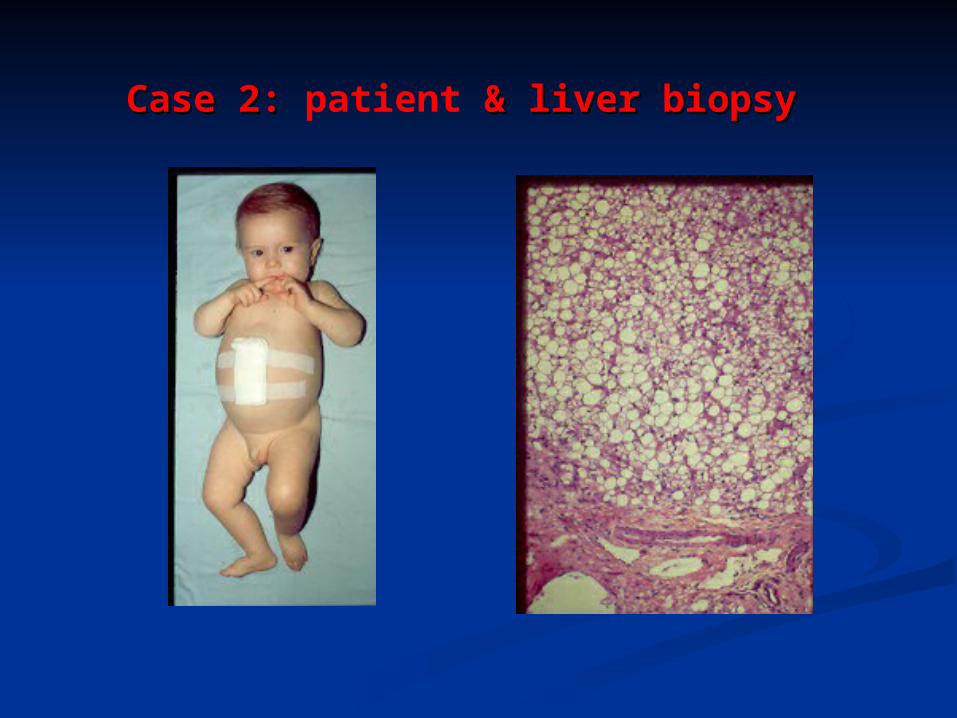
Case 2: Case 2: patient & liver biopsy & liver biopsy

Questions
1. What types of disorders might cause this appearance?
2. What further historical information may be of help?
3. What further studies should you request from the pathologist?

Questions1.1. What types of disorders might cause this appearance?What types of disorders might cause this appearance?
• Glycogen storage disorders (types 1a & 1b, 3, 6 lysosmal storage disorders (Gaucher, Niemann-Pick, MPS, oligosaccharidoses, tyrosinemia, B-oxidation disorders (MCAD, LCHAD, VLCAD)
2.2. What further historical information may be of help?What further historical information may be of help?• Symptoms of hypoglycemia (relationship to fasting
including timing)• Mother indicates baby can only go about 2-4 hours
without a “bottle”
3.3. What further studies should you request from the What further studies should you request from the pathologist?pathologist?
• PAS staining +/- pretreatment with diastase• Electon microscopy

Diagnostic testingDiagnostic testing
Fasting challenge +/- feeding challenges
Enzyme assays Need fresh liver Need to choose specific enzymes to
target based on history Molecular testing
Now have bank of mutations but expensive
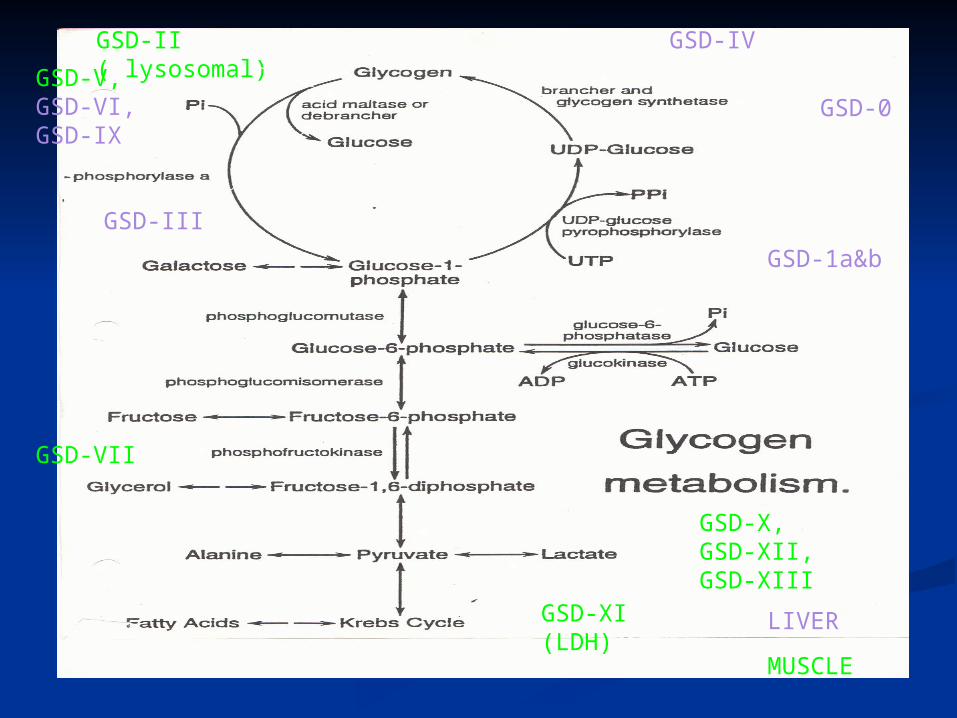
GSD-0
GSD-IV
GSD-1a&b
GSD-V, GSD-VI, GSD-IX
GSD-II ( lysosomal)
GSD-III
GSD-VII
GSD-X, GSD-XII, GSD-XIII
GSD-XI (LDH)
LIVER
MUSCLE
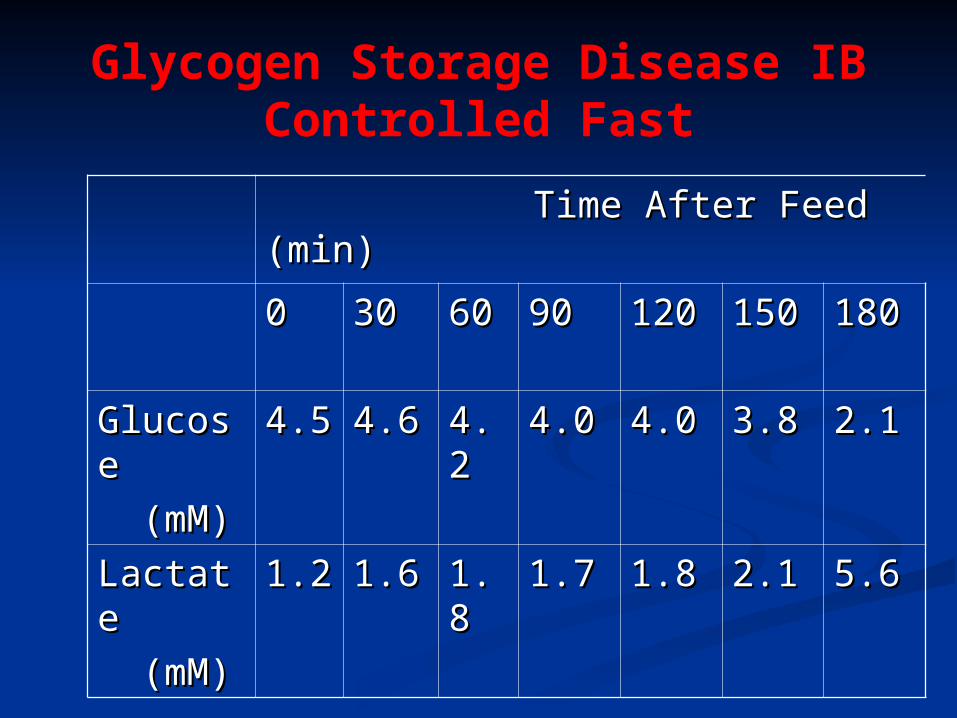
Glycogen Storage Disease IBControlled Fast
Time After Feed (min)Time After Feed (min)
00 3030 6060 9090 120120 150150 180180
GlucosGlucosee
(mM)(mM)
4.54.5 4.64.6 4.24.2 4.04.0 4.04.0 3.83.8 2.12.1
LactateLactate
(mM)(mM)1.21.2 1.61.6 1.81.8 1.71.7 1.81.8 2.12.1 5.65.6

GSD IA &IBClinical features
early onset hypoglycemia
lactic acidosis hepatomegaly Fanconi syndrome hyperuricemia hyperlipidemia
Diagnosis controlled fast (test
BS & LA) enzyme (liver biopsy) DNA testing
Therapy provide 5 - 10 mg
glucose/kg/min continuous .nocturnal
infusion of CHO as polycose or formula
frequent meals during days
corn starch days &/or nights
don’t over treat with CHO
Neutropenia in Type IB prophylactic antibiotics GCSFEmergency protocols for
illness, surgery etc.

Case 3: 18 month boy with hepatomegaly and
obtundationHistory: stuporous on admission found pale & sweaty and unarousable by parents Had been ill for about 18 hours with no
significant intake Seizure in ambulance
Initial studies: Blood sugar = 0.2 mM/l, Na+=145, K+ =3.5, Cl- =104,
TCO2 = 10 Urinalysis = Normal All other testing including lactate, NH4 &
LFE’s normal

Key observationsKey observations
Severe hypoglycemia with hepatomegaly and no ketonuria on setting of history of prolonged fasting
Needs urgent treatment of hypoglycemia Route? How much glucose?
? Significance of no ketones in urine ?diagnostic testing
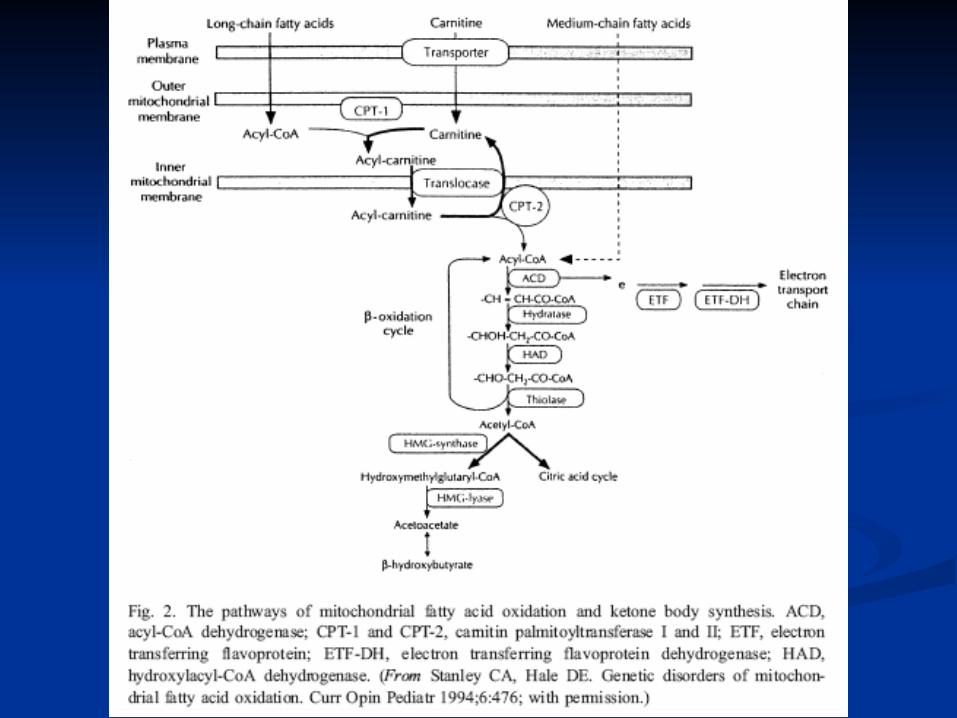

Trifunctional protein
VLCAD,MCAD, SCAD

Diagnostic InvestigationsDiagnostic Investigations
Plasma acylcarnitnes suggest Medium Plasma acylcarnitnes suggest Medium Chain Dehydrogenase deficiency (MCAD)Chain Dehydrogenase deficiency (MCAD)
Plasma free carnitine levels low while Plasma free carnitine levels low while acylcarnitines highacylcarnitines high
1414C- palmitic acid oxidation in leucocutes C- palmitic acid oxidation in leucocutes quite reducedquite reduced
Molecular diagnosis indicates Molecular diagnosis indicates homozygosity for the common caucasian homozygosity for the common caucasian mutation.mutation.
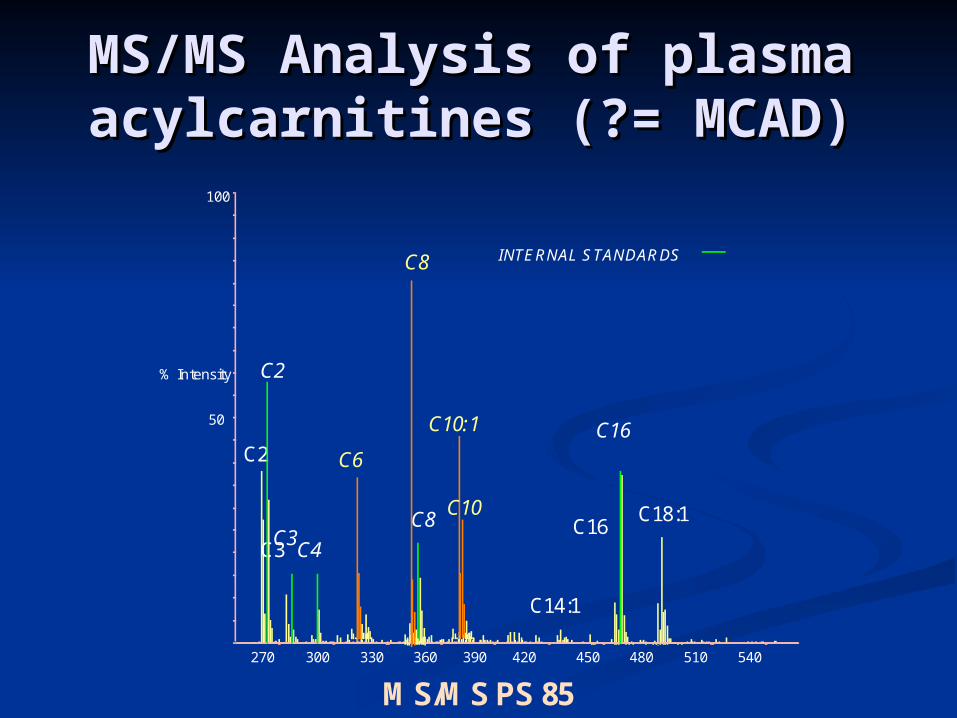
MS/MS Analysis of plasma MS/MS Analysis of plasma acylcarnitines (?= MCAD)acylcarnitines (?= MCAD)
270 300 330 360 450 480 510 540
50
100
% Intensity C2
INTERNAL STANDARDS
C3C4
C8
C16
C18:1C16
C3
C2
C14:1
390 420
C8
C6
C10
C10:1
MS/MS PS 85

Phases of Glucose HomeostasisPhases of Glucose Homeostasis
1.1.Glucose absorptive phaseGlucose absorptive phase: 3 - 4 hrs after : 3 - 4 hrs after glucose ingestion (high insulin) glucose ingestion (high insulin)
2.2.Post absorptive/early starvationPost absorptive/early starvation: 3-12 hrs: 3-12 hrs
glucose (from hepatic glycogen) to brain, glucose (from hepatic glycogen) to brain, RBC, renal medullaRBC, renal medulla
3.3.Early / Intermediate StarvationEarly / Intermediate Starvation: 14+ hrs: 14+ hrs
gluconeogenesis & (later) lipolysis gluconeogenesis & (later) lipolysis

Treatment
Avoid fasting L-carnitine if free carnitine low Emergency protocol & letter
Sick day management Admission to ER/hospital to maintain
blood glucose with IV infusion to prevent excessive lipolysis the would overload the B-oxidation pathway

Case 4: Neonatal PresentationCase 4: Neonatal Presentation
5 d.o. male5 d.o. male Well for 72 hrs then Well for 72 hrs then
became lethargic, fed became lethargic, fed poorly, began vomiting & poorly, began vomiting & developed alternating developed alternating flaccidity & opisthotonic flaccidity & opisthotonic posturing.posturing.
Became comatoseBecame comatose Developed hyperpnea and Developed hyperpnea and
respiratoy alkalosis respiratoy alkalosis progressing to respiratory progressing to respiratory failurefailure
O/E: hepatomegaly, O/E: hepatomegaly, hypothermiahypothermia
Test ResultsTest Resultso NormalNormal: CBC, ‘lytes’, : CBC, ‘lytes’,
bld glucose, lactic bld glucose, lactic acid, urinalysisacid, urinalysiso not acidoticnot acidotico not ketoticnot ketotico not hypoglycemicnot hypoglycemic
o NH3 (350 umole/l) NH3 (350 umole/l)
? Differential ? Differential DiagnosisDiagnosis
? Further testing? Further testing

Case 4: Investigation ResultsCase 4: Investigation Results
5 d.o. male5 d.o. male Well for 72 hrs then Well for 72 hrs then
became lethargic, fed became lethargic, fed poorly, began vomiting & poorly, began vomiting & developed alternating developed alternating flaccidity & opisthotonic flaccidity & opisthotonic posturing.posturing.
Became comatoseBecame comatose Developed hyperpnea and Developed hyperpnea and
respiratoy alkalosis respiratoy alkalosis progressing to respiratory progressing to respiratory failurefailure
O/E: hepatomegaly, O/E: hepatomegaly, hypothermiahypothermia
Test ResultsTest Resultso NormalNormal: urine amino : urine amino
acids & organic acidsacids & organic acidso LowLow: urea, arginine, : urea, arginine,
ornithine, ornithine, o HighHigh: citrulline (1.21 : citrulline (1.21
mM) mM) glutamine (1.4 glutamine (1.4 mM) mM) asparagine asparagine
? Probable Diagnosis? Probable Diagnosis
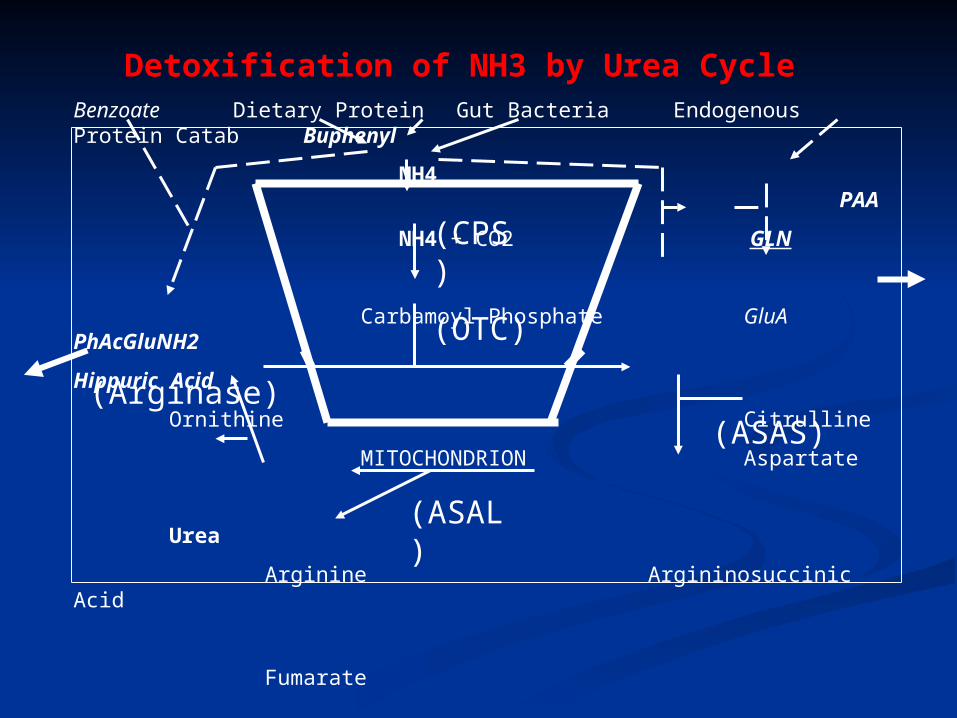
Detoxification of NH3 by Urea Cycle Benzoate Dietary Protein Gut Bacteria Endogenous Protein Catab Buphenyl
NH4 PAA
NH4 + CO2 GLN
Carbamoyl Phosphate GluA PhAcGluNH2
Hippuric Acid
Ornithine Citrulline
MITOCHONDRION Aspartate
Urea
Arginine Argininosuccinic Acid
Fumarate
(CPS)
(OTC)
(ASAS)
(ASAL)
(Arginase)
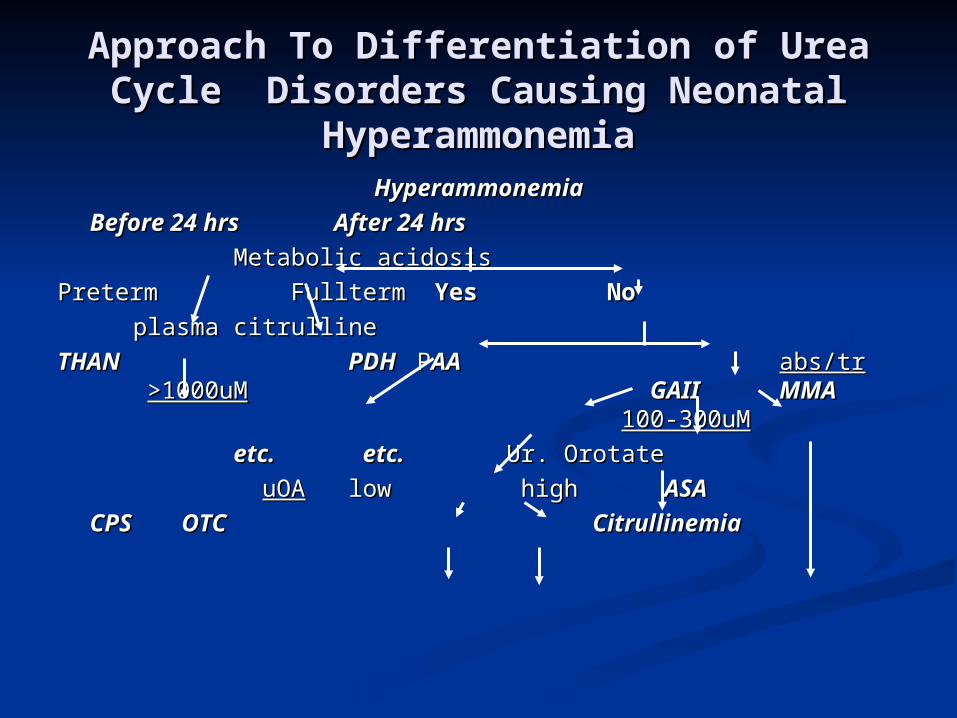
Approach To Differentiation of Urea Approach To Differentiation of Urea Cycle Disorders Causing Neonatal Cycle Disorders Causing Neonatal
HyperammonemiaHyperammonemiaHyperammonemiaHyperammonemia
Before 24 hrsBefore 24 hrs After 24 hrsAfter 24 hrs
Metabolic acidosisMetabolic acidosis
PretermPreterm Fullterm Fullterm YesYes NoNo
plasma citrullineplasma citrulline
THANTHAN PDH PDH P PAA AA abs/trabs/tr >1000uM>1000uM GAIIGAII MMA MMA 100-300uM100-300uM
etc.etc. etc.etc. Ur. Orotate Ur. Orotate
uOAuOA low highlow high ASAASA
CPS OTCCPS OTC CitrullinemiaCitrullinemia

Approaches to Therapy of Urea Cycle Disorders
Acute Mgmt(based on NH3 level)
NPO Dialysis ( prefer.
Hemodialysis) IV: CHO (6–8 mg
Glc/kg/min) Lipid (3 gm / kg)
Alternate Pathway Therapy Oral (Phenylbutyrate +
L-Arg) IV (Phenylacetate +
benzoate + L-arginine
Chronic Mgmt Low protein diet
–1.0 to 1.5 gm/kg/d
-Cyclinex (ess. AA’s)
(up to 50 % of prot)
Phenylbutyrate (Buphenyl)
(450-650mg/kg/d) Arg / ornith /
citrulline Regular monitoring
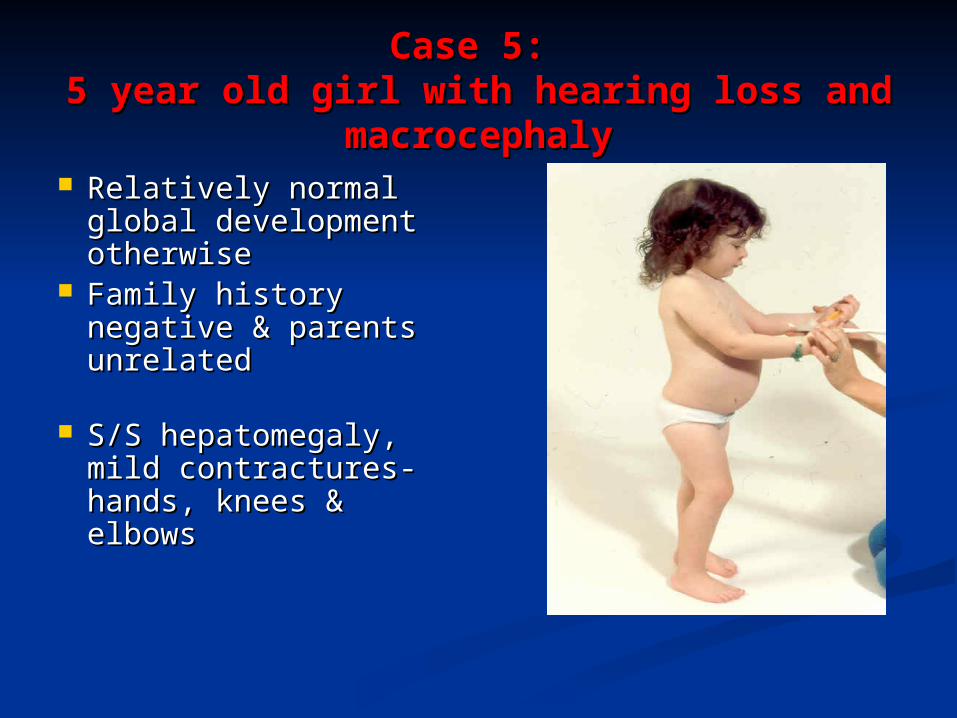
Case 5: Case 5: 5 year old girl with hearing loss and 5 year old girl with hearing loss and
macrocephalymacrocephaly Relatively normal Relatively normal
global development global development otherwiseotherwise
Family history Family history negative & parents negative & parents unrelated unrelated
S/S hepatomegaly, S/S hepatomegaly, mild contractures-mild contractures-hands, knees & hands, knees & elbowselbows
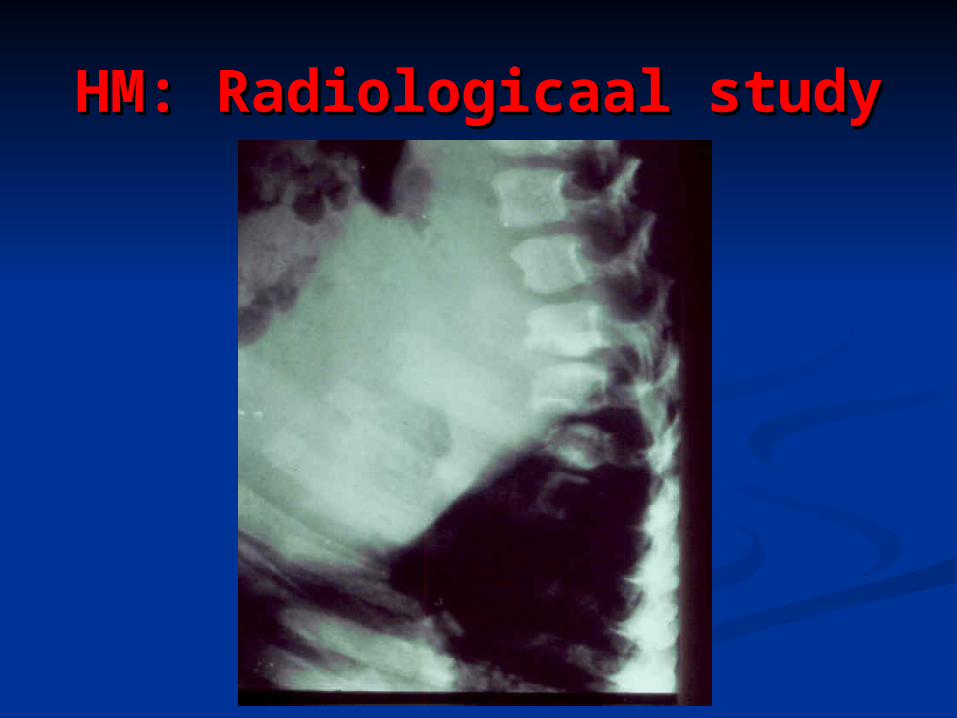
HM: Radiologicaal studyHM: Radiologicaal study

Case History: HMCase History: HM
You were asked to see this girl for You were asked to see this girl for assessment regarding macrocephaly assessment regarding macrocephaly and hearing lossand hearing loss
Initial investigations showed an Initial investigations showed an abnormal urine metabolic screen that abnormal urine metabolic screen that positive for both CTAB & Toluidine bluepositive for both CTAB & Toluidine blue
All other initial metabolic studies All other initial metabolic studies normalnormal
What would you do next?What would you do next?
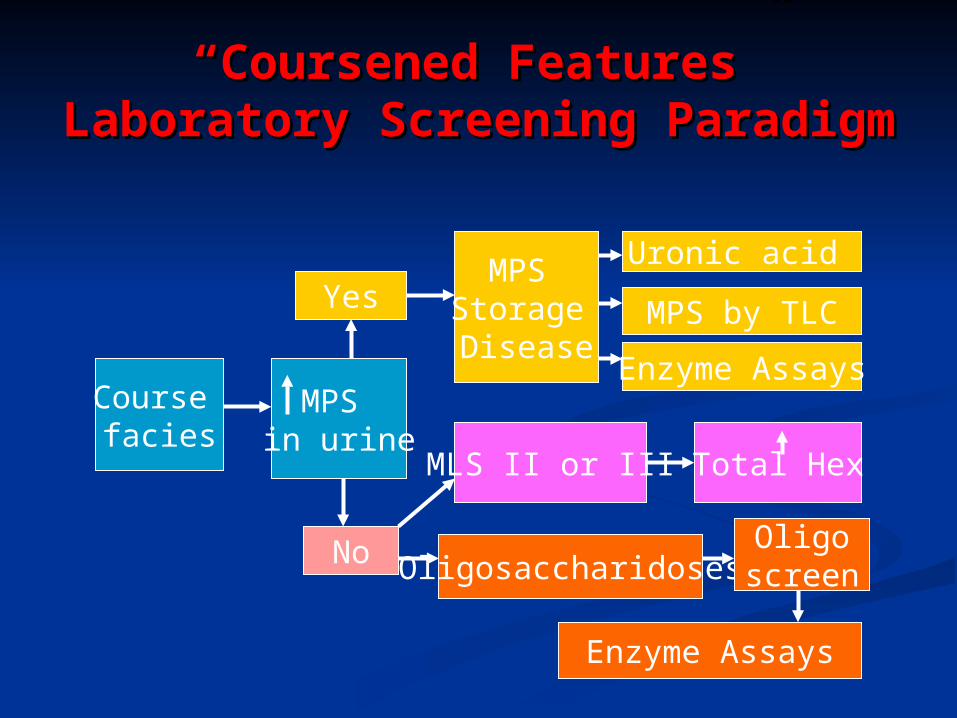
““Coursened Features”Coursened Features”Laboratory Screening Laboratory Screening
ParadigmParadigm
Course facies
MPS in urine
Yes
No
MPS Storage Disease
Uronic acid
MPS by TLC
Enzyme Assays
Total HexMLS II or III
Oligosaccharidoses
Enzyme Assays
Oligoscreen
How do you measure How do you measure mucopolysaccharide mucopolysaccharide
excretion?excretion?Uronic acidUronic acid GAG GAG
electrophoresielectrophoresiss
What are the useful GAGS in diagnosing a
particular MPS disorder?
How is this testing done?

How do you measure How do you measure mucopolysaccharide mucopolysaccharide
excretion?excretion?Uronic acidUronic acid GAG electrophoresisGAG electrophoresis
Acid Hydrolysis to free
monosaccharide subunits
+
Colorimetric detection
of the uronic acid (A or B)
A B
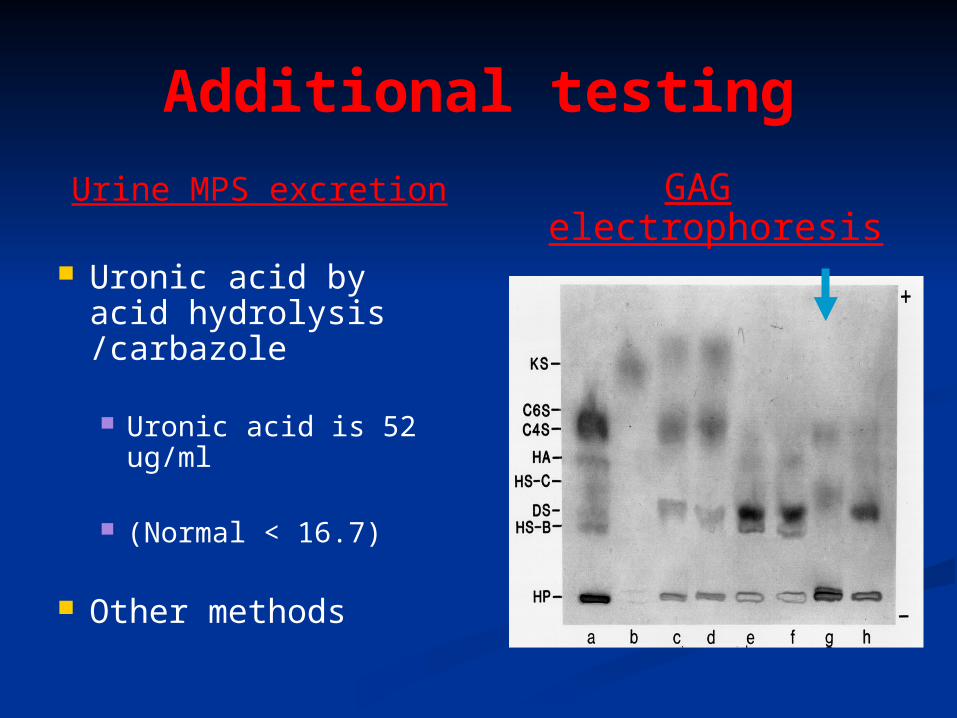
Additional testing
Urine MPS excretion
Uronic acid by acid hydrolysis /carbazole
Uronic acid is 52 ug/ml
(Normal < 16.7)
Other methods
GAG electrophoresis ( HM
spec)

Diagnosis & Management
Mucopolysaccharidosis Type III San Filippo syndrome Four different enzyme deficiencies all leading to
inadequate breakdown of heparin sulfate She had deficiency of heparin-N-sulfatase (MPS-
IIIA)
No definitive treatment at present extensive social/psychological/educational
support appropriate for child with neurodegenerative
disorder

Case 6:Recurrent Abdominal Pain in 6
yo Male

History The mother of a six year old boy tells you that
her son has had three episodes of abdominal pain without any flu-like symptoms or other systemic problems.
During the last episode, blood but no bacteria or “pus” cells were seen in his urine. The urine did not smell, look cloudy or have an unusual color.
Her husbands had had similar episodes as a child but these had stopped once he followed the advice of a pediatrician to ”drink lots of water” at nights..

Your initial investigations showed that this boy had a normal physical exam, normal CBC.
‘lytes, urea & creatinine and aside from microscopic hematuria, a normal microscopic
& chemical urinalysis
What do you think is causing his abdominal pain & microscopic hematuria?
What genetic / metabolic disorders should you consider in your differential diagnosis list?
What specific testing would you suggest to investigate these possibilities?

Differential Diagnostic List
Cystinuria (basic aminoaciduria) Partial HGPRTase deficiency (uricosuria) GSD I (uricosuria) Primary hyperoxaluria (oxalic aciduria +/-
glycolic or glyceric acid) Idiop. Hypercalciuria (calcium oxalate or
urate) Hyperparathyroidism (calcium oxalate / urate) Adenosine phosporibosyl transferase
deficiency (dihydroxyadenosinuria)

Results of Special Results of Special TestingTesting
Unable to isolate characteristic stones in random urine specimen
Urine oxalic acid, calcium, uric acid / creatinine ratio all normal
urine oxypurine profile normal cystine, lysine, ornithine & arginine but
no other amino acids elevated in urine Plasma amino acid profile normal
What is your Diagnosis?

Cystinuria disorder of basic amino
acid transport involving renal tubule & GI mucosa
urolithiasis:poor
solubility of cystine when: urine concentrated
(cystine> 1200 uM/l) urine acidic
autosomal recessive 1: 7000
Types I, II & III (newer classification refers to type I as classical & others as non-classical)
Heterozygotes (type II) may excrete Cyst. Lys & Orn +/- arg but in reduced amts
Renal immaturity (< 1 yr) may cause “apparent” cystinuria in Type III carrier

Dibasic Amino Aciduria Normal: Cystine filtered at glomerulus but over 98
% reabsorbed in proximal renal tubule
Common renal tubular reuptake mechanism for dibasic amino acids (cystine,lysine, ornithine, arginine). All increased in urine but not blood if transporter is deficient
Nephrolithiasis: onset by 10 yrs (25-30 %) to 20 yrs (50-60 %) Cystine precipitates out in urine / renal filtrate at
concentrations above 1200 uM/l stones: hexagonal, golden-brown, grain size to staghorn size account for 1-3 % of all stones in adults (kids ?)
Subtypes based on amt excretion of cys/lys/orn but not arg in heterozygotes. Type III has similar defect in mucosal uptake

Molecular Genetics
Locus 1: 2p16.3-p21
Type I cystinuria rBAT protein SLC3A1 gene 40+ mutations Transmembrane
protein
Locus 2: 19q13.1
Non-Type 1 cystinuria Bo, +AT protein SLC7A9 gene 30 + mutations Complexes with rBAT
protein to form dibasic amino acid transporter

Phenotype/Genotype CorrelationCorrelation
Cystinuria I/I: two fully recessive SLC3A1 mutations no gut absorption kidney - high risk for nephrolithiasis
Cystinuria III/III two incompletely recessive SLC7A9 mutations stones in adults some gut absorption
Mixed types: I/III(A), I/III(B), II/II,

Treatment of Cystinuria dilution of urine:
drink up to 4.0 liters fluid / day (1.75 - 2.0 l/m2/24 hr) important to drink during the night time (water at
bedside) monitor urine cystine concentrations morning &
evening
“alkalinization” of urine: NaHCO 3 ( 1.5 - 2.0 mEq/kg/24 hr)
Medication: D-penicillamine, Thiola R (tiopronin) Surgical: lithotripsy etc.