cancer neutrophil extracellular traps produced during ... · research article summary cancer...
TRANSCRIPT

RESEARCH ARTICLE SUMMARY◥
CANCER
Neutrophil extracellular trapsproduced during inflammationawaken dormant cancer cells in miceJean Albrengues, Mario A. Shields, David Ng, Chun Gwon Park, Alexandra Ambrico,Morgan E. Poindexter, Priya Upadhyay, Dale L. Uyeminami, Arnaud Pommier,Victoria Küttner, Emilis Bružas, Laura Maiorino, Carmelita Bautista, Ellese M. Carmona,Phyllis A. Gimotty, Douglas T. Fearon, Kenneth Chang, Scott K. Lyons, Kent E. Pinkerton,Lloyd C. Trotman, Michael S. Goldberg, Johannes T.-H. Yeh, Mikala Egeblad*
INTRODUCTION: Most cancer patients diefrom cancer that recurs after spreading to adifferent tissue, rather than from their originaltumor. After successful treatment of the orig-inal tumor, cancer cells that have disseminatedto other sites can undergo dormancy, remainingviable but not proliferating. In breast, prostate,and other cancers, cancer cells can remain dor-mant and clinically undetectable for years andeven decades before recurring, or awakening,as metastatic cancer. Little is known aboutwhatmight initiate cancer awakening, and this
in turn reduces our opportunities to preventmetastasis.
RATIONALE: Epidemiological studies havesuggested that inflammation is linked to ahigher risk of breast cancer recurrence after aperiod of clinical dormancy. Smoking, whichcauses chronic lung inflammation, is also as-sociated with a higher risk of recurrence.However, whether inflammation can causeawakening is not clear. Inflammatory cells,such as neutrophils, can provide many differ-
ent signals that promote cancer progression.Neutrophils can kill harmful microorganismsby the release of neutrophil extracellular traps(NETs) into the extracellular space. NETs arescaffolds of DNA with associated cytotoxicproteins and proteases [e.g., neutrophil elastase(NE) and matrix metalloproteinase 9 (MMP9)].NETs induced by bacteria or by cancer cellscan promote metastasis, but the mechanismby which this occurs is not known. In thisstudy, we tested whether NETs formed duringlung inflammation could induce awakening.
RESULTS: We found that sustained experi-mental lung inflammation—induced by either
tobacco smoke exposureor nasal instillation of li-popolysaccharide (LPS)—converted dormant cancercells to aggressive lungmetastases in mice. Bothtypes of sustained inflam-
mation also caused the formation of NETs. In-hibiting NET formation or digesting the NETs’DNA scaffold prevented conversion of singledisseminated cancer cells to growing metastasesin mouse models of breast and prostate cancer.The NET DNA bound to the extracellular matrix(ECM) protein laminin, thus bringing two NET-associated proteases, NE and MMP9, to theirsubstrate. This in turn facilitated a sequentialcleavage of laminin, first by NE and then byMMP9. The NET-mediated proteolytic remod-eling of laminin revealed an epitope that trig-gered proliferation of dormant cancer cellsthrough integrin activation and FAK/ERK/MLCK/YAP signaling. We generated a blockingantibody against NET-remodeled laminin, andthis antibody prevented or reduced tobaccosmoke exposure– or LPS-induced inflammationfrom awakening dormant cancer cells in mice.
CONCLUSION:Our data implicate NETs andNET-mediated ECM remodeling as critical me-diators of inflammation-induced awakeningin mouse models of dormancy. We propose thatNETs awaken cancer by concentrating neutro-phil proteases at the ECM protein laminin,allowing for sequential proteolytic remodelingof laminin and leading to integrin-mediatedsignaling in the cancer cells. Our findings setthe stage for epidemiological studies to testpossible links among inflammation or smok-ing, NETs, and recurrence after dormancy inhuman patients. If such links can be estab-lished, we envision that approaches similar tothe ones used in mouse models in our studycould be used to target NETs and their down-stream effectors to reduce the risk of cancerrecurrence in human patients. ▪
RESEARCH
Albrengues et al., Science 361, 1353 (2018) 28 September 2018 1 of 1
The list of author affiliations is available in the full article online.*Corresponding author. Email: [email protected] this article as J. Albrengues et al., Science 361, eaao4227(2018). DOI: 10.1126/science.aao4227
NETs formed during lung inflammation awaken dormant cancer cells. Lunginflammation—caused, for instance, by tobacco smoke exposure—leads to NETformation. Two proteases on the NETs sequentially cleave the extracellular matrixprotein laminin. This cleavage generates an epitope that activates integrin signalingand proliferation of the previously dormant cancer cells. Antibodies blocking theNET-generated laminin epitope prevent inflammation-induced awakening.
ON OUR WEBSITE◥
Read the full articleat http://dx.doi.org/10.1126/science.aao4227..................................................
on January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from

RESEARCH ARTICLE◥
CANCER
Neutrophil extracellular trapsproduced during inflammationawaken dormant cancer cells in miceJean Albrengues1, Mario A. Shields1, David Ng1, Chun Gwon Park2,3, Alexandra Ambrico1,Morgan E. Poindexter4, Priya Upadhyay4, Dale L. Uyeminami4, Arnaud Pommier1,Victoria Küttner1, Emilis Bružas1,5, Laura Maiorino1,5, Carmelita Bautista1,Ellese M. Carmona2,3, Phyllis A. Gimotty6, Douglas T. Fearon1,7,8, Kenneth Chang1,Scott K. Lyons1, Kent E. Pinkerton4, Lloyd C. Trotman1, Michael S. Goldberg2,3,Johannes T.-H. Yeh1, Mikala Egeblad1*
Cancer cells from a primary tumor can disseminate to other tissues, remaining dormantand clinically undetectable for many years. Little is known about the cues that cause thesedormant cells to awaken, resume proliferating, and develop into metastases. Studyingmouse models, we found that sustained lung inflammation caused by tobacco smokeexposure or nasal instillation of lipopolysaccharide converted disseminated, dormant cancercells to aggressively growing metastases. Sustained inflammation induced the formationof neutrophil extracellular traps (NETs), and these were required for awakening dormantcancer. Mechanistic analysis revealed that two NET-associated proteases, neutrophilelastase and matrix metalloproteinase 9, sequentially cleaved laminin. The proteolyticallyremodeled laminin induced proliferation of dormant cancer cells by activating integrina3b1 signaling. Antibodies against NET-remodeled laminin prevented awakening ofdormant cells. Therapies aimed at preventing dormant cell awakening could potentiallyprolong the survival of cancer patients.
Most cancer patients die not from their orig-inal primary tumor but from metastasesthat arise in distant tissues. Often, meta-static disease occurs after a prolongedperiod of dormancy, when disseminated
cancer cells are present but clinically undetect-able (1). Disseminated cancer cells can remaindormant for years or even decades before recur-ring, or awakening, as metastatic cancer. T cellsand natural killer cells can eliminate the dissemi-nated cancer cells as they start proliferating, pre-venting them from reaching clinically detectablelevels (2–5). In contrast, increased extracellularmatrix (ECM) deposition and sprouting angio-genesis have been shown to trigger awakeningand metastasis in experimental models (6, 7).It is still unclear what triggers a change in
the balance between signals that keep dissem-
inated tumor cells from growing and those thatcause awakening and outright metastases. Inbreast cancer survivors, elevated plasma levelsof C-reactive protein, a nonspecific marker ofchronic inflammation, are associated with re-duced disease-free survival (8), suggesting thatinflammation may play a role in the switch be-tween dormancy and metastasis. Inflammationhas many causes: For example, smoking induceschronic inflammation in the lung, but the as-sociation between smoking and breast cancerrisk has been controversial. Nevertheless, two re-cent, large, pooled analysis studies showed thatcurrent smoking or prior heavy smoking wassignificantly associated with an elevated risk ofbreast cancer recurrence and death from breastcancer (9, 10). In mice, tobacco smoke exposureincreased lung metastasis by a factor of 2 (11).Inflammation is commonly mediated by neu-
trophils [also called polymorphonuclear leuko-cytes (PMNs)], and these cells are critical for cancercell awakening in experimental models (12).Still, it remains unclear how neutrophils causeawakening.Neutrophils are well known for their ability
to kill harmful microorganisms. They do so via(i) phagocytosis, whereby bacteria or fungi areengulfed and digested; (ii) degranulation of cy-totoxic enzymes and proteases into the extracel-lular space; or (iii) the formation of neutrophilextracellular traps (NETs)—scaffolds of chroma-
tin with associated cytotoxic enzymes and pro-teases that are released into the extracellularspace where they can trap microorganisms (13).NETs are generated through a signaling processthat involves citrullination of histones by theprotein arginine deiminase (PAD) 4 enzyme, chro-matin decondensation, and disintegration of thenuclear membrane. Contents from the neutro-phil’s secretory granules—including neutrophilelastase (NE), cathepsin G (CG), and matrix metal-loproteinase 9 (MMP9)—associate with the decon-densed chromatin. Finally, the plasma membraneruptures, and the protease-associated chromatinfibers are released into the extracellular space(14, 15). A growing body of evidence indicates arole for NETs, not just in infections but also innoninfectious inflammatory diseases (13), throm-bosis (16, 17), and impaired wound healing indiabetes (18). NETs formed in response to sys-temic bacterial infection or after surgical stresspromote cancer dissemination (19, 20). Usingmouse models, we set out to identify how NETstructures facilitate metastasis after a periodof dormancy.
Inflammation-activated neutrophilsdrive cancer cell awakening
To determine whether local inflammation in thelung could directly drive awakening of dissem-inated, dormant cancer cells, we studied twomodels of dormancy. We injected luciferase- andmCherry-expressing breast cancer cells (murineD2.0R and human MCF-7 cell lines) intravenous-ly into syngeneic BALB/c or nude mice, respec-tively. Tumors did not form, even 240 days afterinjection. Instead, single, nonproliferative can-cer cells were found in the lungs (Fig. 1A andfig. S1, A and B) (7, 21). To explore the effect oflung inflammation on dormancy, we nasally in-stilled lipopolysaccharide [(LPS), also called en-dotoxin, a potent inducer of inflammation] intomice bearing dormant cancer cells. One LPS in-stillation, which models a short infection, didnot awaken the dormant D2.0R and MCF-7cancer cells; however, three injections, whichmodel sustained, bacterially induced lung in-flammation, led to aggressive lung metastasis(Fig. 1B and fig. S1, C to H). Small clusters ofcancer cells appeared between the second andthird instillations of LPS, indicating their escapefrom dormancy, whereas cancer cells remainedas single, nonproliferative cells in control mice(Fig. 1, A to C, and fig. S1, A and B). The sameresults were observed when starting LPS in-stillation 1 month, instead of 7 days, after in-travenous injection of D2.0R and MCF-7 cells(fig. S1, I to N), showing that dormant cells re-mained sensitive to external stimuli even at latertime points.LPS instillation caused marked neutrophil
recruitment (movies S1 and S2), after both oneand three instillations (fig. S2, A to C). Still, therelationship between the awakening of D2.0Rproliferation and neutrophil recruitment andactivation remained unclear. To assess these dy-namics, we used confocal intravital lung imag-ing (22) with a fluorescence ubiquitination–based
RESEARCH
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 1 of 13
1Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724,USA. 2Department of Cancer Immunology and Virology,Dana-Farber Cancer Institute, Boston, MA 02215, USA.3Department of Microbiology and Immunobiology, HarvardMedical School, Boston, MA 02215, USA. 4Center for Healthand the Environment, University of California, Davis, Davis,CA 95616, USA. 5Watson School of Biological Sciences,Cold Spring Harbor, NY 11724, USA. 6Department ofBiostatistics, Epidemiology and Informatics, Perelman Schoolof Medicine, University of Pennsylvania, Philadelphia,PA 19104, USA. 7Cancer Research UK Cambridge Institute,University of Cambridge, Li Ka Shing Centre, CambridgeCB2 0RE, UK. 8Meyer Cancer Center, Weill Cornell MedicalCollege, New York, NY 10021, USA.*Corresponding author. Email: [email protected]
on January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 2 of 13
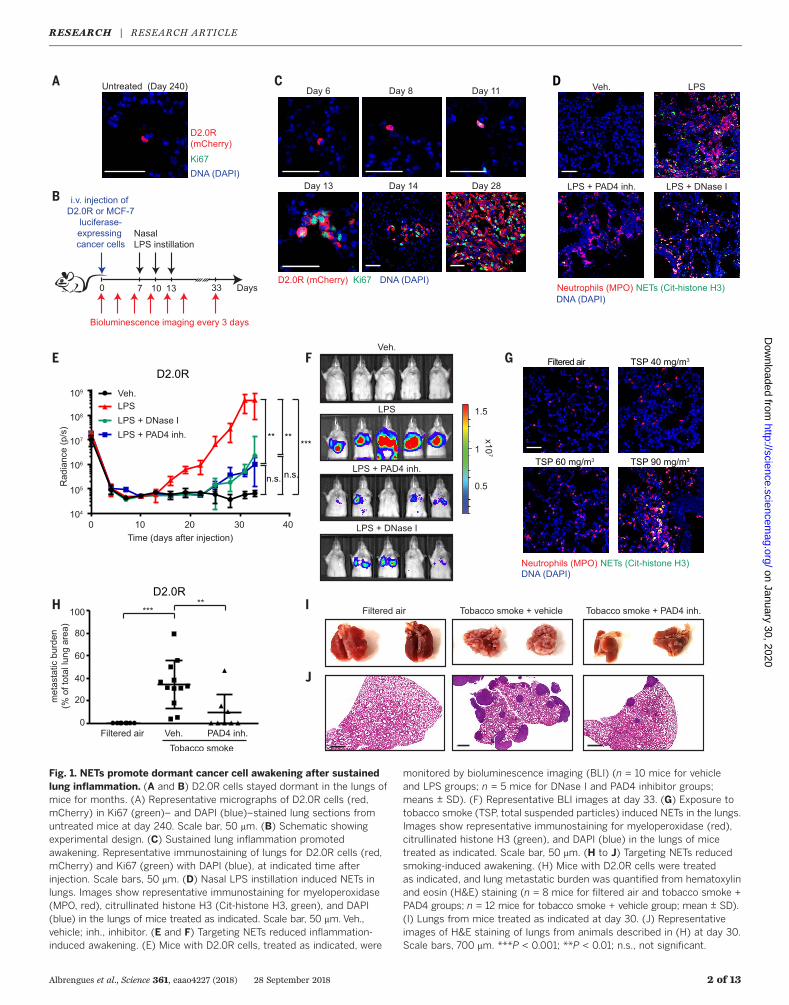
Fig. 1. NETs promote dormant cancer cell awakening after sustainedlung inflammation. (A and B) D2.0R cells stayed dormant in the lungs ofmice for months. (A) Representative micrographs of D2.0R cells (red,mCherry) in Ki67 (green)– and DAPI (blue)–stained lung sections fromuntreated mice at day 240. Scale bar, 50 mm. (B) Schematic showingexperimental design. (C) Sustained lung inflammation promotedawakening. Representative immunostaining of lungs for D2.0R cells (red,mCherry) and Ki67 (green) with DAPI (blue), at indicated time afterinjection. Scale bars, 50 mm. (D) Nasal LPS instillation induced NETs inlungs. Images show representative immunostaining for myeloperoxidase(MPO, red), citrullinated histone H3 (Cit-histone H3, green), and DAPI(blue) in the lungs of mice treated as indicated. Scale bar, 50 mm. Veh.,vehicle; inh., inhibitor. (E and F) Targeting NETs reduced inflammation-induced awakening. (E) Mice with D2.0R cells, treated as indicated, were
monitored by bioluminescence imaging (BLI) (n = 10 mice for vehicleand LPS groups; n = 5 mice for DNase I and PAD4 inhibitor groups;means ± SD). (F) Representative BLI images at day 33. (G) Exposure totobacco smoke (TSP, total suspended particles) induced NETs in the lungs.Images show representative immunostaining for myeloperoxidase (red),citrullinated histone H3 (green), and DAPI (blue) in the lungs of micetreated as indicated. Scale bar, 50 mm. (H to J) Targeting NETs reducedsmoking-induced awakening. (H) Mice with D2.0R cells were treatedas indicated, and lung metastatic burden was quantified from hematoxylinand eosin (H&E) staining (n = 8 mice for filtered air and tobacco smoke +PAD4 groups; n = 12 mice for tobacco smoke + vehicle group; mean ± SD).(I) Lungs from mice treated as indicated at day 30. (J) Representativeimages of H&E staining of lungs from animals described in (H) at day 30.Scale bars, 700 mm. ***P < 0.001; **P < 0.01; n.s., not significant.
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from

cell cycle indicator (FUCCI) system, which flu-orescently labels cells red in G0 and G1 phasesand green in S, G2, and M phases (fig. S2D) (23).Neutrophil infiltration and activation were trackedusing the LysM–enhanced green fluorescent pro-tein (EGFP) transgene and the NE 680 FASTprobe, which fluoresces after NE cleavage.In control mice instilled with phosphate-
buffered saline (PBS), NE activity was low, andall D2.0R cells were red in the G0 and G1 cellcycle phases at days 8, 11, and 21 (movies S3 toS5). In contrast, beginning 5 days after the firstLPS instillation (day 11), D2.0R cells became yel-low, indicating that they had entered the G1/Stransition of the cell cycle, and this transition cor-related with high neutrophil recruitment and NEactivity (movie S6 and fig. S2, E and F). We de-tected small clusters of proliferating cells at day 14and established proliferative lung metastasis atday 21 (movies S7 and S8 and fig. S2F).We next investigated whether neutrophils con-
tributed to awakening of dormant cancer cells.Indeed, neutrophil depletion completely preventedinflammation-induced awakening of dormantD2.0R and MCF-7 cancer cells (fig. S3, A to I).Thus, sustained inflammation induced by LPScauses dormant cancer cells to reenter the cellcycle, and this effect requires neutrophils.
NETs awaken dormant cancercells in mice
After LPS instillation, we detected numerousNETs in the lungs within 4 hours, persisting24 hours later; in contrast, NETs were absent innormal lung tissue (Fig. 1D and fig. S3, J and K).NETs were also present in the plasma after LPStreatment, as assessed by double-stranded DNA(dsDNA) assays or an enzyme-linked immuno-sorbent assay (ELISA) against DNA-bound NE(fig. S3, L and M).To determine whether NETs contributed to
cancer cell awakening, we blocked NET forma-tion with a PAD4 inhibitor or digested the NETDNA scaffold with deoxyribonuclease (DNase) I(free or coated on nanoparticles) (Fig. 1D andfig. S4, A to F). Both treatments prevented ordecreased LPS-induced awakening of dormantD2.0R and MCF-7 cancer cells (Fig. 1, E and F,and fig. S4, G to M). Results were similar whenLPS instillation was started 1 month after intra-venous injection of the cancer cells (fig. S1, Ito N), showing that NETs remained a powerfulexternal stimulus of awakening, even at latertime points. DNase I was most effective as a pre-ventative treatment, immediately before LPS in-stillation, but also reduced end-point metastaticburden when administered at day 14, after theappearance of small clusters of cancer cells inthe lungs (fig. S4N). In all cases, the NET-targetingtreatments reduced neutrophil recruitment (fig.S4, O and P), suggesting that NETs promote fur-ther inflammation.The cancer cell dormancy models described
above were generated by intravenous injectionof cancer cells, so we used two additional ap-proaches to explore whether NETs could alsoinduce proliferation after natural dissemina-
tion of cancer cells. First, we used the RapidCaPprostate cancer model (24), where prostate can-cer develops from normal epithelial tissues withinan intact organ. The model has a low incidenceof lung metastasis (24), and we observed no lungmetastasis in control mice during the observa-tion period, despite the presence of single, dis-seminated cancer cells in the lungs. In contrast,LPS instillations resulted in lung metastasis inthree of five mice. However, zero of five mice de-veloped macroscopic metastasis when NET for-mation was blocked with the PAD4 inhibitor,even after three additional LPS instillations (fig.S5, A to C; P = 0.02). In the second approach, weallowed MCF-7 cells to form primary mammarytumors and spontaneously disseminate. We thenresected those primary tumors. Under these con-ditions also, LPS-induced NETs awakened thecancer cells (fig. S5, D to H).Repeated LPS instillation models a sustained
bacterially induced inflammation. Smoking sim-ilarly induces chronic lung inflammation andhas been associated with increased risk of breastcancer recurrence (9, 10). To examine NETs andcancer cell dormancy under these conditions, weexposed mice to three different concentrationsof tobacco smoke for 3 weeks. This resulted in adose-dependent increase in neutrophil infiltra-tion in the lungs. NETs formed in the lungs atthe highest tobacco smoke exposure level (a levelalmost equivalent to moderate, active smokingand well above secondhand exposure to tobaccosmoke) (Fig. 1G and fig. S5, I and J). NETs, aswell as LPS, were also found in the plasma of thetobacco smoke–exposed mice (fig. S5, K to M),suggesting that tobacco exposure causes systemicexposure to both agents.We next tested whether the tobacco smoke
could awaken dormant D2.0R cells. Indeed,aggressive metastasis developed when micewere exposed to tobacco smoke at the levelthat induced NET formation, and this wasprevented by PAD4 inhibitor treatment. Cellsremained dormant in mice exposed to filteredair (Fig. 1, H to J, and fig. S5N). Thus, NETsformed during inflammation induced theawakening of dormant cancer cells in multiplemouse models.
NETs awaken slow-cycling cells in vitroin ECM models
To determine how NETs induced awakeningof dormant cancer cells, we turned to three-dimensional (3D) culture systems, where theisolated effect of NETs could be tested in theabsence of the many cell types present in lungs.Both D2.0R and MCF-7 cells become slow cy-cling when cultured on basement membranematrix (matrigel) (25), whereas metastatic D2.A1 cells, isolated from the same mammary le-sion as the D2.0R cells, proliferated (fig. S6, Aand B). To generate NETs, we cultured freshlyisolated neutrophils and stimulated them withLPS, phorbol 12-myristate 13-acetate (PMA), orN-formyl-methionyl-leucyl-phenylalanine (fMLP)(Fig. 2A and fig. S6, C and D). We found thatNETs were also induced by coculturing neutro-
phils with metastatic D2.A1 cells but not withthe dormant D2.0R cells NETs (fig. S6C). This issimilar to our previous findings using anotherpair of cell lines (22), the metastatic and NET-inducing 4T1 and the nonmetastatic and non–NET-inducing 4T07 cell lines. Finally, to comparethe effects of NETs with that of neutrophil de-granulation, we induced degranulation by cul-turing with complement 5a (c5a). We next testedthe effect of the NET-containing conditioned me-dia (CM) from the different neutrophil cultureconditions on luciferase-expressing D2.0R andMCF-7 cancer cells cultured on matrigel (Fig. 2A).NET-containing CM led to the awakening andproliferation of dormant cancer cells, whereasCM from degranulated or inactivating conditionshad no effect (Fig. 2, B to D, and figs. S6E andS7, A to C). LPS, PMA, fMLP, and c5a added di-rectly to the cancer cells had no effect on awak-ening in our 3D culture model systems, nor didCM from D2.A1 or D2.0R cancer cells (fig. S7, Dand E). Treatment of D2.0R cells with LPS be-fore intravenous injection also did not lead tometastasis (fig. S7, F and G), further suggestingthat it was the NETs in the CM and not LPSthat activated the slow-cycling cells. When theneutrophil cultures were treated with a PAD4inhibitor or DNase I during NET-activating con-ditions, NETs did not form or were digested (fig.S6, C and D), and the CM no longer inducedawakening of the dormant cancer cells (Fig. 2Dand fig. S7B).Given that nicotine can induce NETs in vitro
(26), we next tested the effect of cigarette smokeextract (CSE) on neutrophils. We found thatCSE induced NETs in vitro in a dose-dependentmanner and that this NET-containing CM couldalso awaken the slow-cycling cells (Fig. 2, E andF, and fig. S7, H and I). Because we had detectedLPS in the plasma of tobacco smoke–exposedmice, we assessed the effect of taurolidine, a com-pound that neutralizes the effect of LPS (27).Taurolidine and PAD4 inhibition both reducedCSE-induced formation of NETs, and the CMwas no longer able to awaken the cancer cells(Fig. 2, E and F, and fig. S7, H and I), suggest-ing that LPS present in CSE drives smoking-induced NETs. CSE had no effect on awakeningwhen it was added directly to the cancer cellsin vitro, and it caused cancer cell death at thehighest concentrations (fig. S7J). Altogether, theseresults show that NETs induced by multiplemeans promote cancer cell awakening in vitroin the presence of an artificial ECM and in theabsence of lung cells, other immune cells, andvasculature.
NET-associated proteases awakencancer cells
Degradation of the ECM, activation of cell sur-face receptors, and the release and activationof cytokines and growth factors by proteasesare necessary steps in metastasis (28, 29). Wehypothesized that NET-associated protease ac-tivity was responsible for the effects of NETs onawakening. Using our 3D culture systems, weanalyzed the effects of inhibiting three major
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 3 of 13
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from
NET-associated proteases: CG, NE, and MMP9.Inhibiting NE or MMP9, but not CG activity, inthe NET-containing CM prevented awakeningof the slow-cycling cancer cells (Fig. 3A and fig.S8, A to D). We also isolated neutrophils fromNE and MMP9 knockout mice and stimulated
them to form NETs. CM from these cultures didnot induce awakening (fig. S8, E and F). Wefound that neutrophils, but not cancer cells,were the main source of these proteases. NET-containing CM did not induce expression ofthe proteases in cancer cells (fig. S9, A to C), and
short hairpin RNA (shRNA)–mediated knock-down of NE and MMP9 in D2.0R cells did notaffect the capacity of these cells to be awakenedby NET-containing CM in vitro (fig. S9, D to F).In vivo, inhibiting NE and MMP9 activity
also prevented LPS-induced cancer recurrence
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 4 of 13
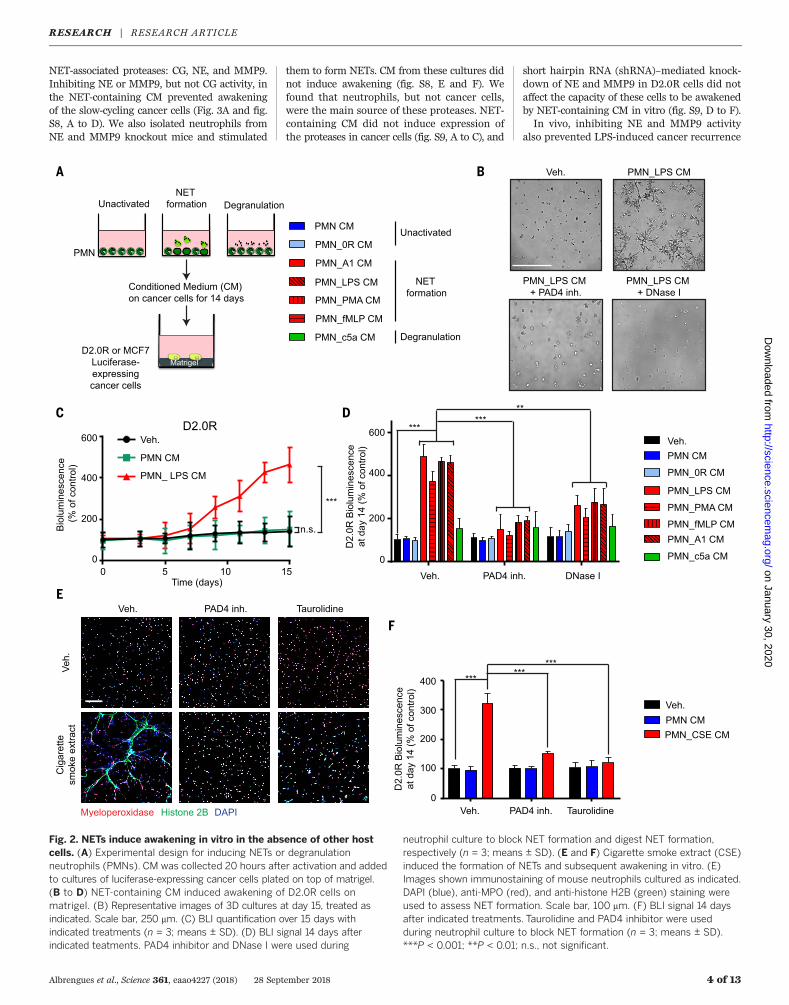
Fig. 2. NETs induce awakening in vitro in the absence of other hostcells. (A) Experimental design for inducing NETs or degranulationneutrophils (PMNs). CM was collected 20 hours after activation and addedto cultures of luciferase-expressing cancer cells plated on top of matrigel.(B to D) NET-containing CM induced awakening of D2.0R cells onmatrigel. (B) Representative images of 3D cultures at day 15, treated asindicated. Scale bar, 250 mm. (C) BLI quantification over 15 days withindicated treatments (n = 3; means ± SD). (D) BLI signal 14 days afterindicated teatments. PAD4 inhibitor and DNase I were used during
neutrophil culture to block NET formation and digest NET formation,respectively (n = 3; means ± SD). (E and F) Cigarette smoke extract (CSE)induced the formation of NETs and subsequent awakening in vitro. (E)Images shown immunostaining of mouse neutrophils cultured as indicated.DAPI (blue), anti-MPO (red), and anti-histone H2B (green) staining wereused to assess NET formation. Scale bar, 100 mm. (F) BLI signal 14 daysafter indicated treatments. Taurolidine and PAD4 inhibitor were usedduring neutrophil culture to block NET formation (n = 3; means ± SD).***P < 0.001; **P < 0.01; n.s., not significant.
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from
(Fig. 3, B and C, and fig. S9, G to J). NE inhibi-tion prevented NET formation in vivo, consistentwith the known requirement for NE activityduring NET formation (30, 31). In contrast, NETsstill formed in vivo after MMP9 inhibition, butcells remained dormant, suggesting that the ac-tivity of this NET protease is critical for awak-ening (Fig. 3, D and E). Similar to the preventionof NET formation by PAD4 inhibition, inhibitionof NE, but not MMP9, also reduced neutrophilrecruitment to the lungs after LPS-induced in-flammation (fig. S9K).
To determine whether the NET proteases in-duced awakening by acting directly on the can-cer cells or by altering their microenvironment,we preincubated matrigel with NET-containingCM before cancer cell plating (fig. S9L). This wassufficient to induce awakening but required bothNE and MMP9 activity (fig. S9M, red bars). If theproteases were inhibited during cancer cell cul-ture, after NET-mediated matrigel remodelinghad already occurred, then awakening was notinhibited (fig. S9M, peach-colored bars). Thesedata suggest that NET-associated NE and MMP9
induce awakening of dormant cancer cells throughECM remodeling.
NETs awaken cancer cellsby laminin remodeling
We next sought to identify the ECM substrate ofNE and MMP9 during NET-mediated awaken-ing of dormant cancer cells. According to themanufacturer, matrigel is composed mainly oflaminin-111 (60%), collagen type IV (30%), andentactin (8%). Proteolysis of laminin-111, whichconsists of the a-1, b-1, and g-1 laminin chains,
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 5 of 13
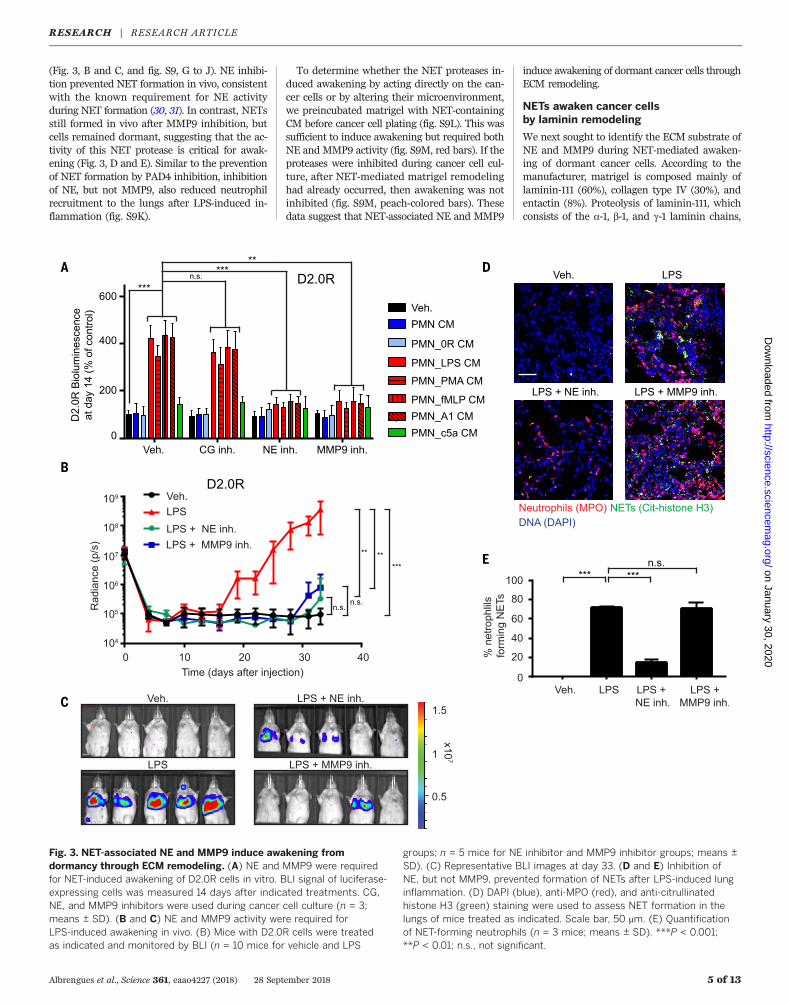
Fig. 3. NET-associated NE and MMP9 induce awakening fromdormancy through ECM remodeling. (A) NE and MMP9 were requiredfor NET-induced awakening of D2.0R cells in vitro. BLI signal of luciferase-expressing cells was measured 14 days after indicated treatments. CG,NE, and MMP9 inhibitors were used during cancer cell culture (n = 3;means ± SD). (B and C) NE and MMP9 activity were required forLPS-induced awakening in vivo. (B) Mice with D2.0R cells were treatedas indicated and monitored by BLI (n = 10 mice for vehicle and LPS
groups; n = 5 mice for NE inhibitor and MMP9 inhibitor groups; means ±SD). (C) Representative BLI images at day 33. (D and E) Inhibition ofNE, but not MMP9, prevented formation of NETs after LPS-induced lunginflammation. (D) DAPI (blue), anti-MPO (red), and anti-citrullinatedhistone H3 (green) staining were used to assess NET formation in thelungs of mice treated as indicated. Scale bar, 50 mm. (E) Quantificationof NET-forming neutrophils (n = 3 mice; means ± SD). ***P < 0.001;**P < 0.01; n.s., not significant.
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from
has previously been linked with regulation ofproliferation and cellular architecture in 3Dcultures (32). Both D2.0R and MCF-7 cells re-mained slow cycling when cultured on puri-fied laminin-111 in vitro (fig. S10, A and B) butwere awakened by NET-containing CM (Fig. 4A,and fig. S10, C to E).
To test whether NET-associated proteases couldcleave laminin, we used commercially availablepurified laminin-111. Whereas NET-containingCM led to laminin-111 cleavage, CM from de-granulating or nonactivating conditions did not(Fig. 4B and fig. S10F). Using recombinant (r)NE and MMP9, we found that both proteases
in combination were necessary to induce can-cer cell awakening (Fig. 4C and fig. S10, G to I).Laminin-111 was cleaved by rNE alone but notby rMMP9; however, when both proteases werepresent, the cleavage fragments were different(Fig. 4D and fig. S10J). Similarly, when laminin-111was treated with NET-containing CM, cleavage
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 6 of 13
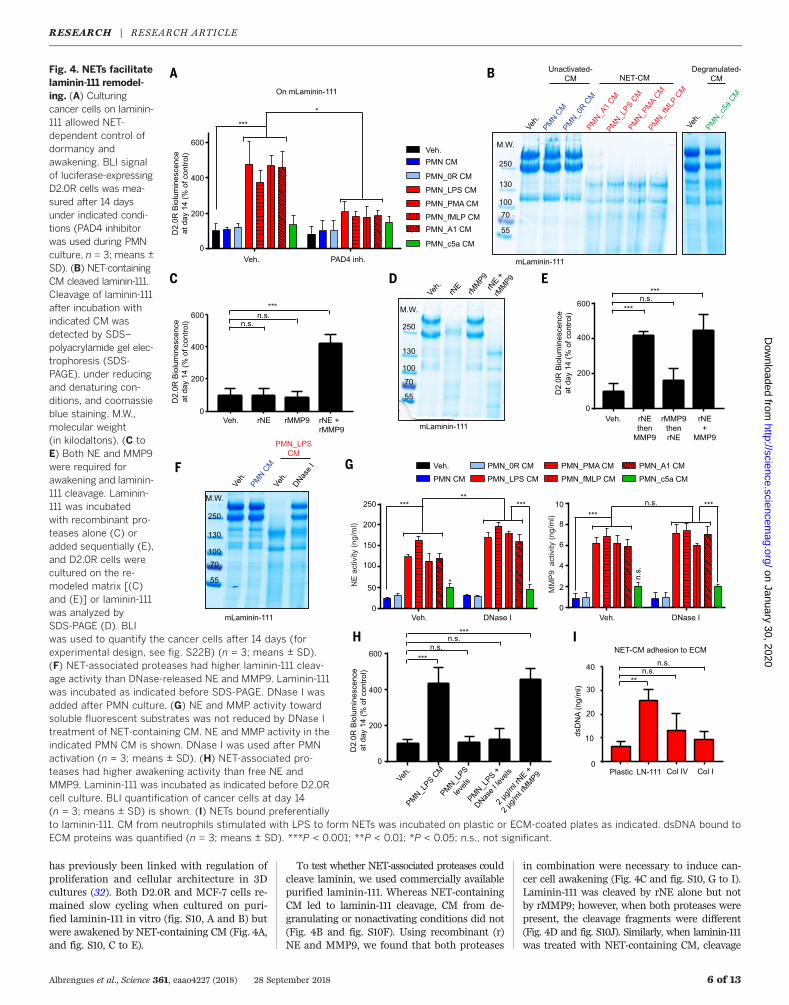
Fig. 4. NETs facilitatelaminin-111 remodel-ing. (A) Culturingcancer cells on laminin-111 allowed NET-dependent control ofdormancy andawakening. BLI signalof luciferase-expressingD2.0R cells was mea-sured after 14 daysunder indicated condi-tions (PAD4 inhibitorwas used during PMNculture, n = 3; means ±SD). (B) NET-containingCM cleaved laminin-111.Cleavage of laminin-111after incubation withindicated CM wasdetected by SDS–polyacrylamide gel elec-trophoresis (SDS-PAGE), under reducingand denaturing con-ditions, and coomassieblue staining. M.W.,molecular weight(in kilodaltons). (C toE) Both NE and MMP9were required forawakening and laminin-111 cleavage. Laminin-111 was incubatedwith recombinant pro-teases alone (C) oradded sequentially (E),and D2.0R cells werecultured on the re-modeled matrix [(C)and (E)] or laminin-111was analyzed bySDS-PAGE (D). BLIwas used to quantify the cancer cells after 14 days (forexperimental design, see fig. S22B) (n = 3; means ± SD).(F) NET-associated proteases had higher laminin-111 cleav-age activity than DNase-released NE and MMP9. Laminin-111was incubated as indicated before SDS-PAGE. DNase I wasadded after PMN culture. (G) NE and MMP activity towardsoluble fluorescent substrates was not reduced by DNase Itreatment of NET-containing CM. NE and MMP activity in theindicated PMN CM is shown. DNase I was used after PMNactivation (n = 3; means ± SD). (H) NET-associated pro-teases had higher awakening activity than free NE andMMP9. Laminin-111 was incubated as indicated before D2.0Rcell culture. BLI quantification of cancer cells at day 14(n = 3; means ± SD) is shown. (I) NETs bound preferentiallyto laminin-111. CM from neutrophils stimulated with LPS to form NETs was incubated on plastic or ECM-coated plates as indicated. dsDNA bound toECM proteins was quantified (n = 3; means ± SD). ***P < 0.001; **P < 0.01; *P < 0.05; n.s., not significant.
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from

was completely blocked after NE inhibition butwas only partially blocked after MMP9 inhi-bition (fig. S10K). Therefore, we hypothesizedthat laminin-111 is cleaved sequentially—firstby NE and subsequently by MMP9—to triggerawakening. Indeed, proteolysis of laminin-111first by rNE and then by rMMP9, but not in thereverse order, led to cancer cell awakening (Fig.4E and fig. S10G).Our in vitro experiments suggest that laminin-
111 is the primary substrate of NET proteasesin 3D culture. However, laminin-111 has not con-sistently been detected in adult lungs. Therefore,we assessed the expression of laminin in lungtissue. We confirmed the presence of mRNAcoding for the three laminin-111 chains by quan-titative polymerase chain reaction (qPCR) andin situ hybridization, and we used multiple anti-bodies for immunofluorescence and Westernblot analyses to detect laminin-111 protein (fig.S11, A to D). Laminin-111 mRNA expression wasnot affected by nasal LPS instillation, but cleavedlaminin-111 was readily detectable in lung tissuelysate after inducing inflammation with LPS(fig. S11, A and D). A polyclonal anti–laminin-111antibody was used for Western blot and im-munofluorescence analysis; thus, we could notexclude the possibility that this reagent recog-nized not only laminin-111 but also other lamininisoforms containing a-1, b-1, or g-1. Hence, wetested whether NET-containing CM also couldawaken cancer cells cultured on other lamininisoforms. We found that laminin-211, -411, and-511 were all sufficient to support NET-inducedawakening (fig. S11E), which suggests that awak-ening can occur in any tissue containing one ofthese laminins—e.g., at the perivascular niche.
NET DNA acts as a proteolysis scaffold
Our data showed that proteolytic remodelingis required for cancer cell awakening; yet, di-gesting NET DNA with DNase I also preventedawakening in vivo and in vitro (Figs. 1, E and F;and 2, B and D; and figs. S4, G to I, L, and M;and S7B). Consistently, laminin-111 was readilycleaved by incubation with NET-containing CM,and this cleavage was prevented by DNase I(Fig. 4F). Nevertheless, DNase I digestion did notreduce the protease activity against soluble flu-orescent substrates (Fig. 4G). However, addingrecombinant proteases at the activities mea-sured in the NET-containing CM (120 ng/ml rNE,6 ng/ml MMP9) or in NET-containing CM afterDNase I treatment (160 ng/ml rNE, 6 ng/mlMMP9) had no effect on awakening. Instead,much higher concentrations of the recombinantproteases (2 mg/ml each, the concentrations usedin Fig. 4, C to E, and fig. S10, H to K, in accord-ance with the literature) were necessary to in-duce cancer cell awakening (Fig. 4H). We foundareas of NE colocalizing with MMP9 on the DNAfibers of NETs (fig. S11F). Furthermore, NET DNApreferentially bound to laminin-111 over othertested ECM proteins (Fig. 4I and fig. S11G). Thesefindings suggested that the NET DNA scaffoldallowed the NET proteases to cleave their sub-strate more efficiently than when the proteases
freely diffused because the DNA binds to lamininand because the two proteases colocalize to thesame DNA scaffold.Thrombospondin-1 (TSP-1), a large glycopro-
tein present in the basement membrane surround-ing mature blood vessels, regulates cancer celldormancy (6). TSP-1 secretion by bone marrow–derived Gr1+ cells (which include neutrophils)generates a metastasis-resistant microenviron-ment, which can be overcome through neutrophil-mediated proteolysis of TSP-1 (33, 34). We foundthat TSP-1 was also a substrate for NET-associatedNE and MMP9 in vitro (fig. S11H) and that it wasdegraded in vivo after LPS-induced lung inflam-mation (fig. S11I). TSP-1 degradation alone didnot convert slow-cycling cells to proliferating cellsin vitro (fig. S11J). However, intact TSP-1 decreasedproliferation caused by cleaved laminin-111 (fig.S11, K and L). Our data suggest that TSP-1 mod-ulates cancer cell awakening caused by proteo-lytic remodeling of laminin-111.
NET-remodeled laminin activatesintegrin a3b1 signaling
We next focused on how NET remodeling oflaminin-111 led to cancer cell awakening. Integrinsare cell surface ECM receptors that recognizeconformational changes in the ECM, and sig-naling through integrin b1 has previously beenshown to induce awakening (7, 35–37). Consist-ently, a marker for cell proliferation, Ki67, wasassociated with integrin b1 activity and reorgani-zation of the actin cytoskeleton after incubatingD2.0R or MCF-7 cells with NET-containing CMor rNE and rMMP9 (Fig. 5A and fig. S12, A andB). The integrin b1 outside-in signaling pathwaycan activate the downstream mediators focaladhesion kinase (FAK), MAP kinase ERK kinase(MEK), extracellular signal–regulated kinase (ERK),myosin light chain kinase (MLCK), myosin lightchain 2 (MLC2), and yes-associated protein (YAP).Accordingly, NET-induced awakening led to ac-tivation of FAK, ERK, and MLC2, which requiredNE and MMP9 activity (Fig. 5B). Moreover, tar-geting the integrin b1 outside-in signaling path-way at different signaling points using chemicalinhibitors or RNA interference inhibited NET-induced awakening in vitro (Fig. 5, C and D, andfig. S12, C and D). Inhibition of integrin b1, FAK,MEK, MLCK, myosin, and YAP activities all ledto the loss of FAK, ERK, and MLC2 phosphoryl-ation upon stimulation with NET-containing CM(fig. S12, E to K), in accordance with the positive-feedback loop within this signaling pathway(38). To identify which b1-containing integrinpair was involved in cancer cell awakening, wetested D2.0R cells for expression of a integrinsknown to associate with integrin b1 (fig. S13A)(39). Through shRNA mediated knockdown (fig.S13B), we found that awakening of dormant can-cer cells by NETs required integrin a3, integrinb1, and YAP in the cancer cells (Fig. 5, E to G,and fig. S13, C and D).LPS-induced lung inflammation resulted in
laminin cleavage that was detectable in lunglysate, and this cleavage depended on neutro-phil recruitment, NET formation, and NE and
MMP9 activities (fig. S14A). Laminin cleavagewas blocked after NE inhibition but was onlypartially blocked after MMP9 inhibition, consist-ent with sequential laminin cleavage by NE andMMP9 in vivo. However, no discernable changesin laminin immunofluorescent staining patternswere observed in lungs after LPS-induced inflam-mation (fig. S10H). Additionally, under nonreduc-ing and nondenaturing conditions, we did notobserve many laminin-111 fragments after rNEand rMMP9 cleavage but rather a reduction inits apparent molecular weight (Fig. 6A). Theseresults suggest that NET-mediated proteolysisinduced a change in the 3D structure of laminin.We hypothesized that a new laminin-111 epitopewas revealed after NET-mediated proteolysis andthat the dormant cancer cells sensed this newepitope through integrin a3b1, leading to cancercell proliferation. To test this idea, we generatedmonoclonal antibodies (mAbs) from rats immu-nized against purified rNE and rMMP9 cleavedlaminin-111 (fig. S14B). Three laminin-111–recognizingantibody clones (Ab19, Ab25, and Ab28) stronglyblocked NET-induced integrin b1 activation andsubsequent cancer cell awakening in vitro (Fig.6B and fig. S14C). Of these antibodies, Ab28 spe-cifically recognized NET-remodeled laminin-111but not intact laminin, whereas Ab19 and Ab25recognized both laminin-111 forms (fig. S14, Dand E). We cannot exclude the possibility thatother laminin isoforms share the antibody epi-topes after NET-mediated remodeling and alsoare recognized by the antibodies.Using Ab28, we detected the NET-remodeled
laminin epitope in inflamed lungs. It was notpresent after neutrophil depletion or after in-hibition of NET formation or NE or MMP9 ac-tivities (Fig. 6C and fig. S14F). The epitope wasalso present in lung tissue from tobacco smoke–exposed mice but only at an exposure level thatinduced NETs (Fig. 6D). Moreover, the NET-remodeled laminin epitope was only detectablein lungs after three LPS instillations, the numberof doses required to awaken cancer cells (fig. S15A).The NET-remodeled laminin epitope was de-
tectable in the same lung regions as the NETs(fig. S15A). Furthermore, using D2.0R cells ex-pressing the FUCCI cell cycle reporter (fig. S2D),we observed that all green cells (indicating S,G2, or M cell cycle phase) or cells that were partof a cluster were located adjacent to remodeledlaminin, whereas cells close to intact laminin allremained red (indicating G0 or G1 cell cycle phase)(fig. S15, B and C). This colocalization patternsuggested that the NET-remodeled laminin epi-tope was driving the awakening of cancer cellsin vivo. To test this, we engineered rat mono-clonal antibodies as chimeric mouse immuno-globulin G 2a (IgG2a) antibodies (chiAbs), whichcan be used in mice without eliciting an im-mune response. ChiAb28 inhibited cancer cellawakening in vitro (fig. S15D) and in both in vivosystems (LPS- and tobacco smoke–induced in-flammation) (Fig. 6, E to I, and fig. S15, E andF). Together, our data identify a mechanism bywhich sustained lung inflammation, induced byeither tobacco smoke or LPS exposure, can lead to
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 7 of 13
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from
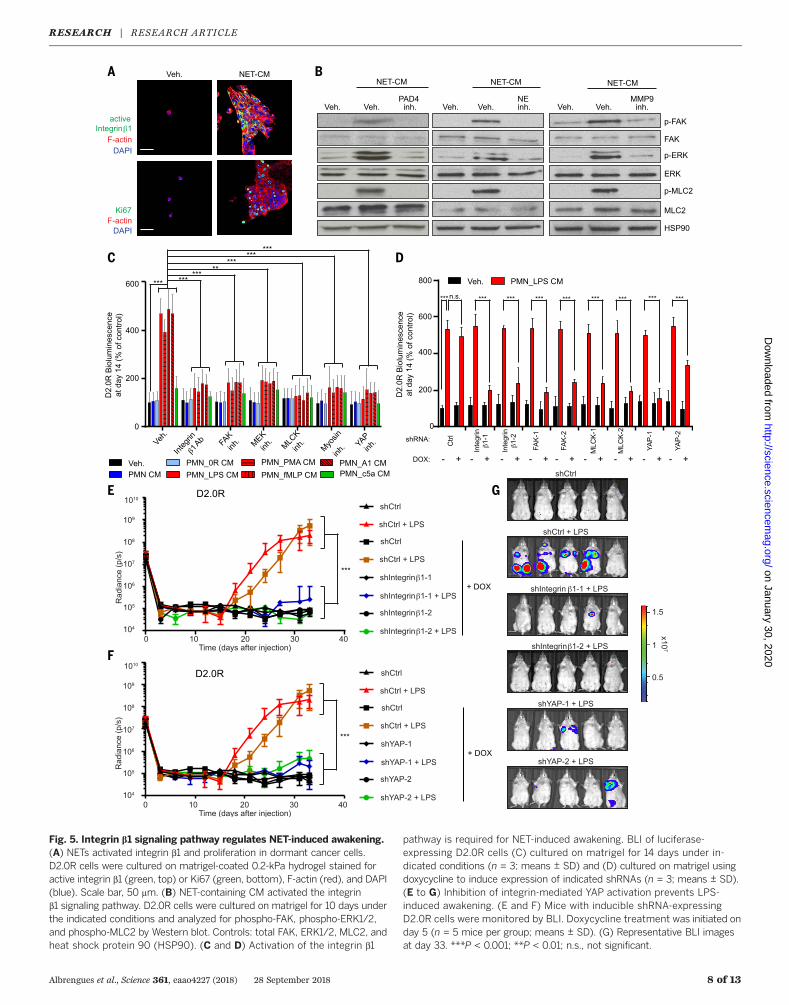
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 8 of 13
Fig. 5. Integrin b1 signaling pathway regulates NET-induced awakening.(A) NETs activated integrin b1 and proliferation in dormant cancer cells.D2.0R cells were cultured on matrigel-coated 0.2-kPa hydrogel stained foractive integrin b1 (green, top) or Ki67 (green, bottom), F-actin (red), and DAPI(blue). Scale bar, 50 mm. (B) NET-containing CM activated the integrinb1 signaling pathway. D2.0R cells were cultured on matrigel for 10 days underthe indicated conditions and analyzed for phospho-FAK, phospho-ERK1/2,and phospho-MLC2 by Western blot. Controls: total FAK, ERK1/2, MLC2, andheat shock protein 90 (HSP90). (C and D) Activation of the integrin b1
pathway is required for NET-induced awakening. BLI of luciferase-expressing D2.0R cells (C) cultured on matrigel for 14 days under in-dicated conditions (n = 3; means ± SD) and (D) cultured on matrigel usingdoxycycline to induce expression of indicated shRNAs (n = 3; means ± SD).(E to G) Inhibition of integrin-mediated YAP activation prevents LPS-induced awakening. (E and F) Mice with inducible shRNA-expressingD2.0R cells were monitored by BLI. Doxycycline treatment was initiated onday 5 (n = 5 mice per group; means ± SD). (G) Representative BLI imagesat day 33. ***P < 0.001; **P < 0.01; n.s., not significant.
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from
NET-induced proteolytic laminin remodeling thatcan drive cancer recurrence in the lungs of mice.
Discussion
Neutrophils recruited during lung inflammationcan initiate awakening of dormant cancer cells,as described here and in previous work (12). Now,we report that NETs formed by neutrophilsduring LPS- or tobacco smoke–induced lung
inflammation are required to awaken dormantcancer cells and cause metastasis in mice. We pro-pose that NETs concentrate neutrophil proteases,NE and MMP9, at their substrate, laminin, al-lowing sequential cleavage and generating anepitope that triggers cancer cell awakening.Previous studies pointed to a central role for
integrin b1 signaling in the awakening of dor-mant cancer cells (7, 21, 35, 40), but it was un-
clear what types of stimuli could activate integrinsignaling in dormant cancer cells. We have foundthat NET-remodeled laminin formed during lunginflammation serves as an integrin a3b1–activatingepitope, leading to FAK/ERK/MLCK/YAP signal-ing in cancer cells and to awakening. On the basisof these results, we developed an inhibitory anti-body against NET-remodeled laminin that pre-vented dormant cancer cells from awakening in
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 9 of 13
Fig. 6. A NET-generatedlaminin epitope promotesawakening. (A) Laminin-111was not fully degraded byNET-associated proteases.Laminin-111 was incubatedwith rNE and rMMP9, andcleavage was assessed bySDS-PAGE and coomassieblue staining under reducingand denaturing or non-reducing and nondenaturingconditions. (B) Antibodiesagainst NE- and MMP9-cleaved laminin-111 preventedNET-mediated awakening.BLI was performed onluciferase-expressing D2.0Rcells cultured on laminin-111,14 days after treatmentwith the indicated antibodyclone (n = 3; means ± SD).(C and D) Lung inflammationand NETs induce productionof cleaved laminin. Immuno-staining of lungs for DAPI(blue), full-length laminin-111(ab11575 from Abcam,green), and cleaved lamininantibody 28 (red), treatedas indicated.The number oftotal suspended particles(TSP) in cigarette smokeexposure conditions is indi-cated. Scale bars, 50 mm.(E to I) Antibodies againstNET-remodeled laminin pre-vented inflammation-inducedawakening in vivo. (E) Micewith D2.0R cells were treatedas indicated and monitoredby BLI (n = 5 mice per group;means ± SD). (F) Represen-tative BLI images at day 33.(G) Metastatic burden inlungs from mice exposed totobacco smoke quantifiedwith H&E staining.(H) Representativephotos of lungs. (I) Rep-resentative H&E imagesof lungs from animalsdescribed in (H) at day 30(n = 7 mice for filteredair + IgG, n = 8 mice fortobacco smoke + ChiAb28 inhibitor, n = 11mice for tobacco smoke + IgG;means ± SD). Scale bars, 700 mm. ***P <0.001; **P <0.01; *P < 0.05; n.s., not significant.
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from

response to LPS- or tobacco smoke–induced lunginflammation. Given that our antibody preservesnormal integrin b1 signaling, it may be a po-tent strategy to prevent cancer recurrence andmore broadly serve as a treatment for other NET-associated pathologies, such as transfusion-relatedacute lung injury (41).Our in vitro experiments identified laminin-111,
-211, -411, and -511 as key ECM proteins that causeNET-induced cancer cell awakening. It is par-ticularly intriguing that NETs target laminin-411and -511, present in the perivascular niche, be-cause this niche has been shown to regulate breastcancer dormancy (6). In previous studies, TSP-1,which is also present in this niche, was identifiedas a regulator of tumor dormancy and metastasis(6, 33, 34). In vitro, NET-mediated degradationof TSP-1 did not induce awakening. Rather, in-tact TSP-1 inhibited NET-induced awakening, andNET-mediated degradation of TSP-1 overcamethe inhibition. Thus, TSP-1 degradation is likelyrequired in parallel with laminin remodeling inthe intact tissue to create a permissive niche forthe awakening of dormant cancer cells.Stiffness of the environment can lead to cell
tension–mediated YAP activity, and we havedemonstrated that YAP activity is critical forinflammation-induced cancer cell awakening.Our results therefore suggest that changes in celltension occur after binding to NET-remodeledlaminin and play a role in the awakening ofdisseminated dormant cancer cells. Consistent-ly, experimental lung fibrosis, which is associatedwith increased ECM stiffness, induces cancercell awakening from dormancy (7). Furthermore,D2.0R and MCF-7 cells are dormant in soft envi-ronments in vitro—such as on matrigel, laminin-111 gel, and 0.2-kPa hydrogel (which mimics thestiffness of the lungs)—but these cells proliferateon stiff 2D cell culture dishes. In vivo, dormant,disseminated tumor cells are preferentially lo-cated within the soft niches of stiff tissues suchas bones—e.g., in the bone marrow (42).NETs drive dormant cancer cells to initiate
proliferation, but T cells and natural killer cellsmay recognize these disseminated cells as theystart to proliferate (2, 3, 43). The role of theadaptive immune system in our mouse modelsis unclear, but LPS increases glucocorticoid lev-els (44), inhibiting adaptive immune cells. There-fore, LPS and smoking may spur the growth ofdormant cells by providing signals to both ini-tiate proliferation, via NETs, and to overcomeimmune control, through glucocorticoids.Chronic inflammation and smoking are well-
known risk factors in metastatic recurrence (8, 9).Obesity is also associated with chronic, low-gradeinflammation and with elevated NET release (45),and neutrophils contribute to obesity-associatedmetastasis in mice (46). Our findings set the stagefor epidemiological studies examining whetherthere are any correlations between inflammation,smoking, and obesity; NETs; and cancer recur-rence after a long period of dormancy in patients.If such correlations exist, we envision that NETsand their downstream effectors could be targetedto reduce the risk of cancer recurrence.
Materials and methods summaryProliferation assayNinety-six-well culture-plates were coated with50 ml of growth factor reduced matrigel (#356231,Corning) or mLN-111 (#3446-005-01, R&D Sys-tems) and incubated at 37°C for 30 min. Themurine D2.0R cell line was used on matrigelor mLN-111 as indicated in the figures, and thehuman MCF-7 cell line was used on matrigel.mCherry-luciferase cells (2 × 103) were resus-pended in 100 ml of DMEM supplemented with1% FCS and 2% matrigel or 2% mLN-111 andgrown on the coated wells. The next day, mediawere replaced with 100 ml of CM from neutro-phils cultured at indicated conditions. The CMwas changed every four days. After 14 days,100 ml of medium containing 5 mg/ml of luci-ferin (#Luck-1G, Goldbio) were added to thewells and proliferation was measured by bio-luminescence imaging (BLI) using a plate reader(SpectraMax i3, Molecular Devices). To measurethe proliferation of D2.0R cells expressing in-tegrin, NE, and MMP9 shRNA, CellTiter 96AQueous One Solution Cell Proliferation Assay(#G3580, Promega) was used, following the man-ufacturer’s instructions.
Animals
Female BALB/c, nude, and C57BL/6J mice werepurchased from Charles River Laboratories.Female NE knockout (KO) (#6112) and MMP9KO (#7084) mice were purchased from TheJackson Laboratory. RapidCaP mice consistedof PtenloxP/loxP; Trp53loxP/loxP; TdTomatoloxP/+
transgenic mice into which a Luc-Cre lentiviralplasmid (Tyler Jacks, #2090, Addgene, purchasedfrom The University of Iowa Viral Vector Core)was injected into the prostate, as previously de-scribed (24). All procedures were approved bythe Cold Spring Harbor Laboratory or Univer-sity of California, Davis Institutional Animal Careand Use Committee and were conducted in ac-cordance with the NIH’s Guide for the Care andUse of Laboratory Animals.
Activation of neutrophilsand NET formation assays
Isolated neutrophils (250,000) were culturedin 24-well plates containing 500 ml of serum-free DMEM and activated overnight with LPS,PMA, fMLP, or CSE to induce NETs. Cancer cellswere also cocultured with neutrophils to induceNETs, as previously described (22), using Trans-well with the two chambers separated by a0.4-mm porous membrane (#353495, Corning).Neutrophils were placed on the bottom of thelower chamber, whereas the cancer cells (100,000D2.0R or D2.A1 cells) were placed on the mem-brane of the upper chamber. To induce degran-ulation, recombinant complement 5a was used.The next day, the neutrophil CM were collected.GSK484 and DNase I were added 30 min beforeneutrophil activation to inhibit PAD4 or to di-gest NET DNA, respectively. To assess NET forma-tion, neutrophils grown on poly-L-lysine-coatedcoverslips (#354085, Corning) were fixed with4% paraformaldehyde (PFA) for 20 min at room
temperature, rinsed twice in PBS, incubated in50 mM of NH4Cl for 10 min and permeabilizedwith 0.5% Triton X-100 (#BB151-500, ThermoFisher Scientific) for 5 min. Cells were next blockedin PBS containing 1% bovine serum albumin(BSA, #A3294, Sigma) for 30 min and incubatedwith anti-H2B (1:200) and anti-myeloperoxidase(1:400) antibodies in blocking buffer overnightat 4°C. After two washes in PBS, cells were incu-bated in the presence of fluorochrome-conjugatedsecondary antibodies (1:250, Invitrogen) for 40 min,rinsed twice in PBS, stained with 4′,6-diamidino-2-phenylindole (DAPI, #D1306, Thermo FisherScientific) for 5 min, rinsed in water, and thecoverslips were mounted onto glass slides usingmounting media (#17985-16, Electron MicroscopySciences).
Analysis of recombinantlaminin-111 degradation
mLN-111 (90 mg) or hLN-111 (1 mg) was incubatedwith neutrophil CM or recombinant proteasesat 37°C for 6 hours before adding lysis buffer(25 mM Tris [pH 6.8], 2% SDS, 5% glycerol, 1%b-mercaptoethanol, 0.01% bromophenol blue).For mLN-111, samples were then loaded on SDS-polyacrylamide gel electrophoresis, and the gelwas stained with coomassie blue in a small plas-tic box. For hLN-111, samples were processedas described in the “Western blot” section ofthe supplementary materials and methods. Foranalysis on native gel, SDS and b-mercaptoethanolwere omitted from the lysis and running buffers.
Effects of experimental inflammationon dormant cancer cells in vivo
To generate dormant disease, we intravenouslyinjected mCherry-luciferase-D2.0R or -MCF-7 cells(500,000 per mouse) in 100 ml of PBS into 8-week-old female BALB/c or nude mice, respectively.Nude mice were implanted with 17b-estradiol0.36 mg/pellet (#SE-121, Innovative Research ofAmerica) 48 hours before MCF-7 cell injection.To determine how lung inflammation reactivatesdormant tumor cells in the lung, we inducedinflammation through intranasal instillation ofLPS derived from Escherichia coli strain 0111:B4(#L4391, Sigma). LPS (50 ml at a concentration of0.25 mg/ml, or PBS as control) was administeredintranasally on days 7, 10, and 13 or on days 30,33, and 36 with a P200 pipette under anesthesia(2.5% isoflurane) into mice that had been in-jected with cancer cells at day 0. Neutrophils weredepleted by administering anti-Ly6G antibodiesintraperitoneally (clone 1A8, #BP0075-1, Bio X Cell,200 mg/mouse). Anti-Ly6G antibodies were firstgiven one day before the first LPS administration,and the treatment continued three times weeklyuntil the end of the experiment on day 33. RatIgG2a isotype was used as a control (clone 2A3,#BP0089, Bio X Cell, 200 mg/mouse). Inhibitorswere intraperitoneally injected, starting on thefirst day of LPS treatment. On days when the micereceived LPS administration, they were treatedthree times with inhibitors (30 min before LPSadministration, three hours after administration,and 6 hours after administration). On all other
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 10 of 13
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from

days, one daily treatment was given, continuinguntil the end of the experiment on day 33. Thedoses of the inhibitors were: 50 mg/kg sivelestat(NE inhibitor, #3535, Tocris); 15,000 units/kgDNase I (#04536282001, Roche); 20mg/kg GSK484(PAD4 inhibitor, #17488, Cayman Chemical),50 mg/kg SB3-CT (MMP9 inhibitor, #HY-12354,Medchem Express). Vehicle consisted of 10%dimethyl sulfoxide (DMSO) in PBS for the micetreated with sivelestat, GSK484, and SB3-CT;and PBS for mice treated with DNase I. Im-mobilization of an enzyme on the surface ofnanoparticles can increase enzyme stability,therefore, we also generated and used DNaseI–coated nanoparticles, as previously described(22). The nanoparticles were suspended suchthat each microliter contained one unit of DNaseI; 75 units of DNase I–coated nanoparticles wereadministered intraperitoneally per mouse. Un-coated nanoparticles were used as control. Tofollow metastatic disease, mice were injectedintraperitoneally with luciferin (5 mg per mouse,#Luck-1G, Goldbio) and imaged using the IVISSpectrum in vivo imaging system (#128201,PerkinElmer) every 3 days.For experiments analyzing the effect of inhib-
itors on lung inflammation, cancer cells werenot injected, and mice were treated with LPSat days 0, 3, and 6. On days when the mice re-ceived LPS administration, they were treatedthree times with inhibitors (30 min before LPSadministration, three hours after administration,and six hours after administration). On all otherdays, one daily treatment was given. Mice weresacrificed at day 7 for tissue processing or at thetimes indicated in the figure legends.
Effects of tobacco smoke exposureon dormant cancer cells in vivo
To generate dormant disease, mCherry-luciferase-D2.0R (500,000 per mouse) in 100 ml of PBS wasintravenously injected into 8-week-old femaleBALB/c mice. To determine how tobacco smoke(TS) exposure reactivates dormant tumor cellsin the lung, the mice were exposed to tobaccosmoke or filtered air for three weeks startingone week after injection using a smoke exposuresystem (Pinkerton laboratory, University of Cal-ifornia, Davis). For the TS exposed group, micewere exposed five days/week for three weeks toan average concentration of 75 ± 11 mg/m3 oftobacco smoke particulate for 6 hours/day using3R4F research cigarettes (Tobacco Research In-stitute, University of Kentucky) that were burnedat a rate of four cigarettes every 10 min with apuff volume of 35 ml over a duration of 2 s, onceper minute. Both side-stream and mainstreamcigarette smoke were collected via a chimneyand passed to a dilution and aging chamber toallow the animals to acclimate to TS exposure.The TS exposure started at an initial exposureconcentration of 60 mg/m3 and was increased,ultimately achieving a final target concentrationof 90 mg/m3. After each 6-hour exposure to TS,the mice were then kept in the same chamberbut in filtered air. For the control group, micewere handled in the same way, but were instead
exposed to filtered air for 24 hours/day, 7 days/week for the duration of the study. The con-centration of carbon monoxide in the exposurechambers was monitored and averaged 215 ± 36ppm over the 3 weeks of exposure. Mice weretreated with vehicle (10% DMSO in PBS) or PAD4inhibitor (GSK484, #17488, Cayman Chemical)intraperitoneally at a dose of 20 mg/kg/day toinhibit NET formation, starting on the first dayof TS exposure and every day until the end of theexperiment. ChiAb28 was administered intra-venously at a dose of 200 mg/mouse to target NET-remodeled laminin in the lungs, starting on thefirst day of environmental TS exposure. ChiAb28was given every 3 days until the end of the ex-periment. Mouse IgG2a isotype was used as acontrol (Clone C1.18.4, #BE0085, Bio X Cell,200 mg/mouse).
Confocal intravital lung imaging
We used a modified version of our previouslydescribed method for lung imaging (22). Micewere anesthetized by intraperitoneal injectionof ketamine/dexdomitor (100 mg/kg; 0.5 mg/kg).Saline (500 ml) was injected peritoneally priorto surgery, then tracheotomies were performed,and mechanical ventilation was initiated (MiniVentventilator for mice, Model 845, Harvard Appa-ratus) with the mice on a heated stage. Fromthis point forward, the mice were kept anesthe-tized at 0.8 to 1.0% isoflurane delivered throughthe ventilator at 80 to 100 breaths per minute,with 25 ml of water maintaining positive end ex-piratory pressure. A thoracotomy was then per-formed by making an incision between the fourthand fifth rib, and a 3D printed thoracic suctionwindow with an 8-mm coverslip was inserted (47).The window was secured to the stage with single-axis translator micromanipulators (#MSH1.5,Thor Labs), and the left lung was held in place byvacuum pressure, which was adjusted to 15 to20 mmHg. Mice were imaged with our custom-built spinning disk confocal microscope mod-ified to upright configuration, as describedpreviously (22). The microscope was controlledby MicroManager software, and data analysisand time-lapse movies were generated usingImaris software.To visualize neutrophil recruitment and neu-
trophil elastase activity, LysM-EGFP mice [onBALB/c background (48)] were administeredintranasally with LPS (50 ml, at a concentrationof 0.25 mg/ml) or PBS as control as indicated andintravenously with neutrophil elastase 680 FASTprobe (4 nmol in 100 ml, #NEV11169, PerkinElmer,3 to 6 hours before imaging). At the time of im-aging, DAPI was administered (5 mg/ml) intra-venously, and images were captured for 2 to8 hours. To monitor cancer cells exiting dor-mancy and undergoing cell cycle progression,D2.0R cells labeled with FUCCI reporter (500,000)were injected intravenously into BALB/c miceat day 0, and the mice were treated with LPS ondays 7, 10, and 13, as described above. On daysof imaging, the neutrophil elastase 680 FASTprobe was administered intravenously 3 to 6hours before imaging.
Enzyme-linked immunosorbent assay(ELISA) for NETs96-well Enzyme ImmunoAssay/Radio Immuno-Assay (EIA/RIA) plates (#3590, Costar) were coatedovernight at 4°C with an anti-elastase antibody(1:250, #sc-9521, Santa Cruz Biotechnology) in15 mM of Na2CO3, 35 mM of NaHCO3, at pH 9.6.The next day, the wells were washed three timeswith PBS, blocked in 5% BSA for two hours atroom temperature, and washed three times withPBS. Then, 50 ml of plasma samples were addedto the wells, incubated for two hours at roomtemperature on a shaker, and plates were washedthree times with wash buffer (1% BSA, 0.05%Tween 20 in PBS). Next, anti-DNA-peroxidaseconjugated antibody (1:50, #11774425001, Roche)in 1% BSA in PBS was added to the wells for2 hours at room temperature, and the wells werewashed five times with wash buffer before theaddition of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS, #37615, Thermo FisherScientific). Optical density was read 40 min laterat 405 nm using a plate reader (SpectraMax i3,Molecular Devices).
Generation of anti–NET-remodeledlaminin-111 antibodies
mLN-111 (6 mg) was incubated with recombi-nant NE and MMP9 (2 mg/ml each) at 37°C for6 hours. To remove the recombinant proteases,the cleaved laminin-111 solution was purified usinga 100-kDa molecular weight cut-off (MWCO)column (#VS0641, Vivaspin 6, Sartorius), follow-ing the manufacturer’s instructions. The pres-ence of laminin-111 and the absence of NE andMMP9 in the upper fraction (>100 kDa) was thenassessed using Western blot analysis. The upperfraction containing the cleaved laminin-111 wasnext dialyzed in PBS using 20,000 MWCO dialy-sis cassettes (#87735, Thermo Fisher Scientific).Three 6-week-old Sprague Dawley rats (Taconics)were immunized with 1.5 mg of cleaved laminin-111 (100 mg per animal per boost, for five weeklyboosts). Immune response was monitored byELISA to measure the serum anti–NET-remodeledlaminin-111 IgG titer from blood samples. Aftera 60-day immunization course, the rat with thestrongest anti-cleaved laminin-111 immune re-sponse was terminated, and 108 splenocytes werecollected for making hybridomas by fusing withthe rat myeloma cell line YB2/0, following thestandard method (49). All procedures were ap-proved by the Cold Spring Harbor LaboratoryInstitutional Animal Care and Use Committee.The antibodies generated were tested and endo-toxin free.
Generation of chimeric antibodies
To make antibodies for in vivo experiments, weengineered the rat mAbs into mouse chimeraantibodies (chiAbs), as this reduces the likeli-hood of the mice developing neutralization anti-bodies. In brief, the rat mAb V-domains fromthe heavy and light chains were cloned into themice IgG2a framework. The resulting chimericIgG2a antibody therefore comprised rat V-domainsin frame with the constant regions of mice IgG2a.
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 11 of 13
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from

The recombinant chimeric antibodies were thenproduced from HEK293 cells transfected withthe chimeric IgG2a expression constructs, fol-lowed by standard purification procedures fromthe culture supernatants.
Statistical analysis
Data from in vivo BLI quantification were ana-lyzed using one-way ANOVA followed by Tukey’sprocedure, except for fig. S7F, where a two-sidedt test was used. Data from in vitro BLI wereanalyzed using two-way ANOVA followed byTukey’s procedure, except for Figs. 4, C and E,and 6B, and figs. S7J and S15D. The data rep-resented in these figures were analyzed usingone-way ANOVA followed by Dunnett’s pro-cedure, with the reference group indicated onthe figure.Data from NE and MMP9 activity in vitro
were analyzed using two-way ANOVA followedby Tukey’s procedure, while data from NE andMMP9 activity in vivo were analyzed using one-way ANOVA followed by Dunnett’s procedure,where the reference group was indicated on thefigure. Assays on dsDNA, plasma NET and LPS,number of DTCs/mm2, metastatic burden, neutro-phils’ number per field, percentage of neutrophilsforming NETs, and percentage of Ki67-positivecancer cells in vitro—except for fig. S7G—wereanalyzed using one-way ANOVA followed byDunnett’s procedure, with the reference groupindicated on the figure. Fig. S7G was analyzedusing a two-sided t test.Data from qPCR quantification were analyzed
using one-way ANOVA followed by Dunnett’sprocedure, where the reference group was in-dicated on the figure, except for fig. S10F, wherea two-sided t test was used. To analyze lungmetastasis-free survival (fig. S5C), Kaplan-Meiercurves of RapidCaP mice were analyzed using alog-rank (Mantel-Cox) test. For fig. S5C, an exactP value was computed using Fisher’s exact testto evaluate the likelihood that the metastatictypes (macrometastasis, micrometastasis, singleDTC) differed among groups. The analyses onfigs. S2E and S15B had the cancer cell as theexperimental unit, with each cell characterizedon the two variables of interest for the figure. Forfig. S2E, cells were characterized based on experi-mental group (vehicle, day 11, day 12, day 13) andby cell feature (G0/G1 single cells, S/G2/M singlecells, clusters of cells). A total of 182 cells werecharacterized and Fisher’s exact test was used toassess whether the cell feature distribution dif-fered among the different experimental groups.The exact test was significant (P < 0.0001): Overtime, G0/G1 single cells and S/G2/M single cellsdecreased and clusters of cells increased. For fig.S15B, cells were characterized based on cell type(PBS and G0/G1 single cells, LPS and G0/G1 sin-gle cells, LPS and S/G2/M single cells, LPS andclusters of cells) and next to Ab28 (yes, no). Atotal of 182 cells were characterized. Fisher’s ex-act test was used to assess whether the percentageof cells that were “next to Ab28” in each cell typegroup were different among the groups. The ex-act test was significant (P < 0.0001). All PBS cells
were G0/G1 single cells and were not “next toAb28.” LPS and G0/G1 single cells were equallylikely to be “next to Ab28” or “not next to Ab28.”All LPS and S/G2/M single cells and all LPS andclusters of cells were “next to Ab28.” Analysis forfigs. S5C, S2E, and S15B were generated usingProc Freq in the SAS/STAT software, version 9.4of the SAS system for Windows.A P value less than 0.05 was considered sig-
nificant, and P values are indicated in figures as***P < 0.001, **P < 0.01, and *P < 0.05. All statis-tical analyses were performed using GraphPadPrism software version 7 unless otherwise stated.
REFERENCES AND NOTES
1. M. S. Sosa, P. Bragado, J. A. Aguirre-Ghiso, Mechanisms ofdisseminated cancer cell dormancy: An awakening field.Nat. Rev. Cancer 14, 611–622 (2014). doi: 10.1038/nrc3793;pmid: 25118602
2. S. Malladi et al., Metastatic latency and immune evasionthrough autocrine inhibition of WNT. Cell 165, 45–60 (2016).doi: 10.1016/j.cell.2016.02.025; pmid: 27015306
3. I. Romero, F. Garrido, A. M. Garcia-Lora, Metastases inimmune-mediated dormancy: A new opportunity for targetingcancer. Cancer Res. 74, 6750–6757 (2014). doi: 10.1158/0008-5472.CAN-14-2406; pmid: 25411345
4. J. D. Farrar et al., Cancer dormancy. VII. A regulatory role forCD8+ T cells and IFN-gamma in establishing and maintainingthe tumor-dormant state. J. Immunol. 162, 2842–2849(1999). pmid: 10072532
5. M. Müller et al., EblacZ tumor dormancy in bone marrow andlymph nodes: Active control of proliferating tumor cells byCD8+ immune T cells. Cancer Res. 58, 5439–5446 (1998).pmid: 9850077
6. C. M. Ghajar et al., The perivascular niche regulates breasttumour dormancy. Nat. Cell Biol. 15, 807–817 (2013).doi: 10.1038/ncb2767; pmid: 23728425
7. D. Barkan et al., Metastatic growth from dormant cells inducedby a col-I-enriched fibrotic environment. Cancer Res. 70,5706–5716 (2010). doi: 10.1158/0008-5472.CAN-09-2356;pmid: 20570886
8. B. L. Pierce et al., Elevated biomarkers of inflammation areassociated with reduced survival among breast cancerpatients. J. Clin. Oncol. 27, 3437–3444 (2009). doi: 10.1200/JCO.2008.18.9068; pmid: 19470939
9. J. P. Pierce et al., Lifetime cigarette smoking and breast cancerprognosis in the After Breast Cancer Pooling Project. J. Natl.Cancer Inst. 106, djt359 (2014). doi: 10.1093/jnci/djt359;pmid: 24317179
10. A. H. Wu et al., The California Breast Cancer SurvivorshipConsortium (CBCSC): Prognostic factors associated withracial/ethnic differences in breast cancer survival.Cancer Causes Control 24, 1821–1836 (2013). doi: 10.1007/s10552-013-0260-7; pmid: 23864487
11. S. Murin, K. E. Pinkerton, N. E. Hubbard, K. Erickson,The effect of cigarette smoke exposure on pulmonarymetastatic disease in a murine model of metastatic breastcancer. Chest 125, 1467–1471 (2004). doi: 10.1378/chest.125.4.1467; pmid: 15078760
12. J. M. De Cock et al., Inflammation Triggers Zeb1-DependentEscape from Tumor Latency. Cancer Res. 76, 6778–6784(2016). doi: 10.1158/0008-5472.CAN-16-0608; pmid: 27530323
13. S. K. Jorch, P. Kubes, An emerging role for neutrophilextracellular traps in noninfectious disease. Nat. Med. 23,279–287 (2017). doi: 10.1038/nm.4294; pmid: 28267716
14. N. Branzk, V. Papayannopoulos, Molecular mechanismsregulating NETosis in infection and disease. Semin.Immunopathol. 35, 513–530 (2013). doi: 10.1007/s00281-013-0384-6; pmid: 23732507
15. C. Carmona-Rivera, W. Zhao, S. Yalavarthi, M. J. Kaplan,Neutrophil extracellular traps induce endothelial dysfunctionin systemic lupus erythematosus through the activation ofmatrix metalloproteinase-2. Ann. Rheum. Dis. 74, 1417–1424(2015). doi: 10.1136/annrheumdis-2013-204837; pmid: 24570026
16. T. A. Fuchs et al., Extracellular DNA traps promote thrombosis.Proc. Natl. Acad. Sci. U.S.A. 107, 15880–15885 (2010).doi: 10.1073/pnas.1005743107; pmid: 20798043
17. A. Brill et al., Neutrophil extracellular traps promote deep veinthrombosis in mice. J. Thromb. Haemost. 10, 136–144 (2012).doi: 10.1111/j.1538-7836.2011.04544.x; pmid: 22044575
18. S. L. Wong et al., Diabetes primes neutrophils to undergoNETosis, which impairs wound healing. Nat. Med. 21, 815–819(2015). doi: 10.1038/nm.3887; pmid: 26076037
19. J. Cools-Lartigue et al., Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis.J. Clin. Invest. 123, 3446–3458 (2013). doi: 10.1172/JCI67484;pmid: 23863628
20. S. Tohme et al., Neutrophil extracellular traps promote thedevelopment and progression of liver metastases aftersurgical stress. Cancer Res. 76, 1367–1380 (2016).doi: 10.1158/0008-5472.CAN-15-1591; pmid: 26759232
21. D. Barkan et al., Inhibition of metastatic outgrowth fromsingle dormant tumor cells by targeting the cytoskeleton.Cancer Res. 68, 6241–6250 (2008). doi: 10.1158/0008-5472.CAN-07-6849; pmid: 18676848
22. J. Park et al., Cancer cells induce metastasis-supportingneutrophil extracellular DNA traps. Sci. Transl. Med. 8,361ra138 (2016). doi: 10.1126/scitranslmed.aag1711;pmid: 27798263
23. S. B. Koh et al., A quantitative FastFUCCI assay defines cellcycle dynamics at a single-cell level. J. Cell Sci. 130, 512–520(2017). doi: 10.1242/jcs.195164; pmid: 27888217
24. H. Cho et al., RapidCaP, a novel GEM model for metastaticprostate cancer analysis and therapy, reveals myc asa driver of Pten-mutant metastasis. Cancer Discov.4, 318–333 (2014). doi: 10.1158/2159-8290.CD-13-0346;pmid: 24444712
25. D. Barkan, J. E. Green, An in vitro system to study tumordormancy and the switch to metastatic growth. J. Vis. Exp.2011, e2914 (2011). pmid: 21860375
26. A. Hosseinzadeh, P. R. Thompson, B. H. Segal, C. F. Urban,Nicotine induces neutrophil extracellular traps. J. Leukoc. Biol.100, 1105–1112 (2016). doi: 10.1189/jlb.3AB0815-379RR;pmid: 27312847
27. R. William, G. Watson, H. P. Redmond, J. M. Carthy,D. Bouchier-Hayes, Taurolidine, an antilipopolysaccharideagent, has immunoregulatory properties that are mediated bythe amino acid taurine. J. Leukoc. Biol. 58, 299–306 (1995).doi: 10.1002/jlb.58.3.299; pmid: 7665985
28. M. Egeblad, Z. Werb, New functions for the matrixmetalloproteinases in cancer progression. Nat. Rev. Cancer 2,161–174 (2002). doi: 10.1038/nrc745; pmid: 11990853
29. K. Kessenbrock, V. Plaks, Z. Werb, Matrix metalloproteinases:Regulators of the tumor microenvironment. Cell 141, 52–67(2010). doi: 10.1016/j.cell.2010.03.015; pmid: 20371345
30. V. Papayannopoulos, K. D. Metzler, A. Hakkim, A. Zychlinsky,Neutrophil elastase and myeloperoxidase regulate theformation of neutrophil extracellular traps. J. Cell Biol. 191,677–691 (2010). doi: 10.1083/jcb.201006052; pmid: 20974816
31. K. D. Metzler, C. Goosmann, A. Lubojemska, A. Zychlinsky,V. Papayannopoulos, A myeloperoxidase-containing complexregulates neutrophil elastase release and actin dynamicsduring NETosis. Cell Reports 8, 883–896 (2014). doi: 10.1016/j.celrep.2014.06.044; pmid: 25066128
32. A. Beliveau et al., Raf-induced MMP9 disrupts tissuearchitecture of human breast cells in three-dimensional cultureand is necessary for tumor growth in vivo. Genes Dev. 24,2800–2811 (2010). doi: 10.1101/gad.1990410; pmid: 21159820
33. R. Catena et al., Bone marrow-derived Gr1+ cells can generatea metastasis-resistant microenvironment via induced secretionof thrombospondin-1. Cancer Discov. 3, 578–589 (2013).doi: 10.1158/2159-8290.CD-12-0476; pmid: 23633432
34. T. El Rayes et al., Lung inflammation promotes metastasisthrough neutrophil protease-mediated degradation of Tsp-1.Proc. Natl. Acad. Sci. U.S.A. 112, 16000–16005 (2015).doi: 10.1073/pnas.1507294112; pmid: 26668367
35. J. A. Aguirre Ghiso, Inhibition of FAK signaling activated byurokinase receptor induces dormancy in human carcinomacells in vivo. Oncogene 21, 2513–2524 (2002). doi: 10.1038/sj.onc.1205342; pmid: 11971186
36. J. A. Aguirre Ghiso, K. Kovalski, L. Ossowski, Tumor dormancyinduced by downregulation of urokinase receptor in humancarcinoma involves integrin and MAPK signaling. J. Cell Biol. 147,89–104 (1999). doi: 10.1083/jcb.147.1.89; pmid: 10508858
37. D. Liu, J. Aguirre Ghiso, Y. Estrada, L. Ossowski, EGFR is atransducer of the urokinase receptor initiated signal that isrequired for in vivo growth of a human carcinoma. Cancer Cell1, 445–457 (2002). doi: 10.1016/S1535-6108(02)00072-7;pmid: 12124174
38. F. Calvo et al., Mechanotransduction and YAP-dependentmatrix remodelling is required for the generation andmaintenance of cancer-associated fibroblasts. Nat. Cell Biol.15, 637–646 (2013). doi: 10.1038/ncb2756; pmid: 23708000
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 12 of 13
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from

39. R. O. Hynes, Integrins: Bidirectional, allosteric signalingmachines. Cell 110, 673–687 (2002). doi: 10.1016/S0092-8674(02)00971-6; pmid: 12297042
40. T. Shibue, M. W. Brooks, R. A. Weinberg, An integrin-linkedmachinery of cytoskeletal regulation that enablesexperimental tumor initiation and metastatic colonization.Cancer Cell 24, 481–498 (2013). doi: 10.1016/j.ccr.2013.08.012;pmid: 24035453
41. A. Caudrillier et al., Platelets induce neutrophil extracellulartraps in transfusion-related acute lung injury. J. Clin. Invest.122, 2661–2671 (2012). doi: 10.1172/JCI61303; pmid: 22684106
42. K. Pantel, C. Alix-Panabières, Bone marrow as a reservoir fordisseminated tumor cells: A special source for liquid biopsy incancer patients. Bonekey Rep. 3, 584 (2014). doi: 10.1038/bonekey.2014.79; pmid: 25419458
43. A. Pommier et al., Unresolved endoplasmic reticulum stressengenders immune-resistant, latent pancreatic cancermetastases. Science 360, eaao4908 (2018). doi: 10.1126/science.aao4908; pmid: 29773669
44. K. Nakano, S. Suzuki, C. Oh, Significance of increased secretionof glucocorticoids in mice and rats injected with bacterialendotoxin. Brain Behav. Immun. 1, 159–172 (1987).doi: 10.1016/0889-1591(87)90018-3; pmid: 3330674
45. V. Delgado-Rizo et al., Neutrophil extracellular traps and itsimplications in inflammation: An overview. Front. Immunol. 8, 81(2017). doi: 10.3389/fimmu.2017.00081; pmid: 28220120
46. D. F. Quail et al., Obesity alters the lung myeloid cell landscapeto enhance breast cancer metastasis through IL5 and GM-CSF.Nat. Cell Biol. 19, 974–987 (2017). doi: 10.1038/ncb3578;pmid: 28737771
47. M. B. Headley et al., Visualization of immediate immuneresponses to pioneer metastatic cells in the lung.
Nature 531, 513–517 (2016). doi: 10.1038/nature16985;pmid: 26982733
48. N. Faust, F. Varas, L. M. Kelly, S. Heck, T. Graf, Insertionof enhanced green fluorescent protein into the lysozyme genecreates mice with green fluorescent granulocytes andmacrophages. Blood 96, 719–726 (2000). pmid: 10887140
49. E. A. Greenfield, Antibodies: A Laboratory Manual (Cold SpringHarbor Laboratory Press, ed. 2, 2014).
ACKNOWLEDGMENTS
We thank J. Green (NIH) for the D2.0R and D2.A1 cells and Z. Werb(University California, San Francisco) for the MMP9 antibody.Funding: This work was supported by the CSHL Cancer CenterSupport P30-CA045508 (to J.T.-H.Y, C.B., K.C., S.K.L., and P.A.G.);the Department of Defense (W81XWH-14-1-0078) and the PershingSquare Sohn Cancer Research Alliance Prize (to M.E.); theAssociation pour la Recherche sur le Cancer (AE20141202131), theTerri Brodeur Breast Cancer Foundation, the European MolecularBiology Organization (ALTF 1425-2015), and the Susan G. KomenFoundation (PDF16376754) (to J.A.); the Lustgarten Foundation,the National Cancer Institute, and the Cedar Hill Foundation (to D.T.F.);R01 NIH Research Grant (5R01CA137050) (to L.C.T. and A.A.);CSHL and Northwell Health (to M.A.S., E.M.C., C.G.P, M.S.G., andM.E.); a Starr Centennial Scholarship and a George A. & MarjorieH. Anderson Scholarship from the Watson School of BiologicalSciences (to E.B. and L.M.); a Boehringer Ingelheim Fonds Ph.D.fellowship (to L.M.); a Deutsche ForschungsGemeinschaft researchfellowship (KU 3264/1-1) (to V.K.); UC Davis NIEHS EnvironmentalHealth Science Center (P30 ES023513) (to K.E.P., M.E.P., P.U., andD.L.U.); and an AACR-Bayer Healthcare Basic Cancer ResearchFellowship (to M.A.S.). Author contributions: J.A. and M.E. designedand directed the project. J.A. and D.N. performed all experiments,
except as follows: M.E.P., P.U., D.L.U., and K.E.P. performed tobaccosmoke exposure experiments on mice; A.A. and L.C.T. designedand performed experiments on RapidCaP mice; C.B. and J.T-H.Y.generated antibodies against cleaved murine laminin-111; C.G.P., E.M.C.,and M.S.G. generated DNase I and free coated nanoparticles; A.P.and D.T.F. designed and performed flow cytometry experiments;K.C. generated the viral particles for shRNA-mediated inducibleknockdown of integrin b1, FAK, MLCK, and YAP; and M.A.S. performedconfocal intravital lung imaging. E.B., L.M., and V.K. generatedreagents and/or helped analyze data. S.K.L. provided the FUCCI cellcycle reporter plasmid. P.A.G., J.A., and M.E. performed statisticalanalysis. J.A. and M.E. wrote the manuscript. Competing interests:D.T.F. is a cofounder of Myosotis LLC (a company developing cancerimmunotherapies) and is on the scientific advisory boards ofiTEOS Therapeutics (a company developing immuno-oncology drugs),IFM Therapeutics, LLC (a company developing therapies targetingthe innate immune system), and Kymab (a company developingtherapeutic antibodies). The authors declare no other competinginterests. Data and materials availability: All data are available inthe main text or the supplementary materials. The Ab28 monoclonalantibody against cleaved laminin is available upon signing amaterial transfer agreement.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/361/6409/eaao4227/suppl/DC1Materials and MethodsFigs. S1 to S15Reference (50)Movies S1 to S8
21 July 2017; accepted 3 August 201810.1126/science.aao4227
Albrengues et al., Science 361, eaao4227 (2018) 28 September 2018 13 of 13
RESEARCH | RESEARCH ARTICLEon January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from

miceNeutrophil extracellular traps produced during inflammation awaken dormant cancer cells in
Goldberg, Johannes T.-H. Yeh and Mikala EgebladPhyllis A. Gimotty, Douglas T. Fearon, Kenneth Chang, Scott K. Lyons, Kent E. Pinkerton, Lloyd C. Trotman, Michael S.Dale L. Uyeminami, Arnaud Pommier, Victoria Küttner, Emilis Bruzas, Laura Maiorino, Carmelita Bautista, Ellese M. Carmona, Jean Albrengues, Mario A. Shields, David Ng, Chun Gwon Park, Alexandra Ambrico, Morgan E. Poindexter, Priya Upadhyay,
DOI: 10.1126/science.aao4227 (6409), eaao4227.361Science
, this issue p. eaao4227; see also p. 1314Scienceagainst the remodeled version of a matrix protein called laminin-111.cells was associated with NET-mediated remodeling of the extracellular matrix and could be prevented by an antibody convert dormant cancer cells to aggressive lung metastases (see the Perspective by Aguirre-Ghiso). Awakening of thesefound that sustained lung inflammation and the accompanying formation of neutrophil extracellular traps (NETs) could
et al.clinically undetectable cells ''wake up'' and form actively growing metastases. Studying mouse models, Albrengues dormancy, where they remain viable but not proliferating. Eventually, by mechanisms that are poorly understood, thesedecades later. This is because cancer cells that have disseminated beyond the primary tumor site enter a state of
Cancer patients who have undergone successful treatment can experience relapse of their disease years or evenWaking up in a trap
ARTICLE TOOLS http://science.sciencemag.org/content/361/6409/eaao4227
MATERIALSSUPPLEMENTARY http://science.sciencemag.org/content/suppl/2018/09/26/361.6409.eaao4227.DC1
CONTENTRELATED
http://stm.sciencemag.org/content/scitransmed/10/436/eaan3464.fullhttp://stm.sciencemag.org/content/scitransmed/10/436/eaan8292.fullhttp://science.sciencemag.org/content/sci/361/6409/1314.full
REFERENCES
http://science.sciencemag.org/content/361/6409/eaao4227#BIBLThis article cites 49 articles, 20 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.ScienceScience, 1200 New York Avenue NW, Washington, DC 20005. The title (print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement ofScience
Science. No claim to original U.S. Government WorksCopyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of
on January 30, 2020
http://science.sciencemag.org/
Dow
nloaded from